

Abstract
Background:
Plasma proteins are critical mediators of cardiovascular processes and are the targets of many drugs. Previous efforts to characterize the genetic architecture of the plasma proteome have been limited by a focus on individuals of European descent and leveraged genotyping arrays and imputation. Here we describe whole genome sequence analysis of the plasma proteome in individuals with greater African ancestry, increasing our power to identify novel genetic determinants.
Methods:
Proteomic profiling of 1,301 proteins was performed in 1852 Black adults from the Jackson Heart Study using aptamer-based proteomics (SomaScan®). Whole genome sequencing association analysis was ascertained for all variants with minor allele count ≥ 5. Results were validated using an alternative, antibody-based, proteomic platform (Olink®) as well as replicated in the Multi-Ethnic Study of Atherosclerosis and the HERITAGE Family Study.
Results:
We identify 569 genetic associations between 479 proteins and 438 unique genetic regions at a Bonferroni-adjusted significance level of 3.8 × 10−11. These associations include 114 novel locus-protein relationships and an additional 217 novel sentinel variant-protein relationships. Novel cardiovascular findings include new protein associations at the APOE gene locus including ZAP70 (sentinel single nucleotide polymorphism [SNP] rs7412-T, β = 0.61±0.05, p-value = 3.27 × 10−30) and MMP-3 (β = −0.60±0.05, p = 1.67 × 10−32), as well as a completely novel pleiotropic locus at the HPX gene, associated with nine proteins. Further, the associations suggest new mechanisms of genetically mediated cardiovascular disease linked to African ancestry; we identify a novel association between variants linked to APOL1 associated chronic kidney and heart disease and the protein CKAP2 (rs73885319-G, β = 0.34±0.04, p = 1.34 × 10−17) as well as an association between ATTR amyloidosis and RBP4 levels in community dwelling individuals without heart failure.
Conclusions:
Taken together, these results provide evidence for the functional importance of variants in non-European populations, and suggest new biological mechanisms for ancestry-specific determinants of lipids, coagulation and myocardial function.
Keywords: Genetics, Cardiovascular Disease, Proteomics, Race and Ethnicity
Introduction
The circulating plasma proteome plays a fundamental role in human biological function and dysfunction. Circulating proteins both mediate and respond to disease, and are frequently the targets of pharmaceutical interventions. Several recent studies have coupled genotyping and proteomic profiling to understand the genetic basis for the individual differences observed in protein levels, which are known to be heritable.1–7 Such work has led to critical advances in our understanding of the genetic architecture of the plasma proteome and its relationship to disease, including factors specifically associated with cardiovascular risk.4,6,7 However, initial findings were derived nearly entirely in European populations such as the Framingham Heart Study using genotyping arrays. Further, individuals with increased African ancestry are known to harbor substantially more genetic diversity than those of European ancestry,8,9 and rare mutations found specifically among persons of African ancestry have been critical in expanding our knowledge of cardiovascular biology, as is the case for PCSK9.10 We hypothesized that coupling whole genome sequence analysis with plasma proteomics in individuals of African ancestry would greatly increase the power to identify novel genetic determinants of the plasma proteome, which would not only inform our understanding of ancestry specific genetic variation, but of human cardiovascular biology in general.
Here we utilize whole genome sequence data and aptamer-based proteomic profiling of 1301 proteins on the SOMAscan™ platform in 1852 self-identified Black individuals from the Jackson Heart Study (JHS)11 to identify novel protein quantitative trait loci (pQTLs) determining protein levels. Associations were replicated in 980 participants from the Multi-Ethnic Study of Atherosclerosis (MESA)12 and 708 from the HERITAGE Family Study (Supplemental Table S1),13 and further validated using an alternate proteomic profiling platform in JHS. These data serve as the basis for an enhanced understanding of proteins highly relevant to cardiovascular homeostasis across diverse human populations.
Methods
Data availability.
Whole genomes for JHS and MESA, generated as part of the NHLBI Trans-Omics for Precision Medicine (TOPMed) program, are available through restricted access via the NHLBI database of Genotypes and Phenotypes (dbGaP). TOPMed accession numbers for JHS and MESA are phs000964/phs002256.v1.p1 and phs001416, respectively. Full GWAS summary statistics for JHS (the discovery cohort) generated in this study will be available for general research use through controlled access at dbGaP accession phs001974: NHLBI TOPMed: Genomic Summary Results for the Trans-Omics for Precision Medicine program. For assistance in accessing the discovery data in JHS prior to full availability on dbGaP, investigators should contact the authors and follow JHS data access procedures (https://www.jacksonheartstudy.org/). GWAS data for the replication studies (MESA and HERITAGE) are fully included in the manuscript. Individual level proteomic and genomic data in the replication datasets are available through application to the respective cohorts.
Study Approval
The JHS study was approved by Jackson State University, Tougaloo College, and the University of Mississippi Medical Center IRBs, and all participants provided written informed consent. All MESA participants provided written informed consent, and the study was approved by the Institutional Review Boards at The Lundquist Institute (formerly Los Angeles BioMedical Research Institute) at Harbor-UCLA Medical Center, University of Washington, Wake Forest School of Medicine, Northwestern University, University of Minnesota, Columbia University, Johns Hopkins University, and University of California Los Angeles. The human study protocols were approved by the Institutional Review Boards of Beth Israel Deaconess Medical Center, University of Washington, and the four clinical centers of HERITAGE.
Cohorts
The JHS, MESA, and the HERITAGE Family Study have all been previously described.11–13 In brief, JHS is a community-based longitudinal cohort study begun in 2000 of 5306 self-identified Black individuals from the Jackson, Mississippi metropolitan statistical area.11 Included in the present study are samples collected at Visit 1 between 2000 and 2004 from 1852 individuals with whole genome sequencing14 and proteomic profiling performed in batches (see below).
MESA began in 2000 with 6814 men and women age 45–84 years recruited at six clinical centers across the US. Participants were identified belonging to four racial/ethnic groups: Black, Hispanic, Asian, or white. Included in the present study are 980 individuals selected randomly across all four racial/ethnic groups with proteomic profiling from Visit 1 between 2000 and 2002 and whole genome sequence analysis.12
HERITAGE enrolled a combination of self-identified white and Black family units, totaling 763 sedentary participants (62% white) between the ages of 17–65 years in a 20-week, graded endurance exercise training study across 4 clinical centers in the US and Canada in 1994–5.13 Included in the present study are a random subset of 708 individuals with baseline plasma samples and genotyping.
Proteomic Profiling.
Proteomic profiling by SomaScan® (aptamer-based affinity platform) and Olink® (antibody-based affinity platform) have been described previously.6,15 Please see Supplemental Methods for further details.
Genotyping and Imputation.
Whole genome sequencing (WGS) in JHS and MESA has been described previously.14,16 Included in the present study are participants included in Freeze 6 of the TOPMed project at the Northwest Genome Center at University of Washington and the Broad Institute. Samples underwent >30× WGS. Genotype calling with vt17 and quality control were performed by the Informatics Resource Center at the University of Michigan.
Genotyping in HERITAGE was performed on the Illumina Infinium Global Screening Array. Genotypes were called using Illumina’s GenCall based on the TOP/BOT strand method. Genotype imputation was performed using the University of Michigan Imputation Server Minimac4 to reference panel TOPMed Freeze5.18 Phasing was performed with Eagle v2.4. Sites were excluded with call rate <90%, mismatched alleles, or invalid alleles (88% of sites retained).
Statistical analysis.
All statistical methods are explained throughout the sections below.
Whole genome sequence association analysis.
Across all three cohorts, proteomic measurements were standardized to a set of control samples (pooled plasma) that were part of each plate. The resulting values were log transformed and scaled to a mean of 0 and standard deviation of 1. In JHS, to account for batch effects, proteins were log-transformed and scaled within batch and then combined. In all cohorts, these log-transformed values were residualized on age, sex, batch, and principal components (PCs) of ancestry 1–10 as determined by GENetic EStimation and Inference in Structured samples (GENESIS).16,19,20 In HERITAGE and MESA, measurements were also residualized on race to account for non-genetic racial effects not captured by genetic ancestry. The resulting residuals were then inverse normalized. The association between these values and genetic variants was tested using linear mixed effects models adjusted for age, sex, the genetic relationship matrix, and PCs 1–10 using the fastGWA model implemented in the GCTA software package (version 1.93.2beta/gcta64).21 Repeat adjustment was implemented to reduce type I error and improve statistical power.22 Variants with a minor allele count less than 5 in a given cohort were excluded from analysis in that cohort. A Bonferroni-adjusted significance threshold of 3.8 × 10−11 (5 × 10−8/1301) was used for discovery in JHS. For variants in cis (<1Mb from the TSS of the coding gene for the associated protein),1 variants with P values of 5 × 10−6 were also considered in a separate analysis, given the biological plausibility of such associations.
Variance explained for each protein.
SNP-based heritability, hSNP2, was estimated using a LD- and MAF-stratified genomic relatedness matrix (GRM) restricted maximum likelihood (GREML-LDMS) model implemented in the GCTA software. This method allows for fitting multiple GRMs with SNPs binned according to their regional LD and MAF.23 It is recommended for heritability estimation on WGS data.23,24 Using this model, we first calculated the segment-based (length of segment: 200Kb) LD scores and partitioned SNPs into four groups based on the quartiles of the regional LD score. GRM for each of the four groups was then computed using SNPs binned into the corresponded group, and jointly fitted into a mixed effect model for estimating the heritability and variance. In our analysis, we allowed for a maximum of 1000 iterations. For all analysis, we adjusted for age, sex and the first 10 principal components of genetic ancestry. Variance explained by the top performing variant (as determined by lowest p-value) was estimated using the equation BETÂ2×(2×AF1× (1-AF1)/VAR) where BETA was the beta estimate for the effect allele, AF1 was the allele frequency of the effect allele, and VAR is the variance of the protein residual used for WGS analysis. Variance explained by clinical covariates was estimated using linear regression of log-transformed protein level regressed on age, sex, systolic blood pressure, diabetes, use of hypertensive medication, current smoking status, and a history of coronary heart disease. Proteins whose total heritability could not be estimated by this model, which often occurs when heritability estimates are low,23 were excluded (N=185, Supplemental Figure S1, Supplemental Table S2).
Defining protein-locus associations and sentinel variants.
