

Abstract
Acute liver failure (ALF) is a rare entity but exhibits a high mortality. The mechanisms underlying ALF are not completely understood. The present study explored the role of the hepatic B cell leukemia-3 (Bcl-3), a transcriptional regulator of nuclear factor-kappa B (NF-κB), in two independent models of ALF. We employed a recently developed transgenic mouse model in a C57BL6/J background comparing wild-type (WT) and transgenic littermates with hepatocyte-specific overexpression of Bcl-3 (Bcl-3Hep) in the ALF model of d-galactosamine (d-GalN) and lipopolysaccharide (LPS). Additionally, the apoptosis-inducing CD95 (FAS/APO-1)-ligand was explored. Bcl-3Hep mice exhibited a significant protection from ALF with decreased serum transaminases, decreased activation of the apoptotic caspases 8, 9, and 3, lower rates of oxidative stress, B-cell lymphoma 2 like 1 (BCL2L1/BCL-XL) degradation and accompanying mitochondrial cytochrome c release, and ultimately a decreased mortality rate from d-GalN/LPS compared to WT mice. d-GalN/LPS treatment resulted in a marked inflammatory cytokine release and stimulated the activation of signal transducer and activator of transcription (STAT) 3, c-Jun N-terminal kinases (JNK) and extracellular signal-regulated kinase (ERK) signaling comparably in the hepatic compartment of Bcl-3Hep and WT mice. However, in contrast to the WT, Bcl-3Hep mice showed a diminished rate of IkappaB kinase-beta (IKK-β) degradation, persistent receptor interacting protein kinase (RIPK) 1 function and thus prolonged cytoprotective nuclear factor-kappa B (NF-κB) p65 signaling through increased p65 stability and enhanced transcription. Likewise, Bcl-3 overexpression in hepatocytes protected from ALF with massive hepatocyte apoptosis induced by the anti-FAS antibody Jo2. The protection was also linked to IKK-β stabilization. Overall, our study showed that Bcl-3 rendered hepatocytes more resistant to hepatotoxicity induced by d-GalN/LPS and FAS-ligand. Therefore, Bcl-3 appears to be a critical regulator of the dynamics in ALF through IKK-β.
Subject terms: Hepatotoxicity, Preclinical research
Introduction
Acute liver failure (ALF) is a rare but lethal entity. The treatment is based on withdrawal or suppression of the causing agent, but once injury and inflammation are initiated, hepatocellular injury is entertained by an intrinsic activation of cell death mechanisms that result in a fatal loss of liver tissue and function. Given that liver transplantation is the only definitive treatment option for these patients [1], there is high unmet need to identify the molecular mechanisms underlying the self-amplifying liver injury spiral, in order to develop biologically plausible treatments for patients with ALF.
Nuclear factor-kappa B (NF-κB) plays a central role in liver tissue homeostasis and regulating inflammation in response to various injurious challenges. In hepatocytes, NF-κB exerts primarily pro-survival effects by increasing the expression of genes encoding anti-apoptotic and anti-oxidant proteins, thus blocking cell death pathways [2–5]. Consequently, dysregulation of hepatocyte NF-κB has been linked to a variety of acute and chronic liver diseases [3–7]. NF-κB is assembled as homo- or heterodimer composed of the subunits p50/p105 (NF-κB1), p52/p100 (NF-κB2), p65 (RelA), c-Rel (Rel), or RelB and forms transcriptional complexes [8]. The primary mechanism regulating NF-κB transcriptional activity is through inhibitory kappa B (IκB) proteins including among others B cell leukemia-3 (Bcl-3) [9, 10].
Bcl-3 was originally identified as a proto-oncogene in leukemia [11] and is unique among the IκB proteins for nuclear localization. It contains two transactivation domains (TAD) that allow selective trans-activation or repression of NF-κB-dependent genes involving heterocomplex formation with p50 or p52 dimers [12, 13]. In addition, Bcl-3 has been reported to interact with other transcriptional regulators besides NF-κB [14–16]. Related to these pleiotropic functions of Bcl-3, we developed a transgenic mouse model overexpressing Bcl-3 selectively in hepatocytes (Bcl-3Hep) to study its abundant role in liver disease [14, 17] and employed two established models of ALF.
Results
Bcl-3 protects from d-GalN/LPS-induced hepatotoxicity
d-GalN/LPS induced a rapid hepatic and systemic inflammatory response in Bcl-3Hep and wild-type (WT) mice at 4 h characterized by inflammatory cytokines including tumor necrosis factor-α (TNF), interleukin (IL)-6, and IL-1α/β, and activation of hepatic signal transducer and activator of transcription 3 (STAT3) (Supplementary Fig. 1A–D). This was comparable between genotypes. Likewise, liver enzymes increased significantly (Fig. 1A, C), however this liver injury pattern was significantly attenuated in Bcl-3Hep mice (Fig. 1A). Blinded histopathological examination of hematoxylin & eosin (H&E)-stained liver sections showed extensive areas of hepatocyte cell death from d-GalN/LPS, that was substantially reduced in Bcl-3Hep mice (Fig. 1B). At 6 h post insult hepatic injury was comparable between the two genotypes. Still 20% of the WT mice succumbed to ALF at 6 h (Fig. 1C), while 20% of Bcl-3Hep mice survived the insult beyond 8 h (Fig. 1D). Taken together, these data suggest that Bcl-3 overexpression slowed the onset of ALF in a TNF-dependent model.
Fig. 1. Conditional overexpression of Bcl-3 in hepatocytes mitigated liver injury from d-GalN/LPS and promoted survival of mice.

Liver injury following treatment with d-GalN/LPS was assessed by measurement of A serum alanine and aspartate aminotransferase (ALT and AST) levels, and B standard H&E staining of liver sections (scale bar: 50 µm, representative histological photomicrographs) in Bcl-3Hep and WT mice at 4 h. C Serum transaminases in Bcl-3Hep and WT mice at 6 h post d-GalN/LPS. D Kaplan–Meier survival plots for Bcl-3Hep and WT mice after d-GalN/LPS injection. Data in A represent means ± SEM from four experiments with a total of n = 21 WT + d-GalN/LPS, n = 21 Bcl-3Hep + d-GalN/LPS, n = 7 WT + PBS and n = 7 Bcl-3Hep + PBS. Data in C represent means ± SEM of n = 10 mice/group, whereby 20% of the WT + d-GalN/LPS group was already dead at this time point. In D survival rate was monitored for 8 h after injection of d-GalN/LPS in a total of n = 12 WT and n = 10 Bcl-3Hep mice. ***p < 0.001 for WT vs. Bcl-3Hep and $p < 0.05, $$$p < 0.001 for PBS vs. d-GalN/LPS using Mann–Whitney U test (A and C) or a log-rank test (D). In C there was no statistical difference between the two groups in respect of the parameters.
Reduction in hepatocellular apoptosis in Bcl-3Hep mice
Next, effector molecules involved in TNF cell death signaling were assessed. Activation of caspase 8 and 3 was significantly lower in Bcl-3Hep mice at 4 h. Both truncated caspase fragments (Fig. 2A, B) as well as caspase activity assays (Fig. 2C) indicated reduced activation in Bcl-3Hep mice. Likewise, enzymatic activity of caspase 9 was only detectable in the WT (Fig. 2C). Caspase 9 activation involves the mitochondrial cell death pathway following cleavage of BH3 interaction domain death agonist (BID) and the subsequent release of mitochondrial cytochrome c. Cytochrome c reduction was detectable in the mitochondrial fractions of WT mice, but not accompanied by increasing cytochrome c in the cytosolic fraction (Fig. 2D). In parallel, translocation of full-length BID protein from the cytosol to the mitochondria fraction was detectable only in the WT 4 h after d-GalN/LPS, but not accompanied by BID cleavage (Fig. 2E). Also, increased pro-apoptotic BCL-2-associated X protein (BAX) was detectable at 4 h in both genotypes (Supplementary Fig. 2A) and BCL-2 antagonist/killer (BAK) expression was unchanged (Supplementary Fig. 2B).
Fig. 2. Hepatoprotection in Bcl-3Hep mice against d-GalN/LPS is associated with a reduction of caspase activation, BCL-XL degradation and mitochondrial ROS formation.

