

Abstract
Double‐stranded DNA is recognized as a danger signal by cyclic guanosine monophosphate‐adenosine monophosphate synthase (cGAS), triggering innate immune responses. Palmitoylation is an important post‐translational modification (PTM) catalyzed by DHHC‐palmitoyl transferases, which participate in the regulation of diverse biological processes. However, whether palmitoylation regulates cGAS function has not yet been explored. Here, we found that palmitoylation of cGAS at C474 restricted its enzymatic activity in the presence of double‐stranded DNA. cGAS palmitoylation was catalyzed mainly by the palmitoyltransferase ZDHHC18 and double‐stranded DNA promoted this modification. Mechanistically, palmitoylation of cGAS reduced the interaction between cGAS and double‐stranded DNA, further inhibiting cGAS dimerization. Consistently, ZDHHC18 negatively regulated cGAS activation in human and mouse cell lines. In a more biologically relevant model system, Zdhhc18‐deficient mice were found to be resistant to infection by DNA viruses, in agreement with the observation that ZDHHC18 negatively regulated cGAS mediated innate immune responses in human and mouse primary cells. In summary, the negative role of ZDHHC18‐mediated cGAS palmitoylation may be a novel regulatory mechanism in the fine‐tuning of innate immunity.
Keywords: cGAS, innate immunity, palmitoylation, ZDHHC18
Subject Categories: Immunology; Microbiology, Virology & Host Pathogen Interaction
ZDHHC18‐mediated palmitoylation of cGAS impedes the interaction between cGAS and double‐stranded DNA, thereby inhibiting cGAS dimerization and cGAS‐mediated innate immune responses.
Introduction
As a marker of pathogen invasion or tissue damage, cytosolic DNA has long been known as a pathogen/danger‐associated molecular pattern (P/DAMP) that triggers robust innate immune responses. Recently, cyclic GMP‐AMP synthase (cGAS) was defined as a pattern‐recognition receptor (PRR) that recognizes and binds to cytosolic double‐stranded DNA (Sun et al, 2013). After binding, cGAS forms a 2:2 complex with DNA and catalyzes the transformation of ATP and GTP into 2′‐3′‐cGAMP. As a cytosolic second messenger, cGAMP binds to stimulator of interferon genes (STING, also known as TMEM173) and causes a 180° rotation of its carboxyl ligand‐binding domain relative to its transmembrane domain, leading to STING activation (Burdette et al, 2011; Gao et al, 2013; Shang et al, 2019; Zhang et al, 2019). Activated STING is then trafficked from the endoplasmic reticulum (ER) to an ER‐Golgi intermediate compartment (ERGIC) and then to the Golgi apparatus. Next, STING recruits and activates the kinase TANK‐binding kinase 1 (TBK1), which in turn phosphorylates STING and IRF3 (Ishikawa et al, 2009; Saitoh et al, 2009; Dobbs et al, 2015). Phosphorylated dimeric IRF3 enters the nucleus and induces the expression of type I interferons (IFNs) (Chen et al, 2016).
Based on the sensing of cytosolic DNA derived from both invaders and self‐DNA, cGAS‐mediated innate immune responses are involved in multiple biological and pathological processes. cGAS recognition of invading DNA exerts a nonredundant role in defending against infection by DNA viruses, including herpes simplex virus 1 (HSV‐1), vaccinia virus (VACV), and retroviruses, such as human immunodeficiency virus 1 (HIV‐1) (Yoh et al, 2015; Lahaye et al, 2018). On the other hand, in the presence of cytosolic self‐DNA, including mitochondrial DNA (mtDNA) and chromatin exposed to the cytosol during mitosis, cGAS function must be well controlled to prevent overactivation. MtDNA released under specific stress is recognized by cGAS and induces the expression of type I IFNs and proinflammatory cytokines, and this process is related to multiple diseases (West et al, 2015; Aarreberg et al, 2019; Huang et al, 2020). In the nucleus, cGAS is tightly bound to nucleosomes, which is important for the restriction of its autoreactivity (Boyer et al, 2020; Kujirai et al, 2020; Michalski et al, 2020; Pathare et al, 2020; Zhao et al, 2020). Moreover, during mitosis, activation and autoreactivity of cGAS are prevented by hyperphosphorylation at its amino (N) terminus and chromatin tethering (Li et al, 2021). However, whether there are other mechanisms that control cGAS activation remains to be further understood.
Post‐translational modifications (PTMs) play critical roles in the regulation of protein functions. Palmitoylation is a post‐translational lipid modification of proteins by which fatty acids, usually 16‐carbon palmitate, are reversibly linked to cysteine residues via a liable thioester bond (Chamberlain & Shipston, 2015). Protein palmitoylation is catalyzed by a series of enzymes known as DHHC‐palmitoyl transferases, which contain a signature DHHC motif (Jiang et al, 2018). Extensive studies have reported that this reversible, dynamic palmitoylation plays important roles in protein subcellular localization and signal transduction by altering their membrane association or conformational state (Baekkeskov & Kanaani, 2009; Zhang et al, 2016, 2020; Lu et al, 2019). For example, different palmitoylation profiles change the conformation of GPCRs (Chini & Parenti, 2009; Zheng et al, 2012). In the cGAS/STING signaling pathway, it has been reported that STING palmitoylation is important for TBK1 recruitment and activation to trigger the innate immune response (Mukai et al, 2016). However, whether cGAS is palmitoylated and whether that modification regulates cGAS function remain unclear.
Here, we found that cGAS is palmitoylated at C474. This palmitoylation restricts cGAS activation in the presence of cytosolic DNA. Moreover, through cell‐based screening, we identified that ZDHHC18 is a major acyltransferase catalyzing cGAS palmitoylation. DNA enhanced the interaction between ZDHHC18 and cGAS and promoted cGAS palmitoylation. cGAS palmitoylation causes a conformational change, subsequently inhibiting cGAS DNA binding and dimerization. Unlike other post‐translational mechanisms of cGAS (e.g., ubiquitination and arginine methylation) (Wang et al, 2017a; Chen & Chen, 2019; Ma et al, 2021), the palmitoylation of cGAS unconventionally impairs its enzymatic activity. ZDHHC18 negatively regulates cGAS activation in human and mouse cell lines. Furthermore, Zdhhc18‐deficient mice are resistant to infection by DNA viruses, in agreement with the fact that Zdhhc18‐deficient macrophages upregulate antiviral gene expression under DNA stimulation. Taken together, our findings elucidate a new mechanism of cGAS regulation, which may imply a novel strategy for therapeutic biotechnology development.
Results
cGAS is palmitoylated at C474
Because palmitoylation is an important PTM involved in the regulation of protein function, we speculated that cGAS was modified by palmitoylation. To verify our hypothesis, we first detected cGAS palmitoylation using an acyl resin‐assisted capture (acyl‐RAC) assay in which the free cysteine thiol groups of overexpressed cGAS were irreversibly blocked by N‐ethylmaleimide (NEM) and the acylated cysteines were then exposed to hydroxylamine; thus, the protein could undergo affinity capture by thiol‐Sepharose beads (Appendix Fig S1A). As shown in Fig 1A, a cGAS band was present in the hydroxylamine addition group, indicating that cGAS was modified by palmitoylation. Given that 2‐bromopalmitate (2‐BP) reportedly effectively inhibits protein palmitoylation (Draper & Smith, 2009), we added 2‐BP to the cells in acyl‐RAC assays to confirm cGAS palmitoylation. The results showed that there was a dramatic reduction in the level of the band pulled down by thiol‐Sepharose beads, indicating that cGAS acylation was indeed due to reversible palmitoylation.
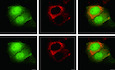
Figure 1. Palmitoylation of cGAS suppresses its activation.
-
AAcyl‐RAC assay of HEK293T cells transfected with the indicated plasmids for 24 h.
-
BAcyl‐RAC assay of HEK293T cells transfected with the indicated plasmids for 24 h.
-
CClick chemistry was applied to detect endogenous cGAS palmitoylation in RAW264.7 cells.
-
D, EIn vitro palmitoylation assay for the indicated proteins. NBD‐Palm‐CoA: (N‐[(7‐nitro‐2‐1,3‐benzoxadiazol‐4‐yl)‐methyl] amino) palmitoyl‐CoA. The recombinant cGAS protein used in the assay was indicated by Coomassie blue staining.
-
FLC‐MS/MS analysis of palmitoylated peptides of cGAS.
-
GAcyl‐RAC assay of HEK293T cells transfected with the indicated plasmids for 24 h.
-
H, ITHP‐1 cells were treated with DMSO or 2‐BP (50 μM) (H) or palmitic acid (100 μM) (I) for 12 h. Six hours after transfection with HT‐DNA (2 μg/ml), cGAMP was extracted and quantified by cGAMP ELISA.
-
JTHP‐1 cells were treated with palmitic acid (0, 50, or 100 μM) for 12 h before a cGAMP bioassay.
-
K–NBMDMs were treated with palmitic acid (0, 100, or 200 μM) for 12 h and transfected with HT‐DNA (2 μg/ml) for 6 h before RT–qPCR analysis of Ifna4 (K), Ifnb1 (L), Cxcl10 (M), and Rantes (N) expression.
-
OL929 cells were treated with BSA and palmitic acid (100 μM) and transfected with HT‐DNA (2 μg/ml) for the indicated times before immunoblotting analysis with the indicated antibodies.
Data information: 17‐ODYA, 17‐octadecanoic acid; 2‐BP, 2‐bromopalmitate; C, C‐terminal domain; EtOH, ethanol; FL, full length; HAM, hydroxylamine; N, N‐terminal domain; PA, palmitic acid. Data are representative of at least two independent experiments. Mean ± SEM from triplicates of technical replicates, unpaired t‐test; ns, not significant; **P < 0.005; ***P < 0.001 (H–N).
Source data are available online for this figure.
cGAS contains three major domains: an N‐terminal domain (1–160 aa), a nucleotidyltransferase domain (161–383 aa) and a Mab‐21 domain (212–522 aa). The carboxy (C)‐terminal domain (161–522 aa) is essential for enzymatic activity, while the N‐terminal domain is important for the formation of phase condensates with DNA (Du & Chen, 2018). To determine which cGAS domain is important for its palmitoylation, we employed the two truncated forms of cGAS, with deletions of the N‐ and C‐terminal domains, in acyl‐RAC assays and found that the palmitoylated band disappeared for both the N‐ and C‐terminal truncated cGAS mutants, indicating that only full‐length cGAS can be palmitoylated (Fig 1B). Moreover, through a click chemistry assay, we found that the palmitoylation level of endogenous cGAS was elevated with the addition of herring testis DNA (HT‐DNA), a long double‐stranded DNA widely used to activate cGAS (Unterholzner, 2013; Fig 1C). To further study the palmitoylation properties of cGAS, we conducted a previously described biophysical palmitoylation assay (Rana et al, 2018) by adding nitrobenzoxadiazolyl (NBD)‐linked palmitoyl‐coenzyme A (CoA) into a solution containing purified recombinant cGAS and found that the NBD fluorescence signal increased over time (Fig 1D). This result suggested that cGAS can bind spontaneously to palmitoyl‐CoA in vitro even in the absence of an acyltransferase. In addition, with the addition of HT‐DNA, the NBD fluorescence signal decreased, indicating that HT‐DNA decreased the binding ability of palmitoyl‐CoA to cGAS. In line with this result, when using the C‐terminal truncated cGAS mutant, which lacks the N‐terminal region responsible for DNA binding, the NBD fluorescence signal did not change in the presence of HT‐DNA, indicating that HT‐DNA regulated cGAS binding to palmitoyl‐CoA through the N terminus (Fig 1E). Moreover, to determine whether the downstream element, for example, STING, was important in cGAS palmitoylation, we overexpressed STING in acyl‐RAC assays and found no dramatical change in cGAS palmitoylation (Appendix Fig S1B and C). Simultaneously, we detected the role of cGAMP by adding cGAMP into cells or using the cGAS C396S mutant, which failed to produce cGAMP, in acyl‐RAC assays. The results showed that cGAS palmitoylation level showed little difference in these conditions, indicating that cGAS palmitoylation is not dependent on cGAMP or STING (Appendix Fig S1B and C).
To further identify which cysteine residue on cGAS is modified by palmitoylation, we performed liquid chromatography‐mass spectrometry analysis. As shown in Fig 1F, we identified C474 as a cGAS palmitoylation site with high confidence. To confirm the palmitoylation site identified from LC‐MS/MS, we employed several systematically engineered mutants in acyl‐RAC assays. We found that compared to wild‐type (WT) cGAS, the C405S, C409S, and C474S mutants had drastically reduced palmitoylation signals (Fig 1G). Since sites C405 and C409 localized toward the inner side of the protein, these two sites were hardly palmitoylated structurally (Appendix Fig S1D–F). The reduced palmitoylation signal of C405S and C409S could be caused by a conformational change, further confirming that the palmitoylated residue of cGAS is C474, which is easily structurally modified (Appendix Fig S1D–F). Taken together, our data suggest that cGAS is palmitoylated at C474.
