

Abstract
The fungal microbiota (mycobiota) is an integral part of the complex multi-kingdom microbial community colonizing the mammalian gastrointestinal tract and plays an important role in immune regulation1–6. Although aberrant mycobiota changes have been linked to several diseases including inflammatory bowel disease (IBD)3–9, it is currently unknown whether fungal species captured by deep sequencing represent living organisms and whether specific fungi have functional consequences for disease development in affected individuals. Here we developed a translational platform for the functional exploration of the mycobiome at a fungal strain- and patient-specific level. Combining high-resolution mycobiota-sequencing, fungal culturomics and genomics, CRISPR/Cas9-based fungal strain editing system, in vitro functional immunoreactivity assays and in vivo models, this platform allows to explore host-fungal crosstalk within the human gut. We discovered a rich genetic diversity of opportunistic Candida albicans strains that dominated the colonic mucosa of IBD patients. Among these human gut-derived isolates, strains with high immune cell-damaging capacity (HD strains) reflect disease features of individual ulcerative colitis patients and aggravated intestinal inflammation in vivo through IL-1β-dependent mechanisms. Niche-specific inflammatory immunity and Th17 antifungal responses by HD strains in the gut were dependent upon the C. albicans secreted peptide toxin candidalysin during the transition from a benign commensal to a pathobiont state. These findings unveil the strain-specific nature of host-fungal interactions in the human gut and highlight new diagnostic and therapeutic targets for diseases of inflammatory origin.
Deep sequencing-based surveys of the gut mycobiome in several disease cohorts provide consistent evidence for “fungal dysbiosis” as a hallmark3–9 of inflammatory bowel disease (IBD), the most prevalent forms of which are Crohn’s disease (CD) and ulcerative colitis (UC), which affect up to 3.5 million individuals10. Antibodies against fungal mannan (ASCA), define IBD subtypes, further linking fungi with intestinal inflammation11. Candida is the most prevalent fungal genus consistently increased in several IBD cohorts based on fecal sequencing3,6,12. Notably gut C. albicans induces an array of antifungal antibodies and acts as an immunogen for ASCA13–15. Candida species associated with the intestinal mucosa are sensed by gut resident-macrophages and thus have the potential to induce protective immunity or trigger inflammation in a context-dependent manner experimentally15,16. Despite this evidence, it is currently unknown whether fungi detected by deep sequencing technologies in the human intestinal mucosa play an essential role in directing mucosal immunity or disease outcomes in individual patients. Consistently, lack of association between changes in mycobiota composition and disease severity have been observed in IBD cohorts, despite a consistent increase in Candida species3,5,6,12. Thus, we hypothesized that niche-specific fungal strain functional diversity among Candida spp. dictates the host-fungal relationship in the human intestinal mucosa.
C. albicans expands in the inflamed gut
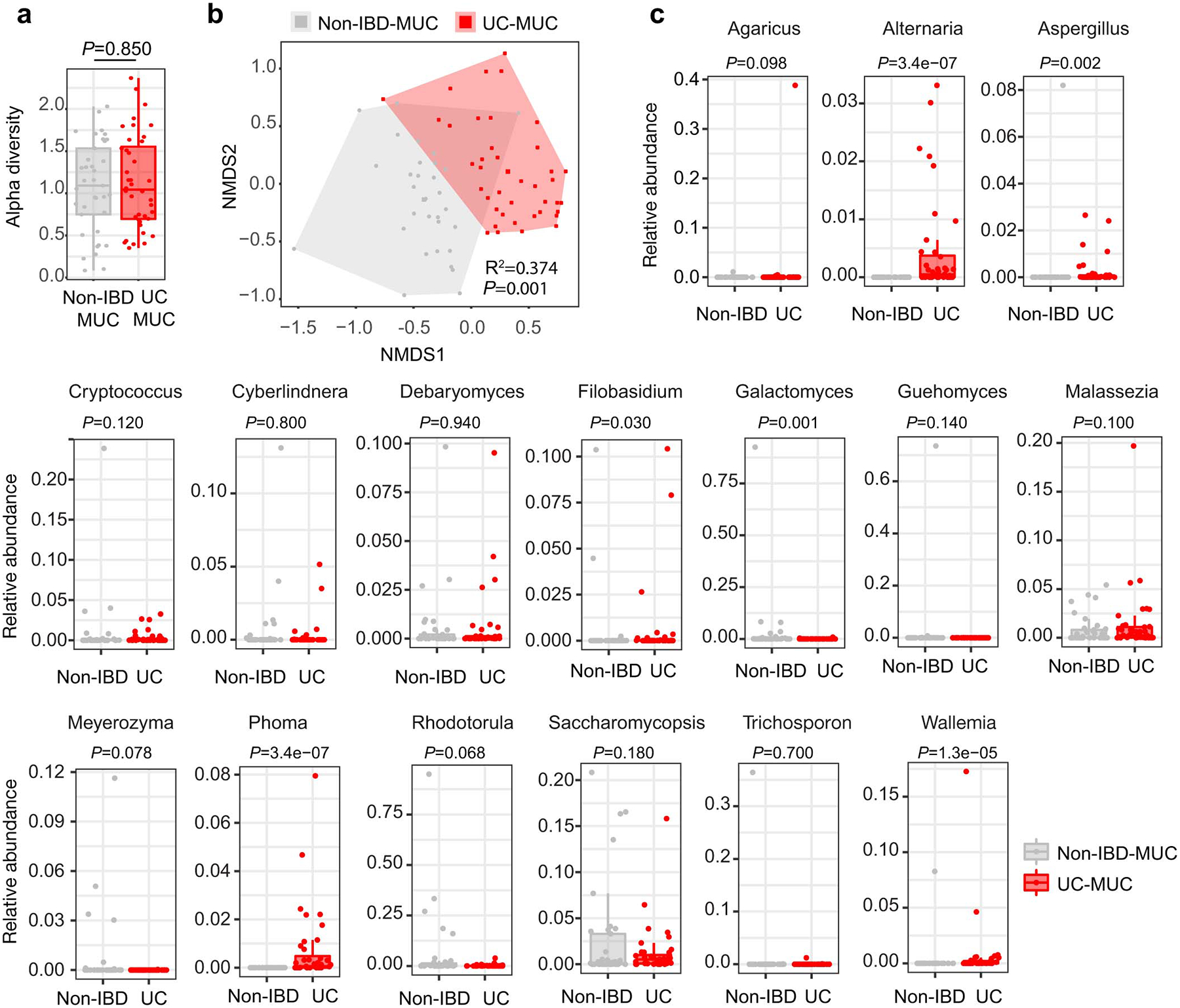
We first focused our analysis on UC which targets the colon: a site where commensal fungi are highly abundant and interact with host immunity5,7,17. To enrich for mucosa-associated immunoreactive mycobiota, we obtained colonic mucosa lavage samples from 38 non-IBD individuals and 40 patients with UC undergoing colon cleansing - a process that removes fecal and other luminal contents in preparation for colonoscopy and performed ITS sequencing of fungal ribosomal DNA (rDNA). This analysis revealed a distinct clustering between non-IBD and UC individuals, while alpha diversity remained similar within each group (Fig.1a and Extended Data Fig. 1a–b). Genus level analysis revealed the presence of 18 highly prevalent fungal genera with > 0.2% average relative abundance across all samples (Fig.1b and Supplementary Table 1). Among those, Candida and Saccharomyces represented the most abundant genera (Fig.1b). with an increase in Candida and a pronounced reduction of Saccharomyces in the mucosa-enriched mycobiome of UC patients when compared to non-IBD individuals (Fig.1c and Extended Data Fig. 1c). In contrast, other fungi8,9, including a rare abundance of Debaromyces spp. were not altered with UC disease status (Fig.1b and Extended Data Fig. 1c). C. albicans was consistently overrepresented in the in the mucosa of UC patients as confirmed by our culture-dependent approach. (Fig.1d). To experimentally determine whether increased C. albicans presence influences intestinal inflammation, we orally gavaged wild-type (WT) C57BL/6 specific pathogen free (SPF) mice with C. albicans SC5314, a laboratory strain and determined its effect on dextran sulfate sodium (DSS)-induced model of colitis. While intestinal inflammation provided a niche for C. albicans overgrowth (Extended Data Fig. 2a), disease severity was not affected by C. albicans in this model (Extended Data Fig. 2b–e), consistent with previous studies8,16. Furthermore, C. albicans did not cause spontaneous colitis following a prolonged colonization (up to 4 months, Extended Data Fig. 2f–g). Thus, while Candida spp. are a disease contributing factor during genetic deficiencies targeting antifungal immunity pathways16, C. albicans does not cause spontaneous intestinal inflammation in a host with intact antifungal immunity, albeit inflammation appeared to be a driver of C. albicans intestinal expansion.
Figure 1. C. albicans expands in UC patient colonic mucosa and promotes inflammation in a murine model of colitis.
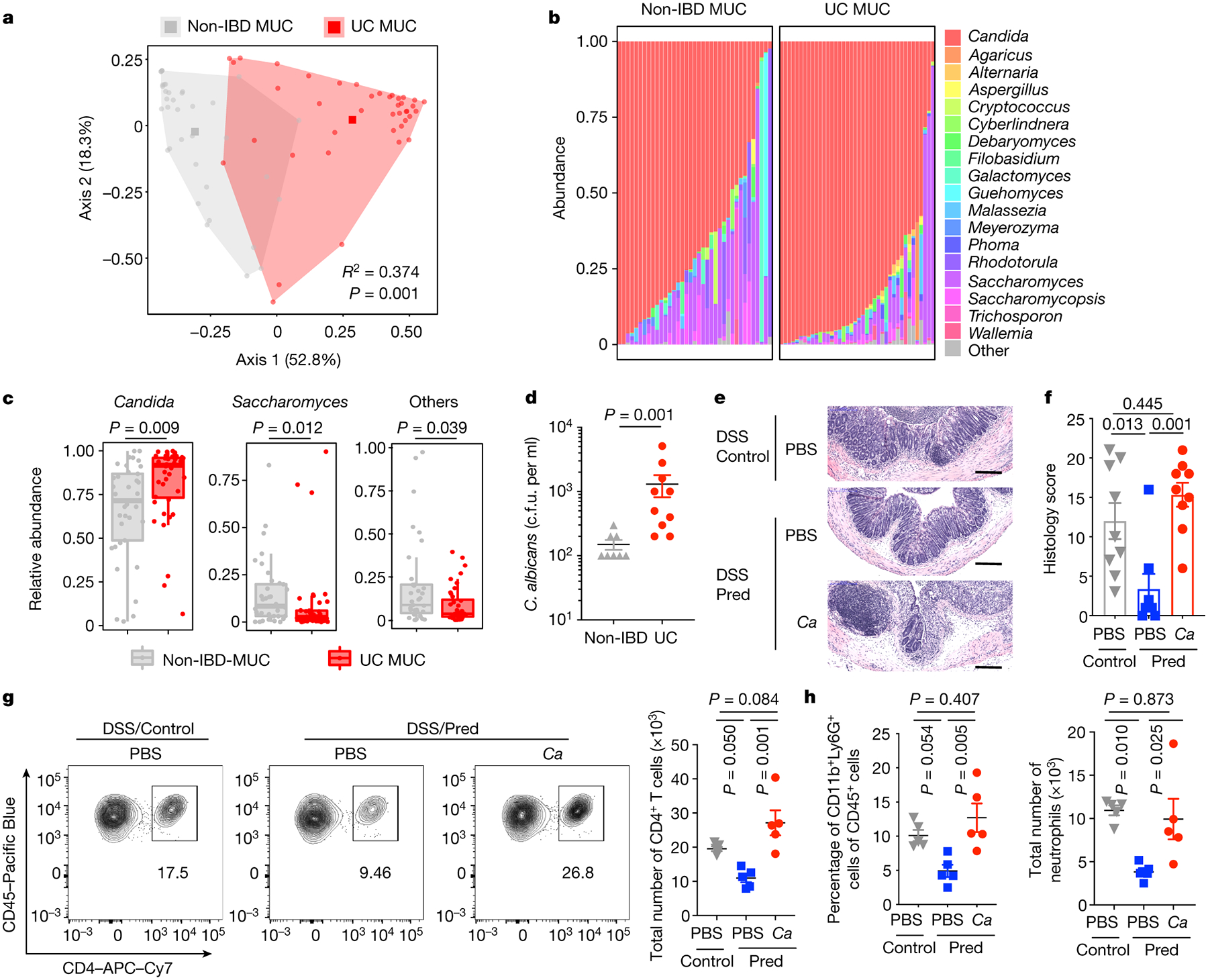
a, Principal coordinate analysis (PCoA) plot of distance ordination for fungal ITS1 OTUs in colonic mucosa (MUC) samples from non-IBD or UC individuals. Analysis of similarities (ANOSIM) statistics. b, Relative abundance of detected fungal genera. c, Relative abundance of Candida spp., Saccharomyces spp. and other less represented fungal genera (“others”); The lower and upper hinges correspond to the first and third quartiles (the 25th and 75th percentiles). The horizontal line shows the median. a-c, Each dot represents an individual human subject, non-IBD (n=37) and UC (n=40) individuals. d, Fungal colonies from each individual subject were identified by MALDI-TOF, and viable C. albicans colony forming units (cfu) per mL of lavage sample were determined. Results are shown as mean ± s.e.m. non-IBD (n=8) and UC (n=10) individuals. e-h, Mice were fed PBS or C. albicans SC5314, then treated with PBS or prednisolone before DSS-mediated induction of colitis. e-f, H&E staining and histology score of colon sections. Ctrl PBS (n=9), Pred+PBS (n=8), Pred+C.a (n=9). Data in f are pooled from two independent experiments with similar results. g-h, Representative flow cytometry plots and quantification of the frequency and total cell number of colonic lamina propria (cLP) CD4+ T cells (g, from left to right) and CD11b+Ly6G+ neutrophils (h, from left to right), n=5 in each group (g and h). f-h, Results are shown as mean ± s.e.m. Each dot represents an individual mouse. Data in g-h are representative of two independent experiments with similar results. Unpaired, two-tailed, Mann-Whitney test (c and d) or one-way ANOVA followed by the Tukey’s post hoc test (f, g and h).
Since Candida spp. influence intestinal inflammation in a context-dependent manner with immunity-related factors being among the key drivers16,18, we utilized a mouse model of DSS-induced inflammation under corticosteroid treatment: a widely used remission induction therapy in UC19(Extended Data Fig. 3a). Treatment with prednisolone allowed the establishment of sustained intestinal fungal colonization, overcoming C. albicans colonization resistance in the murine gut20 (Extended Data Fig. 3b). While WT SPF mice only developed a mild disease, mice colonized with C. albicans displayed enhanced histopathology, as characterized by increased mucosal erosion, crypt destruction, and inflammatory cell infiltration in the colon (Fig. 1e–h). Further characterization revealed increased CD4+ T cell and neutrophil infiltration in the colon (Fig. 1g–h). In contrast, colonization with Pichia kudriavzevii, which is present in healthy individuals8, did not contribute to intestinal pathology (Extended Data Fig. 3c–e). This indicates that commensal C. albicans negatively influences disease outcomes under corticosteroid treatment.
