

Abstract
The COVID-19 posed a serious threat to human life and health, and SARS-CoV-2 Mpro has been considered as an attractive drug target for the treatment of COVID-19. Herein, we report 2-(furan-2-ylmethylene)hydrazine-1-carbothioamide derivatives as novel inhibitors of SARS-CoV-2 Mpro developed by in-house library screening and biological evaluation. Similarity search led to the identification of compound F8–S43 with the enzymatic IC50 value of 10.76 μM. Further structure-based drug design and synthetic optimization uncovered compounds F8–B6 and F8–B22 as novel non-peptidomimetic inhibitors of Mpro with IC50 values of 1.57 μM and 1.55 μM, respectively. Moreover, enzymatic kinetic assay and mass spectrometry demonstrated that F8–B6 was a reversible covalent inhibitor of Mpro. Besides, F8–B6 showed low cytotoxicity with CC50 values of more than 100 μM in Vero and MDCK cells. Overall, these novel SARS-CoV-2 Mpro non-peptidomimetic inhibitors provide a useful starting point for further structural optimization.
Keywords: 2-(furan-2-ylmethylene)hydrazine-1-carbothioamide derivatives, SARS-CoV-2, Main protease, Non-peptidomimetic inhibitors
Graphical abstract

1. Introduction
The outbreak of coronavirus infectious disease 2019 (COVID-19), caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), has been recognized as a serious threat to human life and health [[1], [2], [3]]. As of March 27, 2021, the disease has caused over 480 million people infections with more than 6 million deaths globally [4]. SARS-CoV-2 is a positive-strand RNA enveloped beta-coronavirus, and similar to severe acute respiratory syndrome coronavirus (SARS-CoV) and Middle East respiratory syndrome coronavirus (MERS-CoV), containing genome encodes non-structural proteins including main protease (Mpro, also known as 3-chymotrypsin-like protease, 3CLpro), papain-like protease (PLpro), helicase, and RNA-dependent RNA polymerase (RdRp) [5,6]. Among them, Mpro is a key enzyme in the viral life cycle, which is involved in the virus’ replication process, and results in the maturation of at least 12 non-structural proteins [[7], [8], [9]]. Furthermore, the highly conserved Mpro in coronavirus and the absence of closely related homologs in humans make Mpro an attractive target for the discovery of broad-spectrum antiviral drugs [10,11].
To date, there is only one Mpro inhibitor on market, although a series of SARS-CoV-2 Mpro inhibitors have been identified (Fig. 1 ) [10,12]. Among them, the organoselenium derivative 1 (Ebselen), was first disclosed as a covalent inhibitor of SARS-CoV-2 Mpro by screening the approved drugs and drug candidates [13]. It is reported that Ebselen could inhibit SARS-CoV-2 Mpro activity with an IC50 value of 0.67 μM, and SARS-CoV-2 in Vero cells with an EC50 value of 4.67 μM13. A variety of significant peptidomimetic inhibitors of SARS-CoV-2 Mpro have been reported, including compound 2 (N3), 3 (13b), 4 (11a), 5 (Boceprevir), 6 (GC-376), 7 (MI-09), and 8 (PF-07321332), which exhibited high SARS-CoV-2 Mpro inhibitory activity and SARS-CoV-2 inhibition at micromolar to sub-micromolar levels [[13], [14], [15], [16], [17], [18], [19], [20], [21]]. Furthermore, PF-07321332 was approved for mild and moderate symptoms caused by SARS-CoV-2 infection by US Food and Drug Administration (FDA), and PF-07321332 was combined with protease inhibitor and cytochrome P450 3A4 (CYP3A4) inactivator ritonavir to reduce its metabolism by CYP3A4 [21].
Fig. 1.

Chemical structures of represented inhibitors of SARS-CoV-2 Mpro.
Although the peptidomimetic inhibitors are highly effective against SARS-CoV-2 Mpro, the stability of hydrolase limited the application of these inhibitors [[21], [22], [23], [24]]. In contrast, the non-peptidomimetic inhibitors are less developed [25,26]. Su et al. and Liu et al. simultaneously reported that compound 9 (Baicalein) was a non-peptidomimetic inhibitor of SARS-CoV-2 Mpro with the enzymatic inhibitory ability and antiviral activity both at micromolar levels [27,28]. Meanwhile, compound 10, an N-substituted isatin derivative, was identified as a potent non-peptidomimetic inhibitor of SARS-CoV-2 Mpro, with an IC50 value of 0.045 μM29. Furthermore, compound 11 (Masitinib), an orally bioavailable tyrosine kinase inhibitor, was revealed as a potent inhibitor of SARS-CoV-2 Mpro, with an IC50 value of 2.50 μM, through screening a library of 1900 clinically safe drugs [30]. Masitinib could block the replication of coronavirus in the lungs and nose of SARS-CoV-2 infected mice [30]. Moreover, Kneller and coworkers reported that compound 12 (HL-3-68) exhibited SARS-CoV-2 Mpro inhibitory activity, with an IC50 value of 0.29 μM, through X-ray/neutron crystallography guided drug design [31]. Additionally, compound 13 (Jun8-76-3A) was discovered as a non-peptidomimetic inhibitor of SARS-CoV-2 Mpro by the modification of known Mpro inhibitor ML188 [32]. Overall, the chemical diversity of the identified non-peptidomimetic inhibitors is still highly limited. Therefore, it is urgent to develop novel non-peptidomimetic inhibitors against SARS-CoV-2 Mpro for the broad-spectrum antiviral drug candidate discovery, especially the new scaffold inhibitors that were not reported in previous coronavirus studies.
Herein, we report the identification of 2-(furan-2-ylmethylene)hydrazine-1-carbothioamide derivatives as novel non-peptidomimetic inhibitors of SARS-CoV-2 Mpro by screening our in-house library and subsequent similarity search. Interestingly, compound F8 was disclosed with the dissociation constant (KD) value of 27.7 μM, by surface plasmon resonance (SPR) assay, which also exhibited an IC50 value of 21.28 μM by enzymatic assay. Further two-dimensional similarity search based on the structure of compound F8 led to the identification of a series of 2-(furan-2-ylmethylene)hydrazine-1-carbothioamide derivatives as novel SARS-CoV-2 Mpro inhibitors. Among them, the most potent compound, F8–S43, exhibited the IC50 value of 10.76 μM against SARS-CoV-2 Mpro. Then, three rounds of optimization based on the structure-based drug design and synthetic modification discovered compounds F8–B6 and F8–B22 as non-peptidomimetic inhibitors of Mpro with IC50 values of 1.57 μM and 1.55 μM, respectively. Moreover, enzymatic kinetic and mass spectrometry studies demonstrated that F8–B6 was a reversible covalent inhibitor of Mpro. Besides, F8–B6 exhibited no obvious cytotoxicity in Vero and MDCK cells with CC50 values over 100 μM. Furthermore, the structure-activity relationship (SAR) of the newly identified scaffold was discussed, which provided useful guidance for further chemical optimization.
2. Results and discussion
2.1. Identification of novel Mpro inhibitors by screening in-house library
To discover novel SARS-CoV-2 Mpro inhibitors, especially the new chemical structures that were not previously reported in coronavirus study, an in-house library was screened by SPR assay (Fig. 2 A). As drawn in Fig. 2B, compound F8 showed the high possibility as a new inhibitor of SARS-CoV-2 Mpro, with the KD value of 27.7 μM. The further enzymatic assay demonstrated that compound F8 inhibited SARS-CoV-2 Mpro activity with an IC50 value of 21.28 μM (Fig. 2C). Similarity analysis of compound F8 showed that it is not similar to any of the previously reported SARS-CoV-2 Mpro inhibitors with the Tanimoto coefficient below 0.14 (Fig. 2D). All of these indicated that the identified non-peptidomimetic inhibitor F8 expanded the chemical space of SARS-CoV-2 Mpro inhibitors and provided a useful starting point for further structural optimization.
Fig. 2.

The discovery of SARS-CoV-2 Mpro inhibitors. (A) Screen the in-house library through SPR assay. (B) The chemical structure and KD value of compound F8. (C) The dose-dependent curve of F8 against SARS-CoV-2 Mpro. (D) The similarity analysis of compound F8 with known SARS-CoV-2 Mpro inhibitors.
2.2. Similarity search and preliminary SAR study
To investigate SAR and find more potent inhibitors of this new scaffold, a two-dimensional similarity search was performed based on the chemical structure of compound F8. A total of 70 compounds were captured and purchased from the ChemDiv and SPECS commercial databases. As exhibited in Table 1 , the replacement of the 3-carboxyl phenyl group of F8 with a 3-trifluoromethyl phenyl group (F8–S1), a 2-carboxyl phenyl group (F8–S2), a 2-nitro-4-methoxy phenyl group (F8–S3), a 4-nitrophenyl group (F8–S4), a 3-methyl-4-nitrophenyl group (F8–S5), or a 2,3-dimethyl-4-nitrophenyl (F8–S6), led to a decrease of potency, indicating that the 3-carboxyl phenyl group of F8 was essential for maintaining SARS-CoV-2 Mpro inhibitory activity. Furthermore, the replacement of the R1 group of F8 with a 10-methylacridin-9(10H)-one moiety (F8–S7 to F8–S16), or a poly-substituted pyridine ring (F8–S17 to F8–S20), the inhibitory ability of Mpro was decreased. Meanwhile, the replacement of the R1 group with a 1,2,5-oxadiazol-3-amine group (F8–S22), or a 1-methyl-1H-tetrazol-5-amine group (F8–S24), produced no significant effect on inhibitory potency. Besides, the introduction of the large hydrophobic substituents group (F8–S25, F8–S26, F8–S28 to F8–S39), was not conducive to the maintenance of inhibitory ability, which indicated that the R1 group of F8 was more suitable for small substituents. Notably, the replacement of a urea linker with a thiourea linker was likely to improve the inhibitory potency (F8–S40 to F8–S43). Among them, the most potent compound, F8–S43, displayed an IC50 value of 10.76 μM against SARS-CoV-2 Mpro (Fig. S1).
Table 1.
The chemical structures and enzymatic activities of the 43 analogs of F8.