Palmitoylation of cGAS restricts its enzymatic activity
Next, we assessed whether cGAS function was regulated by palmitoylation. To do so, we measured the level of cGAMP, the enzymatic product of cGAS that is essential for downstream signal transduction, after adding 2‐BP to THP‐1 cells to block cGAS palmitoylation. As shown by the results of an enzyme‐linked immunosorbent assay (ELISA), 2‐BP addition increased the cGAMP production by cGAS stimulated by HT‐DNA, indicating a negative role of palmitoylation on cGAS activation (Fig 1H). Given that palmitic acid reportedly promotes protein palmitoylation (Chamberlain & Shipston, 2015; Chen et al, 2017), we then focused on whether palmitic acid affected the activation of cGAS production of cGAMP. As shown by Fig 1I, addition of palmitic acid, which promoted cGAS palmitoylation, reduced the cGAMP production. Moreover, using a previously developed bioassay (Wu et al, 2013), we transfected THP‐1 cells with HT‐DNA to stimulate cGAMP production by cGAS. Cell lysates were digested with DNase and boiled to remove DNA and proteins, and the supernatant containing cGAMP was added to RAW264.7 cells with digitonin, followed by measurement of Ifn‐β expression, which indirectly represents the cGAMP level. As shown in Fig 1J, the mRNA level of Ifn‐β in RAW264.7 cells increased by approximately 60,000‐fold with the addition of THP‐1 cell lysates, indicating that cGAS was activated to produce a large amount of cGAMP. However, palmitic acid addition significantly reduced Ifn‐β expression in a dose‐dependent manner, suggesting that palmitic acid restricted cGAS activity and cGAMP production (Fig 1J). Next, we sought to clarify the regulatory effects of cGAS palmitoylation in primary cells by adding palmitic acid to mouse bone marrow‐derived macrophages (BMDMs). After transfection with HT‐DNA, the expression of antiviral genes, including Ifna, Ifnb, Rantes, and Cxcl10, in BMDMs was dramatically induced, while their expression was decreased in palmitic acid‐treated cells compared to that in control cells, indicating that promoting cGAS palmitoylation negatively regulates the innate immune response (Fig 1K–N). The same results were obtained when we used mouse L929 cells (Appendix Fig S1G–I). Consistently, palmitic acid addition inhibited the cGAS/STING‐mediated induction of IFN‐β promoter expression in a reporter assay (Appendix Fig S1J). Since STING phosphorylation is an important event in cGAS/STING signal transduction, we assessed whether palmitic acid affected the phosphorylation level of STING. As shown in Fig 1O, STING was apparently phosphorylated 9 and 12 h after transfection of HT‐DNA, whereas palmitic acid addition markedly slowed STING phosphorylation, confirming the negative role of palmitic acid in cGAS/STING signal transduction. Taken together, our findings suggest that palmitoylation suppresses cGAS activation.
ZDHHC18 is a major acyltransferase for cGAS palmitoylation
Protein palmitoylation is catalyzed by DHHC‐palmitoyl transferases, which include 23 family members. To identify which palmitoyl transferase is important for cGAS palmitoylation, we first examined the relative expression of 23 ZDHHC protein S‐acyl transferases in human HEK293T and THP‐1 cells and found that several proteins, including ZDHHC3, ZDHHC5, ZDHHC6, ZDHHC7, ZDHHC9, ZDHHC12, ZDHHC13, ZDHHC16, ZDHHC17, and ZDHHC18, were highly expressed in both HEK293T and THP‐1 cells (Appendix Fig S2A and B). To determine which one is important for cGAS palmitoylation, we adopted short heparin RNA (shRNA)‐mediated knockdown followed by an acyl‐RAC assay in HEK293T cells. The mRNA levels of the individual ZDHHC family proteins were downregulated after shRNA transfection (Appendix Fig S2C), among which ZDHHC18 knockdown significantly abrogated the cGAS palmitoylation signal, indicating that ZDHHC18 is important for cGAS palmitoylation (Fig 2A and B). Furthermore, to confirm the role of ZDHHC18 in cGAS palmitoylation, we co‐overexpressed ZDHHC18 and cGAS in the acyl‐RAC assay and observed an increase in the cGAS palmitoylation signal, indicating that ZDHHC18 promotes cGAS palmitoylation in mammalian cells (Fig 2C). Given that cGAS is palmitoylated at C474, we next sought to investigate whether ZDHHC18 affects cGAS function through this palmitoylation site. Interestingly, as shown in the results of an IFN‐β promoter reporter assay, we found that the cGAS C474S mutant showed no difference from the WT control with respect to the induction of IFN‐β promoter expression when coexpressed with STING in HEK293T cells. However, with ZDHHC18 expression, WT cGAS‐induced IFN‐β expression was dramatically reduced, whereas expression of the cGAS C474S mutant rescued the inhibitory effects of ZDHHC18 on WT cGAS, suggesting that ZDHHC18 specifically regulates cGAS at C474 (Fig 2D). Taken together, these results indicate that ZDHHC18 is a major acyltransferase mediating cGAS palmitoylation.
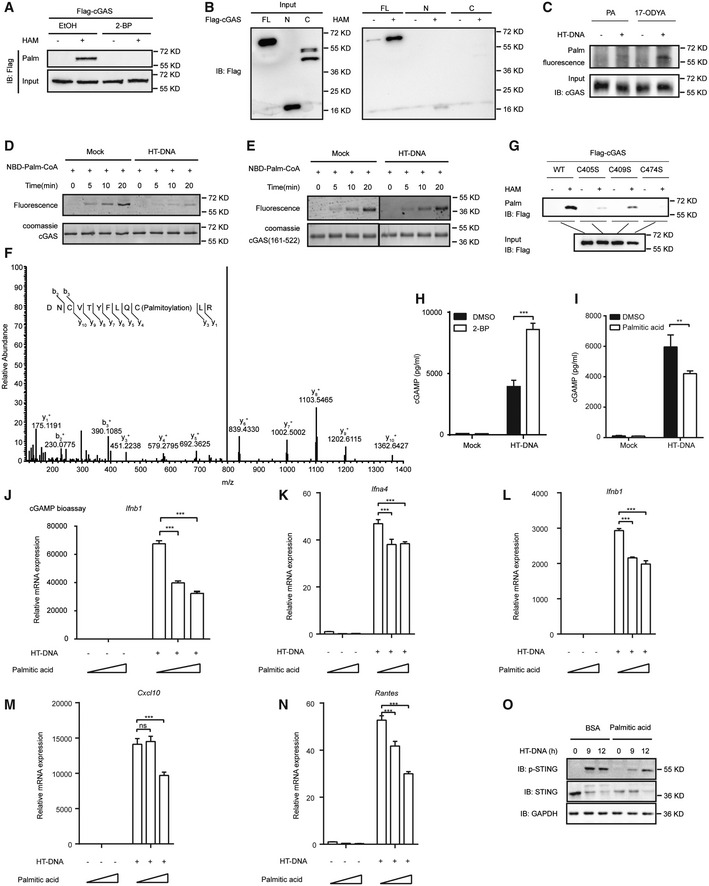
Figure 2. ZDHHC18 is a major acyltransferase catalyzing cGAS palmitoylation.
-
AAcyl‐RAC assay of HEK293T cells transfected with Flag‐cGAS 24 h after transfection with the indicated shRNA.
-
BQuantification of palmitoylation levels of Flag‐cGAS in (A).
-
CAcyl‐RAC assay of HEK293T cells transfected with the indicated plasmids for 24 h. HAM: hydroxylamine.
-
DHEK293T cells were transfected with HA‐cGAS (WT or C474S mutant) (200 ng), Flag‐STING (200 ng), and Myc‐ZDHHC18 (200 ng) expression plasmids for 24 h before luciferase reporter assays.
-
E, FImmunoprecipitation (with an anti‐Flag antibody) and immunoblot analysis of HEK293T cells transfected with the indicated plasmids for 24 h.
-
GSchematic diagrams of ZDHHC18 and its truncation mutants.
-
HImmunoprecipitation (with an anti‐Flag antibody) and immunoblot analysis of HEK293T cells transfected with plasmids encoding HA‐cGAS and Flag‐ZDHHC18 truncations or GFP for 24 h.
-
ISchematic diagrams of cGAS and its truncation mutants.
-
JImmunoprecipitation (with an anti‐Flag antibody) and immunoblot analysis of HEK293T cells transfected with plasmids encoding HA‐ZDHHC18 and Flag‐cGAS truncates or GFP for 24 h.
-
KImmunofluorescence analysis of HA‐cGAS (green) and Flag‐ZDHHC18 (red) in HT‐DNA‐stimulated (or not) HeLa cells. Scale bars: 7 μm.
-
LColocalization (merged volume of total cGAS signal) of cGAS and ZDHHC18 in (K).
-
M, NComparison of the binding free energy (M) and binding energy decomposition of a per‐residue to the binding affinity between ZDHHC18 and cGAS (N) in different complexes.
Data information: FL, full length; IP, immunoprecipitation; NC, negative control; TM, transmembrane domain; WCL, whole‐cell lysate. Data are representative of at least two independent experiments. Mean ± SEM from triplicates of technical replicates, unpaired t‐test; **P < 0.005; ***P < 0.001 (D and L).
Source data are available online for this figure.
ZDHHC18 interacts with cGAS
Given that ZDHHC18 promotes cGAS palmitoylation, we next investigated whether ZDHHC18 directly interacts with cGAS under physiological conditions. We conducted coimmunoprecipitation (co‐IP) experiments to examine whether cGAS physically interacts with ZDHHC18 in cultured mammalian cells. The results showed that epitope tagged ZDHHC18 reciprocally coimmunoprecipitated with cGAS in transfected HEK293T cells (Fig 2E and F).
Previous reports have shown that ZDHHC18 is a membrane‐associated protein (De & Sadhukhan, 2018). To further clarify the subcellular localization of ZDHHC18, we performed cell staining of endogenous ZDHHC18 in HeLa cells. The results showed that endogenous ZDHHC18 showed a dispersed pattern in cells (Appendix Fig S2D). Moreover, we engineered two truncated constructs: an M truncation referring to the middle region consisting of the four predicted transmembrane domains and the linker region and a ΔM truncation with a deletion of the middle region (Fig 2G). We detected the contribution of these truncated regions to the subcellular localization of ZDHHC18. Through confocal imaging, we found that the Pearson’s correlation coefficient of transiently overexpressed ZDHHC18 and the Golgi apparatus reached approximately 0.6, while this value was less than 0.05 between ZDHHC18 and the ER (Appendix Fig S2E and F). These data indicate that transiently overexpressed ZDHHC18 mainly localized at the Golgi apparatus but not the ER, which is consistent with previous reports (De & Sadhukhan, 2018). Moreover, the Pearson’s correlation coefficient of the ΔM truncation and Golgi apparatus decreased to less than 0.2, with no significant difference between the M truncation and WT control, suggesting that the membrane‐associated middle region is essential for ZDHHC18 localization to the Golgi apparatus (Appendix Fig S2E and F). Interestingly, we found that stably overexpressed ZDHHC18 overlapped with ER but not Golgi, indicating the expression level of ZDHHC18 might change its subcellular localization pattern (Appendix Fig S2G). Next, we sought to determine which domain of ZDHHC18 is important for its interactions with cGAS by employing these truncations in co‐IP experiments. As shown in Fig 2H, the middle region truncation showed a weak interaction with cGAS, while the nonmembrane region consisting of the N‐ and C‐terminal portions of ZDHHC18 was predominantly responsible for its association with cGAS.
To clarify which domain is important for cGAS binding to ZDHHC18, we employed the previously described truncations in co‐IP experiments (Fig 2I). As shown in Fig 2J, deletion of the C‐terminal domain of cGAS completely abolished the ZDHHC18‐cGAS interaction, whereas the enzyme core of cGAS with the N‐terminus deleted still interacted with ZDHHC18, indicating that the C‐terminal domain is crucial for the cGAS‐ZDHHC18 interaction.
In line with this result, through immunofluorescence assays, we found that membrane‐associated ZDHHC18 colocalized with cGAS, which was indicated by the superimposition accounting for 10% of the total cGAS signal (Fig 2K and L). Moreover, the superimposition ratio increased to approximately 25% in the presence of HT‐DNA, suggesting that the interaction between cGAS and ZDHHC18 was enhanced in the DNA‐bound activated state (Fig 2K and L). The same results were obtained when we overexpressed ZDHHC18 and endogenous cGAS (Appendix Fig S2H).
To confirm this result, molecular dynamics (MD) simulations were performed on human ZDHHC18/cGAS/DNA and ZDHHC18/cGAS complexes to detect the effect of DNA on the interaction between ZDHHC18 and cCAS (Appendix Fig S2I–L). The simulation results showed that DNA caused the binding energy between ZDHHC18 and cCAS to drop from −33.88 kcal/mol to H −48.38 kcal/mol and increased the contribution of residues to the binding energy (Fig 2M and N), insinuating that the presence of DNA enhances the interaction of the two proteins. Then, the structures of cGAS in different complexes were compared to further analyze the molecular mechanism of DNA affecting the interaction between ZDHHC18 and cCAS (Appendix Fig S2M). The comparison results indicated that DNA as a scaffold led to the cGAS protein being more stable and expanded, which may enhance the binding effect between ZDHHC18 and cGAS. In addition, compared to the absence of DNA, the relative position of the residues of cGAS protein involved in the interaction with DNA exhibited an obvious difference in the presence of DNA, which was consistent with the results of the conformational change of cGAS protein caused by DNA (Appendix Fig S2N–Q). Collectively, these data suggest that cGAS interacts with ZDHHC18 and that DNA promotes their interactions.
