

Abstract
SARS-CoV-2 is the causative agent of COVID-19 pandemic. The approval of vaccines and small molecule antivirals is vital in combating the pandemic. The viral polymerase inhibitors remdesivir and molnupiravir and the viral main protease inhibitor nirmatrelvir/ritonavir, have been approved by FDA. However, emergence of variants of concern/interest strains calls for additional antivirals with a novel mechanism of action. The SARS-CoV-2 papain-like protease (PLpro) mediates the cleavage of viral polyprotein as well as modulates the host innate immune response upon viral infection, rending it a promising antiviral drug target. This perspective highlights major achievements in structure-based design and high-throughput screening of SARS-CoV-2 PLpro inhibitors since the beginning of the pandemic. Encouraging progress includes the design of non-covalent PLpro inhibitors with favorable pharmacokinetic properties and the first-in-class covalent PLpro inhibitors. In addition, we offer our opinion of the knowledge gaps that need to be filled to advance PLpro inhibitors to clinic.
Graphical Abstract

1. INTRODUCTION
Coronaviruses (CoV) are enveloped, positive-sense, and single-stranded RNA(+ssRNA) viruses. CoVs belong to the subfamily Orthocoronavirinae, family Coronaviridae, and order Nidovirales. Seven coronaviruses are known to infect humans including four common human coronaviruses HCoV-229E, HCoV-NL63, HCoV-OC43, and HCoV-HKU1 that cause mild symptoms,1 and three coronaviruses SARS-CoV, MERS-CoV, and SARS-CoV-2 that cause severe acute respiratory tract infections.2, 3 Although humans around the world are commonly infected with HCoV-229E, HCoV-NL63, HCoV-OC43, or HCoV-HKU1, the infection generally only causes mild symptoms that do not require medical treatments.4, 5 Accordingly, no major efforts have been devoted to developing vaccines and antiviral drugs against these viruses. Nonetheless, the 21st century witnessed several coronavirus outbreaks that raised the alarm of this virus family. In late 2002, SARS-CoV emerged in Guangdong, China, and caused approximately 8,000 cases with the fatality rate of 9.6%.6 In 2012, MERS-CoV emerged in Saudi Arabia and South Korea, caused approximately 2,400 cases in the following 8 years with a fatality rate of 34%.7 Notably, in 2019, SARS-CoV-2 emerged in Hubei, China, and quickly ramped to the coronavirus disease 2019 (COVID-19) pandemic.8, 9 The clinical outcomes of COVID-19 range from non-symptomatic, mild to severe respiratory tract infections, influenza-like illness, to lung injuries, organ failure, and death.10 To date, SARS-CoV-2 has spread all over the world and is the most severe pandemic in recent history. As of May 3rd, 2022, 511 million cases and 6.23 million deaths have been reported worldwide, among which United States has 80.5 million cases and 986,298 deaths.11
Given the devastating impact of COVID19 on social life, public health, and global economy, researchers around the globe are working relentlessly to develop countermeasures. This effort has led to the development of vaccines and antiviral drugs in record-breaking speed.12, 13 Vaccine mainly targets the viral surface spike protein and rely on the production of antibodies to block the viral entry through inhibiting the interaction between the viral spike protein and the host cell angiotensin converting enzyme 2 (ACE2) receptor.14 Three vaccines received FDA approval including two mRNA vaccines from Pfizer/BioNTech (Comirnaty) and Moderna (Spikevax), and one adenovirus-based vaccine from Johnson & Johnson/Janssen. In addition, several vaccines from China and Russia have been approved by the World Health Organization.15
For small molecule antivirals, major progress has been made in targeting the SARS-CoV-2 RNA-dependent RNA polymerase (RdRp), the main protease (Mpro or 3CLpro), and the papain-like protease (PLpro).16, 17 The first RdRp inhibitor, remdesivir (1) (Figure 1A), was identified from a drug repurposing approach, and approved for the treatment of severe SARS-CoV-2 infection by intravenous (i.v.) administration.18 Remdesivir acts as a chain terminator during viral RNA synthesis.19 Similarly, the second RdRp inhibitor molnupiravir (2) (Figure 1A) was originally developed as an influenza antiviral and was later shown to have broad-spectrum antiviral activity against several viruses including SARS-CoV-2.20, 21 Molnupiravir (2) is a mutagen, and when incorporated into the RNA chain, it increases the mutation rate of the virus.22 Molnupiravir (2) is a prodrug and has the advantage of oral administration.23 The main protease inhibitor Paxlovid developed by Pfizer is a combination of nirmatrelvir (3) (Figure 1A) and ritonavir.13 Nirmatrelvir (3) is a Mpro inhibitor, and ritonavir is included as a boosting agent to increase the half-life of nirmatrelvir. A similar approach that was explored in the HIV drug combination Kaletra (lopinavir+ritonavir). Ritonavir is an inhibitor of cytochrome P450 3A4 (CYP3A4) and coadministration of ritonavir is required to increase the in vivo concentration of nirmatrelvir (3) to the target therapeutic range.
Figure 1.
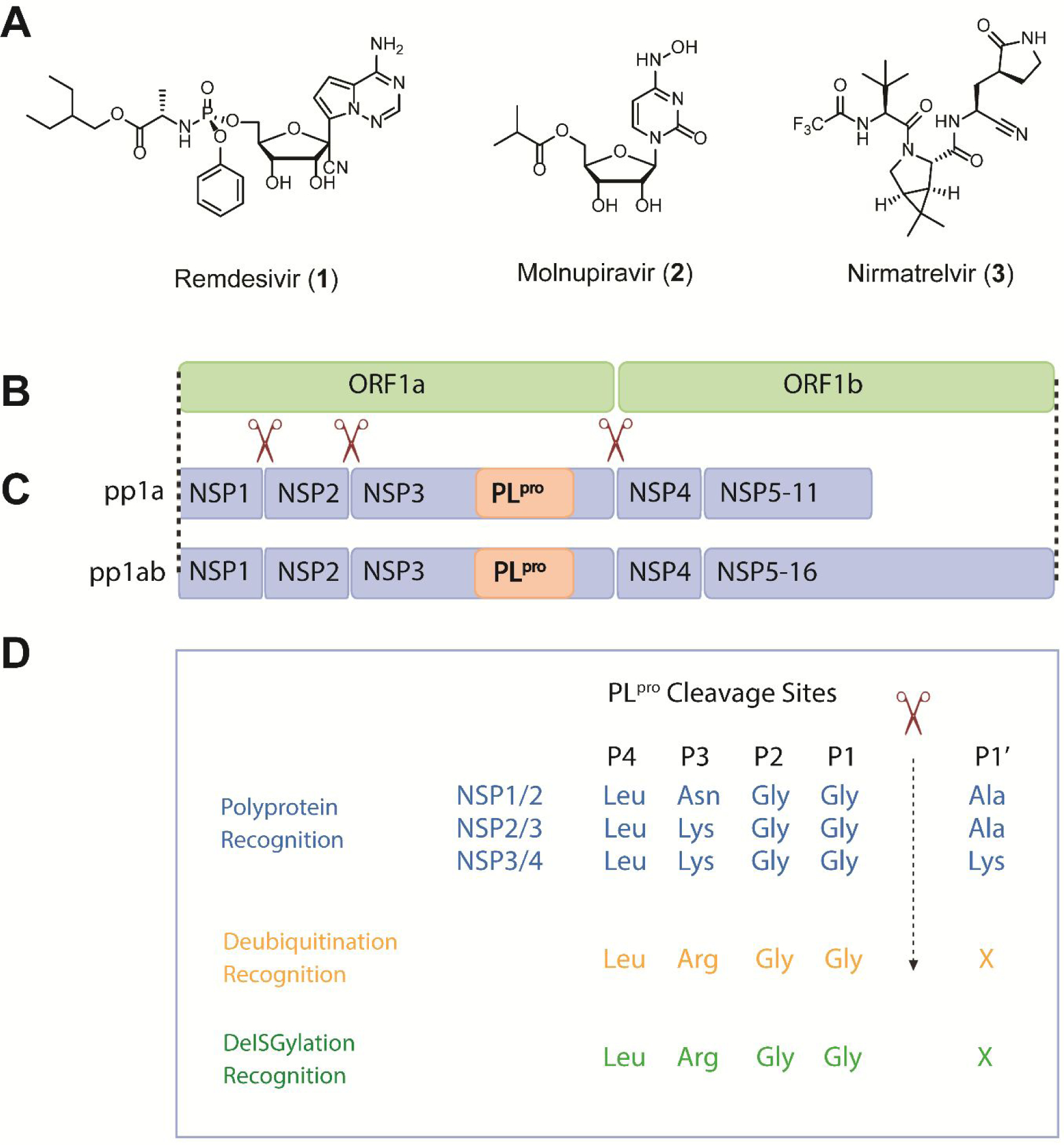
Chemical structures of FDA-approved COVID-19 antiviral drugs (A) and the schematic representation of the SARS-CoV and SARS-CoV-2 Open Reading Frame (B), the polyprotein replicase (C), and the recognition motifs of PLpro (D). The genome contains two open reading frames, ORF1a and ORF1b, which are directly translated into polyprotein pp1a and pp1ab due to the ribosomal frameshift between the two ORFs. pp1a contains 11 NSPs and pp1ab contains 16 NSPs. The PLpro is located within the NSP3. The polyproteins are processed into functional NSP units through cleavage by PLpro and Mpro, and the cleavage sites of PLpro are shown in (C). The substrate amino acid sequence alignment of P4-P1’ recognized by PLpro is shown in (D).
The approval of vaccines and RdRp and Mpro inhibitors are encouraging signs to combat the COVID-19 pandemic and possibly return to the pre-pandemic normalcy.24 However, the emergence of SARS-CoV-2 variants of concern (VOC) and variants of interests (VOI) poses a pressing need for additional vaccines and antiviral drugs.25 Multiple studies have shown the reduced efficacy of vaccines against Omicron VOC.26, 27 Drug resistant mutations have been evolved against remdesivir (1) in cell culture through serial passage experiments28, 29 as well as in an immunocompromised patient.30 In addition, the therapeutic benefits of remdesivir (1) are still under debate from several clinical trials.31, 32 Molnupiravir (2) has the potential risk of inducing mutations in the host, which is pending validation.33, 34 Molnupiravir (2) was shown to be positive in the Ames test,35 which is a standard assay to measure mutagenic potential of drug candidates in bacteria. NHC (β-d-N4-hydroxycytidine), the active metabolite of molnupiravir (2), displayed host mutational activity in mammalian cell culture.34 Multiple mutations have been identified in Mpro among the SARS-CoV-2 VOC and VOI including the Omicron Mpro P132H mutant.36 Although the currently identified Mpro mutants remain sensitive to nirmatrelvir (3),36–38 the scientific community is on high alert for future mutations such as H172Y and S144A that might lead to drug resistance.39 Genetic barrier to resistance for protease inhibitors is generally moderate to low as shown by HIV and HCV protease inhibitors.40 Resistance to Paxlovid is expected to rise with the increasing prescription. In addition, nirmatrelvir (3) is used in combination with ritonavir in clinics to prolong its half-life. Ritonavir is a potent inhibitor of the CYP3A4 isoenzyme and thus poses the risk of drug-drug interactions.41 As such, additional antivirals with a novel mechanism of action are clearly needed to combat emerging variants and drug resistant viruses. In this regard, the SARS-CoV-2 PLpro stands out as one of the next in line high-profile drug targets.
PLpro and the Mpro are the two essential proteases encoded by the SARS-CoV-2 genome. Both PLpro and Mpro cleave the peptide bonds in the viral polyprotein to release functional non-structural proteins (NSPs) for viral transcription and replication. In addition, PLpro is involved in antagonizing the host immune response upon viral infection. PLpro has deubiquitinating and deISGylating activities, and removes ubiquitin and ISG15 modifications from host proteins, leading to suppression of innate immune response and promotion of viral replication.42–44 The deubiquitinating and deISGylating activities of PLpro are indispensable in antagonizing host immune response.45, 46 Recent studies showed that SARS-CoV-2 infection of human macrophages triggers the release of extracellular free ISG15 through the viral PLpro, leading to the subsequent secretion of proinflammatory cytokines and chemokines, which recapitulates the cytokine storm of COVID-19.47, 48 This finding suggests that inhibiting the PLpro activity might alleviate the hyper-inflammation in COVID patients. Thus, targeting PLpro is expected to not only suppress viral replication but also restore antiviral immunity in the host.45
There are two types of PLpros, PL1pro and PL2pro.49, 50 The HCoV-229E, HCoV-NL63, HCoV-HKU1, and HCoV-OC43 encode both PL1pro and PL2pro. PL1pro and PL2pro have distinct substrate specificities in different coronaviruses.51 In contrast, SARS-CoV, MERS-CoV, and SARS-CoV-2 comprise only one functional PL2pro.
PLpro is part of the nsp3, a 215-KDa multidomain viral protein. SARS-CoV-2 PLpro specifically recognizes a consensus cleavage motif, LXGG↓(A/K/X), which is present in between nsp1/2, nsp2/3 and nsp3/4 at the viral polyprotein as well as the C-terminal sequences of ubiquitin and ISG15 with an isopeptide bond (Figure 1B–D).
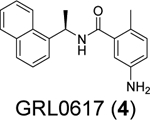
The SARS-CoV-2 PLpro contains four domains: the thumb, palm, zinc-finger domain, and a N-terminal ubiquitin-like domain (Figure 2). The catalytic triad consists of Cys111, His272, and Asp286, which are located at the interface of the palm and thumb domains. The zinc finger motif comprises four cysteines coordinating with a zinc ion and is vital for the structural integrity and the protease activity of PLpro. The flexible BL2 loop undergoes conformational changes from open to closed upon substrate binding (Figure 2A).52 This site is also the drug-binding site for GRL0617 (4) and its analogues.16 The X-ray crystal structures for the apo SARS-CoV-2 PLpro, drug-bound form,52–55 and complex forms with ubiquitin (Figure 2A) and ISG15 (Figure 2B) have been solved,56 paving the way for structure-based drug design and the understanding of the virology of PLpro.
Figure 2.
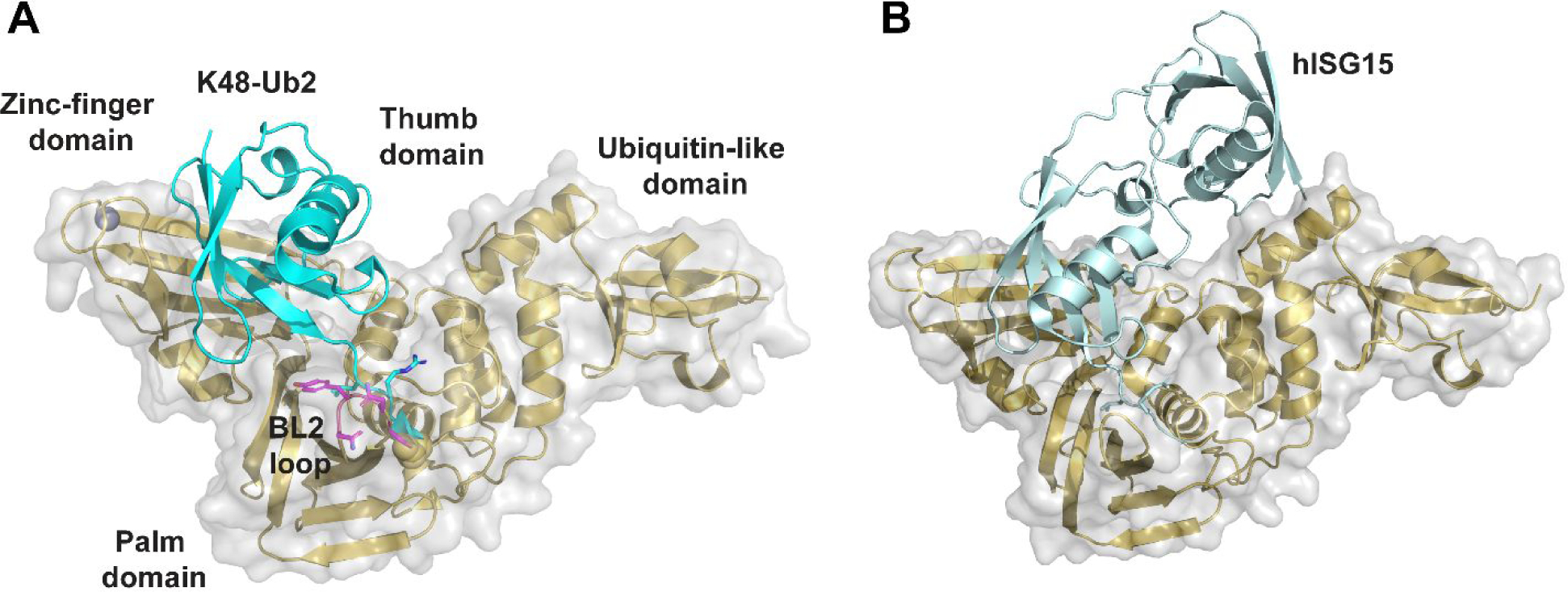
X-ray crystal structures of SARS-CoV-2 PLpro. (A) X-ray crystal structure of SARS-CoV-2 PLpro C111S mutant with K48-linked Ub2 (PDB: 7RBR). The BL2 loop is colored in magenta. (B) X-ray crystal structure of SARS-CoV-2 PLpro C111S mutant with human ISG15 (PDB: 7RBS).56
SARS-CoV-2 PLpro shares a sequence identity of 82.9% with SARS-CoV PLpro and to a lesser extent of 32.9% identify with the MERS-CoV PLpro. Despite the high sequence similarity, SARS-CoV-2 PLpro has enhanced deISGylating activity and reduced deubiquitinating activity compared to SARS-CoV PLpro.45, 46, 57 PLpro is a conserved drug target among SARS-CoV-2 variants (Figure 3). Although mutations have been identified, top high frequency mutations are all located distal from the drug binding site (Figure 3C). Nonetheless, it remains to be experimentally validated whether these mutations will alter drug sensitivity. In addition, resistance might emerge under drug selection pressure.