Strain features affect gut immunity
Current mechanistic studies on the role of fungi in intestinal immunity and disease are limited by the use of laboratory fungal strains of non-intestinal origin8,16,18,21. Since fungal strain-specific features can dramatically influence infectious disease outcomes22–25, we reasoned that important insights could be gained through studies of fungal strains isolated from the human intestine. The results we obtained through ITS sequencing informed the development of culture-based methods that permitted the detection, isolation and collection of live C. albicans isolates from the colonic mucosa of UC patients (Fig.1b–d). Since intestinal macrophages are key to initiation and induction of antifungal immunity in the colonic mucosa16 and virulent C. albicans strains can damage these cells26–29, we next tested the macrophage-damaging capacity of human C. albicans isolates. Notably, gut C. albicans isolates could be ranked into two major categories: a high-damaging (HD/C.a) group consisting of isolates with an increased or similar ability to inflict macrophage damage as compared with C. albicans SC5314, and low-damaging isolates with a decreased cell-damaging capacity (LD/C.a) (Fig. 2a). The hyphal morphogenesis program in C. albicans is key to virulence factor production and pathogenesis at several mucosal surfaces30–32. Conversely, mutations in transcriptional regulators of hyphal morphogenesis can arise during a commensal lifestyle23,26,33,34. Thus, we randomly selected three C. albicans isolates per human subject to assess phenotypic features in vitro using a filamentation phenotypic assay (Supplementary Table 2). Contrary to the prediction that gut-adapted C. albicans strains would be poorly filamentous26, a considerable number (~60%) (Extended Data Fig. 4a) of the isolates formed filaments regardless of the host disease status (Fig. 2a and Extended Data Fig. 4b–i). Furthermore, some C. albicans isolates originating from the same host showed slightly different filamentation phenotypes (Extended Data Fig. 4c and d). Notably, C. albicans isolates that responded to filament-inducing stimuli induced more damage of bone marrow-derived macrophages (mBMDM) compared to non-filamentous isolates (Fig. 2a–b), demonstrating that the cell-damaging capacity of gut C. albicans cells is linked to filamentation ability.
Figure 2. Cell damage and proinflammatory immunity induced by human gut-derived C. albicans is strain-dependent.
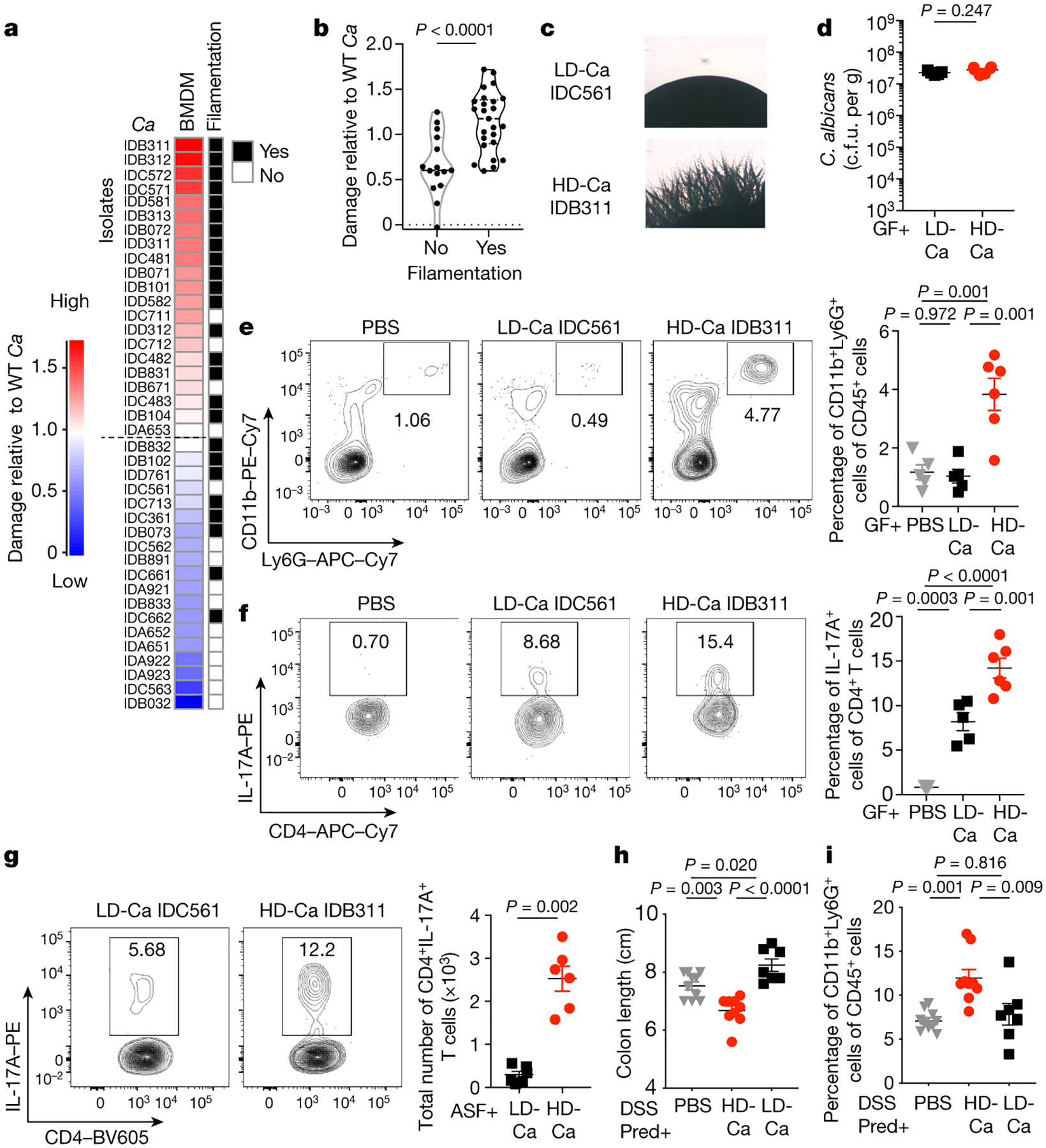
a, mBMDM were infected with C. albicans (C.a) isolates (three isolates per individual; MOI=5) for 16 hours, and cytotoxicity (LDH release as compared to SC5314) was measured. Data are shown as an average value of three technical repeats. b, Filamentation phenotype of each isolate was determined in a spider agar assay and compared the capacity of C. a isolates to induce damage of mBMDM (LDH assay). Results are shown as mean ± s.e.m. a-b, Data are representative of three independent experiments with similar results. c, Representative images of LD/C.a; IDC561 and HD/C.a; IDB311. d-f, WT germ-free (GF) mice were colonized with C.a strains for three weeks. PBS (n=5), LD/C.a (IDC561, n=5), HD/C.a (IDB311, n=6). d, Fecal C. albicans burden was measured at day 21. e-f, Representative flow cytometry plots and quantification of CD11b+Ly6G+ neutrophils and CD4+IL-17A+ Th17 cells in the colon. g-i, ASF mice were colonized with C.a strains for three weeks. PBS (n=6), LD/C.a (IDC561, n=6), and HD/C.a (IDB311, n=6). g, Total numbers of colonic CD4+IL-17A+ Th17 cells. h-i, SPF WT mice were with C.a strains colonized and treated with prednisolone followed by DSS-induced murine colitis. PBS (n=10), HD/C.a IDB311 (n=9) and LD/C.a IDC561 (n=7). h, Colon length. i, Frequencies of CD11b+Ly6G+ neutrophils in the colon. Data in d-i are shown as mean ± s.e.m. Each dot represents an individual mouse. Data are representative of three (c-f) or two (g-i) independent experiments with similar results. Unpaired, two-tailed, Mann-Whitney test (b, d and g) or one-way ANOVA followed by the Tukey’s post hoc test (e, f, h and i).
We next assessed immune responses to HD/C.a and LD/C.a isolates (Fig. 2c) in vivo. To explore the direct effect of these isolates on the host, we first colonized germ-free (GF) mice with a HD/C.a (HD/C.a IDB311) or LD/C.a (LD/C.a IDC561) strains. HD/C.a induced robust proinflammatory responses in the colon including neutrophil infiltration and Th17 induction as compared to the LD/C.a strain (Fig. 2e–f and Extended Data Fig. 5a–c). The effect was independent of fungal load as each strain stably colonized the murine gut at similar levels (Fig. 2d). Fungi and bacteria co-exist in a complex web of interactions in the gut where bacteria can alter C. albicans colonization in mice20,35. To avoid the confounding effect on immunity of commensal bacteria or fungi across colonies of SPF mice35, we utilized altered Schaedler flora (ASF)-colonized mice that are mycobiota-free, and carry a defined community of 8 bacteria35–37. While HD/C.a IDB311 and LD/C.a IDC561 stably colonized ASF mice at a similar level (Extended Data Fig. 5d), only HD/C.a induced strong proinflammatory immune response (Extended Data Fig. 5e–f). These findings were confirmed for multiple HD/C.a (IDB101, IDC711) and LD/C.a (IDB891, IDC662) isolates (Extended Data Fig. 5g–j). Consistent with their ability to induce strong proinflammatory immunity, HD/C.a contributed to severe intestinal inflammation in murine model of DSS induced-colitis under prednisolone therapy when compared to LD/C.a (Fig. 2l–n and Extended Data Fig. 5k). Thus, the striking capacity of specific human-gut derived C. albicans isolates to induce Th17 and proinflammatory immunity is strain-dependent and correlates directly with their ability to inflict cell damage in a manner resilient to the influence of intestinal bacteria.
Candidalysin promotes intestinal inflammation
Having observed a strong correlation between filamentation, cell damage and intestinal immunity activation, we reasoned that transcriptional regulation of C. albicans morphogenesis program might play an important role in the observed phenotypes. In addition to its role in the yeast-to-hyphal transition, the transcription factor enhanced filamentous growth protein 1 (Efg1) and its related pathways have been linked to the C. albicans commensalism34,38. Initial assessment of gut C. albicans strains revealed a dramatic decrease in EFG1 expression in LD compared to HD strains that was not associated with specific genetic variation in EFG1 or other filamentation-related genes (Extended Data Figs. 6a–b, and 7a–c). To investigate the functional role of EFG1 among clinical isolates, we adopted a CRISPR-based technique for gene targeting in Candida spp. This system39,40 consists of a Candida-compatible Cas9 nuclease and a synthetic guide RNA (sgRNA) that directs Cas9-mediated cleavage in the target regions and uses a marker-free repair template for homology directed repair (HDR) (Fig. 3a). The approach allows for achieving homozygous recessive mutations in the genetic target across multiple gut isolates of C. albicans circumventing limitations posed by genetic and chromosome variations among strains41. EFG1 ablation in HD strains abrogated their filamentation and cell-damaging abilities, and in vivo led to substantial decrease in Th17 cell accumulation in the colon of mice, suggesting a role of C. albicans Efg1 in the induction of Th17- and neutrophil-mediated intestinal immunity (Fig. 3b, and Extended Data Fig. 6c–f). The strain dependent immune phenotype was independent of gut fungal expansion (ablation of EFG1 led to intestinal overgrowth of HD/C.a, Fig. 3c). While no direct evidence of hyphal C. albicans adhesion/penetration into the intestinal epithelium was observed (Fig. 3e), mixed yeast and hyphae C. albicans morphotypes for both HD and LD strains were still present in the mucosa (Fig. 3e). Together, these data suggest that a specific fungal factor rather than hyphal morphology per se was the cause of the observed immune activation phenotypes in the gut.
Figure 3. HD C. albicans promotes intestinal pro-inflammatory immunity through the Efg1-Ece1-dependent factor candidalysin.
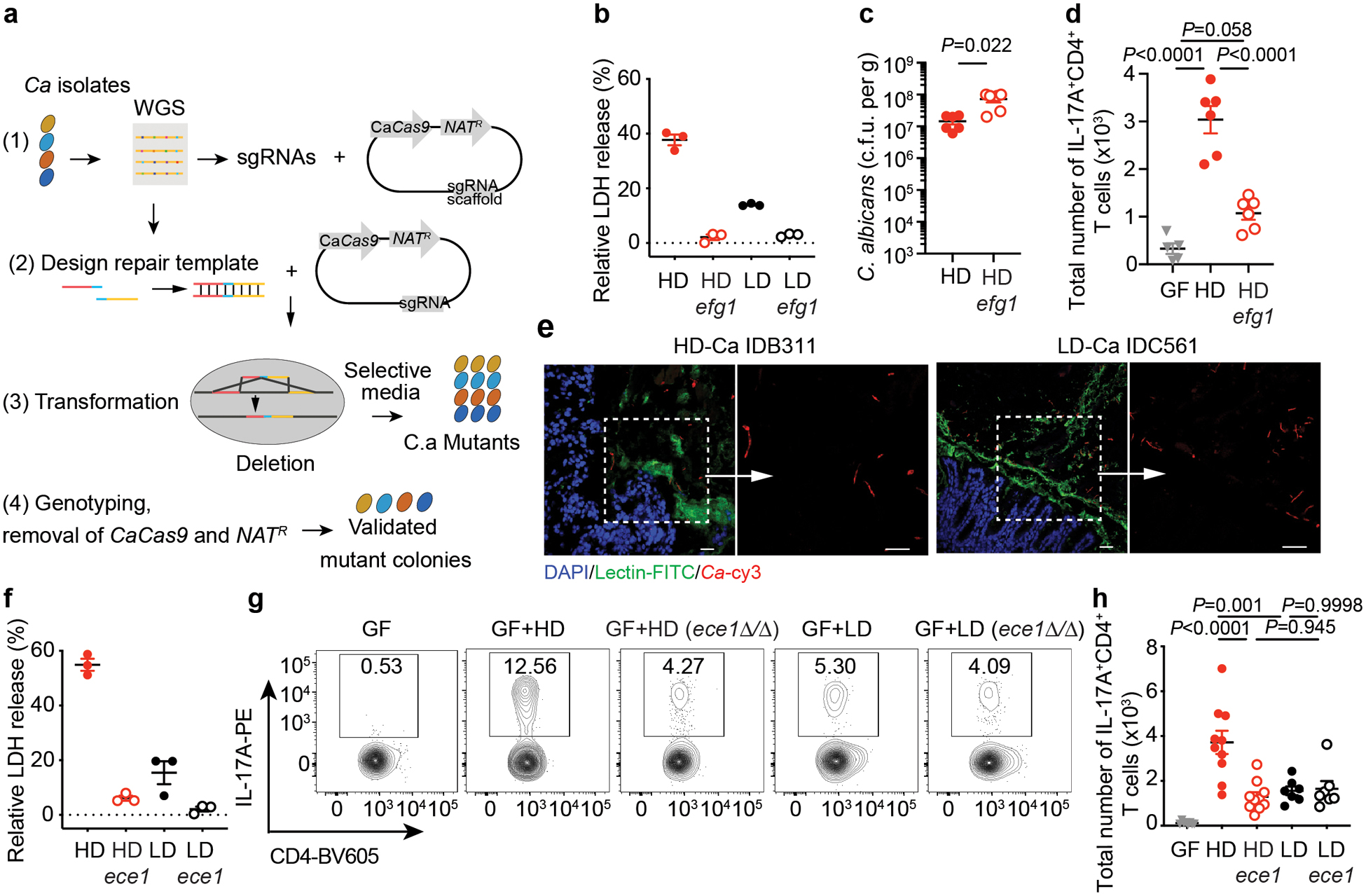
a, Schematic view of CRISPR/Cas9-mediated mutagenesis in C. albicans (C.a) isolates as described in Methods. b, Caco2 cells were infected with C.a for 12 hours, and the LDH release was assessed. HD and LD strains, and respective efg1Δ/Δ (efg1) knockouts were used. c-d, GF mice were colonized with C.a strains for three weeks. PBS GF (n=5), HD/C.a IDB311 (HD, n=6), and HD/C.a IDB311 efg1 (HD efg1, n=6). c, Fecal C. a burden was assessed at day 21. d, Total cell numbers of IL-17A+CD4+ T cell in the colon. e, GF mice were colonized with HD/C.a IDB311 (n=10) and LD/C.a IDC561 (n=7) strains. FISH-stained C.a in the colon. Left, DAPI-stained colonic epithelial cell (blue), FITC-UEA-1-stained mucus layers (green), and rRNA Cy3-probe-stained C.a (red). Right, C.a morphotypes. Scale bar 25 μm. f, LDH release from Caco2 cells infected with C.a was assessed. HD/C.a IDB311, LD/C.a IDC561 strains, and respective ECE1 mutant strains were used. g-h, GF mice were colonized with C.a strains for three weeks. PBS GF (n=8), HD/C.a IDB311 (HD, n=10), HD ece1 (n=10), LD/C.a IDC561 (LD, n=7), and LD ece1 (n=7). g-h, Representative flow cytometry plots and total cell numbers of IL-17A+CD4+ T cells in the colon. Data in b and f are shown as mean ± s.d. Data points indicate technical well replicates (n=3) and are representative of three independent experiments with similar results. Data in c, d and g-h are shown as mean ± s.e.m. Each dot represents an individual mouse. Data are representative of three (c-d) or two (e-h) independent experiments with similar results. Unpaired, two-tailed, Mann-Whitney test (b, c, and f) or one-way ANOVA followed by the Tukey’s post hoc test (d, and h).
