

Abstract
Altered RNA expression of repetitive sequences and retrotransposition are frequently seen in colorectal cancer (CRC) implicating a functional importance of repeat activity in cancer progression. We show the nucleoside reverse transcriptase inhibitor 3TC targets activities of these repeat elements in CRC pre-clinical models with a preferential effect in P53 mutant cell lines linked with direct binding of P53 to repeat elements. We translate these findings to a human Phase 2 trial of single agent 3TC treatment in metastatic CRC with demonstration of clinical benefit in 9 of 32 patients. Analysis of 3TC effects on CRC tumorspheres demonstrates accumulation of immunogenic RNA:DNA hybrids linked with induction of interferon response genes and DNA damage response. Epigenetic and DNA damaging agents induce repeat RNAs and have enhanced cytotoxicity with 3TC. These findings identify a vulnerability in CRC by targeting the viral mimicry of repeat elements.
Keywords: Repeat RNA, Colorectal Cancer, Reverse Transcription
INTRODUCTION
The repeat transcriptome or “repeatome” consists of the expression of a variety of repeat RNA species including long interspersed nuclear element-1 (LINE-1) retrotransposons, human endogenous retroviruses (HERVs), and satellite repeats. In normal development, repeat RNAs are expressed in the setting of embryogenesis (1,2) and are epigenetically suppressed in differentiated tissues. They are awakened in the setting of tumorigenesis and aberrantly expressed in a spectrum of cancers (3–5), which has been shown to engage the innate (6–9) and adaptive (10–12) immune response through pathogen-associated sequence features found in immunostimulatory viral RNA and DNA (13). Analogous to retroviruses, repeat RNAs are also frequently reverse transcribed into DNA (14–16) that can alter the distribution and composition of these immunostimulatory nucleic acids. The presence of repeat DNA products from reverse transcription (RT) can provide a source of cytosolic DNA in cancer cells that has been shown to lead to increased metastatic behavior in cancers through cGAS-STING activation (17,18). This suggests that inhibition of the reverse transcriptase activity of these retroelements with nucleoside reverse transcriptase inhibitors (NRTIs) (16,19,20), a class of agents commonly used in HIV and HBV, would affect tumor progression and metastatic dissemination. Our prior work demonstrated the anti-cancer effects of the NRTIs ddC and d4T in vitro and in vivo in colorectal cancer (CRC) cell lines (16), but these early NRTIs are known to have significant side effects in patients. Looking towards clinical feasibility of repeat RT inhibition, we chose lamivudine (3TC) given previous literature demonstrating potency against endogenous RT activity (19–23) and the well-established clinical tolerability in HIV and HBV patients unlike ddC and d4T. Here, we present the use of 3TC in preclinical functional CRC models, mechanistic studies to understand the effects of 3TC on cytosolic DNA, and the first clinical trial of 3TC in metastatic CRC patients.
RESULTS
3TC Functional Effects on CRC Pre-clinical Models
We evaluated 10 CRC cell lines (DLD1, HCT15, HT29, C2BBe1, LS123, SW948, HCT8, HCT116, LOVO, RKO) with 3TC in transwell migration and in soft agar colony formation as in vitro functional assays of motility and anoikis resistance that are essential components of the metastatic cascade. Boyden chamber transwell migration assays (Fig. 1A) noted marked effects of 3TC on migratory capability in DLD1, HCT15, C2Bbe1, LS123, and RKO cell lines (Fig. 1B-E and Supplementary Fig. S1A-B). Notably, we found that 3TC functional effects on migration were more prevalent amongst P53-Mutant (P53-Mut: DLD1, HCT15, HT29, C2BBe1, LS123, SW948) compared to P53-Wild Type (P53-WT: HCT8, HCT116, LOVO, RKO) cell lines. Similarly, treatment with 3TC demonstrated significant reduction in anchorage independent growth in soft agar in P53-Mut cell lines (Fig. 1F-H and Supplementary Fig. S2A-B) compared to P53-WT cell lines (Fig. 1I-K). We extended these in vitro findings to xenograft tumors using a sensitive (SW620) and resistant (HCT116) cell line. The P53-Mut SW620 xenograft demonstrated significant response to 3TC alone (ANOVA p value <0.0001) compared to PBS vehicle control treated tumors (Fig. 1L), while the P53-WT HCT116 xenograft did not respond to single agent 3TC (Fig. 1M).
Figure 1. Reverse transcriptase inhibitor 3TC has preclinical efficacy in colorectal cancer.

A, Schema illustrating experimental procedure for measuring cell migration. B-E, Colon cancer cell line migration data with representative images and quantification of area covered by stained cells in (B, C) P53-Mut and (D, E) P53-WT colon cancer cell lines treated with DMSO or 3TC across 8-μm pore Transwell after 24~72 hr following fixation and staining with crystal violet. Statistical significance calculated by student’s two tailed t-test: * p < 0.05, ** p < 0.01, *** p < 0.001. F-K, Response to 3TC vs DMSO control in soft agar colony assay in (F-H) P53-Mut cell lines SW620 and DLD1 and (I-K) P53-WT cell lines HCT116 and HCT8. (F, I) Representative images of soft agar colonies of SW620 and HCT116 (left) with image quantification markup (right). Scale bar = 2 mm (left), 1 mm (right). Quantification of colony size by digital image analysis shown with violin plot with median and interquartile range in (G-H) P53-Mut cells and (J-K) P53-WT cells treated with DMSO or 3TC at 5 µM and 100 µM. Statistical significance calculated by student’s two tailed t-test: * p < 0.05, ** p < 0.01, *** p < 0.001. L-M, P53-Mut (SW620) and P53-WT (HCT116) CRC xenograft tumors treated with 3TC vs PBS (Control) treatment. Luciferase-expressing (L) SW620 or (M) HCT116 cells were subcutaneously implanted in immunocompromised Nu/Nu mice and grown for 2 weeks after which mice were treated with PBS or 3TC at 50 mg/kg administered by intraperitoneal injection 3 times a week. Tumor luminescence was measured using IVIS imaging every 5 days. Graph represents relative luminescence units (RLU) normalized to Day 0. Significance determined by two-way ANOVA test: **** p < 0.0001.
P53 is a Direct Repressor of Repeat RNA Expression
Given the observed differential response to 3TC based on P53 mutation status, we wanted to better understand the relationship of repeat RNA expression levels with P53. We evaluated if P53 has direct interactions with genomic repeats, given the association of P53 mutation or loss with de-repression of repeat RNA expression (6,7,24) and LINE-1 retrotransposition (15,25). We performed P53 immunoprecipitation followed by DNA sequencing (P53-IP-seq) in P53-Mut (SW620, DLD1) and P53-WT (HCT116, HCT8) cell lines done in biological duplicates (Fig. 2A-C, Supplementary Fig. S3A-B, and Data S1 and S2). Differential enrichment analysis of P53 bound repeat elements (FDR < 0.2) demonstrated markedly different proportions of specific repeat classes with notable higher satellite (SAT 34% of repeats) and LINE-1 (L1 31% of repeats) elements in P53-WT compared to P53-Mut cell lines (Fig. 2C). Notably, the HSATII satellite repeat was enriched, which we have previously shown to be reverse transcribed in CRC (16). Both P53-Mut cell lines have DNA binding domain mutations (SW620 - R273H; DLD1 - S241F), which suggests loss of function of DNA binding is associated with diminished P53 interaction with these repeat DNA sequences. Total RNA-seq of CRC cell lines (n=3 biological replicates) comparing P53-Mut (SW620, DLD1) and P53-WT (HCT116, HCT8) cells noted significant differential expression of the L1PA2 element (Fig. 2D and Supplementary Fig. S3B) that was also seen in ChIP-seq analysis indicating this particular L1 repeat subfamily is directly regulated by P53. The major active human LINE-1 retrotransposon (L1HS) was also expressed at significantly higher levels in P53-Mut cell lines (Fig. 2D), though we did not see differential enrichment from P53-IP sequencing. To use an alternative method to quantify HSATII repeat expression given the potential heterogeneity of expression within each cell line, we used RNA in situ hybridization (RNA-ISH) and utilized quantitative digital image analysis of multiple areas of each cell line (Fig. 2E). The HSATII repeat had significantly higher expression in the P53-Mut cell lines (Fig. 2E and Supplementary Fig. S4A-B). Dysregulation of SAT and L1 repeats were previously shown to be highly expressed in a broad set of cancers (3,6,26), and the higher expression of HSATII and L1PA2 paired with loss of P53 binding in these cell lines is consistent with a direct suppression of these repeats by P53. In addition, the active L1HS retrotransposon was significantly higher in P53-Mut cell lines, but there was no indication of changes in enrichment in P53-IP-seq analysis. This suggests L1HS de-repression is due to secondary effects of a P53 mutation. Although we cannot rule out potential P53 mutation gain of function effects (27) on repeat expression, the loss of DNA binding to repeat sequences in P53-Mut cell lines would point towards a loss of function effect of P53 on repeat expression including L1HS. Overall, these findings in CRC are consistent with other models demonstrating a direct role of P53 in regulating repeat RNAs (24,28), which would indicate P53 mutation is a potential genetic marker of CRC tumors sensitive to 3TC.
Figure 2. P53 is a direct repressor of Repeat RNA expression.

A, Schematic demonstrating P53-ChIP-seq conducted on colorectal cancer tumorspheres to identify repeatome P53 binding. B, Distribution of significantly enriched repeats (FDR < 0.2) from P53 ChIP-seq in P53-Mut and P53-WT cell lines (SAT = satellite, L1 = LINE-1, ERV = endogenous retrovirus). P53-WT cells show significantly enriched P53 binding to repeat elements compared to P53-Mut cells. Statistical significance is calculated by Chi-squared test. C, Differential expression of repetitive genomic elements measured by RNA-seq of P53-Mut (SW620, DLD1) vs P53-WT (HCT116, HCT8) tumorspheres grown for 14 days represented as volcano plot (y-axis -Log10(p-value) and x-axis Log2(Fold difference)). Highlighted are satellite (SAT), LINE, and ERV repeats. Biological triplicates of RNA-seq were used for each cell line. D, Baseline levels of LINE element RNA L1PA2 (top), and L1HS (bottom) in P53-Mut, and P53-WT cell lines as measured by RNA-seq. Significance determined by student’s two tailed t-test with Welch’s correction calculated pairwise individually for HCT8 and HCT116 cells compared to SW620 and DLD1: * p < 0.05, ** p < 0.01, **** p < 0.0001. Red stars indicate significant difference compared to SW620. Black stars indicate significant difference compared to DLD1. E, HSATII RNA expression in P53-Mut (SW620, DLD1) and P53-WT (HCT8, HCT116) tumorspheres grown for 14 days, measured by RNA in situ hybridization (RNA-ISH). Signal is quantified as percentage of HSATII RNA positive signal per area in tumorspheres across 20 fields. Significance determined by student’s two tailed t-test with Welch’s correction calculated pairwise individually for HCT8 and HCT116 cells compared to SW620 and DLD1: * p < 0.05, ** p < 0.01, **** p < 0.0001. Red stars indicate significant difference compared to SW620. Black stars indicate significant difference compared to DLD1.
Phase 2 Clinical Trial of 3TC in Metastatic CRC
These preclinical data provided evidence to support the initiation of a single-arm Phase 2 clinical trial (NCT03144804) of 3TC in patients with P53 mutant metastatic CRC. Eligibility included patients with TP53 mutant refractory mCRC with progression on or intolerance to 5FU, oxaliplatin and irinotecan and anti-EGFR therapy if RAS WT. Eligible patients were age ≥18 years, with histologically or cytologically documented, advanced (metastatic and/or unresectable) disease that was incurable and had disease progressed on at least two-lines of systemic therapy (Fig. 3A and Supplementary Tables S1-3). See methods for details of inclusion and exclusion criteria. Primary endpoint of the study was to describe overall response rate with the null hypothesis that the response rate is <=1% versus the alternative that response rate is >=10%. Secondary endpoint included median progression-free survival (PFS), overall disease control rate, and median overall survival (OS). The first 9 patients received 150 mg orally twice daily for 28-day cycles, the maximum FDA approved dose of 3TC for HIV. After indication of safety in the first 9 patients, an IRB amendment was approved allowing increased dosing to 600 mg orally twice daily for 28-day cycles, which was the highest dose evaluated in HIV patients in Phase 1 trials. Tumor assessments were performed every 8 weeks until documented disease progression by RECIST criteria or drug intolerance. The median age was 59 years (range 27–83) with 18 males and 14 females. The median number of prior therapies was 3. The study did not meet the primary endpoint of response. However, remarkably, stable disease (SD) was seen in 8 of 32 (25%; Fig. 3B) patients (Pt 7, 8, 11, 15, 20, 28, 31, 34) on single agent 3TC with a median progression free survival of 149 days (Fig. 3C) with Pt 20 remaining on therapy for 230 days. Notably, one patient had mixed response with some reduction in tumor size of target lesions with a concordant 34% drop of the colon cancer serum marker carcinoembryonic antigen (CEA) but had developed new metastases (Pt 21). This patient had the largest maximal CEA response of all patients on trial and continued with 3TC treatment despite new metastases and remained on drug for 131 days given partial disease stability and limited adverse effects. The best CEA response was relatively unchanged (0–10%) or decreased from baseline in all patients with stable disease (Fig. 3D). There were no grade 5 adverse events and the majority of adverse events for patients on trial were grade 3 (18/32; 56.3%), which were mostly not attributable to treatment (Supplementary Tables S4 and S5). There were very few treatment related grade 3 adverse events which included anemia (1/32; 3.1%) and diarrhea (2/32; 6.3%). Altogether, these preclinical and clinical data supported the promising single agent activity of 3TC with minimal adverse effects as a novel class of agents for chemorefractory metastatic CRC.