Figure 3.
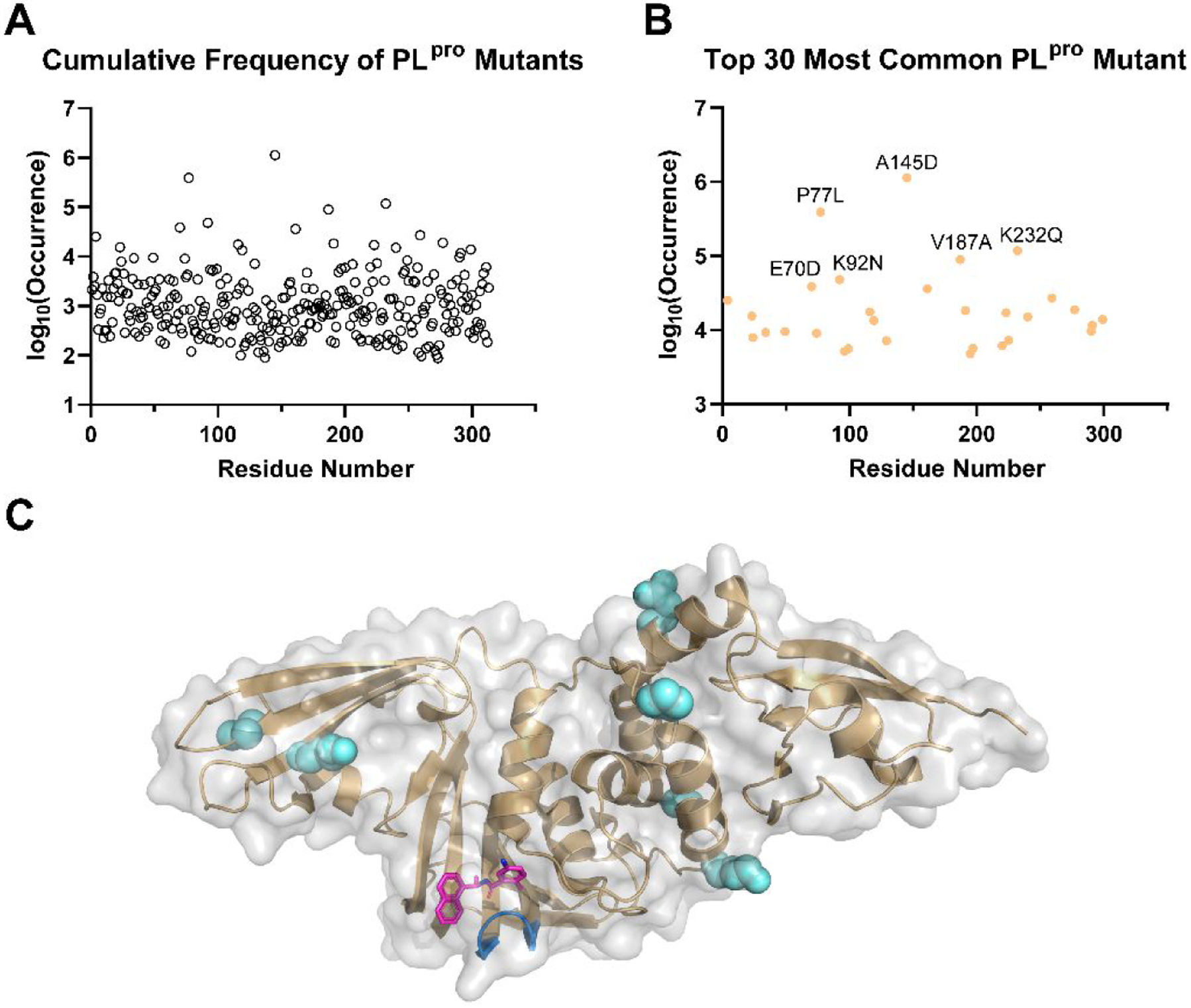
Analysis of SARS-CoV-2 PLpro mutations. Based on the data retrieved from GISAID (www.gisaid.org/epiflu-applications/covsurver-mutations-app), 2,487,047 sequences that contains mutations on PLpro have been identified, which fall into 5,754 different types of mutations on various positions of PLpro. All numbers shown are accurate as of Jan 25, 2022. (A) Cumulative frequency of SARS-CoV-2 PLpro mutants. (B) Top 30 most common SARS-CoV-2 PLpro mutants. Among these mutants, A145 has most frequent mutation to D with 1,131,252 occurrences (99.8% on 145); P77L with 372,993 occurrences (95.7% on 77); K232Q with 117,247 occurrences (99.1% on 232); V187A with 87,861 occurrences (97.9% on 187); and K92N with 47,110 occurrences (98.2%). (C) Mapping of top six SARS-CoV-2 PLpro mutants to the X-ray crystal structure of PLpro in complex with GRL0617 (4) (PDB: 7JRN). The residues are shown as spheres. The BL2 loop in the drug binding site is colored in marine, and the drug GRL0617 (4) is colored in magenta.
The knowledge accumulated through studying the SARS-CoV PLpro provides the foundation for the understanding of the virology of SARS-CoV-2 PLpro and the development of SARS-CoV-2 PLpro inhibitors. For excellent reviews of the structure, function, and inhibition of SARS-CoV PLpro, please refer to previous publications.16, 58–62 This perspective covers recent advances in the development of SARS-CoV-2 PLpro inhibitors and their mechanism of action. We also discuss the knowledge gaps that need to be filled to advance PLpro inhibitors to clinic.
It is not the objective of this perspective to enumerate all SARS-CoV-2 PLpro inhibitors reported in the literature, instead the focus is on highlighting several well-characterized examples. Non-specific PLpro inhibitors will also be discussed with the intention to alert the scientific community.
2. SARS-CoV-2 PLpro ASSAYS
Vigorous pharmacological characterization is vital in triaging non-specific inhibitors at the early stage and prioritizing hits with translational potential for further development. For this, we provide a brief introduction of the commonly used assays for the pharmacological characterization of PLpro inhibitors (Figure 4A).
Figure 4.
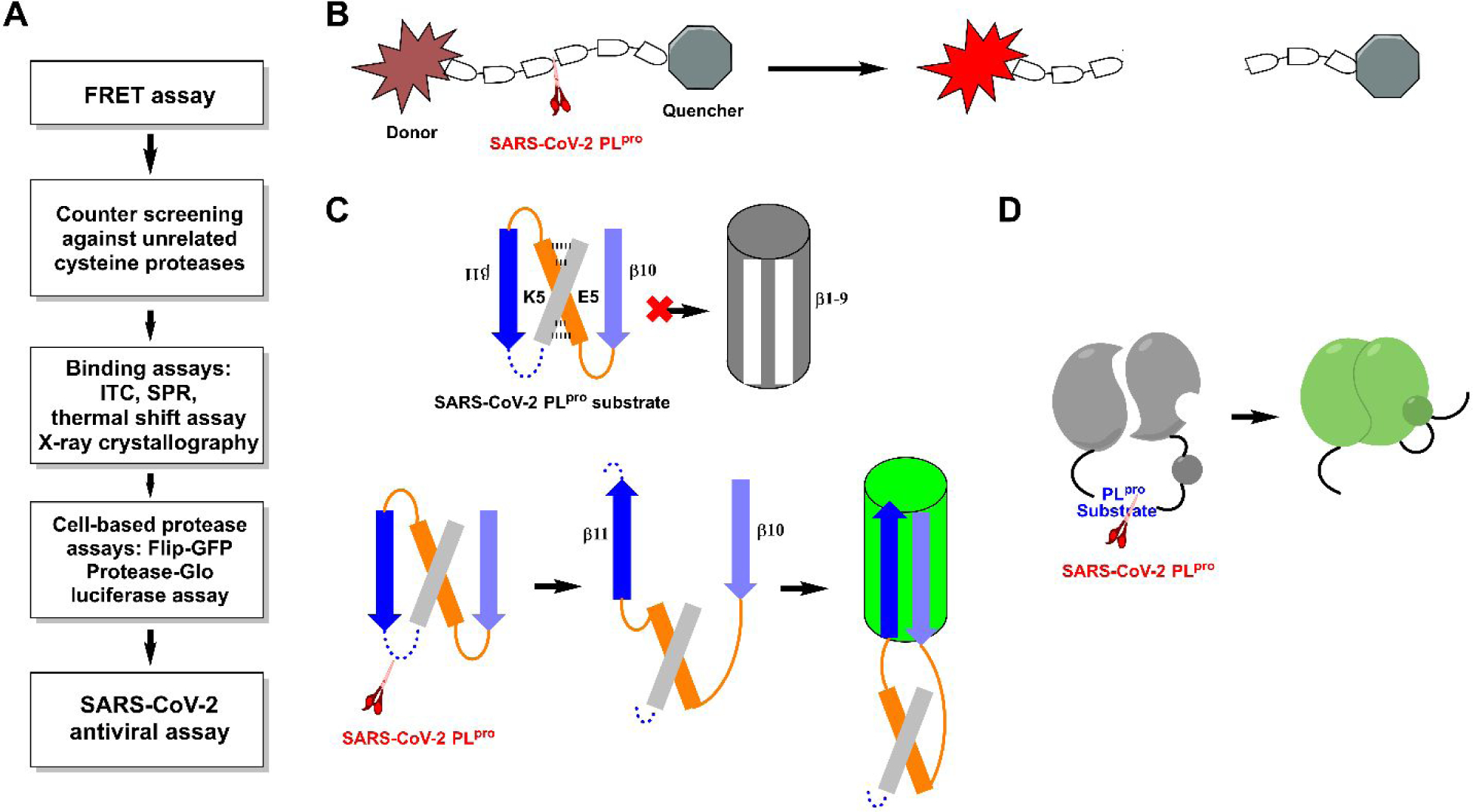
SARS-CoV-2 PLpro assays. (A) General flow chart for the pharmacological characterization of PLpro inhibitors. (B) Assay principle for the FRET-based enzymatic assay. (C) Assay principle for the cell based FlipGFP PLpro assay. (D) Assay principle for the Protease-Glo luciferase PLpro assay.
The gold standard assay for protease is the FRET (fluorescence resonance energy transfer)-based enzymatic assay, which is typically used as a primary assay for compound testing. In the FRET assay, a peptide corresponding to the protease substrate is designed with a fluorophore donor and a quencher at the two ends (Figure 4B). Upon cleavage by the protease, an increase in fluorescence signal is observed. However, the enzymatic assay condition varies among different labs in terms of enzyme concentration, FRET substrate sequence, pH, the addition of detergent (to rule out aggregates), bovine serum albumin (to rule out non-specific hydrophobic interactions) and reducing reagent (to prevent non-specific modification of catalytic Cys111). For this reason, the IC50 values from different studies should be interpreted with caution and not be used for direct comparison. Instead, positive controls such as GRL0617 (4) need to be included as a refence to normalize the results. The assay guidance manual compiled by Eli Lilly & Company and the National Center for Advancing Translational Sciences offer detailed guidance for assay optimization, which might help limit the variations between individual labs.63 In addition, counter screening against unrelated cysteine proteases should be conducted to rule out non-specific inhibitors. Furthermore, compounds that either quench the fluorophore or have overlapping absorbance/emission with the fluorophore will lead to false positive/negative results.
Our studies have shown that reducing reagents such as dithiothreitol (DTT) or glutathione are essential in the FRET enzymatic buffer to rule out promiscuous compounds that have non-specific inhibition against cystine proteases. Our recent studies of validation and invalidation of reported Mpro and PLpro inhibitors demonstrated that the FRET IC50 values obtained in the absence of reducing reagent DTT had poor correlation with the antiviral activity.64–66 We therefore urge the scientific community to be cautious in interpreting the PLpro assay IC50 values obtained in the absence of reducing reagent.
Several binding assays are also commonly used to determine the binding affinity between inhibitors and the PLpro, the thermal shift assay,54 surface plasma resonance (SPR) assay67 and isothermal titration calorimetry (ITC)67, 68. Thermal shift assay measures protein stability, and ligand binding typically leads to the increase of the melting temperature Tm. Nevertheless, decrease in protein stability is also observed for certain ligand-protein interactions. Compared to the thermal shift assay, SPR is more quantitative and binding kinetics kon, koff, and KD can be derived from the binding curves. ITC can determine the thermodynamic binding parameters ΔG, ΔH, and ΔS in a single experiment without a need to modify the protein. To gain molecular level understanding of the PLpro-inhibitor interactions, co-crystal structure needs to be solved.
It is expected that the cell free enzymatic assay or binding assay results can be used to faithfully predict the cellular antiviral activity. However, SARS-CoV-2 is a biological safety level 3 (BSL-3) pathogen, which limits the number of compounds to be tested in the antiviral assay given the paucity of the resources. In this regard, there is a need for cell-based protease assay to help predict the antiviral activity at the BSL-1/2 setting. The cell-based protease assay not only reveals intracellular target engagement but also can rule out compounds that are cell membrane impermeable or cytotoxic. The FlipGFP and Protease-Glo luciferase assays are two representative cell-based protease assays that have been applied for the screening and validation of SARS-CoV-2 PLpro inhibitors.54, 69 In the FlipGFP assay, cells are transfected with two plasmids, one expressing the PLpro and another expressing the GFP reporter (Figure 4C).70–72 The reporter plasmid encodes three proteins including the GFP β1–9 template, the β10–11 fragment, and the mCherry. The β10–11 fragment was restrained in the parallel orientation through the K5/E5 coiled coil, therefore cannot associate with the β1–9 template. Upon cleavage of the PLpro substrate linker, β10 and β11 become antiparallel and can associate with the β1–9 template, leading to the restoration of the GFP signal. mCherry serves as an internal control to normalize the transfection efficiency. As such, the GFP/mCherry ratio correlates to the enzymatic activity of PLpro. Results from us as well as others have shown that the FlipGFP assay is a valuable assay in characterizing the cellular Mpro and PLpro inhibition without the need of the infectious SARS-CoV-2 virus.54, 64, 69, 70, 72, 73 A positive correlation between the FlipGFP IC50 values and the antiviral EC values was observed for the PLpro inhibitors,54 50 suggesting FlipGFP assay can be used as a surrogate assay to prioritize lead compounds for antiviral testing.
The Protease-Glo luciferase assay is designed in an analogous way as the FlipGFP assay in which the luciferase activity depends on the cleavage of the substrate linker by the protease.64 Specifically, the firefly luciferase is engineered with a protease substrate cleavage sequence (Figure 4D). Before cleavage, firefly luciferase is in the permuted circular inactive conformation. Upon protease cleavage, a conformational change leads to the association of the two domains and the restoration of the luciferase activity. The Protease-Glo luciferase assay can be performed either in live cells or in cell lysates.69, 74, 75 As the readout is luminescence, the Protease-Glo luciferase assay can help rule out compounds that have fluorescence interference properties. Other cell-based assays including the GFP ER translocation assay, bioluminescence resonance energy transfer (BRET) assay, and the cell cytotoxicity assay can be similarly engineered for PLpro.75–77
3. SARS-CoV-2 PLpro INHIBITORS
We group SARS-CoV-2 PLpro inhibitors into two categories, the non-covalent inhibitors and covalent inhibitors. The non-covalent inhibitors are further divided into GRL0617 (4) analogues and non-GRL0617 inhibitors (Table 1).
Table 1.