Recent studies revealed strong upregulation of genes encoding for hypha-associated virulence factors upon C. albicans intestinal colonization, including extent of cell elongation 1 gene (ECE1), secreted aspartic proteases genes SAP6, hyphally regulated gene 1 (HYR1) and agglutinin-like protein 3 precursor gene (ALS3), with the ECE1 gene among the top hits34. Deletion in EFG1 gene almost fully inhibits ECE1 expression in an HD strain in vitro (Extended Data Fig. 6g). Consistently, the analysis of a recently published dataset38 revealed that ECE1 is among the top genes regulated by EFG1 during C. albicans colonization of the large intestine (Extended Data Fig. 6h). Thus, we focused our attention on the ECE1-encoded peptide toxin, candidalysin, as a secreted factor with the potential to drive immunity and cell damage27,30,31. We first utilized a C. albicans ece1Δ/Δ mutant, which forms normal hyphae30, and demonstrated a significantly lower level of macrophage and epithelial cell damage in vitro (Extended Data Fig. 8a–b). Upon intestinal colonization, the ece1Δ/Δ strain induced significantly reduced colonic neutrophil infiltration and Th17 responses in ASF-colonized mice in comparison with the C. albicans parental strain (Extended Data Fig. 8c–e), suggesting a role for candidalysin in triggering proinflammatory immunity in the gut, despite comparable levels of gut colonization by both strains (Extended Data Fig. 8e). Similarly, in the murine model of DSS-induced inflammation under prednisolone treatment, SPF mice intestinally colonized with C. albicans ece1Δ/Δ also exhibited significantly reduced intestinal inflammation, characterized by decreased infiltration of neutrophils and proinflammatory Th17 cells in the colon (Extended Data Fig. 8g–j), despite similar levels of colonization by both strains (Extended Data Fig. 8f). Therefore, the hypha secreted toxin candidalysin is a critical factor driving intestinal inflammation by C. albicans.
To investigate the importance of candidalysin in clinical isolates, we generated ECE1 homozygous knockouts of HD and LD strains using CRISPR-based approach (Fig. 3a and Fig. 3f). Notably, knockout of ECE1 in HD/C.a (IDB131 ece1Δ/Δ and IDB101 ece1Δ/Δ) led to consistent reduction of cell damage in vitro (Fig. 3f and Extended Data Fig. 6i). Gut colonization by HD/C.a ece1Δ/Δ resulted in a reduced Th17 recruitment and proinflammatory immunity in the colons of mice, similar to the phenotypes induced by the LD strain and the respective ece1 strain (Fig. 3g–h, and Extended Data Fig. 6j–k). These properties were specific to ECE1 deletion and were not due to differences in fungal survival or morphology in vivo (Extended Data Figs. 6l–m). These findings indicate that C. albicans-mediated Th17 and proinflammatory intestinal immunity is strain-dependent, and that candidalysin in HD strains plays a key role in this process through Efg1-ECE1-dependent regulation.
Since phenotypic characterization allowed us to categorize C. albicans gut isolates into two broad groups (LD/Ca and HD/Ca), we hypothesized that isolates with high immunoreactivity would be closely related as previously reported42. Thus, we sequenced 18 human gut C. albicans isolates, each obtained from a colonic mucosa-enriched sample of distinct individuals, and compared their whole genomes with the previously sequenced collection43 of C. albicans strains that were assigned into specific C. albicans clades (Supplementary Table 4)23,43,44. This analysis revealed a high diversity among gut C. albicans isolates with multiple clades represented irrespective of disease status (non-IBD or UC) (Fig 4a). Surprisingly, gut C. albicans isolates from different individuals represented different strains, despite the close geographical location of our patient population (Fig 4a), suggesting a wide genetic variety of gut C. albicans across individuals.
Figure 4. Capacity of patient-specific C. albicans strains to induce IL-1β reflects UC disease severity and anti-IL-1R antibody treatment ameliorates disease in mice.
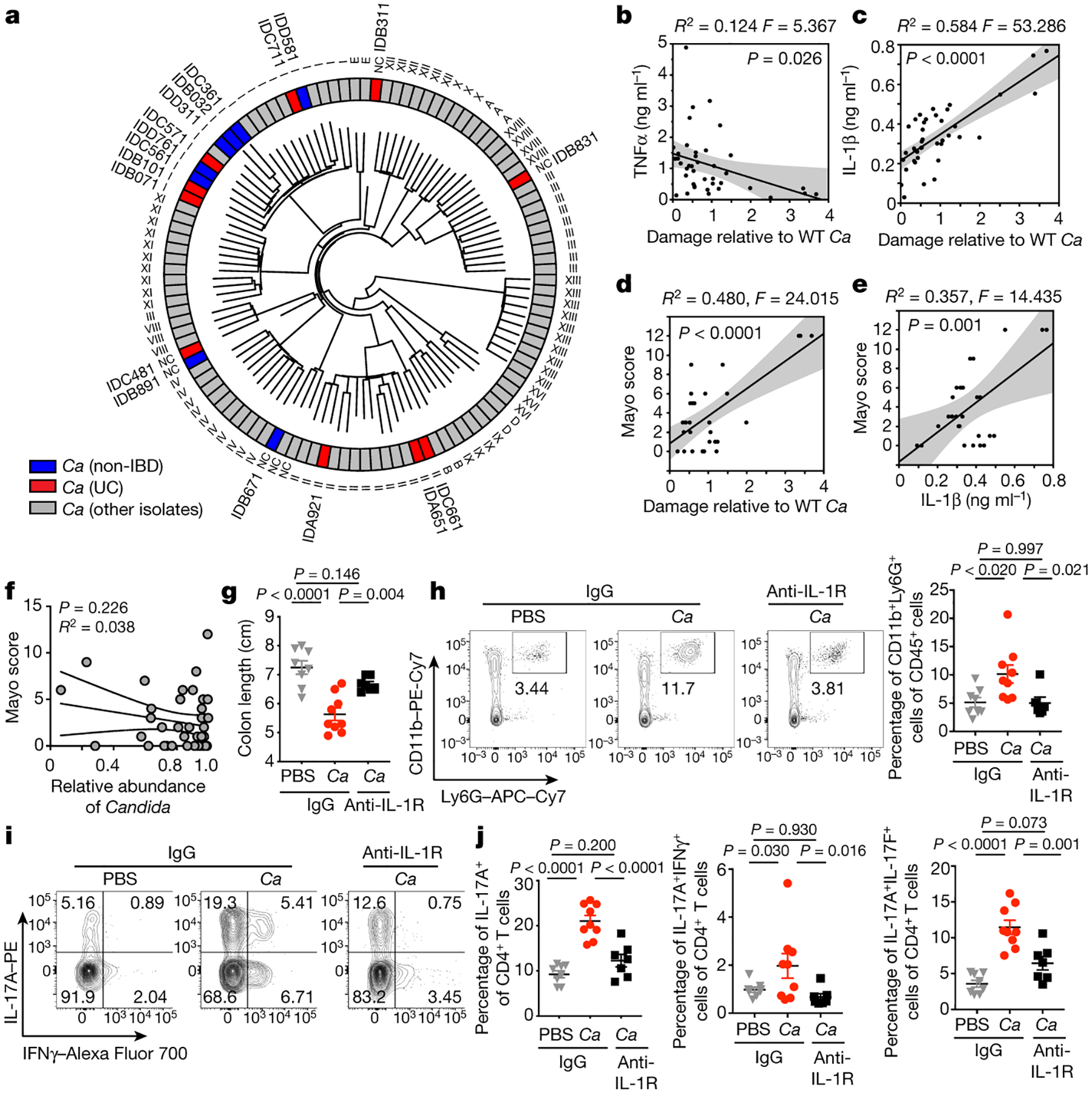
a, Dendrogram based on whole genome sequence similarity of 18 newly sequenced gut C. albicans (C.a) isolates, together with 94 C. a strains collected worldwide43. Strains isolated from non-IBDs (blue), UC (red), or other isolates (gray) are color labeled. Clade labels are shown as defined in Ropars et al.43 or labelled “NC” (no clade assigned). b-c, TNF-α and IL-1β release in culture supernatants of unprimed (b) or LPS-primed (c) hMDMs after incubation with C.a isolates (MOI=5). Cytokine release was measured by ELISA. hMDM LDH release was correlated with cytokine release (b-c). d, Correlation between hMDM damage by patient-specific gut C.a and Mayo score (UC, n=10). e, Correlation between the IL-1α production in hMDMs incubated with patient-specific C. albicans and Mayo score (n=10). f, Correlation between the relative abundance of Candida in UC patient samples and Mayo score. UC (n=40) individuals. b-e, Dot is shown as an average value of three technical repeats, Data are representative of three independent experiments with similar results. The simple linear regression was performed, where P-value calculated by a F test. g-j, Mice colonized with or without HD/C.a IBD311 were treated with prednisolone (Pred) followed by DSS-induced murine colitis, and further treated with anti-IL-1R1 IgG (〈IL1R) or isotype IgG (IgG). IgG+PBS (n=8), IgG+C.a (n=9) and anti-IL-1R1+C.a (n=7). g, Colon length, h, Frequencies of CD11b+Ly6G+ neutrophils in the colon, i-j, Representative flow cytometry plots and frequencies of IL-17A+, IL-17A+IFNγ+ and IL-17A+IL-17F+CD4+ T cells in the colon. Results are shown as mean ± s.e.m. Each dot represents an individual mouse. Data in b-e and g-j are representative of three independent experiments with similar results. One-way ANOVA followed by the Tukey’s post hoc test (g, h and j). One-way ANOVA followed by the Tukey’s post hoc test.
Gut C. albicans undergoes clonal expansion and microevolution (generation-to-generation small-scale genetic changes in a population): two key genetic events reported by experimental studies using a model C. albicans strain26. Phylogenetic and heterozygous SNP density analysis revealed that several strains isolated from the gut of two individual patients (IDB10, IDB83) are derived from an ancestral strain of intestinal C. albicans, suggesting the possibility that microevolutionary events are also occurring in the human gut. Interestingly, we also found that clonal expansion is a common genetic event that occurred across the patient population (IDB31, IDA62, IDA92, IDC48, IDB07, IDD58) (Extended Data Fig. 9a–b). Additionally, we found four isoforms of candidalysin across strains, but no apparent association between specific candidalysin isoforms and cell-damaging properties of the strains (Extended Data Fig. 7d and 9c). While clonal expansion is a typical occurrence in a variety of invasive fungal diseases, these findings imply that parallel mechanisms of fungal expansion could be occurring during both classical fungal infections and inflammatory intestinal disease.
Strain-induced IL-1β reflect UC severity
Since C. albicans-produced candidalysin is a key factor in inflicting damage to human monocyte derived macrophages (hMDM) (Extended Data Fig. 10a), we next sought to define pro-inflammatory mediators that are dependent on candidalysin. Among a panel of hMDM-produced cytokines, TNF-α, IL-6, IL-1β and IL-10 were strongly induced by C. albicans infection, albeit only IL-1β production was candidalysin dependent (Extended Data Fig. 10b–c) confirming the role of C. albicans a driver of NLRP3-dependent IL-1β production by macrophages27,45. Notably, IL-1β, but not TNF-α or IL-6 strongly correlated with the capacity of specific gut C. albicans strains to inflict immune cell damage (Fig. 4b–c and Extended Data Fig. 10d). Further analysis revealed a strong correlation between the capacity of C. albicans strains to induce macrophage cell damage, IL-1β production and disease severity in individual UC patients (Fig. 4d–e). The observed phenomenon was specific to IL-1β and correlated with UC severity (Fig. 4e and Extended Data Fig. 10e–f). In contrast, ITS-based compositional analysis failed to link the relative abundance of Candida with disease severity in individual patients (Fig. 4f), indicating that the functional characteristics of the strain, rather than fungal composition, determines Candida-related proinflammatory characteristics in individual patients. Thus, the capability of specific C. albicans strains to induce cell damage and IL-1β in phagocytes, but not the relative abundance of Candida, reflected the disease severity in the patient of origin.
IL-1β is an important driver of inflammation and is consistently increased in inflamed rectal biopsies from patients with UC2, suggesting that IL-1β might be involved in the pathogenesis of UC2,46,47. Notably, IL-1R blockade dramatically reduced neutrophil recruitment, Th17 cell accumulation and colonic inflammation in HD/C.a-colonized mice (Fig. 4g–j, and Extended Data Fig. 10g–h). Although the cellular sources of IL-1β production and regulation during intestinal fungal colonization need further investigation, this data indicates that IL-1 signaling pathways play a critical role in C. albicans-induced proinflammatory immune cell accumulation in the colon. Remarkably, the presence of gut C. albicans strains with high ability to induce IL-1β production in phagocytes positively correlates with increased disease severity in their UC host, suggesting that C. albicans strains isolated from these patients with severe UC have acquired or retained the ability to secrete candidalysin, which aggravates cell damage and inflammation.
While ITS-based sequencing has allowed for exploration of mycobiota composition, a major shortcoming of this approach is the inability to uncover the functional role of observed fungal species/strains changes. To overcome these shortcomings, we developed a customized functional mycobiome platform. The application of this platform to mucosal samples uncovered a rich C. albicans strain-level diversity in the human gut and an unexpected domination of strains with high cell-damaging capacity in the gut of specific UC patients. Our findings shed light on why species level mycobiome composition of individual patients rarely correlates with disease characteristics3–6 and suggest that only fungal strains with “proinflammatory” properties can contribute to disease etiology. Although the mechanisms by which such strains emerge and evolve across individuals is not addressed by our study, our data suggest that fungal strains with “proinflammatory” properties can be present in any individual (healthy or diseased) but aggravate disease only in susceptible immunologically affected hosts.
While a recent in vitro cell-based study revealed that additional C. albicans derived-factors may be involved in the candidalysin-mediated cell damaging process48, our findings suggest that candidalysin is a key virulence determinant to fuel proinflammatory immunity by C. albicans in the gut, and that strains with high damaging capacity propel intestinal inflammation through IL-1β-dependent mechanisms. In light of the growing therapeutic interest in targeting the lL-1 axis in UC2,46,49 and other inflammatory diseases, patients carrying high-damaging strains might represent a target population for IL-1-blocking therapy and/or antifungal co-therapy.
Methods
Mucosa-enriched lavage samples
Seventy-eight colonic mucosa-enriched lavage samples (38 non-IBD individuals and 40 patients with ulcerative colitis) were obtained following Institutional Review Board-approved protocols from the Center for Advanced Digestive Diseases and the JRI IBD Live Cell Bank Consortium at Weill Cornell Medicine. Samples were collected, processed and stored as previously described50,51. All subjects from which the samples originated were free of anti-fungal treatment.
Mice
8–12-week-old wild-type (WT) C57BL/6J mice were purchased from Jackson laboratory with an approximately equal gender ratio. All animals were kept under specific pathogen-free conditions (Helicobacter pylori free) and raised on a 12-hour light/dark cycle with access to water and food (5053 Rodent Diet 20, PicoLab) ad libitum. Germ-free (GF) WT mice were bred and maintained within sterile vinyl isolators at Weill Cornell Medical College Gnotobiotic Mouse Facility. Altered Schaedler flora (ASF) mice were generated from germ-free WT C57BL/6J mice upon inoculation with ASF community35, bred for at least 5 generations for fully immunocompetent progeny and maintained within sterile vinyl isolators at Weill Cornell Medical College Gnotobiotic Mouse Facility. All mice used in these experiments were housed with a 12-hr light/dark cycle per day at a temperature of 72±2°F, and 30–70% relative humidity. All animal experiments were approved and are in accordance with the Institutional Animal Care and Use Committee guidelines at Weill Cornell Medicine.
Fungal colonization in vivo
8–10-week-old GF and ASF WT C57BL/6J mice were orally gavaged with C. albicans (1 × 108 CFU/mouse) or Pichia kudriavzevii (1 × 108 CFU/mouse, pkID01) at day 1 and day 2. After fungal colonization, mice were maintained within sterile vinyl isolators for 21 days and then sacrificed at day 23. All C. albicans strains used in these experiments were cultured in Sabouraud dextrose broth (SDB) and washed by sterile PBS twice before use. Fecal samples were collected to confirm the C. albicans colonization by plating on Sabouraud dextrose agar (SDA).
