

SUMMARY
Resolving glial contributions to Alzheimer’s disease (AD) is necessary because changes in neuronal function, like reduced synaptic density, altered electrophysiological properties, and degeneration, are not entirely cell autonomous. To improve understanding of transcriptomic heterogeneity in glia during AD, we used single nuclei RNA sequencing (snRNA-seq) to characterize astrocytes and oligodendrocytes from APOE ε2/3 human AD and age- and genotype-matched non-symptomatic (NS) brains. We enriched for astrocytes before sequencing and characterized pathology from the same location as the sequenced material. We characterized baseline heterogeneity in both astrocytes and oligodendrocytes, and identified global and subtype-specific transcriptomic changes between AD and NS astrocytes and oligodendrocytes. We also take advantage of recent human and mouse spatial transcriptomics resources to localize heterogeneous astrocyte subtypes to specific regions in the healthy and inflamed brain. Finally, we integrated our data with published AD snRNA-seq datasets, highlighting the power of combining datasets to resolve previously unidentifiable astrocyte subpopulations.
eTOC Blurb
Transcriptomic and functional changes in glia are hallmarks of Alzheimer’s disease. In this issue of Neuron, Sadick, O’Dea, et al. define transcriptomic differences in astrocytes and oligodendrocytes in Alzheimer’s disease at the single nuclei level. They also localize human Alzheimer’s-associated transcription profiles to strategic location in the inflamed mouse brain.
INTRODUCTION
Alzheimer’s disease (AD) is the most common age-related neurodegenerative disorder in the world and accounts for about 70% of the 50 million people worldwide with dementia, with an estimated 10 million new cases each year, or close to 20 new cases every minute (Alzheimer’s Association, 2021). With an aging population, greater understanding of AD pathogenesis is imperative for development of effective therapies. Defined by progressive memory and cognitive loss, AD is a disease of proteopathic stress associated with abnormal beta-amyloid (Aβ) and tau aggregation, and neuron death (De Strooper and Karran, 2016; Ginsberg et al., 2006; Mattsson et al., 2016; Yue and Jing, 2015). More recently, research highlights non-neuronal central nervous system (CNS) cells, namely glia, as active contributors to AD pathophysiology. Increasing evidence supports a pathological role of ‘reactive’ astrocytes in acute and chronic diseases, like AD (Carter et al., 2012; Owen et al., 2009; Sadick and Liddelow, 2019; Schipper et al., 2006). This includes changes in normal physiological functions like synapse maintenance (Hong et al., 2016), blood-brain barrier (BBB) integrity (de la Torre, 2004; Farkas and Luiten, 2001; Viswanathan and Greenberg, 2011), metabolism (Gonzalez-Reyes et al., 2017), and structural support (Mohamed et al., 2016; Teaktong et al., 2003; Wu et al., 2015). Many studies implicate reactive astrocytes induced by immune cell dysfunction and inflammatory responses to pathogenic proteins (Aβ and tau) and dying cells (Hasel et al., 2021; Liddelow et al., 2017; Shi et al., 2017; Zamanian et al., 2012). Oligodendrocytes are also of interest in AD pathology given their role in myelin production and axon support (Funfschilling et al., 2012; Saab et al., 2016). White matter abnormalities (e.g. lesions, decreased volume, microstructural deterioration), and demyelination are well documented in AD (Lee et al., 2016; Radanovic et al., 2013). Beyond these broad physiological changes, many questions remain, such as how subtypes of astrocytes and oligodendrocytes are altered in AD.
Single nuclei RNA sequencing (snRNA-seq) has been used to assess cellular heterogeneity at the transcriptomic level in human AD postmortem brains (Del-Aguila et al., 2019; Gerrits et al., 2021; Grubman et al., 2019; Lau et al., 2020; Leng et al., 2021; Mathys et al., 2019; Zhou et al., 2020). In most studies however, astrocytes are underrepresented, making up only ~3-18% of nuclei. Therefore, we posit that these studies may not provide large enough samples to confidently define biologically important, but lowly abundant, astrocyte subpopulations - a hypothesis supported by the fact that similar subpopulations are not identified across published datasets. In contrast, oligodendrocytes are well-represented in snRNA-seq studies, with some agreement in identified subpopulations. In addition, given the important spatially-confined responses of astrocytes to AD pathology (e.g. surrounding amyloid plaques, or near degenerated brain regions), no studies have combined pathological characterization with sequencing efforts on the same tissue samples. Several studies provided pathological analysis of contralateral or adjacent brain regions, but these may be far-removed from local pathology-induced microenvironments (Gerrits et al., 2021; Leng et al., 2021; Mathys et al., 2019). A lack of concordance may also be reflective of variability with respect to pathological load and/or underlying genetic variance across donors.
