

Abstract

Despite tremendous efforts in the past two years, our understanding of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), virus–host interactions, immune response, virulence, transmission, and evolution is still very limited. This limitation calls for further in-depth investigation. Computational studies have become an indispensable component in combating coronavirus disease 2019 (COVID-19) due to their low cost, their efficiency, and the fact that they are free from safety and ethical constraints. Additionally, the mechanism that governs the global evolution and transmission of SARS-CoV-2 cannot be revealed from individual experiments and was discovered by integrating genotyping of massive viral sequences, biophysical modeling of protein–protein interactions, deep mutational data, deep learning, and advanced mathematics. There exists a tsunami of literature on the molecular modeling, simulations, and predictions of SARS-CoV-2 and related developments of drugs, vaccines, antibodies, and diagnostics. To provide readers with a quick update about this literature, we present a comprehensive and systematic methodology-centered review. Aspects such as molecular biophysics, bioinformatics, cheminformatics, machine learning, and mathematics are discussed. This review will be beneficial to researchers who are looking for ways to contribute to SARS-CoV-2 studies and those who are interested in the status of the field.
1. Introduction
Since its first case was identified in Wuhan, China, in December 2019, coronavirus disease 2019 (COVID-19), caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), has expeditiously spread to as many as 226 countries and territories worldwide and led to over 433 million confirmed cases and over 5.9 million fatalities as of February 2022. This pandemic has also brought a massive economic recession globally. Countries all around the world have implemented a variety of policies to tackle the COVID-19 pandemic (https://stip.oecd.org/covid/).
Many SARS-CoV-2 vaccines and monoclonal antibodies (mAbs) have already obtained use authorization worldwide (https://www.nytimes.com/interactive/2020/science/coronavirus-vaccine-tracker.html). Additionally, the U.S. Food and Drug Administration (FDA) has given emergency use authorization to the oral SARS-CoV-2 Mpro inhibitor PAXLOVID (PF-07321332) developed by Pfizer1,2 (https://www.pfizer.com/news/press-release/press-release-detail/pfizer-receives-us-fda-emergency-use-authorization-novel). However, COVID-19 has a high infection rate, high prevalence, long incubation period,3 asymptomatic transmission,4−6 and potential seasonal patterns.7 SARS-CoV-2 keeps evolving into new infectious and antibody resistant variants.8−10 Therefore, it is imperative to understand the viral molecular mechanism,11 to track its genetic evolution,12 and to continuously improve the efficacy of its antiviral drugs and antibody therapies.
Belonging to the β-coronavirus genus and coronaviridae family, SARS-CoV-2 is an unsegmented positive-sense single-stranded RNA (+ssRNA) virus with a compact 29,903 nucleotide-long genome, and the diameter of each SARS-CoV-2 virion is about 50–200 nm.14 In the first 20 years of the 21st century, β-coronaviruses have triggered three major outbreaks of deadly pneumonia: SARS-CoV (2002), Middle East respiratory syndrome coronavirus (MERS-CoV) (2012), and SARS-CoV-2 (2019).15 Like SARS-CoV and MERS-CoV, SARS-CoV-2 also causes respiratory infections, but at a much higher infection rate.16,17 The complete genome of SARS-CoV-2 comprises 15 open reading frames (ORFs), which encodes 29 structural and nonstructural proteins (nsps), illustrated in Figure 1. The 16 nonstructural proteins nsp1–nsp16 get expressed by protein-coding genes ORF1a and ORF1b, while four canonical 3′ structural proteins, spike (S), envelope (E), membrane (M), and nucleocapsid (N) proteins, as well as accessory factors, are encoded by another four major ORFs, namely ORF2, ORF4, ORF5, and ORF9 (see Figure 1).18−21
Figure 1.

Genomics organization and proteins of SARS-CoV-2. Adapted with permission from ref (13). Copyright 2021 John Wiley and Sons.
The viral structure of SARS-CoV-2 can be found at the upper right corner of Figure 2. This structure is formed by the four structural proteins: the N protein holds the RNA genome, the S protein helps the virus enter into the host cell, and the M and E proteins define the shape of the viral envelope.22 The studies on SARS-CoV-2 as well as previous SARS-CoV and other coronaviruses have mostly identified the functions of these structural proteins, nonstructural proteins, as well as accessory proteins, which are summarized in Table 1. Their 3D structures are also largely known from experiments or predictions, which can be found in Figure 1.
Figure 2.

Six stages of the SARS-CoV-2 life cycle. Stage I: Virus entry. I(a): Virus can enter the host cell via plasma membrane fusion. I(b): Virus can enter the host cell via endosomes. Stage II: Translation of viral replication. Stage III: Replication. Here, nsp12 (RdRp) and nsp13 (helicase) cooperate to perform the replication of the viral genome. Stage IV: Translation of viral structure proteins. Stage V: Virion assembly. Stage VI: Release of a virus.
Table 1. Descriptions of SARS-CoV-2 Proteins and 3D Structures.
| proteins | 3D structures |
|---|---|
| nsp1 (180 residues) inhibits host innate immune functions by interaction with the human 40S subunit in ribosomal complexes. Its carboxyl terminus (C-terminus) binds to and obstructs the ribosomal mRNA entry tunnel, thereby inhibiting the antiviral response triggered by innate immunity or interferons. The nsp1–40S ribosome complex further induces an endonucleolytic cleavage near the 5′ untranslated region (5′ UTR) of host mRNAs, targeting them for degradation. By suppressing host gene expression, nsp1 facilitates efficient viral gene expression in infected cells and evasion from the host immune response.27 | nsp1: 7K7P (X-ray 1.77 Å),28 7K3N (X-ray 1.65 Å),29 7EQ4 (X-ray 1.25 Å),30 7OPL (cryo-EM 4.12 Å);31 nsp1–40S: 6ZOK (cryo-EM 2.80 Å), 6ZOL (cryo-EM 2.80 Å), 6ZOJ (cryo-EM 2.80 Å),27 etc. |
| nsp2 (638 residues) may play a role in the modulation of the host cell survival-signaling pathway by interacting with the host factors, prohibitin 1 and prohibitin 2, which are involved in maintaining the functional integrity of the mitochondria and protecting cells from various stresses. It appears that nsp2 could change the intracellular milieu and perturb host intracellular signaling.32 | 7MSX (cryo-EM 3.15 Å), 7MSW (cryo-EM 3.76 Å),32 etc. |
| nsp3 (1945 residues) includes the papain-like protease (PLpro) and some multipass membrane proteins. PLpro is responsible for cleaving and releasing nsp1, nsp2, and nsp3. PLpro also possesses a deubiquitinating/delSGylating activity and processes both “Lys-48”- and “Lys-63”-linked polyubiquitin chains from cellular substrates. It cleaves preferentially interferon stimulated gene 15 (ISG15) from substrates in vitro, which can play a role in host ADP-ribosylation by binding ADP-ribose. In addition, nsp3 participates together with nsp4 in the assembly of virally induced cytoplasmic double-membrane vesicles necessary for viral replication and antagonizes innate immune induction of type I interferon by blocking the phosphorylation, dimerization, and subsequent nuclear translocation of host interferon regulatory factor 3 (IRF3). It also prevents host NF-κB signaling.33,34 | Residues 819–929: 7KAG (X-ray 3.21 Å). Residues 1024–1192 (ADP-ribose phosphatase domain): 6WEY (X-ray, 0.95 Å),35 6WCF (X-ray 1.06 Å).36 Residues 1570–1877 (PLpro): 6W9C (X-ray 2.70 Å).37 Etc. |
| nsp4 (500 residues) is a multipass membrane protein. Together with nsp3, it participates in the assembly of virally induced cytoplasmic double-membrane vesicles, which is necessary for viral replication.38 | AlphaFold prediction.39 |
| nsp5 (306 residues) is the main protease (Mpro/3CLpro) of SARS-CoV-2. It takes charge of cleaving and releasing nsp4–nsp16. Additionally, it recognizes substrates containing the core sequence [ILMVF]-Q-|-[SGACN] and is also able to bind an ADP-ribose-1″-phosphate. Moreover, it plays a role in nsp maturation.22 | 6LU7 (X-ray 2.16 Å),40 etc. |
| nsp6 (290 residues) is a multipass membrane protein. Working with nsp3 and nsp4, it induces double-membrane vesicles (autophagosomes) in infected cells from their endoplasmic reticulum. It also limits the expansion of these autophagosomes that are no longer able to deliver viral components to lysosomes.41,42 | AlphaFold prediction.39 |
| nsp7 (83 residues) plays a role in viral RNA synthesis. It forms a hexadecamer with nsp8 that may participate in viral replication by acting as a primase. Alternatively, it may synthesize substantially longer products than oligonucleotide primers.43 | 6M5I (X-ray 2.50 Å), 7BV1 (cryo-EM 2.80 Å), 7BV2 (cryo-EM 2.50 Å)44 |
| nsp8 (198 residues) plays a role in viral RNA synthesis. It forms a hexadecamer with nsp7 that may participate in viral replication by acting as a primase. Alternatively, it may synthesize substantially longer products than oligonucleotide primers.43 | 6M5I (X-ray 2.50 Å), 7BV1 (cryo-EM 2.80 Å), 7BV2 (cryo-EM 2.50 Å).44 |
| nsp9 (113 residues) functions in viral replication as a dimeric ssRNA-binding protein.45 | 6W4B (X-ray 2.95 Å),46 6WXD (X-ray 2.00 Å),47 etc. |
| nsp10 (139 residues) plays a pivotal role in viral transcription. It forms a dodecamer and interacts with both nsp14 and nsp16 to stimulate their respective 3′-5′ exoribonuclease and 2′-O-methyltransferase activities in viral mRNAs that cap methylation.45 | 6ZCT (X-ray 2.55 Å),48 6ZPE (X-ray 1.58 Å),48 etc. |
| nsp11 (13 residues) is a pp1a cleavage product at the nsp10/11 boundary. For pp1ab, it is a frameshift product that becomes the N-terminal of nsp12. Its function, if any, is currently unknown.45 | None |
| nsp12 is the RNA-dependent RNA polymerase (RdRp) performing both replication and transcription of the viral genome. Specifically, it catalyzes the synthesis of the RNA strand complementary to a given RNA template. The RdRp of SARS-CoV-2 can be inhibited by the nucleoside analogue Remdesivir.45 | 6M71 (cryo-EM 2.90 Å),49 6YYT (cryo-EM 2.90 Å),50 etc. |
| nsp13 (Helicase) (932 residues) is a multifunctional superfamily 1 helicase capable of using both double-stranded DNA (dsDNA) and double-stranded RNA (dsRNA) as substrates with 5′-3′ polarity. In addition to working with nsp12 in viral genome replication, it is also involved in viral mRNA capping. It associates with nucleoprotein in membranous complexes.51 | 5RLH (X-ray 2.38 Å),49 6ZSL (X-ray 1.94 Å),52 etc. |
| nsp14 (601 residues) possesses two different activities: (1) an exoribonuclease activity on both single-strand RNA (ssRNA) and dsRNA in a 3′ to 5′ direction; (2) a N7-guanine methyltransferase (viral mRNA capping) activity. It acts as a proofreading exoribonuclease for RNA replication, thereby lowering the sensitivity of the virus to RNA mutagens.53 It always interacts with nsp10.45 | Exoribonuclease domain: 7DIY (X-ray 2.69 Å),54 7QGI (X-ray 1.65 Å), 7QIF (X-ray 2.53 Å), etc. |
| nsp15 (346 residues) is the nidoviral RNA uridylate-specific endoribonuclease (NendoU) that favors the cleavage of RNA at the 3′-ends of uridylates. The loss of nsp15 affects both viral replication and pathogenesis. It is also required for the evasion of host cell dsRNA sensors.55 | 6WLC (X-ray 1.82 Å),56 etc. |
| nsp16 (298 residues) is activated by and interacts with nsp10. Its 2′-O-methyltransferase activity mediates mRNA cap 2′-O-ribose methylation to the 5′-cap structure of viral mRNAs. Since N7-methyl guanosine cap is a prerequisite for binding of nsp16, it plays an important role in viral mRNAs cap methylation which is essential to evade the immune system. It may also work against host cell antiviral sensors.45 | 6W4H (X-ray 1.80 Å),57 etc. |
| ORF2 (Spike (S) protein) (1273 residues) binds to the host ACE2 receptor and internalizes the virus into the endosomes of host cells with the usage of human TMPRSS2 for priming in human lung cells. S protein consists of three chains and its stalk domain contains three hinges, giving the head unexpected orientational freedom. One single chain can be cleaved into two subunits, S1 and S2. S1 interacts with the host receptor, initiating the infection. S2 mediates fusion of the virion and cellular membranes by acting as a class I viral fusion protein. The current model of S protein has at least three conformational states: prefusion native state, prehairpin intermediate state, and postfusion hairpin state. During viral and target cell membrane fusion, the coil regions (heptad repeats) assume a trimer-of-hairpins structure, positioning the fusion peptide in close proximity to the C-terminal region of the ectodomain. The formation of this structure appears to drive apposition and subsequent fusion of viral and target cell membranes.25 | Spike protein: 6VSB (cryo-EM 3.46 Å),17 etc. RBD-ACE2 complex: 6VW1 (X-ray 2.68 Å),58 etc. |
| ORF3a (275 residues) is a multipass membrane protein that forms homotetrameric potassium sensitive ion channels (viroporin). It upregulates expression of fibrinogen subunits fibrinogen alpha (FGA), fibrinogen beta (FGB), and fibrinogen gamma (FGG) in host lung epithelial cells. It also induces apoptosis in cell culture. Additionally, ORF3a downregulates the type 1 interferon receptor by inducing serine phosphorylation within the interferon (IFN) alpha-receptor subunit 1 (IFNAR1) degradation motif and increases IFNAR1 ubiquitination. More importantly, it activates both NF-κB and NLRP3 inflammasome and contributes to the generation of the cytokine storm. It may also modulate viral release.59 | 6XDC (cryo-EM 2.9 Å),60 7KJR (cryo-EM 2.08 Å).60 |
| ORF3b (22 residues) along with nucleocapsid protein and ORF6 and ORF3b (22 residues) appears to block induction of type I interferons (IFN-I). This 22-residue variant is also present in SARS-CoV-2-related viral genomes in bats and pangolins.61 | None |
| ORF4 (Envelope (E) protein) (75 residues) is a single-pass type III membrane protein playing a central role in virus morphogenesis and assembly; it acts as a viroporin and self-assembles in host membranes forming pentameric protein–lipid pores that allow ion transport. It is also involved in the induction of apoptosis.62 | Residues 8–38: 7K3G (solid-state NMR)63 |
| ORF5 (Membrane protein) (222 residues) is the most abundant structural component of the virion and very conserved. It mediates morphogenesis, assembly, and budding of viral particles through the recruitment of other structural proteins to the ER-Golgi-intermediate compartment (ERGIC). It also interacts with N for RNA packaging into the virion.64 | AlphaFold prediction39 |
| ORF6 (61 residues) appears to be a virulence factor. It disrupts cell nuclear import complex formation by tethering karyopherin alpha 2 and karyopherin beta 1 to the membrane. Retention of import factors at the ER/Golgi membrane leads to a loss of transport into the nucleus, thereby preventing STAT1 (signal transducer and activator of transcription 1) nuclear translocation in response to interferon signaling and, thus, blocking the expression of ISGs that display multiple antiviral activities.65 | 7VPH (X-ray 2.8 Å)66 |
| ORF7a (121 residues) is a type I membrane protein that plays a role as an antagonist of bone marrow stromal antigen 2 (BST-2), disrupting its antiviral effect. As BST-2 tethers virions to the host’s plasma membrane, ORF7a binding inhibits BST-2 glycosylation and interferes with this restriction activity. ORF7a may suppress small interfering RNA (siRNA) and also may bind to host integrin, alpha L (ITGAL), thereby playing a role in attachment or modulation of leukocytes.67 | Residues 16–82: 6w37 (X-ray 2.90 Å) |
| ORF7b (43 residues) is a type III integral transmembrane protein in the Golgi apparatus. In SARS-CoV-2, it appears to be a viral attenuation factor. It may be involved in the human infectivity of SARS-CoV-2.68 | None |
| ORF8 (43 residues) might be a luminal ER membrane-associated protein. It may trigger the activating transcription factor 6 (ATF6) activation and affect the unfolded protein response (UPR). Like ORF7b, it may be involved in the human infectivity of SARS-CoV-2.68−70 | 7JTL (X-ray 2.04 Å),71 etc. |
| ORF9a (N protein) (419 residues) packages the positive strand viral genome RNA into a helical ribonucleocapsid (RNP) and plays a fundamental role during virion assembly through its interactions with the viral genome and membrane protein. It also plays an important role in enhancing the efficiency of subgenomic viral RNA transcription as well as viral replication. It may modulate transforming growth factor-beta signaling by binding host smad3.21 | Residues 41–174: 6M3M (X-ray 2.70 Å),72 etc.. Residues 247–364: 6ZCO (X-ray 1.36 Å).73 Etc. |
| ORF9b (97 residues) plays a role in the inhibition of the host’s innate immune response by targeting the mitochondrial antiviral-signaling protein (MAVS). Mechanistically, it usurps the itchy E3 ubiquitin protein ligase (ITCH) to trigger the degradation of MAVS, TNF receptor associated factor 3 (TRAF3), and TRAF6. In addition, it can cause mitochondrial elongation by triggering ubiquitination and proteasomal degradation of dynamin-1-like (DNM1L) protein.74 | 6Z4U (X-ray 1.95 Å) |
| ORF9c (70 residues) is located in the N-coding region and interacts with various host proteins including Sigma receptors, implying involvement in lipid remodeling and the ER stress response. It might also target NF-κB signaling.75 | None |
| ORF10 (38 residues) interacts with factors in the Cullin 2 (CUL2) RING E3 ligase complex and thus may modulate ubiquitination.75 | 7BYF (X-ray 2.5 Å)76 |
With these SARS-CoV-2 proteins, the intracellular viral life cycle of SARS-CoV-2 can be realized.23 This life cycle has six stages as shown in Figure 2. The first stage is the entry of the virus. SARS-CoV-2 enters the host cell via either endosomes or plasma membrane fusion. In both ways, the S protein of SARS-CoV-2 first attaches to the host cell-surface protein, angiotensin converting enzyme 2 (ACE2). Then, the cell’s protease, TMPRSS2, cuts and opens the S protein of the virus, exposing a fusion peptide in the S2 subunit of S protein.24 After fusion, an endosome forms around the virion, separating it from the rest of the host cell. The virion escapes when the pH of the endosome drops or when cathepsin, a host cysteine protease, cleaves it. The virion then releases its RNA into the cell.25 After the RNA release, polyproteins pp1a and pp1ab are translated. Notably, facilitated by viral papain-like protease (PLpro), nsp1, nsp2, nsp3, and the amino terminus of nsp4 from the pp1a and pp1ab are released. Moreover, nsp5– nsp16 are also cleaved proteolytically by the main protease.26 The next stage of the life cycle is the replication process, where nsp12 (RdRp) and nsp13 (helicase) cooperate to perform the replication of the viral genome. Stages IV and V are the translation of viral structural proteins and the virion assembly process. In these stages, structural proteins S, E, and M are translated by ribosomes and then present on the surface of the endoplasmic reticulum (ER) and are transported from the ER through the Golgi apparatus for the preparation of the virion assembly. Meanwhile, multiple copies of the N protein package the genomics RNA in the cytoplasm, which interacts with another three structural proteins to direct the assembly of virions. Finally, virions will be secreted from the infected cell through exocytosis.
Since the initial outbreak of COVID-19, the raging pandemic caused by SARS-CoV-2 has lasted over two years. We do have many promising vaccines, but they might have side effects and their full side effects, particularly, long-term side effects, remain unknown. To make things worse, nearly 29208 unique mutations have been recorded for SARS-CoV-2 as shown by Mutation Tracker (https://users.math.msu.edu/users/weig/SARS-CoV-2_Mutation_Tracker.html). All of these reveal the sad reality that our current understanding of life science, virology, epidemiology, and medicine is severely limited. Ultimately, the core of the challenges is the lack of molecular mechanistic understanding of many aspects, namely coronavirus RNA proofreading, virus–host cell interactions, antibody–antigen interactions, protein–protein interactions, protein–drug interactions, viral regulation of host cell functions, including autophagocytosis and apoptosis, and irregular host immune response behavior such as cytokine storm and antibody-dependent enhancement. Molecular-level experiments on SARS-CoV-2 are both expensive and time-consuming and require heavy safety measures. Moreover, disparities among reported experimental binding affinities can be more than 100-fold for the receptor-binding domain (RBD) of S protein binding to ACE2 or antibodies (see Table 1 of ref (77)). All these complicated realities make the understanding of the viral evolution and transmission mechanism one of the most challenging tasks.
On the other hand, computational tools provide alternative approaches in understanding viral evolution and transmission with higher efficiency and lower costs. The increasing computer power, the accumulation of molecular data, the availability of artificial intelligence (AI) algorithms, and the development of new mathematical tools have paved the road for mechanistic understanding from molecular modeling, simulations, and predictions. RBD residues 452 and 501 were predicted to “have very high chances to mutate into significantly more infectious COVID-19 strains” in summer 202078 and were later confirmed in the prevailing SARS-CoV-2 variants Alpha, Beta, Gamma, Delta, Theta, Epsilon, Kappa, Lambda, Mu, and Omicron. These predictions,78 achieved via the integration of deep learning, biophysics, genotyping, and advanced mathematics, are some of the most remarkable events. Additionally, 3,696 possible RBD mutations were classified into three categories with different appearance likelihoods, namely, 1149 most likely, 1912 likely, and 625 unlikely.78 The predicted “most likely” partition successfully contained all the newly observed RBD mutations, until the recent appearance of S371L from Omicron BA.1. Most remarkably, the mechanism governing SARS-CoV-2 evolution and transmission, i.e., natural selection via mutation-strengthened infectivity, was discovered in July 202078 when there were only 89 RBD mutations with the highest observed frequency of merely 50 globally.78 In April 2021, this mechanism was confirmed beyond any doubt. By using 506,768 sequences isolated from patients, the authors demonstrated that the predicted binding free energy (BFE) changes of the 100 most observed RBD mutations out of 651 existing RBD mutations are all above the BFE change of −0.28 kcal/mol, indicating evolution favors variants having higher infectivity.79 Moreover, using network-based modeling for drug repurposing, Baricitinib was found to be a potential treatment for COVID-19.80 These extraordinary results prove that computational approaches spearhead the discovery of new drugs and the mechanisms of SARS-CoV-2 evolution and transmission.
Considering intensive research activities in molecular modeling, simulations, and predictions of SARS-CoV-2, it has become essentially impossible for experts and researchers to go through the literature. It is important to present a methodology-centered review to enable readers to grasp the current status of SARS-CoV-2 modeling, simulations, and predictions. In this review, the purpose is to provide a general introduction of molecular-level methodologies for SARS-CoV-2 modeling, simulations, and predictions from the aspects of biophysics, mathematical approaches, and machine learning, including deep learning, bioinformatics, and cheminformatics. A wide variety of molecular-level methodologies is described, followed by their applications to SARS-CoV-2. Comments and discussions are presented. Future perspectives are provided in the Concluding Remarks.
2. Methods and Approaches
2.1. Biophysics
The molecular modeling of viruses and their interactions with host cells involves a variety of aspects of biology, biophysics, biochemistry, virology, immunology, computer science, statistics, and mathematics. This section starts with thermodynamics and electrostatics, followed by discussions on molecular dynamics, normal-mode analysis, Monte Carlo methods, molecular docking, and binding free energy analysis, and ends with density-functional theory and quantum mechanics/molecular mechanics methods.
2.1.1. Thermodynamics
Thermodynamics is a foundation of biological science. The laws of thermodynamics are basic principles of biology that govern biological, chemical, and physical processes in all living organisms as well as viruses. The relations among internal energy, Helmholtz free energy, Gibbs free energy, enthalpy, entropy, temperature, volume, and pressure underpin biophysics. The Gibbs–Helmholtz equation describes the thermodynamics calculating changes in the Gibbs energy of a system as a function of temperature. It is a separable differential equation that is given as
| 1 |
where ΔG is the change in Gibbs free energy, ΔH is the enthalpy change, T is the absolute temperature, and P is the constant pressure.
In the study of the N protein of SARS-CoV, it was shown that the N protein shows its maximum conformational stability near pH 9.0. The oligomer dissociation and protein unfolding occur simultaneously.81 In the denaturation of the N protein by chemicals, the Gibbs free energy change (ΔG) of unfolding at temperature (T) is calculated by the solution of the Gibbs–Helmholtz equation
| 2 |
where Tm is the transition temperature, ΔHm is the enthalpy of unfolding at Tm, and ΔCp is the heat capacity change.
2.1.2. Electrostatic Modeling
In biomolecular studies, electrostatic interactions are important due to their ubiquitous existence in solvation, molecular recognition, molecular interactions, protein–ligand binding, antibody–antigen binding, intramolecular interactions, etc. Electrostatics can be computed using explicit solvent or implicit solvent models as shown in Figure 3. However, including explicit solvent models in free energy calculation is computationally expensive due to their detailed description of solvent molecules. Using an atomic description of the solute molecule, implicit solvent models describe the solvent as a dielectric continuum.82−86 A wide variety of two-scale implicit solvent models has been developed for electrostatic analysis, including Poisson–Boltzmann (PB),83,87 generalized Born (GB),88−91 polarized continuum,92,93 and differential geometry-based models.94,95 GB models give an efficient approximation of PB models but provide only heuristic estimates for electrostatic energies, while PB methods offer more accurate methods for electrostatic analysis.86,89,95−99
Figure 3.
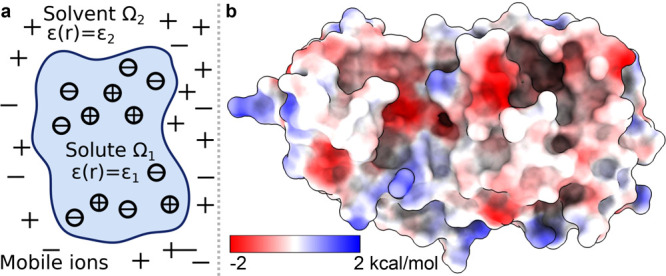
(a) Illustration of the PB model, in which the molecular surface separates the computational domain into the solute region Ω1 and solvent region Ω2. (b) Electrostatic potential of the SARS-CoV-2 Mpro based on the PB model.
2.1.2.1. Generalized Born Model
The GB model is devised to offer a relatively simple and computationally efficient approach to calculate electrostatic solvation free energy.88−91 Under an appropriate parametrization for a given system, a GB solver can achieve accuracy as a PB solver.100 The GB approximation of electrostatic solvation free energy can be given as
| 3 |
where Ri is the effective Born radius of the ith atom, dij is the distance between atoms i and j, ϵ1 is the dielectric constant of the solute, ϵ2 is the dielectric constant of the solvent, qi is the partial charge of atom i, β = ϵ1/ϵ2, α = 0.571412, and A is the electrostatic size of the molecule. Additionally, the function fij is given as
| 4 |
The effective Born radius Ri is calculated via a boundary integral:
| 5 |
where ∂Ω1 is the molecular surface, such as the solvent-excluded surface, dS is the infinitesimal surface element vector, ri represents the position of atom i, and r shows the position of the infinitesimal surface.
2.1.2.2. Poisson–Boltzmann Model
As illustrated in Figure 3, the PB model describes a two-scale treatment of electrostatics. The interior domain Ω1 contains the solute biomolecule with fixed charges, and the exterior domain Ω2 contains the solvent and dissolved ions separated by the interface Γ. While various surface models are available, the most commonly used ones are the molecular surface or solvent excluded surface. A biomolecule in domain Ω1 consists of a set of atomic charges qk located at atomic centers rk for k = 1, ..., Nc, with Nc as the total number of charges. On the other hand, domain Ω2 contains the mobile ions, whose charge source density is approximated by the Boltzmann distribution. The linearized PB model is always applied:
| 6 |
where ϕ(r) is the electrostatic potential, ϵ(r) is a dielectric constant given by ϵ(r) = ϵ1 for r ∈ Ω1 and ϵ(r) = ϵ2 for r ∈ Ω2, and κ is the inverse Debye length representing the ionic effective length. Interface conditions are involved on ∂Ω1, which are given as
| 7 |
where ϕ1 and ϕ2 are the limit values when approaching the interface from inside or outside the solute domain and n is the outward unit normal vector on ∂Ω1. For the far-field boundary condition of the PB model, lim|r|→∞ϕ(r) = 0 is implied. Therefore, the electrostatic solvation free energy can be calculated by
| 8 |
where ϕ0(rk) is the solution of the PB equation as if there were no solvent–solute interface.
Due to their success in describing biomolecular systems, the PB and GB models have attracted wide attention in both the mathematical and biophysical communities.101−103 Meanwhile, much effort has been given to the development of accurate, efficient, reliable, and robust PB solvers. A large number of methods have been proposed in the literature, including the finite difference method (FDM),104 finite element method (FEM),105 and and boundary element method (BEM).106 The emblematic solvers in this category include Poisson–Boltzmann surface area (PBSA),107,108 Delphi,109,110 adaptive Poisson–Boltzmann solver (ABPS),97,111 matched interface and boundary-based Poisson–Boltzmann (MIBPB),98,102,112 chemistry at Harvard macromolecular mechanics (CHARMM) PBEQ-Solver,104 and treecode-accelerated boundary integral (TABI) PB solver.113,114
The PB and GB models have been applied to SARS-CoV-2 studies including protein–ligand binding and protein–protein binding energetics. The surface electrostatic potential values of S protein and Mpro were calculated for SARS-CoV and SARS-CoV-2 with almost the same values for both viruses,115 as well as the SARS-unique domain.116 When focusing on the RBD of S protein binding to ACE2, slightly higher binding energy was revealed for SARS-CoV-2 compared to SARS-CoV because of enhanced electrostatic interactions with the negative electrostatic potential of ACE2 and positive electrostatic potential of RBD.117 For the fusion cleavage site on S protein, mutations near the cleavage site caused changes in the electrostatic distribution of the S protein surface.118 Antigens targeting SARS-CoV-2 from T cells were studied using the electrostatic surface potentials.119 By studying the surface potential of S protein, it was shown that the pH and salt concentration changed dramatically in terms of scale and sign for electrostatic interactions.120 Recently, Dung et al. proposed a theoretical model to elucidate intermolecular electrostatic interactions between a virus and a substrate. Their model treats the virus as a homogeneous particle having surface charge and the polymer fiber of the respirator as a charged plane. The electric potentials surrounding the virus and fiber are influenced by the surface charge distribution of the virus. The PB equation was used to calculate the electric potentials. Then, Derjaguin’s approximation and a linear superposition of the potential function were extended to determine the electrostatic force.121
2.1.3. Molecular Dynamics (MD) Simulation
Macromolecular structures are highly dynamic rather than static. X-ray crystallography and nuclear magnetic resonance (NMR) reveal that even the same molecule can adopt multiple conformations.122,123 On the other hand, conformational change plays a significant role in biomolecular functions. While NMR is limited to small biomolecules and X-ray crystallography can only provide static structures, MD simulation is an effective way to investigate biomolecular conformational changes.124,125 The Poisson–Boltzmann-based MD is also studied.126 Furthermore, thanks to high-performance computing platforms such as graphical processing units (GPUs), current MD simulations can reveal conformational changes of biomacromolecules such as proteins, DNA, and RNA in the time scale of milliseconds (ms).127
MD simulation is becoming an invaluable computational method commonly used for understanding the biomolecular structure and dynamics of atoms in macromolecules (proteins and RNA). Describing internal forces in the structure with simple mathematical functions, the motions are determined by using Newton’s second law.128 In Figure 4a, a general MD algorithm is demonstrated, where potential energy functions (force field), energy minimization, environment settings, ensembles, and solvation are included. In the prediction of atom positions and velocities, equations are given by a standard Taylor expansion. Classical interatomic potentials or quantum mechanisms are applied to calculate forces, which is followed by the correction of positions and velocities with some functions f, g of a, and Δt by energy minimization. Among all their effects on MD simulations, a collection of suitable force field functions is of fundamental importance to all other dynamics methodologies, which will be introduced first.
Figure 4.
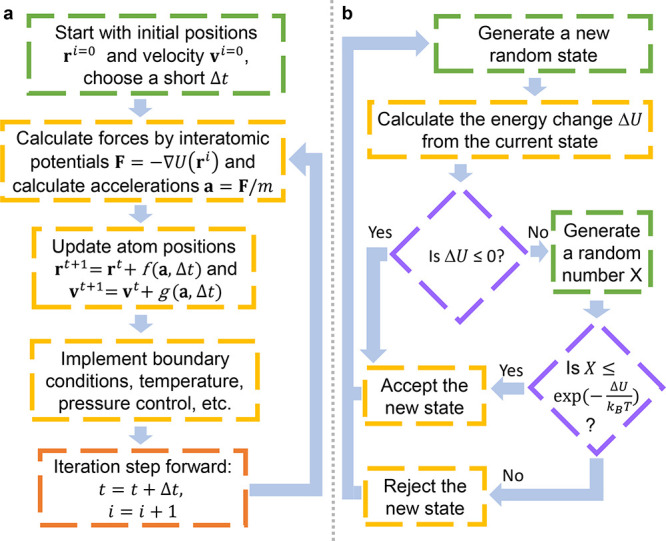
(a) Workflow of molecular dynamics simulations. (b) Workflow of the metropolis Monte Carlo method.
Molecular Mechanics Force Field. A molecular mechanics force field is a set of functions equipped with an associated set of parameters, describing the interactions between atoms. The energy function for nonbonded interactions is associated with simple pairwise additive functions, van der Waals and electrostatic forces, of nuclear coordinates only, while for bonded groups, that is the forms of chemical bonds, bond angles, and bond dihedrals.129 For example, the functional form of a typical force field such as AMBER130 is given as
 |
9 |
where kr and kθ are the force constants for the bond lengths and bond angles, respectively. Here, r and θ are a bond length and a bond angle, r0 and θ0 are the equilibrium bond length and bond angle, ω is the dihedral angle, kn is the corresponding force constant, n is the multiplicity, and phase angle γn takes values of either 0° or 180°. The nonbonded part of the potential is represented by the Lennard-Jones repulsive Aij and attractive Bij terms for Coulomb interactions between partial atomic charges (qi and qj). Here, Rij is the distance between atoms i and j. Finally, ϵ is the dielectric constant that considers the medium effect that is not explicitly represented and usually equals 1.0 in a typical solvated environment where the solvent is represented explicitly. The nonbonded terms are calculated for atom pairs that are either separated by more than three bonds or not bonded. CHARMM is another popular force field.131 Polarizable models, such as the AMOEBA force field,132 have been developed to achieve higher accuracy. To tackle systems with an excessive number of atoms, coarse-grained models are also developed. In these models, a group of atoms is represented by a “pseudo-atom”, so the number of atoms is largely reduced.133 Popular coarse-grained models are the Go̅ model,134 MARTINI force field,135 united-residue (UNRES) force field,136 etc.
Energy Minimization. Energy minimization methods are applied to efficiently optimize molecular structures. In a complex system of N atoms, the potential energy function, such as eq 9U(rN), has its global minimum. It is extremely computationally expensive to provably locate the global minimum. With an unrefined molecular structure equipped with a force field, energy minimization methods can be iteratively applied to molecular systems. The steepest descent method is one of the most popular iterative descent methods, which uses derivatives of various orders and points the path toward the nearest energy minimum. Thus, the force is calculated by
| 10 |
where r is the vector of the atomic coordinates.
Solvation. In an aqueous environment, a protein is solvated in a pure or ion-containing water environment, and explicit solvent models are computationally expensive. To avoid using water explicitly in modeling, numerous implicit solvent models have been developed.82−93 Two implicit solvent models, the Poisson–Boltzmann method and the generalized Born model, have been described in previous sections. In addition, there are explicit solvent models such as the transferable intermolecular potential with 5 points model,137 the extended simple point charge model,138 and the flexible simple point charge model water models.
Since MD simulations can provide many samplings, one can calculate the free energy change between different states from these samplings. Typical binding free energy calculation methods based on MD simulations are the molecular mechanics energies combined with Poisson–Boltzmann or generalized Born and surface area continuum solvation (MM/PBSA and MM/GBSA),108,139,140 free energy perturbation (FEP),141 thermodynamic integration,142 metadynamics,143 and steered MD simulations.144 Recently, a method that is more efficient than normal-mode analysis, called WSAS (work and social adjustment scale),145 was developed to estimate the entropic effect in the free energy calculation.
2.1.3.1. MD Simulations Revealing Conformational Changes
The most important application of MD simulations is to investigate the dynamical properties of SARS-CoV-2 proteins and the interactions between proteins and inhibitors. Moreover, Mpro and S protein are the two main targets of SARS-CoV-2 proteins to investigate, while some studies focused on PLpro. For larger systems, coarse-grained MD simulations are implemented.
Mpro. To enhance sampling of conformational space, a microsecond-scale Gaussian accelerated MD simulation to SARS-CoV-2 Mpro was performed,146 where the simulations identified cryptic pockets within Mpro, including some regions far from the active site. The 2 μs MD trajectories of the apo form of the SARS-CoV-2 Mpro indicated that the long loops, which connect domains II and III and provide access to the binding site and the catalytic dyad, carried out large conformational changes.147 Additionally, MD simulations were applied to compare the dynamical properties of the SARS-CoV-2 Mpro and SARS-CoV Mpro, which suggests that the SARS-CoV Mpro has a larger binding cavity and more flexible loops148 and reveals the key interactions and pharmacophore models between the Mpro and its inhibitors.149 Recently, Sanjeev et al. used MD simulations to study the impact of a crowded environment on drug–Mpro complexes, suggesting that crowding enhances the difference in the dynamics of apo- vs drug-bound complexes.150 Lamichhane et al. not only ran MM/PBSA calculation but also used the analysis by dihedral angle distribution and radial distribution functions to confirm the strong interactions between inhibitor N3 and Mpro.151
S Protein. MD simulations were performed to study the binding of S protein and ACE2,152−156 which were the most important studies of S protein. Notably, a 100 ns MD simulation of the complexes of human ACE2 and S protein from SARS-CoV-2 and SARS-CoV showed that the SARS-CoV-2 complex was more stable.156 Grishin et al.’s MD simulation suggests disulfide bonds play a critical role in S protein–ACE2 binding and the flexibility of the surface loops increases when the four disulfide bonds of the domain are reduced.157 As for temperature, MD simulations at different temperatures suggested S protein had a stronger binding at a low temperature.158 Abdalla et al. investigated the effects of mutations on S protein stability and solubility through MD simulations in a 100 ns period.159 Inhibitor and antibody binding to ACE2 was studied by MD simulations.160−162 One of the studies suggested that the SARS-CoV-2 S protein can interact with a nicotinic acetylcholine receptors (nAChRs) inhibitor.161 Moreover, ACE2-Fc fusion proteins with the SARS-CoV-2 S protein RBD were simulated by a glycosylated molecular model.163 In a steered MD model, a semiopen intermediate state was observed of the transition between closed and open states of S protein.164 Further study was about the motion of glycans in S protein by a 1 μs MD simulation, uncovering the detail of the S protein glycan shield.165 A recent interesting study by Lupala et al. performed an MD simulation of the SARS-CoV-2 S protein with ACE2 from different species. Their findings suggest that the ACE2 proteins of bovine, cat, and panda form strong binding interactions with RBD, while in the cases of rat, least horseshoe bat, horse, pig, mouse, and civet, the ACE2 proteins interact weakly with RBD.166
PLpro and Other Proteins. MD simulations were also used to investigate the conformational changes of other SARS-CoV-2 proteins. For PLpro, researchers performed pH replica-exchange CpHMD (constant pH molecular dynamics) simulations to estimate the pKa values of Asp/Glu/His/Cys/Lys side chains and assessed possible proton-coupled dynamics in SARS-CoV, SARS-CoV-2, and MERS-CoV PLpros.167 They also suggested a possible conformational-selection mechanism by which inhibitors bind to the PLpro. Supervised MD simulations were employed to investigate the unbinding pathways of GRL0617 and its derivates from PLpro.168 Sun et al. applied MD simulations and topological and electrostatic analyses to study the effects of palmitoylation on an E protein pentamer. Their results indicated that the structure of the palmitoylated E protein pentamer was more stable while the loss of palmitoylation caused the pore radius reduced and even collapsed, which might help the drug design for the treatment of COVID-19.169
2.1.3.2. Coarse-Grained MD Simulations
Modeling the whole SARS-CoV-2 in a fine grid is extremely time-consuming, if not impossible. To study the behavior of SARS-CoV-2, a coarse-grained model based on the data from a combination of cryo-electron microscopy and X-ray crystallography was employed in a complete virion model.170 More importantly, the binding between S protein and ACE2 or antibodies can be studied by coarse-grained MD simulations.171 Bai et al. also used their own coarse-grained models to predict mutation-induced binding affinity changes between Mpro and human ACE2.172 With replica-exchange umbrella sampling MD simulations, a comparison of the binding of SARS-CoV-2 S protein and SARS-CoV S protein to human ACE2 revealed that the SARS-CoV-2 binding to human ACE2 is stronger than that of SARS-CoV.173 The infectivity induced by mutations on S protein is another problem after studying the binding of ACE2 and S protein, where coarse-grained MD simulations were employed to reveal the dynamics impact of mutations T307I and D614G174 or SARS-CoV-2 variants B.1.1.7 and P1.175 The impacts of mutations to antibodies CR3022 and CB6 were also predicted.176 Lastly, glycan shield effects on drug binding are also studied via multiscale coarse-grained MD simulations.177
2.1.4. Normal-Mode Analysis
Compared to molecular dynamics simulations, normal-mode analysis (NMA) has its advantages in dealing with the flexible motions accessible to a protein system at a steady-state position. Based on the equation of motion, normal-mode analysis studies molecular structure conformation by a restoring force acting on a vibrational system perturbed slightly at its equilibrium. Achieving its efficiency for large proteins and protein complexes, NMA is widely applied in large molecules and homology modeling studies, as well as evolutionary and stability analysis of proteins. Additionally, for computational efficiency, the elastic network model (ENM), Gaussian network model (GNM), and anisotropic network model (ANM) are developed and applied to similar problems.
The derivation of normal-mode analysis starts from the equations
of motion. It is formulated from Lagrange’s second kind equation,
with the Lagrangian  , where Ek and U are the kinetic and potential energies of the molecular
system, respectively.178−180 The potential energy U is
calculated by eq 9. The
system is defined to be in a potential minimum of equilibrium where
the generalized forces acting on it are eliminated. In this Lagrangian
mechanical system
, where Ek and U are the kinetic and potential energies of the molecular
system, respectively.178−180 The potential energy U is
calculated by eq 9. The
system is defined to be in a potential minimum of equilibrium where
the generalized forces acting on it are eliminated. In this Lagrangian
mechanical system 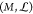 , M is a configuration
space and Lagrangian
, M is a configuration
space and Lagrangian 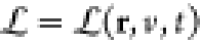 , where r ∈ M, v is the velocity vector at position r, and t is the time. They thus define the
generalized coordinates r̂ and apply the Taylor
expansion to the potential energy. The equation can be formulated
as
, where r ∈ M, v is the velocity vector at position r, and t is the time. They thus define the
generalized coordinates r̂ and apply the Taylor
expansion to the potential energy. The equation can be formulated
as
| 11 |
where ri is the ith component of the instantaneous configuration and ηi = ri – r̂i. Here, the Einstein summation convention is used. Note that eq 11 studies the mechanism system at equilibrium, and therefore, the first term can be set to zero in terms of the minimum value of the potential with the second term zero at any minimum. We can rewrite the potential energy to be
| 12 |
where Uij is the Hessian matrix. As for the kinetic energy Ek, it is given as
| 13 |
where M is a diagonal matrix of the mass of each particle. By applying Lagrange’s equation, the equations of motion can be given as
| 14 |
with one oscillatory solution ηi = aij cos(ωjt + δj), where aij is the amplitude of oscillations, ωj is the frequency, and δj is a factor. Here, it is easy to obtain the eigenvalue problem UA = λA, where A is an eigenvector of the aforementioned Hessian matrix and λ is an eigenvalue which is the square of frequency ωk. The eigenvector is also referred to as a normal-mode vector for a particle’s movement in terms of direction and distance with a certain frequency.
2.1.4.1. Elastic Network Model
Though NMA has computational efficiency compared to MD simulation, its computation is still expensive, especially for proteins containing tens of thousands of atoms. Additionally, one assumption of NMA requires energy minimization to ensure the starting conformation is at equilibrium. This minimization process might distort the structure, leading to a different structure from the experimental one. Therefore, a variety of coarse-grained approximate algorithms has been developed to overcome these limitations.181,182 Among them, the elastic network model (ENM) is widely applied.
The ENM simplifies the force fields used in standard NMA by a harmonic potential183
| 15 |
where dij is the distance between the ith and jth atoms, dij0 stands for the distance at the initial structure, and k mimics the spring constant in Hooke’s law. RC is a cutoff and usually is set between 7.0–8.0 Å, according to the distances between nonbonded atoms.184 More studies consider the Cα atoms only, for their sufficiency in backbone motion investigation. Many generalizations implemented the ENM’s idea to reformulate potential functions.
2.1.4.2. Gaussian Network Model and Its Generalization
One of the generalizations is the Gaussian network model (GNM),185 which is considered the most efficient one, using the discrete Laplacian matrix instead of the Hessian matrix. The expected residue fluctuations constructed by the GNM are in great agreement with the Debye–Waller factor (a.k.a. B factor). More precisely, the B factor of the ith α carbon atom (Cα) in an N-particle coarse-grained representation of a biomolecule can be obtained by the generalized GNM (gGNM) method186,187
| 16 |
where agGNM is a fitting parameter and (Γ–1)ii is the ith diagonal element of the matrix inverse Γ–1. Here, Γ is the generalized Kirchhoff matrix
 |
17 |
where ri are positions of Cα, the kernel functions Φ can be generalized exponential functions or generalized Lorentz functions, and ηj are the characteristic distances.186,187 If the kernel functions are set to be {0, 1} with distance ∥ri – rj∥ outside or inside a fixed cutoff distance, then the Kirchhoff matrix becomes the Laplacian matrix and the GNM is recovered.
2.1.4.3. Anisotropic Network Model and Its Generalization
Another popular method is the anisotropic network model (ANM),183,188 which gives extra information about the directionality of the fluctuations. The B factor of ith Cα in an N-particle coarse-grained biomolecule can be displayed by the generalized ANM method186,187
| 18 |
where agANM is a fitting parameter and (H–1)ii is the ith diagonal element of the matrix inversion of Hessian matrices.
2.1.4.4. Applications to SARS-CoV-2
The SARS-CoV-2 Mpro is used as one of the most popular target proteins for drug repurposing in applications. According to the stability analysis by NMA, the inhibitor repurposing of SARS for COVID-19 may be challenging.148 With further investigations of Mpro by the ENM, possible noncompetitive inhibiting binding sites were suggested189 (see Figure 5). Moreover, the SARS-COV-2 S protein is another target for inhibitor repurposing. In the comparison of the S proteins of SARS-CoV-2, SARS-CoV, and MERS-CoV, the ANM was employed to study the dynamic modes of the S proteins, revealing that the receptor binding motif had high vertically upward motion.190 In ref (191) ENM-based analysis tools were applied for the allosteric modulation region of the S protein with ACE2, and it was indicated that hepcidin induces an inhibitory effect on the binding affinity of the S protein and ACE2. The stability and flexibility of mutations can be examined by normal-mode analysis, especially the mutations on RBD or high-frequency mutation D614G of the S protein175,192,193 and on other SARS-CoV-2 proteins.193,194
Figure 5.
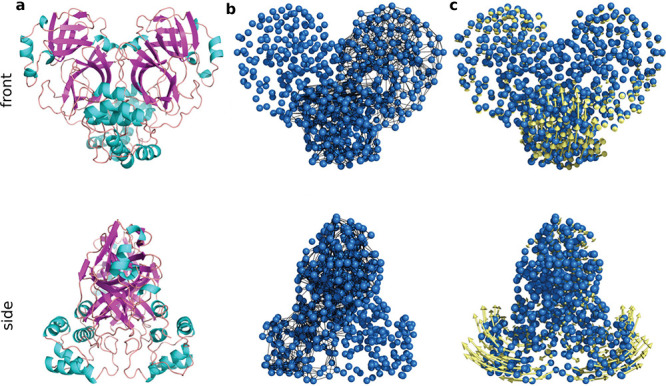
Illustration of the ENM on SARS-CoV-2 Mpro.189 Reproduced with permission from ref (189). Copyright 2021 Dubanevics and McLeish under Creative Commons Attribution 4.0 International License https://creativecommons.org/licenses/by/4.0/. (a) Mpro secondary structure. (b) Elastic model of Mpro. Cα atoms are in blue, and node-connecting springs are in black. (c) The first real vibrational mode eigenvectors are in yellow.
2.1.5. Monte Carlo Methods
Monte Carlo (MC) methods rely on repeated random sampling to obtain optimized numerical results. In principle, Monte Carlo methods can be used to solve any problems having a probabilistic distribution.195 When the probability distribution of the variable is parametrized, researchers often use a Markov chain Monte Carlo (MCMC) sampler,196 whose central idea is to design a judicious Markov chain model with a prescribed stationary probability distribution. By the ergodic theorem, the stationary distribution is approximated by the empirical measures of the random states of the MCMC sampler. Recently, a machine-learning-based implicit solvent Monte Carlo method was developed to predict the molecular structure.197
Importantly, metropolis Monte Carlo methods198 are popular in molecular modeling. As shown in Figure 4b, the essential idea is that, if the energy of a trial conformation is lower than or equal to the current energy, it will always be accepted. If the energy of a trial conformation is higher than the current energy, then it will be accepted with a probability determined by the Boltzmann (energy) distribution,
 |
19 |
where j is the current conformation, i is the new conformation, 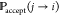 is the probability to accept the
new conformation,
ΔUij is the energy
difference between i and j, kB is the Boltzmann constant,
and T is temperature. Therefore, the evolution of
molecular conformations can be simulated. There are several aspects
of MC applications regarding SARS-CoV-2. An MC simulation of ionizing
radiation damage to the SARS-CoV-2 found that γ-rays produced
significant S protein damage but much less membrane damage.199 Thus, the γ-rays were proposed as a new
effective tool to develop inactivated vaccines. A metropolis MC sampling
process was applied to simulate a pharmacokinetic model of the human
immunodeficiency virus (HIV) drug darunavir against SARS-CoV-2.200 MC modeling was also implemented in the analysis
of SARS-CoV-2 PLpro201 and N protein.202 Studies focusing on Mpro and S protein are
introduced as follows.
is the probability to accept the
new conformation,
ΔUij is the energy
difference between i and j, kB is the Boltzmann constant,
and T is temperature. Therefore, the evolution of
molecular conformations can be simulated. There are several aspects
of MC applications regarding SARS-CoV-2. An MC simulation of ionizing
radiation damage to the SARS-CoV-2 found that γ-rays produced
significant S protein damage but much less membrane damage.199 Thus, the γ-rays were proposed as a new
effective tool to develop inactivated vaccines. A metropolis MC sampling
process was applied to simulate a pharmacokinetic model of the human
immunodeficiency virus (HIV) drug darunavir against SARS-CoV-2.200 MC modeling was also implemented in the analysis
of SARS-CoV-2 PLpro201 and N protein.202 Studies focusing on Mpro and S protein are
introduced as follows.
Mpro. Liang et al. used protein energy landscape exploration (PELE) Monte Carlo simulations for a blind binding site search and the best binding poses for these binding sites.203 Their simulations found that compounds such as cyanidin-3-O-glucoside and hypericin have the strongest interactions with the active sites. Their PELE also identified additional binding sites for hypericin with comparable interaction energies.203 A coarse-grained Monte Carlo simulation was integrated with other computational methods to reveal the relationship between the rigidity and enzymatic function for Mpro,204 while the Mpro inhibitory activity of aromatic disulfide compounds was studied by the weight search of MC simulations for the QSAR (quantitative structure–activity relationship) model.205
S Protein. Two major directions of the S protein are mutation studies,206,207 namely G614D and N501Y, and binding problems about ACE2,117,208 antibodies,171 and peptide-based inhibitors.171,208,209 To estimate the density of states of the S protein system, the Wang and Landau Monte Carlo method was applied to study the human ACE2 complexes with SARS-CoV-2 and SARS-CoV S protein RBDs,208 while the difference between SARS-CoV-2 and SARS-CoV was studied.210 In the study of the heterogeneity of glycosylation on the S protein trimer make up, the MC approach was applied to calculate the glycan mass distribution because of the enormous number of possible glycoforms.211
2.1.6. Molecular Docking
As shown in Figure 6a, molecular docking, which can predict the binding conformation of a ligand on its binding site, is one of the most popular methods in the structure-based drug design.213,214 A typical docking program includes two key components: a scoring function to calculate the binding energies of different conformations and a search algorithm to sample the conformational degrees of freedom and locate the global energy minimum from all the sampled conformations.215 Traditional scoring functions are derived from physical models such as molecular mechanism force fields. In recent years, more and more machine-learning-based scoring functions have been developed, which outperform traditional scoring functions in many cases.216−219 In addition to regular docking, ensemble docking220 considers the dynamics of the receptor and docks a ligand to various receptor conformations (often yielded from molecular dynamics simulation). Molecular docking is well-established in early stage drug discovery. As a result, during the pandemic, to seek drug leads, docking studies have been performed targeting a variety of SARS-CoV-2 proteins.
Figure 6.

(a) Procedure of molecular docking simulation. (b) Procedure of quantum mechanics/molecular mechanics (QM/MM) calculation.212 Reproduced with permission from ref (212). Copyright 2017 Royal Society of Chemistry.
Mpro. One important source for searching for SARS-CoV-2 treatments is existing drugs. In a quite extensive drug repurposing work, 7173 purchasable drugs, including 4574 unique compounds and stereoisomers, were docked and their binding affinities to Mpro were predicted.221 As a result, diosmin, hesperidin, and MK-3207, with a docking score of −10.1 kcal/mol, were suggested as the most potent inhibitors. A collection of 8625 drugs or compounds from FDA, drugbank, and Zinc data sets were docked to Mpro,222 and seven drugs such as metyrapone could maintain key interactions within the active site of the enzyme suggested by the crystallographic complex structures, revealing their repurposing potential. Additionally, Sencanski et al.223 and Gurung et al.224 both screened about 1400 FDA-approved drugs with docking, predicting that dihydroergotamine has a promising affinity. Docking was used to evaluate the potency of around 100 approved protease inhibitors, and it suggested that faldaprevir has the strongest binding affinity.225 In a screening of roughly 7100 molecules, several natural molecules such as δ-viniferin, myricitrin, taiwanhomoflavone A, lactucopicrin 15-oxalate, nympholide A, afzelin, biorobin, hesperidin, and phyllaemblicin B were identified.226 Many other studies222,227−236 have also docked and repurposed existing drugs against SARS-CoV-2 Mpro.
Natural products are popular inhibition candidates. In a study of 1000 active phytochemicals from Indian medicinal plants by molecular docking, rhein and aswagandhanolide were predicted to have binding affinities over −8.0 kcal/mol.237 In a study of 100 natural and nature-inspired products from an in-house library to Mpro, leopolic acid A is predicted to have the highest affinity of −12.22 kcal/mol.238 From mushrooms and other herbal or natural compounds, colossolactone VIII239 and eugenin240 were identified as having a high affinity to Mpro. It is predicted that from Amphilophium paniculatum leaves, luteolin 7-O-b-glucopyranoside (cynaroside) has the highest affinity of −9.54 kcal/mol.241 All of them also predicted ADMET (absorption, distribution, metabolism, excretion, and toxicity) properties of their compounds. Moreover, a variety of natural products were also investigated by docking-based virtual screening, including syzygium aromaticum, cassia acutifoliaaloe vera, rhus spp., moroccan medicinal plants, fungal metabolite, millet, tannins, neem leaves, nigella sativa, etc.242−256
Some researchers focus on peptides and small compounds from other sources. Tsuji et al.257 screened compounds from the ChEMBL (chemical database of bioactive molecules with drug-like properties) database258 to Mpro via docking, suggesting that the compound CHEMBL1559003 with a binding affinity of −10.6 kcal/mol is the most potent. In a set of 13535 Mpro compounds, the consensus of the docking scores from the five different pieces of docking software evaluates the potency.259 Udrea et al.260 predicted the binding affinities of 15 phenothiazines, where the compound sulphoridazine (SPZ) was reported as the most effective. Ghaleb et al.261 also studied some pyridine N-oxide compounds, indicating the most potent one with a predicted pIC50 (the negative log of the IC50 when converted to molar) value of 5.294. More similar works can be found in the literature.262−270
S Protein. Many researchers focus on the binding interactions of S protein and the host ACE2 or antibodies. A docking calculation was applied to construct the complexes of SARS-CoV-2 S protein and SARS-CoV S protein binding to ACE2,271 which suggested that there were more residue interactions between SARS-CoV-2 S protein and ACE2 than that of SARS-CoV S protein. It also leads to a higher binding affinity and is consistent with a recent experiment.272 In a study of ACE2s from cats, tigers, hamsters, dogs, and ferrets via homology modeling and docking, it was shown that the cat and tiger ACE2s could potentially interact with S protein RBD and share the same virus-binding interface with ACE2, whereas the dog, ferret, and hamster ACE2 were not predicted to establish stable interactions with S protein RBD.273 A docking simulation suggested ACE2 polymorphisms from different human races could change the ACE2 affinity to SARS-CoV-2 S protein.274 For other receptors, S protein cannot strongly bind to the human dipeptidyl-peptidase 4 (DPP4) receptor,275 while S protein RBD was able to bind to many amyloidogenic proteins, initiating aggregation of these proteins and leading to neurodegeneration in the brain.276 Interestingly, a recent docking study by Kazybay et al. suggested that the Omicron variant EGFR (epidermal growth factor receptor) was one of the potential binary partners of the S RBD that binds almost with equal affinity as the RBD–hACE2 complex.277 Hanai et al. used docking studies to suggest that Omicron’s binding to ACE2 was stronger than Delta’s and Alpha’s.278
More investigations were carried out to repurpose existing drugs to S protein or find inhibitors for S protein. In a study of screening FDA-approved drugs, iron oxide nanoparticles were suggested for COVID-19 treatment.279 Existing drugs, such as amentoflavone, ledipasvir, tenofovir, levodopa, lopinavir, and ubrogepant, against S protein were investigated.280−283 For natural products inhibiting S protein, studies focusing on indigenous food additives, herbal constituents, antioxidants, traditional medicinal plants, tea, and others284−289 suggested potent inhibitors such as phycocyanobilin, phycoerythrobilin, phycourobilin, folic acid, hinokiflavone, and phytochemicals. Lastly, some other compounds are repurposed to inhibit the S protein. Mohebbi et al.290 screened more than 1 billion compounds from the databases ZINC Pharmer and Pharmit in silicon. The docking of dermaseptin-based antiviral peptides to the S protein was studied.291 Some drugs were identified with high binding potential against the ACE2–S protein interaction pocket, such as Atazanavir, Grazoprevir, Saquinavir, Simeprevir, Telaprevir, and Tipranavir.292
RdRp. RdRp is another target for docking inhibitors from existing drugs or traditional medicines. In a data set of 7922 approved or experimental drugs, Nacartocin has the highest binding affinity.293 Beg et al.294 screened 70 anti-HIV (human immunodeficiency virus) or anti-HCV (hepatitis C virus) drugs, reporting that the drug paritaprevir has the highest binding affinity. Aftab et al.295 studied 10 antiviral drugs and revealed that Remdesivir’s docking score was the highest, but Padhi et al.194 showed that the docking affinity of remdesivir is relatively low. Meanwhile, many researchers screened potential drugs in traditional medicinal compounds. Theaflavin was reported to have the highest binding affinity among a data set of 83 traditional Chinese medicinal compounds plus their similar structures from the ZINC15 database.296 Pandeya et al.297 also investigated some biologically active alkaloids of argemone mexicana. Other RdRp drug repurposing works can be found in the literature.298−301
Other Targets. Some docking studies selected the SARS-CoV-2 PLpro as their targets. Choudhury et al.302 docked 27 existing drugs to PLpro and predicted stallimycin to be the best inhibitor. Similarly, Li et al.303 repurposed 21 drugs to inhibit SARS-CoV-2 PLpro and reported neobavaisoflavone as the most potent candidate. Mohideen et al.304 revealed that the binding affinity of the natural product thymoquinone to the E protein is −9.01 kcal/mol. Borgio et al.305 screened 23 FDA-approved drugs to target the helicase of SARS-CoV-2 and reported vapreotide having a binding affinity of −11.58 kcal/mol as the most potent candidate. Mahmud et al.306 showed that drugs such as valrubicin, aprepitant, and saquinair have excellent docking scores to SARS-CoV-2 nsp15. Khan et al.307 used docking to study the interaction between N protein and nsp3.
Multiple Targets. Many researchers studied the whole SARS-CoV-2 or multiple SARS-CoV-2 proteins for more potent inhibitors. In an analysis of therapeutic targets for SARS-COV-2 involving homology modeling and molecular docking, a data set of 78 commonly used antiviral drugs for SARS-CoV-2 proteins was selected.22 By using molecular docking on 2631 US FDA-approved small molecules, five drugs (avapritinib, bictegravir, ziprasidone, capmatinib, and pexidartinib) were suggested as candidates against SARS-CoV-2 proteins. In a study of docking 11 antiviral drugs to Mpro, S protein, PLpro, nsp10, nsp16, and nsp9, ritonavir, lopinavir, and remdesivir were selected as drug candidates against SARS-CoV-2.308 Other approved structural analogs, such as telbivudine, tenofovir, amprenavir, and fosamprenavir, were identified as potent drugs for SARS-CoV-2 by molecular docking.309 On a data set consisting of 2285 FDA-approved drugs and 1478 Taiwan National Health Insurance-approved drugs (https://covirus.cc/drugs/), a virtual screening targeting S protein, Mpro, PLpro, RdRp, N protein, hACE2, and human cellular TMPRSS2 were conducted.310 Chandel et al.311 repurposed about 2000 FDA-approved compounds targeting S protein and nsp9, reporting that Tegobuvir was the most potent candidate to S protein and Conivaptan was the most potent candidate to nsp9. Many works considered two different protein targets. Elmezayen et al.312 virtually screened 4500 approved or experimental drugs against Mpro and human TMPRSS2, finding out that ZINC000103558522 has the highest binding affinity to Mpro and ZINC000012481889 has the highest binding affinity to the TMPRSS2. Via the DockThor-VS platform, Guedes et al.313 predicted the binding affinities of over 40 approved drugs to SARS-CoV-2 Mpro, S protein, PLpro, RdRp, N protein, and nsp15. The binding affinities of the compounds protoporphyrin IX, verteporfin, and chlorin e6 to Mpro, S protein, ORF3a, ORF9b, and ORF7a were studied.314 In addition, some components such as essential oil components,315 organosulfur compounds,316 and methisazones317 were investigated. It was exhibited that rutin has some inhibitory effect on SARS-CoV-2 proteins.318 Phytochemicals from the traditional medicines were also investigated by docking, such as those from traditional Himalayan medicinal plants,319 Indian traditional medicinal plants,320−333 Chinese traditional medicines,334 and Brazilian herbal medicines.335
Targeting Mpro, S protein, and RdRp, Parvez et al.298 studied some plant metabolites. Maurya et al.336 investigated yashtimadhu (glycyrrhiza glabra) active phytochemicals, and Alexpandi et al.337 simulated quinoline-based inhibitors. Flavonoids might inhibit Mpro, S protein and RdRp as well.338,339 Against the S protein and nsp15S, Sinha et al.340 screened 23 saikosaponins and reported that saikosaponin V was potent to both targets. Montelukast was predicted to be potent to both Mpro and RdRp341 as well as Camptotecin.287 Srikanth et al.342 and Agrawal et al.343 virtually explored the potential of andrographolide as well as other antivirals, antibiotics, antiparasitics, flavonoids, and vitamins in inhibiting S protein and RdRp. Another molecular docking of compounds such as coumarins, porphyrins, propolis, or existing drugs can be found in refs (344−349).
2.1.7. Binding Free Energy Calculations
In the study of SARS-CoV-2, protein–protein and protein–ligand interaction processes are essential. Some of these processes are investigated through molecular biophysics, such as the binding free energies for protein–ligand and protein–protein complexes. To estimate the binding free energies, classical methods such as FEP and thermodynamic integration (TI) methods are computationally expensive, while many other methods are developed considering efficiency, such as the molecular mechanics/Poisson–Boltzmann surface area (MM/PBSA) method,350,351 the molecular mechanics/generalized Born surface area (MM/GBSA) method,351,352 the linear interaction energy (LIE) method,353 the chemical Monte Carlo/molecular dynamics (CMC/MD) method,354,355 the pictorial representation of free energy components (PRO-FEC) method,356 etc. Among these methods, MM/PBSA and MM/GBSA are widely applied for their accuracy and efficiency.
2.1.7.1. MM/PBSA and MM/GBSA
In the MM/PBSA and MM/GBSA approaches,108 the binding free energy ΔΔGbind for binding between a ligand and a protein receptor in the form of a protein–ligand complex can be calculated by
| 20 |
where ΔGcomplex is the total free energy of the complex and ΔGprotein and ΔGligand are the total free energies of the protein and ligand in solvent, respectively (see Figure 7). The free energy for each individual body can be calculated by
| 21 |
where G* are referring to the total free energies of the complex, protein, and ligand, ⟨VMM⟩ is the average molecular mechanical potential energy in a vacuum of eq 9, and TS is the entropic contribution to the free energy with the temperature T and the entropy S in a vacuum. ΔGsol is the free energy of solvation
| 22 |
The polar solvation energy ΔGsolpolar is calculated by solving the PB equation or the GB equation, and the nonpolar solvation energy ΔGsol is evaluated by cavity formation in the solvent and van der Waals interactions between solvent and solute. More details about the PB and GB models can be found in section 2.1.2.
Figure 7.