To identify the broadest genomic region associated with a protein, we applied the following previously described algorithm:1 a 1Mb region around each SNP associated with a given protein was defined. Beginning with the region containing the variant with the lowest p-value, overlapping regions were merged together. This was repeated until no more overlapping regions existed for the given protein. The variant with the lowest P value in each region was identified as the sentinel variant. To describe regions associated with multiple proteins, regions with sentinel variants in linkage disequilibrium (LD) with r2 ≥ 0.8 were described as the same region, exclusively for descriptive purposes. LD was determined using SNPClip, using data from individuals of African ancestry.25,26 Any variants within 1Mb of the TSS for the cognate gene of a protein were considered ‘cis’.
Replication in MESA/HERITAGE.
Associations between sentinel variants and proteins from JHS were evaluated in MESA and HERITAGE separately, if associated statistics were available (if minor allele count was < 5 in either cohort, that variant was not considered in that cohort). Where association statistics were available in both cohorts, the two cohorts were meta-analyzed using the inverse-variance weighted method using fixed effects. Validation threshold was set at p < 0.05 with consistent direction of effect.
Meta-analysis.
Results from JHS, MESA, and HERITAGE were meta-analyzed together using mixed effects models in the `metà package of R4.0.5. Only variants with a p-value for association with a given protein < 1 × 10−5 in at least two of the studies were included.
Comparing to previous pQTLs.
To determine whether pQTLs were novel, we utilized the PhenoScanner package (version 2) for R.27,28 For each protein-locus association identified above, we divided the locus into 1MB or less segments (maximum permitted by PhenoScanner API) if needed. The resulting region or regions were then passed to the phenoscanner function in R, with the following arguments: build was set to ‘38’, p-value to 1 × 10−5, catalogue to ‘pQTL’, proxies set to ‘None’ (query date June 28, 2020). To supplement PhenoScanner, we reviewed the literature for additional studies using SomaScan or Olink to identify the genetic architecture of the plasma proteome and identified three not in PhenoScanner.2,6,7 Results from these studies were considered using the same criteria as above. If the protein linked to that region in our analysis was found to be previously associated with any variants in the region, this was considered a “previous” protein-locus association. For the subset of protein-locus associations that were previously described, we secondarily looked to see whether the sentinel SNP in JHS represented a novel genetic determinant. Sentinel SNPs were queried against both PhenoScanner and the three other studies to look for any variants associated with the same protein and in linkage disequilibrium with the new sentinel SNP. Again the phenoscanner function in R was used with the following arguments: build was set to ‘38’, p-value to 1 × 10−5, catalogue to ‘pQTL’, proxies set to “EUR” (as these variants were discovered in European populations), and r2 set to “0.5” (query date October 1, 2020).
Comparing to previous GWAS results.
To determine overlap between clinical GWAS analyses and pQTLs in this analysis, we utilized the PhenoScanner package for R. All 569 sentinel SNPs as identified above were passed to the phenoscanner function in R with the following arguments: build was set to ‘38’, p-value to ‘1 × 10-’5, catalogue to ‘GWAS’, r2 was set to ‘0.5’, proxies set to ‘AFR’ (query date October 15, 2020).
Comparing results to ClinVar data.
The entirety of the ClinVar database was downloaded from the NCBI FTP site (https://ftp.ncbi.nlm.nih.gov/pub/clinvar/tab_delimited/variant_summary.txt.gz, Access Date: 9/3/20).29 These data were merged to all variants associated with any protein in the JHS at a p-value < 5 × 10−6.
Variant annotations.
Reference allele frequencies from gnomAD30 and variant category from GENCODE31 were obtained from the Functional Annotation of Variants - Online Resource (available favor.genohub.org, download date July 20, 2020).32
Results
Whole genome association analysis of proteomic profiling
We performed whole genome association analysis between 28.1 million variants with an allele count in JHS of at least 5 and 1,301 plasma protein measures in 1852 self-identified Black individuals (61% women). Proteins exhibited a wide range of estimated total heritability (median heritability = 0.33, IQR 0.22 to 0.48, Supplemental Figure S1, Supplemental Table S2). Imputing proteins with non-converged heritability estimates to 0 resulted in a median heritability of 0.29 (See Methods).
At a Bonferroni adjusted significance cut off (5 × 10−8/1301 = 3.8 × 10−11), we identified 569 associations with 479 proteins encompassing 438 unique genetic loci (Figure 1, Supplemental Table S3). Each locus is a genomic region containing at least one variant associated with a protein but often summarizing multiple nearby variants in varying degrees of linkage disequilibrium (See Methods). The variant with the lowest p-value for association in each locus is considered the sentinel variant. Using this method, we identify 114 locus-protein associations not previously described. For previously described locus-protein associations, we identified novel sentinel variants in 217 loci.
Figure 1. Chromosomal locations of 569 protein quantitative trait loci.

The locations of the protein quantitative trait loci are indicated on the x-axis while location of the gene encoding that protein is indicated on the y-axis. Locations of genes associated with many proteins are indicated above the plot. Cis associations align along the identity line, while trans associations are off the line.
Across these 569 associations, 329 (58%) of the sentinel variants for the given locus are within 1MB of the transcription start site (TSS) of the cognate gene for that protein (termed cis), 240 are non-local (termed trans). We identified an additional 183 suggestive cis associations when the P value threshold was lowered to 5 × 10−6 (Supplemental Table S4). The majority of cis protein QTLs (pQTLs) were close to the TSS of the cognate gene, with 90% falling within 100kb of the TSS (Figure 2a).
Figure 2. Summary of protein quantitative trait loci.
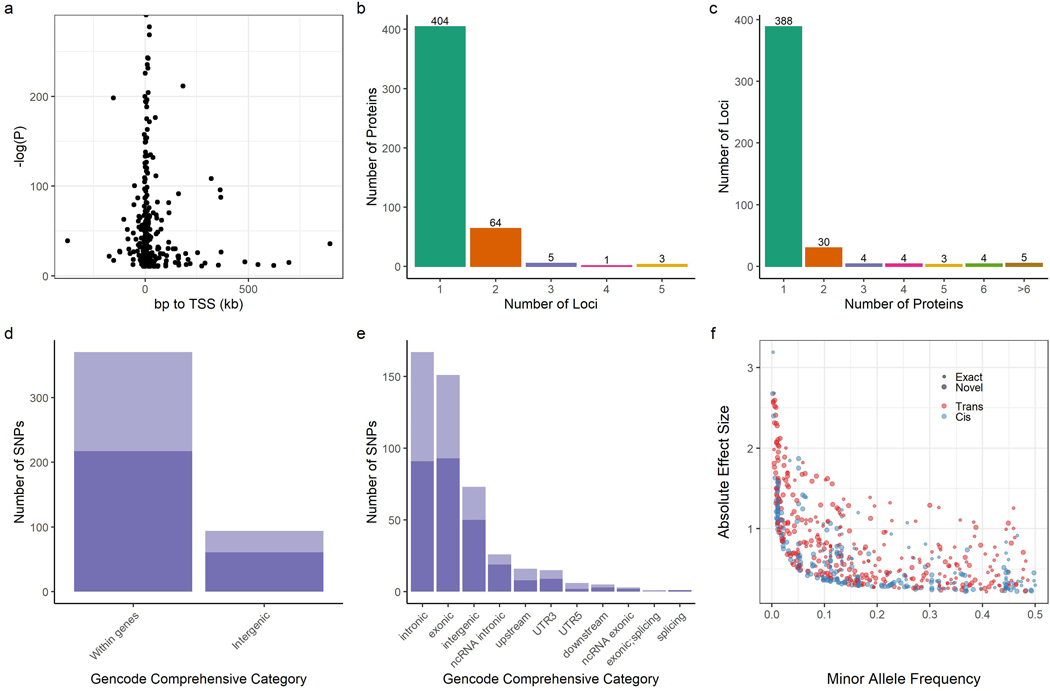
a, Significance level of cis associations according to distance from transcription start site for the cognate gene. b, Number of loci associated with each protein. c, Number of proteins associated with each locus. d,e, Proportion of pQTLs within and between genes, and by GENCODE comprehensive category for each pQTL, darker bars represent novel variant-protein associations. f, Absolute effect size versus minor allele frequency. Small circles indicate known sentinel variant-protein associations, large circles are novel associations. SNPs: single nucleotide polymorphisms. “Exact” indicates that the variant-protein association has been previously identified. “Novel” indicates that the variant-protein associations is novel.
The majority of proteins (70%) with a significant pQTL were associated with a single locus. Three proteins were associated with 5 different loci: Ck-beta-8–1, Cyclin-dependent kinase inhibitor 1B, and apolipoprotein L1 (Figure 2b).
Patterns observed in previous studies were replicated here: most loci (388, 89%) were associated with only one protein, though there were several pleiotropic loci including regions near the VTN, ABO, and APOE genes (Figure 2c), all of which have been implicated in cardiovascular disease.1,3,33–36 Sentinel variants were largely proximate to coding genes, with only 20% in intergenic regions (Figure 2d,e). There was a strong inverse relationship between effect size and minor allele frequency (MAF), consistent with previous protein QTL (pQTL) studies (Figure 2f).1
In contrast to previous studies, a significant number of sentinel variants had allele frequencies that varied substantially from those observed in European populations: 166 (36%) of the 464 identified sentinel variants in JHS had MAF < 1%, while 65 (14%) of the variants had MAF < 0.0001% among Non-Finnish Europeans in gnomAD.30 Many of these variants were much more common in JHS: among the 166 variants with MAF < 1% in Non-Finnish Europeans, 71 had MAF > 5% in JHS. Figure 3 illustrates the wide disparity between allele frequencies of all 569 sentinel variants in African vs Non-Finnish European populations in gnomAD.30
Figure 3. Ancestry specific reference allele frequencies for each sentinel variant.
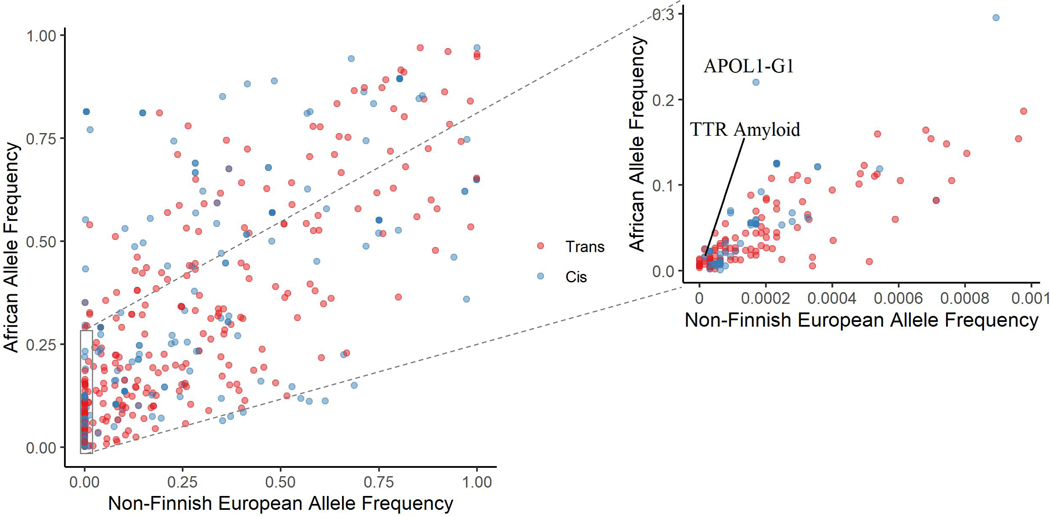
Allele frequency in gnomAD among those of African ancestry compared to Non-Finnish European (NFE) ancestry for all 464 unique sentinel variants. As many variants are rare among NFE individuals, a zoomed in subset is provided with African ancestry disease specific variants labeled. TTR = transthyretin.