Palmitoylation does not affect the subcellular localization of cGAS
Palmitoylation is mostly targeted to membrane‐associated proteins to regulate their subcellular localization. To determine whether palmitoylation affects the subcellular membrane association of cGAS, we constructed a HeLa cell line stably overexpressing GFP‐tagged cGAS to detect its subcellular localization. cGAS was predominantly localized in the nucleus but formed puncta in the cytoplasm when the cells were transfected with double‐stranded DNA (Fig 3A), in agreement with previously reported results showing that cGAS exhibits liquid‐liquid phase separation (LLPS) with DNA (Du & Chen, 2018). Most of the cGAS signal was still localized in the nucleus after treatment with 2‐BP, indicating that palmitoylation did not affect the cGAS distribution between the cytoplasm and nucleus. Although cGAS has been reported to be localized at the plasma membrane, we did not observe this phenomenon (Fig 3A). Moreover, we examined the association of cGAS with membrane‐associated organelles. cGAS showed no colocalization with calnexin, GM130, or EEA1 in either 2‐BP‐treated or untreated cells, indicating that cGAS did not localize to the ER, Golgi apparatus, or early endosomes (Fig 3A). Taken together, our data suggest that palmitoylation does not affect the subcellular localization of cGAS.
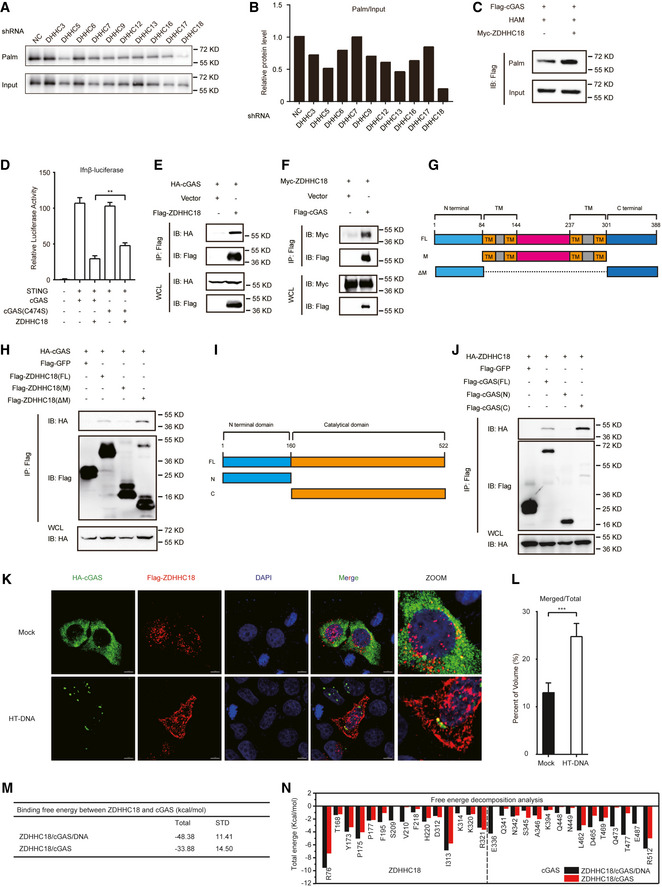
Figure 3. Palmitoylation inhibits DNA binding of cGAS.
-
AImmunofluorescence analysis of GFP‐cGAS, the nucleus (DAPI), the ER (calnexin), the Golgi apparatus (GM130), and early endosomes (EEA1) in HeLa cells. 2‐BP, 2‐bromopalmitate; ISD, IFN‐stimulatory DNA. Arrows indicate cGAS puncta. Scale bars: 10 μm.
-
B, CAtomic MD simulations of the human cGAS‐DNA 2:2 complex in the resting and palmitoylation states. Comparison of the relative positions of S329‐S345, F433‐P450, and E478‐S493 in the WT and palmitoylated cGAS proteins.
-
D, EThe binding modes of the human cGAS‐DNA 2:2 complex in the resting and palmitoylation states. Monomer A, monomer B, and DNA‐1 in the human cGAS‐DNA 2:2 complex in the resting state are shown with pale cyan, light pink, and cyan cartoons, respectively, while these components in the human cGAS‐DNA 2:2 complex in the palmitoylation state are exhibited with light blue, yellow and slate cartoons, respectively. The C474 and S‐palmitoylated C474 residues are highlighted by ruby sticks, while the dominant residues in the human cGAS‐DNA 2:2 complex in both the resting and palmitoylation states are shown in sticks.
-
F, GHEK293T cells were transfected with the indicated plasmids. Biotin‐conjugated ISD was transfected into cells 6 h before harvesting. Lysates were coprecipitated with streptavidin beads and then assessed by immunoblot analysis as shown.
-
HConfocal microscopy of BMDMs (Zdhhc18 +/+ or Zdhhc18 −/−) transfected with FITC‐conjugated ISD for 6 h (left). Scale bar: 20 μm.
Data information: IP, immunoprecipitation; WCL, whole‐cell lysate. Data are representative of at least two independent experiments.
Source data are available online for this figure.
Palmitoylation inhibits DNA binding to cGAS
To further analyze the effect of palmitoylation of cGAS, we performed MD simulations to dissect the molecular basis of cGAS function regulation by palmitoylation. In previous reports, the formation of a 2:2 complex with DNA was shown to be important for cGAS activation (Zhang et al, 2014). Although the structure of the mouse cGAS‐DNA 2:2 complex and the human cGAS‐DNA 1:1 complex have been reported, the conformation of the human cGAS‐DNA 2:2 complex has not been released (Kranzusch et al, 2013, 2014). Therefore, we first used the mouse cGAS‐DNA 2:2 complex as the template to construct the human cGAS‐DNA 2:2 complex based on the human cGAS‐DNA 1:1 complex (Appendix Fig S3A and B). Subsequently, the palmitic acid chain was introduced at C474 to obtain a palmitoylated human cGAS‐DNA 2:2 complex (Appendix Fig S3C–F). Then, MD simulations were performed for resting and palmitoylated cGAS. The results of the MD simulations showed that the palmitic acid tail stretched into the inner space of cGAS. Residue E481 interacted with K479, whereas palmitoylation on C474 caused rotation of the E481 side chain, resulting in the interaction of E481 with R339 and H444 but not K479 (Fig 3B and C). The newly formed interactions of E481 and R339 with H444 caused further tightening of the secondary structure in the S329‐L354, F433‐P450, and E478‐S493 regions (Appendix Fig S3G–K). Among these regions, S329‐L354 is important for cGAS monomer binding with DNA, and the palmitoylation‐mediated secondary structure tightening of cGAS was accompanied by weakening of the binding between DNA and several residues in cGAS, including R236, R255, K327, S329, K347, R349, K350, R353, and L354 (Fig 3D and E). In summary, MD analysis suggested that palmitoylation weakens the binding affinity between DNA and cGAS by enhancing the interactions of residues S329‐L354, F433‐P450, and E478‐S493.
To further validate the MD analysis results, we sought to determine how ZDHHC18‐mediated palmitoylation affects DNA binding to cGAS. We first synthesized 45‐bp nucleotides known as IFN‐stimulatory DNA (ISD), a commonly used cGAS stimulator (Sun et al, 2013), and performed an ISD pull‐down assay in HEK293T cells. After overexpressing HA epitope‐tagged cGAS, the cells were lysed and treated with biotin‐labeled ISD, followed by the addition of streptavidin agarose beads to capture biotin‐labeled ISD. As shown in Fig 3F, a cGAS band was present in the group treated with biotin‐labeled ISD, in contrast to the group without biotin‐ISD, indicating the formation of an ISD‐cGAS complex. Following ZDHHC18 overexpression, the cGAS band progressively disappeared in a dose‐dependent manner, suggesting that ZDHHC18 abrogated cGAS‐DNA complex formation (Fig 3F). To further determine whether the inhibitory functions of ZDHHC18 were relevant to cGAS palmitoylation, we constructed a cysteine‐to‐serine substitution in the DHHC motif of ZDHHC18 (subsequently referred to as ZDHHC18(CS)). We employed both WT and ZDHHC18(CS) expression constructs in the ISD pull‐down assay. As shown in Fig 3G, compared to the WT control, overexpressed ZDHHC18(CS) markedly rescued the inhibitory effects of the cGAS‐ISD complex (Fig 3G), indicating that ZDHHC18‐mediated palmitoylation plays an essential role in attenuating the binding affinity of cGAS to double‐stranded DNA. To further confirm this observation, we attempted to assess the affinity of ISD and endogenous cGAS in mouse primary BMDMs by immunostaining. To do so, ISD was labeled with FITC and transfected into BMDMs, followed by observation by confocal microscopy. FITC‐ISD aggregated into puncta with cGAS, in agreement with a previous report that cGAS undergoes LLPS with DNA (Du & Chen, 2018). The size and quantity were notably greater in ZDHHC18‐deficient BMDMs than in WT control cells under the same conditions, suggesting that ZDHHC18 antagonizes the formation of the double‐stranded DNA‐endogenous cGAS complex (Fig 3H). Given that ZDHHC18 interacted with cGAS through its nonmembrane fragments, we then investigated whether these regions are important for ZDHHC18 inhibition of cGAS‐DNA complex formation. We employed both the M and ΔM truncations along with full‐length ZDHHC18 in ISD pull‐down assays and found that neither truncation was sufficient to mimic the effects of full‐length ZDHHC18 on the inhibition of the ISD‐cGAS complex (Appendix Fig S3L). A simple explanation for this result is that the nonmembrane region is also crucial for ZDHHC18 binding to cGAS, perhaps by pulling it close to the membrane‐associated core region of ZDHHC18 for palmitoylation, which conversely inhibits DNA binding to cGAS. Collectively, these findings suggest that ZDHHC18‐mediated palmitoylation inhibits DNA binding to cGAS.
ZDHHC18‐mediated palmitoylation reduces cGAS dimerization
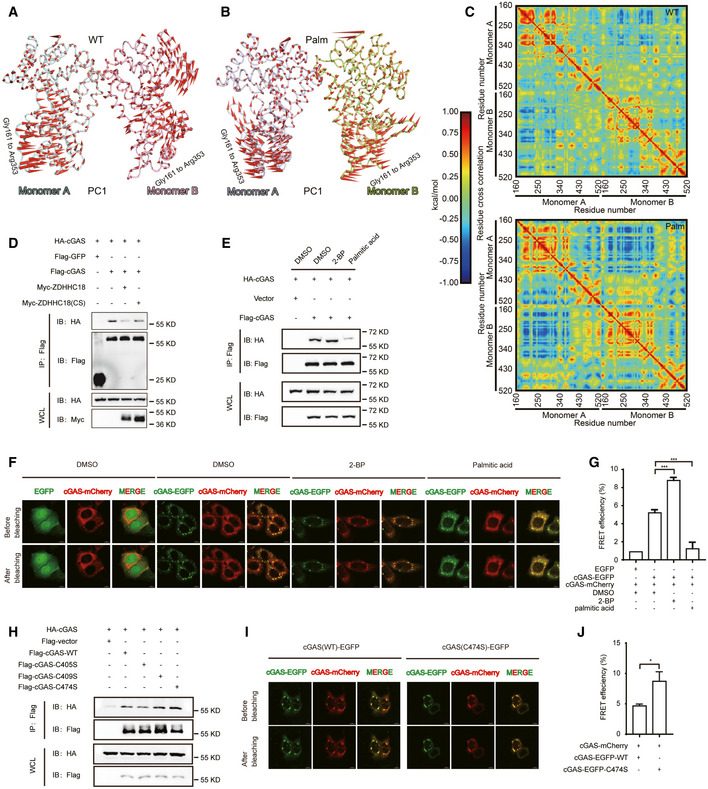
cGAS binds with DNA in a 2:2 complex. Our MD analysis results indicated that the distance between the two monomers was increased under palmitoylation conditions (Appendix Fig S4A). Based on this observation, we speculated that palmitoylation affected cGAS dimerization. To test this hypothesis, principal component analysis (PCA) and residue cross‐correlation analysis were performed to detect the dynamic motions of cGAS proteins during the simulations. Overall, the two cGAS monomers seemed to move in opposite directions in the palmitoylated state (Fig 4A–C), indicating that palmitoylation antagonized the association between the two cGAS monomers in the cGAS‐DNA 2:2 complex. Moreover, the radius of gyration also increased after palmitoylation, suggesting that palmitoylation leads to a less compact state of the cGAS dimer (Appendix Fig S4B). Next, we sought to determine whether palmitoylation affected the binding free energy of cGAS monomers. The dominant residues that contribute to the binding free energy between two cGAS‐DNA complexes are R236, R255, K327, S329, K347, R349, K350, R353, L354, K392, S393, and K394 (Appendix Fig S4C and D). Notably, the contribution of these residues to the binding affinity in the palmitoylated state was significantly lower than that in the resting state. In line with this result, the total binding free energy of the two cGAS monomers was also reduced after palmitoylation (Appendix Fig S4E). Taken together, the MD analysis results reveal that cGAS dimerization is inhibited by palmitoylation.
