

Abstract
The tail of replication-dependent histone H3.1 varies from that of replication-independent H3.3 at the amino acid located at position 31 in plants and animals, but no function has been assigned to this residue to demonstrate a unique and conserved role for H3.1 during replication. Here, we show that TONSOKU (TSK/TONSL), which rescues broken replication forks, specifically interacts with H3.1 via recognition of alanine 31 by its tetratricopeptide repeat domain. Our results indicate that genomic instability in the absence of ATXR5/ATXR6-catalyzed H3K27me1 in plants depends on H3.1, TSK and DNA polymerase theta (Pol θ). Overall, this work reveals an H3.1-specific function during replication and the common strategy used in multicellular eukaryotes for regulating post-replicative chromatin maturation and TSK, which relies on histone mono-methyltransferases and reading the H3.1 variant.
One Sentence Summary:
The TPR domain of TSK reads the histone H3.1 variant to maintain genome stability.
Chromatin replication requires multiple regulatory mechanisms to ensure the maintenance of genome integrity. One of these mechanisms relies on TONSOKU-LIKE (TONSL), a key player in initiating homologous recombination (HR) when replication forks encounter double-stranded DNA breaks (DSB) (1–8). In animals, TONSL is recruited to chromatin via its ankyrin repeat domain (ARD), which specifically interacts with unmethylated histone H4 lysine 20 (H4K20me0) (1, 9). Post-replicative maturation of chromatin is accomplished via SET8/PR-Set7/SETD8 (10–12), which mono-methylates H4K20 (H4K20me1) and thus prevents TONSL from binding chromatin and initiating HR-based DNA repair outside of DNA replication and the G2 phase of the cell cycle (9). Comparative analysis shows that plants contain a TONSL ortholog (TSK/BRUSHY1/MGOUN3) (7, 8, 13), but are lacking SET8. In line with this, the ARD domain of TONSL in animals is not conserved in TSK orthologs (Fig. 1A) (1), thus indicating that post-replicative chromatin maturation in plants is unlikely to depend on the methylated state of H4K20.
Figure 1. The TPR domain of TSK specifically interacts with the N-terminal tail of the H3.1 variant.
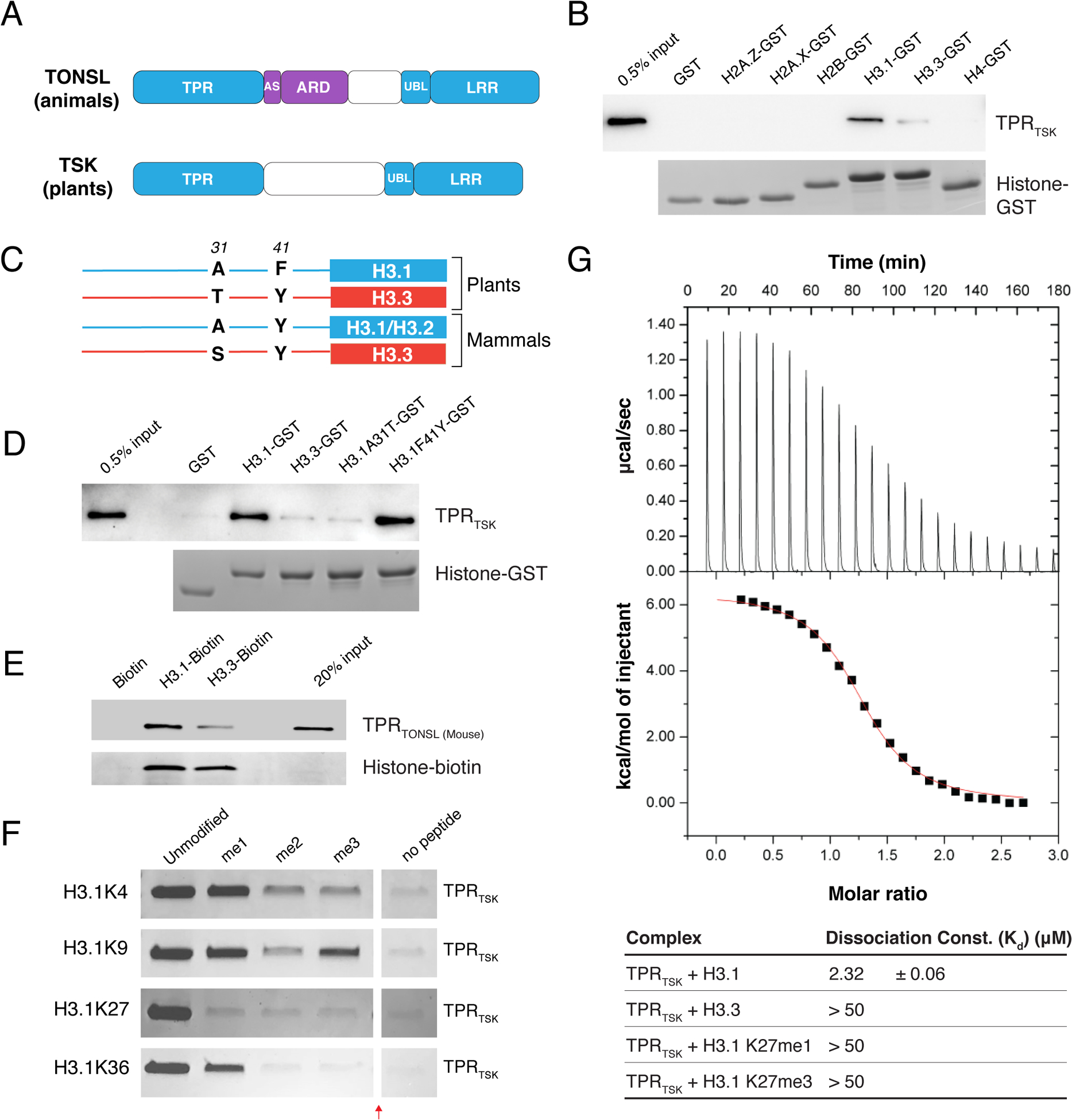
(A) Domain architecture of animal and plant TONSL/TSK. TPR: Tetratricopeptide Repeats, AS: Acidic Sequence, ARD: Ankyrin Repeat Domain, UBL: Ubiquitin-like, LRR: Leucine-Rich Repeats. Conserved domains are shown in blue. (B) Pull-down assay using TPRTSK and GST tagged with the N-terminal tails of histones H2A.Z, H2A.X, H2B, H3.1, H3.3 and H4 from plants. (C) Representation of plant and mammalian H3.1/H3.2 (blue) and H3.3 (red) H3 variants. Thin lines and blocks represent the histone tails and cores, respectively, and numbers indicate amino acid positions in H3. (D) Peptide pull-down assay using plant TPRTSK and GST tagged with the tails of histones H3.1, H3.3, H3.1A31T and H3.1F41Y. (E) Peptide pull-down assay using mouse TPRTONSL and biotin-tagged histones H3.1 and H3.3 (full-length proteins) from mammals. (F) Peptide pull-down assay using plant TPRTSK and methylated peptides at K4, K9, K27 and K36 of H3.1 (a.a. 1–45). The red arrow indicates a gel lane that was removed. (G) ITC assay using plant TPRTSK and different H3 peptides.