SARS-CoV-2 PLpro inhibitors.
| Structure | Enzymatic inhibition IC50 (μM) | Antiviral EC50/CC50 (μM) | Structure | Enzymatic inhibition IC50 (μM) | Antiviral EC50/CC50 (μM) |
|---|---|---|---|---|---|
| Non-covalent SARS-CoV-2 PLpro inhibitors - GRL0617 analogues | |||||
|
1.39 ± 0.2680 1.78981 | 3.18 ± 0.71/~50080 32.6481 > 2067 |
|
0.59 ± 0.0467 Kd = 0.963 μM (SPR) | N.T. |
|
0.56 ± 0.0367 Kd = 0.372 μM (SPR) PDB:7LBS | 1.2 ± 0.2 |
|
0.39 ± 0.0567 Kd = 0.235 μM (SPR) | 1.4 ± 0.1 |
|
0.44 ± 0.0579 Kd = 2.60 ± 0.39 μM (SPR) | 0.18 ± 0.10 CC50 > 10 |
|
2.69 ± 0.3479 PDB: 7E35 | N.T. |
|
7.29 ± 1.03 | N.T. |
|
6.67 ± 0.55 | N.T. |
|
0.67 ± 0.0854 PDB: 7SDR | 6.62 ± 1.31 (Vero) 7.90 ± 2.40 (Caco-2 hACE2) |
|
0.67 ± 0.1454 PDB: 7SQE | 8.31 ± 2.68 (Vero) 11.99 ± 4.52 (Caca-2 hACE2) |
|
5.1 ± 0.752 GRL0617 (4) (2.3 ± 0.2) PDB: 7JIT | Not active |
|
6.4 ± 0.652 PDB: 7JIV, 7JIW | Not active |

|
7.0 ± 0.652 | Not active |
|
12.7 ± 1.352 | 5.2 ± 4.2 (Vero E6) |
|
0.8153 | < 11 μM |
|
11 ± 368 GRL0617 (4) (2.1 ± 0.2) | N.T. |
| Non-covalent SARS-CoV-2 PLpro inhibitors – Non-GRL0617 analogues | |||||
|
3.76 ± 1.1355 (ISG15-Rh substrate) PDB: 7OFU | 0.13 (qRTPCR) 10 (CPE) |
|
6.68 ± 1.2055 (ISG15-Rh substrate) PDB: 7OFS | 1 (qRTPCR) Not active (CPE) |
|
3.99 ± 1.3355 (ISG15-Rh substrate) PDB: 7OFT | Not active (CPE) |
|
1.66 (RLRGG-AMC)82 1.46 (ISG15-AMC) PDB: 7NT4 | A549/ACE 2 EC50 = 86 nM, CC50 = 3.1 μM SI = 36 Vero EC50 = 64 nM, CC50 = 3.4 μM SI = 53 |
|
0.5 (TAP-nsp123) 1.0 (TAP-nap23) 0.1 (de-ISGylation) 72 ± 1268 | 2.13 ± 1.16/35.5 ± 9.45 (Vero E6) |
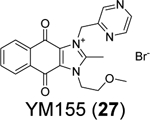
|
2.47 ± 0.4680 Assay condition: 50 mM HEPES, pH 7.5, 2 mM DTT PDB: 7D7L | 0.17 ± 0.02/~40080 |
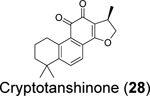
|
5.63 ± 1.4580 1.33681 | 0.70 ± 0.09/>30080 >20081 |
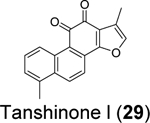
|
2.21 ± 0.1080 | 2.26 ± 0.11/>20080 |
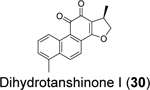
|
0.5981 | 8.1581 |
|
1.57181 | >20081 |
|
0.2681 | N.T. |
|
0.5381 | 20 |
|
0.3981 | cytotoxic |
|
0.6181 | cytotoxic |
|
1.65 ± 0.1383 Kd = 61.0 ± 12.1 μM (BLI assay) | N.T. |
|
7.24 ± 0.6883 (FP assay) Kd = 4.6 ± 0.29 μM (BLI assay) | N.T. |
|
1.784 | Not active |
|
2.284 | Not active |
|
3.184 Mpro (IC50 = 66 μM) | Not active |
|
0.56 ± 0.1685 | N.T. |
|
13.1686 15.68 (DBU) | N.T. CC50 > 100 μM (Vero E6) |
|
16.0886 17.51 (DBU) | N.T. CC50 > 100 μM (Vero E6) |
| Covalent SARS-CoV-2 PLpro inhibitors | |||||
|
Near complete inhibition at 100 μM57 PDB: 6WUU | N.T. |
|
Near complete inhibition at 100 μM57 PDB: 6WX4 | N.T. |
|
0.09487 kinact/Ki=10,0 00 M−1S−1 | 1.1 |
|
0.23087 kinact/Ki=14,00 0 M−1S−1 | Not active |
|
0.09887 kinact/Ki=4,80 0 M−1S−1 | Cytotoxic |
|
5.487 kinact/Ki=103 M−1S−1 | 34 |
|
8.087 | Not active |
|
> 20087 | Cytotoxic |
|
7.40 ± 0.3788 | N.T. |
|
8.63 ± 0.5588 | N.T. |
|
0.6789 Mpro (IC50 = 1.72 μM) | 0.32 (UC1074) 1.37 (UC1075) | |||
| Ebselen analogues | |||||
|
2.02 ± 1.0290 | N.T. |
|
0.236 ± 0.10790 | N.T. |
|
0.256 ± 0.03590 | N.T. |
|
0.339 ± 0.10990 | N.T. |
|
0.263 ± 0.12190 | N.T. |
|
PLpro IC50 = 1.255 ± 0.095 μM91 Mpro IC50 = 25.69 ± 2.64 nM | N.T. |
|
PLpro IC50 = 0.578 ± 0.040 μM Mpro 91 IC50 = 49.55 ± 2.95 nM | N.T. |
|
PLpro IC50 = 1.885 ± 0.098 μM Mpro 91 IC50 = 27.95 ± 5.10 nM | N.T. |
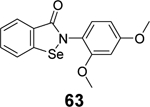
|
PLpro IC50 = 0.990 ± 0.058 μM Mpro 91 IC50 = 52.50 ± 4.51 nM | N.T. |
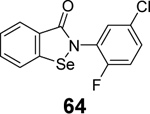
|
PLpro IC50 = 2.067 ± 0.078 μM Mpro 91 IC50 = 15.24 ± 4.58 nM | N.T. |
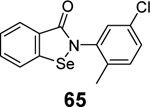
|
PLpro IC50 = 1.038 ± 0.083 μM Mpro 91 IC50 = 37.81 ± 3.28 nM | N.T. |
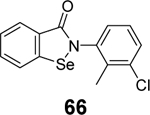
|
PLpro IC50 = 1.288 ± 0.052 μM Mpro 91 IC50 = 27.37 ± 2.35 nM | N.T. |
| Zinc ejectors | |||||
|
7.52 ± 2.13100 | N.T. |
|
N.T. | N.T. |
|
N.T. | N.T. |
|
N.T. | N.T. |
Reactive warheads are colored in red; N.T. = not tested.
3.1.1. Non-covalent SARS-CoV-2 PLpro inhibitors – GRL0617 analogues
The naphthalene containing GRL0617 (4) was a well-characterized SARS-CoV PLpro inhibitor. It was originally developed through lead optimization based on a high-throughput screening hit.62 Several follow up studies have been conducted with the aim of improving the potency of enzymatic inhibition and antiviral activity as well as pharmacokinetic (PK) properties. However, no significant improvement has been achieved.60, 61 As the SARS-CoV-2 PLpro is 83% identical and 90% similar to SARS-CoV PLpro, GRL0617 (4) became a top candidate as the SARS-CoV-2 PLpro inhibitor. Several groups independently showed the potent inhibition of SARS-CoV-2 PLpro by GRL0617 (4).45, 53, 54, 68, 78 However, the moderate to weak antiviral activity of GRL0617 (4) prevents it from advancing to animal model studies.54, 67 Since the beginning of the COVID-19 pandemic, encouraging progress has been made in re-designing GRL0617 analogues as potent SARS-CoV-2 PLpro inhibitors. The X-ray crystal structure of SARS-CoV-2 PLpro in complex with GRL0617 (4) has also been solved by multiple groups,53, 54, 56, 57, 78 paving the way for structure-based lead optimization.
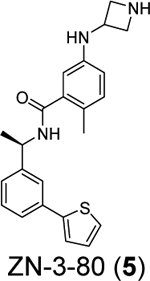
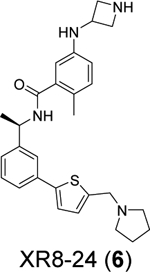
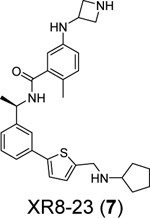
A recent elegant structure-based drug design led to the discovery of potent PLpro inhibitors with favorable PK properties.67 One of the major contributions of this study is the conversion of naphthalene to 2-phenylthiophene, which leads to improved PK properties. In addition, the thiophene substitution extends further into the BL2 groove (Figure 5A), and when coupled with additional substitutions on the aniline amine to engage interaction with Glu167 (Figure 5B, 5C), multiple nanomolar PLpro inhibitors have been identified. Among the more than 100 analogues tested, compounds ZN-3–80 (5), XR8–24 (6), and XR8–23 (7) were the most potent ones with IC50 values of 0.59, 0.56, and 0.39 μM, respectively (Table 1). Compounds 6 and 7 also showed a significantly improved antiviral activity against SARS-CoV-2 in A549-hACE2 cells with EC50 values of 1.2 and 1.4 μM, respectively. In comparison, GRL0617 (4) was not active in the virus yield reduction antiviral assay (EC50 > 20 μM). The complex structure with compound XR8–24 (6) (PDB: 7LBS) revealed several key hydrogen bonds/electrostatic interactions including the water mediated hydrogen bonds between the pyrrolidine NH+ and the main chain carbonyl oxygen of Tyr264 (not shown), the electrostatic interaction between the NH2+ from the azetidine ring and side chain carboxylate from Glu167 (Figure 5C), and the hydrogen bond between the amide NH from compound XR8–24 (6) with the Asp164 side chain carboxylate. When dosed in male C57BL/6 mice at 50 mg/kg by intraperitoneal injection (i.p.), compound XR8–23 (7) and XR8–24 (6) reached the Cmax of 6130 ng/mL and 6403 ng/mL, respectively, indicating favorable in vivo bioavailability. Further optimization might lead to candidates that are suitable for the in vivo antiviral efficacy study.
Figure 5.
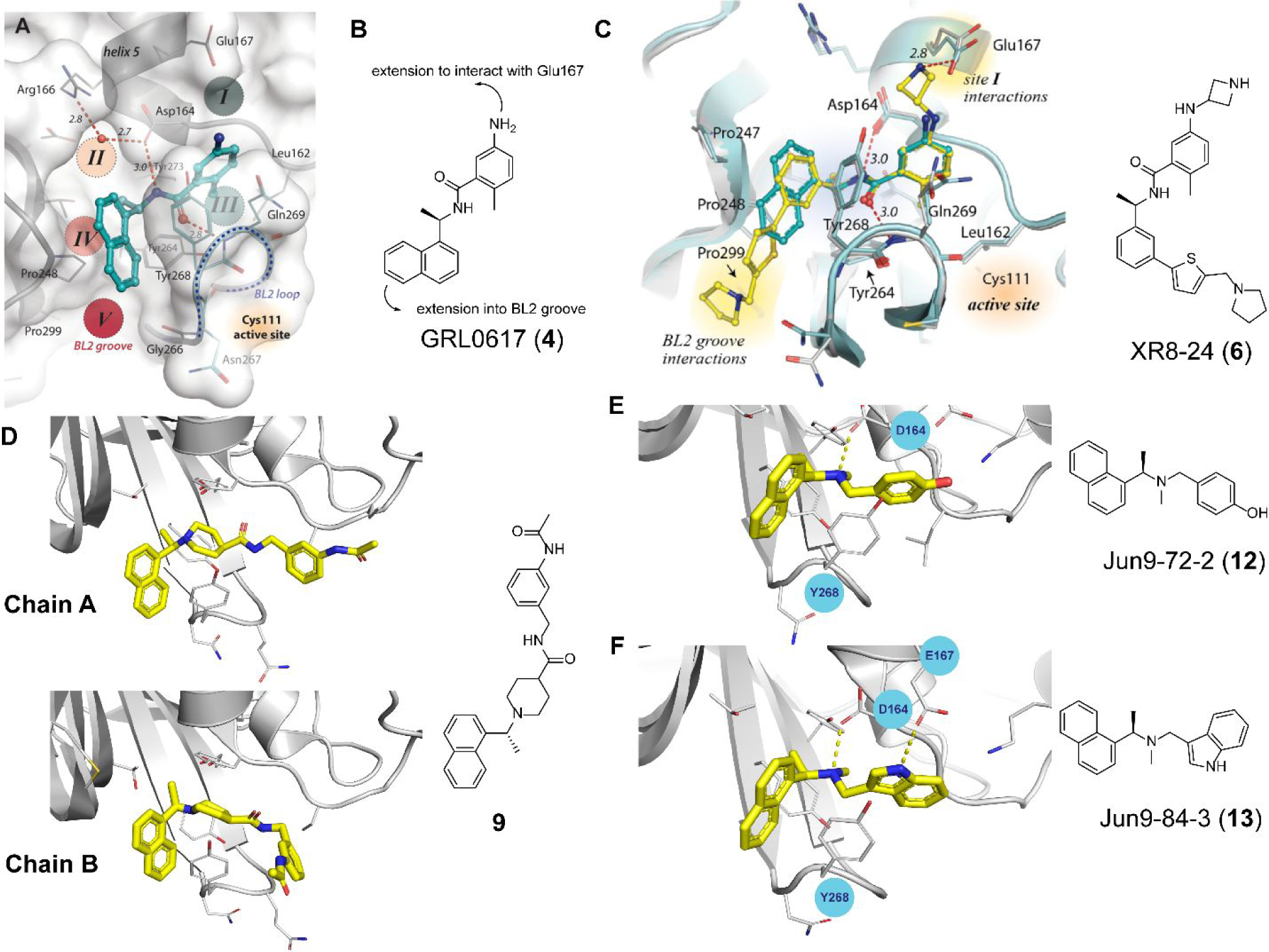
GRL0617-based SARS-CoV-2 PLpro inhibitors. (A) X-ray crystal structure of SARS-CoV-2 PLpro with GRL0617 (4) (PDB: 7JRN). (B) Design hypothesis for the 2-phenylthiophene series of PLpro inhibitors based on GRL0617 (4). (C) X-ray crystal structure of SARS-CoV-2 PLpro with compound XR8–24 (6) (PDB: 7LBS). (D) X-ray crystal structure of SARS-CoV-2 PLpro with compound 9 (PDB: 7E35). (E) X-ray crystal structure of SARS-CoV-2 PLpro with Jun9-72-2 (12) (PDB: 7SDR). (F) X-ray crystal structure of SARS-CoV-2 PLpro with Jun9-84-3 (13) (PDB: 7SQE). Panels A and C were adapted with permission from Shen, Z.; Ratia, K.; Cooper, L.; Kong, D.; Lee, H.; Kwon, Y.; Li, Y.; Alqarni, S.; Huang, F.; Dubrovskyi, O.; Rong, L.; Thatcher, G. R. J.; Xiong, R. Design of SARS-CoV-2 PLpro inhibitors for COVID-19 antiviral therapy leveraging binding cooperativity. J. Med. Chem. 2022, 65, 2940–2955.67 copyright 2022, American Chemical Society).
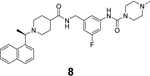
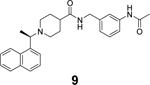
In another study, Shan et al. reported the structure-based design of SARS-CoV-2 PLpro inhibitors based on the GRL0617 scaffold.79 The most potent lead compound 8 inhibited PLpro and SARS-CoV-2 viral replication with IC50 of 0.44 μM and EC50 of 0.18 μM, respectively (Table 1). The Kd was 2.60 μM for compound 8 in the SPR assay, compared to the Kd of 10.79 μM for GRL0617 (4). In the counter screening against 10 deubiquitinases (DUBs) or DUB-like proteases, compound 8 was highly selective towards PLpro and did not show significant inhibition towards a panel of host DUBs and DUB-like proteases. The X-ray crystal structure of PLpro with an analogue 9 showed that compound 9 binds to PLpro in a similar mode as GRL0617 (4) (Figure 5D). It is noted that compound 9 adapts different binding poses in the two monomers (Figure 5D).
