

Abstract
Sulfated polysaccharides and their oligosaccharide components play an important biological role in physiological and pathophysiological processes. Glycosaminoglycans, such as heparin, are currently used as pharmaceutical agents to control coagulation. Moreover, there arc intensive efforts in developing rapid approaches for the analysis of such bioactive carbohydrates to assess their pharmacological activity and in glycomic applications. Soft ionization methods of mass spectrometry have been applied with recent success in the analysis of sulfated carbohydrates. This review will examine the application of mass spectrometry in the compositional analysis and structural characterization of these important highly charged molecules.
Keywords: Mass spectrometry, sulfated carbohydrates, glycosaminoglycan, oligosaccharide, structure, sequence
1. INTRODUCTION
Sulfated polysaccharides and their oligosaccharide components play an important biological role in physiological and pathophysiological processes. In proteoglycan (PC) biosynthesis, sulfated polysaccharides are present as side chains through covalent attachment to a protein core, PGs are found in all animals from C. elegans to man, in the extracellular matrix and basement membranes as a structural scaffold [1]. These biomolecules also play an essential role in regulating biochemical pathways, cellular communication and cell signaling [2–5]. The great structural diversity of the glycosaminoglycan (GAG) portion of PGs associated with variations in polysaccharide type, chain length and composition, the degree of functional group substitution and distribution of polysaccharide chains within the core protein. This structural diversity contributes into the complexity of PGs and it is also critical for the appropriate functioning of PGs [6–9].
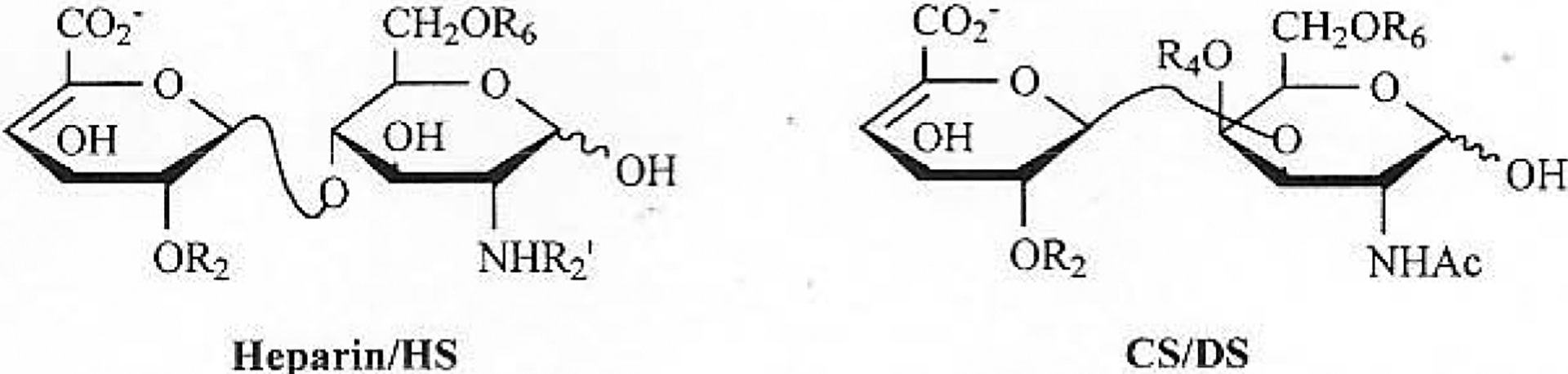
GAGs are a family of highly sulfated, complex, polydisperse linear polysaccharides that display a variety of important biological roles. Based on the difference of repeating disaccharide units comprising GAGs, they can be categorized into four main groups: heparin/heparan sulfate (HS) group (including the invertebrate-derived acharan sulfate (AS)), chondroitin sulfate (CS)/dermatan sulfate (DS) groups, keratin sulfate (KS) and hyaluronan (HA) (Fig. 1).
Fig. (1).

Major and minor disaccharide sequences of heparin/HS and major disaccharide sequences of CS/DS, KS, HA.
Heparin and HS a mixture of sulfated linear polysaccharides, having a molecular weight (MW) range from 5, 000 to 40, 000 with an MW average of 10, 000 to 25, 000. The major repeating disaccharide sequence of heparin (75–90%) is [→4)-2-sulfo-α-L-idopyranosyluronic acid (1→4)-2-amino, 2-deoxy-2, 6-sulfo-α-D-N-glucopyranosylsamine (1→], while minor sequences contain β-D-glucopyranosyluronic acid (GlcAp) residues, a reduced content of sulfo groups as well as N-acetylation. Heparin is O-linked to serine residues of the core protein serglycin and is found intra cellularly in the granules of mast cells. HS is O-linked to serine residues of a number, of core proteins resulting in a number of PGs including glypican and syndecan. HS has the similar structure with heparin, primarily containing nonsulfated disaccharides [→4) β-D-GlcAp (1→4)-α-D-GlcNpAc (1→] and monosulfated disaccharide such as [→4) β-D-GlcAp(1→4)-6-sulfo-α-D-N-GlcNpAc] [10–12].
Heparin is commonly used as a clinical anticoagulant [3, 5]. It also has other biological activities with potential clinical applications, such as effects on lipoprotein lipase, effects on smooth muscle proliferation, inhibition of complement activation, anti-inflammatory activity, angiogenic and antiangiogenic activities, anticancer activity and antiviral activity [2, 4, 9]. HS has a number of important biological activities. It has polysaccharide components that bind antithrombin, supporting blood flow across the vascular endothelium [5]. It is also important in cell-cell interaction involving adhesion proteins, cell-cell communication involving chemokines and cell signaling involving growth factors [2, 4, 13].
The CS/DS family of GAGs is comprised of alternating 1→3, 1→4 linked 2-amino, 2-deoxy-α-D-galactopyranose (GalNpAc) and uronic acid (either β-D-GlcAp or α-L-IdoAp) residue [10, 14]. The molecular weights of CS/DS range from 2 to 50 kD. There are multiple forms of chondroitin sulfate named A, B, C, D, E and K, difference based on sulfo group substitution and the type of ureic acid that each contains [15]. This family is the most common type of GAG, found in extracellular matrix proteoglycans. Some of these PGs are small, containing one or two GAG chains, such as decorin and biglycan, while others, such as aggrecan, are extremely large having many GAG chains. Chondroitin sulfate is important in cell-cell interaction and communication, and dermatan sulfate exhibits important venous antithrombotic activity [16, 17]. Recent research revealed that CS and DS have intriguing functions in infection, inflammation, growth factor signaling, morphogenesis and cell division [15, 18].
KS has two different forms, KS I and II [19]. In the large PGs of the cartilage KS II is present, O-linked to core protein through serine, while in small KS PGs are N-linked to core protein through asparagine of cartilage and cornea, it is type I. KS is comprised primarily of 6-sulfo-GlcNpAc and galactose (which may contain 6-O-sulfo groups). The functional importance of KS is still not clear. But it is clear that structure changes of KS chains in articular cartilage are age-related [20].
Hyaluronic acid (HA) is a homocopolymer made of repeating disaccharide units of [→3)-D-GlcNpAc(1→4)-β-D-GlcAp(1→] [21]. It is an unsulfated glycosaminoglycan, with very high MW of up to 2000 kDa. HA is widely distributed in cartilage, skin, eye, and most body liquids. HA is not found covalently linked to core protein and can interact with a variety of proteins including link protein, cell surface receptors RHAMM and CD44.
GAGs perform a variety of biological functions and play an important role in a number of different diseases. Their activities are mainly triggered by interactions with a wide range of proteins [5]. Experiments have shown that specific sequences within heparin and HS, act as protein binding sites [22]. This suggests that polysaccharides may work as informational molecules and suggests the importance of developing analytical techniques, to sequence GAG molecules [23, 24]. The primary structure determination of biopolymers, such as proteins and nucleic acids, are commonly solved by automated sequencing of amino acid residues and nucleotide residues, respectively. In contrast with current state of the art methods, the sequencing of the polysaccharides having a high level of structural complexity is still extremely challenging.
MS analysis provides a spectrum showing ion abundance as a function of mass-to-charge ratio (m/z), from which the molecular weight of an analyte can be precisely determined. With the development of soft-ionizing sources, such as fast atom bombardment (FAB), matrix-assisted laser-desorption-ionization (MALDI) and electro spray ionization (ESI), MS has become one of the most important technique to sequence biopolymers. MS has most recently been applied to analyze sulfated oligosaccharides and polysaccharides. However, the high acidity of sulfo groups and their liability towards fragmentation, have made such analyses problematic. Another difficulty associated with the MS analysis of sulfated carbohydrate natural products is the enormous structure complexity of their repeating disaccharide units. There are hundreds of different possible disaccharide units that can comprise GAGs, in contrast to four bases that make up DNA and RNA, and the twenty amino acids that make up proteins.
In addition to their sequence microheterogeneity, GAGs are polydisperse mixtures, further complicating their direct analysis by MS. To reduce this structural complexity, GAGs are often first partially depolymerizcd into smaller oligosaccharide chains using either chemical or enzymatic methods. Enzymatic methods offer milder conditions and are uncomplicated by the competing side-reactions, often observed using chemical depolymerization methods. Specific polysaccharide lyases are often used to partially depolymerize GAGs to oligosaccharides from which, sequence information can be obtained [25–27]. Several commercially available polysaccharide lyases: their action pattern and specificity are presented in Table 1. These lyases act on the linkage between hexosamine (HexNp) and uronic acid (UAp) through eliminative cleavage affording an unsaturated uronate residue ΔUAp at the non-reducing end of the newly-formed product.
Table 1.
Polysaccharide Lyases used in GAGs Analysis
| Enzyme | Action Patterna | Spccificityb |
|---|---|---|
| Chondroitin lyase ACI | CS (endo) | →3)-α-D-GalNpAc(4S/OH,6S/OH)(1→4)-β-D-GlcAp(1→ |
| Chondroitin lyase ACII | CS (exo) | →3)-α-D-GalNpAc(4S/OH,6S/OH)(1→4)-β-D-GlcAp(1→ |
| Chondroitin lyase B | DS (endo) | →3)-α-D-GalNpAc(4S/OH)(1→4)- α-L-IdoAp(1→ |
| Chondroitin lyase ABC | CS/DS/HA (endo) | →3)-α-D-GalNpAc(4S/6S/OH)(1→4)HexUAp(1→ |
| Chondroitin lyase ABC | CS/DS (exo) | →3)-α-D-GalNpAc(4S/6S/OH)(1→4)HexUAp(1→ |
| Heparin lyase I | H/HS (endo) | →4)-α-D-GlcNpS(6S/OH)(1→4)-α-L-IdoAp2S(1→ |
| Heparin lyase II | H/HS (endo) | →4)-α-D-GlcNp(S/Ac)(6S/OH)(1→4)-α-L-IdoAp/-β-D-GIcAp(2S/OH)(1→ |
| Heparin lyase III | HS (endo) | →4)-α-D-GlcNp(Ac/S)(6OH/S)(1→4) -β-D-GlcAp/-α-L-IdoAp(1→ |
| Hyaluronate lyase | HA (endo) | →3)- α-D-GlcNpAc(1→4)-β-D-GlcAp(1→ |
endo, random endolytic; exo, random exolytic90
GalNp, 2-amino,2-deoxygalactopyranose; Ac, acetyl; S, sulfo; GlcAp, glucopyranosyluronic acid; GlcNp, 2-amino,2-deoxyglucopyranose; IdoAp, idopyranosyluronic acid; UAp is GlcAp, or IdoAp.
Despite the difficulties associated with the MS analysis of highly sulfated carbohydrates, progress has been made in both MS methodology and the application of MS for the sequencing and analysis of these important highly charged natural products. This article reviews recent research advance on the MS analysis of highly sulfated carbohydrates.
2. MS DETERMINATION OF MOLECULAR WEIGHT
The primary application of MS analysis of highly sulfated carbohydrates, has been made for the determination of molecular weight. From these data the size of the chain, degree of sulfation and the presence of other substitution, such as N-acetylation, can be obtained. To observe molecular-ions, soft-ionization methods including FAB, MALDI and ESI, have been applied, which facilitate the transfer of these highly charged ions to the gas phase with a low level of accompanying fragmentation.
