

Abstract
Single-molecule localization microscopy (SMLM) describes a family of powerful imaging techniques that dramatically improve spatial resolution over standard, diffraction-limited microscopy techniques and can image biological structures at the molecular scale. In SMLM, individual fluorescent molecules are computationally localized from diffraction-limited image sequences and the localizations are used to generate a super-resolution image or a time course of super-resolution images, or to define molecular trajectories. In this Primer, we introduce the basic principles of SMLM techniques before describing the main experimental considerations when performing SMLM, including fluorescent labelling, sample preparation, hardware requirements and image acquisition in fixed and live cells. We then explain how low-resolution image sequences are computationally processed to reconstruct super-resolution images and/or extract quantitative information, and highlight a selection of biological discoveries enabled by SMLM and closely related methods. We discuss some of the main limitations and potential artefacts of SMLM, as well as ways to alleviate them. Finally, we present an outlook on advanced techniques and promising new developments in the fast-evolving field of SMLM. We hope that this Primer will be a useful reference for both newcomers and practitioners of SMLM.
The spatial resolution of standard optical microscopy techniques is limited to roughly half the wavelength of light. As a result of diffraction1, the image of an arbitrarily small source of light imaged using a lens-based microscope is not a point but a point spread function (PSF), usually an Airy pattern, with a central peak approximately ~200–300 nm in width (FIG. 1a), resulting in a blurring of structures below this spatial scale. This diffraction limit restricts the ability of optical microscopy techniques to resolve the subcellular organization of individual molecules or molecular complexes, which are smaller than this limit; for example, the structure of a nuclear pore complex, which is made up of hundreds of individual proteins, with a diameter of only ~120 nm, remains obscured by conventional microscopy (FIG. 1b).
Fig. 1 |. Principle of single-molecule fluorescence microscopy.

a | A single fluorescent molecule (green dot) imaged through a microscope appears on the camera as a fuzzy spot ~200 nm wide known as the point spread function (PSF), which extends over multiple pixels. b | PSFs from simultaneously emitting molecules overlap if they are separated by a distance smaller than the PSF, blurring the structure. c | x and y coordinates of a single molecule (xm, ym) can be computed with high precision because subpixel displacements, here by 0.5 pixels in x and y, lead to predictable changes in pixel values, shown by the greyscale image (bottom) and corresponding 2D histogram (centre) (simulated data). The mesh surface (top) shows a Gaussian model of the PSF centred on (xm, ym). d | Higher photon counts (N) give a better signal to noise ratio and allow more precise localizations. Scatter plots show photon impacts on camera pixels. Pixel values in images on the right are photon counts. e | Single-molecule fluorescence microscopy (SMLM) usually exploits the fact that fluorophores stochastically switch between an active (‘ON’) state and one or more inactive (‘OFF’) states. f | An experimental, diffraction-limited image of nuclear pores, with all fluorophores ON. g | A sequence of diffraction-limited images of the same area as part f, where only few molecules are ON simultaneously. h,i | In each frame, single molecules are computationally detected (part h) and localized (part i). j | SMLM results in a localization table, where each row represents a distinct localization event and columns indicate x, y coordinates and additional information such as frame number and N. There are usually multiple localizations per frame, and the same molecule can be localized in multiple frames. k,l | Accumulated localizations can be visualized as a scatter plot (part k) or a 2D histogram (part l), with subpixel-sized bins. Raw image pixels are shown by the dashed grid, and bins are shown as a 10 × 10 grid inside a single pixel in part k). This ‘super-resolution’ image reveals the ring-like structure of nuclear pores. NPC, nuclear pore complex.
Diffraction.
The bending of light waves at the edges of an obstacle such as an aperture.
Point spread function.
(PSF). The image of an infinitesimally small light source through the optical system.
Airy pattern.
A pattern of light featuring a central bright disc surrounded by increasingly dimmer concentric rings formed by diffraction from a circular aperture.
Microscopy methods have emerged over the past two decades that can overcome the diffraction limit and enable the imaging of biological structures such as nuclear pores, viruses, chromatin complexes and cytoskeletal filaments at resolutions close to the molecular scale2. The most well known of these super-resolution microscopy methods fall into three main categories: stimulated emission depletion3, structured illumination microscopy4,5 and single-molecule localization microscopy (SMLM)6–9, which is the focus of this Primer. SMLM methods usually employ conventional wide-field excitation and achieve super-resolution by localizing individual molecules6–15. They have become broadly adopted in the life sciences owing to their high spatial resolution — typically ~20–50 nm or better — and relative ease of implementation, although each super-resolution method has its unique advantages and limitations and is optimally suited for different applications (discussed elsewhere2).
SMLM is fundamentally based on the fact that the spatial coordinates of single fluorescent molecules (also called fluorophores, or emitters) can be determined with high precision if their PSFs do not overlap. Subpixel shifts in the coordinates of a fluorophore lead to predictable changes in pixel intensities that can be used to compute its precise location (FIG. 1c). The localization precision reflects the scatter of localizations that would be obtained if a molecule was imaged and localized many times, and is fundamentally limited by the signal to noise ratio (SNR) and not by the wavelength of light or the pixel size (FIG. 1d). To avoid overlaps between the PSFs of individual molecules, fluorescent emissions of distinct molecules are separated in time; the most common approach to obtain this temporal separation exploits the phenomenon of photoswitching, where fluorescent molecules can switch between an active ‘ON’ or ‘bright’ state, where they emit fluorescent light when excited, and one or more inactive ‘OFF’ or ‘dark’ state in which they do not fluoresce (FIG. 1e).
Photoswitching for a particular molecule is a stochastic event; however, switching probabilities can be modulated using laser irradiation or by controlling the chemical environment, among other methods. Under suitable conditions, only a small number of molecules will be ON and, hence, appear as spatially isolated, non-overlapping PSFs. The OFF/ON/OFF switching leads to fluorophore ‘blinking’. Many thousands of wide-field image frames of the same field of view (FOV) are then acquired sequentially so that many — ideally all — fluorescent molecules are ON in at least one frame of the sequence (FIG. 1f,g). Images are then processed computationally to detect all ON molecules (FIG. 1h) and determine their coordinates (FIG. 1i). Finally, all of these localizations are accumulated to assemble a single image (FIG. 1j–l). The resolution of this image is determined by the accuracy and precision with which individual molecules are localized and by the distance between localized fluorophores, which itself depends on the density of molecules and the percentage of molecules that are localized (BOX 1).
Box 1 |. Resolution of single-molecule localization microscopy images.
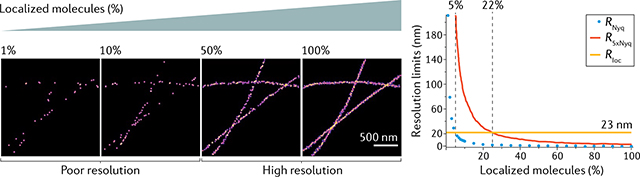
Determining the resolution of a single-molecule localization microscopy (SMLM) image is not straightforward. The resolution, R, cannot be better than Rloc = 2.3 σloc, where σloc is the localization precision (R ≥ Rloc). One approach to estimate σloc, and thus Rloc, is to calculate the Cramér–Rao lower bound (CRLB) (see BOX 4), although this estimate is generally too optimistic. More realistic estimates can be obtained from the standard deviations of coordinates in small localization clusters likely originating from single molecules. Both estimates ignore potential localization biases or linkage errors. A common alternative is to calculate the full-width half-maximum of the dispersion of coordinates across thin structures such as cytoskeletal filaments. This provides a conservative upper limit to 2.3 σloc as the full-width half-maximum accounts for precision and the width of the labelled structure, including linkage errors.
A critical and often overlooked requirement for achieving a resolution of R = Rloc is good sampling; specifically, having a sufficiently high percentage of localized molecules. An early study introduced a Nyquist sampling criterion to calculate the sampling limit to resolution (RNyq) based on the average nearest-neighbour distances between localizations34. Reanalysis suggested that fivefold higher sampling (R5xNyq) is actually required to allow a given resolution R89. In the simulated images (see the figure), where σloc = 10 nm, this criterion implies that 22% of molecules must be localized to achieve a resolution of R = Rloc = 23 nm.
A Fourier ring correlation criterion265 — which measures the correlation between subsets of localizations — simultaneously accounts for localization precision and sampling, but requires a user-defined threshold and can hide spatial variations in resolution. More recently, an alternative, threshold-free approach based on phase correlation has been proposed266.
Although useful, such measures are insufficient to rigorously determine the resolution. Compelling evidence that an image has resolution R or better is to clearly distinguish distinct structures at distances ≤R. In biological samples, this is done most convincingly on complexes where molecules are located at known distances from each other such as nuclear pores, which have become a gold standard to evaluate resolution113.
Sequentially imaging random subsets of fluorophores and computing their positions is common to most forms of SMLM, although there are many variations based on the choice of fluorophore and how these are induced to switch between ON and OFF states. Fluorescence photoactivated localization microscopy (PALM)6,7,16 was initially demonstrated using fluorescent proteins that can be activated by UV illumination, whereas stochastic optical reconstruction microscopy (STORM) and closely related methods8,9,17 use synthetic fluorophores (dyes) that can photoswitch in the presence of suitable buffers. Point accumulation in nanoscale topography10 (PAINT) is an SMLM approach that does not require photoswitching but, instead, relies on fluorophores that switch between free diffusion and immobilization by binding to a target. The most prominent variant of PAINT is DNA-PAINT18, where transient immobilization is achieved by hybridization of DNA strands. Further, similar experimental approaches to SMLM can be used to track large numbers of single molecules in live cells in an approach known as single particle tracking PALM (spt-PALM)19.
Technical choices, including the choice of fluorophore and the optical system, will impact the spatial and temporal resolution and the ability to perform imaging in two or three dimensions, in one or multiple colours, or in fixed or live cells. Other parameters that can be adjusted are labelling strategies, sample preparation workflows (for example, whether and how samples are chemically fixed and what buffer is used to promote photoswitching), the method for acquiring imaging sequences, the laser power and the use of appropriate software to computationally detect, localize and, possibly, track single molecules with high fidelity. SMLM can suffer from numerous artefacts such as sample drift or overlapping PSFs that must be addressed to ensure high-quality images. This Primer aims to provide an introduction to all main facets of SMLM and some closely related approaches. The Experimentation section discusses experimental strategies including fluorophores, labelling, sample preparation, optics and image acquisition. The Results section discusses how to computationally reconstruct and quantify SMLM images. The Applications section highlights examples of biological studies made possible by SMLM. The remaining sections address issues of reproducibility and data deposition, explore limitations and optimizations of current SMLM techniques and, finally, provide an outlook on advanced and promising new approaches.
Experimentation
Molecules imaged in SMLM techniques must be fluorescently labelled. In an ideal SMLM experiment, the function, location and number of target molecules should be preserved by the fluorescent labelling strategy used and each target molecule should be tagged by exactly one fluorophore of minimal physical size (although some advanced studies label multiple sites of the same protein or DNA molecule to determine their 3D conformation20,21). To separate the fluorescent signals of individual fluorophores in time, each fluorophore should ideally have a high contrast ratio by being active in one frame and inactive otherwise, emitting a large number of photons relative to the background to enable a high SNR and localization precision. For live-cell super-resolution imaging, active fluorophores should become inactive very quickly to enable rapid turnover and sampling, and imaging conditions should be compatible with maintaining cellular physiology. Below, we discuss some of the main fluorophores and labelling approaches for SMLM relative to these ideal requirements. A selection of recommended ingredients and protocols for newcomers to SMLM is included in the Supplementary information.
Nyquist sampling.
Sampling a continuous signal at twice the highest frequency, which allows its reconstruction without loss of information.
Fourier ring correlation.
A measure of image resolution computed from the cross-correlation between two independent noisy versions of the image in Fourier (frequency) space.
Fluorophores for SMLM
SMLM fluorophores fall into one of five classes, depending on how they switch between ON and OFF states (FIG. 2a–e). Some of these classes include both synthetic dyes and fluorescent proteins. Compared with fluorescent proteins, synthetic dyes generally have higher photon counts and therefore allow for shorter imaging times and better localization precision. Fluorescent proteins are generally more suited for live-cell applications, although premature bleaching or incomplete labelling can limit the structural resolution (BOX 1). Note that for many fluorophores, the exact molecular mechanisms underlying photoswitching are still under investigation. We detail these five types of fluorophores and how to use them in SMLM below.
Fig. 2 |. Fluorophore types and labelling strategies in single-molecule localization microscopy.

a–e | Fluorophores compatible with single-molecule localization microscopy (SMLM) can be divided into five classes: photoswitchable (part a), photoactivatable (part b), photoconvertible (part c), spontaneously blinking (part d) or temporarily binding (part e), which includes the techniques of point accumulation in nanoscale topography (PAINT) and DNA-PAINT. Bullet points indicate types or properties of fluorophores in each class and an example fluorophore is shown. Activated fluorophores are shown as red dots, freely diffusing fluorophores as pale pink discs. f | Different fluorescent labelling approaches in order of decreasing linkage error (from left to right): immunolabelling with primary (1st) and secondary (2nd) antibodies (yellow); labelling with a small camelid antibody (nanobody, light green), often in combination with green fluorescent protein (dark green), as shown, or when available directly binding to the protein of interest (not shown); labelling with a protein tag or genetically encoded protein, such as Eos (orange); direct labelling with a dye-conjugated ligand (such as the microtubule-binding compound docetaxel in this example); and incorporation of unnatural amino acids such as TCO*-lysine through genetic code expansion, which enables rapid labelling using functionalized synthetic dyes. hνmax, irradiation at the absorption maximum; λ shape, target structure imaged by SMLM.
Photoswitchable fluorophores.
Photoswitchable fluorophores (FIG. 2a) undergo reversible transitions between ON and OFF states and a single fluorophore can therefore ‘blink’ many times. Photoswitchable dyes include the carbocyanine dyes Cy5 and Alexa Fluor 647, rhodamine and oxazine dyes such as most Alexa Fluor dyes and ATTO dyes22. Switching between ON and OFF states occurs upon irradiation at appropriate wavelengths and through using a photoswitching buffer consisting of phosphate-buffered saline (PBS), 10–100 mM of a thiol such as mercaptoethylamine and an enzymatic oxygen scavenger if necessary.
At the beginning of a typical SMLM experiment using photoswitchable dyes, all fluorophores reside in their ON state so cells can easily be visualized and brought into focus and standard wide-field images can be recorded. In order to allow single-molecule localization, the vast majority of fluorophores are then turned OFF, which usually requires high excitation intensities of ~10–30 kW cm−2 at an adequate wavelength λON→OFF for a few seconds. The excitation intensity can then be decreased to ~1–3 kW cm−2 to pump any active dyes to dark, inactive triplet states — generating single-molecule fluorescence in the process. These dark states are reduced by the buffer thiol at pH >7.0 to produce a reduced OFF state that is stable for several seconds before the fluorophore returns to the singlet ground state (ON) upon reaction with residual oxygen23. Direct irradiation of the sample residing in the OFF state with UV light at λOFF→ON = 405 nm can also be used to increase the activation and localization rate for low-density target molecules. Photoswitching can occur either in the presence24 or absence25 of an activator dye — corresponding to STORM8 and direct STORM (dSTORM)9 experiments, respectively.
The ON-state lifetime can be adjusted by changing irradiation intensities (λOFF→ON and λON→OFF), the thiol concentration or the pH of the photoswitching buffer. The ON-state lifetime is typically 5–20 ms, although it can be anywhere between 1 and 500 ms (REF.26). It is recommended to vary the oxygen and thiol concentrations to optimize the blinking performance for new dyes in dSTORM experiments; indeed, some oxazine dyes such as ATTO 655 exhibit photoswitching in the presence of oxygen and generally require lower thiol concentrations than carbocyanine dyes, which perform better in the absence of oxygen. For novices, we recommend starting with Alexa Fluor 647 or Cy5 for dSTORM experiments. Note that cells contain the thiol glutathione at millimolar concentrations and therefore some synthetic dyes can be used for dSTORM in live cells27.
Photoswitchable fluorescent proteins such as Dronpa28 can be toggled reversibly from an OFF state to an ON state upon irradiation at different excitation wavelengths (λOFF→ON = 405 nm, λON→OFF = 488 nm) without requiring any special buffer. Some standard fluorescent proteins such as yellow fluorescent protein (YFP) can undergo a spontaneous recovery from OFF to ON states after partial photobleaching, although their use leads to the localization of only a small subset of molecules and limits resolution (BOX 1). In all experiments, the use of low-intensity excitation radiation and long exposure times can minimize photobleaching and improve resolution29.
Photoactivatable fluorophores.
Photoactivatable fluorophores (FIG. 2b) irreversibly switch from an OFF to an ON state either spontaneously or upon activation by UV light (λOFF→ON = 405 nm) under aqueous conditions. Because fluorophores are initially OFF, activation with UV light is usually required to focus the microscope. To allow single-molecule localization, photoactivated fluorophores must be photobleached by irradiation at longer wavelength λON→OFF, prior to activation of new fluorophores. Synthetic photoactivatable dyes include photochromic rhodamine amides30, the azetidinyl rhodamine PA Janelia Fluor® 549, the silicon rhodamine PA Janelia Fluor® 646 (REF.31) and the bridged carbocyanine Cy5B (REF.32). The Janelia Fluor® dyes give longer ON-state lifetimes and higher photon yields and localization precisions than the rhodamine dyes they are derived from and are highly membrane permeable — enabling live-cell SMLM — but require longer total acquisitions times, typically exceeding several hours.
Photoactivatable fluorescent proteins include PAmCherry and PA- TagRFP (λOFF→ON = 405 nm, λON→OFF = 564 nm), PAmKate (λOFF→ON = 405 nm, λON→OFF = 586 nm) and PA-GFP33 (λOFF→ON = 405 nm, λON→OFF = 488 nm) and have been used in the first demonstration of PALM in fixed cells6,7 as well as in live cells19,34. For most live-cell applications, photoactivatable fluorescent proteins such as PAmCherry or PA-TagRFP are labels of choice because they enable stoichiometric labelling.
Photobleaching.