| Compounds | R [1] | X | R [2] | Inhibition% (50 μM) a | IC50 ± SD (μM) a |
|---|---|---|---|---|---|
| Tideglusib | – | – | – | 98.6 | 0.30 ± 0.02 |
| F8 | 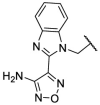 |
O |  |
65.0 | 21.28 ± 0.89 |
| F8–S1 | 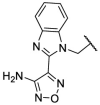 |
O |  |
25.5 | N.T. b |
| F8–S2 | 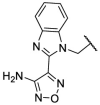 |
O | 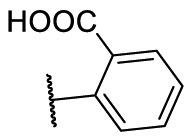 |
41.7 | N.T. |
| F8–S3 | 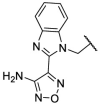 |
O | 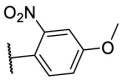 |
<20.0 | N.T. |
| F8–S4 | 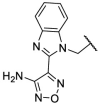 |
O | 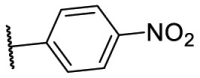 |
30.1 | N.T. |
| F8–S5 | 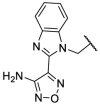 |
O | 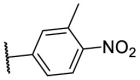 |
42.9 | N.T. |
| F8–S6 | 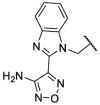 |
O | 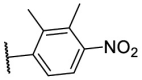 |
<20.0 | N.T. |
| F8–S7 | 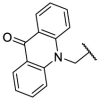 |
O |  |
21.3 | N.T. |
| F8–S8 | 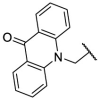 |
O | 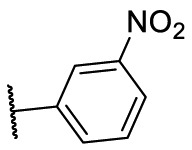 |
26.8 | N.T. |
| F8–S9 | 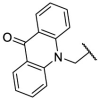 |
O | 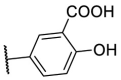 |
54.5 | N.T. |
| F8–S10 | 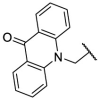 |
O | 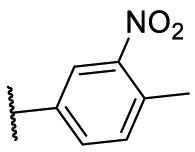 |
55.4 | N.T. |
| F8–S11 | 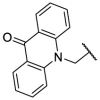 |
O | 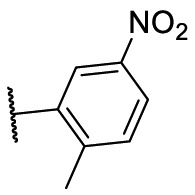 |
22.5 | N.T. |
| F8–S12 | 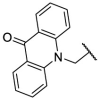 |
O | 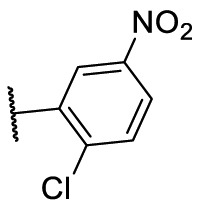 |
<20.0 | N.T. |
| F8–S13 | 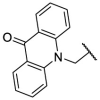 |
O | 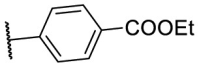 |
<20 | N.T. |
| F8–S14 | 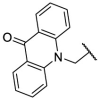 |
O | 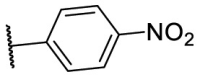 |
30.0 | N.T. |
| F8–S15 | 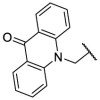 |
O | 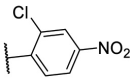 |
24.1 | N.T. |
| F8–S16 | 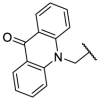 |
O | 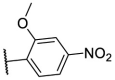 |
26.0 | N.T. |
| F8–S17 | 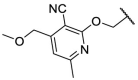 |
O | 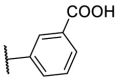 |
37.3 | N.T. |
| F8–S18 | 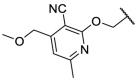 |
O | 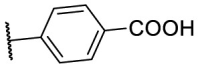 |
36.2 | N.T. |
| F8–S19 | 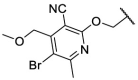 |
O | 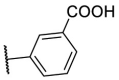 |
32.9 | N.T. |
| F8–S20 | 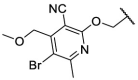 |
O | 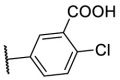 |
38.3 | N.T. |
| F8–S21 | 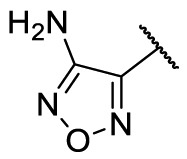 |
O | 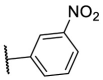 |
31.2 | N.T. |
| F8–S22 | 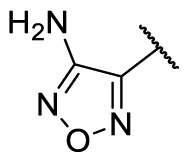 |
O |  |
70.9 | 25.02 ± 1.11 |
| F8–S23 | 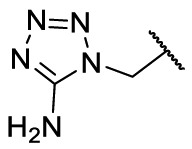 |
O |  |
34.5 | N.T. |
| F8–S24 | 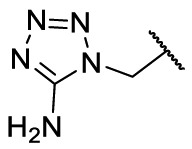 |
O | 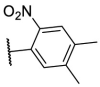 |
86.0 | 17.50 ± 1.65 |
| F8–S25 |  |
O |  |
45.3 | N.T. |
| F8–S26 | 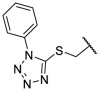 |
O | 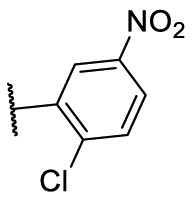 |
22.4 | N.T. |
| F8–S27 | 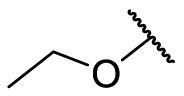 |
O | 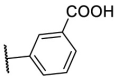 |
29.4 | N.T. |
| F8–S28 | 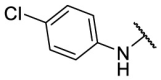 |
O | 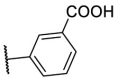 |
44.6 | N.T. |
| F8–S29 | 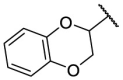 |
O | 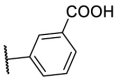 |
33.7 | N.T. |
| F8–S30 | 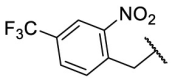 |
O | 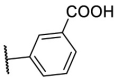 |
35.1 | N.T. |
| F8–S31 | 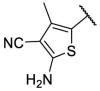 |
O | 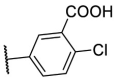 |
35.8 | N.T. |
| F8–S32 | 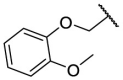 |
O | 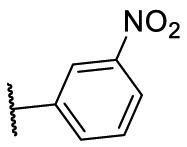 |
24.2 | N.T. |
| F8–S33 | 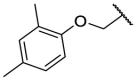 |
O | 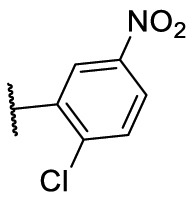 |
<20.0 | N.T. |
| F8–S34 |  |
O | 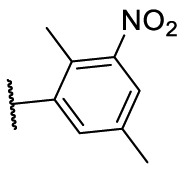 |
<20.0 | N.T. |
| F8–S35 |  |
O | 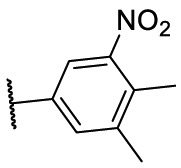 |
62.8 | N.T. |
| F8–S36 | 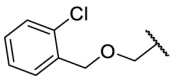 |
O | 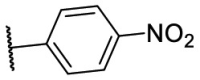 |
23.3 | N.T. |
| F8–S37 | 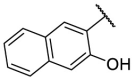 |
O | 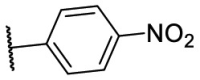 |
<20.0 | N.T. |
| F8–S38 | 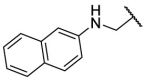 |
O | 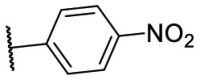 |
23.0 | N.T. |
| F8–S39 | 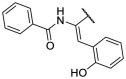 |
O | 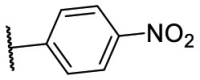 |
38.3 | N.T. |
| F8–S40 | 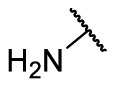 |
S | 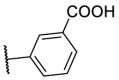 |
85.1 | 10.88 ± 0.16 |
| F8–S41 | 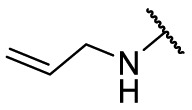 |
S | 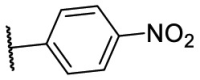 |
51.4 | N.T. |
| F8–S42 | 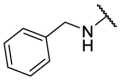 |
S | 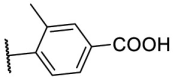 |
55.9 | N.T. |
| F8–S43 | 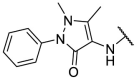 |
S | 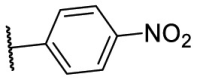 |
95.0 | 10.76 ± 0.48 |
Data are presented as geometric mean values of at least two independent runs.
Not tested.
As shown in Table S1, the replacement of the thiourea linker of F8–S43 with a hydrazine linker (F8–S44 to F8–S48), an imine linker (F8–S49 to F8–S52), or a double bond linker (F8–S53 to F8–S65) led to the decrease of inhibitory potency, suggesting that the thiourea linker might be a good skeleton for these newly identified inhibitors. As for the right moiety of F8, the removal of the benzene ring resulted in the loss of potency (F8–S66 to F8–S70), indicating that the benzene ring of F8 was likely to play a hydrophobic role in the binding pocket of SARS-CoV-2 Mpro. As displayed in Table 2 , the replacement of the 4-nitrophenyl group of F8–S43 with a 4-bromobenzenyl group (F8–S43–S1), led to the remarkable loss of inhibitory activity, and the reduction of the volume of the R1 group was beneficial to the preservation of inhibitory activity (F8–S43–S2, IC50 = 8.08 μM; F8–S43–S3, IC50 = 9.69 μM). Notably, the replacement of the thiourea linker to urea or guanidine linker decreased the inhibitory potency (F8–S43–S4 to F8–S43–S6), indicating that the thiourea linker was very important for the maintenance and improvement of the potency. To study the SAR of the right moiety, the hydrophobic substituent of the left moiety and the thiourea linker were fixed according to the structure of F8–S43. As drawn in Table S2, the decrease of the hydrophobic volume of the right moiety was not conducive to the maintenance of activity (F8–S43–S11 to F8–S43–S32). All in all, these results indicate that the newly identified scaffold expanded the chemical diversity of SARS-CoV-2 Mpro inhibitors, which could serve as a starting point for subsequent structural optimization.
Table 2.
The chemical structures and enzymatic activities of the 10 analogs of F8–S43.
| Compounds | R [1] | X | R [2] | Inhibition% (50 μM) a | IC50 ± SD (μM) a |
|---|---|---|---|---|---|
| Tideglusib | – | – | – | 98.6 | 0.30 ± 0.02 |
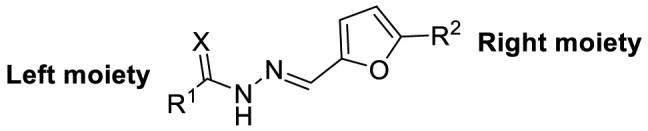
| F8–S43 | 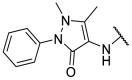 |
S | 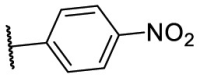 |
95.0 | 10.76 ± 0.48 |
| F8–S43–S1 | 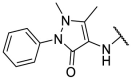 |
S | 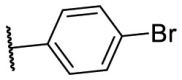 |
27.7 | N.T. b |
| F8–S43–S2 | NH2 | S | 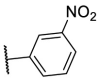 |
81.6 | 8.08 ± 0.38 |
| F8–S43–S3 | NH2 | S | 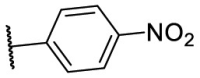 |
81.1 | 9.69 ± 0.45 |
| F8–S43–S4 | NH2 | O | 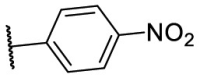 |
<20.0 | N.T. |
| F8–S43–S5 | 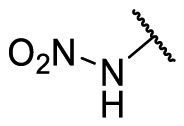 |
NH | 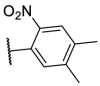 |
20.1 | N.T. |
| F8–S43–S6 | 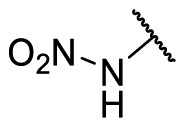 |
NH | 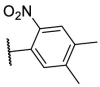 |
28.7 | N.T. |
| F8–S43–S7 | 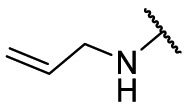 |
S | 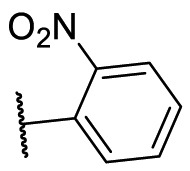 |
28.4 | N.T. |
| F8–S43–S8 | 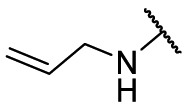 |
S | 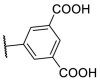 |
34.2 | N.T. |
| F8–S43–S9 | 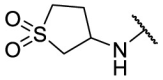 |
S | 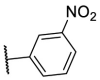 |
30.2 | N.T. |
| F8–S43–S10 | 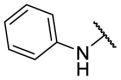 |
S | 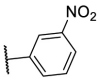 |
<20.0 | N.T. |
Data are presented as geometric mean values of at least two independent runs.
Not tested.
2.3. Rational design and synthetic optimization
2.3.1. Structure-based design of newly identified scaffold
To guide the structural optimization of the newly identified scaffold, the representative compound F8–S43 was docked into the catalytic site of the SARS-CoV-2 Mpro (PDB ID, 7JU7) [30]. As shown in Fig. 3 A, compound F8–S43 was embedded into the catalytic site of SARS-CoV-2 Mpro and occupied the S1 and S2 sites, while the S1′, S3, and S4 sites were not filled with F8–S43. Furthermore, the 4-nitrophenyl moiety of F8–S43 was located at the S1 site and formed a hydrogen bond with His163, and the furan ring was deeply buried in the S2 site and had a π-π stacking interaction with the imidazole of His41 (Fig. 3B and Fig. S2). Meanwhile, the thiourea linker of F8–S43 formed two hydrogen bonds to the backbone carbonyl of Cys44, so the removal or replacement of the thiourea linker led to the loss of inhibitory ability. Besides, the right moiety of F8–S43 was located at the solvent-exposed area and formed a hydrogen bond with Ser46. As drawn in Fig. 3C, the removal of the dihydro-2H-pyrazol-2′-one moiety of F8–S43 had no impact on enzymatic inhibition in the aforementioned similarity search study, so the solvent-exposed region of this scaffold was structurally modified to improve the inhibitory potency of SARS-CoV-2 Mpro, followed by S1 and S2 pockets occupied moieties.
Fig. 3.

Predicted binding mode of compound F8–S43. (A) The binding pattern of compound F8–S43 with SARS-CoV-2 Mpro in surface and 2D diagram (B). Hydrogen bonds are represented by yellow lines. Images depicting the proposed binding modes were generated using PyMOL software. (C) The design of newly identified SARS-CoV-2 Mpro inhibitors.
2.3.2. Synthesis of target compounds
The target compounds F8-A1 to F8-A9 were synthesized as illustrated in Scheme 1 . Briefly, we synthesized the intermediates (15) from appropriate aryl boronic acid (14) with 5-bromofuran-2-carbaldehyde through Pd(PPh3)4 catalyzed Suzuki-Miyaura cross-coupling reaction [33]. Then, corresponding intermediates (15) reacted with substituted thiosemicarbazide to produce imine linkage (F8-A1 to F8-A9) through modification of published procedures or known methods [34,35].
Scheme 1.

Synthetic Route of Compounds F8-A1 to F8-A9. Reagents and conditions: (a) 5-bromofuran-2-carbaldehyde, Pd(PPh3)4, K2CO3, PhMe/EtOH/H2O, 90 °C, overnight; (b) R1NH2CSNHNH2, MeOH, 50 °C, 4 h.
The synthetic routes of the target compounds F8–B1 to F8–B13 were described in Scheme 2 . The building blocks (17 and 19) were prepared following previously described procedures [33]. Shortly, the starting material 5-iodofuran-2-carbaldehyde (16) or (5-formylfuran-2-yl)boronic acid (18) reacted with (4-carbamoylphenyl)boronic acid or substituted bromobenzene to form corresponding intermediates (17 and 19) through Suzuki-Miyaura cross-coupling reaction. Then, the aldehyde group of intermediates (17 and 19) condensed with the amino group of thiosemicarbazide to form the imine linkage giving the target compounds (F8–B1 to F8–B13).
Scheme 2.

Synthetic Route of Compounds F8–B1 to F8–B13. Reagents and conditions: (a) (4-carbamoylphenyl)boronic acid, Pd(PPh3)4, K2CO3, PhMe/EtOH/H2O, 90 °C, overnight; (b) Thiosemicarbazide, MeOH, 50 °C, 4 h; (c) Substituted bromobenzene, Pd(PPh3)2Cl2, Na2CO3, MeCN/H2O, 90 °C, overnight; or substituted bromobenzene, Pd(PPh3)4, K2CO3, PhMe/EtOH/H2O, 90 °C, overnight; or Substituted bromobenzene, Pd(PPh3)2Cl2, 2 M Na2CO3, DME/EtOH, 60 °C, overnight.
As drawn in Scheme 3 , the synthetic methods of F8–B14 to F8–B21 were similar to the aforementioned synthesis of compounds F8–B1 to F8–B13. Simply, 5-bromofuran-2-carbaldehyde (20) reacted with corresponding aryl boronic acid to form intermediates (21), then the newly prepared intermediates condensed with thiosemicarbazide to give target compounds F8–B14 to F8–B21. Moreover, the intermediate (23), 5-benzylfuran-2-carbaldehyde, was synthesized by the starting material 5-(hydroxymethyl)furan-2-carbaldehyde (22) reacted with benzene through substitution reaction following previously described procedures [36]. Then, the intermediate (23) was condensed with thiosemicarbazide to give the target compound F8–B22.
Scheme 3.