We also completed proteomic profiling in two smaller cohorts, MESA (N=980, 53% women, 19% Black) and the HERITAGE Family Study (N=708, 56% women, 36% Black), each containing a subset of self-identified Black individuals, which were meta-analyzed (when possible) to validate the results. Consistent associations were observed for 90% of the 569 sentinel variants at a p-value < 0.05 with matching direction of effect. If a significance threshold adjusted for multiple corrections is used (p < 0.05/569 = 8.8 × 10−5), 72% replicate. Variants that did not replicate in some cases had lower MAF, falling below the minor allele count threshold of 5 in one of the two replication cohorts, reducing overall replication power. (Supplemental Table S3, Supplemental Figure S2). Results from JHS, MESA, and HERITAGE were also meta-analyzed together. This analysis yielded 13 additional pQTLs: 9 trans and 4 cis (Supplemental Table S5).
In a limited subsample of JHS participants (N=488), plasma samples were also profiled using the Olink® Explore platform, which utilizes a completely distinct, immunoassay-based approach for protein measurement, which generally rely on polyclonal antibody conjugates.37 Of the 569 sentinel variant-protein associations, 318 could be compared on the Olink® platform. These associations showed a consistent effect across the two platforms (correlation of effect = 0.82 [95% confidence interval: 0.78 to 0.85], Supplemental Figure S3). Across all 318 comparisons, the median Soma-Olink correlation was 0.62 (IQR 0.35 to 0.74). The direction of effect matched in 86%, and 51% of associations were confirmed at a Bonferroni (0.05/318) level of significance (Supplemental Table S3). There were a small number of discordant associations where effects as measured by SOMA and Olink were significant but with opposing directions of effect, such as the association between rs5744204 and Lipopolysaccharide-binding protein. These may indicate platform specific binding effects, but still support a genetic effect on protein levels as the most likely explanation, save for the unlikely possibility of opposing effects on just the binding of reagents from each platform.
While all pQTLs are listed in Supplemental Table S3, a subset of the results and information discussed in the following sections is highlighted in Table 1.
Table 1.
Selected pQTL Results fromSupplemental Table S3
| Target Full Name | Target | Sentinel SNP | SNP (hg38) | JHS AF | Non-Finish European AF gnomAD | African AF gnomAD | consequence | cis/trans | Nearest gene | Beta | SE | P-Value |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Thrombin | Thrombin | rs1801020 | 5:177409531:A:G | 0.551 | 0.750 | 0.551 | 5 prime UTR | trans | F12 | 0.46 | 0.03 | 3.8E-43 |
| Plasma serine protease inhibitor | PCI | rs1801020 | 5:177409531:A:G | 0.551 | 0.750 | 0.551 | 5 prime UTR | trans | F12 | −0.32 | 0.03 | 1.1E-26 |
| Neutrophil collagenase | MMP-8 | rs429358 | 19:44908684:T:C | 0.221 | 0.138 | 0.214 | missense | trans | APOE | 0.45 | 0.04 | 2.4E-28 |
| Kelch-like ECH-associated protein 1 | KEAP1 | rs769455 | 19:44908783:C:T | 0.019 | 0.000 | 0.021 | missense | trans | APOE | 1.20 | 0.12 | 9.1E-25 |
| Stromelysin-1 | MMP-3 | rs7412 | 19:44908822:C:T | 0.111 | 0.080 | 0.105 | missense | trans | APOE | −0.60 | 0.05 | 1.7E-32 |
| Beta-endorphin | b-Endorphin | rs7412 | 19:44908822:C:T | 0.111 | 0.080 | 0.105 | missense | trans | APOE | 0.35 | 0.05 | 2.2E-11 |
| Sonic hedgehog protein | Sonic Hedgehog | rs7412 | 19:44908822:C:T | 0.111 | 0.080 | 0.105 | missense | trans | APOE | 0.34 | 0.05 | 2.8E-11 |
| Tyrosine-protein kinase ZAP-70 | ZAP70 | rs7412 | 19:44908822:C:T | 0.111 | 0.080 | 0.105 | missense | trans | APOE | 0.61 | 0.05 | 3.3E-30 |
| Bone morphogenetic protein receptor type-2 | BMP RII | rs12117 | 11:6440254:G:A | 0.051 | 0.000 | 0.055 | missense | trans | HPX | 0.81 | 0.07 | 6.4E-31 |
| Natural killer cell receptor 2B4 | CD244 | rs12117 | 11:6440254:G:A | 0.051 | 0.000 | 0.055 | missense | trans | HPX | 0.53 | 0.07 | 1.5E-13 |
| Glial cell line-derived neurotrophic factor | GDNF | rs12117 | 11:6440254:G:A | 0.051 | 0.000 | 0.055 | missense | trans | HPX | 0.65 | 0.07 | 1.6E-19 |
| GTPase KRas | K-ras | rs12117 | 11:6440254:G:A | 0.051 | 0.000 | 0.055 | missense | trans | HPX | −1.31 | 0.07 | 2.5E-70 |
| Tumor necrosis factor | TNF-a | rs12117 | 11:6440254:G:A | 0.051 | 0.000 | 0.055 | missense | trans | HPX | 1.75 | 0.07 | 3.6E-126 |
| Tumor necrosis factor ligand superfamily member 18 | TNFSF18 | rs12117 | 11:6440254:G:A | 0.051 | 0.000 | 0.055 | missense | trans | HPX | 1.87 | 0.07 | 1.0E-145 |
| Nicotinamide phosphoribosyltransferase | PBEF | rs2066702 | 4:99307860:G:A | 0.191 | 0.002 | 0.189 | missense | trans | ADH1B | 0.28 | 0.04 | 5.6E-12 |
| Plasminogen | Plasminogen | rs576753655 | 4:173168604:C:T | 0.011 | 0.000 | 0.011 | upstream | trans | GALNT7 | 1.58 | 0.16 | 2.1E-23 |
| Angiostatin | Angiostatin | rs545617673 | 4:173168618:G:T | 0.011 | 0.000 | 0.011 | upstream | trans | GALNT7 | 1.58 | 0.16 | 1.0E-23 |
| Retinol-binding protein 4 | RBP | rs76992529 | 18:31598655:G:A | 0.018 | 0.000 | 0.016 | missense | trans | TTR | −0.91 | 0.13 | 2.5E-13 |
| Cytoskeleton-associated protein 2 | CKAP2 | rs73885319 | 22:36265860:A:G | 0.232 | 0.000 | 0.220 | missense | trans | APOL1 | 0.34 | 0.04 | 1.3E-17 |
| Protein S100-A9 | calgranulin B | rs10430455 | 1:157733448:T:A | 0.105 | 0.551 | 0.119 | intergenic | trans | FCRL2 | 0.36 | 0.05 | 2.2E-11 |
| Bactericidal permeability-increasing protein | BPI | rs2814778 | 1:159204893:T:C | 0.837 | 0.004 | 0.814 | 5 prime UTR | trans | ACKR1 | −0.30 | 0.04 | 9.3E-12 |
| C-X-C motif chemokine 11 | I-TAC | rs2814778 | 1:159204893:T:C | 0.837 | 0.004 | 0.814 | 5 prime UTR | trans | ACKR1 | −0.36 | 0.04 | 3.2E-16 |
| C-X-C motif chemokine 16 | CXCL16, soluble | rs2234355 | 3:45946488:G:A | 0.441 | 0.002 | 0.433 | missense | trans | CXCR6 | 0.52 | 0.03 | 5.7E-54 |
Novel genetic determinates of plasma proteins related to thrombosis, lipid biology and myocardial disease
To determine the novelty of the wide genomic regions identified as pQTLs by our analysis, we queried pQTL data available in PhenoScanner, a database of GWAS findings.27,28 Of the 569 protein-locus associations, 114 (20%) had not been previously identified (Figure 1, Supplemental Table S3) at a P value < 1 × 10−5. Of these 114 novel associations, 84 (74%) were trans associations. Sixty-two (54%) of the sentinel variants for these loci were uncommon (i.e., MAF < 1%) in Non-Finnish European populations, but had a median MAF in JHS of 5% (IQR 2% to 12%). Novel pQTLs provide the opportunity to better understand biological pathways. As an example, a variant in the 5-prime untranslated region of F12, the gene for clotting factor XII, is observed to be a novel pQTL for thrombin and plasma serine protease inhibitor. This variant has previously been shown to affect thrombin generation and the coagulation cascade.38
Similar to previous studies, we identify multiple pleiotropic genetic loci, which affect the levels of multiple proteins. The APOE locus is one such well-established locus, which is known to be associated with hypercholesterolemia, atherosclerotic heart disease, and Alzheimer’s disease,.39 Our analysis reveals six new proteins associated with this gene at three distinct (r2<0.1) missense variants: rs7412, rs769455, rs42935 (Figure 4a). These six proteins: b-Endorphin, matrix metalloproteinase-3 (MMP-3), Sonic Hedgehog, Zeta chain of T Cell receptor associated protein kinase 70 (ZAP70), Kelch-like ECH-associated protein 1, and matrix metalloproteinase-8 implicate new targets in understanding how APOE may mediates its effects. Indeed, APOE knockout mice, which develop atherosclerotic lesions that mimic human plaques, have shown reduced Zap70 activation.40 Further, MMP-3 levels have been shown to be elevated in affected areas of the brain among those with Alzheimer’s disease.41
Figure 4. In trans associations for novel pleiotropic protein quantitative trait loci.