Liver tissue of d-GalN/LPS-challenged Bcl-3Hep and WT mice was harvested after 4 h for immunodetection of A activated caspase 8 by immunoblotting, B activated caspase 3 by immunhistochemical staining, and C activated caspase 8, 9, and 3 by caspase enzyme assays (means of n = 10 mice/group ± SEM). Activation of the mitochondrial apoptotic pathway was further evaluated by immunoblotting of D cytochrome c and E BID in cytosolic (cyt.) and mitochondrial (mit.) protein fractions, F BCL-XL in whole liver tissue lysates, and G determination of MDA content (means of n = 10 WT + d-GalN/LPS, n = 6 Bcl-3Hep + d-GalN/LPS, n = 2 PBS-treated controls per genotype ± SEM). In A and D–F representative immunoblots with densitometric analysis are shown. GAPDH and COX IV served as protein loading controls. In B representative histological photomicrographs (scale bar: 2000 µM) are depicted. *p < 0.05, **p < 0.01, ***p < 0.001 for WT vs. Bcl-3Hep and $$ p < 0.01 for PBS vs. d-GalN/LPS using unpaired, two-tailed Student’s t-test (A, F, and G) or Mann–Whitney U test (C).
To address the mechanism by which hepatocellular Bcl-3 protects from ALF, we investigated gene expression of key regulators of apoptosis including cellular inhibitor of apoptosis protein (cIAP)-1, X-linked inhibitor of apoptosis (XIAP), the anti-apoptotic BCL-2 family members B-cell lymphoma 2 (BCL-2), B-cell lymphoma 2 like 1 (BCL2L1/BCL-XL), and myeloid cell leukemia-1 (MCL-1), cellular FLICE (FADD-like IL-1β-converting enzyme)-inhibitory protein long/short (cFLIPL/cFLIPS), and A20 (Table 1A). All of these share the regulation through NF-κB but did not show differences at 4 h. The most striking differences between the genotypes were observed for BCL-XL, which was downregulated only in the WT from d-GalN/LPS (0.6-fold in WT vs. 1.7-fold in Bcl-3Hep, p = n.s.), and c-FLIPS, which was upregulated in Bcl-3Hep livers, while unaffected in WT animals (1.0-fold in WT vs. 1.4-fold in Bcl-3Hep, p < 0.05). cFLIPs protein levels were not detectable by immunoblotting (Supplementary Fig. 2C). Protein expression of BCL-XL in WT was significantly lower compared to Bcl-3Hep mice following d-GalN/LPS (Fig. 2F). Additionally, increasing malondialdehyde (MDA) suggestive of reactive oxygen species (ROS) were detectable in WT only (Fig. 2G). Levels of the anti-apoptotic XIAP protein did not follow this pattern and were increased in the WT early after d-GalN/LPS, and declined in Bcl-3Hep mice (Supplementary Fig. 2D). In summary, Bcl-3 overexpression suppresses TNF-induced hepatocyte apoptosis involving decreased caspase and mitochondrial cell death pathway activation, as well as lower rates of oxidative stress.
Table 1.
Hepatic gene expression of (A) anti-apoptotic proteins and (B) NF-κB subunits in Bcl-3Hep and WT mice at 4 h post d-GalN/LPS challenge and in control animals.
| (A) | ||||
|---|---|---|---|---|
| WT + PBS | WT + d-GalN/LPS | Bcl-3Hep + PBS | Bcl-3Hep + d-GalN/LPS | |
| BCL-2 | 1.00 ± 0.21 | 41.46 ± 8.58 ($) | 0.88 ± 0.08 | 23.39 ± 1.72 ($$) |
| BCL-XL | 1.00 ± 0.12 | 0.59 ± 0.03 ($) | 0.57 ± 0.02 | 0.95 ± 0.43 |
| cIAP1 | 1.00 ± 0.11 | 4.10 ± 0.48 ($) | 0.83 ± 0.10 | 3.00 ± 0.35 ($$) |
| c-FLIPL | 1.00 ± 0.07 | 0.89 ± 0.05 | 0.92 ± 0.04 | 0.81 ± 0.03 |
| c-FLIPS | 1.00 ± 0.06 | 0.96 ± 0.06 | 0.78 ± 0.05 | 1.06 ± 0.06 |
| MCL-1 | 1.00 ± 0.04 | 0.60 ± 0.08 | 1.17 ± 0.24 | 0.66 ± 0.12 |
| A20 | 1.00 ± 0.06 | 5.41 ± 0.80 ($) | 0.76 ± 0.06 | 4.29 ± 0.10 ($) |
| XIAP | 1.00 ± 0.04 | 0.81 ± 0.03 ($) | 0.95 ± 0.1 | 0.74 ± 0.06 |
| (B) | ||||
| p50/p105 | 1.00 ± 0.09 | 2.44 ± 0.25 ($) | 0.83 ± 0.05 | 1.90 ± 0.1 ($$) |
| c-Rel (Rel) | 1.00 ± 0.17 | 1.03 ± 0.13 | 0.77 ± 0.06 | 1.70 ± 0.55 ($) |
| p65 (RelA) | 1.00 ± 0.25 | 0.39 ± 0.13 | 0.19 ± 0.02 (*) | 0.53 ± 0.08 ($) |
| RelB | 1.00 ± 0.08 | 2.84 ± 0.53 ($) | 0.99 ± 0.13 | 2.62 ± 0.02 ($) |
Data in (A) and (B) are shown as mean of n = 3-6 mice/group ± SEM. *p < 0.05 for WT vs. Bcl-3Hep and $p < 0.05, $$p < 0.01 for PBS vs. d-GalN/LPS using Mann–Whitney U test (A and B). There was no statistically significant difference between WT + d-GalN/LPS vs. Bcl-3Hep + d-GalN/LPS in respect of the parameters.
Bcl-3 stabilizes hepatic NF-κB p65
In hepatocytes, TNF-induced apoptosis is regulated by mitogen-activated protein kinases (MAPK) and NF-κB signaling pathways. Phosphorylation of both c-Jun N-terminal kinases (JNK) and extracellular signal-regulated kinase (ERK) increased irrespective of the genotype at 4 h after d-GalN/LPS (Fig. 3A). Coincident with MAPK activation, increased NF-κB p50, p52, and p65 DNA-binding activity were detected in whole liver lysates following d-GalN/LPS treatment (Fig. 3B). Differences between the two genotypes related to increased activity of p50 and p65 in the WT compared to Bcl-3Hep mice (p50: 2.1-fold in WT vs. 1.5-fold in Bcl-3Hep, p < 0.01, p65: 2.2-fold in WT vs. 1.3-fold in Bcl-3Hep, p < 0.05), pointing to a stronger activation of the classical (or canonical) NF-κB pathway. To explore the role of cytoprotective p65 NF-κB, we visualized p65 expression by confocal microscopy. At baseline, the p65 signal was predominantly located in the cytosol of hepatocytes, translocating to the nuclear compartment upon d-GalN/LPS treatment (Supplementary Fig. 3). Compared to the WT, Bcl-3Hep mice displayed reduced basal levels as well as translocation of NF-κB p65 during ALF.
Fig. 3. Activation of hepatic JNK, ERK and NF-κB in Bcl-3Hep and WT mice in response to d-GalN/LPS.

A Immunodetection of phospho-JNK (Thr183/Tyr185), phospho-ERK (Thr202/Tyr204) and total JNK/ERK protein in whole liver tissue lysates from Bcl-3Hep and WT mice at 4 h post d-GalN/LPS challenge. B NF-κB p65, p50 and p52 activity was quantified by a functional binding assay. Ratio of relative NF-κB p65/p50/p52 activity in d-GalN/LPS-challenged Bcl-3Hep vs. WT mice is shown (means of n = 7 WT + d-GalN/LPS, n = 8 Bcl-3Hep + d-GalN/LPS and n = 7 PBS-treated controls per genotype ± SEM). C Immunoblotting of phospho-NF-κB p65 (Ser536) and total NF-κB p65 in whole liver tissue lysates and D after cytosolic vs. nuclear cell fractionation. E Relative hepatic NF-κB p65 gene expression in d-GalN/LPS-challenged Bcl-3Hep vs. WT mice as fold change over PBS-treated controls (means of n = 6 mice per genotype ± SEM). In A, C and D representative immunoblots with densitometric analysis are shown. *p < 0.05, **p < 0.01 for WT vs. Bcl-3Hep and $p < 0.05 for PBS vs. d-GalN/LPS using unpaired, two-tailed Student’s t-test (A–C) or Mann–Whitney U test (E).