Figure 4. Palmitoylation dampens cGAS dimerization.
-
A, BView of the first normal modes of monomer A and monomer B in the human cGAS‐DNA 2:2 complex in the resting and palmitoylation states during the simulation. The colors of monomer A and monomer B in (A) and (B) are consistent with those in Fig 3D and E. PC1: the first principal components in PCA.
-
CCross‐correlated motions of the Cα atoms of the WT and palmitoylated cGAS proteins.
-
DImmunoprecipitation (with an anti‐Flag antibody) and immunoblot analysis of HEK293T cells transfected with plasmids encoding Flag‐cGAS, HA‐cGAS, and Myc‐ZDHHC18 (WT or CS mutant) for 24 h.
-
EImmunoprecipitation (with an anti‐Flag antibody) and immunoblot analysis of HEK293T cells transfected with the indicated plasmids followed by the addition of DMSO, 2‐BP (50 μM) or palmitic acid (100 μM).
-
FFRET assay of HeLa cells transfected with the indicated plasmids with the addition of DMSO, 2‐BP (50 μM) or palmitic acid (100 μM). Scale bar: 8 μm.
-
GFRET efficiency of cGAS‐GFP and cGAS‐mCherry in (F).
-
HImmunoprecipitation (with an anti‐Flag antibody) and immunoblot analysis of HEK293T cells transfected with Flag‐cGAS and HA‐cGAS (WT, C405S, C409S, and C474S) expression plasmids.
-
IFRET assay of HeLa cells transfected with the indicated plasmids. Scale bar: 8 μm.
-
JFRET efficiency of cGAS‐GFP and cGAS‐mCherry in (I).
Data information: 2‐BP, 2‐bromopalmitate; IP, immunoprecipitation; WCL, whole‐cell lysate. Data are representative of at least two independent experiments. Mean ± SEM from triplicates of technical replicates, unpaired t‐test; *P < 0.01; ***P < 0.001 (G and J).
Source data are available online for this figure.
To validate the simulation results, we next examined whether ZDHHC18 affects cGAS dimerization in cells. First, we performed co‐IP assays using Flag and HA epitope‐tagged cGAS in HEK293T cells. HA‐tagged cGAS was specifically present in the immunoprecipitates of Flag‐tagged cGAS, implying cGAS dimerization (Fig 4D and Appendix Fig S4F). In contrast, coexpression of ZDHHC18, but not its catalytically inactive mutant, markedly reduced the amount of HA‐tagged cGAS, indicating that attenuation of cGAS dimerization by ZDHHC18 is dependent on the acyltransferase activity of ZDHHC18 (Fig 4D and Appendix Fig S4F). Moreover, we used palmitic acid and 2‐BP, which effectively promoted and inhibited cGAS palmitoylation, respectively, and performed an additional co‐IP assay. The addition of palmitic acid indeed reduced the association between the cGAS monomers, whereas 2‐BP enhanced this interaction, albeit at a low level (Fig 4E). To further confirm these observations, we performed a Förster resonance energy transfer (FRET) experiment to assess cGAS dimer formation. The FRET efficiency between GFP fusion cGAS and mCherry fusion cGAS was apparently higher than that of GFP, indicating the association of cGAS monomers (Fig 4F and G). Treatment with 2‐BP and palmitic acid increased and decreased the FRET efficiency, respectively, indicating a negative impact of palmitoylation on cGAS dimerization (Fig 4F and G). Furthermore, the C474S mutation of cGAS, which abolished cGAS palmitoylation, resulted in a stronger association affinity with another copy of the cGAS monomer than that of the WT control (Fig 4H). In addition, C474S mutation or ZDHHC18 knockdown using shRNA promoted FRET efficiency between cGAS monomers (Fig 4I and J, and Appendix Fig S4G and H). Taken together, these findings suggest that ZDHHC18‐mediated palmitoylation of cGAS attenuates cGAS dimerization.
ZDHHC18 overexpression suppresses cGAS signaling
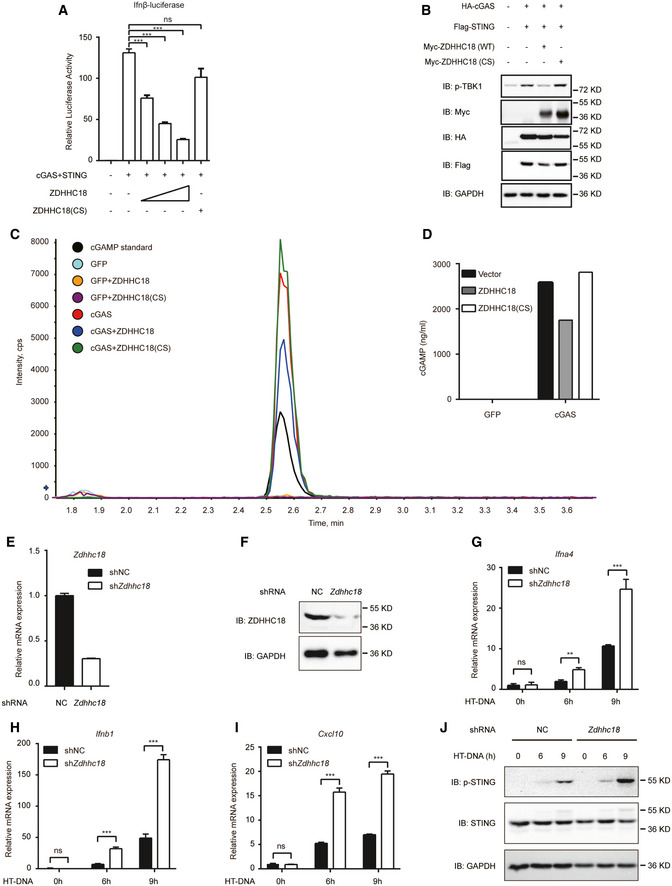
Next, we sought to determine the biological role of ZDHHC18 in cGAS activation. We first tested whether ZDHHC18 overexpression affects type I IFN promoter activation in a dose‐dependent manner. As shown in Fig 5A, cGAS and STING coexpression triggered IFN‐β promoter activation in transfected HEK293T cells. In contrast, ZDHHC18 expression significantly reduced the cGAS/STING‐mediated IFN‐β luciferase activity in a dose‐dependent manner, whereas ZDHHC18(CS) rescued the negative phenotype of ZDHHC18, indicating that ZDHHC18 suppresses cGAS function in an acyltransferase activity‐dependent manner (Fig 5A). Since TBK1 phosphorylation is an important event during cGAS/STING signal transduction that activates the expression of type I interferons, we next assessed whether ZDHHC18 overexpression affected TBK1 phosphorylation. As shown in Fig 5B, cGAS and STING overexpression in HEK293T cells markedly induced TBK1 phosphorylation, whereas coexpression of ZDHHC18 reduced IRF3 phosphorylation levels, which suggested the negative role of ZDHHC18 in cGAS signaling. When we coexpressed the ZDHHC18 (CS) mutant, the negative effect of ZDHHC18 recovered, further confirming that the enzymatic activity of ZDHHC18 was important in regulating cGAS/STING signaling (Fig 5B).
Figure 5. ZDHHC18 negatively regulates cGAS activation.
-
AHEK293T cells (5 × 105) were transfected with HA‐cGAS (200 ng), Flag‐STING (200 ng), and Myc‐ZDHHC18 (WT or CS mutant) expression plasmids (0, 50, 100, or 200 ng) for 24 h before luciferase reporter assays were conducted.
-
BHEK293T cells (1 × 106) were transfected with HA‐cGAS (500 ng), Flag‐STING (500 ng), and Myc‐ZDHHC18 (500 ng) expression plasmids for 24 h. TBK1 phosphorylation was detected by immunoblotting.
-
C, DHEK293T cells were transfected with the indicated plasmids, and the amount of cGAMP in the lysates was quantified by LC‐MS/MS.
-
E–IL929 cells stably transfected with control shRNA or ZDHHC18 shRNA were transfected with HT‐DNA (2 μg/ml) for the indicated times before RT–qPCR analysis of Ifna4 (G), IFNb1 (H), and Cxcl10 (I) expression. The knockdown efficiency of ZDHHC18 is shown by RT‐qPCR (E) and Western blotting (F) results.
-
JL929 cells stably transfected with control shRNA or ZDHHC18 shRNA were transfected with HT‐DNA (2 μg/ml) for the indicated times before immunoblotting analysis with the indicated antibodies.
Data information: HT‐DNA, herring testis DNA; LC‐MS/MS, liquid chromatography‐tandem mass spectrometry; NC, negative control; ZDHHC18(CS), a catalytic mutant with a cysteine‐to‐serine substitution in the DHHC motif of ZDHHC18. All data are representative of at least two independent experiments. Mean ± SEM from triplicates of technical replicates, unpaired t‐test; ns, not significant; **P < 0.01; ***P < 0.001 (A, E, G–I).
Source data are available online for this figure.
Furthermore, we dissected the direct influence of ZDHHC18 on cGAS activation in mammalian cells. As shown in Fig 5C and D, cGAS overexpression in HEK293T cells resulted in the production of a large amount of cGAMP, as measured by LC‐MS/MS, whereas coexpression of ZDHHC18 reduced the amount of cGAMP produced. Consistent with the results from the IFN‐β reporter assay, ZDHHC18(CS) rescued the influence of ZDHHC18 on cGAS, confirming that ZDHHC18 regulates the function of cGAS through its palmitoylation. Collectively, our data suggested that ZDHHC18 overexpression suppresses cGAS signaling.
ZDHHC18 knockdown reinforces cGAS activation
To better understand the function of ZDHHC18, we determined whether knockdown of endogenous ZDHHC18 affects cGAS function. First, we used small interfering RNA (siRNA) oligonucleotides to knock down ZDHHC18 expression in THP‐1 cells. The siRNA oligonucleotides were a cocktail of three different siRNAs designed to specifically target nonoverlapping regions in the coding frame of ZDHHC18. The siRNA oligonucleotides efficiently reduced the expression of endogenous ZDHHC18 in THP‐1 cells (Appendix Fig S5A). We assessed the effects of ZDHHC18 knockdown on innate immune signaling stimulated by ISD. As shown in the Appendix Fig S5B, ISD‐induced IFN‐β transcription was strongly increased after ZDHHC18 knockdown. To confirm this result, we knocked down endogenous ZDHHC18 in mouse L929 cells using lentivirus‐coupled shRNA. Both the mRNA and protein levels of ZDHHC18 were apparently reduced after infection with the shRNA‐lentivirus (Fig 5E and F). The transcription levels of antiviral genes, such as Ifn‐α, Ifn‐β, and Cxcl10, induced by HT‐DNA were significantly increased in ZDHHC18 knockdown L929 cells (Fig 5G–I). Moreover, cGAMP production after ZDHHC18 knockdown also increased, indicating the direct influence of endogenous ZDHHC18 on cGAS activation (Appendix Fig S5C). Next, we assessed whether ZDHHC18 affects STING phosphorylation. As shown in Fig 5J, after ZDHHC18 knockdown, the phosphorylation level of STING dramatically increased, indicating that ZDHHC18 played a negative role in the activation of the cGAS/STING pathway. To determine whether ZDHHC18 is specific for cGAS, we used Sendai virus (SeV), R848, and lipopolysaccharide (LPS) to activate the RIG‐I, TLR7/8, and TLR4 signaling pathways in ZDHHC18 knockout mouse embryo fibroblasts (MEFs), respectively. The data showed that ZDHHC18 did not affect RIG‐I, TLR7/8 or TLR4 activation (Appendix Fig S5D and E). Collectively, our results demonstrate that ZDHHC18 knockdown reinforces cGAS signaling.