Synthetic Route of Compounds F8–B14 to F8–B22. Reagents and conditions: (a) Corresponding aryl boronic acid, Pd(PPh3)4, K2CO3, PhMe/EtOH/H2O, 90 °C, overnight; (b) Thiosemicarbazide, MeOH, 50 °C, 4 h; (c) benzene, trifluoromethanesulfonic acid, r.t.
As shown in Scheme 4 , the synthetic methods of F8–C1 to F8–C4 were similar to the aforesaid synthesis of compounds F8–B1 to F8–B13. Briefly, the appropriate aryl bromides (24) reacted with (4-carbamoylphenyl)boronic acid to form intermediates (25) through Suzuki-Miyaura cross-coupling reaction. Then, the corresponding intermediates condensed with thiosemicarbazide to give target compounds F8–C1 to F8–C4. All of the synthesized target compounds were characterized by 1H NMR, 13C NMR, and high-resolution mass spectrometry (HRMS) experiments as single substances.
Scheme 4.

Synthetic Route of Compounds F8–C1 to F8–C4. Reagents and conditions: (a) (4-carbamoylphenyl)boronic acid, Pd(PPh3)4, K2CO3, PhMe/EtOH/H2O, 90 °C, overnight; (b) Thiosemicarbazide, MeOH, 50 °C, 4 h.
2.3.3. Biological evaluation and SAR study
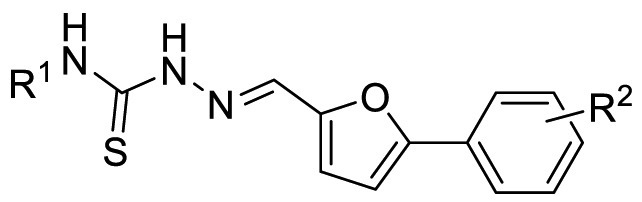
The synthesized target compounds were evaluated through aforesaid enzymatic assay, and the results were summarized in Table 3 . To investigate the SAR of the solvent-exposed region of the newly identified scaffold, the other regions were fixed with the same substituents. Notably, with the replacement of the 3-carboxyl phenyl group of F8-A1 with a 4-carboxyl phenyl group (F8-A2), the inhibitory potency was maintained. Furthermore, the introduction of hydrophobic substituents, including methyl, 4-chlorophenyl, 3-trifluoromethyl phenyl, and cyclohexyl amino groups (F8-A3 to F8-A9), led to decreased inhibitory ability. Thus, the R1 group of this scaffold was fused with a hydrogen atom, and the R2 group was further structural modified to enhance the inhibitory ability and explore explicit SAR.
Table 3.
The chemical structures and enzymatic activities of F8-A1 to F8-A9.
| Compounds | R [1] | R [2] | Inhibition% (50 μM) a | IC50 ± SD (μM) a |
|---|---|---|---|---|
| F8-A1 (F8–S40) | H | 3-COOH | 85.1 | 10.88 ± 0.16 |
| F8-A2 | H | 4-COOH | 89.4 | 17.53 ± 3.98 |
| F8-A3 | 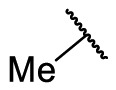 |
3-COOH | 22.2 | N.T. b |
| F8-A4 | 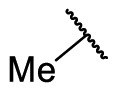 |
4-COOH | 39.5 | N.T. |
| F8-A5 | 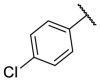 |
3-COOH | 43.8 | N.T. |
| F8-A6 | 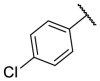 |
4-COOH | 27.5 | N.T. |
| F8-A7 | 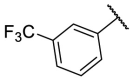 |
3-COOH | 43.0 | N.T. |
| F8-A8 | 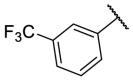 |
4-COOH | 38.6 | N.T. |
| F8-A9 | 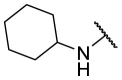 |
4-COOH | 37.3 | N.T. |
Data are presented as geometric mean values of at least two independent runs.
Not tested.
As for the phenyl moiety of the newly identified scaffold (Table 4 ), the replacement of 4-carboxyl phenyl group of F8-A2 with a 4-amide phenyl group (F8–B1), a 4-sulfonyl phenyl group (F8–B2), or a phenyl group (F8–B3), resulted in moderate improve in inhibitory activity. When the benzene ring had multiple substituents (F8–B4 to F8–B14), the polar substituents, including carboxyl group, hydroxyl group, and methoxy group, were favorable to maintain or promote the inhibitory ability. Among them, the 3-hydroxy-4-carboxyl phenyl group substituted compound F8–B6 exhibited good activity, with the IC50 value of 1.57 μM (Fig. S1). Whereas the replacement of the 4-carboxyl phenyl group of F8-A2 with a 3, 4-dichlorobenzyl group (F8–B15), led to the loss of inhibitory potency, which meant that the phenyl moiety of the newly identified scaffold was not suitable to be replaced by a completely hydrophobic substituent. Furthermore, this moiety was compatible with pyridine or substituted pyridine rings (F8–B16 to F8–B17). Intriguingly, the naphthalene ring substitution was not conducive to the preservation of inhibitory activity (F8–B18 to F8–B19), while the quinoline ring did not affect the potency (F8–B20 to F8–B21). Therefore, the aforementioned moiety might be more suitable for nitrogen heterocyclic replacement. Notably, the replacement of the 4-carboxyl phenyl group of F8-A2 with a benzyl group (F8–B22), led to the enhancement of potency, and the compound F8–B22 displayed an IC50 value of 1.55 μM (Fig. S1), which potency was 13-fold more than the initial compound F8.
Table 4.
The chemical structures and enzymatic activities of F8–B1 to F8–B22.
| Compounds | R [1] | Inhibition% (50 μM) a | IC50 ± SD (μM) a |
|---|---|---|---|
| F8-A2 | 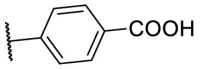 |
89.4 | 17.53 ± 3.98 |
| F8–B1 | 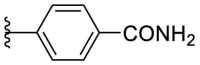 |
94.2 | 4.00 ± 0.25 |
| F8–B2 | 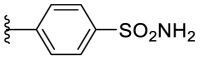 |
98.5 | 5.39 ± 0.14 |
| F8–B3 | 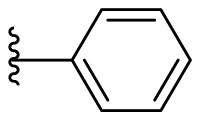 |
100.0 | 4.05 ± 0.26 |
| F8–B4 | 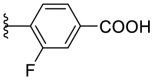 |
100.6 | 8.14 ± 1.14 |
| F8–B5 | 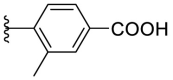 |
98.5 | 4.69 ± 0.34 |
| F8–B6 | 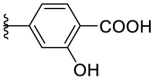 |
102.2 | 1.57 ± 0.08 |
| F8–B7 | 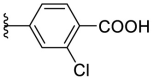 |
89.0 | 32.53 ± 2.33 |
| F8–B8 | 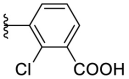 |
84.4 | 40.59 ± 1.06 |
| F8–B9 | 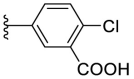 |
93.6 | 25.39 ± 2.05 |
| F8–B10 | 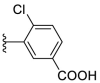 |
97.2 | 10.0 ± 0.50 |
| F8–B11 | 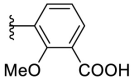 |
79.8 | N.T. b |
| F8–B12 | 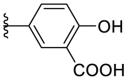 |
114.8 | 9.09 ± 1.05 |
| F8–B13 | 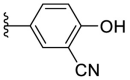 |
106.6 | 4.14 ± 0.16 |
| F8–B14 | 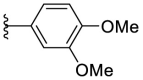 |
102.5 | 5.81 ± 0.07 |
| F8–B15 | 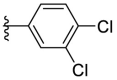 |
49.8 | N.T. |
| F8–B16 | 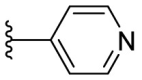 |
95.4 | 7.09 ± 0.51 |
| F8–B17 | 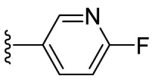 |
97.4 | 8.76 ± 0.75 |
| F8–B18 | 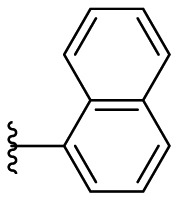 |
73.2 | N.T. |
| F8–B19 | 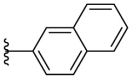 |
54.1 | N.T. |
| F8–B20 | 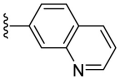 |
92.3 | 10.40 ± 2.20 |
| F8–B21 | 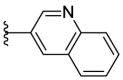 |
88.9 | 10.30 ± 0.70 |
| F8–B22 | 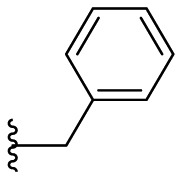 |
100.1 | 1.55 ± 0.08 |
Data are presented as geometric mean values of at least two independent runs.
Not tested.
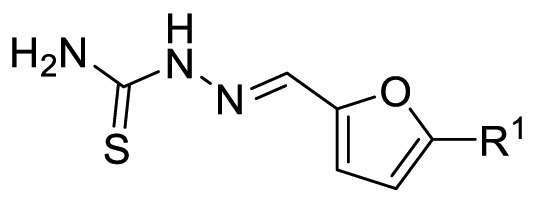
To investigate the furan ring moiety of the newly identified scaffold, the benzene ring moiety was designed to fix with the benzyl group, as the compound F8–B22 displayed an IC50 value of 1.55 μM. However, the designed compounds were difficult to synthesize, so the furan ring moiety was optimized with the 4-carboxyl phenyl group replacement. As drawn in Table 5 , the replacement of 2,5-position substituted furan ring of F8-A2 with a 2,4-position substituted furan ring (F8–C1), a 2,5-position substituted thiophene ring (F8–C2), or a 1,3-position substituted benzene ring (F8–C3), let to maintain or improve inhibitory potency. Whereas, the 1,4-position substituted benzene ring (F8–C4) was not conducive to the preservation of inhibitory activity, indicating that the furan ring moiety acted as an important connecting linker and prefer to three atomic lengths. It is noteworthy that the introduction of a substituent to the furan, thiophene, or benzene ring might be beneficial to the improvement of activity, however the synthesis was too difficult to achieve. Thus, we are trying to perform scaffold hopping based on this scaffold to improve the synthetic accessibility and inhibitory potency in the future.
Table 5.
The chemical structures and enzymatic activities of F8–C1 to F8–C4.
| Compounds | Ar | Inhibition% (50 μM) a | IC50 ± SD (μM) a |
|---|---|---|---|
| F8-A2 | 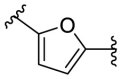 |
89.4 | 17.53 ± 3.98 |
| F8–C1 | 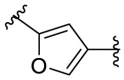 |
90.9 | 24.56 ± 0.70 |
| F8–C2 | 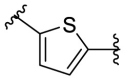 |
95.1 | 5.78 ± 0.30 |
| F8–C3 | 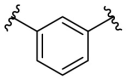 |
99.2 | 5.44 ± 0.16 |
| F8–C4 | 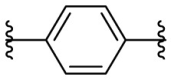 |
64.0 | N.T. b |
Data are presented as geometric mean values of at least two independent runs.
Not tested.
Meanwhile, the mechanism of action of these newly identified inhibitors was investigated through enzymatic kinetic and mass spectrometry assays. As exhibited in Fig. 4 A and B, the Lineweaver-Burk plot with different F8–B6 concentrations yielded an intercept at the X-axis, indicating that F8–B6 is a non-competitive inhibitor of SARS-CoV-2 Mpro. Moreover, prolonged incubation of SARS-CoV-2 Mpro with F8–B6 exhibited a time-dependent increase of inhibition activity (Figs. S3A–3C). The addition of dithiothreitol (DTT) could reverse the inhibitory effect of F8–B6 (Fig. S3D). Further liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis demonstrated that F8–B6 covalently bonded to SARS-CoV-2 Mpro (Fig. 4C), which was similar to the previously reported covalent inhibitor Ebselen [13], with more than one F8–B6 can be covalently bonded to the dimer of Mpro. Notably, thiacetazone moiety was identified as a covalent warhead to cystine in previous work [37,38], and this was consistent with the above SAR that the introduction of thiacetazone moiety led to the increase of inhibitory activity. However, the specific covalent mechanism still needed to be further studied. In addition, reversibility assay of F8–B6 to SARS-CoV-2 Mpro revealed that the ultrafiltration of inhibitor could recover enzymatic activity to a certain extent (Fig. 4D), which indicated that F8–B6 is a reversible inhibitor. Overall, the enzymatic kinetic and mass spectrometry studies demonstrated that F8–B6 is a reversible covalent inhibitor of SARS-CoV-2 Mpro. Interestingly, during the review process of this manuscript, Xu and coworkers reported that thiosemicarbazone is a promising scaffold for the inhibition of SARS-CoV-2 Mpro activity [39]. Besides, thiosemicarbazones are also well-known inhibitors of human Cathepsins. Pandey and coworkers reported that thiosemicarbazones derivatives are inhibitors of Cathepsin B, H, and L through a multi-target approach [40]. Notably, Cathepsin L is a key host cysteine protease utilized by coronaviruses for cell entry and is a promising drug target for novel antivirals against [41,42]. Thus, the selectivity of compounds F8–B6 and F8–B22 against Cathepsin L was investigated through previously reported method [43], and the results demonstrated that compounds F8–B6 and F8–B22 exhibited moderate inhibition of Cathepsin L, with the IC50 values of 16.33 μM and 8.09 μM, respectively (Fig. S4).
Fig. 4.

Michaelis-Menten kinetics analysis (A) and Lineweaver-Burk plot (B) of SARS-CoV-2 Mpro in presence or absence of F8–B6. (C) The liquid chromatograph-mass spectrometer of SARS-CoV-2 Mpro with F8–B6. (D) Reversibility assay of the inhibition ability of F8–B6 against SARS-CoV-2 Mpro.