Modification of a fluorophore due to irradiation, as a result of which it loses its ability to fluoresce.
Photoconvertible fluorophores.
Photoconvertible fluorescent proteins (FIG. 2c) including Eos and its derivatives35,36, Dendra2 (REF.35) and mMaple37, can switch irreversibly from one spectral state (colour) to another upon irradiation. For example, Eos can be switched from green to red by UV irradiation at λG→R = 405 nm and is excited at λG→OFF = 488 nm and λR→OFF = 561 nm in the green and red state, respectively. For Dendra2, these wavelengths are λG→R = 405 nm, λG→OFF = 488 nm and λR→OFF = 520 nm. Fluorescence obtained at the shorter wavelength can be used to focus the sample and obtain a diffraction-limited image before photoconversion. Activated fluorophores must be photobleached after conversion to avoid PSF overlap with newly converted fluorophores and the photobleaching time can be controlled by irradiation intensity. Photoconvertible fluorescent proteins are also good choices for live-cell SMLM.
Spontaneously blinking dyes.
Spontaneously blinking dyes (FIG. 2d) exploit a reversible, pH-dependent chemical reaction to enable SMLM at a defined pH in an aqueous solution38 without requiring a photoswitching buffer. These dyes include the silicon rhodamine dye HMSiR39, HEtetTFER40 and FRD41. These fluorophores exhibit excellent photon yield and are partially membrane permeable, enabling their use in live-cell SMLM experiments. Spontaneously blinking fluorophores show immediate blinking from the onset of an SMLM experiment and the ON-state lifetimes vary in the range of 10–300 ms (REF.38). Blinking kinetics are independent of the irradiation intensity and mainly controlled by the pH of the aqueous buffer.
Temporarily binding dyes.
Unlike PALM and STORM/dSTORM, PAINT does not require fluorophore ON/OFF switching, instead using dyes10 or dye-labelled ligands42 that freely diffuse until they interact with targets of interest by either permanent or transient binding (FIG. 2e). Because free dyes diffuse rapidly over many pixels during acquisition of a single image frame, they only appear as a blurred background and are not localized, whereas bound dyes appear as a PSF and are localized. This strategy effectively decouples the switching between ON and OFF states from dye photophysics. PAINT is not compromised by photobleaching as the fluorophore reservoir can be replenished and bright and photostable fluorescent dyes can be used. Any synthetic dye that exhibits a sufficient fluorescence quantum yield (the ratio of emitted to absorbed photons), such as Cy3B, is compatible with PAINT.
DNA-PAINT43, a variation of PAINT, uses short — typically six to ten nucleotides long — dye-labelled DNA oligonucleotides known as ‘imager strands’ that transiently and specifically interact with their complements, called ‘docking strands’, which are attached to the molecule of interest (FIG. 2e). In DNA-PAINT, the ON times, or bright times (τb), can be tuned based on the stability of the DNA duplex and are related to the dissociation rate (kOFF) of the imager–docking duplex according to τb = 1/kOFF. Typical τb values are ~500 ms for an 8-bp duplex consisting of two CG and six AT pairs. Increasing the length of the imager strands by 1 bp increases τb by roughly an order of magnitude43. Fluorescence OFF times, or dark times (τd), are linked to DNA hybridization rates (kON) and imager concentration (ci) through τd = 1/(ci • kON) and can be tuned by either changing ci or kON. Bright times can be tuned to extract the highest number of photons per binding event, enabling high localization precisions of ~1 nm (REFS18,44).
Fluorescent labelling strategies
Several options are available to permanently or transiently link SMLM- compatible fluorophores to molecules of interest. The main options are genetic fusion of a fluorescent protein to a target protein; immunolabelling using dye-conjugated antibodies; genetic fusion of the target protein with a tag that can bind to synthetic dyes; direct binding of synthetic dyes to specific structures; and using transiently binding labels with or without oligonucleotides. The choice of labelling strategy is critical as it affects the achievable resolution via the linkage error (Fig. 2f) and the labelling density, and determines whether the target molecule retains its physiological localization and whether live-cell imaging is possible.
Encoding fluorescent proteins.
Genetically encoded fluorescent proteins are the most prominent labels for live-cell fluorescence microscopy and have been used for PALM in fixed6 and live19,45 cells. One labelling approach is to transfect cells with an expression vector containing DNA constructs with a gene encoding the target protein genetically fused to a fluorescent protein46. For transient transfection, the number of expressed target proteins can vary widely. In stably transfected cell lines, expression levels can be controlled through construct optimization, although the endogenous protein is left unlabelled. CRISPR knock-ins allow for homozygous labelling of the endogenous genes such that protein abundance is not altered and all target proteins are labelled. Fluorescent proteins are ~2–5 nm in size and can also perturb the function of the target protein47. Ideally, each protein of interest should carry a single fluorescent protein that preserves protein functionality. For more details, see REFS33,45,48,49.
Immunolabelling.
Synthetic dyes cannot be genetically encoded into biomolecules and must be coupled to compounds that can bind the target molecule. Immunolabelling is the standard method for labelling endogenous proteins in fixed cells. In direct immunolabelling, an antibody that binds specifically to the protein of interest is chemically coupled to a synthetic dye. Indirect immunolabelling uses a dye-coupled secondary antibody to target an unlabelled primary antibody; the advantages of this strategy are that the binding of multiple secondary antibodies to a single primary antibody amplifies the fluorescent signal and unlabelled primary antibodies often have better epitope binding affinity than primary antibodies modified for fluorescent labelling, hence reducing the background. An important drawback of immunolabelling for SMLM is that commonly used IgG antibodies are large, causing high linkage errors (~10 nm, or up to ~20 nm for indirect immunolabelling). Nanobodies are substantially smaller than antibodies (~2.5–4 nm), and are therefore a good alternative where highly specific, fluorescent dye-coupled nanobodies are available, as for GFP50.
Linkage error.
The distance between the fluorophore and the molecule of interest, which compounds localization errors.
As immunostaining of intracellular proteins requires permeabilization of cell membranes, it is generally incompatible with live-cell imaging unless labelling extracellular or membrane proteins, and can modulate biological functions in the latter case. However, the development of electroporation methods51 and cell-permeable nanobodies52 may facilitate live-cell immunolabelling in the future. It is important to check that immunolabelling, permeabilization and fixation conditions allow for specific labelling of the target proteins without artefacts53.
Protein tags.
Biorthogonal peptide motifs or self-labelling protein tags, such as FlAsH tags, lipoic acid ligase, SNAP-tags and Halo-tags54–56, can allow for specific labelling with minimal linkage error. These tags are genetically fused to the target protein and covalently bind their respective reagent or ligand, which is directly coupled to a fluorophore. These labelling methods combine genetic expression with the excellent photophysical properties of synthetic dyes, while reducing the linkage error to a few nanometres. Further, they can be used for live-cell imaging experiments as long as the dye substrate exhibits membrane permeability56,57.
Direct labelling.
Some biological structures can be labelled using dye-conjugated small peptides or drugs, such as the bicyclic heptapeptide phalloidin or the taxane paclitaxel, which target actin and microtubule filaments, respectively58,59. Despite their small size and minimal linkage error, such labels often impair biophysical function and act as biological modulators. Additionally, some modified fluorophores such as organelle trackers or modified lipids can be directly integrated into the biological structure itself60. The smallest linkage error associated with peptide labelling can be achieved using site-specific labelling of a single amino acid; genetic code expansion can enable incorporation of synthetic amino acids such as TCO★-lysine, which can be directly coupled to functionalized synthetic dyes through fluorogenic click reactions without affecting biological functionality and enable live-cell imaging61–63.
PAINT and DNA-PAINT labelling.
PAINT was initially restricted to imaging organelles for which transiently binding fluorescent molecules exist10,42,64; for example, PAINT was originally demonstrated by imaging large unicellular vesicles with the fluorophore Nile Red — which is only weakly fluorescent in water but becomes brighter in a lipid environment10 — and has been applied to imaging DNA using dyes such as Hoechst65. DNA-PAINT, by contrast, can be used with any target molecule that can be linked to a docking oligonucleotide strand. The most common DNA-PAINT method involves indirect immunolabelling, using primary antibodies targeting the protein of interest and secondary antibodies conjugated to DNA docking strands18,66. To overcome the large linkage errors of antibodies, DNA-PAINT has recently been combined with smaller and more efficient labelling reagents including nanobodies66–68, genetically encoded tags67, affimers69 and novel aptamer probes called SOMAmers70.
Sample preparation
Cells and tissue samples are fixed for most SMLM experiments as dense intracellular structures such as actin filaments must be imaged over long acquisition times to allow sufficient sampling. Nevertheless, sparse or slowly moving cellular structures such as focal adhesions34 can be investigated using live-cell SMLM. Below, we discuss considerations for fixed samples and live-cell imaging.
Fixed samples.
Chemical fixation is needed to effectively cross-link proteins, preserve their binding sites and preserve molecular interactions. Aldehyde-based fixatives such as paraformaldehyde (PFA) and glutaraldehyde are widely used in fluorescence imaging applications and are the most common fixation reagents for SMLM. Alcohol-based alternatives, such as ice-cold methanol, show efficient fixation for cytoskeletal components, although they are unsuited to preserve the ultrastructure and molecular organization of cellular membranes or organelles. Glyoxal has been suggested as an alternative fixative with improved preservation of cellular morphology for super-resolution microscopy71. For the investigation of dynamic biological processes or protein–protein interactions in membranes, the residual mobility of membrane components must be considered as it can induce antibody cluster formation and other artefacts. To avoid residual mobility, membrane components should be fixed using 4% PFA and 0.2% glutaraldehyde for 30 min or longer72,73. For immunolabelling, cells must be permeabilized after fixation and a blocking buffer such as bovine serum albumin (BSA) in PBS or normal goat serum (NGS) should be used before labelling to reduce non-specific binding and background signals23.
Total internal reflection fluorescence.
A configuration in which a strongly inclined laser beam is reflected by the coverslip–sample interface, leaving only a thin (~200 nm) layer in the sample illuminated, strongly reducing the background.
Samples can also be fixed by cooling them to cryogenic temperatures (below −150 °C) under high pressure to prevent the formation of ice crystals. Although cryofixation methods are technically demanding, are mostly restricted to thin sections6 and can affect photoswitching properties, they achieve optimal preservation of cellular ultrastructure and are compatible with both SMLM and electron microscopy techniques74,75.
Highly inclined and laminated optical sheet illumination.
A configuration in which the laser beam enters the sample at a sharp angle, allowing reduction of the background when imaging at a distance from the coverslip.
Live cells.
In live-cell experiments, cells are imaged in phenol red-free medium or PBS after labelling. Phototoxicity is a major limitation of live-cell SMLM, and typically more pronounced when using higher laser intensities or shorter excitation wavelengths. A useful test for phototoxicity is to grow cells in medium overnight at 37 °C with 5% CO2 after imaging and check their survival using cell proliferation assays. A more sensitive viability parameter is whether the irradiated cells undergo cell division in the 24-h period after the SMLM experiment76. The illumination mode, irradiation wavelength and sample temperature should be adjusted to minimize phototoxic effects76.
Optics of SMLM
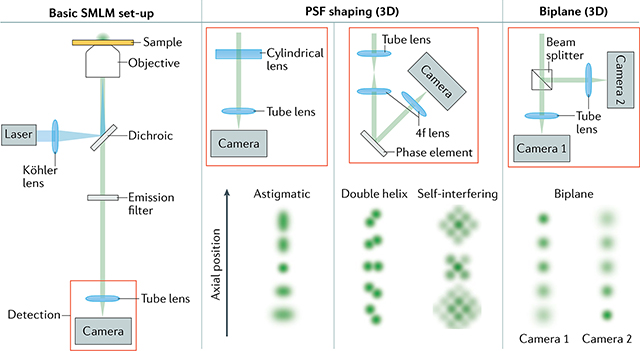
Unlike other super-resolution methods, SMLM requires only a wide-field microscope equipped with standard continuous wave lasers for excitation and activation and a camera able to detect single molecules, such as an electron-multiplying charge-coupled device (EM-CCD) or a scientific complementary metal-oxide semiconductor (sCMOS) camera (FIG. 3a,b). As a result, numerous commercial, open-source hardware and homebuilt implementations exist77–79. EM-CCDs allow detection of single photons with negligible read-out noise and are particularly suited for low photon counts. Modern sCMOS cameras are somewhat less sensitive for weak signals and produce inhomogeneous read-out noise that requires extra correction, but have higher frame rates and enable a larger FOV than EM-CCDs and achieve a similar SNR for bright dyes. Most commercial or custom microscopes for SMLM include automated systems to keep the specimen in focus. For multicolour microscopy (see also BOX 2), multiple laser lines can be combined and co-aligned using dichroic mirrors. Several simple approaches can enable SMLM in three dimensions, for example PSF engineering through inserting a cylindrical lens into the optical path80 (BOX 3; FIG. 3c–e). Wide-field illumination of the sample can be generated by expanding a laser beam’s circular intensity profile, and then cropping the FOV to keep only the relatively uniformly illuminated central area.
Fig. 3 |. Single-molecule localization microscopy hardware.

a | Basic single-molecule localization microscopy (SMLM) set-up consisting of an illumination source (here, a laser) and a Köhler lens (blue box), an objective lens and a stage for placing the sample (yellow box) and a detector with a tube lens and a camera (red box). Dichroic mirrors are used to separate excitation and emission wavelengths, and can be combined with additional emission filters to reject autofluorescence. b | 2D direct stochastic optical reconstruction microscopy (dSTORM) image of microtubules after secondary immunolabelling with Alexa-647-conjugated antibodies, obtained from 60,000 raw frames, with the corresponding wide-field image shown partially on the left. Estimated resolution (Fourier ring correlation) ≈44 nm. c | 3D SMLM system obtained simply by adding an optical component to engineer the point spread function (PSF), such as a cylindrical lens, which generates astigmatism (see BOX 3). d | Z-stack of a fluorescent bead showing the axial variations of an astigmatic PSF. Scale bar: 500 nm. e | 3D dSTORM image obtained by analysis of 2D single-molecule images, displayed here in two dimensions with colour indicating axial (Z) coordinates. PSF calibration and image reconstruction performed with ZOLA-3D (REF.102). Part b courtesy of M. Singh.
Box 2 |. Multicolour single-molecule localization microscopy.
The investigation of biomolecular interactions in cells requires super-resolution imaging of multiple target molecules in a single experiment. Multicolour single-molecule localization microscopy (SMLM) using synthetic dyes can be accomplished using the classical stochastic optical reconstruction microscopy (STORM) concept and probes with both an activator and a reporter fluorophore8. In this approach, various activator–reporter dye pairs attached to a single antibody, such as Alexa Fluor 405/Cy5, Cy2/Cy5 and Cy3/Cy5, enable multicolour STORM using three different activation lasers and spectrally selective activation of the reporter fluorophore (in this case Cy5)267.
Activator-free SMLM using synthetic dyes (direct STORM, or dSTORM) requires fluorophores that exhibit similar blinking efficiency under identical photoswitching buffer conditions. Suitable pairs for two-colour dSTORM include Alexa Fluor 532/Alexa Fluor 647, ATTO 520/Alexa Fluor 647, CF 568/Alexa Fluor 647, and combinations involving dyes that absorb at longer wavelengths such as DyLight 750 (REFS152,268).
Although multicolour dSTORM is susceptible to chromatic aberrations, these can be avoided by spectral demixing. In this approach, synthetic dyes are used that exhibit good photoswitching performance in the same thiol switching buffer and can be efficiently excited with the same laser wavelength, but exhibit different emission maxima. The emission light is spectrally separated by a dichroic beam splitter and imaged onto two cameras or separate parts of the same camera chip. Fluorophores are then classified by their emission ratio. Alternatively, the emission signal of different synthetic dyes can be split into two detection paths and recorded simultaneously on two cameras (or two regions of the same camera), with one recording emission spectra and the other recording localizations269–271. Combinations of fluorescent proteins — for example, Dronpa/mEos — have been used successfully for multicolour SMLM, as have combinations of proteins with synthetic fluorophores, including mEos2/Alexa Fluor 647 and Dronpa/Alexa Fluor 647 (REFS176,272).
Box 3 |. 3D single-molecule localization microscopy.
Biological structures are 3D and should therefore be imaged in three dimensions with axial resolutions similar to those that can be achieved laterally. Innovations in optical set-ups exploiting point spread function (PSF) engineering80,273 or multiplane274,275 detection have enabled precise localization of molecules in three dimensions, in samples up to several micrometres thick102. Single-molecule localization microscopy (SMLM) methods with engineered PSFs exploit the fact that the 2D image of a fluorescent molecule depends not only on its lateral (x, y) coordinates but also on its axial coordinate z. By analysing the 2D image pattern, the z coordinate can be estimated in addition to x and y. In this way, 3D super-resolution images can be reconstructed from 2D images by changing the microscope focus. The most common approach exploits an astigmatic PSF, generated by inserting a single cylindrical lens into the imaging path79,80,102 (see the figure). Other PSFs such as a double helix (figure), tetrapod or saddle point, which can be obtained using phase masks, deformable mirrors or other optical devices, can achieve a larger axial range102,273,276,277.
In set-ups with two focal planes (see the figure) or more274,275,278, analysing the relative intensities in different images of the same molecule also allows one to compute z in addition to (x, y). In multifocal microscopy, a specialized diffractive grating is used to image up to nine focal planes simultaneously on a single camera275. Combining multifocal microscopy with SMLM allowed fast volumetric imaging of mitochondria in whole cells with high resolution276.
PSF shaping or multiplane detection diverts photons for 3D encoding, resulting in a loss of lateral localization precision relative to basic 2D set-ups. The theoretically achievable localization precision and axial range depend on the PSF shape and can be calculated. Some PSFs have even been engineered to achieve theoretically optimal precisions over a given axial range99,277. Many software packages are now available for 3D SMLM with PSF engineering99 (see TABLE 1).