ZDHHC18 deficiency promotes antiviral activity against DNA viruses in vivo
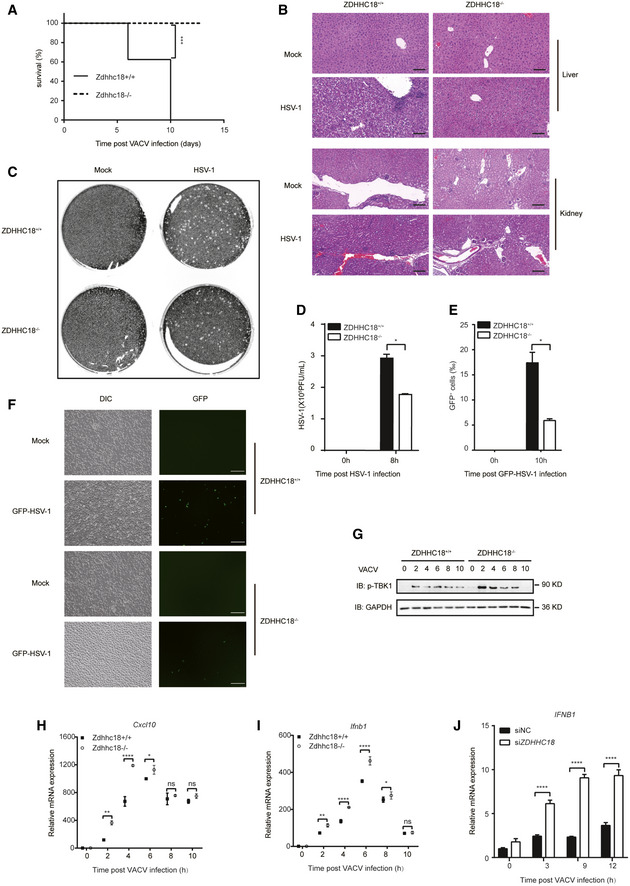
To further understand the biological importance of ZDHHC18‐mediated cGAS palmitoylation in antiviral signaling, we investigated the phenotype in Zdhhc18‐deficient mice created by CRISPR/Cas9‐mediated genome editing (Appendix Fig S6A). Zdhhc18‐deficient mice were born at a Mendelian ratio, developed normally compared to their WT littermates, and were fertile. To examine the role of ZDHHC18 in the regulation of immune defense against DNA virus infection in vivo, we infected Zdhhc18 +/+ and Zdhhc18 −/− mice with VACV intranasally and monitored their survival. We found that mice deficient in Zdhhc18 exhibited prolonged survival and were less vulnerable to VACV infection than their WT littermates (Fig 6A). Moreover, we infected Zdhhc18 −/− mice with HSV‐1. WT mice displayed hepatocyte swelling, extensive hydropic degeneration and obvious inflammatory cell infiltration in the liver, glomerular edema and increased neutrophil counts in the kidney, an enlarged germinal center of the splenic corpuscle and increased lymphocyte counts in the spleen, indicating pathological inflammation. However, all these pathological phenotypes were alleviated in Zdhhc18 −/− mouse organs (Fig 6B and Appendix Fig S6B).
Figure 6. ZDHHC18 deficiency promotes antiviral activity against DNA viruses in mice.
-
ASurvival analysis of Zdhhc18 +/+ and Zdhhc18 −/− mice (n = 8 each, 6–8 weeks old) intranasally injected with VACV (5 × 107 PFU per mouse) and monitored daily for survival for 13 days using Kaplan–Meier curves and a log‐rank (Mantel–Cox) test. ***P < 0.001.
-
BRepresentative photomicrographs of H&E‐stained liver and kidney tissue sections from mice (Zdhhc18 +/+ or Zdhhc18 −/−) intravenously injected with HSV‐1‐FS (5 × 107 PFU per mouse) or not after 2 days. Scale bars: 100 μm (liver), 200 μm (kidney).
-
C, DBMDMs (Zdhhc18 +/+ or Zdhhc18 −/−) were infected with HSV‐1 (MOI = 2) for 8 h, and viral titers in the supernatants were quantified by plaque assay.
-
E, FMicroscopy of BMDMs (Zdhhc18 +/+ or Zdhhc18 −/−) infected with GFP‐HSV‐1 (MOI = 1) for 10 h. Scale bar: 200 μm. Frequency of GFP+ cells among infected cells (E).
-
GMEFs (Zdhhc18 +/+ or Zdhhc18 −/−) were infected with VACV (1:200) for the indicated times before immunoblot analysis.
-
H, IMEFs (Zdhhc18 +/+ or Zdhhc18 −/−) were infected with VACV (1:200) for the indicated times before RT–qPCR analysis of Cxcl10 (H) and Ifnb1 (I) expression.
-
JPBMCs from healthy people were transfected with scrambled siRNA or ZDHHC18 siRNA (cocktail) for 48 h and then infected with VACV (1:200) for the indicated times before RT–qPCR analysis of IFNB1 expression.
Data information: PFU: plaque‐forming units; H&E: hematoxylin–eosin. Data are representative of at least two independent experiments. Mean ± SEM from triplicates of technical replicates, unpaired t‐test; Ns, no significance; *P < 0.05; **P < 0.01; ****P < 0.0001 (D, E, H–J).
Source data are available online for this figure.
Next, we sought to assess the innate immune response against DNA virus infection in BMDMs from Zdhhc18 −/− mice. First, we infected BMDMs with HSV‐1 and detected virus proliferation post‐HSV‐1 infection with a plaque assay. As expected, the virus titers of the cell supernatants decreased in Zdhhc18 −/− BMDMs compared with WT BMDMs (Fig 6C and D). Similarly, in Zdhhc18 −/− BMDMs infected with GFP‐HSV‐1, GFP‐HSV‐1 replication was also restricted (Fig 6E and F). Furthermore, by studying the kinetics of TBK1 phosphorylation level and mRNA expression of Ifn‐β and Cxcl10 during VACV infection in MEFs, we found that at early phase of VACV infection (approximately 2–6 h post‐infection), the TBK1 phosphorylation and mRNA expression of Ifn‐β and Cxcl10 was dramatically high; at later phase of VACV infection (approximately 8–10 h post‐infection), the TBK1 phosphorylation level and mRNA expression of Ifn‐β and Cxcl10 came down (Fig 6G–I). After deletion of ZDHHC18, the TBK1 phosphorylation level and mRNA expression of Ifn‐β and Cxcl10 were upregulated at the early phase but showed no apparent difference at the late phase of VACV infection (Fig 6G–I). Next, we determined whether the expression of type I interferons was boosted in Zdhhc18 −/− BMDMs after DNA induction. We found that the mRNA levels of antiviral genes, including Ifn‐α, Ifn‐β, Cxcl10, and Il6, also increased significantly in Zdhhc18 −/− BMDMs (Appendix Fig S6C–F).
Since cGAS was reported to play roles in inflammatory responses in rheumatoid arthritis (RA) (Wang et al, 2015; Willemsen et al, 2021), we isolated peripheral blood mononuclear cells (PBMCs) from healthy people and rheumatoid arthritis (RA) patients and measured the expression of ZDHHC18 and cGAS. Both the mRNA levels of ZDHHC18 and cGAS were higher in RA patients, indicating the association between ZDHHC18 and cGAS in human cell systems (Appendix Fig S6G and H). Furthermore, to determine whether ZDHHC18 regulated cGAS‐mediated innate immune responses in human cell systems, we first used siRNA to knock down ZDHHC18 in PBMCs (Appendix Fig S6I). When infected with VACV, the IFN‐β expression level in the ZDHHC18 knockdown group was dramatically higher than that in the control group, indicating the negative role of ZDHHC18 in regulating cGAS activation in human cell systems (Fig 6J). Collectively, these results confirm the negative role of ZDHHC18 in regulating the innate immune response to DNA viruses in vivo.
Discussion
cGAS is a nonredundant cytosolic DNA sensor involved in signal transduction via the STING/TBK1/IRF3 axis to produce type I interferons and proinflammatory cytokines. Extensive studies have revealed the essential roles of cGAS in multiple biological processes, including pathogen invasion and autoimmune diseases. cGAS activity must be well regulated to maintain homeostasis, preventing both overinhibition, which leads to silenced innate immune responses and pathogen invasion, and overactivation, which may lead to autoimmune or chronic inflammatory diseases. However, it is still unclear how cGAS function is dynamically regulated in physiologically relevant environments.
As a PTM, palmitoylation usually occurs on membrane‐associated proteins to regulate their subcellular localization or conformational state. cGAS reportedly localizes in the cytoplasm or nucleus, with one exception: cGAS was observed to localize at the plasma membrane (Barnett et al, 2019). We confirmed the cytosolic and nuclear localization of cGAS, but not the plasma membrane. However, cGAS palmitoylation did not affect its subcellular localization. Through MD simulation and biochemical verification, we found that palmitoylation altered the interaction between specific amino acid residues and caused conformational changes, leading to inhibition of cGAS‐DNA binding and dimerization. The regulatory mechanism of palmitoylation on cGAS is similar to that for GPCRs (Chini & Parenti, 2009; Zheng et al, 2012), which implies palmitoylation as a common regulatory mechanism in conformational change. Furthermore, we identified that the palmitoylation site of cGAS was C474. Overall, the identification of cGAS as a palmitoylated protein expands the knowledge of the role of palmitoylation in the regulation of protein function.
ZDHHC18 is a DHHC‐palmitoyl transferase, but its substrate has not yet been well documented. A genome‐wide association study (GWAS) revealed its connection with schizophrenia, whereas other biological roles of ZDHHC18 are ambiguous (Zhao et al, 2018). Our data revealed that ZDHHC18 is a major palmitoyl transferase for cGAS. ZDHHC18‐mediated palmitoylation of cGAS appears when cGAS recognizes DNA. Moreover, the association between cGAS and ZDHHC18 was enhanced by DNA. Based on its inhibitory effect on binding with DNA and dimerization, cGAS palmitoylation may function as a brake in cGAS‐mediated immune signaling to prevent its overactivation. Therefore, this study indicated a fine‐tuned regulatory effect on cGAS activation by ZDHHC18‐mediated palmitoylation of cGAS. Aberrant expression of type I IFNs induced by cGAS activation is closely related to autoimmune diseases (Gao et al, 2015; Chen et al, 2016). For example, DNA repair exonuclease 1 (TREX1) is an exonuclease that digests DNA in the cytoplasm; mouse models show that Trex1 deletion leads to severe cGAS‐dependent autoimmunity, and loss‐of‐function TREX1 mutations are linked to autoimmune diseases in humans (Thomas et al, 2017; Gregg et al, 2019). Therefore, modulation of ZDHHC18‐mediated inhibition of cGAS signaling might also have great application potential in treating TREX1 mutation‐related diseases.
The C‐terminal domain is essential for cGAS to produce cGAMP. However, previous findings from reports on the function of the N‐terminal domain of cGAS are controversial. One group reported that the N‐terminal domain is essential for the formation of LLPS with DNA and the production of cGAMP (Du & Chen, 2018). Another group reported that cGAS is associated with the plasma membrane through the interaction of the N‐terminal domain and phosphoinositide as a result of distinguishing self‐DNA and invading viral DNA (Barnett et al, 2019). Inflammasome activation was found to induce caspase‐1 cleavage of cGAS at the N‐terminal domain to regulate responses to DNA virus infection (Wang et al, 2017b). Moreover, in the nucleus, cGAS was shown to be hyperphosphorylated at its N‐terminal domain to block chromatin sensing (Li et al, 2021). Beyond these reports, our data from acyl‐RAC assays showed that deletion of the N‐terminal domain abolished cGAS palmitoylation. An in vitro palmitoylation assay showed that the catalytic domain of cGAS is sufficient to bind palmitoyl‐CoA. Moreover, the addition of double‐stranded DNA blocked cGAS palmitoylation, which is dependent on the N‐terminal domain. Therefore, our data revealed a mechanism by which the N‐terminal domain regulates the balance between cGAS palmitoylation and DNA binding.
Our results reveal that endogenous cGAS is not palmitoylated in the resting state, while cGAS palmitoylation appears under stimulation by cytosolic double‐stranded DNA. This finding is in agreement with the observation that the association between cGAS and ZDHHC18 is increased in the presence of double‐stranded DNA. Through a kinetics study of DNA‐induced phosphorylation of TBK1 and expression of IFNb1 and Cxcl10 in wt and Zdhhc18 KO cells, we found that the signal transduction was higher in Zdhhc18 deficient cells as soon as cGAS was recognized and activated by DNA. Based on these results, we proposed an elaborate regulatory mechanism of why and how cGAS was palmitoylated: in the resting state, cGAS is not palmitoylated, which primes cGAS for rapid activation to initiate immune responses; as soon as cGAS recognizes and binds DNA, ZDHHC18 closely interacts with cGAS and promotes its palmitoylation, functioning as a brake to shut down cGAS signaling and prevent overactivation.
In summary, we identified cGAS as a palmitoylated protein. Palmitoylation on C474 suppresses cGAS activation. As a DHHC family protein, ZDHHC18 interacts with and promotes palmitoylation of cGAS. Mechanistically, cGAS palmitoylation alters the interactions between specific amino acid residues and causes a conformational change, leading to inhibition of cGAS DNA binding and dimerization. ZDHHC18 inhibits cGAS‐mediated signal transduction. In a more biologically relevant model system, Zdhhc18‐deficient mice were resistant to HSV‐1 infection, in agreement with the observation that Zdhhc18 depletion enhanced innate immune responses under DNA stimulation in macrophages. Therefore, our findings identified palmitoylation as a new PTM of cGAS and a new inhibitory mechanism of the innate immune response. This new understanding of cGAS regulation might provide potential targets for drug development against viral infections and autoimmune diseases.
Materials and Methods
The materials used are listed in Appendix Table S1.