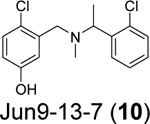
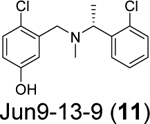
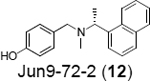
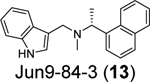
Our group recently conducted a high-throughput screening against SARS-CoV-2 PLpro using the FRET-based enzymatic assay.54 Two closely related compounds Jun9-13-7 (10) and Jun9-13-9 (11) were identified as potent hits with IC50 values of 7.9 and 6.67 μM, respectively (Table 1). Subsequent lead optimization led to the discovery of several compounds with IC50 values in the sub-micromolar range including Jun9-72-2 (12) (IC50 = 0.67 ± 0.08 μM) and Jun9-84-3 (13) (IC50 = 0.67 ± 0.14 μM). In the cell-based FlipGFP reporter assay, Jun9-72-2 (12) and Jun9-84-3 (13) showed dose dependent inhibition with EC50 values of 7.93 and 17.07 μM, respectively, suggesting both compounds are cell membrane permeable and can inhibit the intracellular protease activity of PLpro. In agreement, both compounds had potent antiviral activity against SARS-CoV-2 in Vero E6 and Caco2-hACE2 cells (Table 1). Significantly, there is a positive correlation between the FlipGFP assay results and the antiviral assay results, validating the FlipGFP as a surrogate assay for the prediction of the antiviral activity of PLpro inhibitors.54 In the X-ray crystal structure of PLpro with Jun9-72-2 (12) (PDB: 7SDR), the tertiary NH+ in the linker electrostatically interacts with the Asp164 carboxylate group (Figure 5E). The X-ray crystal structure of PLpro with Jun9-84-3 (13) (PDB: 7SQE) revealed an additional hydrogen bond between the indole NH with the Glu167 side chain carboxylate (Figure 5F).
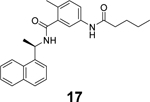
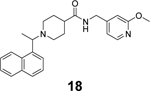
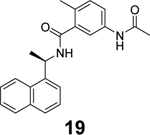
Additional GRL0617 analogues including 14, 15, 16, 17, 18, and 19 have been reported as SARS-CoV-2 PLpro inhibitors (Table 1),52, 53, 68 however, no significant improvement has been made.
3.1.2. Non-covalent SARS-CoV-2 PLpro inhibitors – non-GRL0617 inhibitors
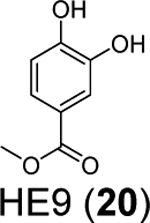
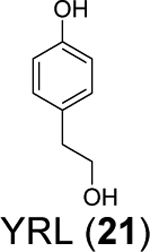
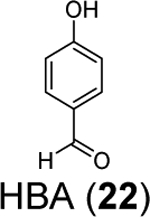
Three phenolic compounds including methyl 3, 4-dihydroxybenzoate (HE9, 20), 4-(2-hydroxyethyl)phenol (YRL, 21), and 4-hydroxybenzaldehyde (HBA, 22) were identified as allosteric SARS-CoV-2 PLpro inhibitors through a high-throughput X-ray crystallization.55 The screened library contains 500 compounds from the ICCBS (International Center for Chemical and Biological Sciences) Molecular Bank. Interestingly, HE9 (20), YRL (21), and HBA (22) all bind to the ISG15/Ub-S2 binding site of PLpro (Figure 6A), an allosteric binding pocket that has not been explored for drug design. The allosteric binding site is located about 30 Å away from the active site residue Cys111. The superimposition structures of PLpro+inhibitors and the PLpro+ISG15 indicate that these compounds might compete with ISG15 for the same binding site. As expected, all three compounds inhibited the deISGylating activity of PLpro with IC50 values of 3.76 ± 1.13 μM (20), 6.68 ± 1.20 μM (21), and 3.99 ± 1.33 μM (22). However, it remains unknown whether these compounds can inhibit the hydrolysis of viral polyprotein by PLpro. HE9 (20) and YRL (21) inhibited SARS-CoV-2 replication in Vero E6 cells in the qRT-PCR assay with EC50 values of 0.13 μM and 1 μM, respectively. However, the antiviral assay results for HBA (22) were not conclusive. In the cytopathic effect (CPE) assay, HE9 (20) had an EC50 of 10.37 μM. In contrast, YRL (21) failed to show inhibition in the CPE assay. The discrepancy of antiviral activity in different assays suggests further validation is needed. Furthermore, these results raise the question of whether inhibiting the deISGlyation activity of PLpro alone is sufficient for the inhibition of viral replication.
Figure 6.
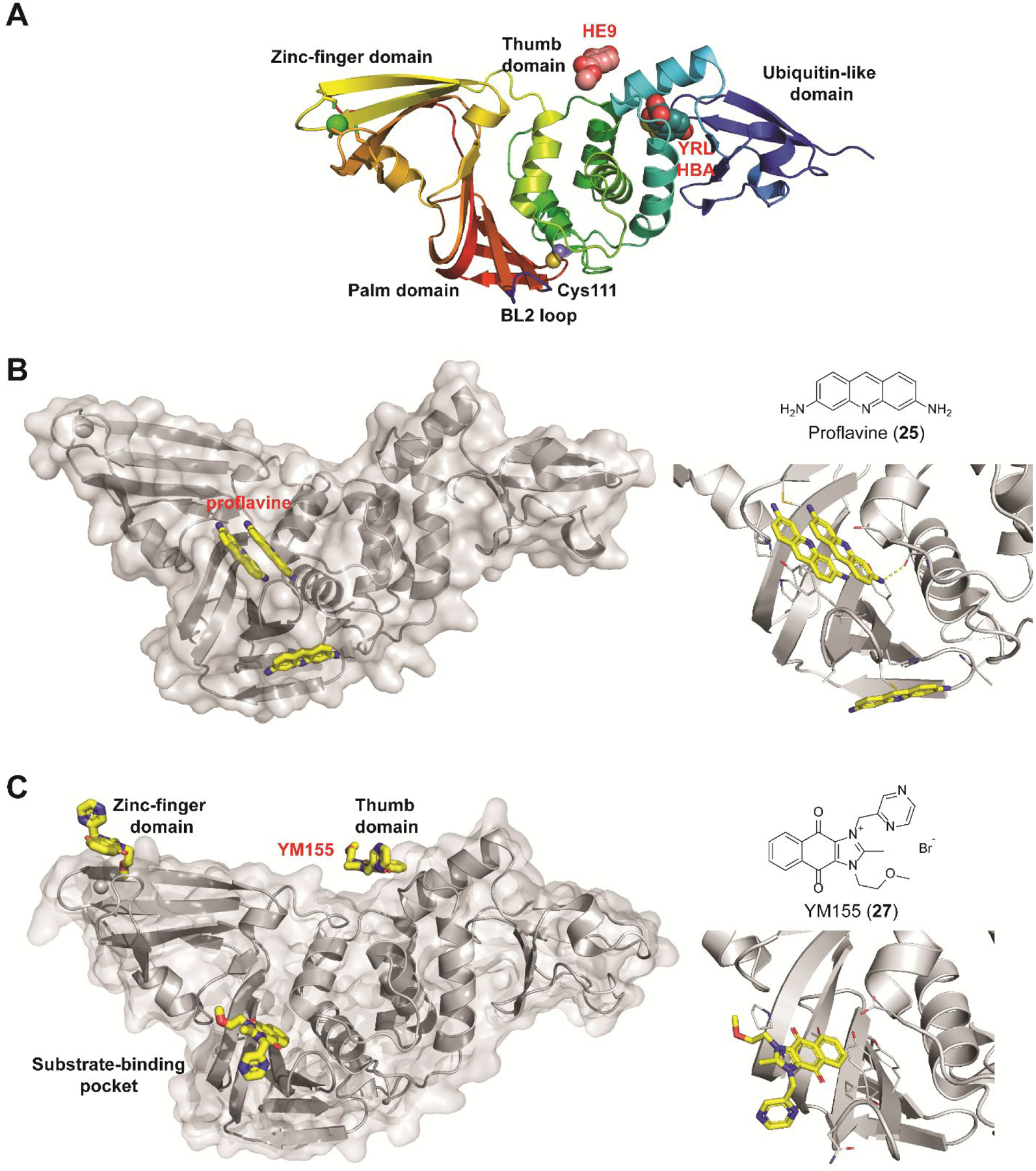
Non-covalent SARS-CoV-2 PLpro inhibitors that do not share structural similarity with GRL0617 (4). (A) X-ray crystal structures of SARS-CoV-2 PLpro in complex with fragments HE9 (20), YRL (21), and HBA (22). (B) X-ray crystal structure of SARS-CoV-2 PLpro in complex with proflavine (25) showing three molecules bind near the BL2 loop (PDB: 7NT4). Two molecules stack on top of each other and fit in the GRL0617 (4) binding pocket, and a third molecule binds at the backside of the BL2 loop. (C) X-ray crystal structures of SARS-CoV-2 PLpro in complex with YM155 (27) (PDB: 7D7L). YM155 (27) binds three different sites located at the zinc-finger domain, thumb domain, and the substrate-binding pocket. Detailed interactions between YM155 and the BL2 loop region residues were shown on the right side.
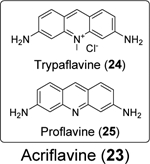
A drug repurposing screening by Napolitano et al. identified acriflavine (23) as a potent inhibitor of SARS-CoV-2 PLpro with in vivo antiviral efficacy.82 Acriflavine (23) is a mixture of trypaflavine (24) and proflavine (25).82 Acriflavine (23) inhibited PLpro with IC50 values of 1.66 and 1.46 μM when RLRGG-AMC and ISG15-AMC were used as substrates, respectively (Table 1). Acriflavine (23) also inhibited the deubiquitylating activity of PLpro in gel-based assay, thus ruling out the potential fluorescence interference effect of acriflavine (23). In addition, acriflavine (23) did not inhibit Mpro. The X-ray crystal structure of PLpro with proflavine (25) was determined (PDB: 7NT4), revealing two molecules of proflavine (25) bind to the S3–S5 pockets of PLpro simultaneously (Figure 6B). The BL2 loop folds inward towards the substrate-recognition cleft, similar to the binding mode of GRL0617 (4). A third proflavine (25) molecule is located at the surface of the protein on the opposite side of the BL2 loop. Acriflavine (23) inhibited SARS-CoV-2 replication in A549-ACE2 and Vero cells with EC50 values of 86 and 64 nM, respectively. However, the selectivity index was low (A549-ACE2 IS = 36; Vero SI = 53). The antiviral activity was further confirmed in human airway epithelial (HAE) cells. Acriflavine (23) also showed potent inhibition against MERS-CoV (IC50 = 21 nM, SI = 162) and HCoV-OC43 (IC50 = 105 nM, SI = 27), but not the alphacoronaviruses including feline infectious peritonitis virus (FIPV) and HCoV-NL63. In the in vivo SARS-CoV-2 infection model with K18-ACE2 mice, acriflavine (23) treatment by either i.p. or intramuscular (i.m.) injection significantly lowered the viral titers in the brain and the lung.
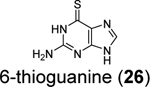
6-thioguanine (6-TG, 26) was previously reported as an inhibitor for the SARS-CoV and MERS-CoV,92, 93 therefore, it was hypothesized that it might also inhibit the SARS-CoV-2 PLpro. Swaim et al. recently demonstrated that 6-TG (26) is a potent inhibitor for SARS-CoV-2 in Vero E6 cells with an EC50 of 2.13 μM (Table 1).94 Next, to confirm the intracellular inhibition of PLpro by 6-TG (26), a TAP-tagged pp1a protein consisting of nsp1, 2, and 3 was expressed. As expected, TAP-nsp1 was the major product due to the self-cleavage pf pp1a polyprotein by PLpro. Treatment with 6-TG (26) led to dose-dependent inhibition of the cleavage with an IC50 of approximately 0.5 μM. In addition, 6-TG (26) showed potent inhibition of the deISGlyation activity of PLpro in HEK293T cells. No in vitro enzymatic assay was performed. In addition, it was proposed that 6-TG (26) might have a secondary mechanism of action by inhibiting the viral RNA synthesis. Nonetheless, in our recently hit validation study, 6-TG (26) did not show inhibition against SARS-CoV-2 PLpro in the enzymatic assay (IC50 > 50 μM), had no binding to PLpro in the thermal shift assay, and did not inhibit the intracellular PLpro activity in the FlipGFP assay.69 Therefore, the antiviral activity of 6-TG (26) may not arise from inhibiting the PLpro.
Through screening a library of 6,000 compounds using the FRET-based enzymatic assay with the Arg-Leu-Arg-Gly-Gly-AMC substrate, Zhao et al. identified YM155 (27) (IC50 = 2.47 ± 0.46 μM), cryptotanshinone (28) (IC50 = 5.63 ± 1.45 μM), tanshinone I (29) (IC50 = 2.21 ± 0.10 μM), and GRL0617 (4) (IC50 = 1.39 ± 0.26 μM) as SARS-CoV-2 PLpro inhibitors (Table 1).80 All four compounds displayed potent antiviral activity against SARS-CoV-2 in Vero E6 cells with the most potent compound being YM155 (27) (EC50 = 0.17 ± 0.02 μM, CC50 ~ 400 μM). The structure of PLpro in complex with YM155 (27) was solved by crystal soaking (PDB: 7D7L). Unexpectedly, YM155 (27) was found in three different binding sites including the orthosteric site, the thumb domain, and the zinc-finger domain (Figure 6C). The binding at the thumb domain is expected to inhibit the binding between PLpro and ISG15. A conformational change was observed at the zinc-finger domain upon YM155 (27) binding, but the physiological relevant of this binding mode has not been validated.
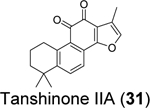
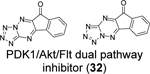
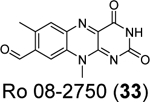
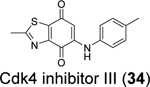
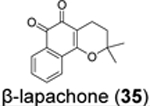
Similarly, cryptotanshinone (28) (IC50 = 1.34 μM), together with two other analogues dihydrotanshinone I (30) (IC50 = 0.59 μM) and tanshinone IIA (31) (IC50 = 1.57 μM), were shown as potent SARS-CoV-2 PLpro inhibitors through a HTS (Table 1).81 In addition, four additional compounds, PKK1/Akt/Flt dual pathway inhibitor (32) (IC50 = 0.26 μM), Ro 08–2750 (33) (IC50 = 0.53 μM), Cdk4 inhibitor III (34) (IC50 = 0.39 μM), and β-lapachone (35) (IC50 = 0.61 μM) were also identified as potent PLpro inhibitors (Table 1). Dihydrotanshinone I (30) inhibited SARS-CoV-2 with an EC50 of 8.15 μM. Unexpectedly, cryptotanshinone (28) and tanshinone IIA (31) had no antiviral activity (EC50 > 200 μM), despite their potent enzymatic inhibition. The antiviral result of cryptotanshinone (28) is also in controversy with the previous study which showed that cryptotanshinone (28) is a potent antiviral with an EC50 of 0.7 μM.80 Further validation is needed to test the antiviral activity of cryptotanshinone (28) against SARS-CoV-2 in multiple cell lines.
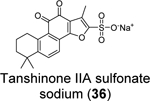
Xu et al. recently reported the discovery of tanshinone IIA sulfonate (36) and chloroxine (37) as SARS-CoV-2 PLpro inhibitors from a drug repurposing screening.83 Tanshinone IIA sulfonate (36) was identified in the fluorogenic assay using the ALKGG-AMC substrate with an IC50 of 1.65 μM (Table 1). Chloroxine (37) was discovered in the fluorescence polarization-based assay using the fluorescein 5-isothiocyanate (FITC) labeled ISG15 with an IC50 of 7.24 μM. Tanshinone IIA sulfonate (36) and chloroxine (37) also showed binding to PLpro in the biolayer interferometry and thermal shift assays. The antiviral activity against SARS-CoV-2 was not reported.
We performed hit validations for YM155 (27), cryptotanshinone (28), tanshinone I (29), dihydrotanshinone I (30), and tanshinone IIA (31).69 Our study found that YM155 (27) (IC50 = 20.13 μM), cryptotanshinone (28) (IC50 = 52.24 μM), tanshinone I (29) (IC50 = 18.58 μM), dihydrotanshinone I (30) (IC50 = 33.01 μM), and tanshinone IIA (31) (IC50 = 15.30 μM) had much higher IC50 values against SARS-CoV-2 PLpro in the FRET assay compared to the previous reports. The intracellular PLpro inhibition by YM155 (27) and cryptotanshinone (28) in the FlipGFP assay was not conclusive due to cell cytotoxicity, while tanshinone I (29), dihydrotanshinone I (30), and tanshinone IIA (31) had no intracellular PLpro inhibition at non-toxic concentrations. Collectively, our results suggest that YM155 (27), cryptotanshinone (28), tanshinone I (29), dihydrotanshinone I (30), and tanshinone IIA (31) are weak PLpro inhibitors and tanshinone I (29), dihydrotanshinone I (30), and tanshinone IIA (31) lack intracellular target engagement.