The sample’s fluorescence emission is imaged through an objective lens onto the detector. Typically, a 60–100× oil immersion objective with a numerical aperture of 1.4 or higher is used to ensure efficient photon collection. When imaging samples close to the coverslip, such as cellular membranes, total internal reflection fluorescence (TIRF)81 or highly inclined and laminated optical sheet illumination (HILO)82 can be used to reduce the out of focus background and improve SNR and localization precision. A dichroic filter is used to separate excitation and emission light; for multicolour imaging, a dichroic filter with multiple pass bands can be chosen to reflect excitation laser lines onto the sample and transmit emission wavelengths to the detector. At the detector, the pixel size should be approximately equal to the PSF standard deviation, typically ~100–150 nm. The optimal magnification can be determined for specific background and photon numbers83,84.
Data acquisition in SMLM
In SMLM, the number of active fluorophores in each frame should be as high as possible to minimize the acquisition time, providing it is low enough to avoid PSF overlaps. For photoswitching dyes, the density of active fluorophores is mediated by a balance between excitation and activation laser intensities, whereas in PAINT it is dependent on binding affinities and binder concentration. Although these parameters are usually determined manually, SMLM can be automated with control software that implements a feedback loop to tune the active fluorophore density85–87.
Structural imaging in fixed cells.
The exposure time of each frame should ideally match the ON-state lifetime of the fluorophore (typically ~10–100 ms, as above). The total number of frames needed to reconstruct an image depends strongly on the structure being imaged and the desired resolution (BOX 1). For higher-dimension structures, the signal from an ON fluorophore will overlap with a larger portion of the structure and will thus require more frames to reconstruct88. An accurate assessment of the number of frames needed to interrogate a specific structure requires detailed analysis34,88,89; however, a rough estimate can be calculated. For example, when imaging the centriole, the structure can be approximated as a hollow cylinder 250 nm in diameter and 400 nm long, for an area of ≈3 × 105 nm2, fitting into a single PSF. To resolve its surface at a scale of 40 nm would require neighbouring localizations to be at most 20 nm from each other34, corresponding to at least 750 evenly spaced molecules. Stochasticity and the need to oversample to build up the image will require at least 5–10 times as many molecules to be localized89 — roughly 5,000–10,000 in this example. With only one molecule ‘ON’ per frame, this implies that at least 10,000–20,000 frames should be collected. For an exposure time of 10–100 ms, this implies an acquisition time of 2–30 min.
Dynamic imaging in live cells.
SMLM studies in live cells can profile structural dynamics or molecular dynamics (FIG. 4). Structural dynamics studies aim to reveal the movement of a structure composed of many molecules, such as a focal adhesion, a clathrin-coated pit or an organelle membrane34,60,90. In these studies, time series of super-resolution images are reconstructed from the localizations obtained in consecutive, non-overlapping sets of frames (FIG. 4a). The use of large numbers of frames for each reconstructed super-resolution image implies diminished temporal resolution and, potentially, motion blur, whereas fewer frames imply sparse sampling of the structure and, hence, worse spatial resolution34 (BOX 1).
Fig. 4 |. Live-cell single-molecule localization microscopy.

a | Structural dynamics of a focal adhesion (tdEos-paxillin) reveals its appearance near the cell edge (top) before maturation and motion towards the interior (bottom). Each super-resolution image is reconstructed from 1,000 raw frames. b | Molecular dynamics of the vesicular stomatitis virus glycoprotein (VSVG-tdEos), a transmembrane protein freely diffusing on the plasma membrane. Motion of each protein traced over multiple frames, with different colours representing different molecules (left). Each trajectory can be analysed to create a map of diffusion coefficients (centre). By contrast, molecules within the actin cytoskeleton (actin-tdEos) show directed motion near the cell’s leading edge and diffusive motion towards the interior (right). D, diffusion coefficient. Part a reprinted from REF.34, Springer Nature Limited. Part b adapted from REF.19, Springer Nature Limited.
Molecular dynamics studies follow the trajectories of single molecules, often considering them as a probe of local environmental properties (FIG. 4b). In an approach first introduced as ‘spt-PALM’, individual molecules are photoactivated or photo-converted, detected and tracked over multiple frames19. The distance between active fluorophores in each frame must be several times larger than the distance they travel between consecutive frames to avoid tracking errors, and the density of active fluorophores must therefore be considerably lower than in structural dynamics studies19,42. The time between frames should be chosen to allow molecules to move a distance greater than their localization precision. SMLM allows for the collection of orders of magnitude more molecular trajectories than single-molecule tracking without photoswitching91,92, and therefore enables more insights into molecular movements and the factors that control them93.
Dark current noise.
A type of noise in electronic devices such as cameras that is independent of incident light, caused by thermal electrons and increases with temperature
Poisson noise.
(Also known as shot noise). A type of statistical noise affecting photon counts and arising from the fact that photons are hitting pixels independently of each other with constant probability per unit time.
Cramér–Rao lower bound.
(CRLB). A fundamental limit to the precision (variance) of any estimator without bias.
Results
In SMLM, super-resolution images are assembled computationally and therefore the quality of the images strongly depends on image processing. Below, we discuss the computational aspects of SMLM data analysis. The majority of SMLM reconstruction methods can be summarized in three steps: single-molecule detection, single-molecule localization and super-resolution image rendering. We discuss these three steps below.
Single-molecule detection
In the detection step (FIG. 1h), each diffraction-limited image is processed to determine the approximate location of active emitters. The image is often preprocessed to remove heterogeneous background, for example using rolling ball algorithms, difference of Gaussians algorithms or wavelet filtering94, or by subtracting the background averaged over the entire image sequence95. Detection is then typically done by extracting local maxima from the background-corrected image. Alternatively, the raw image can be searched for PSF-like patterns by computing the correlation of this image with a model PSF derived using a Gaussian approximation of the Airy function or a more elaborate model for engineered PSFs in 3D SMLM (see BOX 3) and thresholding the correlation image. The result is usually a set of pixel regions likely to contain single molecules. Any detection algorithm will make false positives and/or false negatives whenever images are corrupted by noise96 and a lower SNR implies more unavoidable errors. Cameras used in SMLM are subject to several sources of noise, including dark current noise, which can be reduced by using cooled cameras, read-out noise (usually negligible) and multiplicative noise from stochastic amplification of photoelectrons, which can be reduced by setting a low amplification gain. A fundamentally inevitable noise comes from the fact that fluorophores (and the background) emit only a finite number of photons, leading to Poisson noise. High photon counts relative to the background imply a high SNR and therefore better detection performance.
Single-molecule localization
In the localization step, the pixel regions determined above are analysed further to compute the subpixel (x, y) coordinates of each molecule (FIG. 1i). We briefly discuss the fundamental limits to localization precision and how localizations are computed.
Fundamental precision limits.
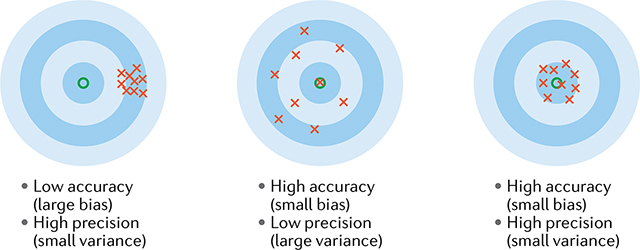
Owing to noise, localization algorithms always make errors. These errors have random and systematic parts as measured by the variance and the bias83,84, which define precision and accuracy, respectively (see BOX 4). Theory shows that the precision of an algorithm without bias is limited by the Cramér–Rao lower bound (CRLB)97. This limit depends on the SNR, with a higher SNR allowing for better precision. A well-known form of the CRLB is:
where σloc is the precision defined as the standard deviation of errors in estimated coordinates, σ0 is the standard deviation of the PSF and N is the number of photons collected by the camera (see BOX 4). Typical values are σ0 ≈ 100 nm and N = 102–104, which would predict precision limits of σloc ≥1–10 nm. However, this simple formula ignores factors such as the non-Gaussian shape of the PSF, read and amplification noise, background signal, finite pixel size and dipole orientation, all of which worsen the precision limit. More complex expressions have been derived that take some or all of these factors into account84,98, for example:
where a is the pixel size and b the background intensity98. The CRLB has become an essential notion in SMLM because it defines a fundamental limit that all localization algorithms should be compared with.
Box 4 |. Localization precision, accuracy and the Cramér–Rao lower bound.
The image I of a single fluorophore is the result of a stochastic process that depends on several unknown parameters, notably the (x, y) coordinates of the molecule and the number of collected photons, N. Localization algorithms attempt to determine (x, y) from I, providing estimates . Algorithms are usually assessed based on their mean squared error (MSE): , where angled brackets denote statistical averaging. The MSE for coordinate x can be rewritten as:
where the variance is the random component of the errors, and the bias is the systematic error (and likewise for y). The variance defines the ‘precision’ of an algorithm, whereas the bias defines the ‘accuracy’. High-precision algorithms tend to provide similar coordinates for different images of the same molecule, but may have a large offset from the true value (that is, a bias). High-accuracy algorithms tend to find the correct position on average, but possibly with high dispersion (low precision), meaning that individual localizations may be far from the true location. The differences between accuracy/bias and precision/variance are shown in the figure, with the true position at the centre and crosses symbolizing independent position estimates.
One usually considers algorithms without bias, so that . The Cramér–Rao lower bound (CRLB) provides a lower limit to the variance (and hence precision) of any unbiased algorithm. Assuming for simplicity that the image depends only on x, the CRLB reads:
where p(I; x) is the probability of observing I if the true coordinate is x (p(I; x) is also called the likelihood). A useful formula can be derived under strongly idealized conditions, where the point spread function (PSF) is modelled as a Gaussian of standard deviation σ0, the background and camera noise are neglected, the exact coordinates of N photons hitting the camera are recorded and pixelation is ignored. Under these assumptions, the CRLB reduces to: and the precision limit is simply:
Localization algorithms.
The gold standard for localization algorithms is maximum likelihood estimation (MLE), which theoretically approaches the CRLB at high SNR84,98–100. MLE algorithms aim to compute the (x, y) coordinates for which the probability p(I; x, y) of obtaining the observed image I — also known as the likelihood — is highest. They do so through an iterative procedure called gradient ascent, which determines the likelihood at an initial position (x0, y0) — for example, the centre of a pixel identified in the detection step — and changes the coordinates by small increments (xn+1 = xn + δxn; yn+1 = yn + δyn) to increase the likelihood until it cannot be increased further. The same approach can be extended to compute 3D coordinates (see BOX 3).
MLE algorithms differ depending on aspects of the mathematical model used to describe how the widefield images are formed, including assumptions on PSF shape, background and noise. MLE algorithms typically reach the CRLB for simulated images, but achieving this in practice requires an accurate model of the PSF. Although Gaussian models are widely used, they are inaccurate as diffraction theory predicts that PSFs follow different shapes such as Airy patterns. More complex functions based on optical theory can provide better PSF models, but may depend on unknown parameters and ignore aberrations caused by imperfect optics and/or induced by the sample, increasing localization errors. It is therefore preferable to calibrate PSF models before each experiment — especially in 3D SMLM imaging — which can be done using cubic spline fitting or phase retrieval algorithms on images of sub-diffraction fluorescent beads101,102. Algorithms that use experimentally measured PSFs outperform those based on idealized PSFs99. Further, modelling or subtraction of the background can reduce localization bias. The iterative nature of MLE generally makes these algorithms slower than less precise, non-iterative algorithms79 such as intensity-weighted centres of mass. However, thanks to the availability of graphics processing units, state-of-the-art MLE algorithms can now analyse SMLM image sequences in real time99.
Post-processing of localizations
After localizations are computed for all detected molecules, post-processing can filter out suboptimal localizations where the model PSF poorly matches the image, or with computed CRLB above some threshold. Although stringent filtering can improve the average localization precision, it will not necessarily improve resolution as rejecting localizations compromises sampling (see BOX 1). Another post-processing step is to merge nearby localizations in consecutive frames that likely arise from the same molecule, resulting in a single, more precise localization without degrading sampling density. In addition, most SMLM experiments require drift correction (see Limitations and optimizations).
Ripley’s K function.
For a given set of points, K(r) is the number of points within the distance r from one point, averaged over all points and normalized by the number of points expected for a uniformly random point distribution.
Super-resolution image rendering
Once all diffraction-limited images have been processed, localizations are rendered as a super-resolution image. Typically, a grid is defined with bin sizes similar to the precision (for example, σloc = 10 nm), localizations in each bin are counted (FIG. 1k) and an image is reconstructed, where counts in each bin are translated to pixel intensities (FIG. 1l). Minor variations exist; for example, more weight can be given to localizations from higher-intensity PSFs or localizations can be shown as Gaussian spots — normalized as a probability density to an integral of one — with standard deviations given by their estimated precision. The resulting image should have sub-diffraction resolution provided that certain conditions are met, as discussed in BOX 1.
Software
More than a hundred independent software packages have been developed for analysing SMLM data. Web resources are available that objectively compare the results of localization software on simulated images and a small set of experimental data99,103. Although not without caveats (analysis results are typically provided by the authors of software themselves, require expert fine-tuning and are contingent on simulation assumptions and the choice of experimental data), these benchmarks provide a good starting point for choosing state-of-the-art SMLM software. See also TABLE 1 for a selection of recommended software packages.
Table 1 |.
Selected SMLM software
| Software name | URL | Comments |
|---|---|---|
| ThunderSTORM94 | https://zitmen.github.io/thunderstorm/ | Widely used, implements multiple 2D and 3D algorithms; strong performance in 2D SMLM challenge |
| SMAP101,264 | https://github.com/jries/SMAP | Handles arbitrary PSFs; strong performance in 3D SMLM challenge |
| SMolPhot | https://bitbucket.org/ardiloot/smolphot-software/wiki/Home | Strong performance in 3D SMLM challenge |
| ZOLA-3D (REF102) | https://github.com/imodpasteur/ZOLA-3D | Handles arbitrary PSFs and refractive index mismatch |
| Picasso18 | https://github.com/jungmannlab/picasso | Software package optimized for DNA-PAINT |
Recommended software packages for single-molecule localization microscopy (SMLM) image reconstruction. All listed software are free, can reconstruct 3D SMLM images and all except SMAP enable drift correction. This selection is partly subjective as it is based on software known to and in part developed by the authors, but also based on objective results from the 2D/3D SMLM localization challenges. For more details and quantitative comparisons of these and other SMLM software, we refer the reader to SMLM software benchmarking resources. DNA-PAINT, DNA point accumulation in nanoscale topography; PSF, point spread function.
Quantitative analyses
SMLM offers distinct possibilities to analyse the density, spatial distribution and, in some cases, numbers of molecules in a sample, but also comes with challenges. We discuss selected quantitative analysis methods for SMLM below.
Identifying clusters and counting molecules.
A common question in SMLM studies is whether proteins form clusters, and, if so, of what size and stoichiometry. Methods from spatial statistics such as Ripley’s K function can help determine whether a distribution of points in an image is clustered and determine cluster length scales. However, applying such tools to SMLM data is complicated by the fact that a single molecule often gives rise to a cluster of localizations owing to stochastic blinking and localization errors. Ignoring this can lead to artefactual clustering104,105. One approach to distinguishing artefactual clusters from real clusters is to determine the statistical distribution of localization errors and apply pair correlation analysis105; however, this method requires careful calibration and provides the average size of clusters, rather than identifying and measuring individual clusters. Analysing clusters individually, therefore, requires segmenting the image into groups of localizations. One proposed clustering solution is a Bayesian method that can adapt to varying localization precisions106, although this requires strong assumptions on cluster shape. Alternative clustering methods include the density-based spatial clustering of applications with noise algorithm (DBSCAN) and tessellation-based approaches107,108.
Once localizations are segmented into clusters, a common goal is to count the underlying target molecules. This is often challenging as target molecules may be localized many times, either because of repeated blinking or because they are labelled by multiple fluorophores (for example, with secondary immunolabelling). Further, some molecules may not be localized at all owing to lack of labelling (as occurs with transfection labelling) or failed photoactivation104,105,109, although these confounding factors can be reduced by using endogenous homozygous labelling with photoactivatable labels and acquisition of long image sequences. Calibration standards can help account for repetitive blinking, failed photoactivation and variations in labelling stoichiometry105,110–114. Another approach to reduce counting errors is kinetic modelling of photoswitching115, which has been demonstrated successfully for photoactivatable proteins and shown to enable accurate estimates of the number of copies of a molecular motor protein in bacteria116. However, modelling photoswitching kinetics can be complicated for fluorophores with multiple dark states. DNA-PAINT offers a particularly promising avenue for quantification as the binding kinetics of imager oligonucleotide probes to corresponding docking strands are well characterized. The DNA-PAINT derivative qPAINT117 exploits the fact that binding frequency scales linearly with the number of docking strands, and thus targets, assuming constant influx of the probes. If calibrated for a single site, the number of targets in an unknown region can be calculated based on the observed blinking frequency and the virtually unlimited sampling of target molecules can minimize statistical error.
Single-particle reconstruction.
When identical copies of a protein complex are imaged in large numbers, information on molecular structure can be derived by superimposing and averaging SMLM images after translations and rotations118,119. Whereas the resolution of reconstructed images is still limited by localization precision, averaging allows measurement of structural features with a precision that is limited by the number of imaged complexes rather than by single-molecule localization precision. This approach has determined the radial position of nucleoporin epitopes within nuclear pore complexes with sub-nanometre precision120 and has been extended to 3D applications, such as analysing the structure of human centrioles119.
Co-localization analyses.
In conventional multicolour fluorescence microscopy, co-localization analyses typically measure the extent to which the diffraction-limited signals from two or more fluorescent channels overlap or correlate121. In multicolour SMLM, co-localization should theoretically never occur as distinct molecules cannot occupy the same physical location. Because of localization errors, however, individual localizations are better viewed as samples of probability densities, which can overlap for different colour channels if molecules are closer than SMLM resolution. In principle, a simple way to assess co-localization is to apply established methods for conventional microscopy121 to pairs of rendered super-resolution images, although results can be strongly affected by stochastic blinking and background noise. Some recent methods therefore measure co-localization based on the localizations themselves, by adapting tools from spatial statistics or using tessellation-based segmentation of probability densities122–124. More work is needed to improve and validate co-localization methods against ground-truth data. Regardless of the method used, chromatic aberrations must be carefully measured and corrected using multicolour beads or fluorospheres.