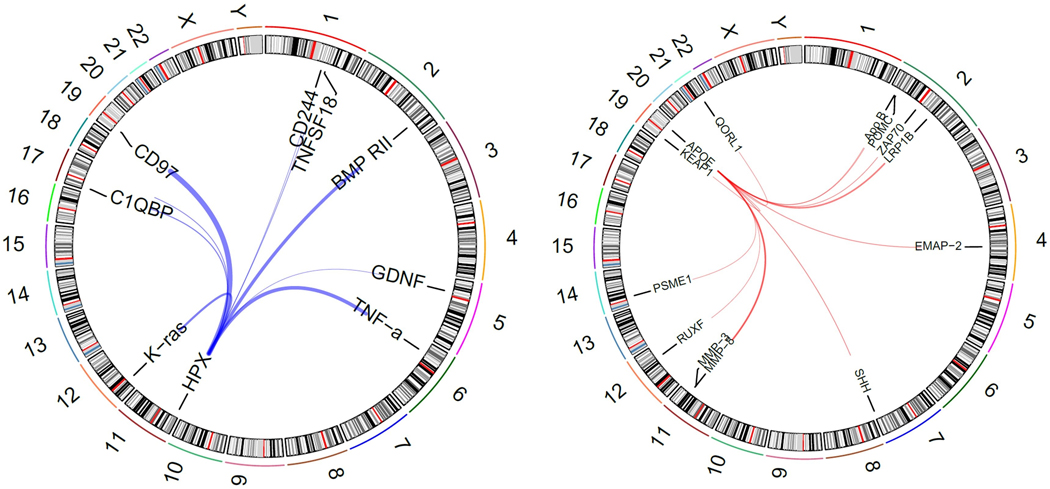
Two pleiotropic loci with new protein associations: HPX (blue) and APOE (red). The thickness of the lines indicates the relative strength of the association.
The analysis also shows a new pleiotropic locus at HPX, the gene for hemopexin. The sentinel variant, rs12117, is nearly monoallelic in European populations, but has a MAF in JHS of 5%. Six proteins are shown to be affected by this variant: Bone morphogenetic protein receptor type-2, Natural killer cell receptor 2B4, K-Ras, Glial cell line-derived neurotrophic factor, Tumor necrosis factor alpha, and Tumor necrosis factor ligand superfamily member 18. Another three proteins are associated with other variants either in or upstream of HPX (Figure 4a). It has been posited that hemopexin protects cells from oxidative stress by clearing heme, and TNFα is known to induce HPX expression in rats as an acute phase response.42 Further, HPX/APOE double knockout mice had accelerated atherosclerosis related to oxidative stress and changes in macrophage function. This role of hemopexin may be particularly important in Black patients with sickle-cell disease: murine models have shown the value of heme-scavenging by hemopexin in reducing inflammation in models of sickle-cell disease.43,44 Our findings suggest specific genetic variation may have a role in the immune functions of hemopexin. While no members of our cohort had sickle-cell disease, 24 individuals did have both the minor allele of rs12117 and sickle-cell trait. However, no definitive interaction between these two variants and any protein could be identified. Unfortunately, given the very low frequency of the variant in European-based GWAS, no clinical implications for rs12117 have been identified, though other variants in HPX have been linked to ulcerative colitis.45 Further data is needed; specifically data from patients with sickle-cell disease would be of value.
Our analysis can implicate new biology related to previously described variants as well. The variant rs2066702 in ADH1B has been identified as a risk locus for alcohol dependence across multiple ancestry specific GWAS.46 The same variant in our analysis is associated with levels of nicotinamide phosphoribosyltransferase (NAMPT, Supplemental Figure S4a), which regulates intracellular NAD+, and plays a role in cardiac hypertrophy and adverse remodeling.47 Importantly, the minor allele of rs2066702 is protective of alcohol dependence, and it is this allele that is associated with higher levels of NAMPT, suggesting that alcohol use may deplete NAMPT in humans. Furthermore, prior murine studies have shown that ethanol administration diminished NAMPT levels, while overexpression of NAMPT was found to protect against steatosis.48
Conversely, the associations between well-described proteins and poorly understood genes can further elucidate biology. Levels of two proteins, plasminogen and angiostatin (itself a fragment of plasminogen) were linked to a variant upstream of GALNT7 (Supplemental Figure S4b). Plasminogen and angiostatin each have a strong cis pQTL, supporting aptamer specificity for their measurement (Supplemental Table S3 & S4). While plasminogen and angiostatin are critical factors in clot dissolution and angiogenesis inhibition,49,50 respectively, the biological role of GALNT7, a glycosyltransferase, has been linked by more limited evidence to cancer proliferation.51 The sentinel SNPs linked to these proteins in our analysis are monoallelic in European populations, so prior GWAS data do not exist. However, other variants at the GALNT7 locus have been linked to vascular disorders in the UK Biobank including “Cause of death: peripheral vascular disease, unspecified” (P = 1 × 10−23), “Cause of death: vascular dementia, unspecified” (P = 8 × 10−20), and “Cause of death: chronic or unspecified with haemorrhage” (P = 2 × 10−17) all three of which are plausibly mediated by plasminogen or angiostatin.27,28
Known ancestry-specific loci highlight ancestry-specific cardiovascular disease pathways
Analysis of samples from individuals of greater African ancestry allows for assessment of specific loci known to be of particular clinical importance in individuals of African descent. We evaluated the proteomic signatures of four such well-described loci.
Transthyretin (TTR) amyloidosis results from the misfolding of the transthyretin tetramer, ultimately resulting in abnormal protein deposition in myocardium and nerve tissue, leading to cardiomyopathy and neuropathy. Protein misfolding is accelerated in the presence of mutations in the TTR gene; specifically, rs76992529 encodes a V122I mutation that is found in 3–4% of Black individuals. In our data we show this variant to be a robust pQTL for retinol-binding protein 4 (RBP4), a binding partner of TTR.52 In individuals with TTR amyloidosis and overt myocardial disease (typically manifested as left ventricular (LV) thickening and diastolic dysfunction), RBP4 levels are known to be diminished – the normal transthyretin tetramer protects RBP4 from renal clearance.53 However, our data show that asymptomatic carriers of this mutation have diminished RBP4 levels as well, even in the absence of reported heart failure (Figure 5a). To further explore this finding, we leveraged extensive metabolite profiling in JHS.54 We found an unknown metabolite feature highly correlated with circulating RBP4, (Pearson correlation 0.64 [CI 0.61 to 0.66]). As expected, the association between this metabolite and the V122I mutation was also quite strong (β = −0.76, P = 4.6 × 10−14). This metabolite feature has a mass-to-charge ratio of 269.226, which strongly suggests its identity as a dehydrated form of retinol, according to Human Metabolome Database, the binding partner of RBP4. These data further complement and validate our proteomic association of RBP4 and TTR. Larger datasets are needed to explore the functional consequences of these proteomic and metabolomic findings.
Figure 5. Ancestry specific genetic disease variants and protein levels.
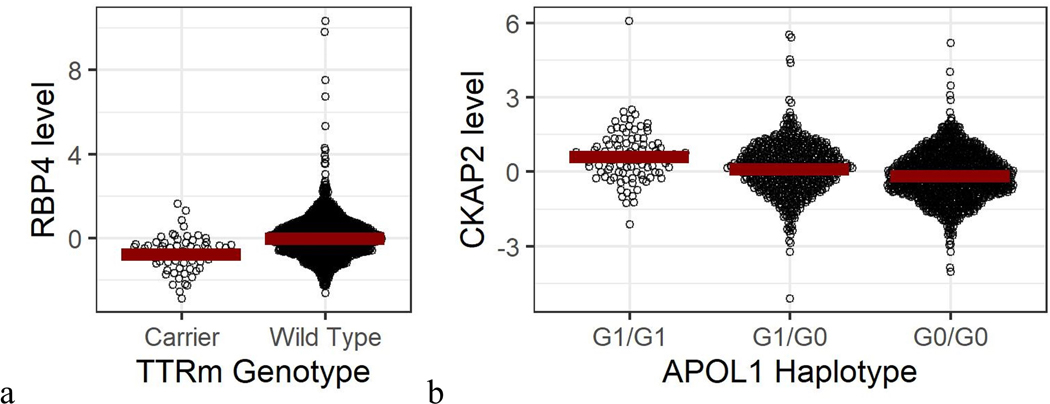
a, TTRm carrier status (rs76992529) and log-scaled RB4 levels. b, APOL1*G1 haplotype status and log-scaled CKAP2 levels
Two alleles in the APOL1 gene (rs73885319/rs60910145 or “G1” and rs71785313 or “G2”) are linked to chronic kidney disease and cardiovascular disease in JHS and are common in individuals with African ancestry.55–57 In JHS, rs73885319 has a MAF of 23%, whereas the variant is not present persons of European ancestry in gnomAD. In addition to being associated with levels of APOL1 in our analysis, it was also the sentinel SNP determining levels of cytoskeleton associated protein 2 (CKAP2, Figure 5b). CKAP2 has been linked to tumor formation as it has a role in mitosis, but has also been observed to be upregulated in renal tubular necrosis.58,59 In models adjusted for age, sex, body mass index, systolic blood pressure, presence of hypertension, presence of diabetes, HbA1c, and proteomic batch/plate, CKAP2 levels as measured by SOMAscan were associated with increased estimated glomerular filtration rate in JHS (β = 1.16, p = 0.002). Since APOL1 risk variants are associated with renal disease, this could point to a protective role for CKAP2 in response to APOL1 genetic risk, requiring further investigation as a therapeutic target.
The Duffy chemokine receptor (DARC) is a binding site crucial to malarial infection with P. vivax, but has also been shown to affect risk for cardiovascular outcomes in JHS.60 Under positive selection in sub-Saharan Africa, the FY*O allele of this gene is thus common in individuals of African descent, though it is present in only 0.4% of individuals of Non-Finnish European descent in gnomAD.30 Levels of CCL14 and Eotaxin have previously been linked to this gene, and to this list we now add protein S100-A9, CXCL11, and bactericidal permeability-increasing protein. Despite being linked to neutropenia, the Duffy-null allele has not been shown to lead to an increased risk of infection.61 However, there is evidence of a slower progression of HIV infection in the Duffy-null state.62 These results expand the list of inflammatory mediators affected by the Duffy-null state.