2.1. FAB-MS
FAB-MS was the first MS technique widely applied to the analysis of sulfated oligosaccharides. A series of heparin oligosaccharides, prepared by chemical synthesis with known structures, were analyzed by FAB-MS in positive-ion and negative-ion modes, using 1-thioglycerol as FAB matrix. Groups of ions related to the molecular weight were observed by FAB-MS. These groups of ions and their dispersion were due to variations in the resident counterations (H+, Na+, K+), as well as to the addition or removal of a proton or alkali-metal cation, required to generate the singly charged ions. Extensive fragmentation related directly to loss of SO3, were observed due to the breakage of the facile S-O bond in the sulfo groups of these oligosaccharides. The negative-ion mode gave better sensitivity than positive-ion mode and oligosaccharides of size up to a pentasaccharide with of mass of 1667, were analyzed. These results suggested that FAB-MS might be useful for the analysis of sulfated glycosaminoglycans but that facile loss of SO3 limited molecular weight analysis, sequence analysis and the maximum size of oligosaccharides, that could be effectively analyzed under these conditions [28].
While acidic oligosaccharides were clearly analyzed in the FAB-MS negative-ion mode, 1-thioglycerol did not aid in the production of structurally significant fragment-ions. Triethanolamine was introduced as FAB matrix for the analysis of a series of heparin-derived oligosaccharides in an effort to enhance the formation of both molecular-ions and structurally significant fragment-ions. The FAB mass spectra of heparin disaccharide, tetrasaccharide and hexasaccharide exhibited molecular ions as well as fragment-ions, produced by glycosidic linkage-fragmentation. For example, the tetrasaccharide ΔIdoAp2S(1→4)-α-D-GlcNp2S6S(1→4)-β-D-GlcNp(1→4)-α-D-GlcNp2S6S lost one sugar residue from its reducing terminus, one sugar residue from its nonreducing terminus, and two sugar residues from its reducing terminus in the FAB ionization process, affording definitive sequence information [29] (Fig. 2).
Fig. (2).

Negative-ion FAB-MS spectrum of a tetrasaccharide. The structure is presented in the polyanionic form (M - 7H). The molecular ion is the hexasodiated, monoanionic species [M + 6Na −7H]- at m/z 1205. (Adapted from ref. 29).
FAB-MS with triethanolamine matrix also facilitated the analysis of a wide range of GAG-derived oligosaccharides. A group of thirteen disaccharides derived from CS or DS, heparin and HS were analyzed to determine whether sulfo groups were lost preferentially from specific positions. Disulfated and trisulfated disaccharides produced much more intense [M + x Na − NaSO3 −x H]- (M represents the fully protonated form of the oligosaccharide) fragment-ions than did monosulfated disaccharides. Also a homologous series of oligosaccharides, with repeating trisulfated disaccharide sequence ΔUAp2S(1[→4)-α-D-GlcNpS6S(1→4)-α-L-IdoAp2S(l]n→GlcNpS6S, n = 0–3), were analyzed to examine the ability of FAB-MS to analyze larger oligosaccharides and to examine similarities in fragmentation patterns. An octasaccharide having molecular-ion of m/z 2637, corresponded to the largest oligosaccharide analyzable at that time. This homologous, series of oligosaccharides showed similar fragmentation [30].
High-resolution MS is particularly important for deriving the molecular formula of unknown compounds. An unknown oligosaccharide, of particular importance for understanding the structural variation in the antithrombin III binding site region responsible for heparin’s anticogulant activity, was analyzed by high resolution FAB-MS. The intense molecular ion at m/z 1306.8112 suggested this oligosaccharide having a molecular formula of C24H30N2S6O38N8. These data, when combined with low resolution FAB-MS fragmentation and two-dimensional COSY and ROESY 1H-NMR spectroscopy, led to the unambiguous assignment of the structure of this oligosaccharide [31].
FAB-MS analysis of additional heparin oligosaccharides of unknown structures, has also been reported. Negative-ion mode FAB-MS of three bacterial lyase-resistant tetrasaccharides gave groups of molecular-ion, from which the sugar and sulfate compositions could be determined. However, again the prevalent fragment-ions corresponded to loss of sulfo groups (SO3), characterized by a series of ions having a difference of 80 amu [32].
The chemical analysis of novel oligosaccharides isolated from chondroitin sulfate K, isolated from king crab, suggested the presence of a novel structure containing 3-O-sulfo GlcA residues. To confirm this proposed structure, four tetrasaccharides and one pentasaccharide were prepared through hyaluronan lyase digestion of chondroitin sulfate K, followed by HPLC purification. The resulting oligosaccharides, analyzed by FAB-MS in the negative-ion mode, exhibited alkali ion-proton heterogeneity, producing a distribution of molecular ions of the type [M − (x+1) H + x Na]- (M represents the fully protonated form of the oligosaccharide), from which molecular weights were obtained. Furthermore, the composition and the maximum number of O-sulfo groups present in each oligosaccharide, could be inferred. The four tetrasaceharides included (UAp)2(HexNpAc)2(OSO3H)2, two isomers of (UAp)2(HexNpAc)2(OSO3H)3, and (UAp)2(HexNpAc)2(OSO3H)4. while the pentasaccharide corresponded to (UAp)3(HexNpAc)2 (OSO3H)4. The structures of these oligosaccharides were definitely established by additional enzymatic digestion, using chondroitin lyase ACII (Table 1) and 1H NMR spectroscopy [33]. Similar methods were used to solve the structures of five tetrasaccharides [34] and five hexasaccharides [35] that had been isolated from squid cartilage-derived chondroitin sulfate E. Negative-ion mode FAB-MS, was used in these studies to determine the molecular weight, composition and the maximum number of O-sulfo groups present in each oligosaccharide.
Sulfated oligosaccharides have been derivatized prior to their FAB-MS analysis to improve sensitivity and to facilitate determination of the position of their sulfo groups. This strategy combines permethylation and peracetylation. Sulfated oligosaccharides are first permethylated leaving sulfo groups intact and then the sulfo groups are removed and replaced with acetyl groups. Positive-ion FAB-MS analysis of the derivatives affords composition and some sequence information [36]. In a second derivatization strategy, the permethylated sample is gradually degraded by partial methanolysis of glycosidic linkages, to determine both sequence and sulfo group positions. This strategy offers an alternative approach of previous derivatization avoiding the incomplete substitution of sulfo groups by acetyl groups [37]. Using these permethylation and peracetylation derivatization strategies, four sulfated trisaccharides, resistant to chondroitin lyase ABC, were examined by FAB-MS. The quality of mass spectra obtained was substantially improved after derivatization. The combination of the FAB-MS of underivatized and derivatized oligosaccharides facilitated structural determination [38].
2.2. MALDI-MS
MALDI-MS represents a second powerful technique to determine molecular weights of sulfated oligosaccharides and polysaccharides. It has the advantages of accuracy, sensitivity (requiring picomoles or subpicomoles of materials), wide dynamic molecular weight ranges and affords singly charged species as major ions, making it well-suited to the analysis of mixtures such as those encountered in the analysis of G AGs. However, sulfated oligosaccharides are difficult to ionize by MALDI due to their high acidity and the propensity to easily adduct cations (such as Na+, K+, etc.), giving rise to broad, unresolved peaks.
Early studies suggested that direct negative-ion mode MALDI analysis of even simple disulfated and trisulfated heparin-derived disaccharides, gave poor mass spectra. To circumvent this limitation, complexes of sulfated oligosaccharides with basic peptides, were used to improve the sensitivity and quality of MALDI mass spectra. A synthetic peptide with sequence (IRRERNKMAAAKSRNRRRELTDTL); for example, was mixed with an octasulfated hexasaccharide and the complex was analyzed in positive-ion mode. The (1:1)+ ion where 1:1 corresponds to the ratio of basic peptide to acidic oligosaccharide as well as fragment-ions corresponding to (1:1-SO3)+, (1:1–2SO3)+, (1:1–3SO3)+, were shown respectively (Fig. 3a). The (1:0)+ ion was observed and it was used to calculate the molecular weight of sulfated oligosaccharides. Sub-picomole sensitivity was achieved using this method. No cation adducts were observed for the peptide-heparin oligosaccharide complexes, even though the sulfated oligosaccharides were used as sodium or ammonium salts, simplifying the mass spectra [39].
Fig. (3).

(a) The complex ion of a heparin octasulfated hexasaccharide with a synthetic peptide in MALDI-MS analysis, where m and n correspond to the moles of peptide and oligosaccharide, respectively. Heparinase treatment of a decasaecharide (AT-10) with sequence ΔUAp2SGlcNpS6SIdoAp2SGlcNpS6SIdoAp2SGlcNpS6SIdoApGlcNpAc6SGlcApGlcNpS3S6S. Those species assignable to the contaminant are marked (*). (b) Incomplete heparinase I treatment of AT−10. Under the conditions used in this study, heparinase I cleaves a glycosidic linkage containing an 12S; (c) MALDI mass spectrum of AT-10 fragments from exhaustive digestion with heparinase I; (d) MALDI mass spectrum of tagged AT-10 treated with heparinase I shows five fragments: one with molecular mass of 576.7 Da (assignable to 6 D), two tetrasaccharides with molecular mass of 1073.9 (*) and 1154.0 Da, and a mass tagged hexasaccharide with a molecular mass of 1671.4 (mass of 1615.3 plus the mass tag of 56.1). Because the coupling efficiency was ‘90%, also seen is unlabeled hexasaccharide (mass of 1615.1). (Revised from ref. 74); (e) MALDI mass spectrum of the protonated complex of AT-10 with(RG)l9R; (f) Direct MALDI-MS analysis of a synthetic highly sulfated oligosaccharide, sucrose octasulfate, using matrices comprised of room temperature ionic liquids.
Additional studies of the utility of non-covalent complexes for MALDI mass spectrometry analysis of heparin-derived oligosaccharides, have also been reported. The molecular weights of heparin oligosaccharides, ranging from disaccharides to hexadecasaccharides, were determined by MALDI-(time of flight)TOF-MS by use of their ionic complexes formed with a basic peptide or protein. Three synthetic peptides were examined, having the sequences RRRRRRRYIL, (RG)10, (RG)15 designated as names SP1, SP2, SP3, respectively. The SP1 was useful for determining the mass of two heparin disaccharides. A high content of arginine was important for efficient complexation with highly sulfated oligosaccharides[40]. However, peptides with contiguous domains of arginines were less effective than peptides having evenly-distributed arginine residues, such as SP2 and SP3 having sequences of alternating arginine and glycine. Ideally, the number of arginine residues on the peptide should be no less than the number of sulfo groups on the oligosaccharides, and the size of peptides should remain as low as possible to give the best accuracy for mass determination. Based on these principles, SP 2 was used to complex a heparin-derived hexasaccharide. The most abundant ion was one assignable to the intact complex and no loss of sulfo groups was observed under optimized conditions. To evaluate the potential of this method for direct analysis of heparin oligosaccharides without isolation and purification, the mixture of a tetrasaccharide, a pentasaccharide and a hexasaccharide was analyzed using SP2 as complexing basic peptide. Molecular weights of all three components could easily be measured in the presence of each other. Larger oligosaccharides were analyzed with SP3 complex and their molecular weights were determined, including a decasaccharide derived from heparin using chemical depolymerization. Angiogenin, a basic protein could be used for complex formation and analysis of large oligosaccharides up to hexadecasaccharides. However, experimental values were different from theoretical values, possibly the result of sulfo group loss in the ionization process. This result demonstrates an important limitation of applying protein and basic peptide components to the analysis of mixtures of sulfated oligosaccharides [41].
Another basic peptide, (RG)19R, applied to the characterization of three synthetic GAG tetrasaccharides demonstrated the ability of MALDI-MS to distinguish oligosaccharides that differ only in degree of sulfation. These results again, suggest the utility of MALDI-MS for analyzing oligosaccharide mixtures. Traditional structural analysis usually requires tedious purification procedures. Using MALDI-MS, it may be possible to use simple size-exclusion chromatography (SEC) for the convenient analysis of heparin oligosaccharides. A hexasaccharide-octasaccharide fraction of a partially 2-O-desulfated heparin were examined with MALDI-MS, using (RG)19R as basic peptide. The experiment identified at least three different families of hexasaccharides, and their molecular weights were in excellent agreement with the theoretical masses calculated for the different oligosaccharides. More complex samples in molecular weight ranges of up to 3000 D, were also characterized [42].