Mice
Zdhhc18 −/− mice were generated by CRISPR/Cas9‐mediated genome editing (Cyagen Biosciences). Briefly, exons 3–7 of ZDHHC18 were selected as the target site, and Cas9 and the guide RNA (gRNA) were coinjected into fertilized eggs to produce knockout mice. The obtained heterozygous mice were intercrossed to generate homozygous mice, which were verified by sequencing. All the mice were genotyped by PCR analysis of tail DNA before use. Six‐ to eight‐week‐old mice were used for all experiments, and littermate WT mice were used as controls. No difference in results between the two sexes was found. All mice were maintained in the specific pathogen‐free animal facility at Tsinghua University, and all animal experiments were conducted in accordance with animal protocols approved by the Institutional Animal Care and Use Committee of Tsinghua University.
The sequences of the gRNA and genotyping primers are shown below:
g1: TTAAATGCATATGACCTCGCTGG
g2: AGCAAGAAACAATCGCTACCTGG
F1: 5′‐CAGCTTGCAGACACTCTTCTAAC‐3′
F2: 5′‐GATAGATGCCACGGTGACTGTTAG‐3′
R1: 5′‐CACTGAACCAAGGACCCATAGATG‐3′
Cell lines
HEK293T, HeLa, RAW264.7, L929, and Vero cells and BMDMs were cultured in Gibco™ Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% (v/v) fetal bovine serum (FBS), 2 mM L‐glutamine, 100 U/ml penicillin and 100 mg/ml streptomycin. THP‐1 cells were cultured in Gibco™ RPMI 1640 medium supplemented with 10% (v/v) FBS, 2 mM L‐glutamine, 100 U/ml penicillin and 100 mg/ml streptomycin. BMDMs were isolated from femurs and tibias of mice and cultured with M‐CSF (20 ng/ml) for differentiation before use. All cultured cells were grown at 37°C in a humidified incubator containing 5% CO2. All cell lines tested negative for mycoplasma contamination.
Plasmid construction
All the expression plasmids were constructed in existing vectors through homologous recombination by using a pEASY‐Uni Seamless Cloning and Assembly Kit (TransGen). Point mutations were achieved by using the Fast Mutagenesis System (TransGen). cDNA was obtained by standard PCR techniques. The accuracy of all constructs was confirmed by sequencing.
Virus preparation and infection
HSV‐1, VACV, HSV‐1‐FS (F strain), and GFP‐HSV‐1 were kindly provided by Zhengfan Jiang’s laboratory (Peking University) and propagated in Vero cells. The virus was inoculated into confluent Vero cells (multiplicity of infection (MOI) = 0.05–0.1) in DMEM supplemented with 2% FBS. The supernatants were collected 48 h later and ultracentrifuged at 20,000 g for 2 h. The pellets were resuspended in PBS, aliquoted and stored at −80°C until use. The virus titer was determined by plaque assay on Vero cells.
For virus infection in vivo, mice were injected intranasally with VACV (5 × 107 PFU) in a volume of 0.02 ml per mouse. For virus infection in vitro, cells seeded in 6‐well plates were incubated with HSV‐1, VACV or GFP‐HSV‐1 (MOI = 2 or 1) for the indicated times, followed by subsequent assays.
RNA interference
Three siRNAs purchased from GenePharma were used in the cocktail, and scrambled siRNA was used as a control. THP‐1 cells were seeded in 6‐well plates and differentiated via the addition of PMA for 24 h before use. siRNAs were transfected with a Lipofectamine 3000 Transfection Kit (used without P3000) at a final concentration of 50 nM. After 48 h of siRNA transfection, the cells were treated with HT‐DNA for the indicated times and harvested for RNA extraction and RT–qPCR.
Generation of knockdown cell lines
PLKO.1 ZDHHC18‐shRNA plasmids were constructed to make stable knockdown cell lines. HEK293T cells were plated in 10‐cm dishes and transfected with the indicated lentiviral expression plasmid (8 μg) together with the packing plasmids psPAX2 (6 μg) and pMD2.G (2 μg). Cell media containing lentiviral particles were harvested at 24 h and 48 h, filtered through a 0.45‐μm membrane filter, and used to infect the indicated target cells in the presence of 8 μg/ml polybrene. After 48 h of culture, transduced cells were selected with puromycin. The shRNA sequences used here are listed in Appendix Table S2.
Immunoblotting
Cells were lysed with RIPA lysis buffer containing protease inhibitor cocktail and phosphatase inhibitor. The lysates were centrifuged at 13,000 rpm for 10 min, and the supernatants were collected to measure the protein concentration by BCA assay. After boiling with SDS‐loading buffer at 95°C for 10 min, the protein samples were separated by 10% SDS–PAGE and transferred to PVDF membranes, and the membranes were blocked with TBST containing 5% (w/v) BSA prior to incubation with primary antibodies at room temperature for 2 h or 4°C overnight and secondary antibodies for 1 h at room temperature. The blots were visualized using a chemiluminescent substrate, and images were observed with an iBright 1500 imager system. Densitometry analysis was performed using ImageJ software.
Immunoprecipitation
Cells were lysed in IP lysis buffer (50 mM Tris–HCl pH 7.5, 150 mM NaCl, 0.2% Triton X‐100, and 10% glycerol) supplemented with protease inhibitor cocktail. Debris was removed by centrifugation at 16,200 g for 10 min. Sixty microliters of supernatant was kept as whole‐cell lysates, and the rest was incubated with a specific antibody (or an equal amount of IgG as a control) at 4°C for 2 h, followed by the addition of Protein A/G Agarose (Santa Cruz) and incubation overnight. For IP with Flag‐M2 or Myc magnetic beads, the lysates were incubated overnight directly. Immunoprecipitates were washed three times with IP lysis buffer. Proteins were eluted from the beads by boiling in SDS–PAGE loading buffer and analyzed by immunoblot.
Acyl resin‐assisted capture (acyl‐RAC) assay
The acyl‐RAC assay was performed as previously described, with minor modifications (Forrester et al, 2011). Briefly, HEK293T cells transfected with Flag‐tagged proteins were homogenized in palmitoylation lysis buffer (10 mM sodium phosphate, 2 mM Na2EDTA, 0.32 M sucrose, 1% Triton X‐100, 50 mM N‐ethylmaleimide and protease inhibitor). Cell lysates were immunoprecipitated by anti‐Flag antibody beads at 4°C for 2 h. Then, the beads were washed 3 times with palmitoylation lysis buffer and incubated with palmitoylation elution buffer (10 mM sodium phosphate, 2 mM Na2EDTA, and 1% SDS) at 50°C for 5 min to release Flag‐tagged proteins. Eluted protein samples were then divided into two equal parts, one treated with 1 M hydroxylamine and the other treated with 1 M Tris (pH = 7.5) in the presence of activated thiol‐Sepharose 6B (Sigma) for 2 h at room temperature. Then, the beads were washed 3 times with palmitoylation washing buffer (10 mM sodium phosphate, 2 mM Na2EDTA, 0.32 M sucrose, 1% Triton X‐100, 500 mM NaCl, and protease inhibitor). The palmitoylation signal was detected by western blotting.
In vitro palmitoylation assay
The in vitro palmitoylation assay was performed as previously described, with minor modifications (Rana et al, 2018). Briefly, purified cGAS or truncated cGAS (50 nM) was incubated with/without HT‐DNA (1 μg/ml) in reaction buffer (50 mM HEPES pH 7.0, 200 mM NaCl, 0.2 μM DTT, 1 mM EDTA, and 0.3 mM n‐dodecyl‐β‐D‐maltoside [DDM]) for 10 min at room temperature. The total reaction volume was 200 μl. The reaction was initiated by preventing light exposure and adding NBD‐palmitoyl‐CoA to a final concentration of 1 μM. After gentle mixing, 50‐μL samples were taken at different time points and quenched with the addition of nonreducing SDS sample buffer. Samples were separated in 10% SDS–PAGE gels, and fluorescence was detected using an iBright 1500 instrument (Invitrogen).
Click chemistry
Click chemistry was applied for 17‐ODYA metabolic labeling of HEK293T cells. Metabolic labeling of RAW264.7 cells was performed as previously described, with a few modifications (Akimzhanov & Boehning, 2015). Briefly, cells were labeled overnight with 50 μM 17‐ODYA (25 mM stock in DMSO, Cayman) or 50 μM palmitic acid (PA) in DMEM supplemented with 10% FBS, collected by centrifugation, washed with cold PBS, and resuspended in lysis buffer (50 mM Tris–HCl pH 7.5, 150 mM NaCl, 0.2% Triton X‐100 and 10% glycerol) supplemented with protease inhibitor, 1 μM palmostatin B and 50 mM NEM. After lysis in a cold room for 30 min, the lysate was clarified at 13,800 g for 10 min at 4°C, and the collected supernatants were incubated with 1 μg of anti‐cGAS antibody (CST) and 20 μl of Protein A/G beads (Santa Cruz). After incubation, the beads were washed three times with lysis buffer, and Cu(I)‐assisted click reactions were performed as previously described. Briefly, the beads were incubated in PBS supplemented with 10 μM IRDye 800 CW azide infrared dye, 1 mM TCEP (50× stock solution: 50 mM TCEP in H2O adjusted to pH 7.0 with 1 mM NaOH), 1 mM CuSO4 (100× stock solution: 100 mM CuSO4 in PBS, prepared immediately before use), 0.1 mM TBTA (50× stock solution: 5 mM TBTA) for 1 h, washed eight times with PBS and resuspended in PBS. The samples were separated in 10% SDS–PAGE gels, and fluorescence was detected using an iBright 1500 instrument (Invitrogen).
FRET assay
HeLa cells were seeded on coverslips in 12‐well plates and transfected with GFP, GFP‐cGAS, and mCherry‐cGAS, or the indicated plasmids using Lipofectamine 3000. Twenty‐four hours later, following removal of the supernatants, coverslips carrying cells were washed with PBS, fixed with 4% paraformaldehyde, and mounted on slides using aqueous mounting medium. A FRET assay was performed on a Zeiss LSM710 confocal microscope with bleaching of the mCherry signal. FRET efficiency was analyzed on a Zeiss LSM710 confocal microscope via receptor bleaching.
Identification of the palmitoylation site using LC‐MS/MS
Proteins were separated by SDS–PAGE, and the gel bands of interest were excised from the gel, reduced with 5 mM DTT, and alkylated with 11 mM iodoacetamide. Then, 1 M hydroxylamine hydrochloride was added to the gel and incubated for 3 h to reduce palmitate, and the gel was alkylated again with 11 mM 13C iodoacetic acid, followed by in‐gel digestion with sequencing‐grade modified trypsin at 37°C overnight. The peptides were extracted twice with 0.1% trifluoroacetic acid in a 50% acetonitrile aqueous solution for 30 min and then dried in a SpeedVac. Peptides were redissolved in 20 μl of 0.1% trifluoroacetic acid, and 6 μl of extracted peptides were analyzed by a Thermo Scientific Q Exactive mass spectrometer.
For LC‐MS/MS analysis, the peptides were separated by 85‐min gradient elution at a flow rate of 0.30 µl/min with a Thermo‐Dionex Ultimate 3000 HPLC system, which was directly interfaced with a Thermo Scientific Q Exactive mass spectrometer. The analytical column was a homemade fused silica capillary column (75 µm ID, 150 mm length; Upchurch, Oak Harbor, WA) packed with C‐18 resin (300 Å, 5 µm; Varian, Lexington, MA). Mobile phase A consisted of 0.1% formic acid, and mobile phase B consisted of 80% acetonitrile and 0.1% formic acid. The Q Exactive mass spectrometer was operated in the data‐dependent acquisition mode using Xcalibur 2.1.2 software, and there was a single full‐scan mass spectrum in the orbitrap (300–1,800 m/z, 70,000 resolution) followed by 20 data‐dependent LC‐MS/MS scans at 27% normalized collision energy (HCD).
The LC‐MS/MS spectra from each LC‐MS/MS run were searched against cGAS.fasta from UniProt using an in‐house Proteome Discoverer program (version PD1.4, Thermo Fisher Scientific, USA). The search criteria were as follows: full tryptic specificity was required; two missed cleavages were allowed; carbamidomethylation (C), iodoacetic acid (I), and oxidation (M) were set as the variable modifications; precursor ion mass tolerances were set at 20 ppm for all MS spectra acquired in the Orbitrap mass analyzer; and the fragment ion mass tolerance was set at 0.02 Da for all MS2 spectra acquired.
DNA pull‐down assay
Cells seeded in 6‐well plates were collected and lysed with IP lysis buffer (with protease inhibitor cocktail). Debris was removed by centrifugation, and 60 μL of supernatant was kept as whole‐cell lysate. Five micrograms of biotin‐ISD or DNA (100 bp) was added to the remaining supernatants (label‐free ISD or DNA as a control) and incubated at 4°C for 4 h, followed by the addition of streptavidin‐Sepharose beads for 2 h. Beads bound with proteins were washed three times with IP lysis buffer (300 mM NaCl), resuspended in SDS–PAGE loading buffer, denatured, and analyzed by immunoblotting.