Illustration of the thermodynamic cycle of MM/PB(GB)SA calculations. ΔGcomplex is the total free energy of the complex, and ΔGprotein and ΔGligand are the total free energies of the protein and ligand in solvent, respectively. ΔGbind,sol and ΔGbind,vac are the total free energies in solvent and in vacuum, respectively.
2.1.7.2. MD-Based Methods
Besides MM/PBSA or MM/GBSA, other binding free energy calculation methods such as FEP, metadynamics, and steered MD simulations were also applied to evaluate the binding affinities of inhibitors to SARS-CoV-2 Mpro or S protein.
FEP. MD simulations and FEP calculations were considered to uncover the mechanism of the stronger binding of SARS-CoV-2 S protein to ACE2 by Wang et al.357 They compared the hydrogen-bonding and hydrophobic interaction networks of SARS-CoV-2 S protein and SARS-CoV S protein to ACE2 and calculated the free energy contribution of each residue mutation from SARS-CoV to SARS-CoV-2. FEP calculations indicated that the N501Y mutation on the SARS-CoV-2 S protein enhanced the binding to host ACE2.358 FEP calculations indicated the E484Q/L452R mutations significantly reduce the binding affinity between the RBD of the Kappa variant and the antibody LY-CoV555.359 Ngo et al.360 first docked about 4600 drugs or compounds to Mpro and then used steered MD simulations to rescore the top 35 compounds. They reevaluated the top three compounds using FEP free energy calculations. Zhang et al.361 docked remdesivir and ATP to RdRp and used FEP to calculate the binding free energy, indicating that the binding of remdesivir was about 100 times stronger than that of ATP and it can inhibit the ATP polymerization process. FEP calculations were performed by Hassan et al.,362 Allam et al.,363 and Alhadrami et al.364
Thermodynamic integration (TI). In a recent work,365 the authors used MD simulations to explore the structural coordination and dynamics associated with the SARS-CoV-2 nsp13 apo enzyme, as well as its complexes with natural ligands. The binding free energy and the corresponding mechanism of action by TI calculations were presented for three small molecules that are revealed as efficient inhibitors of the previous SARS-CoV nsp13 enzyme.
Metadynamics. Researchers docked 16 artificial-intelligence generated compounds by Bung366 to Mpro and then ran metadynamics to calculate their binding affinity and predicted some potential inhibitors.367 Metadymics simulations were used to predict the affinities of three neem tree extracts to S protein.368
Steered MD Simulations. Steered MD simulations were used to dock drugs and marine compounds to Mpro, to infer the top inhibitors by Tam et al.369,370 MM/PBSA and steered MD simulations suggest that the adsorption of the ACE2 on specific silane monolayers could increase its affinity toward the S protein RBD, which could help develop biosensing tools efficient toward any variants of the SARS-CoV-2 S protein.371
Semiempirical Free Energy Force Field Methods. A semiempirical free energy force field372 was also adopted to calculate the affinities of 16 drugs to S protein.373
In most applications, MM/PBSA or MM/GBSA calculations are used to select the trajectories or rank the drugs. The applications of MM/PBSA or MM/GBSA on SARS-CoV-2 proteins are discussed below.
Mpro. Docking, MD simulations, and MM/PBSA binding free energy calculations were applied to investigate around 10000 drugs or experimental drugs from the DrugBank.374 These compounds were first screened by docking, and then MD-based MM/PBSA binding free energy calculations were performed on the top 36 compounds. The reported binding data were the consensus of docking and MM/PBSA prediction, and leuprolide was the top one. Very similar work also screened thousands of compounds from the DrugBank by docking and MM/GBSA MD simulations.375 Gahlawat376 implemented an MM/GBSA procedure on 2454 FDA-approved or experimental drugs, 138 natural products, and 144 other inhibitors to predict the MM/GBSA binding free energy of the top ones screened by docking. MM/PB(GB)SA was also implemented for trajectory selection.377,378 In addition, Sharma et al.379 also implemented MM/PBSA to screen 2100 drugs as well as 400 other compounds and reported that cobicistat had the highest binding affinity of −11.42 kcal/mol. Cobicistat, flavin adenine dinucleotide, and simeprevir were suggested through drug ranking according to binding energy by MM/PB(GB)SA calculations. Other assessments based on docking and MM/PB(GB)SA calculations focused on specific drugs or compounds such as ravidasvir, lopinavir, ritonavir, saquinavir, teicoplanin, GC-376, calpain XII, calpain II, anti-HIV drugs, doxorubicin, chloroquine, quinoline, hydroxychloroquine, noscapine, echinocandins, coumarins, and their derivatives.380−417
MM/PBSA and MM/GBSA methods were also applied to predict the potency of natural products to Mpro. Ibrahim et al.418 virtually screened the MolPort database containing 113,756 natural or natural-like products (https://www.molport.com) by docking. The top 5,000 compounds were selected and subjected to MD simulations combined with MM/GBSA binding affinity calculations, and the compound MolPort-004-849-765 was predicted to have the highest binding free energy. Kapusta et al.419 also performed docking on 13,496 natural or natural-like products from MolPort, and the top 15 were chosen for rescoring by MM/GBSA calculations. The authors reported MolPort-039–338–330 as the most potent one. Prajapati et al.420 investigated 1830 secondary metabolites of fungal via docking, and additional MM/GBSA calculations were performed on the top from compounds. Mahmud et al.421 screened 1480 natural plant products from the literature initially by docking scores, and then the best 10% were rescored by MM/GBSA. Other natural product sources screened by docking and MM/PB(GB)SA against Mpro were flavonoid-based phytochemical constituents of calendula officinalis, phytochemicals in Indian ginseng, food compounds, marine natural polyketides, malaria-box compounds, cressa cretica compounds, strychnos nux-vomica products, ayurvedic compounds, moringa oleifera compounds, withania sp. products, stilbenolignans from plants, acridinedione analogs, alkaloids from justicia adhatoda, tea plant products, neem compounds, turmeric compounds, echinacea angustifolia products, Withania somnifera (ashwagandha) products, cyperus rotundus Linn products, salvia plebeia products, lichen compounds, curcuma longa products, and polyphenols from broussonetia papyrifera.422−458
There are more compounds screened by docking and MM/PBSA or MM/GBSA against Mpro. Andrianov et al.459 first virtually screened over 213.5 million chemical structures from http://pharmit.csb.pitt.edu/ to select the ones satisfying the pharmacophore model from the known X77 potent main protease inhibitor. Then they docked them to Mpro and ran MM/GBSA simulations of the docking complexes to calculate binding free energy. Through this procedure, the authors reported some potent inhibitors such as Pub-chem-22029441. Jimenez et al.460 docked 4858 flavonoids to Mpro, and MM/PBSA calculations were performed on the top six compounds. The pharmacophore procedure was also performed where the top three by docking scores were subjected to MM/GBSA calculations, reporting macimorelin acetate as the best one.461 In a docking of the 15754 compounds in their in-house data set to Mpro, compounds were rescored by MM/GBSA calculations, reporting the most potent one, dimethyl lithospermate.462 Khan et al.463 used docking to screen approximately 8000 compounds in their in-house database and applied MM/GBSA MD simulations to calculate the binding affinities of the top five inhibitors, with remdesivir being the best. Fakhar et al.464 screened 3435 anthocyanin substructure compounds by docking and MM/GBSA calculations, reporting the best compound to be 44256921. Some other compounds, such as α-ketoamide covalent inhibitors, macrolactin compounds, echinocandins, essential oil compounds, glucocorticoids, angucycline compounds, hydroxychloroquine derivatives, aminoglycosides, imidazole derivatives, Se-containing heterocyclic compounds, circadian clock modulating compounds, oxazine substituted 9-anilinoacridines, nitric oxide donor furoxan, nitric oxide donor heterocyclic vasodilators, withanone caffeic acid phenethyl ester, tetracycline, β-glutamyl-S-allylcysteine peptides, and others, were also screened by docking and MM/PB(GB)SA simulations.465−484
S Protein. Both SARS-CoV and SARS-CoV-2 infect humans through S protein binding to the human ACE2, and many investigations focused on the interaction between the S protein and the ACE2. In the comparison of the binding affinities of S protein from SARS-CoV and SARS-CoV-2 to the human ACE2 by MM/PB(GB)SA,485,486 the calculations indicated that SARS-CoV-2 S protein bound to ACE2 much more tightly than SARS-CoV S protein. The mechanism of tighter binding of the SARS-CoV-2 S-protein was studied by using MD simulations and MM/GBSA or MM/PBSA calculations by Xue et al.,487 Jafary et al.,488 Spinello et al.,489 and Bhattacharyya et al.490 Interestingly, MM/PBSA calculations at different temperatures suggested that the SARS-CoV-2 RBD was more resistant to temperature changes than the SARS-CoV RBD.491 Researchers ran MM/PBSA calculations and found that some mutations on S protein could facilitate stronger interactions with human ACE2.492−496
MD simulations and MM/PBSA calculations revealed that the formation of disulfide bonds, prevalent during oxidative stress, created a conformation more ready to bind to the receptor, which offered future clues for alternate therapeutic possibilities.497 Similar work was also performed by Ghasemitarei et al.498 One interesting study performed MM/PBSA calculations to reveal the binding affinities of SARS-CoV-2 S protein to the ACE2s from different species.499,500 This study showed that chimpanzees’ binding affinity was even higher than humans, cats, pangolin, dogs, and monkeys, and chimpanzees had a similar affinity to humans, which suggested some mammals were also vulnerable to SARS-CoV-2. Additionally, a recent MM/GBSA study suggested SARS-CoV-2 Omicron RBD shows weaker binding affinity than the Delta variant to human ACE2.501
Drug repurposing against S protein was also implemented by MM/GBSA or MM/PBSA. De Oliveira et al.502 docked 9091 approved or experimental drugs to S protein and selected the top three to perform MM/PBSA calculation, revealing that suramin sodium had the highest binding affinity. Following a similar scheme, a study repurposed 8770 approved or experimental drugs by docking and MM/GBSA, identifying 31h-phthalocyanine as the most potent candidate.503 Padhi et al.504 performed docking and MM/PBSA calculation studies on the inhibition of umifenovir (Arbidol) to the RBD/ACE2 complex. Moreover, MM/GBSA or MM/PBSA approaches were also applied to calculate the efficacy of other compounds to S protein. For example, a study performed docking to 330 galectin inhibitors against S protein and ran MM/GBSA calculations to some active ones, revealing that ligand No.213 had the highest binding free energy.505 Rane,506 Singh et al.,507 and Li et al.508 calculated the potency of diaryl pyrimidine derivatives, some bioactive molecules, and the MERS-CoV receptor DPP4, respectively. Lastly, some MM/GBSA or MM/PBSA investigations were about the use of natural products against S protein. For example, docking and MM/PBSA calculations were performed for 11 phytochemicals, suggesting that quercetin had the highest affinity.509 Other studies that applied MM/PB(GB)SA calculations on natural compounds to block S protein were from Indian medicinal plants,510 the NPACT (naturally occurring plant-based anticancer compound-activity-target) database,511 luteolin,512 and curry.513
RdRp. In the study of 7496 approved or experimental drugs against both SARS-CoV-2 and SARS-CoV RdRp, lonafarnib, tegobuvir, olysio, filibuvir, and cepharanthine were screened with high potency by docking and MM/GBSA calculations.514 Doharey et al.515 predicted the affinities of amodiaquine, hydroxychloroquine, chloroquine, as well as ATP to RdRp by docking and MM/GBSA calculations, suggesting that these three drugs have higher affinities than ATP. Pirzada et al.516 and Arba et al.517 studied the binding mechanism of remdesivir, ledipasvir, and paritaprevir to S protein through docking, MD, and MM/PBSA simulations. Furthermore, as to natural products and other compounds, Khan et al.518 screened 6842 South African natural products against RdRp using docking and selected the top four for further investigation by MD simulations and MM/GBSA calculations. Their most potent one was Genkwanin 8-C-beta-glucopyranoside ranked by MM/GBSA calculations. In another study of 100 natural polyphenols by docking, the leading eight compounds were used in MD simulations and MM/GBSA calculations, showing that the compound TF3 was the best.519 Nakinadine B and ormycalamide A were subjected to MM/GBSA studies in docking of 51 marine sponge metabolites to RdRp.520 Sonousi et al.521 and Jena et al.522 evaluated the efficacy of adenosine derivatives and synthetic nucleotides to RdRp via docking and MM/GBSA calculations. Molecular docking, MD simulations, and MM/GBSA approaches have also been used to examine the role of several short ionic peptides in inhibiting RdRp.523 Other similar studies include refs (524−531).
PLpro. In a repurposing of 1697 approved drugs against PLpro by docking, the top 10 were studied by MD simulations and MM/GBSA, with the drug phenformin being their best one.532 Mitra et al.533 screened tens of natural compounds from Vitex negundo L. by docking and ADMET predictions and performed MM/GBSA calculations on the top four compounds. By docking, MM/GBSA calculations, and interaction analysis, the prediction of the potency of six fungal metabolites to PLpro found GRL0617 is the only potent one.534 Via a similar procedure, a binding free energy analysis suggests that human ub-like interferon-stimulated gene product 15 binds more strongly with SARS-CoV-2 PLpro compared to SARS-CoV or MERS-CoV.535 Pitsillou et al.536 studied dietary compounds and naphthalene-based inhibitors. Bosken et al.537 assessed the potential effectiveness of one naphthalene-based inhibitor 3k and one thiopurine inhibitor 6MP through docking, MD simulations, and MM/PBSA calculations.
Other Targets. Many MM/GBSA or MM/PBSA investigations focused on the SARS-CoV-2 N protein. For instance, Khan et al.538 studied the mechanism of RNA recognition by the N-terminal RNA-binding domain of the SARS-CoV-2 N protein as well as mutation-induced binding affinity changes by docking, MD simulations, and MM/GBSA calculations. In a collection of 8987 compounds from the Asinex and PubChem databases, one study was targeting against the N protein and assessed the potency of the top 10 by MM/GBSA calculations.539 Meanwhile, SARS-CoV-2 helicase (nsp13) was another attractive target.540 Vivek-Ananth et al.541 estimated the docking scores of 10510 drug-like phytochemicals from PubChem to helicase, and the top five compounds were further evaluated by MM/PBSA calculations. In another work, 131 compounds were docked to helicase. More importantly, via MM/GBSA and MM/PBSA calculations, the best one from docking, nilotinib, was used as a probe to detect its affinities to different binding sites of helicase.542 Research about targets such as nsp16 or nsp10 can be found.543,544 Chandra et al.545 studied 2895 approved or experimental drugs against NendoU (nsp15) and selected the top three compounds from docking results and ran MM/PBSA calculations for these three, identifying glisoxepide, with a MM/PBSA binding free energy, as the most potent one. The docking of 123 antiviral drugs to NendoU found simeprevir had the highest binding energy, where the MM/PBSA calculations also confirmed this finding.546 Encinar et al.547 used docking to screen 8696 approved or experimental drugs against the nsp16(methyltransferase)/nsp10 protein complex and, through MM/PBSA calculations, discovered that the presence of nsp10 strengthens the ligand binding to nsp16. In the repurposing against nsp16 involving 4200 drugs or compounds, the best one predicted from MM-PBSA was Carba-nicotinamide-adenine-dinucleotide.548 The potency of hundreds of bioactive compounds to methyltransferase was also studied via docking and MM/PBSA calculations.549,550 Moreover, El Hassab et al.551 designed a new methyltransferase inhibitor AP-20 based on fragments and calculated the binding affinity using MM/PBSA. Since the MMLFA-1/SARS-CoV-2 Orf7a complex contributes to SARS-CoV-2 infectivity and pathogenicity, Ongaro et al.552 used MM/GBSA calculations to study the interactions inside the MMLFA-1/SARS-CoV-2 Orf7a complex. Other SARS-CoV-2 targets under MM/PB(GB)SA studies also include nsp1553 and nsp14.554
Multiple Targets. Some researchers adopted MM/PB(GB)SA calculations to screen drugs or compounds against multiple targets of SARS-CoV-2. Nunes et al.555 docked 24 approved drugs to SARS-CoV-2 Mpro, PLpro, and the ADP ribose phosphatases of nsp3, nsp9, nsp12, nsp15, and nsp16. The MM/GBSA calculations were further performed on the top three drugs from docking tests. 131 quinoline-based drugs were targeted to Mpro, S protein RBD, PLpro, RdRp, and N protein, and the best drug for each target was further evaluated by MM/PBSA calculations.556 Famotidine was shown to have a high binding affinity to PLpro in similar studies.557 Targeting SARS-CoV-2 Mpro, S protein, RdRp, PLpro, nsp14, Mpro, N protein, human ACE2, and TMPRSS2, Eweas et al.558 in silico screened chloroquine, hydroxychloroquine, ivermectin, remdesivir, favipiravir, lopinavir, and camostat via docking. Their docking simulations identified that ivermectin and remdesivir were potent to all nine targets, and the MM/PBSA calculations confirmed it. Targeting Mpro and PLpro, Jade et al.559 screened 4182 drugs and 321 other compounds through ADMET and docking predictions. Many works aimed to repurpose compounds from other sources to inhibit Mpro and S protein. Panda et al.560 screened 640 compounds through docking and MD simulations and identified that PC786 had high docking scores to both S protein and Mpro. Moreover, their MD simulations and MM/PBSA calculations revealed that the binding of PC786 can change the conformation of the S protein and weaken the S protein’s binding interactions to ACE2. The MM/PBSA binding free energy was calculated on five PLpro–compound and 6 Mpro–compound complexes. Similarly, Naidoo et al.561 investigated the potency of cyanobacterial metabolites against Mpro and S protein. Thurakkal et al.562 predicted the binding affinities of tens of organosulfur compounds to Mpro, S protein, PLpro, RdRp, and helicase. The top six compounds were further investigated by MM/PBSA. The repurposing potential of 34 bioactive terpenes and their derivatives to Mpro and PLpro was also predicted by docking and MM/PBSA calculations.563 Other MM/PB(GB)SA-based drug repurposing works considering two or more targets include studies in the literature.564−580
Natural compounds are another source of drug repurposing. Targeting Mpro, PLpro, and RdRp, a collection of 14492 marine-derived natural bioactive compounds by the criteria of Lipinski’s RO5, predicted ADMET properties, and docking scores, the best 14 compounds were subjected to MM/PBSA calculations.581 In a similar studying, Al-Sanea et al.582 performed docking simulations on about 30 strawberry and ginger silver nanoparticles, and the top four were selected to be studied by MM/GBSA calculations. In ref (583) 56 licorice major components and metabolites were docked to Mpro, S protein RBD, PLpro, RdRp, nsp15, and human ACE2. MM/GBSA calculations were performed on the top six compounds. In the investigation of four compounds targeting four different proteins in SARS-CoV-2, i.e., Mpro, S protein–ACE2 complex, RdRp, and PLpro, through docking, MD simulations, and MM/GBSA, it was found that AGP-3 had potency for all four targets.584 In a collection of 100,000 natural compounds against SARS-CoV-2 proteins, compounds were investigated by MD simulations and MM/PBSA, reporting that Baicalin was potent against RdRp, nsp4, and NendoU.585 Kar et al.586 studied Mpro, S protein, and RdRp. Their ligands were natural products from Clerodendrum spp. After docking and rescoring the top ones by MM/GBSA, these authors found taraxerol to be effective to all three targets. Using docking and MM/GBSA or MM/PBSA calculations, Alajmi et al.587 and Sasidharan et al.588 evaluated the potency of around 40 compounds, including some existing drugs and the protein azurin secreted by the bacterium Pseudomonas aeruginosa as well as its derived peptides, against Mpro, PLpro, and S protein. Prasanth et al.589 studied 48 isolated compounds from cinnamon by docking and MD-simulation-based MM/PBSA calculations, suggesting that the compounds tenufolin and pavetannin C1 were potent to both Mpro and S protein. Other similar works about natural products are reported in the literature.590−604
2.1.8. Density-Functional Theory (DFT) and Quantum Mechanism (QM) Methods
Density-functional theory (DFT) is utilized whenever the electronic structure is important, which is the typical case for chemical reactions. DFT is a computational quantum mechanics modeling method widely used in computational physics, computational chemistry, and computational material science to investigate the electronic structure of atoms, molecules, and condensed phases.605−607 Using this theory, the properties of a many-electron system are represented by functionals (functions of another function) of the spatially dependent electron density.605 Because of the development of DFT, Walter Kohn won the Nobel Prize in Chemistry in 1998.608 DFT is constructed on the total electronic charge density ρ(r) given as
| 23 |
of an N-electron problem, where ri are positions and Ψ(r, r2, ..., rN) is the wave function satisfying the many-electron time-independent Schrödinger equation. Here, Ψ* is the complex conjugate of Ψ. DFT, as a successor of the Schrödinger equation and the Thomas–Fermi model, studies a representative of the N-electron problem as a set of N one-electron problems, whose foundations are the Hohenberg–Kohn and Kohn–Sham theorems. For the Hohenberg–Kohn theorem, if the density function ρ(r) of a quantum system is known at the ground state ρ0, then the wave function is determined as Ψ0 = Ψ[ρ0]. The Kohn–Sham equation for the orbitals φi(r) can be written as
| 24 |
Here ℏ is the reduced Planck constant. The Kohn–Shan potential is given as
| 25 |
where Vext(r) is the external potential, the second term is the Hartree term of the electron–electron Coulomb repulsion, and Vxc[ρ(r)] is the exchange-correlation potential which is
| 26 |
where Exc is the exchange-correlation energy.
DFT calculations were employed to simulate the attack of cysteine to covalent inhibitors by Nogara et al.,609 Madabeni et al.,610 Wang et al.,611 and Shekh et al.612 For example, DFT simulations by Nogara et al. and Madabeni et al. suggested the mechanism of cysteine attacking, and the energy barrier of its attacking to ebselen was around 30 kcal/mol. Many studies applied DFT to predict electronic properties and chemical reactivity. For instance, via DFT calculations, Khan et al.613 suggested the electrophilicity ranking of their six isolated compounds is apigenin > luteolin > diosmetin > quercetin > spinacetin > eriodictoyl. Yadav et al.614 suggested the higher reactivity of favipiravir toward Mpro since the presence of electron donor and receptor groups in favipiravir displayed the capability of forming a complex with the external target molecule. Other similar studies include refs (443 and 615−632). DFT calculations by Aitouna et al.633,634 indicated the epoxidation reaction of parthenolide and himachalene derivatives presented high chemoselectivity. This can explain how parthenolide and himachalene derivatives form. More studies used DFT to preoptimize the structures of compounds.609,635−640 Additionally, the PM7 semiempirical quantum-chemical method was applied to binding calculations of Mpro,641,642 RdRp,643 PLpro, and S protein.642
2.1.9. Quantum Mechanics/Molecular Mechanics (QM/MM)
As illustrated in Figure 6b, the QM/MM approach is a hybrid molecular simulation method that combines the accuracy of QM and the speed of MM. For a biological system, the computational region where the chemical process takes place is treated at an appropriate level of QM,644 while the remainder is described by a MM force field.645 This approach can be used to study chemical processes in solution and proteins. The Nobel Prize in Chemistry in 2013 was awarded to Arieh Warshel and Michael Levitt for the introduction of QM/MM. There are two different ways to calculate the energy of the combined system. The subtractive scheme calculates the energy of the entire system by using a molecular mechanics force field added by the energy of the QM system and subtracted by the MM energy of the QM system
| 27 |
where VMM(QM) is the energy of the quantum mechanics region using molecular mechanics. More widely, the additive scheme is applied and given as
| 28 |
where V(QM) is the QM energy of the QM region, V(MM) is the MM energy in the molecular mechanics region, and V(QM/MM) is the energy of interactions between the two systems given as
| 29 |
where each term is calculated as a similar formula as eq 9 and Vbond(QM/MM), Velec(QM/MM), and VVDW(QM/MM) are calculated in both systems.
The covalent binding of PF-07321332 to Mpro was elaborately investigated via QM/MM calculations, suggesting the reaction energy barrier is −16.3 kcal/mol.646 Ramos-Guzmán et al.647 and Arafet et al.648 also performed QM/MM-based simulations to reveal the mechanism of the Michael reaction to Mpro, and Ramos et al. suggested some strategies to improve inhibitor design. Ramos et al.649 considered aldehyde derivatives. The covalent binding of inhibitor PX-12 and peptides to Mpro was simulated by the QM/MM method.650−655 Regarding the S protein, the interactions of human ACE2 and hydroxychloroquine to S protein RBD were analyzed through QM/MM calculations.656,657
2.2. Mathematical Approaches
Various mathematical tools, including different geometries,94,658−660 algebraic topology,219,661 and graph theory,662 have been applied to the modeling and prediction of biomolecules.663 In this review, network analysis, the flexibility–rigidity index, and topological data analysis are discussed. These approaches become very powerful when paired with deep learning.
2.2.1. Graph Network Analysis
A network is a graph in which vertices represent objects and edges represent relationships between objects. Networks can be used to represent biological systems as sets of biological objects and interactions between biological objects. For example, protein–protein interactions (PPIs) give rise to both protein-scale networks and atom-scale networks. In the protein-scale PPI networks, proteins are regarded as vertices and protein–protein interactions as edges. In the atom-scale PPI networks, such as a RBD-ACE2 complex, the atoms in the RBD and ACE2 can be regarded as vertices and the interactions between atoms in the RBD and atoms in the ACE2 form edges. Other biological systems, such as atomic interactions, drug–target interactions, disease–protein associations, and drug–disease relations, can also be represented as networks. Thanks to the recent development of biological technologies, such as high-throughput affinity purification combined with mass spectrometry and the yeast two-hybrid assay, interactome data are increasing rapidly, and a large number of interactome networks can be constructed. Understanding biology from the perspective of networks is important for many purposes. For instance, the knowledge of a PPI network can shed light on the putative roles of uncharacterized proteins. Graph theory is a well-established mathematical field that is readily applicable to the study of biological networks. In this section, we first recapitulate some basic notions of graph theory; then we introduce several network measures, which are proposed to characterize the local or global properties of a network, such as “irregularity”, “centrality”, and “communicability”.
An undirected simple graph (V, E) consists of a set V of vertices and a set of edges E connecting pairs of vertices, without self-loops or multiple edges between vertices. We denote the number of vertices and the number of edges as nv and ne, respectively. The edge connecting vertices i and j is denoted as eij. A simple graph with V = {v1, ..., vn} can be represented by its adjacency matrix A, where Aij is 1 if there is an edge connecting vi and vj and is 0 otherwise. The degree of a vertex i, sometimes denoted as ki, is the number of edges that are incident to the vertex i. It is clear that the average degree can be calculated by the formula 2ne/nv. A regular graph is a graph where each vertex has the same degree. A walk of length k is a series of vertices i1, i2, ..., ik+1 such that for all 1 ≤ l ≤ k there is an edge connecting il and il+1. A path is a walk in which all vertices are distinct. It is well-known that the (i, j) entry of the kth power of the adjacency matrix, (Ak)ij, is equal to the number of walks of length k starting at vertex i and ending at vertex j. A graph is connected if there is a path between every pair of two vertices, and a tree is an undirected graph in which every pair of two vertices is connected by exactly one path.
Degree Heterogeneity. The degree heterogeneity664,665 measures how heterogeneous the degrees of vertices are. Considering a star graph Sk, its only internal node has degree k and each of its external node has degree 1. The degree heterogeneity of a graph such as Sk reflects its “irregularity”. It is defined as
| 30 |
where  is
the set of edges, ki is
the number of neighbors of vertex i, and eij is
the edge connecting vertices i and j. The degree heterogeneity of a regular graph is 0, since each vertex
has the same degree. The degree heterogeneity of the star Sk is
is
the set of edges, ki is
the number of neighbors of vertex i, and eij is
the edge connecting vertices i and j. The degree heterogeneity of a regular graph is 0, since each vertex
has the same degree. The degree heterogeneity of the star Sk is  .
.
Edge Density. The edge density is defined as
| 31 |
where ne is the number of edges and nv is the number of nodes. For a complete network in which each pair of network vertices is connected, the edge density is equal to one. A noncomplete network has an edge density smaller than one. If ne ≈ (nv)k with 1 < k < 2, we say that this graph is dense. If ne ≈ (nv)k with k ≤ 1, we say that this graph is sparse.
Average Path Length. The average path length can be seen as a measure of the efficiency of information transport and is typically used to characterize the “small-worldness” of a network.666 A network with shorter average path length facilitates quicker transfer of information. Let d(i, j) denote the shortest path length between vertices i and j, then the average path length ⟨L⟩664 is defined as
| 32 |
Betweenness Centrality. If a vertex v falls on the shortest paths between two vertices i and j, by control of the vertex v one can control the transmission of information between vertices i and j. The notion of the betweenness centrality illustrates this potential.667 The betweenness centrality of a vertex v is defined as a sum over all (unordered) pairs of vertices i and j such that i ≠ v ≠ j
| 33 |
where σij is the number of shortest
paths between vertices i and j and
σij(v) is the number
of those paths that passes the
vertex v (v is not an end point).
The probability that the vertex v falls on a randomly
chosen shortest path connecting vertices i and j is  . The average betweeness
centrality is defined
as the average of betweenness centralities over all vertices.
. The average betweeness
centrality is defined
as the average of betweenness centralities over all vertices.
Eigenvector Centrality. The eigenvector centrality664 takes account of not only the shortest paths but also any path connecting two vertices. Let Nl(v) be the number of walks of length l that start at v and end elsewhere. If the given network is not bipartite, one can define the eigenvector centrality of a vertex
| 34 |
which can be regarded as the ratio of the number of infinite length walks starting at v over the number of all infinite length walks. One can define the average eigenvector centrality as the average over all vertices.
Subgraph Centrality. Let A be the adjacency matrix and G = exp(A); then the subgraph centrality664,668 of the ith vertex is defined as
| 35 |
The subgraph centrality is closely related to closed walks. To see this, rewrite Gii as ∑l = 0∞(Al)ii/k!. As (Al)ii is the number of closed walks of length l starting and ending at the same ith vertex, the subgraph centrality is indeed a weighted sum of closed walks of all lengths starting and ending at the same node, in which shorter closed walks are given more weight. To get a global characterization of a network, one may also consider the average subgraph centrality.
Communicability. There are many different ways to measure the communicability of two vertices. Estrada and Hatano669 proposed to define the communicability between vertices i and j as Gij, which is indeed a weighted sum of walks of all lengths starting at vertex i and ending at vertex j. This definition of communicability is justified because communication between two vertices can take place through nonshortest paths. Estrada and Hatano670 also defined the communicability angle between the ith and jth vertices
| 36 |
Taking the average over all pairs of vertices, one can define the average communicability and the average communicability angle. The average communicability angle evaluates the efficiency of a network transmitting information between its pairs of vertices with all possible paths.
Closeness Centrality. The closeness centrality671 measures vertices’ connecting efficiency through the network. In a connected graph, the closeness centrality of the ith vertex is defined as
| 37 |
The sum ∑j≠id(i, j), also referred to as the farness of a vertex i, is the sum of the shortest path distance to the ith vertex over all nv – 1 reachable vertices. The normalized form of the closeness
centrality is given by 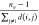 . If
a vertex has a larger closeness centrality,
it has a greater “centrality” in the sense of being
more independent of other vertices.667
. If
a vertex has a larger closeness centrality,
it has a greater “centrality” in the sense of being
more independent of other vertices.667
Topological Coefficient. The topological coefficient672Cit measures the extent to which the ith vertex shares neighbors with other vertices, which is defined as
| 38 |
where J(i, j) is the number of joint neighbors of the ith and jth vertices (plus one if there is an edge between i and j), oi is the degree of the ith vertex, and ⟨J(i, j)⟩ is the average over all vertices that share a neighbor with the ith vertex.
2.2.1.1. Network-Based Biomolecular Structure Analysis
Using networks to analyze the structural similarities is important for drug repurposing and understanding functional mechanisms. Estrada applied the aforementioned network measures to analyze the interaction networks between SARS-CoV-2 Mpro and various inhibitors.664 Chen et al.78 applied a similar strategy to predict binding affinity changes induced by mutations. A variety of studies using the network measures on protein residue/atom networks followed the same path.6,10,77,673−675 Moreover, Chen et al. employed the network analysis of antibody–antigen complexes on Cα atoms77 as illustrated in Figure 8. Lata and Akif676 implemented network analysis on 3CLpro from SARS-CoV and SARS-CoV-2. Amamuddy et al.677 used five independent criteria of network centrality to study the allosteric effects of potential allosteric modulators for the SARS-CoV-2 3CL protein. Focusing on the correlations between the RBD and residues distant to it in the S protein, Ray et al.678 built a protein graph connectivity network and calculated the betweenness centrality. A modification of the average shortest path length was used in ref (679). Saha et al.680 employed various network measures to identify the spreader nodes in the SARS-CoV-human protein–protein interaction network, hoping to find possible lineage with the disease propagation pattern of the COVID-19 pandemic.
Figure 8.

Cα network analysis of three antibody–antigen complexes. Here, circle markers represent antigen (S protein RBD), and cube markers represent antibody or ACE2. The PDB IDs of the three antibody–antigen complexes are 3D0G, 6M0J, and 6W41. The rows represent (a) betweenness centrality, (b) eigencentrality, and (c) subgraph centrality.77 (d) Illustration of the S protein and ACE2 interaction. The RBD is displayed in green, the ACE2 is given in pink, and mutation D614G is highlighted in red. (e) Difference of FRI of the S protein between the network with wild type and the network with mutant type. (f) Difference of the subgraph centrality between the network with wild type and the network with mutant type.10
2.2.2. Flexibility–Rigidity Index (FRI)
The FRI is a geometric graph-based method that utilizes weighted graphs to model molecular interactions.187,681 The multiscale FRI,682 the colored (i.e., element-specific) FRI,683 and their algebraic graph counterpart186 have also been proposed. The atomic rigidity index at position ri is defined as a summation of all the weighted edges around it:
| 39 |
where rj is the position of the ith atom, wij is a weight, Nc′ is the number of atoms in the neighborhood of ri, and η is a characteristic scale. Element-specific rigidity683 and molecular rigidity681 can be obtained by an appropriate collection of atomic rigidity indices. The FRI has been applied to protein and nucleic acid flexibility and fluctuation analysis681 and protein–ligand binding affinity prediction.684 Protein–protein interactions, such as the elasticity between antibody and antigen, especially long-range impacts, were studied by calculating the FRI of the network consisting of Cα atoms. The FRI is an important feature for machine learning models to predict the binding affinity changes on mutations685 and the protein folding energy changes on mutations.686
Some studies applied the machine learning models based on the FRI to study the SARS-CoV-2 proteins combined with network analysis. Wang et al.10 calculated the FRI and investigated the folding stability changes of the S protein (see Figure 8a, e, and f) and other proteins caused by mutations. The FRI-based binding affinity change between the S protein and human ACE2 due to mutations was also calculated by Chen et al. and Wang et al.10,77−79,687
2.2.3. Topological Data Analysis (TDA)
Recent years have witnessed rapid development
in TDA and its applications
to a wide variety of scientific and engineering problems.689,690 The main workhorse of TDA is persistent homology,691,692 a new branch of algebraic topology. This approach has been applied
to characterize biomolecular systems.661,693,694 More powerful methods that provide simultaneous topological
persistence and spectral analysis have been proposed.660,695−697 In TDA, molecular atoms can be treated as
a point cloud and a filtration of simplicial complexes can be constructed.
We first recall some basic notions of algebraic topology. A simplicial
complex is a finite collection of simplices σ, where a k-simplex σk = [v0, ..., vk] is a convex hull of k + 1 points {v0, ..., vk} in  (n ≥ k). A simplicial complex K is valid if any face τ
of a simplex σ in K is also in K, and the nonempty intersection of any two simplices is a face for
both. Given a simplicial complex K, a k-chain is a finite formal sum of k-simplices ∑iαiσik with coefficients in a ring (usually a field such
as
(n ≥ k). A simplicial complex K is valid if any face τ
of a simplex σ in K is also in K, and the nonempty intersection of any two simplices is a face for
both. Given a simplicial complex K, a k-chain is a finite formal sum of k-simplices ∑iαiσik with coefficients in a ring (usually a field such
as 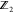 ). The set of all k-chains
forms an abelian group Ck(K). The boundary operator ∂k:Ck(K) → Ck–1(K) is a group homomorphism defined by ∂kσk = ∑i = 0(−1)i[v0,...,v̂i,...,vk], where [v0,...,v̂i,...,vk] is a (k –
1)-simplex excluding vi. A k-cycle is a k-chain whose
image is 0 under the boundary operator ∂k. An important property of boundary operators is that ∂k–1∂k = 0, so we have the following chain complex
). The set of all k-chains
forms an abelian group Ck(K). The boundary operator ∂k:Ck(K) → Ck–1(K) is a group homomorphism defined by ∂kσk = ∑i = 0(−1)i[v0,...,v̂i,...,vk], where [v0,...,v̂i,...,vk] is a (k –
1)-simplex excluding vi. A k-cycle is a k-chain whose
image is 0 under the boundary operator ∂k. An important property of boundary operators is that ∂k–1∂k = 0, so we have the following chain complex
| 40 |
and the kth homology group Hk is defined as Hk = Zk/Bk where Zk = ker ∂k = {c ∈ Ck|∂kc = 0} and Bk = im ∂k+1 = {∂k+1c|c ∈ Ck+1}. The kth Betti number is defined by the rank of the kth homology group Hk which counts the k-dimensional holes. In particular, β0 = rank(H0) reflects the number of connected components, β1 = rank(H1) reflects the number of loops, and β2 = rank(H2) reveals the number of voids or cavities. Together, the set of Betti numbers {β0, β1, β2, ...} indicates the topology of a simplicial complex.
Persistent homology is devised to track the multiscale topological information along a filtration.698 A filtration of simplicial complex K is a nested sequence of subcomplexes {Kt}t=t0,...,tm of K such that
| 41 |
Moreover, the inclusion map Xti ⊆ Xtj induces a homomorphism fkti,tj between homology groups Hk(Kti) → Hk(Ktj) for each dimension k. The p-persistent kth homology group of Kt is defined by
| 42 |
where Zkt = ker ∂k and Bkt + p = im ∂k+1. Intuitively, this homology group records the k-dimensional homology classes of Xt that are persistent at least until Xt+p. The birth and death of homology classes can be encoded by a barcode, a set of intervals. Given a molecule, a filtration can be constructed, and hence, a barcode can be calculated. Feature vectors can be constructed from barcodes for machine learning models.699 One can use the persistent barcode to distinguish the randomness from the structure of the growing graphs. For instance, as illustrated in Figure 9, a filtration was introduced to a graph G to create multiple simplex complexes. We can distinguish the topological differences among complexes along filtration by analyzing the persistent barcode and Betti curves.
Figure 9.

Illustration of persistent homology filtration. Reused with permission from ref (688). Copyright 2020 Anand et al. under Creative Commons Attribution 4.0 International License https://creativecommons.org/licenses/by/4.0/. (a) Simplicial complex at radius 1.2 has 0-simplexes (black dots), 1-simplexes (red edges), 2-simplexes (yellow triangles), and 3-simplexes (purple tetrahedral). The barcode shows β0 and β1. (b) Persistent homology filtration at radius 0.6, 0.8, and 1.2. (c) 0-, 1-, 2-, and 3-simplex. (d) Topological invariants of three examples.
Since the first integration of persistent homology and machine learning,700 topology-based approaches have found much success in biomolecular modeling and prediction.219,663,699,701 Combined with large datasets and machine learning algorithms, TDA is a powerful tool for predicting biomolecular properties such as protein–ligand binding affinity219,699 and drug discovery.702 According to the biomolecular properties, complexes are constructed as an atomic-specific strategy or bipartition graph. For instance, when studying the protein folding energy of the ACE2 and SARS-CoV-2 S protein, one can use element-specific and/or site-specific persistent homology to simplify the structural complexity of the protein structure and encode vital biological information into topological invariants.687,699 Wang et al.6 applied topological features to protein folding studies on the energy changes on mutations of the SARS-CoV-2 nsp6 protein. Moreover, in the complex formed in a bipartite graph, the features of the protein–protein interaction can be studied where the atoms of the antibody and antigen consist of two disjointed and independent sets. Chen et al.77 used this idea to predict the binding free energy changes on mutations of the protein–protein interactions between the S protein and antibodies. Nguyen et al.703 studied the potency and molecular mechanism of the main protease inhibition from 137 crystal structures by integrating mathematics, deep learning methods, and applied persistent homology. Topological data analysis is not only applied to studying protein–protein interactions. Chen et al.10,78,79,687 further studied the mutations that strengthened SARS-CoV-2 infectivity where persistent homology plays a key role in analyzing the interactions between the S protein and human ACE2. Moreover, Pérez-Moraga et al. applied TDA to identify the drug repurposing targeting SARS-CoV-2 proteins (3CLpro, nsp15, and nsp12).704
2.3. Machine Learning
Machine learning (ML), including deep learning (DL), is a transformative technique in artificial intelligence (AI). ML and DL can be categorized into four major tasks, namely regression, classification, clustering, and dimensionality reduction. The first two involve supervised learning using labeled data, and the last two rely on unsupervised learning using unlabeled data. All of these methods are widely used in computational biology, computational chemistry, and computational biophysics.
2.3.1. Dimensionality Reduction
As raw data often exist in high-dimensional space, dimensionality reduction techniques can be applied to transform raw data into a low-dimensional representation, making it easy for visualization and analysis. Various dimensionality reduction algorithms can be divided into two categories, namely matrix factorization and neighbor graphs. The matrix factorization maintains the pairwise distance among the data samples. Techniques such as principal component analysis (PCA),705 multidimensional scaling (MDS),706 linear autoencoder,707 Sammon mapping,708 latent Dirichlet allocation,709 non-negative matrix factorization,710 etc. fall into this category. Neighbor graphs seek to preserve the global distance among the data sample, which includes Laplacian eigenmaps,711,712 Hessian eigenmaps,713 local tangent space alignment,714 Isomap,715 t-distributed stochastic neighbor embedding (t-SNE),716,717 uniform manifold approximation and projection (UMAP),718 etc. In the following, popular methods, such as PCA, t-SNE, and UMAP, are briefly introduced.
PCA. PCA705 aims to find an orthogonal linear transformation such that the projection of transformed data on the first coordinate has the largest variance, the projection on the second coordinate has the second largest variance, and so on. We can take the first several coordinates in the new coordinate system to get a low-dimensional projection of the original data.
t-SNE. t-SNE716,717 is a nonlinear dimensionality reduction technique. It first calculates the pairwise distribution over pairs of high-dimensional data such that a pair of near data points is assigned with a higher probability, while a pair of dissimilar points is assigned with a lower probability. Second, a similar probability distribution over the low-dimensional data is calculated as well. Last, the minimization of the Kullback–Leibler (KL) divergence is carried out between the two distributions to map the high-dimensional data to a low-dimensional space appropriately.
UMAP. Another nonlinear dimensionality reduction technique is UMAP,718 which is computationally faster than t-SNE. UMAP first constructs a weighted graph from the original data, where edge weights represent distances and then projects the weighted graph to low-dimensional space. UMAP is motivated by category theory and Riemannian geometry.
Many dimensionality reduction techniques have been applied in SARS-CoV-2 research. Some research employed PCA to analyze the dynamics of proteins. For instance, Islam et al. examined PCA as a part of the techniques to seek the best candidates that can be used as potent inhibitors against the main protease of SARS-CoV-2. They first selected candidates with strong binding affinities and interactions between the main protease and the phytochemicals with AutoDock Vina and GOLD. Next, MD simulations are applied to validate five top-ranked inhibitors. Among them, three inhibitors, baicalin, cyanidin 3-glucoside, and a-ketoamide-11r, are selected by applying PCA, which has structural similarity with the apo-form of the main protease.719 In addition, PCA was also used to access the chemical space of a given SARS-CoV-2 data set. For example, PCA was set along the SARS-CoV-2 molecular fingerprint descriptors to show that the SARS-CoV-2 chemical space is well distributed with inactive and active molecules.720 Moreover, PCA was also applied to study the motions of the protein during the binding of the ligand by Prasad et al.565 In the initial process of the SARS-CoV-2 entering the host cell, TMPRSS2 and Cathepsins B/L activate the S protein and enable SARS-CoV-2 to invade the host cell through two independent pathways. Therefore, seeking a simultaneous target to both entry pathways would be a good idea to block the virus from entering host cells. Prasad et al. applied PCA techniques to study the significant motions of the drug candidates during the binding of TMPRSS2 and Cathepsins B/L. The results showed that cyclosporin A (CsA), one of the drug candidates, is quite stable with TMPRSS2 in the complex when the dynamics of this structural conformation is increased. A similar pattern can also be observed in the cathepsins L (CTSL)-CsA complex. Furthermore, due to the capacity of PCA to reduce the dimensionality to maximize the data set variance, PCA is used as a metric for analyzing conformational the diversity from Gaussian accelerated MD (GaMD) and conventional MD simulations of the SARS-CoV-2 main protease by Sztain et al.146
Dimensionality reduction techniques are often coupled with K-means clustering. Gussow et al.721 tried to identify genomic determinants of coronavirus that are related to high case fatality rates (CFRs). They performed multiple sequence alignment for 944 human coronavirus genomes and recoded aligned sequences as sequences consisting of 0 and 1. Then they applied PCA and t-SNE on recoded aligned sequences and performed K-means clustering, identifying 11 regions of nucleotide alignments as predictive of the high CFRs of coronaviruses.
UMAP is often used for visualizing gene expression data (e.g., refs (722 and 723)). Zhang et al.723 tried to identify the susceptible cell types and potential infection routes of SARS-CoV-2, since the coexpression of ACE2 and TMPRSS2 is critical for viral entry. Therefore, they analyzed five data sets with single-cell transcriptomes of human tissues to study the coexpression pattern of ACE2 and TMPRSS2 in five different cell types, consisting of esophageal cells, gastric mucosa cells, ileal cells, colon cells, and lung cells. First, UMAP is applied to get the landscape of five different types of cells. Next, the expression of ACE2 (blue) and TMPRSS2 (red) was marked on the UMAP plots. Meanwhile, the UMAP plots were merged as well to show the coexpression of ACE2 and TMPRSS2. Such single-cell analysis indicated that ACE2 and TMPRSS2 may have coexpressed not only in lung alveolar type 2 cells but also in upper epithelial and gland cells from esophageal and absorptive enterocytes from the ileum and colon.
Moreover, a rich reference data set that describes the transcriptional landscape at the single-cell level of the lung and subsegmental bronchial in a total of 16 individuals was established by Lukassen et al.724 The gene expression patterns of ACE2, TMPRSS2, and FURIN in the lung can also be observed by UMAP (see Figure 10a). Furthermore, a work by Ravindra et al. applied UMAP to analyze the gene expression levels of the ACE2, CTSL, TMPRSS2, and TMPRSS4 protease of human bronchial epithelial cell (HBEC) samples as illustrated in Figure 10b and c.
Figure 10.

(a) Expression level of ACE2 in the lung plotted on top of the UMAP coordinates. Expression level of TMPRSS2 in the lung plotted on top of the UMAP coordinates. Expression level of FURIN in the lung plotted on top of the UMAP coordinates. Reproduced with permission from ref (724). Copyright 2020 Lukassen et al. (b and c) UMAP visualization of HBEC samples, colored by expression (normalized and square-root transformed counts) of the ACE2 receptor, CTSL, TMPRSS2, and TMPRSS4 proteases. Reproduced with permission from ref (725). Copyright 2021 Ravindra et al. Under Creative Commons Attribution 4.0 International License https://creativecommons.org/licenses/by/4.0/.
2.3.2. Linear Regression
Linear regression
is one of the basic algorithms in machine learning and can be used
to solve the regression problem. Assume the training set is 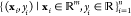 . Here, n is the number
of samples, and m represents the number of features.
Then, the prediction corresponding to xi is defined as
. Here, n is the number
of samples, and m represents the number of features.
Then, the prediction corresponding to xi is defined as
| 43 |
where  represents the
weights,
represents the
weights,  is the bias, and wT represents the transpose of w. The
loss function of the linear regression model is
is the bias, and wT represents the transpose of w. The
loss function of the linear regression model is
| 44 |
The aim of the linear regression is to minimize the loss function, which is demonstrated as eq 45.
| 45 |
Additionally, a regularization term can also be taken into account in the case of overfitting:
| 46 |
where λ represents a penalty constant.
Applications are involved in applying linear regression to calculate the correlation coefficient and predict different types of dependent variable values, where most applications study the experimental data. As for theoretical analysis, studies focus on implementations of QSAR, which will be discussed later. To study the SARS-CoV-2 fatality rates, the CFRs of SARS-CoV-2 variants were compared by linear regression of worldwide data between wild-type and mutant-type virus on the S protein, resulting in G614 being shown to be a more pathogenic strain.206 In the study of gene evolution of SARS-CoV-2, linear regression was applied to the line between the number of viral variants and the gene length, suggesting that the mutation frequency was proportional to the length of the gene.726 A regression model was designed and related the the experimental binding affinity for antibodies by applying structural features, where several mutations at the S binding motif were identified.727 Moreover, a linear regression model was applied by Israel et al. to quantify the association between the logarithm of antibody levels and the elapsed time, in both fully vaccinated and convalescent individuals.728 This study suggests that individuals who received the Pfizer-BioNTech mRNA vaccine had higher initial antibody levels but a much faster exponential decrease compared to patients who had been infected by SARS-CoV-2.
2.3.3. Logistic Regression
Logistic regression
is an algorithm designed for solving classification problems. Assume
the training set is  . Here, n is the number
of samples, m represents the number of features,
and
. Here, n is the number
of samples, m represents the number of features,
and  represents different categories. Then,
the prediction of the logistic regression corresponding to the point xi is
represents different categories. Then,
the prediction of the logistic regression corresponding to the point xi is
| 47 |
where  represents the
weights,
represents the
weights, 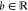 is the bias, and wT represents the transpose of w. When yi ∈ [0, 1], the loss
function can be defined by
is the bias, and wT represents the transpose of w. When yi ∈ [0, 1], the loss
function can be defined by
| 48 |
Ayouba et al.729 used logistic regression to represent the dynamics of the immunoglobulin G (IgG) response to the S protein, N protein, or both antigens at the same time since onset of symptoms. In addition, researchers stated that the publicly shared CD8+ (Cytotoxic T cells with CD8 surface protein) might be used as a potential biomarker of SARS-CoV-2 infection at high specificity and sensitivity by applying the logistic regression.730 In addition, only subtle differences were observed from the initial MD simulations of the two RBD-ACE2 complexes by Pavlova et al. Later, logistic regression was used to successfully identify the individual residues with the most distinctive ACE2 interactions, many of which have been highlighted in previous experimental studies.731
2.3.4. k-Nearest Neighbors
The k-nearest neighbors algorithm (k-NN) is a nonparametric technique proposed by Thomas Cover and P.
Hart in 1967.732k-NN
can be used for solving both regression and classification problems,733 and it is sensitive to the local structure
of the data. The flow chart of the k-NN algorithm
can be found in Figure 11. The features of the training set are {xi}i=1n with  , k shows the number of
the nearest neighbors, and
, k shows the number of
the nearest neighbors, and  is a feature representation of the training
set. Different distance metrics can be employed in the k-NN algorithm, such as Euclidean distance, Manhattan distance, Minkowski
distance, Chebyshev distance, natural log distance, generalized exponential
distance, generalized Lorentzian distance, Canberra distance, quadratic
distance, and Mahalanobis distance.
is a feature representation of the training
set. Different distance metrics can be employed in the k-NN algorithm, such as Euclidean distance, Manhattan distance, Minkowski
distance, Chebyshev distance, natural log distance, generalized exponential
distance, generalized Lorentzian distance, Canberra distance, quadratic
distance, and Mahalanobis distance.
Figure 11.

Flow chart of the k-NN
algorithm. The features
of the training set are {xi}i=1n with  , k shows the number of
nearest neighbors, and
, k shows the number of
nearest neighbors, and  is a feature representation of the training
set.
is a feature representation of the training
set.
The classifier can be built by using the k-NN algorithm. Setting classifiers as k-NN models, an automated system can distinguish the SARS-CoV-2 genome from the SARS-CoV genome and MERS genome by using the genomic sequences from the National Center for Biotechnology Information (NCBI) GenBank for accelerating the diagnosis process and improving the accuracy of disease detection.734 Moreover, AllerTOP v.2.0 classified allergens and nonallergens based on the k-NN method with an accuracy of 88.7%.735 Furthermore, the k-NN algorithm has been employed to classify the human protein sequences of COVID-19 according to country.736 Moreover, k-NN was also applied on the transcriptomics data. The recently reported transcriptomics data of upper airway tissue with acute respiratory illnesses is integrated with some machine learning algorithms such as the k-NN algorithm to identify effective qualitative biomarkers and quantitative rules for the distinction of SARS-CoV-2 infection from other infectious diseases.737 Furthermore, with the implementation of the k-NN algorithm, as well as the GRU (gated recurrent unit) neural networks and LSTM (long short-term memory) autoencoder models by Liang et al.,738 the analysis of the nanosecond backbone root-mean-square deviation (RMSD) of the S protein assisted in the prediction of the long-term properties of SARS-CoV-2 S proteins.
2.3.5. K-Means Clustering
K-means clustering is an unsupervised learning algorithm, aiming to partition a set of observations into K subsets or clusters. It typically begins with selecting K observations as centroids of clusters and assigning observations to clusters according to their distances with centroids. Then, the algorithm recursively updates centroids by minimizing the within-cluster sum of squares.
In the study of SARS-CoV-2, a common method for discovering evolution patterns and transmission pathways is to cluster mutations. Wang et al. retrieved 31,421 genome samples from GISAID and rearranged them according to the reference SARS-CoV-2 genome.9 Then they computed the Jaccard distance matrix (Jaccard distance measures the dissimilarity between two genomes) and clustered genomes using the Jaccard distance matrix. In this work, the authors also analyzed how mutations would impact the efficacy of certain COVID-19 diagnostic kits. As directly applying K-means clustering is often time-consuming, it is often coupled with dimensionality reduction techniques. In refs (12 and 739) the authors followed a similar method as in ref (9) and used PCA or UMAP to reduce the dimensionality of the Jaccard distance matrix before applying K-means clustering. K-means clustering can also be found in works such as ref (721). Additionally, in ref (740) by Assis et al., the authors first constructed a coronavirus antigen microarray (COVAM). Such a model included 11 SARS-CoV-2, 5 SARS1, 5 MERS, and 12 seasonal coronavirus recombinant proteins, which could cluster COVID-19 convalescent plasma (CCP) based on their antibody reactivity patterns against 11 SARS-CoV-2 antigens. Then, K-means analysis, gap statistics, and hierarchical clustering were applied, revealing three main clusters with distinct reactivity intensities and patterns as illustrated in Figure 12.
Figure 12.

Group 1 cluster analysis and PCA demonstrate two subgroups. Reproduced with permission from ref (740). Copyright 2021 Assis et al. under Creative Commons Attribution 4.0 International License https://creativecommons.org/licenses/by/4.0/. (a) Reactivity to the SARS-CoV-2 antigens. Samples were clustered using hierarchical clustering analysis. (b) Bar plot of the mean reactivity and the standard error of each cluster to each individual SARS-CoV-2 antigen. (c) Distribution of the samples that were clustered into three groups by PCA.
Moreover, K-means clustering was also used to select conformations that represent the overall conformational heterogeneity of molecular dynamics simulation data. For instance, a team led by Albert Y. Lau developed a machine learning algorithm called TACTICS (trajectory-based analysis of conformations to identify cryptic sites), aiming to address the difficulties in seeking druggable sites.741 First, by applying the K-means clustering algorithm in TACTICS on multiple molecular dynamics simulation data, a small number of conformations was found. Next, such conformational data was integrated into a random forest model in TACTICS to identify possible druggable sites in each conformation based on its protein motion and geometry. Last, the scores of potential binding pockets were given based on the fragment docking analysis. This approach provided a way to predict the locations of binding sites that cannot be viewed in the experimentally determined structures.
2.3.6. Support Vector Machine
The support
vector machine (SVM) was developed by Vapnik and his colleagues and
can be used for both classification and regression analysis.742,743 For the classification problem, assume the training set is  . The prediction of the SVM at point xi will be ŷ = wTxi + b. Here,
. The prediction of the SVM at point xi will be ŷ = wTxi + b. Here,  is the weights and b is
the bias. If the training set is linearly separable, the aim is to
minimize ∥w∥ subject to yi(wTxi – b) ≥ 1. If the training set is not linearly separable,
then the hinge loss function max(0, 1 – yi(wTxi – b)) will be involved. The aim of the SVM is to minimize
is the weights and b is
the bias. If the training set is linearly separable, the aim is to
minimize ∥w∥ subject to yi(wTxi – b) ≥ 1. If the training set is not linearly separable,
then the hinge loss function max(0, 1 – yi(wTxi – b)) will be involved. The aim of the SVM is to minimize
| 49 |
where λ is the regularization term (a.k.a. penalty). For the regression problem, the aim is to minimize ∥w∥ subject to |yi – ⟨wT, xi⟩ – b| ≤ ϵ.
The SVM mentioned above is a linear classifier. To design a nonlinear classifier, the kernel trick is employed to maximize margin hyperplanes. The feature of the kernel SVM will be Φ(x, z), where the commonly used kernels are the linear kernel Φ(x, z) = xTz, the polynomial kernel defined by Φ(x,z) = (αxTz + r)d, the radial basis function kernel (RBF) Φ(x,z) = e–(∥x–z∥/σ)μ, and the sigmoid kernel denoted as Φ(x,z) = 1/(1 + e–γxTz), where r, α, σ, γ, and μ are constants.
There is a large number of SVM applications on SARS-CoV-2, and some of these focus on the biomolecular level and are summarized as follows. A collection of 100,000 FDA-registered chemicals and approved drugs, as well as about 14 million other purchasable chemicals against multiple SARS-CoV-2 targets, was screened for drug repurposing,744 where SVM was applied for identifying the top features. Additionally, Dutta et al.745 predicted a novel peptide analog of the S protein using SVM models implemented by the AVPred antiviral peptide prediction server.
The SVM was applied to identify inhibitors for Mpro as well. Mekni et al.746 applied the SVM on a data set with two million commercially available compounds to classify two hundred novel chemotypes that are potentially active against the viral protease. Sun et al.747 implemented a hybrid support vector machine classification model to find the viral entry inhibitors using a collection of publicly available SARS-CoV-2 pseudotyped particle entry assay repurposing screen data. An SVM-based Web server called CellPDD was applied to determine the cell penetrating peptides (CPPs) in the proteome of SARS-CoV-2, such as S protein, M glycoprotein, N phosphoprotein, E protein, ORF1ab polyprotein, ORF3a protein, ORF6 protein, ORF7a protein, ORF8 protein, and ORF10 protein. The results showed that CPPs were not found in E protein, one CPP was identified in ORF6, and one CPP was primarily found in the proteome of ORF1ab. Such work may be valuable to some studies in the nuclear localization sequence (NLS) for vaccine development or drug discovery.748
2.3.7. Decision Trees, Random Forest, and Gradient Boosting Decision Trees
The decision tree (DT) method is a basic ML method, which is used to perform both classification and regression models by representing the attribute of the data using a flow-chart-like structure. As the fundamental architecture of tree structure methods, decision trees are further developed to a series of ensemble methods, such as the random forest method, extremely randomized tree method, AdaBoost methods, and the gradient boosting decision tree method. Among them, random forest and gradient boosting decision trees are widely applied. Random forest (RF)749 is an ensemble learning method, which is designed to reduce the overfitting in the original decision trees. Both classification and regression problems are suitable for random forest models. Gradient boosting decision tree (GBDT) is a machine learning technique for regression and classification problems, which produces a prediction model in the form of an ensemble of decision trees.750 This ensemble of decision trees is built in a stagewise fashion like other boosting methods. That is, algorithms optimize a cost function over function space by choosing a function that points in the negative gradient direction iteratively. These methods can be applied using packages such as scikit-learn in Python751 or R packages.749
In applications, DT, RF, and GBDT were used commonly in the diagnosis of COVID-19 or the analysis of virus spreading. In this review, we focus on molecule-based studies. Decision trees and the ensemble methods can handle the small-size data set well and, therefore, were implemented widely at the early stage of the SARS-CoV-2 pandemic, when databases were not well-established. Investigations applying neural networks on large databases are booming and will be introduced in the next section. In ref (752) the authors used GBDT to predict which proteins would likely make up an effective vaccine for COVID-19. A GBDT model repurposed 8565 approved or experimental drugs targeting Mpro, suggesting that 20 FDA-approved drugs could be effective.753 Wang et al.6 used topology-based features and GBDT models to analyze the nsp6 protein stability upon mutation. Bocci et al.754 constructed a machine learning platform to estimate anti-SARS-CoV-2 activities, where a total of 22 feature types are created according to chemical and biophysical information such as topological fingerprints, molecular weights, etc. Another RF classification was conducted for geographic-specific SARS-CoV-2 mutations using genetic sequences.755 In addition, an RF classifier was applied to the analysis of multiple isotype-specific responses to identify infected individuals.756 Rola et al.757 studied different docking protocols and applied structures from 2D and 3D at the molecular mechanics level as features for the random forest for docking studies of the SARS-CoV-2 S protein binding to ACE2. RF models were also applied to identify SARS-CoV-2 drug inhibitors and antibodies for SARS-CoV-2 S protein and N protein.758,759
2.3.8. Artificial Neural Network (ANN)
ANN or deep neural network (DNN) is a ML model inspired by biological
neural networks that constitute animal brains.760 ANN can be viewed as a weighted directed graph in which
artificial neurons can be considered as nodes and weights can be considered
as the links between input and output nodes. ANN is designed for both
regression and classification problems. We assume the training set
is  . Here, n is the number
of samples, m represents the number of features,
and
. Here, n is the number
of samples, m represents the number of features,
and  shows the number of classes. If l = 1, then the training set is designed for the regression
problem. If l > 1, then we say the ANN model is
designed
for a classification problem. There are two main procedures in the
ANN algorithm, the feed-forward the back-propagation procedures. Assume
shows the number of classes. If l = 1, then the training set is designed for the regression
problem. If l > 1, then we say the ANN model is
designed
for a classification problem. There are two main procedures in the
ANN algorithm, the feed-forward the back-propagation procedures. Assume 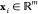 is a feature
representation in the training
set; then the feed-forward starts from the input layer to the first
hidden layer that is defined as
is a feature
representation in the training
set; then the feed-forward starts from the input layer to the first
hidden layer that is defined as
| 50 |
where 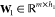 represents the weights from the input layer
to the first hidden layer,
represents the weights from the input layer
to the first hidden layer, 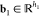 represents
the bias from the input layer
to the first hidden layer. Here h1 is
the number of the neurons in the first hidden layer and function f represents the activation functions such as the ReLu or
Sigmoid function. Next, from the first hidden layer to the second
hidden layer, a similar function is defined as
represents
the bias from the input layer
to the first hidden layer. Here h1 is
the number of the neurons in the first hidden layer and function f represents the activation functions such as the ReLu or
Sigmoid function. Next, from the first hidden layer to the second
hidden layer, a similar function is defined as
| 51 |
where 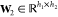 and
and  . Here, h2 is
the number of neurons in the second hidden layer. A similar procedure
goes until it gets to the output layer. The predictor from the last
hidden layer (the jth hidden layer) to the output
layer is
. Here, h2 is
the number of neurons in the second hidden layer. A similar procedure
goes until it gets to the output layer. The predictor from the last
hidden layer (the jth hidden layer) to the output
layer is
| 52 |
where  ,
, 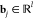 , and hj is the number of neurons
in the last hidden layer in the ANN.
The cross-entropy loss describes the cost function, which is defined
as
, and hj is the number of neurons
in the last hidden layer in the ANN.
The cross-entropy loss describes the cost function, which is defined
as
| 53 |
The ANN algorithm obtains the prediction via the feed-forward procedure and then minimizes the cross-entropy loss through the back-propagation procedure. The back-propagation procedure applies the loss function evaluated at the output layer and propagates it back through the network to update the weights and bias. In the calculation of the gradient for back-propagation, the stochastic gradient descent (SGD) with momentum method is one of the most popular approaches which evaluates a small part of the training data and contributes to the next iteration with different weights. The process of the SGD with momentum can be expressed as
| 54 |
where η is the learning rate and β ∈ [0, 1] is a scalar coefficient for the momentum. Fully connected layers inducing a large number of degrees of freedom cause an overfitting issue in the training process. The dropout technique can prevent the network overfitting,761 which randomly sets partial hidden units zero values to their connected neurons in the next layer. ANN has numerous applications in molecular biology.762 For example, deep learning methods were used to predict the mutation-induced changes of protein stability,763 protein–ligand binding affinity,219 and protein–protein binding affinity.685
Two directions of ANN/DNN application on SARS-CoV-2 are studying the infectivity of SARS-CoV-2 and the efficacy of SARS-CoV-2 antibodies and repurposing existing drugs and compounds or even generating new ones to treat SARS-CoV-2. The former focuses on the binding energy of protein–protein interactions or protein–ligand interactions. Chen et al. applied a GBDT and neural network-integrated method to calculate the BFE changes between SARS-CoV-2 S protein- and ACE2-induced by mutations.78 Assuming that SARS-CoV-2 infectivity is proportional to the BFE of S protein and ACE2, one can quantitatively predict mutation-induced impacts on the infectivity of the SARS-CoV-2 virus. Similar work can be found to study the mutation impacts on the efficacy of SARS-CoV-2 antibodies.764 Moreover, considering the spreading of SARS-CoV-2 variants, Wang et al.79,765 applied the DNN model results about BFE changes induced by mutations of bindings of SARS-CoV-2 S protein and antibodies and discovered the escape mutations and emerging vaccine-breakthrough variants. A deep docking (DD) model provided a fast prediction of docking scores from Glide or any other docking program, hence, enabling structure-based virtual screening of billions of purchasable molecules in a short time.766 The DD model relies on a deep neural network trained with docking scores of small random samples of molecules extracted from a large database to predict the scores of remaining molecules. A deep neural network method was developed and validated by using a docking algorithm on existing drugs for drug repurposing.767 A pretrained deep learning-based drug–target interaction model called molecule transformer–drug target interaction (MT-DTI) identified commercially available drugs that can act on viral proteins of SARS-CoV-2.768 Another deep neural network model was designed to find the protein–ligand interactions for drug repurposing.769 With a deep neural network model, 33 potential compounds were identified as ideal inhibitors against Mpro,366 as well as a similar work for SARS-CoV-2 inhibitors.770 Moreover, Izumi et al.771 used the FASTA file for a deep neural network to predict sequence-based super secondary structure codes. Furthermore, to identify essential physicochemical and structural characters for SARS-CoV-2 Mpro inhibition, a nonlinear QSAR model assisted by ANN and SVM was designed with 69 structurally diverse chemicals with potential SARS-CoV-2 Mpro inhibitory property as descriptors.772 Furthermore, ComboNet was designed to predict (1) the interaction between a drug and multiple biological targets, (2) the intrinsic antiviral activity of a drug, and (3) the synergy of drugs.773 ComnoNet is composed of two subnetworks: a drug–target interaction (DTI) and a target–disease association network, which enabled an effective in silico search for synergistic combinations against SARS-CoV-2 (see Figure 13).
Figure 13.

Structure of ComboNet. ComboNet consists of two networks: a DTI and a target–disease association network. Reused with permission from the authors.773 Copyright 2021 Jin at el. under Creative Commons Attribution 4.0 International License https://creativecommons.org/licenses/by/4.0/. (a) Workflow of ComnoNet for single-drug synergy. First, a single drug is fed into the DTI network to get its molecular representation zA. Then, such a molecular representation will be the input of the target–disease association network, and its output will be the predicted antiviral effect of a single drug. (b) Workflow of ComnoNet for drug combination synergy. First, drugs will be fed into the DTI network to get their molecular representations zA and zB. Then, the combination of such molecular representations zAB, as well as zA and zB, will be fed into the target–disease association network to get the predicted antiviral effect of a combination of drugs.
2.3.9. Convolutional Neural Network (CNN)
CNN774 is a specialized type of neural network model originally designed to analyze visual imagery, but it can also be applied to many areas. Since the first successful CNN was developed in the late 1990s, CNN has achieved much success in image and video recognition, natural language processing, etc. CNN has had success in biophysics such as protein structure prediction and protein–ligand binding.219,699 The core of CNN is the convolutional layer where its name comes from (see Figure 14). In the context of CNN, convolution is a linear operation that involves the multiplication of a set of weights with the input. This multiplication is always called a filter or a kernel. Using a filter smaller than the input is intentional as it allows the same filter to be multiplied by the input array multiple times at different points on the input. Specifically, the filter is applied systematically to each overlapping part or filter-sized patch of the input data, left to right and top to bottom, which allows the filter an opportunity to discover that feature anywhere in the input.
Figure 14.
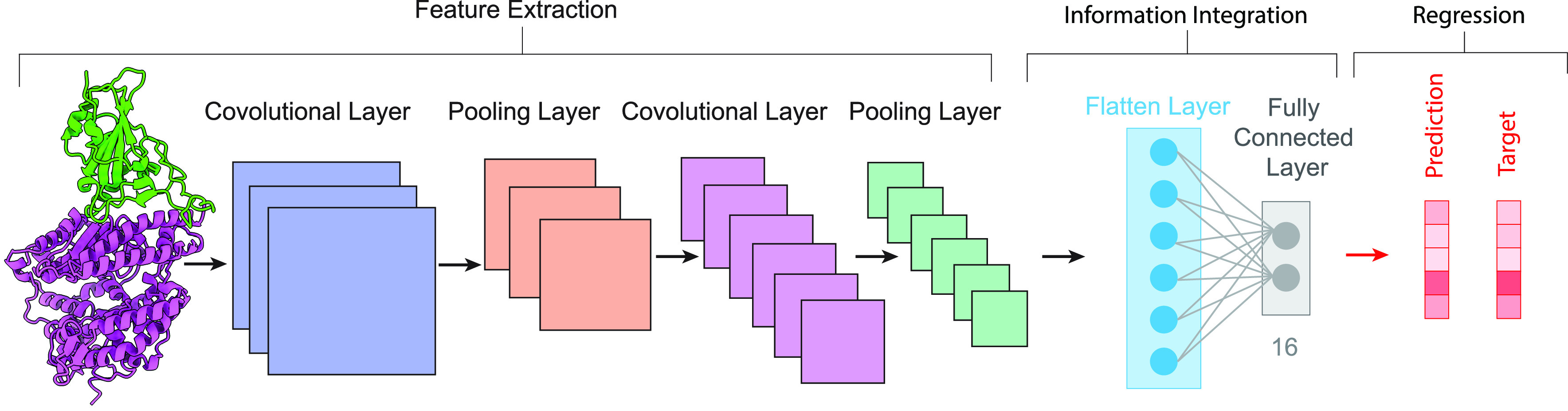
Structure of 2D CNN. The feature extraction process includes multiple convolutional layers and pooling layers. The convolutional layer extracts the local features of the initial input, and the average pooling layer increases the translational invariances of the network and reduces the parameters that need to be trained. The output of the last pooling layer is a 2D array. Next, the flattened layer reshapes a 2D array to a 1D array to feed the feature into a fully connected layer. Last, the integrated information will be fed into a regressor for the final prediction.
In the antibody and vaccine research, algebraic-topology-based features were used to build a CNN-GBT hybrid model for predicting mutation-induced binding affinity change, investigating the impact of S protein mutations on the ACE210,78 and 27 antibodies (see Figure 15),77 as well as suggesting some highly risky ones to vaccine design.221 In the inhibitor research, Nguyen et al.703 used algebraic-topology-based features and CNN models to predict the potency of ligands from the 137 crystal structures of Mpro. Critical assessment of protein structure prediction (CASP) also proved a domain for the application of powerful CNN methods in protein structure prediction. For example, CNN-based AlphaFold by Google DeepMind obtained the highest accuracy in CASP13.783,784 During this epidemic, DeepMind applied the AlphaFold to predict the 3D structures of SARS-CoV-2 Mpro, PLpro, nsp2, nsp4, and nsp6.39 Meanwhile, the CNN-based C-I-TASSER algorithm developed by the Zhang lab was implemented to predict as many as 24 SARS-CoV-2 proteins.785 Yang et al.786 implemented the CNN model on secondary structure information with other biophysics properties to construct a multiepitope vaccine. Lastly, the graph convolutional neural network is considered as one of the graph neural network (GNN) variants,787 which applied convolutional ideas on a graph as the network structure and is a recursive direct-contacting aggregation algorithm.788 In ref (789) Haneczok and Delijewski applied the graph convolutional neural network to study the potential SARS-CoV-2 Mpro inhibitors.
Figure 15.

3D alignment of the available unique 3D structures of SARS-CoV-2 S protein RBD in binding complexes with 19 antibodies as well as ACE2. (a) ACE2 (6XDG775), CT-P59 (7CM4776), and CB6 (7C01777). (b) C135 (7K8Z778), C110 (7K8 V778), REGN10933 (6XDG779), and REGN10987 (6XDG779). (c) C119 (7K8W778), C144 (7K90778), and C121 (7K8Y778). (d) LY-CoV481 (7KMI780), LY-CoV555 (7KMG780), and LY-CoV488 (7KMH780). (e) C002 (7K8T778), C104 (7K8U778), C105 (6XCM778), and C102 (7K8M778). (f) S309 (6WPS781), AZD1061 (7L7E782), and ACD8895 (7L7E782).
2.3.10. Natural Language Processing (NLP) Methods
The recurrent neural network (RNN) (see Figure 16a) is a class of artificial neural networks where connections between nodes form a directed graph along a temporal sequence,790 which allows it to exhibit temporal dynamic behavior in the data. Derived from the feed-forward neural network, RNN can use its internal state (memory) to process variable-length sequences of inputs. RNN was originally designed for language processing tasks, but it can also be applied to other circumstances, such as molecular sequence data.
Figure 16.
(a) Workflow of a RNN cell. Here, t represents an object at time-step t. xt, yt, and at denote the input x, output y, and activation at time-step t, respectively. ŷt represents the prediction at time-step t. (b) Workflow of a LSTM cell. t represents an object at time-step t. xt, yt, at, and ct denote the input x, output y, activation, and cell state at time-step t, respectively. ŷt represents the prediction at time-step t. ft, ut, ot, ct, and c̃t denote the forget gate state, update gate state, output gate state, cell state, and previous cell state at time-step t. σ is the activation function such as tanh function. (c) Workflow of a GRU cell. Here, t represents an object at time-step t. xt, yt, and at denote the input x, output y, and activation state at time-step t, respectively. ŷt represents the prediction at time-step t. rt, ot, and ut denote the reset gate state, output gate state, and update gate state at time-step t. (d) Illustration of the generative network complex.793 SMILES strings are encoded into latent vector space through a gated recurrent neural network (GRU)-based encoder.
The long short-term memory (LSTM) shown in Figure 16b and the gated recurrent unit (GRU) in Figure 16c are two popular variants of RNN. LSTM791 is designed to avoid the vanishing gradient problem and is normally augmented by recurrent gates called “forget gates”, and so errors can flow backward through unlimited numbers of virtual layers unfolded in space. GRU is a gating mechanism in recurrent neural networks introduced in 2014.792 Its performance was found to be similar to that of LSTM. However, as it lacks an output gate, its parameters are fewer than LSTM, so it is easier and faster to train.
Their application to SARS-CoV-2 includes the following: Hofmarcher et al.770 utilized “ChemAI” to screen and rank around one billion molecules from the ZINC database for favorable effects against SARS-CoV-2. In more detail, the network is of the type SMILES LSTM.794 Bung et al.366 employed RNN-based generative and predictive models for de novo design of new small molecules capable of inhibiting SARS-CoV-2 Mpro.
COVID-DeepPredictor is another work using long short-term memory as a recurrent neural network to identify unknown sequences of virus pathogens. Hie et al.795 designed a bidirectional LSTM (BiLSTM) to predict structural escape patterns of influenza hemagglutinin, HIV-1 envelope glycoprotein (HIV Env), and SARS-CoV-2 S proteins.
2.3.11. Autoencoder and Transformer
Autoencoder is an advanced DL model built from RNN, GRU, or LSTM to learn efficient codings of unlabeled data. In molecular sciences, Autoencoders are designed to generate effective low-dimensional molecular representations. As shown in Figure 16d, a generative network complex796 is an autoencoder-based technique designed to automatically generate new drug candidates with desirable properties. It consists of an encoder, a latent space, a latent-space molecular generator, a decoder, and a 2D fingerprint-binding predictor (2DFP-BP). Gao et al.793 used this AI technology to generate some potential Mpro inhibitors judged by the consensus of a latent-space prediction and the 2DFP-BP.
Transformer is one of the frequently used models in the field of natural language processing, which was introduced in 2017 for sequential data analysis.797 A transformer model is equipped with an encoder–decoder structure. Typically, a stack of multiple identical layers consists of a transformer encoder. Each layer is constructed by a multihead self-attention pooling and a position-wise feed-forward network. Similarly, a transformer decoder is also a stack of multiple identical layers. Such a layer is called the encoder–decoder attention. Specifically, the encoder processes the input literately and generates encodings that contain information from the input, and the decoder then takes all the encodings as input and generates decoded sequences as output.
Transformer has been widely applied to seek potential drug candidates. For instance, Beck et al., designed a pretrained model called transformer–drug target interaction (MT-DTI) to find commercially available drugs that target SARS-CoV-2 viral proteins.768 Such a model used the BindingDB database798 as a training set and Ki, Kd, and IC50 as evaluation metrics to predict the interaction between viral proteins and antiviral drugs. Besides, transformer is also integrated into a model called AlphaFold/AlphaFold2799 to predict highly accurate protein structures such as Mpro, OFR8 protein, and ORF3a protein of SARS-CoV-2.800−802
2.4. Topics in Bioinformatics and Cheminformatics
Bioinformatics and cheminformatics are of paramount importance in modeling and analysis of SARS-CoV-2. Bioinformatic and cheminformatic approaches are integrated with experiments, biochemistry, biophysics, ML, statistics, and mathematics. In this section, we illustrate several topics from the methodology-centered perspective. Sequence alignment, homology modeling, and network-based bioinformatics are discussed, followed by cheminformatics methods, namely QSAR and pharmacophore models.
2.4.1. Sequence Alignment
Sequence alignment is a method in which one can arrange DNA, RNA, or amino acid sequences to identify their similar regions.803 Such similar regions may arise from functional, structural, geometrical, or evolutionary similarities. Though sequence alignment offers the best accuracy, it is not practical to be used for a large sample size. There are two main categories of sequence alignment, namely pairwise sequence alignment and multiple sequence alignment. The former only compares two sequences at a time, while the latter compares many sequences. There are many popular tools for sequence alignment such as BLAST (basic local alignment search tool), for pairwise alignment, and MAFFT, Clustal Omega, ClustalW, and MUSCLE, for multiple sequence alignment. The following section describes BLAST first, followed by several multiple sequence alignment tools.
2.4.1.1. Pairwise Sequence Alignment
One of the popular pairwise sequence alignment tools is BLAST. BLAST is a local similarity search tool that is commonly used to find similar DNA, RNA, and amino acid sequences to the sequence in question. BLAST was created in 1990 based on the k-tuple method and has since been implemented in the GenBank and had numerous updates to increase efficiency and accuracy. The k-tuple method804 is a fast heuristic method for pairwise alignment and is commonly used as an initial step for a large sample size. The similarity score Sij between sequences i and j is defined as the number of k-tuple matches in the best pairwise alignment minus a fixed gap penalty term. For DNA and RNA, k usually ranges from 2 to 4, and for amino acids, k is 1 or 2. Sij is calculated as the number of identities divided by the number of residues compared between i and j. The distance is defined as
| 55 |
Note that this method does not guarantee optimal alignment, but it is a fast heuristic method and can be used for the initialization of BLAST and multiple sequence alignment.
BLAST begins by first creating a list of k-letter words. It then searches for possible matching k-letter words in the databank and scores them, and any words that score above a threshold are kept. The high-scoring words are kept in a search tree. This process is then extended to high-scoring pairs (HSPs), which also looks for similar words, rather than only looking at exact matching words. After searching for HSPs, the significance of the HSPs score is considered by utilizing Gumbel extreme value distribution (EVD). Further details can be found in the literature.805,806 The GenBank tutorial can be found in ref (807). As a basic tool for sequence alignment, it is utilized to detect, identify, or search sequences in a database. For example, similar coronavirus strands in other organisms, such pangolins808,809 and bats,810 were found. This tool is also used to detect SARS-CoV-2 virus in the environment811,812 such as in wastewaters.813,814
2.4.1.2. Multiple Sequence Alignment (MSA)
Unlike pairwise sequence alignment, MSA arranges three or more DNA, RNA, or protein sequences by identical regions. Through multiple sequence alignment, one can further analyze sequence homology to find evolutionary origins. In many cases, one uses a reference sequence, which is the first sequenced data, to observe mutation in the SARS-CoV-2 genome.815 There are several popular tools, Clustal,816 MUSCLE,817 MAFFT,818,819 etc.
Clustal. Clustal is a series of multiple sequence alignment tools for sequence analysis. With the first version Clustal released in 1988,816 its package has been developed for several generations based on different methods. ClustalW is the third generation and is updated to ClustalW2 currently, which aligns sequences with the best similarity score first and progressively aligns more distant scores.820,821 This is achieved by first obtaining a rough pairwise sequence alignment using the k-tuple method,804 followed by a neighbor-joining method,822 which uses midpoint rooting to create a guided tree. ClustalW2 is used as the basis for global alignment.
As for Clustal Omega, unlike ClustalW, it uses a guided tree approach, rather than a progressive alignment method. Clustal Omega begins with first producing a pairwise alignment using the k-tuple method. This, however, does not guarantee finding optimal alignment, but it is time-efficient. Then, the sequences are clustered using the mBed method,823 which calculates pairwise distance using the embedding method. Afterward, K-means clustering is used to further cluster the sequence. Then, a guided tree is formed utilizing the UPGMA method.824 Lastly, MSA is produced using the HHAlign package from HH-Suite.824 Clustal Omega’s advantage comes from the large-scale MSA. The accuracy and time complexity are average for a low number of samples. For a large number of samples with a long sequence, Clustal Omega produces high accuracy and is time-efficient. ClustalW is the updated version of the original Clustal MSA tool.
Multiple Alignment Using Fast Fourier Transform (MAFFT). MAFFT is a MSA package based on fast Fourier transform (FFT). Given two sequences v1 and v2, the correlation cv(s) of volume between the two sequences with positional lag of s sites can be defined as
where v̂1 and v̂2 are the FFT of the two sequences. If homologous regions exist, through Fourier analysis, there will be a peak in similar regions. For amino acid sequences, MAFFT also calculates correlation between polarity:
where ρ(s) is the polarity of each amino acid, N is the length of v1, and M is the length of v2. Then, a scoring function can be calculated through the sum of the two correlations
To reduce the computational complexity, only peaks above some threshold are considered. Note that the peak does not tell the location of the homologous region directly and only shows the lag. Therefore, neighboring regions at the peak must be analyzed carefully. Further details of MAFFT can be found in the literature.818,819
Multiple sequence comparison by Log-Expectation (MUSCLE). MUSCLE is an MSA tool that utilizes progressive alignment. There are three stages in MUSCLE: draft progressive, improved progressive, and refinement.817 During the draft progressive stage, a distance matrix is constructed by computing the pairwise distance of each sequence through using k-mer counting or by constructing global alignment of pairs and determining the fractional identity. Then, a tree is constructed using UPGMA (unweighted pair group method with arithmetic mean)824 or neighbor-joining822 in which the root of the tree is determined. Lastly, a progressive alignment is built by tracing the branches of the tree, yielding the first MSA of the sequence. In the improved progressive stage, a new progressive alignment is constructed from iteratively refining the previous tree. First, a new similarity matrix is constructed from their mutual alignment in the current multiple alignments. Then, a new tree is constructed, similar to the draft progressive stage. Each tree is compared to identify any changes in the nodes or branching pathway. These steps are repeated until conversion or a maximum iteration is reached. In the progressive alignment stage, a new alignment is computed for only the set of changed nodes. More details can be found in refs (817 and 825).
Sequence alignment methods are widely used in SARS-CoV-2 analysis. Many applications focus on identifying mutations and comparing virus sequences from species and organisms. Yin used sequence alignment to understand the evolution and transmission of SARS-CoV-2.8 Wang et al. employed Clustal Omega to decode asymptomatic COVID-19 infection and transmission6 and to study mutational impacts on COVID-19 diagnosis, vaccines, and medicine.12 Sequence alignment is an indispensable approach for SARS-CoV-2 modeling and analysis.826−829
2.4.2. Homology Modeling
Homology modeling constructs an atomic-resolution model of the target protein from its amino acid sequence based on experimental 3D structures of related homologous proteins (i.e., templates).830 Homology modeling relies on identifying one or more known protein structures likely to resemble the structure of the query sequence and producing an alignment that maps residues in the query sequence to residues in the template sequence.831
Because the 3D experimental structures of SARS-CoV-2 proteins were largely unknown at the early stage of the epidemic, homology modeling was widely applied to predict 3D structures of SARS-CoV-2 proteins, such as Mpro,221,602,832−838 S protein or variants,271,839−861 RdRp,294−296,299,361,514,517,529,862−864 PLpro,865 E protein,61,304,866−869 N protein,869 and others.22,305,554,602,838,870−886 Some host proteins that interact with SARS-CoV-2 were also predicted, such as ACE2,273,887,888 TMPRSS2,889−894 B cell epitopes,895 and CD147.896 Some 3D structures of vaccine proteins897−899 were also built by homology modeling. Due to the worldwide attention on this virus, experimental structures of various SARS-CoV-2 proteins and their variants can be found in the Protein Data Bank as discussed in Table 1.
2.4.3. Network-Based Bioinformatics
Drug repurposing methods require comparing the unique features, such as chemical components or proteomic, metabolomic, or transcriptomic data, of a drug candidate with existing drugs, diseases, or clinical phenotypes. Compared to de novo drug discovery being time-consuming and costly, drug repurposing methods are considered as a more effective drug discovery strategy, which could shorten the time and reduce the cost. One idea of drug repurposing is that one drug currently working for one disease may also work for other diseases if these diseases share some similar protein targets.901,902 Thus, integrated disease–human–drug interactions could form a network with nodes such as drugs, diseases, and proteins, with weighted edges referring to interactions between them, e.g., the number of drugs with a certain target. Novel drug usage can be discovered based on shared treatment profiles from disease connections, and the weight between two disease connections determines the possibility of repurposing drugs.901 Common pathways between different viruses or diseases are already identified on a large scale.903 Meanwhile, another way to define drug repurposing is based on the structural similarities of two drugs: two drugs may work on the same therapeutic target if the two drugs have similar structures. Causal network models of SARS-CoV-2 expression and aging have been applied to drug repurposing900 (see Figure 17). Synthetic analysis of drug repurposing is carried out by network analysis, which provides the relationships within biological data sets such as protein–protein interaction networks and genomic and/or phenotypic data sets. These biological networks integrate many different data types from different resources such as experiments or in silico methods. Computational drug repurposing can be concluded as four parts: network constructions, computational analyses, validations, and applications, where the network constructions and computational analyses are illustrated as follows.
Figure 17.