Figure 3. Phase II clinical trial of Lamivudine (3TC) for colorectal cancer.
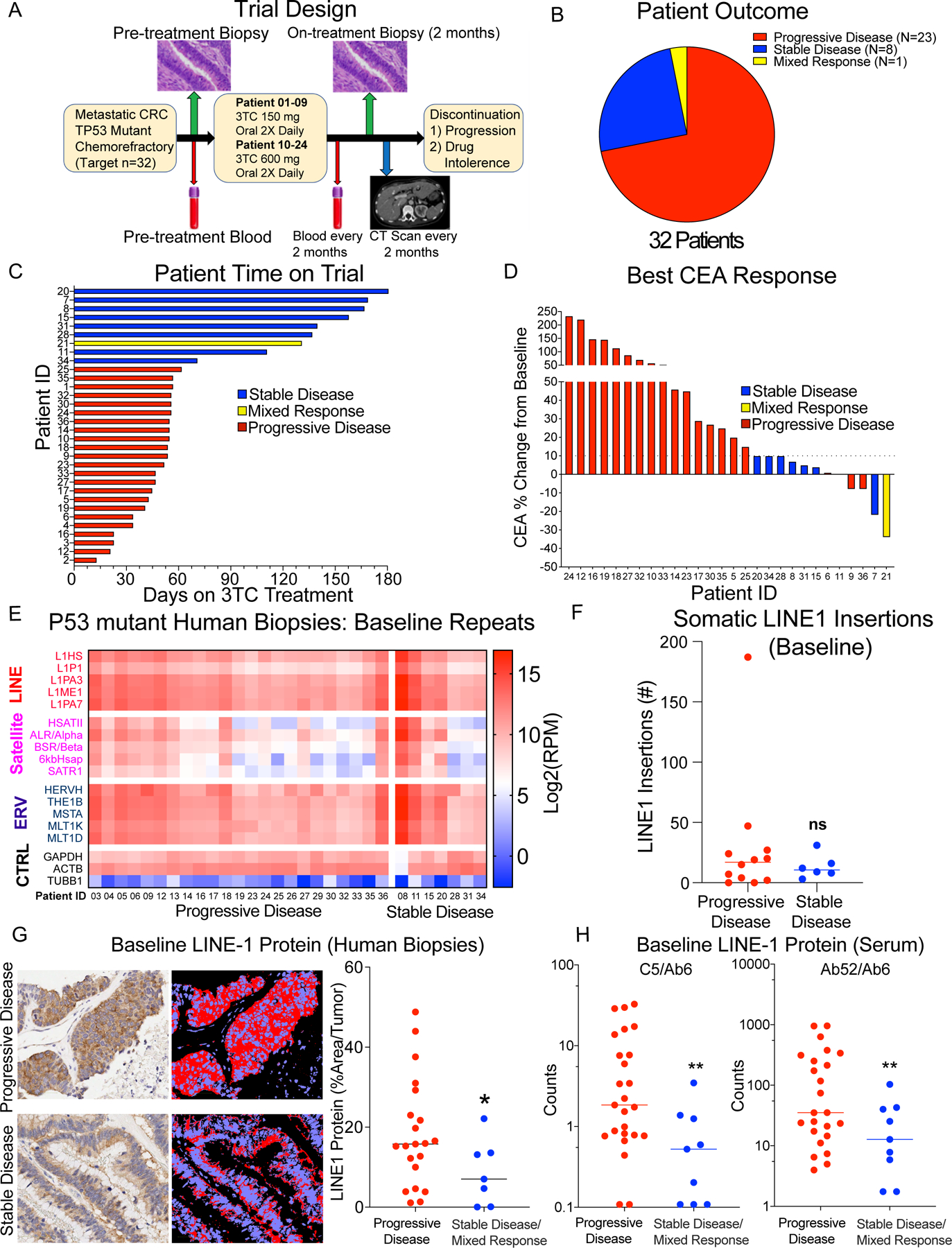
A, Schematic of trial of 3TC single agent in P53 mutant CRC with correlative blood, biopsy, and staging scans. B, Representation of patient response to 3TC. 8 out of 32 patients achieved stable disease after treatment, and 1 patient achieved mixed response. C, Swimmer plot of time on 3TC treatment (x-axis days) for 32 patients (y-axis patient ID). D, Best serum Cancer Embryonic Antigen (CEA) response in patients on the clinical trial. Patients with stable disease (SD: blue: 7, 8, 11, 15, 20, 28, 31, 34) had unchanged or decreased serum CEA levels, while most patients with progression (PD: red) had increased CEA. Pt 21 had a mixed response (yellow) with the largest CEA response but had a new metastasis at restaging scans and for grouped with SD for comparative analysis. E, RNA-seq repeat RNA expression of SAT, L1, and ERV and housekeeping genes in pre-treatment biopsy specimens from patients on 3TC trial. Expression shown in Log2(RPM+1). Pre-treatment biopsies have high expression repeats, without significant differences in baseline repeat expression between patients with stable disease and progressive disease. F, Somatic L1 genomic DNA insertions in pre-treatment biopsies. No significant difference is seen in genomic L1 insertions between patients with SD and PD. G, L1 ORF1 protein (L1 ORF1p) expression in pre-treatment biopsy specimens from patients on 3TC trial. Representative image of L1 ORF1p IHC (left) in SD and PD specimen with image analysis markup (right). Quantification shown for each pre-treatment biopsy in 28 patients. Patients with stable disease have significantly lower baseline L1 ORF1p. Significance determined by student’s two tailed t-test with Welch’s correction: * p < 0.05. H, Serum pre-treatment L1 ORF1p levels in patients with progressive disease and stable disease measured with two different antibodies (C5/Ab6 and Ab52/Ab6). Patients with stable disease have significantly lower baseline serum L1 ORF1p. Significance determined by student’s two tailed t-test with Welch’s correction: ** p < 0.01.
We then evaluated our pre-treatment biopsies from the clinical trial to determine the patterns of repeat RNA, L1 protein expression, and repeat genomic retrotransposition in our patients. Pre-treatment biopsies were obtained on 30 of 32 patients that were processed for RNA-seq, 28 of 32 were evaluable for L1 ORF1 protein (L1 ORF1p) immunohistochemistry (IHC) (Supplementary Fig. S4C-D), and 19 of 32 for whole genome sequencing (WGS) with paired germline from peripheral blood mononuclear cells. RNA-seq demonstrated high expression of multiple repeat RNA classes across all patients as expected given all patients on trial had a P53 mutation in their tumor (Fig. 3E). We looked for differentially expressed repeat RNA from patients with stable disease and Pt 21 mixed response (SD) compared to progressive disease (PD) on treatment, but we did not identify either any consistent repeat RNA difference in pre-treatment biopsies or any clear differences in expression of coding genes after multiple hypothesis correction (Supplementary Fig. S5A-B and Data S3 and S4). Analysis of tumor mutation with clinical targeted gene panels did not reveal any particular mutations enriched in patients with SD compared to PD (Supplementary Table S3). Analysis of LINE-1 retrotransposition using the xTea platform (29) on whole genome sequencing of paired baseline tumor and peripheral blood germline DNA samples also did not reveal any significant differences in transposition frequency between SD and PD tumors (Fig. 3F and Supplementary Fig. S5C-D). However, L1 ORF1p levels determined by quantitative analysis of IHC noted significantly lower L1 levels in patients with SD compared to PD (Fig. 3G; t-test p <0.05), suggestive of patients with PD having sufficient L1 RT activity to overcome 3TC at the doses given in this trial. Given L1 ORF1p immunostaining was done on a biopsy of a single lesion, we wanted to see if L1 ORF1p could be found systemically in our patients using a highly sensitive microbead immunoassay (Simoa®) technology to quantify circulating L1 ORF1p levels (30). Using two independent L1 ORF1p antibodies, we found significantly lower levels of plasma L1 ORF1p in our patients with SD compared to PD (Fig. 3H). This may indicate differences in disease burden or may suggest single agent 3TC may not have the potency to block tumors with higher RT capacity. In summary, disease stability on 3TC is associated with lower baseline levels of L1 ORF1 protein, but not expression of specific repeat RNAs or somatic retrotransposition that points to 3TC effects on the repeat RNA lifecycle as a proximal event before genomic insertion.
3TC Decreases Cytoplasmic DNA Levels and Increases RNA:DNA Hybrids
To better understand the broader effects of 3TC on CRC tumors, we evaluated all available paired pre-treatment and on treatment biopsies from the trial. Analysis of paired Total RNA-seq (n=13) found a consistent pattern of significantly decreased expression of most repeats (Fig. 4A-C). The same pattern of repeat downregulation was seen in both tumorspheres and xenografts treated with 3TC (Supplementary Fig. S6A-F, Supplementary Fig. S7A-E and Data S5). Although we did not find individual coding genes that were statistically different between pre and on treatment biopsies, gene ontology analysis noted significant upregulation of interferon response genes as has been reported to be associated with repeat RNA (7,8,10,11,13) and DNA (22,23) sensing (Fig. 4D and Supplementary Table S6). We found no changes in retrotransposition between pre- and on- treatment biopsies (Supplementary Fig. S7). Altogether, these findings indicate that repeat RNA levels decrease in patients and our cell line models from 3TC treatment with no detectable changes in LINE-1 retrotransposition. Given the decreased repeat RNA seen, we hypothesized that these changes might represent alterations in complementary DNA (cDNA) or RNA:DNA hybrids generated from RT since PCR based sequencers cannot differentiate native cDNA and RNA species from RNA-seq preparation using exogenous RT. To evaluate the potential effects of 3TC on RT products, we analyzed cytoplasmic DNA and RNA:DNA hybrids in our CRC model system. We performed quantitative immunofluorescence (IF) for cytoplasmic double stranded DNA (dsDNA) (Supplementary Fig. S8A) using an anti-dsDNA antibody as previously reported (17) and RNA:DNA hybrids using the S9.6 antibody (Supplementary Fig. S8B). Notably, we found significant reduction in cytoplasmic dsDNA in SW620 and DLD1 cells treated with 3TC compared to DMSO control (Fig. 4E; t-test p-value <0.0001), but not in HCT116 or HCT8 (Supplementary Fig. S9A). RNA:DNA hybrid quantification showed a significant increase with 3TC treatment in both P53-Mut (SW620; DLD1) and P53-WT (HCT116; HCT8) cells treated with 3TC compared to DMSO control (Fig. 4F and Supplementary Fig. S9B). Given the alterations in cytoplasmic RT products from 3TC, we evaluated the potential impact of STING, a known receptor for cytoplasmic DNA (17) and RNA:DNA hybrids (31), in our cell line models. Interestingly, siRNA mediated reduction in STING partially rescued the anti-migratory effects of 3TC in the DLD1 cell line (Fig. 4G-I). This indicated that 3TC reduces cytoplasmic dsDNA and increase RNA:DNA hybrids that block migratory function in P53-Mut CRC cell lines partially through STING signaling. To evaluate potential other mechanisms for anti-migratory effects of 3TC, we analyzed coding genes differentially expressed between CRC tumorspheres treated with 3TC compared to DMSO (Supplementary Fig. S10A and Data S6). This revealed a number of genes downregulated (t-test FDR < 0.15) and we noted S100A4 as a gene that is well known to be a negative prognostic marker in CRC (32–34) and demonstrated functional effects on metastatic potential in preclinical models (35–37). Suppression of S100A4 using siRNA noted reduced migration of P53-Mut CRC cell lines (DLD1, HCT15) relative to P53-WT cell lines (HCT116, HCT8) when compared to mock siRNA controls (Supplementary Fig. S10B-C). Altogether, these findings support a combination of 3TC effects on cytosolic nucleic acid species and S100A4 expression that reduce migratory function in CRC cell lines.