Single-molecule tracking.
The two main approaches for quantifying molecular dynamics in spt-PALM19 are Lagrangian and Eulerian methods. Lagrangian methods characterize the dynamics of individual molecules by following them individually, for example by computing mean-squared displacements to estimate diffusion coefficients or transport states for each molecule19,125 (FIG. 4b). Eulerian methods focus on distinct regions in the sample and use the dynamics of molecules passing through these regions to infer local properties that characterize or affect molecule dynamics, for example to generate maps of diffusivity or energy potentials126. In both approaches, it is important to take into account the fundamental uncertainties discussed above, as localization errors can introduce biases into molecular dynamics data such as apparent sub-diffusion or spurious energy potentials127,128. More extensive discussions of single-molecule tracking methods can be found in REFS93,129.
Other quantification methods.
Many more methods have been developed to analyse SMLM point clouds, including segmentation methods to differentiate structures from background and analyse their morphologies. Segmentation methods have been used to measure the orientation, length and curvature of dendritic spines in neurons108 and cytoskeletal filaments130,131. Further, a recent preprint article has described how iterative hierarchical clustering methods can classify the morphology of protein aggregates132. Given the variety of techniques and the current lack of comparative assessments, careful validation of each quantification method on simulated and experimental data is critical. For a more complete discussion of SMLM data analysis, see REFS107,133,134.
Applications
Compartmentalization of cells is crucial for regulating cell function. In recent years, SMLM has enabled us to visualize the spatial organization of proteins and nucleic acids within subcellular compartments at nanoscale resolution. Below, we discuss major biological insights emerging from these studies, focusing on specific subcellular compartments including the nucleus, cytoplasm and plasma membrane, and also highlight SMLM studies on neurons and microorganisms.
Nucleus
Advances in SMLM have facilitated the study of intranuclear structures and nuclear pores. We discuss these advances below.
Chromatin organization.
Eukaryotic genomes are folded into a complex 3D chromatin organization across length scales ranging from the ~10 nm diameter of nucleosomes to micrometre-sized chromosome territories135. SMLM imaging of histones has enabled the visualization of ‘nucleosome clutches’ consisting of tens to hundreds of nucleosomes along the chromatin fibre in the interphase nuclei of somatic cells136 (FIG. 5a). Similar results showing that nucleosomes pack into nanodomains of varying sizes were obtained by live-cell PALM137 and recently recapitulated at specific genomic loci using mesoscale modelling138. Clutch size may be underestimated by these approaches owing to imperfect labelling, especially in compact chromatin regions; however, these results suggest that chromatin packing in the nucleus is more heterogeneous than previously thought136. Clutch size is cell type-specific and depends on histone modifications139,140, suggesting that it may be a regulator of gene activity. Labelling histones relies on immunostaining or overexpressing a tagged histone and labelling efficiency depends on accessibility of the epitope to the antibody or the incorporation of tagged histones into nucleosomes; however, DNA itself can be labelled very efficiently using modified nucleotides and click chemistry139,141,142. Dual-colour SMLM imaging shows a high level of co-localization between DNA and histones, suggesting that SMLM histone images are representative of chromatin organization within the nucleus139.
Fig. 5 |. Major discoveries enabled by single-molecule localization microscopy.

a | Stochastic optical reconstruction microscopy (STORM) image of histone H2B in human fibroblast cells with progressively higher zoomed insets. b | Top: direct STORM (dSTORM) image of nuclear pore complexes (NPCs) labelled with antibodies to the nucleoporin Nup133. Three individual NPCs are shown on the right and an average image of 4,171 aligned NPCs on the lower right. Bottom left: coloured circles show radial positions of different nucleoporins in the plane of the nuclear envelope, determined from averaged dSTORM images, with the inferred position of the Y-shaped scaffold complex overlaid. Circle thickness reflects 95% confidence intervals of average radial distances. Bottom centre and bottom right: side and frontal views of the electron microscopy structure (grey), with the radial positions of nucleoporins shown in colour, and two positions of the Y complex consistent with the dSTORM data overlaid. c | Interferometric photoactivated localization microscopy (iPALM) image of a human U2OS cell expressing integrin αν-tdEos (left) and actin-mEos2 (right) with colour-coded zoomed insets of boxed regions. Colours represent the z position relative to the substrate (z = 0 nm). d | 3D STORM image of actin in a neuronal axon with zoomed y/z insets of boxed regions showing actin rings. Part a adapted with permission from REF.136, Elsevier. Part b reprinted with permission from REF.120, AAAS. Part c adapted from REF.168, Springer Nature Limited. Part d reprinted with permission from REF.58, AAAS.
SMLM has also been used to study higher-order chromatin structures including topologically associating domains. These are sub-megabase-long regions of DNA (~100–500 nm in size) determined by genome-wide chromosome conformation capture (Hi-C), within which DNA sequences interact more frequently with each other than with neighbouring DNA sequences135. 3D STORM in combination with multiplexed DNA fluorescence in situ hybridization methods showed that the boundaries of topologically associating domains are more variable in single cells than previously imagined20,143,144, highlighting the importance of visualizing these small domains with super-resolution in single cells. For recent reviews of high-resolution imaging of chromatin, see REFS145,146.
Transcriptional machinery and nuclear proteins.
Live-cell SMLM can visualize the dynamics and spatial organization of the transcriptional machinery147. Such studies showed that RNA Pol II assembles into transient nuclear clusters with lifetimes that correlate with their mRNA output, suggesting that they are transcriptionally active148. Follow-up work showed that some RNA Pol II clusters are phase-separated, liquid-droplet condensates that are particularly common in genomic regions known as super-enhancers149. SMLM further showed that several nuclear proteins, including architectural proteins such as CCCTC-binding factor and the Polycomb group proteins, form nanosized clusters within the nucleus150,151. Hence, clustering of nuclear proteins may be a general phenomenon for both shaping genome organization and regulating transcriptional output. Live-cell SMLM will be important to interrogate whether other nuclear proteins form phase-separated clusters, as these cannot be resolved using conventional microscopy techniques.
Nuclear pore complexes.
Nuclear pore complexes are excellent reference structures for characterizing and optimizing super-resolution microscopy modalities113,152,153 and SMLM studies have conversely helped to characterize their structure. As previously mentioned, the arrangement of individual subunits within the nuclear pore complex was elucidated by combining SMLM with single-particle averaging120,152 (FIG. 5b). This approach promises to be a powerful method for elucidating the subunit architecture of other large multi-protein complexes118,119.
Cytoplasmic contents
Microtubules.
Microtubules are tubulin polymers ~25 nm wide that form part of the cytoskeleton and facilitate several important cellular functions, including long-range intracellular transport of organelles and cell division. Microtubules were an early target for validating SMLM methods owing to the ease of labelling α-tubulin and β-tubulin subunits and because their shape and size were already well described79,80,154. Combining super-resolution imaging of microtubules with single-particle tracking of subcellular compartments allowed visualization of how these compartments are transported by the microtubule network in the cell cytoplasm155–157. These studies showed that a special subset of detyrosinated microtubule tracks play a crucial role in transporting and spatially organizing lysosomal and autophagosomal compartments to regulate autophagy157.
Mitochondria, lysosomes and the endoplasmic reticulum.
Common membrane probes specific to the mitochondria (for example, MitoTracker Red), endoplasmic reticulum (ER-Tracker Red) and lysosomes (LysoTracker Red) were found to be photoswitchable and used to characterize the dynamics of these organelles at high spatial (30–60 nm) and temporal (1–2 s) resolution, including fusion and fission dynamics in individual mitochondria60. SMLM techniques including spt-PALM, STORM and DNA-PAINT have resolved the organization of mitochondrial sub-compartments, including the mitochondrial inner and outer membranes, cristae and nucleoids158. Early studies have used PALM and interferometric PALM (iPALM), an advanced form of SMLM (see Outlook), to show that mitochondrial nucleoids are heterogeneous in shape and size and closely associated with the inner mitochondrial membrane, and that the mitochondrial DNA inside nucleoids is more condensed than previously thought159. Low-resolution images of the endoplasmic reticulum suggest that it consists of tubules and sheets, whereas a combination of lattice light sheet microscopy (see Outlook) and PAINT showed that endoplasmic reticulum sheets are actually very dense tubular structures160.
Plasma membrane
SMLM is ideally suited to study the molecular organization of the plasma membrane because it can be imaged with TIRF81, which affords improved resolution. Molecules in the plasma membrane can be imaged with fluorescently labelled antibodies, toxins or ligands. SMLM studies have shown that membrane receptors often cluster in small, functional nanodomains too small to be resolved with conventional light microscopy and investigated their organization. One early PALM study showed that clusters of glycosylphosphatidylinositol (GPI)-anchored proteins are disrupted by cholesterol depletion105. Recent studies used dSTORM to investigate the clustering of tumour necrosis factor receptor 1 (TNFR1)161 and Toll-like receptor 4 (TLR4)162 upon activation by their respective ligands, and detected an antigen on the surface of myeloma cells with implications for cancer immunotherapy163. Earlier SMLM studies found that T cell receptor (TCR) and linker for activation of T cells (LAT) are found in separate protein islands that merge after T cell activation164, with subsequent studies suggesting that TCR nanoclusters have a functional role in antigen recognition and signal initiation165. The latter studies have recently been challenged following results from dSTORM and live-cell PALM with varying label density and quantitative reanalysis that attribute apparent TCR clustering to overcounting artefacts166, emphasizing the importance of careful quantitative analyses of SMLM data. More applications of SMLM for investigating plasma membrane organization and associated challenges are discussed in REF.167.
The plasma membrane interacts with the extracellular matrix through focal adhesions, which are crucial both for cell adhesion and migration. iPALM was used to study the 3D organization of proteins within focal adhesions168. This work revealed that integrins and actin in focal adhesions are vertically separated by a 40-nm focal adhesion core region, consisting of a membrane-apposed layer containing integrin tails, paxillin and focal adhesion kinase, an intermediate layer with talin and vinculin, and an upper layer with actin regulators such as zyxin, α-actinin and vasodilator-stimulated phosphoprotein (FIG. 5c). Further SMLM studies identified talin — not vinculin — as the primary determinant of focal adhesion organization, as it diagonally spans the focal adhesion core region and acts as a molecular ruler169.
Neuronal synapses and axons
Neurons are highly compartmentalized cells with a polarized and extended morphology, in which the cell body is separated from axonal, dendritic and synaptic compartments. The size of a synapse is below the diffraction limit as the distance between presynaptic and postsynaptic structures is ~20–30 nm. SMLM has therefore been particularly useful in elucidating synaptic organization as it can resolve presynaptic and postsynaptic markers and determine the molecular architecture of proteins within synapses170. For example, SMLM revealed the nanoscopic organization of Bruchpilot (Brp) proteins within the synaptic active zone, a structure found within the presynaptic terminal in Drosophila171. Quantitative analysis estimated the precise stoichiometry of the Brp proteins within nanoscopic complexes, showing that there are hundreds of these proteins within the active zone, arranged in several heptameric clusters171. Moreover, SMLM has allowed the detection of AMPA and NMDA receptor nanoclusters within the postsynaptic region — a finding with important implications for the regulation of synaptic transmission172–174 — and a recent dSTORM study characterized glutamate receptor clustering at presynaptic active zones of mouse cerebellum175.
Neuronal axons are densely packed with proteins and therefore challenging to image with conventional microscopy. 3D STORM imaging showed that actin rings wrap around the circumference of neuronal axons, with each ring separated by <200 nm (REF.58) (FIG. 5d). This study showed that spectrin interconnects with the actin rings to form a quasi-1D lattice structure, also known as the membrane-associated periodic scaffold.
Microorganisms
SMLM is well adapted to study the inner structure of microorganisms and how they can subvert host cellular machinery for their own purposes. SMLM is especially useful for studying viruses, most of which are smaller than the diffraction limit. The HIV virion, for example, has a diameter of only ~100 nm. SMLM methods including spt-PALM have been used to characterize all aspects of the viral replication cycle from cellular entry, intracellular transport, replication, assembly, release and maturation for viruses such as HIV, herpes simplex virus and respiratory syncytial virus176–182. Further, SMLM approaches are expected to shed light on interactions of SARS-CoV-2 with host cells.
SMLM has been used to characterize the internal architecture of bacteria, for example to reveal the helical organization of the cytoskeletal protein FtsZ183 and the organization of nucleoid-associated proteins in Escherichia coli184. SMLM has also helped to clarify how bacteria interact with host cells, for example by visualizing how septin cages entrap Shigella as part of a cellular defence mechanism with dSTORM185. For more applications of super-resolution imaging in studying bacterial pathogens, see REF.186.
Reproducibility and data deposition
SMLM involves a much larger number of experimental and analytical parameters than conventional microscopy studies, and the intrinsic stochasticity of single-molecule blinking makes SMLM images particularly noisy. These factors pose a challenge when reproducing SMLM experiments and it is therefore especially important to carefully document experimental protocols and computational analyses and to make data and software available. Many free and open-source software tools for SMLM are now available and have been benchmarked99, but there is still a need to record and share the analysis parameters used, such as adjustable thresholds. Competitions and platforms that benchmark downstream analyses such as cluster identification or single-particle reconstruction will be useful to complement the existing challenges on localization, and to increase the reproducibility of quantitative analyses.
Image deposition is increasingly recognized as important, leading to data-sharing initiatives in the microscopy community, such as archiving data on Zenodo, FigShare, BioStudies and the Image Data Resource187. However, the large volume of images generated by SMLM experiments is an impediment to raw data sharing. Therefore, we propose that a good first step is to share localization data together with metadata detailing the target molecule or molecules and/or epitopes; the fluorescent labelling technique used, with details of antibody concentrations and the functionality of fluorescent protein tagging; the fixation protocol, if relevant; microscope parameters such as the laser wavelength, laser power density, camera type, exposure time, pixel size, objective magnification, numerical aperture and immersion medium; the reconstruction software and parameters used; and a localization table including x and y coordinates, frame numbers and, preferably, photon counts. Currently, no such resource exists, but a community platform that allows users to catalogue data with this information appended would improve data sharing and reuse, and improve reproducibility in SMLM.
Limitations and optimizations
Current SMLM techniques face numerous limitations, including susceptibility to reconstruction artefacts, difficulties in imaging thick samples or tissues, low throughput and limited applicability to live-cell imaging. In this section, we highlight some of these limitations and optimizations to address them.
Reconstruction artefacts and solutions
Several types of artefact can affect the quality of reconstructed super-resolution images, including drift, overlapping PSFs and localization biases.
Sample drift.
A very common issue in SMLM is sample drift relative to the objective during image sequence acquisition, which is often unavoidable even when using autofocus systems. If ignored, small drifts can result in blurring of the reconstructed image or the generation of artefactual features such as shadow microtubules79 (FIG. 6a). Drift can be measured by tracking fiducial markers, such as fluorescent beads — which should ideally remain fluorescent for the entire experiment — or gold particles, and localizations can then be corrected by subtracting the estimated drift (FIG. 6a). An interesting recent alternative is to estimate the drift using speckle patterns generated by backscattered light188. For highly redundant structures such as microtubules, lateral drift can be estimated directly from single-molecule localizations alone using image cross-correlation, although this requires caution189,190. Axial drift can also deteriorate 2D SMLM images; therefore, 3D drift correction methods should be used whenever possible102,188,191. Many SMLM software packages provide drift correction tools (TABLE 1). Unlike drift, high-frequency vibrations that affect localization precision by blurring individual, diffraction-limited frames cannot be corrected computationally. Therefore, it is important to isolate the microscope from sources of vibration. The best way to avoid residual vibrations and drift artefacts is to eliminate them using active, real-time adjustment of the stage position190,192,193; for example, fiducial markers added to the coverslip can be tracked with a second camera and a closed loop system to drive piezoelectrical actuators that precisely reposition the stage194. Although technically more demanding than correcting drift computationally, such approaches can reduce drift to single nanometres or less194–196.
Fig. 6 |. Limitations of single-molecule localization microscopy techniques.

a | Single-molecule localization microscopy (SMLM) image of microtubules before and after drift correction. Arrows show a fluorescent bead used to estimate the drift. b,c | Artefacts caused by point spread function (PSF) overlaps in simulated images. Ground truth image without localization errors, shown as a scatter plot (part b, left). Corresponding SMLM image for a low density of activated fluorophores (10 localizations per square micrometre (locs μm−2), no PSF overlaps) (part b, middle). Corresponding SMLM image for a high activation density (50 locs μm−2); overlapping PSFs cause artefactual localizations near the intersection of filaments at the centre (part b, right). Simulated molecular clusters, with a 10-fold higher density for the top cluster (part c). Simulated ground truth shown as a scatter plot (part c, left). Corresponding SMLM image without filtering (part c, middle). Corresponding SMLM image after filtering out poor localizations caused by overlapping PSFs (part c, right). After filtering, the high-density cluster is barely visible. d | Artefacts in SMLM images of microtubules resulting from subpixel localization bias. Left: without bias. Right: with bias caused by using localization software with an incorrect PSF model. Because of the bias, the reconstructed image shows a grid pattern. Insets show the entire field of view. The localization bias is readily apparent in the histogram of x coordinates relative to the centre of camera pixels. Drift correction was not applied to these data to better highlight the effect of localization bias.
Overlapping PSFs.
The basic principle of SMLM assumes spatially isolated PSFs; however, avoiding PSF overlaps entirely is difficult, especially in high-density regions or when a high activation probability is needed for rapid imaging. Standard localization algorithms tend to treat overlapping PSFs as a single molecule and compute an incorrect localization between the molecules. This can lead to artificial blurring occurring, for example, at intersecting filaments (FIG. 6b). In principle, the occurrence of such artefacts can be reduced by filtering out localizations for which the PSF model does not match the image well. However, stringent filtering can reject most localizations (hence reducing sampling and resolution) and cause high-density regions to appear dimmer than low-density regions in the reconstructed image (FIG. 6c). A simple but effective approach for detecting such artefacts is to verify whether a blurred version of the super-resolution image is consistent with the wide-field image. This approach yields a map that can highlight reconstruction errors197.