Illustration of the drug repurposing. Reproduced with permission from ref (900). Copyright 2021 Belyaeva et al. under Creative Commons Attribution 4.0 International License https://creativecommons.org/licenses/by/4.0/. (a) Hypothesis of the relation between SARS-CoV-2 and aging of individuals. Ciliated cells are in blue, stromal/fibroblast cells are in orange, and SARS-CoV-2 viral cells are in red. (b) RNA-seq/GTEx, protein–protein interactions network, drug–target data, and CMap are integrated as a data set. (c and d) Pipeline of the drug discovery/repurposing platform. First is mining relevant drugs by using an autoencoder with blue and orange points in the latent space representing data from the drug screen and the SARS-CoV-2 infection studies. Second is identifying the disease interactome within the protein–protein interaction network by implementing Steiner tree analysis. Last is investigating the drug mechanism from the first step (green diamond).900
2.4.3.1. Network Constructions
Biological networks have a variety of types or formats such as protein–protein interaction networks, knowledge graphs, or transcriptomic databases. Each of them is a large repository of medical information constructed via dry and wet laboratories.
Protein–Protein Interaction (PPI) Networks. To build the protein–protein interactome, the detection methods are summarized into three types: in vitro, in vivo, and in silico methods. The in vitro methods are performed outside a living organism. There are tremendous methods for PPI networks, such as affinity purification–mass spectrometry (AP-MS) methods, coimmunoprecipitation, affinity chromatography, protein arrays, protein fragment complementation, NMR spectroscopy, viral protein pull-down assay, and X-ray crystallography. For example, the AP-MS experiment is one of the most popular methods to build PPI networks starting to select interesting proteins, called baits, for the coassociating “prey” proteins to build PPI networks.904,905 Then, a 2D bait–prey matrix is generated for analyzing and drug repurposing. Published sample data sets are available such as the host and HIV proteins906 and yeast protein–protein interaction network.907 Gordon et al.75 implemented AP-MS to identify 332 high-confidence protein–protein interactions between SARS-CoV-2 and human proteins based on SARS-CoV-2 proteins in human cells. The in vivo methods are done in a living organism, such as yeast two-hybrid (Y2H) and synthetic lethality, where the Y2H method studies a bait against a random library of potential prey proteins and synthetic lethality studies functional interactions. In the work of drug repurposing by Zhou et al.,908 methods such as Y2H and AP-MS are implemented to build the virus–host and protein–protein interactome. Popular methods are sequence- or structure-based analysis, gene fusion, phylogenetic tree, gene expression analysis, and in silico 2 hybrid.
Knowledge Graphs. A knowledge graph is a large repository including the syntheses of small organic molecules, retrosynthetic steps, pathophysiology, and other biological information from the scientific literature collected via traditional computer-aided search methods or machine learning methods. Several knowledge graphs regarding COVID-19 have been built, including the CovidGraph (https://covidgraph.org/) and the Blender Lab COVID-KG (http://blender.cs.illinois.edu/covid19/).909 Many knowledge graphs were created by extracting entities and their associations from scientific publications. For example, Domingo-Fernández et al.910 retrieved scientific literature related to COVID-19 and manually encode information in the format of biological expression language (BEL). Knowledge graphs can also be constructed via other methods. For example, Monte Carlo tree search was applied and combined with neural networks to guide chemical synthesis.911 Later, this knowledge graph was applied to search approved drugs for COVID-19, resulting in Baricitinib as a potential treatment.80
Genome and Phenome. Genomics gives large volumes of biological data such as disease samples, animal models, cell lines, tissue samples, etc., while the phenome is the collection of phenotypic information.912 Genomic and genetic profiles have been studied for drug repurposing such as the connectivity map (CMap),913 drug versus disease (DvD),914 the database for annotation, visualization, and integrated discovery (DAVID),915 etc. Meanwhile, transcriptome (noncoding RNAs) data sets are recently developed for drug repurposing.916 Data sets of microRNAs are used as predictive biomarkers and therapeutic targets in prostate cancer.917 At the early stage of the COVID-19 pandemic, a high-resolution map of the SARS-CoV-2 transcriptome and epitranscriptome was constructed, which gave a better understanding of the life cycle and pathogenicity of SARS-CoV-2.23 Based on transcriptome data, differentially expressed genes (DEGs) were screened out between SARS-CoV-2-infected cells and the control group and analyzed the changes of the relevant molecular pathway, and it turned out that 136 of 145 genes were upregulated and 9 of 145 genes were downregulated.918 Other transcriptome data sets are applied to study the SARS-CoV-2 infection.737,919−923
2.4.3.2. Computational Analyses
Many computational methods regarding networks have been applied. In this section we briefly describe the most popular ones.
Mass Spectrometry Interaction Statistics (MiST). MiST906 was invented to identify protein–protein interactions that have biological significance from AP-MS data. Roughly speaking, a MiST score is a weighted sum of three features (abundance, reproducibility, and specificity). There are multiple ways to determine the weights, and in ref (906) the weights are determined by performing PCA. MiST was applied to identify high-confidence PPI between SARS-CoV-2 proteins and human proteins by Gordon et al.75 They identified 332 high-confidence PPIs and revealed how SARS-CoV-2 interacts with human immune pathways and host translation machinery. They also performed a cheminformatics search for drugs that modulate human proteins and disrupt the SARS-CoV-2 interactome. Similar applications of MiST can also be found in the literature924 (see Figure 18).
Figure 18.

Illustration of the discovery of PPI inhibitors for SARS-CoV-2. Reproduced with permission from ref (924). Copyright 2021 Elsevier.
Centrality. Different notions of centrality have already been described in section 2.2.1. To analyze the network of the differentially expressed genes (DEGs), degree centrality, closeness centrality, and betweenness centrality are computed for each node in ref (925). Fagone et al.925 studied the high infectivity of SARS-CoV-2 and observed gender differences from the clinical data. They identified a gene signature that characterizes SARS-CoV-2 infection and compared it with the gene signature induced by SARS-CoV infection. To predict drugs, they carried out an antisignature perturbation analysis based on the DEGs identified for SARS-CoV-2. Network analysis via closeness centrality and betweenness centrality is applied to data analysis.925 Maroli et al. implemented three measures, closeness centrality, betweenness centrality, and eigenvector centrality for the network analysis and examined potential drugs.603 Sheik Amamuddy et al. targeted Mpro and used the betweenness centrality to analyze Mpro,204 while Ghorbani et al. targeted S protein.165 Similar notions of centrality also appear in the literature.926,927
Proximity. Supposing C is a set of host genes associated with a HCoV and T is a set of drug targets, one version of network proximity of C with T can be calculated by
where d(c, t) is the shortest distance between c and t in the human protein interactome.908 Zhou et al.908 retrieved HCoV-associated host protein from scientific publications and calculated network proximity between drug targets and HCoV-associated proteins to search for drug candidates. They also sought possible drug combinations, guided by the principle that each drug in a drug combination should target separate neighborhoods in the human interactome network. Notions of network proximity are also used in papers such as ref (928).
Random Walk with Restart (RWR). Given a network, simulating a particle walking from a node to a randomly chosen nearby node can reveal the topology of the network. If one repeatedly starts a random walk from the same node or a set of nodes called seed(s), one can obtain nodes that are proximal to the seed(s). In the study of PPI networks, RWR is a method for identifying the most likely interactions. RWR is used by Messina et al.929 to better understand the SARS-CoV-2 pathogenesis. In their paper, since the SARS-CoV-2 genome is similar to the SARS-CoV genome, they assumed that the SARS-CoV-2 interactome and the SARS-CoV interactome share several interactions. They extracted virus–host interactomes of SARS-CoV, MERS-CoV, and HCoV-229E from public databases and scientific publications, assembling a large PPI network. S-glycoproteins of SARS-CoV, MERS-CoV, and HCoV-229E were taken as seeds for RWR, discovering 200 closest proteins to S-glycoproteins. With a similar assumption, an unbalanced birandom walk with Laplacian regularized least-squares was implemented on a virus–drug association network930 called VDA-RWLRLS. Compared to other state-of-the-art prediction models, their methods showed better VDA prediction performance.
Tremendous network-based applications were performed on SARS-CoV-2. Many of them provide an essential PPI network dataset between SARS-CoV-2 and humans,75 an architecture of SARS-CoV-2 transcriptome,23 or effective candidates.75,80,908 Gordon et al.75 screened drugs targeting the human proteins in the SARS-CoV-2 human interactome based on the PPI network data set and considering the features of drugs such as drug status, drug selectivity, drug availability, and the statistical calculations of the protein interactions. They identified 29 drugs already approved by the United States Department of Agriculture (USDA), 12 investigational new drugs, and 28 preclinical compounds. Zhou et al.908 studied the antiviral drug repurposing methodology targeting SARS-CoV-2. A systematic pharmacology-based network medicine platform was implemented to identify the interplay between the virus–host interactome and drug targets where they investigated the network proximity of the SARS-CoV-2 host and drug target interaction. Based on that, they reported three potential drug combinations. In the study by Sadegh et al.,931 CoVex was developed for SARS-CoV-2 host interactome exploration and drug (target) identification, which also explored the virus–host interactome and potential drug target. The network was constructed based on PPIs, drug–protein–protein interactions, etc. for repurposing drug candidates. Additionally, Srinivasan et al.932 developed a network of the comprehensive structural gene and interactome of SARS-CoV-2. Messina et al.929 investigated the host–pathogen interaction model through the PPI network. Das et al. explored the host protein for SARS-CoV-2 by observing central protein associations in the PPI network.926 Analyses were done via PPI networks between viral and host protein for host biological responses.933,934 Kumar et al. studied the SARS-CoV-2 pathogenesis through the network.927 Bellucci et al. studied the meta-interactome of SARS-CoV-2 via the network analysis.935 Drug repurposing of SARS-CoV-2 is one of the hottest topics in the applications of the network of SARS-CoV-2. In addition to the aforementioned works, other similar works were done for identifying drug repurposing of SARS-CoV-2.310,928,936−940
2.4.4. Quantitative Structure–Activity Relationship (QSAR) Models
QSAR models refer to regression or classification models to predict the physicochemical, biological, and environmental properties of compounds from the knowledge of their chemical structures.941 In QSAR modeling, the predictors consist of physicochemical properties and theoretical molecular descriptors of chemicals. The QSAR response variable can be the biological activity of the chemicals. There are two steps to build a QSAR model. First, the relationship between chemical structures and biological activity is summarized from a data set of chemicals. Second, QSAR models are implemented to predict the activities of new chemicals.942 After features are extracted from a data set, the learning method of QSAR can be the ML methods mentioned in section 2.3, such as linear regression, logistic regression, support vector machine, and random forest.
To identify potential Mpro inhibitors, Ghaleb et al.261 and Acharya et al.943 applied 3D QSAR models where Ghaleb et al.’s 3D model was based on comparative molecular similarity indices analysis (CoMSIA) and Acharya et al.’s 3D model was based on pharmacophores. More works used 2D QSAR models, such as Alves et al.944 using the random forest algorithm to build the model. Kumar et al.263 and Masand et al.945,946 used genetic algorithms. SVM models were applied to a classification-based QSAR model for structural and physicochemical interpretation analysis to identify potential Mpro inhibitors.947 Ghosh et al.947 built a Monte Carlo-based classification model involving classification QSAR-based data mining. Under the framework of QSAR,942 to identify potential SARS-CoV-2 main protease inhibitors, other works adopted multiple linear regression with QSAR models,205,719,948−954 where Borquaye et al.955 used multiple linear regressions. Moreover, QSAR models that predict inhibitors to other SARS-CoV-2 proteins were also constructed. Targeting S protein, Khaldan et al.956 built a 3D-QSAR model. Against PLpro, QSAR models based on Monte Carlo classification and multiple linear regression were constructed by Amin et al.201,957 Blocking both Mpro and RdRp, Ahmed et al.573 built a QSAR model following partial-least-squares regression. The QSAR model of Ahmed et al.573 was based on a partial-least-squares regression.
2.4.5. Pharmacophore Models
A pharmacophore is an abstract description of molecular features that are necessary for the molecular recognition of a ligand by a biological macromolecule. A pharmacophore model represents the binding patterns of bioactive molecules with the target binding site, by a distinct 3D arrangement of abstract interaction features accounting for different types of noncovalent interactions.958 Thus, a pharmacophore model explains the process of structurally diverse ligands binding to a common receptor site and the identification of ligands binding to the same receptor.
In the work of searching COVID-19 therapeutics, most pharmacophore models focused on Mpro.390,415,461,879,959−962 Pharmacophore models were used to screen inhibitor compounds to SARS-CoV-2 from FDA-approved drugs,461,959 DrugBank,879 or HIV inhibitors.960,961 A fragment-based pharmacophore model was built from 22 noncovalent fragments cocrystallized with Mpro.963 More pharmacophore models were created for Mpro,964−967 PLpro,968 and nsp16.551
2.5. Miscellaneous
2.5.1. Molecular Modeling of Peptides, Proteins, or Graphene Binding to SARS-CoV-2 Targets
According to the large RBD of S protein, small-molecule drugs may not efficiently block the entire RBD. The entire RBD of S protein needs to be blocked by peptides.969 In a study of 1070 peptide-based drugs docking to S protein, one high binding affinity was Sar9Met (O2) 11-Substance P.970 Basit et al.971 also designed a truncated version of the ACE2 receptor covering the binding residues. They performed protein–protein docking and MD simulations to analyze its binding affinity to RBD and complex stability. One study predicted the affinities of the peptide analogues Seq12, Seq12m, and Seq13m to S protein through molecular docking, MD simulation, and MM-PB/GBSA calculations.745 Other docking and MD studies of peptides to S protein include refs (876 and 972−981). In addition, potency of peptides to other targets was also anticipated. The 37 peptides from the antimicrobial peptide database were docked to N protein, and the peptides with the highest docking scores were further studied by MD simulations.982 Porto et al.983 exhaustively performed docking of over 70000 peptides to Mpro. Yathisha et al.’s MM/GBSA studies suggested three angiotensin-I converting enzyme (ACE-I) inhibitory peptides are potent to Mpro.984 Zhao et al.’s docking studies identified GSRY among lactoferrin-derived peptides is potent to Mpro.985 Behzadipour et al. evaluated the SARS-CoV-2 Mpro inhibitory activity of 326 di- and tripeptides from the proteolysis of bovine milk proteins by docking.986 Some works974,987,988 aimed to repurpose peptides from seed proteins to targets Mpro, S protein, and PLpro. Some molecular modeling studies of the protein–protein binding were also contributed to treating SARS-CoV-2. Shaheer et al.989 employed protein–protein docking and MD simulations to design degraders of SARS-CoV-2. They first docked Mpro to E3 ligase and predicted the possible complementarity between them. Then, they generated the ternary complexes of Mpro, E3 ligase, and possible linkers and ran MD simulations on these complexes. Recently, Wang et al. studied graphene interacting with SARS-CoV-2 Mpro via MD simulations. They showed that Mpro can be absorbed onto the surfaces of investigated materials and defective graphene and graphene oxide interact with Mpro more intensely.990 Similar works were also performed in the literature.991,992 Through MM/GBSA calculations, Mehranfar et al. also suggested phosphorene can be a good candidate for designing new nanomaterials for selective detection of SARS-CoV-2.993
2.5.2. Molecular Modeling Studies on Vaccines
Inspired by the composition of mRNA-based COVID-19 vaccines as a lipid mixture, Paloncýová et al.994 first studied the behavior of the lipids and their mixtures in preassembled bilayers and then modeled the self-assembly of IL-containing (interleukin-containing) lipid nanoparticles (LNPs) for mRNA delivery by MD simulations with and without the presence of an RNA fragment. Additionally, they investigated the effect of the IL’s charge (i.e., the pH) on the stability of the lipid phase. In other MD simulation studies, the effect of glycan microheterogeneity was evaluated, which could impact the epitope exposure of the S protein.995 This study indicated that glycans shield approximately 40% of the underlying protein surface of the S protein from epitope exposure. Another two studies898,996 generated tens of epitode vaccine candidates originating from S protein by the online servers NetCTL, IEDB, and FNepitope. The tertiary structures of these epitodes were predicted and docked to the toll-like receptor 3, where MD simulations were run for these complexes and the immune reactions also were simulated. Sikora et al.162 performed microsecond-scale MD simulations of a 4.1 million atom system containing a patch of viral membrane with four full-length, fully glycosylated and palmitoylated S proteins. By mapping steric accessibility, structural rigidity, sequence conservation, and generic antibody binding signatures, they recovered known epitopes on S protein and predicted promising epitope candidates for vaccine design. They also discovered that the flexible glycan coat shielded a surface area larger than expected from static structures. Through docking and MD simulations, De Moura et al.997 identified epitopes from the S protein that were able to elicit an immune response mediated by the most frequent MHC-I alleles in the Brazilian population. Both epitodes from S protein and epitodes from other targets were investigated. Rahman et al.998 also investigated some epitodes from the S protein, Mpro, and E proteins. Ezaj et al.,999 Chauhan et al.,1000 Kalita et al.,1001 Waqas et al.,1002 Ranga et al.,1003 and Sarkar et al.1004 designed and simulated epitodes from other proteins of SARS-CoV-2. Other similar reports can be found in the literature.899,1005−1008
2.5.3. Molecular Modeling of Blocking Host Targets with Controversy
Some researchers investigated potential damages of inhibition aimed at host receptor proteins. Parts of molecular modeling studies aimed to block host proteins relating to SARS-CoV-2 attachment, such as ACE2,315,324,876,1009−1029 TMPRSS2,479,892,1019,1030−1032 furin,1015,1031 and glucose regulated protein 78 (GRP78).1033 However, according to some reports, it is controversial to design SARS-CoV-2 inhibitors targeting host ACE2s or related proteins. For instance, ACE2 is an important enzyme attached to cell membranes in the lungs, arteries, heart, kidney, and intestines. It is critical for lowering blood pressure in a human body.1034 It is unclear whether drugs that can inhibit ACE2 or related targets are more beneficial than harmful. Further investigation is needed.1035
2.5.4. Combination of Docking and MD Simulation
Many works combine docking and MD simulation. For example, molecule docking predicts binding poses, and MD simulation further optimize and stabilize the confirmations of complexes. Other researchers follow an ensemble docking procedure to dock compounds to multiple conformations of the protein extracted from MD simulations.
Ensemble Docking. An ensemble docking of the SARS-CoV-2 Mpro was performed by Sztain et al.146 They docked almost 72000 compounds to over 80 conformations of the main protease generated from MD simulations and screened these compounds through the ensemble docking strategy. To obtain extensive conformational samplings of Mpro, a Gaussian accelerated MD simulation1036 was run. Another ensemble docking work of Mpro was implemented by Koulgi.1037 They carried out long-time MD simulations on the apo form of Mpro. Sixteen representative conformations were collected from these MD simulations by clustering analysis and Markov state modeling analysis.1038 Targeting these 16 conformations, ensemble docking was performed on some FDA-approved drugs and other drug leads, suggesting some potent candidates such as Tobramycin. Additionally, Novak et al.391 first screened 8756 approved or experimental drugs by regular docking; then the best 10 drugs from regular docking were further evaluated by an ensemble docking strategy. The 10 drugs were docked to 5 conformation representatives of Mpro obtained from MD simulations and cluster analysis. Other ensemble docking studies were implemented on the targets Mpro1039−1043 and RdRp.1044 Other investigations focused on docking and optimizing by MD simulations.
Mpro. Conivaptan and azelastine were suggested as potential repurposed drugs after applied docking to systematically predict the binding affinities of 1615 FDA-approved drugs to Mpro.1045 Then, MD simulations were performed on these two drugs for revealing their interactions with Mpro. In the docking studies of 18 derivatives of hydroxychloroquine (HCQ), remdesivir, and tetrahydrocannabinol, two derivatives give excellent docking scores and higher stability than the parent molecules. Their strong inhibition toward Mpro was validated by MD simulations.1046 With the same strategy, Cardoso et al.1047 in silico repurposed 10 different HIV protease inhibitors to Mpro, and the binding free energy surfaces of the best 3 drugs to Mpro were depicted by long-time MD simulations. Other existing drugs, such as lopinavir, oseltamivir, ritonavir, atazanavir, darunavir, plitidepsin, testosterone, progesterone, hydroxychloroquine, tetracyclines, flaviolin, hydroxyethylamine analogs, buriti oil (mauritia flexuosa L.) like inhibitors, etc., were also investigated specifically by docking and MD simulations.397,614,1048−1073
Another important inhibitor source is natural products. Jairajpuri et al.1074 performed the most extensive virtual screening of natural products. They screened about 90000 compounds by ADMET, drug-like, and docking score predictions. The best one, ZINC02123811, was further studied by MD simulations. Following the workflow of ligand docking, MD optimization, and rescoring, the library including 14064 marine natural products was screened.961 The best one, heptafuhalol A, was predicted to have a docking score as high as −18.0 kcal/mol. Ul Qamar et al.1075 used docking and MD simulation to screen a medicinal plant library containing 32,297 potential antiviral phytochemicals/traditional Chinese medicinal compounds. Potent inhibitors such as 5,7,3′,4′-tetrahydroxy-2′-(3,3-dimethylallyl)isoflavone with a docking score of −16.35 kcal/mol were predicted. Virtual screening was also performed toward other natural products such as marine products, Indian medicinal herbs, and plant products.238,612,613,620,621,719,1048,1075−1090
Other small molecules were also screened to inhibit the SARS-CoV-2 Mpro. Ton et al.766 identified potential Mpro inhibitors by docking 1.3 billion compounds and suggested that compound ZINC000541677852 had the highest binding affinity of −11.32 kcal/mol. Its interactions with Mpro were studied by MD simulations. The docking and MD simulations tested 4384 molecules from the Zinc data set,1091 and some of them are FDA-approved drugs. Among them, the best one was bisoctrizole.833 Additionally, some diaminobenzophenone derivatives, prototypical-ketoamide inhibitors, HIV protease inhibitors, leucoefdin, nutraceuticals, and others were also studied.619,625,636,1047,1092−1107 Notably, Mohammad et al.1108 optimized some complexes of Mpro with ligands in the protein data bank (PDB) and rescored them by AutoDock Vina, with the best PDB structure being 6M2N.
S Protein. By docking and MD simulations to identify potential inhibitors against SARS-CoV-2 S protein from 1582 FDA-approved drugs, lumacaftor was predicted to have the highest binding affinity.1109 Moreover, to evaluate the binding interactions between S protein and compounds, steered MD simulations1110 were performed on the top compounds. Bharath et al.1111 and Choudhary et al.851 screened 4015 and 1280 small compounds, respectively, and predicted that phytic acid and GR 127935 hydrochloride hydrate had the highest energies. Shoemark et al.1112 applied docking and MD simulations to bind some vitamins, retinoids, and steroids as to the free fatty acid pocket of S protein. Gupta et al. studied the interactions between some functional spike-derived peptides and S protein by docking and MD simulations.1113 Heparin has been found to have antiviral activity against SARS-CoV-2. Paiardi et al. investigated the binding of heparin to the SARS-CoV-2 spike glycoprotein by docking and MD simulations. Their results suggest that heparin can inhibit SARS-CoV-2 infection by three mechanisms: by allosterically hindering binding to the host cell receptor, by directly competing with binding to host heparan sulfate proteoglycan coreceptors, and by preventing S cleavage by furin.1114 Other works evaluating potential inhibitors against S protein include refs (630, 634, 975, and 1115−1123).
RdRp. RdRp is another important target of SARS-CoV-2. Nagar et al.1124 performed ADMET and docking-based screening on 267324 compounds, and the best two of them were subjected to MD-based further study. Another extensive screening based on ADMET, docking, and MD simulations was implemented by Ghazwani et al., which covers 13840 compounds.1125 Singh et al.,1126 Mondal et al.,301 Begum et al.,1127 and Pokhrel et al.1128 evaluated the potency of thousands of existing drugs via docking and MD simulations. Tens of fungal secondary metabolites were docked to RdRp by Ebrahimi et al., and MD simulations were performed on the top five compounds.1129 Elghoneimy’s docking and MD simulations1130 aimed to repurpose some HCV NS5B palm subdomain binders to inhibit SARS-CoV-2 RdRp.
PLpro. Targeting PLpro, in a study aiming to repurpose all current FDA-approved drugs, the best drug, ergotamine, among their predictions was reevaluated by MD simulations.1131 Baildya et al.1132 predicted the potency of 19 compounds from Neem, and MD simulation was performed on the best one, desacetylgedunin. Other similar studies can be found in refs (537 and 1133).
Other Targets. Dihydroergotamine and irinotecan were predicted to be the best drugs of the study of 3000 FDA-approved and experimental drugs against 2′-O-methyltransferase in nsp16 of SARS-CoV-2.1134 De Lima Menezes et al.1135 screened 8694 approved and experimental drugs from DrugBank against nsp1 and predicted that tirilazad was the most potent one. Tazikeh-Lemeski et al.1136 docked 1516 FDA-approved drugs from DrugBank to nsp16 and investigated the drug–protein interactions by MD simulation, finding that raltegravir had the best predicted binding affinity. Following a similar scheme, a collection of 22122 Chinese traditional medicines from TCM Database@Taiwan against nsp14 was screened,884 with the best one being TCM57025. Liang et al.1137 predicted the affinities of 300 potential molecule inhibitors to the nsp10–nsp16 methyltransferase complex via docking and MD simulations. Sixto-López et al.1138 and Kumar et al.1139 predicted the potency of more than 2000 ligands to nsp15. Tatar et al.1140 aimed to repurpose 34 antiviral compounds to inhibit N protein.1140 Following the workflow of ADMET screening, docking, optimizing of the top predictions by MD simulations, and redocking, Rampogu et al.1141 screened potential nsp16 inhibitors from 59619 natural compounds.
Multiple Targets. In many reports, the same drugs were tested against multiple targets. In the work of El-Demerdash et al.,1142 the potentials of 15 marine polycyclic guanidine alkaloids to block Mpro, S protein, N protein, M protein, and nsp15 were evaluated by docking and MD simulations. Many works cover three or two targets. Dwarka et al.1143 docked 14 South African medicinal plants to Mpro, RdRp, and S protein RBD. They also investigated the dynamics and interactions inside the complexes using MD simulations. However, no potent inhibitors were found. Bhowmik et al.1144 virtually screened more than 200 antiviral natural compounds and 348 antiviral drugs targeting the E, M, and N proteins responsible for envelope formation and virion assembly. Molavi et al.1145 repurposed 1760 FDA-approved drugs to Mpro and RdRp, and Rajpoot et al.1146 repurposed 291 drugs to Mpro and PLpro. More similar works include refs (599, 609, 622, 624, 628, 632, 637, 838, 875, 889, and 1147−1173).
2.5.5. Accuracy Tests of Molecular Modeling Methods on SARS-CoV-2 Targets
To assess the predictive power of molecular modeling methods on SARS-CoV-2 targets, accuracy tests were performed on some SARS-CoV-2 inhibitors with known experimental binding affinities. Ngo et al.1174 systematically evaluated four popular binding-affinity calculation approaches, namely, FEP, steered MD, LIE, and MM/PBSA. They tested 20 Mpro inhibitors with available affinity values and found FEP was the most accurate with a correlation of 0.85, while the correlations of steered MD, LIE, and MM/PBSA are 0.74, 0.73, and 0.32, respectively. A test of the affinity prediction of 19 Mpro inhibitors revealed docking can achieve a correlation of 0.50 and steered MD can raise the correlation to 0.65.1175 Another test of the docking poses predictions from several leading docking programs, namely, Glide, DOCK, AutoDock, AutoDock Vina, FRED, and EnzyDock.1176 In the scope of 193 experimental Mpro–inhibitor complexes, the best performance is that 26% of noncovalently bound ligands (from glide) and 45% of covalently bound ligands (from EnzyDock) can reach RMSAs < 2 Å. According to their tests, the work suggested good poses may be provided but the affinity predicted for each pose may not be reliable.1177 Fan et al.1178 added some structural restraints deduced from experiments that could improve the accuracy of docking predictions.
2.5.6. Combined MD Simulation and Deep Learning
Gupta1179 first selected 92 potential Mpro inhibitors from FDA-approved drugs by docking and then further evaluated their potency by MD simulations and MM/PBSA calculations using the hybrid of the Accurate NeurAl networK engINe for molecular energies (ANAKIN-ME) deep learning force field1180 and a conventional molecular mechanics force field. Their results suggested that targeting was the most potent drug against the main protease. A similar work integrated a deep neural network model with docking and MD simulations.1181 Moreover, AI-driven multiscale simulations provided analyses of the spike’s full glycan shield elucidation, the role of spike glycans in the viral infection, the interactions between the S and the human ACE2, etc.1182
2.5.7. Combined MD Simulation and Experiment
The MD simulations revealed three flexible hinges within the stalk, coined hip, knee, and ankle of the S protein, which were consistent with tomographic experiments.1183 In the implementation of virtual screening of 6218 drugs targeting Mpro and RdRp,1147 the best ones and some of their combinations were verified by cell-based assays. Combining experiments and calculation, Zaidman et al.1184 developed a fragment-based and automatic pipeline to design potent Mpro inhibitors. Dwivedi et al.’s docking and MD simulation confirmed their experimental results that holothurian sulfated glycans show potential effects against SARS-CoV-2 S protein RBD.1185 Similar works include refs (1093, 1153, 1154, and 1186−1213). Other studies applied experiments to identify potent compounds and used docking to reveal interactions.1214−1218
2.5.8. Combined MD Simulation and Data Analysis
Some of the popular methods to analyze dynamics characteristics in MD simulation are principal component analysis (PCA) and normal-mode analysis, which can extract the principal modes of motion from MD simulations.1219,1220 Toward SARS-CoV-2, some works applied PCA to reveal the internal motions of Mpro.407,421,481,719,1061,1074 Rane et al.506 and Dehury et al.1221 used such analysis to investigate the dynamics of the S protein. Bera et al.,569 Henderson et al.,167 and Chandra et al.545 used PCA to elucidate the motions of the PLpro and NendoU, respectively. Bhattacharya et al.1005 applied NMA to display the mobility of the human TLR4/5 protein and SARS-CoV-2 vaccine component complex. Shaikh et al.1222 used elastic network models to identify the residue cross-correlation matrix.
2.5.9. Combinations of Multiple Molecular Modeling Methods
In a comprehensive study in predicting potent Mpro inhibitors, it first screened their compounds by molecular docking, and then the top four compounds from the docking prediction were more accurately calculated by MM/PBSA simulation.1223 In the third step, the binding of the top two compounds to Mpro was further studied by QM/MM simulations and the chemical properties of these two compounds, such as highest the occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO), were predicted by DFT calculations. These studies also involve multiple molecular modeling methods.1224−1226
2.5.10. Molecular Modeling of Mutational Impacts
Through docking and MD simulations, the drug candidate PF-0083523, under clinical trial now, was predicted, which was potent to four reported Mpro mutants.1227 Muhammad et al.’s1228 virtual screening suggested that some potent phytochemical inhibitors were effective to two Asian mutants of Mpro. Sheik Amamuddy et al.204 studied the impact of Mpro mutations on its 3D conformation as well. More studies focused on the mutations of S protein. Some works applied docking, MD simulations, MM/PBSA, or MM/GBSA calculations to investigate S protein mutation impacts on the interactions and affinities between S protein RBD and ACE2 protein.1229−1234 Additionally, via MD simulations, it was found that mutations on the distal polybasic cleavage sites of S protein could weaken the binding between S protein and human ACE2, which means these distal polybasic cleavage sites were critical to the binding.1235 Virtual alanine scanning mutagenesis by FEP MD simulations was also performed to uncover key S protein residues in its ACE2 binding.1236 Dehury et al.1221 compared the interactions of mutated S proteins to the wild type of S protein binding to ACE2. Calcagnile et al.274 and Hadi-Alijanvand et al.1237 assessed impacts of ACE2 polymorphisms to their affinities to S protein by docking and MM/GBSA calculations. Furthermore, the impact of S protein mutations on inhibitor and antibody efficacy was also simulated. For example, the docking and MD simulations indicated the Alpha variant did not compromise the efficacy of catechins.1238 However, mutations H49Y, D614G, and T573I were suggested to considerably affect the binding of cepharanthine, nelfinavir, and hydroxychloroquine into their respective binding sites.1239 Wu et al.’s MM/GBSA calculation suggested that most antibodies (about 85%) have weaker binding affinities to the E484 K mutated S protein than to the WT, indicating the high risk of immune evasion of the mutated virus from most current antibodies.1240 Lastly, Hossain et al.1241 performed docking to investigate the impacts of the mutations on nsp1, nsp3, and PLpro. Wu et al.1242 systematically predicted mutation impacts to the conformations of multiple SARS-CoV-2 proteins and the interactions between them.
2.5.11. Protein Pocket Detection
The detection and characterization of protein pockets and cavities are critical issues in molecular biology studies. Pocket detection algorithms can be classified as grid-based and grid-free approaches.1243,1244 Grid-based approaches embed proteins in 3D grids and then search for grid points that satisfy some conditions. Grid-free ones include methods based on the probe (sphere) or the concepts of Voronoi diagrams.
The plug-in Pockets 2.0 combined the pocket-detecting algorithms Fpocket1243 and PLANTS1245 to characterize the SARS-CoV-2 druggable binding pockets.1246 Zimmerman et al.1247 launched large-scale Folding@home simulations from their FAST-pockets adaptive sampling to aid in the discovery of cryptic pockets on various SARS-CoV-2 proteins. Another server, CavityPlus (www.pkumdl.cn/cavityplus), was implemented to search druggable cavities.290 Vithani et al.1248 adopted FAST sampling algorithms to quickly explore cryptic pockets on nsp16 and the nsp10/nsp16 complex. Manfredonia et al.1249 predicted the 3D structure of SARS-CoV-2 RNA by coarse-grained modeling and detected potential pockets. Sheik Amamuddy et al.204 predicted potential binding pockets of Mpro.
2.5.12. AlphaFold
Chains of amino acids spontaneously fold to form biologically active proteins with their native 3D structures. However, it is challenging to understand how amino acid sequences determine the 3D structures of the proteins, and such a topic is called the “protein folding problem”. Protein structures can be identified through multiple techniques such as cryo-electron microscopy, X-ray crystallography, and NMR. However, such techniques are expensive and time-consuming, and only 0.085% (170 K out of 200M) of the 3D structures of proteins have been determined over the past 60 years.784 Therefore, seeking a more efficient and accurate way to predict the 3D structure of proteins is crucial. Starting from 1994, a worldwide experiment for protein structure prediction called CASP has taken place every two years. Researchers worldwide have put efforts into this experiment to help achieve high correlations between the experimental structures and the 3D structures predicted by theoretical-based methods. However, such predicted structures had accuracy without experimental precision, which limited their utility for many biological applications.
Developed by Google’s DeepMind in 2018, an AI-based program called AlphaFold that was designed to predict protein structures achieved high accuracy competitive with experiments in CASP13 assessment.799 As a result, AlphaFold was ranked first among 98 teams. Specifically, AlphaFold made the best prediction for 25 out of 43 targets. It was the first time that AlphaFold drew the attention of scientists worldwide. In 2020, AlphaFold2, a new version of the AI-based model, entered into the CASP14 assessment.799 The predicted protein structures by AlphaFold2 were vastly more accurate than other competing methods. Overall, 88 out of 97 protein structures predicted by AlphaFold2 achieved the most precise structure for targets, which was a significant improvement compared to AlphaFold in 2018.
AlphaFold2 consists of two major modules: the evoformer module and the structure module. First, by applying the genetic database search and structure database search, the primary amino acid sequences are encoded to embed MSAs and pairwise features, which can be treated as the inputs of the evoformer module. The evoformer module consists of attention-based and nonattention-based transformers. Such a module produces a processed MSA array with shape Nseq × Nres and a processed residue pair feature with an array size of Nres × Nres, which are then fed into the structure module to get the explicit predicted 3D structures. Here, Nseq and Nres represent the number of sequences and the number of residues, respectively. Moreover, an iterative refinement called “recycling” is involved in AlphaFold2 to minimize the final loss and achieve higher accuracy as shown in Figure 19.
Figure 19.

AplhaFold2 architecture. Reproduced with permission from ref (799). Copyright 2021 Jumper et al. under Creative Commons Attribution 4.0 International License https://creativecommons.org/licenses/by/4.0/. Arrows show the information flow among the various components described in this paper. Array shapes are (s, r, c), where s shows the number of sequences (Nseq in the main text), r represents the number of residues (Nres in the main text), and c is the number of channels.
AlphaFold2 has been used to predict highly accurate SARS-CoV-2 protein structures since 2020. For instance, the AlphaFold-predicted structure of ORF8 was used as a search model, which provided a phase solution by molecular replacement (MR) by Flower et al.801 Pandey et al. applied AlphaFold to predict the secondary structure of nsp6, which showed 91.7% and 8.3% of residues are located in the most favored and additionally allowed regions, respectively.1250 Slavin et al. employed AlphaFold2 to generate a single consistent all-atom model of SARS-CoV-2 nsp2, which indicated three putative metal binding sites and further suggested that nsp2 may have a role in zinc regulation.1251 Furthermore, a team led by Ad Bax introduced three models to evaluate their concordance with residual dipolar couplings (RDCs) measured in 2021. They are (1) a standard AlphaFold2 model; (2) an implementation of AlphaFold2 that excluded all candidate template X-ray structures after Jan. 1, 2020; and (3) an implementation of the AlphaFold2 model that excluded structures homologous to coronaviral Mpro.800
3. Discussion
Since the outbreak of the COVID-19 epidemics in December 2019, enormous effort has been devoted to scientific research relating to SARS-CoV-2, leading to significant breakthroughs, such as the development of vaccines and experimental determination of protein structures. Vaccines, drugs, and antibody therapies are in emergency use authorization. Along with the rapid development of high-performance computers, biophysical methods, and AI algorithms in recent decades, plenty of theoretical and computational studies were carried out against SARS-CoV-2. Theoretical and computational studies are significant for combating urgent epidemics and pandemics since they are faster and cheaper.1252 This review strives to summarize the existing SARS-CoV-2 related theoretical and computational works and inspire future ones. SARS-CoV-2 protein structure predictions also play an important role, especially at the early stage of the epidemics when experimental structures were largely unavailable. At this point, besides traditional homology modeling, a more popular solution is the high-level deep-learning-based models such as AlphaFold39 and C–I-TASSER,785 both making use of deep CNNs.
3.1. Drug Repurposing
Many research efforts covered by this review are about repurposing current drugs or inhibitors to target SARS-CoV-2 because the drug development has been one of the most urgent issues in combating COVID-19. A variety of drug repurposing approaches has been applied, from molecular docking and MD simulation to network analysis and machine/deep learning, as summarized below. (1) The most straightforward approach is molecular docking, which provides both binding poses and corresponding scores. (2) In many studies, docking poses were further optimized by MD simulations, and these optimized poses were rescored by docking programs. (3) More accurate binding free energies can be achieved by MD-simulation-based or even QM-based calculations, such as MM/PB(GB)SA, free energy perturbation, metadynamics, QM/MM, and DFT. (4) Other than traditional molecular docking and MD simulations, the development of AI, machine/deep learning technologies opens a new approach to discover SARS-CoV-2 drugs, as well as network analysis. With existing drugs as training sets, machine/deep learning can predict the potency of a large number of potential SARS-CoV-2 inhibitors in a short time.753 3D models also provided binding poses.703 Moreover, AI has the potential to create new drugs to combat COVID-19.796,1253 For example, Bung et al.366 employed RNN-based networks, and Gao et al.793 used GRU-based generative networks to design new potential main protease inhibitors. (5) Network-based approaches were also performed in SARS-CoV-2 drug repurposing, promising solutions to identify drugs associated with certain diseases. These networks can be based on proteomic, transcriptomic, or metabolomic data. The basic idea is that one drug currently curing one disease may also work for other diseases if sharing some similar protein targets.75,80,913 Thus, integrated disease–human–drug interactions form a network connecting drugs, diseases, and targets. Novel drug usage can be discovered based on shared treatment profiles from disease connections. At the level of proteomic networks, Gordon et al.75 and Zhou et al.908 systematically explored the host dependencies of the SARS-CoV-2 virus to identify host proteins that are already targeted by existing drugs. Therapies that target the host–virus interface could potentially present durable, broad-spectrum treatments. At the level of transcriptomic networks, Belyaeva et al.900 used the transcriptomic data from the CMap database to search for compounds that may cause genomic changes opposite to the changes caused by SARS-CoV-2, so as to identify novel and potentially effective drugs with antiviral properties. Ahmed1254 adopted a transcriptomic network to reveal the mechanism of Vitamin D treating the cytokine storm caused by SARS-CoV-2. (6) Traditional QSAR approaches were implemented in many calculations for drug rediscovery.
3.2. Mutational Impacts on SARS-CoV-2 Infectivity
SARS-CoV-2 infectivity plays a paramount important role in SARS-CoV-2 transmission and COVID-19 prevention and control. The experimental methods to evaluate viral infectivity are expensive and time-consuming considering the rapid spread of COVID-19. For all SARS-CoV-2 variants around the world, it is even more challenging to experimentally measure infectivity. However, a computational platform based on theoretical analysis, which quantitatively predicts the BFE changes of the RBD-ACE2 complex induced by mutations on the S protein RBD, will deliver a consistent measurement of SARS-CoV-2 infectivity. A computational platform of RBD-ACE2 BFE changes induced by RBD mutations is given in ref (78). It integrates a series of well-established methods, including the genotyping of SARS-CoV-2 genetic sequences,12 protein sequence alignment,78 the biophysics of PPIs, the algebraic topology representation of proteins,219,663 the deep learning modeling of RBD-ACE2 BFE changes induced by mutations,695 and the training with existing experimental mutational data sets. According to the genotyping, the authors theoretically revealed that the SARS-CoV-2 diagnostic target mutations had false-negative test results.9 Experimentally, this finding is confirmed.1255 Similarly, the impacts of mutations on vaccines and antibody therapies can be decoded.12 In addition, by applying the large data set of SARS-CoV-2 spreading around the world, the top eight mutations in the United States are from two groups in which one group of five concurrent mutations is prevailing and another group has three concurrent mutations that faded out gradually.10 Additionally, many of the MD-based studies are devoted to the mutation-induced conformational changes of S protein and its corresponding binding affinity changes to the human ACE2.485−490
3.3. Mechanisms of SARS-CoV-2 Evolution
The understanding of the mechanism of SARS-CoV-2 evolution is a prerequisite for the prediction of emerging variants and essential for combating COVID-19. SARS-CoV-2 evolution is driven by competing mutations at molecular and organism scales. Molecular-scale mutations are mainly caused by a series of random errors in replications, transcriptions, and translations.1256 Unlike other RNA viruses such as the flu virus and HIV, SARS-CoV-2 has a genetic proofreading mechanism1257 and has more fidelity. Additionally, at the organism scale, host gene editing is found to be the dominating source for mutations.11 Moreover, viral genetic recombination may happen at the organism scale as well. In Figure 20a, it is shown that SARS-CoV-2 had over 28,912 unique mutations by November 2021, where each SARS-CoV-2 nucleotide had one known mutation on average. Nonetheless, most mutations have little to do with virus evolution, which is regulated by a population-scale mechanism. In summer 2020, Chen et al. hypothesized that “natural selection favors those mutations that enhance the viral transmission”.78 The authors further hypothesized that mutations that strengthen the binding of the RBD-ACE2 complex will enhance the SARS-CoV-2 infectivity and transmission.78 To investigate this natural selection mechanism of evolution from a theoretical perspective, the authors applied the single-nucleotide polymorphism calling of over 15,000 SARS-CoV-2 genomes from GISAID and identified 725 nondegenerated mutations on the SARS-CoV-2 S protein at the early stage of the pandemic (i.e., May 27, 2020). Among them, 89 RBD mutations are important to the binding of SARS-CoV-2 S protein and ACE2. Despite the highest frequency of 89 mutations being only about 50, the theoretical model resulted in Figure 20c, showing the first evidence of natural selection. There was a dramatic increase in the frequencies of a few infectivity-strengthening RBD mutations as the pandemic unfolded in the following months.79Figure 20d shows that all the 100 most observed RBD mutations have their predicted BFE changes above the average values of −0.28 kcal/mol.79 The chance for this to occur accidentally is one in 2100, which undoubtedly confirms the natural selection mechanism of SARS-CoV-2 evolution.
Figure 20.