The capability of MALDI-MS to analyze oligosaccharides mixtures, has made this technique very useful in elucidating enzyme mechanisms and enzyme-based GAG sequencing. The analysis of enzyme digestion products of specific oligosaccharides has the potential to play an important role in the enzymatic sequencing of GAGs (Fig. 3b)[43–46].
Direct MALDI-MS without the presence of a complexing basic peptide, has been used for the analysis of non-sulfated GAGs or GAG oligosaccharides, containing low levels of sulfo groups. Chondroitin ABC lyase and hyaluronate lyase degradation products of chondroitin sulfate and hyaluronic acid were examined with direct MALDI-MS in both positive-ion and negative-ion modes. Some expected ions corresponding up to the hexasaccharide of CS and the octasaccharide of HA, were observed. Loss of sulfo groups and sodiated ions complicated these mass spectra [47].
Recently unpublished studies in our laboratory show that it is possible to obtain direct MALDI-MS data on synthetic, highly sulfated oligosaccharides using matrices comprised of room temperature ionic liquids, first described for the analysis of polypeptides (Fig. 3c) [48]. Studies are currently underway to extend this approach to the analysis of GAG oligosaccharides.
2.3. ESI-MS
ESI, a relatively soft ionization method, has been particularly promising for the mass spectral analysis of highly sulfated oligosaccharides. The application of negative-ion ESI-MS for the analysis of sulfated carbohydrates complexed to basic peptides was based on the pioneering research of similar complexes by positive-ion MALDI-MS. Pharmaceutically important polysulfated compounds including sucrose octasulfate (SOS) and cyclodextrin sulfates, were examined as complexes with a series of basic peptides, polyamines and the basic protein ubiquitin. Stable (1:1) complexes could be formed that resulted in molecular-ions. Fragmentation, resulting from loss of sulfo groups, could be suppressed by controlling the experimental conditions [49].
Direct ESI-MS has also been applied for the analysis of sulfated oligosaccharides from various GAG families. ESI-MS analysis of dermatan sulfate, heparin, heparan sulfate, chondroitin sulfate, acharan sulfate and carrageenan derived oligosaccharides, have been reported [11, 50–54].
Acharan sulfate is an unusual GAG purified from the giant African snail, Achatina fulica. Seven different oligosaccharides were obtained by heparin lyase digestion using heparin lyase II and a recently discovered, Bacteroides heparin lyase [55]. These oligosaccharides were purified by strong anion exchange (SAX)-HPLC and analyzed by ESI-MS to afford molecular-ion information for each oligosaccharide. Multiply charged and sodiated-ions were prevalent in the form [M-mH+nNa](m-n)− (m > n ≥ 0, where the value of n indicates the degree of sodiation and the value of (m-n) indicates the charge states of ions). Based on these ESI-MS data, the sizes of these oligosaccharides were assigned from disaccharides to octasaccharides. The sulfation degree of each oligosaccharide, could be easily assigned. The sensitivity of ESI-MS detection was determined to be 20 picomoles based on an experiment on a fully sulfated acharan sulfate tetrasaccharide [11].
ESI-MS analysis of DS oligosaccharides prepared using chondroitin lyase ABC, gave similar mass spectra to those obtained on acharan sulfate derived oligosaccharides. DS oligosaccharides were fractionated by SEC and SAX-HPLC to obtain six homogeneous oligosaccharides, ranging from a disaccharide to a dodecasaccharide. A trisaccharide and a pentasaccharide were prepared by removal of the non-reducing terminal ΔUAp from the tetrasaccharide and the hexasaccharide using Hg(OAc)2. These eight oligosaccharides, analyzed with ESI-MS, showed groups of molecular-ions having different charge states. At each charge stale, a distribution of sodiated species was observed. The presence of these multiple molecular-ions afforded the molecular mass of each oligosaccharide with a high level of accuracy. The largest DS oligosaccharide determined by ESI-MS, was a dodecasaccharide (Fig. 4) [50].
Fig. (4).

ESI-MS analysis of DS dodccasaccharide. Six ion-groupings are marked with charges ranging from −7 to −2. The number of protons lost (x) ranged from 1 to 12 and the number of sodium atoms added (y) ranged from 0 to 9. (Adapted from ref. 50).
A study of some of the experimental parameters affecting the ESI-MS analysis of heparin oligosaccharides, was undertaken. Sodium salts and ammonium salts of tetrasaccharides, were analyzed. The ESI-MS of tetrasaccharides sodium salts showed quasimolecular ions with various degrees of sodiation, while ammonium salts gave a much simpler mass spectrum with [M − nH]n− as the major peak. The sensitivity of analysis of the ammonium salt was also better than that of the sodium salt form. These comparisons suggested the use of the ammonium salt, for the characterization of heparin oligosaccharides. SEC of a heparin lyase digestion mixture in aqueous NH4Cl eluent afforded fractions from disaccharide to decasaccharide as the ammonium salts. The hexasaccharide, octasaccharide and decasaccharide fractions were next analyzed with ESI-MS. Six major components of hexasaccharides were identified and their compositions were deduced based on the molecular masses obtained by ESI-MS. The six compositions covered all known heparin hexasaccharide structures, indicating that the ESI-MS spectrum could give a close representation of the components in the fraction. The ESI-MS analysis of octasaccharide fractions identified components with compositions consistent with two described structures, fully sulfated octasaccharide and an undersulfated octasaccharide. Four octasaccharides with compositions corresponding to previously undescribed structures were also identified. The decasaccharide fractions gave two known compositions and seven undescribed compositions. Decasaccharides were further fractionated by HPLC and the resulting ESI-MS analysis of these fractions showed a much simpler pattern than did the mixture. In summary, ESI-MS afforded both oligosaccharide chain length and oligosaccharide composition in terms of sulfo group and acetyl group substitution [52].
As previously described, direct analysis of sulfated oligosaccharides with sodium salts form gives a heterogeneous mixture of sodiated ions in ESI-MS analysis, decreasing the sensitivity and complicating the mass spectra. Ammonium salts of heparin oligosaccharides reportedly simplify ESI-MS analysis and improve the sensitivity. A systematic study was undertaken to evaluate the counterions for in the ESI-MS analysis of the highly sulfated synthetic oligosaccharides, sucrose octasulfate (SOS). SOS was selected as model compound based upon its commercial availability in high purity and its high degree of sulfation. Primary, secondary, tertiary and quaternary ammonium counter ions containing an identical number of carbon atoms, were initially examined. Next, quaternary counter ions of increasing carbon number were examined. Finally, phosphonium counter ions, polyamine counter ions and a basic peptide counter ion were studied using positive-ion mode and negative-ion modes. These experiments surprisingly demonstrated that positive ion ESI-MS gave better quality mass spectra than negative-ion mode. Tetraethylammonium was found to be an optional counterion, giving a high quality mass spectrum with high detection sensitivity [56].
2.4. Online Chromatography Mass Spectrometry
Direct analysis of intact GAGs is limited because of GAG polydispersity and high molecular weight. Usually enzymatic or chemical depolymerization is required to obtain oligosaccharides suitable for structural analysis. However, isolation and purification of oligosaccharides involves tedious work because of the large number of oligosaccharide sequences possible for each chain length [57, 58]. Because multiple separation steps are required for GAG oligosaccharide purification, it is often not practical to isolate components from the sub-microgram quantities of GAGs recovered from biological sources. Thus, on-line chromatography mass spectrometry greatly enhances the analysis, making this a powerful technique for characterizing small quantities GAG oligosaccharides in complex mixtures. HPLC and capillary electrophoresis (CE) are suitable chromatographic methods to separate complex mixtures that can be interfaced ESI-MS. ESI is an ideal ionization method for on-line use, since it introduces a flowing stream of analyte into the mass spectrometer.
Reversed-phase ion-pairing (RPIP) HPLC offers a high-resolution method for the separation of GAG oligosaccharides [59]. RPIP-HPLC ESI-MS was established to analyze CS oligosaccharides. Tetrapropylammonium (TPA), ion-pairing reagent, and its deuterium-labeled derivative d28TPA were used to elucidate the ionization process. Purified CS disaccharides, prepared using chondroitin lyase, were initially analyzed with ESI-MS using continuous infusion with a syringe pump. The results of ion-pairing reagent on ionization were compared. For the nonsulfated CS disaccharide, ESI-MS analysis showed only [M-H]- ions with no fragmentation or adduction with TPA or d28TPA. Monosulfated CS disaccharide (ΔUAp(1→3)-D-GalNpAc4S) afforded a molecular-ion [M-H]-, a single sodiated species as well as a fragment-ion in the absence of TPA. In the presence of ion-pairing reagents, the mass spectra showed no sodiated species, no fragments, only a molecular-ion associated with the adduction of one TPA. In the absence of ion-pairing reagent, the disulfated disaccharide (ΔUAp2S(1→3)-D-GalNpAc6S) and trisulfated disaccharides (ΔUAp2S(1→3)-D-GalNpAc4S6S) afforded heterogeneous mixtures of multiply sodiated molecular-ions having a single or multiple charges, as well as fragments caused from loss of sulfo groups. In the presence of ion-pairing reagent, their mass spectra were much simpler, showing only TPA adducted molecular-ions. The inclusion of d28TPA facilitated the interpretation of these adducted ions and permitted the assignment of charge state. Based on these observations, the application of ion-pairing reagents could facilitate the determination of the molecular masses of these disaccharides. Separation with TPA as an ion-pairing reagent was next performed on a reversed-phase C6 HPLC column and combined with ESI-MS to analyze heparin lyase digested heparin. If TPA was removed using a cation membrane suppressor before MS analysis, the corresponding mass spectrum was extremely complex and impossible to interpret. In the presence of TPA and d28TPA, a much simpler mass spectrum was obtained, in which ions were observed corresponding to intact oligosaccharides adducted with TPA or d28TPA. By combining results obtained with TPA and d28TPA, unambiguous identification of the ions in these spectra ranging from a disaccharide to a hexasaccharide, could be determined [60].
The correct choice of an ion-pairing reagent and its concentration are also critical for the successful LC/MS analysis of oligosaccharides. Ion-pairing reagents need to be in sufficient concentration to provide retention of highly sulfated oligosaccharides on a reversed-phase column, but at the same time, high concentrations of ion-pairing reagents also decrease the sensitivity of LC/MS. Furthermore, commonly used ion-pairing reagents, such as tetraalkylammonium salts are not volatile, contaminating the interface between the LC and the mass spectrometer. To establish a new LC/MS methodology, different ion-pairing reagents have been compared [60]. The use of non-volatile tetraalkylammonium salts having a MS signal-suppressing effect, need to be avoided and replaced with trialkylammonium acetates or dibutylammonium acetates. Dibutylammonium acetate was initially selected for LC/MS analysis of HS-like oligosaccharides because it gave good chromatographic resolution and shorter time. A miniaturized LC method, using capillary HPLC combining with microelectrospray ionization, was evaluated [60]. Detection sensitivities could be improved dramatically by this miniaturization. Heparosan-derived oligosaccharides were analyzed to determine the applicability of this methodology. It should be noted that heparosan is nonsulfated heparan sulfate precursor with a simple redundant repeating unit, →4)-α-D-GlcNpAc(1→4)-α-D-GlcAp(1→, making its analysis considerably simpler than sulfo group containing GAGs, including HS and heparin. Heparosan oligosaccharides of sizes up to tetracontasaccharide were resolved by capillary HPLC and their molecular masses were determined by ESI-MS. Next, a biologically important antithrombin III binding pentasaccharide, associated with heparin’s anticoagulant activity, and several of its precursors, differing by sulfation pattern and/or degree of sulfation, were analyzed. These pentasaccharides were well-resolved and characterized by this LC/MS system with enzymatically incorporated 34SO4 as a mass spectral isotopic probe [61]. This method was used to examine critical biological motifs and map heparan sulfate biosynthetic pathways. HS isolated from various tissues and cells were completely digested with heparin lyases into disaccharides. LC/MS analysis of these disaccharides delineated the fine structural modifications of these HS [62].