Here we present a snRNA-seq dataset characterizing astrocytes and oligodendrocytes isolated from human postmortem prefrontal cortex samples from AD and age-matched non-symptomatic (NS) patients. All individuals were genotyped and controlled for apolipoprotein (APOE) genotype ε2/3. We chose this patient population as it is under-represented in other sequencing studies, and individuals present with AD dementia onset at later ages (onset ~80 years (Reiman et al., 2020)), making age matching between NS and AD patients more similar. In addition, APOEε2/3 individuals have a low odd-risk ratio of contracting AD compared to individuals that carry an APOEε4 allele (Goldberg et al., 2020; Reiman et al., 2020).
In order to improve astrocyte capture for snRNA-seq, we enriched for astrocytes by sorting nuclei based on LIM Homeobox 2 (LHX2)-positive/NeuN-negative staining followed by 10X Genomics-based snRNAseq. This method enriches for astrocytes while maintaining capture rates of oligodendrocytes and depleting neurons. We characterized the pathology of donor tissue from the same location that nuclei were isolated for sequencing. We identified global and subtype-specific transcriptomic changes between AD and NS astrocytes and oligodendrocytes. Additionally, we localized heterogeneous astrocyte subtypes to specific cortical regions in the healthy and inflamed brain using published human and mouse spatial transcriptomics datasets. Last, we integrated our data with published AD snRNA-seq datasets and resolve unique astrocyte subpopulations present across datasets. Overall, we provide a paired sequencing and pathology assessment resource that can be used to further explore the breadth of astrocyte and oligodendrocyte transcriptomic changes in AD.
RESULTS
Defining a well-controlled patient cohort is key for defining AD-associated gene expression profiles
We first decided to reanalyze and compare three recent studies (Grubman et al., 2019; Mathys et al., 2019; Zhou et al., 2020) and found that astrocytes made up on average 15% ± 8.7% of total nuclei captured (2,300-23,000 per study – on average less than 400 astrocytes per donor; Figure S1A–C, Table S1). Given astrocytes are one of the most abundant CNS cell types (Nedergaard et al., 2003; von Bartheld et al., 2016), this low capture rate was surprising. In contrast, other cell populations were well represented. In addition, donors varied in APOE genotype, which may confound resulting profiles of astrocytes given they highly express APOE (Zhang et al., 2016). Therefore we focused on APOE ε2/3 donors, a population of patients that has been understudied. Our approach enabled us to: (1) conduct a highly stringent evaluation of our donor cohort, and (2) optimize enrichment methods for astrocytes prior to sequencing for improved astrocyte capture rates.
We characterized our patient cohort by verifying APOE genotype (Figure S1D) and evaluating pathological load of donor tissue for Aβ (4G8), phosphorylated tau (AT8), glial fibrillary acidic protein (GFAP), and Bielschowsky’s silver staining (Figure 1A, Table S2). Each stain was quantified for normalized staining density, total count of features (i.e., number of 4G8+ Aβ plaques or AT8+ cell bodies), and average cross-sectional area of each feature (Table S2). We evaluated diffuse plaques, neuritic plaques, and neurofibrillary tangle loads (Table S2). In line with clinical diagnoses, phosphorylated tau staining density and total number of AT8+ cell bodies were significantly higher in AD donors compared to NS donors (p = 0.0047). In contrast, we did not detect major differences in Aβ plaque staining density or area or GFAP+ staining density or area between disease conditions. Interestingly, individual AT8+ cell bodies were significantly larger in NS donors than AD donors (p = 0.022) – likely driven by differences in the number of AT8+ cell bodies detected between disease conditions (AT8+ cell bodies counted: 141 NS versus 57,829 AD). Based on Bielschowsky’s silver staining, we classified NS donors as having no or low pathology loads, while AD donors had moderate or severe pathology loads.
Figure 1. Defining a well-controlled patient cohort is key for defining AD-associated gene expression profiles.