(a) Illustration of SARS-CoV-2 mutations given by Mutation Tracker. The interactive version is available at the Web site: https://users.math.msu.edu/users/weig/SARS-CoV-2_Mutation_Tracker.html. (b) Illustration of the analysis of SARS-CoV-2 mutations given by interactive Mutation Analyzer (https://weilab.math.msu.edu/MutationAnalyzer/). (c) Reproduction of Figure 3 of ref (78). The time evolution of 89 SARS-CoV-2 S protein RBD mutations. The green lines represent the mutations that strengthen the infectivity of SARS-CoV-2, and the red lines represent the mutations that weaken the infectivity of SARS-CoV-2. Many mutations overlap their trajectories. Here, the collection date of each genome sequence deposited in GISAID was applied according to the information recorded in June 2020.78 (d) Reproduction of Figure 2 of ref (79). Illustration of SARS-CoV-2 mutation-induced BFE changes for the complexes of S protein and ACE2. Here, the 100 most observed mutations out of 651 mutations on S protein RBD and their frequencies are illustrated as recorded in April 2021.79 The highest frequency was 168,801, while the lowest frequency was 28. Therefore, the frequencies of the rest of the 551 mutations were lower than 28. (e) Reproduction of the right chart of Figure 11 of ref (764). Illustration of SARS-CoV-2 RBD mutation-induced binding free energy changes for the complexes of S protein and antibody LY-CoV555. Here, mutations L452R, V483F/A, E484K/Q, F486L, F490L/S, Q493K/R, and S494P could potentially disrupt the binding of antibodies and S protein RBD.
Currently, the prevailing SARS-CoV-2 variants are known as Alpha, Beta, Gamma, Delta, Epsilon, Theta, Kappa, Lambda, and Mu, involving RBD (co)mutations [N501Y], [K417N, E484K, N501Y], [K417T, E484K, N501Y], [L452R, T478K], [L452R], [E484K, N501Y], [L452R, E484Q], [L452Q, F490S], and [R346K, E484K, N501Y], respectively. More recently, the Omicron variant, starting in late November 2021, includes 15 RBD mutations [G339D, S371L, S373P, S375F, K417N, N440K, G446S, S477N, T478K, E484A, Q493R, G496S, Q498R, N501Y, Y505H]. Remarkably, two common RBD residues, 452 and 501, for these variants were predicted to “have high chances to mutate into significantly more infectious COVID-19 strains” in May 2020,78 many months before any variants were identified.
Meanwhile, efforts from researchers worldwide provide more genome sequence information of SARS-CoV-2 and experimental data on ACE2/antibodies binding to S protein. Some deep mutational data on RBD and ACE2 were available in late 2020,1258,1259 which can be used to improve the mathematical model in terms of accuracy.687 A computational workflow was presented to predict impacts induced by mutations on the binding between RBD and ACE2.1260 Computational mutagenesis work suggested mutations L455D/W, F456 K/W, Q493K, N501T, and Y505W on RBD enhance the ACE2/RBD binding.1261 By docking, Omicron and Delta variants were studied, whose mutations Q493R, N501Y, S371L, S373P, S375F, Q498R, and T478K contribute significantly to the high binding affinity with human ACE2.1262
3.4. Mutational Impacts on SARS-CoV-2 Antibodies and Vaccines
As the vaccination rate increasing and SARS-CoV-2 variants erupting, a more intriguing question is whether vaccination and antibody therapies protect us from the SARS-CoV-2 variants. More precisely, it is imperative to understand how these variants affect vaccines and antibody therapies. Therefore, a statistical analysis of a variety of antibodies can guide the efficacy analysis of vaccination in a population. As the collection of antibody–RBD complexes accumulates, a library of 130 antibody–RBD complex structures765 was used as a statistical ensemble to analyze the RBD mutation impacts on antibodies and vaccines. Additionally, the theoretical model can be applied to study these emerging variant impacts on mAbs, especially the mAbs obtained EUA from the FDA such as Regeneron and Eli Lilly antibodies.764Figure 20e illustrates the BFE changes induced by the 100 most observed RBD mutations of the complex binding of RBD and antibody LY-CoV555. One noted that mutations L452R, V483F/A, E484K/Q, F486L, F490L/S, Q493K/R, and S494P could weaken the binding ability of Eli Lilly mAbs. Coincidentally, LY-CoV555 was taken off from the EUA (Emergency Use Authorization) in March 2021 due to its low efficacy on the Beta [K417N, E484K, N501Y] and Gamma [K417T, E484K, N501Y] variants. Soon afterward, it was allowed to return to the market in September 2021 for its efficacy on the Delta variant [L452R, T478K]. The prediction in Figure 20e is a good explanation behind these events. More results for other mAbs as well as a series of confirmations by experimental data were given in the literature.764 A complete analysis of all RBD mutations is provided at the Web site Mutation Analyzer (https://weilab.math.msu.edu/MutationAnalyzer/) as shown in Figure 20b.
Some computational investigations were devoted to vaccine design. MD simulations were employed to simulate vaccine-related immune reactions, such as the binding of the MCH (major histocompatibility complex) II–epitope complexes.1008 Liu et al.’s359 FEP calculations suggested the E484Q and L452R mutations significantly reduce the binding affinity between the RBD of the Kappa variant and the antibody LY-CoV555 as well. Accurate computational predictions of Omicron’s vaccine breakthrough and antibody resistance were made available before any experiments and were all confirmed by experimental data.1263
4. Concluding Remarks
Since the first COVID-19 case was reported in December 2019, this pandemic has led to five waves of infections, over 400 million reported cases globally, and near 6 million deaths. Despite the exciting progress in the developments of vaccines and monoclonal antibodies, their potential side effects, such as allergic reactions to COVID-19 vaccines, are not very clear. Additionally, the latest Omicron variant is able to evade current vaccines and compromise essentially all monoclonal antibodies. Although the Omicron variant may be less deadly than the original virus, there is no guarantee that future variants will be less virulent. Our present understanding of SARS-CoV-2 and COVID-19 is still quite poor.
Molecular modeling, simulation, and prediction of SARS-CoV-2 have had tremendous contributions to the development of effective vaccines, drugs, and antibody therapies. Their role in combating COVID-19 is indispensable. For example, thanks to an approach that integrates genotyping, biophysics, artificial intelligence, advanced mathematics, and experiment data, it is now well-understood that the SARS-CoV-2 evolution and transmission are governed by natural selection.78 This means the next SARS-CoV-2 variant will be increasingly more transmissible through high infectivity, robust vaccine breakthrough, and strong antibody resistance.765,1264 This understanding cannot be achieved through individual experiments. Therefore, it is imperative to provide a literature review for the study of the molecular modeling, simulation, and prediction of SARS-CoV-2. Since the related literature is huge and varies in quality, we cannot collect all of the existing literature for the topic. However, we try to put forward a methodology-centered review in which we emphasize the methods used in various studies. To this end, we gather the existing theoretical and computational studies of SARS-CoV-2 concerning aspects such as molecular modeling, biophysics, bioinformatics, cheminformatics, machine learning including deep learning, and mathematical approaches, aiming to provide a comprehensive, systematic, and indispensable component for the understanding of the molecular mechanism of SARS-CoV-2 and its interactions with host cells. This review provides a methodology-centered description of the status of the molecular model, simulation, and prediction of SARS-CoV-2. We discuss the traditional molecular theories, models, and methods and emergent machine learning algorithms and mathematical approaches.
Although various vaccines have been approved and in use, vaccine-breakthrough mutations have become a serious problem. Even with the promising news of new vaccines, COVID-19 as a global health crisis may still last for years before it is fully stopped globally.
The research on SARS-CoV-2 will also last for many years. It will take researchers many more years to fully understand the molecular mechanism of coronaviruses, such as RNA proofreading, virus–host cell interactions, antibody–antigen interactions, protein–protein interactions, protein–drug interactions, viral regulation of host cell functions, and immune response. Even if we could control the transmission of SARS-CoV-2 in the future, newly emergent coronaviruses may still cause similar pandemic outbreaks. Therefore, the coronaviral studies will continue even after the current pandemic is fully under control.
Currently, epidemiologists, virologists, biologists, medical scientists, pharmacists, pharmacologists, chemists, biophysicists, mathematicians, computer scientists, and many others are called to investigate various aspects of COVID-19 and SARS-CoV-2. This trend of a joint effort on COVID-19 investigations will continue beyond the present pandemic.
The urgent need for the molecular mechanistic understanding of SARS-CoV-2 and COVID-19 will further stimulate the development of computational biophysical, artificial intelligence, and advanced mathematical methods. The theoretical, computational, and mathematical communities will benefit from this endeavor against the pandemic.
The year 2020 has witnessed the birth of human mRNA vaccines for the first time—a remarkable accomplishment in science and technology. Although there are more dark days ahead of us, humanity will prevail in a post-COVID-19 world. Science will emerge stronger against all pathogens and diseases in the future.
Acknowledgments
This work was supported in part by NIH grant GM126189, NSF Grants DMS-1721024, DMS-1761320, and IIS1900473, NASA grant 80NSSC21M0023, Michigan Economic Development Corporation, George Mason University award PD45722, Bristol Myers Squibb, and Pfizer. The authors thank The IBM TJ Watson Research Center, The COVID-19 High Performance Computing Consortium, NVIDIA, MSU Foundation, and MSU HPCC for computational assistance.
Biographies
Kaifu Gao obtained his Ph.D. degree in physical chemistry from the Chinese Academy of Sciences and completed his postdoctoral studies at the University of California, San Diego, under the guidance of Prof. Michael Gilson. His Ph.D. and postdoctoral studies focused on molecular dynamics (MD) simulations of protein conformational transitions and binding free energy calculations. Now, he is a Research Associate in Prof. Guo-Wei Wei’s group at Michigan State University. His current research concerns the application of deep learning to biological science and drug discovery, especially automated druglike compound generation and drug property prediction. His deep learning models have already been applied to the design and screening of SARS-CoV-2 Mpro inhibitors.
Rui Wang received her B.S. degree in mathematics from Xian Jiaotong University in 2017. Later, she joined Dr. Guo-Wei Wei’s group at Michigan State University. Currently, she is a fifth-year Ph.D. candidate in the Department of Mathematics, and she expects to finish her Ph.D. study in July 2022. Her methodological research focuses on developing mathematical tools for the descriptive and predictive modeling of biomolecules. She has also worked on genomics analysis and mathematical modeling of infectious diseases. She is highly interested in integrating AI, mathematics, genomics, biophysics, bioinformatics, and experimental data to tackle challenges in biological sciences, human diseases, and infectious pathogens.
Jiahui Chen received his Ph.D. degree in computational and applied mathematics from Southern Methodist University, where his research focused on the implementation of mathematical methods for biophysics, including the Poisson–Boltzmann equation, boundary element method, Treecode method, fast multipole method, and parallel computing. After graduation, he joined the Department of Mathematics at Michigan State University as a Research Associate in Prof. Guo-Wei Wei’s group. His current research focuses on topological and geometrical data analysis and machine learning algorithms with their modeling of and application to biomolecules. His model studies were applied to the prediction of mutation-induced binding free energy changes of the SARS-CoV-2 spike protein binding to ACE2 and antibodies.
Limei Cheng is an accomplished scientist with 15+ years of experience in developing novel algorithms, modeling, and simulations for healthcare applications such as quantitative systems pharmacology (QSP). She is currently the QSP-CV Lead of Clinical Pharmacology and Pharmacometrics at (CP&P) at Bristol Myers Squibb (BMS) in New Jersey. In her position, Dr. Cheng uses model-based QSP approaches to integrate clinical and nonclinical data in a quantitative and mechanistic way to generate actionable predictions. Dr. Cheng has numerous recent presentations and publications demonstrating the utility of Systems Pharmacology in Pharmaceutical R&D. Dr. Cheng earned a B.E. in Biomedical Electronic Engineering from Xi’an Jiao Tong University, an M.S. in Electrical Engineering from the University of California, Los Angeles, and a Ph.D. in Biomedical Engineering from the University of Southern California. Her key areas of interest include QSP modeling, virtual population simulation, novel algorithm development, and personalized precision medicine.
Jaclyn Frishcosy is an undergraduate student from the Professorial Assistantship Program at Michigan State University. She will be graduating in May 2022 with a B.S. degree in Data Science. She will then start work at Epic Careers in Verona, Wisconsin.
Yuta Huzumi received his B.S. in Applied Mathematics in 2018 from Case Western Reserve University, where he worked under the mentorship of Dr. Weihong Guo and Dr. Michael Hinczewski. He is currently a Ph.D. candidate at Michigan State University under Dr. Guo-Wei Wei, where he is studying machine learning and its application towards biological science. His current research concerns the dimensional reduction of high-dimensional biological data, such as mutation and gene expression datasets.
Yuchi Qiu obtained his Ph.D. degree in mathematics from the University of California, Irvine. Currently, he is a research associate in the Department of Mathematics at Michigan State University. Now, he uses topological data analysis and machine learning models to study protein mutations and evolutions.
Thomas Schluckbier was born in Schaumburg, Illinois, in 2001. He is currently pursuing B.S. degrees in Mathematics and Computer Science at Michigan State University, working under Professor Guo-Wei Wei.
Xiaoqi Wei was born in Heilongjiang, China, in 1993. He received his B.S. in Mathematics in 2015 from Jilin University and his M.S. in Mathematics in 2018 from ETH Zurich. He is currently a Ph.D. candidate in Prof. Guo-Wei Wei’s group at Michigan State University. He conducted research on using advanced mathematics to encode biological data.
Guo-Wei Wei earned his Ph.D. degree from the University of British Columbia in 1996. He was awarded a fellowship from the NSERC of Canada to pursue his postdoctoral work at the University of Houston. In 1998, he joined the faculty of the National University of Singapore and was promoted to Associate Professor in 2001. In 2002, he relocated to Michigan State University, where he is an MSU Foundation Professor of Mathematics, Electrical and Computer Engineering, and Biochemistry and Molecular Biology. His current research interests include mathematical biosciences, deep learning, drug discovery, and computational geometry, topology, and graphing. He has advised over 150 research students, postdoctoral associates, and visiting scientists. Dr. Wei has served extensively on a wide variety of national and international panels, committees, and journal editorships.
Author Contributions
⊥ K.G. and R.W. contributed equally to this paper.
The authors declare no competing financial interest.
References
- Owen D. R.; Allerton C. M.; Anderson A. S.; Aschenbrenner L.; Avery M.; Berritt S.; Boras B.; Cardin R. D.; Carlo A.; Coffman K. J.; et al. An oral SARS-CoV-2 Mpro inhibitor clinical candidate for the treatment of COVID-19. Science 2021, 374, 1586–1593. 10.1126/science.abl4784. [DOI] [PubMed] [Google Scholar]
- Gao K.; Wang R.; Chen J.; Tepe J. J.; Huang F.; Wei G.-W. Perspectives on SARS-CoV-2 Main Protease Inhibitors. J. Med. Chem. 2021, 64, 16922–16955. 10.1021/acs.jmedchem.1c00409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin M. D.; Shukla S.; Chung Y. H.; Beiss V.; Chan S. K.; Ortega-Rivera O. A.; Wirth D. M.; Chen A.; Sack M.; Pokorski J. K.; et al. COVID-19 vaccine development and a potential nanomaterial path forward. Nat. Nanotechnol. 2020, 15, 646–655. 10.1038/s41565-020-0737-y. [DOI] [PubMed] [Google Scholar]
- Day M. COVID-19: four fifths of cases are asymptomatic, China figures indicate. BMJ 2020, 369, m1375. 10.1136/bmj.m1375. [DOI] [PubMed] [Google Scholar]
- Long Q.-X.; Tang X.-J.; Shi Q.-L.; Li Q.; Deng H.-J.; Yuan J.; Hu J.-L.; Xu W.; Zhang Y.; Lv F.-J.; et al. Clinical and immunological assessment of asymptomatic SARS-CoV-2 infections. Nat. Med. 2020, 26, 1200–1204. 10.1038/s41591-020-0965-6. [DOI] [PubMed] [Google Scholar]
- Wang R.; Chen J.; Hozumi Y.; Yin C.; Wei G.-W. Decoding asymptomatic COVID-19 infection and transmission. J. Phys. Chem. Lett. 2020, 11, 10007–10015. 10.1021/acs.jpclett.0c02765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kissler S. M.; Tedijanto C.; Goldstein E.; Grad Y. H.; Lipsitch M. Projecting the transmission dynamics of SARS-CoV-2 through the postpandemic period. Science 2020, 368, 860–868. 10.1126/science.abb5793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin C. Genotyping coronavirus SARS-CoV-2: methods and implications. Genomics 2020, 112, 3588–3596. 10.1016/j.ygeno.2020.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R.; Hozumi Y.; Yin C.; Wei G.-W. Mutations on COVID-19 diagnostic targets. Genomics 2020, 112, 5204–5213. 10.1016/j.ygeno.2020.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R.; Chen J.; Gao K.; Hozumi Y.; Yin C.; Wei G.-W. Analysis of SARS-CoV-2 mutations in the United States suggests presence of four substrains and novel variants. Commun. Biol. 2021, 4, 1–14. 10.1038/s42003-021-01867-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R.; Hozumi Y.; Zheng Y.-H.; Yin C.; Wei G.-W. Host immune response driving SARS-CoV-2 evolution. Viruses 2020, 12, 1095. 10.3390/v12101095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R.; Hozumi Y.; Yin C.; Wei G.-W. Decoding SARS-CoV-2 Transmission and Evolution and Ramifications for COVID-19 Diagnosis, Vaccine, and Medicine. J. Chem. Inf Model 2020, 60, 5853. 10.1021/acs.jcim.0c00501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubin J. H.; Zardecki C.; Dolan E. M.; Lu C.; Shen Z.; Dutta S.; Westbrook J. D.; Hudson B. P.; Goodsell D. S.; Williams J. K.; et al. Evolution of the SARS-CoV-2 proteome in three dimensions (3D) during the first 6 months of the COVID-19 pandemic. Proteins 2021, 10.1002/prot.26250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen N.; Zhou M.; Dong X.; Qu J.; Gong F.; Han Y.; Qiu Y.; Wang J.; Liu Y.; Wei Y.; et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet 2020, 395, 507–513. 10.1016/S0140-6736(20)30211-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu R.; Zhao X.; Li J.; Niu P.; Yang B.; Wu H.; Wang W.; Song H.; Huang B.; Zhu N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. 10.1016/S0140-6736(20)30251-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walls A. C.; Park Y.-J.; Tortorici M. A.; Wall A.; McGuire A. T.; Veesler D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 2020, 181, 281–292. 10.1016/j.cell.2020.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrapp D.; Wang N.; Corbett K. S.; Goldsmith J. A.; Hsieh C.-L.; Abiona O.; Graham B. S.; McLellan J. S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. 10.1126/science.abb2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel C. J.; Mayer C.; Poch O.; Thompson J. D. Characterization of accessory genes in coronavirus genomes. Virol. J. 2020, 17, 1–13. 10.1186/s12985-020-01402-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmy Y. A.; Fawzy M.; Elaswad A.; Sobieh A.; Kenney S. P.; Shehata A. A. The COVID-19 pandemic: a comprehensive review of taxonomy, genetics, epidemiology, diagnosis, treatment, and control. J. Clin. Med. 2020, 9, 1225. 10.3390/jcm9041225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naqvi A. A. T.; Fatima K.; Mohammad T.; Fatima U.; Singh I. K.; Singh A.; Atif S. M.; Hariprasad G.; Hasan G. M.; Hassan M. I. Insights into SARS-CoV-2 genome, structure, evolution, pathogenesis and therapies: Structural genomics approach. Biochim Biophys Acta Mol. Basis Dis 2020, 1866, 165878. 10.1016/j.bbadis.2020.165878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu J.; Fang Y.; Yang Q.; Shu T.; Wang A.; Huang M.; Jin L.; Deng F.; Qiu Y.; Zhou X. SARS-CoV-2 N protein antagonizes type I interferon signaling by suppressing phosphorylation and nuclear translocation of STAT1 and STAT2. Cell Discov 2020, 6, 1–4. 10.1038/s41421-020-00208-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C.; Liu Y.; Yang Y.; Zhang P.; Zhong W.; Wang Y.; Wang Q.; Xu Y.; Li M.; Li X.; et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B 2020, 10, 766–788. 10.1016/j.apsb.2020.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D.; Lee J.-Y.; Yang J.-S.; Kim J. W.; Kim V. N.; Chang H. The architecture of SARS-CoV-2 transcriptome. Cell 2020, 181, 914–921. 10.1016/j.cell.2020.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuyama S.; Nao N.; Shirato K.; Kawase M.; Saito S.; Takayama I.; Nagata N.; Sekizuka T.; Katoh H.; Kato F.; et al. Enhanced isolation of SARS-CoV-2 by TMPRSS2-expressing cells. Proc. Natl. Acad. Sci. U.S.A. 2020, 117, 7001–7003. 10.1073/pnas.2002589117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann M.; Kleine-Weber H.; Schroeder S.; Krüger N.; Herrler T.; Erichsen S.; Schiergens T. S.; Herrler G.; Wu N.-H.; Nitsche A.; et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. cell 2020, 181, 271–280. 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- V'kovski P.; Kratzel A.; Steiner S.; Stalder H.; Thiel V. Coronavirus biology and replication: implications for SARS-CoV-2. Nat. Rev. Microbiol. 2021, 19, 155–170. 10.1038/s41579-020-00468-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert K.; Karousis E. D.; Jomaa A.; Scaiola A.; Echeverria B.; Gurzeler L.-A.; Leibundgut M.; Thiel V.; Mühlemann O.; Ban N. SARS-CoV-2 Nsp1 binds the ribosomal mRNA channel to inhibit translation. Nat. Struct. Mol. Biol. 2020, 27, 959–966. 10.1038/s41594-020-0511-8. [DOI] [PubMed] [Google Scholar]
- Clark L. K.; Green T. J.; Petit C. M. Structure of nonstructural protein 1 from SARS-CoV-2. J. Virol. 2021, 95, e02019–20. 10.1128/JVI.02019-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semper C.; Watanabe N.; Savchenko A. Structural characterization of nonstructural protein 1 from SARS-CoV-2. iScience 2021, 24, 101903. 10.1016/j.isci.2020.101903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao K.; Ke Z.; Hu H.; Liu Y.; Li A.; Hua R.; Guo F.; Xiao J.; Zhang Y.; Duan L.; et al. Structural basis and function of the N terminus of SARS-CoV-2 nonstructural protein 1. Microbiol. Spectr. 2021, 9, e00169–21. 10.1128/Spectrum.00169-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny M. L.; Veale C. E.; Guppy A.; Hardwick S. W.; Chirgadze D. Y.; Rzechorzek N. J.; Maman J. D.; Pellegrini L. Structural basis for the interaction of SARS-CoV-2 virulence factor nsp1 with DNA polymerase α–primase. Protein Sci. 2022, 31, 333–344. 10.1002/pro.4220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta M.; Azumaya C. M.; Moritz M.; Pourmal S.; Diallo A.; Merz G. E.; Jang G.; Bouhaddou M.; Fossati A.; Brilot A. F.; et al. CryoEM and AI reveal a structure of SARS-CoV-2 Nsp2, a multifunctional protein involved in key host processes. bioRxiv 2021, 10.1101/2021.05.10.443524. [DOI] [Google Scholar]
- Lei J.; Kusov Y.; Hilgenfeld R. Nsp3 of coronaviruses: Structures and functions of a large multi-domain protein. Antivir. Res. 2018, 149, 58–74. 10.1016/j.antiviral.2017.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin D.; Mukherjee R.; Grewe D.; Bojkova D.; Baek K.; Bhattacharya A.; Schulz L.; Widera M.; Mehdipour A. R.; Tascher G.; et al. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature 2020, 587, 657–662. 10.1038/s41586-020-2601-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frick D. N.; Virdi R. S.; Vuksanovic N.; Dahal N.; Silvaggi N. R. Molecular basis for ADP-ribose binding to the Mac1 domain of SARS-CoV-2 nsp3. Biochemistry 2020, 59, 2608–2615. 10.1021/acs.biochem.0c00309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalska K.; Kim Y.; Jedrzejczak R.; Maltseva N. I.; Stols L.; Endres M.; Joachimiak A. Crystal structures of SARS-CoV-2 ADP-ribose phosphatase: from the apo form to ligand complexes. IUCrJ. 2020, 7, 814–824. 10.1107/S2052252520009653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osipiuk J.; Jedrzejczak R.; Tesar C.; Endres M.; Stols L.; Babnigg G.; Kim Y.; Michalska K.; Joachimiak A. 6W9C: The crystal structure of papain-like protease of SARS CoV-2. RSCB PDB 2020, 10. 10.2210/pdb6W9C/pdb. [DOI] [Google Scholar]
- Sakai Y.; Kawachi K.; Terada Y.; Omori H.; Matsuura Y.; Kamitani W. Two-amino acids change in the nsp4 of SARS coronavirus abolishes viral replication. Virology 2017, 510, 165–174. 10.1016/j.virol.2017.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jumper J.; Tunyasuvunakool K.; Kohli P.; Hassabis D.. The AlphaFold Team Computational predictions of protein structures associated with COVID-19, Version 3. DeepMind website, 4 August 2020, https://deepmind.com/research/open-source/computational-predictions-of-protein-structures-associated-with-COVID-19, 2020.
- Jin Z.; Du X.; Xu Y.; Deng Y.; Liu M.; Zhao Y.; Zhang B.; Li X.; Zhang L.; Peng C.; et al. Structure of M pro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- Angelini M. M.; Akhlaghpour M.; Neuman B. W.; Buchmeier M. J.. Severe acute respiratory syndrome coronavirus nonstructural proteins 3, 4, and 6 induce double-membrane vesicles. MBio 2013, 4, 10.1128/mBio.00524-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cottam E. M.; Whelband M. C.; Wileman T. Coronavirus NSP6 restricts autophagosome expansion. Autophagy 2014, 10, 1426–1441. 10.4161/auto.29309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchdoerfer R. N.; Ward A. B. Structure of the SARS-CoV nsp12 polymerase bound to nsp7 and nsp8 co-factors. Nat. Commun. 2019, 10, 1–9. 10.1038/s41467-019-10280-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin W.; Mao C.; Luan X.; Shen D.-D.; Shen Q.; Su H.; Wang X.; Zhou F.; Zhao W.; Gao M.; et al. Structural basis for inhibition of the RNA-dependent RNA polymerase from SARS-CoV-2 by remdesivir. Science 2020, 368, 1499–1504. 10.1126/science.abc1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snijder E.; Decroly E.; Ziebuhr J. Adv. Virus Res. Elsevier 2016, 96, 59–126. 10.1016/bs.aivir.2016.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar M.; Sodhi K. K.; Singh D. K. Addressing the potential role of curcumin in the prevention of COVID-19 by targeting the Nsp9 replicase protein through molecular docking. Arch. Microbiol. 2021, 203, 1691–1696. 10.1007/s00203-020-02163-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Littler D. R.; Gully B. S.; Colson R. N.; Rossjohn J. Crystal structure of the SARS-CoV-2 non-structural protein 9, Nsp9. iScience 2020, 23, 101258. 10.1016/j.isci.2020.101258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogstam A.; Nyblom M.; Christensen S.; Sele C.; Talibov V. O.; Lindvall T.; Rasmussen A. A.; André I.; Fisher Z.; Knecht W.; et al. Crystal Structure of Non-Structural Protein 10 from Severe Acute Respiratory Syndrome Coronavirus-2. Int. J. Mol. Sci. 2020, 21, 7375. 10.3390/ijms21197375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y.; Yan L.; Huang Y.; Liu F.; Zhao Y.; Cao L.; Wang T.; Sun Q.; Ming Z.; Zhang L.; et al. Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science 2020, 368, 779–782. 10.1126/science.abb7498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillen H. S.; Kokic G.; Farnung L.; Dienemann C.; Tegunov D.; Cramer P. Structure of replicating SARS-CoV-2 polymerase. Nature 2020, 584, 154–156. 10.1038/s41586-020-2368-8. [DOI] [PubMed] [Google Scholar]
- Jang K.-J.; Jeong S.; Kang D. Y.; Sp N.; Yang Y. M.; Kim D.-E. A high ATP concentration enhances the cooperative translocation of the SARS coronavirus helicase nsP13 in the unwinding of duplex RNA. Sci. Rep. 2020, 10, 1–13. 10.1038/s41598-020-61432-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman J. A.; Douangamath A.; Yadzani S.; Yosaatmadja Y.; Aimon A.; Brandão-Neto J.; Dunnett L.; Gorrie-Stone T.; Skyner R.; Fearon D.; et al. Structure, mechanism and crystallographic fragment screening of the SARS-CoV-2 NSP13 helicase. Nat. Commun. 2021, 12, 1–11. 10.1038/s41467-021-25166-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferron F.; Subissi L.; De Morais A. T. S.; Le N. T. T.; Sevajol M.; Gluais L.; Decroly E.; Vonrhein C.; Bricogne G.; Canard B.; et al. Structural and molecular basis of mismatch correction and ribavirin excision from coronavirus RNA. Proc. Natl. Acad. Sci. U.S.A. 2018, 115, E162–E171. 10.1073/pnas.1718806115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S.; Chen H.; Chen Z.; Yang F.; Ye F.; Zheng Y.; Yang J.; Lin X.; Sun H.; Wang L.; et al. Crystal structure of SARS-CoV-2 nsp10 bound to nsp14-ExoN domain reveals an exoribonuclease with both structural and functional integrity. Nucleic Acids Res. 2021, 49, 5382–5392. 10.1093/nar/gkab320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng X.; Hackbart M.; Mettelman R. C.; O'Brien A.; Mielech A. M.; Yi G.; Kao C. C.; Baker S. C. Coronavirus nonstructural protein 15 mediates evasion of dsRNA sensors and limits apoptosis in macrophages. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, E4251–E4260. 10.1073/pnas.1618310114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y.; Wower J.; Maltseva N.; Chang C.; Jedrzejczak R.; Wilamowski M.; Kang S.; Nicolaescu V.; Randall G.; Michalska K.; et al. Tipiracil binds to uridine site and inhibits Nsp15 endoribonuclease NendoU from SARS-CoV-2. Commun. Biol. 2021, 4, 1–11. 10.1038/s42003-021-01735-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosas-Lemus M.; Minasov G.; Shuvalova L.; Inniss N. L.; Kiryukhina O.; Brunzelle J.; Satchell K. J. High-resolution structures of the SARS-CoV-2 2’-O-methyltransferase reveal strategies for structure-based inhibitor design. Sci. Signal. 2020, 13, 13. 10.1126/scisignal.abe1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang J.; Ye G.; Shi K.; Wan Y.; Luo C.; Aihara H.; Geng Q.; Auerbach A.; Li F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224. 10.1038/s41586-020-2179-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siu K.-L.; Yuen K.-S.; Castano-Rodriguez C.; Ye Z.-W.; Yeung M.-L.; Fung S.-Y.; Yuan S.; Chan C.-P.; Yuen K.-Y.; Enjuanes L.; et al. Severe acute respiratory syndrome Coronavirus ORF3a protein activates the NLRP3 inflammasome by promoting TRAF3-dependent ubiquitination of ASC. FASEB J. 2019, 33, 8865–8877. 10.1096/fj.201802418R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern D. M.; Sorum B.; Mali S. S.; Hoel C. M.; Sridharan S.; Remis J. P.; Toso D. B.; Kotecha A.; Bautista D. M.; Brohawn S. G. Cryo-EM structure of SARS-CoV-2 ORF3a in lipid nanodiscs. Nat. Struct. Mol. Biol. 2021, 28, 573–582. 10.1038/s41594-021-00619-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alam I.; Kamau A. A.; Kulmanov M.; Jaremko Ł.; Arold S. T.; Pain A.; Gojobori T.; Duarte C. M. Functional pangenome analysis shows key features of e protein are preserved in SARS and SARS-CoV-2. Front. Cell. Infect. Microbiol. 2020, 10, 405. 10.3389/fcimb.2020.00405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoeman D.; Fielding B. C. Coronavirus envelope protein: current knowledge. Virol. J. 2019, 16, 1–22. 10.1186/s12985-019-1182-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandala V. S.; McKay M. J.; Shcherbakov A. A.; Dregni A. J.; Kolocouris A.; Hong M. Structure and drug binding of the SARS-CoV-2 envelope protein transmembrane domain in lipid bilayers. Nat. Struct. Mol. Biol. 2020, 27, 1202–1208. 10.1038/s41594-020-00536-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voß D.; Pfefferle S.; Drosten C.; Stevermann L.; Traggiai E.; Lanzavecchia A.; Becker S. Studies on membrane topology, N-glycosylation and functionality of SARS-CoV membrane protein. Virol. J. 2009, 6, 1–13. 10.1186/1743-422X-6-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S.-H.; Lee T.-Y.; Lin Y.-J.; Wan L.; Lai C.-H.; Lin C.-W. Phage display technique identifies the interaction of severe acute respiratory syndrome coronavirus open reading frame 6 protein with nuclear pore complex interacting protein NPIPB3 in modulating Type I interferon antagonism. J. Microbiol Immunol Infect 2017, 50, 277–285. 10.1016/j.jmii.2015.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T.; Wen Y.; Guo H.; Yang T.; Yang H.; Ji X. Molecular Mechanism of SARS-CoVs Orf6 Targeting the Rae1–Nup98 Complex to Compete With mRNA Nuclear Export. Front. Mol. Biosci 2022, 8, 8. 10.3389/fmolb.2021.813248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor J. K.; Coleman C. M.; Postel S.; Sisk J. M.; Bernbaum J. G.; Venkataraman T.; Sundberg E. J.; Frieman M. B. Severe acute respiratory syndrome coronavirus ORF7a inhibits bone marrow stromal antigen 2 virion tethering through a novel mechanism of glycosylation interference. J. Virol. 2015, 89, 11820–11833. 10.1128/JVI.02274-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfefferle S.; Krähling V.; Ditt V.; Grywna K.; Mühlberger E.; Drosten C. Reverse genetic characterization of the natural genomic deletion in SARS-Coronavirus strain Frankfurt-1 open reading frame 7b reveals an attenuating function of the 7b protein in-vitro and in-vivo. Virol. J. 2009, 6, 131. 10.1186/1743-422X-6-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muth D.; Corman V. M.; Roth H.; Binger T.; Dijkman R.; Gottula L. T.; Gloza-Rausch F.; Balboni A.; Battilani M.; Rihtarič D.; et al. Attenuation of replication by a 29 nucleotide deletion in SARS-coronavirus acquired during the early stages of human-to-human transmission. Sci. Rep. 2018, 8, 1–11. 10.1038/s41598-018-33487-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung S.-C.; Chao C.-Y.; Jeng K.-S.; Yang J.-Y.; Lai M. M. The 8ab protein of SARS-CoV is a luminal ER membrane-associated protein and induces the activation of ATF6. Virology 2009, 387, 402–413. 10.1016/j.virol.2009.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flower T. G.; Buffalo C. Z.; Hooy R. M.; Allaire M.; Ren X.; Hurley J. H.. Structure of SARS-CoV-2 ORF8, a rapidly evolving immune evasion protein. Proc. Natl. Acad. Sci. U.S.A. 2021, 118, 10.1073/pnas.2021785118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang S.; Yang M.; Hong Z.; Zhang L.; Huang Z.; Chen X.; He S.; Zhou Z.; Zhou Z.; Chen Q.; et al. Crystal structure of SARS-CoV-2 nucleocapsid protein RNA binding domain reveals potential unique drug targeting sites. Acta Pharm. Sin. B 2020, 10, 1228–1238. 10.1016/j.apsb.2020.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinzula L.; Basquin J.; Bohn S.; Beck F.; Klumpe S.; Pfeifer G.; Nagy I.; Bracher A.; Hartl F. U.; Baumeister W. High-resolution structure and biophysical characterization of the nucleocapsid phosphoprotein dimerization domain from the COVID-19 severe acute respiratory syndrome coronavirus 2. Biochem. Biophys. Res. Commun. 2021, 538, 54–62. 10.1016/j.bbrc.2020.09.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi C.-S.; Qi H.-Y.; Boularan C.; Huang N.-N.; Abu-Asab M.; Shelhamer J. H.; Kehrl J. H. SARS-coronavirus open reading frame-9b suppresses innate immunity by targeting mitochondria and the MAVS/TRAF3/TRAF6 signalosome. J. Immunol. 2014, 193, 3080–3089. 10.4049/jimmunol.1303196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon D. E.; Jang G. M.; Bouhaddou M.; Xu J.; Obernier K.; White K. M.; O'Meara M. J.; Rezelj V. V.; Guo J. Z.; Swaney D. L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468. 10.1038/s41586-020-2286-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng H.; Tian H.; Wang Y.; Zhang Q.; Lin N.; Liu S.; Yu Y.; Deng H.; Gao P. Molecular mechanism underlying selective inhibition of mRNA nuclear export by herpesvirus protein ORF10. Proc. Natl. Acad. Sci. U.S.A. 2020, 117, 26719–26727. 10.1073/pnas.2007774117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.; Gao K.; Wang R.; Nguyen D. D.; Wei G.-W. Review of COVID-19 antibody therapies. Annu. Rev. 2021, 50, 1–30. 10.1146/annurev-biophys-062920-063711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.; Wang R.; Wang M.; Wei G.-W. Mutations strengthened SARS-CoV-2 infectivity. J. Mol. Biol. 2020, 432, 5212–5226. 10.1016/j.jmb.2020.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R.; Chen J.; Gao K.; Wei G.-W. Vaccine-escape and fast-growing mutations in the United Kingdom, the United States, Singapore, Spain, India, and other COVID-19-devastated countries. Genomics 2021, 113, 2158–2170. 10.1016/j.ygeno.2021.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson P.; Griffin I.; Tucker C.; Smith D.; Oechsle O.; Phelan A.; Rawling M.; Savory E.; Stebbing J. Baricitinib as potential treatment for 2019-nCoV acute respiratory disease. Lancet 2020, 395, e30 10.1016/S0140-6736(20)30304-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo H.; Ye F.; Sun T.; Yue L.; Peng S.; Chen J.; Li G.; Du Y.; Xie Y.; Yang Y.; et al. In vitro biochemical and thermodynamic characterization of nucleocapsid protein of SARS. Biophys. Chem. 2004, 112, 15–25. 10.1016/j.bpc.2004.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis M. E.; McCammon J. A. Electrostatics in biomolecular structure and dynamics. Chem. Rev. 1990, 90, 509–521. 10.1021/cr00101a005. [DOI] [Google Scholar]
- Sharp K. A.; Honig B. Electrostatic interactions in macromolecules: theory and applications. Annu. Rev. Biophys. Biophys. Chem. 1990, 19, 301–332. 10.1146/annurev.bb.19.060190.001505. [DOI] [PubMed] [Google Scholar]
- Honig B.; Nicholls A. Classical electrostatics in biology and chemistry. Science 1995, 268, 1144–1149. 10.1126/science.7761829. [DOI] [PubMed] [Google Scholar]
- Roux B.; Simonson T. Implicit solvent models. Biophys. Chem. 1999, 78, 1–20. 10.1016/S0301-4622(98)00226-9. [DOI] [PubMed] [Google Scholar]
- Rocchia W.; Alexov E.; Honig B. Extending the applicability of the nonlinear Poisson- Boltzmann equation: multiple dielectric constants and multivalent ions. J. Phys. Chem. B 2001, 105, 6507–6514. 10.1021/jp010454y. [DOI] [Google Scholar]
- Fogolari F.; Brigo A.; Molinari H. The Poisson–Boltzmann equation for biomolecular electrostatics: a tool for structural biology. J. Mol. Recognit. 2002, 15, 377–392. 10.1002/jmr.577. [DOI] [PubMed] [Google Scholar]
- Dominy B. N.; Brooks C. L. Development of a generalized Born model parametrization for proteins and nucleic acids. J. Phys. Chem. B 1999, 103, 3765–3773. 10.1021/jp984440c. [DOI] [Google Scholar]
- Bashford D.; Case D. A. Generalized born models of macromolecular solvation effects. Annu. Rev. Phys. Chem. 2000, 51, 129–152. 10.1146/annurev.physchem.51.1.129. [DOI] [PubMed] [Google Scholar]
- Onufriev A.; Case D. A.; Bashford D. Effective Born radii in the generalized Born approximation: the importance of being perfect. J. Comput. Chem. 2002, 23, 1297–1304. 10.1002/jcc.10126. [DOI] [PubMed] [Google Scholar]
- Mongan J.; Simmerling C.; McCammon J. A.; Case D. A.; Onufriev A. Generalized Born model with a simple, robust molecular volume correction. J. Chem. Theory Comput. 2007, 3, 156–169. 10.1021/ct600085e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasi J.; Mennucci B.; Cammi R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3094. 10.1021/cr9904009. [DOI] [PubMed] [Google Scholar]
- Cossi M.; Barone V.; Cammi R.; Tomasi J. Ab initio study of solvated molecules: a new implementation of the polarizable continuum model. Chem. Phys. Lett. 1996, 255, 327–335. 10.1016/0009-2614(96)00349-1. [DOI] [Google Scholar]
- Wei G.-W. Differential geometry based multiscale models. Bull. Math. Biol. 2010, 72, 1562–1622. 10.1007/s11538-010-9511-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z.; Baker N. A.; Wei G.-W. Differential geometry based solvation model I: Eulerian formulation. J. Comput. Phys. 2010, 229, 8231–8258. 10.1016/j.jcp.2010.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onufriev A.; Bashford D.; Case D. A. Modification of the generalized Born model suitable for macromolecules. J. Phys. Chem. B 2000, 104, 3712–3720. 10.1021/jp994072s. [DOI] [Google Scholar]
- Jurrus E.; Engel D.; Star K.; Monson K.; Brandi J.; Felberg L. E.; Brookes D. H.; Wilson L.; Chen J.; Liles K.; et al. Improvements to the APBS biomolecular solvation software suite. Protein Sci. 2018, 27, 112–128. 10.1002/pro.3280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D.; Chen Z.; Chen C.; Geng W.; Wei G.-W. MIBPB: a software package for electrostatic analysis. J. Comput. Chem. 2011, 32, 756–770. 10.1002/jcc.21646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y.; Zhao S.; Feig M.; Wei G.-W. High order matched interface and boundary method for elliptic equations with discontinuous coefficients and singular sources. J. Comput. Phys. 2006, 213, 1–30. 10.1016/j.jcp.2005.07.022. [DOI] [Google Scholar]
- Forouzesh N.; Izadi S.; Onufriev A. V. Grid-based surface generalized Born model for calculation of electrostatic binding free energies. J. Chem. Inf Model 2017, 57, 2505–2513. 10.1021/acs.jcim.7b00192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilson M. K.; Sharp K. A.; Honig B. H. Calculating the electrostatic potential of molecules in solution: method and error assessment. J. Comput. Chem. 1988, 9, 327–335. 10.1002/jcc.540090407. [DOI] [Google Scholar]
- Zhou Y.; Feig M.; Wei G.-W. Highly accurate biomolecular electrostatics in continuum dielectric environments. J. Comput. Chem. 2008, 29, 87–97. 10.1002/jcc.20769. [DOI] [PubMed] [Google Scholar]
- Ren P.; Chun J.; Thomas D. G.; Schnieders M. J.; Marucho M.; Zhang J.; Baker N. A. Biomolecular electrostatics and solvation: a computational perspective. Q. Rev. Biophys. 2012, 45, 427–491. 10.1017/S003358351200011X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo S.; Vargyas M.; Vasko-Szedlar J.; Roux B.; Im W. PBEQ-Solver for online visualization of electrostatic potential of biomolecules. Nucleic Acids Res. 2008, 36, W270–W275. 10.1093/nar/gkn314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker N. A.; Sept D.; Holst M. J.; McCammon J. A. The adaptive multilevel finite element solution of the Poisson-Boltzmann equation on massively parallel computers. IBM J. Res. Dev 2001, 45, 427–438. 10.1147/rd.453.0427. [DOI] [Google Scholar]
- Juffer A.; Botta E. F.; van Keulen B. A.; van der Ploeg A.; Berendsen H. J. The electric potential of a macromolecule in a solvent: A fundamental approach. J. Comput. Phys. 1991, 97, 144–171. 10.1016/0021-9991(91)90043-K. [DOI] [Google Scholar]
- Wang J.; Tan C.; Tan Y.-H.; Lu Q.; Luo R. Poisson-Boltzmann solvents in molecular dynamics simulations. Commun. Comput. Phys. 2008, 3, 1010–1031. [Google Scholar]
- Wang E.; Sun H.; Wang J.; Wang Z.; Liu H.; Zhang J. Z.; Hou T. End-point binding free energy calculation with MM/PBSA and MM/GBSA: strategies and applications in drug design. Chem. Rev. 2019, 119, 9478–9508. 10.1021/acs.chemrev.9b00055. [DOI] [PubMed] [Google Scholar]
- Li L.; Li C.; Sarkar S.; Zhang J.; Witham S.; Zhang Z.; Wang L.; Smith N.; Petukh M.; Alexov E. DelPhi: a comprehensive suite for DelPhi software and associated resources. BMC Biophys 2012, 5, 9. 10.1186/2046-1682-5-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocchia W.; Sridharan S.; Nicholls A.; Alexov E.; Chiabrera A.; Honig B. Rapid grid-based construction of the molecular surface and the use of induced surface charge to calculate reaction field energies: Applications to the molecular systems and geometric objects. J. Comput. Chem. 2002, 23, 128–137. 10.1002/jcc.1161. [DOI] [PubMed] [Google Scholar]
- Baker N. A.; Sept D.; Joseph S.; Holst M. J.; McCammon J. A. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 10037–10041. 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng W.; Yu S.; Wei G. Treatment of charge singularities in implicit solvent models. J. Chem. Phys. 2007, 127, 114106. 10.1063/1.2768064. [DOI] [PubMed] [Google Scholar]
- Geng W.; Krasny R. A treecode-accelerated boundary integral Poisson–Boltzmann solver for electrostatics of solvated biomolecules. J. Comput. Phys. 2013, 247, 62–78. 10.1016/j.jcp.2013.03.056. [DOI] [Google Scholar]
- Chen J.; Geng W. On preconditioning the treecode-accelerated boundary integral (TABI) Poisson–Boltzmann solver. J. Comput. Phys. 2018, 373, 750–762. 10.1016/j.jcp.2018.07.011. [DOI] [Google Scholar]
- Shu C.; Huang X.; Huang T.; Chen L.; Yao B.; Zhou J.; Deng C. Potential inhibitors for targeting Mpro and Spike of SARS-CoV-2 based on sequence and structural pharmacology analysis. STEMedicine 2020, 1, e41–e41. 10.37175/stemedicine.v1i2.41. [DOI] [Google Scholar]
- Zhang R.; Xiao K.; Gu Y.; Liu H.; Sun X. Whole genome identification of potential G-quadruplexes and analysis of the G-quadruplex binding domain for SARS-CoV-2. Front. Genet. 2020, 11, 1430. 10.3389/fgene.2020.587829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin M.; Sorour M. K.; Kasry A. Comparing the binding interactions in the receptor binding domains of SARS-CoV-2 and SARS-CoV. J. Phys. Chem. Lett. 2020, 11, 4897–4900. 10.1021/acs.jpclett.0c01064. [DOI] [PubMed] [Google Scholar]
- Jin X.; Xu K.; Jiang P.; Lian J.; Hao S.; Yao H.; Jia H.; Zhang Y.; Zheng L.; Zheng N.; et al. Virus strain from a mild COVID-19 patient in Hangzhou represents a new trend in SARS-CoV-2 evolution potentially related to Furin cleavage site. Emerg. Microbes Infect. 2020, 9, 1474–1488. 10.1080/22221751.2020.1781551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nerli S.; Sgourakis N. G. Structure-based modeling of SARS-CoV-2 peptide/HLA-A02 antigens. Front. Med. Technol. 2020, 2, 9. 10.3389/fmedt.2020.553478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javidpour L.; Božič A.; Naji A.; Podgornik R. Electrostatic interactions between the SARS-CoV-2 virus and a charged electret fibre. Soft Matter 2021, 17, 4296–4303. 10.1039/D1SM00232E. [DOI] [PubMed] [Google Scholar]
- Dung D.; Phan A. D.; Nguyen T. T.; Lam V. D. Effects of surface charge and environmental factors on the electrostatic interaction of fiber with virus-like particle: A case of coronavirus. AIP Adv. 2021, 11, 105008. 10.1063/5.0065147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstein M.; Krebs W. A database of macromolecular motions. Nucleic Acids Res. 1998, 26, 4280–4290. 10.1093/nar/26.18.4280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goh C.-S.; Milburn D.; Gerstein M. Conformational changes associated with protein–protein interactions. Curr. Opin. Struct. Biol. 2004, 14, 104–109. 10.1016/j.sbi.2004.01.005. [DOI] [PubMed] [Google Scholar]
- Karplus M.; McCammon J. A. Molecular dynamics simulations of biomolecules. Nat. Struct. Biol. 2002, 9, 646–652. 10.1038/nsb0902-646. [DOI] [PubMed] [Google Scholar]
- Hospital A.; Goñi J. R.; Orozco M.; Gelpí J. L. Molecular dynamics simulations: advances and applications. Adv. Appl. Bioinform. Chem. 2015, 8, 37. 10.2147/AABC.S70333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng W.; Wei G.-W. Multiscale molecular dynamics using the matched interface and boundary method. J. Comput. Phys. 2011, 230, 435–457. 10.1016/j.jcp.2010.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw D. E.; Maragakis P.; Lindorff-Larsen K.; Piana S.; Dror R. O.; Eastwood M. P.; Bank J. A.; Jumper J. M.; Salmon J. K.; Shan Y.; et al. Atomic-level characterization of the structural dynamics of proteins. Science 2010, 330, 341–346. 10.1126/science.1187409. [DOI] [PubMed] [Google Scholar]
- Adcock S. A.; McCammon J. A. Molecular dynamics: survey of methods for simulating the activity of proteins. Chem. Rev. 2006, 106, 1589–1615. 10.1021/cr040426m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen M. P.; et al. Introduction to molecular dynamics simulation. Comput. Soft Matter 2004, 23, 1–28. [Google Scholar]
- Cornell W. D.; Cieplak P.; Bayly C. I.; Gould I. R.; Merz K. M.; Ferguson D. M.; Spellmeyer D. C.; Fox T.; Caldwell J. W.; Kollman P. A. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J. Am. Chem. Soc. 1995, 117, 5179–5197. 10.1021/ja00124a002. [DOI] [Google Scholar]
- MacKerell A. D. Jr; Bashford D.; Bellott M.; Dunbrack R. L. Jr; Evanseck J. D.; Field M. J.; Fischer S.; Gao J.; Guo H.; Ha S.; et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 1998, 102, 3586–3616. 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- Ponder J. W.; Wu C.; Ren P.; Pande V. S.; Chodera J. D.; Schnieders M. J.; Haque I.; Mobley D. L.; Lambrecht D. S.; DiStasio R. A. Jr; et al. Current status of the AMOEBA polarizable force field. J. Phys. Chem. B 2010, 114, 2549–2564. 10.1021/jp910674d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kmiecik S.; Gront D.; Kolinski M.; Wieteska L.; Dawid A. E.; Kolinski A. Coarse-grained protein models and their applications. Chem. Rev. 2016, 116, 7898–7936. 10.1021/acs.chemrev.6b00163. [DOI] [PubMed] [Google Scholar]
- Ueda Y.; Taketomi H.; Go̅ N. Studies on protein folding, unfolding, and fluctuations by computer simulation. II. A. Three-dimensional lattice model of lysozyme. Biopolymers 1978, 17, 1531–1548. 10.1002/bip.1978.360170612. [DOI] [Google Scholar]
- Marrink S. J.; Risselada H. J.; Yefimov S.; Tieleman D. P.; De Vries A. H. The MARTINI force field: coarse grained model for biomolecular simulations. J. Phys. Chem. B 2007, 111, 7812–7824. 10.1021/jp071097f. [DOI] [PubMed] [Google Scholar]
- Maisuradze G. G.; Senet P.; Czaplewski C.; Liwo A.; Scheraga H. A. Investigation of protein folding by coarse-grained molecular dynamics with the UNRES force field. J. Phys. Chem. A 2010, 114, 4471–4485. 10.1021/jp9117776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahoney M. W.; Jorgensen W. L. A five-site model for liquid water and the reproduction of the density anomaly by rigid, nonpolarizable potential functions. J. Chem. Phys. 2000, 112, 8910–8922. 10.1063/1.481505. [DOI] [Google Scholar]
- Berendsen H.; Grigera J.; Straatsma T. The missing term in effective pair potentials. J. Phys. Chem. 1987, 91, 6269–6271. 10.1021/j100308a038. [DOI] [Google Scholar]
- Massova I.; Kollman P. A. Computational alanine scanning to probe protein- protein interactions: a novel approach to evaluate binding free energies. J. Am. Chem. Soc. 1999, 121, 8133–8143. 10.1021/ja990935j. [DOI] [Google Scholar]
- Genheden S.; Ryde U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin Drug Discov 2015, 10, 449–461. 10.1517/17460441.2015.1032936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu N.; Kofke D. A. Accuracy of free-energy perturbation calculations in molecular simulation. I. Modeling. J. Chem. Phys. 2001, 114, 7303–7311. 10.1063/1.1359181. [DOI] [Google Scholar]
- Straatsma T.; Berendsen H. Free energy of ionic hydration: Analysis of a thermodynamic integration technique to evaluate free energy differences by molecular dynamics simulations. J. Chem. Phys. 1988, 89, 5876–5886. 10.1063/1.455539. [DOI] [Google Scholar]
- Laio A.; Parrinello M. Escaping free-energy minima. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 12562–12566. 10.1073/pnas.202427399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izrailev S.; Stepaniants S.; Isralewitz B.; Kosztin D.; Lu H.; Molnar F.; Wriggers W.; Schulten K.. Computational molecular dynamics: challenges, methods, ideas; Springer, 1999; pp 39–65. [Google Scholar]
- Wang J.; Hou T. Develop and test a solvent accessible surface area-based model in conformational entropy calculations. J. Chem. Inf Model 2012, 52, 1199–1212. 10.1021/ci300064d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sztain T.; Amaro R.; McCammon J. A. Elucidation of cryptic and allosteric pockets within the SARS-CoV-2 main protease. J. Chem. Inf Model 2021, 61, 3495–3501. 10.1021/acs.jcim.1c00140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grottesi A.; Bešker N.; Emerson A.; Manelfi C.; Beccari A. R.; Frigerio F.; Lindahl E.; Cerchia C.; Talarico C. Computational Studies of SARS-CoV-2 3CLpro: Insights from MD Simulations. Int. J. Mol. Sci. 2020, 21, 5346. 10.3390/ijms21155346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bzówka M.; Mitusińska K.; Raczyńska A.; Samol A.; Tuszyński J. A.; Góra A. Structural and Evolutionary Analysis Indicate That the SARS-CoV-2 Mpro Is a Challenging Target for Small-Molecule Inhibitor Design. Int. J. Mol. Sci. 2020, 21, 3099. 10.3390/ijms21093099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshino R.; Yasuo N.; Sekijima M. Identification of key interactions between SARS-CoV-2 main protease and inhibitor drug candidates. Sci. Rep. 2020, 10, 10. 10.1038/s41598-020-69337-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjeev B.; Chitara D.; Madhumalar A. Physiological models to study the effect of molecular crowding on multi-drug bound proteins: insights from SARS-CoV-2 main protease. J. Biomol. Struct 2021, 1–17. 10.1080/07391102.2021.1993342. [DOI] [PubMed] [Google Scholar]
- Lamichhane T. R.; Ghimire M. P. Evaluation of SARS-CoV-2 main protease and inhibitor interactions using dihedral angle distributions and radial distribution function. Heliyon 2021, 7, e08220 10.1016/j.heliyon.2021.e08220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deganutti G.; Prischi F.; Reynolds C. A. Supervised molecular dynamics for exploring the druggability of the SARS-CoV-2 spike protein. J. Comput. Aided Mol. Des. 2021, 35, 195–207. 10.1007/s10822-020-00356-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghuvamsi P. V.; Tulsian N. K.; Samsudin F.; Qian X.; Purushotorman K.; Yue G.; Kozma M. M.; Hwa W. Y.; Lescar J.; Bond P. J.; et al. SARS-CoV-2 S protein: ACE2 interaction reveals novel allosteric targets. Elife 2021, 10, e63646 10.7554/eLife.63646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casalino L.; Gaieb Z.; Goldsmith J. A.; Hjorth C. K.; Dommer A. C.; Harbison A. M.; Fogarty C. A.; Barros E. P.; Taylor B. C.; McLellan J. S.; et al. Beyond shielding: the roles of glycans in the SARS-CoV-2 spike protein. ACS Cent. Sci. 2020, 6, 1722–1734. 10.1021/acscentsci.0c01056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barros E. P.; Casalino L.; Gaieb Z.; Dommer A. C.; Wang Y.; Fallon L.; Raguette L.; Belfon K.; Simmerling C.; Amaro R. E. The flexibility of ACE2 in the context of SARS-CoV-2 infection. Biophys. J. 2021, 120, 1072–1084. 10.1016/j.bpj.2020.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veeramachaneni G. K.; Thunuguntla V.; Bobbillapati J.; Bondili J. S. Structural and simulation analysis of hotspot residues interactions of SARS-CoV 2 with human ACE2 receptor. J. Biomol. Struct. Dyn. 2021, 39, 4015–4025. 10.1080/07391102.2020.1773318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grishin A. M.; Dolgova N. V.; Landreth S.; Fisette O.; Pickering I. J.; George G. N.; Falzarano D.; Cygler M. Disulfide bonds play a critical role in the structure and function of the receptor-binding domain of the SARS-CoV-2 Spike antigen. J. Mol. Biol. 2022, 434, 167357. 10.1016/j.jmb.2021.167357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z.; Yang Z.; Ou J.; Zhang H.; Zhang Q.; Dong M.; Zhang G. Temperature dependence of the SARS-CoV-2 affinity to human ACE2 determines COVID-19 progression and clinical outcome. Comput. Struct. Biotechnol. J. 2021, 19, 161–167. 10.1016/j.csbj.2020.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdalla M.; Eltayb W. A.; El-Arabey A. A.; Singh K.; Jiang X. Molecular dynamic study of SARS-CoV-2 with various S protein mutations and their effect on thermodynamic properties. Comput. Biol. Med. 2022, 141, 105025. 10.1016/j.compbiomed.2021.105025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y.; Král P. Computational design of ACE2-based peptide inhibitors of SARS-CoV-2. ACS Nano 2020, 14, 5143–5147. 10.1021/acsnano.0c02857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira A. S. F.; Ibarra A. A.; Bermudez I.; Casalino L.; Gaieb Z.; Shoemark D. K.; Gallagher T.; Sessions R. B.; Amaro R. E.; Mulholland A. J. A potential interaction between the SARS-CoV-2 spike protein and nicotinic acetylcholine receptors. Biophys. J. 2021, 120, 983–993. 10.1016/j.bpj.2021.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikora M.; von Bülow S.; Blanc F. E.; Gecht M.; Covino R.; Hummer G. Computational epitope map of SARS-CoV-2 spike protein. PLoS Comput. Biol. 2021, 17, e1008790 10.1371/journal.pcbi.1008790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardi A.; Huang Y.; Harris B.; Xiong Y.; Nandi S.; McDonald K. A.; Faller R. Development and simulation of fully glycosylated molecular models of ACE2-Fc fusion proteins and their interaction with the SARS-CoV-2 spike protein binding domain. PLoS One 2020, 15, e0237295 10.1371/journal.pone.0237295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gur M.; Taka E.; Yilmaz S. Z.; Kilinc C.; Aktas U.; Golcuk M. Conformational transition of SARS-CoV-2 spike glycoprotein between its closed and open states. J. Chem. Phys. 2020, 153, 075101. 10.1063/5.0011141. [DOI] [PubMed] [Google Scholar]
- Ghorbani M.; Brooks B. R.; Klauda J. B. Exploring dynamics and network analysis of spike glycoprotein of SARS-COV-2. Biophys. J. 2021, 120, 2902. 10.1016/j.bpj.2021.02.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupala C. S.; Kumar V.; Su X.-d.; Wu C.; Liu H. Computational insights into differential interaction of mammalian angiotensin-converting enzyme 2 with the SARS-CoV-2 spike receptor binding domain. Comput. Biol. Med. 2022, 141, 105017. 10.1016/j.compbiomed.2021.105017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson J. A.; Verma N.; Harris R. C.; Liu R.; Shen J. Assessment of proton-coupled conformational dynamics of SARS and MERS coronavirus papain-like proteases: Implication for designing broad-spectrum antiviral inhibitors. J. Chem. Phys. 2020, 153, 115101. 10.1063/5.0020458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohraby F.; Aryapour H. Unraveling the unbinding pathways of SARS-CoV-2 Papain-like proteinase known inhibitors by Supervised Molecular Dynamics simulation. PLoS One 2021, 16, e0251910 10.1371/journal.pone.0251910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S.; Karki C.; Aguilera J.; Lopez Hernandez A. E.; Sun J.; Li L. Computational Study on the Function of Palmitoylation on the Envelope Protein in SARS-CoV-2. J. Chem. Theory Comput. 2021, 17, 6483–6490. 10.1021/acs.jctc.1c00359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu A.; Pak A. J.; He P.; Monje-Galvan V.; Casalino L.; Gaieb Z.; Dommer A. C.; Amaro R. E.; Voth G. A. A multiscale coarse-grained model of the SARS-CoV-2 virion. Biophys. J. 2021, 120, 1097–1104. 10.1016/j.bpj.2020.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giron C. C.; Laaksonen A.; da Silva F. L. B. On the interactions of the receptor-binding domain of SARS-CoV-1 and SARS-CoV-2 spike proteins with monoclonal antibodies and the receptor ACE2. Virus Res. 2020, 285, 198021. 10.1016/j.virusres.2020.198021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai C.; Wang J.; Chen G.; Zhang H.; An K.; Xu P.; Du Y.; Ye R. D.; Saha A.; Zhang A.; et al. Predicting Mutational Effects on Receptor Binding of the Spike Protein of SARS-CoV-2 Variants. J. Am. Chem. Soc. 2021, 143, 17646–17654. 10.1021/jacs.1c07965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen H. L.; Lan P. D.; Thai N. Q.; Nissley D. A.; O’Brien E. P.; Li M. S. Does SARS-CoV-2 Bind to Human ACE2 Stronger Than SARS-CoV?. J. Phys. Chem. B 2020, 124, 7336–7347. 10.1021/acs.jpcb.0c04511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkhivker G. M.; Agajanian S.; Oztas D.; Gupta G. Computational analysis of protein stability and allosteric interaction networks in distinct conformational forms of the SARS-CoV-2 spike D614G mutant: reconciling functional mechanisms through allosteric model of spike regulation. J. Biomol. Struct 2021, 1–18. 10.1080/07391102.2021.1933594. [DOI] [PubMed] [Google Scholar]
- Teruel N.; Mailhot O.; Najmanovich R. J. Modelling conformational state dynamics and its role on infection for SARS-CoV-2 Spike protein variants. PLoS Comput. Biol. 2021, 17, e1009286 10.1371/journal.pcbi.1009286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkhivker G. M.; Di Paola L. Integrated Biophysical Modeling of the SARS-CoV-2 Spike Protein Binding and Allosteric Interactions with Antibodies. J. Phys. Chem. B 2021, 125, 4596–4619. 10.1021/acs.jpcb.1c00395. [DOI] [PubMed] [Google Scholar]
- Seitz C.; Casalino L.; Konecny R.; Huber G.; Amaro R. E.; McCammon J. A. Multiscale simulations examining glycan shield effects on drug binding to influenza neuraminidase. Biophys. J. 2020, 119, 2275–2289. 10.1016/j.bpj.2020.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks B.; Karplus M. Harmonic dynamics of proteins: normal modes and fluctuations in bovine pancreatic trypsin inhibitor. Proc. Natl. Acad. Sci. U.S.A. 1983, 80, 6571–6575. 10.1073/pnas.80.21.6571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Go N.; Noguti T.; Nishikawa T. Dynamics of a small globular protein in terms of low-frequency vibrational modes. Proc. Natl. Acad. Sci. U.S.A. 1983, 80, 3696–3700. 10.1073/pnas.80.12.3696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinsen K. Analysis of domain motions by approximate normal mode calculations. Proteins 1998, 33, 417–429. . [DOI] [PubMed] [Google Scholar]
- Skjaerven L.; Hollup S. M.; Reuter N. Normal mode analysis for proteins. J. Mol. Struct. THEOCHEM 2009, 898, 42–48. 10.1016/j.theochem.2008.09.024. [DOI] [Google Scholar]
- Mahajan S.; Sanejouand Y.-H. On the relationship between low-frequency normal modes and the large-scale conformational changes of proteins. Arch. Biochem. Biophys. 2015, 567, 59–65. 10.1016/j.abb.2014.12.020. [DOI] [PubMed] [Google Scholar]
- Tirion M. M. Large amplitude elastic motions in proteins from a single-parameter, atomic analysis. Phys. Rev. Lett. 1996, 77, 1905. 10.1103/PhysRevLett.77.1905. [DOI] [PubMed] [Google Scholar]
- Miyazawa S.; Jernigan R. L. Estimation of effective interresidue contact energies from protein crystal structures: quasi-chemical approximation. Macromolecules 1985, 18, 534–552. 10.1021/ma00145a039. [DOI] [Google Scholar]
- Bahar I.; Atilgan A. R.; Erman B. Direct evaluation of thermal fluctuations in proteins using a single-parameter harmonic potential. Fold. Des. 1997, 2, 173–181. 10.1016/S1359-0278(97)00024-2. [DOI] [PubMed] [Google Scholar]
- Xia K.; Opron K.; Wei G.-W. Multiscale Gaussian network model (mGNM) and multiscale anisotropic network model (mANM). J. Chem. Phys. 2015, 143, 204106. 10.1063/1.4936132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen D. D.; Xia K.; Wei G.-W. Generalized flexibility-rigidity index. J. Chem. Phys. 2016, 144, 234106. 10.1063/1.4953851. [DOI] [PubMed] [Google Scholar]
- Hinsen K. The molecular modeling toolkit: a new approach to molecular simulations. J. Comput. Chem. 2000, 21, 79–85. . [DOI] [Google Scholar]
- Dubanevics I.; McLeish T. C. Computational analysis of dynamic allostery and control in the SARS-CoV-2 main protease. J. R. Soc. Interface 2021, 18, 20200591. 10.1098/rsif.2020.0591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumder S.; Chaudhuri D.; Datta J.; Giri K. Exploring the intrinsic dynamics of SARS-CoV-2, SARS-CoV and MERS-CoV spike glycoprotein through normal mode analysis using anisotropic network model. J. Mol. Graph. Model. 2021, 102, 107778. 10.1016/j.jmgm.2020.107778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Paola L.; Hadi-Alijanvand H.; Song X.; Hu G.; Giuliani A. The discovery of a putative allosteric site in the SARS-CoV-2 spike protein using an integrated structural/dynamic approach. J. Proteome Res. 2020, 19, 4576–4586. 10.1021/acs.jproteome.0c00273. [DOI] [PubMed] [Google Scholar]
- Singh A.; Steinkellner G.; Köchl K.; Gruber K.; Gruber C. C. Serine 477 plays a crucial role in the interaction of the SARS-CoV-2 spike protein with the human receptor ACE2. Sci. Rep. 2021, 11, 1–11. 10.1038/s41598-021-83761-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta A. M.; Chakrabarti J.; Mandal S. Non-synonymous mutations of SARS-CoV-2 leads epitope loss and segregates its variants. Microbes Infect 2020, 22, 598–607. 10.1016/j.micinf.2020.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padhi A. K.; Shukla R.; Saudagar P.; Tripathi T. High-throughput rational design of the remdesivir binding site in the RdRp of SARS-CoV-2: implications for potential resistance. Iscience 2021, 24, 101992. 10.1016/j.isci.2020.101992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroese D. P.; Brereton T.; Taimre T.; Botev Z. I. Why the Monte Carlo method is so important today. Wiley Interdiscip. Rev. Comput. Stat. 2014, 6, 386–392. 10.1002/wics.1314. [DOI] [Google Scholar]
- Hastings W. K. Monte Carlo sampling methods using Markov chains and their applications. Biometrika 1970, 57, 97–109. 10.1093/biomet/57.1.97. [DOI] [Google Scholar]
- Chen J.; Geng W.; Wei G.-W. MLIMC: Machine Learning-based implicit-solvent Monte Carlo. Chin. J. Chem. Phys. 2021, 34, 683. 10.1063/1674-0068/cjcp2109150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metropolis N.; Rosenbluth A. W.; Rosenbluth M. N.; Teller A. H.; Teller E. Equation of state calculations by fast computing machines. J. Chem. Phys. 1953, 21, 1087–1092. 10.1063/1.1699114. [DOI] [Google Scholar]
- Francis Z.; Incerti S.; Zein S. A.; Lampe N.; Guzman C. A.; Durante M. Monte Carlo simulation of SARS-CoV-2 radiation-induced inactivation for vaccine development. Radiat. Res. 2021, 195, 221–229. 10.1667/RADE-20-00241.1. [DOI] [PubMed] [Google Scholar]
- Cojutti P. G.; Londero A.; Della Siega P.; Givone F.; Fabris M.; Biasizzo J.; Tascini C.; Pea F. Comparative Population Pharmacokinetics of Darunavir in SARS-CoV-2 Patients vs. HIV Patients: The Role of Interleukin-6. Clin. Pharmacokinet. 2020, 59, 1251–1260. 10.1007/s40262-020-00933-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin S. A.; Ghosh K.; Gayen S.; Jha T. Chemical-informatics approach to COVID-19 drug discovery: Monte Carlo based QSAR, virtual screening and molecular docking study of some in-house molecules as papain-like protease (PLpro) inhibitors. J. Biomol. Struct. Dyn. 2021, 39, 4764. 10.1080/07391102.2020.1780946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cubuk J.; Alston J. J.; Incicco J. J.; Singh S.; Stuchell-Brereton M. D.; Ward M. D.; Zimmerman M. I.; Vithani N.; Griffith D.; Wagoner J. A.; et al. The SARS-CoV-2 nucleocapsid protein is dynamic, disordered, and phase separates with RNA. Nat. Commun. 2021, 12, 1–17. 10.1038/s41467-021-21953-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang J.; Pitsillou E.; Ververis K.; Guallar V.; Hung A.; Karagiannis T. C. Small molecule interactions with the SARS-CoV-2 main protease: In silico all-atom microsecond MD simulations, PELE Monte Carlo simulations, and determination of in vitro activity inhibition. J. Mol. Graph. Model. 2022, 110, 108050. 10.1016/j.jmgm.2021.108050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheik Amamuddy O.; Verkhivker G. M.; Tastan Bishop O. Impact of early pandemic stage mutations on molecular dynamics of SARS-CoV-2 Mpro. J. Chem. Inf Model 2020, 60, 5080–5102. 10.1021/acs.jcim.0c00634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toropov A. A.; Toropova A. P.; Veselinović A. M.; Leszczynska D.; Leszczynski J. SARS-CoV Mpro inhibitory activity of aromatic disulfide compounds: QSAR model. J. Biomol. Struct. Dyn. 2022, 40, 780. 10.1080/07391102.2020.1818627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becerra-Flores M.; Cardozo T. SARS-CoV-2 viral spike G614 mutation exhibits higher case fatality rate. Int. J. Clin. Pract. 2020, 74, e13525 10.1111/ijcp.13525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali F.; Kasry A.; Amin M. The new SARS-CoV-2 strain shows a stronger binding affinity to ACE2 due to N501Y mutant. Med. Drug Dis. 2021, 10, 100086. 10.1016/j.medidd.2021.100086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polydorides S.; Archontis G. Computational optimization of the SARS-CoV-2 receptor-binding-motif affinity for human ACE2. Biophys. J. 2021, 120, 2859–2871. 10.1016/j.bpj.2021.02.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X.; Pearce R.; Zhang Y. De novo design of protein peptides to block association of the SARS-CoV-2 spike protein with human ACE2. Aging (Albany NY) 2020, 12, 11263. 10.18632/aging.103416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai C.; Warshel A. Critical Differences Between the Binding Features of the Spike Proteins of SARS-CoV-2 and SARS-CoV. J. Phys. Chem. B 2020, 124, 5907–5912. 10.1021/acs.jpcb.0c04317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller L. M.; Barnes L. F.; Raab S. A.; Draper B. E.; El-Baba T. J.; Lutomski C. A.; Robinson C. V.; Clemmer D. E.; Jarrold M. F. Heterogeneity of Glycan Processing on Trimeric SARS-CoV-2 Spike Protein Revealed by Charge Detection Mass Spectrometry. J. Am. Chem. Soc. 2021, 143, 3959–3966. 10.1021/jacs.1c00353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero-Rivera A.; Garcia-Borràs M.; Osuna S. Computational tools for the evaluation of laboratory-engineered biocatalysts. Chem. Commun. 2017, 53, 284–297. 10.1039/C6CC06055B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen R.; Christensen M. H. MolDock: a new technique for high-accuracy molecular docking. J. Med. Chem. 2006, 49, 3315–3321. 10.1021/jm051197e. [DOI] [PubMed] [Google Scholar]
- Kitchen D. B.; Decornez H.; Furr J. R.; Bajorath J. Docking and scoring in virtual screening for drug discovery: methods and applications. Nat. Rev. Drug Discovery 2004, 3, 935–949. 10.1038/nrd1549. [DOI] [PubMed] [Google Scholar]
- Brooijmans N.; Kuntz I. D. Molecular recognition and docking algorithms. Annu. Rev. Biophys. Biomol. Struct. 2003, 32, 335–373. 10.1146/annurev.biophys.32.110601.142532. [DOI] [PubMed] [Google Scholar]
- Li H.; Sze K.-H.; Lu G.; Ballester P. J. Machine-learning scoring functions for structure-based virtual screening. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2021, 11, e1478 10.1002/wcms.1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wójcikowski M.; Ballester P. J.; Siedlecki P. Performance of machine-learning scoring functions in structure-based virtual screening. Sci. Rep. 2017, 7, 1–10. 10.1038/srep46710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ain Q. U.; Aleksandrova A.; Roessler F. D.; Ballester P. J. Machine-learning scoring functions to improve structure-based binding affinity prediction and virtual screening. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2015, 5, 405–424. 10.1002/wcms.1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cang Z.; Wei G.-W. TopologyNet: Topology based deep convolutional and multi-task neural networks for biomolecular property predictions. PLoS Comput. Biol. 2017, 13, e1005690 10.1371/journal.pcbi.1005690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaro R. E.; Baudry J.; Chodera J.; Demir Ö.; McCammon J. A.; Miao Y.; Smith J. C. Ensemble docking in drug discovery. Biophys. J. 2018, 114, 2271–2278. 10.1016/j.bpj.2018.02.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y. W.; Yiu C.-P. B.; Wong K.-Y. Prediction of the SARS-CoV-2 (2019-nCoV) 3C-like protease (3CL pro) structure: virtual screening reveals velpatasvir, ledipasvir, and other drug repurposing candidates. F1000Research 2020, 9, 129. 10.12688/f1000research.22457.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Federico L. B.; Silva G. M.; da Silva Hage-Melim L. I.; Gomes S. Q.; Barcelos M. P.; Galindo Francischini I. A.; Tomich de Paula da Silva C. H. Identification of known drugs as potential SARS-CoV-2 Mpro inhibitors using ligand-and structure-based virtual screening. Future Med. Chem. 2021, 13, 1353–1366. 10.4155/fmc-2021-0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sencanski M.; Perovic V.; Pajovic S. B.; Adzic M.; Paessler S.; Glisic S. Drug Repurposing for Candidate SARS-CoV-2 Main Protease Inhibitors by a Novel in Silico Method. Molecules 2020, 25, 3830. 10.3390/molecules25173830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurung A. B.; Ali M. A.; Lee J.; Farah M. A.; Al-Anazi K. M. In silico screening of FDA approved drugs reveals ergotamine and dihydroergotamine as potential coronavirus main protease enzyme inhibitors. Saudi J. Biol. Sci. 2020, 27, 2674–2682. 10.1016/j.sjbs.2020.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eleftheriou P.; Amanatidou D.; Petrou A.; Geronikaki A. In Silico Evaluation of the Effectivity of Approved Protease Inhibitors against the Main Protease of the Novel SARS-CoV-2 Virus. Molecules 2020, 25, 2529. 10.3390/molecules25112529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi R. S.; Jagdale S. S.; Bansode S. B.; Shankar S. S.; Tellis M. B.; Pandya V. K.; Chugh A.; Giri A. P.; Kulkarni M. J. Discovery of potential multi-target-directed ligands by targeting host-specific SARS-CoV-2 structurally conserved main protease. J. Biomol. Struct. Dyn. 2021, 39, 3099–3114. 10.1080/07391102.2020.1760137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alaaeldin R.; Mustafa M.; Abuo-Rahma G. E.-D. A.; Fathy M. In vitro inhibition and molecular docking of a new ciprofloxacin chalcone against SARS-CoV-2 main protease. Fundam. Clin. Pharmacol. 2022, 36, 160. 10.1111/fcp.12708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes M. S.; da Silva F. S.; Freitas A. C. S.; de Melo E. B.; Trossini G. H.; Paula F. R. Insights on 3D structures of potential drug-targeting proteins of SARS-CoV-2: application of cavity search and molecular docking. Mol. Inform. 2021, 40, 2000096. 10.1002/minf.202000096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu H.; Zheng Y.; Wang Y.; Li H.; Liu X.; Xiong X.; Zhang L.; Gu J.; Yang G.; Zhu Z.; et al. The potential effects of clinical antidiabetic agents on SARS-CoV-2. J. Diabetes 2021, 13, 243–252. 10.1111/1753-0407.13135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikanovic T.; Sehercehajic E.; Saric B.; Tomic N.; Hadziselimovic R.. New Technologies, Development and Application IV; Springer, 2021; pp 897–903. [Google Scholar]