We reasoned that TSK might directly interact with histones in plants through a different domain. Sequence alignment of TSK orthologs shows extensive similarity in the N-terminal tetratricopeptide repeat (TPR) domain (fig. S1), which is conserved in animals (Fig. 1A) (1, 4). Many TPR domains have been shown to bind long peptides (>20 a.a.) adopting an extended conformation (14). We therefore hypothesized that one of the N-terminal unstructured tails of histones could specifically interact with the TPR domain of TSK (TPRTSK). To assess this, we performed in vitro binding assays with Arabidopsis thaliana TPRTSK and the tails of different histones. We detected binding of TPRTSK with H3 variants, with stronger binding for H3.1 compared to H3.3 (Fig. 1B). A preference for TPRTSK to bind H3.1 over H3.3 was also observed using nucleosomes and in A. thaliana protoplasts (fig. S2A–C). In vascular plants, amino acids 31 and 41 vary between the N-terminal tails of H3.1 and H3.3 (Fig. 1C) (15). We created hybrid H3.1/H3.3tail-GST fusion proteins based on these differences and determined that only alanine at position 31 of H3.1 (H3.1A31) is required for the H3.1-binding specificity of TPRTSK (Fig. 1D). Variation at position 31 of H3 is also observed between replication-dependent H3.1/H3.2 variants and the replication-independent H3.3 in mammals (Fig. 1C), and similarly to plant TSK orthologs, the TPR domain of mouse TONSL also interacts preferentially with H3.1 compared to H3.3 (Fig. 1E). We then assessed the impact of TPRTSK binding to H3.1 in the context of methylation at different lysine residues in the N-terminal tail of H3.1. We found that increasing levels of methylation at K4, K9, K27 and K36 negatively impact the interaction of TPRTSK to H3.1, with binding being most sensitive to methylation at K27 (Fig. 1F–G and fig. S2D). The binding profile of TSK on histone H3 suggests a preference for binding newly synthesized H3.1 variants.
To gain mechanistic insights into how TSK discriminates H3.1 from H3.3, we solved the crystal structure of the TPRTSK-H3.1(1–45) complex at 3.17 Å resolution by using the TSK ortholog from Citrus unshiu (CuTSK) (fig. S1 and Table S1). TPRTSK folds as eleven TPR motifs placed in tandem, which collectively form a hollow solenoid tube (Fig. 2A–B and fig. S3A). The C-terminal lobe of the tube is composed of TPR 6–11 and generates a wide channel in which two segments of H3.1 (K4-K9 and K18-A24) are found along opposite sides of its wall (Fig. 2A and C). In the center lobe, TPR 3–7 form a narrow tunnel that encircles A25 to P30 of H3.1 (Fig. 2A and C). H3.1K27 is located inside a polar pocket where its ε-amine is surrounded by the side chains of Asp234, Cys238, Ser208 and the backbone carbonyl groups of Asp234 and Gly246 (Fig. 2D). The polarity of this pocket makes it non-conducive for the binding of hydrophobic moieties such as methyl groups, thus explaining the large decrease in binding affinity of TPRTSK to H3.1 when K27 is mono- or tri-methylated (Fig. 1G and fig. S2D). TPR 1–3 make up the N-terminal lobe of TPRTSK, which forms an open channel that accommodates P30 to R40 of H3.1 (Fig. 2A and C). A deep pocket formed between α-helices 2–4 (TPR 1 and 2) is occupied by the side chain of H3.1K36, where its ε-amine is in close proximity to the carboxyl group of Asp54 (Fig. 2A, C, and E). The side chain of H3.1A31 is oriented towards the aliphatic portion of three residues (Arg109, Gln113 and Gln72) strictly conserved among plant TSK orthologs (Fig. 2F and fig. S3A). These residues form a shallow pocket in which Gln113 and Gln72 also likely interact with the H3.1 backbone via hydrogen bonds with the carbonyl group of G34 and the amide group of A31, respectively (Fig. 2F–G). Consistent with our binding assays (Fig. 1B, D, G and fig. S2D), modeling an A31T substitution in H3.1 generates van der Waals clashes between the Cγ methyl group of T31 and the aliphatic chain of Gln113, and similarly between the hydroxyl group of T31 and Arg109 (fig. S3B). We mutated various amino acids of TPRTSK from different H3.1 binding pockets and validated that they contribute to the TPRTSK-H3.1 interaction (fig. S3C). Overall, the structure of the TPRTSK-H3.1 complex supports our finding that TSK preferentially binds the replication-dependent H3.1 variant.