To analyse images with high activation density without generating these artefacts, ‘multi-emitter fitting’ algorithms have been developed that explicitly allow for PSF overlaps99,103 or compute differences between consecutive images to create images closer to single-molecule conditions198, which can then be processed to localize single molecules. Some related methods dispense of the notion of localizing single molecules altogether199, but can still produce super-resolution images by computationally exploiting temporal and spatial correlations of fluorescence fluctuations. Nevertheless, regardless of improvements in data analysis, overlapping PSFs lead to markedly poorer resolution than low-density SMLM regimes99,103,200.
Other biases.
Other artefacts can occur in the absence of drift or PSF overlaps. Localization bias within camera pixels can arise from poor convergence of MLE algorithms, for example caused by a mismatch between the model PSF and the actual PSF. This type of bias results in images with apparent grid patterns201 and can be easily identified by a non-uniform histogram of localizations relative to pixel centres (FIG. 6d). Spatially varying biases can be caused by a non-uniform background and are more difficult to identify, but can be reduced by background correction as described in the Results section above. A common issue in 3D SMLM when using engineered PSFs is aberrations due to the mismatch of refractive indexes between the sample and immersion oil, which can cause distortions along the axial dimension. Such distortions can be detected and prevented by careful calibration, typically by imaging fluorescent markers at known axial positions in the sample and adjusting the PSF model to fit them102,202,203 (BOX 3).
A fundamental and often overlooked localization bias stems from the fact that a PSF can depend on the orientation of a molecule’s dipole, which is generally unknown. If the dipole remains fixed or constrained to certain angles during exposure, ignoring dipole orientation can bias estimated molecule positions by up to ~125 nm (REF.204). Recent work has shown that molecular positions and dipole orientations can be simultaneously estimated in advanced set-ups that use polarizers and multiple cameras205. In standard SMLM systems, localization artefacts can be avoided by using fluorescent labels that rotate rapidly during exposure so that all possible dipole orientations are equally sampled and biases are averaged205.
Finally, non-specific detections can arise from autofluorescence or impurities on the coverslip. Autofluorescence is stronger at shorter wavelengths and can hence be alleviated using fluorophores with longer emission wavelengths and/or appropriate filters. Impurities can be reduced, although not entirely removed, by appropriate cleaning of coverslips, for example using plasma cleaners206.
Limitations for imaging tissues
Applying SMLM to tissues poses several challenges, some of which can be addressed by adapting sample preparation methods. Tissues are thicker than cells and must be sufficiently permeabilized to be labelled properly. However, extensive permeabilization can compromise tissue structure. Additionally, fixed thick tissues are more likely to have background fluorescence and increased light scattering. One way to overcome this problem is to cut tissues into thin slices and reconstruct a 3D SMLM image from many slices207. Cutting thin tissue slices often requires paraffin embedding of the tissue, which can make epitopes unavailable for antibody binding and decrease labelling efficiency. Performing antibody labelling prior to embedding and using specific resins for the embedding step, such as epoxy resin, can overcome this problem and maintain high photon output of fluorophores207. Tissue-clearing approaches can reduce the fluorescent background208 and, together with other optimizations, allowed for high-resolution imaging of chromatin using dSTORM in pathological tissue relevant for cancer diagnosis209. In this context, dSTORM was applied successfully to samples obtained by scalpel excision from tumour tissue210. Finally, SMLM puts additional demands on the way tissue samples are mounted as sample drift must be avoided and specialized aqueous imaging buffers are required. To address these issues, an imaging chamber was developed consisting of a permeable agarose pad and a custom-built stainless steel imaging adapter to keep tissue slices flat, immobile and bathed in imaging buffer during image acquisition208. Optical systems adapted to imaging thick samples and tissue without slicing will be discussed in the Outlook section.
Throughput limitations
A major limitation of SMLM techniques is their limited throughput, resulting from the small FOV required for single-molecule localization and the fact that it often takes several minutes or longer to obtain a high-quality super-resolution image of a single animal cell. These issues severely restrict the ability of SMLM to image large areas or tissue volumes, or to reveal rare cellular phenotypes. Furthermore, SMLM approaches are typically limited to a few colours because of the small number of spectrally distinguishable fluorophores available. Nevertheless, several efforts have been deployed to address these limitations, described below.
Larger fields of view.
Initial SMLM studies were restricted to a FOV of ~25 × 25 μm2 as they relied on EM-CCD cameras with 512 × 512 pixel arrays and because non-uniform laser illumination resulted in spatially varying photoswitching and photon emission rates, requiring cropping of images to a central region where illumination was approximately constant. Much larger FOVs can now be obtained using sCMOS cameras with larger arrays (2024 × 2024 pixels) and methods to homogenize illumination211–213. For example, a microlens array-based epi-illumination system known as flat illumination for field-independent imaging achieved a FOV of 100 × 100 μm2 (REF.212), allowing the imaging of about two to five mammalian cells simultaneously at high resolution.
Multiplexing.
Several multiplexing approaches have been developed to image many molecular species in the same sample214,215. DNA-PAINT is particularly amenable to multiplexing because DNA sequences are highly programmable and specific. In Exchange-PAINT215, different molecular targets are simultaneously labelled with orthogonal DNA docking strands, followed by sequential delivery, imaging and washing steps using complementary imager strands (FIG. 7). Each imager species can carry the same dye as the target identity is encoded by the respective sequence and imaging round, as opposed to the dye colour216. Similar approaches can sequentially label proteins using different primary antibodies and the same dye-conjugated secondary antibody; for example, multiplexed automated serial staining STORM, or maS3TORM, enabled localization of 15 target proteins in single cells214.
Fig. 7 |. Multiplexed single-molecule localization microscopy with Exchange-PAINT.

a | Exchange-PAINT implements sequential imaging of multiple targets by DNA point accumulation in nanoscale topography (DNA-PAINT) with different imager strands labelled with the same dye. Sample is labelled with orthogonal docking strands P1, P2, Pn, before the first imager strand species P1* — complementary to docking strand P1 — is introduced and a DNA-PAINT image of P1 is acquired. Next, the P1* imager strands are washed out, imager strands P2* are introduced and a DNA-PAINT image of P2 is acquired. This goes on for n cycles. Each DNA-PAINT image is assigned a distinct pseudocolour and n images are then superposed. b | Pseudocolour DNA-PAINT images of origami structures displaying the digits 0–9. Part a reprinted from REF.18, Springer Nature Limited. Part b reprinted from REF.215, Springer Nature Limited.
To increase throughput in multiplexed DNA-PAINT experiments, target molecules can be engineered to blink at a precise, adjustable frequency and duration, providing a distinct ‘kinetic barcode’ and allowing the simultaneous detection of hundreds of unique molecular species217. In a related approach, known as frequency multiplexing DNA-PAINT, excitation lasers are modulated between different frequencies and Fourier transformation is used to unmix fluorescent responses, enabling simultaneous imaging of several fluorophores218. Highly multiplexed approaches to visualize thousands of distinct RNA species or DNA loci in single cells are increasingly used, including multiplexed error-robust fluorescence in situ hybridization (MERFISH) or sequential barcoded fluorescence in situ hybridization (seqFISH+). Although they rely on sequential labelling rather than photoswitching, these methods give super-resolution information regarding the 3D structure of the genome and transcriptome. For more information, see REFS146,219.
Automation.
Performing high-throughput or multiplexed imaging for more than a few conditions or target proteins requires a fully automated microscope solution that allows fluid exchange and can perform staining, sample positioning and image acquisition. Various approaches have addressed these issues. High-content screening SMLM approaches using an automated microscope and automated data analysis workflow enabled 3D imaging of an entire 96-well plate220. Issues with fluid exchange have been tackled using a fluidics system composed of low-cost Lego® hardware controlled by ImageJ software221. Further, maS3TORM uses a fully automated and coordinated 3D SMLM microscope with a pipetting robot to perform staining experiments in situ214.
Faster SMLM.
The speed of SMLM is typically low. Nevertheless, tremendous progress in optimizing speed has been made since the first PALM study, which required ~2–12 h to image a single FOV. Using sCMOS cameras with high (>1 kHz) frame rates can dramatically increase the rate of localization, although high laser powers (>50 kW cm−2) are required to maintain good SNR and localization precision26,222. Such optimizations have allowed for two-colour 3D SMLM of more than 10,000 mammalian cells in ~26 h, as published in a recent preprint article223.
DNA-PAINT has traditionally been a particularly slow SMLM technique as the influx rate of dye-labelled imagers to their targets ultimately determines the achievable acquisition speed and is limited by the overall DNA association rate kON (~106 M−1 s−1), which leads to a blinking event every 100 s for typical imager concentrations of 10 nM. Increasing the imager concentration can speed up DNA-PAINT in principle, but concentrations in excess of a few tens of nanomoles increase background fluorescence, reducing the SNR and precision. Fluorescence resonance energy transfer (FRET)-based approaches have been implemented to address this issue224–226, effectively suppressing fluorescence caused by unbound imager strands. Orthogonal approaches such as rational sequence design and buffer optimizations can increase kON by an order of magnitude, therefore increasing the imaging speed227. Another approach to increase kON is based on preloading DNA-PAINT imager strands with Argonaute proteins228. Recently, the use of repeated, concatenated sequence motifs in rationally designed sequences has increased the speed of DNA-PAINT 100-fold over classical implementations229.
Cheaper SMLM.
Although the cost of SMLM hardware, especially custom-built set-ups, is moderate compared with other advanced microscopy systems, typical systems cost ~€100,000 or more. Efforts to replace expensive components using cheaper alternatives, for example replacing EM-CCDs with sCMOS cameras, replacing scientific-grade lasers with LEDs or leveraging smartphone technology have resulted in prototypes costing less than ~€10,000 (REFS77,230–232). Although these systems do not always achieve the spatial resolution of more sophisticated microscopes, such approaches may prove sufficient for many applications and could also enable large-scale parallelization and subsequent gains in throughput.
Structural dynamics limitations
Perhaps the most severe limitation of standard SMLM is its restricted applicability for studying structural dynamics in live cells. Live-cell structural studies are rare as collecting enough localizations to provide a super-resolution snapshot of a biological structure before it rearranges is challenging, when not impossible. Further, the high laser power required for fast turnover and rapid imaging is problematic for live cells because of rapid photobleaching and phototoxicity, especially when using UV activation34. Because of these constraints, structural SMLM studies in live cells have mostly been limited to proofs of principle on relatively slowly moving structures with few reconstructed snapshots34,85,90,222. As discussed above, methods designed for higher activation densities can overcome this limitation, to some extent, at the cost of a marked reduction in spatial resolution.
Outlook
We conclude this Primer with a look at advanced SMLM techniques and particularly promising ongoing developments. Although their implementation is often challenging, these advances can address some of the main remaining challenges in SMLM. These include imaging the ultrastructural context, moving towards true molecular resolution, imaging thick samples and tissues, imaging live samples without phototoxicity or photobleaching and more.
SMLM with light sheets
SMLM has been combined with light sheet fluorescence microscopy to overcome the challenges of imaging deep inside thick tissues when imaging live233 and fixed89 tissues. In light sheet fluorescence microscopy, the sample is illuminated with a thin light sheet perpendicular to the optical axis234. This optical sectioning approach is advantageous for living tissues as they are exposed to reduced irradiation intensity and less stress during imaging. Further, background fluorescence is significantly reduced and contrast is improved. In individual molecule localization-selective plane illumination microscopy, a cylindrical lens is used to create activation and read out light sheets to photoactivate single fluorescent molecules in thick tissue233. More recently, lattice light sheet microscopy was combined with 3D dSTORM to image plasma membrane receptors on the basal and apical membrane and perform 3D single-particle tracking235. Further, combining lattice light sheet microscopy with PAINT and newly developed membrane probes89 achieved multicolour 3D super-resolution imaging of samples up to 20 μm thick and whole-cell imaging of intracellular membranes. Light sheet SMLM can further be combined with adaptive optics to correct for PSF aberrations induced by scattering in the tissue, to improve the imaging depth and spatial resolution when imaging thick samples236.
Combining electron microscopy and SMLM
Electron microscopy generally offers even better spatial resolution than SMLM and also provides a global view of the cellular ultrastructure, which SMLM does not. Conversely, electron microscopy lacks the molecular specificity afforded by fluorescence imaging. Visualization of specific molecules within an ultrastructural context is possible by combining electron microscopy with optical microscopy in the same sample, a technique known as correlative light and electron microscopy237, which can also be extended to SMLM. Platinum replica electron microscopy — a technique in which the sample is coated with a heavy metal to increase its contrast in electron microscopy — was combined with iPALM and STORM to characterize distinct structural zones inside clathrin-coated structures238,239. Similarly, SMLM and platinum replica electron microscopy showed that actin rings found in neurons consist of long, intertwined actin filaments, revealing an unexpected braid-like organization240. Correlative cryogenic SMLM and electron tomography were used to study the subcellular localization of important regulatory proteins in Caulobacter crescentus241. In a recent tour de force, fluorescently labelled, high pressure-frozen cells were preserved in vitreous ice and imaged with cryogenic SMLM and 3D-focused ion beam scanning electron microscopy, and the images resulting from the two modalities were registered to nanoscale precision. This approach achieved ~40 nm resolution 3D visualization of the protein distribution within frozen whole cells in the context of their ultrastructure242, and revealed unexpected relationships between different cellular compartments, including the existence of endoplasmic reticulum proteins inside the nucleus. These correlative methods hold great promise for visualizing molecular complexes in their ultrastructural context.
SMLM with opposing objectives
Localization precision critically depends on the SNR; therefore, one way to improve resolution is to collect more photons from single fluorophores. This can be done using 4 Pi set-ups consisting of two opposing objectives that collect twice as many photons from single fluorophores as single objective systems, thus improving the localization precision by a factor of . Such systems enable lateral resolutions of <10 nm and an axial resolution of <20 nm in 3D STORM and have been used to visualize individual actin filaments, revealing two distinct layers of actin networks243. iPALM244 also uses two opposing objectives, but allows laser beams to interfere in a three-way beam splitter and propagate to three CCD cameras, allowing determination of the 3D coordinates of fluorescent molecules. In contrast to PSF shaping and multiplane systems (see BOX 3), iPALM does not trade off localization precision for axial range and has achieved isotropic, 3D resolution <20 nm using fluorescent proteins. More recently, an interferometric system has also been used to achieve 10–20 nm resolution imaging of the endoplasmic reticulum, mitochondria, nuclear pore complexes, bacteriophages and other structures in whole mammalian cells245. Careful calibration of refractive index variations will be needed to extend this approach to whole nuclei.
SMLM with minimum fluxes
The dominant paradigm to increase localization precision is to collect more photons from single fluorophores. However, in MINFLUX246, single fluorophores are localized according to the lowest emission fluxes that arise from a local minimum in excitation. In the original implementation, a doughnut-shaped excitation beam in combination with a three-point estimator was used to precisely determine emitter positions (FIG. 8a). In combination with photoswitching, MINFLUX attains single-nanometre resolution with considerably lower photon counts than standard SMLM (FIG. 8b). This allows for very high localization precision — even with relatively dim fluorescent proteins — and >100-fold faster single-molecule tracking than previous methods246.
Fig. 8 |. New directions in single-molecule localization microscopy.

a | MINFLUX excitation concept for precisely probing emitter positions using minimal photon fluxes. A doughnut-shaped excitation beam (green) is moved sequentially to four probing positions r0, r1, r2 and r3 (coloured circles; probing range L) in the vicinity of a single fluorophore (orange star). If the doughnut centre coincides perfectly with the fluorophore position, no photons are emitted. The position of the fluorophore can be calculated with very high precision from the fluorescence photon counts (shown below). b | Example nuclear pore complexes imaged by MINFLUX. c | In expansion microscopy, samples are embedded in a gel that expands upon hydration. Immunolabelling of epitopes can be performed before or after gelation and expansion using linkers that bind to the gel and to a fluorescent dye. Full or partial protein digestion is commonly used to enable isotropic expansion. In order to enable direct stochastic optical reconstruction microscopy (dSTORM) imaging in photoswitching buffer, the sample is re-embedded in an uncharged polyacrylamide gel after expansion. d | Left: 3D post-labelling expansion dSTORM image of a 3.2× expanded and re-embedded sample showing 9-fold symmetry of the procentriole. Scale bar: 500 nm. Right: 3.1-fold expanded and re-embedded tubulin filaments, with magnified view of highlighted region. An xz side-view cross-section of a tubulin filament (bottom right) shows its hollow structure. Scale bars: 500 nm (vertical rectangle), 200 nm (small square). e | Deep learning accelerates single-molecule localization microscopy (SMLM) image acquisition (ANNAPALM). A wide-field image (WF) and a sparse SMLM image obtained from only 300 frames are fed as inputs to an artificial neural network (ANN) that was previously trained on high-quality (long acquisition) SMLM images of microtubules. The ANN outputs a super-resolution image that is in good agreement with an SMLM image obtained from 30,000 frames (‘ground truth’), suggesting that ANNAPALM can reduce the acquisition time 100-fold without compromising spatial resolution. Part a adapted with permission from REF.246, AAAS. Part b reprinted from REF.249, Springer Nature Limited. Part d adapted from REF.255, CC BY 4.0 (https://creativecommons.org/licenses/by/4.0/). Part e adapted from REF.259, Springer Nature Limited.
Recently, two independent groups have used a standing wave instead of a doughnut-shaped excitation beam to spatially modulate excitation intensities, either using diffraction gratings (SIMFLUX247) or optical interference (repetitive optical selective exposure248). In both implementations, localization precision is improved ~2-fold over standard SMLM methods. MINFLUX can theoretically achieve arbitrarily high localization precision, although SIMFLUX and repetitive optical selective exposure currently offer faster acquisition speeds as image acquisition times do not scale with the FOV in these methods. With further advances in 3D, multicolour and live-cell imaging249 and hardware and software optimizations, these methods are poised to set a new standard for molecular-resolution SMLM.