- Marinho E. M.; de Andrade Neto J. B.; Silva J.; da Silva C. R.; Cavalcanti B. C.; Marinho E. S.; Júnior H. V. N. Virtual screening based on molecular docking of possible inhibitors of COVID-19 main protease. Microb. Pathog. 2020, 148, 104365. 10.1016/j.micpath.2020.104365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi A. J.; Rupareliya J. D.; Shukla V.; Donga S. B.; Acharya R. An Ayurvedic Perspective along with in Silico Study of the Drugs for the Management of SARS-CoV-2. J. Ayurveda Integr Med. 2020, 100343. 10.1016/j.jaim.2020.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usman A.; Uzairu A.; Uba S.; Shallangwa G. A. Molecular Docking Analysis of Chloroquine and Hydroxychloroquine and Design of Anti SARS-CoV2 Protease. SSRN J. 2020, 14. 10.2139/ssrn.3601295. [DOI] [Google Scholar]
- Das S.; Sarmah S.; Lyndem S.; Singha Roy A. An investigation into the identification of potential inhibitors of SARS-CoV-2 main protease using molecular docking study. J. Biomol. Struct. Dyn. 2020, 1–18. 10.1080/07391102.2020.1763201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamsi A.; Mohammad T.; Anwar S.; AlAjmi M. F.; Hussain A.; Rehman M.; Islam A.; Hassan M.; et al. Glecaprevir and Maraviroc are high-affinity inhibitors of SARS-CoV-2 main protease: possible implication in COVID-19 therapy. Biosci. Rep. 2020, 40, 40. 10.1042/BSR20201256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mithilesh S.; Raghunandan D.; Suresh P. In-Silico Identification of Natural Compounds from Traditional Medicine as Potential Drug Leads against SARS-CoV-2 Through Virtual Screening. Proc. Natl. Acad. Sci. India Sect B Biol. Sci. 2022, 92, 81. 10.1007/s40011-021-01292-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar D.; Chandel V.; Raj S.; Rathi B. In silico identification of potent FDA approved drugs against Coronavirus COVID-19 main protease: A drug repurposing approach. Chem. Biol. Lett. 2020, 7, 166–175. [Google Scholar]
- Mazzini S.; Musso L.; Dallavalle S.; Artali R. Putative SARS-CoV-2 Mpro Inhibitors from an In-House Library of Natural and Nature-Inspired Products: A Virtual Screening and Molecular Docking Study. Molecules 2020, 25, 3745. 10.3390/molecules25163745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangsinth P.; Sillapachaiyaporn C.; Nilkhet S.; Tencomnao T.; Ung A. T.; Chuchawankul S. Mushroom-derived bioactive compounds potentially serve as the inhibitors of SARS-CoV-2 main protease: An in silico approach. J. Tradit. Complement. Med. 2021, 11, 158–172. 10.1016/j.jtcme.2020.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saraswat J.; Singh P.; Patel R. A computational approach for the screening of potential antiviral compounds against SARS-CoV-2 protease: Ionic liquid vs herbal and natural compounds. J. Mol. Liq. 2021, 326, 115298. 10.1016/j.molliq.2021.115298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samy M. N.; Attia E. Z.; Shoman M. E.; Khalil H. E.; Sugimoto S.; Matsunami K.; Fahim J. R. Phytochemical investigation of Amphilophium paniculatum; an underexplored Bignoniaceae species as a source of SARS-CoV-2 Mpro inhibitory metabolites: isolation, identification, and molecular docking study. S. Afr. J. Bot. 2021, 141, 421–430. 10.1016/j.sajb.2021.05.023. [DOI] [Google Scholar]
- Vicidomini C.; Roviello V.; Roviello G. N. In Silico Investigation on the Interaction of Chiral Phytochemicals from Opuntia ficus-indica with SARS-CoV-2 Mpro. Symmetry 2021, 13, 1041. 10.3390/sym13061041. [DOI] [Google Scholar]
- Umar H. I.; Josiah S. S.; Saliu T. P.; Jimoh T. O.; Ajayi A.; Danjuma J. B. In-silico analysis of the inhibition of the SARS-CoV-2 main protease by some active compounds from selected African plants. J. Taibah Univ. Medical Sci. 2021, 16, 162–176. 10.1016/j.jtumed.2020.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surya U. R.; Praveen N. A molecular docking study of SARS-CoV-2 main protease against phytochemicals of Boerhavia diffusa Linn. for novel COVID-19 drug discovery. VirusDisease 2021, 32, 46–54. 10.1007/s13337-021-00683-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Aissouq A.; Chedadi O.; Bouachrine M.; Ouammou A. Identification of Novel SARS-CoV-2 Inhibitors: A Structure-Based Virtual Screening Approach. J. Chem. 2021, 2021, 1901484. 10.1155/2021/1901484. [DOI] [Google Scholar]
- Vijayaraj R.; Altaff K.; Rosita A. S.; Ramadevi S.; Revathy J. Bioactive compounds from marine resources against novel corona virus (2019-nCoV): in silico study for corona viral drug. Nat. Prod. Res. 2021, 35, 5525–5529. 10.1080/14786419.2020.1791115. [DOI] [PubMed] [Google Scholar]
- Mpiana P. T.; Tshibangu D. S.; Kilembe J. T.; Gbolo B. Z.; Mwanangombo D. T.; Inkoto C. L.; Lengbiye E. M.; Mbadiko C. M.; Matondo A.; Bongo G. N.; et al. Identification of potential inhibitors of SARS-CoV-2 main protease from Aloe vera compounds: a molecular docking study. Chem. Phys. Lett. 2020, 754, 137751. 10.1016/j.cplett.2020.137751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aanouz I.; Belhassan A.; El-Khatabi K.; Lakhlifi T.; El-Ldrissi M.; Bouachrine M. Moroccan Medicinal plants as inhibitors against SARS-CoV-2 main protease: Computational investigations. J. Biomol. Struct. Dyn. 2021, 39, 2971. 10.1080/07391102.2020.1758790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enmozhi S. K.; Raja K.; Sebastine I.; Joseph J. Andrographolide as a potential inhibitor of SARS-CoV-2 main protease: an in silico approach. J. Biomol. Struct. Dyn. 2020, 1–7. 10.1080/07391102.2020.1760136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampangi-Ramaiah M. H.; Vishwakarma R.; Shaanker R. U. Molecular docking analysis of selected natural products from plants for inhibition of SARS-CoV-2 main protease. Curr. Sci. 2020, 118, 1087–1092. 10.18520/cs/v118/i7/1087-1092. [DOI] [Google Scholar]
- Rehman M.; AlAjmi M. F.; Hussain A.; et al. Natural compounds as inhibitors of SARS-CoV-2 main protease (3CLpro): A molecular docking and simulation approach to combat COVID-19. Curr. Pharm. Des. 2021, 27, 3577–3589. 10.2174/1381612826999201116195851. [DOI] [PubMed] [Google Scholar]
- Khalifa I.; Zhu W.; Mohammed H. H. H.; Dutta K.; Li C. Tannins inhibit SARS-CoV-2 through binding with catalytic dyad residues of 3CLpro: An in silico approach with 19 structural different hydrolysable tannins. J. Food Biochem. 2020, 44, e13432 10.1111/jfbc.13432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojo O. A.; Ojo A. B.; Taiwo O. A.; Oluba O. M. Novel Coronavirus (SARS-CoV-2) Main Protease: Molecular docking of Puerarin as a Potential inhibitor. Malays. J. Biochem. Mol. 2021, 24, 108–114. [Google Scholar]
- Abd El-Mordy F. M.; El-Hamouly M. M.; Ibrahim M. T.; El-Rheem G. A.; Aly O. M.; Abd El-kader A. M.; Youssif K. A.; Abdelmohsen U. R. Inhibition of SARS-CoV-2 main protease by phenolic compounds from Manilkara hexandra (Roxb.) Dubard assisted by metabolite profiling and in silico virtual screening. RSC Adv. 2020, 10, 32148–32155. 10.1039/D0RA05679K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaerunnisa S.; Kurniawan H.; Awaluddin R.; Suhartati S.; Soetjipto S. Potential inhibitor of COVID-19 main protease (Mpro) from several medicinal plant compounds by molecular docking study. Preprints 2020, 20944, 1–14. 10.20944/preprints202003.0226.v1. [DOI] [Google Scholar]
- Santos-Filho O. A. Identification of Potential Inhibitors of Severe Acute Respiratory Syndrome-Related Coronavirus 2 (SARS-CoV-2) Main Protease from Non-Natural and Natural Sources: A Molecular Docking Study. J. Braz. Chem. Soc. 2020, 31, 2638–2643. 10.21577/0103-5053.20200139. [DOI] [Google Scholar]
- Tsuji M. Potential anti-SARS-CoV-2 drug candidates identified through virtual screening of the ChEMBL database for compounds that target the main coronavirus protease. FEBS Open Bio 2020, 10, 995–1004. 10.1002/2211-5463.12875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaulton A.; Bellis L. J.; Bento A. P.; Chambers J.; Davies M.; Hersey A.; Light Y.; McGlinchey S.; Michalovich D.; Al-Lazikani B.; et al. ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012, 40, D1100–D1107. 10.1093/nar/gkr777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manelfi C.; Gossen J.; Gervasoni S.; Talarico C.; Albani S.; Philipp B. J.; Musiani F.; Vistoli G.; Rossetti G.; Beccari A. R.; et al. Combining Different Docking Engines and Consensus Strategies to Design and Validate Optimized Virtual Screening Protocols for the SARS-CoV-2 3CL Protease. Molecules 2021, 26, 797. 10.3390/molecules26040797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udrea A.-M.; Avram S.; Nistorescu S.; Pascu M.-L.; Romanitan M. O. Laser irradiated phenothiazines: New potential treatment for COVID-19 explored by molecular docking. J. Photochem. Photobiol. B: Biol. 2020, 211, 111997. 10.1016/j.jphotobiol.2020.111997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghaleb A.; Aouidate A.; Ayouchia H. B. E.; Aarjane M.; Anane H.; Stiriba S.-E. In silico molecular investigations of pyridine N-Oxide compounds as potential inhibitors of SARS-CoV-2:3D QSAR, molecular docking modeling, and ADMET screening. J. Biomol. Struct. Dyn. 2022, 40, 143–153. 10.1080/07391102.2020.1808530. [DOI] [PubMed] [Google Scholar]
- Scior T.; Abdallah H. H.; Mustafa S. F. Z.; Guevara-García J. A.; Rehder D. Are vanadium complexes druggable against the main protease Mpro of SARS-CoV-2?–A computational approach. Inorg. Chim. Acta 2021, 519, 120287. 10.1016/j.ica.2021.120287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar V.; Roy K. Development of a simple, interpretable and easily transferable QSAR model for quick screening antiviral databases in search of novel 3C-like protease (3CLpro) enzyme inhibitors against SARS-CoV diseases. SAR QSAR Environ. Res. 2020, 31, 511–526. 10.1080/1062936X.2020.1776388. [DOI] [PubMed] [Google Scholar]
- Cherrak S. A.; Merzouk H.; Mokhtari-Soulimane N. Potential bioactive glycosylated flavonoids as SARS-CoV-2 main protease inhibitors: A molecular docking and simulation studies. PLoS One 2020, 15, e0240653 10.1371/journal.pone.0240653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owis A. I.; El-Hawary M. S.; El Amir D.; Aly O. M.; Abdelmohsen U. R.; Kamel M. S. Molecular docking reveals the potential of Salvadora persica flavonoids to inhibit COVID-19 virus main protease. RSC Adv. 2020, 10, 19570–19575. 10.1039/D0RA03582C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chhetri A.; Chettri S.; Rai P.; Sinha B.; Brahman D. Exploration of inhibitory action of Azo imidazole derivatives against COVID-19 main protease (Mpro): A computational study. J. Mol. Struct. 2021, 1224, 129178. 10.1016/j.molstruc.2020.129178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P.; Liu H.; Sun Q.; Liang H.; Li C.; Deng X.; Liu Y.; Lai L. Potent inhibitors of SARS-CoV-2 3C-like protease derived from N-substituted isatin compounds. Eur. J. Med. Chem. 2020, 206, 112702. 10.1016/j.ejmech.2020.112702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad V. A Molecular Docking Study against COVID-19 Protease with a Pomegranate Phyto-Constituents’ Urolithin’and Other Repurposing Drugs: From a Supplement to Ailment. J. Pharm. Res. Int. 2020, 51–62. 10.9734/jpri/2020/v32i1130545. [DOI] [Google Scholar]
- Llanes A.; Cruz H.; Nguyen V. D.; Larionov O. V.; Fernández P. L. A computational approach to explore the interaction of semisynthetic nitrogenous heterocyclic compounds with the SARS-CoV-2 main protease. Biomolecules 2021, 11, 18. 10.3390/biom11010018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soulère L.; Barbier T.; Queneau Y. Docking-based virtual screening studies aiming at the covalent inhibition of SARS-CoV-2 MPro by targeting the cysteine 145. Comput. Biol. Chem. 2021, 92, 107463. 10.1016/j.compbiolchem.2021.107463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega J. T.; Serrano M. L.; Pujol F. H.; Rangel H. R. Role of changes in SARS-CoV-2 spike protein in the interaction with the human ACE2 receptor: An in silico analysis. EXCLI J. 2020, 19, 410–417. 10.17179/excli2020-1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrapp D.; De Vlieger D.; Corbett K. S.; Torres G. M.; Wang N.; Van Breedam W.; Roose K.; van Schie L.; COVID V.-C.; Team R.; et al. Structural basis for potent neutralization of betacoronaviruses by single-domain camelid antibodies. Cell 2020, 181, 1004–1015. 10.1016/j.cell.2020.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rendon-Marin S.; Martinez-Gutierrez M.; Whittaker G. R.; Jaimes J. A.; Ruiz-Saenz J. SARS CoV-2 Spike protein in silico interaction with ACE2 receptors from wild and domestic species. Front. Genet. 2021, 12, 27. 10.3389/fgene.2021.571707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calcagnile M.; Forgez P.; Iannelli A.; Bucci C.; Alifano M.; Alifano P. Molecular docking simulation reveals ACE2 polymorphisms that may increase the affinity of ACE2 with the SARS-CoV-2 Spike protein. Biochimie 2021, 180, 143–148. 10.1016/j.biochi.2020.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron K.; Rozano L.; Falasca M.; Mancera R. L. Does the SARS-CoV-2 Spike Protein Receptor Binding Domain Interact Effectively with the DPP4 (CD26) Receptor? A Molecular Docking Study. Int. J. Mol. Sci. 2021, 22, 7001. 10.3390/ijms22137001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Idrees D.; Kumar V. SARS-CoV-2 spike protein interactions with amyloidogenic proteins: Potential clues to neurodegeneration. Biochem. Biophys. Res. Commun. 2021, 554, 94–98. 10.1016/j.bbrc.2021.03.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazybay B.; Ahmad A.; Mu C.; Mengdesh D.; Xie Y. Omicron N501Y mutation among SARS-CoV-2 lineages: In-silico analysis of potent binding to tyrosine kinase and hypothetical repurposed medicine. Travel Med. Infect Dis 2022, 45, 102242. 10.1016/j.tmaid.2021.102242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanai T. Quantitative in silico analysis of SARS-CoV-2 S-RBD omicron mutant transmissibility. Talanta 2022, 240, 123206. 10.1016/j.talanta.2022.123206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abo-Zeid Y.; Ismail N. S.; McLean G. R.; Hamdy N. M. A molecular docking study repurposes FDA approved iron oxide nanoparticles to treat and control COVID-19 infection. Eur. J. Pharm. Sci. 2020, 153, 105465. 10.1016/j.ejps.2020.105465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miroshnychenko K. V.; Shestopalova A. V. Combined use of the hepatitis C drugs and amentoflavone could interfere with binding of the spike glycoprotein of SARS-CoV-2 to ACE2: the results of a molecular simulation study. J. Biomol. Struct. Dyn. 2021, 1–15. 10.1080/07391102.2021.1914168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toor H. G.; Banerjee D. I.; Rath S. L.; Darji S. A. Computational drug re-purposing targeting the spike glycoprotein of SARS-CoV-2 as an effective strategy to neutralize COVID-19. Eur. J. Pharmacol. 2021, 890, 173720. 10.1016/j.ejphar.2020.173720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baig A. M.; Khaleeq A.; Syeda H.; Bibi N. Docking Prediction of Levodopa in the Receptor Binding Domain of Spike Protein of SARS-CoV-2. ACS Pharmacol. Transl. Sci. 2021, 4, 406–409. 10.1021/acsptsci.1c00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaikh V. S.; Shaikh Y.; Ahmed K.. Lopinavir as a Potential Inhibitor for SARS-CoV-2 Target Protein: A Molecular Docking Study; SSRN, 2020; DOI: 10.2139/ssrn.3596820. [DOI] [Google Scholar]
- Subbaiyan A.; Ravichandran K.; Singh S. V.; Sankar M.; Thomas P.; Dhama K.; Malik Y. S.; Singh R. K.; Chaudhuri P. In silico molecular docking analysis targeting SARS-CoV-2 spike protein and selected herbal constituents. J. Pure Appl. Microbiol 2020, 14, 989–998. 10.22207/JPAM.14.SPL1.37. [DOI] [Google Scholar]
- Mondal S.; Karmakar A.; Mallick T.; Begum N. A. Exploring the efficacy of naturally occurring biflavone based antioxidants towards the inhibition of the SARS-CoV-2 spike glycoprotein mediated membrane fusion. Virology 2021, 556, 133–139. 10.1016/j.virol.2021.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallah M. S.; Bayati M.; Najafi A.; Behmard E.; Javad S. Molecular Docking Investigation of Antiviral Herbal Compounds as Potential Inhibitors of SARS-CoV-2 Spike Receptor. Biointerface Res. Appl. Chem. 2021, 11, 12916–12924. 10.33263/BRIAC115.1291612924. [DOI] [Google Scholar]
- Devasia R. M.; Altaf M.; Alrefaei A. F.; Manoharadas S. Enhanced production of camptothecin by immobilized callus of Ophiorrhiza mungos and a bioinformatic insight into its potential antiviral effect against SARS-CoV-2. J. King Saud Univ. Sci. 2021, 33, 101344. 10.1016/j.jksus.2021.101344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiti S.; Banerjee A. Epigallocatechin gallate and theaflavin gallate interaction in SARS-CoV-2 spike-protein central channel with reference to the hydroxychloroquine interaction: bioinformatics and molecular docking study. Drug Dev. Res. 2021, 82, 86–96. 10.1002/ddr.21730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jomhori M.; Mosaddeghi H. Molecular modeling of natural and synthesized inhibitors against SARS-CoV-2 spike glycoprotein. Res. Biomed. Eng. 2021, 1–16. 10.1007/s42600-020-00122-3. [DOI] [Google Scholar]
- Mohebbi A.; Askari F. S.; Sammak A. S.; Ebrahimi M.; Najafimemar Z. Druggability of cavity pockets within SARS-CoV-2 spike glycoprotein and pharmacophore-based drug discovery. Future Virol 2021, 16, 389–397. 10.2217/fvl-2020-0394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fakih T. M. Dermaseptin-Based Antiviral Peptides to Prevent COVID-19 through In Silico Molecular Docking Studies against SARS-CoV-2 Spike Protein. Pharm. Sci. Res. 2020, 7, 65–70. 10.7454/psr.v7i4.1079. [DOI] [Google Scholar]
- Patil S.; Hofer J.; Ballester P. J.; Fattakhova E.; DiFlumeri J.; Campbell A.; Oravic M. Drug repurposing for COVID-19: Discovery of potential small-molecule inhibitors of spike protein-ACE2 receptor interaction through virtual screening and consensus scoring. ChemRxiv 2020, 10.26434/chemrxiv.12482435.v1. [DOI] [Google Scholar]
- Ahmad J.; Ikram S.; Ahmad F.; Rehman I. U.; Mushtaq M. SARS-CoV-2 RNA Dependent RNA polymerase (RdRp)–A drug repurposing study. Heliyon 2020, 6, e04502 10.1016/j.heliyon.2020.e04502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beg M.; Athar F. Anti-HIV and Anti-HCV drugs are the putative inhibitors of RNA-dependent-RNA polymerase activity of NSP12 of the SARS CoV-2 (COVID-19). Pharm. Pharmacol Int. J. 2020, 8, 163–172. 10.15406/ppij.2020.08.00292. [DOI] [Google Scholar]
- Aftab S. O.; Ghouri M. Z.; Masood M. U.; Haider Z.; Khan Z.; Ahmad A.; Munawar N. Analysis of SARS-CoV-2 RNA-dependent RNA polymerase as a potential therapeutic drug target using a computational approach. J. Transl. Med. 2020, 18, 1–15. 10.1186/s12967-020-02439-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lung J.; Lin Y.-S.; Yang Y.-H.; Chou Y.-L.; Shu L.-H.; Cheng Y.-C.; Liu H. T.; Wu C.-Y. The potential chemical structure of anti-SARS-CoV-2 RNA-dependent RNA polymerase. J. Med. Virol. 2020, 92, 693–697. 10.1002/jmv.25761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandeya K.; Ganeshpurkar A.; Mishra M. K. Natural RNA dependent RNA polymerase inhibitors: Molecular docking studies of some biologically active alkaloids of Argemone mexicana. Med. Hypotheses 2020, 144, 109905. 10.1016/j.mehy.2020.109905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parvez M. S. A.; Karim M. A.; Hasan M.; Jaman J.; Karim Z.; Tahsin T.; Hasan M. N.; Hosen M. J. Prediction of potential inhibitors for RNA-dependent RNA polymerase of SARS-CoV-2 using comprehensive drug repurposing and molecular docking approach. Int. J. Biol. Macromol. 2020, 163, 1787–1797. 10.1016/j.ijbiomac.2020.09.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elfiky A. A. Ribavirin, Remdesivir, Sofosbuvir, Galidesivir, and Tenofovir against SARS-CoV-2 RNA dependent RNA polymerase (RdRp): A molecular docking study. Life Sci. 2020, 253, 117592. 10.1016/j.lfs.2020.117592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heydargoy M. H. Investigation of Antiviral Drugs with Direct Effect on RNA Polymerases and Simulation of Their Binding to SARS-CoV-2 (COVID-19) RNA-Dependent RNA Polymerase by Molecular Docking Method. Iran J. Med. Microbiol 2020, 14, 342–347. 10.30699/ijmm.14.4.342. [DOI] [Google Scholar]
- Mondal S. K.; Mukhoty S.; Kundu H.; Ghosh S.; Sen M. K.; Das S.; Brogi S. In silico analysis of RNA-dependent RNA polymerase of the SARS-CoV-2 and therapeutic potential of existing antiviral drugs. Comput. Biol. Med. 2021, 135, 104591. 10.1016/j.compbiomed.2021.104591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhury S.; Moulick D.; Borah A.; Saikia P.; Mazumder M. K. In search of drugs to alleviate suppression of the host’s innate immune responses against SARS-CoV-2 using a molecular modeling approach. In Silico Pharmacol 2021, 9, 1–9. 10.1007/s40203-021-00085-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D.; Luan J.; Zhang L. Molecular docking of potential SARS-CoV-2 papain-like protease inhibitors. Biochem. Biophys. Res. Commun. 2021, 538, 72–79. 10.1016/j.bbrc.2020.11.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohideen A. K. S. Molecular docking analysis of phytochemical thymoquinone as a therapeutic agent on SARS-CoV-2 envelope protein. Biointerface Res. Appl. Chem. 2021, 11, 8389–8401. 10.33263/BRIAC111.83898401. [DOI] [Google Scholar]
- Borgio J. F.; Alsuwat H. S.; Al Otaibi W. M.; Ibrahim A. M.; Almandil N. B.; Al Asoom L. I.; Salahuddin M.; Kamaraj B.; AbdulAzeez S. State-of-the-art tools unveil potent drug targets amongst clinically approved drugs to inhibit helicase in SARS-CoV-2. Arch Med. Sci. 2020, 16, 508. 10.5114/aoms.2020.94567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmud S.; Elfiky A. A.; Amin A.; Mohanto S. C.; Rahman E.; Acharjee U. K.; Saleh A. Targeting SARS-CoV-2 nonstructural protein 15 endoribonuclease: an in silico perspective. Future Virol 2021, 16, 467–474. 10.2217/fvl-2020-0233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan M. T.; Zeb M. T.; Ahsan H.; Ahmed A.; Ali A.; Akhtar K.; Malik S. I.; Cui Z.; Ali S.; Khan A. S.; et al. SARS-CoV-2 nucleocapsid and Nsp3 binding: an in silico study. Arch. Microbiol. 2021, 203, 59–66. 10.1007/s00203-020-01998-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande R. R.; Tiwari A. P.; Nyayanit N.; Modak M. In silico molecular docking analysis for repurposing therapeutics against multiple proteins from SARS-CoV-2. Eur. J. Pharmacol. 2020, 886, 173430. 10.1016/j.ejphar.2020.173430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman M. R.; Banik A.; Chowdhury I. M.; Sajib E. H.; Sarkar S. Identification of potential antivirals against SARS-CoV-2 using virtual screening method. Inform. Med. Unlocked 2021, 23, 100531. 10.1016/j.imu.2021.100531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T.-F.; Chang Y.-C.; Hsiao Y.; Lee K.-H.; Hsiao Y.-C.; Lin Y.-H.; Tu Y.-C. E.; Huang H.-C.; Chen C.-Y.; Juan H.-F. DockCoV2: a drug database against SARS-CoV-2. Nucleic Acids Res. 2021, 49, D1152–D1159. 10.1093/nar/gkaa861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandel V.; Sharma P. P.; Raj S.; Choudhari R.; Rathi B.; Kumar D. Structure-based drug repurposing for targeting Nsp9 replicase and spike proteins of severe acute respiratory syndrome coronavirus 2. J. Biomol. Struct. Dyn. 2022, 40, 249–262. 10.1080/07391102.2020.1811773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmezayen A. D.; Al-Obaidi A.; Şahin A. T.; Yelekçi K. Drug repurposing for coronavirus (COVID-19): in silico screening of known drugs against coronavirus 3CL hydrolase and protease enzymes. J. Biomol. Struct. Dyn. 2021, 39, 2980–2992. 10.1080/07391102.2020.1758791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guedes I. A.; Costa L. S.; Dos Santos K. B.; Karl A. L.; Rocha G. K.; Teixeira I. M.; Galheigo M. M.; Medeiros V.; Krempser E.; Custódio F. L.; et al. Drug design and repurposing with DockThor-VS web server focusing on SARS-CoV-2 therapeutic targets and their non-synonym variants. Sci. Rep. 2021, 11, 1–20. 10.1038/s41598-021-84700-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koifman O. I.; Lebedeva N. S.; Gubarev Y. A.; Koifman M. O. Modeling the binding of protoporphyrin IX, verteporfin, and chlorin e6 to SARS-CoV-2 proteins. Chem. Heterocycl. Compd. 2021, 57, 423–431. 10.1007/s10593-021-02920-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Da Silva J. K. R.; Figueiredo P. L. B.; Byler K. G.; Setzer W. N. Essential Oils as Antiviral Agents, Potential of Essential Oils to Treat SARS-CoV-2 Infection: An In-Silico Investigation. Int. J. Mol. Sci. 2020, 21, 3426. 10.3390/ijms21103426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurakkal L.; Singh S.; Roy R.; Kar P.; Sadhukhan S.; Porel M. An in-silico study on selected organosulfur compounds as potential drugs for SARS-CoV-2 infection via binding multiple drug targets. Chem. Phys. Lett. 2021, 763, 138193. 10.1016/j.cplett.2020.138193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkhatip A. A. A. M. M.; Georgakis M.; Montero Valenzuela L. R.; Hamza M.; Farag E.; Hodgkinson J.; Hosny H.; Kamal A. M.; Wagih M.; Naguib A.; et al. Metal-Bound Methisazone; Novel Drugs Targeting Prophylaxis and Treatment of SARS-CoV-2, a Molecular Docking Study. Int. J. Mol. Sci. 2021, 22, 2977. 10.3390/ijms22062977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman F.; Tabrez S.; Ali R.; Alqahtani A. S.; Ahmed M. Z.; Rub A. Molecular docking analysis of rutin reveals possible inhibition of SARS-CoV-2 vital proteins. J. Tradit. Complement. Med. 2021, 11, 173–179. 10.1016/j.jtcme.2021.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natesh J.; Mondal P.; Kaur B.; Salam A. A. A.; Kasilingam S.; Meeran S. M. Promising phytochemicals of traditional Himalayan medicinal plants against putative replication and transmission targets of SARS-CoV-2 by computational investigation. Comput. Biol. Med. 2021, 133, 104383. 10.1016/j.compbiomed.2021.104383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nallusamy S.; Mannu J.; Ravikumar C.; Angamuthu K.; Nathan B.; Nachimuthu K.; Ramasamy G.; Muthurajan R.; Subbarayalu M.; Neelakandan K. Exploring Phytochemicals of Traditional Medicinal Plants Exhibiting Inhibitory Activity Against Main Protease, Spike Glycoprotein, RNA-dependent RNA Polymerase and Non-Structural Proteins of SARS-CoV-2 Through Virtual Screening. Front. Pharmacol. 2021, 12, 1704. 10.3389/fphar.2021.667704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ameen F.; Mamidala E.; Davella R.; Vallala S. Rilpivirine inhibits SARS-CoV-2 protein targets: A potential multi-target drug. J. Infect. Public Health 2021, 14, 1454–1460. 10.1016/j.jiph.2021.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vardhan S.; Sahoo S. K. In silico ADMET and molecular docking study on searching potential inhibitors from limonoids and triterpenoids for COVID-19. Comput. Biol. Med. 2020, 124, 103936. 10.1016/j.compbiomed.2020.103936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimgampalle M.; Devanathan V.; Saxena A. Screening of Chloroquine, Hydroxychloroquine and its derivatives for their binding affinity to multiple SARS-CoV-2 protein drug targets. J. Biomol. Struct. Dyn. 2021, 39, 4949–4961. 10.1080/07391102.2020.1782265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laksmiani N. P. L.; Larasanty L. P. F.; Santika A. A. G. J.; Prayoga P. A. A.; Dewi A. A. I. K.; Dewi N. P. A. K. Active Compounds Activity from the Medicinal Plants Against SARS-CoV-2 using in Silico Assay. Biomed. Pharmacol. J. 2020, 13, 873–881. 10.13005/bpj/1953. [DOI] [Google Scholar]
- Hiremath S.; Kumar H. V.; Nandan M.; Mantesh M.; Shankarappa K.; Venkataravanappa V.; Basha C. J.; Reddy C. L. In silico docking analysis revealed the potential of phytochemicals present in Phyllanthus amarus and Andrographis paniculata, used in Ayurveda medicine in inhibiting SARS-CoV-2. 3 Biotech 2021, 11, 1–18. 10.1007/s13205-020-02578-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nag A.; Banerjee R.; Chowdhury R. R.; Venkatesh C. K. Phytochemicals as potential drug candidates for targeting SARS CoV 2 proteins, an in silico study. VirusDisease 2021, 32, 98–107. 10.1007/s13337-021-00654-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adegbola P. I.; Semire B.; Fadahunsi O. S.; Adegoke A. E. Molecular docking and ADMET studies of Allium cepa, Azadirachta indica and Xylopia aethiopica isolates as potential anti-viral drugs for Covid-19. VirusDisease 2021, 32, 85–97. 10.1007/s13337-021-00682-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tallei T. E.; Tumilaar S. G.; Niode N. J.; Kepel B. J.; Idroes R.; Effendi Y.; Sakib S. A.; Emran T. B.; et al. Potential of plant bioactive compounds as SARS-CoV-2 main protease (Mpro) and spike (S) glycoprotein inhibitors: a molecular docking study. Scientifica 2020, 6307457. 10.1155/2020/6307457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narkhede R. R.; Cheke R. S.; Ambhore J. P.; Shinde S. D. The molecular docking study of potential drug candidates showing anti-COVID-19 activity by exploring of therapeutic targets of SARS-CoV-2. EMJO 2020, 4, 185–195. 10.14744/ejmo.2020.31503. [DOI] [Google Scholar]
- Cubuk H.; Özbİl M. Comparison of clinically approved molecules on SARS-CoV-2 drug target proteins: a molecular docking study. Turkish J. Chem. 2021, 45, 35–41. 10.3906/kim-2008-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harisna A. H.; Nurdiansyah R.; Syaifie P. H.; Nugroho D. W.; Saputro K. E.; Prakoso C. D.; Rochman N. T.; Maulana N. N.; Noviyanto A.; Mardliyati E.; et al. In silico investigation of potential inhibitors to main protease and spike protein of SARS-CoV-2 in propolis. Biochem. Biophys. Rep. 2021, 26, 100969. 10.1016/j.bbrep.2021.100969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messaoudi O.; Gouzi H.; El-Hoshoudy A. N.; Benaceur F.; Patel C.; Goswami D.; Boukerouis D.; Bendahou M. Berries anthocyanins as potential SARS-CoV–2 inhibitors targeting the viral attachment and replication; molecular docking simulation. Egypt. J. Pet. 2021, 30, 33–43. 10.1016/j.ejpe.2021.01.001. [DOI] [Google Scholar]
- Kamaz Z.; Al-Jassani M. J.; Haruna U. Screening of Common Herbal Medicines as Promising Direct Inhibitors of SARS-CoV-2 in Silico. Annu. Res. Rev. Biol. 2020, 53–67. 10.9734/arrb/2020/v35i830260. [DOI] [Google Scholar]
- Yu R.; Chen L.; Lan R.; Shen R.; Li P. Computational screening of antagonists against the SARS-CoV-2 (COVID-19) coronavirus by molecular docking. Int. J. Antimicrob. Agents 2020, 56, 106012. 10.1016/j.ijantimicag.2020.106012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amparo T. R.; Seibert J. B.; Almeida T. C.; Costa F. S.; Silveira B. M.; da Silva G. N.; Dos Santos O. D.; de Souza G. H. In silico approach of secondary metabolites from Brazilian herbal medicines to search for potential drugs against SARS-CoV-2. Phytother Res. 2021, 35, 4297–4308. 10.1002/ptr.7097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurya D. K. Evaluation of Yashtimadhu (Glycyrrhiza glabra) active Phytochemicals Against Novel Coronavirus (SARS-CoV-2). Austin J. Pharmacol. Ther. 2021, 9, 1153. 10.21203/rs.3.rs-26480/v1. [DOI] [Google Scholar]
- Alexpandi R.; De Mesquita J. F.; Pandian S. K.; Ravi A. V. Quinolines-Based SARS-CoV-2 3CLpro and RdRp Inhibitors and Spike-RBD-ACE2 Inhibitor for Drug-Repurposing Against COVID-19: An in silico Analysis. Front. Microbiol. 2020, 11, 1796. 10.3389/fmicb.2020.01796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rameshkumar M. R.; Indu P.; Arunagirinathan N.; Venkatadri B.; El-Serehy H. A.; Ahmad A. Computational selection of flavonoid compounds as inhibitors against SARS-CoV-2 main protease, RNA-dependent RNA polymerase and spike proteins: A molecular docking study. Saudi J. Biol. Sci. 2021, 28, 448–458. 10.1016/j.sjbs.2020.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijayakumar B. G.; Ramesh D.; Joji A.; Kannan T.; et al. In silico pharmacokinetic and molecular docking studies of natural flavonoids and synthetic indole chalcones against essential proteins of SARS-CoV-2. Eur. J. Pharmacol. 2020, 886, 173448. 10.1016/j.ejphar.2020.173448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha S. K.; Shakya A.; Prasad S. K.; Singh S.; Gurav N. S.; Prasad R. S.; Gurav S. S. An in-silico evaluation of different Saikosaponins for their potency against SARS-CoV-2 using NSP15 and fusion spike glycoprotein as targets. J. Biomol. Struct. Dyn. 2020, 39, 3244–3255. 10.1080/07391102.2020.1762741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copertino D. C. Jr; Duarte R. R.; Powell T. R.; de Mulder Rougvie M.; Nixon D. F. Montelukast drug activity and potential against severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). J. Med. Virol. 2021, 93, 187–189. 10.1002/jmv.26299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srikanth L.; Sarma P. V. G. K. Andrographolide binds to spike glycoprotein and RNA-dependent RNA polymerase (NSP12) of SARS-CoV-2 by in silico approach: a probable molecule in the development of anti-coronaviral drug. J. Genet. Eng. Biotechnol. 2021, 19, 1–7. 10.1186/s43141-021-00201-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agrawal L.; Poullikkas T.; Eisenhower S.; Monsanto C.; Bakku R. K.; Chen M.-H.; Kalra R. S. Viroinformatics-based analysis of SARS-CoV-2 core proteins for potential therapeutic targets. Antibodies 2021, 10, 3. 10.3390/antib10010003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adegbola P. I.; Fadahunsi O. S.; Adegbola A. E.; Semire B. In silico studies of Potency and safety assessment of selected trial drugs for the treatment of COVID-19. In Silico Pharmacol 2021, 9, 1–12. 10.1007/s40203-021-00105-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdelmohsen U. R.; Albohy A.; Abdulrazik B. S.; Bayoumi S. A.; Malak L. G.; Khallaf I. S.; Bringmann G.; Farag S. F. Natural coumarins as potential anti-SARS-CoV-2 agents supported by docking analysis. RSC Adv. 2021, 11, 16970–16979. 10.1039/D1RA01989A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gubarev Y. A.; Lebedeva N. S.; Yurina E. S.; Syrbu S. S.; Kiselev A. N.; Lebedev M. A. Possible therapeutic targets and promising drugs based on unsymmetrical hetaryl-substituted porphyrins to combat SARS-CoV-2. J. Pharm. Anal. 2021, 11, 691–698. 10.1016/j.jpha.2021.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Masoudi N. A.; Elias R. S.; Saeed B. Molecular Docking Studies of some Antiviral and Antimalarial Drugs Via Bindings to 3CL-Protease and Polymerase Enzymes of the Novel Coronavirus (SARS-CoV-2). Biointerface Res. Appl. Chem. 2020, 10, 6444–6459. 10.33263/BRIAC105.64446459. [DOI] [Google Scholar]
- Yosri N.; El-Wahed A.; Aida A.; Ghonaim R.; Khattab O. M.; Sabry A.; Ibrahim M. A.; Moustafa M. F.; Guo Z.; Zou X.; et al. Anti-Viral and Immunomodulatory Properties of Propolis: Chemical Diversity, Pharmacological Properties, Preclinical and Clinical Applications, and In Silico Potential against SARS-CoV-2. Foods 2021, 10, 1776. 10.3390/foods10081776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iftikhar H.; Ali H. N.; Farooq S.; Naveed H.; Shahzad-ul-Hussan S. Identification of potential inhibitors of three key enzymes of SARS-CoV2 using computational approach. Comput. Biol. Med. 2020, 122, 103848. 10.1016/j.compbiomed.2020.103848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilson M. K.; Given J. A.; Bush B. L.; McCammon J. A. The statistical-thermodynamic basis for computation of binding affinities: a critical review. Biophys. J. 1997, 72, 1047–1069. 10.1016/S0006-3495(97)78756-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan J.; Cheatham T. E.; Cieplak P.; Kollman P. A.; Case D. A. Continuum solvent studies of the stability of DNA, RNA, and phosphoramidate- DNA helices. J. Am. Chem. Soc. 1998, 120, 9401–9409. 10.1021/ja981844+. [DOI] [Google Scholar]
- Dill K. A. Additivity principles in biochemistry. J. Biol. Chem. 1997, 272, 701–704. 10.1074/jbc.272.2.701. [DOI] [PubMed] [Google Scholar]
- Almlöf M.; Brandsdal B. O.; Åqvist J. Binding affinity prediction with different force fields: examination of the linear interaction energy method. J. Comput. Chem. 2004, 25, 1242–1254. 10.1002/jcc.20047. [DOI] [PubMed] [Google Scholar]
- Eriksson M. A.; Pitera J.; Kollman P. A. Prediction of the binding free energies of new TIBO-like HIV-1 reverse transcriptase inhibitors using a combination of PROFEC, PB/SA, CMC/MD, and free energy calculations. J. Med. Chem. 1999, 42, 868–881. 10.1021/jm980277y. [DOI] [PubMed] [Google Scholar]
- Pitera J. W.; Kollman P. A. Exhaustive mutagenesis in silico: multicoordinate free energy calculations on proteins and peptides. Proteins 2000, 41, 385–397. . [DOI] [PubMed] [Google Scholar]
- Radmer R. J.; Kollman P. A. The application of three approximate free energy calculations methods to structure based ligand design: Trypsin and its complex with inhibitors. J. Comput. Aided Mol. Des. 1998, 12, 215–227. 10.1023/A:1007905722422. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Liu M.; Gao J. Enhanced receptor binding of SARS-CoV-2 through networks of hydrogen-bonding and hydrophobic interactions. Proc. Natl. Acad. Sci. U.S.A. 2020, 117, 13967–13974. 10.1073/pnas.2008209117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luan B.; Wang H.; Huynh T. Enhanced binding of the N501Y-mutated SARS-CoV-2 spike protein to the human ACE2 receptor: insights from molecular dynamics simulations. FEBS Lett. 2021, 595, 1454–1461. 10.1002/1873-3468.14076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S.; Huynh T.; Stauft C. B.; Wang T. T.; Luan B. Structure–Function Analysis of Resistance to Bamlanivimab by SARS-CoV-2 Variants Kappa, Delta, and Lambda. J. Chem. Inf Model 2021, 61, 5133–5140. 10.1021/acs.jcim.1c01058. [DOI] [PubMed] [Google Scholar]
- Ngo S. T.; Quynh Anh Pham N.; Thi Le L.; Pham D.-H.; Vu V. V. Computational Determination of Potential Inhibitors of SARS-CoV-2 Main Protease. J. Chem. Inf Model 2020, 60, 5771–5780. 10.1021/acs.jcim.0c00491. [DOI] [PubMed] [Google Scholar]
- Zhang L.; Zhou R. Structural basis of the potential binding mechanism of remdesivir to SARS-CoV-2 RNA-dependent RNA polymerase. J. Phys. Chem. B 2020, 124, 6955–6962. 10.1021/acs.jpcb.0c04198. [DOI] [PubMed] [Google Scholar]
- Hassan A. R.; Sanad I. M.; Allam A. E.; Abouelela M. E.; Sayed A. M.; Emam S. S.; El-Kousy S. M.; Shimizu K. Chemical constituents from Limonium tubiflorum and their in silico evaluation as potential antiviral agents against SARS-CoV-2. RSC Adv. 2021, 11, 32346–32357. 10.1039/D1RA05927K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allam A. E.; Amen Y.; Ashour A.; Assaf H. K.; Hassan H. A.; Abdel-Rahman I. M.; Sayed A. M.; Shimizu K. In silico study of natural compounds from sesame against COVID-19 by targeting M pro, PL pro and RdRp. RSC Adv. 2021, 11, 22398–22408. 10.1039/D1RA03937G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alhadrami H. A.; Sayed A. M.; Al-Khatabi H.; Alhakamy N. A.; Rateb M. E. Scaffold Hopping of α-Rubromycin Enables Direct Access to FDA-Approved Cromoglicic Acid as a SARS-CoV-2 MPro Inhibitor. Pharmaceuticals 2021, 14, 541. 10.3390/ph14060541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Lemus G. R.; Menéndez C. A.; Alvarado W.; Byléhn F.; de Pablo J. J. Toward wide-spectrum antivirals against coronaviruses: Molecular characterization of SARS-CoV-2 NSP13 helicase inhibitors. Sci. Adv. 2022, 8, eabj4526 10.1126/sciadv.abj4526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bung N.; Krishnan S. R.; Bulusu G.; Roy A. De novo design of new chemical entities for SARS-CoV-2 using artificial intelligence. Future Med. Chem. 2021, 13, 575–585. 10.4155/fmc-2020-0262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namsani S.; Pramanik D.; Khan M. A.; Roy S.; Singh J. Potential drug candidates for SARS-CoV-2 using computational screening and enhanced sampling methods. ChemRxiv 2020, 10.26434/chemrxiv.12254135.v3. [DOI] [Google Scholar]
- Shadrack D. M.; Vuai S. A.; Sahini M. G.; Onoka I. In silico study of the inhibition of SARS-COV-2 viral cell entry by neem tree extracts. RSC Adv. 2021, 11, 26524–26533. 10.1039/D1RA04197E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam N. M.; Pham M. Q.; Ha N. X.; Nam P. C.; Phung H. T. T. Computational estimation of potential inhibitors from known drugs against the main protease of SARS-CoV-2. RSC Adv. 2021, 11, 17478–17486. 10.1039/D1RA02529E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam N. M.; Pham M. Q.; Nguyen H. T.; Hong N. D.; Hien N. K.; Quang D. T.; Phung H. T. T.; Ngo S. T. Potential inhibitors for SARS-CoV-2 Mpro from marine compounds. RSC Adv. 2021, 11, 22206–22213. 10.1039/D1RA03852D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecot S.; Chevolot Y.; Phaner-Goutorbe M.; Yeromonahos C. Curious Binding Energy Increase between the Receptor-Binding Domain of the SARS-CoV-2 Spike Protein and Angiotensin-Converting Enzyme 2 Adsorbed on a Silane Monolayer from Molecular Dynamics Simulations. J. Phys. Chem. B 2021, 125, 11078–11090. 10.1021/acs.jpcb.1c06050. [DOI] [PubMed] [Google Scholar]
- Huey R.; Morris G. M.; Olson A. J.; Goodsell D. S. A semiempirical free energy force field with charge-based desolvation. J. Comput. Chem. 2007, 28, 1145–1152. 10.1002/jcc.20634. [DOI] [PubMed] [Google Scholar]
- Hu X.; Zhou Z.; Li F.; Xiao Y.; Wang Z.; Xu J.; Dong F.; Zheng H.; Yu R. The study of antiviral drugs targeting SARS-CoV-2 nucleocapsid and spike proteins through large-scale compound repurposing. Heliyon 2021, 7, e06387 10.1016/j.heliyon.2021.e06387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorucci D.; Milletti E.; Orofino F.; Brizzi A.; Mugnaini C.; Corelli F. Computational drug repurposing for the identification of SARS-CoV-2 main protease inhibitors. J. Biomol. Struct. Dyn. 2021, 39, 6242–6248. 10.1080/07391102.2020.1796805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim M. A.; Abdelrahman A. H.; Hegazy M.-E. F. In-silico drug repurposing and molecular dynamics puzzled out potential SARS-CoV-2 main protease inhibitors. J. Biomol. Struct. Dyn. 2021, 39, 5756–5767. 10.1080/07391102.2020.1791958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gahlawat A.; Kumar N.; Kumar R.; Sandhu H.; Singh I. P.; Singh S.; Sjöstedt A.; Garg P. Structure-based virtual screening to discover potential lead molecules for the SARS-CoV-2 main protease. J. Chem. Inf Model 2020, 60, 5781–5793. 10.1021/acs.jcim.0c00546. [DOI] [PubMed] [Google Scholar]
- Avti P.; Chauhan A.; Shekhar N.; Prajapat M.; Sarma P.; Kaur H.; Bhattacharyya A.; Kumar S.; Prakash A.; Sharma S.; et al. Computational basis of SARS-CoV 2 main protease inhibition: an insight from molecular dynamics simulation based findings. J. Biomol. Struct. Dyn. 2021, 1–11. 10.1080/07391102.2021.1922310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Khafaji K.; AL-Duhaidahawi D.; Taskin Tok T. Using integrated computational approaches to identify safe and rapid treatment for SARS-CoV-2. J. Biomol. Struct. Dyn. 2021, 39, 3387–3395. 10.1080/07391102.2020.1764392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P.; Vijayan V.; Pant P.; Sharma M.; Vikram N.; Kaur P.; Singh T.; Sharma S. Identification of potential drug candidates to combat COVID-19: a structural study using the main protease (mpro) of SARS-CoV-2. J. Biomol. Struct. Dyn. 2021, 39, 6649–6659. 10.1080/07391102.2020.1798286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marimuthu P.; Gorle S.; Karnati K. R. Mechanistic Insights into SARS-CoV-2 Main Protease Inhibition Reveals Hotspot Residues. J. Chem. Inf Model 2021, 61, 6053–6065. 10.1021/acs.jcim.1c00928. [DOI] [PubMed] [Google Scholar]
- Nath V.; Rohini A.; Kumar V. Identification of Mpro inhibitors of SARS-CoV-2 using structure based computational drug repurposing. Biocatal. Agric. Biotechnol. 2021, 37, 102178. 10.1016/j.bcab.2021.102178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamed M. I.; Darwish K. M.; Soltane R.; Chrouda A.; Mostafa A.; Shama N. M. A.; Elhady S. S.; Abulkhair H. S.; Khodir A. E.; Elmaaty A. A.; et al. β-Blockers bearing hydroxyethylamine and hydroxyethylene as potential SARS-CoV-2 Mpro inhibitors: rational based design, in silico, in vitro, and SAR studies for lead optimization. RSC Adv. 2021, 11, 35536–35558. 10.1039/D1RA04820A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahato R. K.; Mahanty A. K.; Kotakonda M.; Prasad S.; Bhattacharyya S.; Biswas B. A hydrated 2, 3-diaminophenazinium chloride as a promising building block against SARS-CoV-2. Sci. Rep. 2021, 11, 1–17. 10.1038/s41598-021-02280-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al Khoury C.; Bashir Z.; Tokajian S.; Nemer N.; Merhi G.; Nemer G. In silico evidence of beauvericin antiviral activity against SARS-CoV-2. Comput. Biol. Med. 2022, 141, 105171. 10.1016/j.compbiomed.2021.105171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhury M.; Dhanabalan A. K.; Goswami N. Understanding the binding mechanism for potential inhibition of SARS-CoV-2 Mpro and exploring the modes of ACE2 inhibition by hydroxychloroquine. J. Cell. Biochem. 2022, 123, 347–358. 10.1002/jcb.30174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray A. K.; Gupta P. S. S.; Panda S. K.; Biswal S.; Bhattacharya U.; Rana M. K. Repurposing of FDA-approved drugs as potential inhibitors of the SARS-CoV-2 main protease: Molecular insights into improved therapeutic discovery. Comput. Biol. Med. 2022, 142, 105183. 10.1016/j.compbiomed.2021.105183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyaz S.; Tasneem A.; Rai G. P.; Bairagya H. R. Investigation of structural analogs of hydroxychloroquine for SARS-CoV-2 main protease (Mpro): A computational drug discovery study. J. Mol. Graph. Model. 2021, 109, 108021. 10.1016/j.jmgm.2021.108021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y.; Wang Z.; Ren J.; Wei Z.; Li J. Potential inhibitors for the novel coronavirus (SARS-CoV-2). Brief. Bioinformatics 2021, 22, 1225–1231. 10.1093/bib/bbaa209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat Z. A.; Chitara D.; Iqbal J.; Sanjeev B.; Madhumalar A. Targeting allosteric pockets of SARS-CoV-2 main protease Mpro. J. Biomol. Struct 2021, 1–16. 10.1080/07391102.2021.1891141. [DOI] [PubMed] [Google Scholar]
- Chowdhury K. H.; Chowdhury M.; Mahmud S.; Tareq A. M.; Hanif N. B.; Banu N.; Reza A.; Emran T. B.; Simal-Gandara J.; et al. Drug repurposing approach against novel coronavirus disease (COVID-19) through virtual screening targeting SARS-CoV-2 main protease. Biology 2021, 10, 2. 10.3390/biology10010002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novak J.; Rimac H.; Kandagalla S.; Pathak P.; Naumovich V.; Grishina M.; Potemkin V. Proposition of a new allosteric binding site for potential SARS-CoV-2 3CL protease inhibitors by utilizing molecular dynamics simulations and ensemble docking. J. Biomol. Struct 2021, 1–14. 10.1080/07391102.2021.1927845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sisakht M.; Solhjoo A.; Mahmoodzadeh A.; Fathalipour M.; Kabiri M.; Sakhteman A. Potential inhibitors of the main protease of SARS-CoV-2 and modulators of arachidonic acid pathway: Non-steroidal anti-inflammatory drugs against COVID-19. Comput. Biol. Med. 2021, 136, 104686. 10.1016/j.compbiomed.2021.104686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy R.; Sk M. F.; Jonniya N. A.; Poddar S.; Kar P. Finding potent inhibitors against SARS-CoV-2 main protease through virtual screening, ADMET, and molecular dynamics simulation studies. J. Biomol. Struct 2021, 1–13. 10.1080/07391102.2021.1897680. [DOI] [PubMed] [Google Scholar]
- Debnath P.; Bhaumik S.; Sen D.; Muttineni R. K.; Debnath S. Identification of SARS-CoV-2 Main Protease Inhibitors Using Structure Based Virtual Screening and Molecular Dynamics Simulation of DrugBank Database. ChemistrySelect 2021, 6, 4991–5013. 10.1002/slct.202100854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim M. A.; Abdelrahman A. H.; Allemailem K. S.; Almatroudi A.; Moustafa M. F.; Hegazy M.-E. F. In silico evaluation of prospective anti-COVID-19 drug candidates as potential SARS-CoV-2 main protease inhibitors. Protein J. 2021, 40, 296–309. 10.1007/s10930-020-09945-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bera K. Binding and inhibitory effect of ravidasvir on 3CLpro of SARS-CoV-2: a molecular docking, molecular dynamics and MM/PBSA approach. J. Biomol. Struct 2021, 1–8. 10.1080/07391102.2021.1896388. [DOI] [PubMed] [Google Scholar]
- Baby K.; Maity S.; Mehta C. H.; Suresh A.; Nayak U. Y.; Nayak Y. Targeting SARS-CoV-2 main protease: A computational drug repurposing study. Arch. Med. Res. 2021, 52, 38–47. 10.1016/j.arcmed.2020.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sajid Jamal Q. M.; Alharbi A. H.; Ahmad V. Identification of doxorubicin as a potential therapeutic against SARS-CoV-2 (COVID-19) protease: a molecular docking and dynamics simulation studies. J. Biomol. Struct 2021, 1–15. 10.1080/07391102.2021.1905551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azam F.; Eid E. E.; Almutairi A. Targeting SARS-CoV-2 main protease by teicoplanin: a mechanistic insight by docking, MM/GBSA and molecular dynamics simulation. J. Mol. Struct. 2021, 1246, 131124. 10.1016/j.molstruc.2021.131124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinzi L.; Tinivella A.; Caporuscio F.; Rastelli G. Drug repurposing and polypharmacology to fight SARS-CoV-2 through inhibition of the main protease. Front. Pharmacol. 2021, 12, 84. 10.3389/fphar.2021.636989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yañez O.; Osorio M. I.; Uriarte E.; Areche C.; Tiznado W.; Pérez-Donoso J. M.; García-Beltrán O.; González-Nilo F. In Silico Study of Coumarins and Quinolines Derivatives as Potent Inhibitors of SARS-CoV-2 Main Protease. Front. Chem. 2021, 8, 1273. 10.3389/fchem.2020.595097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P.; Bhardwaj T.; Kumar A.; Garg N.; Giri R. One microsecond MD simulations of the SARS-CoV-2 main protease and hydroxychloroquine complex reveal the intricate nature of binding. J. Biomol. Struct. Dyn. 2021, 1–8. 10.1080/07391102.2021.1948447. [DOI] [PubMed] [Google Scholar]
- Swain S. S.; Singh S. R.; Sahoo A.; Hussain T.; Pati S. Anti-HIV-drug and phyto-flavonoid combination against SARS-CoV-2: a molecular docking-simulation base assessment. J. Biomol. Struct. Dyn. 2021, 1–14. 10.1080/07391102.2021.1885495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh V. K.; Chaurasia H.; Kumari P.; Som A.; Mishra R.; Srivastava R.; Naaz F.; Singh A.; Singh R. K. Design, synthesis, and molecular dynamics simulation studies of quinoline derivatives as protease inhibitors against SARS-CoV-2. J. Biomol. Struct. Dyn. 2021, 1–24. 10.1080/07391102.2021.1946716. [DOI] [PubMed] [Google Scholar]
- Vergoten G.; Bailly C. In silico analysis of echinocandins binding to the main proteases of coronaviruses PEDV (3CLpro) and SARS-CoV-2 (Mpro). In Silico Pharmacol 2021, 9, 1–10. 10.1007/s40203-021-00101-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basha G. M.; Parulekar R. S.; Al-Sehemi A. G.; Pannipara M.; Siddaiah V.; Kumari S.; Choudhari P. B.; Tamboli Y. Design and in silico investigation of novel Maraviroc analogues as dual inhibition of CCR-5/SARS-CoV-2 Mpro. J. Biomol. Struct. Dyn. 2021, 1–16. 10.1080/07391102.2021.1955742. [DOI] [PubMed] [Google Scholar]
- De Souza A. S.; de Souza R. F.; Guzzo C. R. Quantitative structure-activity relationships, molecular docking and molecular dynamics simulations reveal drug repurposing candidates as potent SARS-CoV-2 main protease inhibitors. J. Biomol. Struct. Dyn. 2021, 1–18. 10.1080/07391102.2021.1958700. [DOI] [PubMed] [Google Scholar]
- Liu W.-S.; Li H.-G.; Ding C.-H.; Zhang H.-X.; Wang R.-R.; Li J.-Q. Screening potential FDA-approved inhibitors of the SARS-CoV-2 major protease 3CLpro through high-throughput virtual screening and molecular dynamics simulation. Aging (Albany NY) 2021, 13, 6258. 10.18632/aging.202703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nutho B.; Mahalapbutr P.; Hengphasatporn K.; Pattaranggoon N. C.; Simanon N.; Shigeta Y.; Hannongbua S.; Rungrotmongkol T. Why are lopinavir and ritonavir effective against the newly emerged Coronavirus 2019? Atomistic insights into the inhibitory mechanisms. Biochemistry 2020, 59, 1769–1779. 10.1021/acs.biochem.0c00160. [DOI] [PubMed] [Google Scholar]
- Bello M.; Martínez-Muñoz A.; Balbuena-Rebolledo I. Identification of saquinavir as a potent inhibitor of dimeric SARS-CoV2 main protease through MM/GBSA. J. Mol. Model. 2020, 26, 1–11. 10.1007/s00894-020-04600-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sang P.; Tian S.-H.; Meng Z.-H.; Yang L.-Q. Anti-HIV drug repurposing against SARS-CoV-2. RSC Adv. 2020, 10, 15775–15783. 10.1039/D0RA01899F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ancy I.; Sivanandam M.; Kumaradhas P. Possibility of HIV-1 protease inhibitors-clinical trial drugs as repurposed drugs for SARS-CoV-2 main protease: a molecular docking, molecular dynamics and binding free energy simulation study. J. Biomol. Struct. Dyn. 2021, 39, 5368–5375. 10.1080/07391102.2020.1786459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan A.; Ali S. S.; Khan M. T.; Saleem S.; Ali A.; Suleman M.; Babar Z.; Shafiq A.; Khan M.; Wei D.-Q. Combined drug repurposing and virtual screening strategies with molecular dynamics simulation identified potent inhibitors for SARS-CoV-2 main protease (3CLpro). J. Biomol. Struct. Dyn. 2021, 39, 4659–4670. 10.1080/07391102.2020.1779128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee S.; Dasgupta S.; Adhikary T.; Adhikari U.; Panja S. S. Structural insight to hydroxychloroquine-3C-like proteinase complexation from SARS-CoV-2: inhibitor modelling study through molecular docking and MD-simulation study. J. Biomol. Struct. Dyn. 2021, 39, 7322–7334. 10.1080/07391102.2020.1804458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beura S.; Chetti P. In-silico strategies for probing chloroquine based inhibitors against SARS-CoV-2. J. Biomol. Struct. Dyn. 2021, 39, 3747–3739. 10.1080/07391102.2020.1772111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar D.; Kumari K.; Jayaraj A.; Kumar V.; Kumar R. V.; Dass S. K.; Chandra R.; Singh P. Understanding the binding affinity of noscapines with protease of SARS-CoV-2 for COVID-19 using MD simulations at different temperatures. J. Biomol. Struct. Dyn. 2021, 39, 2659–2672. 10.1080/07391102.2020.1752310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayeem S. M.; Reddy M. S. Target SARS-CoV-2: Computation of Binding energies with drugs of Dexamethasone/Umifenovir by Molecular Dynamics using OPLS-AA force field. Res. Biomed. Eng. 2021, 1–10. 10.1007/s42600-020-00119-y. [DOI] [Google Scholar]
- Ibrahim M. A.; Abdeljawaad K. A.; Abdelrahman A. H.; Hegazy M.-E. F. Natural-like products as potential SARS-CoV-2 Mpro inhibitors: in-silico drug discovery. J. Biomol. Struct. Dyn. 2021, 39, 5722–5734. 10.1080/07391102.2020.1790037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapusta K.; Kar S.; Collins J. T.; Franklin L. M.; Kolodziejczyk W.; Leszczynski J.; Hill G. A. Protein reliability analysis and virtual screening of natural inhibitors for SARS-CoV-2 main protease (Mpro) through docking, molecular mechanic & dynamic, and ADMET profiling. J. Biomol. Struct. Dyn. 2021, 39, 6810–6827. 10.1080/07391102.2020.1806930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prajapati J.; Patel R.; Goswami D.; Saraf M.; Rawal R. M. Sterenin M as a potential inhibitor of SARS-CoV-2 main protease identified from MeFSAT database using molecular docking, molecular dynamics simulation and binding free energy calculation. Comput. Biol. Med. 2021, 135, 104568. 10.1016/j.compbiomed.2021.104568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmud S.; Uddin M. A. R.; Zaman M.; Sujon K. M.; Rahman M. E.; Shehab M. N.; Islam A.; Alom M. W.; Amin A.; Akash A. S.; et al. Molecular docking and dynamics study of natural compound for potential inhibition of main protease of SARS-CoV-2. J. Biomol. Struct. Dyn. 2021, 39, 6281–6289. 10.1080/07391102.2020.1796808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Demerdash A.; Al-Karmalawy A. A.; Abdel-Aziz T. M.; Elhady S. S.; Darwish K. M.; Hassan A. H. Investigating the structure–activity relationship of marine natural polyketides as promising SARS-CoV-2 main protease inhibitors. RSC Adv. 2021, 11, 31339–31363. 10.1039/D1RA05817G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastav A. K.; Jaiswal J.; Kumar U. In silico bioprospecting of antiviral compounds from marine fungi and mushroom for rapid development of nutraceuticals against SARS-CoV-2. J. Biomol. Struct 2021, 1–12. 10.1080/07391102.2021.2023048. [DOI] [PubMed] [Google Scholar]
- Patel C. N.; Jani S. P.; Jaiswal D. G.; Kumar S. P.; Mangukia N.; Parmar R. M.; Rawal R. M.; Pandya H. A. Identification of antiviral phytochemicals as a potential SARS-CoV-2 main protease (Mpro) inhibitor using docking and molecular dynamics simulations. Sci. Rep. 2021, 11, 1–13. 10.1038/s41598-021-99165-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aminah N. S.; Abdjan M. I.; Wardana A. P.; Kristanti A. N.; Siswanto I.; Rakhman K. A.; Takaya Y. The dolabellane diterpenes as potential inhibitors of the SARS-CoV-2 main protease: molecular insight of the inhibitory mechanism through computational studies. RSC Adv. 2021, 11, 39455–39466. 10.1039/D1RA07584E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra A.; Khan W. H.; Rathore A. S. Synergistic Effects of Natural Compounds Toward Inhibition of SARS-CoV-2 3CL Protease. J. Chem. Inf Model 2021, 61, 5708–5718. 10.1021/acs.jcim.1c00994. [DOI] [PubMed] [Google Scholar]
- Zackria A. A.; Pattabiraman R.; Murthy T.; Kumar S. B.; Mathew B. B.; Biju V. G. Computational screening of natural compounds from Salvia plebeia R. Br. for inhibition of SARS-CoV-2 main protease. Vegetos 2021, 1–15. 10.1007/s42535-021-00304-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohapatra P. K.; Chopdar K. S.; Dash G. C.; Mohanty A. K.; Raval M. K. In silico screening and covalent binding of phytochemicals of Ocimum sanctum against SARS-CoV-2 (COVID 19) main protease. J. Biomol. Struct 2021, 1–10. 10.1080/07391102.2021.2007170. [DOI] [PubMed] [Google Scholar]
- Joshi T.; Bhat S.; Pundir H.; Chandra S. Identification of Berbamine, Oxyacanthine and Rutin from Berberis asiatica as anti-SARS-CoV-2 compounds: An in silico study. J. Mol. Graph. Model. 2021, 109, 108028. 10.1016/j.jmgm.2021.108028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakshit M.; Muduli S.; Srivastav P. P.; Mishra S. Pomegranate peel polyphenols prophylaxis against SARS-CoV-2 main protease by in-silico docking and molecular dynamics study. J. Biomol. Struct 2021, 1–15. 10.1080/07391102.2021.1979427. [DOI] [PubMed] [Google Scholar]
- Jha R. K.; Khan R. J.; Parthiban A.; Singh E.; Jain M.; Amera G. M.; Singh R. P.; Ramachandran P.; Ramachandran R.; Sachithanandam V.; et al. Identifying the natural compound Catechin from tropical mangrove plants as a potential lead candidate against 3CLpro from SARS-CoV-2: An integrated in silico approach. J. Biomol. Struct 2021, 1–20. 10.1080/07391102.2021.1988710. [DOI] [PubMed] [Google Scholar]
- Tolah A. M.; Altayeb L. M.; Alandijany T. A.; Dwivedi V. D.; El-Kafrawy S. A.; Azhar E. I. Computational and In Vitro Experimental Investigations Reveal Anti-Viral Activity of Licorice and Glycyrrhizin against Severe Acute Respiratory Syndrome Coronavirus 2. Pharmaceuticals 2021, 14, 1216. 10.3390/ph14121216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingwan M.; Shagun S.; Pahwa F.; Kumar A.; Verma D. K.; Pant Y.; Kamatam L. V.; Kumari B.; Nanda R. K.; Sunil S.; et al. Phytochemical rich Himalayan Rhododendron arboreum petals inhibit SARS-CoV-2 infection in vitro. J. Biomol. Struct 2021, 1–11. 10.1080/07391102.2021.2021287. [DOI] [PubMed] [Google Scholar]
- De Oliveira O. V.; Cristina Andreazza Costa M.; Marques da Costa R.; Giordano Viegas R.; Paluch A. S.; Miguel Castro Ferreira M. Traditional herbal compounds as candidates to inhibit the SARS-CoV-2 main protease: an in silico study. J. Biomol. Struct 2022, 1–14. 10.1080/07391102.2021.2023646. [DOI] [PubMed] [Google Scholar]
- Mahmud S.; Uddin M. A. R.; Paul G. K.; Shimu M. S. S.; Islam S.; Rahman E.; Islam A.; Islam M. S.; Promi M. M.; Emran T. B.; et al. Virtual screening and molecular dynamics simulation study of plant-derived compounds to identify potential inhibitors of main protease from SARS-CoV-2. Brief. Bioinformatics 2021, 22, 1402–1414. 10.1093/bib/bbaa428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah S.; Chaple D.; Arora S.; Yende S.; Mehta C.; Nayak U. Prospecting for Cressa cretica to treat COVID-19 via in silico molecular docking models of the SARS-CoV-2. J. Biomol. Struct 2021, 1–10. 10.1080/07391102.2021.1872419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahamad S.; Kanipakam H.; Birla S.; Ali M. S.; Gupta D. Screening Malaria-box compounds to identify potential inhibitors against SARS-CoV-2 Mpro, using molecular docking and dynamics simulation studies. Eur. J. Pharmacol. 2021, 890, 173664. 10.1016/j.ejphar.2020.173664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma S.; Patel C. N.; Chandra M. Identification of novel inhibitors of SARS-CoV-2 main protease (Mpro) from Withania sp. by molecular docking and molecular dynamics simulation. J. Comput. Chem. 2021, 42, 1861. 10.1002/jcc.26717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen D.; Bhaumik S.; Debnath P.; Debnath S. Potentiality of Moringa oleifera against SARS-CoV-2: identified by a rational computer aided drug design method. J. Biomol. Struct 2021, 1–18. 10.1080/07391102.2021.1898475. [DOI] [PubMed] [Google Scholar]
- Mathpal S.; Sharma P.; Joshi T.; Joshi T.; Pande V.; Chandra S. Screening of potential bio-molecules from Moringa olifera against SARS-CoV-2 main protease using computational approaches. J. Biomol. Struct 2021, 1–12. 10.1080/07391102.2021.1936183. [DOI] [PubMed] [Google Scholar]
- Masand V. H.; Sk M. F.; Kar P.; Rastija V.; Zaki M. E. Identification of Food Compounds as inhibitors of SARS-CoV-2 main protease using molecular docking and molecular dynamics simulations. Chemom. Intell. Lab. Syst. 2021, 217, 104394. 10.1016/j.chemolab.2021.104394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar B.; Parasuraman P.; Murthy T. P. K.; Murahari M.; Chandramohan V. In silico screening of therapeutic potentials from Strychnos nux-vomica against the dimeric main protease (Mpro) structure of SARS-CoV-2. J. Biomol. Struct 2021, 1–19. 10.1080/07391102.2021.1902394. [DOI] [PubMed] [Google Scholar]
- Gogoi B.; Chowdhury P.; Goswami N.; Gogoi N.; Naiya T.; Chetia P.; Mahanta S.; Chetia D.; Tanti B.; Borah P.; et al. Identification of potential plant-based inhibitor against viral proteases of SARS-CoV-2 through molecular docking, MM-PBSA binding energy calculations and molecular dynamics simulation. Mol. Divers. 2021, 25, 1963–1977. 10.1007/s11030-021-10211-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudrapal M.; Issahaku A. R.; Agoni C.; Bendale A. R.; Nagar A.; Soliman M. E.; Lokwani D. In silico screening of phytopolyphenolics for the identification of bioactive compounds as novel protease inhibitors effective against SARS-CoV-2. J. Biomol. Struct 2021, 1–17. 10.1080/07391102.2021.1944909. [DOI] [PubMed] [Google Scholar]
- Mahmud S.; Mita M. A.; Biswas S.; Paul G. K.; Promi M. M.; Afrose S.; Hasan R.; Shimu S. S.; Zaman S.; Uddin S.; et al. Molecular docking and dynamics study to explore phytochemical ligand molecules against the main protease of SARS-CoV-2 from extensive phytochemical datasets. Expert Rev. Clin. Pharmacol. 2021, 14, 1305–1315. 10.1080/17512433.2021.1959318. [DOI] [PubMed] [Google Scholar]
- Bhardwaj V. K.; Singh R.; Das P.; Purohit R. Evaluation of acridinedione analogs as potential SARS-CoV-2 main protease inhibitors and their comparison with repurposed anti-viral drugs. Comput. Biol. Med. 2021, 128, 104117. 10.1016/j.compbiomed.2020.104117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cetin A. In silico studies on stilbenolignan analogues as SARS-CoV-2 Mpro inhibitors. Chem. Phys. Lett. 2021, 771, 138563. 10.1016/j.cplett.2021.138563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bharadwaj S.; El-Kafrawy S. A.; Alandijany T. A.; Bajrai L. H.; Shah A. A.; Dubey A.; Sahoo A. K.; Yadava U.; Kamal M. A.; Azhar E. I.; et al. Structure-Based Identification of Natural Products as SARS-CoV-2 Mpro Antagonist from Echinacea angustifolia Using Computational Approaches. Viruses 2021, 13, 305. 10.3390/v13020305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushwaha P. P.; Singh A. K.; Prajapati K. S.; Shuaib M.; Gupta S.; Kumar S. Phytochemicals present in Indian ginseng possess potential to inhibit SARS-CoV-2 virulence: A molecular docking and MD simulation study. Microb. Pathog. 2021, 157, 104954. 10.1016/j.micpath.2021.104954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ram T. S.; Munikumar M.; Raju V. N.; Devaraj P.; Boiroju N. K.; Hemalatha R.; Prasad P.; Gundeti M.; Sisodia B. S.; Pawar S.; et al. In silico evaluation of the compounds of the ayurvedic drug, AYUSH-64, for the action against the SARS-CoV-2 main protease. J. Ayurveda Integr Med. 2022, 13, 100413. 10.1016/j.jaim.2021.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das P.; Majumder R.; Mandal M.; Basak P. In-Silico approach for identification of effective and stable inhibitors for COVID-19 main protease (Mpro) from flavonoid based phytochemical constituents of Calendula officinalis. J. Biomol. Struct. Dyn. 2021, 39, 6265. 10.1080/07391102.2020.1796799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhardwaj V. K.; Singh R.; Sharma J.; Rajendran V.; Purohit R.; Kumar S. Identification of bioactive molecules from Tea plant as SARS-CoV-2 main protease inhibitors. J. Biomol. Struct. Dyn. 2021, 39, 3449. 10.1080/07391102.2020.1766572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi M. K.; Singh P.; Sharma S.; Singh T. P.; Ethayathulla A.; Kaur P. Identification of bioactive molecule from Withania somnifera (Ashwagandha) as SARS-CoV-2 main protease inhibitor. J. Biomol. Struct. Dyn. 2021, 39, 5668. 10.1080/07391102.2020.1790425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A.; Choudhir G.; Shukla S. K.; Sharma M.; Tyagi P.; Bhushan A.; Rathore M. Identification of phytochemical inhibitors against main protease of COVID-19 using molecular modeling approaches. J. Biomol. Struct. Dyn. 2021, 39, 3760. 10.1080/07391102.2020.1772112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh R.; Chakraborty A.; Biswas A.; Chowdhuri S. Evaluation of green tea polyphenols as novel corona virus (SARS CoV-2) main protease (Mpro) inhibitors–an in silico docking and molecular dynamics simulation study. J. Biomol. Struct. Dyn. 2020, 1–13. 10.1080/07391102.2020.1779818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi T.; Sharma P.; Joshi T.; Pundir H.; Mathpal S.; Chandra S. Structure-based screening of novel lichen compounds against SARS Coronavirus main protease (Mpro) as potentials inhibitors of COVID-19. Mol. Divers. 2021, 25, 1665–1677. 10.1007/s11030-020-10118-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S.; Singh A. K.; Kushwaha P. P.; Prajapati K. S.; Shuaib M.; Senapati S.; Kumar S. Identification of potential natural inhibitors of SARS-CoV2 main protease by molecular docking and simulation studies. J. Biomol. Struct. Dyn. 2021, 39, 4334–4345. 10.1080/07391102.2020.1776157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh R.; Chakraborty A.; Biswas A.; Chowdhuri S. Identification of polyphenols from Broussonetia papyrifera as SARS CoV-2 main protease inhibitors using in silico docking and molecular dynamics simulation approaches. J. Biomol. Struct. Dyn. 2021, 39, 6747–6760. 10.1080/07391102.2020.1802347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrianov A. M.; Kornoushenko Y. V.; Karpenko A. D.; Bosko I. P.; Tuzikov A. V. Computational discovery of small drug-like compounds as potential inhibitors of SARS-CoV-2 main protease. J. Biomol. Struct. Dyn. 2021, 39, 5779–5791. 10.1080/07391102.2020.1792989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paula Vargas Ruiz A.; Jimenez Avalos G.; Delgado N. E.; Olivos Ramirez G.; Sheen P.; Quiliano M.; Zimic M. Comprehensive virtual screening of 4.8 k flavonoids reveals novel insights into allosteric inhibition of SARS-CoV-2 MPRO. Sci. Rep. 2022, 121, 337a. 10.1016/j.bpj.2021.11.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arun K.; Sharanya C.; Abhithaj J.; Francis D.; Sadasivan C. Drug repurposing against SARS-CoV-2 using E-pharmacophore based virtual screening, molecular docking and molecular dynamics with main protease as the target. J. Biomol. Struct. Dyn. 2021, 39, 4647–4658. 10.1080/07391102.2020.1779819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary M. I.; Shaikh M.; tul-Wahab A.; ur-Rahman A. In silico identification of potential inhibitors of key SARS-CoV-2 3CL hydrolase (Mpro) via molecular docking, MMGBSA predictive binding energy calculations, and molecular dynamics simulation. PLoS One 2020, 15, e0235030 10.1371/journal.pone.0235030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan S. A.; Zia K.; Ashraf S.; Uddin R.; Ul-Haq Z. Identification of chymotrypsin-like protease inhibitors of SARS-CoV-2 via integrated computational approach. J. Biomol. Struct. Dyn. 2021, 39, 2607. 10.1080/07391102.2020.1751298. [DOI] [PubMed] [Google Scholar]
- Fakhar Z.; Faramarzi B.; Pacifico S.; Faramarzi S. Anthocyanin derivatives as potent inhibitors of SARS-CoV-2 main protease: An in-silico perspective of therapeutic targets against COVID-19 pandemic. J. Biomol. Struct. Dyn. 2021, 39, 6171. 10.1080/07391102.2020.1801510. [DOI] [PubMed] [Google Scholar]
- Sharma S.; Sharma A.; Bhattacharyya D.; Chauhan R. S. Computational identification of potential inhibitory compounds in Indian medicinal and aromatic plant species against major pathogenicity determinants of SARS-CoV-2. J. Biomol. Struct 2021, 1–19. 10.1080/07391102.2021.2000500. [DOI] [PubMed] [Google Scholar]
- Hassab M. A. E.; Fares M.; Amin M. K.; Al-Rashood S. T.; Alharbi A.; Eskandrani R. O.; Alkahtani H. M.; Eldehna W. M. Toward the Identification of Potential α-Ketoamide Covalent Inhibitors for SARS-CoV-2 Main Protease: Fragment-Based Drug Design and MM-PBSA Calculations. Processes 2021, 9, 1004. 10.3390/pr9061004. [DOI] [Google Scholar]
- Kumar D.; Kumari K.; Jayaraj A.; Kumar V.; Kumar R. V.; Dass S. K.; Chandra R.; Singh P. Understanding the binding affinity of noscapines with protease of SARS-CoV-2 for COVID-19 using MD simulations at different temperatures. J. Biomol. Struct 2021, 39, 2659–2672. 10.1080/07391102.2020.1752310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Sehemi A. G.; Parulekar R. S.; Pannipara M.; PP M. A.; Zubaidha P. K.; Bhatia M. S.; Mohanta T. K.; Al-Harrasi A. In silico evaluation of NO donor heterocyclic vasodilators as SARS-CoV-2 Mpro protein inhibitor. J. Biomol. Struct 2021, 1–18. 10.1080/07391102.2021.2005682. [DOI] [PubMed] [Google Scholar]
- Elmaaty A. A.; Alnajjar R.; Hamed M. I.; Khattab M.; Khalifa M. M.; Al-Karmalawy A. A. Revisiting activity of some glucocorticoids as a potential inhibitor of SARS-CoV-2 main protease: theoretical study. RSC Adv. 2021, 11, 10027–10042. 10.1039/D0RA10674G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed M. Z.; Zia Q.; Haque A.; Alqahtani A. S.; Almarfadi O. M.; Banawas S.; Alqahtani M. S.; Ameta K. L.; Haque S. Aminoglycosides as potential inhibitors of SARS-CoV-2 main protease: an in silico drug repurposing study on FDA-approved antiviral and anti-infection agents. J. Infect. Public Health 2021, 14, 611–619. 10.1016/j.jiph.2021.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Bustany H. A.; Ercan S.; Ince E.; Pirinccioglu N. Investigation of angucycline compounds as potential drug candidates against SARS Cov-2 main protease using docking and molecular dynamic approaches. Mol. Divers. 2022, 26, 293. 10.1007/s11030-021-10219-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fakhar Z.; Khan S.; AlOmar S. Y.; Alkhuriji A.; Ahmad A. ABBV-744 as a potential inhibitor of SARS-CoV-2 main protease enzyme against COVID-19. Sci. Rep. 2021, 11, 1–15. 10.1038/s41598-020-79918-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson T. O.; Adegboyega A. E.; Iwaloye O.; Eseola O. A.; Plass W.; Afolabi B.; Rotimi D.; Ahmed E. I.; Albrakati A.; Batiha G. E. Computational study of the therapeutic potentials of a new series of imidazole derivatives against SARS-CoV-2. JPS 2021, 147, 62. 10.1016/j.jphs.2021.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noorbakhsh A.; Askandar R. H.; Alhagh M. S.; Farshadfar C.; Seyedi S. H.; Ahmadizad M.; Rahimi A.; Ardalan N.; Koushki E. H. Prevention of SARS-CoV-2 Proliferation with a Novel and Potent Main Protease Inhibitor by Docking, ADMET, MM-PBSA, and Molecular Dynamics Simulation. J. Comput. Biophys. Chem. 2021, 20, 305–322. 10.1142/S2737416521500149. [DOI] [Google Scholar]
- Rakib A.; Nain Z.; Sami S. A.; Mahmud S.; Islam A.; Ahmed S.; Siddiqui A. B. F.; Babu S. O. F.; Hossain P.; Shahriar A.; et al. A molecular modelling approach for identifying antiviral selenium-containing heterocyclic compounds that inhibit the main protease of SARS-CoV-2: An in silico investigation. Brief. Bioinformatics 2021, 22, 1476–1498. 10.1093/bib/bbab045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sultan A.; Ali R.; Sultan T.; Ali S.; Khan N. J.; Parganiha A. Circadian clock modulating small molecules repurposing as inhibitors of SARS-CoV-2 Mpro for pharmacological interventions in COVID-19 pandemic. Chronobiol. Int. 2021, 38, 971. 10.1080/07420528.2021.1903027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopal K.; Varakumar P.; Aparna B.; Byran G.; Jupudi S. Identification of some novel oxazine substituted 9-anilinoacridines as SARS-CoV-2 inhibitors for COVID-19 by molecular docking, free energy calculation and molecular dynamics studies. J. Biomol. Struct. Dyn. 2021, 39, 5551. 10.1080/07391102.2020.1798285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Sehemi A. G.; Pannipara M.; Parulekar R. S.; Patil O.; Choudhari P. B.; Bhatia M.; Zubaidha P.; Tamboli Y. Potential of NO donor furoxan as SARS-CoV-2 main protease (Mpro) inhibitors: in silico analysis. J. Biomol. Struct. Dyn. 2021, 39, 5804. 10.1080/07391102.2020.1790038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar V.; Dhanjal J. K.; Bhargava P.; Kaul A.; Wang J.; Zhang H.; Kaul S. C.; Wadhwa R.; Sundar D. Withanone and withaferin-A are predicted to interact with transmembrane protease serine 2 (TMPRSS2) and block entry of SARS-CoV-2 into cells. J. Biomol. Struct. Dyn. 2022, 40, 1–27. 10.1080/07391102.2020.1775704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pant S.; Singh M.; Ravichandiran V.; Murty U.; Srivastava H. K. Peptide-like and small-molecule inhibitors against COVID-19. J. Biomol. Struct. Dyn. 2021, 39, 2904. 10.1080/07391102.2020.1757510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sk M. F.; Roy R.; Jonniya N. A.; Poddar S.; Kar P. Elucidating biophysical basis of binding of inhibitors to SARS-CoV-2 main protease by using molecular dynamics simulations and free energy calculations. J. Biomol. Struct. Dyn. 2021, 1–21. 10.1080/07391102.2021.1897680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Shar'i N. A. Tackling COVID-19: identification of potential main protease inhibitors via structural analysis, virtual screening, molecular docking and MM-PBSA calculations. J. Biomol. Struct. Dyn. 2021, 39, 6689. 10.1080/07391102.2020.1800514. [DOI] [PubMed] [Google Scholar]