Figure 2. Crystal structure of plant TPRTSK bound to the H3.1 tail.
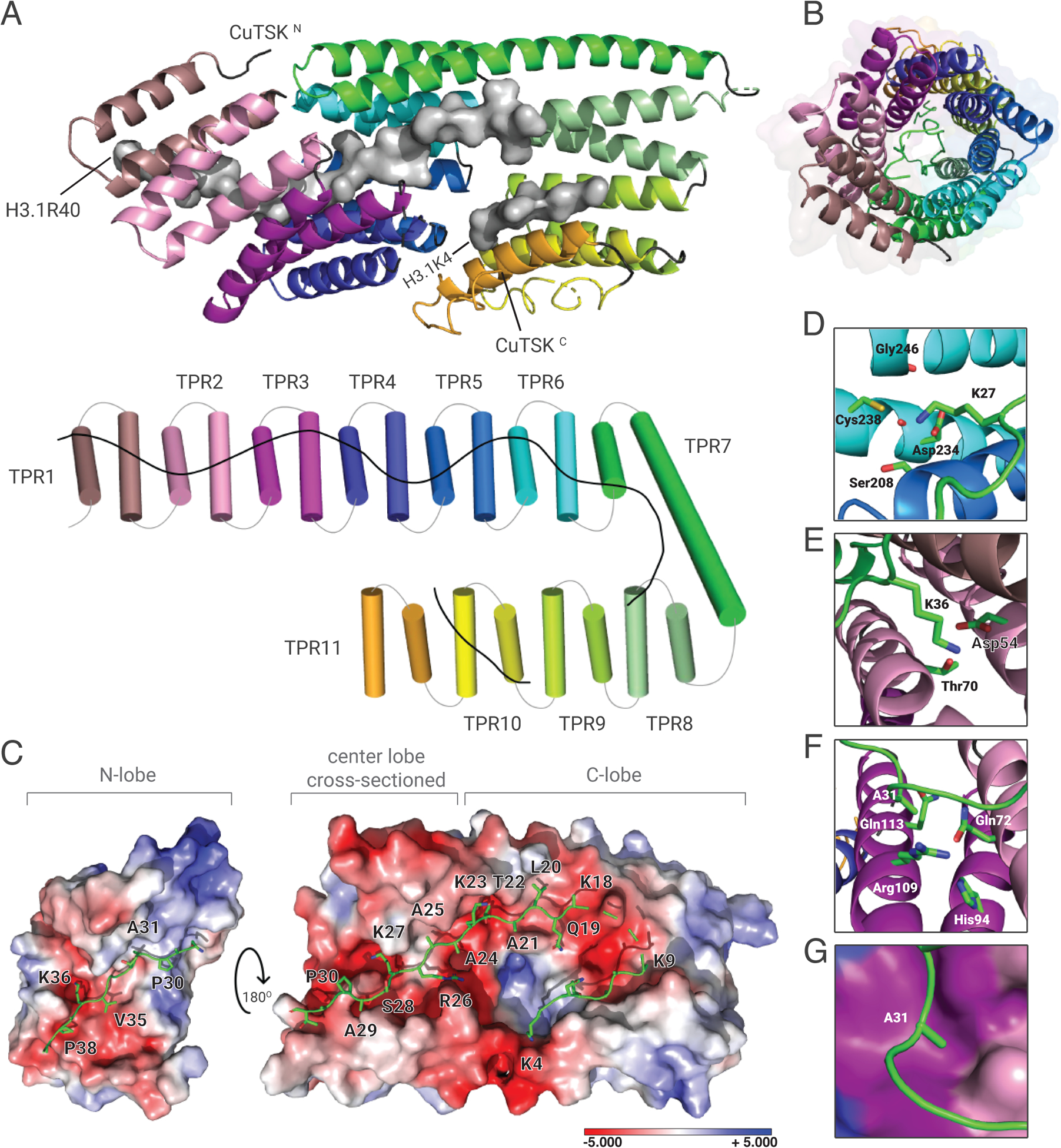
(A) The TPR domain is depicted as a cartoon (top) or a cylinder (bottom) with individual TPR motifs as distinct colors. H3.1 is shown as surfaces (top) or line (bottom). (B) Channel view of the TPR solenoid tube showing the space inside the tube where H3.1 is extended (represented as a green line). (C) Surface representation of the TPR domain shown as electrostatic potential gradients contoured from +5.000 kBTe−1 (blue) to −5.000 kBTe−1 (red), where e is the electron, T is temperature and kB is the Boltzmann constant. H3.1 is depicted as sticks. The N-terminal lobe (N-lobe) is rotated 180° along the horizontal axis relative to the center lobe and the C-terminal lobe (C-lobe). The surface of the center lobe is sectioned off to reveal the underlying segment of H3.1. (D, E, and F) Amino acid residues from TPRTSK (3-letter code) interacting with H3.1 residues (1-letter code) in their binding pockets are shown for D) K27, E) K36, and F) A31. (G) Surface representation of the H3.1A31 binding pocket. Surface colors correspond to that of TPR helices shown in panel F.