In agreement with our results, Brewitz et al. applied mass spectrometry assay to monitor PLpro-mediated cleavage of the nsp 2/3 substrate.95 Among the list of compounds tested, YM155 (27), tanshinone I (29), tanshinone IIA sulfonate sodium (36) were not active (IC50 > 50 μM), while cryptotanshinone (28) showed moderate activity with an IC50 of 19.4 μM.
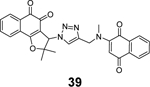
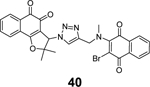
Through virtual screening of a library of naphthoquinoidal compounds followed by enzymatic assay validation, Santos et al. identified three compounds 38, 39, and 40 as potent SARS-CoV-2 PLpro inhibitors with IC50 values of 1.7 μM, 2.2 μM, and 3.1 μM, respectively (Table 1).84 Among the three hits, compound 40 had moderate inhibition against Mpro with an IC50 of 66 μM, therefore it was considered as a dual inhibitor for further optimization. MD simulations predicted that compound 39 binds non-covalently to the S3 and S4 subsites in PLpro. However, the detailed mechanism of action remains to be characterized. When tested in the antiviral assay against SARS-CoV-2 in two different cell lines Vero E6 and HeLa-ACE2, none of the identified Mpro and PLpro inhibitors had antiviral activity, suggesting these naphthoquinoidal compounds might have off-target effects. It is noted that no reducing agent such as dithiothreitol (DTT) was added in the Mpro enzymatic assay, however, 0.1 mM DTT was included in the PLpro assay. Therefore, the observed PLpro inhibition might not be due to non-specific modification of the PLpro C111 residue. Further validation studies are warranted to confirm their enzymatic inhibition.
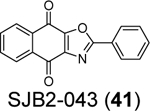
Cho et al. reported SJB2–043 (41) as a SARS-CoV-2 PLpro inhibitor with an apparent IC50 of 0.56 μM.85 However, no complete inhibition was achieved at high drug concentration. Therefore, it remains to be validated whether SJB2–043 (41) is a specific PLpro inhibitor.
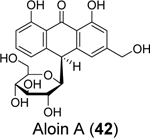
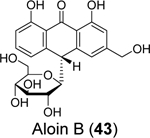
Commercial mouth rinses are known to inactivate SARS-CoV-2,96, 97 but the detailed mechanism remains elusive. Lewis et al. tested the active ingredients of mouth rinses against the SARS-CoV-2 Mpro and PLpro.86 Although none of the compounds were active against Mpro, two compounds, aloin A (42) and aloin B (43), inhibited PLpro with IC50 values of 13.16 and 16.08 μM, respectively in the enzymatic assay. Aloin A (42) and B (43) also inhibited the deubiquitinating activity of PLpro with IC50 values of 15.68 and 17.51 μM. Molecular dynamics simulations suggest that aloin A (42) and B (43) bind to the GRL0617 (4) binding site and mainly interact with Glu167, Tyr268, and Glu269.
3.2.1. Specific covalent PLpro inhibitors
The cleavage of PLpro substrate occurs after the second glycine in the Leu-X-Gly-Gly sequence.57 As a result, the binding pockets for the S2 and S1 subsites are absent, which leaves the S4 and S3 subsites for inhibitor binding. Accordingly, to develop covalent inhibitor to react with the catalytic C111, a linker is needed to conjugate the S4/S3 pocket binder with a reactive warhead.53, 57
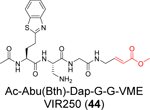
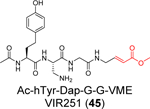
A positional scanning was conducted to identify the optimal substrate of SARS-CoV-2 PLpro.57 A total of 19 natural and 109 nonproteinogenic amino acids were screened at each position. It was found that the P2 and P4 positions have high preference for glycine and hydrophobic residues, respectively, while the P3 position can tolerate both charged residues including Phe(guan), Dap, Dab, Arg, Lys, Orn, and hArg and hydrophobic residues including hTyr, Phe(F5), Cha, Met, Met(O), Met(O)2, D-hPhe. Leveraging this information, two covalent inhibitors VIR250 (44) (Ac-Abu(Bth)-Dap-Gly-Gly-VME) and VIR251 (45) (Ac-hTyr-Dap-Gly-Gly-VME) were designed by incorporating the optimal P3 and P4 substitutions with the vinylmethyl ester (VME) reactive warhead (Table 1). VIR250 (44) and VIR251 (45) showed dose-dependent inhibition against both SARS-CoV-2 and SARS-CoV PLpros, however the IC50 values were not quantified. The X-ray crystal structures of SARS-CoV-2 PLpro in complex with VIR250 (44) (PDB: 6WUU) and VIR251 (45) (PDB: 6WX4) were solved (Figure 7), revealing covalent thioether linkage of the C111 thiol and the β carbon of the vinyl group. Although no antiviral assay results were reported, this is an elegant rational design that led to the first covalent SARS-CoV-2 PLpro inhibitors.
Figure 7.
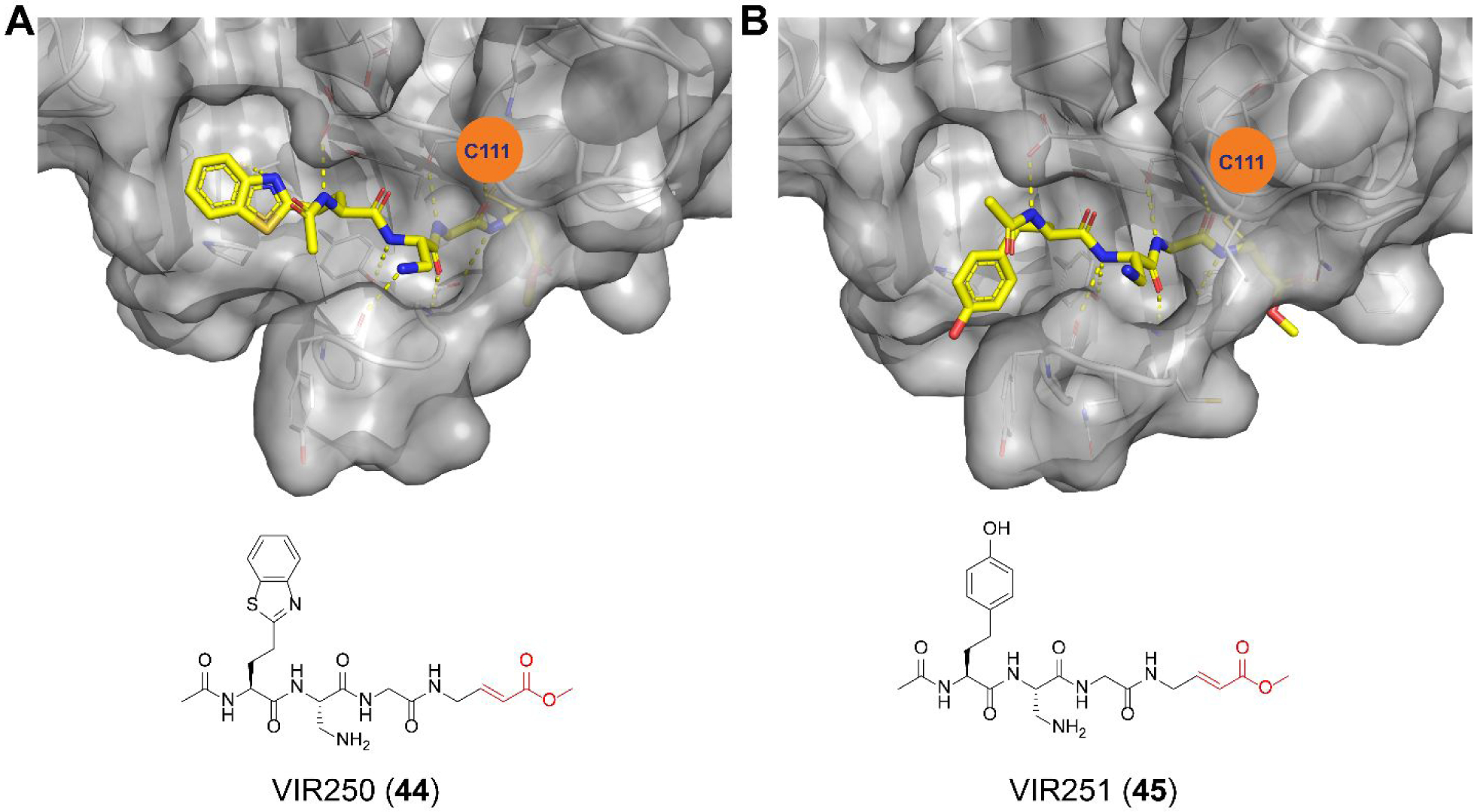
X-ray crystal structures of SARS-CoV-2 PLpro in complex with peptidomimetic covalent inhibitors VIR250 (44) (PDB: 6WUU) (A) and VIR251 (45) (PDB: 6WX4) (B).
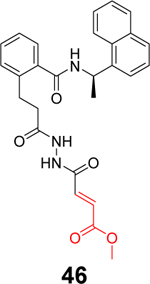
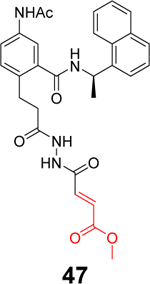
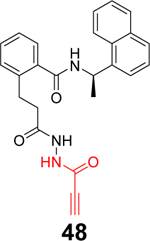
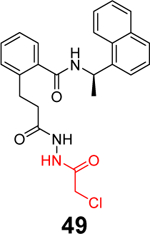
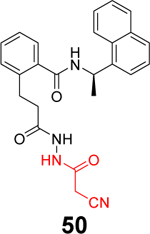
Sanders et al. recently reported the rational design of the first-in-class drug-like covalent SARS-CoV-2 PLpro inhibitors.87 An N, N’-diacetylhydrazine linker was designed as a mimetic of the Gly-Gly to conjugate the GRL0617 methyl group with different reactive warheads. A series of commonly used cysteine reactive warheads including fumarate methyl ester, chloroacetamide, propiolamide, cyanoacetamide, and α-cyanoacrylamide have been exploited. Among the designed covalent PLpro inhibitors, compounds 46 and 47 with the fumarate methyl ester, and compound 48 with the propiolamide showed significantly improved potency with IC50 values of 0.094, 0.230, and 0.098 μM, respectively (Table 1). Compound 49 with the chloroacetamide and compound 50 with the cyanoacetamide were less active and the IC50 values were 5.4 and 8.0 μM, respectively. In contrast, compound 51 with the α-cyanoacrylamide was not active (IC50 > 200 μM). As expected, covalent protein adduct with inhibitors were observed for compounds 46, 47, 48, 49, and 50 in electrospray ionization (ESI) mass spectrometry. The X-ray crystal structure of PLpro with compound 46 was solved at 3.10 Å resolution (PDB: not released), showing a covalent adduct between the C111 thiol and the C1 of compound 46. The N, N’-diacetylhydrazine linker forms four hydrogen bonds with Gly163 and Gly271, highlighting the importance of this rationally designed linker. In SARS-CoV-2 infected Vero E6 cells, compound 46 had an EC50 of 1.1 μM, which is comparable to the potency of remdesivir (EC50 = 0.74 μM). Surprisingly, compound 47 had insignificant cytoprotective effects, despite its potent enzymatic inhibition. Compound 48 was cytotoxic; therefore, its antiviral activity was not conclusive. Similar to GRL0617 (4), compound 46 also inhibited the deubiquitinating and the deISGylating activities with IC50 values of 76 and 39 nM, respectively. Selectivity screening against a panel of DUBs showed that compound 46 is highly selective and no inhibition was observed at up to 30 μM. In vitro pharmacokinetic profiling showed that compound 46 is stable in human liver S9 and microsomes with T1/2 of 60 and 50 mins, respectively. This study represents the first rational design of drug-like covalent PLpro inhibitors with potent antiviral activity, and the X-ray crystal structure is invaluable in guiding the lead optimization.
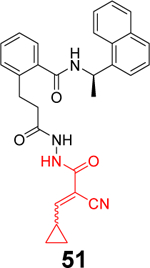
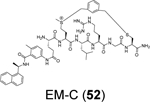
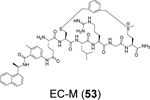
Liu et al. reported the design of peptide-drug conjugates (PDCs) as covalent inhibitors of SARS-CoV-2 PLpro.88 The PDCs consist of GRL0617 and cyclic sulfonium-containing peptides derived from PLpro substrate Leu-Arg-Gly-Gly (Table 1). The sulfonium serves as a warhead and is designed to react with the C111. Among the examined PDCs, EM-C (52) and EC-M (53) were the most potent against SARS-CoV-2 PLpro with IC50 values of 7.40 ± 0.37 and 8.63 ± 0.55 μM, respectively (Table 1). Both conjugates were cell membrane permeable and inhibited the deISGylating activity of PLpro. In-gel digestion of the PLpro+PDC mixture followed by MS/MS analysis confirmed that C111 is the enriched conjugation site. No antiviral assay results were presented. Although the results presented convincingly demonstrated the covalent labeling of PLpro C111, it remains unknown about their binding mode. The EC-M (52) and EM-C (53) PDCs contain the GRL0617 and the Leu-Arg dipeptide sequence, both of which are S3 and S4 subsite binders. It is not clear why the design contains duplicate binding elements. The x-ray crystal structure might solve the puzzle.
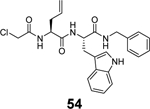
A tryptophane containing dipeptide, compound 54, was recently reported as a dual inhibitor of SARS-CoV-2 Mpro and PLpro.89 Compound 54 inhibited Mpro and PLpro with IC50 values of 1.72 and 0.67 μM, respectively, while had no binding to the viral spike protein (KD > 25 μM). In the antiviral assay, compound 54 inhibited two SARS-CoV-2 clinical isolates UC-1074 and UC-1075 with EC50 values of 0.32 and 1.37 μM, respectively. Given the lack of structural similarities between Mpro and PLpro, coupled with the high reactivity of α-chloroacetamide warhead in 54, it remains to be investigated whether the inhibition of Mpro and PLpro by compound 54 is specific. Nevertheless, the potent antiviral activity of compound 54 is encouraging, which warrants further optimization.
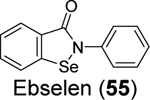
3.2.2. Non-specific covalent PLpro inhibitors – Ebselen analogues
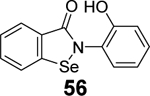
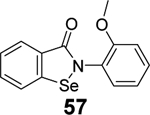
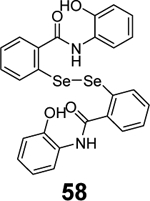
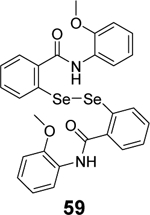
Given the broad-spectrum antiviral activity of ebselen against several viruses, WeglarzTomczak- et al. explored ebselen and its analogues as SARS-CoV-2 PLpro inhibitors.90 Ebselen (55) inhibited PLpro with an IC50 of 2.02 ± 1.02 μM, and dialysis experiment showed that no enzymatic activity was recovered, suggesting irreversible inhibition. Subsequently, a library of analogues was designed, among which two ebselen derivatives 56 (IC50 = 236 ± 107 nM) and 57 (IC50 = 256 ±35 nM), and two diselenide orthologs 58 (IC50 = 339 ±109 nM) and 59 (IC50 = 263 ± 121 nM), had improved enzymatic inhibition against SARS-CoV-2 PLpro compared to ebselen (55) (IC50 = 2.02 ± 1.02 μM) (Table 1). In this study, 2 mM DTT was added in the enzymatic assay buffer. However, our previous studies showed that ebselen (55) only inhibited SARS-CoV-2 PLpro in the absence of DTT but not with DTT.65 This discrepancy needs to be further validated.
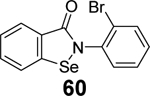
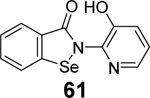
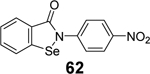
In another study, a similar strategy has been exploited for the development of dual inhibitors targeting both SARS-CoV-2 Mpro and PLpro based on the ebselen scaffold.91 Among the 23 ebselen analogs, seven showed dual inhibition with the Mpro IC50 values in the nanomolar range and the PLpro IC50 values in the single digit to submicromolar range (60-66, Table 1). No reducing reagent was added in either the Mpro or the PLpro enzymatic assay. The antiviral activity of the potent hits was not reported. Nonetheless, ebselen (55) was previously reported to inhibit SARS-CoV-2 replication with an EC50 value of 4.67 μM in the plaque assay, albeit the proposed mechanism of action is through Mpro inhibition.98
The inconsistent PLpro enzymatic inhibitory activity of ebselen (55) and its analogues from several groups, coupled with their antiviral activity against SARS-CoV-2, suggest that further characterizations are needed to confirm their cellular PLpro target engagement and additional targets that might contribute to the antiviral activity.