Murine models of colitis and fungal colonization
Wild-type C57BL/6 (WT) SPF mice were orally gavaged with C. albicans (1 × 108 CFU/mouse) or Pichia kudriavzevii (1 × 108 CFU/mouse) twice per week for the duration of the experiments. In a one-cycle of DSS-induced murine model of colitis, WT C57BL/6 SPF mice received prednisolone daily (10 mg/kg/day) or control PBS via intraperitoneal injection for a total of 10 days. After prednisolone treatment, mice were rested for 4 days prior to Dextran sulfate sodium salt (DSS, MP Biomedicals) exposure. To induce colitis, 3% DSS water (w/v) was provided to the mice for 7 days. At day 4 of DSS treatment, mice received another 4 days of daily prednisolone (10mg/kg/day) treatment. Three or four days after DSS withdrawal, mice were sacrificed. In the IL-1R blockade experiments, 1 mg of InVivoMab anti-IL-1R1 IgG (JAMA147; BioXCell) or 1 mg of InVivoMAb Armenian hamster IgG (BioXcell) were administered via intraperitoneal injection every 4 days for the duration of the experiment.
Human gut fungal strains isolation and culture conditions
Fecal colonic mucosa-enriched lavages collected from non-IBD or UC-affected subjects were diluted in sterile PBS and plated onto SDA, supplemented with penicillin/streptomycin (Sigma) and inhibitory mold agar (Hardy Diagnostics). SDA plates were incubated at 37 °C for 48 hours. Inhibitory mold agar plates were incubated at 30 °C for 72 hours. Fungal colonies were picked up from both cultures (37 °C, overnight). Isolated fungal colonies from each individual subject were identified by matrix-assisted laser desorption/ionization-time of flight (MALDI-TOF) mass spectrometer.
DNA isolation, mycobiome library generation and sequencing
Mucosa-enriched lavage samples (300 μL) from non-IBD or UC-affected subjects were centrifuged at 400 g. Pellets were collected for fungal DNA isolation. Pellets were treated with 200 U/mL lyticase (Sigma) followed by bead beating, and processing using the Quick-DNA Fungal/Bacterial Kit (Zymo Research) as previously described15. Fungal DNA presence was validated by RT-PCR for fungal 18S. Based on this approach one low quality sample (out of 78) was excluded and did not proceed for further mycobiome sequencing and analysis. Fungal ITS1–2 regions were amplified by PCR using primers with sample barcodes and sequencing adaptors.
Fungal primers: ITS1F-CTTGGTCATTTAGAGGAAGTAA
ITS2R-GCTGCGTTCTTCATCGATGC
Forward overhang: 5’ TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG-[locus-specific sequence]
Reverse overhang: 5’ GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAG-[locus-specific sequence]
ITS amplicons were generated with 35 cycles using Invitrogen AccuPrime PCR reagents (Carlsbad). Amplicons were then used in the second PCR reaction, using Illumina Nextera XT v2 (Illumina) barcoded primers to uniquely index each sample. 2×300 paired-end sequencing was then performed on the Illumina MiSeq (Illumina). DNA was amplified using the following PCR protocol: Initial denaturation at 94°C for 10 min, followed by 40 cycles of denaturation at 94°C for 30 s, annealing at 55°C for 30 s, and elongation at 72°C for 2 min, followed by an elongation step at 72°C for 30 min. All libraries were subjected to quality control using DNA 1000 Bioanalyzer (Agilent), and Qubit (Life Technologies) to validate and quantify library construction prior to preparing a Paired-End flow cell. Samples were randomly divided among flow cells to minimize sequencing bias. Clonal bridge amplification (Illumina) was performed using a cBot (Illumina). 2 × 250 bp sequencing-by-synthesis was performed on Illumina MiSeq platform (Illumina).
ITS1 fungal reads
Raw FASTQ ITS1 sequencing data were filtered to enrich for high quality reads, removing the adapter sequence by cutadapt v1.4.1 or any reads that did not contain the proximal primer sequence52. Sequence reads were then quality-trimmed by truncating reads not having an average quality score of 20 over a 3-base pair sliding window and removing reads shorter than 100 bp52. These high-quality reads were then aligned to Targeted Host Fungi (THF) ITS1 database, using BLAST v2.2.22 and the pick_otus.py pipeline in the QIIME v1.6 wrapper with an identity percentage ≥97% for OTU picking53. The alignment results were subsequently tabulated across all reads, using the accession identifier of the ITS reference sequences as surrogate OTUs and using a Perl script16. Among the analyzed samples, one sample was excluded due to insufficient quality size (ITS reads < 200). Shannon index at the OTU level, NMDS scaling of Bray-Curtis dissimilarities, and relative abundances at various taxonomic levels were analyzed with the R packages Phyloseq (1.26.1) and Vegan (2.5–5). Boxplots of graphs were calculated using the geom_boxplot function of ggplot2 (v3.3.3) in R with default arguments. The lower and upper hinges correspond to the first and third quartiles (the 25th and 75th percentiles). The horizontal line shows the median. Mann-Whitney test was performed to test the significance of the difference in the relative abundance of fungal genera between the two groups with “Benjamini-Hochberg” (BH) correction. Analyses were performed in R (v3.5.2).
Isolation of cells from intestinal lamina propria of mice
Colonic lamina propria (cLP) cells were isolated as previously described16. Briefly, colons were excised, opened longitudinally, washed of fecal contents and then cut into 1 cm pieces. Intestinal pieces were transferred into Hank’s Balanced Salt Solution (HBSS) medium (Thermo Fisher Scientific), supplemented with 2 mM EDTA, and were shaken for 8 min at 37°C. The remaining tissue was washed, minced and subsequently incubated in digestion medium consisting of RPMI 1640 (Thermo Fisher Scientific), 5% FBS, 0.5 mg/ml collagenase type VIII (Sigma-Aldrich), 5 U/ml DNase (Sigma-Aldrich), 100 IU/ml penicillin and 100 μg/ml streptomycin (Thermo Fisher Scientific), for 40 min at 37°C by gentle shaking at a speed of 150 rpm. The cell suspensions were filtered through a 100 μm mesh and centrifuged at 1700 rpm. The obtained cells were filtered through a 70 μm filter, washed twice with PBS and used as cLP cells.
Flow cytometry
The staining antibodies for flow cytometry were purchased from Thermo Fisher Scientific, Biolegend or BD Biosciences. Dead cells were excluded with eBioscience Fixable Viability Dye eFluor 506 (Thermo Fisher Scientific) during surface staining. For cell surface staining, cells were incubated with antibodies at 4°C for 20 min. All antibodies used at 1:200 unless otherwise noted. Fluorophore-conjugated antibodies against mouse antigens: anti-CD16/CD32 (92), anti-CD45 (30-F11), anti-I-A/I-E (M5/114.15.2), anti-CD11b (M1/70), anti-Ly6G (1A8-Ly6g), anti-CD11c (N418), anti-CD4 (RM4–5), anti-TCRβ (H57–597), and anti-Ly6C (HK1.4). The FoxP3 Fix/Perm kit (Thermo Fisher Scientific) was used for intracellular transcription factor staining in accordance with the manufacturer’s instructions. For cytokine staining, cells were re-stimulated with 20 nM phorbol 12-myristate 13- acetate (Sigma-Aldrich), 1.3 uM ionomycin (Sigma-Aldrich) and brefeldin A Solution (Thermo Fisher Scientific) in RPMI-1640 medium (Corning) supplemented with 10% FBS and penicillin/streptomycin for 4 hours. Intracellular staining for indicated cytokines was fixed with a Cytofix/Cytoperm kit (BD Biosciences) according to manufacturer’s instructions. All antibodies used at 1:200 unless otherwise noted. The following antibodies for intracellular staining were used: anti-FOXP3 (FJK-16S, 1:150), anti-IL-17A (eBio 17B7,1:100), anti-IFNγ (XMG1.2, 1:100), anti-RORγt (B2D), and anti-IL-17F (9D3.1C8, 1:100). Flow cytometry was performed using a BD LSRFortessa cell analyzer (BD Biosciences), and flow cytometry data were collected by FACS Diva Software V9.0, and data was analyzed with FlowJo V10 (TreeStar).
In vitro cell-damaging and cell stimulation assays
Caco2 cells were seeded into were seeded in 24-well tissue culture treated plates at a concentration of 0.2 ×105/ml in DMEM Medium (10% fetal bovine serum, 1% penicillin/streptomycin solution, these reagents were purchased from Corning or Gibco) for 3 days at 37 °C. Then Caco2 were co-incubated with live C. albicans at MOI 1 or 5 in DMEM Medium (serum free, 1% penicillin/streptomycin solution) for 24 hours. Supernatants were obtained for lactate dehydrogenase (CytoTox 96® Non-Radioactive Cytotoxicity Assay kit, Promega) and various cytokine measurements. RNA was extracted from C. albicans co-infected with Caco2 cells after 6–7 hours using the YeaStar RNA kit. RT-PCR was performed with C. albicans ACTIN as the control (Primers listed in the Supplementary Table 3). Murine bone marrow cells were harvested and seeded in 150 mm non-tissue culture treated plates at a concentration of 2 × 106/ml in RPMI Medium (10% fetal bovine serum, 10 mM HEPES solution, 1% L-glutamine, 1 mM Sodium Pyruvate, 1% MEM non-essential amino acids, 55 μM β-mercaptoethanol, 1% penicillin/streptomycin solution, these reagents were purchased from Corning or Gibco) supplemented with 50 ng/ml macrophage colony stimulating factor (M-CSF) (PeproTech) for 7 days at 37 °C. On day 7, after removing the medium and washing the cells with PBS, adherent cells were incubated with cell dissociation buffer (Invitrogen) for 5 min at 37 °C. Murine bone marrow-derived macrophages (mBMDMs) were seeded in 96-well plates to a final number of 1 × 105/well in serum-free medium for 12 hours. Then mBMDMs were co-incubated with C. albicans at MOI 5 for 16 hours. Human peripheral blood mononuclear cells (hPBMCs) were isolated from healthy volunteers’ buffy coats requested from NYC blood center with Ficoll density gradient centrifugation media. Then CD14+ positive monocytes were selected by CD14 positive selection kit (Miltenyi Biotec). To differentiate monocytes into human monocyte-derived macrophages, 6 ×104 CD14+ monocytes were seeded in each well of a 96-well plate in RPMI 1640 media with 2 mM L-glutamine (Thermo Fisher Scientific) containing 10% heat-inactivated fetal bovine serum (FBS; Bio&SELL) (RPMI + FBS) and 50 ng/mL recombinant human M-CSF (ImmunoTools) and incubated for seven days at 37 °C and 5% CO2. Medium was changed with serum-free medium overnight, and cells were stimulated with LPS (50 ng/mL). In both human and murine in vitro assays, live C. albicans (MOI=5) was added to each well, cultures were incubated for 16 hours and supernatants were obtained for lactate dehydrogenase (LDH, CytoTox 96® Non-Radioactive Cytotoxicity Assay kit, Promega) and various cytokine measurements. Cell-damage was calculated using the following equation: Cell-damage (%) =100*(Experimental absorbance (OD492)-Average of media-only absorbance (OD492))/Average of C. albicans SC5314 or parental C. albicans control strain absorbance (OD492). Murine and human IL-1β, TNF-α and IL-6 were measured by ELISA kit (Biolegend). A larger panel of human cytokines was assessed by LEGENDplex™ Human Macrophage/Microglia Panel kit following the manufacturer’s instructions.
Filamentation assays
C. albicans strain was cultured in Sabouraud dextrose broth at 28°C for 24 hours26. C. albicans strain was plated on Spider agar (2% Nutrient broth, 2% Mannitol, 0.4% potassium phosphate, 2.7% Bacto Agar, PH 7.2) and incubated at 37°C for 5 days followed by filamentation assessment of the edge of wrinkled and smooth colonies with bright field microscopy.
CRISPR- mediated genome editing of the human gut-derived Candida albicans isolates
All our plasmid construction were based on plasmid pV1524, a gift from Gerald Fink (http://n2t.net/addgene:111431; RRID:Addgene_111431)39. Guide RNAs (gRNAs) and plasmids: gRNAs for specific gene were either immediately adjacent to or within 15 bp of the desired mutagenesis point. Phosphorylated and annealed guide sequence–containing primers were ligated into CIP (calf intestinal phosphatase)–treated BsmBI-digested parent vectors. Correct clones with the right gRNAs were identified by sequencing. Repair templates were generated with 60-bp bases with homology to the sequences upstream or downstream from the target gene and containing 20-bp overlaps at their 3′ ends centered at the mutation point for the target gene, which consisted of an open reading frame (ORF) deletion of the target gene. Primers were extended to generated repair templates by thermocycling performed with Ex Taq (TaKaRa). Plasmids and Oligonucleotide sequences used in this study are listed in Supplementary Table 3. Transformation: For clinical C. albicans isolates, we achieved efficient transformation with a hybrid lithium acetate/electroporation protocol was described previously39. 5 mL gut-derived C. albcians isolates were grown in YPD medium overnight at 30°C in an incubator with a 150-rpm shaking speed to achieve saturation. C. albicans cells were quickly pelleted by centrifugation and resuspended in 2 ml of freshly prepared TE/lithium acetate solution (100 mM lithium acetate–10 mM Tris (pH 8.0)–1 mM EDTA (pH 8.0) containing 50 μl 1 M dithiothreitol (DTT). C. albicans cells were incubated for 6 hours on a 30°C roller drum. The cells were then washed twice in ice-cold sterile water, once in ice-cold 1 M sorbitol, and then resuspended in 500 μL of ice-cold 1 M sorbitol. 40 ul C. albicans cells were mixed with 5 μg digested plasmid DNA and with 6 ug purified repair templates in a pre-chilled electroporation cuvette (#9140–2002, USA scientific) and kept on ice for 5–10 minutes before electroporation. C. albicans cells was electroporated on a Bio-Rad Gene pulser with a capacitance of 25 μF and resistance of 200 Ω at 1.8 kV. The cuvette was immediately filled with 600 mL cold YPD medium, and the cell mixture was incubated for 12 hours at 30°C before plating onto YPD selective media (1% Difco Bacto yeast extract, 2% Difco Bacto peptone, 2% glucose 200 μg nourseothricin) and selected with nourseothricin (Nat) at 200 μg/ml. In 2–3 days, colonies appeared. Colonies were picked up and re-plated with a selective YPD agar plate. Flipout of the Natr gene from C. albicans vectors was induced by overnight growth in YP maltose medium (1% Difco Bacto yeast extract, 2% Difco Bacto peptone, 2% maltose). Episomal plasmid loss experiments were performed by overnight growth in nonselective liquid YPD medium. Drug-sensitive isolates, which had either flipped out the cassette or lost the plasmid, were identified by plating for single colonies on nonselective media and subsequent identification by replica plating to selective media. Colony PCR flanking the deleted region was used to select CRISPR/Cas9-mutagenized ECE1 or EFG1 mutants, which were then confirmed using sequence analysis colony PCR products. Oligonucleotide sequences for PCR/sequencing used in this study are listed in Supplementary Table 3. Whole-genome sequencing of the resulting C. albicans mutants was utilized to confirm single-gene mutagenesis in the EFG1 or ECE1 genes. All strains were stored as glycerol stocks at −80°C.