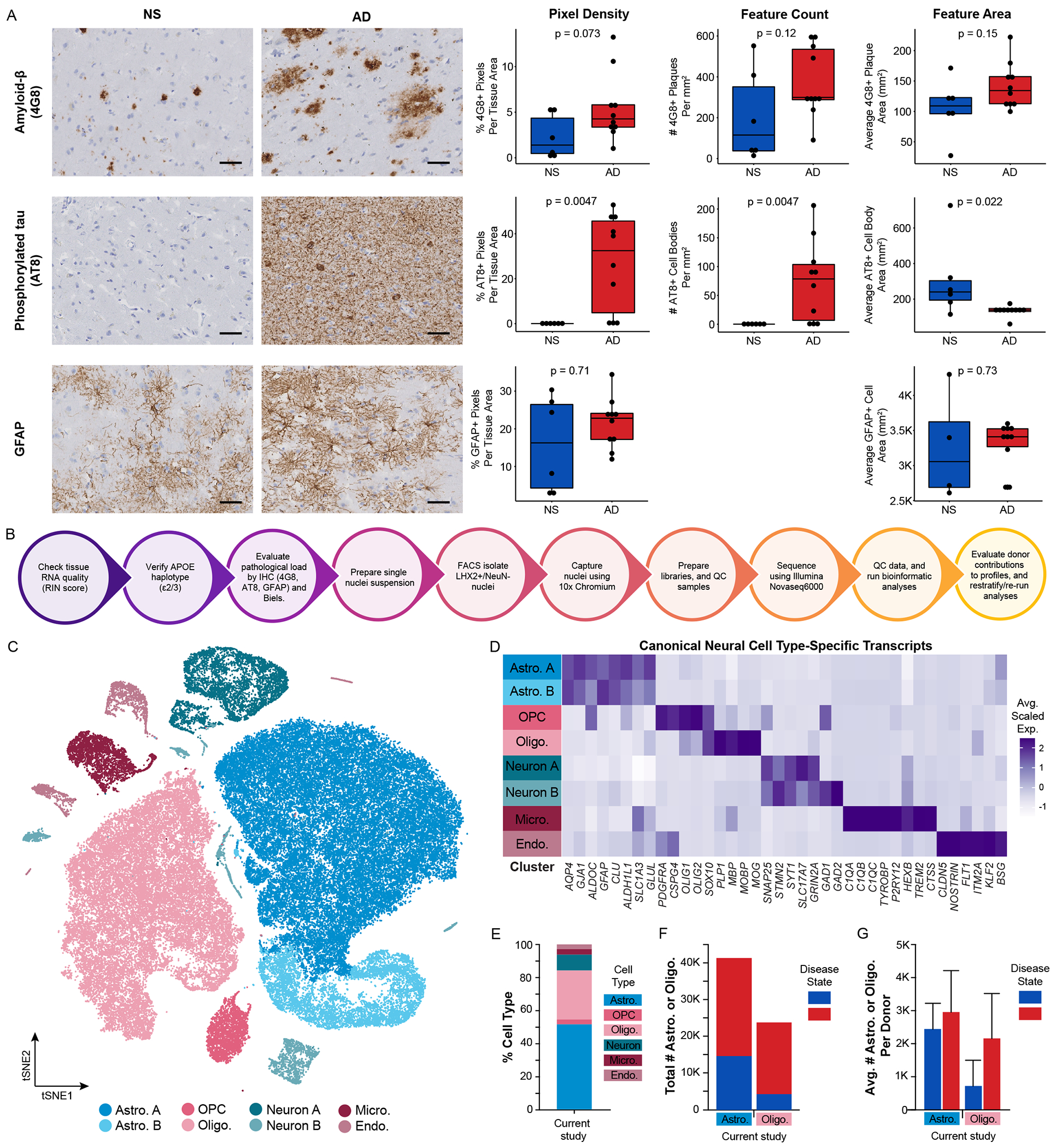
(A) Representative micrographs and corresponding quantification in non-symptomatic (NS) and AD donors of immunohistochemistry for amyloid-β plaques (4G8), phosphorylated tau (AT8), and GFAP. Scale bars are 50 μm. Raw quantification values are displayed as well as mean ± s.d. (B) Workflow for donor quality control and astrocyte enrichment strategy. (C) tSNE plot of total nuclei (N = 80,247) and (D) corresponding average scaled expression heatmap of cell type-specific transcripts by cluster. (E) Cell type proportions of total nuclei captured, (F) total numbers of astrocytes and oligodendrocytes captured split by disease state, and (G) average number of astrocytes and oligodendrocytes captured per donor split by disease state: NS (blue), AD (red). Abbreviations: AD, Alzheimer’s disease; Astro., astrocyte; Endo., endothelial cell; FACS, fluorescence-activated cell sorting; GFAP, glial fibrillary acidic protein; IHC, immunohistochemistry; Micro., microglia; NS, non-symptomatic; Oligo., oligodendrocyte; OPC, oligodendrocyte precursor cell; RIN, RNA Integrity Number. See also Figures S1–S3.