Figure 4. Reverse transcriptase inhibition leads to decreased cytoplasmic DNA, and increased RNA:DNA hybrid intermediates.
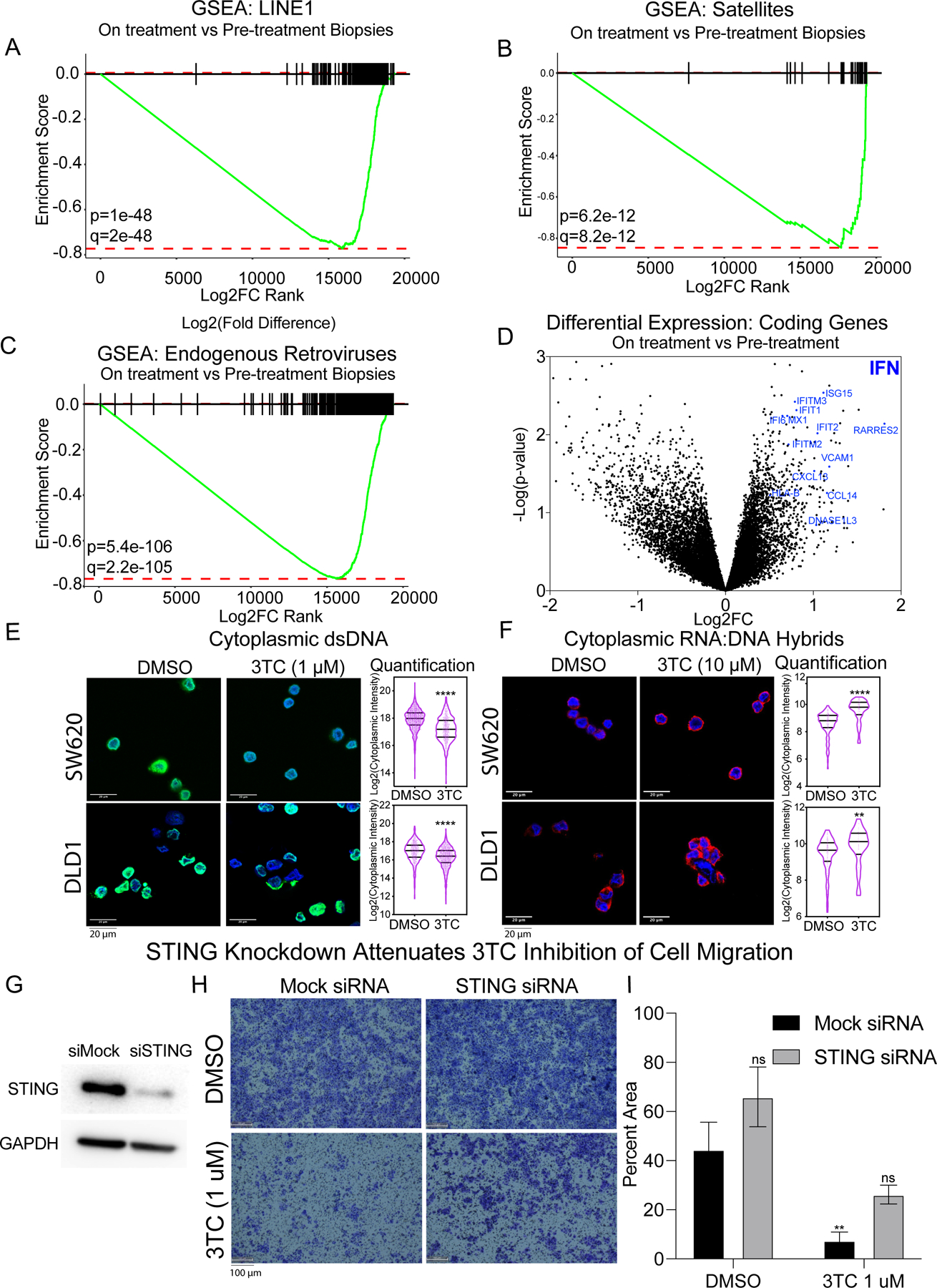
A-C, Gene set enrichment analysis (GSEA) comparing 3TC patient pre-treatment and on treatment biopsy RNA-seq (A) L1, (B) Satellite, and (C) ERV repeats. Repeats were ranked by Log2(Fold Change, on-treatment vs pre-treatment tumor sample) for GSEA. Significant decrease in L1, Satellite, and ERV repeat levels after treatment with 3TC shown in patient biopsies with p value and corrected q value shown (See Supplementary Methods). D, Volcano plot representing differential expression (Log2FC) vs Significance (-LogP) of coding genes in tumor biopsies before treatment, and after treatment with 3TC. Interferon response genes (blue) are significantly upregulated after treatment with 3TC. E, Immunofluorescence (IF) of cytoplasmic dsDNA in P53-Mut tumorspheres treated with DMSO, or 3TC (1 µM) for 7 days. Quantification of cytoplasmic IF signal indicates significant decrease in cytoplasmic dsDNA in response to 3TC. Significance determined by two tailed t-test with Welch’s correction: **** p < 0.0001. F, IF of cytoplasmic RNA:DNA hybrids in P53-Mut tumorspheres treated with DMSO or 3TC (10 µM) for 7 days. Quantification of cytoplasmic IF signal indicates significant increase in cytoplasmic RNA:DNA in response to 3TC. Significance determined by two tailed t-test with Welch’s correction: **** p < 0.0001. G-I, Rescue of effects of 3TC on cell migration by STING knockdown. (G) Western blot analysis confirms knockdown of STING (TMEM173) protein using pooled siRNA compared to non-targeting control in DLD1 cells. (H) Representative images of DLD1 cell migration with siRNA against STING (right) or Mock (left) and treated with DMSO or 3TC. (I) Quantification of DLD1 migrated cells in siRNA STING and 3TC experiments in (H). Statistical significance determined by student’s two tailed t-test as compared to non-targeting control siRNA treated with DMSO: ** p < 0.01.
Combination Repeatome Targeting Induces Cancer Cell Death Through Necroptosis
Given the effect of 3TC on the cytosolic pool of nucleic acid species, we explored the possibility of combining NRTIs with the DNA hypomethylating agent 5-azacitidine (AZA), which has been shown to derepress a wide range of repeat RNAs and activate an interferon response (3,7,8). We evaluated a broad panel of 9 NRTIs and combined with low dose AZA (300 nM) to achieve epigenetic repeatome effects, which resulted in significant reduction in tumorsphere growth across both P53-Mut and P53-WT CRC cell lines (Fig. 5A-B). The efficacy of AZA in our cell lines is consistent with analogous work in mouse cancer cell lines demonstrating significant induction of repeat species triggering and IFN response linked with p53 status (7). Evaluation of all NRTIs revealed that ddC had the most consistent cytotoxic effect across our CRC cell lines, and in general, the C analogues (FTC, 3TC, ddC) and G analogue (entecavir: ETV) had the most toxicity in our CRC cell lines. This indicates that not all NRTIs have the same functional effects, which may be driven by differences in CpG sequence motif biases in repeat RNAs (13). We next investigated the mechanism of cancer cell toxicity induced by repeat dysregulation and focused on necroptosis given recent work indicating that mutant P53 primes epithelial cells to necroptosis (38). Inhibition of necroptosis effectors RIPK1 by the small molecule inhibitor Necrostatin-1 was sufficient to rescue multiple CRC cell lines from ddC, AZA, and combination ddC+AZA mediated toxicity (Fig. 5C).
Figure 5. Combination repeatome targeting induces cancer cell death through Necroptosis.

A-B, Efficacy of combination of 9 reverse-transcriptase inhibitors (5 μM) with DNA demethylating agent Azacitidine (AZA) at 300 nM in (A) P53-Mut and (B) P53-WT CRC tumorspheres measured by CellTiter-Glo viability assay after 7 days of treatment. C, Necroptosis pathway inhibition by treatment with RIPK1 inhibitor Necrostatin-1 (Nec-1) at 10 μM attenuates effect of NRTI ddC (5 μM) and DNA demethylating agent AZA (300 nM) as measured by CellTiter-Glo assay after 7 days of treatment. Significance determined by student’s two tailed t-test: * p < 0.05, ** p < 0.01, ***p < 0.001. D, Treatment with antisense locked nucleic acids (LNA) targeting HSATII in P53-Mut cell lines (SW620, DLD1) and P53-WT (HCT8, HCT116) cell lines grown as tumorspheres compared to scrambled LNA, as measured by CellTiter-Glo assay. Significance determined by two way ANOVA test: ** p < 0.01. E, Necroptosis pathway inhibition with Nec-1 (10 μM) rescues effect of anti-HSATII LNA in SW620 tumorspheres. Significance determined by student’s two tailed t-test: *** p < 0.001. F, Schematic showing multiple avenues for repeatome targeting in cancer.
We next wanted to determine if the effects seen with ddC and AZA were linked specifically to repeat mediated effects. We chose to focus on the HSATII satellite repeat given the consistent loss of HSATII binding by mutant P53 (Fig. 2C), the associated higher HSATII RNA expression (Fig. 2E), and our previous success in targeting HSATII with locked nucleic acids (LNAs) in CRC cell lines (16). We modified these original HSATII LNAs with phosphorothioate to enhance cellular uptake and LNA stability to maximize cancer cell toxicity (see methods). We found specific inhibition of P53-Mut (SW620, DLD1) compared to P53-WT (HCT8, HCT116) cell line growth with HSATII LNAs compared to scrambled LNA controls (Fig. 5D). Necrostatin was able to rescue cell toxicity in the SW620 cell line, which supports necroptosis as the mechanism of cytotoxicity induced with HSATII modulation (Fig. 5E). Collectively, these data support a model of repeat mediated IFN response and necroptotic cell death induced by agents that disrupt genomic repeat epigenetic suppression, directly target the repeat RNA, or inhibit repeat reverse transcription (Fig. 5F).
Reverse Transcriptase Inhibition Results in Increased DNA Damage
Emerging data has recently demonstrated a relationship between genomic instability with satellite RNA expression (39–41) and LINE-1 activity (42,43), but the effects of blocking repeat RNA reverse transcription in the setting of high retrotransposon activity has not been fully characterized. To understand the acute effects of 3TC, we analyzed Total RNA-seq data from P53-Mut CRC cell lines treated for 1 and 7 days. We noted significant upregulation (t-test FDR < 0.15) of innate immunity and interferon response genes including EGR1, IFI6, HLA-B, TLR2, and APOBEC3B (Fig. 6A), consistent with our findings in paired patient biopsies (Fig. 4D). Unbiased GO analysis of upregulated genes (t-test FDR < 0.15) was highly enriched for gene signatures involved with DNA damage response including the genes BRCA1, BRCA2, BRIP1, FANCI, and FANCD2 (Fig. 6A and Supplementary Table S7 and Data S6). Western blot analysis for gamma-H2AX confirmed increased double strand DNA breaks in cell lines treated with 3TC (Supplementary Fig. S11A). Downregulated genes were enriched for genes involved with amino acid metabolism and endoplasmic reticulum stress including multiple aminoacyl-tRNA synthetase genes, which implies effects on translational machinery (Supplementary Table S8 and Data S6). Temporal analysis of post-3TC exposure demonstrated DNA damage and interferon/innate immune genes starting at Day 1 and increasing expression at Day 7 post-treatment in the P53-Mut cell lines (Fig. 6B). As LINE-1 RT can act at DNA damage breakpoints (15,42,43), the accumulation of DNA damage and interferon signatures in cell lines and patient biopsies suggests a model of 3TC mediated disruption of the retroviral-like life cycle of repeats leading to RNA:DNA hybrid accumulation that enhances the ability of the innate immune system to detect their presence and leads to unresolved DNA damage. Given the enhanced DNA damage response signatures in CRC cell lines, we wanted to determine the relationship of repeat RNA expression in the setting of DNA damaging agents. We treated CRC cell lines with the standard combination chemotherapy 5FU/Oxaliplatin (5FU/Oxa) for 14 days and detected marked elevation of HSATII RNA by RNA-ISH (Fig. 6C and Supplementary Fig. S11B). We then applied HSATII RNA-ISH to 160 human primary CRC tumors that were untreated or pre-treated (neoadjuvant) with chemoradiation before resection, which demonstrated significant enrichment of HSATII repeat RNAs in tumors that received cytotoxic therapy (Fig. 6D). To determine if this had therapeutic implications, we treated CRC lines with 5FU/Oxa +/− 3TC, which showed significantly increased cytotoxicity in all 4 cell lines with the combination compared to 5FU/Oxa alone (Fig. 6E). These combination approaches we present boost repeat RNA expression and inhibit RT activity that leads to accumulation of immunostimulatory RNA:DNA hybrids. Overall, these data support a model of RT inhibition leading to enhanced DNA damage due to unresolved, chain terminated intermediates of retrotransposition or secondary DNA damaging effects from alterations in the balance of cytosolic RT products (Fig. 6F).