Whole genomic sequencing (WGS) and analysis
Each C. albicans isolate was cultured in SDB for 24 hours. Genomic C. albicans DNA was extracted from colonies using the QiaAmp DNA Mini Kit (Qiagen). Quality control, library preparation and deep sequencing on Illumina HiSeqX platform were performed at Novogene Co., Ltd. Raw sequencing data for each isolate was obtained. For comparison, raw sequencing data for representative C. albicans strains belonging to 17 previously established SNP-based clades43 were downloaded from the NCBI Sequence Read Archive (BioProject ID PRJNA432884). Ten strains from each SNP-based clade were selected (or when fewer than 10 strains were available, all available strains were selected) for comparison with our human gut C. albicans isolates. Both downloaded and newly sequenced WGS raw sequence data were processed according with the following workflow: Raw reads were aligned using the Burrows-Wheeler Aligner (BWA)54 to the haplotype A chromosomes of the C. albicans strain SC5314 assembly 22 obtained from the Candida Genome Database (www.candidagenome.org). Aligned reads were processed using the Genome Analysis Toolkit version 4 (GATK4)55. Specifically, duplicate reads were marked using the GATK4 MarkDuplicates command and variants were called using the HaplotypeCaller command. Variants were then filtered using the VariantFiltration command with filters “QD < 2.0”, “ReadPosRankSum < −8.0”, “FS > 60.0”, “MQRankSum < −12.5”, and “MQ < 40.0”. Further analysis included only variants marked “PASS” by VariantFiltration. The biallelic SNPs from the resulting VCF files were used to compute a sample-sample distance matrix and dendrogram using the SNPRelate R package56. The resulting dendrogram was plotted using the circlize R package57. Similar dendrograms were generated for genomic subregions using region-specific VCF files extracted using the GATK4 command SelectVariants. SNP-based clade assignments for the SRA-downloaded samples were obtained from Ropars et al.43. Clade labels in the dendrogram were propagated to newly sequenced isolates according to the following rule: if the latest common ancestor of all strains labeled clade X does not have descendants with other labels, then all its descendants receive the label for clade X. Individual VCF files were extracted from the joint VCF file using GATK4 SelectVariants. Heterozygous SNPs were extracted using bcftools58 and heterozygous SNPs were counted in each 10kbp of each genome using bedtools59. Genome assemblies were generated from raw FASTQ files using Spades60 version 3.10 with options -k 21,33,55,77–careful. Nucleotide sequences of interest (SK1, ECE1, EFG1, CPH1, UME6, FLO8) in the genome assembly graphs (fastg files) were extracted using the querypaths command of Bandage61. The corresponding amino acid sequences were generated with the transeq command of EMBOSS2 using translation table 12 (Alternative Yeast Nuclear).
Fluorescence In Situ Hybridization (FISH)
8–10-week-old GF mice were orally gavaged with C. albicans (1 × 108 CFU/mouse) at day 1 and day 2. After fungal colonization, mice were maintained within sterile vinyl isolators for 21 days and then sacrificed at day 23. Colon sections were collected along with feces and fixed in a methacarn solution (60% methanol, 30% chloroform, and 10% glacial acetic acid) at room temperature (RT) overnight. The following procedures were used to wash the colon sections: 2 × 30 minutes in methanol, 2 × 20 minutes in ethanol, 2 × 20 minutes in xylene substitute. All chemical reagents are purchased from Sigma-Aldrich. Colon sections were then immersed for two hours at 70°C in melted paraffin wax. Tissues were embedded and further cut at 10 μm by the histology facility at WCMC. The following procedures were used to dewax colon sections: 2×10 minutes at 60°C in xylene substitution, 2×5 minutes at RT in ethanol. For C. albicans detection, DNA hybridization was performed for three hours at 50°C using a pan-fungal probe conjugated to Cy3 (/5Cy3/CTCTGGCTTCACCCTATTC, Integrated DNA Technologies, 0.25 μg per sample)34, and mucus layers were stained with FITC conjugated UEA-1 (Sigma, L9006–1MG) for overnight incubation at 4°C in a sealed chamber. All sections were further stained for 4 minutes at room temperature with 4′,6-diamidino-2-phenylindole (DAPI). The sections were then mounted with ProLong™ Gold Antifade Mountant (Thermo Fisher Scientific) and imaged on Zeiss LSM880. Images were merged by Fiji ImageJ 2.1.0/1.53c software with the scale bar indicating 25 μm.
Histological staining
Distal colon sections were collected and fixed in 10% neutral buffered formalin for 24 hours at room temperature and then were transferred to 70% ethanol. Complete colon tissue was embedded in paraffin, sectioned and stained with hematoxylin and eosin by IDEXX BioAnalytics. Blinded histological evaluation was conducted by a board-certificated pathologist (C. Y.). Samples were scored according to the following criteria: area involved (0–4), erosion/ulceration (0–4), follicles (0–3), edema (0–3), fibrosis (0–3), crypt loss (0–4), granulocytes (0–3), mononuclear cells (0–3), and crypt damage/apoptosis (0–4). Scores were summed to give a total inflammation score.
Statistical analysis
The investigators were blinded during colonic mucosa-enriched lavage sample and data collection. The investigators were blinded for the colon histological evaluation. As noted, based on quality control, one non-IBD sample was excluded from mycobiome sequencing and analysis. No data or mice were excluded for other analysis. No statistical analysis was used to determine the appropriate sample size. n indicated in the figure legends means the number of mice or human subjects in the experiments. All attempts at replication of experiments were successful and were performed at least two to three times. The simple linear regression was performed with JMP Data analysis software v16.1. The Prob > F value measures the probability of obtaining an F Ratio as large as what is observed, where P-values indicate the probability that the slope of the regression line is zero. Unless otherwise specified, P values were calculated using unpaired nonparametric Mann-Whitney test two-tailed t-test with a 95% confidence interval or one-way ANOVA followed by the Tukey’s post hoc test by GraphPad Prism (GraphPad Software v9) as indicated in the figure legends. P values of less than 0.05 were considered to be significant.
Reporting summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this manuscript.
Extended Data
Extended Data Fig. 1. Relative abundance of intestinal fungal genera in non-IBD and patients with UC.
a, Alpha diversity analysis was analyzed using the Shannon diversity index among fungi communities at the fungal OTU level in colonic mucosa (MUC) enriched samples from non-IBD or ulcerative colitis-affected (UC) individuals. Based on quality control one non-IBD sample was excluded from further mycobiome sequencing and analysis. b, Non-metric multidimensional scaling (NMDS) plot of distance ordination based on Bray–Curtis dissimilarities for fungal ITS1 OTUs in colonic mucosa (MUC) enriched samples from non-IBD or ulcerative colitis-affected (UC) individuals. Analysis of similarities (ANOSIM) statistics. c, Relative genus abundance of intestinal fungal genera. Boxplots in a and c, the lower and upper hinges correspond to the first and third quartiles (the 25th and 75th percentiles). The horizontal line shows the median. a-c, Each dot represents an individual human subject, non-IBD (n=37) or ulcerative colitis-affected (UC, n=40) individuals. Analysis performed with two-sided, non-paired, Mann-Whitney test, Benjamini-Hochberg (BH) corrected for data in a and c.
Extended Data Fig. 2. Intestinal colonization by C. albicans does not cause spontaneous colitis during homeostasis nor does it aggravate DSS-induced colitis.
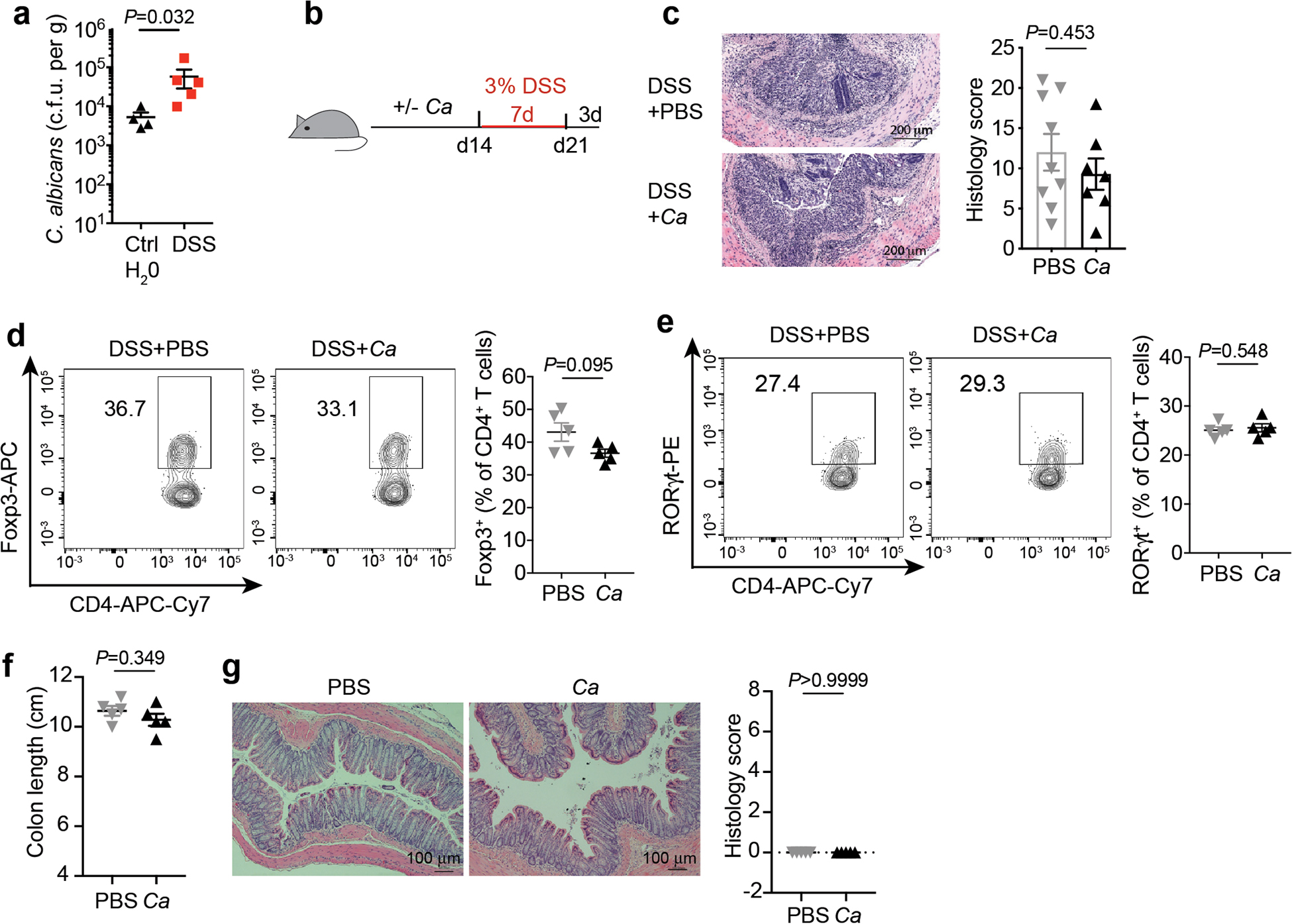
a, Fecal C. albicans burdens were assessed after 3 days of C. albicans colonization in mice that received either control feeding water (n=4) or DSS water (n=5) for 4 days. Dots represent individual mice. b-g, Mice, after being gavaged with PBS (n=5) or C. albicans (n=5) for 14 days, were induced by 3% DSS water for 7 days. b, Schematic figure of DSS-induced colitis model of mice with intestinal colonization by C. albicans. c, H&E staining and histology score of colon sections. n=9 (DSS+ control PBS) and n=7 (DSS+ C. albicans). The scale bar 200μm. Data in c are pooled from two independent experiments with similar results. d-e, Representative flow cytometry plot and quantification of Foxp3+ (d) and RORγt+ CD4+ T (e) cells. f-g, Mice were gavaged twice per week with PBS (n=5) or C. albicans WT SC5314 (n=5). Mice were sacrificed four months later for colon length (f) and histology evaluation, the scale bar 100μm (g). Each dot represents an individual mouse. Data in a-e are representative of three independent experiments with similar results. Data in f-g representative of two independent experiments with similar results. Data in a, c, and d-g are shown as mean ± s.e.m., unpaired, two-tailed, Mann-Whitney test.
Extended Data Fig. 3. C. albicans expands and promotes intestinal inflammation under immunosuppression therapy for UC.
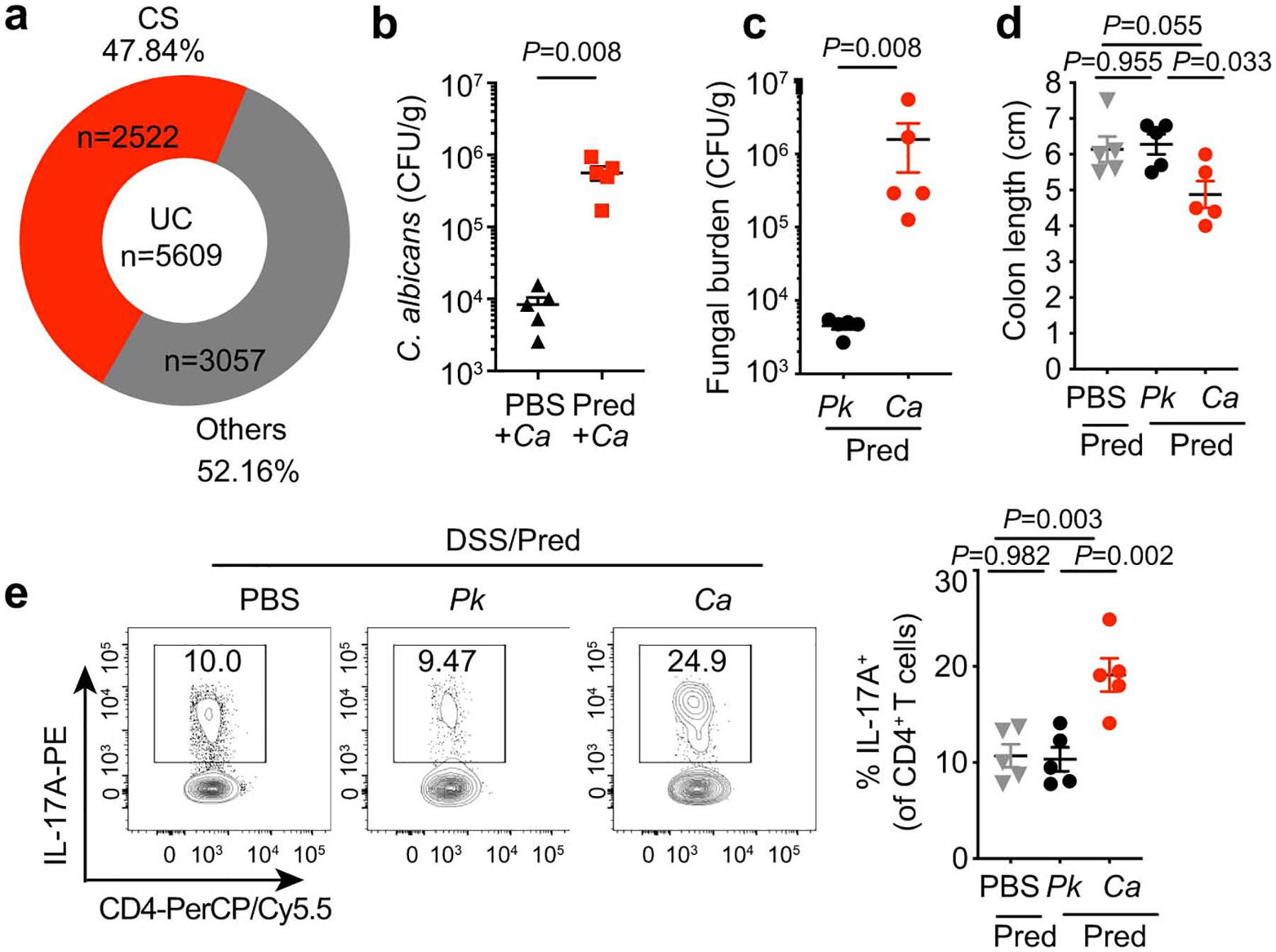
a, Medication data summary for UC patients (n=5609) who visited New York-Presbyterian Hospital from 2016–2018. Corticosteroids (CS, n=2522); Others include Mercaptopurine, Azathioprine, Methotrexate, Tacrolimus, Cyclosporine and Biologics (n=3057). Some individuals were being treated with two or more treatments. b, Fecal C. albicans burdens were measured after 3 days of C. albicans colonization in mice that received either PBS (n=5) or prednisolone daily (10 mg/kg/day, n=5). c-e, WT SPF mice were fed PBS (n=5), Pichia kudriavzevii (P.k, n=5), or Candida albicans (C.a, n=5) while receiving prednisolone treatment (Pred+PBS, Pred+P.k, or Pred+C.a). 3% DSS drinking water was used to induce colitis for 7 days. Mice were sacrificed three days after the DSS water was removed. c, Fecal fungal burdens upon sacrifice. d, Colon length was assessed. e, Representative flow cytometry plots and quantification of the frequency IL-17A+CD4+ T cells in the colons. Results in b-f are shown as mean ± s.e.m. Each dot represents an individual mouse. Data in b-e are representative of three independent experiments with similar results. Unpaired, two-tailed, Mann-Whitney test (b and c) or one-way ANOVA followed by the Tukey’s post hoc test (d and e).