Post-translational modifications including phosphorylation at serine 536 (Ser536) are required to increase p65 transcriptional activity. Ser536 phosphorylation will lead to nuclear localization of p65 and enhance its binding to coactivators and transcription factors [18]. Immunoblots of liver tissue lysates demonstrated significant lower expression of total and phospho-Ser536 p65 protein - in particular in WT mice at 4 h of D-GalN/LPS (Fig. 3C). We observed significant different phospho-Ser536 p65 levels between WT and Bcl-3Hep mice following d-GalN/LPS (p < 0.05). Cell fractionation and separate analysis of cytosolic and nuclear extracts confirmed a reduction of the cytosolic amounts of both total and phospho-Ser536 p65 protein in the WT group that was 2-fold stronger compared to the Bcl-3Hep group (Fig. 3D). These changes were attributable to degradation rather than increased nuclear translocation in response to d-GalN/LPS. On contrast, a relatively stable, albeit lower, expression of total NF-κB p65 was observed in the nuclear fraction of Bcl-3Hep mice compared to those of WT mice (Fig. 3D). In parallel, we did not observe Ser536-phosphorylated p65 in the nuclear fractions at 4 h, suggesting that the Bcl-3’s effects did not depend on S536-phosphorylation status of nuclear p65 (Fig. 3D). In contrast to the WT, Bcl-3Hep mice displayed a relatively low basal mRNA expression level of p65 (p < 0.05, Table 1B), but exhibited increasing p65 mRNA expression following D-GalN/LPS (0.4-fold in WT vs. 2.8-fold in Bcl-3Hep, p < 0.01, Fig. 3E), reflecting an increased stability of the transcript resp. an increase in gene transcription. Levels of NF-κB p50 and RelB did not significantly differ between the genotypes. The Rel family member c-Rel is involved in liver regeneration and was significantly upregulated in the hepatic compartment of Bcl-3Hep mice 4 h after d-GalN/LPS (Table 1B).
Cell death in Bcl-3Hep hepatocytes from ActD/TNF is depending on caspase, but not JNK activation
In order to explore the mechanisms related to the anti-apoptotic effect observed with Bcl-3, we employed small molecular inhibitors ex vivo. Primary hepatocytes isolated from Bcl-3Hep mice were treated with TNF and the transcriptional inhibitor actinomycin D (ActD). Caspase-dependent cell death was comparable in both genotypes at 24 h (Fig. 4A) and responsive to caspase inhibition by zVAD. Remarkably, while JNK inhibition using SP600125 completely prevented cell death from ActD/TNF in WT hepatocytes, the inhibitory effect of SP600125 was less potent in Bcl-3Hep hepatocytes. In Bcl-3 knockout (KO) hepatocytes, treatment with SP600125 or zVAD was able to efficiently abrogate cell death from ActD/TNF (Fig. 4A). Blockade of NF-κB using the IkappaB kinase (IKK) inhibitor BAY-11-7082 had no impact on ActD/TNF-induced cell death in cultured Bcl-3Hep, WT, or Bcl-3 KO hepatocytes at 24 h. A time course analysis of ActD/TNF-induced cell death in Bcl-3Hep hepatocytes, however, showed a delay compared to WT cells (Cell death in WT vs. Bcl-3Hep hepatocyte cultures: 24.1% vs. 14.7% at 6 h, 33.3% vs. 17.6% at 12 h, and 56.0% vs. 46.0% at 18 h, p < 0.05, Fig. 4B). ActD/TNF-induced cell death ex vivo was prevented by zVAD, but not by SP600125. Remarkably, the protective effect of Bcl-3 overexpression in primary hepatocytes was lost by blocking the IKK/NF-κB pathway with BAY-11-7082.
Fig. 4. Rate of TNF-induced hepatocyte cell death in vitro.
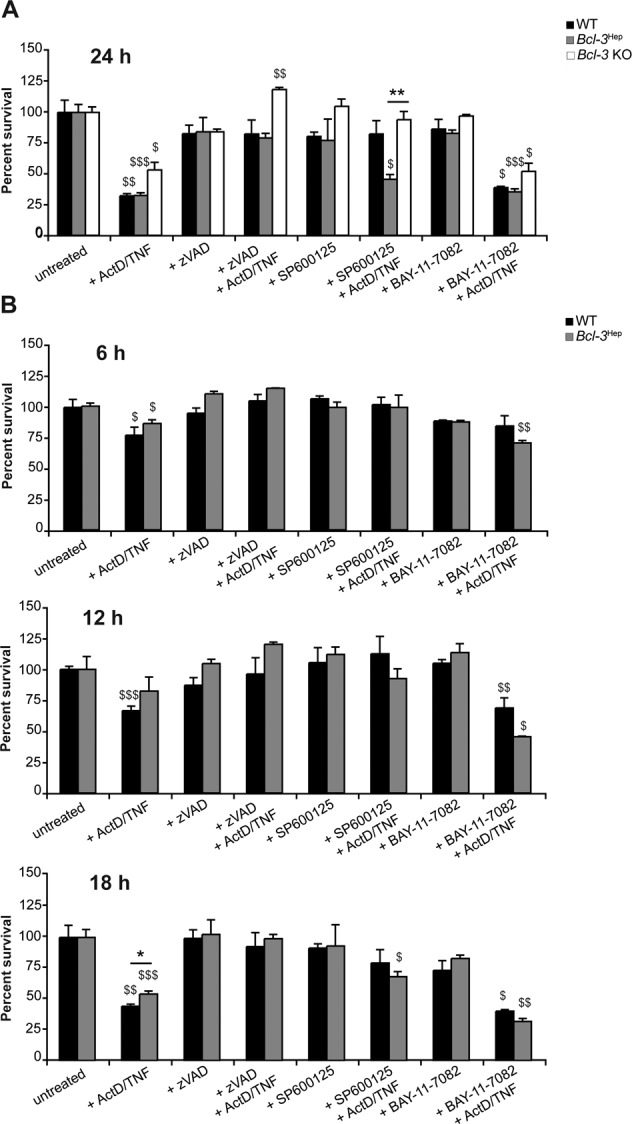
A Primary hepatocytes derived from Bcl-3Hep, WT, and Bcl-3 KO mice were treated ex vivo with ActD (200 ng/mL) and TNF (10 ng/mL) to induce TNF-R driven cell death. Pan caspase inhibitor zVAD (50 µM), JNK inhibitor SP600125 (100 µM), or IKK inhibitor BAY-11-7082 (10 µM), where ever indicated, were added 1 h before ActD/TNF-treatment to examine the activity of caspases, JNK and NF-κB signaling in TNF-induced cell death in Bcl-3Hep, WT, and Bcl-3 KO hepatocytes. After 24 h cell viability was assessed by MTT colorimetric assay relative to untreated samples. B Time course analysis of ActD/TNF-induced cell death in Bcl-3Hep and WT hepatocytes determined by MTT assays after 6, 12, and 18 h. Numerical data in mean ± SEM of A three or B two independent experiments performed at least in duplicate readings. *p < 0.05, **p < 0.01 for WT vs. Bcl-3Hep or Bcl-3Hep vs. Bcl-3 KO and $p < 0.05, $$p < 0.01, $$$p < 0.001 for untreated vs. treated hepatocytes according to an unpaired, two-tailed Student’s t-test (A and B).
IKK-β protects Bcl-3 overexpressing hepatocytes from apoptosis in vivo
The regenerative response in primary hepatocytes is largely dependent on activation of IKK-β (also called IKK2), which is required for the TNF-induced pro-survival transcriptional activity of the NF-κB subunit RelA/p65 and also protects against apoptosis through a NF-κB-independent axis [19–21]. Hence, we examined IKK-β expression in d-GalN/LPS-induced acute liver injury. Immunoblotting showed significantly lower IKK-β in WT liver tissue at 4 h post d-GalN/LPS compared to Bcl-3Hep (Fig. 5A). Thus, this data supports that Bcl-3 overexpression stabilizes IKK-β during TNF-induced apoptosis.
Fig. 5. Reduction of hepatic IKK-β, cleavage and Ser166-dephosphorylation of RIPK1 during apoptosis induced by d-GalN/LPS does not occur in Bcl-3Hep mice.

A Immunoblotting was performed with a specific antibody against the C terminus of IKK-β in whole liver tissue lysates from Bcl-3Hep and WT mice at 4 h after d-GalN/LPS challenge and corresponding controls. B To test the involvement of RIPK1 in d-GalN/LPS-induced liver injury, RIPK1 protein levels were analyzed in cytosolic (cyt.) and mitochondrial (mit.) protein fractions from two representative Bcl-3Hep and WT mice treated with d-GalN/LPS. Phospho-RIPK1 (Ser166) levels were determined in whole liver homogenates from Bcl-3Hep and WT mice at 4 h post D-GalN/LPS or PBS injection. In A and B representative western blots with densitometric analysis are shown. Tubulin and GAPDH served as protein loading controls. **p < 0.01 for WT vs. Bcl-3Hep according to an unpaired, two-tailed Student’s t-test (A and B).