Meanwhile, two compounds F8–B6 and F8–B22, with good SARS-CoV-2 Mpro inhibitory abilities, were further evaluated for their cytotoxic activities in Vero and MDCK cells by MTT assay. As drawn in Fig. 5 A and B, both compounds F8–B6 and F8–B22 exhibited low cytotoxicity, and F8–B6 showed the CC50 values more than 100 μM in Vero and MDCK cells, while F8–B22 displayed the CC50 values around 100 μM in Vero cells, and above 100 μM in MDCK cells.
Fig. 5.

The cytotoxicity of compounds F8–B6 and F8–B22 in Vero (A) and MDCK cells (B).
3. Conclusion
In the current work, a series of 2-(furan-2-ylmethylene)hydrazine-1-carbothioamide derivatives were identified as non-peptidomimetic inhibitors of SARS-CoV-2 Mpro through screening an in-house library by SPR and enzymatic assays. Further similarity search led to the identification of compound F8–S43, which exhibited an IC50 value of 10.76 μM against SARS-CoV-2 Mpro. Then, three rounds of optimization based on the structure-based drug design and synthetic modification discovered compounds F8–B6 and F8–B22 as non-peptidomimetic inhibitors of Mpro with IC50 values of 1.57 μM and 1.55 μM, respectively. Moreover, enzymatic kinetic and mass spectrometry analysis demonstrated that F8–B6 was a reversible covalent inhibitor of Mpro. Besides, F8–B6 displayed no obvious cytotoxicity in Vero and MDCK cells. Taken together, this chemical series may serve as a good starting point for the further optimization of SARS-CoV-2 Mpro non-peptidomimetic inhibitors.
4. Experimental section
4.1. Chemistry
General method. Synthesis reagents and solvents were obtained from commercial suppliers and used without further purification. Characterizations of compounds are provided in the Supporting Information. 1H and 13C NMR spectra were recorded on Bruker (400 MHz) instruments, using dimethyl sulfoxide (DMSO‑d 6) as solvents. Chemical shifts are given in parts per million (ppm) downfield from tetramethylsilane (δ) as the internal standard in deuterated solvent and coupling constants (J) are in Hertz (Hz). Data are reported as follows: chemical shift, integration, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, dd = doublet of doublet, ddd = doublet of doublet of doublet, dt = doublet of triplet, m = multiplet, bs = broad signal), and coupling constants. High-resolution mass spectra were recorded on a Bruker Apex IV FTMS mass spectrometer using electrospray ionization (ESI). All compounds tested in biological assays were >95% pure.
4.1.1. General procedure A
A schlenk tube was charged with 5-bromofuran-2-carbaldehyde (1 mmol), corresponding boronic acid (1.2 mmol), tetrakis(triphenylphosphine)palladium (0.05 mmol), and potassium carbonate (3 mmol). The vessel was evacuated and backfilled with argon. A mixed solvent of Toluene/EtOH/H2O (5/5/2 mL) was added. Then the schlenk tube was heated to 90 °C overnight. After cooling to room temperature, the reaction mixture was concentrated and diluted with water, filtered and the filtrate was adjusted to pH 2 with 2 N HCl. The precipitate was filtered and dried in vacuo to give the crude products without further purification.
4.1.2. General procedure B
A schlenk tube was charged with (5-formylfuran-2-yl)boronic acid (1.5 mmol), substituted bromobenzene (1 mmol), Pd(PPh3)2Cl2 (0.05 mmol), and sodium carbonate (2 mmol). The vessel was evacuated and backfilled with argon. A mixed solvent of MeCN/H2O (4/1.3 mL) was added. Then the schlenk tube was heated to 60 °C overnight. After cooling to room temperature, the reaction mixture was concentrated and diluted with water, filtered and the filtrate was adjusted to pH 2 with 2 N HCl. The precipitate was filtered and dried in vacuo to give the crude products without further purification.
4.1.3. General procedure C
A mixture of (5-formylfuran-2-yl)boronic acid (1.5 mmol), substituted bromobenzene (1 mmol), Pd(PPh3)2Cl2 (0.05 mmol) in DME/EtOH (3/3 mL), and 2 M aqueous Na2CO3 (3 mL, 6 mmol of Na2CO3) was flushed with nitrogen for 3 min and heated at 60 °C overnight under nitrogen atmosphere. The solvents were removed under reduced pressure, the residue was dissolved in water, the mixture obtained was filtered through Celite, and the filtrate was adjusted to pH 2 with 2 N HCl. The precipitate was filtered and dried in vacuo to give the crude products without further purification.
4.1.4. General procedure D
To a solution of the corresponding aldehyde (1.0 mmol) in MeOH (5.0 mL) was added thiosemicarbazide or substituted thiosemicarbazide (1.05 mmol). The mixture was heated to 50 °C and stirred for 4 h. The residue was recrystallized from ethanol, and the precipitate was filtered and dried in vacuo to give target compounds.
4.1.5. (E)-N-(1,5-dimethyl-3-oxo-2-phenyl-2,3-dihydro-1H-pyrazol-4-yl)-2-((5-(4-nitrophenyl)furan-2-yl)methylene)hydrazine-1-carbothioamide (F8–S43)
Compound F8–S43 was purchased from commercial database. Reddish brown solid. 1H NMR (400 MHz, DMSO‑d 6) δ 12.05 (s, 1H), 9.21 (s, 1H), 8.27 (d, J = 8.9 Hz, 2H), 8.09 (s, 2H), 8.07 (s, 1H), 7.52 (t, J = 7.8 Hz, 2H), 7.47 (d, J = 3.6 Hz, 1H), 7.38 (dt, J = 8.4, 1.1 Hz, 2H), 7.36–7.30 (m, 1H), 7.24 (d, J = 3.7 Hz, 1H), 3.12 (s, 3H), 2.19 (s, 3H). 13C NMR (100 MHz, DMSO‑d 6) δ 178.70, 162.73, 154.95, 152.84, 151.43, 146.73, 135.70, 135.67, 132.34, 129.57, 126.67, 125.12, 124.85, 123.94, 116.30, 113.06, 109.85, 36.36, 11.66. HRMS (ESI) [M + H] + calcd for C23H21N6O4S: 477.1345; found: 477.1341.
4.1.6. (E)-3-(5-((2-carbamothioylhydrazono)methyl)furan-2-yl)benzoic acid (F8-A1)
Following the general procedures A and D, compound F8-A1 was obtained in 42% yield. Yellowish solid. 1H NMR (400 MHz, DMSO‑d 6) δ 13.18 (s, 1H), 11.53 (s, 1H), 8.29 (s, 2H), 8.07 (d, J = 7.6 Hz, 1H), 8.00 (s, 1H), 7.89 (d, J = 7.6 Hz, 1H), 7.79 (s, 1H), 7.57 (t, J = 7.6 Hz, 1H), 7.24 (d, J = 3.6 Hz, 1H), 7.11 (d, J = 3.6 Hz, 1H). 13C NMR (100 MHz, DMSO‑d 6) δ 178.18, 167.43, 153.97, 149.98, 132.40, 132.11, 130.33, 129.84, 129.22, 128.54, 124.82, 115.54, 109.70. HRMS (ESI) [M − H]- calcd for C13H10N3O3S−: 288.0448; found: 288.0442.
4.1.7. (E)-4-(5-((2-carbamothioylhydrazono)methyl)furan-2-yl)benzoic acid (F8-A2)
Following the general procedures A and D, compound F8-A2 was obtained in 59% yield. Yellowish solid. 1H NMR (400 MHz, DMSO‑d 6) δ 13.00 (s, 1H), 11.56 (s, 1H), 8.33 (s, 1H), 8.02–7.90 (m, 5H), 7.83 (s, 1H), 7.27 (d, J = 3.6 Hz, 1H), 7.10 (d, J = 3.6 Hz, 1H). 13C NMR (100 MHz, DMSO‑d 6) δ 178.25, 167.35, 153.85, 150.44, 133.67, 132.14, 130.44, 130.22, 124.29, 115.78, 110.93. HRMS (ESI) [M − H]- calcd for C13H10N3O3S−: 288.0448; found: 288.0442.
4.1.8. (E)-3-(5-((2-(methylcarbamothioyl)hydrazono)methyl)furan-2-yl)benzoic acid (F8-A3)
Following the general procedures A and D, compound F8-A3 was obtained in 62% yield. Yellowish solid. 1H NMR (400 MHz, DMSO‑d 6) δ 13.19 (s, 1H), 11.56 (s, 1H), 8.40–8.32 (m, 1H), 8.30 (s, 1H), 8.07 (d, J = 7.6 Hz, 1H), 8.01 (s, 1H), 7.89 (d, J = 7.6 Hz, 1H), 7.59 (t, J = 7.8 Hz, 1H), 7.25 (d, J = 3.6 Hz, 1H), 7.08 (d, J = 3.6 Hz, 1H), 3.03 (d, J = 4.4 Hz, 3H). 13C NMR (100 MHz, DMSO‑d 6) δ 177.93, 167.42, 153.94, 150.10, 132.08, 132.05, 130.35, 129.83, 129.22, 128.54, 124.84, 115.41, 109.77, 31.37. HRMS (ESI) [M − H]- calcd for C14H12N3O3S−: 302.0605; found: 302.0601.
4.1.9. (E)-4-(5-((2-(methylcarbamothioyl)hydrazono)methyl)furan-2-yl)benzoic acid (F8-A4)
Following the general procedure, compound F8-A4 was obtained in 51% yield. Yellowish solid. 1H NMR (400 MHz, DMSO‑d 6) δ 13.01 (s, 1H), 11.61 (s, 1H), 8.34 (q, J = 4.6 Hz, 1H), 8.03–7.90 (m, 5H), 7.28 (d, J = 3.6 Hz, 1H), 7.09 (d, J = 3.6 Hz, 1H), 3.05 (d, J = 4.4 Hz, 3H). 13C NMR (100 MHz, DMSO‑d 6) δ 177.97, 167.33, 153.79, 150.56, 133.66, 131.75, 130.43, 130.22, 124.24, 115.54, 110.97, 31.37. HRMS (ESI) [M − H]- calcd for C14H12N3O3S−: 302.0605; found: 302.0600.
4.1.10. (E)-3-(5-((2-((4-chlorophenyl)carbamothioyl)hydrazono)methyl)furan-2-yl)benzoic acid (F8-A5)
Following the general procedures A and D, compound F8-A5 was obtained in 74% yield. Yellowish solid. 1H NMR (400 MHz, DMSO‑d 6) δ 12.97 (s, 1H), 12.01 (s, 1H), 10.05 (s, 1H), 8.33 (s, 1H), 8.12 (s, 1H), 8.08 (d, J = 7.8 Hz, 1H), 7.90 (d, J = 7.8 Hz, 1H), 7.65 (d, J = 8.4 Hz, 2H), 7.59 (t, J = 7.6 Hz, 1H), 7.43 (d, J = 8.4 Hz, 2H), 7.28 (d, J = 3.6 Hz, 1H), 7.24 (d, J = 3.6 Hz, 1H). 13C NMR (100 MHz, DMSO‑d 6) δ 176.09, 167.40, 154.29, 149.87, 138.48, 133.14, 132.09, 130.28, 129.87, 129.72, 129.31, 128.61, 128.49, 127.54, 124.91, 116.16, 109.87. HRMS (ESI) [M − H]- calcd for C19H13ClN3O3S−: 398.0372; found: 398.0366.
4.1.11. (E)-4-(5-((2-((4-chlorophenyl)carbamothioyl)hydrazono)methyl)furan-2-yl)benzoic acid (F8-A6)
Following the general procedures A and D, compound F8-A6 was obtained in 51% yield. Yellowish solid. 1H NMR (400 MHz, DMSO‑d 6) δ 13.02 (s, 1H), 12.03 (s, 1H), 10.03 (s, 1H), 8.12 (s, 1H), 8.00 (d, J = 8.4 Hz, 2H), 7.95 (d, J = 8.4 Hz, 2H), 7.64 (d, J = 8.8 Hz, 2H), 7.43 (d, J = 8.8 Hz, 2H), 7.33 (d, J = 3.6 Hz, 1H), 7.25 (d, J = 3.6 Hz, 1H). 13C NMR (100 MHz, DMSO‑d 6) δ 176.22, 167.32, 154.19, 150.36, 138.47, 133.60, 132.95, 130.47, 130.32, 129.86, 128.49, 127.86, 124.35, 116.32, 111.12. HRMS (ESI) [M − H]- calcd for C19H13ClN3O3S−: 398.0372; found: 398.0361.
4.1.12. (E)-3-(5-((2-((3-(trifluoromethyl)phenyl)carbamothioyl)hydrazono)methyl)furan-2-yl)benzoic acid (F8-A7)
Following the general procedures A and D, compound F8-A7 was obtained in 35% yield. Yellowish solid. 1H NMR (400 MHz, DMSO‑d 6) δ 12.11 (s, 1H), 10.23 (s, 1H), 8.33 (s, 1H), 8.15 (s, 1H), 8.12–8.03 (m, 2H), 7.98–7.87 (m, 2H), 7.65–7.53 (m, 3H), 7.31–7.21 (m, 2H). 13C NMR (100 MHz, DMSO‑d 6) δ 176.15, 167.40, 154.41, 149.86, 140.33, 133.51, 132.12, 130.26, 129.84, 129.71, 129.64, 129.33, 129.29 (d, J = 31.9 Hz), 128.60, 124.95, 124.54 (q, J = 273.3 Hz), 122.11 (d, J = 4.0 Hz), 122.03, 116.24, 109.87. 19F NMR (376 MHz, DMSO‑d 6) δ −61.11. HRMS (ESI) [M − H]- calcd for C20H13F3N3O3S−: 432.0635; found: 432.0623.