Extended Data Fig. 4. Gut-derived C. albicans isolates exhibit different phenotypic responses in a filamentation assay.

a, Gut C. albicans isolates were cultured on Spider agar at 37°C for 5 days followed by assessment of the edge of wrinkled and smooth colonies. Percentage of the filamentation phenotypes of gut C. albicans isolates in Fig. 2a on spider agar. b-i, Filamentation phenotype of representative gut C. albicans isolates used in Fig 2a. b, C. albicans IDB311, IDB312, and IDB313 isolates from UC patient IDB31. c, C. albicans IDB831, IDB832, and IDB833 isolates from UC patient IDB83. d, C. albicans IDB101, IDB102, and IDB104 isolates from UC patient IDB10. e, C. albicans IDC481, IDC482, and IDC483 isolates from UC patient IDC48. f, C. albicans IDB071, IDB072, and IDB073 isolates from UC patient IDB07. g, C. albicans IDA651, IDA652, and IDA653 isolates from UC patient IDA65. h, C. albicans IDA921, IDA922, and IDA923 isolates from UC patient IDA92. i, C. albicans IDC561, IDC562, and IDC563 isolates from non-IBD individual IDC56. Data are representative of three independent experiments with same results.
Extended Data Fig. 5. High-damaging strains induce greater proinflammatory immune responses.
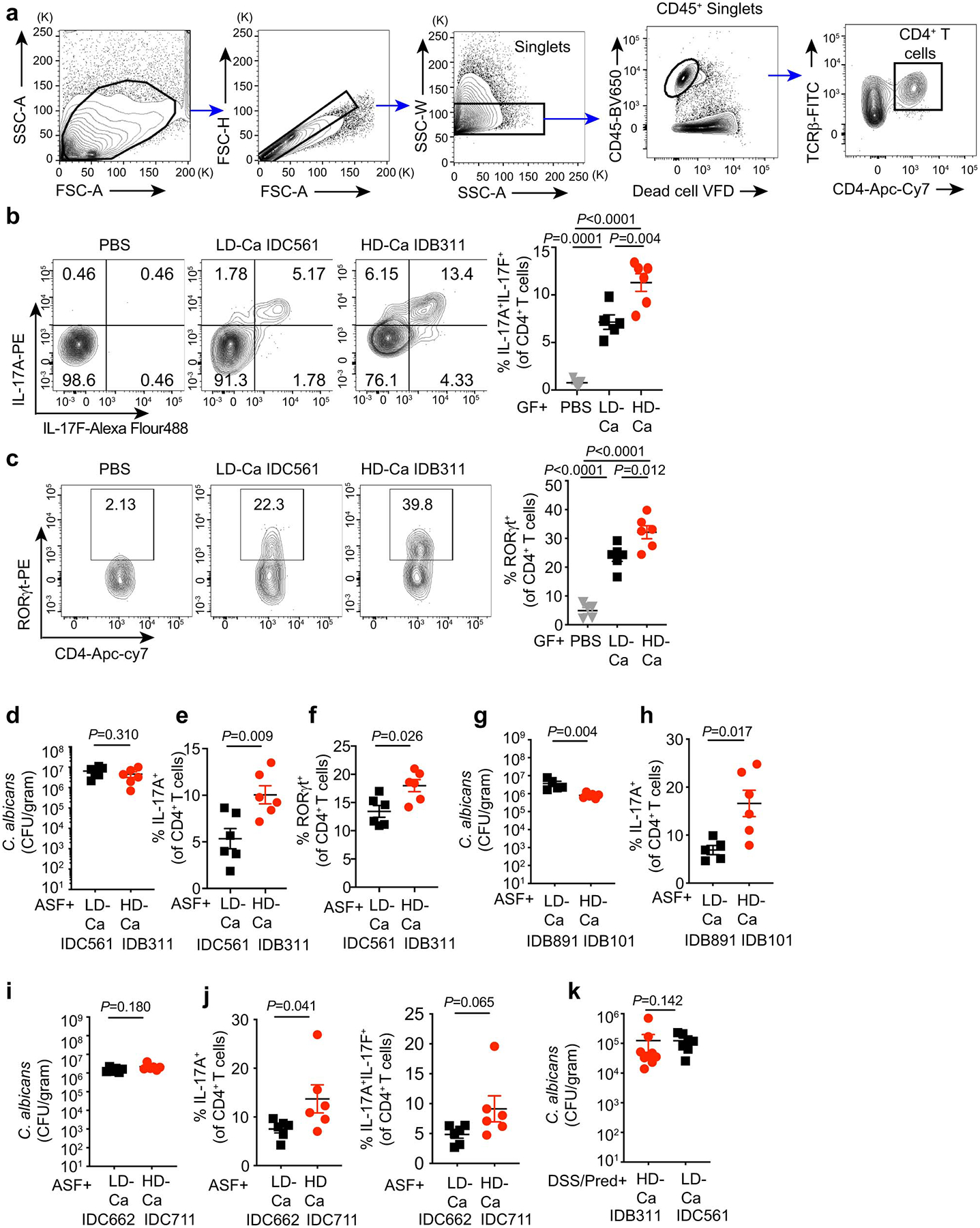
a-c, WT germ-free (GF) mice were colonized with C.a strains for three weeks. PBS (n=5), LD/C.a (IDC561, n=5), HD/C.a (IDB311, n=6). a, Gating strategy to analyze CD4+ T cells in colonic lamina propria cells. Representative flow cytometry plots and quantification of CD4+IL-17A+IL-17F+ (b, from left to right) and CD4+RORγt+ cells (c, from left to right) in the colon. d-f, ASF mice were colonized with C.a strains for three weeks. PBS (n=6), LD/C.a (IDC561, n=6), and HD/C.a (IDB311, n=6). d, C. albicans burden in the feces at day 21. e, Frequency of colonic CD4+IL-17A+ Th17 cells. f, Frequency of colonic CD4+RORγt+ cells. g-h, ASF mice were colonized with LD/C.a (IDB891, n=5) or HD/C.a (IDB101, n=6) for three weeks. g, C. albicans burden in the feces at day 21. h, Frequency of CD4+IL-17A+ Th17 cells in the colon were assessed. i-j, ASF mice were colonized with LD/C.a (IDC662, n=6) or HD/C.a (IDC711, n=6) for three weeks. i, C. albicans burden in the feces at day 21. j, Frequencies of CD4+IL-17A+ and CD4+IL-17A+ IL-17F+ Th17 cells in the colon were assessed. k, SPF WT mice were with C.a strains colonized and treated with prednisolone followed by DSS-induced murine colitis. PBS (n=10), HD/C.a IDB311 (n=9) and LD/C.a IDC561 (n=7). Fecal C. albicans burdens were assessed upon sacrifice. Results in b-k are shown as mean ± s.e.m. Each dot represents an individual mouse. Data in b-c are representative of three independent experiments with similar results. Data in d-k are representative of three independent experiments with similar results. Unpaired, two-tailed, Mann-Whitney test (d-k) or one-way ANOVA followed by the Tukey’s post hoc test (b and c).
Extended Data Fig. 6. EFG1-dependent candidalysin is required for cell damage in high-damaging C. albicans strains from the human gut.
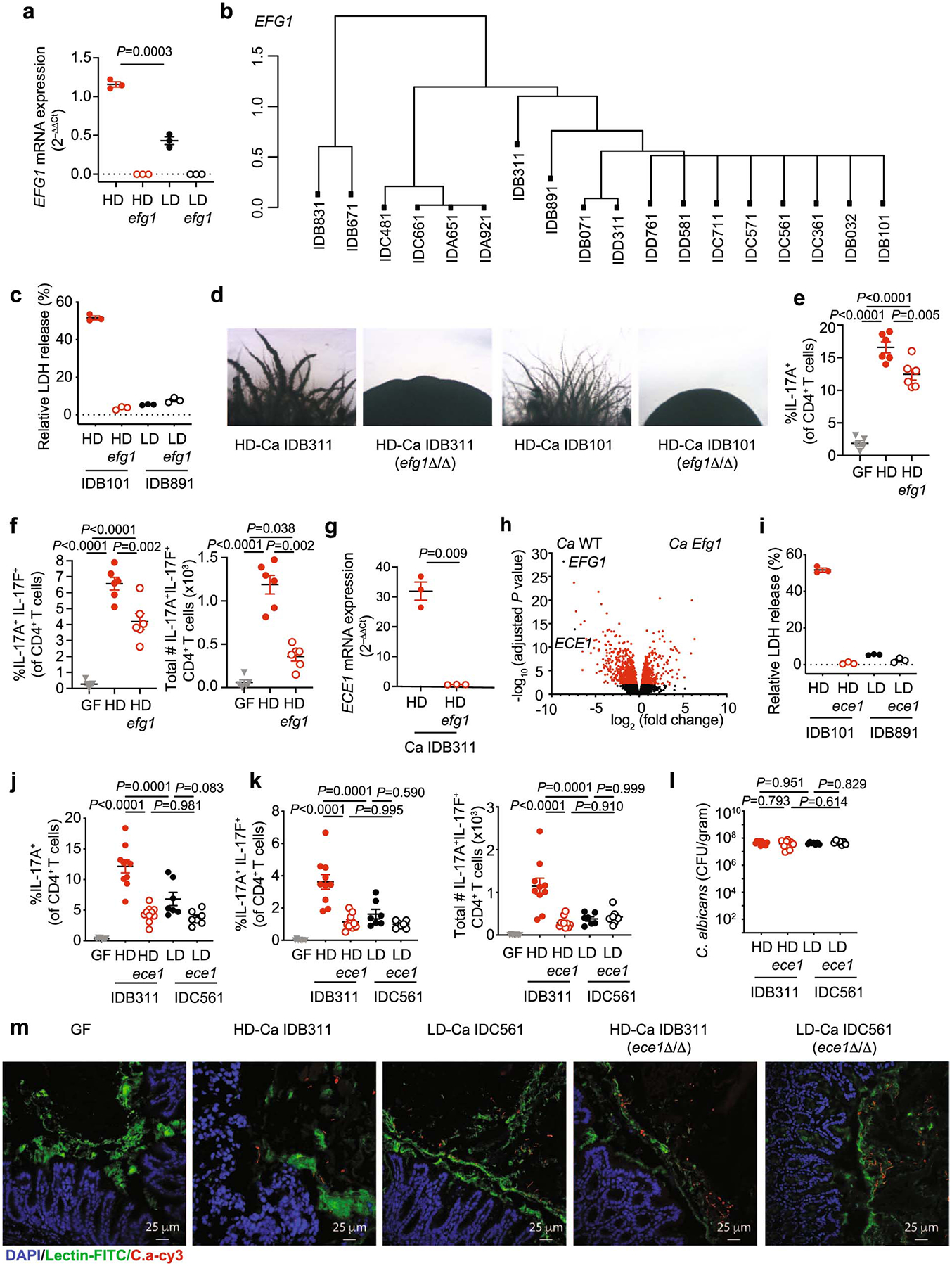
a, Caco2 cells were infected with live C. albicans for 12 hours, and both the EFG1 expression of C. albicans. HD/C.a IDB311 and LD/C.a IDC561 strains, and respective efg1Δ/Δ (efg1) strains were used in this experiment. Data shown as mean ± s.d. Data points indicate technical well replicates (n=3). Data are representative of three independent experiments with similar results. b, Dendrogram showing SNP-based distances between EFG1 sequences of isolates. Each isolate was obtained from an individual subject from the non-IBD (n=8) or UC patient (n=10) group. c, Caco2 cells were infected with C. albicans and LDH release of Caco2 cells was assessed. HD/C.a IDB101, HD/C.a IDB101 efg1Δ/Δ mutant (efg1), LD/C.a IDB891, and LD/C.a IDB891 efg1Δ/Δ mutant (efg1) were used. d, C. albicans HD/C.a IDB311, HD/C.a IDB101 and the respective EFG1 mutants were cultured on Spider agar at 37°C for 5 days followed by assessment of the filamentation phenotype. Data are representative of two independent experiments with same results. e-f, GF mice were colonized with PBS (GF, n=5), HD/C.a IDB311 (HD, n=6) or HD/C.a IDB311 efg1 (HD efg1, n=6) strains for three weeks. e, Frequencies of IL-17A+CD4+ T cells; f, Frequencies, and total cell numbers of IL-17A+IL-17F+CD4+ T cells, in the colon were assessed by flow cytometry. g, ECE1 gene expression in C. albicans cells were assessed upon EFG1 gene deletion. Data shown as mean ± s.d. Data points indicate technical well replicates (n=3). Data are representative of three independent experiments with similar results. h, Log2 transformed ratio of gene expression between EFG1 mutant and WT C. albicans strain upon colonization of the large intestine represented as a volcano plot. Data were obtained and analyzed from a recent resource dataset from Witchley et al. 2021. i, LDH release were assessed from Caco2 cells. HD/C.a IDB101, LD/C.a IDB891 and respective ECE1 mutant strains were used. Data shown as mean ± s.d. Data points indicate technical well replicates (n=3). Data are representative of three independent experiments with similar results. j-m, GF mice were colonized with C.a strains for three weeks. PBS GF (n=8), HD/C.a IDB311 (HD, n=10), HD ece1 (n=10), LD/C.a IDC561 (LD, n=7), and LD ece1 (n=7). j, Quantification of the frequencies of IL-17A+CD4+ T cells; k, Quantification of the frequencies, and total cell numbers of IL-17A+IL-17F+CD4+ T cells in the colon. l, Fecal C. albicans burden were measured at day 21. Each dot represents an individual mouse. m, HD and LD C. albicans strains both colonize the intestine and form a mixture of yeast and hyphal morphotypes. Fluorescence in situ hybridization (FISH) was utilized to visualize the morphology of C. albicans HD/C.a IDB311, LD/C.a IDC561, and respective ECE1 mutant strains (HD/C.a IDB311 ece1Δ/Δ and LD/C.a IDC561 ece1Δ/Δ) in the colon tissue after 21 days of mono-colonization of C. albicans. The nuclei of colonic epithelial cells were stained with DAPI (blue), colonic mucin was stained with a FITC- conjugated Ulex europaeus agglutinin (UEA-I, lectin) (green), and C. albicans was stained with a Cy3-coupled pan-fungal-specific probe (red). The colon tissue of germ-free mice was used as a control. The scale bar 25 μm. Results in e-f and j-l are shown as mean ± s.e.m.. Data in e-f are representative of three independent experiments with similar results. Data in j-m are representative of two independent experiments with similar results. Unpaired, two-tailed, Mann-Whitney test (a, g), One-way ANOVA followed by the Tukey’s post hoc test (e, f, j, k, l).
Extended Data Fig. 7. Genomic comparative analysis of genetic polymorphisms in CPH1, UME6, FLO8 and ECE1 among high- and low-damaging strains.