Figure 6. Reverse transcriptase inhibition induces DNA damage in colon cancer.
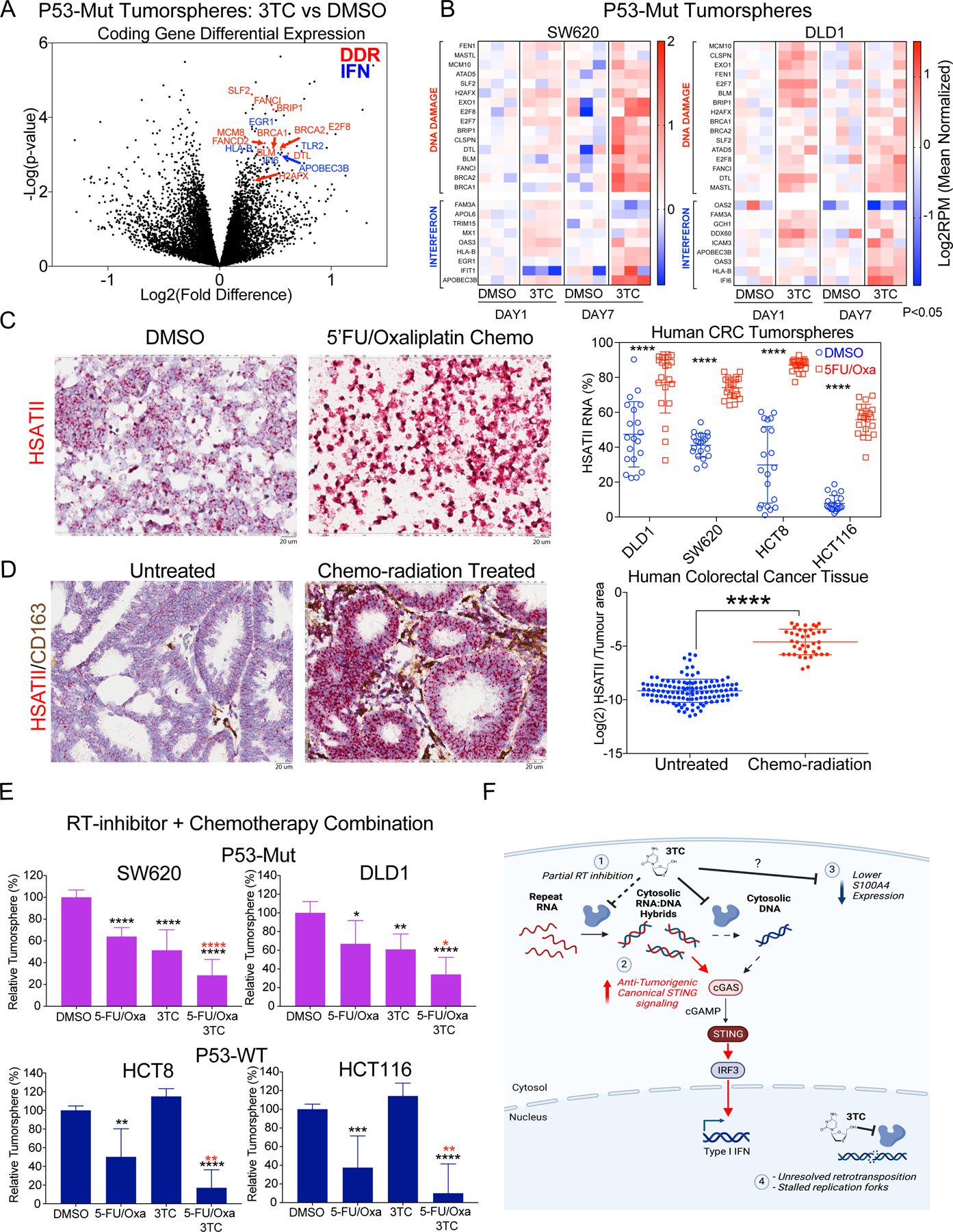
A, Volcano plot of coding gene RNAs with 3TC (5 µM) or DMSO treatment for 7 days in P53-Mut cell lines (DLD1 and SW620; n = 3 for 2 independent experiments per cell line). Highlighted are DNA Damage Response (DDR) and interferon/innate response (IFN) genes B, Tumorsphere RNA-seq expression heatmap of P53-Mut CRC cell lines for DDR and IFN genes at Day 1 and Day 7 post-3TC vs DMSO treatment. Expression is Log2RPM normalized to mean of DMSO per timepoint and cell line. Genes represented have significantly increased expression in response to 3TC at either Day 1, or Day 7, or both (P<0.05 student’s two-tailed t-test). C, HSATII expression in HCT8 tumorspheres grown in the presence of DMSO (control), or 50 μM 5FU + 1.25 μM Oxaliplatin (5FU/Oxa) for 2 weeks. Scale bar = 20 μm. HSATII RNA-ISH in DLD1, SW620, HCT8, and HCT116 tumorspheres treated with DMSO or 5FU/Oxa for 2 weeks. Plots represent HSATII staining as a percentage of tumor area across 20 fields. Significance determined by two tailed t-test with Welch’s correction: **** p < 0.0001. D, HSATII RNA-ISH on human CRC tumors from untreated patients (Left) and patients who received neoadjuvant chemoradiation (Right). Scale bar = 20 μm. HSATII RNA levels quantified as a percentage of tumor area. Significance determined by student’s two tailed t-test with Welch’s correction: **** p < 0.0001. E, Cell viability of CRC tumorspheres treated with either DMSO or 5 μM 3TC for 7 days in the presence of 5FU/Oxa. Significance determined by student’s two tailed t-test: * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001. Black stars indicate statistical significance compared to DMSO alone. Red stars indicate statistical significance compared to 5FU/Oxa. F, Schematic of multiple effects of 3TC on colorectal cancer including alterations in cytosolic nucleic acids (1 & 2), S100A4 expression (3), and DNA damage (4).
DISCUSSION
The combination of preclinical and clinical results provides a path for the evaluation of NRTIs as a new class of anti-cancer therapeutics. We have shown 3TC is able to decrease migration of mutant TP53 CRC cell lines partially through a STING dependent nucleic acid mechanism and reduction in S100A4 levels. Reduction of cytoplasmic dsDNA species and increased RNA:DNA hybrids from 3TC appears to trigger STING signaling and induce DNA damage. These combined effects of 3TC (Fig. 6F) had anti-tumorigenic effects in mouse models and potential benefit to patients in a clinical trial. This unexpected RNA:DNA elevation with 3TC suggests incomplete RT inhibition with potential re-engagement of RT for multiple rounds of synthesis or delayed chain termination after initial RT of the polyA tail, however, these mechanisms require further elucidation. Cytoplasmic L1 DNA has been found elevated in mouse models of senescence, and notably, 3TC has been shown to reduce L1 cytoplasmic DNA in senescence model systems (22,23). Although the effects we have observed could be from L1 repeat species, we acknowledge that 3TC effects on RNA:DNA hybrids and cytoplasmic DNA species likely affect a broad range of RNA species that can be processed by L1 (44). The associated accumulation of immunogenic RNA:DNA species drives an interferon response potentially through cGAS-STING (31), DDX41 (45), or other nucleic acid cytoplasmic sensor. We demonstrate in our CRC cell lines that STING is likely involved in RNA:DNA detection that attenuates migratory capability. Notably, others have shown that increased cytoplasmic DNA levels are linked with genomic instability and increased metastatic potential in cancers through non-canonical chronic STING signaling (17). Our findings are complementary to this work by demonstrating the importance of the nuances in STING signaling on cancer cell phenotype and the use of 3TC and other NRTIs providing a therapeutic modality to decrease the pro-tumorigenic effects of cytoplasmic DNA.
Cytosolic nucleic acid species have been shown to trigger IFN response and adaptive immunity across mouse model and human tumors (6–13) with recent papers demonstrating a relationship with response to immune checkpoint response (46–48). Similarly, we found increased IFN response from 3TC affecting the repeat life cycle, which was also seen in recent work demonstrating the association of IFN response gene activation of normal colon samples from non-HIV patients on NRTIs for anti-viral prevention (49). These findings may explain in part the apparent decreased incidence of breast, prostate, and colorectal cancers in patients living with HIV who are on stable anti-HIV regimens that include NRTIs (50). Finally, we speculate that incomplete RT creates DNA lesions that are difficult to repair or prevents the retrotransposition-mediated repair of other forms of DNA damage. Altogether, our preclinical and proof of concept clinical trial provide the foundation to evaluate combination reverse transcriptase inhibitors to obtain more potent effects of disrupting the cancer repeatome, a strategy that clearly changed the course of HIV treatment (51,52). The lack of significant dose limiting toxicity of 3TC in metastatic CRC patients affords the use of 3TC or related compounds in combination with both existing and novel cancer therapies in future clinical trials to augment the efficacy of these drugs as we have shown with 5FU/Oxaliplatin and AZA in CRC cell line models. Interestingly, a recent paper has also shown that re-activation of P53 with a novel MDM2 inhibitors can also induce HERV expression through epigenetic targets that triggers an anti-tumoral immune response (53). These intricate relationships between tumor suppressors, epigenetics, and repeat RNA expression will need to be further elucidated to fully understand the impacts of these drugs alone and in combination. The inherent massive de-repression of repeat RNAs in cancer provides a rich opportunity to target this genomic “dark matter” as a therapeutic strategy in human disease.
MATERIALS AND METHODS
Cell Culture:
Cell lines were originally obtained from ATCC. Cell lines were cultured in either RPMI-1640 (SW620, HCT116, HCT8, DLD1, HCT15, LOVO), DMEM (GP5D), or DMEM/F12 (SW948, C2bbe1, RKO) media supplemented with 10% FBS + 1% Pen/Strep (Gibco/Life Technologies). Cell lines were maintained in adherent 2D culture on standard tissue culture plates (Corning). For subculturing, media was aspirated and cells were washed in cell culture grade PBS (pH 7.4, ThermoFisher). Cells were passaged using 0.25% Trypsin-EDTA (ThermoFisher) for 5 minutes, and reaction was inhibited using media with serum. Cells were centrifuged and resuspended in media for subculture. For 3D culture (tumorspheres), serum-free media (RPMI, DMEM, or DMEM/F12) was supplemented with 20 μl/ml B27+ 20 ng/ml EGF + 20 ng/ml bFGF (ThermoFisher). Cells were plated in ultra-low attachment plates (Corning). All cell lines were routinely tested for Mycoplasma on a monthly basis or earlier if there were indications of slower cell growth or changes in morphology. All cell lines were used within 2 months of thawing for experiments. Cell line authentication was not performed on all cell lines. All cell lines will be provided upon request with appropriate MTA approvals.
Drug Treatment:
Nucleoside analog reverse transcriptase inhibitors (NRTIs) Lamivudine (3TC), Zalcitbine (ddC), Emtricitabine (FTC), Entecavir (ETV), Abacavir (ABC), ddA, Tenofovir (Teno), Zidovudine (ZDV), and Stavudine (d4T), and DNA demethylating agent Azacitidine (AZA) were purchased from Selleck Chemicals. Drugs were reconstituted in tissue culture grade DMSO (Sigma). The RIPK1/Necroptosis inhibitor Necrostatin-1 was obtained from Sigma Aldrich and reconstituted in DMSO. Chemotherapy drug 5-Fluorouracil (5-FU) was obtained from Invivogen. Oxaliplatin was obtained from Selleck Chemicals.
Migration Assays:
Colon cancer cells were pre-incubated with or without 3TC into ultra-low attachment plates for 24 hr. Then, cells were dissociated to single cell suspensions and seeded onto 8-μm pore Transwell membranes (CELLTREAT® Scientific Products, Cat# 230639) in serum-free media at 50,000~100,000 cells/ well in a 24-well plate containing complete growth media (10% FBS) in the bottom chamber. 3TC was treated both upper chamber and bottom chamber during migration. After 24~96 h at 37 °C, Transwell chambers were fixed with methanol for 10 min and stained with 0.1% of Crystal Violet (Sigma-Aldrich, Cat# V5265) and cells on top of the chamber were stripped off with cotton swabs. Images were acquired and the total crystal violet-stained area was quantitated using HALO software.