In plants, the histone H3K27 mono-methyltransferases ATXR5 and ATXR6 (ATXR5/6) maintain genome stability by specifically methylating the H3.1 variant (H3.1K27me1) during DNA replication (16–18). Loss of H3.1K27me1 in atxr5/6 double mutants results in genomic amplification of heterochromatin, transposon (TE) de-repression and disruption of heterochromatin structure (17, 19). Additional work has shown that heterochromatin amplification in atxr5/6 mutants is dependent on DNA repair (20). Therefore, ATXR5/6 may play a role analogous to the mammalian H4K20 mono-methyltransferase SET8 in regulating TONSL/TSK activity, with the difference that H3.1K27me1, not H4K20me1, is the key histone modification used in plants to prevent TSK from interacting with chromatin and initiating DNA repair. To validate this model, we generated an atxr5/6 tsk triple mutant in A. thaliana (fig. S4A). Flow cytometry analyses of atxr5/6 tsk mutants showed suppression of heterochromatin amplification induced by the absence of H3.1K27me1, as represented by the loss of the broad peaks corresponding to 8C and 16C endoreduplicated nuclei in atxr5/6 mutants (Fig. 3A and fig. S4B). This result was confirmed by genome sequencing of 16C nuclei from leaf tissue (Fig. 3B). We also observed transcriptional suppression of the genome instability marker BRCA1, which is highly expressed in atxr5/6 but not in atxr5/6 tsk (fig. S4C) (21). In addition, the number of chromocenters adopting a hollowed sphere conformation characteristic of atxr5/6 mutants is decreased when TSK is inactivated (Fig. 3C and D) (20). Similarly, transcriptional de-repression in heterochromatin of atxr5/6 mutants is reduced when TSK is inactivated (Fig. 3E and fig. S4D–F) (Table S2). These results indicate that the heterochromatic defects caused by the loss of H3.1K27me1 in plants are dependent on TSK.
Figure 3. Mutations in TSK suppress heterochromatin amplification of atxr5/6 mutants.
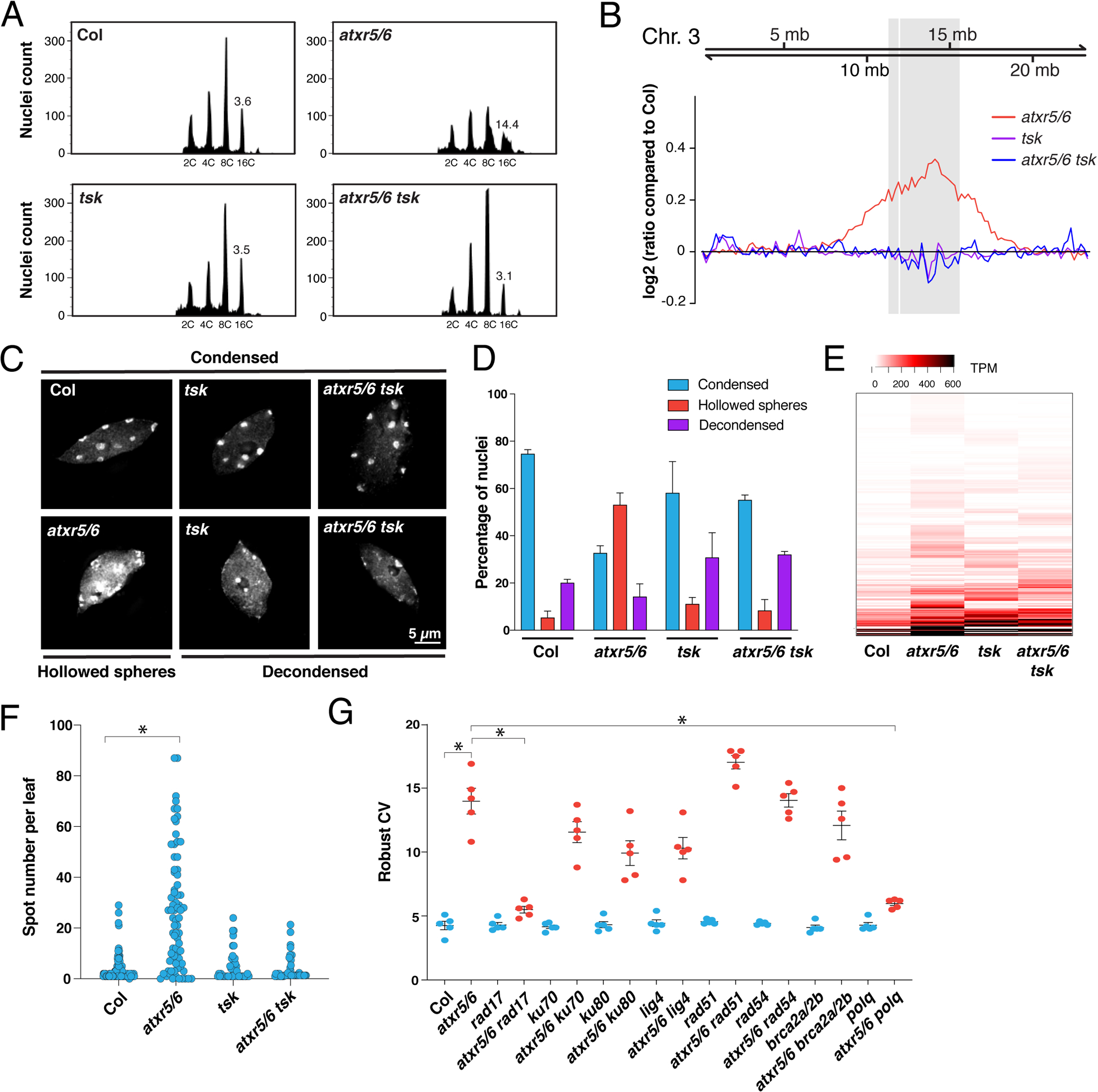
(A) Flow cytometry profiles of Col, atxr5/6, tsk and atxr5/6 tsk leaf nuclei. The numbers below the peaks indicate ploidy levels of the nuclei. The numbers above the 16C peaks indicate the robust coefficient of variation (rCV). (B) Chromosomal view (Chromosome 3 of A. thaliana) of DNA sequencing reads from sorted 16C nuclei. The pericentromeric region is highlighted in gray. (C) Leaf interphase nuclei of Col, atxr5/6, tsk and atxr5/6 tsk stained with DAPI. (D) Quantification of nuclei from experiment shown in panel C. Error bars indicate SEM. (E) Heat map showing the relative expression levels of atxr5/6-induced TEs as measured by TPM (transcripts per million). (F) Average number of blue spots per leaf in Col