IKK kinases maintain receptor interacting protein kinase 1 (RIPK1) in a pro-survival mode during TNF signaling, while their inhibition can sensitize cells to death through RIPK1-dependent apoptosis or necroptosis via RIPK3 [21, 22]. No significant changes in RIPK1 or RIPK3 gene expression levels were seen (Supplementary Table 1A). RIPK1 protein was cleaved in the liver cytosolic fractions from d-GalN/LPS-challenged WT mice, whereas no RIPK1 degradation occurred in Bcl-3Hep mice (Fig. 5B). These data are aligned with a role of caspase-dependent RIPK1 inhibition of pro-survival IKK/NF-κB signaling [23]. Dephosphorylation of RIPK1 at Ser166 in WT livers in response to d-GalN/LPS (Fig. 5B) pointed to an inactivation of RIPK1 enzyme activity [24, 25]. By contrast, Bcl-3Hep mice showed relative stable phospho-Ser166 RIPK1 protein levels at a level above the controls. The use of a small molecule inhibitor of RIPK1 kinase activity necrostatin (Nec-1) aggravated liver injury from d-GalN/LPS in both genotypes (Supplementary Table 1B). This was paralleled by a significant induction of RIPK1 and RIPK3 transcripts (Supplementary Table 1A). These data suggest that inhibition of RIPK1 kinase activity with Nec-1 is detrimental in this model in line with previous observations across different models and cell types [26–28], and support that stable RIPK1 functioning in Bcl-3Hep mice in response to TNF is protective for hepatocyte survival.
Hepatocyte-specific Bcl-3 overexpression protects mice from FAS-mediated hepatocyte apoptosis
Finally, we employed a second model of death-receptor mediated hepatocellular injury and ALF. Administration of the murine CD95 (FAS/APO-1)-stimulating antibody Jo2 resulted in massive liver cell apoptosis in WT mice accompanied by ALT and AST enzymes and a significant increase in caspase 3-positive apoptotic hepatocytes at 4 h. Interestingly, liver injury in Bcl-3Hep mice from Jo2 at this time point was minimal (Fig. 6A, B). FAS stimulation by Jo2 activated the hepatic JNK pathway, but there was no significant difference between Bcl-3Hep and WT mice (Fig. 6C and Supplementary Fig. 4), indicating that JNK activation was not required for FAS-mediated caspase activation and liver injury [29, 30]. Previous studies have suggested that FAS stimulation does not activate NF-κB in hepatocytes, while inhibition of NF-κB promotes apoptosis induced through CD95 [31]. FAS-induced apoptosis also requires proteolysis of IKK-β. Remarkably, in response to Jo2, IKK-β was clearly reduced in WT liver tissue at 4 h, whereas hepatic IKK-β expression remained unaffected in Bcl-3Hep mice (Fig. 6D). This suggests that Bcl-3 overexpression is linked to persistence of IKK-β activity over time in this model. Ex vivo stimulation of primary Bcl-3Hep and WT hepatocytes recapitulated lower levels of Jo2-induced caspase-dependent apoptosis in Bcl-3Hep-derived hepatocytes (Cell death in WT vs. Bcl-3Hep hepatocyte cultures: 13.6% vs. 0% at 12 h, p = 0.07, and 34.8% vs. 11.9% at 18 h, p < 0.01), which was independent of JNK (SP600125) and NF-κB (BAY-11-7082) inhibition (Fig. 7). By contrast, the hepatoxic effects of Jo2 in WT hepatocytes were augmented by NF-κB inhibition.
Fig. 6. Agonistic anti-FAS (CD95/APO-1) antibody Jo2-induced acute liver injury is attenuated in Bcl-3Hep mice.

Liver injury was assessed by measurement of A serum ALT and AST levels and B caspase 3 activity on liver histology at 4 h post Jo2. C Hepatic JNK activity and D IKK-β expression were analyzed by immunoblotting in whole liver tissue lysates. Data in A represent means ± SEM from two experiments with a total of n = 8 WT + Jo2, n = 8 Bcl-3Hep + Jo2, n = 7 WT + PBS, and n = 7 Bcl-3Hep + PBS. In B representative histological photomicrographs (scale bar: 2000 µM) are depicted. In C and D representative western blots with densitometric analysis are shown. **p < 0.01, ***p < 0.001 for WT vs. Bcl-3Hep and $$p < 0.01, $$$p < 0.001 for PBS vs. Jo2 using Mann–Whitney U test (A: ALT) or unpaired, two-tailed Student’s t-test (A: AST and B–D). In C there was no statistical difference between the two groups in respect of JNK activation.
Fig. 7. Rate of Jo2-induced hepatocyte cell death in vitro.
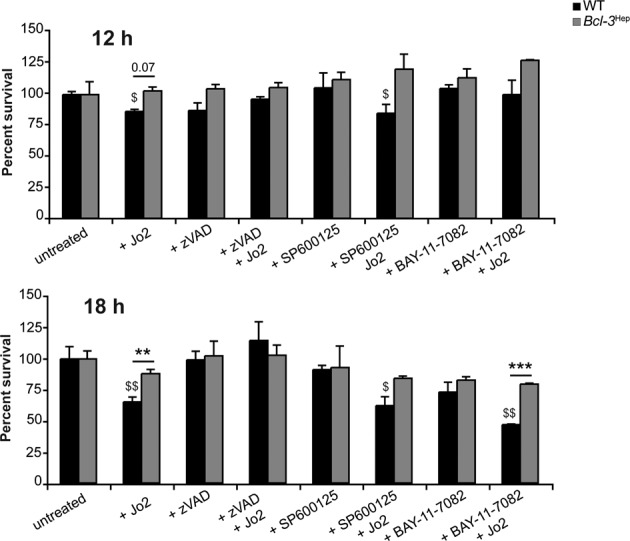
Primary Bcl-3Hep and WT hepatocytes were treated ex vivo with Jo2 (50 ng/mL). Pan caspase inhibitor zVAD (50 µM), JNK inhibitor SP600125 (100 µM), or IKK inhibitor BAY-11-7082 (10 µM), where ever indicated, were added 1 h before Jo2-treatment. After 12 and 18 h cell survival was assessed by MTT assay. Untreated hepatocytes were used as baseline controls. Numerical data in mean ± SEM of two independent experiments performed at least in duplicate readings. **p < 0.01, ***p < 0.001 for WT vs. Bcl-3Hep and $p < 0.05, $$p < 0.01 for untreated vs. treated hepatocytes according to an unpaired, two-tailed Student’s t-test.
Discussion
In order to develop novel therapeutic interventions in ALF, the molecular mechanisms underlying hepatocyte survival or death need to be clearly understood. Bcl-3 has been identified as a key survival factor for various cell types, but also as an anti-inflammatory regulator of immune cells related to its regulatory function on NF-κB activity. We and others have previously shown that Bcl-3 interacts also with non-NF-κB proteins and exerts important effects on cell homeostasis and metabolism [14, 16, 32]. The current analysis explored mice with a hepatocyte-specific overexpression of Bcl-3 in two models of experimental ALF in vivo and in a hepatocyte-based ex vivo culture system. Our results indicate that Bcl-3 overexpression in hepatocytes reduced the sensitivity towards TNF-R and FAS-mediated apoptosis in vivo and ex vivo resulting in different dynamics of cell death with protection from Bcl-3 and increased overall survival. This is in contrast to a recently published report from Hu and colleagues, which employed global Bcl-3 KO mice in the TNF/D-GalN model and the hepatic cell lines HepG2 and LO2 for in vitro analyses [33].
Here, we show that Bcl-3 acts as a pro-survival factor in hepatocytes by a mechanism located upstream of the mitochondrial cell death pathway and the activation of caspases. In fact, only WT mice showed a clear reduction of BCL-XL protein, leading to the loss of mitochondrial integrity through a decrease in mitochondrial membrane potential and ROS production [34] and through BAD, truncated (t)BID and BAX [35]. Likewise, enhanced oxidative stress as measured by increasing MDA levels was evident in livers of d-GalN/LPS-challenged WT mice compared to Bcl-3Hep mice. This was accompanied by caspase 8 activation, translocation of the pro-apoptotic BID protein to mitochondria, cytochrome c release and activation of the downstream caspases 9 and 3 in the WT. Interestingly, we did not detect proteolytic cleavage of BID, which has been indicated as a trigger of cytochrome c release [36]. Kinetics of BID cleavage and very low levels of tBID at 4 h post d-GalN/LPS could be related to this. On the other hand, full-length BID was also described to exhibit apoptotic and cytochrome c releasing activity [37]. In neuronal cells even in the absence of detectable BID cleavage, the MAPK JNK phosphorylated full-length BID, which as a consequence translocated to mitochondria and potentiated extrinsic TNF-induced apoptosis signaling [38]. In addition, non-BID-mediated mechanisms of cytochrome c release have been described [39]. Further expression analyses of proteins involved in the mitochondrial apoptosis pathway, however, revealed that Bcl-3 did not influence the levels of pro-apoptotic BAX, BAK, BCL-2, or MCL1, nor the anti-apoptotic cIAP1, c-FLIP or A20 proteins.