- Zhao T. Y.; Patankar N. A. Tetracycline as an inhibitor to the SARS-CoV-2. J. Cell. Biochem. 2021, 122, 752–759. 10.1002/jcb.29909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimeno A.; Mestres-Truyol J.; Ojeda-Montes M. J.; Macip G.; Saldivar-Espinoza B.; Cereto-Massagué A.; Pujadas G.; Garcia-Vallvé S. Prediction of Novel Inhibitors of the Main Protease (M-pro) of SARS-CoV-2 through Consensus Docking and Drug Reposition. Int. J. Mol. Sci. 2020, 21, 3793. 10.3390/ijms21113793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassanzadeh K.; Perez Pena H.; Dragotto J.; Buccarello L.; Iorio F.; Pieraccini S.; Sancini G.; Feligioni M. Considerations around the SARS-CoV-2 Spike Protein with particular attention to COVID-19 brain infection and neurological symptoms. ACS Chem. Neurosci. 2020, 11, 2361–2369. 10.1021/acschemneuro.0c00373. [DOI] [PubMed] [Google Scholar]
- He J.; Tao H.; Yan Y.; Huang S.-Y.; Xiao Y. Molecular mechanism of evolution and human infection with sars-cov-2. Viruses 2020, 12, 428. 10.3390/v12040428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Q.; Liu X.; Pan W.; Zhang A.; Fu J.; Jiang G. Computational Insights on Allosteric Effect and Dynamic Structural Feature of SARS-COV-2 Spike Protein. Chem.—Eur. J. 2022, 28, e202104215. 10.1002/chem.202104215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jafary F.; Jafari S.; Ganjalikhany M. R. In silico investigation of critical binding pattern in SARS-CoV-2 spike protein with angiotensin-converting enzyme 2. Sci. Rep. 2021, 11, 1–13. 10.1038/s41598-021-86380-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spinello A.; Saltalamacchia A.; Magistrato A. Is the rigidity of SARS-CoV-2 spike receptor-binding motif the hallmark for its enhanced infectivity? Insights from all-atom simulations. J. Phys. Chem. Lett. 2020, 11, 4785–4790. 10.1021/acs.jpclett.0c01148. [DOI] [PubMed] [Google Scholar]
- Hati S.; Bhattacharyya S. Impact of Thiol–Disulfide Balance on the Binding of COVID-19 Spike Protein with Angiotensin-Converting Enzyme 2 Receptor. ACS omega 2020, 5, 16292–16298. 10.1021/acsomega.0c02125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan F.-F.; Gao F. Comparison of the binding characteristics of SARS-CoV and SARS-CoV-2 RBDs to ACE2 at different temperatures by MD simulations. Brief. Bioinformatics 2021, 22, 1122–1136. 10.1093/bib/bbab044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah M.; Ahmad B.; Choi S.; Woo H. G. Sequence variation of SARS-CoV-2 spike protein may facilitate stronger interaction with ACE2 promoting high infectivity. Comput. Struct. Biotechnol. J. 2020, 18, 3402–3414. 10.1016/j.csbj.2020.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhseen Z. T.; Kadhim S.; Yahiya Y. I.; Alatawi E. A.; Aba Alkhayl F. F.; Almatroudi A. Insights into the Binding of Receptor-Binding Domain (RBD) of SARS-CoV-2 Wild Type and B. 1.620 Variant with hACE2 Using Molecular Docking and Simulation Approaches. Biology 2021, 10, 1310. 10.3390/biology10121310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Istifli E. S.; Netz P. A.; Sihoglu Tepe A.; Sarikurkcu C.; Tepe B. Understanding the molecular interaction of SARS-CoV-2 spike mutants with ACE2 (angiotensin converting enzyme 2). J. Biomol. Struct 2021, 1–12. 10.1080/07391102.2021.1975569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan A.; Hussain S.; Ahmad S.; Suleman M.; Bukhari I.; Khan T.; Rashid F.; Azad A. K.; Waseem M.; Khan W.; et al. Computational modelling of potentially emerging SARS-CoV-2 spike protein RBDs mutations with higher binding affinity towards ACE2: A structural modelling study. Comput. Biol. Med. 2022, 141, 105163. 10.1016/j.compbiomed.2021.105163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kullappan M.; Mary U.; Ambrose J. M.; Veeraraghavan V. P.; Surapaneni K. M. Elucidating the role of N440K mutation in SARS-CoV-2 spike–ACE-2 binding affinity and COVID-19 severity by virtual screening, molecular docking and dynamics approach. J. Biomol. Struct 2021, 1–18. 10.1080/07391102.2021.2014973. [DOI] [PubMed] [Google Scholar]
- Fossum C. J.; Laatsch B. F.; Lowater H. R.; Narkiewicz-Jodko A. W.; Lonzarich L.; Hati S.; Bhattacharyya S. Pre-Existing Oxidative Stress Creates a Docking-Ready Conformation of the SARS-CoV-2 Receptor-Binding Domain. ACS Bio & Med. Chem. Au 2022, 2, 84–93. 10.1021/acsbiomedchemau.1c00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghasemitarei M.; Privat-Maldonado A.; Yusupov M.; Rahnama S.; Bogaerts A.; Ejtehadi M. R. Effect of Cysteine Oxidation in SARS-CoV-2 Receptor-Binding Domain on Its Interaction with Two Cell Receptors: Insights from Atomistic Simulations. J. Chem. Inf. Model. 2022, 62, 129. 10.1021/acs.jcim.1c00853. [DOI] [PubMed] [Google Scholar]
- Piplani S.; Singh P. K.; Winkler D. A.; Petrovsky N. In silico comparison of SARS-CoV-2 spike protein-ACE2 binding affinities across species and implications for virus origin. Sci. Rep. 2021, 11, 1–13. 10.1038/s41598-021-92388-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P.; Wang J.; Xu X.; Li Y.; Zhu Y.; Li X.; Li M.; Hao P. Molecular dynamic simulation analysis of SARS-CoV-2 spike mutations and evaluation of ACE2 from pets and wild animals for infection risk. Comput. Biol. Chem. 2022, 96, 107613. 10.1016/j.compbiolchem.2021.107613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L.; Zhou L.; Mo M.; Liu T.; Wu C.; Gong C.; Lu K.; Gong L.; Zhu W.; Xu Z. SARS-CoV-2 Omicron RBD shows weaker binding affinity than the currently dominant Delta variant to human ACE2. Signal Transduct. Target. Ther. 2022, 7, 1–3. 10.1038/s41392-021-00863-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Oliveira O. V.; Rocha G. B.; Paluch A. S.; Costa L. T. Repurposing approved drugs as inhibitors of SARS-CoV-2 S-protein from molecular modeling and virtual screening. J. Biomol. Struct. Dyn. 2021, 39, 3924–3933. 10.1080/07391102.2020.1772885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romeo A.; IaCoVelli F.; Falconi M. Targeting the SARS-CoV-2 spike glycoprotein prefusion conformation: virtual screening and molecular dynamics simulations applied to the identification of potential fusion inhibitors. Virus Res. 2020, 286, 198068. 10.1016/j.virusres.2020.198068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padhi A. K.; Seal A.; Khan J. M.; Ahamed M.; Tripathi T. Unraveling the mechanism of Arbidol binding and inhibition of SARS-CoV-2: Insights from atomistic simulations. Eur. J. Pharmacol. 2021, 894, 173836. 10.1016/j.ejphar.2020.173836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sethi A.; Sanam S.; Munagalasetty S.; Jayanthi S.; Alvala M. Understanding the role of galectin inhibitors as potential candidates for SARS-CoV-2 spike protein: in silico studies. RSC Adv. 2020, 10, 29873–29884. 10.1039/D0RA04795C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rane J. S.; Pandey P.; Chatterjee A.; Khan R.; Kumar A.; Prakash A.; Ray S. Targeting virus–host interaction by novel pyrimidine derivative: an in silico approach towards discovery of potential drug against COVID-19. J. Biomol. Struct. Dyn. 2021, 39, 5768–5778. 10.1080/07391102.2020.1794969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R.; Bhardwaj V. K.; Sharma J.; Kumar D.; Purohit R. Identification of potential plant bioactive as SARS-CoV-2 Spike protein and human ACE2 fusion inhibitors. Comput. Biol. Med. 2021, 136, 104631. 10.1016/j.compbiomed.2021.104631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.; Zhang Z.; Yang L.; Lian X.; Xie Y.; Li S.; Xin S.; Cao P.; Lu J. The MERS-CoV receptor DPP4 as a candidate binding target of the SARS-CoV-2 spike. Iscience 2020, 23, 101160. 10.1016/j.isci.2020.101160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey P.; Rane J. S.; Chatterjee A.; Kumar A.; Khan R.; Prakash A.; Ray S. Targeting SARS-CoV-2 spike protein of COVID-19 with naturally occurring phytochemicals: an in silico study for drug development. J. Biomol. Struct. Dyn. 2021, 39, 6306–6316. 10.1080/07391102.2020.1796811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umashankar V.; Deshpande S. H.; Hegde H. V.; Singh I.; Chattopadhyay D. Phytochemical moieties from Indian traditional medicine for targeting dual hotspots on SARS-CoV-2 spike protein: an integrative in-silico approach. Front. Med. 2021, 8, 545. 10.3389/fmed.2021.672629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel C. N.; Goswami D.; Sivakumar P. K.; Pandya H. A. Repurposing of anticancer phytochemicals for identifying potential fusion inhibitor for SARS-CoV-2 using molecular docking and molecular dynamics (MD) simulations. J. Biomol. Struct 2021, 1–18. 10.1080/07391102.2021.1902393. [DOI] [PubMed] [Google Scholar]
- Alvarado W.; Perez-Lemus G. R.; Menéndez C. A.; Byléhn F.; de Pablo J. J. Molecular characterization of COVID-19 therapeutics: luteolin as an allosteric modulator of the spike protein of SARS-CoV-2. MSDE 2022, 7, 58. 10.1039/D1ME00119A. [DOI] [Google Scholar]
- Patel C. N.; Goswami D.; Jaiswal D. G.; Parmar R. M.; Solanki H. A.; Pandya H. A. Pinpointing the potential hits for hindering interaction of SARS-CoV-2 S-protein with ACE2 from the pool of antiviral phytochemicals utilizing molecular docking and molecular dynamics (MD) simulations. J. Mol. Graph. Model. 2021, 105, 107874. 10.1016/j.jmgm.2021.107874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan Z.; Liu C.; Guo Y.; He Z.; Huang X.; Jia X.; Yang T. SARS-CoV-2 and SARS-CoV: Virtual screening of potential inhibitors targeting RNA-dependent RNA polymerase activity (NSP12). J. Med. Virol 2021, 93, 389–400. 10.1002/jmv.26222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doharey P. K.; Singh V.; Gedda M. R.; Sahoo A. K.; Varadwaj P. K.; Sharma B. In silico study indicates antimalarials as direct inhibitors of SARS-CoV-2-RNA dependent RNA polymerase. J. Biomol. Struct 2021, 1–18. 10.1080/07391102.2021.1871956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirzada R. H.; Haseeb M.; Batool M.; Kim M.; Choi S. Remdesivir and Ledipasvir among the FDA-Approved Antiviral Drugs Have Potential to Inhibit SARS-CoV-2 Replication. Cells 2021, 10, 1052. 10.3390/cells10051052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arba M.; Wahyudi S. T.; Brunt D. J.; Paradis N.; Wu C. Mechanistic insight on the remdesivir binding to RNA-Dependent RNA polymerase (RdRp) of SARS-Cov-2. Comput. Biol. Med. 2021, 129, 104156. 10.1016/j.compbiomed.2020.104156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan A.; Khan M.; Saleem S.; Babar Z.; Ali A.; Khan A. A.; Sardar Z.; Hamayun F.; Ali S. S.; Wei D.-Q. Phylogenetic analysis and structural perspectives of RNA-dependent RNA-polymerase inhibition from SARS-CoV-2 with natural products. Interdiscip. Sci. 2020, 12, 335–348. 10.1007/s12539-020-00381-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh S.; Sk M. F.; Sonawane A.; Kar P.; Sadhukhan S. Plant-derived natural polyphenols as potential antiviral drugs against SARS-CoV-2 via RNA-dependent RNA polymerase (RdRp) inhibition: An in-silico analysis. J. Biomol. Struct. Dyn. 2021, 39, 6249. 10.1080/07391102.2020.1796810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali H. S. M.; Altayb H. N.; Firoz A.; Bayoumi A. A. M.; El Omri A.; Chaieb K. Inhibitory activity of marine sponge metabolites on SARS-CoV-2 RNA dependent polymerase: virtual screening and molecular dynamics simulation. J. Biomol. Struct 2021, 1–12. 10.1080/07391102.2021.1940283. [DOI] [PubMed] [Google Scholar]
- Sonousi A.; Mahran H. A.; Ibrahim I. M.; Ibrahim M. N.; Elfiky A. A.; Elshemey W. M. Novel adenosine derivatives against SARS-CoV-2 RNA-dependent RNA polymerase: an in silico perspective. Pharmacol. Rep. 2021, 73, 1754. 10.1007/s43440-021-00300-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jena N.; Pant S.; Srivastava H. K. Artificially expanded genetic information systems (AEGISs) as potent inhibitors of the RNA-dependent RNA polymerase of the SARS-CoV-2. J. Biomol. Struct 2021, 1–17. 10.1080/07391102.2021.1883112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pant S.; Jena N. Inhibition of the RNA-dependent RNA Polymerase of the SARS-CoV-2 by Short Peptide Inhibitors. Eur. J. Pharm. Sci. 2021, 167, 106012. 10.1016/j.ejps.2021.106012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R.; Bhardwaj V. K.; Purohit R. Potential of turmeric-derived compounds against RNA-dependent RNA polymerase of SARS-CoV-2: An in-silico approach. Comput. Biol. Med. 2021, 139, 104965. 10.1016/j.compbiomed.2021.104965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanachai K.; Mahalapbutr P.; Sanghiran Lee V.; Rungrotmongkol T.; Hannongbua S. In Silico Elucidation of Potent Inhibitors and Rational Drug Design against SARS-CoV-2 Papain-like Protease. J. Phys. Chem. B 2021, 125, 13644–13656. 10.1021/acs.jpcb.1c07060. [DOI] [PubMed] [Google Scholar]
- Bhardwaj V. K.; Singh R.; Sharma J.; Rajendran V.; Purohit R.; Kumar S. Bioactive molecules of Tea as potential inhibitors for RNA-dependent RNA polymerase of SARS-CoV-2. Front. Med. 2021, 8, 645. 10.3389/fmed.2021.684020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koulgi S.; Jani V.; VN M. U.; Sonavane U.; Joshi R. Structural insight into the binding interactions of NTPs and nucleotide analogues to RNA dependent RNA polymerase of SARS-CoV-2. J. Biomol. Struct 2021, 1–15. 10.1080/07391102.2021.1894985. [DOI] [PubMed] [Google Scholar]
- Aktaş A.; Tüzün B.; Aslan R.; Sayin K.; Ataseven H. New anti-viral drugs for the treatment of COVID-19 instead of favipiravir. J. Biomol. Struct. Dyn. 2021, 39, 7263. 10.1080/07391102.2020.1806112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkateshan M.; Muthu M.; Suresh J.; Kumar R. R. Azafluorene derivatives as inhibitors of SARS CoV-2 RdRp: Synthesis, physicochemical, quantum chemical, modeling and molecular docking analysis. J. Mol. Struct. 2020, 1220, 128741. 10.1016/j.molstruc.2020.128741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad S.; Abbasi H. W.; Shahid S.; Gul S.; Abbasi S. W. Molecular Docking, Simulation and MM-PBSA Studies of Nigella Sativa Compounds: A Computational Quest to identify Potential Natural Antiviral for COVID-19 Treatment. J. Biomol. Struct. Dyn. 2021, 39, 4225. 10.1080/07391102.2020.1775129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasan M. K.; Kamruzzaman M.; Manjur O. H. B.; Mahmud A.; Hussain N.; Mondal M. S. A.; Hosen M. I.; Bello M.; Rahman A. Structural analogues of existing anti-viral drugs inhibit SARS-CoV-2 RNA dependent RNA polymerase: A computational hierarchical investigation. Heliyon 2021, 7, e06435 10.1016/j.heliyon.2021.e06435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandeel M.; Abdelrahman A. H.; Oh-Hashi K.; Ibrahim A.; Venugopala K. N.; Morsy M. A.; Ibrahim M. A. Repurposing of FDA-approved antivirals, antibiotics, anthelmintics, antioxidants, and cell protectives against SARS-CoV-2 papain-like protease. J. Biomol. Struct. Dyn. 2021, 39, 5129. 10.1080/07391102.2020.1784291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra D.; Verma D.; Mahakur B.; Kamboj A.; Srivastava R.; Gupta S.; Pandey A.; Arora B.; Pant K.; Panneerselvam P.; et al. Molecular docking and simulation studies of natural compounds of Vitex negundo L. against papain-like protease (PLpro) of SARS CoV-2 (coronavirus) to conquer the pandemic situation in the world. J. Biomol. Struct 2021, 1–22. 10.1080/07391102.2021.1873185. [DOI] [PubMed] [Google Scholar]
- Rao P.; Patel R.; Shukla A.; Parmar P.; Rawal R. M.; Saraf M.; Goswami D. Identifying structural–functional analogue of GRL0617, the only well-established inhibitor for papain-like protease (PLpro) of SARS-CoV2 from the pool of fungal metabolites using docking and molecular dynamics simulation. Mol. Divers. 2022, 26, 309. 10.1007/s11030-021-10220-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy R.; Jonniya N. A.; Poddar S.; Sk M. F.; Kar P. Unraveling the Molecular Mechanism of Recognition of Human Interferon-Stimulated Gene Product 15 by Coronavirus Papain-Like Proteases: A Multiscale Simulation Study. J. Chem. Inf Model 2021, 61, 6038–6052. 10.1021/acs.jcim.1c00918. [DOI] [PubMed] [Google Scholar]
- Pitsillou E.; Liang J.; Hung A.; Karagiannis T. C. Inhibition of interferon-stimulated gene 15 and lysine 48-linked ubiquitin binding to the SARS-CoV-2 papain-like protease by small molecules: In silico studies. Chem. Phys. Lett. 2021, 771, 138468. 10.1016/j.cplett.2021.138468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosken Y. K.; Cholko T.; Lou Y.-C.; Wu K.-P.; Chang C.-e. A. Insights Into Dynamics of Inhibitor and Ubiquitin-Like Protein Binding in SARS-CoV-2 Papain-Like Protease. Front. Mol. Biosci 2020, 7, 174. 10.3389/fmolb.2020.00174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan A.; Khan M. T.; Saleem S.; Junaid M.; Ali A.; Ali S. S.; Khan M.; Wei D.-Q. Structural Insights into the mechanism of RNA recognition by the N-terminal RNA-binding domain of the SARS-CoV-2 nucleocapsid phosphoprotein. Comput. Struct. Biotechnol. J. 2020, 18, 2174–2184. 10.1016/j.csbj.2020.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav R.; Imran M.; Dhamija P.; Suchal K.; Handu S. Virtual screening and dynamics of potential inhibitors targeting RNA binding domain of nucleocapsid phosphoprotein from SARS-CoV-2. J. Biomol. Struct. Dyn. 2021, 39, 4433–4448. 10.1080/07391102.2020.1778536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abidi S. H.; Almansour N. M.; Amerzhanov D.; Allemailem K. S.; Rafaqat W.; Ibrahim M. A.; la Fleur P.; Lukac M.; Ali S. Repurposing potential of posaconazole and grazoprevir as inhibitors of SARS-CoV-2 helicase. Sci. Rep. 2021, 11, 1–11. 10.1038/s41598-021-89724-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivek-Ananth R.; Krishnaswamy S.; Samal A. Potential phytochemical inhibitors of SARS-CoV-2 helicase Nsp13: a molecular docking and dynamic simulation study. Mol. Divers. 2022, 26, 429–442. 10.1007/s11030-021-10251-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad S.; Waheed Y.; Ismail S.; Bhatti S.; Abbasi S. W.; Muhammad K. Structure-based virtual screening identifies multiple stable binding sites at the RecA domains of SARS-CoV-2 helicase enzyme. Molecules 2021, 26, 1446. 10.3390/molecules26051446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sk M. F.; Jonniya N. A.; Roy R.; Poddar S.; Kar P. Computational investigation of structural dynamics of SARS-CoV-2 methyltransferase-stimulatory factor heterodimer nsp16/nsp10 bound to the cofactor SAM. Front. Mol. Biosci. 2020, 7, 353. 10.3389/fmolb.2020.590165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma J.; Bhardwaj V. K.; Singh R.; Rajendran V.; Purohit R.; Kumar S. An in-silico evaluation of different bioactive molecules of tea for their inhibition potency against non structural protein-15 of SARS-CoV-2. Food Chem. 2021, 346, 128933. 10.1016/j.foodchem.2020.128933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra A.; Gurjar V.; Qamar I.; Singh N. Identification of Potential Inhibitors of SARS-CoV-2 Endoribonuclease (EndoU) from FDA Approved Drugs: A Drug Repurposing Approach to find Therapeutics for COVID19. J. Biomol. Struct. Dyn. 2021, 39, 4201. 10.1080/07391102.2020.1775127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan R. J.; Jha R. K.; Singh E.; Jain M.; Amera G. M.; Singh R. P.; Muthukumaran J.; Singh A. K. Identification of promising antiviral drug candidates against non-structural protein 15 (NSP15) from SARS-CoV-2: an in silico assisted drug-repurposing study. J. Biomol. Struct. Dyn. 2022, 40, 438. 10.1080/07391102.2020.1814870. [DOI] [PubMed] [Google Scholar]
- Encinar J. A.; Menendez J. A. Potential drugs targeting early innate immune evasion of SARS-coronavirus 2 via 2′-O-methylation of viral RNA. Viruses 2020, 12, 525. 10.3390/v12050525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijayan V.; Pant P.; Vikram N.; Kaur P.; Singh T.; Sharma S.; Sharma P. Identification of promising drug candidates against NSP16 of SARS-CoV-2 through computational drug repurposing study. J. Biomol. Struct. Dyn. 2021, 39, 6713. 10.1080/07391102.2020.1802349. [DOI] [PubMed] [Google Scholar]
- Chandra A.; Chaudhary M.; Qamar I.; Singh N.; Nain V. In silico identification and validation of natural antiviral compounds as potential inhibitors of SARS-CoV-2 methyltransferase. J. Biomol. Struct 2021, 1–11. 10.1080/07391102.2021.1886174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R.; Bhardwaj V. K.; Sharma J.; Purohit R.; Kumar S. In-silico evaluation of bioactive compounds from tea as potential SARS-CoV-2 nonstructural protein 16 inhibitors. J. Tradit. Complement. Med. 2022, 12, 35. 10.1016/j.jtcme.2021.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Hassab M. A.; Ibrahim T. M.; Shoun A. A.; Al-Rashood S. T.; Alkahtani H. M.; Alharbi A.; Eskandrani R. O.; Eldehna W. M. In silico identification of potential SARS COV-2 2’-O-methyltransferase inhibitor: fragment-based screening approach and MM-PBSA calculations. RSC Adv. 2021, 11, 16026–16033. 10.1039/D1RA01809D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ongaro A.; Oselladore E.; Memo M.; Ribaudo G.; Gianoncelli A. Insight into the LFA-1/SARS-CoV-2 Orf7a Complex by Protein–Protein Docking, Molecular Dynamics, and MM-GBSA Calculations. J. Chem. Inf Model 2021, 61, 2780–2787. 10.1021/acs.jcim.1c00198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R.; Bhardwaj V. K.; Das P.; Purohit R. A computational approach for rational discovery of inhibitors for non-structural protein 1 of SARS-CoV-2. Comput. Biol. Med. 2021, 135, 104555. 10.1016/j.compbiomed.2021.104555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C.; Zhu X.; Lu Y.; Zhang X.; Jia X.; Yang T. Potential treatment with Chinese and Western medicine targeting NSP14 of SARS-CoV-2. Journal of pharmaceutical analysis 2021, 11, 272–277. 10.1016/j.jpha.2020.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunes V. S.; Paschoal D. F.; Costa L. A. S.; Santos H. F. D. Antivirals virtual screening to SARS-CoV-2 non-structural proteins. J. Biomol. Struct 2021, 1–15. 10.1080/07391102.2021.1921033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anju A.; Chaturvedi S.; Chaudhary V.; Pant P.; Hussain F.; Mishra A. K. Virtual screening of quinoline derived library for SARS-COV-2 targeting viral entry and replication. J. Biomol. Struct 2021, 1–30. 10.1080/07391102.2021.1913228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen Gupta P. S.; Biswal S.; Singha D.; Rana M. K. Binding insight of clinically oriented drug famotidine with the identified potential target of SARS-CoV-2. J. Biomol. Struct. Dyn. 2021, 39, 5327. 10.1080/07391102.2020.1784795. [DOI] [PubMed] [Google Scholar]
- Eweas A. F.; Alhossary A. A.; Abdel-Moneim A. S. Molecular docking reveals Ivermectin and Remdesivir as potential repurposed drugs against SARS-CoV-2. Front. Microbiol. 2021, 11, 3602. 10.3389/fmicb.2020.592908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jade D.; Ayyamperumal S.; Tallapaneni V.; Nanjan C. M. J.; Barge S.; Mohan S.; Nanjan M. J. Virtual high throughput screening: Potential inhibitors for SARS-CoV-2 PLPRO and 3CLPRO proteases. Eur. J. Pharmacol. 2021, 901, 174082. 10.1016/j.ejphar.2021.174082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panda P. K.; Arul M. N.; Patel P.; Verma S. K.; Luo W.; Rubahn H.-G.; Mishra Y. K.; Suar M.; Ahuja R. Structure-based drug designing and immunoinformatics approach for SARS-CoV-2. Sci. Adv. 2020, 6, eabb8097 10.1126/sciadv.abb8097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naidoo D.; Roy A.; Kar P.; Mutanda T.; Anandraj A. Cyanobacterial metabolites as promising drug leads against the Mpro and PLpro of SARS-CoV-2: an in silico analysis. J. Biomol. Struct. Dyn. 2021, 39, 6218. 10.1080/07391102.2020.1794972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurakkal L.; Singh S.; Roy R.; Kar P.; Sadhukhan S.; Porel M. An in-silico study on selected organosulfur compounds as potential drugs for SARS-CoV-2 infection via binding multiple drug targets. Chem. Phys. Lett. 2021, 763, 138193. 10.1016/j.cplett.2020.138193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diniz L. R. L.; Perez-Castillo Y.; Elshabrawy H. A.; de Sousa D. P.; et al. Bioactive terpenes and their derivatives as potential SARS-CoV-2 proteases inhibitors from molecular modeling studies. Biomolecules 2021, 11, 74. 10.3390/biom11010074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuesta S. A.; Mora J. R.; Márquez E. A. In Silico Screening of the DrugBank Database to Search for Possible Drugs against SARS-CoV-2. Molecules 2021, 26, 1100. 10.3390/molecules26041100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad K.; Ahamad S.; Kanipakam H.; Gupta D.; Kumar V. Simultaneous Inhibition of SARS-CoV-2 Entry Pathways by Cyclosporine. ACS Chem. Neurosci. 2021, 12, 930–944. 10.1021/acschemneuro.1c00019. [DOI] [PubMed] [Google Scholar]
- Kumar S.; Sharma P. P.; Upadhyay C.; Kempaiah P.; Rathi B.; et al. Multi-targeting approach for nsp3, nsp9, nsp12 and nsp15 proteins of SARS-CoV-2 by Diosmin as illustrated by molecular docking and molecular dynamics simulation methodologies. Methods 2021, 195, 44–56. 10.1016/j.ymeth.2021.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tejera E.; Pérez-Castillo Y.; Toscano G.; Noboa A. L.; Ochoa-Herrera V.; Giampieri F.; Álvarez-Suarez J. M. Computational modeling predicts potential effects of the herbal infusion ′horchata′ against COVID-19. Food Chem. 2022, 366, 130589. 10.1016/j.foodchem.2021.130589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta T.; Baildya N.; Khan A. A.; Ghosh N. N. Inhibitory effect of anti-HIV compounds extracted from Indian medicinal plants to retard the replication and transcription process of SARS-CoV-2: an insight from molecular docking and MD-simulation studies. Netw. Model. Anal. Health Inform. Bioinform. 2021, 10, 1–11. 10.1007/s13721-021-00309-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bera K.; Reeda V. J.; Babila P.; Dinesh D. C.; Hritz J.; Karthick T. An in silico molecular dynamics simulation study on the inhibitors of SARS-CoV-2 proteases (3CLpro and PLpro) to combat COVID-19. Mol. Simul 2021, 47, 1168–1184. 10.1080/08927022.2021.1957884. [DOI] [Google Scholar]
- Mandal M.; Chowdhury S. K.; Khan A. A.; Baildya N.; Dutta T.; Misra D.; Ghosh N. N. Inhibitory efficacy of RNA virus drugs against SARS-CoV-2 proteins: an extensive study. J. Mol. Struct. 2021, 1234, 130152. 10.1016/j.molstruc.2021.130152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahdian S.; Zarrabi M.; Panahi Y.; Dabbagh S. Repurposing FDA-approved drugs to fight COVID-19 using in silico methods: Targeting SARS-CoV-2 RdRp enzyme and host cell receptors (ACE2, CD147) through virtual screening and molecular dynamic simulations. Inform. Med. Unlocked 2021, 23, 100541. 10.1016/j.imu.2021.100541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gul S.; Ozcan O.; Asar S.; Okyar A.; Barıs I.; Kavakli I. H. In silico identification of widely used and well-tolerated drugs as potential SARS-CoV-2 3C-like protease and viral RNA-dependent RNA polymerase inhibitors for direct use in clinical trials. J. Biomol. Struct. Dyn. 2021, 39, 6772–6791. 10.1080/07391102.2020.1802346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed S.; Mahtarin R.; Ahmed S. S.; Akter S.; Islam M. S.; Mamun A. A.; Islam R.; Hossain M. N.; Ali M. A.; Sultana M. U.; et al. Investigating the binding affinity, interaction, and structure-activity-relationship of 76 prescription antiviral drugs targeting RdRp and Mpro of SARS-CoV-2. J. Biomol. Struct. Dyn. 2021, 39, 6290–6305. 10.1080/07391102.2020.1796804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra K.; Ghanta P.; Acharya S.; Chakrapani G.; Ramaiah B.; Doble M. Dual inhibitors of SARS-CoV-2 proteases: pharmacophore and molecular dynamics based drug repositioning and phytochemical leads. J. Biomol. Struct. Dyn. 2021, 39, 6324–6337. 10.1080/07391102.2020.1796802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snoussi M.; Redissi A.; Mosbah A.; De Feo V.; Adnan M.; Aouadi K.; Alreshidi M.; Patel M.; Kadri A.; Noumi E. Emetine, a potent alkaloid for the treatment of SARS-CoV-2 targeting papain-like protease and non-structural proteins: pharmacokinetics, molecular docking and dynamic studies. J. Biomol. Struct 2021, 1–14. 10.1080/07391102.2021.1946715. [DOI] [PubMed] [Google Scholar]
- Kwofie S. K.; Broni E.; Asiedu S. O.; Kwarko G. B.; Dankwa B.; Enninful K. S.; Tiburu E. K.; Wilson M. D. Cheminformatics-Based Identification of Potential Novel Anti-SARS-CoV-2 Natural Compounds of African Origin. Molecules 2021, 26, 406. 10.3390/molecules26020406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra G. P.; Bhadane R. N.; Panigrahi D.; Amawi H. A.; Asbhy C. R. Jr; Tiwari A. K. The interaction of the bioflavonoids with five SARS-CoV-2 proteins targets: An in silico study. Comput. Biol. Med. 2021, 134, 104464. 10.1016/j.compbiomed.2021.104464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borkotoky S.; Banerjee M. A computational prediction of SARS-CoV-2 structural protein inhibitors from Azadirachta indica (Neem). J. Biomol. Struct. Dyn. 2021, 39, 4111–4121. 10.1080/07391102.2020.1774419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhankhar P.; Dalal V.; Singh V.; Tomar S.; Kumar P. Computational guided identification of novel potent inhibitors of N-terminal domain of nucleocapsid protein of severe acute respiratory syndrome coronavirus 2. J. Biomol. Struct. Dyn. 2020, 1–16. 10.1080/07391102.2020.1852968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyebi G. A.; Ogunyemi O. M.; Ibrahim I. M.; Ogunro O. B.; Adegunloye A. P.; Afolabi S. O. SARS-CoV-2 host cell entry: an in silico investigation of potential inhibitory roles of terpenoids. J. Genet Eng. Biotechnol 2021, 19, 1–22. 10.1186/s43141-021-00209-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar V.; Parate S.; Yoon S.; Lee G.; Lee K. W. Computational Simulations Identified Marine-Derived Natural Bioactive Compounds as Replication Inhibitors of SARS-CoV-2. Front. Microbiol. 2021, 12, 583. 10.3389/fmicb.2021.647295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Sanea M. M.; Abelyan N.; Abdelgawad M. A.; Musa A.; Ghoneim M. M.; Al-Warhi T.; Aljaeed N.; Alotaibi O. J.; Alnusaire T. S.; Abdelwahab S. F.; et al. Strawberry and Ginger Silver Nanoparticles as Potential Inhibitors for SARS-CoV-2 Assisted by In Silico Modeling and Metabolic Profiling. Antibiotics 2021, 10, 824. 10.3390/antibiotics10070824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddah M.; Bahramsoltani R.; Yekta N. H.; Rahimi R.; Aliabadi R.; Pourfath M. Proposing high-affinity inhibitors from Glycyrrhiza glabra L. against SARS-CoV-2 infection: Virtual screening and computational analysis. New J. Chem. 2021, 45, 15977. 10.1039/D1NJ02031E. [DOI] [Google Scholar]
- Murugan N. A.; Pandian C. J.; Jeyakanthan J. Computational investigation on Andrographis paniculata phytochemicals to evaluate their potency against SARS-CoV-2 in comparison to known antiviral compounds in drug trials. J. Biomol. Struct. Dyn. 2021, 39, 4415. 10.1080/07391102.2020.1777901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alazmi M.; Motwalli O. In silico virtual screening, characterization, docking and molecular dynamics studies of crucial SARS-CoV-2 proteins. J. Biomol. Struct. Dyn. 2021, 39, 6761. 10.1080/07391102.2020.1803965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kar P.; Sharma N. R.; Singh B.; Sen A.; Roy A. Natural compounds from Clerodendrum spp. as possible therapeutic candidates against SARS-CoV-2: An in silico investigation. J. Biomol. Struct. Dyn. 2021, 39, 4774. 10.1080/07391102.2020.1780947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alajmi M. F.; Azhar A.; Owais M.; Rashid S.; Hasan S.; Hussain A.; Rehman M. T. Antiviral potential of some novel structural analogs of standard drugs repurposed for the treatment of COVID-19. J. Biomol. Struct. Dyn. 2020, 39, 6676–6688. 10.1080/07391102.2020.1799865. [DOI] [PubMed] [Google Scholar]
- Sasidharan S.; Selvaraj C.; Singh S. K.; Dubey V. K.; Kumar S.; Fialho A. M.; Saudagar P. Bacterial protein azurin and derived peptides as potential anti-SARS-CoV-2 agents: insights from molecular docking and molecular dynamics simulations. J. Biomol. Struct. Dyn. 2021, 39, 5706–5721. 10.1080/07391102.2020.1787864. [DOI] [PubMed] [Google Scholar]
- Prasanth D.; Murahari M.; Chandramohan V.; Panda S. P.; Atmakuri L. R.; Guntupalli C. In silico identification of potential inhibitors from Cinnamon against main protease and spike glycoprotein of SARS-CoV-2. J. Biomol. Struct. Dyn. 2021, 39, 4618–4632. 10.1080/07391102.2020.1779129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma D.; Mitra D.; Paul M.; Chaudhary P.; Kamboj A.; Thatoi H.; Janmeda P.; Jain D.; Panneerselvam P.; Shrivastav R.; et al. Potential inhibitors of SARS-CoV-2 (COVID 19) proteases PLpro and Mpro/3CLpro: molecular docking and simulation studies of three pertinent medicinal plant natural components. CRPHAR 2021, 2, 100038. 10.1016/j.crphar.2021.100038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chikhale R. V.; Gurav S. S.; Patil R. B.; Sinha S. K.; Prasad S. K.; Shakya A.; Shrivastava S. K.; Gurav N. S.; Prasad R. S. SARS-CoV-2 host entry and replication inhibitors from Indian ginseng: an in-silico approach. J. Biomol. Struct. Dyn. 2021, 39, 4510–4521. 10.1080/07391102.2020.1778539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gogoi M.; Borkotoky M.; Borchetia S.; Chowdhury P.; Mahanta S.; Barooah A. K. Black tea bioactives as inhibitors of multiple targets of SARS-CoV-2 (3CLpro, PLpro and RdRp): a virtual screening and molecular dynamic simulation study. J. Biomol. Struct 2021, 1–24. 10.1080/07391102.2021.1897679. [DOI] [PubMed] [Google Scholar]
- Giofrè S. V.; Napoli E.; Iraci N.; Speciale A.; Cimino F.; Muscarà C.; Molonia M. S.; Ruberto G.; Saija A. Interaction of selected terpenoids with two SARS-CoV-2 key therapeutic targets: An in silico study through molecular docking and dynamics simulations. Comput. Biol. Med. 2021, 134, 104538. 10.1016/j.compbiomed.2021.104538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsbaey M.; Ibrahim M. A.; Bar F. A.; Elgazar A. A. Chemical constituents from coconut waste and their in silico evaluation as potential antiviral agents against SARS-CoV-2. S. Afr. J. Bot. 2021, 141, 278–289. 10.1016/j.sajb.2021.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shady N. H.; Hayallah A. M.; Mohamed M. F.; Ghoneim M. M.; Chilingaryan G.; Al-Sanea M. M.; Fouad M. A.; Kamel M. S.; Abdelmohsen U. R. Targeting 3CLpro and SARS-CoV-2 RdRp by Amphimedon sp. Metabolites: A Computational Study. Molecules 2021, 26, 3775. 10.3390/molecules26123775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma A. K.; Kumar V.; Singh S.; Goswami B. C.; Camps I.; Sekar A.; Yoon S.; Lee K. W. Repurposing potential of Ayurvedic medicinal plants derived active principles against SARS-CoV-2 associated target proteins revealed by molecular docking, molecular dynamics and MM-PBSA studies. Biomed. Pharmacother. 2021, 137, 111356. 10.1016/j.biopha.2021.111356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naik B.; Gupta N.; Ojha R.; Singh S.; Prajapati V. K.; Prusty D. High throughput virtual screening reveals SARS-CoV-2 multi-target binding natural compounds to lead instant therapy for COVID-19 treatment. Int. J. Biol. Macromol. 2020, 160, 1–17. 10.1016/j.ijbiomac.2020.05.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhseen Z. T.; Hameed A. R.; Al-Hasani H. M.; Ahmad S.; Li G. Computational Determination of Potential Multiprotein Targeting Natural Compounds for Rational Drug Design Against SARS-COV-2. Molecules 2021, 26, 674. 10.3390/molecules26030674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pushkaran A. C.; Melge A. R.; Puthiyedath R.; Mohan C. G. A phytochemical-based medication search for the SARS-CoV-2 infection by molecular docking models towards spike glycoproteins and main proteases. RSC Adv. 2021, 11, 12003–12014. 10.1039/D0RA10458B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skariyachan S.; Gopal D.; Muddebihalkar A. G.; Uttarkar A.; Niranjan V. Structural insights on the interaction potential of natural leads against major protein targets of SARS-CoV-2: Molecular modelling, docking and dynamic simulation studies. Comput. Biol. Med. 2021, 132, 104325. 10.1016/j.compbiomed.2021.104325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quimque M. T. J.; Notarte K. I. R.; Fernandez R. A. T.; Mendoza M. A. O.; Liman R. A. D.; Lim J. A. K.; Pilapil L. A. E.; Ong J. K. H.; Pastrana A. M.; Khan A.; et al. Virtual Screening-Driven Drug discovery of SARS-CoV2 Enzyme Inhibitors Targeting Viral Attachment, Replication, Post-Translational Modification and Host Immunity Evasion Infection Mechanisms. J. Biomol. Struct. Dyn. 2021, 39, 4316–4333. 10.1080/07391102.2020.1776639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirza M. U.; Froeyen M. Structural elucidation of SARS-CoV-2 vital proteins: Computational methods reveal potential drug candidates against main protease, Nsp12 polymerase and Nsp13 helicase. J. Pharm. Anal. 2020, 10, 320–328. 10.1016/j.jpha.2020.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroli N.; Bhasuran B.; Natarajan J.; Kolandaivel P. The potential role of procyanidin as a therapeutic agent against SARS-CoV-2: a text mining, molecular docking and molecular dynamics simulation approach. J. Biomol. Struct. Dyn. 2020, 1–16. 10.1080/07391102.2020.1823887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parida P. K.; Paul D.; Chakravorty D. The natural way forward: Molecular dynamics simulation analysis of phytochemicals from Indian medicinal plants as potential inhibitors of SARS-CoV-2 targets. Phytother Res. 2020, 34, 3420–3433. 10.1002/ptr.6868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parr R. G.Materials Horizons; Springer, 1980; pp 5–15. [Google Scholar]
- Lee C.; Yang W.; Parr R. G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785. 10.1103/PhysRevB.37.785. [DOI] [PubMed] [Google Scholar]
- Yu Q.; Hammes-Schiffer S. Nuclear-Electronic Orbital Multistate Density Functional Theory. J. Phys. Chem. Lett. 2020, 11, 10106–10113. 10.1021/acs.jpclett.0c02923. [DOI] [PubMed] [Google Scholar]
- Kohn W. Nobel Lecture: Electronic structure of matter′wave functions and density functionals. Rev. Mod. Phys. 1999, 71, 1253. 10.1103/RevModPhys.71.1253. [DOI] [Google Scholar]
- Nogara P. A.; Omage F. B.; Bolzan G. R.; Delgado C. P.; Aschner M.; Orian L.; Teixeira Rocha J. B. In silico Studies on the Interaction Between Mpro and PLpro From SARS-CoV-2 and Ebselen, its Metabolites and Derivatives. Mol. Inform. 2021, 40, 2100028. 10.1002/minf.202100028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madabeni A.; Nogara P. A.; Omage F. B.; Rocha J. B. T.; Orian L. Mechanistic Insight into SARS-CoV-2 Mpro Inhibition by Organoselenides: The Ebselen Case Study. Appl. Sci. 2021, 11, 6291. 10.3390/app11146291. [DOI] [Google Scholar]
- Wang Y.; Murlidaran S.; Pearlman D. A. Quantum simulations of SARS-CoV-2 main protease Mpro enable high-quality scoring of diverse ligands. J. Comput. Aided Mol. Des. 2021, 35, 963. 10.1007/s10822-021-00412-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shekh S.; Reddy K. K. A.; Gowd K. H. In silico allicin induced S-thioallylation of SARS-CoV-2 main protease. J. Sulphur Chem. 2021, 42, 109–120. 10.1080/17415993.2020.1817457. [DOI] [Google Scholar]
- Khan J.; Sakib S. A.; Mahmud S.; Khan Z.; Islam M. N.; Sakib M. A.; Emran T. B.; Simal-Gandara J. Identification of potential phytochemicals from Citrus Limon against main protease of SARS-CoV-2: molecular docking, molecular dynamic simulations and quantum computations. J. Biomol. Struct 2021, 1–12. 10.1080/07391102.2021.1947893. [DOI] [PubMed] [Google Scholar]
- Yadav P.; Rana M.; Chowdhury P. DFT and MD Simulation Investigation of Favipiravir as an Emerging Antiviral Option against Viral Protease (3CLpro) of SARS-CoV-2. J. Mol. Struct. 2021, 1246, 131253. 10.1016/j.molstruc.2021.131253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu Quy P.; Thi Ai My T.; Thi Thanh Hai N.; Bui T. Q.; Tuan Quang D.; Thanh Triet N.; Phuoc Hien P.; Thi Ai Nhung N. A Computational Screening on Inhibitability of Piper Betle Essential Oil Chemical Structures against Spike Proteins of Mutated SARS-CoV-2-variants D614G, N501Y, and S477N. Smart Sci. 2021, 1–18. 10.1080/23080477.2021.2003948. [DOI] [Google Scholar]
- Singh P.; Kumar D.; Pal S.; Kumari K.; Bahadur I. L-amino-acids as immunity booster against COVID-19: DFT, molecular docking and MD simulations. J. Mol. Struct. 2022, 1250, 131924. 10.1016/j.molstruc.2021.131924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Divya K.; Savitha D.; Krishna G. A.; Dhanya T.; Mohanan P. Crystal structure, DFT studies, Hirshfeld surface and energy framework analysis of 4-(5-nitro-thiophen-2-yl)-pyrrolo [1, 2-a] quinoxaline: A potential SARS-CoV-2 main protease inhibitor. J. Mol. Struct. 2022, 1251, 131932. 10.1016/j.molstruc.2021.131932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye N.; Caruso F.; Rossi M. Mechanistic insights into the inhibition of SARS-CoV-2 main protease by clovamide and its derivatives: in silico studies. Biophysica 2021, 1, 377–404. 10.3390/biophysica1040028. [DOI] [Google Scholar]
- de Sa E. R. A.; Costa A. N.; Costa R. K. M.; Souza J. L.; Ramos R. M.; Lima F. d. C. A. In silico study of the interactions of Pilocarpus microphyllus imidazolic alkaloids with the main protease (Mpro) of SARS-CoV-2. Mol. Simul 2021, 47, 74–87. 10.1080/08927022.2021.1873321. [DOI] [Google Scholar]
- Erdogan T. DFT, molecular docking and molecular dynamics simulation studies on some newly introduced natural products for their potential use against SARS-CoV-2. J. Mol. Struct. 2021, 1242, 130733. 10.1016/j.molstruc.2021.130733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sepay N.; Sekar A.; Halder U. C.; Alarifi A.; Afzal M. Anti-COVID-19 terpenoid from marine sources: A docking, admet and molecular dynamics study. J. Mol. Struct. 2021, 1228, 129433. 10.1016/j.molstruc.2020.129433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkhimova L. E.; Babashkina M. G.; Safin D. A. α-Aminophosphonates 4-XC6H4–NH–CH (4-BrC6H4)–P (O)(OiPr) 2 (X= H, Br, MeO): Crystal structures, Hirshfeld surface analysis, computational studies and in silico molecular docking with the SARS-CoV-2 proteins. Tetrahedron 2021, 97, 132376. 10.1016/j.tet.2021.132376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadela-Tomanek M.; Jastrzebska M.; Marciniec K.; Chrobak E.; Bebenek E.; Boryczka S. Lipophilicity, Pharmacokinetic Properties, and Molecular Docking Study on SARS-CoV-2 Target for Betulin Triazole Derivatives with Attached 1, 4-Quinone. Pharmaceutics 2021, 13, 781. 10.3390/pharmaceutics13060781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karagoz Genç Z.; Genç M.; Çoşut B.; Turgut M. The novel tetrahydropyrimidine derivative as inhibitor of SARS CoV-2: synthesis, modeling and molecular docking analysis. J. Biomol. Struct 2021, 1–12. 10.1080/07391102.2021.1938230. [DOI] [PubMed] [Google Scholar]
- Mohapatra R. K.; Perekhoda L.; Azam M.; Suleiman M.; Sarangi A. K.; Semenets A.; Pintilie L.; Al-Resayes S. I. Computational investigations of three main drugs and their comparison with synthesized compounds as potent inhibitors of SARS-CoV-2 main protease (Mpro): DFT, QSAR, molecular docking, and in silico toxicity analysis. J. King Saud Univ. Sci. 2021, 33, 101315. 10.1016/j.jksus.2020.101315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgon N. H.; Grandini G. S.; Yoguim M. I.; Porto C. M.; Santana L. C.; Biswas S.; de Souza A. R. Potential activity of Linezolid against SARS-CoV-2 using electronic and molecular docking study. J. Mol. Model. 2021, 27, 1–11. 10.1007/s00894-021-04828-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faria S. H.; Teleschi J. G. Computational search for drug repurposing to identify potential inhibitors against SARS-COV-2 using Molecular Docking, QTAIM and IQA methods in viral Spike protein–Human ACE2 interface. J. Mol. Struct. 2021, 1232, 130076. 10.1016/j.molstruc.2021.130076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Sayed D. S.; El-sayed M. A. Computational details of molecular structure, spectroscopic properties, topological studies and SARS-Cov-2 enzyme molecular docking simulation of substituted triazolo pyrimidine thione heterocycles. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2021, 261, 120006. 10.1016/j.saa.2021.120006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zia M.; Muhammad S.; Bibi S.; Abbasi S. W.; Al-Sehemi A. G.; Chaudhary A. R.; Bai F. Q.; et al. Exploring the potential of novel phenolic compounds as potential therapeutic candidates against SARS-CoV-2, using quantum chemistry, molecular docking and dynamic studies. Bioorg. Med. Chem. Lett. 2021, 43, 128079. 10.1016/j.bmcl.2021.128079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majeed A.; Hussain W.; Yasmin F.; Akhtar A.; Rasool N. Virtual Screening of Phytochemicals by Targeting HR1 Domain of SARS-CoV-2 S Protein: Molecular Docking, Molecular Dynamics Simulations, and DFT Studies. Biomed Res. Int. 2021, 2021, 6661191. 10.1155/2021/6661191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alagumuthu M.; Rajpoot S.; Baig M. S. Structure-based design of novel peptidomimetics targeting the SARS-CoV-2 spike protein. Cell Mol. Bioeng 2021, 14, 177–185. 10.1007/s12195-020-00658-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhammad S.; Hassan S. H.; Al-Sehemi A. G.; Shakir H. A.; Khan M.; Irfan M.; Iqbal J. Exploring the new potential antiviral constituents of Moringa oliefera for SARS-COV-2 pathogenesis: An in silico molecular docking and dynamic studies. Chem. Phys. Lett. 2021, 767, 138379. 10.1016/j.cplett.2021.138379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aitouna A. O.; Belghiti M.; Eşme A.; Zeroual A.; Salah M.; Chakroun A.; El Abdallaoui H. E. A.; Benharref A.; Mazoir N.; et al. Chemical Reactivities and Molecular Docking Studies of Parthenolide with the main protease of HEP-G2 and SARS-CoV-2. J. Mol. Struct. 2021, 1243, 130705. 10.1016/j.molstruc.2021.130705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aitouna A. O.; Belghiti M.; Eşme A.; Aitouna A. O.; Salah M.; Chekroun A.; El Abdallaoui H. E. A.; Benharref A.; Mazoir N.; Zeroual A.; et al. Divulging the Regioselectivity of Epoxides in the Ring-Opening Reaction, and Potential Himachalene Derivatives Predicted to Target the Antibacterial Activities and SARS-CoV-2 Spike Protein with Docking Study. J. Mol. Struct. 2021, 1244, 130864. 10.1016/j.molstruc.2021.130864. [DOI] [Google Scholar]
- Sheena Mary Y.; Shyma Mary Y.; Yadav R.; Celik I.; Rad A. S.; Sarala S. MD DFT Investigations and Inhibition of the Novel SARS-CoV-2 Mainprotease in Three Cocrystals of Hydrochloro-thiazide. Anal. Chem. Lett. 2021, 11, 450–468. 10.1080/22297928.2021.1934538. [DOI] [Google Scholar]
- Raj V.; Park J. G.; Cho K.-H.; Choi P.; Kim T.; Ham J.; Lee J. Assessment of antiviral potencies of cannabinoids against SARS-CoV-2 using computational and in vitro approaches. Int. J. Biol. 2021, 168, 474–485. 10.1016/j.ijbiomac.2020.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadela-Tomanek M.; Jastrzebska M.; Marciniec K.; Bebenek E.; Chrobak E.; Boryczka S. Spectroscopic Investigations, Computational Analysis and Molecular Docking to SAR-Cov-2 Targets Studies of 5, 8-Quinolinedione Attached to Betulin Derivatives. Crystals 2021, 11, 76. 10.3390/cryst11010076. [DOI] [Google Scholar]
- Bui T. Q.; Loan H. T. P.; My T. T. A.; Quang D. T.; Thuy B. T. P.; Nhan V. D.; Quy P. T.; Van Tat P.; Dao D. Q.; Trung N. T.; et al. A density functional theory study on silver and bis-silver complexes with lighter tetrylene: are silver and bis-silver carbenes candidates for SARS-CoV-2 inhibition? Insight from molecular docking simulation. RSC Adv. 2020, 10, 30961–30974. 10.1039/D0RA05159D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatfaoui S.; Sagaama A.; Issaoui N.; Roisnel T.; Marouani H. Synthesis, experimental, theoretical study and molecular docking of 1-ethylpiperazine-1, 4-diium bis (nitrate). Solid State Sci. 2020, 106, 106326. 10.1016/j.solidstatesciences.2020.106326. [DOI] [Google Scholar]
- Hagar M.; Ahmed H. A.; Aljohani G.; Alhaddad O. A. Investigation of Some Antiviral N-Heterocycles as COVID 19 Drug: Molecular Docking and DFT Calculations. Int. J. Mol. Sci. 2020, 21, 3922. 10.3390/ijms21113922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulimov A.; Ilin I.; Kutov D.; Stolpovskaya N.; Shikhaliev K. S.; Sulimov V. Supercomputing, Docking and Quantum Mechanics in Quest for Inhibitors of Papain-like Protease of SARS-CoV-2. Lobachevskii J. Math. 2021, 42, 1571–1579. 10.1134/S1995080221070222. [DOI] [Google Scholar]
- Cavasotto C. N.; Di Filippo J. I. In Silico drug repurposing for COVID-19: Targeting SARS-CoV-2 proteins through docking and consensus ranking. Mol. Inform. 2021, 40, 2000115. 10.1002/minf.202000115. [DOI] [PubMed] [Google Scholar]
- Ahmed S.; Mahtarin R.; Islam M. S.; Das S.; Al Mamun A.; Ahmed S. S.; Ali M. A. Remdesivir analogs against SARS-CoV-2 RNA-dependent RNA polymerase. J. Biomol. Struct 2021, 1–14. 10.1080/07391102.2021.1955743. [DOI] [PubMed] [Google Scholar]
- Hammes-Schiffer S.; Tully J. C. Proton transfer in solution: Molecular dynamics with quantum transitions. J. Chem. Phys. 1994, 101, 4657–4667. 10.1063/1.467455. [DOI] [Google Scholar]
- Warshel A.; Levitt M. Theoretical studies of enzymic reactions: dielectric, electrostatic and steric stabilization of the carbonium ion in the reaction of lysozyme. J. Mol. Biol. 1976, 103, 227–249. 10.1016/0022-2836(76)90311-9. [DOI] [PubMed] [Google Scholar]
- Ramos-Guzmán C. A.; Ruiz-Pernía J. J.; Tuñón I. Computational Simulations on the Binding and Reactivity of a Nitrile Inhibitor of SARS-CoV-2 Main Protease. ChemComm 2021, 57, 9096. 10.1039/D1CC03953A. [DOI] [PubMed] [Google Scholar]
- Ramos-Guzmán C. A.; Ruiz-Pernía J. J.; Tuñón I. A microscopic description of SARS-CoV-2 main protease inhibition with Michael acceptors. Strategies for improving inhibitor design. Chem. Sci. 2021, 12, 3489–3496. 10.1039/D0SC04978F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arafet K.; Serrano-Aparicio N.; Lodola A.; Mulholland A. J.; González F. V.; Świderek K.; Moliner V. Mechanism of inhibition of SARS-CoV-2 M pro by N3 peptidyl Michael acceptor explained by QM/MM simulations and design of new derivatives with tunable chemical reactivity. Chem. Sci. 2021, 12, 1433–1444. 10.1039/D0SC06195F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos-Guzmán C. A.; Ruiz-Pernía J. J.; Tuñón I. Multiscale simulations of SARS-CoV-2 3CL protease inhibition with aldehyde derivatives. Role of protein and inhibitor conformational changes in the reaction mechanism. ACS Catal. 2021, 11, 4157–4168. 10.1021/acscatal.0c05522. [DOI] [PubMed] [Google Scholar]
- Monajemi H.; Zain S. M. Strong inhibition of M-Protease activity of Coronavirus by using PX-12 inhibitor based on ab initio ONIOM calculations. J. Chem. Res. 2021, 45, 136–140. 10.1177/1747519820938025. [DOI] [Google Scholar]
- Fernandes H. S.; Sousa S. F.; Cerqueira N. M. New insights into the catalytic mechanism of the SARS-CoV-2 main protease: an ONIOM QM/MM approach. Mol. Divers. 2021, 1–9. 10.1007/s11030-021-10259-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Díaz N.; Suárez D. Influence of charge configuration on substrate binding to SARS-CoV-2 main protease. ChemComm 2021, 57, 5314–5317. 10.1039/D1CC01449H. [DOI] [PubMed] [Google Scholar]
- Khrenova M. G.; Tsirelson V. G.; Nemukhin A. V. Dynamical properties of enzyme–substrate complexes disclose substrate specificity of the SARS-CoV-2 main protease as characterized by the electron density descriptors. Phys. Chem. Chem. Phys. 2020, 22, 19069–19079. 10.1039/D0CP03560B. [DOI] [PubMed] [Google Scholar]
- Świderek K.; Moliner V. Revealing the molecular mechanisms of proteolysis of SARS-CoV-2 M pro by QM/MM computational methods. Chem. Sci. 2020, 11, 10626–10630. 10.1039/D0SC02823A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos-Guzmán C. A.; Ruiz-Pernia J. J.; Tuñón I. Unraveling the SARS-CoV-2 Main Protease Mechanism Using Multiscale Methods. ACS Catal. 2020, 10, 12544–12554. 10.1021/acscatal.0c03420. [DOI] [PubMed] [Google Scholar]
- Gómez S. A.; Rojas-Valencia N.; Gómez S.; Egidi F.; Cappelli C.; Restrepo A. Binding of SARS-CoV-2 to cell receptors: a tale of molecular evolution. ChemBioChem. 2021, 22, 724–732. 10.1002/cbic.202000618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santra S.; Giri S.; Jana M. Unraveling the origin of interactions of hydroxychloroquine with the receptor-binding domain of SARS-CoV-2 in aqueous medium. Chem. Phys. Lett. 2021, 764, 138280. 10.1016/j.cplett.2020.138280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao R.; Wang M.; Chen J.; Tong Y.; Wei G.-W. The de Rham–Hodge analysis and modeling of biomolecules. Bull. Math. Biol. 2020, 82, 1–38. 10.1007/s11538-020-00783-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wee J.; Xia K. Forman persistent Ricci curvature (FPRC)-based machine learning models for protein–ligand binding affinity prediction. Brief. Bioinformatics 2021, 22, bbab136. 10.1093/bib/bbab136. [DOI] [PubMed] [Google Scholar]
- Chen J.; Zhao R.; Tong Y.; Wei G.-W. Evolutionary de rham-hodge method. Discrete Continuous Dyn Syst. Ser. B 2021, 26, 3785. 10.3934/dcdsb.2020257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia K.; Wei G.-W. Persistent homology analysis of protein structure, flexibility, and folding. Int. J. Numer. Methods Biomed. Eng. 2014, 30, 814–844. 10.1002/cnm.2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen D. D.; Wei G.-W. AGL-score: algebraic graph learning score for protein–ligand binding scoring, ranking, docking, and screening. J. Chem. Inf Model 2019, 59, 3291–3304. 10.1021/acs.jcim.9b00334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen D. D.; Cang Z.; Wei G.-W. A review of mathematical representations of biomolecular data. Phys. Chem. Chem. Phys. 2020, 22, 4343–4367. 10.1039/C9CP06554G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estrada E. Topological analysis of SARS CoV-2 main protease. Chaos 2020, 30, 061102. 10.1063/5.0013029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estrada E. Quantifying network heterogeneity. Phys. Rev. E 2010, 82, 066102. 10.1103/PhysRevE.82.066102. [DOI] [PubMed] [Google Scholar]
- Watts D. J.; Strogatz S. H. Collective dynamics of ‘small-world’networks. nature 1998, 393, 440–442. 10.1038/30918. [DOI] [PubMed] [Google Scholar]
- Freeman L. C. Centrality in Soc Networks conceptual clarification. Soc. Networks 1978, 1, 215–239. 10.1016/0378-8733(78)90021-7. [DOI] [Google Scholar]
- Estrada E.; Rodriguez-Velazquez J. A. Subgraph centrality in complex networks. Phys. Rev. E 2005, 71, 056103. 10.1103/PhysRevE.71.056103. [DOI] [PubMed] [Google Scholar]
- Estrada E.; Hatano N. Communicability in complex networks. Phys. Rev. E 2008, 77, 036111. 10.1103/PhysRevE.77.036111. [DOI] [PubMed] [Google Scholar]
- Estrada E.; Hatano N. Communicability angle and the spatial efficiency of networks. SIAM Rev. 2016, 58, 692–715. 10.1137/141000555. [DOI] [Google Scholar]
- Bavelas A. Communication patterns in task-oriented groups. J. Acoust. Soc. Am. 1950, 22, 725–730. 10.1121/1.1906679. [DOI] [Google Scholar]
- Stelzl U.; Worm U.; Lalowski M.; Haenig C.; Brembeck F. H.; Goehler H.; Stroedicke M.; Zenkner M.; Schoenherr A.; Koeppen S.; et al. A human protein-protein interaction network: a resource for annotating the proteome. Cell 2005, 122, 957–968. 10.1016/j.cell.2005.08.029. [DOI] [PubMed] [Google Scholar]
- Griffin J. W. SARS-CoV and SARS-CoV-2 main protease residue interaction networks change when bound to inhibitor N3. J. Struct. Biol. 2020, 211, 107575. 10.1016/j.jsb.2020.107575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Díaz J. SARS-CoV-2 Molecular Network Structure. Front. Physiol. 2020, 11, 870. 10.3389/fphys.2020.00870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karathanou K.; Lazaratos M.; Bertalan É.; Siemers M.; Buzar K.; Schertler G. F.; Del Val C.; Bondar A.-N. A graph-based approach identifies dynamic H-bond communication networks in spike protein S of SARS-CoV-2. J. Struct. Biol. 2020, 212, 107617. 10.1016/j.jsb.2020.107617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lata S.; Akif M. Comparative protein structure network analysis on 3CLpro from SARS-CoV-1 and SARS-CoV-2. Proteins 2021, 89, 1216–1225. 10.1002/prot.26143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amamuddy O. S.; Boateng R. A.; Barozi V.; Nyamai D. W.; Bishop Ö. T. Novel dynamic residue network analysis approaches to study allosteric modulation: SARS-CoV-2 Mpro and its evolutionary mutations as a case study. Comput. Struct. Biotechnol. J. 2021, 19, 6431–6455. 10.1016/j.csbj.2021.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray D.; Le L.; Andricioaei I.. Distant residues modulate conformational opening in SARS-CoV-2 spike protein. Proc. Natl. Acad. Sci. U.S.A. 2021, 118, 10.1073/pnas.2100943118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahbar M. R.; Jahangiri A.; Khalili S.; Zarei M.; Mehrabani-Zeinabad K.; Khalesi B.; Pourzardosht N.; Hessami A.; Nezafat N.; Sadraei S.; et al. Hotspots for mutations in the SARS-CoV-2 spike glycoprotein: a correspondence analysis. Sci. Rep. 2021, 11, 1–17. 10.1038/s41598-021-01655-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha S.; Chatterjee P.; Nasipuri M.; Basu S. Detection of spreader nodes in human-SARS-CoV protein-protein interaction network. PeerJ. 2021, 9, e12117 10.7717/peerj.12117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia K.; Opron K.; Wei G.-W. Multiscale multiphysics and multidomain models–Flexibility and rigidity. J. Chem. Phys. 2013, 139, 194109. 10.1063/1.4830404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opron K.; Xia K.; Wei G.-W. Communication: Capturing protein multiscale thermal fluctuations. J. Chem. Phys. 2015, 142, 211101. 10.1063/1.4922045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bramer D.; Wei G.-W. Multiscale weighted colored graphs for protein flexibility and rigidity analysis. J. Chem. Phys. 2018, 148, 054103. 10.1063/1.5016562. [DOI] [PubMed] [Google Scholar]
- Nguyen D. D.; Xiao T.; Wang M.; Wei G.-W. Rigidity strengthening: A mechanism for protein–ligand binding. J. Chem. Inf Model 2017, 57, 1715–1721. 10.1021/acs.jcim.7b00226. [DOI] [PubMed] [Google Scholar]
- Wang M.; Cang Z.; Wei G.-W. A topology-based network tree for the prediction of protein–protein binding affinity changes following mutation. Nat. Mach. Intell. 2020, 2, 116–123. 10.1038/s42256-020-0149-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cang Z.; Wei G.-W. Analysis and prediction of protein folding energy changes upon mutation by element specific persistent homology. Bioinformatics 2017, 33, 3549–3557. 10.1093/bioinformatics/btx460. [DOI] [PubMed] [Google Scholar]
- Chen J.; Gao K.; Wang R.; Wei G.-W. Prediction and mitigation of mutation threats to COVID-19 vaccines and antibody therapies. Chem. Sci. 2021, 12, 6929–6948. 10.1039/D1SC01203G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand D. V.; Meng Z.; Xia K.; Mu Y. Weighted persistent homology for osmolyte molecular aggregation and hydrogen-bonding network analysis. Sci. Rep. 2020, 10, 1–17. 10.1038/s41598-020-66710-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaczynski T.; Mischaikow K.; Mrozek M.. Computational homology; Springer Science & Business Media, 2006; Vol. 157. [Google Scholar]
- Carlsson G. Topology and data. Bull. Am. Math. Soc. 2009, 46, 255–308. 10.1090/S0273-0979-09-01249-X. [DOI] [Google Scholar]
- Zomorodian A.; Carlsson G. Computing persistent homology. Discrete Comput. Geom 2005, 33, 249–274. 10.1007/s00454-004-1146-y. [DOI] [Google Scholar]
- Edelsbrunner H.; Harer J. Persistent homology-a survey. Contemp. Math. 2008, 453, 257–282. 10.1090/conm/453/08802. [DOI] [Google Scholar]
- Kovacev-Nikolic V.; Bubenik P.; Nikolić D.; Heo G. Using persistent homology and dynamical distances to analyze protein binding. Stat Appl. Genet Mol. 2016, 15, 19–38. 10.1515/sagmb-2015-0057. [DOI] [PubMed] [Google Scholar]
- Xia K.; Zhao Z.; Wei G.-W. Multiresolution persistent homology for excessively large biomolecular datasets. J. Chem. Phys. 2015, 143, 134103. 10.1063/1.4931733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R.; Nguyen D. D.; Wei G.-W. Persistent spectral graph. Int. J. Numer. Methods Biomed. Eng. 2020, 36, e3376 10.1002/cnm.3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng Z.; Xia K. Persistent spectral–based machine learning (PerSpect ML) for protein-ligand binding affinity prediction. Sci. Adv. 2021, 7, eabc5329 10.1126/sciadv.abc5329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wee J.; Xia K. Persistent spectral based ensemble learning (PerSpect-EL) for protein–protein binding affinity prediction. Brief. Bioinformatics 2022, 10.1093/bib/bbac024. [DOI] [PubMed] [Google Scholar]
- Edelsbrunner H.; Letscher D.; Zomorodian A.. Proceedings of the 41st annual symposium on foundations of computer science; 2000; pp 454–463.
- Cang Z.; Mu L.; Wei G.-W. Representability of algebraic topology for biomolecules in Mach. Learn. based scoring and virtual screening. PLoS Comput. Biol. 2018, 14, e1005929 10.1371/journal.pcbi.1005929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cang Z.; Mu L.; Wu K.; Opron K.; Xia K.; Wei G.-W. A topological approach for protein classification. Mol. Based Math. Biol. 2015, 3, 140–162. 10.1515/mlbmb-2015-0009. [DOI] [Google Scholar]
- Cang Z.; Wei G.-W. Integration of element specific persistent homology and machine learning for protein-ligand binding affinity prediction. Int. J. Numer. Methods Biomed. Eng. 2018, 34, e2914 10.1002/cnm.2914. [DOI] [PubMed] [Google Scholar]
- Nguyen D. D.; Cang Z.; Wu K.; Wang M.; Cao Y.; Wei G.-W. Mathematical deep learning for pose and binding affinity prediction and ranking in D3R Grand Challenges. J. Comput. Aided Mol. Des. 2019, 33, 71–82. 10.1007/s10822-018-0146-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen D. D.; Gao K.; Chen J.; Wang R.; Wei G.-W. Unveiling the molecular mechanism of SARS-CoV-2 main protease inhibition from 137 crystal structures using algebraic topology and deep learning. Chem. Sci. 2020, 11, 12036–12046. 10.1039/D0SC04641H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Moraga R.; Forés-Martos J.; Suay-García B.; Duval J.-L.; Falcó A.; Climent J. A COVID-19 Drug Repurposing Strategy through Quantitative Homological Similarities Using a Topological Data Analysis-Based Framework. Pharmaceutics 2021, 13, 488. 10.3390/pharmaceutics13040488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotelling H. Analysis of a complex of statistical variables into principal components. J. Educ. Psychol. 1933, 24, 417. 10.1037/h0071325. [DOI] [Google Scholar]
- Kruskal J. B. Multidimensional scaling by optimizing goodness of fit to a nonmetric hypothesis. Psychometrika 1964, 29, 1–27. 10.1007/BF02289565. [DOI] [Google Scholar]
- Hinton G. E.; Salakhutdinov R. R. Reducing the dimensionality of data with neural networks. Science 2006, 313, 504–507. 10.1126/science.1127647. [DOI] [PubMed] [Google Scholar]
- Sammon J. W. A. A Nonlinear Mapping for Data Structure Analysis. IEEE Trans. Comput. 1969, C-18, 401–409. 10.1109/T-C.1969.222678. [DOI] [Google Scholar]
- Blei D. M.; Ng A. Y.; Jordan M. I. Latent Dirichlet Allocation. J. Mach. Learn. Res. 2003, 3, 993–1022. [Google Scholar]
- Lee D. D.; Seung H. S. Learning the parts of objects by non-negative matrix factorization. Nature 1999, 401, 788–791. 10.1038/44565. [DOI] [PubMed] [Google Scholar]
- Belkin M.; Niyogi P. Laplacian Eigenmaps and Spectral Techniques for Embedding and Clustering. NIPS 2001 2001, 14, 585–591. [Google Scholar]
- Belkin M.; Niyogi P. Laplacian eigenmaps for dimensionality reduction and data representation. Neural Comput 2003, 15, 1373–1396. 10.1162/089976603321780317. [DOI] [Google Scholar]
- Donoho D. L.; Grimes C. Hessian eigenmaps: Locally linear embedding techniques for high-dimensional data. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 5591–5596. 10.1073/pnas.1031596100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z.; Zha H. Principal manifolds and nonlinear dimensionality reduction via tangent space alignment. SIAM J. Sci. Comput 2004, 26, 313–338. 10.1137/S1064827502419154. [DOI] [Google Scholar]
- Tenenbaum J. B.; De Silva V.; Langford J. C. A global geometric framework for nonlinear dimensionality reduction. Science 2000, 290, 2319–2323. 10.1126/science.290.5500.2319. [DOI] [PubMed] [Google Scholar]
- Van Der Maaten L. Accelerating t-SNE using tree-based algorithms. J. Mach. Learn. Res. 2014, 15, 3221–3245. [Google Scholar]
- Van der Maaten L.; Hinton G. Visualizing data using t-SNE. J. Mach. Learn. Res. 2008, 9, 2579–2605. [Google Scholar]
- McInnes L.; Healy J.; Saul N.; Großberger L. Umap: Uniform manifold approximation and projection for dimension reduction. JOSS 2018, 3, 861. 10.21105/joss.00861. [DOI] [Google Scholar]
- Islam R.; Parves M. R.; Paul A. S.; Uddin N.; Rahman M. S.; Mamun A. A.; Hossain M. N.; Ali M. A.; Halim M. A. A molecular modeling approach to identify effective antiviral phytochemicals against the main protease of SARS-CoV-2. J. Biomol. Struct 2021, 39, 3213–3224. 10.1080/07391102.2020.1761883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gawriljuk V. O.; Zin P. P. K.; Puhl A. C.; Zorn K. M.; Foil D. H.; Lane T. R.; Hurst B.; Tavella T. A.; Costa F. T. M.; Lakshmanane P.; et al. Machine learning models identify inhibitors of SARS-CoV-2. J. Chem. Inf Model 2021, 61, 4224–4235. 10.1021/acs.jcim.1c00683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gussow A. B.; Auslander N.; Faure G.; Wolf Y. I.; Zhang F.; Koonin E. V. Genomic determinants of pathogenicity in SARS-CoV-2 and other human coronaviruses. Proc. Natl. Acad. Sci. U. S. A. 2020, 117, 15193–15199. 10.1073/pnas.2008176117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bost P.; Giladi A.; Liu Y.; Bendjelal Y.; Xu G.; David E.; Blecher-Gonen R.; Cohen M.; Medaglia C.; Li H.; et al. Host-viral infection maps reveal signatures of severe COVID-19 patients. Cell 2020, 181, 1475–1488. 10.1016/j.cell.2020.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H.; Kang Z.; Gong H.; Xu D.; Wang J.; Li Z.; Li Z.; Cui X.; Xiao J.; Zhan J.; et al. Digestive system is a potential route of COVID-19: an analysis of single-cell coexpression pattern of key proteins in viral entry process. Gut 2020, 69, 1010–1018. 10.1136/gutjnl-2020-320953. [DOI] [Google Scholar]
- Lukassen S.; Chua R. L.; Trefzer T.; Kahn N. C.; Schneider M. A.; Muley T.; Winter H.; Meister M.; Veith C.; Boots A. W.; et al. SARS-CoV-2 receptor ACE 2 and TMPRSS 2 are primarily expressed in bronchial transient secretory cells. EMBO J. 2020, 39, e105114 10.15252/embj.2020105114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravindra N. G.; Alfajaro M. M.; Gasque V.; Huston N. C.; Wan H.; Szigeti-Buck K.; Yasumoto Y.; Greaney A. M.; Habet V.; Chow R. D.; et al. Single-cell longitudinal analysis of SARS-CoV-2 infection in human airway epithelium identifies target cells, alterations in gene expression, and cell state changes. PLoS Biol. 2021, 19, e3001143 10.1371/journal.pbio.3001143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jary A.; Leducq V.; Malet I.; Marot S.; Klement-Frutos E.; Teyssou E.; Soulié C.; Abdi B.; Wirden M.; Pourcher V.; et al. Evolution of viral quasispecies during SARS-CoV-2 infection. Clin. Microbiol. Infect. 2020, 26, 1560.e1. 10.1016/j.cmi.2020.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma D.; Rawat P.; Janakiraman V.; Gromiha M. M. Elucidating important structural features for the binding affinity of spike-SARS-CoV-2 neutralizing antibody complexes. Proteins 2022, 90, 824–834. 10.1002/prot.26277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Israel A.; Shenhar Y.; Green I.; Merzon E.; Golan-Cohen A.; Schäffer A. A.; Ruppin E.; Vinker S.; Magen E. Large-scale study of antibody titer decay following BNT162b2 mRNA vaccine or SARS-CoV-2 infection. Vaccines 2022, 10, 64. 10.3390/vaccines10010064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayouba A.; Thaurignac G.; Morquin D.; Tuaillon E.; Raulino R.; Nkuba A.; Lacroix A.; Vidal N.; Foulongne V.; Le Moing V.; et al. Multiplex detection and dynamics of IgG antibodies to SARS-CoV2 and the highly pathogenic human coronaviruses SARS-CoV and MERS-CoV. J. Clin. Virol. 2020, 129, 104521. 10.1016/j.jcv.2020.104521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levantovsky R.; van der Heide V. Shared CD8+ T cell receptors for SARS-CoV-2. Nat. Rev. Immunol. 2020, 20, 591–591. 10.1038/s41577-020-00433-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlova A.; Zhang Z.; Acharya A.; Lynch D. L.; Pang Y. T.; Mou Z.; Parks J. M.; Chipot C.; Gumbart J. C. Machine Learning Reveals the Critical Interactions for SARS-CoV-2 Spike Protein Binding to ACE2. J. Phys. Chem. Lett. 2021, 12, 5494–5502. 10.1021/acs.jpclett.1c01494. [DOI] [PubMed] [Google Scholar]
- Cover T.; Hart P. Nearest neighbor pattern classification. IEEE Trans. Inf. Theory 1967, 13, 21–27. 10.1109/TIT.1967.1053964. [DOI] [Google Scholar]
- Altman N. S. An introduction to kernel and nearest-neighbor nonparametric regression. Am. Stat 1992, 46, 175–185. 10.2307/2685209. [DOI] [Google Scholar]
- Naeem S. M.; Mabrouk M. S.; Marzouk S. Y.; Eldosoky M. A. A diagnostic genomic signal processing (GSP)-based system for automatic feature analysis and detection of COVID-19. Brief. Bioinformatics 2021, 22, 1197–1205. 10.1093/bib/bbaa170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee S.; Tworowski D.; Detroja R.; Mukherjee S. B.; Frenkel-Morgenstern M. Immunoinformatics and Structural Analysis for Identification of Immunodominant Epitopes in SARS-CoV-2 as Potential Vaccine Targets. Vaccines 2020, 8, 290. 10.3390/vaccines8020290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afify H. M.; Zanaty M. S. Computational predictions for protein sequences of COVID-19 virus via Mach. Learn. algorithms. Med. Biol. Eng. Comput 2021, 59, 1723–1734. 10.1007/s11517-021-02412-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.-H.; Li H.; Zeng T.; Chen L.; Li Z.; Huang T.; Cai Y.-D. Identifying transcriptomic signatures and rules for SARS-CoV-2 infection. Front. Cell Dev. Biol. 2021, 8, 1763. 10.3389/fcell.2020.627302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang D.; Song M.; Niu Z.; Zhang P.; Rafailovich M.; Deng Y. Supervised machine learning approach to molecular dynamics forecast of SARS-CoV-2 spike glycoproteins at varying temperatures. MRS Adv. 2021, 6, 362. 10.1557/s43580-021-00021-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hozumi Y.; Wang R.; Yin C.; Wei G.-W. UMAP-assisted K-means clustering of large-scale SARS-CoV-2 mutation datasets. Comput. Biol. Med. 2021, 131, 104264. 10.1016/j.compbiomed.2021.104264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assis R.; Jain A.; Nakajima R.; Jasinskas A.; Khan S.; Davies H.; Corash L.; Dumont L. J.; Kelly K.; Simmons G.; et al. Distinct SARS-CoV-2 antibody reactivity patterns in coronavirus convalescent plasma revealed by a coronavirus antigen microarray. Sci. Rep. 2021, 11, 1–12. 10.1038/s41598-021-87137-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans D. J.; Yovanno R. A.; Rahman S.; Cao D. W.; Beckett M. Q.; Patel M. H.; Bandak A. F.; Lau A. Y. Finding Druggable Sites in Proteins Using TACTICS. J. Chem. Inf Model 2021, 61, 2897–2910. 10.1021/acs.jcim.1c00204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortes C.; Vapnik V. Support-vector networks. Mach. Learn. 1995, 20, 273–297. 10.1007/BF00994018. [DOI] [Google Scholar]
- Drucker H.; Burges C. J.; Kaufman L.; Smola A. J.; Vapnik V. Support Vector Regression Machines. Adv. Neural Inf. Process. Syst. 1997, 155–161. [Google Scholar]
- Kowalewski J.; Ray A. Predicting novel drugs for SARS-CoV-2 using maching learning from a >10 million chemical space. Heliyon 2020, 6, e04639 10.1016/j.heliyon.2020.e04639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta K.; Elmezayen A. D.; Al-Obaidi A.; Zhu W.; Morozova O. V.; Shityakov S.; Khalifa I. Seq12, Seq12m, and Seq13m, peptide analogues of the spike glycoprotein shows antiviral properties against SARS-CoV-2: An in silico study through molecular docking, molecular dynamics simulation, and MM-PB/GBSA calculations. J. Mol. Struct. 2021, 1246, 131113. 10.1016/j.molstruc.2021.131113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mekni N.; Coronnello C.; Langer T.; Rosa M. D.; Perricone U. Support Vector Machine as a Supervised Learning for the Prioritization of Novel Potential SARS-CoV-2 Main Protease Inhibitors. Int. J. Mol. Sci. 2021, 22, 7714. 10.3390/ijms22147714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H.; Wang Y.; Chen C. Z.; Xu M.; Guo H.; Itkin M.; Zheng W.; Shen M. Identification of SARS-CoV-2 viral entry inhibitors using machine learning and cell-based pseudotyped particle assay. Bioorg. Med. Chem. 2021, 38, 116119. 10.1016/j.bmc.2021.116119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kardani K.; Bolhassani A. Exploring novel and potent cell penetrating peptides in the proteome of SARS-COV-2 using bioinformatics approaches. PLoS One 2021, 16, e0247396 10.1371/journal.pone.0247396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breiman L. Random forests. Mach Learn 2001, 45, 5–32. 10.1023/A:1010933404324. [DOI] [Google Scholar]
- Mason L.; Baxter J.; Bartlett P. L.; Frean M. R. Boosting Algorithms as Gradient Descent. Adv. Neural Inf. Process. Syst. 2000, 512–518. [Google Scholar]