SMLM in expanded samples
Whereas almost all super-resolution methods aim to diminish the size of the effective PSF2, expansion microscopy improves resolution by physically expanding the sample following embedding in a polyelectrolyte hydrogel250 (FIG. 8c). This approach can achieve super-resolution using conventional microscopes250,251, but combining it with SMLM can potentially improve resolution to single nanometres. An important challenge is that targeted molecules can lose their label after free-radical polymerization of the hydrogel, compromising labelling density and resolution252. Further, addition of the photoswitching buffer required for dSTORM can lead to hydrogel shrinking. Finally, expansion of pre-labelled samples results in further displacement of the fluorophore from the target molecule, increasing the linkage error. Recently, trifunctional linkers have been developed that are inert to polymerization, digestion and denaturation, and enable direct labelling and covalent linking of target molecules and functional groups to the hydrogel253,254. Re-embedding of the expanded hydrogel can prevent shrinking and post-expansion labelling preserves the ultrastructure of multi-protein complexes, improving labelling efficiency and reducing the linkage error255. The potential of these new methods has been demonstrated through imaging of clathrin-coated pits, microtubules and centrioles at ~5 nm resolution254,255 (FIG. 8d). Expansion SMLM thus provides another promising avenue for achieving true molecular resolution.
Enhancing SMLM using deep learning
In recent years, machine learning methods based on multilayer artificial neural networks (deep learning)256 have been used in SMLM data processing257. Deep learning has been used to extract single-molecule localizations from diffraction-limited images, including in conditions of high activation density, which can dramatically reduce the processing time compared with using MLE258. Deep learning can also help accelerate image acquisition itself by reconstructing high-quality super-resolution images from much fewer localizations than commonly believed necessary259 after learning structural redundancies from similar images (FIG. 8e). Such approaches could facilitate super-resolution imaging in live cells while minimizing phototoxicity, as has been recently demonstrated for 3D light sheet microscopy260. Deep learning will certainly also make inroads into the quantitative analysis of SMLM data, such as the segmentation of biological features261,262.
The power of neural networks comes from their ability to learn complex features from training data. However, this carries the risk that neural network outputs might be biased towards training data, miss novel phenotypes and generate artefacts257,259,263. Use of deep learning techniques for analysing SMLM data therefore requires careful validation, retraining of existing deep learning frameworks on new imaging data and the development of methods that are aware of or robust to mismatches between testing and training data. If these challenges are addressed, deep learning methods will likely play a key role in overcoming some of the major limitations of SMLM.
Concluding remarks
SMLM represents a powerful yet accessible family of imaging techniques that combines fluorophore chemistry, optics and computation to image and analyse biological structures and dynamics at increasingly high resolutions. In this Primer, we attempted to introduce all of the main basic aspects of SMLM, highlighting biological applications that illustrate the maturity of SMLM methods, while covering advanced techniques and promising recent developments. As the SMLM field continues to evolve, it brings us nearer to a future where all molecules within a cell, tissue or organism will be individually localized, counted and tracked. We hope that this Primer will encourage more investigators to adopt and further improve SMLM to explore the innermost architecture and mechanisms of living systems.
Supplementary Material
Acknowledgements
The authors apologize to the authors of numerous papers that could not be cited owing to limited space. M.Le. and C.Z. thank B. Lelandais for excellent comments on the manuscript and M. Singh for acquiring the image shown in Fig. 3b. C.Z. acknowledges funding by Institut Pasteur, Fondation pour la Recherche Médicale (grant DEQ 20150331762), Région Ile de France, Agence Nationale de la Recherche and Investissement d’Avenir grant ANR-16-CONV-0005. M.La. acknowledges funding from the National Institutes of Health/National Institutes of General Medical Sciences (NIH/NIGMS) under grant RO1 GM133842–01. G.B. and M.S. acknowledge funding by the German Research Foundation (DFG) (SA829/19–1) and the European Regional Development Fund (EFRE project ‘Center for Personalized Molecular Immunotherapy’). F.S. and R.J. acknowledge support by the DFG through SFB1032 (project A11) and the Max Planck Society. J.G. and S.M. acknowledge funding by the European Union’s H2020 programme under the Marie Skłodowska-Curie grant BALTIC (to J.G.) and ERC Piko (to S.M.).
Footnotes
Competing interests
R.J. is cofounder of Ultivue, Inc. and Massive Photonics GmbH. All other authors declare no competing interests.
Peer review information
Nature Reviews Methods Primers thanks T. Jovanovic-Talisman, A. Saftics, J. Ries, H. Zhang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Supplementary information
The online version contains supplementary material available at https://doi.org/10.1038/s43586-021-00038-x.
RELATED LINkS
BioStudies: https://www.ebi.ac.uk/biostudies/
FigShare: https://figshare.com/
Image Data Resource: https://idr.openmicroscopy.org/
SMLM software benchmarking: http://bigwww.epfl.ch/smlm
Zenodo: https://zenodo.org/
References
- 1.Abbe E Beiträge zur Theorie des Mikroskops und der mikroskopischen Wahrnehmung [German]. Arch. für Mikroskopische Anatomie 9, 413–468 (1873). [Google Scholar]
- 2.Schermelleh L et al. Super-resolution microscopy demystified. Nat. Cell Biol. 21, 72–84 (2019). [DOI] [PubMed] [Google Scholar]
- 3.Donnert G et al. Macromolecular-scale resolution in biological fluorescence microscopy. Proc. Natl Acad. Sci. USA 103, 11440–11445 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gustafsson MGL Surpassing the lateral resolution limit by a factor of two using structured illumination microscopy. J. Microsc. 198, 82–87 (2000). [DOI] [PubMed] [Google Scholar]
- 5.Heintzmann R, Jovin TM & Cremer C Saturated patterned excitation microscopy — a concept for optical resolution improvement. J. Opt. Soc. Am. A Opt Image Sci. Vis. 19, 1599–1609 (2002). [DOI] [PubMed] [Google Scholar]
- 6.Betzig E et al. Imaging intracellular fluorescent proteins at nanometer resolution. Science 313, 1642–1645 (2006). [DOI] [PubMed] [Google Scholar]
- 7.Hess ST, Girirajan TP & Mason MD Ultra-high resolution imaging by fluorescence photoactivation localization microscopy. Biophys. J. 91, 4258–4272 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rust MJ, Bates M & Zhuang X Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat. Methods 3, 793–795 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heilemann M et al. Subdiffraction-resolution fluorescence imaging with conventional fluorescent probes. Angew. Chem. Int. Ed. 47, 6172–6176 (2008). [DOI] [PubMed] [Google Scholar]
- 10.Sharonov A & Hochstrasser RM Wide-field subdiffraction imaging by accumulated binding of diffusing probes. Proc. Natl Acad. Sci. USA 103, 18911–18916 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lidke K, Rieger B, Jovin T & Heintzmann R Superresolution by localization of quantum dots using blinking statistics. Opt. Express 13, 7052–7062 (2005). [DOI] [PubMed] [Google Scholar]
- 12.van Oijen AM, Kohler J, Schmidt J, Muller M & Brakenhoff GJ 3-Dimensional super-resolution by spectrally selective imaging. Chem. Phys. Lett. 292, 183–187 (1998). [Google Scholar]
- 13.Gordon MP, Ha T & Selvin PR Single-molecule high-resolution imaging with photobleaching. Proc. Natl Acad. Sci. USA 101, 6462–6465 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heilemann M et al. High-resolution colocalization of single dye molecules by fluorescence lifetime imaging microscopy. Anal. Chem. 74, 3511–3517 (2002). [DOI] [PubMed] [Google Scholar]
- 15.Qu X, Wu D, Mets L & Scherer NF Nanometer-localized multiple single-molecule fluorescence microscopy. Proc. Natl Acad. Sci. USA 101, 11298–11303 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Biteen JS et al. Super-resolution imaging in live Caulobacter crescentus cells using photoswitchable EYFP. Nat. Methods 5, 947–949 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Folling J et al. Fluorescence nanoscopy by ground-state depletion and single-molecule return. Nat. Methods 5, 943–945 (2008). [DOI] [PubMed] [Google Scholar]
- 18.Schnitzbauer J, Strauss MT, Schlichthaerle T, Schueder F & Jungmann R Super-resolution microscopy with DNA-PAINT. Nat. Protoc. 12, 1198–1228 (2017). [DOI] [PubMed] [Google Scholar]
- 19.Manley S et al. High-density mapping of single-molecule trajectories with photoactivated localization microscopy. Nat. Methods 5, 155–157 (2008). [DOI] [PubMed] [Google Scholar]
- 20.Bintu B et al. Super-resolution chromatin tracing reveals domains and cooperative interactions in single cells. Science 362, 6413 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Weisenburger S et al. Cryogenic optical localization provides 3D protein structure data with Angstrom resolution. Nat. Methods 14, 141–144 (2017). [DOI] [PubMed] [Google Scholar]
- 22.Klein T, Proppert S & Sauer M Eight years of single-molecule localization microscopy. Histochem. Cell Biol. 141, 561–575 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van de Linde S et al. Direct stochastic optical reconstruction microscopy with standard fluorescent probes. Nat. Protoc. 6, 991–1009 (2011). [DOI] [PubMed] [Google Scholar]
- 24.Bates M, Blosser TR & Zhuang X Short-range spectroscopic ruler based on a single-molecule optical switch. Phys. Rev. Lett. 94, 108101 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Heilemann M, Margeat E, Kasper R, Sauer M & Tinnefeld P Carbocyanine dyes as efficient reversible single-molecule optical switch. J. Am. Chem. Soc. 127, 3801–3806 (2005). [DOI] [PubMed] [Google Scholar]
- 26.Lin Y et al. Quantifying and optimizing single-molecule switching nanoscopy at high speeds. PLoS ONE 10, e0128135 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wombacher R et al. Live-cell super-resolution imaging with trimethoprim conjugates. Nat. Methods 7, 717–719 (2010). [DOI] [PubMed] [Google Scholar]
- 28.Andresen M et al. Structural basis for reversible photoswitching in Dronpa. Proc. Natl Acad. Sci. USA 104, 13005 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Diekmann R et al. Optimizing imaging speed and excitation intensity for single-molecule localization microscopy. Nat. Methods 17, 909–912 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bossi M et al. Multicolor far-field fluorescence nanoscopy through isolated detection of distinct molecular species. Nano Lett. 8, 2463–2468 (2008). [DOI] [PubMed] [Google Scholar]
- 31.Grimm JB et al. Bright photoactivatable fluorophores for single-molecule imaging. Nat. Methods 13, 985–988 (2016). [DOI] [PubMed] [Google Scholar]
- 32.Michie MS et al. Cyanine conformational restraint in the far-red range. J. Am. Chem. Soc. 139, 12406–12409 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lukyanov KA, Chudakov DM, Lukyanov S & Verkhusha VV Photoactivatable fluorescent proteins. Nat. Rev. Mol. Cell Biol. 6, 885–890 (2005). [DOI] [PubMed] [Google Scholar]
- 34.Shroff H, Galbraith CG, Galbraith JA & Betzig E Live-cell photoactivated localization microscopy of nanoscale adhesion dynamics. Nat. Methods 5, 417–423 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Turkowyd B, Virant D & Endesfelder U From single molecules to life: microscopy at the nanoscale. Anal. Bioanal. Chem. 408, 6885–6911 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang S, Moffitt JR, Dempsey GT, Xie XS & Zhuang X Characterization and development of photoactivatable fluorescent proteins for single-molecule-based superresolution imaging. Proc. Natl Acad. Sci. USA 111, 8452–8457 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McEvoy AL et al. mMaple: a photoconvertible fluorescent protein for use in multiple imaging modalities. PLoS ONE 7, e51314 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Uno SN et al. A spontaneously blinking fluorophore based on intramolecular spirocyclization for live-cell super-resolution imaging. Nat. Chem. 6, 681–689 (2014). [DOI] [PubMed] [Google Scholar]
- 39.Takakura H et al. Long time-lapse nanoscopy with spontaneously blinking membrane probes. Nat. Biotechnol. 35, 773–780 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Uno SN, Kamiya M, Morozumi A & Urano Y A green-light-emitting, spontaneously blinking fluorophore based on intramolecular spirocyclization for dual-colour super-resolution imaging. Chem. Commun. 54, 102–105 (2017). [DOI] [PubMed] [Google Scholar]
- 41.Macdonald PJ et al. Rhodamine-derived fluorescent dye with inherent blinking behavior for super-resolution imaging. Anal. Chem. 90, 9165–9173 (2018). [DOI] [PubMed] [Google Scholar]
- 42.Giannone G et al. Dynamic superresolution imaging of endogenous proteins on living cells at ultra-high density. Biophys. J. 99, 1303–1310 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jungmann R et al. Single-molecule kinetics and super-resolution microscopy by fluorescence imaging of transient binding on DNA origami. Nano Lett. 10, 4756–4761 (2010). [DOI] [PubMed] [Google Scholar]
- 44.Dai M, Jungmann R & Yin P Optical imaging of individual biomolecules in densely packed clusters. Nat. Nanotechnol. 11, 798–807 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shcherbakova DM, Sengupta P, Lippincott-Schwartz J & Verkhusha VV Photocontrollable fluorescent proteins for superresolution imaging. Annu. Rev. Biophys. 43, 303–329 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thorn K Genetically encoded fluorescent tags. Mol. Biol. Cell 28, 848–857 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rodriguez EA et al. The growing and glowing toolbox of fluorescent and photoactive proteins. Trends Biochem. Sci. 42, 111–129 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li H & Vaughan JC Switchable fluorophores for single-molecule localization microscopy. Chem. Rev. 118, 9412–9454 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fernandez-Suarez M & Ting AY Fluorescent probes for super-resolution imaging in living cells. Nat. Rev. Mol. Cell Biol. 9, 929–943 (2008). [DOI] [PubMed] [Google Scholar]
- 50.Ries J, Kaplan C, Platonova E, Eghlidi H & Ewers H A simple, versatile method for GFP-based super-resolution microscopy via nanobodies. Nat. Methods 9, 582–584 (2012). [DOI] [PubMed] [Google Scholar]
- 51.Conic S et al. Imaging of native transcription factors and histone phosphorylation at high resolution in live cells. J. Cell Biol. 217, 1537–1552 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Herce HD et al. Cell-permeable nanobodies for targeted immunolabelling and antigen manipulation in living cells. Nat. Chem. 9, 762–771 (2017). [DOI] [PubMed] [Google Scholar]
- 53.Schnell U, Dijk F, Sjollema KA & Giepmans BN Immunolabeling artifacts and the need for live-cell imaging. Nat. Methods 9, 152–158 (2012). [DOI] [PubMed] [Google Scholar]
- 54.Yan Q & Bruchez MP Advances in chemical labeling of proteins in living cells. Cell Tissue Res. 360, 179–194 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liss V, Barlag B, Nietschke M & Hensel M Self-labelling enzymes as universal tags for fluorescence microscopy, super-resolution microscopy and electron microscopy. Sci. Rep. 5, 17740 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lelek M, Di Nunzio F & Zimmer C in Exocytosis and Endocytosis Methods in Molecular Biology (ed. Ivanov AI) 183–193 (Springer, 2014). [DOI] [PubMed] [Google Scholar]
- 57.Klein T et al. Live-cell dSTORM with SNAP-tag fusion proteins. Nat. Methods 8, 7–9 (2011). [DOI] [PubMed] [Google Scholar]
- 58.Xu K, Zhong G & Zhuang X Actin, spectrin, and associated proteins form a periodic cytoskeletal structure in axons. Science 339, 452–456 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wijesooriya CS et al. A photoactivatable BODIPY probe for localization-based super-resolution cellular imaging. Angew. Chem. Int. Ed. 57, 12685–12689 (2018). [DOI] [PubMed] [Google Scholar]
- 60.Shim SH et al. Super-resolution fluorescence imaging of organelles in live cells with photoswitchable membrane probes. Proc. Natl Acad. Sci. USA 109, 13978–13983 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Neubert F et al. Bioorthogonal click chemistry enables site-specific fluorescence labeling of functional NMDA receptors for super-resolution imaging. Angew. Chem. Int. Ed. 57, 16364–16369 (2018). [DOI] [PubMed] [Google Scholar]
- 62.Beliu G et al. Bioorthogonal labeling with tetrazine-dyes for super-resolution microscopy. Commun. Biol. 2, 261 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lang K & Chin JW Cellular incorporation of unnatural amino acids and bioorthogonal labeling of proteins. Chem. Rev. 114, 4764–4806 (2014). [DOI] [PubMed] [Google Scholar]
- 64.Lew MD et al. Three-dimensional superresolution colocalization of intracellular protein superstructures and the cell surface in live Caulobacter crescentus. Proc. Natl Acad. Sci. USA 108, E1102–1110 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Spahn CK et al. A toolbox for multiplexed super-resolution imaging of the E. coli nucleoid and membrane using novel PAINT labels. Sci. Rep. 8, 14768 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Agasti SS et al. DNA-barcoded labeling probes for highly multiplexed Exchange-PAINT imaging. Chem. Sci. 8, 3080–3091 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schlichthaerle T et al. Direct visualization of single nuclear pore complex proteins using genetically-encoded probes for DNA-PAINT. Angew. Chem. Int. Ed. 58, 13004–13008 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sograte-Idrissi S et al. Circumvention of common labelling artefacts using secondary nanobodies. Nanoscale 12, 10226–10239 (2020). [DOI] [PubMed] [Google Scholar]
- 69.Schlichthaerle T et al. Site-specific labeling of affimers for DNA-PAINT microscopy. Angew. Chem. Int. Ed. 57, 11060–11063 (2018). [DOI] [PubMed] [Google Scholar]
- 70.Strauss S et al. Modified aptamers enable quantitative sub-10-nm cellular DNA-PAINT imaging. Nat. Methods 15, 685–688 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Richter KN et al. Glyoxal as an alternative fixative to formaldehyde in immunostaining and super-resolution microscopy. EMBO J. 37, 139–159 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tanaka KA et al. Membrane molecules mobile even after chemical fixation. Nat. Methods 7, 865–866 (2010). [DOI] [PubMed] [Google Scholar]