Sustained activation of JNK plays a critical role in hepatoxicity by d-GalN/LPS and ROS [40, 41]. Remarkably, both genotypes exhibited comparable early hepatic JNK activation, suggesting a JNK-independent effect. Supporting this, in vitro studies in primary hepatocytes indicated that cell death from ActD/TNF-treatment could be prevented by JNK inhibition only in WT hepatocytes. We can only speculate on the role of Bcl-3 counteracting the adverse effects of JNK signaling and switching TNF signaling to a protective response. One potential link relates to the activating protein-1 (AP-1) complex, which is transcriptionally regulated by Bcl-3 [42]. In hepatocytes, c-Jun/ AP-1 functionally antagonizes the cell death-promoting functions of JNK [43]. In previous studies using RNA-seq we identified JunB to be differentially expressed in Bcl-3Hep mice [14]. JunB is able to inhibit the transcriptional activity of c-Jun [44] and was significantly downregulated in Bcl-3Hep liver tissue (p < 0.01, hit: 456) [14]. Thus, transcriptional activity of c-Jun or other members of the AP-1 family might exert a protective signal through Bcl-3 overexpression.
Activation of NF-κB p65 antagonizes hepatocyte cell death induced by TNF, by increasing the expression of genes encoding anti-apoptotic and anti-oxidant proteins, which block the activity of either death receptors or the mitochondrial pathway of apoptosis, and additionally by repression of JNK activity [2]. While Bcl-3Hep livers exhibited reduced NF-κB 65 and p50 DNA-binding activities and p65 translocation in response to d-GalN/LPS, WT mice showed a stronger degradation of total and Ser536-phosphorylated NF-κB p65 protein - in particular in the cytosol - after d-GalN/LPS and no upregulation of p65 gene expression.
NF-κB is regulated by multiple mechanisms. Phosphorylation of p65 at Ser536 by IKK-β promotes the nuclear translocation and its transcription activity, but also accelerates its ubiquitination and proteasomal degradation [45] - a process that is involved in terminating the NF-κB protection during TNF signaling. Given that IKK-β is the predominant kinase to phosphorylate NF-κB p65 in its TAD in hepatocytes [46], the relatively stable IKK-β expression in Bcl-3Hep mice compared to the WT indicates that this could be a crucial difference. At this point it remains undefined if stabilization of IKK-β is an effect of Bcl-3 overexpression or depends on caspase 3-related caspases as previously observed [47].
Deletion or inactivation of IKK-β increases the sensitivity of rat hepatocytes [48] towards TNF-induced apoptosis. In a hepatocyte-specific Ikk2 knockout mouse model (IkkβΔhep), Maeda and colleagues demonstrated - in agreement with our data - that lack of functional IKK-β in hepatocytes resulted in massive hepatocyte apoptosis and higher lethality in mice given d-GalN/LPS - likely through substantial inhibition of survival gene expression [4]. Interestingly, IkkβΔhep mice were also highly susceptible to liver injury following ConA-induced activation of T cells, that express cell-bound TNF, through induction of prolonged JNK activation [4]. Also, soluble TNF induced substantial apoptosis in IkkβΔhep hepatocytes, accompanied by sustained JNK activation [4]. These findings indicate that the anti-apoptotic function of IKK-β might be related to cell-bound and/or high levels of circulating TNF and is partially dependent on JNK activity. Given the comparable levels of circulating TNF in both genotypes, one hypothesis on the slower emergence of liver injury in Bcl-3Hep is related to IKK-β. This is supported by the observation that Bcl-3 exerted a strong protective effect against FAS-induced hepatocyte apoptosis and liver injury, which was linked to IKK-β stabilization. The hepatoprotective function of IKK-β against FAS-mediated apoptosis has previously been shown [49].
IKK-α and IKK-β - in addition to their known function in NF-κB activation - can also directly phosphorylate RIPK1 at distinct regions, thereby regulating cell viability independent of NF-κB [22]. Any regulator of IKK-α/IKK-β activation results in defective phosphorylation of RIPK1 in complex I leading to pro-death complex IIb assembly and RIPK1 kinase-dependent necroptosis [50]. Furthermore, this is also observed when TNF-induced transcription is inhibited and RIPK1 is de-ubiquitinated by enzymes such as cylindromatosis (CYLD). Interestingly, CYLD was recently identified as a physical interaction partner of Bcl-3 [33, 51]. In the current analysis, we observed that cleavage of RIPK1 occurred in parallel to lower levels of Ser166 posphorylation of RIPK1 in the WT. This suggests that caspase-dependent RIPK1 cleavage shuts down protective IKK/NF-κB signaling pathways. It remains to be shown if RIPK1 cleavage was a direct cause for the dissociation of RIPK1 from complex I related to reduced Ser166 phosphorylation [24]. By contrast, the slight increase of phospho-Ser166 RIPK1 levels in Bcl-3Hep mice following d-GalN/LPS challenge points to autophosphorylation of RIPK1 upon TNF-R1 stimulation in complex I, which is not by itself sufficient to induce cell death [24]. Although further research is required to elucidate the involvement of RIPK1, increased sensitivity of hepatocytes lacking RIPK1 to TNF-mediated apoptosis has been reported in several models [52, 53].
In summary, our results suggest that Bcl-3 exerts robust hepatoprotective effects early during ALF. The complexity around NF-κB signaling makes therapeutic modulation of Bcl-3 in the context of ALF a difficult target. However, the current analysis expands the field and provides the rational to further study Bcl-3 both as a target of pharmacotherapy or as a predictive biomarker in ALF.
Materials and methods
Animals
Transgenic mice with hepatocyte-specific overexpression of Bcl-3 (Alfp-cre:bcl-3, Bcl-3Hep mice) were generated as previously described [14]. RNA-sequencing (RNA-seq) analysis revealed that under 981 genes differentially expressed (@ FDR p < 0.05) in liver between naïve 8-week old Bcl-3Hep and WT mice no significant differential expression of Tnfrsf1a encoding TNF-R1 was detected. However, gene expression of Tnfrsf1b (logFC = 0.63, p < 0.05, hit: 893) and Fas (logFC = 0.63, p < 0.01, hit: 485), which encode TNF-R2 and FAS (CD95/APO-1), resp., was significantly increased in Bcl-3Hep livers [14]. Bcl-3 knockout mice (Bcl-3 KO) were kindly provided by Dr. Hana Algül (Technical University Munich, Germany). All animals were bred at the animal facility of the University Medical Center Mainz according to the criteria outlined by the “Guide for the Care and Use of Laboratory Animals”. Studies were approved by the Landesuntersuchungsamt Rheinland-Pfalz (Koblenz, Germany, G-18-1-066).
Models of acute liver injury
Acute liver injury was induced in Bcl-3Hep mice and WT littermates aged 10–12 weeks and of mixed sex by intraperitoneal (i.p.) injection of d-GalN (0.75 mg/g bodyweight, from d-(+)-galactosamine hydrochloride G1639, Carl Roth, Karlsruhe, Germany) and LPS (2.5 μg/g bodyweight, from Escherichia coli Serotype 026:B6, L-8274, Sigma-Aldrich, Hamburg, Germany) according to published protocols (average weight of the mice: 22 ± 1 g) [54, 55]. Age-matched controls received phosphate-buffered saline (PBS) injections. If indicated, necrostatin (Nec-1) was injected i.p. at a dose of 2 mg/kg body weight 1 h before d-GalN/LPS challenge. Blood and liver tissue were harvested at 4 or 6 h after d-GalN/LPS administration and preserved for evaluation of liver injury using established standard operating procedures. In separate experiments, survival rates were monitored up to 8 h after i.p. injection of d-GalN/LPS. In a second model of acute liver injury, female Bcl-3Hep and WT mice aged 10–12 weeks were treated with agonistic FAS (CD95/APO-1) receptor antibody Jo2 (0.15 μg/g bodyweight, BD Pharmingen, Heidelberg, Germany, Cat# 554254) using intravenous (i.v.) tail vein injection [30]. Bcl-3Hep and WT mice were randomly divided into the experimental and control groups.
Serological analysis
Serum was obtained by cardiac puncture from anesthetized mice and serum ALT and AST levels were measured using a standard clinical analyzer (Hitachi 917, Roche, Mannheim, Germany). Serum cytokine levels were measured by BD cytometric bead array (CBA) Mouse Flex Sets (BD Biosciences, Heidelberg, Germany) using a BD FACS Canto II flow cytometer (BD Biosciences). Analysis was performed by FCAP ArrayTMv3 analysis software (Soft Flow, St. Louis Park, MN, USA).