3.2.3. Non-specific covalent PLpro inhibitors – Zinc ejector
PLpro contains a zinc-binding domain in which the zinc ion is coordinated by four conserved cysteine residues Cys189, Cys192, Cys224, Cys226. The Zinc-binding domain is essential for the structural integrity and hence the enzymatic activity of PLpro. As such, the cysteine rich zinc-binding domain (ZBD) was also proposed as a putative drug target.99
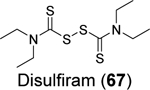
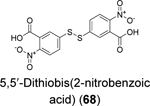
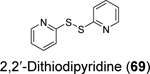
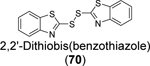
Disulfiram (67), ebselen (55), together with 5,5’ -dithiobis(2-nitrobenzoic acid) (DTNB, 68), 2,2’ -dithiodipyridine (69), and 2,2’ -dithiobis(benzothiazole) (70) were found to eject zinc from PLpro as shown by the increase in fluorescence emission signal from the zinc-specific fluorophore, FluoZin-3.100 Matrix-assisted laser desorption/ionization-time of flight (MALDI-TOF) mass spectrum further confirmed the covalent adduct formation between disulfiram and ebselen with PLpro and nsp10. LC-MS/MS experiment mapped the ebselen and disulfiram conjugation sites to C189 and C192, both of which are involved in zinc chelation in the ZBD of PLpro. In the FRET-based enzymatic assay, disulfiram (67) and ebselen (55) inhibited PLpro with IC50 values of 7.52 and 2.36 μM, respectively. It is noted that the enzymatic inhibition might be a combined effect of targeting both the catalytic C111 and the cysteines in the ZBD. Combination experiment showed that ebselen and disulfiram had synergistic antiviral effect when combined with hydroxychloroquine. This study suggested that clinically safe zinc-ejectors could potentially target the conserved ZBD in multiple viral proteins and could potentially be exploited as broad-spectrum antiviral drug candidates. Following studies from the same group further showed that disulfiram (67) and ebselen (55) are zinc-ejectors of the SARS-CoV-2 nsp13 and nsp14 and consequently inhibit nsp13 ATPase and nsp14 exoribonuclease activities.101 The antiviral activity of ebselen (55) and disulfiram (67) against SARS-CoV-2 was synergistic with remdesivir.
As discussed above, ebselen analogs have also been extensively exploited as Mpro and PLpro inhibitors by targeting the active site cysteine.102, 103 Combined with the zinc ejecting property, the antiviral activity of ebselen (55) and its derivatives might be due to its polypharmacology in targeting the ZBD, PLpro, Mpro, and others.
4. Perspectives of targeting the SARS-CoV-2 PLpro
The COVID-19 pandemic is a timely call for the immediate need of antivirals. As the SARS-CoV and MERS-CoV epidemics subsided, the interest of developing coronavirus inhibitors unfortunately waned, and no significant efforts were devoted to optimizing the hits identified from early high-throughput screening campaigns. Nevertheless, the COVID-19 pandemic reignited the interest in PLpro drug discovery and the past two years have seen encouraging progress in the field. Although drug repurposing largely failed to identify potent and selective PLpro inhibitors, rational design based on the X-ray crystal structures led to major breakthroughs including the design of 2-phenylthiophene PLpro inhibitors with favorable PK properties and the fist-in-class covalent PLpro inhibitors since the pandemic. In light of this encouraging progress, we hereby share our opinions in the further development of SARS-CoV-2 PLpro inhibitors and hope to clarify some of the confusions in the field based on our experience.
First, there is a need to broaden the antiviral spectrum of PLpro inhibitors to target MERS-CoV. The BL2 loop located at the drug binding site is poorly conserved among SARS-CoV and MERS-CoV,104 explaining the lack of activity of GRL0617 (4) series of compounds against MERS-CoV PLpro. No potent and specific MERS-CoV PLpro inhibitors have been reported till now. In search of PLpro inhibitors with a broader spectrum of antiviral activity, it is worthwhile to include MERS-CoV PLpro in the secondary assays. It might be possible to identify allosteric inhibitors with dual inhibitions against both SARS-CoV-2 PLpro and MERS-CoV PLpro. Alternatively, PLpro inhibitors can be developed specifically for SARS-CoV-2 and SARS-CoV, and MERS-CoV PLpro inhibitors can be pursued separately.
Second, structurally disparate PLpro inhibitors are needed to advance PLpro inhibitors to clinic. Compared to PLpro, Mpro is a more amenable drug target and structurally disparate inhibitors have been identified from HTS as potent Mpro inhibitors. In contrast, several recent HTS failed to identify additional potent and selective SARS-CoV-2 PLpro inhibitors other than GRL0617 analogues.67, 85 GRL0617 (4) contains the naphthalene ring, which is a known metabolic labile group, and a possible toxicophore.105 Therefore, it might present a challenge in PK optimization. To increase the chances of success, additional structurally disparate PLpro inhibitors are needed as backups. The recent elegant design of 2-phenylthiophene and the covalent PLpro inhibitors are prominent examples in this direction.67, 87
Third, target selectivity needs to be addressed at the early stage of development. Although there is a lack of sequence or structural similarity between PLpro and human DUBs, both PLpro and human DUBs bind ubiquitin at the extended C-terminus with the consensus sequence Leu-X-Gly-Gly, raising the potential concern of the off-target effects of PLpro inhibitors against human DUBs.106 Consequently, it is important to conduct counter screening of PLpro inhibitors against a panel related human DUBs to avoid potential toxicity. Along this line, counter screening should also be conducted with other cysteine proteases like the Mpro, cathepsin L, calpains and etc to rule out promiscuous inhibitors that non-specifically inhibit unrelated proteases.
Fourth, be aware of promiscuous inhibitors and compounds with polypharmacology. Promiscuous compounds are defined as compounds that lack a defined mechanism of action or compounds that showed inconsistent results in different assays. PLpro is a cysteine protease that is prone to non-specific inhibition by redox cycling compounds (quinone, arylsulfonamide, tolyl-hydrazide, etc)107, 108, alkylating reagents, and other pan-assay interference compounds (PAINS).109–111 In addition, compounds such as acriflavine and YM155 are cationic amphiphilic drugs (CADs), which could cause phospholipidosis and disturb endosome/lysosome functions. This effect may explain the improved antiviral potency over biochemical potency. In this regard, the antiviral activity of acriflavine and YM155 might be a combined effect of PLpro inhibition and endosome/lysosome disruption. Furthermore, it is better to perform the antiviral assays in different cell lines, especially in physiologically relevant cell lines such as Calu3 or normal human airway epithelial cells. This eliminates the cell-type dependent antiviral activity of certain compounds.
Fifth, for translational drug discovery, we need to differentiate chemical probes from drug candidates. Compounds such as ebselen and disulfiram having non-specific inhibition against PLpro and Mpro as well as other unrelated cysteine proteases should not be classified as PLpro inhibitors. Nevertheless, this does not indicate that these promiscuous compounds should not be further pursued as SARS-CoV-2 antivirals. Instead, they should be defined as chemical probes for mechanistic studies. The aforementioned cell-based protease assays such as the FlipGFP and Protease-Glo luciferase assays are valuable tools to help rule out promiscuous compounds like ebselen and disulfiram and delineate the cellular target engagement of the specific PLpro inhibitors.
In summary, despite the encouraging progress in the past two years, there is still a long journey to advance PLpro inhibitors to clinic. No rationally designed drug-like PLpro inhibitors have been shown to have in vivo antiviral efficacy against SARS-CoV-2 infection in animal models yet. In addition to the RdRp and Mpro inhibitors, PLpro inhibitors are expected to enrich our armamentarium in flighting the current COVID-19 pandemic and future unforeseeable coronavirus outbreaks. Combination experiments need to be planned to characterize the combination therapy potential of PLpro inhibitors with RdRp or Mpro inhibitors. Furthermore, the knowledge accumulated in developing SARS-CoV-2 PLpro inhibitors can be similarly applied to MERS-CoV PLpro.
ACKNOWLEDGMENTS
This work was supported by the National Institute of Allergy and Infectious Diseases of Health (NIH-NIAID) grants AI147325, AI157046, and AI158775.
ABBREVIATIONS USED
- ACE2
angiotensin converting enzyme 2
- BRET
bioluminescence resonance energy transfer
- BSL-3
biological safety level 3
- COVID-19
coronavirus disease 2019
- DTT
dithiothreitol
- DUBs
deubiquitinases
- ESI
electrospray ionization
- FITC
fluorescein 5-isothiocyanate
- FRET
fluorescence resonance energy transfer
- HBA
4-hydroxybenzaldehyde
- HE9
methyl 3, 4-dihydroxybenzoate
- i.m.
intramuscular
- i.p.
intraperitoneal
- ITC
isothermal titration calorimetry
- i.v.
intravenous
- MALDI-TOF
matrix-assisted laser desorption/ionization-time of flight
- Mpro
main protease
- NSP
non-structural protein
- PAINS
pan-assay interference compounds
- PDC
peptide-drug conjugate
- PK
pharmacokinetic
- PLpro
papain-like protease
- RdRp
RNA-dependent RNA polymerase
- SPR
surface plasma resonance
- 6-TG
6-thioguanine
- VOC
variants of concern
- VOI
variants of interests
- YRL
4-(2-hydroxyethyl)phenol
- ZBD
zinc-binding domain
Biographies
Jun Wang earned his Ph.D. degree from the University of Pennsylvania in 2010. He continued the postdoctoral training with Dr. William F. DeGrado at the University of California, San Francisco. In 2014 he started an independent career as an assistant professor at the college of pharmacy at the University of Arizona. He was promoted to associate professor in 2020. In 2022, he relocated his lab to the Ernest Mario School of Pharmacy at Rutgers University. His current research interests include antiviral drug discovery, assay development, drug resistance, and combination therapy. Dr. Wang serves as the associate editor and editorial board member for multiple journals including Journal of Medical Virology, Acta Pharmaceutica Sinica B, Medicinal Research Reviews, and European Journal of Pharmaceutical Sciences.
Haozhou Tan earned his BS degree from the Hunan Agricultural University (2016, Changsha, China) and the master’s degree from Northeastern University (2018, Boston, Massachusetts). In 2020, he joined Dr. Jun Wang Lab as graduate student at the College of Pharmacy at the University of Arizona. In 2022, he relocated to the Ernest Mario School of Pharmacy at the Rutgers University with his PI Dr. Wang. His current study focuses on antiviral drug research for Influenza and coronavirus.
Yanmei Hu earned her Ph.D. degree from the University of Arizona in August 2021. She continued the postdoctoral training with Dr. Jun Wang and relocated with the Wang laboratory to the Ernest Mario School of Pharmacy at the Rutgers University in 2022. Her current research interests include antiviral drug discovery, assay development, drug resistance mechanism study, and combination therapy targeting enteroviruses and coronaviruses.
Prakash Jadhav obtained the M.Sc. in Chemistry from Pune University, India in 2011, and then joined Dr. D. S. Reddy’s research group as a research assistant in CSIR-National Chemical Laboratory, Pune. He received his Ph.D. in 2019 from the National Tsing-Hua University, Taiwan, under the supervision of Dr. R. S. Liu and continued his postdoctoral studies at the same research group. After completion of his postdoctoral work in the group of Dr. S. C. Hung at the Genomics Research Center, Academia Sinica, Taiwan, he joined Dr. Jun Wang group as a postdoctoral researcher in January 2022. His research interests include the design of SARS-CoV-2 PLpro inhibitors.
Bin Tan received his bachelor’s and master’s degrees from the China Pharmaceutical University in 2018 and 2020, respectively. After graduation, he worked for one year as a research assistant in Dr. Jing Xu’s group in Southern University of Science and Technology. He is now a first-year PhD student in Dr. Jun Wang’s lab at Rutgers University and his research interests include drug discovery targeting SARS-CoV-2 Mpro and PLpro.
Footnotes
Dr. Jun Wang is an inventor of a patent filed for the SARS-CoV-2 PLpro inhibitors.
REFERENCES
- (1).Liu DX; Liang JQ; Fung TS Human coronavirus-229E, -OC43, -NL63, and -HKU1 (Coronaviridae). Encyclopedia of Virology 2021, 428–440. [Google Scholar]
- (2).Chen B; Tian EK; He B; Tian L; Han R; Wang S; Xiang Q; Zhang S; El Arnaout T; Cheng W Overview of lethal human coronaviruses. Signal. Transduct. Target Ther 2020, 5, 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Cui J; Li F; Shi ZL Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol 2019, 17, 181–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Pyrc K; Berkhout B; van der Hoek L The novel human coronaviruses NL63 and HKU1. J. Virol 2007, 81, 3051–3057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Gaunt ER; Hardie A; Claas EC; Simmonds P; Templeton KE Epidemiology and clinical presentations of the four human coronaviruses 229E, HKU1, NL63, and OC43 detected over 3 years using a novel multiplex real-time PCR method. J. Clin. Microbiol 2010, 48, 2940–2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Stadler K; Masignani V; Eickmann M; Becker S; Abrignani S; Klenk HD; Rappuoli R SARS--beginning to understand a new virus. Nat. Rev. Microbiol 2003, 1, 209–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Chafekar A; Fielding BC MERS-CoV: Understanding the latest human coronavirus threat. Viruses 2018, 10, 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Zhu N; Zhang D; Wang W; Li X; Yang B; Song J; Zhao X; Huang B; Shi W; Lu R; Niu P; Zhan F; Ma X; Wang D; Xu W; Wu G; Gao GF; Tan W; China Novel Coronavirus Investigating and Research Team. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med 2020, 382, 727–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Wu F; Zhao S; Yu B; Chen YM; Wang W; Song ZG; Hu Y; Tao ZW; Tian JH; Pei YY; Yuan ML; Zhang YL; Dai FH; Liu Y; Wang QM; Zheng JJ; Xu L; Holmes EC; Zhang YZ A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Hu B; Guo H; Zhou P; Shi ZL Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol 2021, 19, 141–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).https://covid19.who.int/. Accessed May 3, 2022.