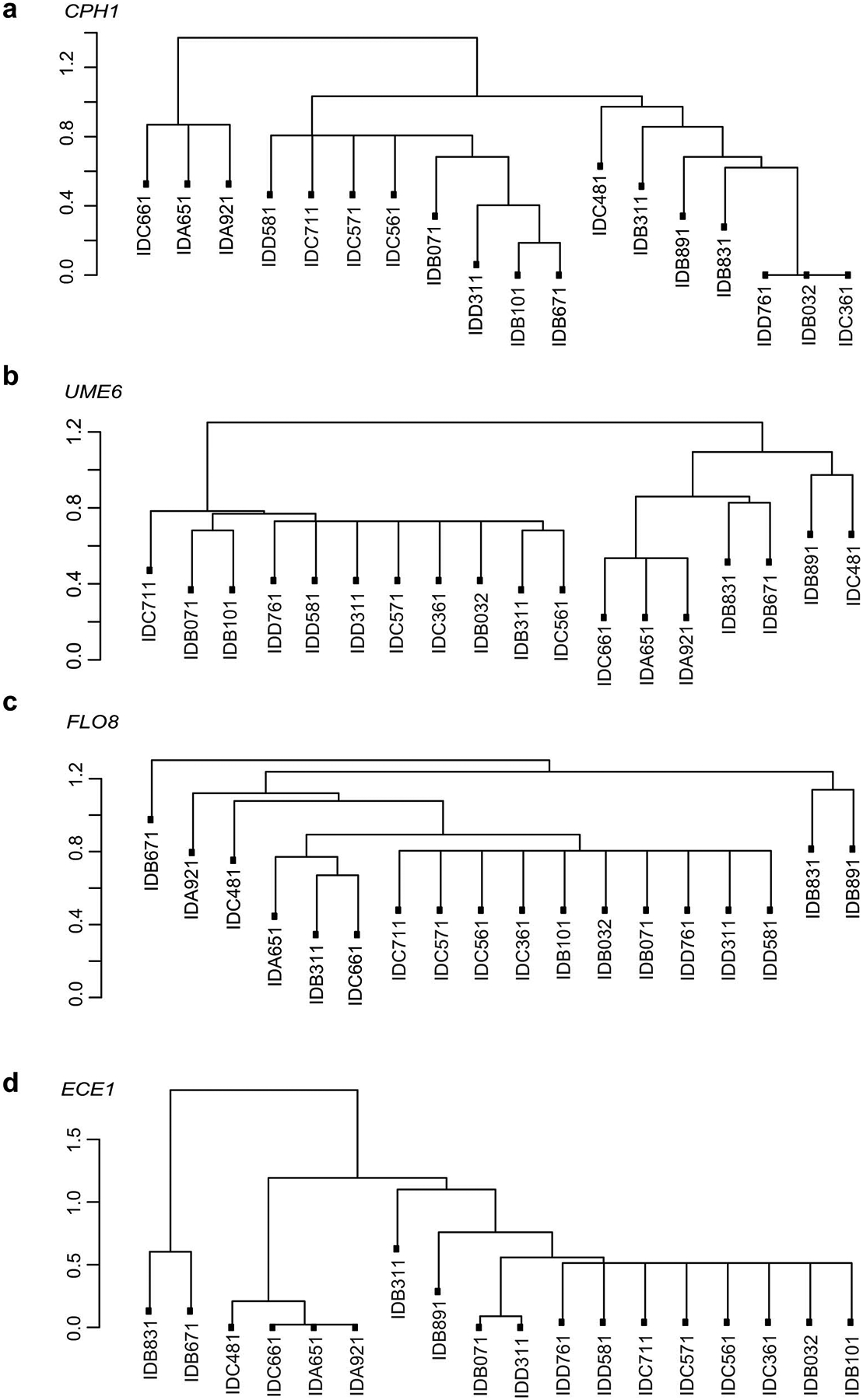
a-c, Dendrogram showing SNP-based distances between CPH1 (a), UME6 (b), FLO8 (c) and ECE1 (d) sequences of isolates. Each isolate was obtained from an individual subject from the non-IBD (n=8) or UC patient (n=10) group.
Extended Data Fig. 8. Gut C. albicans promotes intestinal pro-inflammatory immunity through candidalysin.
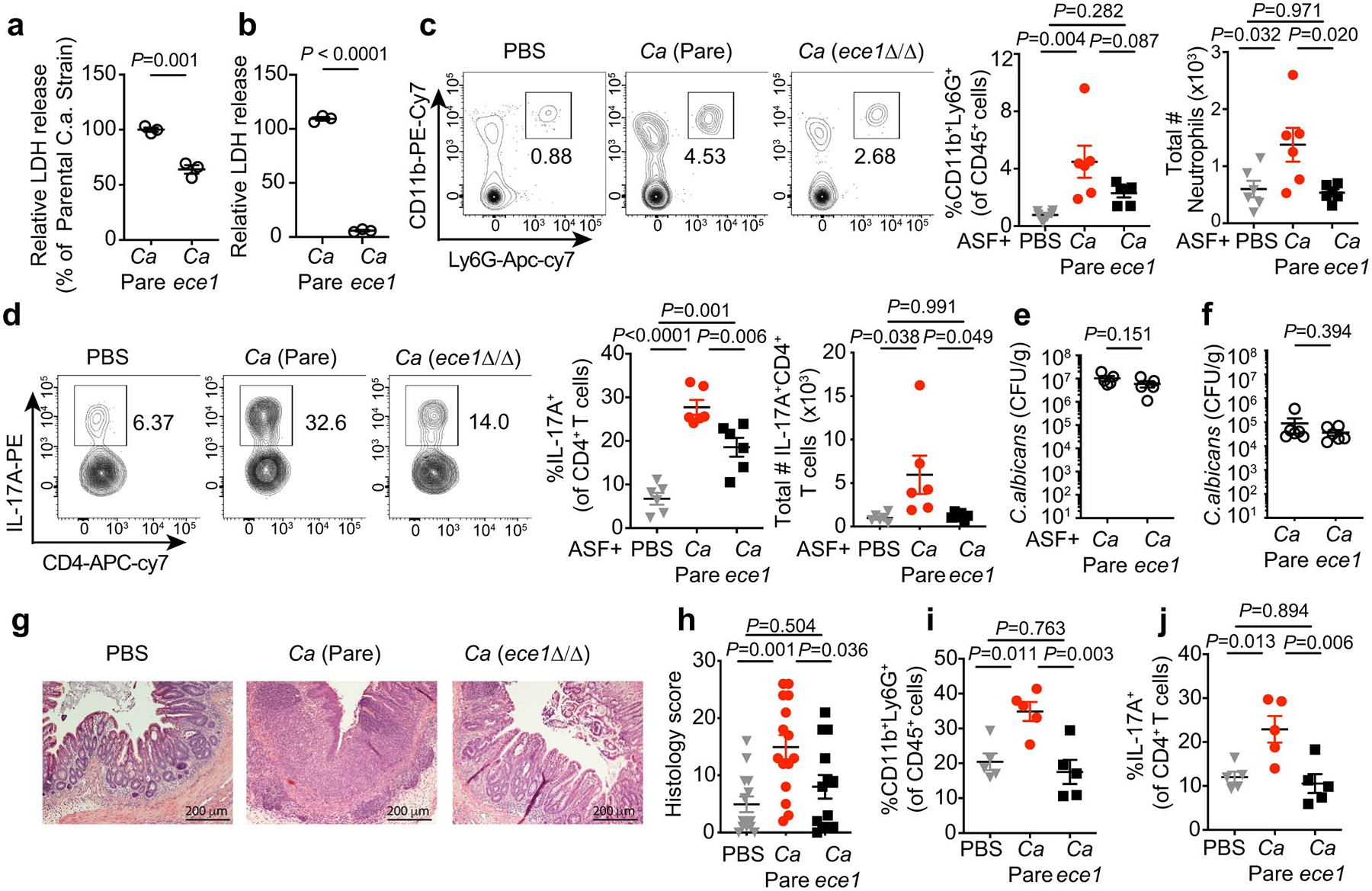
a-b, Cell damage by C. albicans. mBMDM (a) and Caco2 cells (b) were incubated with live C. albicans (MOI=5) wild-type parental (C.a Pare) or C. albicans ece1Δ/Δ (C.a ece1) strains for 16 hours and LDH release in the supernatants was measured. Results are shown as mean ± s.d. Data points indicate technical well replicates (n=3). Data are representative of three independent experiments. Unpaired, two-tailed, student test. c-e, ASF mice were colonized with C.a. Pare or C.a ece1 strains for three weeks. n=6 mice in each group. c-d, Frequency and total cell numbers of CD11b+Ly6G+ neutrophils (c, from left to right) and IL-17A+CD4+ T cells (d, from left to right) in the colon. e, Fecal C. albicans burdens were measured at day 21. f-j, WT SPF mice were colonized with or without C.a Pare or C.a ece1 strain, DSS colitis was induced followed by treatment with prednisolone. f, Fecal C. albicans burden were measured upon sacrifice. g, representative H&E colon section. The scale bar 200μm. h, Histology scores. PBS (n=14), C.a Pare (n=15), and C.a ece1 (n=13). Data are pooled from two independent experiment with similar results. i, Frequency of cLP neutrophils. j, IL-17A+CD4+ T cells were assessed upon sacrifice. Results in c-f and h-j are shown as mean ± s.e.m. Each dot represents an individual mouse. Data in c-f and i-j are representative of two independent experiments with similar results. Unpaired, two-tailed, Mann-Whitney test (e, f) or one-way ANOVA followed by the Tukey’s post hoc test (c, d, h, i, j).
Extended Data Fig. 9. C. albicans clonal expansion, and microevolution occur in the human gut.
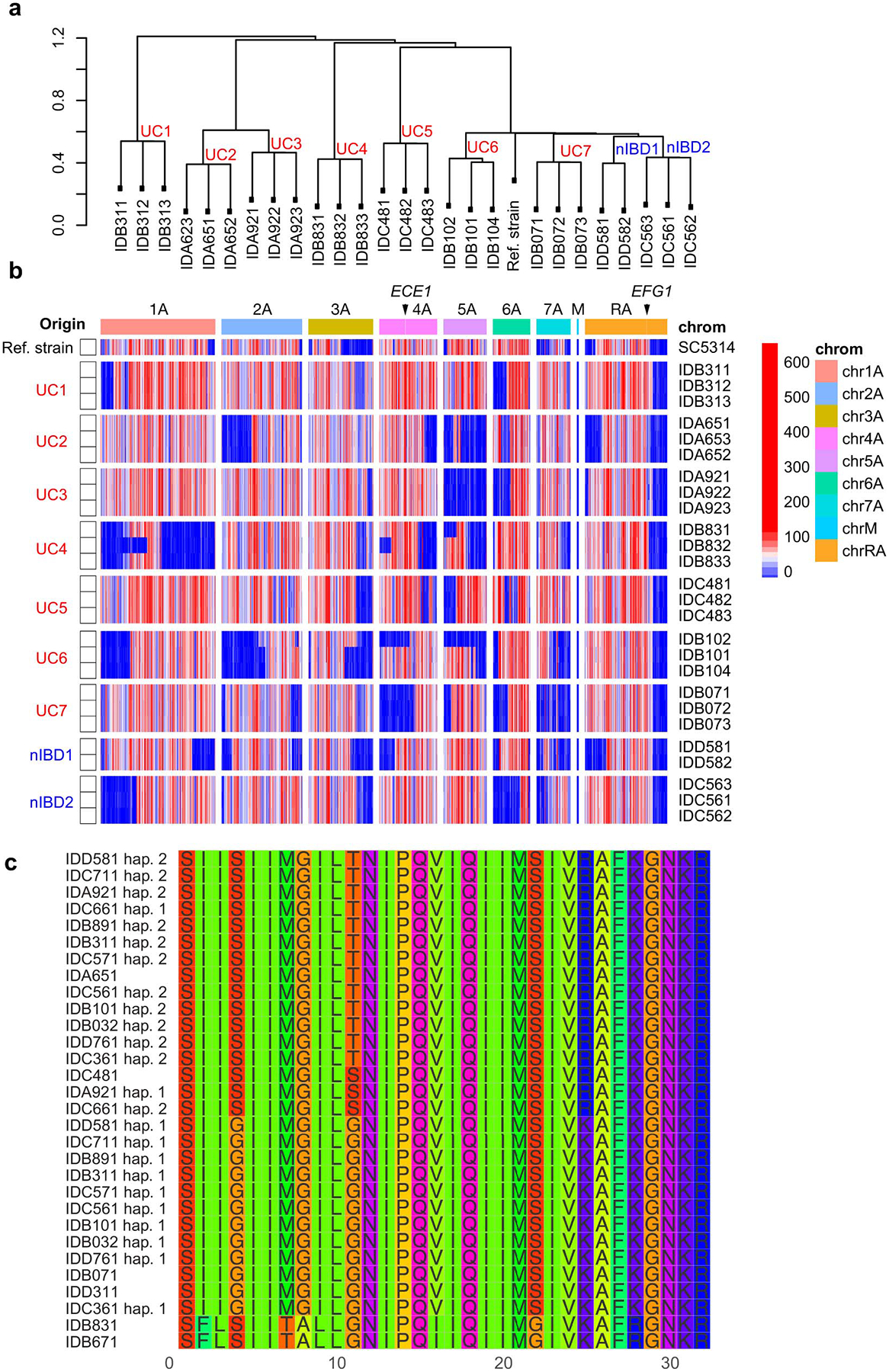
a, Dendrogram showing genome-wide SNP-based distances among two or three human gut C. albicans isolates obtained from the same individual subject. Strains isolated from non-IBD subjects (nIBD), UC patients (UC), and refence strain C. albicans SC5314 (Ref. Strain). b, Heatmap showing genome-wide density of heterozygous SNPs from two or three human gut C. albicans isolates obtained from the same individual subject. Strains isolated from non-IBD subjects (nIBD), UC patients (UC), and refence strain C. albicans SC5314 (Ref. Strain) are labeled. Color density indicates the number of heterozygous SNPs detected in each 10 kbp window of an isolate’s genome. Arrows point to the genomic locations of ECE1 and EFG1 genes. c, Multiple sequence alignment of candidalysin (SK1 peptide) amino acid sequences across multiple isolates of C. albicans. Each isolate was obtained from an individual subject from the non-IBD or UC patient group. Four isoforms of candidalysin across strains show no association/clustering of specific isoform with HD or LD strain. Two haplotypes are shown for isolates that are heterozygous in this region.
Extended Data Fig. 10. Immune mediators released by macrophage upon infection with gut -derived C. albicans strains.