Finally, the variant that causes sickle cell trait, rs334, has an allele frequency of 4% in JHS. This variant was associated with fractalkine (P-value = 2.5 × 10−6). Previous work has linked fractalkine, an inflammatory cytokine, to incident heart failure, specifically in Black individuals.63
Protein associations for clinically relevant variants
Among the other 435 protein-locus pairs with previously identified pQTLs in the same region, 44 of the previous pQTLs were at P values > 5 × 10−8, and 177 of the previous pQTLs differed from the sentinel variants identified in JHS (r2 < 0.5). Thus, even in genetic regions previously linked to a given protein, many sentinel variants identified in this analysis may point to novel genetic effects when combined with existing genetic databases (Supplemental Tables S6 and S7). As an example, the variant rs2234355 in the CXCR6 gene is nearly monoallelic in European populations, but is common among African populations, and thus well represented in JHS (MAF 44%). The variant has been previously shown to be protective against Pneumocystis jiorvecii infection in HIV infected individuals, and was more common in those achieving viremic control.64,65 Interactions between CXCR6 and its ligand CXCL16 have been posited as a potential mechanism; we show this variant to be a strong (p = 5.7 × 10−54) sentinel pQTL for CXCL16, supporting this hypothesis. The relationship may also have cardiovascular consequences, as CXCL16 levels have been associated acute coronary syndromes.66
Discussion
Our data represent a comprehensive effort to understand the genetic determinants of the circulating plasma proteome using whole genome sequence analysis in individuals with greater genetic diversity than those in prior analyses. We identify numerous novel genetic determinants of a wide range of circulating proteins, many of which are important in vascular and cardiac biology. Many of these genetic variants have known clinical implications, in which case our data delineate novel biology potentially linking genetic variation to disease. As an example, the genetic mutation associated with TTR amyloidosis in persons of African ancestry, rs76992529, is shown here to be associated with RBP4 levels in persons without overt cardiomyopathy. A very recent study from the BioMe database found a similar difference among persons with this mutation and without cardiomyopathy.67 Our findings extend the small case-control biobank study to a large, well-defined prospective cohort, advancing RBP4 levels as a potential pre-clinical biomarker. Further studies are needed to determine if there is an interaction between this mutation, RBP4 levels, and incident cardiomyopathy.
In other cases, the proteomic associations identified represent the first meaningful annotation of a given genetic variant. Such is the case for rs12117, a missense variant in the gene for hemopexin. Despite a MAF of ~2.6% in persons of African ancestry, little is known about this variant. Here, we describe it as a pleiotropic locus, affecting the levels of multiple inflammatory proteins. Given hemopexin’s role in heme-scavenging, identifying additional carriers, particularly those with sickle-cell disease, may offer critical insights, and the proteins identified here would be useful starting points. The paucity of genome-wide association data in diverse populations limits our ability to interrogate associations, such as rs12117, with tools such as Mendelian randomization but hopefully highlights the need for greater inclusion of diverse populations in genetic research going forward. Greater diversity in genetic association studies will not only increase our understanding of functional genomics but may also help delineate gene-environment interactions that affect individuals of diverse ancestry. Indeed, our analysis identifies novel variants which are not particularly rare in Europeans, but are only now described in a cohort of Black Americans. This finding suggests the possibility of gene-environment interactions, including, importantly, the effects of social and structural differences which have biological/health effects at multiple levels (healthcare access, stress response, environmental toxins, etc).68 Such future work is important not only for the populations themselves, but also for optimum understanding of the genomic basis of biological variability and disease susceptibility.
Future work leveraging these data may also center around the intriguing finding of genetic variants that produce opposing findings on the Soma platform compared to the Olink platform. These variants, often protein altering, likely affect binding of one platform, but the significant opposing effects suggest they are true pQTLs. Understanding the implications of such variants on a genome-wide scale may identify functionally important gene-regions and inform interpretation of binding data.
Our study has several strengths: as mentioned it is the largest analysis of its kind in a Black population which gives it the power to detect many novel variants. The results are compared to two multi-ethnic populations and an alternate profiling platform. Our study also has several important limitations. While this is the largest pQTL analysis in a Black population, the sample size for genome-wide association is relatively modest compared to many GWAS. This also informs a second limitation, the use of multi-ancestry cohorts for validation rather than a population of similar ancestry to JHS. This fact is related to limited availability of proteomic data in Black persons, and the desire to maintain an adequate sample size for validation of our original findings. For example, all 980 MESA participants with proteomics are included, regardless of their racial or ethnic identification in the hopes that statistical validation can be performed on as many variants as possible. Limiting MESA to only the Black participants would have left only 190 individuals. A further limitation is aptamer specificity on the SomaScan platform. While cis pQTLs (both from this study and others) and validation on the Olink platform can confirm aptamer specificity, off target effects may be falsely attributed as trans-pQTLs, though we expect most cases off non-specificity to bias toward the null. Aptamer validation efforts beyond those included here are ongoing across many groups.1,2,69,70
Taken together, our work highlights the importance of extending proteomics, genomics, and likely other -omics studies, to diverse populations, both to identify important potential biomarkers and disease pathways in those populations, but also in the human population at large.
Supplementary Material
Clinical Perspective.
What is new?
First study to look to examine the genetic architecture of the plasma proteome using whole genome sequencing in persons of African ancestry, providing a chance to look at rare, ancestry-specific variation.
Adds 114 novel genomic loci associated with protein levels in human samples
Clinical Implications
Genetic variant associated with amyloidosis in persons of African ancestry shown to be associated with RBP4 levels, even in those without cardiomyopathy, implicating it as a potential biomarker
Acknowledgements
JHS
The authors wish to thank the staff and participants of the JHS.
HERITAGE
We thank Drs. Arthur S. Leon, D.C. Rao, James S. Skinner, Tuomo Rankinen, Jacques Gagnon, and the late Jack H. Wilmore for contributions to the planning, data collection, and conduct of the HERITAGE project.
Funding Sources
Dr. Katz is supported by National Heart, Lung and Blood Institute T32 postdoctoral training grant (T32HL007374–40). Dr. Tahir is supported by the Ruth L. Kirchstein post-doctoral individual National Research Award (F32HL150992). Dr. Bick is supported by NIH DP5-OD029586–01 and is a recipient of a Career Award for Medical Scientists from the Burroughs Wellcome Foundation. Dr. Cruz is supported by the KL2/Catalyst Medical Research Investigator Training award from Harvard Catalyst (NIH/National Center for Advancing Translational Sciences Award TR002542). Dr. Robbins is supported by the John S. LaDue Memorial Fellowship in Cardiology at Harvard Medical School. Dr. Benson is supported by National Heart, Lung and Blood Institute K08HL145095 award. Dr. Natarajan is supported by NIH R01HL142711. Drs. Gerszten, Wang and Wilson are supported by NIH R01 DK081572. Drs. Gerszten, Wang, and Vasan are supported by NIH R01 HL132320. Drs. Gerszten and Vasan are supported by National Institute on Aging Grant RF1AG063507.
Jackson Heart Study
The Jackson Heart Study (JHS) is supported and conducted in collaboration with Jackson State University (HHSN268201800013I), Tougaloo College (HHSN268201800014I), the Mississippi State Department of Health (HHSN268201800015I/HHSN26800001) and the University of Mississippi Medical Center (HHSN268201800010I, HHSN268201800011I and HHSN268201800012I) contracts from the National Heart, Lung, and Blood Institute and the National Institute for Minority Health and Health Disparities.
Molecular data for the Trans-Omics in Precision Medicine (TOPMed) program was supported by the National Heart, Lung and Blood Institute (NHLBI). Genome sequencing for “NHLBI TOPMed: The Jackson Heart Study” (phs000964.v1.p1) was performed at the Northwest Genomics Center (HHSN268201100037C). Core support including centralized genomic read mapping and genotype calling, along with variant quality metrics and filtering were provided by the TOPMed Informatics Research Center (3R01HL-117626–02S1; contract HHSN268201800002I). Core support including phenotype harmonization, data management, sample-identity QC, and general program coordination were provided by the TOPMed Data Coordinating Center (R01HL-120393; U01HL-120393; contract HHSN268201800001I). We gratefully acknowledge the studies and participants who provided biological samples and data for TOPMed.
TOPMed MESA Multi-Omics/MESA Study Acknowledgement
Whole genome sequencing (WGS) for the Trans-Omics in Precision Medicine (TOPMed) program was supported by the National Heart, Lung and Blood Institute (NHLBI). WGS for “NHLBI TOPMed: Multi-Ethnic Study of Atherosclerosis (MESA)” (phs001416.v1.p1) was performed at the Broad Institute of MIT and Harvard (3U54HG003067–13S1). Centralized read mapping and genotype calling, along with variant quality metrics and filtering were provided by the TOPMed Informatics Research Center (3R01HL-117626–02S1). Phenotype harmonization, data management, sample-identity QC, and general study coordination, were provided by the TOPMed Data Coordinating Center (3R01HL-120393–02S1). The MESA projects are conducted and supported by the National Heart, Lung, and Blood Institute in collaboration with MESA investigators.
Support for the Multi-Ethnic Study of Atherosclerosis (MESA) projects are conducted and supported by the National Heart, Lung, and Blood Institute in collaboration with MESA investigators. Support for MESA is provided by contracts 75N92020D00001, HHSN268201500003I, N01-HC-95159, 75N92020D00005, N01-HC-95160, 75N92020D00002, N01-HC-95161, 75N92020D00003, N01-HC-95162, 75N92020D00006, N01-HC-95163, 75N92020D00004, N01-HC-95164, 75N92020D00007, N01-HC-95165, N01-HC-95166, N01-HC-95167, N01-HC-95168, N01-HC-95169, UL1-TR-000040, UL1-TR-001079, and UL1-TR-001420. Also supported in part by the National Center for Advancing Translational Sciences, CTSI grant UL1TR001881, and the National Institute of Diabetes and Digestive and Kidney Disease Diabetes Research Center grant DK063491 to the Southern California Diabetes Endocrinology Research Center.
HERITAGE
This research was partially funded by National Heart, Lung, and Blood Institute Grants HL-45670, HL-47317, HL-47321, HL-47323, and HL-47327, all in support of the HERITAGE Family Study. C. B. is partially funded by the John W. Barton Sr. Chair in Genetics and Nutrition, and NIH Centers of Biomedical Research Excellence grant (NIH P30GM118430–01). Dr. Sarzynski is supported by R01HL146462.