Soft Agar Assays:
Cells were seeded at a density of 5,000 cells per well in a 6 well plate of 0.3% agarose (Seaplaque, Lonza) in RPMI containing 10% (vol/vol) FBS in the presence of DMSO or Lamivudine (3TC). After 14 days of growth cells were stained in 0.005% crystal violet solution in 4% PFA/PBS solution for 30 minutes followed by 3X washes in water. Crystal violet stained colonies were imaged using a dissection microscope and quantified using HALO software.
Cell viability assays:
Cell viability assays were conducted on both adherent cells (2D) colorectal cancer cell lines, as well as cells grown as tumorspheres (3D) in response to drug treatment. 3D media. Cells were plated at a concentration of 5000 cells per well of 96 well plates (Corning). Cells were treated for the indicated concentrations as described for the indicated number of days, and cell viability was measured using the Cell titer Glo reagent (Promega) according to manufacturer recommendations, and luminescence was measured using the Spectramax i3X (Molecular Devices) plate reader. Percent cell/tumorsphere viability was represented as luminescence units normalized to DMSO control.
siRNA knockdown:
STING:
Colorectal cancer cells were transfected with pooled siRNA targeting STING (Horizon Discovery, TMEM173 SMARTPool L-024333–00-0005) or Non-targeting siRNA (Horizon Discovery, D-001810–01-05) with Lipofectamine 2000 at a concentration of 100 nM. Knockdown was assessed 48 hours post-transfection using Immunoblotting for STING (TMEM173).
S100A4:
Transfection with siRNA was performed with Lipofectamine 2000 reagent (Thermo Fisher Scientific, Cat# 11668019) according to manufacturer’s instructions. 100 nM siRNA targeting human S100A4 ON-TARGET plus SMARTpool (GE Healthcare Dharmacon™), mixture of 4 siRNAs targeting the following sequences: 5’-GAUGUGUAACGAAUUCUUU-3’, 5’-CCACAAGUACUCGGGCAAA-3’, 5’-GUGACAAGUUCAAGCUCAA-3’, 5’-GAAAACUCCUCUGAUGUGG-3’ or ON-TARGETplus Non-targeting siRNA as negative control (GE Healthcare Dharmacon™) were used. Knockdown efficiency was assessed by qRT-PCR and migration assay was performed for 48 hrs.
Quantitative reverse transcriptase – polymerase chain reaction (qRT-PCR)
RNA was isolated using the miRNEasy Mini Kit (QIAGEN, Cat# 217004) according to the manufacturer’s instructions including on-column DNase treatment (QIAGEN, Cat# 79254). The total RNA (1 µg) was reverse transcribed using TaqMan™ Reverse Transcription Reagents (Invitrogen™, Cat# N8080234). qRT-PCR was conducted using the PowerUp™ SYBR™ Green Master mix (Applied Biosystems, Cat# A25742) according to the manufacturer’s instructions. Primers used include the following: GAPDH Forward 5′-ACATCATCCCTGCCTCTACT-3′, Reverse 5′-TCCACCACTGACACGTTG-3′; S100A4 Forward 5′-CAGAACTAAAGGAGCTGCTGACC-3′, Reverse 5′-CTTGGAAGTCCACCTCGTTGTC-3’ Reactions were run on a QuantStudio 3 (Applied Biosystems) thermocycler. The level of gene expression was calculated by the 2–ΔΔCT method and normalized to the Ct value for GAPDH.
Antisense Locked Nucleic Acid Treatment:
Colorectal cancer cells were transfected with antisense locked nucleic acids (Exiquon, Scrambled LNA: GATTCCATTCGATGAT, anti-HSATII LNA: +A*+T*+g*+G*A*A*T*C*A*T*C*A*T*+C*+G*+A*+A) with Lipofectamine 2000 at a concentration of 500 nM. Cell viability assay was conducted 24 hours post LNA treatment.
Animal Studies:
Mouse xenograft studies were performed according to an animal protocol approved by the MGH Institutional Animal Care and Use Committee (IACUC) under protocol 2014N000321. SW620 or HCT116 cell lines stably transduced with Luciferase (1 × 106) were injected in the flank of six-week old female nude mice (Charles River Laboratories) at a 1:1 ratio with Matrigel (Sigma Aldrich). Tumors were allowed to grow for 2 weeks post-implantation. At 2 weeks before beginning drug treatment mice were intraperitoneally injected with Luciferin (Promega) to visualize Luciferase expression. Tumors were visualized through Luciferase Luminescence measurement with the IVIS Spectrum Imaging System (PerkinElmer). Mice were randomized and divided into treatment arms: Control (PBS), and Lamivudine (3TC). 3TC was solubilized in PBS and administered intraperitoneally at a dosage of 50 mg/kg 3 times a week. In the control group, PBS was administered intraperitoneally. Mice were treated for 20 days, and tumor volume was measured every 5 days through IVIS imaging. At 20 days mice were euthanized according to IACUC guidelines. Animals did not show any sign of systemic toxicity upon drug administration. Tumor growth curves were calculated using relative luminescence units normalized to Day 0 of treatment. After euthanasia, tumors were dissected and used for RNA isolation using Trizol, or standard FFPE processing.
Clinical Trial:
This is a single-arm Phase 2 clinical trial of 3TC in patients who have progressed on systemic therapy for metastatic CRC with TP53 mutations (NCT03144804). The trial was reviewed and approved by the Dana-Farber Harvard Cancer Center (DFHCC) IRB (Protocol 17–044). Eligibility included patients with TP53 mutant refractory mCRC with progression on or intolerance to 5FU, oxaliplatin and irinotecan and anti-EGFR therapy if RAS WT. Eligible patients were age ≥18 years, with histologically or cytologically documented, advanced (metastatic and/or unresectable) disease that was incurable and had disease progressed on at least two-lines of systemic therapy. Patients were required to provide an evaluable tissue sample for biomarker analysis from a tumor lesion not previously irradiated; had radiologically measurable disease per RECIST version 1.1 as assessed by independent central radiologic review; had an Eastern Cooperative Oncology Group performance status of 0,1 or 2; and had adequate organ function. Key exclusion criteria included uncontrolled intercurrent illness, HIV-positive patients on combination antiretroviral therapy as well as patients with Hepatitis B. The study was designed as a two-stage disease with target accrual of 20 patients in stage 1 and a total of 36 patients with continuation beyond stage I allowed if at least one patient demonstrated a response. Primary endpoint of the study was to describe overall response rate with the null hypothesis that the response rate is <=1% versus the alternative that response rate is >=10%. Secondary endpoint included median progression-free survival (PFS), overall disease control rate, and median overall survival (OS). The first 9 patients received a dose of 150 mg orally twice daily for 28 day cycles, the maximum FDA approved dose of 3TC for HIV. The subsequent patients received 600 mg orally twice daily for 28 day cycles. Tumor assessments were performed every 8 weeks until documented disease progression by RECIST criteria or drug intolerance. CEA was obtained as part of clinical care.
RNA-sequencing:
RNA was isolated using Trizol reagent (ThermoFisher) or the AllPrep DNA/RNA/protein mini kit (Qiagen #80004) according to manufacturer recommendations. RNA-sequencing was performed using the two different sequencing library construction methods. For cell lines and xenografts, we performed Illumina TruSeq stranded RNA-seq Ribo-zero TM Gold (Illumina RS-122–2301) library construction per protocol for coding gene transcriptional profiling. Given known issues with repeat RNA quantification with standard RNA-seq protocols, we utilized the Clontech/Takara SMARTer Stranded Total RNA-seq PICO v2 kit (Clontech/Takara 634414) for repeatome profiling. Given limited sample material in biopsies only the Clontech/Takara SMARTer Stranded Total RNA-seq PICO v2 kit (Clontech/Takara 634414) was performed on samples. All libraries then had 75 bp paired end sequencing on the NextSeq500 using a 150 cycle high output kit.
RNA-seq computational analysis:
Illumina reads were mapped to the human or mouse genomes (or their union for xenograft samples), build 38 using STAR aligner (54). Aligned reads were assigned to genes using the featureCounts function of Rsubread package with the external Gencode annotations (55,56). Counts for repeat elements were obtained using RepeatMasker annotation (https://www.repeatmasker.org). This produced the raw read counts for each gene and repeat element. Gene expression in terms of log2-CPM (counts per million reads) was computed and normalized across samples using the trimmed mean of M-values method (TMM), as implemented in the calcNormFactors function of edgeR package (57,58).
Differential expression: For comparisons with a small number (typically two or three) of samples per phenotype we used DESeq2 (59), which employs moderated estimate of dispersion useful for a small sample size. For comparisons with a larger (typically ten or more) samples we computed the normalized log transformed CPM values and then performed the t-test between the conditions. Where applicable, we subtracted the mean within each group of samples (e.g., each cell line is a separate group when comparing the treated and untreated samples using multiple cell lines). This was done using linear model approach.
Gene set enrichment analysis (GSEA) was performed using repeats of various classes as gene sets (Satellite repeats, LINEs, SINEs, ERVs). We used the pre-ranked GSEA as implemented in fgsea R package (60). We performed two types of analysis ranking the genes and repeat elements either by the fold change or the t-statistic from the differential expression analysis.
Whole Genome Sequencing:
DNA and RNA were extracted from samples using the AllPrep DNA/RNA/protein mini kit (Qiagen #80004). DNA was quantified fluorometrically using the Quant-iT Picogreen dsDNA assay kit (Invitrogen #P7589) and 100 ng DNA input was used for library generation. The Nextera DNA library prep kit (Illumina #20020188) was used in combination with the Nextera DNA CD Indexes (Illumina # 20018708) for library preparation. In brief, DNA samples were fragmented using Illumina transposon-based technology. After tagmentation, right-sided size selection was carried out using AMPure-XP beads (Beckman Coulter # A63881; 1:0.4, followed by 1:1.1) to remove insufficiently-tagmented gDNA. Unique 5’ and 3’ index adapters were added to each sample by PCR using five cycles of amplification. Amplified and barcoded libraries were captured during AMPure-XP bead clean-up (1:0.8). Libraries were quantified using the KAPA-library quantification kit for Illumina platform (Roche # 07960140001). Library size was evaluated with a High Sensitivity DNA kit (Agilent # 5067–4626) on an Agilent 2100 Bioanalyzer. Samples were pooled and sequenced on the Novaseq 6000 platform.
Whole Genome Sequencing Analysis:
For each patient, both the case and control samples were sequenced with Illumina paired-end reads in length 150bp. BWA-mem (61) was used to align the reads to the human reference genome hg38. GATK (62) was used to mark duplicates and recalibrate base quality scores. Then, SAMtools (63) was used to sort and index the alignment files. In addition, NGSCheckMate (64) was used to make sure the case-ctrl samples are well matched.
With the preprocessed case-control bams, we ran the xTea (29) “--case_ctrl” mode on each pair of bams to identify somatic L1, Alu and SVA insertions. We used the xTea default parameters, where the cutoffs are automatically adjusted based on the average read depth.
Measurement of circulating ORF1p with Single Molecule Arrays (Simoa):
Affinity reagents for the Simoa assays were obtained from John LaCava (capture nanobody) and Abcam (ab246317 and ab246320). Recombinant ORF1p was obtained from Dr. Martin Taylor and Dr. Kathleen Burns. All buffers, beads, and consumables used for Simoa reagent preparation and assay running were purchased from Quanterix Corporation.
Preparation of antibody-coated paramagnetic beads. 7×108 carboxylated 2.7-µm paramagnetic beads were washed three times with 400 µL Bead Wash Buffer, followed by two times with 400 µL Bead Conjugation Buffer, before being resuspended in 390 µL of cold Bead Conjugation Buffer. 10 µL of freshly dissolved 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC) (Thermo Fisher Scientific) in cold Bead Conjugation Buffer (10 mg/mL) was then added to the beads, which were shaken at 4 °C for 30 minutes. After activation with EDC, the beads were washed once with 400 µL cold Bead Conjugation Buffer, resuspended in 400 µL of the capture antibody (0.25 mg/mL) or nanobody (0.025 mg/mL), and shaken at 4 °C for two hours for conjugation to the nanobody. The conjugated beads were then washed two times with 400 µL Bead Wash Buffer, resuspended in 400 µL Bead Blocking Buffer, and shaken at room temperature for 30 minutes. After blocking, the beads were washed once each with 400 µL Bead Wash Buffer and 400 µL Bead Diluent, before resuspending in 400 µL Bead Diluent for storage at 4 °C. The conjugated beads were counted using a Beckman Coulter Z1 Particle Counter.