and atxr5/6 mutants as determined using a GUS reporter for homologous recombination. Error bars represent SEM. Welch’s ANOVA followed by Dunnett’s T3 test: * p < 0.0001. (G) rCV values for 16C nuclei obtained by flow cytometry analyses. Each dot represents an independent biological replicate. Horizontal bars indicate the mean. Error bars represent SEM. Welch’s ANOVA followed by the Dunnett’s T3 test: * p < 0.05.
In mammals, TONSL is recruited to newly replicated chromatin and promotes DNA repair via HR at broken replication forks (1, 3–6, 9). Cell-cycle expression analysis in synchronized tobacco cells indicates that TSK is specifically expressed in S phase (22), which supports a conserved role for TSK during replication. To assess if H3.1K27me1 suppresses HR activity in plants, we used a reporter system for HR based on intra-chromosomal recombination restoring activity at a colorimetric GUS transgene (23). Our results show that GUS activity is much stronger in atxr5/6 mutants compared to wild-type plants, but not in atxr5/6 tsk mutants (Fig. 3F and fig. S5), thus indicating a role for H3.1K27me1 in preventing TSK-mediated HR in plants.
The protein kinases ATM and ATR, which participate in the early signaling steps leading to HR-mediated DNA repair (24), were previously shown to be required for inducing heterochromatin amplification in atxr5/6 mutants (20). We therefore tested the contributions of different DNA repair pathways to the phenotypes observed in atxr5/6 mutants. Mutating non-homologous end joining (Ku70, Ku80, and LIG4) or HR (RAD51, RAD54, and BRCA2A/BRCA2b) genes did not have a major effect on heterochromatin amplification in atxr5/6 mutants (Fig. 3G), although eliminating the HR recombinase RAD51 enhances the morphological phenotypes of atxr5/6 mutants (fig. S6A). In contrast, RAD17 plays an important role in inducing heterochromatin amplification, loss of chromatin structure and transcriptional de-repression in atxr5/6 mutants (Fig. 3G and fig. S6B–C). RAD17 is responsible for loading the MRE11-RAD50-NBS1 complex that mediates DNA resection, one of the initial steps of HR (25). In animals, DNA resection can also lead to substrates that are repaired in an error-prone manner by Pol θ via polymerase theta-mediated end-joining (TMEJ), which can create large tandem duplications of 1 kb to 1 Mb (26). We introduced a mutant allele of the A. thaliana POLQ/TEBICHI gene coding for Pol θ in the atxr5/6 background and observed strong suppression of heterochromatin amplification and related phenotypes (Fig. 3G and fig. S7A–D). Taken together, these results show that genomic instability in atxr5/6 mutants is caused by a TSK-dependent pathway involving TMEJ.