Histological analyses and immunohistochemistry
For histological examination representative liver sections were cut, fixed in 4% paraformaldehyde–PBS, embedded in paraffin, and stained with H&E using standard protocols and evaluated blinded by an expert hepatopathologist (BKS). Immunohistochemistry for activated (cleaved) caspase 3 (antibody from Cell Signaling Technology Inc., Danvers, MA, USA, Cat# 9664) was performed and analyzed as previously described [17, 30]. Immunocytochemistry using an NF-κB p65 antibody (Cell Signaling, Cat# 4764) was performed according to published protocols [3]. Representative pictures were obtained using an Olympus BX45 microscope (Olympus Deutschland, Hamburg, Germany) with a Jenoptik PROGRES GRYPHAX camera (Micro Optimal, Meerbusch, Germany) and the Olympus Image Analysis Software analySIS docu (Olympus Deutschland).
Immunoblotting
Proteins were isolated and separated as previously described [56]. Primary antibodies included: BAK, BAX, BCL-XL, total and phospho-STAT3 (all Santa Cruz Biotechnology, Santa Cruz, CA, USA, Cat# sc-7873, sc-7480, sc-8392, sc-483, sc-8059), BID, c-FLIP, total and cleaved caspase 8, COX IV, cytochrome c, IKK-β, NF-κB p65, phospho-NF-κB p65 (Ser536), p44/42 MAPK (ERK1/2), phospho-p44/42 MAPK (ERK1/2) (Thr202/Tyr204), total and phospho-RIPK1 (Ser166), SAPK/JNK, phospho-SAPK/JNK (Thr183/Tyr185), XIAP (all Cell Signaling Technology Inc., Cat #2003, 3210, 4927, 9429, 4844, 4272, 2370, 4764, 3033, 9102, 9101, 3493, 53286, 9252, 4668, 2042), GAPDH (EnoGene, New York, NY, USA, Cat #E1C603) and tubulin (Abcam, Cambridge, MA, USA, Cat #ab4074). Membranes were exposed to anti-mouse (DAKO Denmark A/S, Glostrup, Denmark, Cat #P0447) or anti-rabbit (Santa Cruz Biotechnology, Cat #sc-2054) secondary antibodies conjugated with horseradish peroxidase. PaperPort Professional software v14.0 (Nuance Communications Germany, München, Germany) was used for image acquisition and the Adobe Acrobat Professional software program (Adobe Systems Incorporated, San Jose, CA, USA) was used to cut immunoblot images to size. No post-processing of images was performed. Densitometric analysis was performed using National Institutes of Health ImageJ software. Original, uncropped blots are presented in Supplementary Figure 5.
Determination of caspase activity
Caspase 3, 8 and 9 activity was determined in whole liver tissue in duplicate experiments (Lysis buffer: 20 mM Tris/HCl pH 8.0, 5 mM EDTA, 0.5 % Triton X, cOmplete Mini protease inhibitor cocktail (Roche, Indianapolis, IN, USA)). 50 µl of tissue lysate (4 mg/ml) were used with an assay mixture containing 50 µl 2x reaction buffer (50 mM HEPES pH 7.5, 100 mM NaCl, 20% glycerol, 0.1% 3-[(cholamidopropyl-)dimethylammonio]-1-propanesulfonate (CHAPS), 10 mM DTT) and 5 µl 4 mM chromogenic peptide substrate (Ac-DEVD-AFC, Ac-IETD-AFC, or Ac-LEHD-AFC for caspase 3, 8, and 9, respectively, all Biomol, Hamburg, Germany) and incubated at 37 °C for 2 h without light. Cleavage was monitored colorimetrically at 405 nm.
Determination of the malondialdehyde (MDA) content
MDA levels in whole liver tissue were detected and quantitated using the Lipid Peroxidation (MDA) Colorimetric Assay Kit (BioVision, Milpitas, CA, USA) according to the manufacturer’s instructions.
Determination of the NF-κB activity
Activity of the NF-κB subunits p50, p52, and p65 was measured in duplicates using the TransAM NF-κB Family Kit (Active Motif, Carlsbad, CA, USA).
Quantitative real-time (qRT-) PCR
Isolation of total RNA from snap frozen liver tissue, cDNA synthesis and qRT-PCR were performed as previously described [30]. All samples were performed in duplicates. Roche LightCycler software (LightCycler 480 Software Release 1.5.0) was used to perform advanced analysis relative quantification using the 2(−ΔΔC(T)) method. Expression data were normalized to the housekeeping gene Gapdh (Qiagen, Hilden, Germany) and the mean of PBS-treated WT mice was considered 1. Primer sequences (all Eurofins Genomics, Ebersberg, Germany) are detailed in Supplementary Table 2.
Isolation of primary hepatocytes and ex vivo stimulation
Hepatocytes were isolated and cultured as previously described [56]. After 24 h, cells were treated with the gene transcription inhibitor actinomycin D (ActD, 200 ng/mL) in combination with murine TNF (10 ng/mL, both from Sigma-Aldrich, Hamburg, Germany) or Jo2 (50 ng/mL, BD Pharmingen), to induce death receptor-mediated apoptosis. Where indicated the pan caspase inhibitor zVAD (50 µM), the JNK inhibitor SP600125 (100 µM, both from Enzo Life Sciences, Lörrach, Germany), or the IKK inhibitor BAY-11-7082 (10 µM, Calbiochem, EMD Chemicals, Inc., San Diego, CA, USA) was added 1 h prior to ActD/TNF or Jo2 treatment. Cell survival was assessed by MTT assay (Sigma-Aldrich) at the indicated time points.
Statistical analysis
All statistical analyses were performed using GraphPad Prism 7 software (GraphPad Software, La Jolla, CA, USA). All results were initially submitted to Shapiro–Wilk normality test for normality and to Levene’s test for homogeneity of variance. Comparisons between experimental groups were carried out using the unpaired, two-tailed Student’s t test or the Mann–Whitney U test to determine statistical significance of differences. The significance-level α was adjusted using Holm’s sequential Bonferroni adjustment in analyses involving multiple comparisons. Results with a p value of < 0.05 were considered to be significant. All data are shown as mean ± standard error of mean (SEM) to determine the precision and differences of means and statistically significant values were assumed with */$p < 0.05, **/$$p < 0.01, ***/$$$p < 0.001. Survival times were analyzed by Kaplan–Meier curves with p values assessed with log-rank (Mantel–Cox) test. Median survival was also calculated.
Supplementary information
Acknowledgements
Christine Waldmann, Irina Wagner, Sonja Hoch-Kraft (I. Department of Medicine) and Claudia Braun (Core Facility Immunohistochemistry, University Medical Center Mainz, Germany) contributed with excellent technical assistance.
Author contributions
NG: acquisition of data, analysis and interpretation of data, statistical analysis, drafting of the manuscript, obtained funding. MAW: acquisition of data, critical revision of the manuscript for important intellectual content. AM: acquisition of data, critical revision of the manuscript for important intellectual content. NH: technical and material support, critical revision of the manuscript for important intellectual content. AW: technical and material support, critical revision of the manuscript for important intellectual content. BKS: histopathological analysis and photomicrographs. PRG: material support, critical revision of the manuscript for important intellectual content. JMS: study concept and design, study supervision, drafting of the manuscript, obtained funding. All authors had access to the study data and had reviewed and approved the final manuscript.
Funding
NG received funding from Mainzer Wissenschaftsstiftung (MWS) and Else Kröner-Fresenius-Stiftung (EKFS). NH and AW were supported by CRC1292. JMS received funding from Gilead Sciences and Boehringer Ingelheim. Open Access funding enabled and organized by Projekt DEAL.
Data availability
All data will be shared upon request.
Competing interests
JMS reports Consultancy: BMS, Boehringer Ingelheim, Echosens, Genfit, Gilead Sciences, Intercept Pharmaceuticals, Madrigal, Merck, Nordic Bioscience, Novartis, Pfizer, Roche, Sanofi, Siemens Healthcare GmbH. Research Funding: Gilead Sciences, Boehringer Ingelheim, Siemens Healthcare GmbH. Speaker Honorarium: Falk Foundation. The other authors declare no competing interests.
Ethics statement
All applicable national and institutional guidelines for the care and use of animals were followed.
Footnotes
Edited by Thomas Brunner
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s41419-022-04946-y.