We next sought to improve astrocyte capture for snRNA-seq by enriching for astrocytic nuclei prior to sequencing (Figure S2). Initially, we attempted astrocyte enrichment via SOX9 sorting (Figure S1E–G, S2A), given SOX9 is a transcription factor highly and uniquely expressed in astrocytes (Zhang et al., 2016). However, this strategy proved ineffective at increasing astrocyte capture yields, likely due to non-specific SOX9 antibodies (Table S3). Instead, we turned to a dual immunolabeling strategy pioneered by Nott and colleagues (Nott et al., 2019) by sorting samples based on LHX2+NeuN− (Figure S1H–M, S2B, Table S3). Using this strategy, we enriched for astrocytes, maintained oligodendrocyte numbers, and depleted neurons (Figure 1C–G). While optimizing this process, we sorted a single donor using both sorting strategies, highlighting significant improvement in total astrocyte capture (Figure S1H–M). Overall, in our final donor cohort, 51.5% of nuclei captured were astrocytes, totaling 41,340 astrocytes and averaging 2,756 ± 1,087 astrocytes per donor (Figure 1E–G). In addition, 29.7% of nuclei captured were oligodendrocytes, totaling 23,840 oligodendrocytes and averaging 1,589 ± 1,342 oligodendrocytes per donor (Figure 1E–G).
Putative loss of critical oligodendrocyte functions in Alzheimer’s disease
We identified five unique oligodendrocyte transcriptomically defined clusters (Figure 2A–B) and evaluated these gene sets on their own and by gene ontology (GO)/pathway analysis (Table S4). We did not identify any singular sample variable, such as disease state, sex, RNA quality, age of donor, or post mortem interval (PMI), that was exclusively associated with a cluster (Figure 2C, Figure S3). While few genes were exclusively expressed by a single cluster, several were enriched in some clusters over others. Oligodendrocyte cluster 1 for example was enriched for transcripts involved in glial cell development (PLP1, CNP, CD9) and apoptotic signaling (SEPTIN4, SERINC3). Oligodendrocyte cluster 2 was enriched for transcripts associated with cholesterol metabolism (MSMO1, FDFT1, LSS). Oligodendrocyte clusters 2, 3, and 4 enriched transcripts were involved in synapse assembly and organization. Additionally, oligodendrocyte cluster 4 expresses transcripts involved in antigen processing/presentation (PSMB1, B2M, HLA-A) and innate immunity-associated pathways, such as interleukin-1 signaling, tumor-necrosis factor signaling, and NFκB signaling. Finally, although we did not identify any statistically significant GO terms associated with oligodendrocyte cluster 0, based on cluster-enriched differentially expressed genes (DEGs), we inferred that this cluster was associated with synapse organization and aspects of metabolism (Table S3).
Figure 2. Oligodendrocytes are minimally heterogeneous but have cluster-specific transcriptomic changes in Alzheimer’s disease.