The LC/MS analysis of highly sulfated and long chain oligosaccharides is more challenging due to their complex structures, high molecular weight and large number of negative charges. Heparin, partially digested with heparin lyase and containing very large oligosaccharides, has been analyzed by reversed-phase ion-pairing LC/MS. Tributylamine was chosen as ion-pairing reagent representing a compromise between low alkyl chain length, affording high volatility required for compatibility with online MS and longer alkyl chain length affording higher on-column retention, giving enhanced resolution. HPLC parameters, such as the addition of organic co-solvent, volatile inorganic salt concentration, solvent pH, temperature and eluent gradient were optimized through extensive offline experiments [59]. Post-column addition of tributylamine/acetonitrile suppressed the Na+/NH4+ adduction and simplified the mass spectra. The analysis of heparin digested with heparin lyase to 30% completion afforded a complex mixture of oligosaccharides detected by ultraviolet (UV) absorption and total ion chromatography (TIC) (Fig. 5). The negative-ion ESI mass spectra of fully sulfated oligosaccharides with degree of polymerization (dp) 2 to dp 14, the major components in heparin-derived oligosaccharides mixture, are shown in (Fig. 6). Multiply-charged ions were dominant in the mass spectra. Their charge states could be determined by high resolution scans and charge states up to −5, could be determined by the resolution of isotopic ions. The smaller oligosaccharides (dp 2-dp 6) exhibited single molecular ions, while larger oligosaccharides (dp 8-dp14) showed peaks corresponding to adducts of TrBA as the form of [M-(x+y)H+yTiBA]x−. Oligosaccharides of dp >14 show prominent peaks corresponding to multiple TrBA+/NH4+/Na+ adducts and the interpretation has proven to be difficult. Interestingly, a group of unpredicted ions were observed. The lack of chromophores that absorb in the UV at 232 nm suggested that they contained a saturated sugar residue at their non-reducing ends in place of an unsaturated uronate ΔUAp residue. The masses of these components were 18 amu larger than corresponding unsaturated products, suggesting that they arise from the original non-reducing end of the heparin starting material. The presence of these saturated oligosaccharides suggests the possibility that sequence information can be obtained using the non-reducing end as a reading frame [63].
Fig. (5).
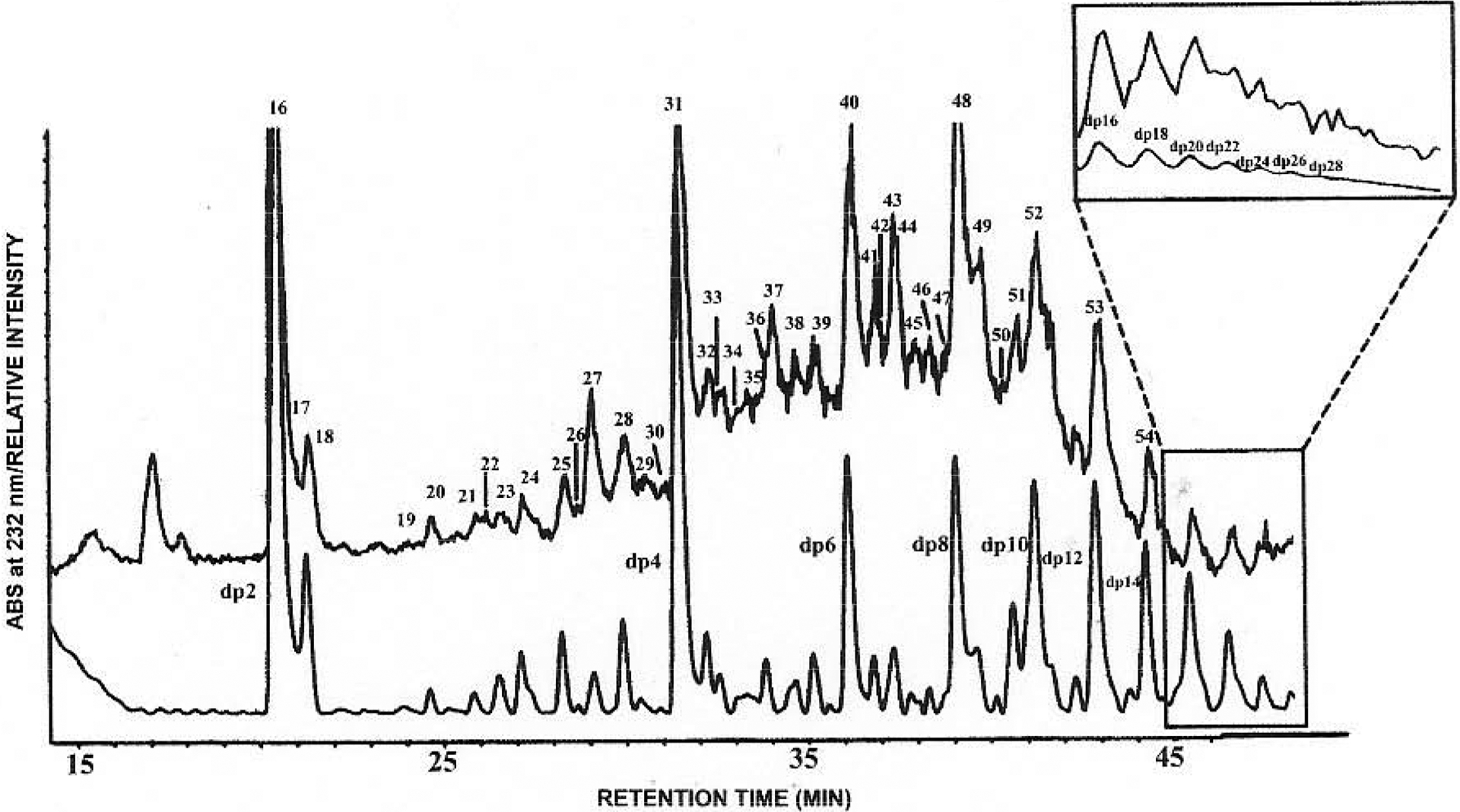
LC/MS analysis of heparin-derived oligosaccharides. The main figure shows the UV trace (lower) and MS-TIC trace (upper with numbered peaks) of RPIP-HPLC separation. Early time in the LC/MS analysis (inset) shows the TIC of mono- and disaccharides using the shallow elution gradient. (Adapted from ref. 63).
Fig. (6).
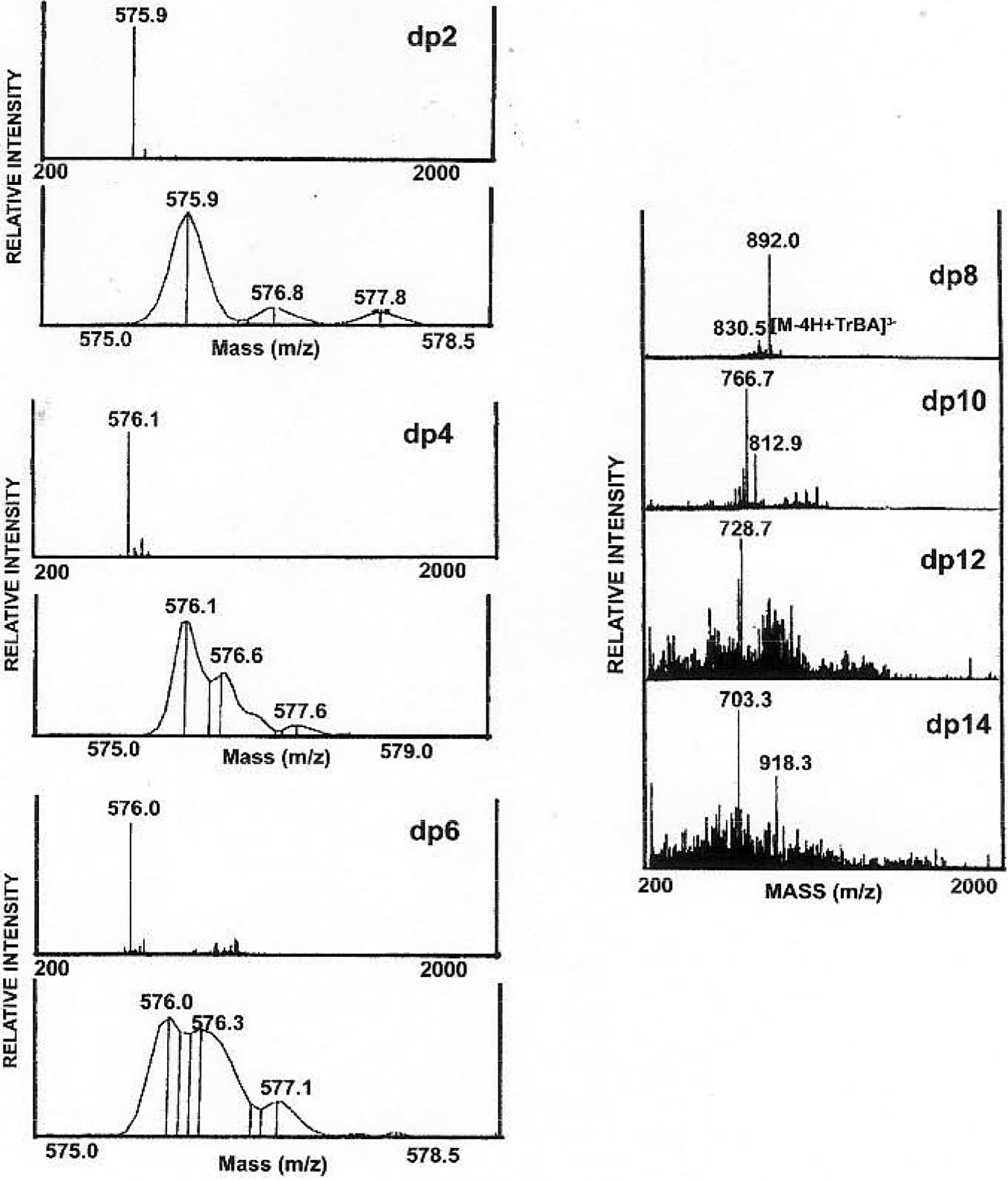
Simplified ESI mass spectra of fully sulfated heparin oligosaccharides ranging in size from disaccharide (dp2) to tetradecasaccharide (dp14). (Adapted from ref. 63).
In addition to reversed-phase chromatography, size-exclusion chromatography was also shown to be amenable to on-line ESI-MS analysis. The composition of eluant used was very simple and no ion-pairing reagent was required. On-line cation exchange was accomplished with a self-regenerating cation-exchange column, to reduce the adduction of oligosaccharide molecular-ions with various cations. The cation exchange column was comprised of two regenerated compartments to which, electrodes were applied and one eluant compartment separated by ion-exchange membranes. When eluant passes through the column, ammonium and sodium ions are exchanged with protons. The results of analysis of heparin trisulfated disaccharide (ΔUAP2S(1→4)-d-GlcNpS6S) with and without the ion-exchange column were compared. The use of ion-exchange column resulted in the preferential formation of [M-3H]3− ions, while [M-H]− ions were dominant with the absence of ion-exchange. The intensity of the [M-3H]3− ions were ~100 times greater than the [M-H]− ions. The ESI-MS conditions were optimized using a cone voltage of 10 V as a compromise between minimal fragmentation and high ionization efficiency. Tinzaparin, a pharmaceutical with low in which more molecular weight heparin, was analyzed with the on-line size-exclusion MS method, than 60 components were detected. The isotopic patterns for components with up to 12 saccharide units were well resolved. Their molecular masses were determined, from which the number of monosaccharide units, sulfo groups and acetyl groups could be deduced. The masses of components with 14 to 18 saccharide units, could be calculated from their measured average mass [64].
Capillary electrophoresis (CE) is an ultra high-resolution analytical technique that has been applied to a wide variety of different types of molecules. When a high voltage is applied, the positive ions in the solvent migrate towards the negative electrode, through a process called electro osmotic flow (EOF). EOF drives analyte flow through the capillary. Combined with the electrophoresis effects, positively charged molecules move at a rate that greater than the EOF, negatively charged molecules move slower than EOF, and neutral molecules move at the same rate as EOF. This gives the separation of both charged and neutral molecules [65]. There are two different modes of CE, normal polarity mode and reversed polarity mode. In normal polarity mode, the sample is injected at the anode and detected at the cathode. Basic or neutral buffers are required for normal polarity mode. In reversed polarity mode, the sample is injected at the cathode and detected at the anode with acidic buffer applied. These modes have been successfully applied in analysis of GAG-derived disaccharides (Table 2) and oligosaccharides [66]. CE has excellent potential as a hyphenated method with ESI-MS analysis, for the analysis of GAGs.