4.1.13. (E)-4-(5-((2-((3-(trifluoromethyl)phenyl)carbamothioyl)hydrazono)methyl)furan-2-yl)benzoic acid (F8-A8)
Following the general procedures A and D, compound F8-A8 was obtained in 51% yield. Yellowish solid. 1H NMR (400 MHz, DMSO‑d 6) δ 13.01 (s, 1H), 12.13 (s, 1H), 10.20 (s, 1H), 8.18–7.89 (m, 7H), 7.68–7.52 (m, 2H), 7.34 (d, J = 3.6 Hz, 1H), 7.27 (d, J = 3.6 Hz, 1H). 13C NMR (100 MHz, DMSO‑d 6) δ 176.21, 167.35, 154.31, 150.32, 140.26, 133.54, 133.29, 130.44, 130.37, 129.79, 129.56, 129.21, 124.50 (q, J = 273.4 Hz), 124.29, 122.27 (d, J = 4.0 Hz), 122.04, 116.31, 110.98. 19F NMR (376 MHz, DMSO‑d 6) δ −61.15. HRMS (ESI) [M − H]- calcd for C20H13F3N3O3S−: 432.0635; found: 432.0627.
4.1.14. (E)-4-(5-((2-(cyclohexylcarbamothioyl)hydrazono)methyl)furan-2-yl)benzoic acid (F8-A9)
Following the general procedures A and D, compound F8-A9 was obtained in 74% yield. Yellowish solid. 1H NMR (400 MHz, DMSO‑d 6) δ 13.01 (s, 1H), 11.58 (s, 1H), 8.13–7.78 (m, 6H), 7.30 (d, J = 3.6 Hz, 1H), 7.16 (d, J = 3.6 Hz, 1H), 4.28–4.08 (m, 1H), 2.01–1.05 (m, 10H). 13C NMR (100 MHz, DMSO‑d 6) δ 175.93, 167.30, 153.89, 150.50, 133.61, 132.04, 130.48, 130.24, 124.22, 115.65, 111.06, 52.97, 32.24, 25.54, 25.24. HRMS (ESI) [M − H]- calcd for C19H20N3O3S−: 370.1231; found: 370.1225.
4.1.15. (E)-4-(5-((2-carbamothioylhydrazono)methyl)furan-2-yl)benzamide (F8–B1)
Following the general procedures B and D, compound F8–B1 was obtained in 44% yield. Yellowish solid. 1H NMR (400 MHz, DMSO‑d 6) δ 11.54 (s, 1H), 8.31 (s, 1H), 8.05 (s, 1H), 8.02–7.81 (m, 6H), 7.43 (s, 1H), 7.24 (d, J = 3.6 Hz, 1H), 7.09 (d, J = 3.6 Hz, 1H). 13C NMR (100 MHz, DMSO‑d 6) δ 178.18, 167.78, 154.16, 150.10, 133.78, 132.32, 132.22, 128.65, 124.08, 115.86, 110.30. HRMS (ESI) [M − H]- calcd for C13H11N4O2S−: 287.0608; found: 287.0604.
4.1.16. (E)-2-((5-(4-sulfamoylphenyl)furan-2-yl)methylene)hydrazine-1-carbothioamide (F8–B2)
Following the general procedures B and D, compound F8–B2 was obtained in 63% yield. Yellowish solid. 1H NMR (400 MHz, DMSO‑d 6) δ 11.57 (s, 1H), 8.34 (s, 1H), 8.09–7.76 (m, 6H), 7.42 (s, 2H), 7.28 (d, J = 3.6 Hz, 1H), 7.11 (d, J = 3.6 Hz, 1H). 13C NMR (100 MHz, DMSO‑d 6) δ 178.23, 153.41, 150.46, 143.45, 132.77, 132.04, 126.82, 124.59, 115.77, 110.99. HRMS (ESI) [M − H]- calcd for C12H11N4O3S2 −: 323.0278; found: 323.0275.
4.1.17. (E)-2-((5-phenylfuran-2-yl)methylene)hydrazine-1-carbothioamide (F8–B3) [44,45]
Following the general procedures C and D, compound F8–B3 was obtained in 24% yield. Yellowish solid. 1H NMR (400 MHz, DMSO‑d 6) δ 11.50 (s, 1H), 8.33–8.25 (m, 1H), 7.97 (s, 1H), 7.87–7.81 (m, 2H) 7.78 (s, 1H), 7.47–7.41 (m, 2H), 7.38–7.30 (m, 1H), 7.12 (d, J = 3.6 Hz, 1H), 7.07 (d, J = 3.6 Hz, 1H).
4.1.18. (E)-4-(5-((2-carbamothioylhydrazono)methyl)furan-2-yl)-3-fluorobenzoic acid (F8–B4)
Following the general procedures A and D, compound F8–B4 was obtained in 51% yield. Orange red solid. 1H NMR (400 MHz, DMSO‑d 6) δ 13.33 (s, 1H), 11.60 (s, 1H), 8.36 (s, 1H), 8.11 (t, J = 8.0 Hz, 1H), 8.01 (s, 1H), 7.93–7.71 (m, 3H), 7.20–7.09 (m, 3H).13C NMR (100 MHz, DMSO‑d 6) δ 178.35, 166.32 (d, J = 2.5 Hz), 158.00 (d, J = 252.1 Hz), 150.58, 147.97 (d, J = 3.4 Hz), 131.95 (d, J = 7.8 Hz), 131.79, 126.75 (d, J = 1.9 Hz), 126.26 (d, J = 3.1 Hz), 121.76 (d, J = 11.9 Hz), 117.16 (d, J = 22.5 Hz), 115.59, 114.91 (d, J = 12.3 Hz). 19F NMR (376 MHz, DMSO‑d 6) δ −113.32. HRMS (ESI) [M − H]- calcd for C13H9FN3O3S−: 306.0354; found: 306.0347.
4.1.19. (E)-4-(5-((2-carbamothioylhydrazono)methyl)furan-2-yl)-3-methylbenzoic acid (F8–B5)
Following the general procedures B and D, compound F8–B5 was obtained in 56% yield. Yellowish solid. 1H NMR (400 MHz, DMSO‑d 6) δ 12.97 (s, 1H), 11.56 (s, 1H), 8.32 (s, 1H), 8.01 (s, 1H), 7.95 (d, J = 8.2 Hz, 1H), 7.89–7.75 (m, 3H), 7.14 (d, J = 3.6 Hz, 1H), 7.04 (d, J = 3.6 Hz, 1H), 2.53 (s, 3H). 13C NMR (100 MHz, DMSO‑d 6) δ 178.23, 167.43, 153.33, 149.90, 134.80, 132.91, 132.76, 132.26, 130.00, 127.58, 127.07, 115.43, 113.80, 22.29. HRMS (ESI) [M − H]- calcd for C14H12N3O3S−: 302.0605; found: 302.0597.
4.1.20. (E)-4-(5-((2-carbamothioylhydrazono)methyl)furan-2-yl)-2-hydroxybenzoic acid (F8–B6)
Following the general procedures A and D, compound F8–B6 was obtained in 73% yield. Yellowish solid. 1H NMR (400 MHz, DMSO‑d 6) δ 11.56 (s, 1H), 8.31 (s, 1H), 7.98 (s, 1H), 7.87 (s, 1H), 7.81 (d, J = 8.0 Hz, 1H), 7.45–7.34 (m, 2H), 7.28 (d, J = 3.6 Hz, 1H), 7.09 (d, J = 3.6 Hz, 1H). 13C NMR (100 MHz, DMSO‑d 6) δ 178.22, 172.05, 161.99, 153.45, 150.60, 136.16, 132.09, 131.42, 115.75, 115.32, 112.49, 112.02, 111.65. HRMS (ESI) [M − H]- calcd for C13H10N3O4S−: 304.0398; found: 304.0394.
4.1.21. (E)-4-(5-((2-carbamothioylhydrazono)methyl)furan-2-yl)-2-chlorobenzoic acid (F8–B7)
Following the general procedures A and D, compound F8–B7 was obtained in 83% yield. Yellowish solid. 1H NMR (400 MHz, DMSO‑d 6) δ 13.36 (s, 1H), 11.57 (s, 1H), 8.31 (s, 1H), 8.06–7.77 (m, 5H), 7.31 (d, J = 3.6 Hz, 1H), 7.08 (d, J = 3.6 Hz, 1H). 13C NMR (100 MHz, DMSO‑d 6) δ 178.27, 166.61, 152.36, 150.78, 133.68, 133.44, 132.32, 132.05, 129.98, 125.85, 122.65, 115.79, 111.76. HRMS (ESI) [M − H]- calcd for C13H9ClN3O5S−: 322.0059; found: 32.0053.
4.1.22. (E)-3-(5-((2-carbamothioylhydrazono)methyl)furan-2-yl)-2-chlorobenzoic acid (F8–B8)
Following the general procedures C and D, compound F8–B8 was obtained in 53% yield. Yellowish solid. 1H NMR (400 MHz, DMSO‑d 6) δ 13.54 (br, 1H), 11.56 (s, 1H), 8.32 (s, 1H), 8.12 (d, J = 8.0 Hz, 1H), 8.00 (s, 1H), 7.82 (s, 1H), 7.63 (dd, J = 8.0, 1.8 Hz, 1H), 7.52 (t, J = 7.6 Hz, 1H), 7.33 (d, J = 3.6 Hz, 1H), 7.14 (d, J = 3.6 Hz, 1H). 13C NMR (100 MHz, DMSO‑d 6) δ 178.30, 167.77, 150.56, 149.94, 135.47, 132.05, 130.65, 129.45, 129.26, 127.96, 127.01, 115.12, 114.65. HRMS (ESI) [M − H]- calcd for C13H9ClN3O3S−: 322.0059; found: 322.0059.
4.1.23. (E)-5-(5-((2-carbamothioylhydrazono)methyl)furan-2-yl)-2-chlorobenzoic acid (F8–B9)
Following the general procedures A and D, compound F8–B9 was obtained in 50% yield. Yellowish solid. 1H NMR (400 MHz, DMSO‑d 6) δ 13.61 (s, 1H), 11.54 (s, 1H), 8.30 (s, 1H), 8.13 (s, 1H), 8.03–7.91 (m, 2H), 7.83 (s, 1H), 7.60 (d, J = 8.4 Hz, 1H), 7.27 (d, J = 3.6 Hz, 1H), 7.09 (d, J = 3.6 Hz, 1H). 13C NMR (100 MHz, DMSO‑d 6) δ 178.22, 166.96, 152.92, 150.19, 132.98, 132.15, 131.65, 130.95, 128.96, 127.81, 125.91, 115.72, 110.31. HRMS (ESI) [M − H]- calcd for C13H9ClN3O3S−: 322.0059; found: 322.0059.
4.1.24. (E)-3-(5-((2-carbamothioylhydrazono)methyl)furan-2-yl)-4-chlorobenzoic acid (F8–B10)
Following the general procedures C and D, compound F8–B10 was obtained in 73% yield. Yellowish solid. 1H NMR (400 MHz, DMSO‑d 6) δ 13.34 (s, 1H), 11.60 (s, 1H), 8.38 (d, J = 2.1 Hz, 1H), 8.33 (s, 1H), 8.04 (s, 1H), 7.85 (dd, J = 8.4, 2.1 Hz, 1H), 7.78 (s, 1H), 7.68 (d, J = 8.4 Hz, 1H), 7.34 (d, J = 3.6 Hz, 1H), 7.20 (d, J = 3.6 Hz, 1H). 13C NMR (100 MHz, DMSO‑d 6) δ 178.32, 166.64, 150.43, 150.05, 133.93, 132.30, 131.82, 130.68, 129.98, 129.03, 128.37, 114.58, 114.26. HRMS (ESI) [M − H]- calcd for C13H9N3O3S−: 322.0059; found: 322.0057.
4.1.25. (E)-3-(5-((2-carbamothioylhydrazono)methyl)furan-2-yl)-2-methoxybenzoic acid (F8–B11)
Following the general procedures C and D, compound F8–B11 was obtained in 59% yield. Yellowish solid. 1H NMR (400 MHz, DMSO‑d 6) δ 13.14 (s, 1H), 11.54 (s, 1H), 8.31 (s, 1H), 8.12 (d, J = 7.6 Hz, 1H), 8.00 (s, 1H), 7.81 (s, 1H), 7.66 (d, J = 7.6 Hz, 1H), 7.29 (t, J = 7.6 Hz, 1H), 7.11 (s, 2H), 3.79 (s, 3H). 13C NMR (100 MHz, DMSO‑d 6) δ 178.17, 167.60, 155.87, 150.57, 149.05, 132.21, 130.84, 130.10, 127.46, 124.60, 124.41, 116.02, 113.14, 61.64. HRMS (ESI) [M − H]- calcd for C14H12N3O4S−: 318.0554; found: 318.0549.
4.1.26. (E)-5-(5-((2-carbamothioylhydrazono)methyl)furan-2-yl)-2-hydroxybenzoic acid (F8–B12)
Following the general procedures A and D, compound F8–B12 was obtained in 67% yield. Yellowish solid. 1H NMR (400 MHz, DMSO‑d 6) δ 11.48 (s, 1H), 8.25 (s, 1H), 8.14 (d, J = 2.4 Hz, 1H), 8.00–7.92 (m, 2H), 7.74 (s, 1H), 7.07–6.98 (m, 3H). 13C NMR (100 MHz, DMSO‑d 6) δ 178.04, 171.94, 161.37, 154.31, 149.03, 132.64, 131.79, 125.98, 121.69, 118.40, 115.84, 113.94, 107.60. HRMS (ESI) [M − H]- calcd for C13H10N3O4S−: 304.0398; found: 304.0394.