References
- 1.Stravitz RT, Lee WM. Acute liver failure. Lancet. 2019;394:869–81. doi: 10.1016/S0140-6736(19)31894-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Geisler F, Algul H, Paxian S, Schmid RM. Genetic inactivation of RelA/p65 sensitizes adult mouse hepatocytes to TNF-induced apoptosis in vivo and in vitro. Gastroenterology. 2007;132:2489–503. doi: 10.1053/j.gastro.2007.03.033. [DOI] [PubMed] [Google Scholar]
- 3.Beraza N, Ludde T, Assmus U, Roskams T, Vander Borght S, Trautwein C. Hepatocyte-specific IKK gamma/NEMO expression determines the degree of liver injury. Gastroenterology. 2007;132:2504–17. doi: 10.1053/j.gastro.2007.03.045. [DOI] [PubMed] [Google Scholar]
- 4.Maeda S, Kamata H, Luo JL, Leffert H, Karin M. IKKbeta couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell. 2005;121:977–90. doi: 10.1016/j.cell.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 5.Luedde T, Beraza N, Kotsikoris V, van Loo G, Nenci A, De Vos R, et al. Deletion of NEMO/IKKgamma in liver parenchymal cells causes steatohepatitis and hepatocellular carcinoma. Cancer Cell. 2007;11:119–32. doi: 10.1016/j.ccr.2006.12.016. [DOI] [PubMed] [Google Scholar]
- 6.Sakurai T, Maeda S, Chang L, Karin M. Loss of hepatic NF-kappa B activity enhances chemical hepatocarcinogenesis through sustained c-Jun N-terminal kinase 1 activation. Proc Natl Acad Sci USA. 2006;103:10544–51. doi: 10.1073/pnas.0603499103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.He G, Yu GY, Temkin V, Ogata H, Kuntzen C, Sakurai T, et al. Hepatocyte IKKbeta/NF-kappaB inhibits tumor promotion and progression by preventing oxidative stress-driven STAT3 activation. Cancer Cell. 2010;17:286–97. doi: 10.1016/j.ccr.2009.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ghosh S, May MJ, Kopp EB. NF-kappa B and Rel proteins: evolutionarily conserved mediators of immune responses. Annu Rev Immunol. 1998;16:225–60. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- 9.Oeckinghaus A, Ghosh S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb Perspect Biol. 2009;1:a000034. doi: 10.1101/cshperspect.a000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tergaonkar V, Correa RG, Ikawa M, Verma IM. Distinct roles of IkappaB proteins in regulating constitutive NF-kappaB activity. Nat Cell Biol. 2005;7:921–3. doi: 10.1038/ncb1296. [DOI] [PubMed] [Google Scholar]
- 11.Ohno H, Takimoto G, McKeithan TW. The candidate proto-oncogene bcl-3 is related to genes implicated in cell lineage determination and cell cycle control. Cell. 1990;60:991–7. doi: 10.1016/0092-8674(90)90347-H. [DOI] [PubMed] [Google Scholar]
- 12.Wulczyn FG, Naumann M, Scheidereit C. Candidate proto-oncogene bcl-3 encodes a subunit-specific inhibitor of transcription factor NF-kappa B. Nature. 1992;358:597–9. doi: 10.1038/358597a0. [DOI] [PubMed] [Google Scholar]
- 13.Bours V, Franzoso G, Azarenko V, Park S, Kanno T, Brown K, et al. The oncoprotein Bcl-3 directly transactivates through kappa B motifs via association with DNA-binding p50B homodimers. Cell. 1993;72:729–39. doi: 10.1016/0092-8674(93)90401-B. [DOI] [PubMed] [Google Scholar]
- 14.Gehrke N, Worns MA, Huber Y, Hess M, Straub BK, Hovelmeyer N, et al. Hepatic B cell leukemia-3 promotes hepatic steatosis and inflammation through insulin-sensitive metabolic transcription factors. J Hepatol. 2016;65:1188–97. doi: 10.1016/j.jhep.2016.06.026. [DOI] [PubMed] [Google Scholar]
- 15.Na SY, Choi HS, Kim JW, Na DS, Lee JWBcl3. an IkappaB protein, as a novel transcription coactivator of the retinoid X receptor. J Biol Chem. 1998;273:30933–8. doi: 10.1074/jbc.273.47.30933. [DOI] [PubMed] [Google Scholar]
- 16.Collins PE, Somma D, Kerrigan D, Herrington F, Keeshan K, Nibbs RJB, et al. The IkappaB-protein BCL-3 controls Toll-like receptor-induced MAPK activity by promoting TPL-2 degradation in the nucleus. Proc Natl Acad Sci USA. 2019;116:25828–38. doi: 10.1073/pnas.1900408116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gehrke N, Worns MA, Mann A, Huber Y, Hoevelmeyer N, Longerich T, et al. Hepatic B cell leukemia-3 suppresses chemically-induced hepatocarcinogenesis in mice through altered MAPK and NF-kappaB activation. Oncotarget. 2017;8:56095–109. doi: 10.18632/oncotarget.10893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang F, Tang E, Guan K, Wang CY. IKK beta plays an essential role in the phosphorylation of RelA/p65 on serine 536 induced by lipopolysaccharide. J Immunol. 2003;170:5630–5. doi: 10.4049/jimmunol.170.11.5630. [DOI] [PubMed] [Google Scholar]
- 19.Schwabe RF, Bennett BL, Manning AM, Brenner DA. Differential role of I kappa B kinase 1 and 2 in primary rat hepatocytes. Hepatology. 2001;33:81–90. doi: 10.1053/jhep.2001.20799. [DOI] [PubMed] [Google Scholar]
- 20.Yan J, Xiang J, Lin Y, Ma J, Zhang J, Zhang H, et al. Inactivation of BAD by IKK inhibits TNFalpha-induced apoptosis independently of NF-kappaB activation. Cell. 2013;152:304–15. doi: 10.1016/j.cell.2012.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dondelinger Y, Jouan-Lanhouet S, Divert T, Theatre E, Bertin J, Gough PJ, et al. NF-kappaB-independent role of IKKalpha/IKKbeta in preventing RIPK1 kinase-dependent apoptotic and necroptotic cell death during TNF signaling. Mol Cell. 2015;60:63–76. doi: 10.1016/j.molcel.2015.07.032. [DOI] [PubMed] [Google Scholar]
- 22.Koppe C, Verheugd P, Gautheron J, Reisinger F, Kreggenwinkel K, Roderburg C, et al. IkappaB kinasealpha/beta control biliary homeostasis and hepatocarcinogenesis in mice by phosphorylating the cell-death mediator receptor-interacting protein kinase 1. Hepatology. 2016;64:1217–31. doi: 10.1002/hep.28723. [DOI] [PubMed] [Google Scholar]
- 23.Kim JW, Choi EJ, Joe CO. Activation of death-inducing signaling complex (DISC) by pro-apoptotic C-terminal fragment of RIP. Oncogene. 2000;19:4491–9. doi: 10.1038/sj.onc.1203796. [DOI] [PubMed] [Google Scholar]
- 24.Laurien L, Nagata M, Schunke H, Delanghe T, Wiederstein JL, Kumari S, et al. Autophosphorylation at serine 166 regulates RIP kinase 1-mediated cell death and inflammation. Nat Commun. 2020;11:1747. doi: 10.1038/s41467-020-15466-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dondelinger Y, Vandenabeele P, Bertrand MJ. Regulation of RIPK1’s cell death function by phosphorylation. Cell Cycle. 2016;15:5–6. doi: 10.1080/15384101.2015.1112688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dannappel M, Vlantis K, Kumari S, Polykratis A, Kim C, Wachsmuth L, et al. RIPK1 maintains epithelial homeostasis by inhibiting apoptosis and necroptosis. Nature. 2014;513:90–4. doi: 10.1038/nature13608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tang Y, Tu H, Zhang J, Zhao X, Wang Y, Qin J, et al. K63-linked ubiquitination regulates RIPK1 kinase activity to prevent cell death during embryogenesis and inflammation. Nat Commun. 2019;10:4157. doi: 10.1038/s41467-019-12033-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Deutsch M, Graffeo CS, Rokosh R, Pansari M, Ochi A, Levie EM, et al. Divergent effects of RIP1 or RIP3 blockade in murine models of acute liver injury. Cell Death Dis. 2015;6:e1759. doi: 10.1038/cddis.2015.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tournier C, Hess P, Yang DD, Xu J, Turner TK, Nimnual A, et al. Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science. 2000;288:870–4. doi: 10.1126/science.288.5467.870. [DOI] [PubMed] [Google Scholar]