Table 2.
Structures of Heparin/HS and CS/DS Disaccharides
| R2’ | R2 | R4 | R6 | ||
|---|---|---|---|---|---|
| 1 | ΔUAp(1→4)-D-GlcNpAc | Ac | H | - | H |
| 2 | ΔUAp(1→4)-D-GlcNpS | SO3 | H | - | H |
| 3 | ΔUAp(1→4)-D-GlcNpAc6S | Ac | H | - | SO3 |
| 4 | ΔUAp2S(l→4)-D-GlcNpAc | Ac | SO3 | - | H |
| 5 | ΔUAp(l→4)-D-GlcNpS6S | SO3 | H | - | SO3 |
| 6 | ΔUAp2S(1→4)-D-GlcNpS | SO3 | SO3 | - | H |
| 7 | ΔUAp2S(l→4)-D-GlcNpAc6S | Ac | SO3 | - | SO3 |
| 8 | ΔUAp2S(1→4)-D-GlcNpS6S | SO3 | SO3 | - | SO3 |
| 9 | ΔUAp2S(1→4)-D-GlcNp6S | H | SO3 | - | SO3 |
| 10 | ΔUAp(1→4)-D-GlcNp6S | H | H | - | SO3 |
| 11 | ΔUAp2S(l→4)-D-GlcNp | H | SO3 | - | H |
| 12 | ΔUAp(1→4)-D-GlcNp | H | H | - | H |
| 13 | ΔUAp(1→3)-D-GalNpAc | - | H | H | H |
| 14 | ΔUAp(1→3)-D-GalNpAc4S | - | H | SO3 | H |
| 15 | ΔUAp(1→3)-D-GalNpAc6S | - | H | H | SO3 |
| 16 | ΔUAp2S(1→3)-D-GalNpAc | - | SO3 | H | H |
CE/MS analysis of CS/DS oligosaccharides from bovine aorta has been studied off-line. Oligosaccharides were separated by CE. Fractions were collected and introduced to ESI-MS. Oligosaccharides differing in chain length and degree of sulfation, could be detected by this method [67].
Direct coupling of CE/MS method was used to identify heparin oligosaccharides. The CE/MS interface consisted of a double coaxial stainless steel tubing, assembly surrounding the fused silica separation capillary. This interface enabled both the addition of a liquid make-up and the delivery of a gas sheath flow. The liquid make-up was used to compensate for the very low electro osmotic flow emerging from CE and to establish the electrical contact required to apply both the separation voltage and the spray voltage. The gas sheath flow was able to assist in the formation of the spray and to ensure its stability. With eight heparin disaccharides standards (Fig. 7 and Table 2), the CE/MS interface parameters; such as flow rate and composition of the sheath liquid, flow rate of the sheath gas and position of the capillary in the needle, were optimized. The behavior of heparin disaccharides under different combinations of positive or negative CE voltage polarity and positive or negative MS ionization modes, was investigated. The results indicated that the use of positive polarity CE with negative MS ionization seemed to be the best approach to analyze complex oligosaccharides. An oligosaccharide mixture prepared from porcine mucosa heparin by depolymerization with heparin lyase II and III, was analyzed by CE/MS. Peaks corresponding to the known eight disaccharides (Table 2), as well as some unknown compounds, were observed corresponding to molecular-ions adducted with the cations, sodium, ammonium and triethylamine. By comparison to calculated molecular masses, their structures were deduced to be a saturated disaccharide and two heparinase-resistant tetrasaccharides [68]. Other CE/MS analysis of sulfated oligosaccharides regarding sequencing analysis by MS/MS or quantification analysis, are discussed later in this article [69, 70].
Fig. (7).
Structures of heparin/HS and CS/DS disaccharides where R = SO3− or H and R’ = SO3−, Ac or H (see Table 2).
2.5. Other MS Techniques
FAB-MS, MALDI-MS and ESI-MS are most popular soft-ionization methods used in the MS analysis of sulfated carbohydrates. The use of other ionization techniques, such as liquid secondary ion MS (LSIMS), plasma desorption MS (PDMS), have also been reported for GAG oligosaccharide analysis.
LSIMS involves the bombardment of a solid analyte and matrix mixture by a fast particle beam (an ion, typically Cs+). Oligosaccharides, may or may not be derivatized before MS analysis. One derivatization approach was conversion of oligosaccharides to neoglycolipid derivatives, by reductive-amination with an amino lipid (dihexadecyl phosphatidylethanolamine, DHPE). The derivatives were separated by TLC and analyzed by LSIMS. Molecular ions were obtained as well as fragment ions and sodiated ions [71]. Underivatized CS oligosaccharides were analyzed with low sensitivity. Oligosaccharides with up to eight sugar units were detected; although, fragment and sodiated ions were prevalent in the mass spectra. PDMS has also been used to analyze heparin oligosaccharides and determine the molecular weight of di-, tetra-, and hexasaccharides [72].
3. MS DETERMINATION OF STRUCTURE
Simple MS analysis of sulfated oligosaccharides can be used to obtain molecular weight information, from which the monosaccharide composition, degree of sulfation, and degree of acetylation can be deduced. However, such data are not sufficient for elucidating the primary structure of oligosaccharides necessary for explaining their structure-activity relationship. Structural information that precisely locates sulfo and acetyl groups, linkage position, and which identities the type of hexosamine (GlcNp or GalNp) and uronic acid (GlcAp or IdoAp), can only be achieved by MS in combination with enzymatic analysis or by tandem MS (MS/MS) experiments.
3.1. MALDI-MS Combined with Enzymatic Digestion
Oligosaccharides complexed with basic peptides can be analyzed by MALDI-MS and their molecular weights can be derived from the resulting mass spectral data. To obtain unambiguous sequence of the analyte, enzymes that cleave specific glycosidic linkages are applied. The resulting enzymatic fragments can be analyzed again by MALDI-MS and the sequence of original oligosaccharide can be obtained by interpretation of the mass spectral data.
To facilitate the sequencing process, a notation system called property encoded nomenclature (PEN), has been developed [23]. Heparin or HS oligosaccharides are comprised of a basic repeating disaccharides unit, UA (IdoAp or GlcAp) linked 1, 4 to GlcN. Four potential sites for chemical modification exist in each disaccharide unit. Within PEN, the possible sulfo-substitutions of a disaccharide are represented by a single hexadecimal digit. And “+” or “−” signs are used to represent GlcAp/IdoAp. All 32 possible disaccharide units are assigned a one-letter code based on its hexadecimal value and its sign. Their theoretical molecular weights can be calculated from this value. With the 32 building blocks, a list of theoretical molecular weights of all possible saccharides sequence can be generated. With the combination of the notation system and the accuracy of MALDI-MS, the length and the number of sulfo and acetyl groups can be readily assigned. A sequence can be restricted to reasonable possibilities. Different heparin lyases can cleave the glycosidic linkage between specific saccharide residues; heparin lyase I is specific for the linkage between GlcNS(6S/OH)→IdoA2S; heparin lyase II is specific for the linkage between GlcN(S/Ac)(6S/OH)→IdoA/GlcA(2S/OH); heparin lyase III is specific for the linkage between GlcN(Ac/S)(6OH/S)→GlcA/IdoA [73]. MALDI-MS analysis following the application of different heparin lyases, can permit a sequence to be unambiguously deduced. Several examples of sequencing heparin oligosaccharides have been described. For an unknown saccharide, MALDI-MS determined its molecular weight to be 2230.2 Da, which corresponded to an octasaccharide with 11 sulfo and 0 acetyl groups. The disaccharide composition was obtained by CE analysis of the completely digested octasaccharide, leaving 32 possible sequences. Heparin lyase I digestion followed by MALDI-MS reduced the number of possible sequences, and finally, treatment with heparin lyase III gave MALDI-MS data and elucidated the sequence to be ΔUAp2S(1→4)-β-D-GlcNpS6S-(1→4)-α-L-IdoAp2S(1→4)-β-D-GlcNpS6S(1→4)-β-D-GlcAp(1→4)-D-GlcNS6S. In other examples, the sequence of a nitrous acid-derived hexasaccharide, a basic fibroblast growth factor binding decasaccharide, and a antithrombin III binding decasaccharide, were successfully sequenced [23].
This methodology was also applied to sequence some unusual 3-O-sulfo groups containing heparin decasaccharide. Using the PEN system and MALDI-MS, and enzymatic or chemical degradation as experimental constraints, the sequence of this decasaccharide was determined to be ΔUAp2S(1→4)-β-D-GlcNpS6S-(1→4)-α-L-IdoAp2S(1→4)-β-D-GlcNpS6S(1→4)-α-L-IdoAp2S(1→4)-β-D-GlcNpS6S(1→4)-α-L-IdoAp(1→4)-β-D-GlcNpAc6S(1→4)-β-D-GlcNpAc(1→4)-D-GlcNpS6S. The sequence was consistent with integral glycan sequencing and 1H-NMR spectroscopy. Furthermore, this approach was shown able to afford sequence information on an oligosaccharide mixture [74].
The successful application of this methodology to sequence HS oligosaccharides, has also been reported. Two novel tetrasaccharides modified by 3-O-sulfotransferase [75] and an octasaccharide [22] binding to herpes simplex virus type 1 glycoprotein D, were analyzed by the PEN MALDI-MS approach and their sequences were deduced.
The PEN MALDI-MS sequencing method can be combined with surface non-covalent affinity (SNA) analysis. SNA-MS made the direct isolation and sequencing of specific protein-binding GAGs possible. This strategy involves protein immobilization on a surface, subsequent addition of an aqueous mixture of oligosaccharides to bind the protein, low salt washing to remove non-specific binders, addition of a basic peptide to form complex with oligosaccharides and MALDI-MS analysis of these complexes. Antithrombin III and basic fibroblast growth factor were each immobilized on a surface and the different oligosaccharides interacting with each protein were determined by MALDI-MS. The results indicate that SNA-MS can provide a foundation for elucidating key polysaccharide-protein interactions [76].
3.2. Tandem MS
Tandem MS (MS/MS) has been widely used for sequencing peptides. Fragments from peptide bond cleavage could be easily obtained by the multiple-stage MS of the precursor-ions. The amino acid sequence can be then deduced by interpretation of the fragment-ions. MS/MS has also more recently been applied to carbohydrate analysis. Glycosidic bond fragmentation or through-ring cleavages can offer important sequencing information. A nomenclature of these carbohydrate fragmentations has been proposed [77]. (Fig. 8) The application of tandem MS sequencing of sulfated carbohydrates is extremely challenging because the S-O in sulfo group is more labile than the glycosidic bond. Major effort has been devoted to applying tandem MS to analysis of sulfated oligosaccharides, but with limited success.
Fig. (8).