4.1.27. (E)-2-((5-(3-cyano-4-hydroxyphenyl)furan-2-yl)methylene)hydrazine-1-carbothioamide (F8–B13)
Following the general procedures A and D, compound F8–B13 was obtained in 52% yield. Pale brown solid. 1H NMR (400 MHz, DMSO‑d 6) δ 11.49 (s, 1H), 11.43 (s, 1H), 8.27 (s, 1H), 8.10 (d, J = 2.4 Hz, 1H), 7.99–7.90 (m, 2H), 7.81 (s, 1H), 7.13–6.97 (m, 3H). 13C NMR (100 MHz, DMSO‑d 6) δ 178.07, 160.43, 153.52, 149.07, 132.16, 130.84, 129.19, 122.12, 117.25, 116.99, 116.13, 107.94, 100.05. HRMS (ESI) [M − H]- calcd for C13H9N4O2S−: 285.0452; found: 285.0448.
4.1.28. (E)-2-((5-(3,4-dimethoxyphenyl)furan-2-yl)methylene)hydrazine-1-carbothioamide (F8–B14)
Following the general procedures A and D, compound F8–B14 was obtained in 71% yield. Yellowish solid. 1H NMR (400 MHz, DMSO‑d 6) δ 11.47 (s, 1H), 8.24 (s, 1H), 7.97 (s, 1H), 7.75 (s, 1H), 7.38 (d, J = 9.2 Hz, 1H), 7.34 (s, 1H), 7.08–6.97 (m, 3H), 3.84 (s, 3H), 3.79 (s, 3H). 13C NMR (100 MHz, DMSO‑d 6) δ 178.01, 155.40, 149.62, 149.52, 148.77, 132.57, 122.98, 117.44, 115.97, 112.47, 108.26, 107.47, 56.17, 56.02. HRMS (ESI) [M + H]+ calcd for C14H16N3O3S+: 306.0907; found: 306.0906.
4.1.29. (E)-2-((5-(3,4-dichlorophenyl)furan-2-yl)methylene)hydrazine-1-carbothioamide (F8–B15)
Following the general procedures A and D, compound F8–B15 was obtained in 63% yield. Yellowish solid. 1H NMR (400 MHz, DMSO‑d 6) δ 11.55 (s, 1H), 8.31 (s, 1H), 8.08 (s, 1H), 7.96 (s, 1H), 7.87 (s, 1H), 7.81 (d, J = 8.4 Hz, 1H), 7.68 (d, J = 8.4 Hz, 1H), 7.28 (d, J = 3.6 Hz, 1H), 7.08 (d, J = 3.6 Hz, 1H). 13C NMR (100 MHz, DMSO‑d 6) δ 178.25, 152.37, 150.29, 132.40, 131.94, 131.60, 130.71, 130.50, 125.90, 124.44, 115.86, 110.73. HRMS (ESI) [M − H]- calcd for C12H8Cl2N3OS−: 311.9771; found: 311.9767.
4.1.30. (E)-2-((5-(pyridin-4-yl)furan-2-yl)methylene)hydrazine-1-carbothioamide (F8–B16)
Following the general procedures A and D, compound F8–B16 was obtained in 72% yield. Yellowish solid. 1H NMR (400 MHz, DMSO‑d 6) δ 11.60 (s, 1H), 8.60 (d, J = 5.2 Hz, 2H), 8.36 (s, 1H), 7.99 (s, 1H), 7.87 (s, 1H), 7.76 (d, J = 5.2 Hz, 2H), 7.41 (d, J = 3.6 Hz, 1H), 7.19 (d, J = 3.6 Hz, 1H) 13C NMR (100 MHz, DMSO‑d 6) δ 178.36, 152.14, 151.11, 150.72, 136.44, 131.84, 118.24, 115.41, 112.49. HRMS (ESI) [M − H]- calcd for C11H9N4OS−: 245.0497; found: 245.0502.
4.1.31. (E)-2-((5-(6-fluoropyridin-3-yl)furan-2-yl)methylene)hydrazine-1-carbothioamide (F8–B17)
Following the general procedures A and D, compound F8–B17 was obtained in 61% yield. Yellowish solid. 1H NMR (400 MHz, DMSO‑d 6) δ 11.54 (s, 1H), 8.73 (d, J = 2.4 Hz, 1H), 8.42–8.27 (m, 2H), 7.97 (s, 1H), 7.92–7.80 (m, 1H), 7.26 (dd, J = 8.8, 2.4 Hz, 1H), 7.20 (d, J = 3.6 Hz, 1H), 7.06 (d, J = 3.6 Hz, 1H). 13C NMR (100 MHz, DMSO‑d 6) δ 178.24, 162.76 (d, J = 234.7 Hz), 151.26, 150.12, 143.68 (d, J = 15.5 Hz), 137.89 (d, J = 7.9 Hz), 131.90, 124.85 (d, J = 4.4 Hz), 115.68, 110.45 (d, J = 37.9 Hz), 109.96. 19F NMR (376 MHz, DMSO‑d 6) δ −69.11. HRMS (ESI) [M − H]- calcd for C11H8FN4OS−: 263.0408; found: 263.0403.
4.1.32. (E)-2-((5-(naphthalen-1-yl)furan-2-yl)methylene)hydrazine-1-carbothioamide (F8–B18)
Following the general procedures A and D, compound F8–B18 was obtained in 79% yield. Yellowish solid. 1H NMR (400 MHz, DMSO‑d 6) δ 11.56 (s, 1H), 8.40 (d, J = 8.0 Hz, 1H), 8.30 (s, 1H), 8.08 (s, 1H), 8.02–7.99 (m, 2H), 7.89 (dd, J = 7.2, 1.2 Hz, 1H), 7.74 (s, 1H), 7.64–7.56 (m, 3H), 7.21 (d, J = 3.2 Hz, 1H), 7.11 (d, J = 3.2 Hz, 1H) 13C NMR (100 MHz, DMSO‑d 6) δ 178.20, 154.42, 149.96, 134.04, 132.70, 129.70, 129.64, 129.19, 127.73, 127.34, 126.87, 126.72, 125.99, 125.30, 115.12, 112.67. HRMS (ESI) [M − H]- calcd for C16H13N3OS−: 294.0701; found: 294.0703.
4.1.33. (E)-2-((5-(naphthalen-2-yl)furan-2-yl)methylene)hydrazine-1-carbothioamide (F8–B19)
Following the general procedures A and D, compound F8–B19 was obtained in 45% yield. Yellowish solid. 1H NMR (400 MHz, DMSO‑d 6) δ 11.57 (s, 1H), 8.37 (s, 1H), 8.34 (s, 1H), 8.06–7.79 (m, 6H), 7.60–7.48 (m, 2H), 7.25 (d, J = 3.6 Hz, 1H), 7.13 (d, J = 3.6 Hz, 1H). 13C NMR (100 MHz, DMSO‑d 6) δ 178.16, 155.05, 149.79, 133.53, 133.00, 132.38, 129.03, 128.59, 128.19, 127.38, 127.29, 126.91, 122.77, 115.91, 109.55. HRMS (ESI) [M − H]- calcd for C16H12N3OS−: 294.0707; found: 294.0705.
4.1.34. (E)-2-((5-(quinolin-7-yl)furan-2-yl)methylene)hydrazine-1-carbothioamide (F8–B20)
Following the general procedures A and D, compound F8–B20 was obtained in 56% yield. Yellowish solid. 1H NMR (400 MHz, DMSO‑d 6) δ 11.62 (s, 1H), 8.86 (dd, J = 4.0, 2.0 Hz, 1H), 8.47–8.34 (m, 3H), 8.17 (dd, J = 8.8, 2.0 Hz, 1H), 8.03 (d, J = 9.8 Hz, 2H), 7.92–7.83 (m, 1H), 7.53 (dd, J = 8.4, 4.0 Hz, 1H), 7.28 (d, J = 3.6 Hz, 1H), 7.13 (d, J = 3.6 Hz, 1H). 13C NMR (100 MHz, DMSO‑d 6) δ 178.21, 154.33, 151.12, 150.16, 147.77, 136.61, 132.29, 130.11, 128.60, 127.82, 126.33, 122.75, 122.66, 115.83, 110.22. HRMS (ESI) [M − H]- calcd for C15H11N4OS−: 295.0659; found: 295.0654.
4.1.35. (E)-2-((5-(quinolin-3-yl)furan-2-yl)methylene)hydrazine-1-carbothioamide (F8–B21)
Following the general procedures A and D, compound F8–B21 was obtained in 71% yield. Yellowish solid. 1H NMR (400 MHz, DMSO‑d 6) δ 11.60 (s, 1H), 9.39 (s, 1H), 8.72 (s, 1H), 8.37 (s, 1H), 8.11–7.97 (m, 3H), 7.89 (s, 1H), 7.75 (t, J = 7.6 Hz, 1H), 7.64 (t, J = 7.6 Hz, 1H), 7.42 (d, J = 3.6 Hz, 1H), 7.17 (d, J = 3.6 Hz, 1H). 13C NMR (100 MHz, DMSO‑d 6) δ 178.25, 152.57, 150.48, 147.60, 147.20, 132.02, 130.20, 129.70, 129.33, 128.81, 127.92, 127.90, 123.38, 115.74, 110.60. HRMS (ESI) [M − H]- calcd for C15H11N4OS−: 295.0659; found: 295.0654.
4.1.36. (E)-2-((5-benzylfuran-2-yl)methylene)hydrazine-1-carbothioamide (F8–B22)
5-hydroxymethyl-2-furfuraldehyde (0.31 g, 2.45 mmol) was added to the mixture of TfOH (4 mL) and benzene (0.28 mL) in an ice bath. The reaction mixture was stirred at room temperature for 2 h. The mixture was poured into water (30 mL), and extracted with chloroform (3 × 30 mL). The combined extracts were washed with water, the saturated aqueous solution of NaHCO3, water again, and dried over Na2SO4. The solvent was removed under reduced pressure, and the residue was subjected to chromatographic separation on silica gel to give intermediate 5-(phenylmethyl)furan-2-carbaldehyde 0.1 g, yielding 21%. Then following the general procedure D, compound F8–B22 was obtained in 42% yield. Yellowish solid. 1H NMR (400 MHz, DMSO‑d 6) δ 11.38 (s, 1H), 8.17 (s, 1H), 7.89 (s, 1H), 7.55 (s, 1H), 7.37–7.19 (m, 5H), 6.88 (d, J = 3.6 Hz, 1H), 6.26 (d, J = 3.6 Hz, 1H), 4.02 (s, 2H). 13C NMR (100 MHz, DMSO‑d 6) δ 177.98, 157.32, 148.87, 138.02, 133.07, 129.08, 129.01, 127.03, 114.52, 109.71, 34.18. HRMS (ESI) [M − H]- calcd for C13H12N3OS−: 258.0707; found: 258.0699.
4.1.37. (E)-4-(5-((2-carbamothioylhydrazono)methyl)furan-3-yl)benzoic acid (F8–C1)
Following the general procedures A and D, compound F8–C1 was obtained in 25% yield. Yellowish solid. 1H NMR (400 MHz, DMSO‑d 6) δ 12.96 (s, 1H), 11.53 (s, 1H), 8.45 (s, 1H), 8.30 (s, 1H), 8.03–7.92 (m, 3H), 7.81–7.65 (m, 3H), 7.53 (s, 1H). 13C NMR (100 MHz, DMSO‑d 6) δ 178.35, 167.45, 151.23, 142.77, 135.97, 132.41, 130.44, 129.88, 127.55, 125.95, 111.02. HRMS (ESI) [M − H]- calcd for C13H10N3O3S−: 288.0448; found: 288.0447.
4.1.38. (E)-4-(5-((2-carbamothioylhydrazono)methyl)thiophen-2-yl)benzoic acid (F8–C2)
Following the general procedures A and D, compound F8–C2 was obtained in 51% yield. Yellowish solid. 1H NMR (400 MHz, DMSO‑d 6) δ 13.02 (s, 1H), 11.56 (s, 1H), 8.27 (s, 1H), 8.22 (s, 1H), 7.98 (d, J = 8.0 Hz, 2H), 7.81 (d, J = 8.0 Hz, 2H), 7.67 (d, J = 4.0 Hz, 1H), 7.64 (s, 1H), 7.50 (d, J = 4.0 Hz, 1H). 13C NMR (100 MHz, DMSO‑d 6) δ 178.09, 167.25, 144.26, 139.89, 137.66, 137.51, 132.33, 130.70, 130.41, 126.43, 125.82. HRMS (ESI) [M − H]- calcd for C13H10N3O2S−: 304.0220; found: 304.0215.
4.1.39. (E)-3'-((2-carbamothioylhydrazono)methyl)-[1,1′-biphenyl]-4-carboxylic acid (F8–C3)
Following the general procedures A and D, compound F8–C3 was obtained in 23% yield. White solid. 1H NMR (400 MHz, DMSO‑d 6) δ 13.00 (br, 1H), 11.52 (s, 1H), 8.27 (s, 1H), 8.22 (s, 1H), 8.18 (s, 1H), 8.13 (s, 1H), 8.03 (d, J = 8.0 Hz, 2H), 7.88 (d, J = 8.0 Hz, 2H), 7.79–7.74 (m, 2H), 7.52 (t, J = 7.4 Hz, 1H) 13C NMR (100 MHz, DMSO‑d 6) δ 178.52, 167.59, 144.13, 142.40, 139.90, 135.50, 130.34, 130.27, 129.88, 128.72, 128.03, 127.48, 125.61. HRMS (ESI) [M − H]- calcd for C15H13N3O2S−: 298.0650; found: 298.0656.