- 30.Schattenberg JM, Zimmermann T, Worns M, Sprinzl MF, Kreft A, Kohl T, et al. Ablation of c-FLIP in hepatocytes enhances death-receptor mediated apoptosis and toxic liver injury in vivo. J Hepatol. 2011;55:1272–80. doi: 10.1016/j.jhep.2011.03.008. [DOI] [PubMed] [Google Scholar]
- 31.Dudley E, Hornung F, Zheng L, Scherer D, Ballard D, Lenardo M. NF-kappaB regulates Fas/APO-1/CD95- and TCR-mediated apoptosis of T lymphocytes. Eur J Immunol. 1999;29:878–86. doi: 10.1002/(SICI)1521-4141(199903)29:03<878::AID-IMMU878>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 32.Na SY, Choi JE, Kim HJ, Jhun BH, Lee YC, Lee JWBcl3. an IkappaB protein, stimulates activating protein-1 transactivation and cellular proliferation. J Biol Chem. 1999;274:28491–6. doi: 10.1074/jbc.274.40.28491. [DOI] [PubMed] [Google Scholar]
- 33.Hu Y, Zhang H, Xie N, Liu D, Jiang Y, Liu Z, et al. Bcl-3 promotes TNF-induced hepatocyte apoptosis by regulating the deubiquitination of RIP1. Cell Death Differ. 2021. 10.1038/s41418-021-00908-07. [DOI] [PMC free article] [PubMed]
- 34.Kim WS, Lee KS, Kim JH, Kim CK, Lee G, Choe J, et al. The caspase-8/Bid/cytochrome c axis links signals from death receptors to mitochondrial reactive oxygen species production. Free Radic Biol Med. 2017;112:567–77. doi: 10.1016/j.freeradbiomed.2017.09.001. [DOI] [PubMed] [Google Scholar]
- 35.Wu G, Tu Z, Yang F, Mai Z, Chen H, Tang Q, et al. Evaluating the inhibitory priority of Bcl-xL to Bad, tBid and Bax by using live-cell imaging assay. Cytometry A. 2021;99:1091–101. doi: 10.1002/cyto.a.24351. [DOI] [PubMed] [Google Scholar]
- 36.Huang K, Zhang J, O’Neill KL, Gurumurthy CB, Quadros RM, Tu Y, et al. Cleavage by Caspase 8 and mitochondrial membrane association activate the BH3-only protein bid during TRAIL-induced apoptosis. J Biol Chem. 2016;291:11843–51. doi: 10.1074/jbc.M115.711051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sarig R, Zaltsman Y, Marcellus RC, Flavell R, Mak TW, Gross A. BID-D59A is a potent inducer of apoptosis in primary embryonic fibroblasts. J Biol Chem. 2003;278:10707–15. doi: 10.1074/jbc.M210296200. [DOI] [PubMed] [Google Scholar]
- 38.Prakasam A, Ghose S, Oleinik NV, Bethard JR, Peterson YK, Krupenko NI, et al. JNK1/2 regulate Bid by direct phosphorylation at Thr59 in response to ALDH1L1. Cell Death Dis. 2014;5:e1358. doi: 10.1038/cddis.2014.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen X, Ding WX, Ni HM, Gao W, Shi YH, Gambotto AA, et al. Bid-independent mitochondrial activation in tumor necrosis factor alpha-induced apoptosis and liver injury. Mol Cell Biol. 2007;27:541–53. doi: 10.1128/MCB.01166-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schattenberg JM, Wang Y, Rigoli RM, Koop DR, Czaja MJ. CYP2E1 overexpression alters hepatocyte death from menadione and fatty acids by activation of ERK1/2 signaling. Hepatology. 2004;39:444–55. doi: 10.1002/hep.20067. [DOI] [PubMed] [Google Scholar]
- 41.Donovan N, Becker EB, Konishi Y, Bonni A. JNK phosphorylation and activation of BAD couples the stress-activated signaling pathway to the cell death machinery. J Biol Chem. 2002;277:40944–9. doi: 10.1074/jbc.M206113200. [DOI] [PubMed] [Google Scholar]
- 42.Rebollo A, Dumoutier L, Renauld JC, Zaballos A, Ayllon V, Martinez AC. Bcl-3 expression promotes cell survival following interleukin-4 deprivation and is controlled by AP1 and AP1-like transcription factors. Mol Cell Biol. 2000;20:3407–16. doi: 10.1128/MCB.20.10.3407-3416.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hasselblatt P, Rath M, Komnenovic V, Zatloukal K, Wagner EF. Hepatocyte survival in acute hepatitis is due to c-Jun/AP-1-dependent expression of inducible nitric oxide synthase. Proc Natl Acad Sci USA. 2007;104:17105–10. doi: 10.1073/pnas.0706272104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chiu R, Angel P, Karin M. Jun-B differs in its biological properties from, and is a negative regulator of, c-Jun. Cell. 1989;59:979–86. doi: 10.1016/0092-8674(89)90754-X. [DOI] [PubMed] [Google Scholar]
- 45.Mao X, Gluck N, Li D, Maine GN, Li H, Zaidi IW, et al. GCN5 is a required cofactor for a ubiquitin ligase that targets NF-kappaB/RelA. Genes Dev. 2009;23:849–61. doi: 10.1101/gad.1748409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schwabe RF, Sakurai H. IKKbeta phosphorylates p65 at S468 in transactivaton domain 2. FASEB J. 2005;19:1758–60. doi: 10.1096/fj.05-3736fje. [DOI] [PubMed] [Google Scholar]
- 47.Tang G, Yang J, Minemoto Y, Lin A. Blocking caspase-3-mediated proteolysis of IKKbeta suppresses TNF-alpha-induced apoptosis. Mol Cell. 2001;8:1005–16. doi: 10.1016/S1097-2765(01)00380-X. [DOI] [PubMed] [Google Scholar]
- 48.Hatano E, Bennett BL, Manning AM, Qian T, Lemasters JJ, Brenner DA. NF-kappaB stimulates inducible nitric oxide synthase to protect mouse hepatocytes from TNF-alpha- and Fas-mediated apoptosis. Gastroenterology. 2001;120:1251–62. doi: 10.1053/gast.2001.23239. [DOI] [PubMed] [Google Scholar]
- 49.Hatano E, Bradham CA, Stark A, Iimuro Y, Lemasters JJ, Brenner DA. The mitochondrial permeability transition augments Fas-induced apoptosis in mouse hepatocytes. J Biol Chem. 2000;275:11814–23. doi: 10.1074/jbc.275.16.11814. [DOI] [PubMed] [Google Scholar]
- 50.Dondelinger Y, Aguileta MA, Goossens V, Dubuisson C, Grootjans S, Dejardin E, et al. RIPK3 contributes to TNFR1-mediated RIPK1 kinase-dependent apoptosis in conditions of cIAP1/2 depletion or TAK1 kinase inhibition. Cell Death Differ. 2013;20:1381–92. doi: 10.1038/cdd.2013.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sun SC. CYLD: a tumor suppressor deubiquitinase regulating NF-kappaB activation and diverse biological processes. Cell Death Differ. 2010;17:25–34. doi: 10.1038/cdd.2009.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Suda J, Dara L, Yang L, Aghajan M, Song Y, Kaplowitz N, et al. Knockdown of RIPK1 markedly exacerbates murine immune-mediated liver injury through massive apoptosis of hepatocytes, independent of necroptosis and inhibition of NF-kappaB. J Immunol. 2016;197:3120–9. doi: 10.4049/jimmunol.1600690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Filliol A, Piquet-Pellorce C, Le Seyec J, Farooq M, Genet V, Lucas-Clerc C, et al. RIPK1 protects from TNF-alpha-mediated liver damage during hepatitis. Cell Death Dis. 2016;7:e2462. 10.1038/cddis.2016.362. [DOI] [PMC free article] [PubMed]
- 54.Urbanik T, Koehler BC, Wolpert L, Elssner C, Scherr AL, Longerich T, et al. CYLD deletion triggers nuclear factor-kappaB-signaling and increases cell death resistance in murine hepatocytes. World J Gastroenterol. 2014;20:17049–64. doi: 10.3748/wjg.v20.i45.17049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gehrke N, Hovelmeyer N, Waisman A, Straub BK, Weinmann-Menke J, Worns MA, et al. Hepatocyte-specific deletion of IL1-RI attenuates liver injury by blocking IL-1 driven autoinflammation. J Hepatol. 2018;68:986–95. doi: 10.1016/j.jhep.2018.01.008. [DOI] [PubMed] [Google Scholar]
- 56.Kohl T, Gehrke N, Schad A, Nagel M, Worns MA, Sprinzl MF, et al. Diabetic liver injury from streptozotocin is regulated through the caspase-8 homolog cFLIP involving activation of JNK2 and intrahepatic immunocompetent cells. Cell Death Dis. 2013;4:e712. doi: 10.1038/cddis.2013.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data will be shared upon request.