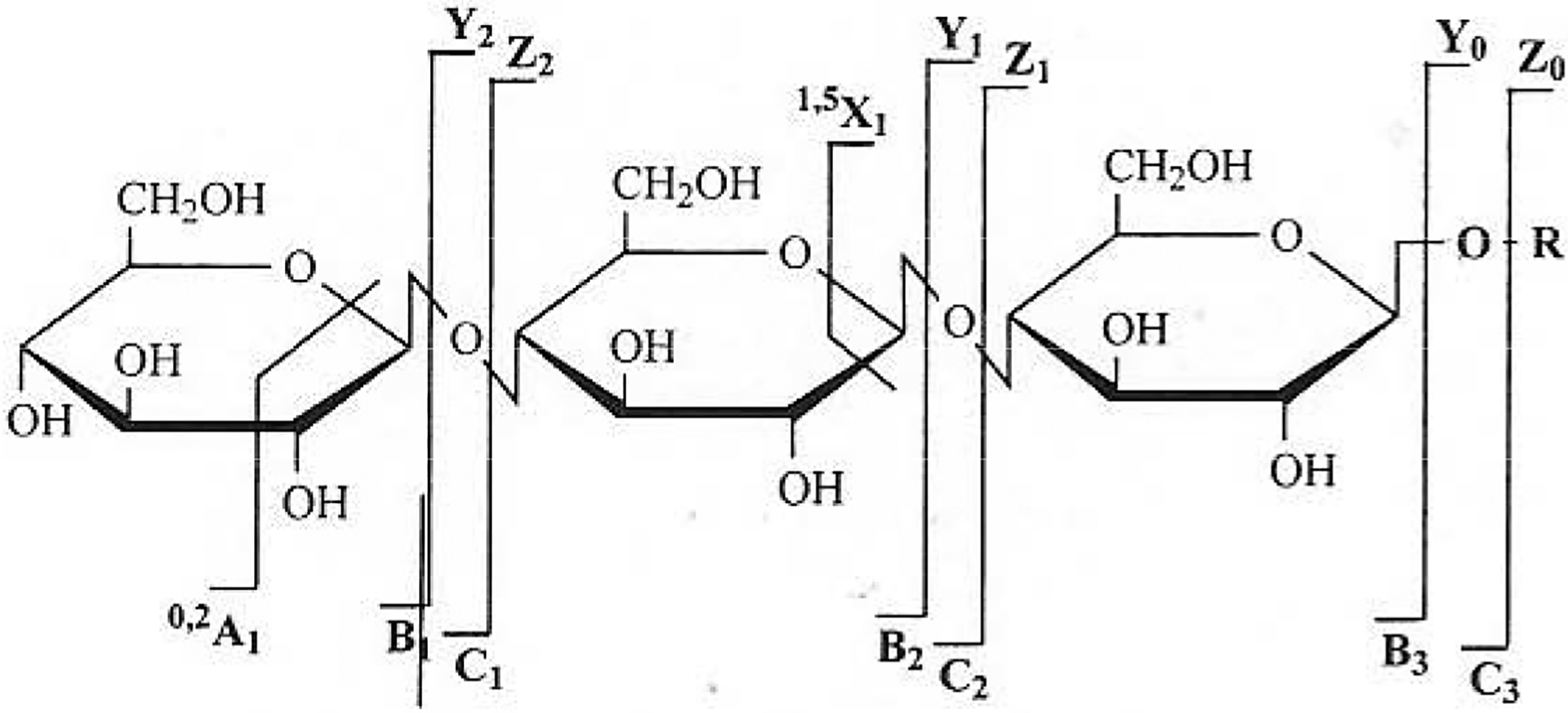
Nomenclature of carbohydrate fragmentation. (Adapted from ref. 77).
GAG derived disaccharides were first examined to develop a tandem MS method due to their relatively simple structures. The application of negative-ion FAB-MS to five monosulfated disaccharides from heparin/HS and CS/DS (Table 2) was reported using collision-induced dissociation (CID) mass-analyzed ionic kinetic energy (MIKE) MS. These disaccharides have same molecular weights, differing only from the positions of their sulfo groups (2S, 4S, or 6S) (Table 2: 3, 4, 14, 15, 16). The negative-ion FAB-MS exhibited intense molecular-ions as well as fragment ions that arise from glycosidic bond cleavage. Isomers having sulfates on the same saccharide unit (i.e., 4S (14) from 6S (3, 15)) gave the same spectrum, which meant they could not be distinguished. When the sulfo groups were in different saccharide unit (i.e., 2S (4, 16) from 4S (14) and 6S (3, 15)), significant difference was observed in their FAB-MS spectra. Peaks at m/z 300 corresponding to loss of [M-ΔUA]− were observed in the 4S (14) and 6S (3, 15) disaccharides, while peaks at m/z 277 corresponding to [M+Na-H−GalNAc]− or [M+Na-H-GlcNAc]− were observed in 2S (4, 16) disaccharides. The secondary ions were generated through CID. Molecular-ions [M-H]− (m/z 458) and [M+Na-2H]− were used as precursor ions respectively, since they were very intense and had long lifetimes in the spectra. The 0.2X2 ions and [HSO4]− ions were common to the [M-H]− generated MS2 spectra for each isomeric disaccharide. The 2S containing disaccharides (4, 16), could be characterized by BI ions corresponding to glycosidic bond cleavage. Both 4S (14) and 6S (3, 15) disaccharides gave abundant YI ions and ZI ions. However, they could be differentiated by the intensity of these ions (Fig. 9). Linkage information could be obtained from tandem MS as well because the 0.2A2 fragment was characteristic of the (1→4) linkage [78].
Fig. (9).
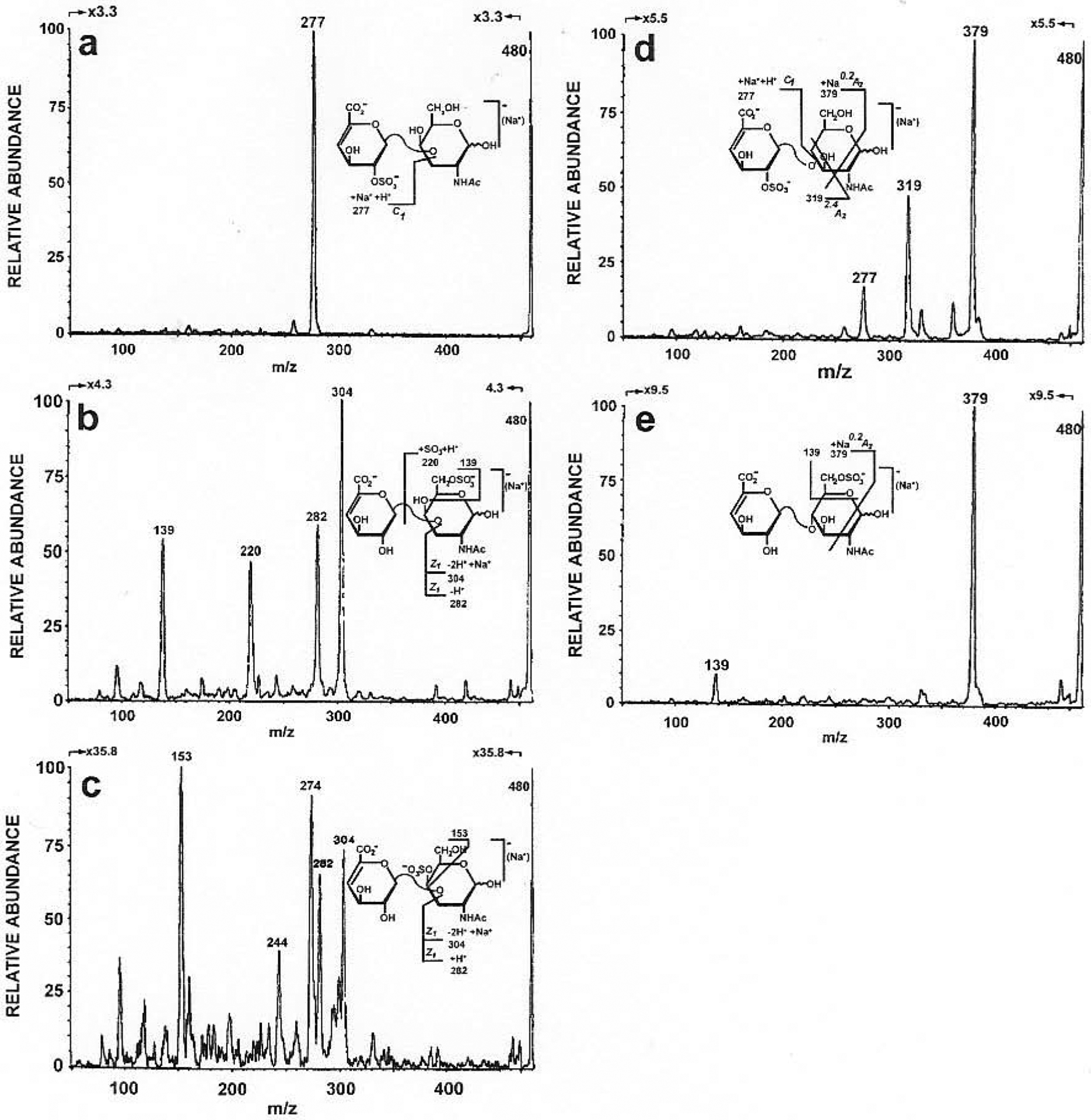
The negative-ion CID-MIKE spectra of m/z 480, ([M + Na − 2H]−) (a) ΔUAP2S(1→3)-D-GalNpAc, (b) ΔUAP (1→3)-D-GalNpAc6S, (c) ΔUAp (1→3)-D-GalNpAc4S, (d) ΔUAP2S(1→4)-D-GlcNpAc and (e) ΔUAP (1→4)-D-GlcNpAc6S. (Adapted from ref. 78)
CS has relatively simple repeating units. Their sulfation occurs at the 4- or 6- position of the N-acetylgalactosamine residues. Determination of the 4S/6S sulfation is the key respect of sequencing chondroitin sulfate oligosaccharides. The mixture of CS oligosaccharides was studied with ESI CID tandem MS. Molecular-ions with charge states where charge equaled the number of sulfo groups, were chosen as precursor ions and abundant odd-numbered Bn and Yn ions were observed. The percent total ion abundances of these ions indicate the position of sulfation. However, the 4S/6S positional information could not be produced on the GalNAc residue adjacent to the nonreducing terminus due to lack of a BI ion [79].
Further study on the influence of charge state on production mass spectra was reported. This time, sustained off-resonance irradiation collision-induced dissociation and infrared multiphoton dissociation were applied to generate product ions in a Fourier transform ion cyclotron resonance mass spectrometer. Ions in which, the charge was less than the number of sulfo groups, dissociated to produce predominantly even-numbered Bn, Cn, Yn, and Zn ions, and that odd-numbered fragment-ions were observed for ions that had charge equaled to the number of sulfo groups [80].
Based on previous work [79, 80]. strategies for complete structural characterization of CS oligosaccharides, in terms of determination of sulfo group position and uronic acid epimerization, were investigated. The analytes were a mixture of CS oligosaccharides, thus, when the charge state equaled the number of sulfo groups, their most abundant peaks overlapped. To solve this problem, and to retain the information on sulfo group position in the reducing end GalNAc residue, GAG derivatives were used to shift the m/z value of most abundant ions. The 2-aminopyridine, 2-aminobenzamide and 3, 5-dimethylpyrazole derivatives were used. The abundances of B and Y ions in the tandem mass spectra reflected the sulfation positions, while the abundances of 0.2X and Y ions, resulting from fragmentation of the non-reducing residue of unsaturated chondroitin oligosaccharides reflected the positions of uronic acid epimerization. These results indicated that GAG oligosaccharides might be completely characterized by using tandem MS [81].
To elucidate the sequence of large oligosaccharides, separation and purification was necessary, prior to tandem MS analysis. On-line sheathless capillary electrophoresis/nanoelcctrospray ionization-tandem mass spectrometry was reported for the analysis of CS/DS oligosaccharides. The total ion chromatogram showed nine electrophoretic peaks with good resolution. The mass accuracy allowed the assignment of the composition of each component. The length of the sugar chain, the number of sulfo groups and the presence/absence of unsaturated non-reducing end could be determined. The largest oligosaccharide detected by this method was an eicosasaccharide. To obtain additional structural information, on line CE-ESI-MS/MS was applied to an oversulfated unsaturated eicosasaccharide. The most abundant pentacharged ion was selected as precursor-ion automatically and the resulted MS/MS spectrum showed a complex series of fragmentation-ions, from which, the positions of sulfo groups could be assigned. Based on these spectral data, a sequence was proposed for this eicosasaccharide [69].
Heparin GAGs have a higher density of sulfo groups and more complex sequences than CS GAGs, making their analysis more difficult by tandem MS. The analysis of heparin oligosaccharides by tandem MS, has been investigated [82]. Three synthetic oligosaccharides were used as model compounds. ESI-MS without degradation and displaying multiple charge states, were observed. The number of sulfo groups gave the possible sites of deprotonation, while charge-charge repulsion limited the abundance of ions having highly deprotonated sulfo groups. For example, the tetrasulfated trisaccharide produced an abundant [M-4H]4− ions, as all sulfo groups were deprotonated. The pentasulfated tetrasaccharide produced 5-, 4- and 3- charge states ions. The fully deprotonated ion, [M-5H]5−, was less abundant because of charge-charge repulsion. The octasulfated pentasaccharide produced charge states where abundance was ordered 6->5->4-≥3-, and with no 7- or 8- charge states. CID generated product-ions arise from glycosidic bonds cleavage as well as loss of sulfo groups. The pattern of glycosidic bond cleavage changed dramatically with the precursor-ion charge state. The complementary Bm and Yn ions resulting from cleavage of the same bond, were most useful for sequencing and cross-ring cleavage was important to determine the position of sulfo group within a sugar residue. Combining the data obtained from fragmentation of different charged states and MS stages offers substantial structural information. To increase the relative abundance of backbone cleavage ions and decrease the abundance of ions produced from loss of sulfo groups, metal complex were used. Calcium ion complexation of heparin-like oligosaccharides was found able to stabilize the sulfo groups [82].