Preparation of biotinylated detector antibody: Detector antibody (Abcam, ab246317) was biotinylated by adding 80-fold molar excess of sulfo-NHS-LC-LC-biotin (ThermoFisher Scientific) and incubating for 30 minutes at room temperature. Excess biotin was removed using an Amicon filter (50 kDa, MilliporeSigma), using Biotinylation Reaction Buffer (Quanterix) as the buffer and centrifuging five times at 14,000 x g for five minutes. The filter was then inverted and centrifuged at 1,000 x g for two minutes to recover the purified antibody. The filter was then washed with 50 µL Biotinylation Reaction Buffer and centrifuged again at 1,000 x g for two minutes.
L1ORF1p Simoa assays: Simoa assays were performed on an HD-X Analyzer (Quanterix Corp.). Beads, detector antibodies, streptavidin-beta-galactosidase (SβG), diluted plasma samples, and necessary consumables were loaded onto the HD-X analyzer according to the manufacturer’s instructions. Conjugated beads were diluted in Bead Diluent, detector antibody was diluted in Homebrew Detector and Sample Diluent to 0.3 µg/mL, and SβG was diluted in SβG Diluent to 150 pM. Plasma samples were diluted fourfold in Homebrew Detector and Sample Diluent with added protease inhibitor (Halt™ Protease Inhibitor Cocktail, Thermo Fisher Scientific). For each assay, 100 µL diluted plasma sample was incubated with 250,000 conjugated beads and 250,000 unconjugated helper beads for 15 minutes, followed by washing and incubation with 100 µL biotinylated detector antibody (0.3 µg/mL) for five minutes. The beads were then washed, incubated with 100 µL SβG (150 pM) for five minutes, and washed again before resuspending in 25 µL of the enzyme substrate, resorufin-β-D-galactopyranoside (RGP), and loaded into a 216,000 microwell array, in which each femtoliter-sized microwell can fit at most one bead. The wells were sealed with oil, with each well containing an enzyme-labeled bead subsequently generating a detectable fluorescent signal for counting of fluorescent “on” and “off” wells. The average enzyme per bead (AEB) was calculated by the HD-X Analyzer software, and calibration curves were fit with four parameter logistic regression. Limits of detection were determined as three standard deviations above the blank AEB.
Gene set enrichment analysis (GSEA):
Coding genes were ranked according to log2(fold change), and enriched gene sets were obtained using the GSEA analysis of the pre-ranked gene list (65,66).
TP53 Chromatin Immunoprecipitation:
HCT116, HCT8, DLD1, and SW620 cells were cross-linked with formaldehyde (1% final) for 10 min at room temperature. Cross-linked cells were quenched with glycine (125 mM final) for 5 min, followed by two washes in cold PBS. Nuclei were then isolated from 20 million cells as previously described (67), and chromatin was sheared to 250-bp average size using a Covaris S220. Immunoprecipitations were performed using 1000 μg of sheared chromatin lysate and 5 μg of p53 antibody (p53 D07; Santa Cruz, sc-47698) preconjugated to protein G beads (Invitrogen). ChIP reactions were incubated for 16 hours at 4°C with rotation and then washed four times in wash buffer [50 mM Hepes-HCl (pH 8), 100 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 0.1% sodium deoxycholate, and 0.5% N-laurylsarcosine], followed by one wash in ChIP final wash buffer (1× tris-EDTA (TE) Buffer and 50 mM NaCl). Immunoprecipitated DNA was eluted from washed beads, reverse cross-linked overnight, purified, and used to construct libraries. Sequencing for ChIP experiments were prepared using NEBNext Ultra reagents (New England Biolabs). All ChIP samples and input were double-end–sequenced on an Illumina NextSeq 550.
ChIP-Seq Computational analysis:
Raw Illumina reads were quality filtered as follows. First ends of the reads were trimmed to remove N’s and bases with quality less than 20. After that the quality scores of the remaining bases were sorted and the quality at the 20th percentile was computed. If the quality at the 20th percentile was less than 15, the whole read was discarded. Reads that are shorter than 40 bases after trimming were discarded as well. If at least one of the reads in the pair failed the quality check and had to be discarded, then the mate was discarded as well. Quality filtered reads were mapped to the human genome (HG38) using Bowtie2 aligner(68). First the reads are mapped to the genome and then the duplicate reads are removed using Picard MarkDuplicates tool. The mapped BAM files containing the reads were used to call peaks using MACS peak calling algorithm (69). The files were also used to get counts of reads for each repeat region annotated using repeatmasker and BEDTools coverage tool (70). These counts for each repeat region were then further analyzed in R to create plots and derive inference.
Histology/Cytology:
Tumorsphere cell block preparation for RNA-ISH/IHC: tumorspheres were cultured in ultralow attachment cell culture flasks (Corning) in 3D culture media. For cell block preparation tumorspheres were transferred to 15 ml Falcon tubes with media and allowed to settle at the bottom of the tube. Media was removed, and tumorspheres were washed 3x in PBS. Tumorspheres were resuspended in human plasma (Sigma Aldrich), reconstituted in MilliQ water. Cell pellet/plasma mixture was coagulated with addition of Bovine Thrombin reconstituted in MilliQ water/Bovine Serum Albumin (Sigma Aldrich). Coaggulated cell pellets were fixed for 3 hours in 10% Formalin/PBS. Pellets were processed using standard FFPE. For mice xenograft tumors, tissue was fixed overnight in 10% Formalin/PBS, and processed using standard FFPE.
Cytospin Preparation for Immunofluorescence: Tumorspheres were transferred grown in ultra-low attachment flasks in 3D media were allowed to settle in 15 ml falcon tubes. Media was removed, and washed 3x in PBS. Tumorspheres were dissociated in trypsin for 5 minutes at 37°C followed by neutralization with media containing 10% FBS. Cells were centrifuged at 150g and resuspended in PBS. Cells were counted and diluted to a concentration of 500,000/1ml of PBS. 300 µl of the cell solution was added to a cytology funnel attached to a slide and centrifuged at 350 rpm for 5 minutes in the Cytospin 4 centrifuge (ThermoFisher).
Human Tissue: For Lamivudine clinical trial human biopsies tissue was fixed in 10% Formalin/PBS and processed using standard FFPE. Standard FFPE processed tissue for untreated and chemo/radiation treated human colorectal cancer tissue was obtained from the MGH Pathology Tissue Bank. All slides were cut at 5 micron thickness.
RNA in situ hybridization (RNA-ISH) and protein immunohistochemistry (IHC):
Automated RNA-ISH assay was performed using the ViewRNA eZ-L Detection 1-Plex Kit from Affymetrix (Catalogue No. 19500) on the BondRx 6.0 platform (Leica Biosystems Inc., Buffalo Grove, IL). 5 μm sections of FFPE tissue (human colorectal cancer tissue, or cell blocks) were mounted on Surgipath X-tra glass slides, baked for 1 hour at 60°C, and placed on the BOND RX for processing. The Bond RX user selectable protocol was as follows: The RNA unmasking conditions for the tissue consisted of a 10-minute incubation at 90°C in Bond Epitope Retrieval Solution 1 (Leica Biosystems). This was followed by incubation with Proteinase K with 1:1000 dilution (Leica Biosystems, Catalogue No. AR9551). Proteinase K incubation on human TMAs and biopsies was done for 10min; and cell blocks for 5 min at 40°C. This was followed by HSATII-Type1 (Catalogue No.VA1–10874) probe hybridization with 1:20 dilution for 3 hours at 40°C. After the run, the slides were rinsed with water and allowed to air dry for 1 hour at RT. The slides were mounted using Micromount (Leica Biosystems, Catalogue No. 3801731) and visualized using a standard bright-field microscope. “Dot like” red color hybridization signals in the tumor cell nuclei and cytoplasm were defined as HSATII positive signals and quantified using Visiopharm software.
HSATII (ACD 512018) & LINE1 (ACD 565098) were also detected using RNAscope 2.5 LS Reagent Kit-Brown from Advanced Cell Diagnostics (ACD) (Catalogue No.322100) on the BondRx platform. On the BOND RX, the staining protocol used was the ACD ISH DAB Protocol. The RNA unmasking conditions for the tissue consisted of a 15-minute incubation at 95°C in Bond Epitope Retrieval Solution 2 (Leica Biosystems) followed by 15-minute incubation with Proteinase K which was provided in the kit. Probe hybridization was done for 2 hours with RNAscope probes which were provided by ACD.
For Immunohistochemistry, FFPE sections were deparaffinized by baking them for 1 hour at 60°C. IHC staining was done on the BondRx using the BOND Polymer Refine Detection kit (Catalogue No. DS9800). Antigen retrieval was carried out for 10 mins for cell blocks, 15 mins for Xenografts, and 20 mins for human tissue using Bond Epitope Retrieval Solution 2 (Leica Biosystems). LINE1 Orf1p antibody was used at a concentration of 1:3000 (EMD Millipore, MABC1152)
Immunofluorescence (IF):
Cells were washed with PBS, resuspended to 50,000 cells/ml and fixed in 4% paraformaldehyde for 15 minutes at room temperature (RT). Cell pellets were washed 3 times in PBS and then cytospun using a volume of 300 μl/spot. Cytospun cells were treated with 0.02% saponin for 5min at RT. Permeabilization of cells using 1% Triton for 4 min at room temperature (RT). Cells were washed 3 × 10 min with standard IF Wash Buffer. Samples blocked in 50 μl blocking buffer for 1hr at RT. Blocking buffer was then aspirated and 50 μl primary Ab was added. Samples were incubated overnight at 4ºC. Cells were then wash 3 × 10 min with IF Wash Buffer and 50 μl secondary antibody (Goat Anti-Mouse IgG H&L Alexa Fluor® 488 (ab150113) used at 1:1000) was added and incubated for 1 hr at RT in the dark. Samples washed 3 × 10 min with IF Wash Buffer. The samples were mounted with Vectashield and coverslipped with clear nail polish at the edges.
IF Wash Buffer
0.5% NP40
0.3% w/v Sodium Azide
In 1x PBS made in MilliQ H2O
IF Block Buffer
10% BSA in IF Wash Buffer
Antibodies
dsDNA (AB27156): mouse mAb; 0.910 mg/ml. 1:1000 dilution
S9.6 (Kerafast): 1:400 dilution
Image Analysis and quantification:
Cell Migration Assay: The migration assay plates stained with crystal violet were imaged in brightfield with a 4X objective on an inverted tissue culture microscope. Migration was quantified as an area percentage using color-deconvolution in the HALO software Immunohistochemistry Area Quantification Module.
Soft Agar Assay (Bright Field): Soft agar plates stained with Crystal violet were imaged in brightfield with a 1X objective on an Olympus MVX10 macro zoom microscope. The size of each colony and colony density for each treatment condition was quantified using the HALO software Immunohistochemistry Module. Colonies were identified based on contrast, intensity and size of the color-deconvolved blue stain. Colony size, and colony number were represented in figures.
Immunohistochemistry/RNA-ISH (Bright Field): Slides were scanned at 40X resolution using Aperio Scanscope slide scanner (Leica Biosystems). The human rectal carcinoma tissue sections and tumorsphere sections stained with HSATII RNA-ISH, LINE-1 RNA-ISH, or LINE-1 IHC were imaged with a Leica Aperio Scanscope CSO (Leica Biosystems, Wetzlar, Germany). Stain quantification was performed with the digital image analysis software VIS (Visiopharm, Hørsholm, Denmark). Tumor regions of the human tissue images were annotated by a pathologist to mark the regions for analysis. Twenty 100um x 100um square regions of interest (ROIs) were manually annotated throughout each tumorsphere image to mark the regions for analysis. The next step of the analysis was the training of classifiers to extract and separate the Hematoxylin, red or brown, and background regions. Representative stain regions were manually selected to constitute the training set. The classification method was chosen to be Bayesian or K-means clustering, depending on visual validation of the resulting classification. The classifiers were trained using multiple image features, including color deconvolution of the individual stains, spatial mean and median filters to remove noise and enhance punctate signals, and the Visiopharm Polynomial Blobs filter to further enhance punctate signals for ISH quantification. The red and Hematoxylin classifier used the following color features: the green band of the RGB color space, red-blue contrast, and red chromaticity. The brown ISH or IHC and Hematoxylin classifier used the following color features: the green and blue bands of the RGB color space, the saturation band of the IHS color model, green chromaticity, and a custom defined Hematoxylin and DAB color model. After classification, post-processing steps were performed to finetune the separation of the Hematoxlyin, red or brown, and background regions. Post-processing steps included hole-filling and minimum thresholds for size and signal intensity. The following metrics were outputted by the analysis algorithm for each image: total red or brown area, total Hematoxylin area, red or brown area percentage of the total stain area, and mean signal intensity of red or brown (per image for human tissue, and per ROI for tumorspheres). The red or brown area percentage of the total stain area was calculated as follows: 100 x red or brown area / (red or brown area + Hematoxylin area).