The specificity of ATXR5/6 and TSK for replication-dependent H3.1 led us to hypothesize that this H3 variant is responsible for inducing TSK-mediated genomic instability in atxr5/6 mutants. To test if A31 of H3.1 is required for heterochromatin amplification in the absence of H3.1K27me1, we used a genetic system based on expression of the H3.1 point mutant H3.1S28A that mimics the phenotypes of atxr5/6 mutants (Fig. 4A–F and fig. S8A) (27). The S28A point mutation prevents H3.1K27 mono-methylation by ATXR5/6 (27), but does not affect the binding of TPRTSK to H3.1 (fig. S8B), thus supporting a role for H3.1K27me1 in preventing the interaction of TSK with H3.1 in vivo. We then transformed A. thaliana with a transgene expressing H3.1S28A A31T (A31 replaced with threonine, as in plant H3.3 variants), and observed suppression of the heterochromatin phenotypes (Fig. 4A–E and fig. S8A), which demonstrates the importance of H3.1A31 in regulating TSK activity in plants. The dependence of TSK on H3.1 explains why plants expressing H3.1A31T do not induce heterochromatin amplification despite losing ATXR5/6-catalyzed H3.1K27me1 (Fig. 4A) (16). A role for H3.1A31 in mediating TSK activity is also supported by the observation that plants expressing H3.1A31T are hypersensitive to genotoxic stress, similarly to tsk and h3.1 mutants (fig. S9A–F). We also used the H3.1S28A genetic system to assess the role of K4, K9 and K36 of H3.1 in contributing to the interaction with the TPR domain of TSK. Our in vivo results show that alanine replacement at K4 and K36 almost completely suppresses genomic instability and transcriptional de-repression mediated by expression of H3.1S28A (Fig. 4F and fig. S10A–D). These results are in line with in vitro experiments showing that H3.1K4A and H3.1K36A strongly disrupt binding of TSK to H3.1, but not H3.1K9A (fig. S10E). Finally, we used CRISPR to create a septuple mutant background, where all five H3.1 genes are inactivated, in addition to mutations in ATXR5/6 (atxr5/6 h3.1) (fig. S11A–B). In atxr5/6 h3.1 septuple mutants, both heterochromatin amplification and transcriptional de-repression are suppressed (fig. S11C–D), thus confirming that the H3.1 variant is required to induce these phenotypes. These results support that TSK makes specific interactions with the N-terminal tail of the H3.1 variant in vivo to disrupt heterochromatin stability and silencing when H3.1K27me1 deposition is impaired.
Figure 4. H3.1 is required to mediate genomic instability in atxr5/6 mutants.
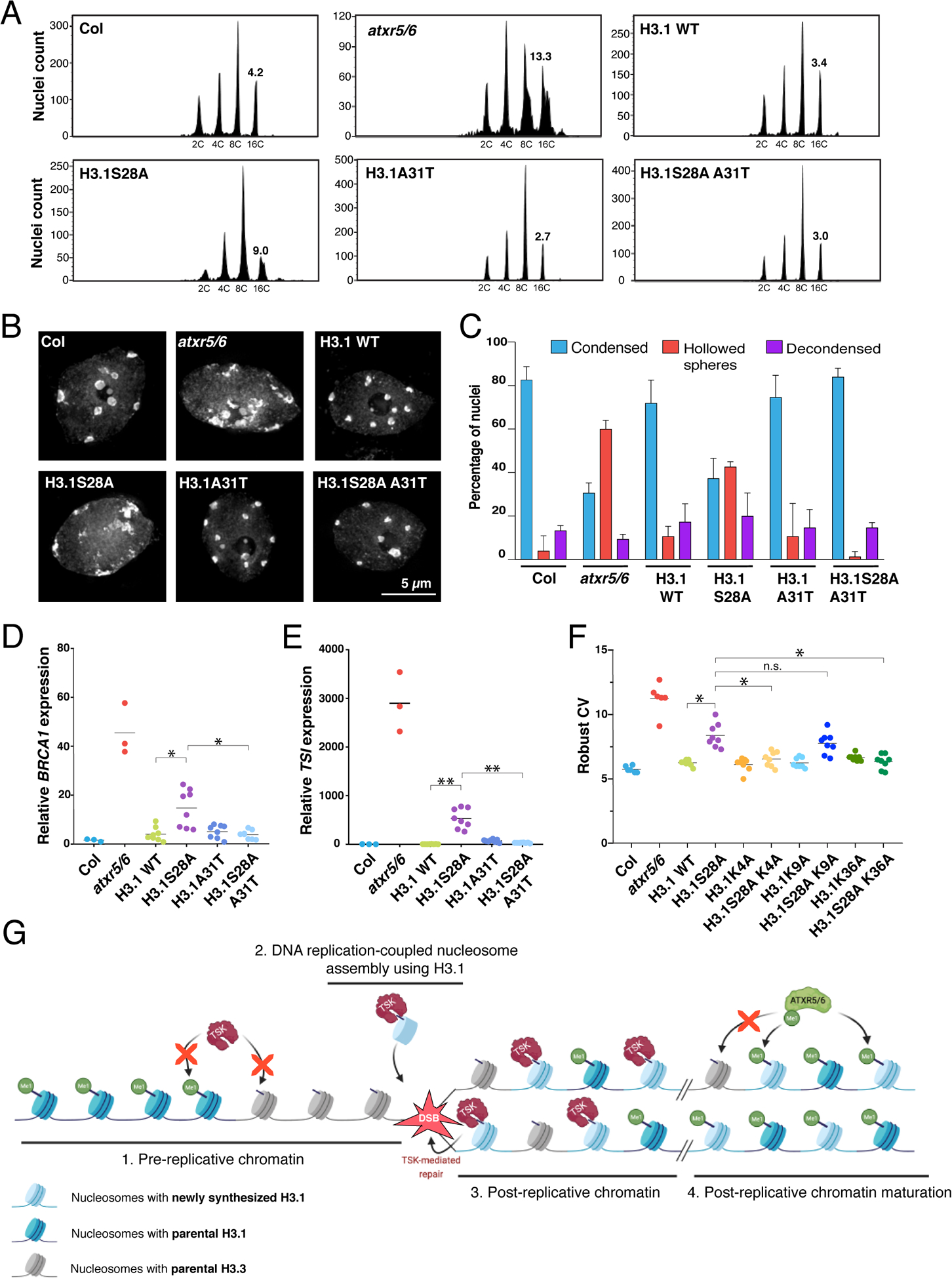
(A) Flow cytometry of leaf nuclei. Numbers below the peaks indicate ploidy, and those above indicate rCV. (B) Leaf nuclei of Col, atxr5/6, and first-generation (T1) H3.1 lines stained with DAPI. (C) Quantification from nuclei in B. Error bars indicate SEM. (D, E) RT-qPCR of BRCA1 and TSI. Horizontal bars indicate the mean. Welch’s ANOVA followed by the Dunnett’s T3 test: * p < 0.05, ** p < 0.001. (F) rCV for 16C nuclei obtained by flow cytometry. For Col and atxr5/6, each dot represents a biological replicate. For the H3.1 lines, each dot represents one T1 plant. Horizontal bars indicate the mean. Welch’s ANOVA followed by the Dunnett’s T3 test: * p < 0.05, n.s. = not significantly different. (G) Model depicting the interplay between H3.1, TSK and ATXR5/6 during replication. Step 1. TSK cannot interact with chromatin containing H3.3K27me0 or H3.1K27me1. Step 2. Newly synthesized H3.1 (H3.1K27me0) in complex with TSK are inserted at replication forks. Step 3. DSBs caused by broken replication forks are repaired by TSK. Step 4. Mono-methylation of newly inserted H3.1 (but not H3.3) at K27 by ATXR5/6 prevents binding of TSK.