The mechanism of dissociation for isomeric heparin disaccharides was studied using isotope labeling and ion trap tandem mass spectrometry. In this process twelve heparin disaccharides were investigated. Eight N-sulfated or N-acetylated disaccharides naturally occur in heparin/HS (Table 2) and four additional N-unsubstituted disaccharides, ΔUAp2S(1→4)-D-GlcNp6S (9), ΔUAp(1→4)-D-GlcNp6S (10), ΔUAp2S(1→4)-D-GlcNp (11), and ΔUAp(1→4)-D-GlcNp (12), were of potential structural importance. The MS of these disaccharides showed molecular-ions with charge states equaling the number of sulfo groups as most abundant ions. Four of the twelve disaccharides, 7, 1, 8, 12, could be distinguished by MS directly due to their characteristic mass-to-charge ratios. Three sets of isomers, 3/4, 5/6/9, and 10/11/2 could not be directly differentiated from the MS1 spectrum. MS/MS was applied to these isomers, respectively. Fragmentations through neutral loss of a small molecule (SO3, H2O or CO2) glycosidic cleavage and cross-ring cleavage, were observed. 2H and 18O-labeling studies were used to assist in identifying the neutral molecule lost in all cases and their dissociation mechanism were postulated. Product-ions generated from glycosidic and through-ring cleavages were diagnosed for each disaccharide, identifying positions of sulfation. The mechanisms of forming B, Y, Z ions from glycosidic cleavage were postulated and consistent with the data obtained from the 18O and 2H-labelling experiments. 0.2A2, and 0.2XI ions from through-ring cleavages were observed for all eight to heparin undergo disaccharides. Their formation mechanisms were postulated to be through a charge-remote process. These results provided a foundation for developments towards sequencing large heparin/HS oligosaccharides, using tandem mass spectrometry [83].
4. QUANTIFICATION ANALYSIS OF GLYCOS-AMINOGLYCAN DISACCHARIDES BY MS
Disaccharide composition analysis, in terms of quantification of different disaccharides arise from complete digestion of GAGs, is one important aspect of GAGs analysis. It is helpful to understand structural variability of GAGs and their biological importance. High sensitivity of MS can afford the analysis of trace GAG samples, obtained from biological samples.
4.1. On-line Chromatography Mass Spectrometry Methods
Disaccharide mixtures can be separated by chromatographic methods, such as HPLC or CE. MS can be used as a detector and quantification can be achieved by integration of total ion chromatography. A LC/MS method for analysis of chondroitin/dermatan sulfate, has been reported. CS/DS disaccharides ΔUAP(1→3)-D-GalNpAc4S (14) and ΔUAP(1→3)-D-GalNpAc6S (15) (Table 2) could be separated by using tetrabutylammonium hydroxide (TBAOH) as an ion-pairing reagent. Turbo ions pray ionization mass spectrometry was used to detect the analytes. Both 4-O-sulfo disaccharide (14) and 6-O-sulfo disaccharide (15) showed [M+TBAOH-H]− at m/z 699.4. The sensitivity was increased by post-column adduction of acetonitrile and the unsaturated disaccharides could be determined in the amounts as low as 0.5 pmol levels. This method was also successfully applied into analysis of tissue samples, prepared from MethA tumor-bearing mice [84].
CE-MS was reported for the analysis heparin disaccharides. All eight heparin unsaturated disaccharides (Table 2: 1–8) were resolved by pressure-assisted capillary electrophoresis and electro spray ion-trap mass spectrometer was used to determine the analytes. To differentiate isomers, MS/MS was applied. MS or MS/MS identified all eight disaccharides and the relative abundance of characteristic ions could be used, to quantify corresponding disaccharides [70].
4.2. Tandem Mass Spectrometry Methods
Direct tandem MS analysis of disaccharide mixtures have been used to determine the ratio of the components. The key to quantification analysis by tandem MS, is to find diagnostic ions for each disaccharide and in this process, ESI-MS and ESI-MS/MS of CS disaccharides were investigated. The isomeric 4-O-sulfo (14) and 6-O-sulfo (15) showed molecular ions with same m/z values. However, their CID MS/MS spectra showed a remarkably different pattern of fragment ions. The ZI ion was absent in the CID mass spectrum of 4-O-sulfo disaccharides while the YI ion was absent in the 6-O-sulfo disaccharide. The abundances of ZI and YI ions was used to monitor 4-O-sulfo and 6-O-sulfo disaccharide, respectively [85].
Further development of quantitative method with high accuracy, has been reported [86, 87]. CID spectra of pure CS disaccharide standards were used to develop a system of equations that relates the abundance of each component to the intensity of ions in the mass spectra. The CID mass spectra of three disaccharides, 2-O-sulfo disaccharide (used as an internal standard), 4-O-sulfo disaccharide and 6-O-sulfo disaccharide, showed three distinguishable product ions with m/z 237, 282 and 300. The contributions of the three ions were measured from the CID spectra of individual disaccharides and their 1:1:1 mixture, from which the amount of each component present in the mixture, could be determined [87].
Quantification of heparin and HS disaccharides is more challenging, because eight disaccharides (Table 2: 1–8) gave more complex structures and different sets of isomers. MS, MS2 and MS3 spectra of these HS disaccharide standards and an internal standard (int), ΔUA2S-GlcNCOEt6S (I-P), were investigated to obtain diagnostic product ions. An equation was developed based on experiments of pure disaccharide standards:
Where Idi· was the intensity of the ion specific for a particular disaccharide, Iint was the intensity of the internal standard ion and R, was response factor. This method was validated using several quality control samples and showed satisfactory accuracy and precision. Using this quantitative analysis procedure, percentages of disaccharide compositions for heparins from porcine and bovine intestinal mucosa and HS from bovine kidney, were determined [88].
CONCLUSION
The utility of MS for analyzing highly charged sulfated polysaccharides, is just beginning to be realized. With modern MS techniques, structural information such as molecular weight, monosaccharide composition, number and position of sulfo groups, composition of disaccharide blocks, and sequence of highly charged sulfated carbohydrates, can be obtained. The extremely high sensitivity of MS techniques made them amenable to analyze micro amounts of carbohydrates from biological samples.
The fragility of sulfo groups during ionization steps and tandem MS dissociation steps makes the MS analysis challenging. Optimization of MS conditions reduces undesired fragmentation. Tandem MS has the potential to become a robust method to completely sequence complex oligosaccharides. Mechanistic studies of oligosaccharide behavior in MS and tandem MS will help advance methods, development. Accumulation of MS data of various oligosaccharides will be useful to establish a library of sulfated oligosaccharides sequences. The progress of structure analysis of GAGs should dramatically improve our understanding of their biological functions and help in the development of structure-activity relationships for these important biopolymers.
ABBREVIATION
- AS
Acharan sulfate
- CE
Capillary electrophoresis
- CID
Collision-induced dissociation
- CS
Chondroitin sulfate
- dp
Degree of polymerization
- DS
Dermatan sulfate
- EOF
Electroosmotic flow
- ESI
Electrospray ionization
- FAB
Fast atom bombardment
- GAG
Glycosaminoglycan
- GalNp
2-amino, 2-deoxygalactopyranose
- GlcAp
Glucopyranosyluronic acid
- GlcNp
2-amino, 2-deoxyglucosopyranose
- HA
Hyaluronic acid or hyaluronan
- HexNp
2-amino, 2-deoxyhexopyranose, GalNp or GlcNp
- HPLC
High performance liquid chromatography
- HS
Heparan sulfate
- IdoAp
Idopyranosyluronic acid
- KS
Keratan sulfate
- LSI
Liquid secondary ion
- MALDI
Matrix-assisted-laser-desorption-ionization
- MIKE
Mass-analyzed ionic kinetic energy
- MS
Mass spectrometry
- MW
Molecular weight
- PD
Plasma desorption
- PEN
Property encoded nomenclature
- PG
Proteoglycan
- RPIP
Reversed-phase ion-pairing
- SAX
Strong anion exchange
- SEC
Size-exclusion chromatography
- SNA
Surface non-covalent affinity
- TIC
Total ion chromatography
- TOF
Time of flight
- TPA
Tetrapropylammonium
- UAp
Uronopyranosyl acid IdoAp or GlcAp
- ΔUAp
Unsaturated uronate residue
- UV
UV
REFERENCES
- [1].Perrimon N; Bernfield M Semin. Cell Dev. Biol, 2001, 12, 65. [DOI] [PubMed] [Google Scholar]
- [2].Linhardt RJ; Toida T Acc. Chem. Res, 2004, 37, 431. [DOI] [PubMed] [Google Scholar]
- [3].Linhardt RJ J. Med. Chem, 2003, 46, 2551. [DOI] [PubMed] [Google Scholar]
- [4].Capila I; Linhardt RJ Angew. Chem. Int. Ed. Engl, 2002, 41, 390. [DOI] [PubMed] [Google Scholar]
- [5].Munoz EM; Linhardt RJ Arterioscler. Thromb. Vasc. Biol, 2004, 24, 1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Carney SL; Muir H Physiol., Rev, 1998, 68, 858. [DOI] [PubMed] [Google Scholar]
- [7].Iozzo RV Annu. Rev. Biochem, 1998, 67, 609. [DOI] [PubMed] [Google Scholar]
- [8].Schwartz NB Front. Biosci, 2000, 5, D649. [DOI] [PubMed] [Google Scholar]
- [9].Toida T; Chaidedgumjorn A; Linhardt RJ Trends Glycosci. Glycotechnol, 2003, 15, 29. [Google Scholar]
- [10].Hileman RE; Siegel MM;Tabel K; Balagurunathan K; Linhardt RJ Electrophoresis, 1998, 19, 2677. [DOI] [PubMed] [Google Scholar]
- [11].Kim YS; Ahn MY; Wu SJ; Kim D; Toida T; Teesch LM; Park Y; Yu G; Lin J; Linhardt RJ Glycobiology, 1998, 8, 869. [DOI] [PubMed] [Google Scholar]