4.1.40. (E)-4'-((2-carbamothioylhydrazono)methyl)-[1,1′-biphenyl]-4-carboxylic acid (F8–C4)
Following the general procedures A and D, compound F8–C4 was obtained in 12% yield. White solid. 1H NMR (400 MHz, DMSO‑d 6) δ 13.01 (s, 1H), 11.52 (s, 1H), 8.26 (s, 1H), 8.10 (d, J = 5.1 Hz, 2H), 8.03 (d, J = 8.4 Hz, 2H), 7.92 (d, J = 8.4 Hz, 2H), 7.85 (d, J = 8.4 Hz, 2H), 7.77 (d, J = 8.4 Hz, 2H). 13C NMR (100 MHz, DMSO‑d 6) δ 178.47, 167.56, 143.92, 142.08, 140.46, 134.60, 130.45, 130.32, 128.44, 127.62, 127.24. HRMS (ESI) [M − H]- calcd for C15H13N3O2S−: 298.0650; found: 298.0653.
4.2. Reagents and compounds
Tideglusib was purchased from Topscience Co. Ltd. SARS-CoV-2 Mpro fluorescent substrate Dabcyl-KTSAVLQSGFRKM-E(Edans)-NH2 was synthesized by GL (Shanghai) Biochem Ltd. (Shanghai, China). Compounds used for SARS-CoV-2 Mpro inhibitors screening were purchased from ChemDiv (https://www.chemdiv.com/) and SPECS (https://www.specs.net/) commercial databases.
4.3. Cloning, expression, and purification of SARS-CoV-2 Mpro
The full-length gene encoding SARS-CoV-2 Mpro was synthesized for Escherichia coli (E. coli) expression (Hienzyme Biotech). The expression and purification of SARS-CoV-2 Mpro were carried out using the reported protocol [29].
4.4. SPR assay
The SPR assay was used to analyze the interaction between SARS-CoV-2 Mpro with compounds on a Biacore 8 K instrument (GE Healthcare). SARS-CoV-2 Mpro was immobilized on a sensor chip (CM5) via Amine Coupling Kit (GE Healthcare, Buckinghamshire, UK) at levels of approximately 10,000 response units (RU). The first flow channel without immobilized protein was set as a reference, and the compounds were injected at the concentration of 50 μM for screening and 0.2 μM–100 μM for binding study in a period of 60 s. Dissociation was measured for 100–200 s at a flow rate of 30 μL/min using the following assay running buffer: 10 mM phosphate buffer containing 2.7 mM KCl, 137 mM NaCl, and 0.05% surfactant P20 (pH 7.5). All of the data were analyzed through Biacore evaluation software (8 K version 1.0), and the curve was fitted with a 1:1 kinetics binding model.
4.5. Enzymatic assay of SARS-CoV-2 Mpro
A fluorescent substrate Dabcyl-KTSAVLQSGFRKM-E(Edans)-NH2 (GL Biochemistry Ltd) and assay buffer (40 mM PBS, 100 mM NaCl, 1 mM EDTA, 0.1% Triton 100, pH 7.3) was used for the inhibition assay. For the preliminary screening and IC50 measurements, 0.5 μM protease was incubated with inhibitor at room temperature for 30 min, and then the reaction was initiated by adding 20 μM substrate. The fluorescence signal generated by the cleavage of the substrate was monitored for 20 min at an emission wavelength of 460 nm with excitation at 360 nm using a plate reader (Synergy, Biotek). IC50 values were fitted with the Hill1 function of Origin 2018. For the enzymatic kinetic assay, 0.5 μM SARS-CoV-2 Mpro was pre-incubated with DMSO or F8–B6 for 3 h and different concentrations of the fluorescent substrate were added to initiate the reaction. Data was collected from three replicates and curve-fitted by Origin 2018. For the DTT assay, 0.5 μM SARS-CoV-2 Mpro was premixed with 400 mM DTT or H2O. Then, the protease solution was co-preincubated with DMSO or inhibitor at various concentrations for 30 min. Enzyme activity was tested and data was collected from three replicates. For time-dependent inhibitory measurement, various concentrations of inhibitors were pre-incubated with SARS-CoV-2 Mpro at a different time at room temperature before the addition of fluorescent substrate.
4.6. Mass spectrometry
2 μM protease was co-incubated with 15 μM F8–B6 on ice for 3 h. Then, the complex solution was analyzed by Quadrupole-TOF LC-MS/MS System (Vion, Waters). Raw data of mass signal was deconvoluted to obtain the total mass of the protein.
4.7. Enzymatic reversibility assay
SARS-CoV-2 Mpro (10 μM) was incubated with 100 μM F8–B6 for 180 min and divided into four parts which were ultra-filtrated for different times. In each time of ultrafiltration, an equal volume of buffer used in the enzyme activity assay was added into the protease solution and ultra-filtrated together for 5 min at 4 °C, 12,000 rpm. Protease left on the upper layer of Millipore was collected and diluted to the final concentration of 0.5 μM and enzyme activity was tested. All data were collected from three replicates to obtain average enzymatic activities and error bars.
4.8. Cathepsin L inhibition assay
The inhibition assay of Cathepsin L was performed as previously reported method [41,43]. Briefly, compounds F8–B6 and F8–B22 were tested using the commercial Cathepsin L Inhibitor Assay Kit (Abcam, Cat# ab197012). And the known Cathepsin L inhibitor FF-FMK was used as the positive compound.
4.9. Cell culture
Vero cells were cultured in (MEM, M&C Gene Technology, Beijing, China) and MDCK cells were cultured in Minimum Essential Medium Dulbecco's modified Eagle's medium (DMEM, M&C Gene Technology, Beijing, China), supplemented with 10% fetal bovine serum (FBS) (PAN Seratech, Aidenbach, Germany) and 100 U/mL penicillin/streptomycin (P/S), and maintained in a humidified atmosphere of 95% air and 5% CO2.
4.10. Cell viability assay
MTT assay was performed to examine the cell viability of the newly identified SARS-CoV-2 Mpro inhibitors. Vero and MDCK cells were seeded in 96-well plates. After 18 h incubation, SARS-CoV-2 Mpro inhibitors were added to each well. Corresponding vehicles were simultaneously added for blank well. Cell viability was determined 48 h after drug treatment.
4.11. Molecular docking
All of the chemical structures were processed using the LigPrep module in Schrödinger 10.2 software (Schrodinger, LLC, NY, USA) [46]. The OPLS3 force field was adopted to perform energy minimization. Default settings were used for all other parameters. The crystallographic structures of the Mpro in complex with inhibitors were retrieved from the RCSB Protein Data Bank (PDB) and crystal structures (PDB IDs: 7JUJ) were prepared by the Protein Preparation Wizard module in Schrödinger 10.2 software. Default settings were used for all parameters based on the OPLS3 force field. The molecular docking was performed using the Glide module with XP (extra precision) in Schrödinger 10.2 software with default settings for all other parameters [47,48].
4.12. Similarity search
The similarity search was performed using Pipeline Pilot 8.5 of Accelrys. The similarities were calculated through the Tanimoto coefficient based on the fingerprint ECFP_6 of each structure. Then, 70 analogs of compound F8 and 32 analogs of compound F8–S43 were captured and purchased for biological evaluation.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
We acknowledge the National Natural Science Foundation of China (Nos. 22131002, 81821004, and 22161142019), Peking University Special Fund for COVID-19, Beijing Xinxi Disruptive Technology Innovation Foundation, and the Tencent Foundation for financial support.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ejmech.2022.114508.
Abbreviations used
- SARS-CoV-2
severe acute respiratory syndrome coronavirus 2
- COVID-19
coronavirus infectious disease 2019
- SARS-CoV
severe acute respiratory syndrome coronavirus
- MERS-CoV
Middle East respiratory syndrome coronavirus
- Mpro
Main protease
- 3CLpro
3-chymotrypsin-like protease
- PLpro
papain-like protease
- RdRp
RNA-dependent RNA polymerase
- FDA
US Food and Drug Administration
- CYP3A4
cytochrome P450 3A4
- KD
dissociation constant
- SPR
surface plasmon resonance
- IC50
half-maximal inhibitory concentration
- SAR
structure-activity relationship;
- S2C
S2 channel
- HRMS
high-resolution mass spectrometry
- PDB
protein data bank
- RU
response units
- MTT
3-(4,5-dimethylthiazolyl-2)- 2,5-diphenyltetrazolium bromide;
- CC50
half-maximal cytotoxic concentration
- log P
octanol/water partition coefficient
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Zhu N., Zhang D., Wang W., Li X., Yang B., Song J., Zhao X., Huang B., Shi W., Lu R., Niu P., Zhan F., Ma X., Wang D., Xu W., Wu G., Gao G.F., Tan W., China Novel Coronavirus I., Research T. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 2020;382:727–733. doi: 10.1056/NEJMoa2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Phan L.T., Nguyen T.V., Luong Q.C., Nguyen T.V., Nguyen H.T., Le H.Q., Nguyen T.T., Cao T.M., Pham Q.D. Importation and human-to-human transmission of a novel coronavirus in Vietnam. N. Engl. J. Med. 2020;382:872–874. doi: 10.1056/NEJMc2001272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang C., Horby P.W., Hayden F.G., Gao G.F. A novel coronavirus outbreak of global health concern. Lancet. 2020;395:470–473. doi: 10.1016/S0140-6736(20)30185-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dong M., Dando E.E., Kotliar I., Su X., Dzikovski B., Freed J.H., Lin H. The asymmetric function of Dph1-Dph2 heterodimer in diphthamide biosynthesis. J. Biol. Inorg. Chem. 2019;24:777–782. doi: 10.1007/s00775-019-01702-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li G., De Clercq E. Therapeutic options for the 2019 novel coronavirus (2019-nCoV) Nat. Rev. Drug Discov. 2020;19:149–150. doi: 10.1038/d41573-020-00016-0. [DOI] [PubMed] [Google Scholar]
- 6.Amin S.A., Jha T. Fight against novel coronavirus: a perspective of medicinal chemists. Eur. J. Med. Chem. 2020;201 doi: 10.1016/j.ejmech.2020.112559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu C., Liu Y., Yang Y., Zhang P., Zhong W., Wang Y., Wang Q., Xu Y., Li M., Li X., Zheng M., Chen L., Li H. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B. 2020;10:766–788. doi: 10.1016/j.apsb.2020.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gil C., Ginex T., Maestro I., Nozal V., Barrado-Gil L., Cuesta-Geijo M.A., Urquiza J., Ramirez D., Alonso C., Campillo N.E., Martinez A. COVID-19: drug targets and potential treatments. J. Med. Chem. 2020;63:12359–12386. doi: 10.1021/acs.jmedchem.0c00606. [DOI] [PubMed] [Google Scholar]
- 9.Xiu S., Dick A., Ju H., Mirzaie S., Abdi F., Cocklin S., Zhan P., Liu X. Inhibitors of SARS-CoV-2 entry: current and future opportunities. J. Med. Chem. 2020;63:12256–12274. doi: 10.1021/acs.jmedchem.0c00502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu Y., Liang C., Xin L., Ren X., Tian L., Ju X., Li H., Wang Y., Zhao Q., Liu H., Cao W., Xie X., Zhang D., Wang Y., Jian Y. The development of Coronavirus 3C-Like protease (3CL(pro)) inhibitors from 2010 to 2020. Eur. J. Med. Chem. 2020;206 doi: 10.1016/j.ejmech.2020.112711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zheng L., Zhang L., Huang J., Nandakumar K.S., Liu S., Cheng K. Potential treatment methods targeting 2019-nCoV infection. Eur. J. Med. Chem. 2020;205 doi: 10.1016/j.ejmech.2020.112687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cannalire R., Cerchia C., Beccari A.R., Di Leva F.S., Summa V. Targeting SARS-CoV-2 proteases and polymerase for COVID-19 treatment: state of the art and future opportunities. J. Med. Chem. 2020 doi: 10.1021/acs.jmedchem.0c01140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jin Z., Du X., Xu Y., Deng Y., Liu M., Zhao Y., Zhang B., Li X., Zhang L., Peng C., Duan Y., Yu J., Wang L., Yang K., Liu F., Jiang R., Yang X., You T., Liu X., Yang X., Bai F., Liu H., Liu X., Guddat L.W., Xu W., Xiao G., Qin C., Shi Z., Jiang H., Rao Z., Yang H. Structure of M(pro) from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582:289–293. doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- 14.Zhang L., Lin D., Sun X., Curth U., Drosten C., Sauerhering L., Becker S., Rox K., Hilgenfeld R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved alpha-ketoamide inhibitors. Science. 2020;368:409–412. doi: 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dai W., Zhang B., Jiang X.M., Su H., Li J., Zhao Y., Xie X., Jin Z., Peng J., Liu F., Li C., Li Y., Bai F., Wang H., Cheng X., Cen X., Hu S., Yang X., Wang J., Liu X., Xiao G., Jiang H., Rao Z., Zhang L.K., Xu Y., Yang H., Liu H. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science. 2020;368:1331–1335. doi: 10.1126/science.abb4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ma C., Sacco M.D., Hurst B., Townsend J.A., Hu Y., Szeto T., Zhang X., Tarbet B., Marty M.T., Chen Y., Wang J. Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Res. 2020;30:678–692. doi: 10.1038/s41422-020-0356-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fu L., Ye F., Feng Y., Yu F., Wang Q., Wu Y., Zhao C., Sun H., Huang B., Niu P., Song H., Shi Y., Li X., Tan W., Qi J., Gao G.F. Both Boceprevir and GC376 efficaciously inhibit SARS-CoV-2 by targeting its main protease. Nat. Commun. 2020;11:4417. doi: 10.1038/s41467-020-18233-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sacco M.D., Ma C., Lagarias P., Gao A., Townsend J.A., Meng X., Dube P., Zhang X., Hu Y., Kitamura N., Hurst B., Tarbet B., Marty M.T., Kolocouris A., Xiang Y., Chen Y., Wang J. Structure and inhibition of the SARS-CoV-2 main protease reveals strategy for developing dual inhibitors against M(pro) and cathepsin L. Sci. Adv. 2020;6 doi: 10.1126/sciadv.abe0751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hoffman R.L., Kania R.S., Brothers M.A., Davies J.F., Ferre R.A., Gajiwala K.S., He M., Hogan R.J., Kozminski K., Li L.Y., Lockner J.W., Lou J., Marra M.T., Mitchell L.J., Jr., Murray B.W., Nieman J.A., Noell S., Planken S.P., Rowe T., Ryan K., Smith G.J., 3rd, Solowiej J.E., Steppan C.M., Taggart B. Discovery of Ketone-based covalent inhibitors of coronavirus 3CL proteases for the potential therapeutic treatment of COVID-19. J. Med. Chem. 2020;63:12725–12747. doi: 10.1021/acs.jmedchem.0c01063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qiao J., Li Y.S., Zeng R., Liu F.L., Luo R.H., Huang C., Wang Y.F., Zhang J., Quan B., Shen C., Mao X., Liu X., Sun W., Yang W., Ni X., Wang K., Xu L., Duan Z.L., Zou Q.C., Zhang H.L., Qu W., Long Y.H., Li M.H., Yang R.C., Liu X., You J., Zhou Y., Yao R., Li W.P., Liu J.M., Chen P., Liu Y., Lin G.F., Yang X., Zou J., Li L., Hu Y., Lu G.W., Li W.M., Wei Y.Q., Zheng Y.T., Lei J., Yang S. SARS-CoV-2 M(pro) inhibitors with antiviral activity in a transgenic mouse model. Science. 2021;371:1374–1378. doi: 10.1126/science.abf1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Owen D.R., Allerton C.M.N., Anderson A.S., Aschenbrenner L., Avery M., Berritt S., Boras B., Cardin R.D., Carlo A., Coffman K.J., Dantonio A., Di L., Eng H., Ferre R., Gajiwala K.S., Gibson S.A., Greasley S.E., Hurst B.L., Kadar E.P., Kalgutkar A.S., Lee J.C., Lee J., Liu W., Mason S.W., Noell S., Novak J.J., Obach R.S., Ogilvie K., Patel N.C., Pettersson M., Rai D.K., Reese M.R., Sammons M.F., Sathish J.G., Singh R.S.P., Steppan C.M., Stewart A.E., Tuttle J.B., Updyke L., Verhoest P.R., Wei L., Yang Q., Zhu Y. An oral SARS-CoV-2 M(pro) inhibitor clinical candidate for the treatment of COVID-19. Science. 2021;374:1586–1593. doi: 10.1126/science.abl4784. [DOI] [PubMed] [Google Scholar]
- 22.Li Y., Li W., Xu Z. Improvement on permeability of cyclic peptide/peptidomimetic: backbone N-Methylation as A useful tool. Mar. Drugs. 2021;19:311. doi: 10.3390/md19060311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Qvit N., Rubin S.J.S., Urban T.J., Mochly-Rosen D., Gross E.R. Peptidomimetic therapeutics: scientific approaches and opportunities. Drug Discov. Today. 2017;22:454–462. doi: 10.1016/j.drudis.2016.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gao K., Wang R., Chen J., Tepe J.J., Huang F., Wei G.-W. Perspectives on SARS-CoV-2 main protease inhibitors. J. Med. Chem. 2021;64:16922–16955. doi: 10.1021/acs.jmedchem.1c00409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Han S.H., Goins C.M., Arya T., Shin W.-J., Maw J., Hooper A., Sonawane D.P., Porter M.R., Bannister B.E., Crouch R.D., Lindsey A.A., Lakatos G., Martinez S.R., Alvarado J., Akers W.S., Wang N.S., Jung J.U., Macdonald J.D., Stauffer S.R. Structure-based optimization of ML300-derived, noncovalent inhibitors targeting the severe acute respiratory syndrome coronavirus 3CL protease (SARS-CoV-2 3CLpro) J. Med. Chem. 2021 doi: 10.1021/acs.jmedchem.1c00598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang C.H., Stone E.A., Deshmukh M., Ippolito J.A., Ghahremanpour M.M., Tirado-Rives J., Spasov K.A., Zhang S., Takeo Y., Kudalkar S.N., Liang Z., Isaacs F., Lindenbach B., Miller S.J., Anderson K.S., Jorgensen W.L. Potent noncovalent inhibitors of the main protease of SARS-CoV-2 from molecular sculpting of the drug perampanel guided by free energy perturbation calculations. ACS Cent. Sci. 2021;7:467–475. doi: 10.1021/acscentsci.1c00039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Su H.X., Yao S., Zhao W.F., Li M.J., Liu J., Shang W.J., Xie H., Ke C.Q., Hu H.C., Gao M.N., Yu K.Q., Liu H., Shen J.S., Tang W., Zhang L.K., Xiao G.F., Ni L., Wang D.W., Zuo J.P., Jiang H.L., Bai F., Wu Y., Ye Y., Xu Y.C. Anti-SARS-CoV-2 activities in vitro of Shuanghuanglian preparations and bioactive ingredients. Acta Pharmacol. Sin. 2020;41:1167–1177. doi: 10.1038/s41401-020-0483-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu H., Ye F., Sun Q., Liang H., Li C., Li S., Lu R., Huang B., Tan W., Lai L. Scutellaria baicalensis extract and baicalein inhibit replication of SARS-CoV-2 and its 3C-like protease in vitro. J. Enzym. Inhib. Med. Chem. 2021;36:497–503. doi: 10.1080/14756366.2021.1873977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu P., Liu H., Sun Q., Liang H., Li C., Deng X., Liu Y., Lai L. Potent inhibitors of SARS-CoV-2 3C-like protease derived from N-substituted isatin compounds. Eur. J. Med. Chem. 2020;206 doi: 10.1016/j.ejmech.2020.112702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Drayman N., DeMarco J.K., Jones K.A., Azizi S.A., Froggatt H.M., Tan K., Maltseva N.I., Chen S., Nicolaescu V., Dvorkin S., Furlong K., Kathayat R.S., Firpo M.R., Mastrodomenico V., Bruce E.A., Schmidt M.M., Jedrzejczak R., Muñoz-Alía M., Schuster B., Nair V., Han K.Y., O'Brien A., Tomatsidou A., Meyer B., Vignuzzi M., Missiakas D., Botten J.W., Brooke C.B., Lee H., Baker S.C., Mounce B.C., Heaton N.S., Severson W.E., Palmer K.E., Dickinson B.C., Joachimiak A., Randall G., Tay S. Masitinib is a broad coronavirus 3CL inhibitor that blocks replication of SARS-CoV-2. Science. 2021;373:931–936. doi: 10.1126/science.abg5827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kneller D.W., Li H., Galanie S., Phillips G., Labbe A., Weiss K.L., Zhang Q., Arnould M.A., Clyde A., Ma H., Ramanathan A., Jonsson C.B., Head M.S., Coates L., Louis J.M., Bonnesen P.V., Kovalevsky A. Structural, electronic, and electrostatic determinants for inhibitor binding to subsites S1 and S2 in SARS-CoV-2 main protease. J. Med. Chem. 2021;64:17366–17383. doi: 10.1021/acs.jmedchem.1c01475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kitamura N., Sacco M.D., Ma C., Hu Y., Townsend J.A., Meng X., Zhang F., Zhang X., Ba M., Szeto T., Kukuljac A., Marty M.T., Schultz D., Cherry S., Xiang Y., Chen Y., Wang J. Expedited approach toward the rational design of noncovalent SARS-CoV-2 main protease inhibitors. J. Med. Chem. 2021 doi: 10.1021/acs.jmedchem.1c00509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Staron J., Kurczab R., Warszycki D., Satala G., Krawczyk M., Bugno R., Lenda T., Popik P., Hogendorf A.S., Hogendorf A., Dubiel K., Matloka M., Moszczynski-Petkowski R., Pieczykolan J., Wieczorek M., Zajdel P., Bojarski A.J. Virtual screening-driven discovery of dual 5-HT6/5-HT2A receptor ligands with pro-cognitive properties. Eur. J. Med. Chem. 2020;185 doi: 10.1016/j.ejmech.2019.111857. [DOI] [PubMed] [Google Scholar]
- 34.Yi W., Dubois C., Yahiaoui S., Haudecoeur R., Belle C., Song H., Hardre R., Reglier M., Boumendjel A. Refinement of arylthiosemicarbazone pharmacophore in inhibition of mushroom tyrosinase. Eur. J. Med. Chem. 2011;46:4330–4335. doi: 10.1016/j.ejmech.2011.07.003. [DOI] [PubMed] [Google Scholar]
- 35.De Monte C., Carradori S., Secci D., D'Ascenzio M., Guglielmi P., Mollica A., Morrone S., Scarpa S., Agliano A.M., Giantulli S., Silvestri I. Synthesis and pharmacological screening of a large library of 1,3,4-thiadiazolines as innovative therapeutic tools for the treatment of prostate cancer and melanoma. Eur. J. Med. Chem. 2015;105:245–262. doi: 10.1016/j.ejmech.2015.10.023. [DOI] [PubMed] [Google Scholar]
- 36.Su Y.-L., Han Z.-Y., Li Y.-H., Gong L.-Z. Asymmetric allylation of furfural derivatives: synergistic effect of chiral Ligand and organocatalyst on stereochemical control. ACS Catal. 2017;7:7917–7922. [Google Scholar]
- 37.Schroder J., Noack S., Marhofer R.J., Mottram J.C., Coombs G.H., Selzer P.M. Identification of semicarbazones, thiosemicarbazones and triazine nitriles as inhibitors of Leishmania mexicana cysteine protease CPB. PLoS One. 2013;8 doi: 10.1371/journal.pone.0077460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Farjallah A., Chiarelli L.R., Forbak M., Degiacomi G., Danel M., Goncalves F., Carayon C., Seguin C., Fumagalli M., Zahorszka M., Vega E., Abid S., Grzegorzewicz A., Jackson M., Peixoto A., Kordulakova J., Pasca M.R., Lherbet C., Chassaing S. A coumarin-based analogue of thiacetazone as dual covalent inhibitor and potential fluorescent Label of HadA in Mycobacterium tuberculosis. ACS Infect. Dis. 2021;7:552–565. doi: 10.1021/acsinfecdis.0c00325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xu Y.S., Chigan J.Z., Li J.Q., Ding H.H., Sun L.Y., Liu L., Hu Z., Yang K.W. Hydroxamate and thiosemicarbazone: two highly promising scaffolds for the development of SARS-CoV-2 antivirals. Bioorg. Chem. 2022;124 doi: 10.1016/j.bioorg.2022.105799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pandey V., Sharma K., Raghav N. Ligand-based modeling of semicarbazones and thiosemicarbazones derivatives as Cathepsin B, H, and L inhibitors: a multi-target approach. J. Mol. Struct. 2022;1257 [Google Scholar]
- 41.Ashhurst A.S., Tang A.H., Fajtova P., Yoon M.C., Aggarwal A., Bedding M.J., Stoye A., Beretta L., Pwee D., Drelich A., Skinner D., Li L., Meek T.D., McKerrow J.H., Hook V., Tseng C.T., Larance M., Turville S., Gerwick W.H., O'Donoghue A.J., Payne R.J. Potent anti-SARS-CoV-2 activity by the natural product Gallinamide A and analogues via inhibition of cathepsin L. J. Med. Chem. 2022;65:2956–2970. doi: 10.1021/acs.jmedchem.1c01494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sacco M.D., Ma C., Lagarias P., Gao A., Townsend J.A., Meng X., Dube P., Zhang X., Hu Y., Kitamura N., Hurst B., Tarbet B., Marty M.T., Kolocouris A., Xiang Y., Chen Y., Wang J. Structure and inhibition of the SARS-CoV-2 main protease reveal strategy for developing dual inhibitors against M(pro) and cathepsin L. Sci. Adv. 2020;6 doi: 10.1126/sciadv.abe0751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vicik R., Busemann M., Gelhaus C., Stiefl N., Scheiber J., Schmitz W., Schulz F., Mladenovic M., Engels B., Leippe M., Baumann K., Schirmeister T. Aziridide-based inhibitors of cathepsin L: synthesis, inhibition activity, and docking studies. ChemMedChem. 2006;1:1126–1141. doi: 10.1002/cmdc.200600106. [DOI] [PubMed] [Google Scholar]
- 44.Oleinik A.F.B., T I., Novitskii K. Yu. Syntheses based on arylfurans. Khimiya Geterotsiklicheskikh Soedin. 1971;7:1011–1013. [Google Scholar]
- 45.Oleinik A.F.N., Yu K., Brattseva L.V., Gus'kova T.A., Pershin G.N., Kravchenko A.I., Chernov V.A. Synthesis and biological properties of aryloxyfuran. Khim. Farm. Zh. 1976;10:65–69. [Google Scholar]
- 46.LigPrep. Schrodinger, LLC; New York: 2015. [Google Scholar]
- 47.Friesner R.A., Banks J.L., Murphy R.B., Halgren T.A., Klicic J.J., Mainz D.T., Repasky M.P., Knoll E.H., Shelley M., Perry J.K., Shaw D.E., Francis P., Shenkin P.S. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 48.Halgren T.A., Murphy R.B., Friesner R.A., Beard H.S., Frye L.L., Pollard W.T., Banks J.L. Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004;47:1750–1759. doi: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.