- Pedregosa F.; Varoquaux G.; Gramfort A.; Michel V.; Thirion B.; Grisel O.; Blondel M.; Prettenhofer P.; Weiss R.; Dubourg V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Ong E.; Wong M. U.; Huffman A.; He Y. COVID-19 coronavirus vaccine design using reverse vaccinology and Mach. Learn. Front. Immunol. 2020, 11, 1581. 10.3389/fimmu.2020.01581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao K.; Nguyen D. D.; Chen J.; Wang R.; Wei G.-W. Repositioning of 8565 existing drugs for COVID-19. J. Phys. Chem. Lett. 2020, 11, 5373–5382. 10.1021/acs.jpclett.0c01579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KC G. B.; Bocci G.; Verma S.; Hassan M. M.; Holmes J.; Yang J. J.; Sirimulla S.; Oprea T. I.; et al. A machine learning platform to estimate anti-SARS-CoV-2 activities. Nat. Mach. Intell. 2021, 3, 527. 10.1038/s42256-021-00335-w. [DOI] [Google Scholar]
- Kandpal M.; Davuluri R. V. Identification of Geographic Specific SARS-CoV-2 Mutations by Random Forest Classification and Variable Selection Methods. Stat. Appl. 2020, 18, 253–268. [PMC free article] [PubMed] [Google Scholar]
- Iyer A. S.; Jones F. K.; Nodoushani A.; Kelly M.; Becker M.; Slater D.; Mills R.; Teng E.; Kamruzzaman M.; Garcia-Beltran W. F.; et al. Dynamics and significance of the antibody response to SARS-CoV-2 infection. MedRxiv 2020, 10.1101/2020.07.18.20155374. [DOI] [Google Scholar]
- Rola M.; Krassowski J.; Górska J.; Grobelna A.; Płonka W.; Paneth A.; Paneth P. Machine Learning augmented docking studies of aminothioureas at the SARS-CoV-2—ACE2 interface. PLoS One 2021, 16, e0256834 10.1371/journal.pone.0256834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauvat A.; Ciccosanti F.; Colavita F.; Di Rienzo M.; Castilletti C.; Capobianchi M. R.; Kepp O.; Zitvogel L.; Fimia G. M.; Piacentini M.; et al. On-target versus off-target effects of drugs inhibiting the replication of SARS-CoV-2. Cell Death Dis 2020, 11, 1–11. 10.1038/s41419-020-02842-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariën J.; Ceulemans A.; Michiels J.; Heyndrickx L.; Kerkhof K.; Foque N.; Widdowson M.-A.; Mortgat L.; Duysburgh E.; Desombere I.; et al. Evaluating SARS-CoV-2 spike and nucleocapsid proteins as targets for antibody detection in severe and mild COVID-19 cases using a Luminex bead-based assay. J. Virol. Methods 2021, 288, 114025. 10.1016/j.jviromet.2020.114025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.-Y.; Lin Y.-H.; Kung C.-C.; Chung M.-H.; Yen I.; et al. Design and implementation of cloud analytics-assisted smart power meters considering advanced artificial intelligence as edge analytics in demand-side management for smart homes. Sensors 2019, 19, 2047. 10.3390/s19092047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava N.; Hinton G.; Krizhevsky A.; Sutskever I.; Salakhutdinov R. Dropout: a simple way to prevent neural networks from overfitting. J. Mach. Learn. Res. 2014, 15, 1929–1958. [Google Scholar]
- Secor M.; Soudackov A. V.; Hammes-Schiffer S. Artificial Neural Networks as Mappings between Proton Potentials, Wave Functions, Densities, and Energy Levels. J. Phys. Chem. Lett. 2021, 12, 2206–2212. 10.1021/acs.jpclett.1c00229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao H.; Wang J.; He L.; Qi Y.; Zhang J. Z. DeepDDG: predicting the stability change of protein point mutations using neural networks. J. Chem. Inf Model 2019, 59, 1508–1514. 10.1021/acs.jcim.8b00697. [DOI] [PubMed] [Google Scholar]
- Chen J.; Gao K.; Wang R.; Wei G.-W. Revealing the threat of emerging SARS-CoV-2 mutations to antibody therapies. J. Mol. Biol. 2021, 433, 167155. 10.1016/j.jmb.2021.167155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R.; Chen J.; Hozumi Y.; Yin C.; Wei G.-W. Emerging vaccine-breakthrough SARS-CoV-2 variants. ACS Infect. Dis. 2022, 8, 546. 10.1021/acsinfecdis.1c00557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ton A.-T.; Gentile F.; Hsing M.; Ban F.; Cherkasov A. Rapid identification of potential inhibitors of SARS-CoV-2 main protease by deep docking of 1.3 billion compounds. Mol. Inform. 2020, 39, 2000028. 10.1002/minf.202000028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karki N.; Verma N.; Trozzi F.; Tao P.; Kraka E.; Zoltowski B. Predicting Potential SARS-COV-2 Drugs–In Depth Drug Database Screening Using Deep Neural Network Framework SSnet, Classical Virtual Screening and Docking. Int. J. Mol. Sci. 2021, 22, 1573. 10.3390/ijms22041573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck B. R.; Shin B.; Choi Y.; Park S.; Kang K. Predicting commercially available antiviral drugs that may act on the novel coronavirus (SARS-CoV-2) through a drug-target interaction deep learning model. Comput. Struct. Biotechnol. J. 2020, 18, 784–790. 10.1016/j.csbj.2020.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuvaraj N.; Srihari K.; Chandragandhi S.; Raja R. A.; Dhiman G.; Kaur A. Analysis of protein-ligand interactions of SARS-Cov-2 against selective drug using deep neural networks. Big Data Min. Anal. 2021, 4, 76–83. 10.26599/BDMA.2020.9020007. [DOI] [Google Scholar]
- Hofmarcher M.; Mayr A.; Rumetshofer E.; Ruch P.; Renz P.; Schimunek J.; Seidl P.; Vall A.; Widrich M.; Hochreiter S.; et al. Large-scale ligand-based virtual screening for SARS-CoV-2 inhibitors using deep neural networks. SSRN J. 2020, 10.2139/ssrn.3561442. [DOI] [Google Scholar]
- Izumi H.; Nafie L. A.; Dukor R. K. SSSCPreds: Deep Neural Network-Based Software for the Prediction of Conformational Variability and Application to SARS-CoV-2. ACS Omega 2020, 5, 30556–30567. 10.1021/acsomega.0c04472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adhikari N.; Banerjee S.; Baidya S. K.; Ghosh B.; Jha T. Ligand-based quantitative structural assessments of SARS-CoV-2 3CLpro inhibitors: An analysis in light of structure-based multi-molecular modeling evidences. J. Mol. Struct. 2022, 1251, 132041. 10.1016/j.molstruc.2021.132041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin W.; Stokes J. M.; Eastman R. T.; Itkin Z.; Zakharov A. V.; Collins J. J.; Jaakkola T. S.; Barzilay R. Deep learning identifies synergistic drug combinations for treating COVID-19. Proc. Natl. Acad. Sci. U.S.A. 2021, 118, 118. 10.1073/pnas.2105070118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krizhevsky A.; Sutskever I.; Hinton G. E. Imagenet classification with deep convolutional neural networks. Commun. ACM 2017, 60, 84–90. 10.1145/3065386. [DOI] [Google Scholar]
- Lan J.; Ge J.; Yu J.; Shan S.; Zhou H.; Fan S.; Zhang Q.; Shi X.; Wang Q.; Zhang L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. 10.1038/s41586-020-2180-5. [DOI] [PubMed] [Google Scholar]
- Kim C.; Ryu D.-K.; Lee J.; Kim Y.-I.; Seo J.-M.; Kim Y.-G.; Jeong J.-H.; Kim M.; Kim J.-I.; Kim P.; et al. A therapeutic neutralizing antibody targeting receptor binding domain of SARS-CoV-2 spike protein. Nat. Commun. 2021, 12, 1–10. 10.1038/s41467-020-20602-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi R.; Shan C.; Duan X.; Chen Z.; Liu P.; Song J.; Song T.; Bi X.; Han C.; Wu L.; et al. A human neutralizing antibody targets the receptor-binding site of SARS-CoV-2. Nature 2020, 584, 120–124. 10.1038/s41586-020-2381-y. [DOI] [PubMed] [Google Scholar]
- Barnes C. O.; Jette C. A.; Abernathy M. E.; Dam K.-M. A.; Esswein S. R.; Gristick H. B.; Malyutin A. G.; Sharaf N. G.; Huey-Tubman K. E.; Lee Y. E.; et al. SARS-CoV-2 neutralizing antibody structures inform therapeutic strategies. Nature 2020, 588, 682–687. 10.1038/s41586-020-2852-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen J.; Baum A.; Pascal K. E.; Russo V.; Giordano S.; Wloga E.; Fulton B. O.; Yan Y.; Koon K.; Patel K.; et al. Studies in humanized mice and convalescent humans yield a SARS-CoV-2 antibody cocktail. Science 2020, 369, 1010–1014. 10.1126/science.abd0827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones B. E.; Brown-Augsburger P. L.; Corbett K. S.; Westendorf K.; Davies J.; Cujec T. P.; Wiethoff C. M.; Blackbourne J. L.; Heinz B. A.; Foster D.; et al. The neutralizing antibody, LY-CoV555, protects against SARS-CoV-2 infection in nonhuman primates. Sci. Transl. Med. 2021, 13, eabf1906 10.1126/scitranslmed.abf1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto D.; Park Y.-J.; Beltramello M.; Walls A. C.; Tortorici M. A.; Bianchi S.; Jaconi S.; Culap K.; Zatta F.; De Marco A.; et al. Cross-neutralization of SARS-CoV-2 by a human monoclonal SARS-CoV antibody. Nature 2020, 583, 290–295. 10.1038/s41586-020-2349-y. [DOI] [PubMed] [Google Scholar]
- Dong J.; Zost S. J.; Greaney A. J.; Starr T. N.; Dingens A. S.; Chen E. C.; Chen R. E.; Case J. B.; Sutton R. E.; Gilchuk P.; et al. Genetic and structural basis for SARS-CoV-2 variant neutralization by a two-antibody cocktail. Nat. Microbiol. 2021, 6, 1233–1244. 10.1038/s41564-021-00972-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senior A. W.; Evans R.; Jumper J.; Kirkpatrick J.; Sifre L.; Green T.; Qin C.; Žídek A.; Nelson A. W.; Bridgland A.; et al. Improved protein structure prediction using potentials from deep learning. Nature 2020, 577, 706–710. 10.1038/s41586-019-1923-7. [DOI] [PubMed] [Google Scholar]
- Wei G.-W. Protein structure prediction beyond AlphaFold. Nat. Mach. Intell. 2019, 1, 336–337. 10.1038/s42256-019-0086-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Lab. Genome-wide structure and function modeling of SARS-CoV-2. https://zhanglab.ccmb.med.umich.edu/COVID-19/ (accessed 12 February 2020).
- Yang Z.; Bogdan P.; Nazarian S. An in silico deep learning approach to multi-epitope vaccine design: a SARS-CoV-2 case study. Sci. Rep. 2021, 11, 1–21. 10.1038/s41598-021-81749-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kipf T. N.; Welling M.. Semi-supervised classification with graph convolutional networks. arXiv 2016, 1609.02907 [Google Scholar]
- Scarselli F.; Gori M.; Tsoi A. C.; Hagenbuchner M.; Monfardini G. The graph neural network model. IEEE Trans. Neural Netw. 2009, 20, 61–80. 10.1109/TNN.2008.2005605. [DOI] [PubMed] [Google Scholar]
- Haneczok J.; Delijewski M. Machine learning enabled identification of potential SARS-CoV-2 3CLpro inhibitors based on fixed molecular fingerprints and Graph-CNN neural representations. J. Biomed Inform 2021, 119, 103821. 10.1016/j.jbi.2021.103821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumelhart D. E.; Hinton G. E.; Williams R. J. Learning representations by back-propagating errors. nature 1986, 323, 533–536. 10.1038/323533a0. [DOI] [Google Scholar]
- Hochreiter S.; Schmidhuber J. Long short-term memory. Neural Comput 1997, 9, 1735–1780. 10.1162/neco.1997.9.8.1735. [DOI] [PubMed] [Google Scholar]
- Cho K.; Van Merriënboer B.; Gulcehre C.; Bahdanau D.; Bougares F.; Schwenk H.; Bengio Y.. Learning phrase representations using RNN encoder-decoder for statistical machine translation. Proceedings of the 2014 Conference on Empirical Methods in Natural Language Processing (EMNLP); Association for Computational Linguistics, 2014; pp 1724–1734 10.3115/v1/D14-1179. [DOI]
- Gao K.; Nguyen D. D.; Wang R.; Wei G.-W. Machine intelligence design of 2019-nCoV drugs. bioRxiv 2020, 10.1101/2020.01.30.927889. [DOI] [Google Scholar]
- Mayr A.; Klambauer G.; Unterthiner T.; Steijaert M.; Wegner J. K.; Ceulemans H.; Clevert D.-A.; Hochreiter S. Large-scale comparison of Mach. Learn. methods for drug target prediction on ChEMBL. Chem. Sci. 2018, 9, 5441–5451. 10.1039/C8SC00148K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hie B.; Zhong E. D.; Berger B.; Bryson B. Learning the language of viral evolution and escape. Science 2021, 371, 284–288. 10.1126/science.abd7331. [DOI] [PubMed] [Google Scholar]
- Gao K.; Nguyen D. D.; Tu M.; Wei G.-W. Generative network complex for the automated generation of druglike molecules. J. Chem. Inf Model 2020, 60, 5682–5698. 10.1021/acs.jcim.0c00599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf T.; Chaumond J.; Debut L.; Sanh V.; Delangue C.; Moi A.; Cistac P.; Funtowicz M.; Davison J.; Shleifer S.; et al. Proceedings of the 2020 Conference on Empirical Methods in Natural Language Processing; System Demonstrations, 2020; pp 38–45.
- Liu T.; Lin Y.; Wen X.; Jorissen R. N.; Gilson M. K. BindingDB: a web-accessible database of experimentally determined protein–ligand binding affinities. Nucleic Acids Res. 2007, 35, D198–D201. 10.1093/nar/gkl999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jumper J.; Evans R.; Pritzel A.; Green T.; Figurnov M.; Ronneberger O.; Tunyasuvunakool K.; Bates R.; Žídek A.; Potapenko A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson A. J.; Courtney J. M.; Shen Y.; Ying J.; Bax A. Concordance of X-ray and AlphaFold2Models of SARS-CoV-2 Main Protease with Residual Dipolar Couplings Measured in Solution. J. Am. Chem. Soc. 2021, 143, 19306–19310. 10.1021/jacs.1c10588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flower T. G.; Hurley J. H. Crystallographic molecular replacement using an in silico-generated search model of SARS-CoV-2 ORF8. Protein Sci. 2021, 30, 728–734. 10.1002/pro.4050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kryshtafovych A.; Moult J.; Billings W. M.; Della Corte D.; Fidelis K.; Kwon S.; Olechnovič K.; Seok C.; Venclovas Č.; Won J.; et al. Modeling SARS-CoV-2 proteins in the CASP-commons experiment. Proteins 2021, 89, 1987–1996. 10.1002/prot.26231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mount D.Bioinformatics: Sequence and Genome Analysis; Cold Spring Harbor Laboratory Press: New York, 2004; 692 pp. ISBN 0-87969-712-1. [Google Scholar]; Rich D. H. Evaluation of Enzyme Inhibitors in Drug Discovery: A Guide for Medicinal Chemists and Pharmacologists. Clin. Chem. 2005, 51, 2219–2219. 10.1373/clinchem.2005.051946. [DOI] [PubMed] [Google Scholar]
- Wilbur W. J.; Lipman D. J. Rapid similarity searches of nucleic acid and protein data banks. Proc. Natl. Acad. Sci. U.S.A. 1983, 80, 726–730. 10.1073/pnas.80.3.726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul S. F.; Gish W.; Miller W.; Myers E. W.; Lipman D. J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Ye J.; McGinnis S.; Madden T. L. BLAST: improvements for better sequence analysis. Nucleic Acids Res. 2006, 34, W6–W9. 10.1093/nar/gkl164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mount D. W. Using the basic local alignment search tool (BLAST). Cold Spring Harb. Protoc. 2007, 2007, pdb.top17. 10.1101/pdb.top17. [DOI] [PubMed] [Google Scholar]
- Zhang T.; Wu Q.; Zhang Z. Probable pangolin origin of SARS-CoV-2 associated with the COVID-19 outbreak. Curr. Biol. 2020, 30, 1578. 10.1016/j.cub.2020.03.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao K.; Zhai J.; Feng Y.; Zhou N.; Zhang X.; Zou J.-J.; Li N.; Guo Y.; Li X.; Shen X.; et al. Isolation of SARS-CoV-2-related coronavirus from Malayan pangolins. Nature 2020, 583, 286. 10.1038/s41586-020-2313-x. [DOI] [PubMed] [Google Scholar]
- Wang H.; Pipes L.; Nielsen R. Synonymous mutations and the molecular evolution of SARS-Cov-2 origins. Virus Evol. 2021, 7, veaa098. 10.1093/ve/veaa098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Rosa G.; Mancini P.; Ferraro G. B.; Veneri C.; Iaconelli M.; Bonadonna L.; Lucentini L.; Suffredini E. SARS-CoV-2 has been circulating in northern Italy since December 2019: Evidence from environmental monitoring. Sci. Total Environ. 2021, 750, 141711. 10.1016/j.scitotenv.2020.141711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sah R.; Rodriguez-Morales A. J.; Jha R.; Chu D. K.; Gu H.; Peiris M.; Bastola A.; Lal B. K.; Ojha H. C.; Rabaan A. A.; et al. Complete genome sequence of a 2019 novel coronavirus (SARS-CoV-2) strain isolated in Nepal. Microbiol. Resour. Announc. 2020, 9, e00169–20. 10.1128/MRA.00169-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Rosa G.; Iaconelli M.; Mancini P.; Ferraro G. B.; Veneri C.; Bonadonna L.; Lucentini L.; Suffredini E. First detection of SARS-CoV-2 in untreated wastewaters in Italy. Sci. Total Environ. 2020, 736, 139652. 10.1016/j.scitotenv.2020.139652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westhaus S.; Weber F.-A.; Schiwy S.; Linnemann V.; Brinkmann M.; Widera M.; Greve C.; Janke A.; Hollert H.; Wintgens T.; et al. Detection of SARS-CoV-2 in raw and treated wastewater in Germany–suitability for COVID-19 surveillance and potential transmission risks. Sci. Total Environ. 2021, 751, 141750. 10.1016/j.scitotenv.2020.141750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The species Severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536. 10.1038/s41564-020-0695-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins D. G.; Sharp P. M. CLUSTAL: a package for performing multiple sequence alignment on a microcomputer. Gene 1988, 73, 237–244. 10.1016/0378-1119(88)90330-7. [DOI] [PubMed] [Google Scholar]
- Edgar R. C. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinform 2004, 5, 113. 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K.; Asimenos G.; Toh H.. Bioinformatics for DNA sequence analysis; Springer: 2009; pp 39–64. [Google Scholar]
- Katoh K.; Misawa K.; Kuma K.-i.; Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson J. D.; Higgins D. G.; Gibson T. J. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin M. A.; Blackshields G.; Brown N. P.; Chenna R.; McGettigan P. A.; McWilliam H.; Valentin F.; Wallace I. M.; Wilm A.; Lopez R.; et al. Clustal W and Clustal X version 2.0. bioinformatics 2007, 23, 2947–2948. 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- Saitou N.; Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- Blackshields G.; Sievers F.; Shi W.; Wilm A.; Higgins D. G. Sequence embedding for fast construction of guide trees for multiple sequence alignment. Algorithms Mol. Biol. 2010, 5, 21. 10.1186/1748-7188-5-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Söding J. Protein homology detection by HMM–HMM comparison. Bioinformatics 2005, 21, 951–960. 10.1093/bioinformatics/bti125. [DOI] [PubMed] [Google Scholar]
- Edgar R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallavilla T.; Bertelli M.; Morresi A.; Bushati V.; Stuppia L.; Beccari T.; Chiurazzi P.; Marceddu G. Bioinformatic analysis indicates that SARS-CoV-2 is unrelated to known artificial coronaviruses. Eur. Rev. Med. Pharmacol Sci. 2020, 24, 4558–4564. 10.26355/eurrev_202004_21041. [DOI] [PubMed] [Google Scholar]
- Trigueiro-Louro J.; Correia V.; Figueiredo-Nunes I.; Gíria M.; Rebelo-de-Andrade H. Unlocking COVID therapeutic targets: A structure-based rationale against SARS-CoV-2, SARS-CoV and MERS-CoV Spike. Comput. Struct. Biotechnol. J. 2020, 18, 2117. 10.1016/j.csbj.2020.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T.; Liu D.; Yang Y.; Guo J.; Feng Y.; Zhang X.; Cheng S.; Feng J. Phylogenetic supertree reveals detailed evolution of SARS-CoV-2. Sci. Rep. 2020, 10, 1–9. 10.1038/s41598-020-79484-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sallam M.; Ababneh N. A.; Dababseh D.; Bakri F. G.; Mahafzah A. Temporal increase in D614G mutation of SARS-CoV-2 in the Middle East and North Africa. Heliyon 2021, 7, e06035 10.1016/j.heliyon.2021.e06035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwede T.; Kopp J.; Guex N.; Peitsch M. C. SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res. 2003, 31, 3381–3385. 10.1093/nar/gkg520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chothia C.; Lesk A. M. The relation between the divergence of sequence and structure in proteins. EMBO J. 1986, 5, 823–826. 10.1002/j.1460-2075.1986.tb04288.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdelrheem D. A.; Ahmed S. A.; Abd El-Mageed H.; Mohamed H. S.; Rahman A. A.; Elsayed K. N.; Ahmed S. A. The inhibitory effect of some natural bioactive compounds against SARS-CoV-2 main protease: insights from molecular docking analysis and molecular dynamic simulation. J. Environ. Sci. Health A 2020, 55, 1373–1386. 10.1080/10934529.2020.1826192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiménez-Alberto A.; Ribas-Aparicio R. M.; Aparicio-Ozores G.; Castelán-Vega J. A. Virtual screening of approved drugs as potential SARS-CoV-2 main protease inhibitors. Comput. Biol. Chem. 2020, 88, 107325. 10.1016/j.compbiolchem.2020.107325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calligari P.; Bobone S.; Ricci G.; Bocedi A. Molecular Investigation of SARS–CoV-2 Proteins and Their Interactions with Antiviral Drugs. Viruses 2020, 12, 445. 10.3390/v12040445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q.; Zhao Y.; Chen X.; Hong A. Virtual screening of approved clinic drugs with main protease (3CLpro) reveals potential inhibitory effects on SARS-CoV-2. J. Biomol. Struct. Dyn. 2022, 40, 685–695. 10.1080/07391102.2020.1817786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milenković D. A.; Dimić D. S.; Avdović E. H.; Marković Z. S. Several coumarin derivatives and their Pd (II) complexes as potential inhibitors of the main protease of SARS-CoV-2, an in silico approach. RSC Adv. 2020, 10, 35099–35108. 10.1039/D0RA07062A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanker A. K.; Bhanu D.; Alluri A.; Gupta S. Whole-genome sequence analysis and homology modelling of the main protease and non-structural protein 3 of SARS-CoV-2 reveal an aza-peptide and a lead inhibitor with possible antiviral properties. New J. Chem. 2020, 44, 9202–9212. 10.1039/D0NJ00974A. [DOI] [Google Scholar]
- Khan R. J.; Jha R. K.; Amera G. M.; Jain M.; Singh E.; Pathak A.; Singh R. P.; Muthukumaran J.; Singh A. K. Targeting SARS-CoV-2: A systematic drug repurposing approach to identify promising inhibitors against 3C-like proteinase and 2’-O-ribose methyltransferase. J. Biomol. Struct 2021, 39, 2679–2692. 10.1080/07391102.2020.1753577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pach S.; Nguyen T. N.; Trimpert J.; Kunec D.; Osterrieder N.; Wolber G. ACE2-Variants Indicate Potential SARS-CoV-2-Susceptibility in Animals: A Molecular Dynamics Study. Mol. Inform. 2021, 40, 2100031. 10.1002/minf.202100031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aatif M.; Muteeb G.; Alsultan A.; Alshoaibi A.; Khelif B. Y. Dieckol and Its Derivatives as Potential Inhibitors of SARS-CoV-2 Spike Protein (UK Strain: VUI 202012/01): A Computational Study. Mar. Drugs 2021, 19, 242. 10.3390/md19050242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parvez M. S. A.; Rahman M. M.; Morshed M. N.; Rahman D.; Anwar S.; Hosen M. J. Genetic analysis of SARS-CoV-2 isolates collected from Bangladesh: Insights into the origin, mutational spectrum and possible pathomechanism. Comput. Biol. Chem. 2021, 90, 107413. 10.1016/j.compbiolchem.2020.107413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall D. C. Jr; Ji H.-F. A search for medications to treat COVID-19 via in silico molecular docking models of the SARS-CoV-2 spike glycoprotein and 3CL protease. Travel Med. Infect Dis 2020, 35, 101646. 10.1016/j.tmaid.2020.101646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morsy S.; Morsy A. Epitope mimicry analysis of SARS-COV-2 surface proteins and human lung proteins. J. Mol. Graph. Model. 2021, 105, 107836. 10.1016/j.jmgm.2021.107836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luan J.; Lu Y.; Jin X.; Zhang L. Spike protein recognition of mammalian ACE2 predicts the host range and an optimized ACE2 for SARS-CoV-2 infection. Biochem. Biophys. Res. Commun. 2020, 526, 165–169. 10.1016/j.bbrc.2020.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain M.; Jabeen N.; Raza F.; Shabbir S.; Baig A. A.; Amanullah A.; Aziz B. Structural variations in human ACE2 may influence its binding with SARS-CoV-2 spike protein. J. Med. Virol. 2020, 92, 1580–1586. 10.1002/jmv.25832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigrist C. J.; Bridge A.; Le Mercier P. A potential role for integrins in host cell entry by SARS-CoV-2. Antivir. Res. 2020, 177, 104759. 10.1016/j.antiviral.2020.104759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baig A. M.; Khaleeq A.; Syeda H. Elucidation of cellular targets and exploitation of the receptor-binding domain of SARS-CoV-2 for vaccine and monoclonal antibody synthesis. J. Med. Virol. 2020, 92, 2792–2803. 10.1002/jmv.26212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaimes J. A.; André N. M.; Chappie J. S.; Millet J. K.; Whittaker G. R. Phylogenetic analysis and structural modeling of SARS-CoV-2 spike protein reveals an evolutionary distinct and proteolytically sensitive activation loop. J. Mol. Biol. 2020, 432, 3309–3325. 10.1016/j.jmb.2020.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling R.; Dai Y.; Huang B.; Huang W.; Yu J.; Lu X.; Jiang Y. In silico design of antiviral peptides targeting the spike protein of SARS-CoV-2. Peptides 2020, 130, 170328. 10.1016/j.peptides.2020.170328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh N.; Decroly E.; Khatib A.-M.; Villoutreix B. O. Structure-based drug repositioning over the human TMPRSS2 protease domain: search for chemical probes able to repress SARS-CoV-2 Spike protein cleavages. Eur. J. Pharm. Sci. 2020, 153, 105495. 10.1016/j.ejps.2020.105495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary S.; Malik Y. S.; Tomar S. Identification of SARS-CoV-2 cell entry inhibitors by drug repurposing using in silico structure-based virtual screening approach. Front. Immunol. 2020, 11, 1664. 10.3389/fimmu.2020.01664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H.; Chen X.; Hu T.; Li J.; Song H.; Liu Y.; Wang P.; Liu D.; Yang J.; Holmes E. C.; et al. A novel bat coronavirus closely related to SARS-CoV-2 contains natural insertions at the S1/S2 cleavage site of the spike protein. Curr. Biol. 2020, 30, 2196–2203. 10.1016/j.cub.2020.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharisma V. D.; Ansori A. N. M. Construction of epitope-based peptide vaccine against SARS-CoV-2: Immunoinformatics study. J. Pure Appl. Microbiol 2020, 14, 999–1005. 10.22207/JPAM.14.SPL1.38. [DOI] [Google Scholar]
- Shanmugarajan D.; Prabitha P.; Kumar B. P.; Suresh B. Curcumin to inhibit binding of spike glycoprotein to ACE2 receptors: computational modelling, simulations, and ADMET studies to explore curcuminoids against novel SARS-CoV-2 targets. RSC Adv. 2020, 10, 31385–31399. 10.1039/D0RA03167D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng S.; Luan X.; Wang Y.; Wang H.; Zhang Z.; Wang Y.; Tian Z.; Liu M.; Xiao Y.; Zhao Y.; et al. Eltrombopag is a potential target for drug intervention in SARS-CoV-2 spike protein. Infect. Genet. Evol. 2020, 85, 104419. 10.1016/j.meegid.2020.104419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W. Delving deep into the structural aspects of a furin cleavage site inserted into the spike protein of SARS-CoV-2: a structural biophysical perspective. Biophys. Chem. 2020, 264, 106420. 10.1016/j.bpc.2020.106420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park T.; Lee S.-Y.; Kim S.; Kim M. J.; Kim H. G.; Jun S.; Kim S. I.; Kim B. T.; Park E. C.; Park D. Spike protein binding prediction with neutralizing antibodies of SARS-CoV-2. BioRxiv 2020, 10.1101/2020.02.22.951178. [DOI] [Google Scholar]
- Lupala C. S.; Li X.; Lei J.; Chen H.; Qi J.; Liu H.; Su X.-d. Computational simulations reveal the binding dynamics between human ACE2 and the receptor binding domain of SARS-CoV-2 spike protein. Quant. Biol. 2021, 9, 61–72. 10.15302/J-QB-020-0231. [DOI] [Google Scholar]
- Parray H. A.; Chiranjivi A. K.; Asthana S.; Yadav N.; Shrivastava T.; Mani S.; Sharma C.; Vishwakarma P.; Das S.; Pindari K.; et al. Identification of an anti–SARS–CoV-2 receptor-binding domain–directed human monoclonal antibody from a naïve semisynthetic library. J. Biol. Chem. 2020, 295, 12814–12821. 10.1074/jbc.AC120.014918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S.; Maurya V. K.; Prasad A. K.; Bhatt M. L.; Saxena S. K. Structural, glycosylation and antigenic variation between 2019 novel coronavirus (2019-nCoV) and SARS coronavirus (SARS-CoV). Virusdisease 2020, 31, 13–21. 10.1007/s13337-020-00571-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Q.-d.; Yi Y.; Zou Y.-n.; Jia Z.-y.; Qiu F.; Wang F.; Yin W.-j.; Zhou W.-t.; Zhang S.; Yu P.-c.; et al. The biological characteristics of SARS-CoV-2 spike protein Pro330-Leu650. Vaccine 2020, 38, 5071–5075. 10.1016/j.vaccine.2020.04.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arya R.; Das A.; Prashar V.; Kumar M. Potential inhibitors against papain-like protease of novel coronavirus (SARS-CoV-2) from FDA approved drugs. ChemRxiv 2020, 10.26434/chemrxiv.11860011.v2. [DOI] [Google Scholar]
- Zhang L.; Zhou R. Structural basis of the potential binding mechanism of remdesivir to SARS-CoV-2 RNA-dependent RNA polymerase. J. Phys. Chem. B 2020, 124, 6955–6962. 10.1021/acs.jpcb.0c04198. [DOI] [PubMed] [Google Scholar]
- Neogi U.; Hill K. J.; Ambikan A. T.; Heng X.; Quinn T. P.; Byrareddy S. N.; Sönnerborg A.; Sarafianos S. G.; Singh K. Feasibility of known rna polymerase inhibitors as anti-SARS-CoV-2 drugs. Pathogens 2020, 9, 320. 10.3390/pathogens9050320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu F.; Jiang J.; Yin P.. Prediction of potential commercially inhibitors against SARS-CoV-2 by multi-task deep model. arXiv 2020, 2003.00728. 10.48550/arXiv.2003.00728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Maio F.; Cascio E. L.; Babini G.; Sali M.; Della Longa S.; Tilocca B.; Roncada P.; ArCoVito A.; Sanguinetti M.; Scambia G.; et al. Improved binding of SARS-CoV-2 Envelope protein to tight junction-associated PALS1 could play a key role in COVID-19 pathogenesis. Microbes Infect 2020, 22, 592–597. 10.1016/j.micinf.2020.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar M.; Saha S. Structural insight into the role of novel SARS-CoV-2 E protein: A potential target for vaccine development and other therapeutic strategies. PloS one 2020, 15, e0237300 10.1371/journal.pone.0237300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oany A. R.; Mia M.; Pervin T.; Junaid M.; Hosen S. Z.; Moni M. A. Design of novel viral attachment inhibitors of the spike glycoprotein (S) of severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) through virtual screening and dynamics. Int. J. Antimicrob. Agents 2020, 56, 106177. 10.1016/j.ijantimicag.2020.106177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azeez S. A.; Alhashim Z. G.; Al Otaibi W. M.; Alsuwat H. S.; Ibrahim A. M.; Almandil N. B.; Borgio J. F. State-of-the-art tools to identify druggable protein ligand of SARS-CoV-2. Arch Med. Sci. 2020, 16, 497. 10.5114/aoms.2020.94046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loureiro C. L.; Jaspe R. C.; D'Angelo P.; Zambrano J. L.; Rodriguez L.; Alarcon V.; Delgado M.; Aguilar M.; Garzaro D.; Rangel H. R.; et al. SARS-CoV-2 genetic diversity in Venezuela: Predominance of D614G variants and analysis of one outbreak. PloS One 2021, 16, e0247196 10.1371/journal.pone.0247196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selvaraj C.; Dinesh D. C.; Panwar U.; Abhirami R.; Boura E.; Singh S. K. Structure-based virtual screening and molecular dynamics simulation of SARS-CoV-2 Guanine-N7 methyltransferase (nsp14) for identifying antiviral inhibitors against COVID-19. J. Biomol. Struct 2021, 39, 4582–4593. 10.1080/07391102.2020.1778535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiqa M. A.; Rao D.; Suvarna G.; Chennamachetty V.; Verma M.; Rao M. In-Silico Drug Designing of Spike Receptor with Its ACE2 Receptor and Nsp10/Nsp16 MTase Complex Against SARS-CoV-2. Int. J. Pept Res. Ther 2021, 27, 1633. 10.1007/s10989-021-10196-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngwe Tun M. M.; Morita K.; Ishikawa T.; Urata S. The Antiviral Effect of the Chemical Compounds Targeting DED/EDh Motifs of the Viral Proteins on Lymphocytic Choriomeningitis Virus and SARS-CoV-2. Viruses 2021, 13, 1220. 10.3390/v13071220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suryawanshi R. K.; Patil C. D.; Koganti R.; Singh S. K.; Ames J. M.; Shukla D. Heparan Sulfate Binding Cationic Peptides Restrict SARS-CoV-2 Entry. Pathogens 2021, 10, 803. 10.3390/pathogens10070803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundar S.; Thangamani L.; Piramanayagam S.; Rahul C. N.; Aiswarya N.; Sekar K.; Natarajan J. Screening of FDA-approved compound library identifies potential small-molecule inhibitors of SARS-CoV-2 non-structural proteins NSP1, NSP4, NSP6 and NSP13: molecular modeling and molecular dynamics studies. J. Proteins Proteom. 2021, 12, 161. 10.1007/s42485-021-00067-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya R.; Gupta A. M.; Mitra S.; Mandal S.; Biswas S. R. A natural food preservative peptide nisin can interact with the SARS-CoV-2 spike protein receptor human ACE2. Virology 2021, 552, 107–111. 10.1016/j.virol.2020.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong S.; Sun J.; Mao Z.; Wang L.; Lu Y.-L.; Li J. A guideline for homology modeling of the proteins from newly discovered betacoronavirus, 2019 novel coronavirus (2019-nCoV). J. Med. Virol. 2020, 92, 1542–1548. 10.1002/jmv.25768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin W. R.; Cheng F. Repurposing of FDA-approved toremifene to treat COVID-19 by blocking the Spike glycoprotein and NSP14 of SARS-CoV-2. J. Proteome Res. 2020, 19, 4670–4677. 10.1021/acs.jproteome.0c00397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culletta G.; Gulotta M. R.; Perricone U.; Zappalà M.; Almerico A. M.; Tutone M. Exploring the SARS-CoV-2 Proteome in the Search of Potential Inhibitors via Structure-Based Pharmacophore Modeling/Docking Approach. Computation 2020, 8, 77. 10.3390/computation8030077. [DOI] [Google Scholar]
- Zhou Z.-J.; Qiu Y.; Pu Y.; Huang X.; Ge X.-Y. BioAider: An efficient tool for viral genome analysis and its application in tracing SARS-CoV-2 transmission. Sustain. Cities Soc. 2020, 63, 102466. 10.1016/j.scs.2020.102466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hijikata A.; Shionyu-Mitsuyama C.; Nakae S.; Shionyu M.; Ota M.; Kanaya S.; Shirai T. Knowledge-based structural models of SARS-CoV-2 proteins and their complexes with potential drugs. FEBS Lett. 2020, 594, 1960–1973. 10.1002/1873-3468.13806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng T.; Cao H.; Zhang H.; Kang Z.; Xu D.; Gong H.; Wang J.; Li Z.; Cui X.; Xu H.; et al. The insert sequence in SARS-CoV-2 enhances spike protein cleavage by TMPRSS. BioRxiv 2020, 10.1101/2020.02.08.926006. [DOI] [Google Scholar]
- Chen L.; Zhong L. Genomics functional analysis and drug screening of SARS-CoV-2. Genes Dis 2020, 7, 542–550. 10.1016/j.gendis.2020.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selvaraj C.; Dinesh D. C.; Panwar U.; Abhirami R.; Boura E.; Singh S. K. Structure-based virtual screening and molecular dynamics simulation of SARS-CoV-2 Guanine-N7 methyltransferase (nsp14) for identifying antiviral inhibitors against COVID-19. J. Biomol. Struct. Dyn. 2021, 39, 4582. 10.1080/07391102.2020.1778535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benvenuto D.; Angeletti S.; Giovanetti M.; Bianchi M.; Pascarella S.; Cauda R.; Ciccozzi M.; Cassone A. Evolutionary analysis of SARS-CoV-2: how mutation of Non-Structural Protein 6 (NSP6) could affect viral autophagy. J. Infect. 2020, 81, e24–e27. 10.1016/j.jinf.2020.03.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cárdenas-Conejo Y.; Liñan-Rico A.; García-Rodríguez D. A.; Centeno-Leija S.; Serrano-Posada H. An exclusive 42 amino acid signature in pp1ab protein provides insights into the evolutive history of the 2019 novel human-pathogenic coronavirus (SARS-CoV-2). J. Med. Virol. 2020, 92, 688–692. 10.1002/jmv.25758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C.; Gong C. ACE2 models of frequently contacted animals provide clues of their SARS-CoV-2 S protein affinity and viral susceptibility. J. Med. Virol 2021, 93, 4469–4479. 10.1002/jmv.26953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekins S.; Mottin M.; Ramos P. R.; Sousa B. K.; Neves B. J.; Foil D. H.; Zorn K. M.; Braga R. C.; Coffee M.; Southan C.; et al. Déjà vu: Stimulating open drug discovery for SARS-CoV-2. Drug Discovery Today 2020, 25, 928–941. 10.1016/j.drudis.2020.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashyap D.; Jakhmola S.; Tiwari D.; Kumar R.; Moorthy N. H. N.; Elangovan M.; Brás N. F.; Jha H. C. Plant derived active compounds as potential anti SARS-CoV-2 agents: an in-silico study. J. Biomol. Struct 2021, 1–22. 10.1080/07391102.2021.1947384. [DOI] [PubMed] [Google Scholar]
- Barge S.; Jade D.; Gosavi G.; Talukdar N. C.; Borah J. In-silico screening for identification of potential inhibitors against SARS-CoV-2 transmembrane serine protease 2 (TMPRSS2). Eur. J. Pharm. Sci. 2021, 162, 105820. 10.1016/j.ejps.2021.105820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escalante D. E.; Ferguson D. M. Structural modeling and analysis of the SARS-CoV-2 cell entry inhibitor camostat bound to the trypsin-like protease TMPRSS2. Med. Chem. Res. 2021, 30, 399–409. 10.1007/s00044-021-02708-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pooja M.; Reddy G. J.; Hema K.; Dodoala S.; Koganti B. Unravelling high-affinity binding compounds towards transmembrane protease serine 2 enzyme in treating SARS-CoV-2 infection using molecular modelling and docking studies. Eur. J. Pharmacol. 2021, 890, 173688. 10.1016/j.ejphar.2020.173688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rensi S.; Altman R. B.; Liu T.; Lo Y.-C.; McInnes G.; Derry A.; Keys A. Homology Modeling of TMPRSS2 Yields Candidate Drugs That May Inhibit Entry of SARS-CoV-2 into Human Cells. ChemRxiv 2020, 10.26434/chemrxiv.12009582.v1. [DOI] [Google Scholar]
- Huggins D. J. Structural analysis of experimental drugs binding to the SARS-CoV-2 target TMPRSS2. J. Mol. Graph. Model. 2020, 100, 107710. 10.1016/j.jmgm.2020.107710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasheed M. A.; Raza S.; Zohaib A.; Riaz M. I.; Amin A.; Awais M.; Khan S. U.; Khan M. I.; Chu Y.-M. Immunoinformatics based prediction of recombinant multi-epitope vaccine for the control and prevention of SARS-CoV-2. Alex. Eng. J. 2021, 60, 3087–3097. 10.1016/j.aej.2021.01.046. [DOI] [Google Scholar]
- Xia P.; Dubrovska A. Tumor markers as an entry for SARS-CoV-2 infection?. FEBS J. 2020, 287, 3677–3680. 10.1111/febs.15499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abraham Peele K.; Srihansa T.; Krupanidhi S.; Ayyagari V. S.; Venkateswarulu T. Design of multi-epitope vaccine candidate against SARS-CoV-2: A in-silico study. J. Biomol. Struct 2021, 39, 3793–3801. 10.1080/07391102.2020.1770127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peele K. A.; Srihansa T.; Krupanidhi S.; Sai A. V.; Venkateswarulu T. Design of multi-epitope vaccine candidate against SARS-CoV-2: a in-silico study. J. Biomol. Struct. Dyn. 2021, 39, 3793. 10.1080/07391102.2020.1770127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samad A.; Ahammad F.; Nain Z.; Alam R.; Imon R. R.; Hasan M.; Rahman M. S. Designing a multi-epitope vaccine against SARS-CoV-2: an immunoinformatics approach. J. Biomol. Struct. Dyn. 2022, 40, 14. 10.1080/07391102.2020.1792347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belyaeva A.; Cammarata L.; Radhakrishnan A.; Squires C.; Dai Yang K.; Shivashankar G.; Uhler C. Causal network models of SARS-CoV-2 expression and aging to identify candidates for drug repurposing. Nat. Commun. 2021, 12, 1–13. 10.1038/s41467-021-21056-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang A. P.; Butte A. J. Systematic evaluation of drug–disease relationships to identify leads for novel drug uses. Clin. Pharmacol. Ther. 2009, 86, 507–510. 10.1038/clpt.2009.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keiser M. J.; Roth B. L.; Armbruster B. N.; Ernsberger P.; Irwin J. J.; Shoichet B. K. Relating protein pharmacology by ligand chemistry. Nat. Biotechnol. 2007, 25, 197–206. 10.1038/nbt1284. [DOI] [PubMed] [Google Scholar]
- Smith S. B.; Dampier W.; Tozeren A.; Brown J. R.; Magid-Slav M. Identification of common biological pathways and drug targets across multiple respiratory viruses based on human host gene expression analysis. PLoS One 2012, 7, e33174 10.1371/journal.pone.0033174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris J. H.; Knudsen G. M.; Verschueren E.; Johnson J. R.; Cimermancic P.; Greninger A. L.; Pico A. R. Affinity purification–mass spectrometry and network analysis to understand protein-protein interactions. Nat. Protoc. 2014, 9, 2539–2554. 10.1038/nprot.2014.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingras A.-C.; Gstaiger M.; Raught B.; Aebersold R. Analysis of protein complexes using mass spectrometry. Nat. Rev. Mol. Cell Biol. 2007, 8, 645–654. 10.1038/nrm2208. [DOI] [PubMed] [Google Scholar]
- Jaeger S.; Cimermancic P.; Gulbahce N.; Johnson J. R.; McGovern K. E.; Clarke S. C.; Shales M.; Mercenne G.; Pache L.; Li K.; et al. Global landscape of HIV–human protein complexes. Nature 2012, 481, 365–370. 10.1038/nature10719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins S. R.; Kemmeren P.; Zhao X.-C.; Greenblatt J. F.; Spencer F.; Holstege F. C.; Weissman J. S.; Krogan N. J. Toward a Comprehensive Atlas of the Physical Interactome of Saccharomyces cerevisiae* S. Mol. Cell. Proteom. 2007, 6, 439–450. 10.1074/mcp.M600381-MCP200. [DOI] [PubMed] [Google Scholar]
- Zhou Y.; Hou Y.; Shen J.; Huang Y.; Martin W.; Cheng F. Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell Discovery 2020, 6, 1–18. 10.1038/s41421-020-0153-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q.; Li M.; Wang X.; Parulian N.; Han G.; Ma J.; Tu J.; Lin Y.; Zhang H.; Liu W.; et al. COVID-19 literature knowledge graph construction and drug repurposing report generation. Proceedings of the 2021 Conference of the North American Chapter of the Association for Computational Linguistics: Human Language Technologies: Demonstrations; 2021; pp 66–77 10.18653/v1/2021.naacl-demos.8. [DOI]
- Domingo-Fernández D.; Baksi S.; Schultz B.; Gadiya Y.; Karki R.; Raschka T.; Ebeling C.; Hofmann-Apitius M.; Kodamullil A. T. COVID-19 Knowledge Graph: a computable, multi-modal, cause-and-effect knowledge model of COVID-19 pathophysiology. Bioinformatics 2021, 37, 1332–1334. 10.1093/bioinformatics/btaa834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segler M. H.; Preuss M.; Waller M. P. Planning chemical syntheses with deep neural networks and symbolic AI. Nature 2018, 555, 604–610. 10.1038/nature25978. [DOI] [PubMed] [Google Scholar]
- Li J.; Zheng S.; Chen B.; Butte A. J.; Swamidass S. J.; Lu Z. A survey of current trends in computational drug repositioning. Brief. Bioinformatics 2016, 17, 2–12. 10.1093/bib/bbv020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb J.; Crawford E. D.; Peck D.; Modell J. W.; Blat I. C.; Wrobel M. J.; Lerner J.; Brunet J.-P.; Subramanian A.; Ross K. N.; et al. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science 2006, 313, 1929–1935. 10.1126/science.1132939. [DOI] [PubMed] [Google Scholar]
- Pacini C.; Iorio F.; Gonçalves E.; Iskar M.; Klabunde T.; Bork P.; Saez-Rodriguez J. DvD: An R/Cytoscape pipeline for drug repurposing using public repositories of gene expression data. Bioinformatics 2013, 29, 132–134. 10.1093/bioinformatics/bts656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis G.; Sherman B. T.; Hosack D. A.; Yang J.; Gao W.; Lane H. C.; Lempicki R. A. DAVID: database for annotation, visualization, and integrated discovery. Genome Biol. 2003, 4, 1–11. 10.1186/gb-2003-4-9-r60. [DOI] [PubMed] [Google Scholar]
- Xing Z.; Li D.; Yang L.; Xi Y.; Su X. MicroRNAs and anticancer drugs. Acta Biochim Biophys Sin 2014, 46, 233–239. 10.1093/abbs/gmu003. [DOI] [PubMed] [Google Scholar]
- Wen X.; Deng F.-M.; Wang J. MicroRNAs as predictive biomarkers and therapeutic targets in prostate cancer. Am. J. Clin. Exp. Urol. 2014, 2, 219–230. [PMC free article] [PubMed] [Google Scholar]
- Chen W.; Fang Z.; Lv X.; Zhou Q.; Yao M.; Deng M. Prediction of potential therapeutic drugs against SARS-CoV-2 by using Connectivity Map based on transcriptome data. Eur. Rev. Med. Pharmacol Sci. 2021, 25, 3122–3131. 10.26355/eurrev_202104_25567. [DOI] [PubMed] [Google Scholar]
- Wyler E.; Mösbauer K.; Franke V.; Diag A.; Gottula L. T.; Arsiè R.; Klironomos F.; Koppstein D.; Hönzke K.; Ayoub S.; et al. Transcriptomic profiling of SARS-CoV-2 infected human cell lines identifies HSP90 as target for COVID-19 therapy. iScience 2021, 24, 102151. 10.1016/j.isci.2021.102151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thair S. A.; He Y. D.; Hasin-Brumshtein Y.; Sakaram S.; Pandya R.; Toh J.; Rawling D.; Remmel M.; Coyle S.; Dalekos G. N.; et al. Transcriptomic similarities and differences in host response between SARS-CoV-2 and other viral infections. iScience 2021, 24, 101947. 10.1016/j.isci.2020.101947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mousavi S. Z.; Rahmanian M.; Sami A. A connectivity map-based drug repurposing study and integrative analysis of transcriptomic profiling of SARS-CoV-2 infection. Infect. Genet. Evol. 2020, 86, 104610. 10.1016/j.meegid.2020.104610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S.; Wu S.; Yu Z.; Huang J.; Zhong X.; Liu X.; Zhu H.; Xiao L.; Deng Q.; Sun W. Transcriptomic analysis reveals novel mechanisms of SARS-CoV-2 infection in human lung cells. Immun. Inflamm. Dis. 2020, 8, 753–762. 10.1002/iid3.366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prokop J. W.; Hartog N. L.; Chesla D.; Faber W.; Love C. P.; Karam R.; Abualkheir N.; Feldmann B.; Teng L.; McBride T.; et al. High-density blood transcriptomics reveals precision immune signatures of SARS-CoV-2 infection in hospitalized individuals. Front. Immunol. 2021, 12, 2844. 10.3389/fimmu.2021.694243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Guo M.; Tian X.; Wang X.; Yang X.; Wu P.; Liu C.; Xiao Z.; Qu Y.; Yin Y.; et al. Virus-host interactome and proteomic survey reveal potential virulence factors influencing SARS-CoV-2 pathogenesis. Med. 2021, 2, 99–112. 10.1016/j.medj.2020.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagone P.; Ciurleo R.; Lombardo S. D.; Iacobello C.; Palermo C. I.; Shoenfeld Y.; Bendtzen K.; Bramanti P.; Nicoletti F. Transcriptional landscape of SARS-CoV-2 infection dismantles pathogenic pathways activated by the virus, proposes unique sex-specific differences and predicts tailored therapeutic strategies. Autoimmun Rev. 2020, 19, 102571. 10.1016/j.autrev.2020.102571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das J. K.; Roy S.; Guzzi P. H. Analyzing host-viral interactome of SARS-CoV-2 for identifying vulnerable host proteins during COVID-19 pathogenesis. Infect. Genet. Evol. 2021, 93, 104921. 10.1016/j.meegid.2021.104921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar N.; Mishra B.; Mehmood A.; Athar M.; Mukhtar M. S. Integrative network biology framework elucidates molecular mechanisms of SARS-CoV-2 pathogenesis. iScience 2020, 23, 101526. 10.1016/j.isci.2020.101526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gysi D. M.; Do Valle Í.; Zitnik M.; Ameli A.; Gan X.; Varol O.; Ghiassian S. D.; Patten J.; Davey R. A.; Loscalzo J.; et al. Network medicine framework for identifying drug-repurposing opportunities for COVID-19. Proc. Natl. Acad. Sci. U.S.A. 2021, 118, 118. 10.1073/pnas.2025581118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messina F.; Giombini E.; Agrati C.; Vairo F.; Bartoli T. A.; Al Moghazi S.; Piacentini M.; Locatelli F.; Kobinger G.; Maeurer M.; et al. COVID-19: viral–host interactome analyzed by network based-approach model to study pathogenesis of SARS-CoV-2 infection. J. Transl. Med. 2020, 18, 1–10. 10.1186/s12967-020-02405-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L.; Liu F.; Huang L.; Liu G.; Zhou L.; Peng L. VDA-RWLRLS: An anti-SARS-CoV-2 drug prioritizing framework combining an unbalanced bi-random walk and Laplacian regularized least squares. Comput. Biol. Med. 2022, 140, 105119. 10.1016/j.compbiomed.2021.105119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadegh S.; Matschinske J.; Blumenthal D. B.; Galindez G.; Kacprowski T.; List M.; Nasirigerdeh R.; Oubounyt M.; Pichlmair A.; Rose T. D.; et al. Exploring the SARS-CoV-2 virus-host-drug interactome for drug repurposing. Nat. Commun. 2020, 11, 1–9. 10.1038/s41467-020-17189-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan S.; Cui H.; Gao Z.; Liu M.; Lu S.; Mkandawire W.; Narykov O.; Sun M.; Korkin D. Structural genomics of SARS-CoV-2 indicates evolutionary conserved functional regions of viral proteins. Viruses 2020, 12, 360. 10.3390/v12040360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messina F.; Giombini E.; Montaldo C.; Sharma A. A.; Zoccoli A.; Sekaly R.-P.; Locatelli F.; Zumla A.; Maeurer M.; Capobianchi M. R.; et al. Looking for pathways related to COVID-19: confirmation of pathogenic mechanisms by SARS-CoV-2–host interactome. Cell Death Dis. 2021, 12, 1–10. 10.1038/s41419-021-03881-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadeau R.; Shahryari Fard S.; Scheer A.; Hashimoto-Roth E.; Nygard D.; Abramchuk I.; Chung Y.-E.; Bennett S. A.; Lavallée-Adam M. Computational Identification of Human Biological Processes and Protein Sequence Motifs Putatively Targeted by SARS-CoV-2 Proteins Using Protein–Protein Interaction Networks. J. Proteome Res. 2020, 19, 4553–4566. 10.1021/acs.jproteome.0c00422. [DOI] [PubMed] [Google Scholar]
- Bellucci G.; Ballerini C.; Mechelli R.; Bigi R.; Rinaldi V.; Reniè R.; Buscarinu M. C.; Baranzini S. E.; Madireddy L.; Matarese G. SARS-CoV-2 meta-interactome suggests disease-specific, autoimmune pathophysiologies and therapeutic targets. F1000Research 2020, 9, 992. 10.12688/f1000research.25593.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muratov E. N.; Amaro R.; Andrade C. H.; Brown N.; Ekins S.; Fourches D.; Isayev O.; Kozakov D.; Medina-Franco J. L.; Merz K. M.; et al. A critical overview of computational approaches employed for COVID-19 drug discovery. Chem. Soc. Rev. 2021, 50, 9121. 10.1039/D0CS01065K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing J.; Paithankar S.; Liu K.; Uhl K.; Li X.; Ko M.; Kim S.; Haskins J.; Chen B. Published anti-SARS-CoV-2 in vitro hits share common mechanisms of action that synergize with antivirals. Brief. Bioinformatics 2021, 22, bbab249. 10.1093/bib/bbab249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith D. P.; Oechsle O.; Rawling M. J.; Savory E.; Lacoste A.; Richardson P. J. Expert-augmented computational drug repurposing identified baricitinib as a treatment for COVID-19. Front. Pharmacol. 2021, 12, 1699. 10.3389/fphar.2021.709856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omolo C. A.; Soni N.; Fasiku V. O.; Mackraj I.; Govender T. Update on therapeutic approaches and emerging therapies for SARS-CoV-2 virus. Eur. J. Pharmacol. 2020, 883, 173348. 10.1016/j.ejphar.2020.173348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morselli Gysi D.; do Valle Í.; Zitnik M.; Ameli A.; Gan X.; Varol O.; Ghiassian S. D.; Patten J.; Davey R. A.; Loscalzo J.; et al. Network medicine framework for identifying drug-repurposing opportunities for COVID-19. Proc. Natl. Acad. Sci. U.S.A. 2021, 118, 118. 10.1073/pnas.2025581118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gramatica P. Principles of QSAR models validation: internal and external. QSAR Comb. Sci. 2007, 26, 694–701. 10.1002/qsar.200610151. [DOI] [Google Scholar]
- Roy K.; Kar S.; Das R. N.. A primer on QSAR/QSPR modeling: fundamental concepts; Springer, 2015; pp 1–121. [Google Scholar]
- Acharya B. N. Kinase Inhibitors Can Inhibit SARS-CoV-2 Mpro: A Theoretical Study. ChemRxiv 2020, 10.26434/chemrxiv.12865016.v1. [DOI] [Google Scholar]
- Alves V. M.; Bobrowski T.; Melo-Filho C. C.; Korn D.; Auerbach S.; Schmitt C.; Muratov E. N.; Tropsha A. QSAR Modeling of SARS-CoV Mpro Inhibitors Identifies Sufugolix, Cenicriviroc, Proglumetacin, and Other Drugs as Candidates for Repurposing against SARS-CoV-2. Mol. Inform. 2021, 40, 2000113. 10.1002/minf.202000113. [DOI] [PubMed] [Google Scholar]
- Masand V.; Rastija V.; Patil M.; Gandhi A.; Chapolikar A. Extending the identification of structural features responsible for anti-SARS-CoV activity of peptide-type compounds using QSAR modelling. SAR QSAR Environ. Res. 2020, 31, 643–654. 10.1080/1062936X.2020.1784271. [DOI] [PubMed] [Google Scholar]
- Masand V. H.; Akasapu S.; Gandhi A.; Rastija V.; Patil M. K. Structure features of peptide-type SARS-CoV main protease inhibitors: Quantitative structure activity relationship study. Chemometr Intell Lab Syst 2020, 206, 104172. 10.1016/j.chemolab.2020.104172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh K.; Amin S. A.; Gayen S.; Jha T. Chemical-informatics approach to COVID-19 drug discovery: Exploration of important fragments and data mining based prediction of some hits from natural origins as main protease (Mpro) inhibitors. J. Mol. Struct. 2021, 1224, 129026. 10.1016/j.molstruc.2020.129026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong J.-B.; Luo D.; Xu H.-Y.; Bian S.; Zhang X.; Xiao X.-C.; Wang J. A computational approach for designing novel SARS-CoV-2 M pro inhibitors: combined QSAR, molecular docking, and molecular dynamics simulation techniques. New J. Chem. 2021, 45, 11512. 10.1039/D1NJ02127C. [DOI] [Google Scholar]
- Amin S. A.; Banerjee S.; Singh S.; Qureshi I. A.; Gayen S.; Jha T. First structure–activity relationship analysis of SARS-CoV-2 virus main protease (Mpro) inhibitors: an endeavor on COVID-19 drug discovery. Mol. Divers. 2021, 25, 1827. 10.1007/s11030-020-10166-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanni R.; Galvez-Llompart M.; Galvez J. Computational analysis of macrolides as SARS-CoV-2 main protease inhibitors: a pattern recognition study based on molecular topology and validated by molecular docking. New J. Chem. 2021, 45, 8654–8675. 10.1039/D0NJ05983H. [DOI] [Google Scholar]
- Zaki M. E.; Al-Hussain S. A.; Masand V. H.; Akasapu S.; Bajaj S. O.; El-Sayed N. N.; Ghosh A.; Lewaa I. Identification of Anti-SARS-CoV-2 Compounds from Food Using QSAR-Based Virtual Screening, Molecular Docking, and Molecular Dynamics Simulation Analysis. Pharmaceuticals 2021, 14, 357. 10.3390/ph14040357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Tat P.; Hoa T. T.; Vo Ky A.; Nu Ngoc Han P. Novel SARS-CoV-2 Inhibitors from Phenethylthiazolethiourea Derivatives Using Hybrid QSAR Models and Docking Simulation. Smart Sci. 2021, 9, 165. 10.1080/23080477.2021.1914967. [DOI] [Google Scholar]
- De P.; Bhayye S.; Kumar V.; Roy K. In silico modeling for quick prediction of inhibitory activity against 3CLpro enzyme in SARS CoV diseases. J. Biomol. Struct. Dyn. 2022, 40, 1010. 10.1080/07391102.2020.1821779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Achutha A.; Pushpa V.; Suchitra S. Theoretical Insights into the Anti-SARS-CoV-2 Activity of Chloroquine and Its Analogs and In Silico Screening of Main Protease Inhibitors. J. Proteome Res. 2020, 19, 4706–4717. 10.1021/acs.jproteome.0c00683. [DOI] [PubMed] [Google Scholar]
- Borquaye L. S.; Gasu E. N.; Ampomah G. B.; Kyei L. K.; Amarh M. A.; Mensah C. N.; Nartey D.; Commodore M.; Adomako A. K.; Acheampong P.; et al. Alkaloids from Cryptolepis sanguinolenta as Potential Inhibitors of SARS-CoV-2 Viral Proteins: An In Silico Study. Biomed Res. Int. 2020, 2020, 5324560. 10.1155/2020/5324560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaldan A.; Bouamrane S.; En-Nahli F.; El-Mernissi R.; Hmamouchi R.; Maghat H.; Ajana M. A.; Sbai A.; Bouachrine M.; Lakhlifi T.; et al. Prediction of potential inhibitors of SARS-CoV-2 using 3D-QSAR, molecular docking modeling and ADMET properties. Heliyon 2021, 7, e06603 10.1016/j.heliyon.2021.e06603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin S. A.; Ghosh K.; Singh S.; Qureshi I. A.; Jha T.; Gayen S. Exploring naphthyl derivatives as SARS-CoV papain-like protease (PLpro) inhibitors and its implications in COVID-19 drug discovery. Mol. Divers. 2022, 26, 215. 10.1007/s11030-021-10198-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhury C.; Sastry G. N.. Structural Bioinformatics: Applications in Preclinical Drug Discovery Process; Springer, 2019; pp 25–53. [Google Scholar]
- Balaramnavar V. M.; Ahmad K.; Saeed M.; Ahmad I.; Kamal M.; Jawed T. Pharmacophore-based approaches in the rational repurposing technique for FDA approved drugs targeting SARS-CoV-2 Mpro. RSC Adv. 2020, 10, 40264–40275. 10.1039/D0RA06038K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshino R.; Yasuo N.; Sekijima M. Identification of key interactions between SARS-CoV-2 main protease and inhibitor drug candidates. Sci. Rep. 2020, 10, 1–8. 10.1038/s41598-020-69337-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentile D.; Patamia V.; Scala A.; Sciortino M. T.; Piperno A.; Rescifina A. Putative inhibitors of SARS-CoV-2 main protease from a library of marine natural products: a virtual screening and molecular modeling study. Mar. Drugs 2020, 18, 225. 10.3390/md18040225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan A.; Arafa R. K. On the search for COVID-19 therapeutics: identification of potential SARS-CoV-2 main protease inhibitors by virtual screening, pharmacophore modeling and molecular dynamics. J. Biomol. Struct 2021, 1–14. 10.1080/07391102.2021.1902399. [DOI] [PubMed] [Google Scholar]
- Franco L. S.; Maia R. C.; Barreiro E. J. Identification of LASSBio-1945 as an inhibitor of SARS-CoV-2 main protease (M PRO) through in silico screening supported by molecular docking and a fragment-based pharmacophore model. RSC Med. Chem. 2021, 12, 110–119. 10.1039/D0MD00282H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohamed N. M.; Ali E. M.; AboulMagd A. M. Ligand-based design, molecular dynamics and ADMET studies of suggested SARS-CoV-2 M pro inhibitors. RSC Adv. 2021, 11, 4523–4538. 10.1039/D0RA10141A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elekofehinti O. O.; Iwaloye O.; Molehin O. R.; Famusiwa C. D. Identification of lead compounds from large natural product library targeting 3C-like protease of SARS-CoV-2 using E-pharmacophore modelling, QSAR and molecular dynamics simulation. In Silico Pharmacol 2021, 9, 1–19. 10.1007/s40203-021-00109-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Said M. A.; Albohy A.; Abdelrahman M. A.; Ibrahim H. S. Importance of glutamine 189 flexibility in SARS-CoV-2 main protease: Lesson learned from in silico virtual screening of ChEMBL database and molecular dynamics. Eur. J. Pharm. Sci. 2021, 160, 105744. 10.1016/j.ejps.2021.105744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dermawan D.; Prabowo B. A.; Rakhmadina C. A. In silico study of medicinal plants with cyclodextrin inclusion complex as the potential inhibitors against SARS-CoV-2 main protease (Mpro) and spike (S) receptor. Inform. Med. Unlocked 2021, 25, 100645. 10.1016/j.imu.2021.100645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stasiulewicz A.; Maksymiuk A. W.; Nguyen M. L.; Bełza B.; Sulkowska J. I. SARS-CoV-2 Papain-Like Protease Potential Inhibitors–In Silico Quantitative Assessment. Int. J. Mol. Sci. 2021, 22, 3957. 10.3390/ijms22083957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Y.; Shang J.; Graham R.; Baric R. S.; Li F. Receptor recognition by the novel coronavirus from Wuhan: an analysis based on decade-long structural studies of SARS coronavirus. J. Virol. 2020, 94, 94. 10.1128/JVI.00127-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egieyeh S.; Egieyeh E.; Malan S.; Christofells A.; Fielding B. Computational drug repurposing strategy predicted peptide-based drugs that can potentially inhibit the interaction of SARS-CoV-2 spike protein with its target (humanACE2). PLoS One 2021, 16, e0245258 10.1371/journal.pone.0245258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basit A.; Ali T.; Rehman S. U. Truncated human Angiotensin Converting Enzyme 2; a potential inhibitor of SARS-CoV-2 spike glycoprotein and potent COVID-19 therapeutic agent. J. Biomol. Struct. Dyn. 2021, 39, 3605. 10.1080/07391102.2020.1768150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batista A. D.; Rajpal S.; Keitel B.; Dietl S.; Fresco-Cala B.; Dinc M.; Groß R.; Sobek H.; Münch J.; Mizaikoff B. Plastic Antibodies Mimicking the ACE2 Receptor for Selective Binding of SARS-CoV-2 Spike. Adv. Mater. Interfaces 2022, 9, 2101925. 10.1002/admi.202101925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devi A.; Chaitanya N. S. Designing of peptide aptamer targeting the receptor-binding domain of spike protein of SARS-CoV-2: an in silico study. Mol. Divers. 2022, 26, 157. 10.1007/s11030-020-10171-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W.; Xu G.; Yu Z.; Li J.; Liu J. Identification of nut protein-derived peptides against SARS-CoV-2 spike protein and main protease. Comput. Biol. Med. 2021, 138, 104937. 10.1016/j.compbiomed.2021.104937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efaz F. M.; Islam S.; Talukder S. A.; Akter S.; Tashrif M. Z.; Ali M. A.; Sufian M. A.; Parves M. R.; Islam M. J.; Halim M. A. Repurposing fusion inhibitor peptide against SARS-CoV-2. J. Comput. Chem. 2021, 42, 2283–2293. 10.1002/jcc.26758. [DOI] [PubMed] [Google Scholar]
- Rajpoot S.; Ohishi T.; Kumar A.; Pan Q.; Banerjee S.; Zhang K. Y.; Baig M. S. A Novel Therapeutic Peptide Blocks SARS-CoV-2 Spike Protein Binding with Host Cell ACE2 Receptor. Drugs R D 2021, 21, 273. 10.1007/s40268-021-00357-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- V. K. P.; Rath S. P.; Abraham P. Computational designing of a peptide that potentially blocks the entry of SARS-CoV, SARS-CoV-2 and MERS-CoV. PloS one 2021, 16, e0251913 10.1371/journal.pone.0251913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padhi S.; Sanjukta S.; Chourasia R.; Labala R. K.; Singh S. P.; Rai A. K. A Multifunctional Peptide From Bacillus Fermented Soybean for Effective Inhibition of SARS-CoV-2 S1 Receptor Binding Domain and Modulation of Toll Like Receptor 4: A Molecular Docking Study. Front. Mol. Biosci. 2021, 8, 198. 10.3389/fmolb.2021.636647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baig M. S.; Alagumuthu M.; Rajpoot S.; Saqib U. Identification of a Potential Peptide Inhibitor of SARS-CoV-2 Targeting its Entry into the Host Cells. Drugs R D 2020, 20, 161–169. 10.1007/s40268-020-00312-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souza P. F.; Lopes F. E.; Amaral J. L.; Freitas C. D.; Oliveira J. T. A molecular docking study revealed that synthetic peptides induced conformational changes in the structure of SARS-CoV-2 spike glycoprotein, disrupting the interaction with human ACE2 receptor. Int. J. Biol. Macromol. 2020, 164, 66–76. 10.1016/j.ijbiomac.2020.07.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panda S. K.; Sen Gupta P. S.; Biswal S.; Ray A. K.; Rana M. K. ACE-2-derived biomimetic peptides for the inhibition of spike protein of SARS-CoV-2. J. Proteome Res. 2021, 20, 1296–1303. 10.1021/acs.jproteome.0c00686. [DOI] [PubMed] [Google Scholar]
- Bansal R.; Mohagaonkar S.; Sen A.; Khanam U.; Rathi B. In-silico study of peptide-protein interaction of antimicrobial peptides potentially targeting SARS and SARS-CoV-2 nucleocapsid protein. In Silico Pharmacol 2021, 9, 1–14. 10.1007/s40203-021-00103-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porto W. F. Virtual screening of peptides with high affinity for SARS-CoV-2 main protease. Comput. Biol. Med. 2021, 133, 104363. 10.1016/j.compbiomed.2021.104363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yathisha U. G.; Srinivasa M. G.; Siddappa BC R.; Mandal S.; Dixit S. R.; Pujar G. V.; Bangera Sheshappa M. Isolation and characterization of ACE-I inhibitory peptides from ribbonfish for a potential inhibitor of the main protease of SARS-CoV-2; an in-silico analysis. Proteins: Struct. Funct. Genet. 2022, 90, 982. 10.1002/prot.26291. [DOI] [PubMed] [Google Scholar]
- Zhao W.; Li X.; Yu Z.; Wu S.; Ding L.; Liu J. Identification of lactoferrin-derived peptides as potential inhibitors against the main protease of SARS-CoV-2. LWT 2022, 154, 112684. 10.1016/j.lwt.2021.112684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behzadipour Y.; Gholampour M.; Pirhadi S.; Seradj H.; Khoshneviszadeh M.; Hemmati S. Viral 3CLpro as a target for antiviral intervention using milk-derived bioactive peptides. Int. J. Pept Res. Ther 2021, 27, 2703–2716. 10.1007/s10989-021-10284-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong F.-C.; Ong J.-H.; Kumar D. T.; Chai T.-T. In Silico Identification of Multi-target Anti-SARS-CoV-2 Peptides from Quinoa Seed Proteins. Int. J. Pept. Res. Ther 2021, 27, 1837. 10.1007/s10989-021-10214-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong F.-C.; Ong J.-H.; Chai T.-T. SARS-CoV-2 spike protein-, main protease-and papain-like-protease-targeting peptides from seed proteins following gastrointestinal digestion: an in silico study. Phytomedicine Plus 2021, 1, 100016. 10.1016/j.phyplu.2020.100016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaheer M.; Singh R.; Sobhia M. E. Protein degradation: a novel computational approach to design protein degrader probes for main protease of SARS-CoV-2. J. Biomol. Struct 2021, 1–13. 10.1080/07391102.2021.1953601. [DOI] [PubMed] [Google Scholar]
- Wang J.; Yu Y.; Leng T.; Li Y.; Lee S.-T. The Inhibition of SARS-CoV-2 3CL Mpro by Graphene and Its Derivatives from Molecular Dynamics Simulations. ACS Appl. Mater. Interfaces 2022, 14, 191. 10.1021/acsami.1c18104. [DOI] [PubMed] [Google Scholar]
- Benková Z.; Cordeiro M. N. D. Structural behavior of monomer of SARS-CoV-2 spike protein during initial stage of adsorption on graphene. Mater. Today Chem. 2021, 22, 100572. 10.1016/j.mtchem.2021.100572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J.; Yang C.; Ma X.; Li Q. Insights into the conformation changes of SARS-CoV-2 spike receptor-binding domain on graphene. Appl. Surf. Sci. 2022, 578, 151934. 10.1016/j.apsusc.2021.151934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehranfar A.; Khavani M.; Izadyar M. A molecular dynamic study on the ability of phosphorene for designing new sensor for SARS-CoV-2 detection. J. Mol. Liq. 2022, 345, 117852. 10.1016/j.molliq.2021.117852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paloncýová M.; Cechová P.; Srejber M.; Kuhrová P.; Otyepka M. Role of Ionizable Lipids in SARS-CoV-2 Vaccines As Revealed by Molecular Dynamics Simulations: From Membrane Structure to Interaction with mRNA Fragments. J. Phys. Chem. Lett. 2021, 12, 11199–11205. 10.1021/acs.jpclett.1c03109. [DOI] [PubMed] [Google Scholar]
- Grant O. C.; Montgomery D.; Ito K.; Woods R. J. Analysis of the SARS-CoV-2 spike protein glycan shield reveals implications for immune recognition. Sci. Rep. 2020, 10, 1–11. 10.1038/s41598-020-71748-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z.; Ruan P.; Wang L.; Nie X.; Ma X.; Tan Y. T and B cell Epitope analysis of SARS-CoV-2 S protein based on immunoinformatics and experimental research. JCMM 2021, 25, 1274–1289. 10.1111/jcmm.16200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Moura R. R.; Agrelli A.; Santos-Silva C. A.; Silva N.; Assunção B. R.; Brandão L.; Benko-Iseppon A. M.; Crovella S. Immunoinformatic approach to assess SARS-CoV-2 protein S epitopes recognised by the most frequent MHC-I alleles in the Brazilian population. J. Clin. Pathol. 2021, 74, 528–532. 10.1136/jclinpath-2020-206946. [DOI] [PubMed] [Google Scholar]
- Rahman M. S.; Hoque M. N.; Islam M. R.; Akter S.; Rubayet-Ul-Alam A.; Siddique M. A.; Saha O.; Rahaman M. M.; Sultana M.; Crandall K. A.; et al. Epitope-based chimeric peptide vaccine design against S, M and E proteins of SARS-CoV-2 etiologic agent of global pandemic COVID-19: an in silico approach. PeerJ. 2020, 8, e9572 10.7717/peerj.9572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezaj M. M. A.; Junaid M.; Akter Y.; Nahrin A.; Siddika A.; Afrose S. S.; Nayeem S. A.; Haque M. S.; Moni M. A.; Hosen S. Z. Whole proteome screening and identification of potential epitopes of SARS-CoV-2 for vaccine design-an immunoinformatic, molecular docking and molecular dynamics simulation accelerated robust strategy. J. Biomol. Struct 2021, 1–26. 10.1080/07391102.2021.1886171. [DOI] [PubMed] [Google Scholar]
- Chauhan V.; Rungta T.; Rawat M.; Goyal K.; Gupta Y.; Singh M. P. Excavating SARS-coronavirus 2 genome for epitope-based subunit vaccine synthesis using immunoinformatics approach. J. Cell. Physiol. 2021, 236, 1131–1147. 10.1002/jcp.29923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalita P.; Padhi A. K.; Zhang K. Y.; Tripathi T. Design of a peptide-based subunit vaccine against novel coronavirus SARS-CoV-2. Microb. Pathog. 2020, 145, 104236. 10.1016/j.micpath.2020.104236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waqas M.; Haider A.; Rehman A.; Qasim M.; Umar A.; Sufyan M.; Akram H. N.; Mir A.; Razzaq R.; Rasool D.; et al. Immunoinformatics and Molecular Docking Studies Predicted Potential Multiepitope-Based Peptide Vaccine and Novel Compounds against Novel SARS-CoV-2 through Virtual Screening. Biomed Res. Int. 2021, 2021, 1. 10.1155/2021/1596834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranga V.; Niemelä E.; Tamirat M. Z.; Eriksson J. E.; Airenne T. T.; Johnson M. S. Immunogenic SARS-CoV-2 Epitopes: In Silico Study Towards Better Understanding of COVID-19 Disease–Paving the Way for Vaccine Development. Vaccines 2020, 8, 408. 10.3390/vaccines8030408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar B.; Ullah M. A.; Johora F. T.; Taniya M. A.; Araf Y. Immunoinformatics-guided designing of epitope-based subunit vaccines against the SARS Coronavirus-2 (SARS-CoV-2). Immunobiology 2020, 225, 151955. 10.1016/j.imbio.2020.151955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya M.; Sharma A. R.; Patra P.; Ghosh P.; Sharma G.; Patra B. C.; Saha R. P.; Lee S.-S.; Chakraborty C. A SARS-CoV-2 vaccine candidate: In-silico cloning and validation. Inform. Med. 2020, 20, 100394. 10.1016/j.imu.2020.100394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanami S.; Zandi M.; Pourhossein B.; Mobini G.-R.; Safaei M.; Abed A.; Arvejeh P. M.; Chermahini F. A.; Alizadeh M. Design of a multi-epitope vaccine against SARS-CoV-2 using immunoinformatics approach. Int. J. Biol. Macromol. 2020, 164, 871–883. 10.1016/j.ijbiomac.2020.07.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman N.; Ali F.; Basharat Z.; Shehroz M.; Khan M. K.; Jeandet P.; Nepovimova E.; Kuca K.; Khan H. Vaccine Design from the Ensemble of Surface Glycoprotein Epitopes of SARS-CoV-2: An Immunoinformatics Approach. Vaccines 2020, 8, 423. 10.3390/vaccines8030423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lizbeth R.-S. G.; Jazmín G.-M.; José C.-B.; Marlet M.-A. Immunoinformatics study to search epitopes of spike glycoprotein from SARS-CoV-2 as potential vaccine. J. Biomol. Struct. Dyn. 2021, 39, 4878. 10.1080/07391102.2020.1780944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Karmalawy A. A.; Dahab M. A.; Metwaly A. M.; Elhady S. S.; Elkaeed E. B.; Eissa I. H.; Darwish K. M. Molecular docking and dynamics simulation revealed the potential inhibitory activity of ACEIs against SARS-CoV-2 targeting the hACE2 receptor. Front. Chem. 2021, 9, 9. 10.3389/fchem.2021.661230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AlAjmi M. F.; Rehman M. T.; Hussain A. Celecoxib, Glipizide, Lapatinib, and Sitagliptin as potential suspects of aggravating SARS-CoV-2 (COVID-19) infection: a computational approach. J. Biomol. Struct 2021, 1–12. 10.1080/07391102.2021.1994013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baildya N.; Ghosh N. N.; Chattopadhyay A. P. Inhibitory capacity of Chloroquine against SARS-COV-2 by effective binding with Angiotensin converting enzyme-2 receptor: an insight from molecular docking and MD-simulation studies. J. Mol. Struct. 2021, 1230, 129891. 10.1016/j.molstruc.2021.129891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ismail N. Z.; Adebayo I. A.; Mohamad Zain N. N.; Arsad H. Molecular docking of compounds from Clinacanthus nutans extract detected by GC-MS analysis with the SARS-CoV-2 main protease and ACE2 protein. Nat. Prod. Res. 2021, 1–5. 10.1080/14786419.2021.1919104. [DOI] [PubMed] [Google Scholar]
- Tanaka S.; Nelson G.; Olson C. A.; Buzko O.; Higashide W.; Shin A.; Gonzalez M.; Taft J.; Patel R.; Buta S.; et al. An ACE2 Triple Decoy that neutralizes SARS-CoV-2 shows enhanced affinity for virus variants. Sci. Rep. 2021, 11, 1–12. 10.1038/s41598-021-91809-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan A. A.; Baildya N.; Dutta T.; Ghosh N. N. Inhibitory efficiency of potential drugs against SARS-CoV-2 by blocking human angiotensin converting enzyme-2: Virtual screening and molecular dynamics study. Microb. Pathog. 2021, 152, 104762. 10.1016/j.micpath.2021.104762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Çakır B.; Okuyan B.; Şener G.; Tunali-Akbay T. Investigation of beta-lactoglobulin derived bioactive peptides against SARS-CoV-2 (COVID-19): in silico analysis. Eur. J. Pharmacol. 2021, 891, 173781. 10.1016/j.ejphar.2020.173781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nath H.; Mallick A.; Roy S.; Sukla S.; Biswas S. Computational modelling supports that dengue virus envelope antibodies can bind to SARS-CoV-2 receptor binding sites: Is pre-exposure to dengue virus protective against COVID-19 severity?. Comput. Struct. Biotechnol. J. 2021, 19, 459–466. 10.1016/j.csbj.2020.12.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirolli D.; Righino B.; De Rosa M. C. Targeting SARS-CoV-2 Spike Protein/ACE2 Protein-Protein Interactions: a Computational Study. Mol. Inform. 2021, 40, 2060080. 10.1002/minf.202060080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge S.; Wang X.; Hou Y.; Lv Y.; Wang C.; He H. Repositioning of histamine H1 receptor antagonist: Doxepin inhibits viropexis of SARS-CoV-2 Spike pseudovirus by blocking ACE2. Eur. J. Pharmacol. 2021, 896, 173897. 10.1016/j.ejphar.2021.173897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baby K.; Maity S.; Mehta C. H.; Suresh A.; Nayak U. Y.; Nayak Y. SARS-CoV-2 entry inhibitors by dual targeting TMPRSS2 and ACE2: An in silico drug repurposing study. Eur. J. Pharmacol. 2021, 896, 173922. 10.1016/j.ejphar.2021.173922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumawat A.; Namsani S.; Pramanik D.; Roy S.; Singh J. K. Integrated docking and enhanced sampling-based selection of repurposing drugs for SARS-CoV-2 by targeting host dependent factors. J. Biomol. Struct 2021, 1–12. 10.1080/07391102.2021.1937319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Z.; Kan R.; Ji H.; Wu S.; Zhao W.; Shuian D.; Liu J.; Li J. Identification of tuna protein-derived peptides as potent SARS-CoV-2 inhibitors via molecular docking and molecular dynamic simulation. Food Chem. 2021, 342, 128366. 10.1016/j.foodchem.2020.128366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi T.; Joshi T.; Sharma P.; Mathpal S.; Pundir H.; Bhatt V.; Chandra S. In silico screening of natural compounds against COVID-19 by targeting Mpro and ACE2 using molecular docking. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 4529–4536. 10.26355/eurrev_202004_21036. [DOI] [PubMed] [Google Scholar]