- [12].Yu G; LeBrun L; Gunay NS; Hoppensteadt D; Walenga JM; Fareed J; Linhardt RJ Thromb. Res, 2000, 100, 549. [DOI] [PubMed] [Google Scholar]
- [13].Toida T; Linhardt RJ Trends Glycosci. Glycotechnol, 1998, 10, 125. [Google Scholar]
- [14].Silbert JE Glycoconj. J 1996, 13, 907. [DOI] [PubMed] [Google Scholar]
- [15].Sugahara K; Mikami T; Uyama T; Mizuguchi S; Nomura K; Kitagawa H Curr. Opin. Struct. Biol, 2003, 13, 612. [DOI] [PubMed] [Google Scholar]
- [16].Hardingham TE; Fosang AJ FASEB J, 1992, 6, 861. [PubMed] [Google Scholar]
- [17].Linhardt RJ; Hileman RE Gen Pharmacol., 1995, 26, 443. [DOI] [PubMed] [Google Scholar]
- [18].Akiyama H; Sakai S; Linhardt RJ; Goda Y; Toida T; Maitani T Biochem. J, 2004, 382, 269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Greiling H; Scott JE Keratan sulphate-chemistry, biology, chemical pathology, The Biochemical Society: London, 1989. [Google Scholar]
- [20].Lauder RM; Huckerby TN; Brown GM; Bayliss MT; Nieduszynski IA Biochem. J, 2001, 358, 523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Toole BP Nat. Rev. Cancer, 2004, 4, 528. [DOI] [PubMed] [Google Scholar]
- [22].Liu J; Shriver Z; Pope RM; Thorp SC; Duncan MB; Copeland RJ; Raska CS; Yoshida K; Eisenberg RJ; Cohen G; Linhardt RJ; Sasisekharan R J. Biol. Chem, 2002, 277, 33456. [DOI] [PubMed] [Google Scholar]
- [23].Venkataraman G; Shriver Z; Raman R; Sasisekharan R Science, 1999, 286, 537. [DOI] [PubMed] [Google Scholar]
- [24].Liu J, Desai UR; Han X-J; Toida T; Linhardt RJ Glycobiology, 1995, 5, 765. [DOI] [PubMed] [Google Scholar]
- [25].Ernst S; Langer R; Cooney CL; Sasisekharan R Crit. Rev. Biochem. Mol. Biol, 1995, 30, 387. [DOI] [PubMed] [Google Scholar]
- [26].LeBrun LA; Linhardt RJ In Methods In Molecular Biology; lozzo RV, Ed.; Humana Press Inc.: Totowa, New Jersey, 2001; Vol. 171, pp. 353–361. [DOI] [PubMed] [Google Scholar]
- [27].Hernaiz MJ; Linhardt RJ In Methods in Molecular Biology, Iozzo RV, Ed.; Humana Press Inc.: Totowa, New Jersey, 2001; Vol. 171, pp. 363–371. [DOI] [PubMed] [Google Scholar]
- [28].Reinhold VN; Carr SA; Green BN; Petitou M; Choay J; Sinay P Carbohydr. Res, 1987, 161, 305. [DOI] [PubMed] [Google Scholar]
- [29].Mallis LM; Wang HM; Loganathan D; Linhardt RJ Anal. Chem, 1989, 61, 1453. [DOI] [PubMed] [Google Scholar]
- [30].Linhardt RJ; Wang HW; Loganathan D; Lamb DJ; Mallis LM Carbohydr. Res, 1992, 225, 137. [DOI] [PubMed] [Google Scholar]
- [31].Loganathan D; Wang HM; Mallis LM; Linhardt RJ Biochemistry, 1990, 29 4362. [DOI] [PubMed] [Google Scholar]
- [32].Yamada S; Yoshida K; Sugiura M; Sugahara K J. Biol. Chem, 1993, 268, 4780. [PubMed] [Google Scholar]
- [33].Kinoshita A; Yamada S; Haslam SM; Morris HR; Dell A; Sugahara K J. Biol. Chem, 1996, 271, 26745. [DOI] [PubMed] [Google Scholar]
- [34].Sugahara K; Tanaka Y; Yamada S; Seno N; Kitagawa H; Haslam SM; Morris HR; Dell A J. Biol. Chem, 1997, 272, 19656. [DOI] [PubMed] [Google Scholar]
- [35].Kinoshita A; Yamada S; Haslam SM; Morris HR; Dell A; Sugahara K Biochemistry, 2001, 40, 12654. [DOI] [PubMed] [Google Scholar]
- [36].Dell A; Rogers ME; Thomas-Oates JE Carbohydr. Res, 1988, 179, 7. [Google Scholar]
- [37].Khoo K; Morris HR; McDowell RA; Dell A; Maccarana M; Lindahl U Carbohydr. Res, 1993, 244, 205. [DOI] [PubMed] [Google Scholar]
- [38].Sugahara K; Takemura Y; Sugiura M; Kohno Y; Yoshida K; Takeda K; Khoo KH; Morris HR; Dell A Carbohydr. Res, 1994, 255, 165. [DOI] [PubMed] [Google Scholar]
- [39].Juhasz P; Biemann K Proc. Natl. Acad. Sci. U.S.A, 1994, 91, 4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Fromm JR; Hileman RE; Caldwell EEO; Weiler JM; Linhardt RJ Arch. Biochem. Biophys, 1995, 323, 279. [DOI] [PubMed] [Google Scholar]
- [41].Juhasz P; Biemann K Carbohydr. Res, 1995, 270, 131. [DOI] [PubMed] [Google Scholar]
- [42].Sturiale L; Naggi A; Torri G Semin. Thromb. Hemost, 2001, 27, 465. [DOI] [PubMed] [Google Scholar]
- [43].Ernst S; Rhomberg AJ; Biemann K; Sasisekharan R Proc. Natl. Acad. Sci. U.S.A, 1998, 95, 4182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Rhomberg AJ; Shriver Z; Biemann K; Sasisekharan R Proc. Natl. Acad. Sci. U.S.A, 1998, 95, 12232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Shriver Z; Sundaram M; Venkataraman G; Fareed J; Linhardt RJ; Biemann K, Sasisekharan R Proc. Natl. Acad. Sci. U.S.A, 2000, 97, 10365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Rhomberg AJ; Ernst S; Sasisekharan R; Biemann K Proc. Natl. Acad. Sci. U.S.A, 1998, 95, 4176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Schiller J; Arnhold J; Benard S; Reichl S; Arnold K Carbohydr. Res, 1999, 318, 116. [DOI] [PubMed] [Google Scholar]
- [48].Armstrong DW; Zhang LK; He L; Gross ML Anal. Chem, 2001, 73, 3679. [DOI] [PubMed] [Google Scholar]
- [49].Siegel MM; Tabei K; Kagan MZ; Vlahov IR; Hilemanm RE; Linhardt RJ J. Mass Spectrom, 1997, 32, 760. [DOI] [PubMed] [Google Scholar]
- [50].Yang HO; Gunnay NS; Toida T; Kuberan B; Yu G; Kim YS; Linhardt RJ Glycobiology, 2000, 10, 1033. [DOI] [PubMed] [Google Scholar]
- [51].Yu G; Guan H; Ioanoviciu AS; Sikkander SA; Thanawiroon C; Tobacman JK; Toida T; Linhardt RJ Carbohydr. Res, 2002, 337, 433. [DOI] [PubMed] [Google Scholar]
- [52].Chai W; Luo J; Lim CK; Lawson AM Anal. Chem, 1998, 70, 2060. [DOI] [PubMed] [Google Scholar]
- [53].Chai W; Beeson JB; Lawson AM J. Biol. Chem, 2002, 277, 22438. [DOI] [PubMed] [Google Scholar]
- [54].Rope RM; Raska CS; Thorp SC; Liu J Glycobiology, 2001, 11, 505. [DOI] [PubMed] [Google Scholar]
- [55].Ahn MY; Shin KH; Kim DH; Jung EA; Toida T; Linhardt RJ; Kim YS Can. J. Microbiol, 1998, 44, 423. [DOI] [PubMed] [Google Scholar]
- [56].Gunay NS; Tadano-Aritomi K; Toida T; Ishizuka I; Linhardt RJ Anal. Chem, 2003, 75, 3226. [DOI] [PubMed] [Google Scholar]
- [57].Pervin A; Gallo C; Jandik KA; Han X-J; Linhardt RJ Glycobiology, 1995, 5, 83. [DOI] [PubMed] [Google Scholar]
- [58].Hileman RE; Smith AE, Toida T; Linhardt RJ Glycobiology, 1997, 7, 231. [DOI] [PubMed] [Google Scholar]
- [59].Col RD; Silvestro L; Naggi A; Torri G; Baiocchi C; Moltrasio D; Cedro A; Viano I J. Chromatogr, 1993, 647, 289. [DOI] [PubMed] [Google Scholar]
- [60].Kuberan B; Lech M; Zhang L; Wu ZL; Beeler DL; Rosenberg RD, J. Am. Chem. Soc, 2002, 124, 8707. [DOI] [PubMed] [Google Scholar]
- [61].Lawrence R; Kuberan B; Lech M; Beeler DL; Rosenberg RD Glycobiology, 2004, 14, 467. [DOI] [PubMed] [Google Scholar]
- [62].Thanawiroon C; Linhardt RJ J. Chromatogr A, 2003, 1014, 215. [DOI] [PubMed] [Google Scholar]
- [63].Thanawiroon C; Rice KG; Toida T; Linhardt RJ, J. Biol. Chem, 2004, 279, 2608. [DOI] [PubMed] [Google Scholar]
- [64].Henriksen J; Ringborg LH; Roepstorrf P J. Mass Spectrom, 2004, 39, 1305. [DOI] [PubMed] [Google Scholar]
- [65].Linhardt RJ; Toida T Science, 2002, 298, 1441. [DOI] [PubMed] [Google Scholar]
- [66].Mao W; Thanawiroon C; Linhardt RJ Biomed. Chromatogr, 2002, 16, 77. [DOI] [PubMed] [Google Scholar]
- [67].Zamfir A; Seidler DG; Kresse H; Peter-Katalinic J Rapid Commun. Mass Spectrom, 2002, 16, 2015. [DOI] [PubMed] [Google Scholar]
- [68].Duteil S; Gareil P; Girault S; Mallet A; Feve C; Siret L Rapid Commun. Mass Spectrom, 1999, 13, 1889. [DOI] [PubMed] [Google Scholar]
- [69].Zamfir A; Seidler DG; Schonherr E; Kresse H; Peter-Katalinic J Electrophoresis, 2004, 25, 2010. [DOI] [PubMed] [Google Scholar]
- [70].Ruiz-Calero V; Moyano E; Puignou L; Galceran MT J. Chromatogr. A, 2001, 914, 277. [DOI] [PubMed] [Google Scholar]
- [71].Chai W; Rosankiewicz JR; Lawson AM Carbohydr. Res, 1995, 269, 111. [DOI] [PubMed] [Google Scholar]
- [72].McNeal CJ; Macfarlane RD Biochem. Biophys. Res. Commun, 1986, 139, 18. [DOI] [PubMed] [Google Scholar]
- [73].Desai UR; Wang HM; Linhardt RJ Arch. Biochem. Biophys, 1993, 306, 461. [DOI] [PubMed] [Google Scholar]
- [74].Shriver Z; Raman R; Venkataraman G; Drummond K; Turnbull J; Toida T; Linhardt RJ; Biemann K; Sasisekharan R Proc. Natl. Acad. Sci. U.S.A, 2000, 97, 10359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Liu J; Shriver Z; Blaiklock P; Yoshida K; Sasisekharan R; Rosenberg RD J. Biol. Chem, 1999, 274, 38155. [DOI] [PubMed] [Google Scholar]
- [76].Keiser N; Venkataraman G; Shriver Z; Sasisekharan R Nat. Med, 2001, 7, 123. [DOI] [PubMed] [Google Scholar]
- [77].Domon B; Costello CE Glycoconj. J, 1988, 5, 397. [Google Scholar]
- [78].Lamb DJ; Wang HM; Mallis LM; Linhardt RJ J. Am. Soc. Mass Spectrom, 1992, 3, 797. [DOI] [PubMed] [Google Scholar]
- [79].Zaia J; McClellan JE; Costello CE Anal. Chem, 2001, 73, 6030. [DOI] [PubMed] [Google Scholar]
- [80].McClellan JE; Costello CE; O’Connor PB; Zaia, J. Anal. Chem, 2002, 74, 3760. [DOI] [PubMed] [Google Scholar]
- [81].Zaia J; Li X; Chan S; Costello CE J. Am. Soc. Mass Spectrom, 2003, 14, 1270. [DOI] [PubMed] [Google Scholar]
- [82].Zaia J; Costello CE Anal. Chem, 2003, 75, 2445. [DOI] [PubMed] [Google Scholar]
- [83].Saad OM; Leary JA J. Am. Soc. Mass Spectrom, 2004, 15, 1274. [DOI] [PubMed] [Google Scholar]
- [84].Oguma T; Toyoda H; Toida T; Imanan T Biomed. Chromatogr, 2001, 15, 356. [DOI] [PubMed] [Google Scholar]
- [85].Zaia J; Costello CE Anal. Chem, 2001, 73, 233. [DOI] [PubMed] [Google Scholar]
- [86].Desaire H; Leary JA J. Am. Soc. Mass Spectrom, 2000, 11, 916. [DOI] [PubMed] [Google Scholar]
- [87].Desaire H; Sirich TL; Leary JA Anal. Chem, 2001, 73, 3513. [DOI] [PubMed] [Google Scholar]
- [88].Saad OM; Leary JA Anal. Chem, 2003, 75, 2985. [DOI] [PubMed] [Google Scholar]
- [89].Zaia J Mass Spectrom. Rev, 2004, 23, 161. [DOI] [PubMed] [Google Scholar]
- [90].Jandik KA; Gu K; Linhardt RJ Glycobiology, 1994, 4, 289. [DOI] [PubMed] [Google Scholar]