- (12).Boras B; Jones RM; Anson BJ; Arenson D; Aschenbrenner L; Bakowski MA; Beutler N; Binder J; Chen E; Eng H; Hammond H; Hammond J; Haupt RE; Hoffman R; Kadar EP; Kania R; Kimoto E; Kirkpatrick MG; Lanyon L; Lendy EK; Lillis JR; Logue J; Luthra SA; Ma C; Mason SW; McGrath ME; Noell S; Obach RS; MN OB; O’Connor R; Ogilvie K; Owen D; Pettersson M; Reese MR; Rogers TF; Rosales R; Rossulek MI; Sathish JG; Shirai N; Steppan C; Ticehurst M; Updyke LW; Weston S; Zhu Y; White KM; Garcia-Sastre A; Wang J; Chatterjee AK; Mesecar AD; Frieman MB; Anderson AS; Allerton C Preclinical characterization of an intravenous coronavirus 3CL protease inhibitor for the potential treatment of COVID19. Nat. Commun 2021, 12, 6055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Owen DR; Allerton CMN; Anderson AS; Aschenbrenner L; Avery M; Berritt S; Boras B; Cardin RD; Carlo A; Coffman KJ; Dantonio A; Di L; Eng H; Ferre R; Gajiwala KS; Gibson SA; Greasley SE; Hurst BL; Kadar EP; Kalgutkar AS; Lee JC; Lee J; Liu W; Mason SW; Noell S; Novak JJ; Obach RS; Ogilvie K; Patel NC; Pettersson M; Rai DK; Reese MR; Sammons MF; Sathish JG; Singh RSP; Steppan CM; Stewart AE; Tuttle JB; Updyke L; Verhoest PR; Wei L; Yang Q; Zhu Y An oral SARS-CoV-2 Mpro inhibitor clinical candidate for the treatment of COVID-19. Science 2021, 374, 1586–1593. [DOI] [PubMed] [Google Scholar]
- (14).Li Y; Tenchov R; Smoot J; Liu C; Watkins S; Zhou Q A comprehensive review of the global efforts on COVID-19 vaccine development. ACS Cent. Sci 2021, 7, 512–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Tregoning JS; Flight KE; Higham SL; Wang Z; Pierce BF Progress of the COVID-19 vaccine effort: viruses, vaccines and variants versus efficacy, effectiveness and escape. Nat. Rev. Immunol 2021, 21, 626–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Ghosh AK; Brindisi M; Shahabi D; Chapman ME; Mesecar AD Drug development and medicinal chemistry efforts toward SARS-coronavirus and Covid-19 therapeutics. ChemMedChem 2020, 15, 907–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Morse JS; Lalonde T; Xu S; Liu WR Learning from the past: possible urgent prevention and treatment options for severe acute respiratory infections caused by 2019-nCoV. Chembiochem 2020, 21, 730–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Beigel JH; Tomashek KM; Dodd LE; Mehta AK; Zingman BS; Kalil AC; Hohmann E; Chu HY; Luetkemeyer A; Kline S; Lopez de Castilla D; Finberg RW; Dierberg K; Tapson V; Hsieh L; Patterson TF; Paredes R; Sweeney DA; Short WR; Touloumi G; Lye DC; Ohmagari N; Oh M.-d.; Ruiz-Palacios GM; Benfield T; Fätkenheuer G; Kortepeter MG; Atmar RL; Creech CB; Lundgren J; Babiker AG; Pett S; Neaton JD; Burgess TH; Bonnett T; Green M; Makowski M; Osinusi A; Nayak S; Lane HC Remdesivir for the treatment of Covid-19 — final report. N. Engl. J. Med 2020, 383, 1813–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Yin W; Mao C; Luan X; Shen DD; Shen Q; Su H; Wang X; Zhou F; Zhao W; Gao M; Chang S; Xie YC; Tian G; Jiang HW; Tao SC; Shen J; Jiang Y; Jiang H; Xu Y; Zhang S; Zhang Y; Xu HE Structural basis for inhibition of the RNA-dependent RNA polymerase from SARS-CoV-2 by remdesivir. Science 2020, 368, 1499–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Cox RM; Wolf JD; Plemper RK Therapeutically administered ribonucleoside analogue MK-4482/EIDD-2801 blocks SARS-CoV-2 transmission in ferrets. Nat. Microbiol 2021, 6, 11–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Jayk Bernal A; Gomes da Silva MM; Musungaie DB; Kovalchuk E; Gonzalez A; Delos Reyes V; Martin-Quiros A; Caraco Y; Williams-Diaz A; Brown ML; Du J; Pedley A; Assaid C; Strizki J; Grobler JA; Shamsuddin HH; Tipping R; Wan H; Paschke A; Butterton JR; Johnson MG; De Anda C; MOVe-OUT Study Group. Molnupiravir for oral treatment of Covid-19 in nonhospitalized patients. N. Engl. J. Med 2022, 386, 509–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Kabinger F; Stiller C; Schmitzova J; Dienemann C; Kokic G; Hillen HS; Hobartner C; Cramer P Mechanism of molnupiravir-induced SARS-CoV-2 mutagenesis. Nat. Struct. Mol. Biol 2021, 28, 740–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Painter WP; Holman W; Bush JA; Almazedi F; Malik H; Eraut N; Morin MJ; Szewczyk LJ; Painter GR Human safety, tolerability, and pharmacokinetics of molnupiravir, a novel broad-spectrum oral antiviral agent with activity against SARS-CoV-2. Antimicrob. Agents Chemother 2021, 65, e02428–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Liu C; Zhou Q; Li Y; Garner LV; Watkins SP; Carter LJ; Smoot J; Gregg AC; Daniels AD; Jervey S; Albaiu D Research and development on therapeutic agents and vaccines for COVID-19 and related human coronavirus diseases. ACS Cent. Sci 2020, 6, 315–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Harvey WT; Carabelli AM; Jackson B; Gupta RK; Thomson EC; Harrison EM; Ludden C; Reeve R; Rambaut A; COVID-19 Genomics UK (COG-UK) Consortium; Peacock SJ; Robertson DL SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol 2021, 19, 409–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Supasa P; Zhou D; Dejnirattisai W; Liu C; Mentzer AJ; Ginn HM; Zhao Y; Duyvesteyn HME; Nutalai R; Tuekprakhon A; Wang B; Paesen GC; Slon-Campos J; Lopez-Camacho C; Hallis B; Coombes N; Bewley KR; Charlton S; Walter TS; Barnes E; Dunachie SJ; Skelly D; Lumley SF; Baker N; Shaik I; Humphries HE; Godwin K; Gent N; Sienkiewicz A; Dold C; Levin R; Dong T; Pollard AJ; Knight JC; Klenerman P; Crook D; Lambe T; Clutterbuck E; Bibi S; Flaxman A; Bittaye M; Belij-Rammerstorfer S; Gilbert S; Hall DR; Williams MA; Paterson NG; James W; Carroll MW; Fry EE; Mongkolsapaya J; Ren J; Stuart DI; Screaton GR Reduced neutralization of SARS-CoV-2 B.1.1.7 variant by convalescent and vaccine sera. Cell 2021, 184, 2201–2211 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).VanBlargan LA; Errico JM; Halfmann PJ; Zost SJ; Crowe JE Jr.; Purcell LA; Kawaoka Y; Corti D; Fremont DH; Diamond MS An infectious SARS-CoV-2 B.1.1.529 Omicron virus escapes neutralization by therapeutic monoclonal antibodies. Nat. Med 2022, 28, 490–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Szemiel AM; Merits A; Orton RJ; MacLean OA; Pinto RM; Wickenhagen A; Lieber G; Turnbull ML; Wang S; Furnon W; Suarez NM; Mair D; da Silva Filipe A; Willett BJ; Wilson SJ; Patel AH; Thomson EC; Palmarini M; Kohl A; Stewart ME In vitro selection of Remdesivir resistance suggests evolutionary predictability of SARS-CoV-2. PLoS Pathog. 2021, 17, e1009929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Stevens LJ; Pruijssers AJ; Lee HW; Gordon CJ; Tchesnokov EP; Gribble J; George AS; Hughes TM; Lu X; Li J; Perry JK; Porter DP; Cihlar T; Sheahan TP; Baric RS; Götte M; Denison MR Mutations in the SARS-CoV-2 RNA dependent RNA polymerase confer resistance to remdesivir by distinct mechanisms. Sci. Transl. Med 2022, eabo0718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Focosi D; Maggi F; McConnell S; Casadevall A Very low levels of remdesivir resistance in SARS-COV-2 genomes after 18 months of massive usage during the COVID19 pandemic: A GISAID exploratory analysis. Antiviral Res. 2022, 198, 105247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Wang Y; Zhang D; Du G; Du R; Zhao J; Jin Y; Fu S; Gao L; Cheng Z; Lu Q; Hu Y; Luo G; Wang K; Lu Y; Li H; Wang S; Ruan S; Yang C; Mei C; Wang Y; Ding D; Wu F; Tang X; Ye X; Ye Y; Liu B; Yang J; Yin W; Wang A; Fan G; Zhou F; Liu Z; Gu X; Xu J; Shang L; Zhang Y; Cao L; Guo T; Wan Y; Qin H; Jiang Y; Jaki T; Hayden FG; Horby PW; Cao B; Wang C Remdesivir in adults with severe COVID-19: a randomised, double-blind, placebo-controlled, multicentre trial. Lancet 2020, 395, 1569–1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Spinner CD; Gottlieb RL; Criner GJ; Arribas Lopez JR; Cattelan AM; Soriano Viladomiu A; Ogbuagu O; Malhotra P; Mullane KM; Castagna A; Chai LYA; Roestenberg M; Tsang OTY; Bernasconi E; Le Turnier P; Chang SC; SenGupta D; Hyland RH; Osinusi AO; Cao H; Blair C; Wang H; Gaggar A; Brainard DM; McPhail MJ; Bhagani S; Ahn MY; Sanyal AJ; Huhn G; Marty FM; GS-US-540-5774 Investigators. Effect of remdesivir vs standard care on clinical status at 11 days in patients with moderate COVID-19: a randomized clinical trial. JAMA 2020, 324, 1048–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Swanstrom R; Schinazi RF Lethal mutagenesis as an antiviral strategy. Science 2022, 375, 497–498. [DOI] [PubMed] [Google Scholar]
- (34).Zhou S; Hill CS; Sarkar S; Tse LV; Woodburn BMD; Schinazi RF; Sheahan TP; Baric RS; Heise MT; Swanstrom R Beta-d-N4-hydroxycytidine inhibits SARS-CoV-2 through lethal mutagenesis but is also mutagenic to mammalian cells. J. Infect. Dis 2021, 224, 415–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).FDA, Fact sheet for healthcare providers: Emergency use authorization for molnupiravir (2021); www.fda.gov/media/155054/download. Accessed May 5, 2022.
- (36).Sacco MD; Hu YM; Gongora MV; Meilleur F; Kemp MT; Zhang XJ; Wang J; Chen Y The P132H mutation in the main protease of Omicron SARS-CoV-2 decreases thermal stability without compromising catalysis or small-molecule drug inhibition. Cell. Res 2022, 32, 498–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Greasley SE; Noell S; Plotnikova O; Ferre RA; Liu W; Bolanos B; Fennell K; Nicki J; Craig T; Zhu Y; Stewart AE; Steppan CM Structural basis for Nirmatrelvir in vitro efficacy against the Omicron variant of SARS-CoV-2. J. Biol. Chem 2022, 101972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Ullrich S; Ekanayake KB; Otting G; Nitsche C Main protease mutants of SARS-CoV-2 variants remain susceptible to nirmatrelvir (PF-07321332). Bioorg. Med. Chem. Lett 2022, 62, 128629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).FDA, Fact sheet for healthcare providers: Emergency use authorization for Paxlovid (2021); https://www.fda.gov/media/155050/download. Accessed May 5, 2022.
- (40).Matthew AN; Leidner F; Lockbaum GJ; Henes M; Zephyr J; Hou S; Rao DN; Timm J; Rusere LN; Ragland DA; Paulsen JL; Prachanronarong K; Soumana DI; Nalivaika EA; Kurt Yilmaz N; Ali A; Schiffer CA Drug design strategies to avoid resistance in direct-acting antivirals and beyond. Chem. Rev 2021, 121, 3238–3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Heskin J; Pallett SJC; Mughal N; Davies GW; Moore LSP; Rayment M; Jones R Caution required with use of ritonavir-boosted PF-07321332 in COVID-19 management. Lancet 2022, 399, 21–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Mielech AM; Kilianski A; Baez-Santos YM; Mesecar AD; Baker SC MERS-CoV papain-like protease has deISGylating and deubiquitinating activities. Virology 2014, 450–451, 64–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Sulea T; Lindner HA; Purisima EO; Menard R Deubiquitination, a new function of the severe acute respiratory syndrome coronavirus papain-like protease? J. Virol 2005, 79, 4550–4551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Bekes M; van der Heden van Noort GJ; Ekkebus R; Ovaa H; Huang TT; Lima CD Recognition of lys48-linked di-ubiquitin and deubiquitinating activities of the SARS coronavirus papain-like protease. Mol. Cell 2016, 62, 572–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Freitas BT; Durie IA; Murray J; Longo JE; Miller HC; Crich D; Hogan RJ; Tripp RA; Pegan SD Characterization and noncovalent inhibition of the deubiquitinase and deISGylase activity of SARS-CoV-2 papain-like protease. ACS Infect. Dis 2020, 6, 2099–2109. [DOI] [PubMed] [Google Scholar]
- (46).Shin D; Mukherjee R; Grewe D; Bojkova D; Baek K; Bhattacharya A; Schulz L; Widera M; Mehdipour AR; Tascher G; Geurink PP; Wilhelm A; van der Heden van Noort GJ; Ovaa H; Müller S; Knobeloch K-P; Rajalingam K; Schulman BA; Cinatl J; Hummer G; Ciesek S; Dikic I Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature 2020, 587, 657–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Munnur D; Teo Q; Eggermont D; Lee HHY; Thery F; Ho J; van Leur SW; Ng WWS; Siu LYL; Beling A; Ploegh H; Pinto-Fernandez A; Damianou A; Kessler B; Impens F; Mok CKP; Sanyal S Altered ISGylation drives aberrant macrophage-dependent immune responses during SARS-CoV-2 infection. Nat. Immunol 2021, 22, 1416–1427. [DOI] [PubMed] [Google Scholar]
- (48).Cao X ISG15 secretion exacerbates inflammation in SARS-CoV-2 infection. Nat. Immunol 2021, 22, 1360–1362. [DOI] [PubMed] [Google Scholar]
- (49).Lei J; Kusov Y; Hilgenfeld R Nsp3 of coronaviruses: structures and functions of a large multi-domain protein. Antiviral Res. 2018, 149, 58–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Woo PC; Huang Y; Lau SK; Yuen KY Coronavirus genomics and bioinformatics analysis. Viruses 2010, 2, 1804–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Chen Z; Wang Y; Ratia K; Mesecar AD; Wilkinson KD; Baker SC Proteolytic processing and deubiquitinating activity of papain-like proteases of human coronavirus NL63. J. Virol 2007, 81, 6007–6018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Osipiuk J; Azizi SA; Dvorkin S; Endres M; Jedrzejczak R; Jones KA; Kang S; Kathayat RS; Kim Y; Lisnyak VG; Maki SL; Nicolaescu V; Taylor CA; Tesar C; Zhang YA; Zhou Z; Randall G; Michalska K; Snyder SA; Dickinson BC; Joachimiak A Structure of papain-like protease from SARS-CoV-2 and its complexes with non-covalent inhibitors. Nat. Commun 2021, 12, 743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Klemm T; Ebert G; Calleja DJ; Allison CC; Richardson LW; Bernardini JP; Lu BG; Kuchel NW; Grohmann C; Shibata Y; Gan ZY; Cooney JP; Doerflinger M; Au AE; Blackmore TR; van der Heden van Noort GJ; Geurink PP; Ovaa H; Newman J; Riboldi-Tunnicliffe A; Czabotar PE; Mitchell JP; Feltham R; Lechtenberg BC; Lowes KN; Dewson G; Pellegrini M; Lessene G; Komander D Mechanism and inhibition of the papain-like protease, PLpro, of SARS-CoV-2. EMBO J 2020, 39, e106275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Ma C; Sacco MD; Xia Z; Lambrinidis G; Townsend JA; Hu Y; Meng X; Szeto T; Ba M; Zhang X; Gongora M; Zhang F; Marty MT; Xiang Y; Kolocouris A; Chen Y; Wang J Discovery of SARS-CoV-2 papain-like protease inhibitors through a combination of high-throughput screening and a FlipGFP-based reporter assay. ACS Cent. Sci 2021, 7, 1245–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Srinivasan V; Brognaro H; Prabhu PR; de Souza EE; Günther S; Reinke PYA; Lane TJ; Ginn H; Han H; Ewert W; Sprenger J; Koua FHM; Falke S; Werner N; Andaleeb H; Ullah N; Franca BA; Wang M; Barra ALC; Perbandt M; Schwinzer M; Schmidt C; Brings L; Lorenzen K; Schubert R; Guaragna Machado RR; Candido ED; Leal Oliveira DB; Durigon EL; Yefanov O; Lieske J; Gelisio L; Domaracky M; Middendorf P; Groessler M; Trost F; Galchenkova M; Saouane S; Hakanpää J; Wolf M; Turk D; Pearson AR; Chapman HN; Hinrichs W; Wrenger C; Meents A; Betzel C SARS-CoV-2 papain-like protease PLpro in complex with natural compounds reveal allosteric sites for antiviral drug design. bioRxiv 2021, 2021.11.17.468943. [Google Scholar]
- (56).Osipiuk J; Wydorski PM; Lanham BT; Tesar C; Endres M; Engle E; Jedrzejczak R; Mullapudi V; Michalska K; Fidelis K; Fushman D; Joachimiak A; Joachimiak LA Dual domain recognition determines SARS-CoV-2 PLpro selectivity for human ISG15 and K48-linked di-ubiquitin. bioRxiv 2021, 2021.09.15.460543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Rut W; Lv Z; Zmudzinski M; Patchett S; Nayak D; Snipas SJ; El Oualid F; Huang TT; Bekes M; Drag M; Olsen SK Activity profiling and crystal structures of inhibitor-bound SARS-CoV-2 papain-like protease: A framework for anti-COVID-19 drug design. Sci Adv 2020, 6, eabd4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Baez-Santos YM; St John SE; Mesecar AD The SARS-coronavirus papain-like protease: structure, function and inhibition by designed antiviral compounds. Antiviral Res. 2015, 115, 21–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Capasso C; Nocentini A; Supuran CT Protease inhibitors targeting the main protease and papain-like protease of coronaviruses. Expert Opin. Ther. Pat 2021, 31, 309–324. [DOI] [PubMed] [Google Scholar]