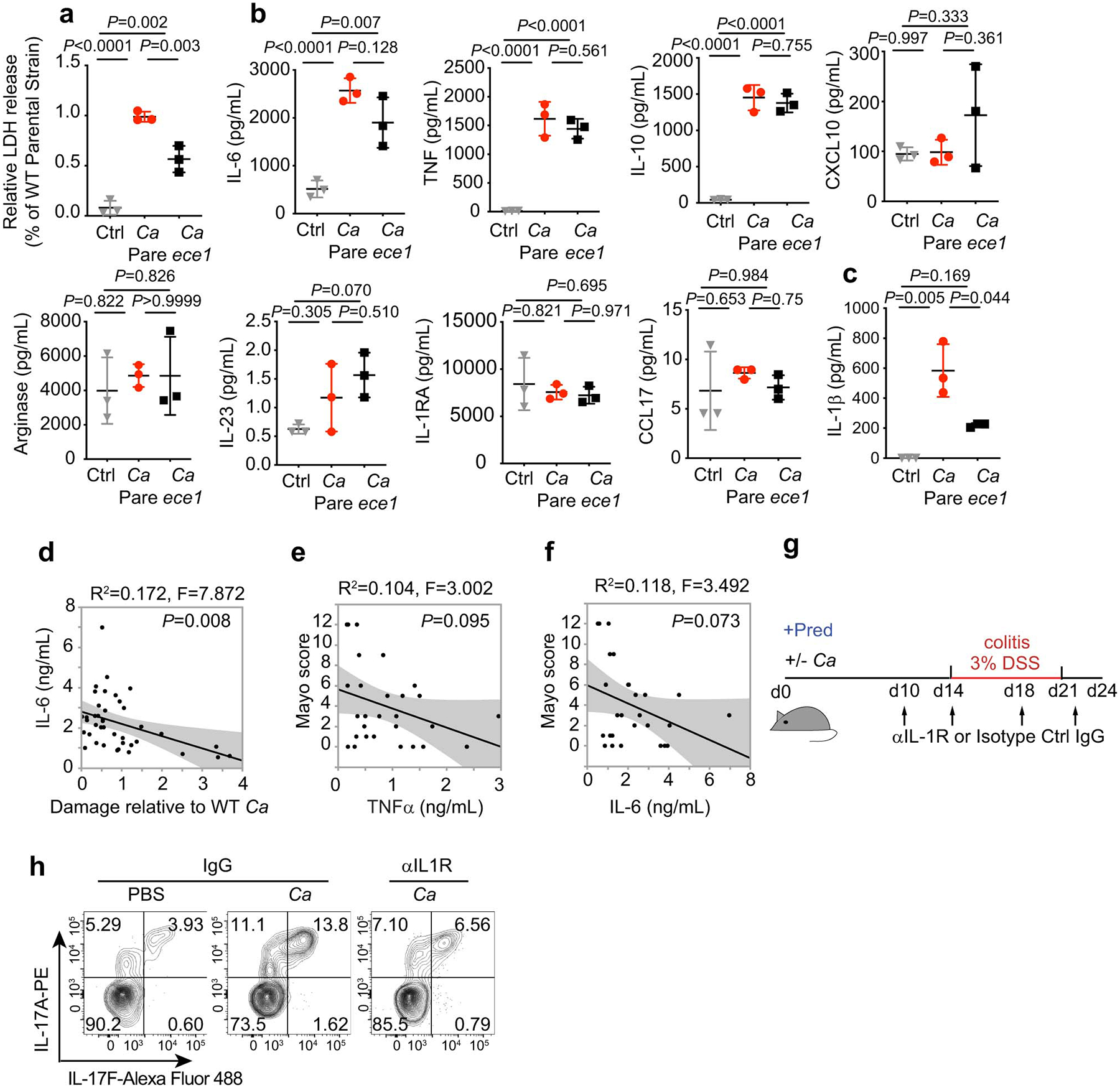
a, LDH release measured in culture supernatants of hMDM after infection with C. albicans parental strain (Pare), C.a ece1D/ece1/ D (C.a ece1) and an untreated group (Ctrl) for 16 hours. b, Macrophage-released mediators measured by cytometric bead assays from cultures in Extended Data Fig. 10a. c, IL-1β release measured in culture supernatants of LPS-primed hMDMs infected with C. albicans parental strain, C.a ece1, and in uninfected group. Parallel experiments were performed with a. Results a-c are shown as mean ± s.d. Data are representative of three independent experiments with similar results. d-f, Cytokine release in culture supernatants of unprimed human monocyte-derived macrophages (hMDMs) after incubation with live gut derived-C. albicans isolates (MOI=5). d, IL-6 cytokine production was measured by ELISA. hMDM damage measured (LDH assay) in the same experiment was correlated with specific cytokine release. e-f, Correlation between TNF-α and IL-6 cytokine from hMDM induced by patient-specific gut C. albicans and Mayo score in corresponding UC patients (n=10). Dot is shown as an average value of three technical repeats, Data are representative of three independent experiments with similar results. The simple linear regression was performed, where P-value calculated by a F test. g-h, Mice colonized with or without HD/C.a IBD311 were treated with prednisolone (Pred) followed by DSS-mediated induction of colitis. Each mouse further treated with 1 mg anti-IL-1R1 IgG (αIL1R) or isotype IgG (IgG) at the time point indicated in the schematic figure of experimental layout (g). IgG+PBS (n=8), IgG+C.a (n=9) and αIL-1R1+C.a (n=7). h, Representative flow cytometry plots of IL-17A+IL-17F+CD4+ T cells. Data in d-h are representative of three independent experiments with similar results. One-way ANOVA followed by the Tukey’s post hoc test (a-c).
Supplementary Material
Acknowledgments
We would like to thank members of the Iliev laboratory for helpful suggestions related to the manuscript, Wenbing Jin and Chunjun Guo who generously shared cloning reagents and equipment, Gerald Fink, Valmik K Vyas and Douglas A Bernstein for providing plasmids and protocols and all contributing members of the JRI IBD Live Cell Bank Consortium, and the Microbiome Core Laboratory of Weill Cornell Medicine. Support for sample acquisition through the JRI IBD Live Cell Bank is provided by the JRI, Jill Roberts Center for IBD, Cure for IBD, the Rosanne H. Silbermann Foundation and Weill Cornell Medicine Division of Pediatric Gastroenterology and Nutrition. X.V.L. is supported by fellowship from the Charles H. Revson Foundation. I.L. is supported by fellowships from the Crohn’s and Colitis Foundation (568319). W.D.F. is supported by a fellowship from the NIH (F32DK120228). J.R.N is supported by the Wellcome Trust (214229_Z_18_Z), NIH (R37-DE022550), and IS-BRC-1215-20006. BH is supported by the German Research Foundation (Deutsche Forschungsgemeinschaft, DFG) within the Cluster of Excellence “Balance of the Microverse”, under Germany’s Excellence Strategy – EXC 2051 – Project-ID 390713860. Research in the Iliev laboratory is supported by US National Institutes of Health (R01DK113136, R01DK121977, R21AI146957 and R01AI163007), the Irma T. Hirschl Career Scientist Award, Kenneth Rainin Foundation, Crohn’s and Colitis Foundation, the Leona M. and Harry B. Helmsley Charitable Trust and the Burrough Welcome Trust PATH Award.
Footnotes
Competing interests
Cornell University has filed a provisional patent application covering inventions described in this manuscript. The authors declare no other competing interests.
Supplementary Information includes Extended Data Figure 1–10, and Supplementary Table 1–4.
Data availability.
ITS sequencing data are publicly available in NCBI Sequence Read Archive (SRA) under the Bioproject ID PRJNA610042 (https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA610042). The data from whole-genome sequencing of human gut-derived C. albicans isolates are publicly available in the NCBI Sequence Read Archive (SRA) under Bioproject ID PRJNA702809 (https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA702809). Raw sequencing data for representative C. albicans strains were downloaded from the NCBI Sequence Read Archive under BioProject ID PRJNA432884 (https://www.ncbi.nlm.nih.gov/bioproject?term=PRJNA432884)43. Other data necessary to evaluate the manuscript are provided whithn the main text or whitin the supplementaty data associated with this manuscript. Source data for Figures 1–4 and Extended Data Figures 2–6, 8 and 10 are provided with this paper.
References:
- 1.Human Microbiome Project, C. Structure, function and diversity of the healthy human microbiome. Nature 486, 207–14 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lloyd-Price J et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature 569, 655–662 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chehoud C et al. Fungal Signature in the Gut Microbiota of Pediatric Patients With Inflammatory Bowel Disease. Inflammatory bowel diseases 21, 1948–56 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hoarau G et al. Bacteriome and Mycobiome Interactions Underscore Microbial Dysbiosis in Familial Crohn’s Disease. mBio 7, e01250–16 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liguori G et al. Fungal Dysbiosis in Mucosa-associated Microbiota of Crohn’s Disease Patients. Journal of Crohn’s & colitis 10, 296–305 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sokol H et al. Fungal microbiota dysbiosis in IBD. Gut 66, 1039–1048 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ott SJ et al. Fungi and inflammatory bowel diseases: Alterations of composition and diversity. Scand J Gastroenterol 43, 831–41 (2008). [DOI] [PubMed] [Google Scholar]
- 8.Limon JJ et al. Malassezia Is Associated with Crohn’s Disease and Exacerbates Colitis in Mouse Models. Cell host & microbe (2019) doi: 10.1016/j.chom.2019.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jain U et al. Debaryomyces is enriched in Crohn’s disease intestinal tissue and impairs healing in mice. Science 371, 1154–1159 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kaplan GG The global burden of IBD: from 2015 to 2025. Nature reviews. Gastroenterology & hepatology 12, 720–7 (2015). [DOI] [PubMed] [Google Scholar]
- 11.Israeli E et al. Anti-Saccharomyces cerevisiae and antineutrophil cytoplasmic antibodies as predictors of inflammatory bowel disease. Gut 54, 1232–6 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lewis JD et al. Inflammation, Antibiotics, and Diet as Environmental Stressors of the Gut Microbiome in Pediatric Crohn’s Disease. Cell host & microbe 18, 489–500 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schaffer T et al. Anti-Saccharomyces cerevisiae mannan antibodies (ASCA) of Crohn’s patients crossreact with mannan from other yeast strains, and murine ASCA IgM can be experimentally induced with Candida albicans. Inflammatory bowel diseases 13, 1339–46 (2007). [DOI] [PubMed] [Google Scholar]
- 14.Standaert-Vitse A et al. Candida albicans colonization and ASCA in familial Crohn’s disease. Am J Gastroenterol 104, 1745–53 (2009). [DOI] [PubMed] [Google Scholar]
- 15.Doron I et al. Human gut mycobiota tune immunity via CARD9-dependent induction of anti-fungal IgG antibodies. Cell 184, 1017–1031.e14 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leonardi I et al. CX3CR1(+) mononuclear phagocytes control immunity to intestinal fungi. Science 359, 232–236 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cohen R, Roth FJ, Delgado E, Ahearn DG & Kalser MH Fungal flora of the normal human small and large intestine. The New England journal of medicine 280, 638–41 (1969). [DOI] [PubMed] [Google Scholar]
- 18.Sovran B et al. Enterobacteriaceae are essential for the modulation of colitis severity by fungi. Microbiome 6, 152 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Danese S & Fiocchi C Ulcerative Colitis. N Engl J Med 365, 1713–1725 (2011). [DOI] [PubMed] [Google Scholar]
- 20.Fan D et al. Activation of HIF-1alpha and LL-37 by commensal bacteria inhibits Candida albicans colonization. Nature medicine 21, 808–14 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jawhara S et al. Colonization of mice by Candida albicans is promoted by chemically induced colitis and augments inflammatory responses through galectin-3. J Infect Dis 197, 972–80 (2008). [DOI] [PubMed] [Google Scholar]
- 22.Marakalala MJ et al. Differential adaptation of Candida albicans in vivo modulates immune recognition by dectin-1. PLoS pathogens 9, e1003315 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hirakawa MP et al. Genetic and phenotypic intra-species variation in Candida albicans. Genome Res 25, 413–25 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schonherr FA et al. The intraspecies diversity of C-albicans triggers qualitatively and temporally distinct host responses that determine the balance between commensalism and pathogenicity. Mucosal Immunol 10, 1335–1350 (2017). [DOI] [PubMed] [Google Scholar]
- 25.Forche A et al. Selection of Candida albicans trisomy during oropharyngeal infection results in a commensal-like phenotype. PLoS Genet 15, e1008137 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tso GHW et al. Experimental evolution of a fungal pathogen into a gut symbiont. Science 362, 589–595 (2018). [DOI] [PubMed] [Google Scholar]
- 27.Kasper L et al. The fungal peptide toxin Candidalysin activates the NLRP3 inflammasome and causes cytolysis in mononuclear phagocytes. Nature communications 9, 4260 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wellington M, Koselny K, Sutterwala FS & Krysan DJ Candida albicans triggers NLRP3-mediated pyroptosis in macrophages. Eukaryot Cell 13, 329–40 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Uwamahoro N et al. The Pathogen Candida albicans Hijacks Pyroptosis for Escape from Macrophages. mBio 5, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moyes DL et al. Candidalysin is a fungal peptide toxin critical for mucosal infection. Nature 532, 64–8 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Verma AH et al. Oral epithelial cells orchestrate innate type 17 responses to Candida albicans through the virulence factor candidalysin. Science immunology 2, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Naglik JR, Gaffen SL & Hube B Candidalysin: discovery and function in Candida albicans infections. Curr Opin Microbiol 52, 100–109 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pierce JV & Kumamoto CA Variation in Candida albicans EFG1 expression enables host-dependent changes in colonizing fungal populations. mBio 3, e00117–12 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Witchley JN et al. Candida albicans Morphogenesis Programs Control the Balance between Gut Commensalism and Invasive Infection. Cell host & microbe 25, 432–443 e6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li X et al. Response to Fungal Dysbiosis by Gut-Resident CX3CR1(+) Mononuclear Phagocytes Aggravates Allergic Airway Disease. Cell host & microbe 24, 847–856 e4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rohde CM et al. Metabonomic evaluation of Schaedler altered microflora rats. Chem Res Toxicol 20, 1388–92 (2007). [DOI] [PubMed] [Google Scholar]
- 37.Schaedler RW, Dubos R & Costello R The Development of the Bacterial Flora in the Gastrointestinal Tract of Mice. The Journal of experimental medicine 122, 59–66 (1965). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Witchley JN, Basso P, Brimacombe CA, Abon NV & Noble SM Recording of DNA-binding events reveals the importance of a repurposed Candida albicans regulatory network for gut commensalism. Cell Host & Microbe 29, 1002–1013.e9 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vyas VK et al. New CRISPR Mutagenesis Strategies Reveal Variation in Repair Mechanisms among Fungi. mSphere 3, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vyas VK, Barrasa MI & Fink GR A Candida albicans CRISPR system permits genetic engineering of essential genes and gene families. Sci Adv 1, e1500248 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Selmecki A, Forche A & Berman J Genomic plasticity of the human fungal pathogen Candida albicans. Eukaryot Cell 9, 991–1008 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.MacCallum DM et al. Property differences among the four major Candida albicans strain clades. Eukaryot Cell 8, 373–87 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ropars J et al. Gene flow contributes to diversification of the major fungal pathogen Candida albicans. Nature communications 9, 2253 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Butler G et al. Evolution of pathogenicity and sexual reproduction in eight Candida genomes. Nature 459, 657–62 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Drummond RA et al. CARD9(+) microglia promote antifungal immunity via IL-1beta- and CXCL1-mediated neutrophil recruitment. Nature immunology 20, 559–570 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shouval DS et al. Interleukin 1beta Mediates Intestinal Inflammation in Mice and Patients With Interleukin 10 Receptor Deficiency. Gastroenterology 151, 1100–1104 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Friedrich M et al. IL-1-driven stromal-neutrophil interaction in deep ulcers defines a pathotype of therapy non-responsive inflammatory bowel disease. http://biorxiv.org/lookup/doi/10.1101/2021.02.05.429804 (2021) doi: 10.1101/2021.02.05.429804. [DOI] [Google Scholar]
- 48.Mogavero S et al. Candidalysin delivery to the invasion pocket is critical for host epithelial damage induced by Candida albicans. Cellular Microbiology 23, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Friedrich M et al. IL-1-driven stromal–neutrophil interactions define a subset of patients with inflammatory bowel disease that does not respond to therapies. Nat Med (2021) doi: 10.1038/s41591-021-01520-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Longman RS et al. CX(3)CR1(+) mononuclear phagocytes support colitis-associated innate lymphoid cell production of IL-22. The Journal of experimental medicine 211, 1571–83 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hepworth MR et al. Immune tolerance. Group 3 innate lymphoid cells mediate intestinal selection of commensal bacteria-specific CD4(+) T cells. Science 348, 1031–5 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tang J, Iliev ID, Brown J, Underhill DM & Funari VA Mycobiome: Approaches to analysis of intestinal fungi. J Immunol Methods 421, 112–121 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Altschul SF, Gish W, Miller W, Myers EW & Lipman DJ Basic local alignment search tool. J Mol Biol 215, 403–10 (1990). [DOI] [PubMed] [Google Scholar]
- 54.Li H & Durbin R Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–60 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McKenna A et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20, 1297–303 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zheng X et al. A high-performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics 28, 3326–8 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gu Z, Gu L, Eils R, Schlesner M & Brors B circlize Implements and enhances circular visualization in R. Bioinformatics 30, 2811–2 (2014). [DOI] [PubMed] [Google Scholar]
- 58.Danecek P et al. Twelve years of SAMtools and BCFtools. Gigascience 10, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Quinlan AR & Hall IM BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bankevich A et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19, 455–477 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wick RR, Schultz MB, Zobel J & Holt KE Bandage: interactive visualization of de novo genome assemblies. Bioinformatics 31, 3350–3352 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
ITS sequencing data are publicly available in NCBI Sequence Read Archive (SRA) under the Bioproject ID PRJNA610042 (https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA610042). The data from whole-genome sequencing of human gut-derived C. albicans isolates are publicly available in the NCBI Sequence Read Archive (SRA) under Bioproject ID PRJNA702809 (https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA702809). Raw sequencing data for representative C. albicans strains were downloaded from the NCBI Sequence Read Archive under BioProject ID PRJNA432884 (https://www.ncbi.nlm.nih.gov/bioproject?term=PRJNA432884)43. Other data necessary to evaluate the manuscript are provided whithn the main text or whitin the supplementaty data associated with this manuscript. Source data for Figures 1–4 and Extended Data Figures 2–6, 8 and 10 are provided with this paper.