Plaque assay
Vero cells were seeded in 6‐well plates, and the supernatants were removed when the cells reached confluency. A 300‐μl sample (supernatants of infected cell cultures or virus stock) was added to infect Vero cells for 1 h in an incubator. The plates were shaken every 10 min to uniformly cover the monolayers with the samples. Then, 2 ml of MEM containing 2% methylcellulose was added to each well, and the plates were incubated for 48 h at 37°C with 5% CO2. The cells were fixed with 4% paraformaldehyde for 30 min and stained with crystal violet staining solution for 30 min before imaging and counting the plaques.
Luciferase reporter assay
HEK293T cells were seeded in 12‐well plates and cotransfected with pGL3 IFN‐β (200 ng), HA‐cGAS (100 ng), Flag‐STING (200 ng), and other indicated plasmids in each well. An empty vector (pCMV‐3Myc) was added to keep the amount of DNA constant. Cells were lysed with Passive Lysis Buffer (Promega) 24 h post‐transfection. The debris was removed by centrifugation at 4°C, and 20 µl of the supernatant was used to measure the luciferase activity using Luciferase Assay Reagent (Promega). Luminescence was read on a Varioskan Flash microplate reader (Thermo Scientific).
Enzyme‐linked immunosorbent assay (ELISA)
BMDMs seeded in 12‐well plates were infected with or without HSV‐1 (MOI = 2) for the indicated times. At the time of collection, supernatants were harvested, and secreted IFN‐β protein levels were quantified using a Mouse IFN Beta ELISA Kit (Abcam) according to the manufacturer's instructions. Generally, absorbance at 450 nm was measured, and the concentration of the target protein in the samples was determined by interpolating the blank control‐subtracted absorbance values against the standard curve.
cGAMP quantification
Cells were plated in 10‐cm dishes and treated with or without HT‐DNA (2 μg/ml) for the indicated times before collection. Collected cells were washed with ice‐cold PBS, lysed with methanol, and quickly scraped into a 1.5‐ml centrifuge tube, followed by five freeze–thaw cycles in liquid nitrogen. The insoluble pellet was removed by centrifugation in a cooled centrifuge (4°C), and the supernatant was transferred to a new tube and evaporated to dryness. Dried extracts were reconstituted in 80 μl of water, vortexed, and centrifuged at 13,000 rpm for 10 min at 4°C, and the supernatant was centrifuged again before being loaded into the LC‐MS/MS instrument for quantification. The LC‐MS/MS system consisted of an ACQUITY UPLC I‐Class (Waters, USA) and a QTRAP 4500 triple quadrupole mass spectrometer (AB SCIEX). An electrospray ionization source and positive ionization mode were applied. Commercial cGAMP was used as a standard sample, and the retention time was 2.56 min. The optimized ion transitions (m/z: 675.2–524.1, 675.2–506.1) were used for quantification.
Immunofluorescence
Cells were seeded on coverslips in 12‐well plates and transfected with the indicated plasmids using Lipofectamine 3000 as indicated. After removing the supernatants, cells were washed with PBS, fixed with 4% paraformaldehyde, permeabilized with 0.2% (v/v) Triton X‐100 and blocked in 3% (w/v) BSA for 1 h. Then, the cells were incubated with the indicated primary antibodies (in 3% (w/v) BSA) overnight at 4°C followed by incubation with fluorescent secondary antibodies and DAPI for 1 h at room temperature. Finally, the coverslips were fixed on slides using aqueous mounting medium, and images were acquired with a Nikon A1RMP or Zeiss LSM710 confocal microscope. Colocalization analysis was performed using Imaris software.
Hematoxylin–eosin staining
Six‐to‐seven‐week‐old mice were intravenously injected with or without HSV‐1‐FS (5 × 107 PFU per mouse), and 2 days later, the mice were euthanized, and the liver, kidney, and spleen were collected. These organs were fixed in 4% paraformaldehyde overnight and embedded in paraffin. Then, the paraffin blocks were sectioned and stained with H&E. Images were acquired using a 3D HISTECH panoramic scanner (Zeiss) and processed with CASviewer 2.2.
RT–qPCR
Cells were seeded in 6‐well plates and treated as indicated. Total RNA was extracted with TRIzol, and the diluted RNA was reverse transcribed using an iScript cDNA synthesis kit and analyzed via qPCR using iTaq Universal SYBR Green Supermix (on a Bio‐Rad T100 thermal cycler). All reagents were used according to the manufacturer's instructions. The primers are listed in Appendix Table S3. GAPDH (for humans) and β‐Actin (for mice) were used as internal controls.
Construction of the human cGAS‐DNA 2:2 complex
To investigate the effect of palmitoylation on human cGAS dimerization, the mouse cGAS‐DNA 2:2 complex (PDB ID: 4O6A) (Zhang et al, 2014) was used as the template to construct the human cGAS‐DNA 2:2 complex (Appendix Fig S3B, WT) by SWISS MODEL (Waterhouse et al, 2018) based on the human cGAS‐DNA 1:1 complex (Appendix Fig S3A, PDB ID: 6CT9) (Zhou et al, 2018). Subsequently, the two C474 residues were modified to S‐palmitoylated cysteines in the human cGAS‐DNA 2:2 complex (Appendix Fig S3C, Palm). The palmitic acid tail was oriented toward the contact interface of the two monomers (Appendix Fig S3D–F).
Molecular dynamics simulations
Atomic MD simulations of the human cGAS‐DNA 2:2 complex in the resting and palmitoylation states were performed using AMBER16 software, applying the Amber ff14SB (Maier et al, 2015) force field for protein and the Amber DNA.bsc1 force field (Ivani et al, 2016) for DNA. The parameters of the S‐palmitoyl tail were obtained from Amber Lipid17 (Gould et al, in preparation). The human cGAS‐DNA 2:2 complex in the resting and palmitoylation states was immersed in a hexahedral solvent box filled with TIP3P water molecules with a distance of 10 Å from the surface of the protein to the edge of the water box, and each system was neutralized with magnesium ions. Energy minimization was conducted by imposing a strong restraint on each resting state and palmitoylation state system and was followed by minimizing the whole system for a few thousand steps. Then, NVT simulations were carried out by heating the whole system slowly from 0 to 300 K, which was followed by a 5‐ns NPT equilibration run. Subsequently, the production run was performed for 1 μs. During the production run, the SHAKE algorithm was employed to constrain all bonds associated with hydrogen atoms. Electrostatic interactions were treated by Particle Mesh Ewald, and the cutoff value of nonbonded interactions was set to 10 Å. Fifteen independent MD simulations for each resting state and palmitoylation state system were employed with different initial velocities, which were randomly generated at the beginning of the simulations.
Molecular dynamics simulations for human ZDHHC18/cGAS/DNA complex
Human ZDHHC18 models were built by AlphaFold v2.0 and the pLDDT values of the M truncation were greater than 90, which indicates that the predicted structure was very credible (https://alphafold.ebi.ac.uk/entry/Q9NUE0). In the docked pose, the irregular structures of the N‐terminus (M1‐G63) and C‐terminus (P348‐P378) in human ZDHHC18 models were removed. Thereafter, the human cGAS‐DNA 1:1 complex (PDB ID: 6CT9) was docked to the human ZDHHC18 models to construct the complex structures of ZDHHC18/cGAS/DNA, which were performed using FRODOCK (https://chaconlab.org/modeling/frodock). The top three docking structures with the highest score were chosen for the next analysis. To investigate the effect of DNA on the interaction of ZDHHC18 and cGAS, the ZDHHC18/cGAS complex was built by removing the DNA from the ZDHHC18/cGAS/DNA complex.
Subsequently, the cysteine 222 residue of human ZDHHC18 was modified to s‐palmitoylated‐cysteine. Then, all the complexes were embedded in a pre‐equilibrated 1‐palmitoyl 2‐oleoyl‐sn glycero‐3‐phosphocholine (POPC) lipid bilayer followed by solvation (TIP3P water model) and neutralization using potassium and chloride ions at 150 mM. Amber ff14SB, Amber DNA.bsc1 force field, and Amber Lipid17 force field were employed for the MD simulations including protein, lipids and DNA respectively, which were constructed using CHARMM‐GUI (https://charmm‐gui.org). The parameters of the S‐palmitoyl tail were obtained from Amber Lipid17. The setting of simulation parameters and time was the same as that of the human cGAS‐DNA 2:2 complex. Similarly, the MM‐GBSA method was used to estimate the effect of palmitoylation on the binding free energy between ZDHHC18 and cGAS.
Free energy decomposition calculation
The molecular mechanics generalized Born surface area (MM‐GBSA) method was used to estimate the effect of palmitoylation on the binding free energy between two cGAS‐DNA 1:1 complexes, as well as the energy contribution of each residue to the binding energy between DNA and protein. In this study, all energy components were calculated using 500 snapshots extracted from the last 100 ns in MD trajectories. The total binding energy (ΔGbind ) was computed using the following equation:
| (1) |
where ΔGbind is the binding free energy between two cGAS‐DNA 1:1 complexes, which was calculated as the difference between the total free energy of the complex (Gcomplex ) and the sum of the free energy of the protein (Gprotein ) and the ligand (Gligand ). The binding energy is represented as:
| (2) |
where EMM is the molecular mechanics energy of the molecule expressed as the sum of the internal energy of the molecule plus electrostatic and van der Waals energies. The solvation free energy is expressed as polar and nonpolar contributions to the solvation energy:
| (3) |
| (4) |
Gnonpolar is calculated from the solvent‐accessible surface area (SASA):
| (5) |
where γ = 0.0072 kcal/mol/Å, and b = 0 kcal/mol.
PCA
PCA uses the covariance matrix of all conformations relative to the average conformation to reduce the dimensionality and extract the related fluctuations from MD trajectories, which provides a highly detailed image of biomolecular motions. PCA provides orthogonal eigenvalues and eigenvectors based on covariance matrix diagonalization. The elements Cij in the matrix are defined as
| (6) |
where Δri (Δrj ) indicates the deviation of the Cα atom of the ith residue from its mean position. The value of Cij fluctuates between −1 and 1. A positive Cij value indicates the correlation motion between the ith residue and the jth residue, while a negative Cij value represents the inverse correlation motion. The eigenvalues indicate the magnitude of motions along a direction. In general, the first few principal components (PCs) describe the most important slow system modes related to the functional motions of a biomolecular system.
Statistical analyses
GraphPad Prism 7 software was applied to perform statistical analyses. All experiments were performed with at least triplicate samples as independent biological replicates. Two‐tailed Student’s t‐test was used to compare data points. A log‐rank (Mantel–Cox) test was adopted for survival analysis. Significance levels are shown with asterisks, as follows: *P < 0.05; **P < 0.01; ***P < 0.001. Data with error bars are presented as the mean values with the SEMs.
Author contributions
Chengrui Shi: Resources; Data curation; Software; Formal analysis; Validation; Investigation; Visualization; Methodology; Writing—original draft; Project administration; Writing—review & editing. Xikang Yang: Data curation; Software; Investigation; Methodology; Writing—original draft. Ye Liu: Data curation; Software; Formal analysis; Validation. Hongpeng Li: Methodology. Huiying Chu: Software. Guohui Li: Supervision; Funding acquisition. Hang, Hubert Yin: Conceptualization; Resources; Supervision; Funding acquisition; Writing—original draft; Project administration; Writing—review & editing.
In addition to the CRediT author contributions listed above, the contributions in detail are:
HY conceptualized the project and supervised all the experiments; CS and XY performed the experiments and interpreted the data; YL, HC and GL performed the MD analysis and HL performed helpful experimental assistance in BMDM isolation; CS and HY wrote the manuscript; XY and YL contributed to the discussion and improvement of the manuscript.
Disclosure and competing interests statement
The authors declare that they have no competing interests.
Supporting information
Appendix
Source Data for Appendix
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Acknowledgments
The authors are grateful to Professor Zhengfan Jiang for sharing the HSV‐1 virus strains; to Dr. Conggang Zhang for helpful discussion and technical support; to Dr. Ying Zhang and Xue Jin for helpful discussion and experiment assistance; to Gaoge Sun and Jiaqi Liang for manuscript editing; to Dr. Conggang Zhang, Yanfei Hou and Yinlong Yang for providing recombinant cGAS protein; and to Meng Han for LC‐MS/MS quantification. We also thank the Center of Pharmaceutical Technology (Tsinghua University) for cGAMP quantification and data collection. This work was supported by National Natural Science Foundation of China Grant (21825702 and 22137004 (H.Y.), 21933010 (G.L.), and 21907094 (H.C.)) and Beijing Outstanding Young Talents Grant (BJJWZYJH01201910003013 (H.Y.)).
The EMBO Journal (2022) 41: e109272.
Contributor Information
Guohui Li, Email: ghli@dicp.ac.cn.
Hang Yin, Email: yin_hang@tsinghua.edu.cn.
Data availability
All data associated with this study are presented in the paper and/or Appendix. Materials that support the findings of this study are available from the corresponding author upon request. This study includes no data deposited in external repositories.