Nonstandard abbreviations
- dbGaP
database of Genotypes and Phenotypes
- GRM
genomic relatedness matrix
- JHS
Jackson Heart Study
- LD
linkage disequilibrium
- LV
left ventricle
- MAF
minor allele frequency
- MESA
Multi-Ethnic Study of Atherosclerosis
- NFE
non-Finnish European
- PC
principle component
- pQTL
protein quantitative trait locus
- TOPMed
Trans-Omics for Precision Medicine
- TSS
transcription start site
- TTR
transthyretin
- WGS
whole genome sequencing
Appendix
Abe, Namiko
Abecasis, Gonçalo
Aguet, Francois
Albert, Christine
Almasy, Laura
Alonso, Alvaro
Ament, Seth
Anderson, Peter
Anugu, Pramod
Applebaum-Bowden, Deborah
Ardlie, Kristin
Arking, Dan
Arnett, Donna K
Ashley-Koch, Allison
Aslibekyan, Stella
Assimes, Tim
Auer, Paul
Avramopoulos, Dimitrios
Ayas, Najib
Balasubramanian, Adithya
Barnard, John
Barnes, Kathleen
Barr, R. Graham
Barron-Casella, Emily
Barwick, Lucas
Beaty, Terri
Beck, Gerald
Becker, Diane
Becker, Lewis
Beer, Rebecca
Beitelshees, Amber
Benjamin, Emelia
Benos, Takis
Bezerra, Marcos
Bielak, Larry
Bis, Joshua
Blackwell, Thomas
Blangero, John
Boerwinkle, Eric
Bowden, Donald W.
Bowler, Russell
Brody, Jennifer
Broeckel, Ulrich
Broome, Jai
Brown, Deborah
Bunting, Karen
Burchard, Esteban
Bustamante, Carlos
Buth, Erin
Cade, Brian
Cardwell, Jonathan
Carey, Vincent
Carrier, Julie
Carson, April
Carty, Cara
Casaburi, Richard
Casas Romero, Juan P
Casella, James
Castaldi, Peter
Chaffin, Mark
Chang, Christy
Chang, Yi-Cheng
Chasman, Daniel
Chavan, Sameer
Chen, Bo-Juen
Chen, Wei-Min
Chen, Yii-Der Ida
Cho, Michael
Choi, Seung Hoan
Chuang, Lee-Ming
Chung, Mina
Chung, Ren-Hua
Clish, Clary
Comhair, Suzy
Conomos, Matthew
Cornell, Elaine
Correa, Adolfo
Crandall, Carolyn
Crapo, James
Cupples, L. Adrienne
Curran, Joanne
Curtis, Jeffrey
Custer, Brian
Damcott, Coleen
Darbar, Dawood
David, Sean
Davis, Colleen
Daya, Michelle
de Andrade, Mariza
de las Fuentes, Lisa
de Vries, Paul
DeBaun, Michael
Deka, Ranjan
DeMeo, Dawn
Devine, Scott
Dinh, Huyen
Doddapaneni, Harsha
Duan, Qing
Dugan-Perez, Shannon
Duggirala, Ravi
Durda, Jon Peter
Dutcher, Susan K.
Eaton, Charles
Ekunwe, Lynette
El Boueiz, Adel
Ellinor, Patrick
Emery, Leslie
Erzurum, Serpil
Farber, Charles
Farek, Jesse
Fingerlin, Tasha
Flickinger, Matthew
Fornage, Myriam
Franceschini, Nora
Frazar, Chris
Fu, Mao
Fullerton, Stephanie M.
Fulton, Lucinda
Gabriel, Stacey
Gan, Weiniu
Gao, Shanshan
Gao, Yan
Gass, Margery
Geiger, Heather
Gelb, Bruce
Geraci, Mark
Germer, Soren
Gerszten, Robert
Ghosh, Auyon
Gibbs, Richard
Gignoux, Chris
Gladwin, Mark
Glahn, David
Gogarten, Stephanie
Gong, Da-Wei
Goring, Harald
Graw, Sharon
Gray, Kathryn J.
Grine, Daniel
Gross, Colin
Gu, C. Charles
Guan, Yue
Guo, Xiuqing
Gupta, Namrata
Haas, David M.
Haessler, Jeff
Hall, Michael
Han, Yi
Hanly, Patrick
Harris, Daniel
Hawley, Nicola L.
He, Jiang
Heavner, Ben
Heckbert, Susan
Hernandez, Ryan
Herrington, David
Hersh, Craig
Hidalgo, Bertha
Hixson, James
Hobbs, Brian
Hokanson, John
Hong, Elliott
Hoth, Karin
Hsiung, Chao (Agnes)
Hu, Jianhong
Hung, Yi-Jen
Huston, Haley
Hwu, Chii Min
Irvin, Marguerite Ryan
Jackson, Rebecca
Jain, Deepti
Jaquish, Cashell
Johnsen, Jill
Johnson, Andrew
Johnson, Craig
Johnston, Rich
Jones, Kimberly
Kang, Hyun Min
Kaplan, Robert
Kardia, Sharon
Kelly, Shannon
Kenny, Eimear
Kessler, Michael
Khan, Alyna
Khan, Ziad
Kim, Wonji
Kimoff, John
Kinney, Greg
Konkle, Barbara
Kooperberg, Charles
Kramer, Holly
Lange, Christoph
Lange, Ethan
Lange, Leslie
Laurie, Cathy
Laurie, Cecelia
LeBoff, Meryl
Lee, Jiwon
Lee, Sandra
Lee, Wen-Jane
LeFaive, Jonathon
Levine, David
Levy, Dan
Lewis, Joshua
Li, Xiaohui
Li, Yun
Lin, Henry
Lin, Honghuang
Lin, Xihong
Liu, Simin
Liu, Yongmei
Liu, Yu
Loos, Ruth J.F.
Lubitz, Steven
Lunetta, Kathryn
Luo, James
Magalang, Ulysses
Mahaney, Michael
Make, Barry
Manichaikul, Ani
Manning, Alisa
Manson, JoAnn
Martin, Lisa
Marton, Melissa
Mathai, Susan
Mathias, Rasika
May, Susanne
McArdle, Patrick
McDonald, Merry-Lynn
McFarland, Sean
McGarvey, Stephen
McGoldrick, Daniel
McHugh, Caitlin
McNeil, Becky
Mei, Hao
Meigs, James
Menon, Vipin
Mestroni, Luisa
Metcalf, Ginger
Meyers, Deborah A
Mignot, Emmanuel
Mikulla, Julie
Min, Nancy
Minear, Mollie
Minster, Ryan L
Mitchell, Braxton D.
Moll, Matt
Momin, Zeineen
Montasser, May E.
Montgomery, Courtney
Muzny, Donna
Mychaleckyj, Josyf C
Nadkarni, Girish
Naik, Rakhi
Naseri, Take
Natarajan, Pradeep
Nekhai, Sergei
Nelson, Sarah C.
Neltner, Bonnie
Nessner, Caitlin
Nickerson, Deborah
Nkechinyere, Osuji
North, Kari
O’Connell, Jeff
O’Connor, Tim
Ochs-Balcom, Heather
Okwuonu, Geoffrey
Pack, Allan
Paik, David T.
Palmer, Nicholette
Pankow, James
Papanicolaou, George
Parker, Cora
Peloso, Gina
Peralta, Juan Manuel
Perez, Marco
Perry, James
Peters, Ulrike
Peyser, Patricia
Phillips, Lawrence S
Pleiness, Jacob
Pollin, Toni
Post, Wendy
Powers Becker, Julia
Preethi Boorgula, Meher
Preuss, Michael
Psaty, Bruce
Qasba, Pankaj
Qiao, Dandi
Qin, Zhaohui
Rafaels, Nicholas
Raffield, Laura
Rajendran, Mahitha
Ramachandran, Vasan S.
Rao, D.C.
Rasmussen-Torvik, Laura
Ratan, Aakrosh
Redline, Susan
Reed, Robert
Reeves, Catherine
Regan, Elizabeth
Reiner, Alex
Reupena, Muagututi’a Sefuiva
Rice, Ken
Rich, Stephen
Robillard, Rebecca
Robine, Nicolas
Roden, Dan
Roselli, Carolina
Rotter, Jerome
Ruczinski, Ingo
Runnels, Alexi
Russell, Pamela
Ruuska, Sarah
Ryan, Kathleen
Sabino, Ester Cerdeira
Saleheen, Danish
Salimi, Shabnam
Salvi, Sejal
Salzberg, Steven
Sandow, Kevin
Sankaran, Vijay G.
Santibanez, Jireh
Schwander, Karen
Schwartz, David
Sciurba, Frank
Seidman, Christine
Seidman, Jonathan
Sériès, Frédéric
Sheehan, Vivien
Sherman, Stephanie L.
Shetty, Amol
Shetty, Aniket
Sheu, Wayne Hui-Heng
Shoemaker, M. Benjamin
Silver, Brian
Silverman, Edwin
Skomro, Robert
Smith, Albert Vernon
Smith, Jennifer
Smith, Josh
Smith, Nicholas
Smith, Tanja
Smoller, Sylvia
Snively, Beverly
Snyder, Michael
Sofer, Tamar
Sotoodehnia, Nona
Stilp, Adrienne M.
Storm, Garrett
Streeten, Elizabeth
Su, Jessica Lasky
Sung, Yun Ju
Sylvia, Jody
Szpiro, Adam
Taliun, Daniel
Tang, Hua
Taub, Margaret
Taylor, Kent D.
Taylor, Matthew
Taylor, Simeon
Telen, Marilyn
Thornton, Timothy A.
Threlkeld, Machiko
Tinker, Lesley
Tirschwell, David
Tishkoff, Sarah
Tiwari, Hemant
Tong, Catherine
Tracy, Russell
Tsai, Michael
Vaidya, Dhananjay
Van Den Berg, David
VandeHaar, Peter
Vrieze, Scott
Walker, Tarik
Wallace, Robert
Walts, Avram
Wang, Fei Fei
Wang, Heming
Wang, Jiongming
Watson, Karol
Watt, Jennifer
Weeks, Daniel E.
Weinstock, Joshua
Weir, Bruce
Weiss, Scott T
Weng, Lu-Chen
Wessel, Jennifer
Willer, Cristen
Williams, Kayleen
Williams, L. Keoki
Wilson, Carla
Wilson, James
Winterkorn, Lara
Wong, Quenna
Wu, Joseph
Xu, Huichun
Yanek, Lisa
Yang, Ivana
Yu, Ketian
Zekavat, Seyedeh Maryam
Zhang, Yingze
Zhao, Snow Xueyan
Zhao, Wei
Zhu, Xiaofeng
Zody, Michael
Zoellner, Sebastian
Footnotes
Disclaimer
The views expressed in this manuscript are those of the authors and do not necessarily represent the views of the National Heart, Lung, and Blood Institute; the National Institutes of Health; or the U.S. Department of Health and Human Services
Disclosures
None.