- Khelfaoui H.; Harkati D.; Saleh B. A. Molecular docking, molecular dynamics simulations and reactivity, studies on approved drugs library targeting ACE2 and SARS-CoV-2 binding with ACE2. J. Biomol. Struct. Dyn. 2021, 39, 7246. 10.1080/07391102.2020.1803967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdelli I.; Hassani F.; Bekkel Brikci S.; Ghalem S. In silico study the inhibition of Angiotensin converting enzyme 2 receptor of COVID-19 by Ammoides verticillata components harvested from western Algeria. J. Biomol. Struct. Dyn. 2020, 1–17. 10.1080/07391102.2020.1763199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakmi M.; Bouricha E. M.; Akachar J.; Lmimouni B.; El Harti J.; Belyamani L.; Ibrahimi A. In silico exploration of small-molecule α-helix mimetics as inhibitors of SARS-CoV-2 attachment to ACE2. J. Biomol. Struct. Dyn. 2022, 40, 1546. 10.1080/07391102.2020.1830175. [DOI] [PubMed] [Google Scholar]
- Zhang X.-Y.; Huang H.-J.; Zhuang D.-L.; Nasser M. I.; Yang M.-H.; Zhu P.; Zhao M.-Y. Biological, clinical and epidemiological features of COVID-19, SARS and MERS and AutoDock simulation of ACE2. Infect. Dis. Poverty 2020, 9, 1–11. 10.1186/s40249-020-00691-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez-Villagomez J. M.; Campos-García T.; Molina-Torres J.; López M. G.; Vázquez-Martínez J. Alkamides and piperamides as potential antivirals against the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). J. Phys. Chem. Lett. 2020, 11, 8008–8016. 10.1021/acs.jpclett.0c01685. [DOI] [PubMed] [Google Scholar]
- Marciniec K.; Chrobak E.; Dabrowska A.; Bebenek E.; Kadela-Tomanek M.; Pecak P.; Boryczka S. Phosphate Derivatives of 3-Carboxyacylbetulin: SynThesis, In Vitro Anti-HIV and Molecular Docking Study. Biomolecules 2020, 10, 1148. 10.3390/biom10081148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P.; Shanavas A. Natural derivatives with dual binding potential against SARS-CoV-2 main protease and human ACE2 possess low oral bioavailability: a brief computational analysis. J. Biomol. Struct. Dyn. 2021, 39, 5819. 10.1080/07391102.2020.1794970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramakrishnan J.; Kandasamy S.; Iruthayaraj A.; Magudeeswaran S.; Chinnasamy K.; Poomani K. Strong Binding of Leupeptin with TMPRSS2 Protease May Be an Alternative to Camostat and Nafamostat for SARS-CoV-2 Repurposed Drug: Evaluation from Molecular Docking and Molecular Dynamics Simulations. Appl. Biochem. Biotechnol. 2021, 193, 1909–1923. 10.1007/s12010-020-03475-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vardhan S.; Sahoo S. K. Virtual screening by targeting proteolytic sites of furin and TMPRSS2 to propose potential compounds obstructing the entry of SARS-CoV-2 virus into human host cells. J. Tradit. Complement. Med. 2022, 12, 6. 10.1016/j.jtcme.2021.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chikhale R. V.; Gupta V. K.; Eldesoky G. E.; Wabaidur S. M.; Patil S. A.; Islam M. A. Identification of potential anti-TMPRSS2 natural products through homology modelling, virtual screening and molecular dynamics simulation studies. J. Biomol. Struct. Dyn. 2021, 39, 6660. 10.1080/07391102.2020.1798813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmeira A.; Sousa E.; Köseler A.; Sabirli R.; Gören T.; Türkçüer İ.; Kurt Ö.; Pinto M. M.; Vasconcelos M. H. Preliminary virtual screening studies to identify GRP78 inhibitors which may interfere with SARS-CoV-2 infection. Pharmaceuticals 2020, 13, 132. 10.3390/ph13060132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keidar S.; Kaplan M.; Gamliel-Lazarovich A. ACE2 of the heart: from angiotensin I to angiotensin (1–7). Cardiovasc. Res. 2007, 73, 463–469. 10.1016/j.cardiores.2006.09.006. [DOI] [PubMed] [Google Scholar]
- Fang L.; Karakiulakis G.; Roth M. Antihypertensive drugs and risk of COVID-19?–Authors’ reply. Lancet Respir. Med. 2020, 8, e32 10.1016/S2213-2600(20)30159-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao Y.; Feher V. A.; McCammon J. A. Gaussian accelerated molecular dynamics: Unconstrained enhanced sampling and free energy calculation. J. Chem. Theory Comput. 2015, 11, 3584–3595. 10.1021/acs.jctc.5b00436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koulgi S.; Jani V.; Uppuladinne M.; Sonavane U.; Nath A. K.; Darbari H.; Joshi R. Drug repurposing studies targeting SARS-CoV-2: an ensemble docking approach on drug target 3C-like protease (3CLpro). J. Biomol. Struct. Dyn. 2021, 39, 5735. 10.1080/07391102.2020.1792344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chodera J. D.; Noé F. Markov state models of biomolecular conformational dynamics. Curr. Opin. Struct. Biol. 2014, 25, 135–144. 10.1016/j.sbi.2014.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ismail M. I.; Ragab H. M.; Bekhit A. A.; Ibrahim T. M. Targeting multiple conformations of SARS-CoV2 Papain-Like Protease for drug repositioning: An in-silico study. Comput. Biol. Med. 2021, 131, 104295. 10.1016/j.compbiomed.2021.104295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elfiky A. A.; Azzam E. B.; Shafaa M. W. The anti-HCV, Sofosbuvir, versus the anti-EBOV Remdesivir against SARS-CoV-2 RNA dependent RNA polymerase in silico. Mol. Divers. 2022, 26, 171. 10.1007/s11030-020-10178-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar V.; Liu H.; Wu C. Drug repurposing against SARS-CoV-2 receptor binding domain using ensemble-based virtual screening and molecular dynamics simulations. Comput. Biol. Med. 2021, 135, 104634. 10.1016/j.compbiomed.2021.104634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawant S.; Patil R.; Khawate M.; Zambre V.; Shilimkar V.; Jagtap S. Computational assessment of select antiviral phytochemicals as potential SARS-Cov-2 main protease inhibitors: molecular dynamics guided ensemble docking and extended molecular dynamics. In Silico Pharmacol 2021, 9, 1–17. 10.1007/s40203-021-00107-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoddard S. V.; Stoddard S. D.; Oelkers B. K.; Fitts K.; Whalum K.; Whalum K.; Hemphill A. D.; Manikonda J.; Martinez L. M.; Riley E. G.; et al. Optimization Rules for SARS-CoV-2 Mpro Antivirals: Ensemble Docking and Exploration of the Coronavirus Protease Active Site. Viruses 2020, 12, 942. 10.3390/v12090942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elfiky A. A. SARS-CoV-2 RNA dependent RNA polymerase (RdRp) targeting: An in silico perspective. J. Biomol. Struct. Dyn. 2020, 1–9. 10.1080/07391102.2020.1761882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odhar H. A.; Ahjel S. W.; Albeer A. A. M. A.; Hashim A. F.; Rayshan A. M.; Humadi S. S. Molecular docking and dynamics simulation of FDA approved drugs with the main protease from 2019 novel coronavirus. Bioinformation 2020, 16, 236. 10.6026/97320630016236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra D.; Maurya R. R.; Kumar K.; Munjal N. S.; Bahadur V.; Sharma S.; Singh P.; Bahadur I. Structurally modified compounds of hydroxychloroquine, remdesivir and tetrahydrocannabinol against main protease of SARS-CoV-2, a possible hope for COVID-19: Docking and molecular dynamics simulation studies. J. Mol. Liq. 2021, 335, 116185. 10.1016/j.molliq.2021.116185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardoso W. B.; Mendanha S. A. Molecular dynamics simulation of docking structures of SARS-CoV-2 main protease and HIV protease inhibitors. J. Mol. Struct. 2021, 1225, 129143. 10.1016/j.molstruc.2020.129143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yalçın S.; Yalçınkaya S.; Ercan F. In silico detection of inhibitor potential of Passiflora compounds against SARS-Cov-2 (Covid-19) main protease by using molecular docking and dynamic analyses. J. Mol. Struct. 2021, 1240, 130556. 10.1016/j.molstruc.2021.130556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akdad M.; Moujane S.; Bouadid I.; Benlyas M.; Eddouks M. Phytocompounds from Anvillea radiata as promising anti-Covid-19 drugs: in silico studies and in vivo safety assessment. J. Environ. Sci. Health A 2022, 1–12. 10.1080/10934529.2021.2020029. [DOI] [PubMed] [Google Scholar]
- Kumar N.; Singh J. V.; Bhagat K.; Gulati H. K.; Sharma A.; Rani A.; Duggal A.; Gulati P.; Singh H.; Bedi P. M. S.; et al. Discovery of potent inhibitors for Mpro enzyme of SARS-COV2 by multi-stage in-silico screening of Alkannin/shikonin. Nat. Prod. Res. 2021, 1–5. 10.1080/14786419.2021.2013212. [DOI] [PubMed] [Google Scholar]
- Pavlova A.; Lynch D. L.; Daidone I.; Zanetti-Polzi L.; Smith M. D.; Chipot C.; Kneller D. W.; Kovalevsky A.; Coates L.; Golosov A. A.; et al. Inhibitor binding influences the protonation states of histidines in SARS-CoV-2 main protease. Chem. Sci. 2021, 12, 1513–1527. 10.1039/D0SC04942E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma S.; Pandey A. K. Factual insights of the allosteric inhibition mechanism of SARS-CoV-2 main protease by quercetin: an in silico analysis. 3 Biotech 2021, 11, 1–10. 10.1007/s13205-020-02630-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vishvakarma V. K.; Pal S.; Singh P.; Bahadur I. Interactions between main protease of SARS-CoV-2 and testosterone or progesterone using computational approach. J. Mol. Struct. 2022, 1251, 131965. 10.1016/j.molstruc.2021.131965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vishvakarma V. K.; Singh M. B.; Jain P.; Kumari K.; Singh P. Hunting the main protease of SARS-CoV-2 by plitidepsin: Molecular docking and temperature-dependent molecular dynamics simulations. Amino acids 2022, 54, 205. 10.1007/s00726-021-03098-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussein R.; Elkhair H. Molecular docking identification for the efficacy of some zinc complexes with chloroquine and hydroxychloroquine against main protease of COVID-19. J. Mol. Struct. 2021, 1231, 129979. 10.1016/j.molstruc.2021.129979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai H.; Barik A.; Singh Y. P.; Suresh A.; Singh L.; Singh G.; Nayak U. Y.; Dubey V. K.; Modi G. Molecular docking, binding mode analysis, molecular dynamics, and prediction of ADMET/toxicity properties of selective potential antiviral agents against SARS-CoV-2 main protease: an effort toward drug repurposing to combat COVID-19. Mol. Divers. 2021, 25, 1905. 10.1007/s11030-021-10188-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baildya N.; Ghosh N. N.; Chattopadhyay A. P. Inhibitory activity of hydroxychloroquine on COVID-19 main protease: An insight from MD-simulation studies. J. Mol. Struct. 2020, 1219, 128595. 10.1016/j.molstruc.2020.128595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S.; Sharma P. P.; Shankar U.; Kumar D.; Joshi S. K.; Pena L.; Durvasula R.; Kumar A.; Kempaiah P.; Poonam; et al. Discovery of New Hydroxyethylamine Analogs Against 3CLpro Protein Target of SARS-CoV-2: Molecular Docking, Molecular Dynamics Simulation and Structure-Activity Relationship Studies. J. Chem. Inf Model 2020, 60, 5754–5770. 10.1021/acs.jcim.0c00326. [DOI] [PubMed] [Google Scholar]
- Muralidharan N.; Sakthivel R.; Velmurugan D.; Gromiha M. M. Computational studies of drug repurposing and synergism of lopinavir, oseltamivir and ritonavir binding with SARS-CoV-2 Protease against COVID-19. J. Biomol. Struct. Dyn. 2021, 39, 2673. 10.1080/07391102.2020.1752802. [DOI] [PubMed] [Google Scholar]
- Huynh T.; Wang H.; Luan B. In silico exploration of the molecular mechanism of clinically oriented drugs for possibly inhibiting SARS-CoV-2’s main protease. J. Phys. Chem. Lett. 2020, 11, 4413–4420. 10.1021/acs.jpclett.0c00994. [DOI] [PubMed] [Google Scholar]
- Kumar N.; Sood D.; van der Spek P. J.; Sharma H. S.; Chandra R. Molecular binding mechanism and pharmacology comparative analysis of noscapine for repurposing against SARS-CoV-2 protease. J. Proteome Res. 2020, 19, 4678–4689. 10.1021/acs.jproteome.0c00367. [DOI] [PubMed] [Google Scholar]
- Khan M. A.; Mahmud S.; Alam A. R. U.; Rahman M. E.; Ahmed F.; Rahmatullah M. Comparative molecular investigation of the potential inhibitors against SARS-CoV-2 main protease: a molecular docking study. J. Biomol. Struct. Dyn. 2021, 39, 6317. 10.1080/07391102.2020.1796813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar Y.; Singh H.; Patel C. N. In silico prediction of potential inhibitors for the main protease of SARS-CoV-2 using molecular docking and dynamics simulation based drug-repurposing. J. Infect. Public Health 2020, 13, 1210–1223. 10.1016/j.jiph.2020.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahanta S.; Chowdhury P.; Gogoi N.; Goswami N.; Borah D.; Kumar R.; Chetia D.; Borah P.; Buragohain A. K.; Gogoi B. Potential anti-viral activity of approved repurposed drug against main protease of SARS-CoV-2: an in silico based approach. J. Biomol. Struct. Dyn. 2021, 39, 3802. 10.1080/07391102.2020.1768902. [DOI] [PubMed] [Google Scholar]
- Bharadwaj S.; Lee K. E.; Dwivedi V. D.; Kang S. G. Computational insights into tetracyclines as inhibitors against SARS-CoV-2 Mpro via combinatorial molecular simulation calculations. Life Sci. 2020, 257, 118080. 10.1016/j.lfs.2020.118080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fintelman-Rodrigues N.; Sacramento C. Q.; Lima C. R.; da Silva F. S.; Ferreira A. C.; Mattos M.; de Freitas C. S.; Soares V. C.; Dias S. d. S. G.; Temerozo J. R.; et al. Atazanavir, alone or in combination with ritonavir, inhibits SARS-CoV-2 replication and proinflammatory cytokine production. Antimicrob. Agents Chemother. 2020, 64, 64. 10.1128/AAC.00825-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razzaghi-Asl N.; Ebadi A.; Shahabipour S.; Gholamin D. Identification of a potential SARS-CoV2 inhibitor via molecular dynamics simulations and amino acid decomposition analysis. J. Biomol. Struct. Dyn. 2021, 39, 6633. 10.1080/07391102.2020.1797536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa A. N.; de Sa E. R. A.; Bezerra R. D. S.; Souza J. L.; Lima F. d. C. A. Constituents of buriti oil (Mauritia flexuosa L.) like inhibitors of the SARS-Coronavirus main peptidase: an investigation by docking and molecular dynamics. J. Biomol. Struct. Dyn. 2021, 39, 4610. 10.1080/07391102.2020.1778538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao P.; Shukla A.; Parmar P.; Rawal R. M.; Patel B. V.; Saraf M.; Goswami D. Proposing a fungal metabolite-flaviolin as a potential inhibitor of 3CLpro of novel coronavirus SARS-CoV-2 identified using docking and molecular dynamics. J. Biomol. Struct. Dyn. 2022, 40, 348. 10.1080/07391102.2020.1813202. [DOI] [PubMed] [Google Scholar]
- De Sousa Silva J. D.; da Costa Leite S.; da Silva M. T. S.; Meirelles L. M. A.; Andrade A. W. L. In silico evaluation of the inhibitory effect of antiretrovirals Atazanavir and Darunavir on the main protease of SARS-CoV-2: docking studies and molecular dynamics. Res., Soc. Dev. 2020, 9, e826986562. 10.33448/rsd-v9i8.6562. [DOI] [Google Scholar]
- Shree P.; Mishra P.; Selvaraj C.; Singh S. K.; Chaube R.; Garg N.; Tripathi Y. B. Targeting COVID-19 (SARS-CoV-2) main protease through active phytochemicals of ayurvedic medicinal plants–Withania somnifera (Ashwagandha), Tinospora cordifolia (Giloy) and Ocimum sanctum (Tulsi)–a molecular docking study. J. Biomol. Struct. Dyn. 2022, 40, 190. 10.1080/07391102.2020.1810778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadil Y.; Mouhcine M.; Filali H. In silico Study of Pharmacological Treatments against SARS-CoV2Main Protease. J. Pure Appl. Microbiol 2020, 14, 1065–1071. 10.22207/JPAM.14.SPL1.45. [DOI] [Google Scholar]
- Alam R.; Imon R. R.; Talukder M. E. K.; Akhter S.; Hossain M. A.; Ahammad F.; Rahman M. M. GC-MS analysis of phytoconstituents from Ruellia prostrata and Senna tora and identification of potential anti-viral activity against SARS-CoV-2. RSC Adv. 2021, 11, 40120–40135. 10.1039/D1RA06842C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jairajpuri D. S.; Hussain A.; Nasreen K.; Mohammad T.; Anjum F.; Rehman M. T.; Hasan G. M.; Alajmi M. F.; Hassan M. I. Identification of natural compounds as potent inhibitors of SARS-CoV-2 main protease using combined docking and molecular dynamics simulations. Saudi J. Biol. Sci. 2021, 28, 2423–2431. 10.1016/j.sjbs.2021.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ul Qamar M. T.; Alqahtani S. M.; Alamri M. A.; Chen L.-L. Structural basis of SARS-CoV-2 3CLpro and anti-COVID-19 drug discovery from medicinal plants. J. Pharm. Anal. 2020, 10, 313–319. 10.1016/j.jpha.2020.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmud S.; Biswas S.; Paul G. K.; Mita M. A.; Promi M. M.; Afrose S.; Hasan M.; Zaman S.; Uddin M.; Dhama K.; et al. Plant-based phytochemical screening by targeting main protease of SARS-CoV-2 to design effective potent inhibitors. Biology 2021, 10, 589. 10.3390/biology10070589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee S.; Sharma D.; Sharma A. K.; Jaiswal S.; Sharma N.; Borah S.; Kaur G. Flavan-based phytoconstituents inhibit Mpro, a SARS-COV-2 molecular target, in silico. J. Biomol. Struct 2021, 1–15. 10.1080/07391102.2021.1960196. [DOI] [PubMed] [Google Scholar]
- Cao T. Q.; Kim J. A.; Woo M. H.; Min B. S. SARS-CoV-2 main protease inhibition by compounds isolated from Luffa cylindrica using molecular docking. Bioorg. Med. Chem. Lett. 2021, 40, 127972. 10.1016/j.bmcl.2021.127972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halder P.; Pal U.; Paladhi P.; Dutta S.; Paul P.; Pal S.; Das D.; Ganguly A.; Dutta I.; Mandal S.; et al. Evaluation of potency of the selected bioactive molecules from Indian medicinal plants with MPro of SARS-CoV-2 through in silico analysis. J. Ayurveda Integr Med. 2022, 13, 100449. 10.1016/j.jaim.2021.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullah A.; Ullah K. Inhibition of SARS-CoV-2 3CL Mpro by Natural and Synthetic Inhibitors: Potential Implication for Vaccine Production Against COVID-19. Front. Mol. Biosci. 2021, 8, 211. 10.3389/fmolb.2021.640819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamsi S.; Anjum H.; Shahbaaz M.; Khan M. S.; Ataya F. S.; Alamri A.; Alhumaydhi F. A.; Husain F. M.; Rehman M. T.; Mohammad T.; et al. A computational study on active constituents of Habb-ul-aas and Tabasheer as inhibitors of SARS-CoV-2 main protease. J. Biomol. Struct 2021, 1–12. 10.1080/07391102.2021.1900920. [DOI] [PubMed] [Google Scholar]
- Zaki A. A.; Ashour A.; Elhady S. S.; Darwish K. M.; Al-Karmalawy A. A. Calendulaglycoside A showing potential activity against SARS-CoV-2 main protease: Molecular docking, molecular dynamics, and SAR studies. J. Tradit. Complement. Med. 2022, 12, 16–34. 10.1016/j.jtcme.2021.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hejazi I. I.; Beg M. A.; Imam M. A.; Athar F.; Islam A. Glossary of phytoconstituents: Can these be repurposed against SARS CoV-2? A quick in silico screening of various phytoconstituents from plant Glycyrrhiza glabra with SARS CoV-2 main protease. FCT 2021, 150, 112057. 10.1016/j.fct.2021.112057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan M. T.; Ali A.; Wang Q.; Irfan M.; Khan A.; Zeb M. T.; Zhang Y.-J.; Chinnasamy S.; Wei D.-Q. Marine natural compounds as potents inhibitors against the main protease of SARS-CoV-2. A molecular dynamic study. J. Biomol. Struct. Dyn. 2021, 39, 3627. 10.1080/07391102.2020.1769733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao P.; Shukla A.; Parmar P.; Rawal R. M.; Patel B.; Saraf M.; Goswami D. Reckoning a fungal metabolite, Pyranonigrin A as a potential Main protease (Mpro) inhibitor of novel SARS-CoV-2 virus identified using docking and molecular dynamics simulation. Biophys. Chem. 2020, 264, 106425. 10.1016/j.bpc.2020.106425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krupanidhi S.; Abraham Peele K.; Venkateswarulu T.; Ayyagari V. S.; Nazneen Bobby M.; John Babu D.; Venkata Narayana A.; Aishwarya G. Screening of phytochemical compounds of Tinospora cordifolia for their inhibitory activity on SARS-CoV-2: an in silico study. J. Biomol. Struct. Dyn. 2021, 39, 5799. 10.1080/07391102.2020.1787226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury P. In silico investigation of phytoconstituents from Indian medicinal herb ‘Tinospora cordifolia (giloy)’against SARS-CoV-2 (COVID-19) by molecular dynamics approach. J. Biomol. Struct. Dyn. 2021, 39, 6792. 10.1080/07391102.2020.1803968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umesh; Kundu D.; Selvaraj C.; Singh S. K.; Dubey V. K. Identification of new anti-nCoV drug chemical compounds from Indian spices exploiting SARS-CoV-2 main protease as target. J. Biomol. Struct. Dyn. 2020, 1–9. 10.1080/07391102.2020.1763202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yepes-Pérez A. F.; Herrera-Calderon O.; Sánchez-Aparicio J.-E.; Tiessler-Sala L.; Maréchal J.-D.; Cardona-G W. Investigating Potential Inhibitory Effect of Uncaria tomentosa (Cat′s Claw) against the Main Protease 3CLpro of SARS-CoV-2 by Molecular Modeling. Evid. Based Complementary Altern. Med. 2020, 2020, 4932572. 10.1155/2020/4932572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peele K. A.; Durthi C. P.; Srihansa T.; Krupanidhi S.; Ayyagari V. S.; Babu D. J.; Indira M.; Reddy A. R.; Venkateswarulu T. Molecular docking and dynamic simulations for antiviral compounds against SARS-CoV-2: A computational study. Inform. Med. 2020, 19, 100345. 10.1016/j.imu.2020.100345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterling T.; Irwin J. J. ZINC 15–ligand discovery for everyone. J. Chem. Inf Model 2015, 55, 2324–2337. 10.1021/acs.jcim.5b00559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouassaf M.; Belaidi S.; Al Mogren M. M.; Chtita S.; Khan S. U.; Htar T. T. Combined docking methods and molecular dynamics to identify effective antiviral 2, 5-diaminobenzophenonederivatives against SARS-CoV-2. J. King Saud Univ. Sci. 2021, 33, 101352. 10.1016/j.jksus.2021.101352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mody V.; Ho J.; Wills S.; Mawri A.; Lawson L.; Ebert M. C.; Fortin G. M.; Rayalam S.; Taval S. Identification of 3-chymotrypsin like protease (3CLPro) inhibitors as potential anti-SARS-CoV-2 agents. Commun. Biol. 2021, 4, 1–10. 10.1038/s42003-020-01577-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singhal S.; Khanna P.; Khanna L. Synthesis, Comparative in vitro Antibacterial, Antioxidant & UV fluorescence studies of bis Indole Schiff bases and Molecular docking with ct-DNA & SARS-CoV-2 Mpro. Luminescence 2021, 36, 1531–1543. 10.1002/bio.4098. [DOI] [PubMed] [Google Scholar]
- Jawaria R.; Khan M. U.; Hussain M.; Muhammad S.; Sagir M.; Hussain A.; Al-Sehemi A. G. Synthesis and characterization of ferrocene-based thiosemicarbazones along with their computational studies for potential as inhibitors for SARS-CoV-2. J. Iran. Chem. Soc. 2022, 19, 839–846. 10.1007/s13738-021-02346-1. [DOI] [Google Scholar]
- Rolta R.; Salaria D.; Sharma P.; Sharma B.; Kumar V.; Rathi B.; Verma M.; Sourirajan A.; Baumler D. J.; Dev K. Phytocompounds of Rheum emodi, Thymus serpyllum, and Artemisia annua Inhibit Spike Protein of SARS-CoV-2 Binding to ACE2 Receptor: In Silico Approach. Curr. Pharmacol. Rep 2021, 7, 135–149. 10.1007/s40495-021-00259-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaral J. L.; Oliveira J. T.; Lopes F. E.; Freitas C. D.; Freire V. N.; Abreu L. V.; Souza P. F. Quantum biochemistry, molecular docking, and dynamics simulation revealed synthetic peptides induced conformational changes affecting the topology of the catalytic site of SARS-CoV-2 main protease. J. Biomol. Struct 2021, 1–13. 10.1080/07391102.2021.1920464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uddin M. N.; Siddique Z. A.; Akter J.; Rahman M. S.; Shumi W.; Nasiruddin M. Synthesis, molecular modeling, and biomedical applications of oxovanadium (IV) complexes of Schiff bases as a good SARS-CoV-2 inhibitor. Inorg. Nano-Met. Chem. 2021, 1–16. 10.1080/24701556.2021.1952261. [DOI] [Google Scholar]
- Domínguez-Villa F. X.; Durán-Iturbide N. A.; Ávila-Zárraga J. G. Synthesis, molecular docking, and in silico ADME/Tox profiling studies of new 1-aryl-5-(3-azidopropyl) indol-4-ones: Potential inhibitors of SARS CoV-2 main protease. Bioorg. Chem. 2021, 106, 104497. 10.1016/j.bioorg.2020.104497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omar A. Z.; Mosa T. M.; El-Sadany S. K.; Hamed E. A.; El-Atawy M. Novel piperazine based compounds as potential inhibitors for SARS-CoV-2 Protease Enzyme: Synthesis and molecular docking study. J. Mol. Struct. 2021, 1245, 131020. 10.1016/j.molstruc.2021.131020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chidambaram S.; El-Sheikh M. A.; Alfarhan A. H.; Radhakrishnan S.; Akbar I. Synthesis of novel coumarin analogues: Investigation of molecular docking interaction of SARS-CoV-2 proteins with natural and synthetic coumarin analogues and their pharmacokinetics studies. Saudi J. Biol. Sci. 2021, 28, 1100–1108. 10.1016/j.sjbs.2020.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang J.; Pitsillou E.; Karagiannis C.; Darmawan K. K.; Ng K.; Hung A.; Karagiannis T. C. Interaction of the prototypical α-ketoamide inhibitor with the SARS-CoV-2 main protease active site in silico: molecular dynamic simulations highlight the stability of the ligand-protein complex. Comput. Biol. Chem. 2020, 87, 107292. 10.1016/j.compbiolchem.2020.107292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battisti V.; Wieder O.; Garon A.; Seidel T.; Urban E.; Langer T. A Computational Approach to Identify Potential Novel Inhibitors against the Coronavirus SARS-CoV-2. Mol. Inform. 2020, 39, 2000090. 10.1002/minf.202000090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodchakorn K.; Poovorawan Y.; Suwannakarn K.; Kongtawelert P. Molecular modelling investigation for drugs and nutraceuticals against protease of SARS-CoV-2. J. Mol. Graph. Model. 2020, 101, 107717. 10.1016/j.jmgm.2020.107717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroylov V. S.; Svitanko I. V. Computational identification of disulfiram and neratinib as putative SARS-CoV-2 main protease inhibitors. Mendeleev Commun. 2020, 30, 419–420. 10.1016/j.mencom.2020.07.004. [DOI] [Google Scholar]
- Singh A.; Mishra A. Leucoefdin a potential inhibitor against SARS CoV-2 Mpro. J. Biomol. Struct. Dyn. 2021, 39, 4427–4432. 10.1080/07391102.2020.1777903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lokhande K. B.; Doiphode S.; Vyas R.; Swamy K. V. Molecular docking and simulation studies on SARS-CoV-2 Mpro reveals Mitoxantrone, LeuCoVorin, Birinapant, and Dynasore as potent drugs against COVID-19. J. Biomol. Struct. Dyn. 2021, 39, 7294–7305. 10.1080/07391102.2020.1805019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammad T.; Shamsi A.; Anwar S.; Umair M.; Hussain A.; Rehman M. T.; AlAjmi M. F.; Islam A.; Hassan M. I. Identification of high-affinity inhibitors of SARS-CoV-2 main protease: Towards the development of effective COVID-19 therapy. Virus Res. 2020, 288, 198102. 10.1016/j.virusres.2020.198102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trezza A.; Iovinelli D.; Santucci A.; Prischi F.; Spiga O. An integrated drug repurposing strategy for the rapid identification of potential SARS-CoV-2 viral inhibitors. Sci. Rep. 2020, 10, 1–8. 10.1038/s41598-020-70863-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isralewitz B.; Baudry J.; Gullingsrud J.; Kosztin D.; Schulten K. Steered molecular dynamics investigations of protein function. J. Mol. Graph. Model. 2001, 19, 13–25. 10.1016/S1093-3263(00)00133-9. [DOI] [PubMed] [Google Scholar]
- BR B.; Damle H.; Ganju S.; Damle L. In silico screening of known small molecules to bind ACE2 specific RBD on Spike glycoprotein of SARS-CoV-2 for repurposing against COVID-19. F1000Research 2020, 9, 663. 10.12688/f1000research.24143.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoemark D. K.; Colenso C. K.; Toelzer C.; Gupta K.; Sessions R. B.; Davidson A. D.; Berger I.; Schaffitzel C.; Spencer J.; Mulholland A. J. Mol Simuls suggest Vitamins, Retinoids and Steroids as Ligands of the Free Fatty Acid Pocket of the SARS-CoV-2 Spike Protein. Angew. Chem. 2021, 133, 7174–7186. 10.1002/ange.202015639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta Y.; Maciorowski D.; Medernach B.; Becker D. P.; Durvasula R.; Libertin C. R.; Kempaiah P. Iron dysregulation in COVID-19 and reciprocal evolution of SARS-CoV-2: Natura nihil frustra facit. J. Cell. Biochem. 2022, 10.1002/jcb.30207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paiardi G.; Richter S.; Oreste P.; Urbinati C.; Rusnati M.; Wade R. C. The binding of heparin to spike glycoprotein inhibits SARS-CoV-2 infection by three mechanisms. J. Biol. Chem. 2022, 298, 101507. 10.1016/j.jbc.2021.101507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jose S.; Gupta M.; Sharma U.; Quintero-Saumeth J.; Dwivedi M. Potential of phytocompounds from Brassica oleracea targeting S2-domain of SARS-CoV-2 spike glycoproteins: Structure and molecular insights. J. Mol. Struct. 2022, 1254, 132369. 10.1016/j.molstruc.2022.132369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar N.; Thakur A.; Ghadge J.; Rath S. L. Computational studies reveal Fluorine based quinolines to be potent inhibitors for proteins involved in SARS-CoV-2 assembly. J. Fluor. Chem. 2021, 250, 109865. 10.1016/j.jfluchem.2021.109865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Shuhaib M. B. S.; Hashim H. O.; Al-Shuhaib J. M. Epicatechin is a promising novel inhibitor of SARS-CoV-2 entry by disrupting interactions between angiotensin-converting enzyme type 2 and the viral receptor binding domain: A computational/simulation study. Comput. Biol. Med. 2022, 141, 105155. 10.1016/j.compbiomed.2021.105155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei J.; Zhou Y.; Yang X.; Zhang F.; Liu X.; Yu B. Active components in Ephedra sinica Stapf disrupt the interaction between ACE2 and SARS-CoV-2 RBD: potent COVID-19 therapeutic agents. J. Ethnopharmacol. 2021, 278, 114303. 10.1016/j.jep.2021.114303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elfiky A. A. Natural products may interfere with SARS-CoV-2 attachment to the host cell. J. Biomol. Struct 2020, 39, 3194–3203. 10.1080/07391102.2020.1761881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulakuntla S.; Lokhande K. B.; Padmavathi P.; Pal M.; Swamy K. V.; Sadasivam J.; Singh S. A.; Aramgam S. L.; Reddy V. D. Mutational analysis in international isolates and drug repurposing against SARS-CoV-2 spike protein: molecular docking and simulation approach. VirusDisease 2021, 32, 690–702. 10.1007/s13337-021-00720-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain A. S.; Sushma P.; Dharmashekar C.; Beelagi M. S.; Prasad S. K.; Shivamallu C.; Prasad A.; Syed A.; Marraiki N.; Prasad K. S. In silico evaluation of flavonoids as effective antiviral agents on the spike glycoprotein of SARS-CoV-2. Saudi J. Biol. Sci. 2021, 28, 1040–1051. 10.1016/j.sjbs.2020.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fantini J.; Chahinian H.; Yahi N. Synergistic antiviral effect of hydroxychloroquine and azithromycin in combination against SARS-CoV-2: What molecular dynamics studies of virus-host interactions reveal. Int. J. Antimicrob. Agents 2020, 56, 106020. 10.1016/j.ijantimicag.2020.106020. [DOI] [PubMed] [Google Scholar]
- Kalathiya U.; Padariya M.; Mayordomo M.; Lisowska M.; Nicholson J.; Singh A.; Baginski M.; Fahraeus R.; Carragher N.; Ball K.; et al. Highly Conserved Homotrimer Cavity Formed by the SARS-CoV-2 Spike Glycoprotein: A Novel Binding Site. J. Clin. Med. 2020, 9, 1473. 10.3390/jcm9051473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagar P. R.; Gajjar N. D.; Dhameliya T. M. Search of SARS CoV-2 Replication Inhibitors: Virtual Screening, Molecular Dynamics Simulations and ADMET Analysis. J. Mol. Struct. 2021, 1246, 131190. 10.1016/j.molstruc.2021.131190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghazwani M. Y.; Bakheit A. H.; Hakami A. R.; Alkahtani H. M.; Almehizia A. A. Virtual Screening and Molecular Docking Studies for Discovery of Potential RNA-Dependent RNA Polymerase Inhibitors. Crystals 2021, 11, 471. 10.3390/cryst11050471. [DOI] [Google Scholar]
- Singh S. K.; Upadhyay A. K.; Reddy M. S. Screening of potent drug inhibitors against SARS-CoV-2 RNA polymerase: an in silico approach. 3 Biotech 2021, 11, 1–13. 10.1007/s13205-020-02610-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begum F.; Srivastava A. K.; Ray U. Repurposing nonnucleoside antivirals against SARS-CoV2 NSP12 (RNA dependent RNA polymerase): In silico-molecular insight. Biochem. Biophys. Res. Commun. 2021, 571, 26–31. 10.1016/j.bbrc.2021.07.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pokhrel R.; Chapagain P.; Siltberg-Liberles J. Potential RNA-dependent RNA polymerase inhibitors as prospective therapeutics against SARS-CoV-2. J. Med. Microbiol. 2020, 69, 864. 10.1099/jmm.0.001203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebrahimi K. S.; Ansari M.; Moghaddam M. S. H.; Ebrahimi Z.; Shahlaei M.; Moradi S.; et al. In silico investigation on the inhibitory effect of fungal secondary metabolites on RNA dependent RNA polymerase of SARS-CoV-II: A docking and molecular dynamic simulation study. Comput. Biol. Med. 2021, 135, 104613. 10.1016/j.compbiomed.2021.104613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elghoneimy L. K.; Ismail M. I.; Boeckler F. M.; Azzazy H. M.; Ibrahim T. M. Facilitating SARS CoV-2 RNA-Dependent RNA polymerase (RdRp) drug discovery by the aid of HCV NS5B palm subdomain binders: In silico approaches and benchmarking. Comput. Biol. Med. 2021, 134, 104468. 10.1016/j.compbiomed.2021.104468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh T.; Cornell W.; Luan B. In silico exploration of inhibitors for SARS-CoV-2’s papain-like protease. Front. Chem. 2021, 8, 1240. 10.3389/fchem.2020.624163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baildya N.; Khan A. A.; Ghosh N. N.; Dutta T.; Chattopadhyay A. P. Screening of potential drug from Azadirachta Indica (Neem) extracts for SARS-CoV-2: an insight from molecular docking and MD-simulation studies. J. Mol. Struct. 2021, 1227, 129390. 10.1016/j.molstruc.2020.129390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivakumar D.; Stein M. Binding of SARS-CoV Covalent Non-Covalent Inhibitors to the SARS-CoV-2 Papain-Like Protease and Ovarian Tumor Domain Deubiquitinases. Biomolecules 2021, 11, 802. 10.3390/biom11060802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma K.; Morla S.; Goyal A.; Kumar S. Computational guided drug repurposing for targeting 2’-O-ribose methyltransferase of SARS-CoV-2. Life Sci. 2020, 259, 118169. 10.1016/j.lfs.2020.118169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Lima Menezes G.; da Silva R. A. Identification of potential drugs against SARS-CoV-2 non-structural protein 1 (nsp1). J. Biomol. Struct. Dyn. 2021, 39, 5657. 10.1080/07391102.2020.1792992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tazikeh-Lemeski E.; Moradi S.; Raoufi R.; Shahlaei M.; Janlou M. A. M.; Zolghadri S. Targeting SARS-CoV-2 non-structural protein 16: a virtual drug repurposing study. J. Biomol. Struct. Dyn. 2021, 39, 4633. 10.1080/07391102.2020.1779133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang J.; Pitsillou E.; Burbury L.; Hung A.; Karagiannis T. C. In silico investigation of potential small molecule inhibitors of the SARS-CoV-2 nsp10-nsp16 methyltransferase complex. Chem. Phys. Lett. 2021, 774, 138618. 10.1016/j.cplett.2021.138618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sixto-López Y.; Martínez-Archundia M. Drug repositioning to target NSP15 protein on SARS-CoV-2 as possible COVID-19 treatment. J. Comput. Chem. 2021, 42, 897–907. 10.1002/jcc.26512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S.; Kashyap P.; Chowdhury S.; Kumar S.; Panwar A.; Kumar A. Identification of phytochemicals as potential therapeutic agents that binds to Nsp15 protein target of coronavirus (SARS-CoV-2) that are capable of inhibiting virus replication. Phytomedicine 2021, 85, 153317. 10.1016/j.phymed.2020.153317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatar G.; Ozyurt E.; Turhan K. Computational drug repurposing study of the RNA binding domain of SARS-CoV-2 nucleocapsid protein with antiviral agents. Biotechnol. Prog. 2021, 37, e3110 10.1002/btpr.3110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rampogu S.; Lee G.; Kulkarni A. M.; Kim D.; Yoon S.; Kim M. O.; Lee K. W. Computational Approaches to Discover Novel Natural Compounds for SARS-CoV-2 Therapeutics. ChemistryOpen 2021, 10, 593. 10.1002/open.202000332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Demerdash A.; Metwaly A. M.; Hassan A.; El-Aziz T. M. A.; Mohamed T.; Elkaeed E. B.; Eissa I. H.; Arafa R. K.; Stockand J. D. Comprehensive virtual screening of the antiviral potentialities of marine polycyclic guanidine alkaloids against SARS-CoV-2 (COVID-19). Biomolecules 2021, 11, 460. 10.3390/biom11030460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwarka D.; Agoni C.; Mellem J. J.; Soliman M. E.; Baijnath H. Identification of potential SARS-CoV-2 inhibitors from South African medicinal plant extracts using molecular modelling approaches. S. Afr. J. Bot. 2020, 133, 273–284. 10.1016/j.sajb.2020.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhowmik D.; Nandi R.; Jagadeesan R.; Kumar N.; Prakash A.; Kumar D. Identification of potential inhibitors against SARS-CoV-2 by targeting proteins responsible for envelope formation and virion assembly using docking based virtual screening, and pharmacokinetics approaches. Infect. Genet. Evol. 2020, 84, 104451. 10.1016/j.meegid.2020.104451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molavi Z.; Razi S.; Mirmotalebisohi S. A.; Adibi A.; Sameni M.; Karami F.; Niazi V.; Niknam Z.; Aliashrafi M.; Taheri M.; et al. Identification of FDA approved drugs against SARS-CoV-2 RNA dependent RNA polymerase (RdRp) and 3-chymotrypsin-like protease (3CLpro), drug repurposing approach. Biomed. Pharmacother. 2021, 138, 111544. 10.1016/j.biopha.2021.111544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajpoot S.; Alagumuthu M.; Baig M. S. Dual targeting of 3clpro and Plpro of Sars-Cov-2: a novel structure-based design approach to treat Covid-19. CRSB 2021, 3, 9–18. 10.1016/j.crstbi.2020.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang W. D.; Jeon S.; Kim S.; Lee S. Y. Drugs repurposed for COVID-19 by virtual screening of 6,218 drugs and cell-based assay. Proc. Natl. Acad. Sci. U.S.A. 2021, 118, e2024302118. 10.1073/pnas.2024302118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bobrovs R.; Kanepe I.; Narvaiss N.; Patetko L.; Kalnins G.; Sisovs M.; Bula A. L.; Grinberga S.; Boroduskis M.; Ramata-Stunda A.; et al. Discovery of SARS-CoV-2 Nsp14 and Nsp16 Methyltransferase Inhibitors by High-Throughput Virtual Screening. Pharmaceuticals 2021, 14, 1243. 10.3390/ph14121243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francés-Monerris A.; García-Iriepa C.; Iriepa I.; Hognon C.; Miclot T.; Barone G.; Monari A.; Marazzi M. Microscopic interactions between ivermectin and key human and viral proteins involved in SARS-CoV-2 infection. Phys. Chem. Chem. Phys. 2021, 23, 22957–22971. 10.1039/D1CP02967C. [DOI] [PubMed] [Google Scholar]
- Gajjar N. D.; Dhameliya T. M.; Shah G. B. In search of RdRp and Mpro inhibitors against SARS CoV-2: Molecular docking, molecular dynamic simulations and ADMET analysis. J. Mol. Struct. 2021, 1239, 130488. 10.1016/j.molstruc.2021.130488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasool N.; Yasmin F.; Sahai S.; Hussain W.; Inam H.; Arshad A. Biological perspective of thiazolide derivatives against Mpro and MTase of SARS-CoV-2: molecular docking, DFT and MD simulation investigations. Chem. Phys. Lett. 2021, 771, 138463. 10.1016/j.cplett.2021.138463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abouelela M. E.; Assaf H. K.; Abdelhamid R. A.; Elkhyat E. S.; Sayed A. M.; Oszako T.; Belbahri L.; Zowalaty A. E. E.; Abdelkader M. S. A. Identification of Potential SARS-CoV-2 Main Protease and Spike Protein Inhibitors from the Genus Aloe: An In Silico Study for Drug Development. Molecules 2021, 26, 1767. 10.3390/molecules26061767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day C. J.; Bailly B.; Guillon P.; Dirr L.; Jen F. E.-C.; Spillings B. L.; Mak J.; von Itzstein M.; Haselhorst T.; Jennings M. P. Multidisciplinary Approaches Identify Compounds that Bind to Human ACE2 or SARS-CoV-2 Spike Protein as Candidates to Block SARS-CoV-2–ACE2 Receptor Interactions. Mbio 2021, 12, e03681–20. 10.1128/mBio.03681-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milani M.; Donalisio M.; Bonotto R. M.; Schneider E.; Arduino I.; Boni F.; Lembo D.; Marcello A.; Mastrangelo E. Combined in silico and in vitro approaches identified the antipsychotic drug lurasidone and the antiviral drug elbasvir as SARS-CoV2 and HCoV-OC43 inhibitors. Antivir. Res. 2021, 189, 105055. 10.1016/j.antiviral.2021.105055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed S. A.; Abdelrheem D. A.; Abd El-Mageed H.; Mohamed H. S.; Rahman A. A.; Elsayed K. N.; Ahmed S. A. Destabilizing the structural integrity of COVID-19 by caulerpin and its derivatives along with some antiviral drugs: An in silico approaches for a combination therapy. Struct Chem. 2020, 31, 2391–2412. 10.1007/s11224-020-01586-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha S. K.; Prasad S. K.; Islam M. A.; Gurav S. S.; Patil R. B.; AlFaris N. A.; Aldayel T. S.; AlKehayez N. M.; Wabaidur S. M.; Shakya A. Identification of bioactive compounds from glycyrrhiza glabra as possible inhibitor of SARS-CoV-2 spike glycoprotein and non-structural protein-15: A pharmacoinformatics study. J. Biomol. Struct. Dyn. 2021, 39, 4686. 10.1080/07391102.2020.1779132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teli D. M.; Shah M. B.; Chhabria M. T. In silico Screening of Natural Compounds as Potential Inhibitors of SARS-CoV-2 Main Protease and Spike RBD: Targets for COVID-19. Front. Mol. Biosci. 2021, 7, 429. 10.3389/fmolb.2020.599079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhury A.; Das N. C.; Patra R.; Bhattacharya M.; Ghosh P.; Patra B. C.; Mukherjee S. Exploring the binding efficacy of ivermectin against the key proteins of SARS-CoV-2 pathogenesis: an in silico approach. Future Virol 2021, 16, 277–291. 10.2217/fvl-2020-0342. [DOI] [Google Scholar]
- Adem Ş.; Eyupoglu V.; Sarfraz I.; Rasul A.; Zahoor A. F.; Ali M.; Abdalla M.; Ibrahim I. M.; Elfiky A. A. Caffeic acid derivatives (CAFDs) as inhibitors of SARS-CoV-2: CAFDs-based functional foods as a potential alternative approach to combat COVID-19. Phytomedicine 2021, 85, 153310. 10.1016/j.phymed.2020.153310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shawan M. M. A. K.; Halder S. K.; Hasan M. A. Luteolin and abyssinone II as potential inhibitors of SARS-CoV-2: An in silico molecular modeling approach in battling the COVID-19 outbreak. Bull. Natl. Res. Cent 2021, 45, 1–21. 10.1186/s42269-020-00479-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y.; Tian Y.; Pan C.; Liang A.; Zhang W.; Sheng Y. Target-Based In Silico Screening for Phytoactive Compounds Targeting SARS-CoV-2. Interdiscip. Sci. Comput. Life Sci. 2022, 14, 64. 10.1007/s12539-021-00461-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mhatre S.; Naik S.; Patravale V. A molecular docking study of EGCG and theaflavin digallate with the druggable targets of SARS-CoV-2. Comput. Biol. Med. 2021, 129, 104137. 10.1016/j.compbiomed.2020.104137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elhady S. S.; Abdelhameed R. F.; Malatani R. T.; Alahdal A. M.; Bogari H. A.; Almalki A. J.; Mohammad K. A.; Ahmed S. A.; Khedr A. I.; Darwish K. M. Molecular Docking and Dynamics Simulation Study of Hyrtios erectus Isolated Scalarane Sesterterpenes as Potential SARS-CoV-2 Dual Target Inhibitors. Biology 2021, 10, 389. 10.3390/biology10050389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thirumalaisamy R.; Aroulmoji V.; Iqbal M. N.; Deepa M.; Sivasankar C.; Khan R.; Selvankumar T. Molecular insights of hyaluronic acid-hydroxychloroquine conjugate as a promising drug in targeting SARS-CoV-2 viral proteins. J. Mol. Struct. 2021, 1238, 130457. 10.1016/j.molstruc.2021.130457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta A.; Ahmad R.; Siddiqui S.; Yadav K.; Srivastava A.; Trivedi A.; Ahmad B.; Khan M. A.; Shrivastava A. K.; Singh G. K. Flavonol morin targets host ACE2, IMP-α, PARP-1 and viral proteins of SARS-CoV-2, SARS-CoV and MERS-CoV critical for infection and survival: a computational analysis. J. Biomol. Struct 2021, 1–32. 10.1080/07391102.2021.1871863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adeoye A. O.; Oso B. J.; Olaoye I. F.; Tijjani H.; Adebayo A. I. Repurposing of chloroquine and some clinically approved antiviral drugs as effective therapeutics to prevent cellular entry and replication of coronavirus. J. Biomol. Struct. Dyn. 2021, 39, 3469. 10.1080/07391102.2020.1765876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega J. T.; Serrano M. L.; Jastrzebska B. Class AG protein-coupled receptor antagonist famotidine as a therapeutic alternative against SARS-CoV2: an in silico analysis. Biomolecules 2020, 10, 954. 10.3390/biom10060954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aouidate A.; Ghaleb A.; Chtita S.; Aarjane M.; Ousaa A.; Maghat H.; Sbai A.; Choukrad M.; Bouachrine M.; Lakhlifi T. Identification of a novel dual-target scaffold for 3CLpro and RdRp proteins of SARS-CoV-2 using 3D-similarity search, molecular docking, molecular dynamics and ADMET evaluation. J. Biomol. Struct. Dyn. 2021, 39, 4522. 10.1080/07391102.2020.1779130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentile D.; Fuochi V.; Rescifina A.; Furneri P. M. New Anti SARS-CoV-2 Targets for Quinoline Derivatives Chloroquine and Hydroxychloroquine. Int. J. Mol. Sci. 2020, 21, 5856. 10.3390/ijms21165856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Leon V. N. O.; Manzano J. A. H.; Pilapil D. Y. H.; Fernandez R. A. T.; Ching J. K. A. R.; Quimque M. T. J.; Agbay J. C. M.; Notarte K. I. R.; Macabeo A. P. G. Anti-HIV reverse transcriptase plant polyphenolic natural products with in silico inhibitory properties on seven non-structural proteins vital in SARS-CoV-2 pathogenesis. J. Genet. Eng. Biotechnol. 2021, 19, 1–17. 10.1186/s43141-021-00206-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grahl M. V.; Alcará A. M.; Perin A. P. A.; Moro C. F.; Pinto É. S.; Feltes B. C.; Ghilardi I. M.; Rodrigues F. V.; Dorn M.; da Costa J. C.; et al. Evaluation of drug repositioning by molecular docking of pharmaceutical resources available in the Brazilian healthcare system against SARS-CoV-2. Inform. Med. Unlocked 2021, 23, 100539. 10.1016/j.imu.2021.100539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosseini M.; Chen W.; Xiao D.; Wang C. Computational molecular docking and virtual screening revealed promising SARS-CoV-2 drugs. Precis. Clin. Med. 2021, 4, 1–16. 10.1093/pcmedi/pbab001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coghi P.; Yang L. J.; Ng J. P.; Haynes R. K.; Memo M.; Gianoncelli A.; Wong V. K. W.; Ribaudo G. A Drug Repurposing Approach for Antimalarials Interfering with SARS-CoV-2 Spike Protein Receptor Binding Domain (RBD) and Human Angiotensin-Converting Enzyme 2 (ACE2). Pharmaceuticals 2021, 14, 954. 10.3390/ph14100954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngo S. T.; Tam N. M.; Pham M. Q.; Nguyen T. H. Benchmark of Popular Free Energy Approaches Revealing the Inhibitors Binding to SARS-CoV-2 Mpro. J. Chem. Inf Model 2021, 61, 2302–2312. 10.1021/acs.jcim.1c00159. [DOI] [PubMed] [Google Scholar]
- Tam N. M.; Nam P. C.; Quang D. T.; Tung N. T.; Vu V. V.; Ngo S. T. Binding of inhibitors to the monomeric and dimeric SARS-CoV-2 Mpro. RSC Adv. 2021, 11, 2926–2934. 10.1039/D0RA09858B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zev S.; Raz K.; Schwartz R.; Tarabeh R.; Gupta P. K.; Major D. T. Benchmarking the Ability of Common Docking Programs to Correctly Reproduce and Score Binding Modes in SARS-CoV-2 Protease Mpro. J. Chem. Inf Model 2021, 61, 2957–2966. 10.1021/acs.jcim.1c00263. [DOI] [PubMed] [Google Scholar]
- Llanos M. A.; Gantner M. E.; Rodriguez S.; Alberca L. N.; Bellera C. L.; Talevi A.; Gavernet L. Strengths and Weaknesses of Docking Simulations in the SARS-CoV-2 Era: the Main Protease (Mpro) Case Study. J. Chem. Inf Model 2021, 61, 3758–3770. 10.1021/acs.jcim.1c00404. [DOI] [PubMed] [Google Scholar]
- Fan W.; Mencius J.; Du W.; Fan H.; Zhu H.; Wei D.; Zhou M.; Quan S. Online bioinformatics teaching practice: Comparison of popular docking programs using SARS-CoV-2 spike RBD–ACE2 complex as a benchmark. Biochem Mol. Biol. Educ 2021, 49, 833–840. 10.1002/bmb.21566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta A.; Zhou H.-X. Profiling SARS-CoV-2 Main Protease (Mpro) Binding to Repurposed Drugs Using Molecular Dynamics Simulations in Classical and Neural Network-Trained Force Fields. ACS Comb. Sci. 2020, 22, 826–832. 10.1021/acscombsci.0c00140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith J. S.; Nebgen B. T.; Zubatyuk R.; Lubbers N.; Devereux C.; Barros K.; Tretiak S.; Isayev O.; Roitberg A. E. Approaching coupled cluster accuracy with a general-purpose neural network potential through transfer learning. Nat. Commun. 2019, 10, 1–8. 10.1038/s41467-019-10827-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi T.; Joshi T.; Pundir H.; Sharma P.; Mathpal S.; Chandra S. Predictive modeling by deep learning, virtual screening and molecular dynamics study of natural compounds against SARS-CoV-2 main protease. J. Biomol. Struct. Dyn. 2020, 1–19. 10.1080/07391102.2020.1802341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casalino L.; Dommer A. C.; Gaieb Z.; Barros E. P.; Sztain T.; Ahn S.-H.; Trifan A.; Brace A.; Bogetti A. T.; Clyde A.; et al. AI-driven multiscale simulations illuminate mechanisms of SARS-CoV-2 spike dynamics. Int. J. High Perform Comput. Appl. 2021, 35, 432–451. 10.1177/10943420211006452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turoňová B.; Sikora M.; Schürmann C.; Hagen W. J.; Welsch S.; Blanc F. E.; von Bülow S.; Gecht M.; Bagola K.; Hörner C.; et al. In situ structural analysis of SARS-CoV-2 spike reveals flexibility mediated by three hinges. Science 2020, 370, 203–208. 10.1126/science.abd5223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidman D.; Gehrtz P.; Filep M.; Fearon D.; Gabizon R.; Douangamath A.; Prilusky J.; Duberstein S.; Cohen G.; Owen C. D.; et al. An automatic pipeline for the design of irreversible derivatives identifies a potent SARS-CoV-2 Mpro inhibitor. Cell Chem. Biol. 2021, 28, 1795–1806. 10.1016/j.chembiol.2021.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwivedi R.; Samanta P.; Sharma P.; Zhang F.; Mishra S. K.; Kucheryavy P.; Kim S. B.; Aderibigbe A. O.; Linhardt R. J.; Tandon R.; et al. Structural and kinetic analyses of holothurian sulfated glycans suggest potential treatment for SARS-CoV-2 infection. J. Biol. Chem. 2021, 297, 297. 10.1016/j.jbc.2021.101207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahun M.; Jukić M.; Oblak D.; Kranjc L.; Bajc G.; Butala M.; Bozovičar K.; Bratkovič T.; Podlipnik Č.; Ulrih N. P. Inhibition of the SARS-CoV-2 3CLpro main protease by plant polyphenols. Food Chem. 2022, 373, 131594. 10.1016/j.foodchem.2021.131594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Power H.; Wu J.; Turville S.; Aggarwal A.; Valtchev P.; Schindeler A.; Dehghani F. Virtual screening and in vitro validation of natural compound inhibitors against SARS-CoV-2 spike protein. Bioorg. Chem. 2022, 119, 105574. 10.1016/j.bioorg.2021.105574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Zhang Y.; Pang H.; Li S. J. Heparin interacts with the main protease of SARS-CoV-2 and inhibits its activity. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2022, 267, 120595. 10.1016/j.saa.2021.120595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur B.; Maity H. S.; Rakshit M.; Srivastav P. P.; Nag A. Cryo-Ground Mango Kernel Powder: Characterization, LC-MS/MS Profiling, Purification of Antioxidant-Rich Gallic Acid, and Molecular Docking Study of Its Major Polyphenols as Potential Inhibitors against SARS-CoV-2 Mpro. Food Sci. Technol. 2021, 1, 1776–1786. 10.1021/acsfoodscitech.1c00179. [DOI] [PubMed] [Google Scholar]
- Elli S.; Bojkova D.; Bechtel M.; Vial T.; Boltz D.; Muzzio M.; Peng X.; Sala F.; Cosentino C.; Goy A.; et al. Enisamium inhibits sars-cov-2 rna synthesis. Biomedicines 2021, 9, 1254. 10.3390/biomedicines9091254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toft-Bertelsen T. L.; Jeppesen M. G.; Tzortzini E.; Xue K.; Giller K.; Becker S.; Mujezinovic A.; Bentzen B. H.; B. Andreas L.; Kolocouris A.; et al. Amantadine inhibits known and novel ion channels encoded by SARS-CoV-2 in vitro. Commun. Biol. 2021, 4, 1–10. 10.1038/s42003-021-02866-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haribabu J.; Garisetti V.; Malekshah R. E.; Srividya S.; Gayathri D.; Bhuvanesh N.; Mangalaraja R. V.; Echeverria C.; Karvembu R. Design and synthesis of heterocyclic azole based bioactive compounds: Molecular structures, quantum simulation, and mechanistic studies through docking as multi-target inhibitors of SARS-CoV-2 and cytotoxicity. J. Mol. Struct. 2022, 1250, 131782. 10.1016/j.molstruc.2021.131782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.; Zhang Y.; Zeng D.; Zhang B.; Ye X.; Zeng Z.; Zhang X.-k.; Wang Z.; Zhou H. Merbromin is a mixed-type inhibitor of 3-chyomotrypsin like protease of SARS-CoV-2. Biochem. Biophys. Res. Commun. 2022, 591, 118–123. 10.1016/j.bbrc.2021.12.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Defant A.; Dosi F.; Innocenti N.; Mancini I. Synthesis of Nucleoside-like Molecules from a Pyrolysis Product of Cellulose and Their Computational Prediction as Potential SARS-CoV-2 RNA-Dependent RNA Polymerase Inhibitors. Int. J. Mol. Sci. 2022, 23, 518. 10.3390/ijms23010518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Li P.; Rajpoot S.; Saqib U.; Yu P.; Li Y.; Li Y.; Ma Z.; Baig M. S.; Pan Q. Comparative assessment of favipiravir and remdesivir against human coronavirus NL63 in molecular docking and cell culture models. Sci. Rep. 2021, 11, 1–13. 10.1038/s41598-021-02972-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iype E.; Pillai U J.; Kumar I.; Gaastra-Nedea S. V.; Subramanian R.; Saha R. N.; Dutta M. In silico and in vitro assays reveal potential inhibitors against 3CL pro main protease of SARS-CoV-2. J. Biomol. Struct 2021, 1–12. 10.1080/07391102.2021.1977181. [DOI] [PubMed] [Google Scholar]
- Speciale A.; Muscarà C.; Molonia M. S.; Cimino F.; Saija A.; Giofrè S. V. Silibinin as potential tool against SARS-Cov-2: In silico spike receptor-binding domain and main protease molecular docking analysis, and in vitro endothelial protective effects. Phytother Res. 2021, 35, 4616–4625. 10.1002/ptr.7107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye M.; Luo G.; Ye D.; She M.; Sun N.; Lu Y.-J.; Zheng J. Network pharmacology, molecular docking integrated surface plasmon resonance technology reveals the mechanism of Toujie Quwen Granules against coronavirus disease 2019 pneumonia. Phytomedicine 2021, 85, 153401. 10.1016/j.phymed.2020.153401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J.; Lin X.; Xing N.; Zhang Z.; Zhang H.; Wu H.; Xue W. Structure-Based Discovery of Novel Nonpeptide Inhibitors Targeting SARS-CoV-2 Mpro. J. Chem. Inf Model 2021, 61, 3917–3926. 10.1021/acs.jcim.1c00355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pendyala B.; Patras A.; Dash C. Phycobilins as Potent Food Bioactive Broad-Spectrum Inhibitors Against Proteases of SARS-CoV-2 and Other Coronaviruses: A Preliminary Study. Front. Microbiol. 2021, 12, 1399. 10.3389/fmicb.2021.645713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawrot-Hadzik I.; Zmudzinski M.; Matkowski A.; Preissner R.; Kesik-Brodacka M.; Hadzik J.; Drag M.; Abel R. Reynoutria Rhizomes as a Natural Source of SARS-CoV-2 Mpro Inhibitors–Molecular Docking and In Vitro Study. Pharmaceuticals 2021, 14, 742. 10.3390/ph14080742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao T.; Cui M.; Zheng C.; Wang M.; Sun R.; Gao D.; Bao J.; Ren S.; Yang B.; Lin J.; et al. Myricetin Inhibits SARS-CoV-2 Viral Replication by Targeting Mpro and Ameliorates Pulmonary Inflammation. Front. Pharmacol. 2021, 12, 1012. 10.3389/fphar.2021.669642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qayyum S.; Mohammad T.; Slominski R. M.; Hassan M. I.; Tuckey R. C.; Raman C.; Slominski A. T. Vitamin D and lumisterol novel metabolites can inhibit SARS-CoV-2 replication machinery enzymes. Am. J. Physiol. Endocrinol. Metab. 2021, 321, E246–E251. 10.1152/ajpendo.00174.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Rabia M. W.; Alhakamy N. A.; Ahmed O. A.; Eljaaly K.; Alaofi A.; Mostafa A.; Asfour H. Z.; Aldarmahi A. A.; Darwish K. M.; Ibrahim T. S.; et al. Repurposing of sitagliptin-melittin optimized nanoformula against sars-cov-2: Antiviral screening and molecular docking studies. Pharmaceutics 2021, 13, 307. 10.3390/pharmaceutics13030307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loschwitz J.; Jäckering A.; Keutmann M.; Olagunju M.; Eberle R. J.; Coronado M. A.; Olubiyi O. O.; Strodel B. Novel inhibitors of the main protease enzyme of SARS-CoV-2 identified via molecular dynamics simulation-guided in vitro assay. Bioorg. Chem. 2021, 111, 104862. 10.1016/j.bioorg.2021.104862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amendola G.; Ettari R.; Previti S.; Di Chio C.; Messere A.; Di Maro S.; Hammerschmidt S. J.; Zimmer C.; Zimmermann R. A.; Schirmeister T.; et al. Lead discovery of SARS-CoV-2 main protease inhibitors through covalent docking-based virtual screening. J. Chem. Inf Model 2021, 61, 2062–2073. 10.1021/acs.jcim.1c00184. [DOI] [PubMed] [Google Scholar]
- Guo S.; Xie H.; Lei Y.; Liu B.; Zhang L.; Xu Y.; Zuo Z. Discovery of novel inhibitors against main protease (Mpro) of SARS-CoV-2 via virtual screening and biochemical evaluation. Bioorg. Chem. 2021, 110, 104767. 10.1016/j.bioorg.2021.104767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee R.; Bhattacharya A.; Bojkova D.; Mehdipour A. R.; Shin D.; Khan K. S.; Cheung H. H.-Y.; Wong K.-B.; Ng W.-L.; Cinatl J.; et al. Famotidine inhibits toll-like receptor 3-mediated inflammatory signaling in SARS-CoV-2 infection. J. Biol. Chem. 2021, 297, 100925. 10.1016/j.jbc.2021.100925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elshamy A. I.; Mohamed T. A.; Ibrahim M. A.; Atia M. A.; Yoneyama T.; Umeyama A.; Hegazy M.-E. F. Two novel oxetane containing lignans and a new megastigmane from Paronychia arabica and in silico analysis of them as prospective SARS-CoV-2 inhibitors. RSC Adv. 2021, 11, 20151–20163. 10.1039/D1RA02486H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuller M.; Correy G. J.; Gahbauer S.; Fearon D.; Wu T.; Díaz R. E.; Young I. D.; Martins L. C.; Smith D. H.; Schulze-Gahmen U.; et al. Fragment binding to the Nsp3 macrodomain of SARS-CoV-2 identified through crystallographic screening and computational docking. Sci. Adv. 2021, 7, eabf8711 10.1126/sciadv.abf8711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitsillou E.; Liang J.; Ververis K.; Hung A.; Karagiannis T. C. Interaction of small molecules with the SARS-CoV-2 papain-like protease: In silico studies and in vitro validation of protease activity inhibition using an enzymatic inhibition assay. J. Mol. Graph. Model. 2021, 104, 107851. 10.1016/j.jmgm.2021.107851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan H.; Liu J.; Shen J.; Dai J.; Xu G.; Lu K.; Han C.; Wang Y.; Xu X.; Tong Y.; et al. Development of potent and selective inhibitors targeting the papain-like protease of SARS-CoV-2. Cell Chem. Biol. 2021, 28, 855–865. 10.1016/j.chembiol.2021.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C.; Sacco M. D.; Xia Z.; Lambrinidis G.; Townsend J. A.; Hu Y.; Meng X.; Szeto T.; Ba M.; Zhang X.; et al. Discovery of SARS-CoV-2 papain-like protease inhibitors through a combination of high-throughput screening and a FlipGFP-based reporter assay. ACS Cent. Sci. 2021, 7, 1245–1260. 10.1021/acscentsci.1c00519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S. K.; Mahanta S.; Tanti B.; Tag H.; Hui P. K. Identification of phytocompounds from Houttuynia cordata Thunb. as potential inhibitors for SARS-CoV-2 replication proteins through GC–MS/LC–MS characterization, molecular docking and molecular dynamics simulation. Mol. Divers. 2022, 26, 365. 10.1007/s11030-021-10226-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto K. Z.; Yasuo N.; Sekijima M. Screening for Inhibitors of Main Protease in SARS-CoV-2: In Silico and In Vitro Approach Avoiding Peptidyl Secondary Amides. J. Chem. Inf. Model. 2022, 62, 350. 10.1021/acs.jcim.1c01087. [DOI] [PubMed] [Google Scholar]
- Khan A.; Heng W.; Wang Y.; Qiu J.; Wei X.; Peng S.; Saleem S.; Khan M.; Ali S. S.; Wei D.-Q. In silico and in vitro evaluation of kaempferol as a potential inhibitor of the SARS-CoV-2 main protease (3CLpro). Phytother Res. 2021, 35, 2841–2845. 10.1002/ptr.6998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai T.; Zhang F.; Haider S.; Kraut D.; Huang Z. An Integrated Computational and Experimental Approach to Identifying Inhibitors for SARS-CoV-2 3CL Protease. Front. Mol. Biosci. 2021, 8, 267. 10.3389/fmolb.2021.661424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y.; Chen K.; Pan J.; Lei Y.; Zhang D.; Fang L.; Tang J.; Chen X.; Ma Y.; Zheng Y. Repurposing clinically approved drugs for COVID-19 treatment targeting SARS-CoV-2 papain-like protease. Int. J. Biol. 2021, 188, 137. 10.1016/j.ijbiomac.2021.07.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skjaerven L.; Martinez A.; Reuter N. Principal component and normal mode analysis of proteins; a quantitative comparison using the GroEL subunit. Proteins: Struct. Funct. Genet. 2011, 79, 232–243. 10.1002/prot.22875. [DOI] [PubMed] [Google Scholar]
- Amadei A.; Linssen A. B.; Berendsen H. J. Essential dynamics of proteins. Proteins: Struct. Funct. Genet. 1993, 17, 412–425. 10.1002/prot.340170408. [DOI] [PubMed] [Google Scholar]
- Dehury B.; Raina V.; Misra N.; Suar M. Effect of mutation on structure, function and dynamics of receptor binding domain of human SARS-CoV-2 with host cell receptor ACE2: a molecular dynamics simulations study. J. Biomol. Struct. Dyn. 2021, 39, 7231. 10.1080/07391102.2020.1802348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar D T.; Shaikh N.; Kumar S U.; Doss C G. P.; Zayed H.; et al. Structure-based virtual screening to identify novel potential compound as an alternative to remdesivir to overcome the RdRp protein mutations in SARS-CoV-2. Front. Mol. Biosci. 2021, 8, 213. 10.3389/fmolb.2021.645216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varadharajan V.; Arumugam G. S.; Shanmugam S. Isatin-based virtual high throughput screening, molecular docking, DFT, QM/MM, MD and MM-PBSA study of novel inhibitors of SARS-CoV-2 main protease. J. Biomol. Struct 2021, 1–16. 10.1080/07391102.2021.1904003. [DOI] [PubMed] [Google Scholar]
- Awoonor-Williams E.; Abu-Saleh A. A.-A. A. Covalent and non-covalent binding free energy calculations for peptidomimetic inhibitors of SARS-CoV-2 main protease. Phys. Chem. Chem. Phys. 2021, 23, 6746–6757. 10.1039/D1CP00266J. [DOI] [PubMed] [Google Scholar]
- Bharadwaj S.; Dubey A.; Yadava U.; Mishra S. K.; Kang S. G.; Dwivedi V. D. Exploration of natural compounds with anti-SARS-CoV-2 activity via inhibition of SARS-CoV-2 Mpro. Brief. Bioinformatics 2021, 22, 1361–1377. 10.1093/bib/bbaa382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarezade V.; Rezaei H.; Shakerinezhad G.; Safavi A.; Nazeri Z.; Veisi A.; Azadbakht O.; Hatami M.; Sabaghan M.; Shajirat Z. The identification of novel inhibitors of human angiotensin-converting enzyme 2 and main protease of Sars-Cov-2: A combination of in silico methods for treatment of COVID-19. J. Mol. Struct. 2021, 1237, 130409. 10.1016/j.molstruc.2021.130409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baig M. H.; Sharma T.; Ahmad I.; Abohashrh M.; Alam M. M.; Dong J.-J. Is PF-00835231 a Pan-SARS-CoV-2 Mpro Inhibitor? A Comparative Study. Molecules 2021, 26, 1678. 10.3390/molecules26061678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhammad I.; Rahman N.; Niaz S.; Basharat Z.; Rastrelli L.; Jayanthi S.; Efferth T.; Khan H.; et al. Screening of potent phytochemical inhibitors against SARS-CoV-2 protease and its two Asian mutants. Comput. Biol. Med. 2021, 133, 104362. 10.1016/j.compbiomed.2021.104362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan A.; Zia T.; Suleman M.; Khan T.; Ali S. S.; Abbasi A. A.; Mohammad A.; Wei D.-Q. Higher infectivity of the SARS-CoV-2 new variants is associated with K417N/T, E484K, and N501Y mutants: An insight from structural data. J. Cell. Physiol. 2021, 236, 7045–7057. 10.1002/jcp.30367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aljindan R. Y.; Al-Subaie A. M.; Al-Ohali A. I.; Kamaraj B.; et al. Investigation of nonsynonymous mutations in the spike protein of SARS-CoV-2 and its interaction with the ACE2 receptor by molecular docking and MM/GBSA approach. Comput. Biol. Med. 2021, 135, 104654. 10.1016/j.compbiomed.2021.104654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakhmola S.; Indari O.; Kashyap D.; Varshney N.; Das A.; Manivannan E.; Jha H. C. Mutational analysis of structural proteins of SARS-CoV-2. Heliyon 2021, 7, e06572 10.1016/j.heliyon.2021.e06572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou J.; Zhou Z.; Dai R.; Zhang J.; Zhao S.; Wu X.; Lan W.; Ren Y.; Cui L.; Lan Q.; et al. V367F mutation in SARS-CoV-2 spike RBD emerging during the early transmission phase enhances viral infectivity through increased human ACE2 receptor binding affinity. J. Virol. 2021, 95, e00617–21. 10.1128/JVI.00617-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahhosseini N.; Babuadze G. G.; Wong G.; Kobinger G. P. Mutation Signatures and In Silico Docking of Novel SARS-CoV-2 Variants of Concern. Microorganisms 2021, 9, 926. 10.3390/microorganisms9050926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega J. T.; Pujol F. H.; Jastrzebska B.; Rangel H. R. Mutations in the SARS-CoV-2 spike protein modulate the virus affinity to the human ACE2 receptor, an in silico analysis. EXCLI J. 2021, 20, 585–600. 10.17179/excli2021-3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao B.; Olvera de la Cruz M. Enhanced binding of SARS-CoV-2 spike protein to receptor by distal polybasic cleavage sites. ACS Nano 2020, 14, 10616–10623. 10.1021/acsnano.0c04798. [DOI] [PubMed] [Google Scholar]
- Zou J.; Yin J.; Fang L.; Yang M.; Wang T.; Wu W.; Bellucci M.; Zhang P. Computational Prediction of Mutational Effects on the SARS-CoV-2 Binding by Relative Free Energy Calculations. J. Chem. Inf Model 2020, 60, 5794–5802. 10.1021/acs.jcim.0c00679. [DOI] [PubMed] [Google Scholar]
- Hadi-Alijanvand H.; Rouhani M. Studying the Effects of ACE2Mutations on the Stability, Dynamics, and Dissociation Process of SARS-CoV-2 S1/hACE2 Complexes. J. Proteome Res. 2020, 19, 4609–4623. 10.1021/acs.jproteome.0c00348. [DOI] [PubMed] [Google Scholar]
- Mhatre S.; Gurav N.; Shah M.; Patravale V. Entry-inhibitory role of catechins against SARS-CoV-2 and its UK variant. Comput. Biol. Med. 2021, 135, 104560. 10.1016/j.compbiomed.2021.104560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sixto-López Y.; Correa-Basurto J.; Bello M.; Landeros-Rivera B.; Garzón-Tiznado J. A.; Montaño S. Structural insights into SARS-CoV-2 spike protein and its natural mutants found in Mexican population. Sci. Rep. 2021, 11, 1–16. 10.1038/s41598-021-84053-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L.; Peng C.; Yang Y.; Shi Y.; Zhou L.; Xu Z.; Zhu W. Exploring the immune evasion of SARS-CoV-2 variant harboring E484K by molecular dynamics simulations. Brief. Bioinformatics 2022, 23, bbab383. 10.1093/bib/bbab383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hossain M. U.; Bhattacharjee A.; Emon M. T. H.; Chowdhury Z. M.; Ahammad I.; Mosaib M. G.; Moniruzzaman M.; Rahman M. H.; Islam M. N.; Ahmed I.; et al. Novel mutations in NSP-1 and PLPro of SARS-CoV-2 NIB-1 genome mount for effective therapeutics. J. Genet. Eng. Biotechnol. 2021, 19, 1–10. 10.1186/s43141-021-00152-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S.; Tian C.; Liu P.; Guo D.; Zheng W.; Huang X.; Zhang Y.; Liu L. Effects of SARS-CoV-2 mutations on protein structures and intraviral protein–protein interactions. J. Med. Virol 2021, 93, 2132–2140. 10.1002/jmv.26597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Guilloux V.; Schmidtke P.; Tuffery P. Fpocket: an open source platform for ligand pocket detection. BMC Bioinform. 2009, 10, 1–11. 10.1186/1471-2105-10-168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao R.; Cang Z.; Tong Y.; Wei G.-W. Protein pocket detection via convex hull surface evolution and associated Reeb graph. Bioinformatics 2018, 34, i830–i837. 10.1093/bioinformatics/bty598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korb O.; Stutzle T.; Exner T. E. Empirical scoring functions for advanced protein- ligand docking with PLANTS. J. Chem. Inf Model 2009, 49, 84–96. 10.1021/ci800298z. [DOI] [PubMed] [Google Scholar]
- Gervasoni S.; Vistoli G.; Talarico C.; Manelfi C.; Beccari A. R.; Studer G.; Tauriello G.; Waterhouse A. M.; Schwede T.; Pedretti A. A Comprehensive Mapping of the Druggable Cavities within the SARS-CoV-2 Therapeutically Relevant Proteins by Combining Pocket and Docking Searches as Implemented in Pockets 2.0. Int. J. Mol. Sci. 2020, 21, 5152. 10.3390/ijms21145152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman M. I.; Porter J. R.; Ward M. D.; Singh S.; Vithani N.; Meller A.; Mallimadugula U. L.; Kuhn C. E.; Borowsky J. H.; Wiewiora R. P.; et al. SARS-CoV-2 simulations go exascale to predict dramatic spike opening and cryptic pockets across the proteome. Nat. Chem. 2021, 13, 651–659. 10.1038/s41557-021-00707-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vithani N.; Ward M. D.; Zimmerman M. I.; Novak B.; Borowsky J. H.; Singh S.; Bowman G. R. SARS-CoV-2 Nsp16 activation mechanism and a cryptic pocket with pan-coronavirus antiviral potential. Biophys. J. 2021, 120, 2880–2889. 10.1016/j.bpj.2021.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manfredonia I.; Nithin C.; Ponce-Salvatierra A.; Ghosh P.; Wirecki T. K.; Marinus T.; Ogando N. S.; Snijder E. J.; van Hemert M. J.; Bujnicki J. M.; et al. Genome-wide mapping of SARS-CoV-2 RNA structures identifies therapeutically-relevant elements. Nucleic Acids Res. 2020, 48, 12436–12452. 10.1093/nar/gkaa1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey P.; Prasad K.; Prakash A.; Kumar V. Insights into the biased activity of dextromethorphan and haloperidol towards SARS-CoV-2 NSP6: in silico binding mechanistic analysis. J. Mol. Med. 2020, 98, 1659–1673. 10.1007/s00109-020-01980-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slavin M.; Zamel J.; Zohar K.; Eliyahu T.; Braitbard M.; Brielle E.; Baraz L.; Stolovich-Rain M.; Friedman A.; Wolf D. G.; et al. Targeted in situ cross-linking mass spectrometry and integrative modeling reveal the architectures of three proteins from SARS-CoV-2. Proc. Natl. Acad. Sci. U.S.A. 2021, 118, 118. 10.1073/pnas.2103554118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estrada E. COVID-19 and SARS-CoV-2. Modeling the present, looking at the future. Phys. Rep. 2020, 869, 1–51. 10.1016/j.physrep.2020.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter R.; Montanari F.; Steffen A.; Briem H.; Noé F.; Clevert D.-A. Efficient multi-objective molecular optimization in a continuous latent space. Chem. Sci. 2019, 10, 8016–8024. 10.1039/C9SC01928F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed F. A network-based analysis reveals the mechanism underlying vitamin D in suppressing cytokine storm and virus in SARS-CoV-2 infection. Front. immunol 2020, 11, 3084. 10.3389/fimmu.2020.590459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artesi M.; Bontems S.; Göbbels P.; Franckh M.; Maes P.; Boreux R.; Meex C.; Melin P.; Hayette M.-P.; Bours V.; et al. A recurrent mutation at position 26340 of SARS-CoV-2 is associated with failure of the E gene quantitative reverse transcription-PCR utilized in a commercial dual-target diagnostic assay. J. Clin. Microbiol. 2020, 58, e01598–20. 10.1128/JCM.01598-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjuán R.; Domingo-Calap P. Mechanisms of viral mutation. Cell. Mol. Life Sci. 2016, 73, 4433–4448. 10.1007/s00018-016-2299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribble J.; Stevens L. J.; Agostini M. L.; Anderson-Daniels J.; Chappell J. D.; Lu X.; Pruijssers A. J.; Routh A. L.; Denison M. R. The coronavirus proofreading exoribonuclease mediates extensive viral recombination. PLoS Pathog 2021, 17, e1009226 10.1371/journal.ppat.1009226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr T. N.; Greaney A. J.; Hilton S. K.; Ellis D.; Crawford K. H.; Dingens A. S.; Navarro M. J.; Bowen J. E.; Tortorici M. A.; Walls A. C.; et al. Deep mutational scanning of SARS-CoV-2 receptor binding domain reveals constraints on folding and ACE2 binding. Cell 2020, 182, 1295–1310. 10.1016/j.cell.2020.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linsky T. W.; Vergara R.; Codina N.; Nelson J. W.; Walker M. J.; Su W.; Barnes C. O.; Hsiang T.-Y.; Esser-Nobis K.; Yu K.; et al. De novo design of potent and resilient hACE2 decoys to neutralize SARS-CoV-2. Science 2020, 370, 1208–1214. 10.1126/science.abe0075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C.; Boorla V. S.; Banerjee D.; Chowdhury R.; Cavener V. S.; Nissly R. H.; Gontu A.; Boyle N. R.; Vandegrift K.; Nair M. S.; et al. Computational prediction of the effect of amino acid changes on the binding affinity between SARS-CoV-2 spike RBD and human ACE2. Proc. Natl. Acad. Sci. U.S.A. 2021, 118, 118. 10.1073/pnas.2106480118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurini E.; Marson D.; Aulic S.; Fermeglia A.; Pricl S. Computational mutagenesis at the SARS-CoV-2 spike protein/angiotensin-converting enzyme 2 binding interface: comparison with experimental evidence. ACS Nano 2021, 15, 6929–6948. 10.1021/acsnano.0c10833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S.; Thambiraja T. S.; Karuppanan K.; Subramaniam G. Omicron and Delta variant of SARS-CoV-2: a comparative computational study of spike protein. J. Med. Virol. 2022, 94, 1641–1649. 10.1002/jmv.27526. [DOI] [PubMed] [Google Scholar]
- Chen J.; Wang R.; Gilby N. B.; Wei G.-W. Omicron variant (B. 1.1. 529): Infectivity, vaccine breakthrough, and antibody resistance. J. Chem. Inf Model 2022, 62, 412. 10.1021/acs.jcim.1c01451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R.; Chen J.; Wei G.-W. Mechanisms of SARS-CoV-2 evolution revealing vaccine-resistant mutations in Europe and America. J. Phys. Chem. Lett. 2021, 12, 11850–11857. 10.1021/acs.jpclett.1c03380. [DOI] [PMC free article] [PubMed] [Google Scholar]