Immunofluorescence (Fluorescent): Slides stained with dsDNA or RNA:DNA Hybrid (S9.6) antibody (Kerafast, ENH001) were imaged in DAPI and AF488 with a Zeiss LSM710 confocal microscope (MGH Cancer Center / Molecular Pathology Confocal Core). dsDNA slides were imaged with a 40x oil immersion objective, and RNA/DNA hybrid slides were imaged with a 63x oil immersion objective. AF488 signal per cell was quantified using digital image analysis, which was performed with the HALO software platform (Indica Labs, Albuquerque, NM) Fluorescence Module. Non-cellular material and cells with staining artifacts were manually excluded from analysis, leaving about 800 cells per treatment condition to be included for analysis. The first step in analysis was segmentation of nuclei based on contrast, intensity and size of the DAPI stain. Next, a distance from the nucleus was chosen to define the cytoplasmic region in the absence of neighboring cells. Average AF488 signal per cell was quantified in three compartments: nucleus, cytoplasm, and the entire cell. Cytoplasmic signal was represented in figures.
Immunoblotting:
For western blot cells were lysed in Laemlli buffer and run on a standard denaturing SDS-PAGE gel (Thermofisher) followed by transfer to PVDF membrane (primed in 10% methanol). Blocking was conducted for 1 hour at room temperature with 5% BSA/PBS-T followed by overnight incubation at 40 C with primary antibody for phospho-histone-h2a-x-ser139 (Cell signaling, 9718S), STING (Cell signaling, 13647) or GAPDH (Cell signaling, 2118S), followed by 3X washes with PBS-T and incubation with anti-rabbit secondary antibody diluted 1:1000 in 5% BSA/PBS-T. Signal was detected using Supersignal WestPico (ThermoFisher).
Supplementary Material
STATEMENT OF SIGNIFICANCE.
Colorectal cancers express abundant repeat elements that have a viral-like life cycle that can be therapeutically targeted with nucleoside reverse transcriptase inhibitors (NRTIs) commonly used for viral diseases. NRTIs induces DNA damage and interferon response that provide a new anti-cancer therapeutic strategy.
Acknowledgments:
We are grateful to Laura Libby for mouse colony care, Emily Silva, and Danielle Bestoso for administrative support. We appreciate productive discussions with Arnold Levine and Junne Kamihara on this work.
Funding:
National Institutes of Health grants R01CA240924 (DTT, BDG), U01CA228963 (BDG, DTT), R01GM130680 (KHB), R01CA240816 (KHB), T32GM007753 (RYE), 1F30CA232407–01 (RYE)
Gateway for Cancer Research G-17–1000 (DTT, ARP)
National Science Foundation NSF PHY-1549535 (DTT, BDG)
Burroughs Wellcome Fund 1010968.01 (DTT)
V Foundation for Cancer Research D2015–034 (MR)
SU2C AACR-PS-17 (DTT, SLB)
SU2C-Lustgarten Foundation 2015–002 (DTT, BDG)
Affymetrix, Inc. (DTT, KSA, ND, MNR, VD)
ACD-Biotechne (DTT, ASK, MNR, VD)
Robert L. Fine Cancer Research Foundation (DTT)
The Pershing Square Sohn Prize—Mark Foundation Fellowship (BDG)
Footnotes
Conflict of Interest:
The following authors have filed patents related to targeting repeat RNAs and their use as novel biomarkers (MR, AS, KSA, MNR, VD, BDG, DTT).
DTT and BDG are co-founders, own equity, and receive consulting fees from ROME Therapeutics, a company developing drugs targeting repetitive RNA expressed in cancers and other diseases. This work was not supported by ROME Therapeutics. DTT’s interests were reviewed and are managed by Mass General Brigham in accordance with their conflict of interest policies. Parts of this work was supported by ACD-Biotechne (ASK, VD, MNR, DTT).
DTT has received consulting fees from Tekla Capital Management, Ikena Oncology, NanoString Technologies, Pfizer, Merrimack Pharmaceuticals, Ventana Roche, Foundation Medicine, Inc., and EMD Millipore Sigma, which are not related to this work. DTT is a founder and has equity in PanTher Therapeutics and TellBio, Inc., which are not related to this work.
BDG is a consultant or received honoraria for Darwin Health, Merck, PMV Pharma, ROME Therapeutics, Merck, Bristol–Meyers Squibb, and Chugai Pharmaceuticals and has research funding from Bristol-Meyers Squibb.
ARP is a consultant/advisory board member for Eli Lilly, Natera, Checkmate Pharmaceuticals, Inivata, and Pfizer; holds equity in C2I; serves on the DSMC for Roche; and has research funding from Puretech, PMV Pharmaceuticals, Plexxikon, Takeda, BMS, Novartis, Genentech, Guardant, Array, and Eli Lilly.
MST is a consultant for ROME therapeutics.
JWC is author for McGraw Hill and UpToDate.
LG is a consultant/advisory board member for Alentis, AstraZeneca, Exelixis, and Sirtex, Genentech, Genentech, H3Biomedicine, Incyte, QED Therapeutics, Servier, and Taiho; and has research funding from Adaptimmune, Bayer, Bristol-Myers Squibb, Eisai, Leap Therapeutics, Loxo Oncology, MacroGenics, Merck, Novartis, Nucana, Relay Therapeutics, Genentech, H3Biomedicine, Incyte, QED Therapeutics, Servier, and Taiho.
DPR is a consultant/advisory board member for MPM Capital, Gritstone Oncology, Oncorus, Maverick Therapeutics, 28/7 Therapeutics, Thrive/Exact Sciences; has equity in MPM Capital, Acworth Pharmaceuticals, and Thrive/Exact Sciences; is a legal consultant for Boeringer Ingelheim; and serves as author for Johns Hopkins University Press, UpToDate, McGraw Hill.
RBC is a consultant/advisory board member for Abbvie, Amgen, Array Biopharma/Pfizer, Asana Biosciences, Astex Pharmaceuticals, AstraZeneca, Avidity Biosciences, BMS, C4 Therapeutics, Chugai, Elicio, Erasca, Fog Pharma, Genentech, Guardant Health, Ipsen, Kinnate Biopharma, LOXO, Merrimack, Mirati Therapeutics, Natera, Navire, N-of-one/Qiagen, Novartis, nRichDx, Remix Therapeutics, Revolution Medicines, Roche, Roivant, Shionogi, Shire, Spectrum Pharmaceuticals, Symphogen, Tango Therapeutics, Taiho, Warp Drive Bio, Zikani Therapeutics; holds equity in Avidity Biosciences, C4 Therapeutics, Erasca, Kinnate Biopharma, nRichDx, Remix Therapeutics, and Revolution Medicines; and has research funding from Asana, AstraZeneca, Lilly, Novartis, and Sanofi.
DRW is an inventor of the Simoa technology and a founder, board member, and equity holder of Quanterix Corporation. Interests of DRW have been reviewed and are managed by Brigham and Women’s Hospital and Mass General Brigham in accordance with their policies on competing interests.
KHB has consulted for Rome Therapeutics, Tessera Therapeutics, Transposon Therapeutics, and is the Scientific Co-founder of Oncolinea Pharmaceuticals.
All other authors declare no competing interests.
Data and materials availability:
All RNA-seq and WGS data from clinical biopsy specimens has been uploaded to NCBI SRA accession number phs002833.v1.p1. All RNA-seq and ChIP-seq from cell lines and xenografts have been uploaded to NCBI GEO accession number GSE199097. All software for WGS, RNAseq, ChIP-seq, and digital image data analysis is described in the methods and all software will be provided upon request.
REFERENCES
- 1.Probst AV, Okamoto I, Casanova M, El Marjou F, Le Baccon P, Almouzni G. A strand-specific burst in transcription of pericentric satellites is required for chromocenter formation and early mouse development. Dev Cell 2010;19(4):625–38 doi 10.1016/j.devcel.2010.09.002. [DOI] [PubMed] [Google Scholar]
- 2.Kano H, Godoy I, Courtney C, Vetter MR, Gerton GL, Ostertag EM, et al. L1 retrotransposition occurs mainly in embryogenesis and creates somatic mosaicism. Genes Dev 2009;23(11):1303–12 doi 23/11/1303 [pii] 10.1101/gad.1803909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ting DT, Lipson D, Paul S, Brannigan BW, Akhavanfard S, Coffman EJ, et al. Aberrant overexpression of satellite repeats in pancreatic and other epithelial cancers. Science 2011;331(6017):593–6 doi science.1200801 [pii] 10.1126/science.1200801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 2015;160(1–2):48–61 doi 10.1016/j.cell.2014.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zapatka M, Borozan I, Brewer DS, Iskar M, Grundhoff A, Alawi M, et al. The landscape of viral associations in human cancers. Nature genetics 2020;52(3):320–30 doi 10.1038/s41588-019-0558-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Desai N, Sajed D, Arora KS, Solovyov A, Rajurkar M, Bledsoe JR, et al. Diverse repetitive element RNA expression defines epigenetic and immunologic features of colon cancer. JCI Insight 2017;2(3):e91078 doi 10.1172/jci.insight.91078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Leonova KI, Brodsky L, Lipchick B, Pal M, Novototskaya L, Chenchik AA, et al. p53 cooperates with DNA methylation and a suicidal interferon response to maintain epigenetic silencing of repeats and noncoding RNAs. Proc Natl Acad Sci U S A 2013;110(1):E89–98 doi 10.1073/pnas.1216922110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roulois D, Loo Yau H, Singhania R, Wang Y, Danesh A, Shen SY, et al. DNA-Demethylating Agents Target Colorectal Cancer Cells by Inducing Viral Mimicry by Endogenous Transcripts. Cell 2015;162(5):961–73 doi 10.1016/j.cell.2015.07.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guler GD, Tindell CA, Pitti R, Wilson C, Nichols K, KaiWai Cheung T, et al. Repression of Stress-Induced LINE-1 Expression Protects Cancer Cell Subpopulations from Lethal Drug Exposure. Cancer cell 2017;32(2):221–37 e13 doi [DOI] [PubMed] [Google Scholar]
- 10.Chiappinelli KB, Strissel PL, Desrichard A, Li H, Henke C, Akman B, et al. Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses. Cell 2015;162(5):974–86 doi 10.1016/j.cell.2015.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sheng W, LaFleur MW, Nguyen TH, Chen S, Chakravarthy A, Conway JR, et al. LSD1 Ablation Stimulates Anti-tumor Immunity and Enables Checkpoint Blockade. Cell 2018;174(3):549–63 e19 doi 10.1016/j.cell.2018.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Griffin GK, Wu J, Iracheta-Vellve A, Patti JC, Hsu J, Davis T, et al. Epigenetic silencing by SETDB1 suppresses tumour intrinsic immunogenicity. Nature 2021;595(7866):309–14 doi 10.1038/s41586-021-03520-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tanne A, Muniz LR, Puzio-Kuter A, Leonova KI, Gudkov AV, Ting DT, et al. Distinguishing the immunostimulatory properties of noncoding RNAs expressed in cancer cells. Proc Natl Acad Sci U S A 2015;112(49):15154–9 doi 10.1073/pnas.1517584112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee E, Iskow R, Yang L, Gokcumen O, Haseley P, Luquette LJ 3rd, et al. Landscape of somatic retrotransposition in human cancers. Science 2012;337(6097):967–71 doi science.1222077 [pii] 10.1126/science.1222077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rodriguez-Martin B, Alvarez EG, Baez-Ortega A, Zamora J, Supek F, Demeulemeester J, et al. Pan-cancer analysis of whole genomes identifies driver rearrangements promoted by LINE-1 retrotransposition. Nature genetics 2020;52(3):306–19 doi 10.1038/s41588-019-0562-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bersani F, Lee E, Kharchenko PV, Xu AW, Liu M, Xega K, et al. Pericentromeric satellite repeat expansions through RNA-derived DNA intermediates in cancer. Proc Natl Acad Sci U S A 2015;112(49):15148–53 doi 10.1073/pnas.1518008112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bakhoum SF, Ngo B, Laughney AM, Cavallo JA, Murphy CJ, Ly P, et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 2018;553(7689):467–72 doi 10.1038/nature25432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kwon J, Bakhoum SF. The Cytosolic DNA-Sensing cGAS-STING Pathway in Cancer. Cancer discovery 2020;10(1):26–39 doi 10.1158/2159-8290.CD-19-0761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dai L, Huang Q, Boeke JD. Effect of reverse transcriptase inhibitors on LINE-1 and Ty1 reverse transcriptase activities and on LINE-1 retrotransposition. BMC biochemistry 2011;12:18 doi 10.1186/1471-2091-12-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Contreras-Galindo RA, Dube D, Fujinaga K, Kaplan MH, Markovitz DM. Susceptibility of Human Endogenous Retrovirus Type-K to Reverse Transcriptase Inhibitors. Journal of virology 2017;91(23):e01309–17 doi 10.1128/JVI.01309-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wood JG, Jones BC, Jiang N, Chang C, Hosier S, Wickremesinghe P, et al. Chromatin-modifying genetic interventions suppress age-associated transposable element activation and extend life span in Drosophila. Proc Natl Acad Sci U S A 2016;113(40):11277–82 doi 10.1073/pnas.1604621113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.De Cecco M, Ito T, Petrashen AP, Elias AE, Skvir NJ, Criscione SW, et al. L1 drives IFN in senescent cells and promotes age-associated inflammation. Nature 2019;566(7742):73–8 doi 10.1038/s41586-018-0784-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Simon M, Van Meter M, Ablaeva J, Ke Z, Gonzalez RS, Taguchi T, et al. LINE1 Derepression in Aged Wild-Type and SIRT6-Deficient Mice Drives Inflammation. Cell Metabolism 2019;29(4):871–85.e5 doi 10.1016/j.cmet.2019.