References
- 1.Sun BB, Maranville JC, Peters JE, Stacey D, Staley JR, Blackshaw J, Burgess S, Jiang T, Paige E, Surendran P, et al. Genomic atlas of the human plasma proteome. Nature. 2018;558:73–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Emilsson V, Ilkov M, Lamb JR, Finkel N, Gudmundsson EF, Pitts R, Hoover H, Gudmundsdottir V, Horman SR, Aspelund T, et al. Co-regulatory networks of human serum proteins link genetics to disease. Science. 2018;361:769–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Suhre K, Arnold M, Bhagwat AM, Cotton RJ, Engelke R, Raffler J, Sarwath H, Thareja G, Wahl A, DeLisle RK, et al. Connecting genetic risk to disease end points through the human blood plasma proteome. Nat Commun. 2017;8:14357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yao C, Chen G, Song C, Keefe J, Mendelson M, Huan T, Sun BB, Laser A, Maranville JC, Wu H, et al. Genome-wide mapping of plasma protein QTLs identifies putatively causal genes and pathways for cardiovascular disease. Nat Commun. 2018;9:3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Folkersen L, Fauman E, Sabater-Lleal M, Strawbridge RJ, Frånberg M, Sennblad B, Baldassarre D, Veglia F, Humphries SE, Rauramaa R, et al. Mapping of 79 loci for 83 plasma protein biomarkers in cardiovascular disease. PLoS Genet. 2017;13:e1006706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Folkersen L, Gustafsson S, Wang Q, Hansen DH, Hedman ÅK, Schork A, Page K, Zhernakova DV, Wu Y, Peters J, et al. Genomic and drug target evaluation of 90 cardiovascular proteins in 30,931 individuals. Nature Metabolism. 2020;2:1135–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Benson MD, Yang Q, Ngo D, Zhu Y, Shen D, Farrell LA, Sinha S, Keyes MJ, Vasan RS, Larson MG, et al. The Genetic Architecture of the Cardiovascular Risk Proteome. Circulation. 2018;137:1158–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.1000 Genomes Project Consortium, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA, et al. A global reference for human genetic variation. Nature. 2015;526:68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McClellan JM, Lehner T, King M-C. Gene Discovery for Complex Traits: Lessons from Africa. Cell. 2017;171:261–264. [DOI] [PubMed] [Google Scholar]
- 10.Kotowski IK, Pertsemlidis A, Luke A, Cooper RS, Vega GL, Cohen JC, Hobbs HH. A spectrum of PCSK9 alleles contributes to plasma levels of low-density lipoprotein cholesterol. Am J Hum Genet. 2006;78:410–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Taylor HA, Wilson JG, Jones DW, Sarpong DF, Srinivasan A, Garrison RJ, Nelson C, Wyatt SB. Toward resolution of cardiovascular health disparities in African Americans: design and methods of the Jackson Heart Study. Ethn Dis. 2005;15:S6–4–17. [PubMed] [Google Scholar]
- 12.Bild DE, Bluemke DA, Burke GL, Detrano R, Diez Roux AV, Folsom AR, Greenland P, Jacob DR, Kronmal R, Liu K, et al. Multi-Ethnic Study of Atherosclerosis: objectives and design. Am J Epidemiol. 2002;156:871–881. [DOI] [PubMed] [Google Scholar]
- 13.Bouchard C, Leon AS, Rao DC, Skinner JS, Wilmore JH, Gagnon J. The HERITAGE family study. Aims, design, and measurement protocol. Med Sci Sports Exerc. 1995;27:721–729. [PubMed] [Google Scholar]
- 14.Taliun D, Harris DN, Kessler MD, Carlson J, Szpiech ZA, Torres R, Taliun SAG, Corvelo A, Gogarten SM, Kang HM, et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature. 2021;590:290–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ngo D, Sinha S, Shen D, Kuhn EW, Keyes MJ, Shi X, Benson MD, O’Sullivan JF, Keshishian H, Farrell LA, et al. Aptamer-Based Proteomic Profiling Reveals Novel Candidate Biomarkers and Pathways in Cardiovascular Disease. Circulation. 2016;134:270–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Raffield LM, Zakai NA, Duan Q, Laurie C, Smith JD, Irvin MR, Doyle MF, Naik RP, Song C, Manichaikul AW, et al. D-Dimer in African Americans: Whole Genome Sequence Analysis and Relationship to Cardiovascular Disease Risk in the Jackson Heart Study. Arterioscler Thromb Vasc Biol. 2017;37:2220–2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tan A, Abecasis GR, Kang HM. Unified representation of genetic variants. Bioinformatics. 2015;31:2202–2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Das S, Forer L, Schönherr S, Sidore C, Locke AE, Kwong A, Vrieze SI, Chew EY, Levy S, McGue M, et al. Next-generation genotype imputation service and methods. Nat Genet. 2016;48:1284–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Polfus LM, Raffield LM, Wheeler MM, Tracy RP, Lange LA, Lettre G, Miller A, Correa A, Bowler RP, Bis JC, et al. Whole genome sequence association with E-selectin levels reveals loss-of-function variant in African Americans. Hum Mol Genet. 2019;28:515–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Conomos MP, Miller MB, Thornton TA. Robust inference of population structure for ancestry prediction and correction of stratification in the presence of relatedness. Genet Epidemiol. 2015;39:276–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jiang L, Zheng Z, Qi T, Kemper KE, Wray NR, Visscher PM, Yang J. A resource-efficient tool for mixed model association analysis of large-scale data. Nature Genetics. 2019;51:1749–1755. [DOI] [PubMed] [Google Scholar]
- 22.Sofer T, Zheng X, Gogarten SM, Laurie CA, Grinde K, Shaffer JR, Shungin D, O’Connell JR, Durazo‐Arvizo RA, Raffield L, et al. A fully adjusted two-stage procedure for rank-normalization in genetic association studies. Genetic Epidemiology. 2019;43:263–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang J, Bakshi A, Zhu Z, Hemani G, Vinkhuyzen AAE, Lee SH, Robinson MR, Perry JRB, Nolte IM, van Vliet-Ostaptchouk JV, et al. Genetic variance estimation with imputed variants finds negligible missing heritability for human height and body mass index. Nature Genetics. 2015;47:1114–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Evans LM, Tahmasbi R, Vrieze SI, Abecasis GR, Das S, Gazal S, Bjelland DW, de Candia TR, Haplotype Reference Consortium, Goddard ME, et al. Comparison of methods that use whole genome data to estimate the heritability and genetic architecture of complex traits. Nat Genet. 2018;50:737–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Machiela MJ, Chanock SJ. LDlink: a web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics. 2015;31:3555–3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Myers TA, Chanock SJ, Machiela MJ. LDlinkR: An R Package for Rapidly Calculating Linkage Disequilibrium Statistics in Diverse Populations. Frontiers in Genetics. 2020;11:157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kamat MA, Blackshaw JA, Young R, Surendran P, Burgess S, Danesh J, Butterworth AS, Staley JR. PhenoScanner V2: an expanded tool for searching human genotype-phenotype associations. Bioinformatics. 2019;35:4851–4853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Staley JR, Blackshaw J, Kamat MA, Ellis S, Surendran P, Sun BB, Paul DS, Freitag D, Burgess S, Danesh J, et al. PhenoScanner: a database of human genotype-phenotype associations. Bioinformatics. 2016;32:3207–3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Landrum MJ, Lee JM, Benson M, Brown GR, Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Jang W, et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018;46:D1062–D1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Frankish A, Diekhans M, Ferreira A-M, Johnson R, Jungreis I, Loveland J, Mudge JM, Sisu C, Wright J, Armstrong J, et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 2019;47:D766–D773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li X, Li Z, Zhou H, Gaynor SM, Liu Y, Chen H, Sun R, Dey R, Arnett DK, Aslibekyan S, et al. Dynamic incorporation of multiple in silico functional annotations empowers rare variant association analysis of large whole-genome sequencing studies at scale. Nat Genet. 2020;52:969–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reilly MP, Li M, He J, Ferguson JF, Stylianou IM, Mehta NN, Burnett MS, Devaney JM, Knouff CW, Thompson JR, et al. Identification of ADAMTS7 as a novel locus for coronary atherosclerosis and association of ABO with myocardial infarction in the presence of coronary atherosclerosis: two genome-wide association studies. Lancet. 2011;377:383–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu O, Bayoumi N, Vickers MA, Clark P. ABO(H) blood groups and vascular disease: a systematic review and meta-analysis. J Thromb Haemost. 2008;6:62–69. [DOI] [PubMed] [Google Scholar]
- 35.Luo M, Ji Y, Luo Y, Li R, Fay WP, Wu J. Plasminogen activator inhibitor-1 regulates the vascular expression of vitronectin. J Thromb Haemost. 2017;15:2451–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dankner R, Ben Avraham S, Harats D, Chetrit A. ApoE Genotype, Lipid Profile, Exercise, and the Associations With Cardiovascular Morbidity and 18-Year Mortality. J Gerontol A Biol Sci Med Sci. 2020;75:1887–1893. [DOI] [PubMed] [Google Scholar]
- 37.Assarsson E, Lundberg M, Holmquist G, Björkesten J, Thorsen SB, Ekman D, Eriksson A, Rennel Dickens E, Ohlsson S, Edfeldt G, et al. Homogenous 96-plex PEA immunoassay exhibiting high sensitivity, specificity, and excellent scalability. PLoS ONE. 2014;9:e95192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Olson NC, Butenas S, Lange LA, Lange EM, Cushman M, Jenny NS, Walston J, Souto JC, Soria JM, Chauhan G, et al. Coagulation factor XII genetic variation, ex vivo thrombin generation, and stroke risk in the elderly: results from the Cardiovascular Health Study. J Thromb Haemost. 2015;13:1867–1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bos MM, Noordam R, Blauw GJ, Slagboom PE, Rensen PCN, van Heemst D. The ApoE ε4 Isoform: Can the Risk of Diseases be Reduced by Environmental Factors? The Journals of Gerontology Series A, Biological Sciences and Medical Sciences. 2019;74:99–107. [DOI] [PubMed] [Google Scholar]
- 40.Chyu K-Y, Lio WM, Dimayuga PC, Zhou J, Zhao X, Yano J, Trinidad P, Honjo T, Cercek B, Shah PK. Cholesterol lowering modulates T cell function in vivo and in vitro. PloS One. 2014;9:e92095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yoshiyama Y, Asahina M, Hattori T. Selective distribution of matrix metalloproteinase-3 (MMP-3) in Alzheimer’s disease brain. Acta Neuropathologica. 2000;99:91–95. [DOI] [PubMed] [Google Scholar]