References
- Aarreberg LD, Esser‐Nobis K, Driscoll C, Shuvarikov A, Roby JA, Gale M Jr (2019) Interleukin‐1 beta induces mtDNA release to activate innate immune signaling via cGAS‐STING. Mol Cell 74: 801–815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akimzhanov AM, Boehning D (2015) Rapid and transient palmitoylation of the tyrosine kinase Lck mediates Fas signaling. Proc Natl Acad Sci U S A 112: 11876–11880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baekkeskov S, Kanaani J (2009) Palmitoylation cycles and regulation of protein function (Review). Mol Membr Biol 26: 42–54 [DOI] [PubMed] [Google Scholar]
- Barnett KC, Coronas‐Serna JM, Zhou W, Ernandes MJ, Cao A, Kranzusch PJ, Kagan JC (2019) Phosphoinositide interactions position cGAS at the plasma membrane to ensure efficient distinction between self‐ and viral DNA. Cell 176: 1432–1446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer JA, Spangler CJ, Strauss JD, Cesmat AP, Liu P, McGinty RK, Zhang Q (2020) Structural basis of nucleosome‐dependent cGAS inhibition. Science 370: 450–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdette DL, Monroe KM, Sotelo‐Troha K, Iwig JS, Eckert B, Hyodo M, Hayakawa Y, Vance RE (2011) STING is a direct innate immune sensor of cyclic di‐GMP. Nature 478: 515–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain LH, Shipston MJ (2015) The physiology of protein S‐acylation. Physiol Rev 95: 341–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Sun L, Chen ZJ (2016) Regulation and function of the cGAS‐STING pathway of cytosolic DNA sensing. Nat Immunol 17: 1142–1149 [DOI] [PubMed] [Google Scholar]
- Chen S, Zhu BO, Yin C, Liu W, Han C, Chen B, Liu T, Li X, Chen X, Li C et al (2017) Palmitoylation‐dependent activation of MC1R prevents melanomagenesis. Nature 549: 399–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Chen Y (2019) Ubiquitination of cGAS by TRAF6 regulates anti‐DNA viral innate immune responses. Biochem Biophys Res Commun 514: 659–664 [DOI] [PubMed] [Google Scholar]
- Chini B, Parenti M (2009) G‐protein‐coupled receptors, cholesterol and palmitoylation: facts about fats. J Mol Endocrinol 42: 371–379 [DOI] [PubMed] [Google Scholar]
- De I, Sadhukhan S (2018) Emerging roles of DHHC‐mediated protein S‐palmitoylation in physiological and pathophysiological context. Eur J Cell Biol 97: 319–338 [DOI] [PubMed] [Google Scholar]
- Dobbs N, Burnaevskiy N, Chen DD, Gonugunta VK, Alto NM, Yan N (2015) STING activation by translocation from the ER is associated with infection and autoinflammatory disease. Cell Host Microbe 18: 157–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draper JM, Smith CD (2009) Palmitoyl acyltransferase assays and inhibitors (Review). Mol Membr Biol 26: 5–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du M, Chen ZJ (2018) DNA‐induced liquid phase condensation of cGAS activates innate immune signaling. Science 361: 704–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forrester MT, Hess DT, Thompson JW, Hultman R, Moseley MA, Stamler JS, Casey PJ (2011) Site‐specific analysis of protein S‐acylation by resin‐assisted capture. J Lipid Res 52: 393–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao D, Li T, Li XD, Chen X, Li QZ, Wight‐Carter M, Chen ZJ (2015) Activation of cyclic GMP‐AMP synthase by self‐DNA causes autoimmune diseases. Proc Natl Acad Sci U S A 112: E5699–5705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao P, Ascano M, Wu Y, Barchet W, Gaffney B, Zillinger T, Serganov A, Liu Y, Jones R, Hartmann G et al (2013) Cyclic [G(2 ',5 ') pA(3 ',5 ')p] is the metazoan second messenger produced by DNA‐activated cyclic GMP‐AMP synthase. Cell 153: 1094–1107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregg RW, Sarkar SN, Shoemaker JE (2019) Mathematical modeling of the cGAS pathway reveals robustness of DNA sensing to TREX1 feedback. J Theor Biol 462: 148–157 [DOI] [PubMed] [Google Scholar]
- Huang LS, Hong ZG, Wu W, Xiong SQ, Zhong M, Gao XP, Rehman J, Malik AB (2020) mtDNA activates cGAS signaling and suppresses the YAP‐mediated endothelial cell proliferation program to promote inflammatory injury. Immunity 52: 475–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa H, Ma Z, Barber GN (2009) STING regulates intracellular DNA‐mediated, type I interferon‐dependent innate immunity. Nature 461: 788–792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivani I, Dans PD, Noy A, Pérez A, Faustino I, Hospital A, Walther J, Andrio P, Goñi R, Balaceanu A et al (2016) Parmbsc1: a refined force field for DNA simulations. Nat Methods 13: 55–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Zhang X, Chen X, Aramsangtienchai P, Tong Z, Lin H (2018) Protein lipidation: occurrence, mechanisms, biological functions, and enabling technologies. Chem Rev 118: 919–988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kranzusch PJ, Lee AS, Berger JM, Doudna JA (2013) Structure of human cGAS reveals a conserved family of second‐messenger enzymes in innate immunity. Cell Rep 3: 1362–1368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kranzusch PJ, Lee ASY, Wilson SC, Solovykh MS, Vance RE, Berger JM, Doudna JA (2014) Structure‐guided reprogramming of human cGAS dinucleotide linkage specificity. Cell 158: 1011–1021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kujirai T, Zierhut C, Takizawa Y, Kim R, Negishi L, Uruma N, Hirai S, Funabiki H, Kurumizaka H (2020) Structural basis for the inhibition of cGAS by nucleosomes. Science 370: 455–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahaye X, Gentili M, Silvin A, Conrad C, Picard L, Jouve M, Zueva E, Maurin M, Nadalin F, Knott GJ et al (2018) NONO detects the nuclear HIV capsid to promote cGAS‐mediated innate immune activation. Cell 175: 488–501 [DOI] [PubMed] [Google Scholar]
- Li T, Huang T, Du M, Chen X, Du F, Ren J, Chen ZJ (2021) Phosphorylation and chromatin tethering prevent cGAS activation during mitosis. Science 371: eabc5386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Zheng Y, Coyaud É, Zhang C, Selvabaskaran A, Yu Y, Xu Z, Weng X, Chen JS, Meng Y et al (2019) Palmitoylation of NOD1 and NOD2 is required for bacterial sensing. Science 366: 460–467 [DOI] [PubMed] [Google Scholar]
- Ma D, Yang M, Wang Q, Sun C, Shi H, Jing W, Bi Y, Shen X, Ma X, Qin Z et al (2021) Arginine methyltransferase PRMT5 negatively regulates cGAS‐mediated antiviral immune response. Sci Adv 7: eabc1834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser KE, Simmerling C (2015) ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J Chem Theory Comput 11: 3696–3713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalski S, de Oliveira Mann CC, Stafford CA, Witte G, Bartho J, Lammens K, Hornung V, Hopfner KP (2020) Structural basis for sequestration and autoinhibition of cGAS by chromatin. Nature 587: 678–682 [DOI] [PubMed] [Google Scholar]
- Mukai K, Konno H, Akiba T, Uemura T, Waguri S, Kobayashi T, Barber GN, Arai H, Taguchi T (2016) Activation of STING requires palmitoylation at the Golgi. Nat Commun 7: 11932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathare GR, Decout A, Gluck S, Cavadini S, Makasheva K, Hovius R, Kempf G, Weiss J, Kozicka Z, Guey B et al (2020) Structural mechanism of cGAS inhibition by the nucleosome. Nature 587: 668–672 [DOI] [PubMed] [Google Scholar]
- Rana MS, Kumar P, Lee CJ, Verardi R, Rajashankar KR, Banerjee A (2018) Fatty acyl recognition and transfer by an integral membrane S‐acyltransferase. Science 359: eaao6326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoh T, Fujita N, Hayashi T, Takahara K, Satoh T, Lee H, Matsunaga K, Kageyama S, Omori H, Noda T et al (2009) Atg9a controls dsDNA‐driven dynamic translocation of STING and the innate immune response. Proc Natl Acad Sci U S A 106: 20842–20846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang G, Zhang C, Chen ZJ, Bai XC, Zhang X (2019) Cryo‐EM structures of STING reveal its mechanism of activation by cyclic GMP‐AMP. Nature 567: 389–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun LJ, Wu JX, Du FH, Chen X, Chen ZJJ (2013) Cyclic GMP‐AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339: 786–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas CA, Tejwani L, Trujillo CA, Negraes PD, Herai RH, Mesci P, Macia A, Crow YJ, Muotri AR (2017) Modeling of TREX1‐dependent autoimmune disease using human stem cells highlights L1 accumulation as a source of neuroinflammation. Cell Stem Cell 21: 319–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unterholzner L (2013) The interferon response to intracellular DNA: why so many receptors? Immunobiology 218: 1312–1321 [DOI] [PubMed] [Google Scholar]
- Wang Q, Huang L, Hong Z, Lv Z, Mao Z, Tang Y, Kong X, Li S, Cui Y, Liu H et al (2017a) The E3 ubiquitin ligase RNF185 facilitates the cGAS‐mediated innate immune response. PLoS Pathog 13: e1006264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Ning X, Gao P, Wu S, Sha M, Lv M, Zhou X, Gao J, Fang R, Meng G et al (2017b) Inflammasome activation triggers caspase‐1‐mediated cleavage of cGAS to regulate responses to DNA virus infection. Immunity 46: 393–404 [DOI] [PubMed] [Google Scholar]
- Wang Y, Su GH, Zhang F, Chu JX, Wang YS (2015) Cyclic GMP‐AMP synthase is required for cell proliferation and inflammatory responses in rheumatoid arthritis synoviocytes. Mediat Inflamm 2015: 1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, Heer FT, de Beer TA P, Rempfer C, Bordoli L et al (2018) SWISS‐MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res 46: W296–W303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AP, Khoury‐Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, Bestwick M, Duguay BA, Raimundo N, MacDuff DA et al (2015) Mitochondrial DNA stress primes the antiviral innate immune response. Nature 520: 553–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willemsen J, Neuhoff M‐T, Hoyler T, Noir E, Tessier C, Sarret S, Thorsen TN, Littlewood‐Evans A, Zhang J, Hasan M et al (2021) TNF leads to mtDNA release and cGAS/STING‐dependent interferon responses that support inflammatory arthritis. Cell Rep 37: 109977 [DOI] [PubMed] [Google Scholar]
- Wu J, Sun L, Chen X, Du F, Shi H, Chen C, Chen ZJ (2013) Cyclic GMP‐AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 339: 826–830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoh S, Schneider M, Seifried J, Soonthornvacharin S, Akleh R, Olivieri K, De Jesus P, Ruan C, de Castro E, Ruiz P et al (2015) PQBP1 is a proximal sensor of the cGAS‐dependent innate response to HIV‐1. Cell 161: 1293–1305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Shang G, Gui X, Zhang X, Bai XC, Chen ZJ (2019) Structural basis of STING binding with and phosphorylation by TBK1. Nature 567: 394–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Zhou L, Xu Y, Yang M, Xu Y, Komaniecki GP, Kosciuk T, Chen X, Lu X, Zou X et al (2020) A STAT3 palmitoylation cycle promotes TH17 differentiation and colitis. Nature 586: 434–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Wu J, Du F, Xu H, Sun L, Chen Z, Brautigam CA, Zhang X, Chen ZJ (2014) The cytosolic DNA sensor cGAS forms an oligomeric complex with DNA and undergoes switch‐like conformational changes in the activation loop. Cell Rep 6: 421–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Zheng Q, Sun C, Song J, Gao L, Zhang S, Munoz A, Read ND, Lu L (2016) Palmitoylation of the cysteine residue in the DHHC Motif of a palmitoyl transferase mediates Ca2+ homeostasis in aspergillus. PLoS Genet 12: e1005977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Xu P, Rowlett CM, Jing T, Shinde O, Lei Y, West AP, Liu WR, Li P (2020) The molecular basis of tight nuclear tethering and inactivation of cGAS. Nature 587: 673–677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, He A, Zhu F, Ding M, Hao J, Fan Q, Li P, Liu LI, Du Y, Liang X et al (2018) Integrating genome‐wide association study and expression quantitative trait locus study identifies multiple genes and gene sets associated with schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry 81: 50–54 [DOI] [PubMed] [Google Scholar]
- Zheng H, Pearsall EA, Hurst DP, Zhang Y, Chu J, Zhou Y, Reggio PH, Loh HH, Law P‐Y (2012) Palmitoylation and membrane cholesterol stabilize μ‐Opioid receptor homodimerization and G protein coupling. BMC Cell Biol 13: 6–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Whiteley AT, de Oliveira Mann CC, Morehouse BR, Nowak RP, Fischer ES, Gray NS, Mekalanos JJ, Kranzusch PJ (2018) Structure of the human cGAS‐DNA complex reveals enhanced control of immune surveillance. Cell 174: 300–311 [DOI] [PMC free article] [PubMed] [Google Scholar]