Overall, this work uncovers a role for the TPR domain of TSK in selectively interacting with the H3.1 variant. Previous work in human cell lines has shown that the TSK ortholog TONSL co-purifies with H3.1 in affinity purification/biochemical fractionation assays (28), and that TONSL-mediated dsDNA break repair depends on the H3.1 chaperone CAF-1 (2). These findings, combined with our identification of the TPR domain of TSK/TONSL acting as an H3.1 reader, point to a model where post-replicative chromatin maturation in plants and animals relies on similar mechanisms involving H3.1 and clade-specific enzymes that mono-methylate histones to prevent TSK/TONSL binding (Fig. 4G). In plants, mono-methylation occurs at H3.1K27 via ATXR5/6 and prevents binding of TSK through the TPR domain. In animals, SET8-mediated mono-methylation at H4K20 interferes with TONSL binding via the ARD domain (9). However, in both plants and animals, recruitment of TSK/TONSL to chromatin likely relies on the ability of the conserved TPR domain to preferentially interact with the H3.1 variant. Thus, our work reveals the importance of selectively incorporating H3.1 variants during DNA replication, as it confers a window of opportunity during the cell cycle for the TSK/TONSL DNA repair pathway to resolve broken replication forks.
Supplementary Material
Acknowledgments:
We want to thank Dr. Xinnian Dong (Duke University) for providing us the seeds for the brca2a mutant (13F-1) and the recombination reporter line Col 1445. We also want to acknowledge specific contributions by support staff at Yale University: Christopher Bolick, Eileen Williams and Nathan Guzzo for help with plant growth and maintenance, Kenneth Nelson for technical help with flow cytometry, and members of the Yale Center for Genome Analysis. We are grateful to the Edinburgh Protein Production Facility (EPPF) for their support. We also thank Dr. Eric Campos (University of Toronto), Yale scientists Wei Liu, Japinder Nijjer and Wenxin Yuan, and members of the Jacob and Couture labs, for discussions, advice and/or materials that contributed to this work.
Funding:
This project was supported by grant #R35GM128661 from the National Institutes of Health to Y.J. J.F.C. is funded by grants from the Natural Science Engineering Research Council and the Canadian Institutes of Health research. Work in the Voigt lab was supported by the Wellcome Trust ([104175/Z/14/Z], Sir Henry Dale Fellowship to P.V.) and the UK Biotechnology and Biological Sciences Research Council (BBS/E/B/000C0421). The Wellcome Centre for Cell Biology received core funding from the Wellcome Trust [203149]. H.D. is thankful for an Ontario Graduate Scholarship and a University of Ottawa Excellence Scholarship. Postdoctoral support was provided to B.M. by a Yale University Brown Fellowship, to V.J. by the Fonds de Recherche du Québec-Nature et Technologies (FRQNT) [272565], and to M.M. by the Strategic Research Postdoctoral Fellowship from the University of Ottawa Heart Institute and the Strategic Research Endowed Funds. The EPPF was supported by the Wellcome Trust through a Multi-User Equipment grant [101527/Z/13/Z].
Footnotes
Data and materials availability:
Sequencing data (DNA-seq and RNA-seq datasets) generated for this study are available at the Gene Expression Omnibus (GEO) under accession code GSE184738. The Protein Data Bank (PDB) accession number for the TPRTSK-H3.1(1–45) structure is 7T7T. All data are available in the main text or the supplementary materials.