- 42.Immenschuh S, Song DX, Satoh H, Muller-Eberhard U. The type II hemopexin interleukin-6 response element predominates the transcriptional regulation of the hemopexin acute phase responsiveness. Biochemical and Biophysical Research Communications. 1995;207:202–208. [DOI] [PubMed] [Google Scholar]
- 43.Vinchi F, Costa da Silva M, Ingoglia G, Petrillo S, Brinkman N, Zuercher A, Cerwenka A, Tolosano E, Muckenthaler MU. Hemopexin therapy reverts heme-induced proinflammatory phenotypic switching of macrophages in a mouse model of sickle cell disease. Blood. 2016;127:473–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Belcher JD, Chen C, Nguyen J, Abdulla F, Zhang P, Nguyen H, Nguyen P, Killeen T, Miescher SM, Brinkman N, et al. Haptoglobin and hemopexin inhibit vaso-occlusion and inflammation in murine sickle cell disease: Role of heme oxygenase-1 induction. PLoS One. 2018;13:e0196455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, Lee JC, Schumm LP, Sharma Y, Anderson CA, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491:119–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gelernter J, Sun N, Polimanti R, Pietrzak RH, Levey DF, Lu Q, Hu Y, Li B, Radhakrishnan K, Aslan M, et al. Genome-wide Association Study of Maximum Habitual Alcohol Intake in >140,000 U.S. European and African American Veterans Yields Novel Risk Loci. Biological Psychiatry. 2019;86:365–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pillai VB, Sundaresan NR, Kim G, Samant S, Moreno-Vinasco L, Garcia JGN, Gupta MP. Nampt secreted from cardiomyocytes promotes development of cardiac hypertrophy and adverse ventricular remodeling. Am J Physiol Heart Circ Physiol. 2013;304:H415–H426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xiong X, Yu J, Fan R, Zhang C, Xu L, Sun X, Huang Y, Wang Q, Ruan H-B, Qian X. NAMPT overexpression alleviates alcohol-induced hepatic steatosis in mice. PloS One. 2019;14:e0212523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Loscalzo J, Braunwald E. Tissue plasminogen activator. N Engl J Med. 1988;319:925–931. [DOI] [PubMed] [Google Scholar]
- 50.Koshida R, Ou J, Matsunaga T, Chilian WM, Oldham KT, Ackerman AW, Pritchard KA. Angiostatin: a negative regulator of endothelial-dependent vasodilation. Circulation. 2003;107:803–806. [DOI] [PubMed] [Google Scholar]
- 51.Li Y, Zeng C, Hu J, Pan Y, Shan Y, Liu B, Jia L. Long non-coding RNA-SNHG7 acts as a target of miR-34a to increase GALNT7 level and regulate PI3K/Akt/mTOR pathway in colorectal cancer progression. Journal of Hematology & Oncology. 2018;11:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zanotti G, Berni R. Plasma Retinol-Binding Protein: Structure and Interactions with Retinol, Retinoids, and Transthyretin [Internet]. In: Vitamins & Hormones. Academic Press; 2004. [cited 2020 Nov 25]. p. 271–295.Available from: http://www.sciencedirect.com/science/article/pii/S0083672904690108 [DOI] [PubMed] [Google Scholar]
- 53.Arvanitis M, Koch CM, Chan GG, Torres-Arancivia C, LaValley MP, Jacobson DR, Berk JL, Connors LH, Ruberg FL. Identification of Transthyretin Cardiac Amyloidosis Using Serum Retinol-Binding Protein 4 and a Clinical Prediction Model. JAMA cardiology. 2017;2:305–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tahir UA, Katz DH, Zhao T, Ngo D, Cruz DE, Robbins JM, Chen Z-Z, Peterson B, Benson MD, Shi X, et al. Metabolomic Profiles and Heart Failure Risk in Black Adults: Insights From the Jackson Heart Study. Circ Heart Fail. 2021;14:e007275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Genovese G, Friedman DJ, Ross MD, Lecordier L, Uzureau P, Freedman BI, Bowden DW, Langefeld CD, Oleksyk TK, Uscinski Knob AL, et al. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science. 2010;329:841–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ito K, Bick AG, Flannick J, Friedman DJ, Genovese G, Parfenov MG, Depalma SR, Gupta N, Gabriel SB, Taylor HA, et al. Increased burden of cardiovascular disease in carriers of APOL1 genetic variants. Circ Res. 2014;114:845–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bick AG, Akwo E, Robinson-Cohen C, Lee K, Lynch J, Assimes TL, DuVall S, Edwards T, Fang H, Freiberg SM, et al. Association of APOL1 Risk Alleles With Cardiovascular Disease in Blacks in the Million Veteran Program. Circulation. 2019;140:1031–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang K, Huang R, Li G, Zeng F, Zhao Z, Liu Y, Hu H, Jiang T. CKAP2 expression is associated with glioma tumor growth and acts as a prognostic factor in high-grade glioma. Oncol Rep. 2018;40:2036–2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lin P, Pan Y, Chen H, Jiang L, Liao Y. Key genes of renal tubular necrosis: a bioinformatics analysis. Transl Androl Urol. 2020;9:654–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kim S, Eliot M, Koestler DC, Wu W-C, Kelsey KT. Association of Neutrophil-to-Lymphocyte Ratio With Mortality and Cardiovascular Disease in the Jackson Heart Study and Modification by the Duffy Antigen Variant. JAMA Cardiol. 2018;3:455–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Legge SE, Christensen RH, Petersen L, Pardiñas AF, Bracher-Smith M, Knapper S, Bybjerg-Grauholm J, Baekvad-Hansen M, Hougaard DM, Werge T, et al. The Duffy-null genotype and risk of infection. Hum Mol Genet. 2020; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kulkarni H, Marconi VC, He W, Landrum ML, Okulicz JF, Delmar J, Kazandjian D, Castiblanco J, Ahuja SS, Wright EJ, et al. The Duffy-null state is associated with a survival advantage in leukopenic HIV-infected persons of African ancestry. Blood. 2009;114:2783–2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Katz DH, Tahir UA, Ngo D, Benson MD, Gao Y, Shi X, Nayor M, Keyes MJ, Larson MG, Hall ME, et al. Multiomic Profiling in Black and White Populations Reveals Novel Candidate Pathways in Left Ventricular Hypertrophy and Incident Heart Failure Specific to Black Adults. Circ Genom Precis Med. 2021;14:e003191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Duggal P, An P, Beaty TH, Strathdee SA, Farzadegan H, Markham RB, Johnson L, O’Brien SJ, Vlahov D, Winkler CA. Genetic influence of CXCR6 chemokine receptor alleles on PCP-mediated AIDS progression among African Americans. Genes & Immunity. 2003;4:245–250. [DOI] [PubMed] [Google Scholar]
- 65.Picton ACP, Paximadis M, Chaisson RE, Martinson NA, Tiemessen CT. CXCR6 gene characterization in two ethnically distinct South African populations and association with viraemic disease control in HIV-1-infected black South African individuals. Clin Immunol. 2017;180:69–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Andersen T, Ueland T, Ghukasyan Lakic T, Åkerblom A, Bertilsson M, Aukrust P, Michelsen AE, James SK, Becker RC, Storey RF, et al. C-X-C Ligand 16 Is an Independent Predictor of Cardiovascular Death and Morbidity in Acute Coronary Syndromes. Arterioscler Thromb Vasc Biol. 2019;39:2402–2410. [DOI] [PubMed] [Google Scholar]
- 67.Kontorovich AR, Abul-Husn NS. Retinol binding protein 4 as a screening biomarker for hereditary TTR amyloidosis in African American adults with TTR V142I. J Card Fail. 2021;S1071–9164(21)00199–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Churchwell K, Elkind MSV, Benjamin RM, Carson AP, Chang EK, Lawrence W, Mills A, Odom TM, Rodriguez CJ, Rodriguez F, et al. Call to Action: Structural Racism as a Fundamental Driver of Health Disparities: A Presidential Advisory From the American Heart Association. Circulation. 2020;142:e454–e468. [DOI] [PubMed] [Google Scholar]
- 69.Raffield LM, Dang H, Pratte KA, Jacobson S, Gillenwater LA, Ampleford E, Barjaktarevic I, Basta P, Clish CB, Comellas AP, et al. Comparison of Proteomic Assessment Methods in Multiple Cohort Studies. Proteomics. 2020;20:e1900278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Benson MD, Ngo D, Ganz P, Gerszten RE. Emerging Affinity Reagents for High Throughput Proteomics: Trust, but Verify. Circulation. 2019;140:1610–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Carpenter MA, Crow R, Steffes M, Rock W, Heilbraun J, Evans G, Skelton T, Jensen R, Sarpong D. Laboratory, reading center, and coordinating center data management methods in the Jackson Heart Study. Am J Med Sci. 2004;328:131–144. [DOI] [PubMed] [Google Scholar]
- 72.Gold L, Ayers D, Bertino J, Bock C, Bock A, Brody EN, Carter J, Dalby AB, Eaton BE, Fitzwater T, et al. Aptamer-based multiplexed proteomic technology for biomarker discovery. PLoS ONE. 2010;5:e15004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Levey AS, Stevens LA, Schmid CH, Zhang YL, Castro AF, Feldman HI, Kusek JW, Eggers P, Van Lente F, Greene T, et al. A new equation to estimate glomerular filtration rate. Ann Intern Med. 2009;150:604–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pandey A, Keshvani N, Ayers C, Correa A, Drazner MH, Lewis A, Rodriguez CJ, Hall ME, Fox ER, Mentz RJ, et al. Association of Cardiac Injury and Malignant Left Ventricular Hypertrophy With Risk of Heart Failure in African Americans: The Jackson Heart Study. JAMA Cardiol. 2019;4:51–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Marwick TH, Gillebert TC, Aurigemma G, Chirinos J, Derumeaux G, Galderisi M, Gottdiener J, Haluska B, Ofili E, Segers P, et al. Recommendations on the Use of Echocardiography in Adult Hypertension: A Report from the European Association of Cardiovascular Imaging (EACVI) and the American Society of Echocardiography (ASE)†. Journal of the American Society of Echocardiography. 2015;28:727–754. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Whole genomes for JHS and MESA, generated as part of the NHLBI Trans-Omics for Precision Medicine (TOPMed) program, are available through restricted access via the NHLBI database of Genotypes and Phenotypes (dbGaP). TOPMed accession numbers for JHS and MESA are phs000964/phs002256.v1.p1 and phs001416, respectively. Full GWAS summary statistics for JHS (the discovery cohort) generated in this study will be available for general research use through controlled access at dbGaP accession phs001974: NHLBI TOPMed: Genomic Summary Results for the Trans-Omics for Precision Medicine program. For assistance in accessing the discovery data in JHS prior to full availability on dbGaP, investigators should contact the authors and follow JHS data access procedures (https://www.jacksonheartstudy.org/). GWAS data for the replication studies (MESA and HERITAGE) are fully included in the manuscript. Individual level proteomic and genomic data in the replication datasets are available through application to the respective cohorts.