- 73.Baumgart F et al. Varying label density allows artifact-free analysis of membrane-protein nanoclusters. Nat. Methods 13, 661–664 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tuijtel MW, Koster AJ, Jakobs S, Faas FGA & Sharp TH Correlative cryo super-resolution light and electron microscopy on mammalian cells using fluorescent proteins. Sci. Rep. 9, 1369 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kopek BG et al. Diverse protocols for correlative super-resolution fluorescence imaging and electron microscopy of chemically fixed samples. Nat. Protoc. 12, 916–946 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Waldchen S, Lehmann J, Klein T, van de Linde S & Sauer M Light-induced cell damage in live-cell super-resolution microscopy. Sci. Rep. 5, 15348 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Holm T et al. A blueprint for cost-efficient localization microscopy. ChemPhysChem 15, 651–654 (2014). [DOI] [PubMed] [Google Scholar]
- 78.Kwakwa K et al. easySTORM: a robust, lower-cost approach to localisation and TIRF microscopy. J. Biophotonics 9, 948–957 (2016). [DOI] [PubMed] [Google Scholar]
- 79.Henriques R et al. QuickPALM: 3D real-time photoactivation nanoscopy image processing in ImageJ. Nat. Methods 7, 339–340 (2010). [DOI] [PubMed] [Google Scholar]
- 80.Huang B, Wang W, Bates M & Zhuang X Three-dimensional super-resolution imaging by stochastic optical reconstruction microscopy. Science 319, 810–813 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mashanov GI, Tacon D, Knight AE, Peckham M & Molloy JE Visualizing single molecules inside living cells using total internal reflection fluorescence microscopy. Methods 29, 142–152 (2003). [DOI] [PubMed] [Google Scholar]
- 82.Tokunaga M, Imamoto N & Sakata-Sogawa K Highly inclined thin illumination enables clear single-molecule imaging in cells. Nat. Methods 5, 159–161 (2008). [DOI] [PubMed] [Google Scholar]
- 83.Thompson RE, Larson DR & Webb WW Precise nanometer localization analysis for individual fluorescent probes. Biophys. J. 82, 2775–2783 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ober RJ, Ram S & Ward ES Localization accuracy in single-molecule microscopy. Biophys. J. 86, 1185–1200 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Holden SJ et al. High throughput 3D super-resolution microscopy reveals Caulobacter crescentus in vivo Z-ring organization. Proc. Natl Acad. Sci. USA 111, 4566–4571 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kechkar A, Nair D, Heilemann M, Choquet D & Sibarita JB Real-time analysis and visualization for single-molecule based super-resolution microscopy. PLoS ONE 8, e62918 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mund M et al. Systematic nanoscale analysis of endocytosis links efficient vesicle formation to patterned actin nucleation. Cell 174, 884–896.e17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Fox-Roberts P et al. Local dimensionality determines imaging speed in localization microscopy. Nat. Commun. 8, 13558–13558 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Legant WR et al. High-density three-dimensional localization microscopy across large volumes. Nat. Methods 13, 359–365 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Jones SA, Shim SH, He J & Zhuang X Fast, three-dimensional super-resolution imaging of live cells. Nat. Methods 8, 499–505 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gelles J, Schnapp BJ & Sheetz MP Tracking kinesin-driven movements with nanometre-scale precision. Nature 331, 450–453 (1988). [DOI] [PubMed] [Google Scholar]
- 92.Sergé A, Bertaux N, Rigneault H & Marguet D Dynamic multiple-target tracing to probe spatiotemporal cartography of cell membranes. Nat. Methods 5, 687–694 (2008). [DOI] [PubMed] [Google Scholar]
- 93.Elf J & Barkefors I Single-molecule kinetics in living cells. Annu. Rev. Biochem. 88, 635–659 (2019). [DOI] [PubMed] [Google Scholar]
- 94.Ovesny M, Kiek P, Borkovec J, Vindrych Z & Hagen GM ThunderSTORM: a comprehensive ImageJ plug-in for PALM and STORM data analysis and super-resolution imaging. Bioinformatics 30, 2389–2390 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cheng C-Y & Hsieh C-L Background estimation and correction for high-precision localization microscopy. ACS Photonics 4, 1730–1739 (2017). [Google Scholar]
- 96.Kay SM Fundamentals of Statistical Signal Processing Vol. 2: Detection Theory (Prentice Hall PTR, 1998). [Google Scholar]
- 97.Kay SM Fundamentals of Statistical Signal Processing Vol. 1: Estimation Theory (Prentice Hall PTR, 1993). [Google Scholar]
- 98.Mortensen KI, Churchman LS, Spudich JA & Flyvbjerg H Optimized localization analysis for single-molecule tracking and super-resolution microscopy. Nat. Methods 7, 377–381 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sage D et al. Super-resolution fight club: assessment of 2D and 3D single-molecule localization microscopy software. Nat. Methods 16, 387–395 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Small A & Stahlheber S Fluorophore localization algorithms for super-resolution microscopy. Nat. Methods 11, 267–279 (2014). [DOI] [PubMed] [Google Scholar]
- 101.Li Y et al. Real-time 3D single-molecule localization using experimental point spread functions. Nat. Methods 15, 367–369 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Aristov A, Lelandais B, Rensen E & Zimmer C ZOLA-3D allows flexible 3D localization microscopy over an adjustable axial range. Nat. Commun. 9, 2409 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sage D et al. Quantitative evaluation of software packages for single-molecule localization microscopy. Nat. Methods 12, 717–724 (2015). [DOI] [PubMed] [Google Scholar]
- 104.Annibale P, Vanni S, Scarselli M, Rothlisberger U & Radenovic A Identification of clustering artifacts in photoactivated localization microscopy. Nat. Methods 8, 527–528 (2011). [DOI] [PubMed] [Google Scholar]
- 105.Sengupta P et al. Probing protein heterogeneity in the plasma membrane using PALM and pair correlation analysis. Nat. Methods 8, 969–975 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Rubin-Delanchy P et al. Bayesian cluster identification in single-molecule localization microscopy data. Nat. Methods 12, 1072–1076 (2015). [DOI] [PubMed] [Google Scholar]
- 107.Khater IM, Nabi IR & Hamarneh G A review of super-resolution single-molecule localization microscopy cluster analysis and quantification methods. Patterns 1, 100038 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Levet F et al. SR-Tesseler: a method to segment and quantify localization-based super-resolution microscopy data. Nat. Methods 12, 1065–1071 (2015). [DOI] [PubMed] [Google Scholar]
- 109.Durisic N, Laparra-Cuervo L, Sandoval-Alvarez A, Borbely JS & Lakadamyali M Single-molecule evaluation of fluorescent protein photoactivation efficiency using an in vivo nanotemplate. Nat. Methods 11, 156–162 (2014). [DOI] [PubMed] [Google Scholar]
- 110.Bohrer CH et al. A pairwise distance distribution correction (DDC) algorithm to eliminate blinking-caused artifacts in super-resolution microscopy. Preprint at bioRxiv 10.1101/768051 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Veatch SL et al. Correlation functions quantify super-resolution images and estimate apparent clustering due to over-counting. PLoS ONE 7, e31457 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zanacchi FC et al. A DNA origami platform for quantifying protein copy number in super-resolution. Nat. Methods 14, 789–792 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Thevathasan JV et al. Nuclear pores as versatile reference standards for quantitative superresolution microscopy. Nat. Methods 16, 1045–1053 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Finan K, Raulf A & Heilemann M A set of homo-oligomeric standards allows accurate protein counting. Angew. Chem. Int. Ed. 54, 12049–12052 (2015). [DOI] [PubMed] [Google Scholar]
- 115.Rollins GC, Shin JY, Bustamante C & Presse S Stochastic approach to the molecular counting problem in superresolution microscopy. Proc. Natl Acad. Sci. USA 112, E110–118 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Lee S-H, Shin JY, Lee A & Bustamante C Counting single photoactivatable fluorescent molecules by photoactivated localization microscopy (PALM). Proc. Natl Acad. Sci. USA 109, 17436–17441 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Jungmann R et al. Quantitative super-resolution imaging with qPAINT. Nat. Methods 13, 439–442 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Salas D et al. Angular reconstitution-based 3D reconstructions of nanomolecular structures from superresolution light-microscopy images. Proc. Natl Acad. Sci. USA 114, 9273–9278 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Sieben C, Banterle N, Douglass KM, Gönczy P & Manley S Multicolor single-particle reconstruction of protein complexes. Nat. Methods 15, 777–780 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Szymborska A et al. Nuclear pore scaffold structure analyzed by super-resolution microscopy and particle averaging. Science 341, 655–658 (2013). [DOI] [PubMed] [Google Scholar]
- 121.Bolte S & Cordelières FP A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 224, 213–232 (2006). [DOI] [PubMed] [Google Scholar]
- 122.Lagache T et al. Mapping molecular assemblies with fluorescence microscopy and object-based spatial statistics. Nat. Commun. 9, 698 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Levet F et al. A tessellation-based colocalization analysis approach for single-molecule localization microscopy. Nat. Commun. 10, 2379 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Malkusch S et al. Coordinate-based colocalization analysis of single-molecule localization microscopy data. Histochem. Cell Biol. 137, 1–10 (2012). [DOI] [PubMed] [Google Scholar]
- 125.Monnier N et al. Bayesian approach to MSD-based analysis of particle motion in live cells. Biophys. J. 103, 616–626 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Beheiry ME, Dahan M & Masson J-B InferenceMAP: mapping of single-molecule dynamics with Bayesian inference. Nat. Methods 12, 594–595 (2015). [DOI] [PubMed] [Google Scholar]
- 127.Martin DS, Forstner MB & Kas JA Apparent subdiffusion inherent to single particle tracking. Biophys. J. 83, 2109–2117 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Masson JB et al. Mapping the energy and diffusion landscapes of membrane proteins at the cell surface using high-density single-molecule imaging and Bayesian inference: application to the multiscale dynamics of glycine receptors in the neuronal membrane. Biophys. J. 106, 74–83 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Manzo C & Garcia-Parajo MF A review of progress in single particle tracking: from methods to biophysical insights. Rep. Prog. Phys. 78, 124601 (2015). [DOI] [PubMed] [Google Scholar]
- 130.Peters R, Griffié J, Burn GL, Williamson DJ & Owen DM Quantitative fibre analysis of single-molecule localization microscopy data. Sci. Rep. 8, 10418 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Zhang Z, Nishimura Y & Kanchanawong P Extracting microtubule networks from superresolution single-molecule localization microscopy data. Mol. Biol. Cell 28, 333–345 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Gyparaki MT et al. Tau forms oligomeric complexes on microtubules that are distinct from tau aggregates. Proc. Natl Acad. Sci. USA 118, e2021461118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Nicovich PR, Owen DM & Gaus K Turning single-molecule localization microscopy into a quantitative bioanalytical tool. Nat. Protoc. 12, 453–460 (2017). [DOI] [PubMed] [Google Scholar]
- 134.Deschout H, Shivanandan A, Annibale P, Scarselli M & Radenovic A Progress in quantitative single-molecule localization microscopy. Histochem. Cell Biol. 142, 5–17 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Parmar JJ, Woringer M & Zimmer C How the genome folds: the biophysics of four-dimensional chromatin organization. Annu. Rev. Biophys. 48, 231–253 (2019). [DOI] [PubMed] [Google Scholar]
- 136.Ricci MA, Manzo C, García-Parajo MF, Lakadamyali M & Cosma MP Chromatin fibers are formed by heterogeneous groups of nucleosomes in vivo. Cell 160, 1145–1158 (2015). [DOI] [PubMed] [Google Scholar]
- 137.Nozaki T et al. Dynamic organization of chromatin domains revealed by super-resolution live-cell imaging. Mol. Cell 67, 282–293.e7 (2017). [DOI] [PubMed] [Google Scholar]
- 138.Gomez-Garcia PA et al. Mesoscale modeling and single-nucleosome tracking reveal remodeling of clutch folding and dynamics in stem cell differentiation. Cell Rep. 34, 108614 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Otterstrom J et al. Super-resolution microscopy reveals how histone tail acetylation affects DNA compaction within nucleosomes in vivo. Nucleic Acids Res. 47, 8470–8484 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Xu J et al. Super-resolution imaging of higher-order chromatin structures at different epigenomic states in single mammalian cells. Cell Rep. 24, 873–882 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Zessin PJ, Finan K & Heilemann M Super-resolution fluorescence imaging of chromosomal DNA. J. Struct. Biol. 177, 344–348 (2012). [DOI] [PubMed] [Google Scholar]
- 142.Hao X et al. Super-resolution visualization and modeling of human chromosomal regions reveals cohesin-dependent loop structures. Genome Biol. 22, 150 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Nir G et al. Walking along chromosomes with super-resolution imaging, contact maps, and integrative modeling. PLoS Genet. 14, e1007872 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Cardozo Gizzi AM et al. Microscopy-based chromosome conformation capture enables simultaneous visualization of genome organization and transcription in intact organisms. Mol. Cell 74, 212–222.e5 (2019). [DOI] [PubMed] [Google Scholar]
- 145.Lakadamyali M & Cosma MP Visualizing the genome in high resolution challenges our textbook understanding. Nat. Methods 17, 371–379 (2020). [DOI] [PubMed] [Google Scholar]
- 146.Boettiger A & Murphy S Advances in chromatin imaging at kilobase-scale resolution. Trends Genet. 36, 273–287 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Cisse II et al. Real-time dynamics of RNA polymerase II clustering in live human cells. Science 341, 664–667 (2013). [DOI] [PubMed] [Google Scholar]
- 148.Cho WK et al. RNA Polymerase II cluster dynamics predict mRNA output in living cells. eLife 5, e13617 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Cho WK et al. Mediator and RNA polymerase II clusters associate in transcription-dependent condensates. Science 361, 412–41 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Hansen AS, Pustova I, Cattoglio C, Tjian R & Darzacq X CTCF and cohesin regulate chromatin loop stability with distinct dynamics. eLife 6, e25776 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Wani AH et al. Chromatin topology is coupled to Polycomb group protein subnuclear organization. Nat. Commun. 7, 1–13 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Löschberger A et al. Super-resolution imaging visualizes the eightfold symmetry of gp210 proteins around the nuclear pore complex and resolves the central channel with nanometer resolution. J. Cell Sci. 125, 570–575 (2012). [DOI] [PubMed] [Google Scholar]
- 153.Broeken J et al. Resolution improvement by 3D particle averaging in localization microscopy. Methods Appl. Fluoresc. 3, 014003–014003 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Zhu L, Zhang W, Elnatan D & Huang B Faster STORM using compressed sensing. Nat. Methods 9, 721–723 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Bálint Š, Vilanova IV, Álvarez ÁS & Lakadamyali M Correlative live-cell and superresolution microscopy reveals cargo transport dynamics at microtubule intersections. Proc. Natl Acad. Sci. USA 110, 3375–3380 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Verdeny-Vilanova I et al. 3D motion of vesicles along microtubules helps them to circumvent obstacles in cells. J. Cell Sci. 130, 1904–1916 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Mohan N, Sorokina EM, Verdeny IV, Alvarez AS & Lakadamyali M Detyrosinated microtubules spatially constrain lysosomes facilitating lysosome–autophagosome fusion. J. Cell Biol. 218, 632–643 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Jakobs S, Stephan T, Ilgen P & Bruser C Light microscopy of mitochondria at the nanoscale. Annu. Rev. biophysics 49, 289–308 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Brown TA et al. Superresolution fluorescence imaging of mitochondrial nucleoids reveals their spatial range, limits, and membrane interaction. Mol. Cell. Biol. 31, 4994–5010 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Nixon-Abell J et al. Increased spatiotemporal resolution reveals highly dynamic dense tubular matrices in the peripheral ER. Science 354, aaf3928 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Karathanasis C et al. Single-molecule imaging reveals the oligomeric state of functional TNFα-induced plasma membrane TNFR1 clusters in cells. Sci. Signal. 13, eaax5647 (2020). [DOI] [PubMed] [Google Scholar]
- 162.Neumann J et al. Nanoscale distribution of TLR4 on primary human macrophages stimulated with LPS and ATI. Nanoscale 11, 9769–9779 (2019). [DOI] [PubMed] [Google Scholar]
- 163.Nerreter T et al. Super-resolution microscopy reveals ultra-low CD19 expression on myeloma cells that triggers elimination by CD19 CAR-T. Nat. Commun. 10, 3137 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Lillemeier BF et al. TCR and Lat are expressed on separate protein islands on T cell membranes and concatenate during activation. Nat. Immunol. 11, 90–96 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Pageon SV et al. Functional role of T-cell receptor nanoclusters in signal initiation and antigen discrimination. Proc. Natl Acad. Sci. USA 113, E5454–E5463 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Rossboth B et al. TCRs are randomly distributed on the plasma membrane of resting antigen-experienced T cells. Nat. Immunol. 19, 821–827 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Stone MB, Shelby SA & Veatch SL Super-resolution microscopy: shedding light on the cellular plasma membrane. Chem. Rev. 117, 7457–7477 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Kanchanawong P et al. Nanoscale architecture of integrin-based cell adhesions. Nature 468, 580–584 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Liu J et al. Talin determines the nanoscale architecture of focal adhesions. Proc. Natl Acad. Sci. USA 112, E4864–E4873 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Dani A, Huang B, Bergan J, Dulac C & Zhuang X Superresolution imaging of chemical synapses in the brain. Neuron 68, 843–856 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Ehmann N et al. Quantitative super-resolution imaging of Bruchpilot distinguishes active zone states. Nat. Commun. 5, 1–12 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Nair D et al. Super-resolution imaging reveals that AMPA receptors inside synapses are dynamically organized in nanodomains regulated by PSD95. J. Neurosci. 33, 13204–13224 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Kellermayer B et al. Differential nanoscale topography and functional role of GluN2-NMDA receptor subtypes at glutamatergic synapses. Neuron 100, 106–119.e7 (2018). [DOI] [PubMed] [Google Scholar]