References and Notes:
- 1.Duro E et al. , Identification of the MMS22L-TONSL complex that promotes homologous recombination. Mol Cell 40, 632–644 (2010). [DOI] [PubMed] [Google Scholar]
- 2.Huang TH et al. , The Histone Chaperones ASF1 and CAF-1 Promote MMS22L-TONSL-Mediated Rad51 Loading onto ssDNA during Homologous Recombination in Human Cells. Mol Cell 69, 879–892 e875 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.O’Connell BC et al. , A genome-wide camptothecin sensitivity screen identifies a mammalian MMS22L-NFKBIL2 complex required for genomic stability. Mol Cell 40, 645–657 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.O’Donnell L et al. , The MMS22L-TONSL complex mediates recovery from replication stress and homologous recombination. Mol Cell 40, 619–631 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Piwko W et al. , The MMS22L-TONSL heterodimer directly promotes RAD51-dependent recombination upon replication stress. Embo J 35, 2584–2601 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Piwko W et al. , RNAi-based screening identifies the Mms22L-Nfkbil2 complex as a novel regulator of DNA replication in human cells. Embo J 29, 4210–4222 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Suzuki T et al. , A novel Arabidopsis gene TONSOKU is required for proper cell arrangement in root and shoot apical meristems. Plant J 38, 673–684 (2004). [DOI] [PubMed] [Google Scholar]
- 8.Takeda S et al. , BRU1, a novel link between responses to DNA damage and epigenetic gene silencing in Arabidopsis. Genes Dev 18, 782–793 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Saredi G et al. , H4K20me0 marks post-replicative chromatin and recruits the TONSL-MMS22L DNA repair complex. Nature 534, 714–718 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fang J et al. , Purification and functional characterization of SET8, a nucleosomal histone H4-lysine 20-specific methyltransferase. Curr Biol 12, 1086–1099 (2002). [DOI] [PubMed] [Google Scholar]
- 11.Nishioka K et al. , PR-Set7 is a nucleosome-specific methyltransferase that modifies lysine 20 of histone H4 and is associated with silent chromatin. Mol Cell 9, 1201–1213 (2002). [DOI] [PubMed] [Google Scholar]
- 12.Rice JC et al. , Mitotic-specific methylation of histone H4 Lys 20 follows increased PR-Set7 expression and its localization to mitotic chromosomes. Genes Dev 16, 2225–2230 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guyomarc’h S, Vernoux T, Traas J, Zhou DX, Delarue M, MGOUN3, an Arabidopsis gene with TetratricoPeptide-Repeat-related motifs, regulates meristem cellular organization. J Exp Bot 55, 673–684 (2004). [DOI] [PubMed] [Google Scholar]
- 14.Perez-Riba A, Itzhaki LS, The tetratricopeptide-repeat motif is a versatile platform that enables diverse modes of molecular recognition. Curr Opin Struct Biol 54, 43–49 (2019). [DOI] [PubMed] [Google Scholar]
- 15.Lu L, Chen X, Qian S, Zhong X, The plant-specific histone residue Phe41 is important for genome-wide H3.1 distribution. Nat Commun 9, 630 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jacob Y et al. , Selective methylation of histone H3 variant H3.1 regulates heterochromatin replication. Science 343, 1249–1253 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jacob Y et al. , ATXR5 and ATXR6 are H3K27 monomethyltransferases required for chromatin structure and gene silencing. Nat Struct Mol Biol 16, 763–768 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Raynaud C et al. , Two cell-cycle regulated SET-domain proteins interact with proliferating cell nuclear antigen (PCNA) in Arabidopsis. Plant J 47, 395–407 (2006). [DOI] [PubMed] [Google Scholar]
- 19.Jacob Y et al. , Regulation of heterochromatic DNA replication by histone H3 lysine 27 methyltransferases. Nature 466, 987–991 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Feng W et al. , Large-scale heterochromatin remodeling linked to overreplication-associated DNA damage. Proc Natl Acad Sci U S A 114, 406–411 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stroud H et al. , DNA methyltransferases are required to induce heterochromatic re-replication in Arabidopsis. PLoS Genet 8, e1002808 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Suzuki T et al. , TONSOKU is expressed in S phase of the cell cycle and its defect delays cell cycle progression in Arabidopsis. Plant Cell Physiol 46, 736–742 (2005). [DOI] [PubMed] [Google Scholar]
- 23.Lucht JM et al. , Pathogen stress increases somatic recombination frequency in Arabidopsis. Nat Genet 30, 311–314 (2002). [DOI] [PubMed] [Google Scholar]
- 24.Williams RM, Zhang X, Roles of ATM and ATR in DNA double strand breaks and replication stress. Prog Biophys Mol Biol 163, 109–119 (2021). [DOI] [PubMed] [Google Scholar]
- 25.Wang Q et al. , Rad17 recruits the MRE11-RAD50-NBS1 complex to regulate the cellular response to DNA double-strand breaks. Embo J 33, 862–877 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kamp JA, van Schendel R, Dilweg IW, Tijsterman M, BRCA1-associated structural variations are a consequence of polymerase theta-mediated end-joining. Nat Commun 11, 3615 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dong J et al. , H3.1K27me1 maintains transcriptional silencing and genome stability by preventing GCN5-mediated histone acetylation. Plant Cell 33, 961–979 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Campos EI et al. , Analysis of the Histone H3.1 Interactome: A Suitable Chaperone for the Right Event. Mol Cell 60, 697–709 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequencing data (DNA-seq and RNA-seq datasets) generated for this study are available at the Gene Expression Omnibus (GEO) under accession code GSE184738. The Protein Data Bank (PDB) accession number for the TPRTSK-H3.1(1–45) structure is 7T7T. All data are available in the main text or the supplementary materials.