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wylie A, Jones AE, D’Brot A, Lu WJ, Kurtz P, Moran JV, et al. p53 genes function to restrain mobile elements. Genes Dev 2016;30(1):64–77 doi 10.1101/gad.266098.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jung H, Choi JK, Lee EA. Immune signatures correlate with L1 retrotransposition in gastrointestinal cancers. Genome Res 2018;28(8):1136–46 doi 10.1101/gr.231837.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Solovyov A, Vabret N, Arora KS, Snyder A, Funt SA, Bajorin DF, et al. Global Cancer Transcriptome Quantifies Repeat Element Polarization between Immunotherapy Responsive and T Cell Suppressive Classes. Cell reports 2018;23(2):512–21 doi 10.1016/j.celrep.2018.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhu J, Sammons MA, Donahue G, Dou Z, Vedadi M, Getlik M, et al. Gain-of-function p53 mutants co-opt chromatin pathways to drive cancer growth. Nature 2015;525(7568):206–11 doi 10.1038/nature15251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Levine AJ, Ting DT, Greenbaum BD. P53 and the defenses against genome instability caused by transposons and repetitive elements. BioEssays : news and reviews in molecular, cellular and developmental biology 2016;38(6):508–13 doi 10.1002/bies.201600031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chu C, Borges-Monroy R, Viswanadham VV, Lee S, Li H, Lee EA, et al. Comprehensive identification of transposable element insertions using multiple sequencing technologies. Nature communications 2021;12(1):3836 doi 10.1038/s41467-021-24041-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cohen L, Cui N, Cai Y, Garden PM, Li X, Weitz DA, et al. Single Molecule Protein Detection with Attomolar Sensitivity Using Droplet Digital Enzyme-Linked Immunosorbent Assay. ACS Nano 2020;14(8):9491–501 doi 10.1021/acsnano.0c02378. [DOI] [PubMed] [Google Scholar]
- 31.Mankan AK, Schmidt T, Chauhan D, Goldeck M, Höning K, Gaidt M, et al. Cytosolic RNA:DNA hybrids activate the cGAS-STING axis. Embo j 2014;33(24):2937–46 doi 10.15252/embj.201488726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boye K, Jacob H, Frikstad KA, Nesland JM, Maelandsmo GM, Dahl O, et al. Prognostic significance of S100A4 expression in stage II and III colorectal cancer: results from a population-based series and a randomized phase III study on adjuvant chemotherapy. Cancer Med 2016;5(8):1840–9 doi 10.1002/cam4.766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Boye K, Nesland JM, Sandstad B, Haugland Haugen M, Mælandsmo GM, Flatmark K. EMMPRIN is associated with S100A4 and predicts patient outcome in colorectal cancer. Br J Cancer 2012;107(4):667–74 doi 10.1038/bjc.2012.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kang YG, Jung CK, Lee A, Kang WK, Oh ST, Kang CS. Prognostic significance of S100A4 mRNA and protein expression in colorectal cancer. J Surg Oncol 2012;105(2):119–24 doi 10.1002/jso.22070. [DOI] [PubMed] [Google Scholar]
- 35.Sack U, Walther W, Scudiero D, Selby M, Aumann J, Lemos C, et al. S100A4-induced cell motility and metastasis is restricted by the Wnt/β-catenin pathway inhibitor calcimycin in colon cancer cells. Mol Biol Cell 2011;22(18):3344–54 doi 10.1091/mbc.E10-09-0739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang H, Shi J, Luo Y, Liao Q, Niu Y, Zhang F, et al. LIM and SH3 protein 1 induces TGFβ-mediated epithelial-mesenchymal transition in human colorectal cancer by regulating S100A4 expression. Clinical cancer research : an official journal of the American Association for Cancer Research 2014;20(22):5835–47 doi [DOI] [PubMed] [Google Scholar]
- 37.Zuo Z, Zhang P, Lin F, Shang W, Bi R, Lu F, et al. Interplay between Trx-1 and S100P promotes colorectal cancer cell epithelial-mesenchymal transition by up-regulating S100A4 through AKT activation. J Cell Mol Med 2018;22(4):2430–41 doi 10.1111/jcmm.13541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Watanabe H, Ishibashi K, Mano H, Kitamoto S, Sato N, Hoshiba K, et al. Mutant p53-Expressing Cells Undergo Necroptosis via Cell Competition with the Neighboring Normal Epithelial Cells. Cell reports 2018;23(13):3721–9 doi 10.1016/j.celrep.2018.05.081. [DOI] [PubMed] [Google Scholar]
- 39.Zhu Q, Hoong N, Aslanian A, Hara T, Benner C, Heinz S, et al. Heterochromatin-Encoded Satellite RNAs Induce Breast Cancer. Molecular cell 2018;70(5):842–53 e7 doi 10.1016/j.molcel.2018.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhu Q, Pao GM, Huynh AM, Suh H, Tonnu N, Nederlof PM, et al. BRCA1 tumour suppression occurs via heterochromatin-mediated silencing. Nature 2011;477(7363):179–84 doi nature10371 [pii] 10.1038/nature10371 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kishikawa T, Otsuka M, Yoshikawa T, Ohno M, Ijichi H, Koike K. Satellite RNAs promote pancreatic oncogenic processes via the dysfunction of YBX1. Nature communications 2016;7(13006):1–12 doi 10.1038/ncomms13006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ardeljan D, Steranka JP, Liu C, Li Z, Taylor MS, Payer LM, et al. Cell fitness screens reveal a conflict between LINE-1 retrotransposition and DNA replication. Nat Struct Mol Biol 2020;27(2):168–78 doi 10.1038/s41594-020-0372-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mita P, Sun X, Fenyo D, Kahler DJ, Li D, Agmon N, et al. BRCA1 and S phase DNA repair pathways restrict LINE-1 retrotransposition in human cells. Nat Struct Mol Biol 2020;27(2):179–91 doi 10.1038/s41594-020-0374-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Taylor MS, Altukhov I, Molloy KR, Mita P, Jiang H, Adney EM, et al. Dissection of affinity captured LINE-1 macromolecular complexes. Elife 2018;7 doi 10.7554/eLife.30094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stavrou S, Aguilera AN, Blouch K, Ross SR. DDX41 Recognizes RNA/DNA Retroviral Reverse Transcripts and Is Critical for In Vivo Control of Murine Leukemia Virus Infection. mBio 2018;9(3):e00923–18 doi 10.1128/mBio.00923-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Parikh AR, Szabolcs A, Allen JN, Clark JW, Wo JY, Raabe M, et al. Radiation therapy enhances immunotherapy response in microsatellite stable colorectal and pancreatic adenocarcinoma in a phase II trial. Nature Cancer 2021;2(11):1124–35 doi 10.1038/s43018-021-00269-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Smith CC, Beckermann KE, Bortone DS, De Cubas AA, Bixby LM, Lee SJ, et al. Endogenous retroviral signatures predict immunotherapy response in clear cell renal cell carcinoma. The Journal of clinical investigation 2018;128(11):4804–20 doi 10.1172/JCI121476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Panda A, de Cubas AA, Stein M, Riedlinger G, Kra J, Mayer T, et al. Endogenous retrovirus expression is associated with response to immune checkpoint blockade in clear cell renal cell carcinoma. JCI Insight 2018;3(16) doi 10.1172/jci.insight.121522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hughes SM, Levy CN, Calienes FL, Stekler JD, Pandey U, Vojtech L, et al. Treatment with Commonly Used Antiretroviral Drugs Induces a Type I/III Interferon Signature in the Gut in the Absence of HIV Infection. Cell Rep Med 2020;1(6):100096 doi 10.1016/j.xcrm.2020.100096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Coghill AE, Engels EA, Schymura MJ, Mahale P, Shiels MS. Risk of Breast, Prostate, and Colorectal Cancer Diagnoses Among HIV-Infected Individuals in the United States. J Natl Cancer Inst 2018;110(9):959–66 doi 10.1093/jnci/djy010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ho DD. Time to hit HIV, early and hard. N Engl J Med 1995;333(7):450–1 doi 10.1056/nejm199508173330710. [DOI] [PubMed] [Google Scholar]
- 52.Perelson AS, Neumann AU, Markowitz M, Leonard JM, Ho DD. HIV-1 dynamics in vivo: virion clearance rate, infected cell life-span, and viral generation time. Science 1996;271(5255):1582–6 doi 10.1126/science.271.5255.1582. [DOI] [PubMed] [Google Scholar]
- 53.Zhou X, Singh M, Sanz Santos G, Guerlavais V, Carvajal LA, Aivado M, et al. Pharmacological activation of p53 triggers viral mimicry response thereby abolishing tumor immune evasion and promoting anti-tumor immunity. Cancer discovery 2021. doi 10.1158/2159-8290.CD-20-1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2012;29(1):15–21 doi 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liao Y, Smyth GK, Shi W. The Subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res 2013;41(10):e108 doi 10.1093/nar/gkt214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Frankish A, Diekhans M, Ferreira AM, Johnson R, Jungreis I, Loveland J, et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res 2019;47(D1):D766–D73 doi 10.1093/nar/gky955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010;26(1):139–40 doi 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Robinson MD, Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol 2010;11(3):R25 doi 10.1186/gb-2010-11-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15(12):550 doi 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Korotkevich G, Sukhov V, Budin N, Shpak B, Artyomov MN, Sergushichev A. Fast gene set enrichment analysis. bioRxiv 2021:060012 doi 10.1101/060012. [DOI] [Google Scholar]
- 61.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009;25(14):1754–60 doi 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics 2013;43:11 0 1–0 33 doi 10.1002/0471250953.bi1110s43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009;25(16):2078–9 doi 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lee S, Lee S, Ouellette S, Park WY, Lee EA, Park PJ. NGSCheckMate: software for validating sample identity in next-generation sequencing studies within and across data types. Nucleic Acids Res 2017;45(11):e103 doi 10.1093/nar/gkx193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102(43):15545–50 doi 0506580102 [pii] 10.1073/pnas.0506580102 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nature genetics 2003;34(3):267–73 doi 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 67.Shah PP, Donahue G, Otte GL, Capell BC, Nelson DM, Cao K, et al. Lamin B1 depletion in senescent cells triggers large-scale changes in gene expression and the chromatin landscape. Genes Dev 2013;27(16):1787–99 doi 10.1101/gad.223834.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods 2012;9(4):357–9 doi 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol 2008;9(9):R137 doi 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 2010;26(6):841–2 doi 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All RNA-seq and WGS data from clinical biopsy specimens has been uploaded to NCBI SRA accession number phs002833.v1.p1. All RNA-seq and ChIP-seq from cell lines and xenografts have been uploaded to NCBI GEO accession number GSE199097. All software for WGS, RNAseq, ChIP-seq, and digital image data analysis is described in the methods and all software will be provided upon request.