- 174.Ladépêche L et al. NMDA receptor autoantibodies in autoimmune encephalitis cause a subunit-specific nanoscale redistribution of NMDA receptors. Cell Rep. 23, 3759–3768 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Siddig S et al. Super-resolution imaging reveals the nanoscale organization of metabotropic glutamate receptors at presynaptic active zones. Sci. Adv. 6, eaay7193 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Lehmann M et al. Quantitative multicolor super-resolution microscopy reveals tetherin HIV-1 interaction. PLoS Pathog. 7, e1002456 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Chojnacki J & Eggeling C Super-resolution fluorescence microscopy studies of human immunodeficiency virus. Retrovirology 15, 41 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Pak AJ et al. Immature HIV-1 lattice assembly dynamics are regulated by scaffolding from nucleic acid and the plasma membrane. Proc. Natl Acad. Sci. USA 114, E10056–E10065 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Lelek M et al. Superresolution imaging of HIV in infected cells with FlAsH-PALM. Proc. Natl Acad. Sci. USA 109, 8564–8569 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Van Engelenburg SB et al. Distribution of ESCRT machinery at HIV assembly sites reveals virus scaffolding of ESCRT subunits. Science 343, 653–656 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Laine RF et al. Structural analysis of herpes simplex virus by optical super-resolution imaging. Nat. Commun. 6, 5980 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Rincheval V et al. Functional organization of cytoplasmic inclusion bodies in cells infected by respiratory syncytial virus. Nat. Commun. 8, 563 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Fu G et al. In vivo structure of the E. coli FtsZ-ring revealed by photoactivated localization microscopy (PALM). PLoS ONE 5, e12682 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Wang W, Li GW, Chen C, Xie XS & Zhuang X Chromosome organization by a nucleoid-associated protein in live bacteria. Science 333, 1445–1449 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Mostowy S et al. Entrapment of intracytosolic bacteria by septin cage-like structures. Cell Host Microbe 8, 433–444 (2010). [DOI] [PubMed] [Google Scholar]
- 186.Singh MK & Kenney LJ Super-resolution imaging of bacterial pathogens and visualization of their secreted effectors. FEMS Microbiol. Rev. 45, 1–12 (2020). [DOI] [PubMed] [Google Scholar]
- 187.Williams E et al. The Image Data Resource: a bioimage data integration and publication platform. Nat. Methods 14, 775–781 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Chen SY, Heintzmann R & Cremer C Sample drift estimation method based on speckle patterns formed by backscattered laser light. Biomed. Opt. Express 10, 6462–6475 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Wang Y et al. Localization events-based sample drift correction for localization microscopy with redundant cross-correlation algorithm. Opt. Express 22, 15982–15991 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.McGorty R, Kamiyama D & Huang B Active microscope stabilization in three dimensions using image correlation. Opt. Nanoscopy 2, 3 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Mlodzianoski MJ et al. Sample drift correction in 3D fluorescence photoactivation localization microscopy. Opt. Express 19, 15009 (2011). [DOI] [PubMed] [Google Scholar]
- 192.Lee SH et al. Using fixed fiduciary markers for stage drift correction. Opt. Express 20, 12177–12183 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Grover G, Mohrman W & Piestun R Real-time adaptive drift correction for super-resolution localization microscopy. Opt. Express 23, 23887–23898 (2015). [DOI] [PubMed] [Google Scholar]
- 194.Carter AR et al. Stabilization of an optical microscope to 0.1 nm in three dimensions. Appl. Opt. 46, 421–427 (2007). [DOI] [PubMed] [Google Scholar]
- 195.Coelho S et al. Ultraprecise single-molecule localization microscopy enables in situ distance measurements in intact cells. Sci. Adv. 6, eaay8271 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Pertsinidis A, Zhang Y & Chu S Subnanometre single-molecule localization, registration and distance measurements. Nature 466, 647–651 (2010). [DOI] [PubMed] [Google Scholar]
- 197.Culley S et al. Quantitative mapping and minimization of super-resolution optical imaging artifacts. Nat. Methods 15, 263–266 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Marsh RJ et al. Artifact-free high-density localization microscopy analysis. Nat. Methods 15, 689–692 (2018). [DOI] [PubMed] [Google Scholar]
- 199.Dertinger T, Colyer R, Iyer G, Weiss S & Enderlein J Fast, background-free, 3D super-resolution optical fluctuation imaging (SOFI). Proc. Natl Acad. Sci. USA 106, 22287–22292 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Ram S, Ward ES & Ober RJ Beyond Rayleigh’s criterion: a resolution measure with application to single-molecule microscopy. Proc. Natl Acad. Sci. USA 103, 4457–4462 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Gould TJ, Verkhusha VV & Hess ST Imaging biological structures with fluorescence photoactivation localization microscopy. Nat. Protoc. 4, 291–308 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Cabriel C, Bourg N, Dupuis G & Lévêque-Fort S Aberration-accounting calibration for 3D single-molecule localization microscopy. Opt. Lett. 43, 174–177 (2018). [DOI] [PubMed] [Google Scholar]
- 203.Li Y, Wu Y-L, Hoess P, Mund M & Ries J Depth-dependent PSF calibration and aberration correction for 3D single-molecule localization. Biomed. Opt. Express 10, 2708–2718 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Engelhardt J et al. Molecular orientation affects localization accuracy in superresolution far-field fluorescence microscopy. Nano Lett. 11, 209–213 (2011). [DOI] [PubMed] [Google Scholar]
- 205.Cruz CAV et al. Quantitative nanoscale imaging of orientational order in biological filaments by polarized superresolution microscopy. Proc. Natl Acad. Sci. USA 113, E820–E828 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Davis JL, Dong B, Sun C & Zhang HF Method to identify and minimize artifacts induced by fluorescent impurities in single-molecule localization microscopy. J. Biomed. Opt. 23, 1–14 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Sigal YM, Speer CM, Babcock HP & Correspondence XZ Mapping synaptic input fields of neurons with super-resolution imaging in brief. Cell 163, 493–505 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.German CL, Gudheti MV, Fleckenstein AE & Jorgensen EM Brain slice staining and preparation for three-dimensional super-resolution microscopy. Methods Mol. Biol. 1663, 153–162 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Xu J et al. Super-resolution imaging reveals the evolution of higher-order chromatin folding in early carcinogenesis. Nat. Commun. 11, 1–17 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Tobin SJ et al. Single molecule localization microscopy coupled with touch preparation for the quantification of trastuzumab-bound HER2. Sci. Rep. 8, 15154 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Archetti A et al. Waveguide-PAINT offers an open platform for large field-of-view super-resolution imaging. Nat. Commun. 10, 1267 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Douglass KM, Sieben C, Archetti A, Lambert A & Manley S Super-resolution imaging of multiple cells by optimized flat-field epi-illumination. Nat. Photonics 10, 705–708 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Stehr F, Stein J, Schueder F, Schwille P & Jungmann R Flat-top TIRF illumination boosts DNA-PAINT imaging and quantification. Nat. Commun. 10, 1268 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Klevanski M et al. Automated highly multiplexed super-resolution imaging of protein nano-architecture in cells and tissues. Nat. Commun. 11, 1–11 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Jungmann R et al. Multiplexed 3D cellular super-resolution imaging with DNA-PAINT and Exchange-PAINT. Nat. Methods 11, 313–318 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Schueder F et al. Universal super-resolution multiplexing by DNA exchange. Angew. Chem. Int. Ed. 56, 4052–4055 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Wade OK et al. 124-Color super-resolution imaging by engineering DNA-PAINT blinking kinetics. Nano Lett. 19, 2641–2646 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Gómez-García PA, Garbacik ET, Otterstrom JJ, Garcia-Parajo MF & Lakadamyali M Excitation-multiplexed multicolor superresolution imaging with fm-STORM and fm-DNA-PAINT. Proc. Natl Acad. Sci. USA 115, 12991–12996 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Zhuang X Spatially resolved single-cell genomics and transcriptomics by imaging. Nat. Methods 18, 18–22 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Beghin A et al. Localization-based super-resolution imaging meets high-content screening. Nat. Methods 14, 1184–1190 (2017). [DOI] [PubMed] [Google Scholar]
- 221.Almada P et al. Automating multimodal microscopy with NanoJ-Fluidics. Nat. Commun. 10, 1–9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Huang F et al. Video-rate nanoscopy using sCMOS camera-specific single-molecule localization algorithms. Nat. Methods 10, 653–658 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Barentine AES et al. 3D multicolor nanoscopy at 10,000 cells a day. Preprint at bioRxiv 10.1101/606954 (2019). [DOI] [Google Scholar]
- 224.Auer A, Strauss MT, Schlichthaerle T & Jungmann R Fast, background-free DNA-PAINT imaging using FRET-based probes. Nano Lett. 17, 6428–6434 (2017). [DOI] [PubMed] [Google Scholar]
- 225.Lee J, Park S & Hohng S Accelerated FRET-PAINT microscopy. Mol. Brain 11, 70 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Lee J, Park S, Kang W & Hohng S Accelerated super-resolution imaging with FRET-PAINT. Mol. Brain 10, 63 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Schueder F et al. An order of magnitude faster DNA-PAINT imaging by optimized sequence design and buffer conditions. Nat. Methods 16, 1101–1104 (2019). [DOI] [PubMed] [Google Scholar]
- 228.Filius M et al. High-speed super-resolution imaging using protein-assisted DNA-PAINT. Nano Lett. 20, 2264–2270 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Strauss S & Jungmann R Up to 100-fold speed-up and multiplexing in optimized DNA-PAINT. Nat. Methods. 17, 789–791 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Diederich B, Then P, Jugler A, Forster R & Heintzmann R cellSTORM-Cost-effective super-resolution on a cellphone using dSTORM. PLoS ONE 14, e0209827 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Ma H, Fu R, Xu J & Liu Y A simple and cost-effective setup for super-resolution localization microscopy. Sci. Rep. 7, 1542 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Auer A et al. Nanometer-scale multiplexed super-resolution imaging with an economic 3D-DNA-PAINT microscope. Chemphyschem 19, 3024–3034 (2018). [DOI] [PubMed] [Google Scholar]
- 233.Cella Zanacchi F et al. Live-cell 3D super-resolution imaging in thick biological samples. Nat. Methods 8, 1047–1050 (2011). [DOI] [PubMed] [Google Scholar]
- 234.Huisken J, Swoger J, Del Bene F, Wittbrodt J & Stelzer EH Optical sectioning deep inside live embryos by selective plane illumination microscopy. Science 305, 1007–1009 (2004). [DOI] [PubMed] [Google Scholar]
- 235.Waldchen F et al. Whole-cell imaging of plasma membrane receptors by 3D lattice light-sheet dSTORM. Nat. Commun. 11, 887 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Mlodzianoski MJ et al. Active PSF shaping and adaptive optics enable volumetric localization microscopy through brain sections. Nat. Methods 15, 583–586 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Bykov YS, Cortese M, Briggs JA & Bartenschlager R Correlative light and electron microscopy methods for the study of virus-cell interactions. FEBS Lett. 590, 1877–1895 (2016). [DOI] [PubMed] [Google Scholar]
- 238.Sochacki KA, Shtengel G, van Engelenburg SB, Hess HF & Taraska JW Correlative super-resolution fluorescence and metal-replica transmission electron microscopy. Nat. Methods 11, 305–308 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Sochacki KA, Dickey AM, Strub MP & Taraska JW Endocytic proteins are partitioned at the edge of the clathrin lattice in mammalian cells. Nat. Cell Biol. 19, 352–361 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Vassilopoulos S, Gibaud S, Jimenez A, Caillol G & Leterrier C Ultrastructure of the axonal periodic scaffold reveals a braid-like organization of actin rings. Nat. Commun. 10, 1–13 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Dahlberg PD et al. Cryogenic single-molecule fluorescence annotations for electron tomography reveal in situ organization of key proteins in Caulobacter. Proc. Natl Acad. Sci. USA 117, 13937–13944 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Hoffman DP et al. Correlative three-dimensional super-resolution and block-face electron microscopy of whole vitreously frozen cells. Science 367, eaaz5357 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Xu K, Babcock HP & Zhuang X Dual-objective STORM reveals three-dimensional filament organization in the actin cytoskeleton. Nat. Methods 9, 185–188 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Shtengel G et al. Interferometric fluorescent super-resolution microscopy resolves 3D cellular ultrastructure. Proc. Natl Acad. Sci. USA 106, 3125–3130 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Huang F et al. Ultra-high resolution 3D imaging of whole cells. Cell 166, 1028–1040 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Balzarotti F et al. Nanometer resolution imaging and tracking of fluorescent molecules with minimal photon fluxes. Science 355, 606–612 (2017). [DOI] [PubMed] [Google Scholar]
- 247.Cnossen J et al. Localization microscopy at doubled precision with patterned illumination. Nat. Methods 17, 59–63 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Gu L et al. Molecular resolution imaging by repetitive optical selective exposure. Nat. Methods 16, 1114–1118 (2019). [DOI] [PubMed] [Google Scholar]
- 249.Gwosch KC et al. MINFLUX nanoscopy delivers 3D multicolor nanometer resolution in cells. Nat. Methods 17, 217–224 (2020). [DOI] [PubMed] [Google Scholar]
- 250.Chen F, Tillberg PW & Boyden ES Expansion microscopy. Science 347, 543–548 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Chang JB et al. Iterative expansion microscopy. Nat. Methods 14, 593–599 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Tillberg PW et al. Protein-retention expansion microscopy of cells and tissues labeled using standard fluorescent proteins and antibodies. Nat. Biotechnol. 34, 987–992 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Wen G et al. Evaluation of direct grafting strategies via trivalent anchoring for enabling lipid membrane and cytoskeleton staining in expansion microscopy. ACS Nano 14, 7860–7867 (2020). [DOI] [PubMed] [Google Scholar]
- 254.Shi X et al. Label-retention expansion microscopy. Preprint at bioRxiv 10.1101/687954 (2019). [DOI] [Google Scholar]
- 255.Zwettler FU et al. Molecular resolution imaging by post-labeling expansion single-molecule localization microscopy (Ex-SMLM). Nat. Commun. 11, 3388 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.LeCun Y, Bengio Y & Hinton G Deep learning. Nature 521, 436–444 (2015). [DOI] [PubMed] [Google Scholar]
- 257.Mockl L, Roy AR & Moerner WE Deep learning in single-molecule microscopy: fundamentals, caveats, and recent developments [Invited]. Biomed. Opt. Express 11, 1633–1661 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Nehme E et al. DeepSTORM3D: dense 3D localization microscopy and PSF design by deep learning. Nat. Methods 17, 734–740 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Ouyang W, Aristov A, Lelek M, Hao X & Zimmer C Deep learning massively accelerates super-resolution localization microscopy. Nat. Biotechnol. 36, 460–468 (2018). [DOI] [PubMed] [Google Scholar]
- 260.Weigert M et al. Content-aware image restoration: pushing the limits of fluorescence microscopy. Nat. Methods 15, 1090 (2018). [DOI] [PubMed] [Google Scholar]
- 261.Falk T et al. U-Net: deep learning for cell counting, detection, and morphometry. Nat. Methods 16, 67–70 (2019). [DOI] [PubMed] [Google Scholar]
- 262.Haberl MG et al. CDeep3M-Plug-and-Play cloud-based deep learning for image segmentation. Nat. Methods 15, 677–680 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Belthangady C & Royer LA Applications, promises, and pitfalls of deep learning for fluorescence image reconstruction. Nat. Methods 16, 1215–1225 (2019). [DOI] [PubMed] [Google Scholar]
- 264.Ries J SMAP: a modular super-resolution microscopy analysis platform for SMLM data. Nat. Methods 17, 870–872 (2020). [DOI] [PubMed] [Google Scholar]
- 265.Nieuwenhuizen RPJ et al. Measuring image resolution in optical nanoscopy. Nat. Methods 10, 557–562 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Descloux A, Grussmayer KS & Radenovic A Parameter-free image resolution estimation based on decorrelation analysis. Nat. Methods 16, 918–924 (2019). [DOI] [PubMed] [Google Scholar]
- 267.Bates M, Huang B, Dempsey GT & Zhuang X Multicolor super-resolution imaging with photoswitchable fluorescent probes. Science 317, 1749–1753 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Flottmann B et al. Correlative light microscopy for high-content screening. Biotechniques 55, 243–252 (2013). [DOI] [PubMed] [Google Scholar]
- 269.Yan R, Moon S, Kenny SJ & Xu K Spectrally resolved and functional super-resolution microscopy via ultrahigh-throughput single-molecule spectroscopy. Acc. Chem. Res. 51, 697–705 (2018). [DOI] [PubMed] [Google Scholar]
- 270.Testa I et al. Multicolor fluorescence nanoscopy in fixed and living cells by exciting conventional fluorophores with a single wavelength. Biophys. J. 99, 2686–2694 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Dong B et al. Super-resolution spectroscopic microscopy via photon localization. Nat. Commun. 7, 12290 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Muranyi W, Malkusch S, Muller B, Heilemann M & Krausslich HG Super-resolution microscopy reveals specific recruitment of HIV-1 envelope proteins to viral assembly sites dependent on the envelope C-terminal tail. PLoS Pathog. 9, e1003198 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Pavani SRP et al. Three-dimensional, single-molecule fluorescence imaging beyond the diffraction limit by using a double-helix point spread function. Proc. Natl Acad. Sci. USA 106, 2995–2999 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Juette MF et al. Three-dimensional sub-100 nm resolution fluorescence microscopy of thick samples. Nat. Methods 5, 527–529 (2008). [DOI] [PubMed] [Google Scholar]
- 275.Abrahamsson S et al. Fast multicolor 3D imaging using aberration-corrected multifocus microscopy. Nat. Methods 10, 60–63 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Hajj B, Beheiry ME, Izeddin I, Darzacq X & Dahan M Accessing the third dimension in localization-based super-resolution microscopy. Phys. Chem. Chem. Phys. 16, 16340–16348 (2014). [DOI] [PubMed] [Google Scholar]
- 277.Shechtman Y, Sahl SJ, Backer AS & Moerner WE Optimal point spread function design for 3D imaging. Phys. Rev. Lett. 10.1103/PhysRevLett.113.133902 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Ram S, Prabhat P, Ward ES & Ober RJ Improved single particle localization accuracy with dual objective multifocal plane microscopy. Opt. Express 17, 6881–6898 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.