- (60).Ghosh AK; Takayama J; Aubin Y; Ratia K; Chaudhuri R; Baez Y; Sleeman K; Coughlin M; Nichols DB; Mulhearn DC; Prabhakar BS; Baker SC; Johnson ME; Mesecar AD Structure-based design, synthesis, and biological evaluation of a series of novel and reversible inhibitors for the severe acute respiratory syndrome-coronavirus papain-like protease. J. Med. Chem 2009, 52, 5228–5240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Ghosh AK; Takayama J; Rao KV; Ratia K; Chaudhuri R; Mulhearn DC; Lee H; Nichols DB; Baliji S; Baker SC; Johnson ME; Mesecar AD Severe acute respiratory syndrome coronavirus papain-like novel protease inhibitors: design, synthesis, protein-ligand X-ray structure and biological evaluation. J. Med. Chem 2010, 53, 4968–4979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Ratia K; Pegan S; Takayama J; Sleeman K; Coughlin M; Baliji S; Chaudhuri R; Fu W; Prabhakar BS; Johnson ME; Baker SC; Ghosh AK; Mesecar AD A noncovalent class of papain-like protease/deubiquitinase inhibitors blocks SARS virus replication. Proc. Natl. Acad. Sci. U. S. A 2008, 105, 16119–16124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Markossian S, G. A., Brimacombe K, et al. , editors. Assay guidance manual [Internet]. Bethesda (MD): Eli Lilly & Company and the National Center for Advancing Translational Sciences; 2004-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK53196/. Accessed May 5, 2022. [PubMed] [Google Scholar]
- (64).Ma C; Tan H; Choza J; Wang Y; Wang J Validation and invalidation of SARS-CoV-2 main protease inhibitors using the Flip-GFP and Protease-Glo luciferase assays. Acta Pharm. Sin. B 2022, 12, 1636–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Ma C; Hu Y; Townsend JA; Lagarias PI; Marty MT; Kolocouris A; Wang J Ebselen, disulfiram, carmofur, PX-12, tideglusib, and shikonin are nonspecific promiscuous SARS-CoV-2 main protease inhibitors. ACS Pharmacol. Transl. Sci 2020, 3, 1265–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Ma C; Wang J Dipyridamole, chloroquine, montelukast sodium, candesartan, oxytetracycline, and atazanavir are not SARS-CoV-2 main protease inhibitors. Proc. Natl. Acad. Sci. U. S. A 2021, 118, e2024420118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Shen Z; Ratia K; Cooper L; Kong D; Lee H; Kwon Y; Li Y; Alqarni S; Huang F; Dubrovskyi O; Rong L; Thatcher GRJ; Xiong R Design of SARS-CoV-2 PLpro inhibitors for COVID-19 antiviral therapy leveraging binding cooperativity. J. Med. Chem 2022, 65, 2940–2955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Fu Z; Huang B; Tang J; Liu S; Liu M; Ye Y; Liu Z; Xiong Y; Zhu W; Cao D; Li J; Niu X; Zhou H; Zhao YJ; Zhang G; Huang H The complex structure of GRL0617 and SARS-CoV-2 PLpro reveals a hot spot for antiviral drug discovery. Nat. Commun 2021, 12, 488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Ma C; Wang J Validation and invalidation of SARS-CoV-2 papain-like protease inhibitors. ACS Pharmacol. Transl. Sci 2022, 5, 102–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Li X; Lidsky PV; Xiao Y; Wu CT; Garcia-Knight M; Yang J; Nakayama T; Nayak JV; Jackson PK; Andino R; Shu X Ethacridine inhibits SARS-CoV-2 by inactivating viral particles. PLoS Pathog 2021, 17, e1009898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Zhang Q; Schepis A; Huang H; Yang J; Ma W; Torra J; Zhang SQ; Yang L; Wu H; Nonell S; Dong Z; Kornberg TB; Coughlin SR; Shu X Designing a green fluorogenic protease reporter by flipping a beta strand of GFP for imaging apoptosis in animals. J. Am. Chem. Soc 2019, 141, 4526–4530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Froggatt HM; Heaton BE; Heaton NS Development of a fluorescence-based, high-throughput SARS-CoV-2 3CL(pro) reporter assay. J. Virol 2020, 94, e01265–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Ma C; Xia Z; Sacco MD; Hu Y; Townsend JA; Meng X; Choza J; Tan H; Jang J; Gongora MV; Zhang X; Zhang F; Xiang Y; Marty MT; Chen Y; Wang J Discovery of di- and trihaloacetamides as covalent SARS-CoV-2 main protease inhibitors with high target specificity. J. Am. Chem. Soc 2021, 143, 20697–20709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Rawson JMO; Duchon A; Nikolaitchik OA; Pathak VK; Hu WS Development of a cell-based luciferase complementation assay for identification of SARS-CoV-2 3CL(pro) inhibitors. Viruses 2021, 13, 173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Gerber PP; Duncan LM; Greenwood EJ; Marelli S; Naamati A; Teixeira-Silva A; Crozier TW; Gabaev I; Zhan JR; Mulroney TE; Horner EC; Doffinger R; Willis AE; Thaventhiran JE; Protasio AV; Matheson NJ A protease-activatable luminescent biosensor and reporter cell line for authentic SARS-CoV-2 infection. PLoS Pathog. 2022, 18, e1010265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Pahmeier F; Neufeldt CJ; Cerikan B; Prasad V; Pape C; Laketa V; Ruggieri A; Bartenschlager R; Cortese M A versatile reporter system to monitor virus-infected cells and its application to dengue virus and SARS-CoV-2. J. Virol 2021, 95, e01715–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Cao W; Cho CD; Geng ZZ; Shaabani N; Ma XR; Vatansever EC; Alugubelli YR; Ma Y; Chaki SP; Ellenburg WH; Yang KS; Qiao Y; Allen R; Neuman BW; Ji H; Xu S; Liu WR Evaluation of SARS-CoV-2 main protease inhibitors using a novel cell-based assay. ACS Cent. Sci 2022, 8, 192–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Gao X; Qin B; Chen P; Zhu K; Hou P; Wojdyla JA; Wang M; Cui S Crystal structure of SARS-CoV-2 papain-like protease. Acta Pharm. Sin. B 2021, 11, 237–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Shan H; Liu J; Shen J; Dai J; Xu G; Lu K; Han C; Wang Y; Xu X; Tong Y; Xiang H; Ai Z; Zhuang G; Hu J; Zhang Z; Li Y; Pan L; Tan L Development of potent and selective inhibitors targeting the papain-like protease of SARS-CoV-2. Cell Chem. Biol 2021, 28, 855–865 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Zhao Y; Du X; Duan Y; Pan X; Sun Y; You T; Han L; Jin Z; Shang W; Yu J; Guo H; Liu Q; Wu Y; Peng C; Wang J; Zhu C; Yang X; Yang K; Lei Y; Guddat LW; Xu W; Xiao G; Sun L; Zhang L; Rao Z; Yang H High-throughput screening identifies established drugs as SARS-CoV-2 PLpro inhibitors. Protein Cell 2021, 12, 877–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Lim CT; Tan KW; Wu M; Ulferts R; Armstrong LA; Ozono E; Drury LS; Milligan JC; Zeisner TU; Zeng J; Weissmann F; Canal B; Bineva-Todd G; Howell M; O’Reilly N; Beale R; Kulathu Y; Labib K; Diffley JFX Identifying SARS-CoV-2 antiviral compounds by screening for small molecule inhibitors of Nsp3 papain-like protease. Biochem. J 2021, 478, 2517–2531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Napolitano V; Dabrowska A; Schorpp K; Mourao A; Barreto-Duran E; Benedyk M; Botwina P; Brandner S; Bostock M; Chykunova Y; Czarna A; Dubin G; Frohlich T; Holscher M; Jedrysik M; Matsuda A; Owczarek K; Pachota M; Plettenburg O; Potempa J; Rothenaigner I; Schlauderer F; Slysz K; Szczepanski A; Greve-Isdahl Mohn K; Blomberg B; Sattler M; Hadian K; Popowicz GM; Pyrc K Acriflavine, a clinically approved drug, inhibits SARS-CoV-2 and other betacoronaviruses. Cell Chem. Biol 2022. doi: 10.1016/j.chembiol.2021.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Xu Y; Chen K; Pan J; Lei Y; Zhang D; Fang L; Tang J; Chen X; Ma Y; Zheng Y; Zhang B; Zhou Y; Zhan J; Xu W Repurposing clinically approved drugs for COVID-19 treatment targeting SARS-CoV-2 papain-like protease. Int. J. Biol. Macromol 2021, 188, 137–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Santos LH; Kronenberger T; Almeida RG; Silva EB; Rocha REO; Oliveira JC; Barreto LV; Skinner D; Fajtova P; Giardini MA; Woodworth B; Bardine C; Lourenco AL; Craik CS; Poso A; Podust LM; McKerrow JH; Siqueira-Neto JL; O’Donoghue AJ; da Silva Junior EN; Ferreira RS Structure-based identification of naphthoquinones and derivatives as novel inhibitors of main protease Mpro and papain-like protease PLpro of SARS-CoV-2. bioRxiv 2022, 2022.01.05.475095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Cho CC; Li SG; Lalonde TJ; Yang KS; Yu G; Qiao Y; Xu S; Ray Liu W Drug repurposing for the SARS-CoV-2 papain-like protease. ChemMedChem 2022, 17, e202100455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Lewis DSM; Ho J; Wills S; Kawall A; Sharma A; Chavada K; Ebert M; Evoli S; Singh A; Rayalam S; Mody V; Taval S Aloin isoforms (A and B) selectively inhibits proteolytic and deubiquitinating activity of papain like protease (PLpro) of SARS-CoV-2 in vitro. Sci. Rep 2022, 12, 2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Sanders B; Pohkrel S; Labbe A; Mathews I; Cooper C; Davidson R; Phillips G; Weiss K; Zhang Q; O’Neill H; Kaur M; Ferrins L; Schmidt J; Reichard W; Surendranathan S; Kumaran D; Andi B; Babnigg G; Moriarty N; Adams P; Joachimiak A; Jonsson C; Wakatsuki S; Galanie S; Head M; Parks J Potent and selective covalent inhibitors of the papain-like protease from SARS-CoV-2. Res Sq 2021. doi: 10.21203/rs.3.rs-906621/v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Liu N; Zhang Y; Lei Y; Wang R; Zhan M; Liu J; An Y; Zhou Y; Zhan J; Yin F; Li Z Design and evaluation of a novel peptide–drug conjugate covalently targeting SARS-CoV-2 papain-like protease. J. Med. Chem 2022, 65, 876–884. [DOI] [PubMed] [Google Scholar]
- (89).Di Sarno V; Lauro G; Musella S; Ciaglia T; Vestuto V; Sala M; Scala MC; Smaldone G; Di Matteo F; Novi S; Tecce MF; Moltedo O; Bifulco G; Campiglia P; Gomez-Monterrey IM; Snoeck R; Andrei G; Ostacolo C; Bertamino A Identification of a dual acting SARS-CoV-2 proteases inhibitor through in silico design and step-by-step biological characterization. Eur. J. Med. Chem 2021, 226, 113863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (90).Weglarz-Tomczak E; Tomczak JM; Talma M; Burda-Grabowska M; Giurg M; Brul S Identification of ebselen and its analogues as potent covalent inhibitors of papain-like protease from SARS-CoV-2. Sci. Rep 2021, 11, 3640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (91).Zmudzinski M; Rut W; Olech K; Granda J; Giurg M; Burda-Grabowska M; Zhang L; Sun X; Lv Z; Nayak D; Kesik-Brodacka M; Olsen SK; Hilgenfeld R; Drag M Ebselen derivatives are very potent dual inhibitors of SARS-CoV-2 proteases - PLpro and Mpro in in vitro studies. bioRxiv 2020, 2020.08.30.273979. [Google Scholar]
- (92).Cheng KW; Cheng SC; Chen WY; Lin MH; Chuang SJ; Cheng IH; Sun CY; Chou CY Thiopurine analogs and mycophenolic acid synergistically inhibit the papain-like protease of Middle East respiratory syndrome coronavirus. Antiviral Res. 2015, 115, 9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (93).Chou CY; Chien CH; Han YS; Prebanda MT; Hsieh HP; Turk B; Chang GG; Chen X Thiopurine analogues inhibit papain-like protease of severe acute respiratory syndrome coronavirus. Biochem. Pharmacol 2008, 75, 1601–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (94).Swaim CD; Dwivedi V; Perng Y-C; Zhao X; Canadeo LA; Harastani HH; Darling TL; Boon ACM; Lenschow DJ; Kulkarni V; Huibregtse JM 6-Thioguanine blocks SARS-CoV-2 replication by inhibition of PLpro. iScience 2021, 24, 103213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (95).Brewitz L; Kamps J; Lukacik P; Strain-Damerell C; Zhao Y; Tumber A; Malla TR; Orville AM; Walsh MA; Schofield CJ Mass spectrometric assays reveal discrepancies in inhibition profiles for the SARS-CoV-2 papain-like protease. ChemMedChem 2022, e202200016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (96).Meister TL; Bruggemann Y; Todt D; Conzelmann C; Muller JA; Gross R; Munch J; Krawczyk A; Steinmann J; Steinmann J; Pfaender S; Steinmann E Virucidal efficacy of different oral rinses against severe acute respiratory syndrome coronavirus 2. J. Infect. Dis 2020, 222, 1289–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Xu C; Wang A; Hoskin ER; Cugini C; Markowitz K; Chang TL; Fine DH Differential effects of antiseptic mouth rinses on SARS-CoV-2 infectivity in vitro. Pathogens 2021, 10, 272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (98).Jin Z; Du X; Xu Y; Deng Y; Liu M; Zhao Y; Zhang B; Li X; Zhang L; Peng C; Duan Y; Yu J; Wang L; Yang K; Liu F; Jiang R; Yang X; You T; Liu X; Yang X; Bai F; Liu H; Liu X; Guddat LW; Xu W; Xiao G; Qin C; Shi Z; Jiang H; Rao Z; Yang H Structure of M(pro) from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [DOI] [PubMed] [Google Scholar]
- (99).Maiti BK Can papain-like protease inhibitors halt SARS-CoV-2 replication? ACS Pharmacol. Transl. Sci 2020, 3, 1017–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (100).Sargsyan K; Lin CC; Chen T; Grauffel C; Chen YP; Yang WZ; Yuan HS; Lim C Multi-targeting of functional cysteines in multiple conserved SARS-CoV-2 domains by clinically safe Zn-ejectors. Chem. Sci 2020, 11, 9904–9909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (101).Chen T; Fei CY; Chen YP; Sargsyan K; Chang CP; Yuan HS; Lim C Synergistic inhibition of SARS-CoV-2 replication using disulfiram/ebselen and remdesivir. ACS Pharmacol. Transl. Sci 2021, 4, 898–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (102).Amporndanai K; Meng X; Shang W; Jin Z; Rogers M; Zhao Y; Rao Z; Liu ZJ; Yang H; Zhang L; O’Neill PM; Samar Hasnain S Inhibition mechanism of SARS-CoV-2 main protease by ebselen and its derivatives. Nat. Commun 2021, 12, 3061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (103).Qiao Z; Wei N; Jin L; Zhang H; Luo J; Zhang Y; Wang K The Mpro structure-based modifications of ebselen derivatives for improved antiviral activity against SARS-CoV-2 virus. Bioorg. Chem 2021, 117, 105455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (104).Lee H; Lei H; Santarsiero BD; Gatuz JL; Cao S; Rice AJ; Patel K; Szypulinski MZ; Ojeda I; Ghosh AK; Johnson ME Inhibitor recognition specificity of MERS-CoV papain-like protease may differ from that of SARS-CoV. ACS Chem. Biol 2015, 10, 1456–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (105).Yang H; Li J; Wu Z; Li W; Liu G; Tang Y Evaluation of Different methods for identification of structural alerts using chemical ames mutagenicity data set as a benchmark. Chem. Res. Toxicol 2017, 30, 1355–1364. [DOI] [PubMed] [Google Scholar]
- (106).Baez-Santos YM; Barraza SJ; Wilson MW; Agius MP; Mielech AM; Davis NM; Baker SC; Larsen SD; Mesecar AD X-ray structural and biological evaluation of a series of potent and highly selective inhibitors of human coronavirus papain-like proteases. J. Med. Chem 2014, 57, 2393–2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (107).Johnston PA Redox cycling compounds generate H2O2 in HTS buffers containing strong reducing reagents--real hits or promiscuous artifacts? Curr. Opin. Chem. Biol 2011, 15, 174–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (108).Soares KM; Blackmon N; Shun TY; Shinde SN; Takyi HK; Wipf P; Lazo JS; Johnston PA Profiling the NIH small molecule repository for compounds that generate H2O2 by redox cycling in reducing environments. Assay Drug Dev. Technol 2010, 8, 152–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (109).Baell JB; Nissink JWM Seven Year Itch: Pan-assay interference compounds (PAINS) in 2017-utility and limitations. ACS Chem. Biol 2018, 13, 36–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (110).Dahlin JL; Walters MA How to triage PAINS-full research. Assay Drug Dev. Technol 2016, 14, 168–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (111).Dahlin JL; Nissink JW; Strasser JM; Francis S; Higgins L; Zhou H; Zhang Z; Walters MA PAINS in the assay: chemical mechanisms of assay interference and promiscuous enzymatic inhibition observed during a sulfhydryl-scavenging HTS. J. Med. Chem 2015, 58, 2091–2113. [DOI] [PMC free article] [PubMed] [Google Scholar]