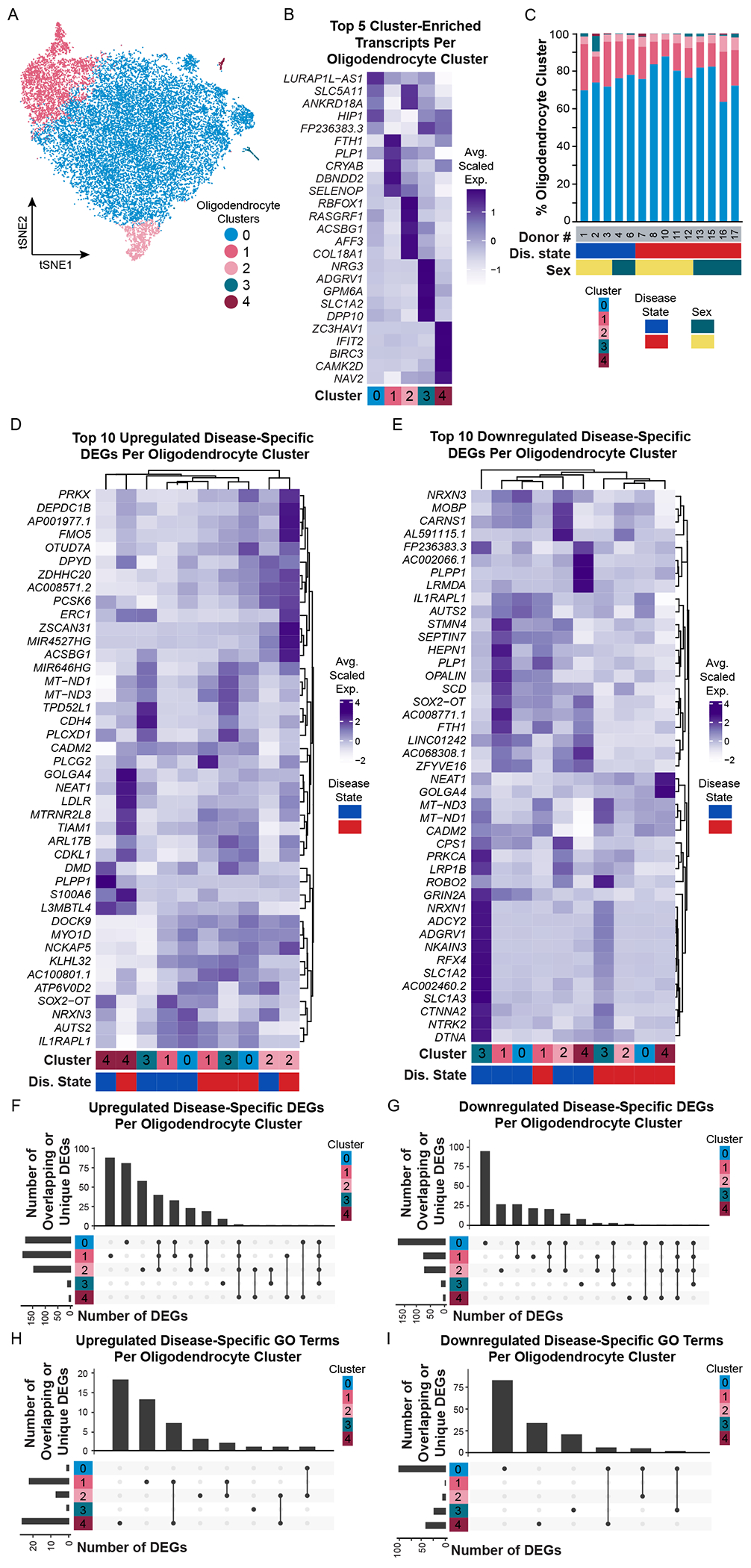
(A) tSNE plot of oligodendrocyte nuclei (N = 23,840) and (B) corresponding average scaled expression heatmap of top 5 enriched/unique transcripts per cluster. (C) Proportion of oligodendrocyte clusters identified in each donor. Additional donor metavariables highlighted include disease state (blue, NS; red, AD) and sex (green, female; yellow, male). Average scaled expression of the top 10 (D) upregulated and (E) downregulated disease-specific differentially expressed genes (DEGs) split by cluster. (F-I) UpSetR plots highlighting upregulated and downregulated DEGs or GO terms that are unique to or shared between clusters. Bars show number of DEGs per cluster (colored at left). Lines between cluster highlight shared DEGs. Abbreviations: AD, Alzheimer’s disease; D#, donor number; DEG, differentially expressed gene; Dis., disease; F, female; GO, gene ontology; M, male; NS, non-symptomatic. See also Figures S4, S5.
We next evaluated differential gene expression between AD and NS oligodendrocytes using edgeR paired with zinbwave-generated observational weights. We identified 358 unique upregulated and 227 unique downregulated DEGs in AD oligodendrocytes (Table S4). We did not find any transcriptomic changes that were conserved/common across all oligodendrocyte subpopulations, but instead find highly cluster-specific transcriptomic changes based on disease state (Figure 2D–I).
We next classified our findings by assigning ‘GO descriptions’ – a manual evaluation of oligodendrocyte cluster-specific or cluster-shared GO terms. This allowed us to summarize multiple identified GO terms associated with either upregulated or downregulated pathways, for single or multiple oligodendrocyte clusters (Figures S4, S5, Table S4). Oligodendrocyte ‘GO descriptions’ associated with AD fell into two broad categories: upregulation of synaptic maintenance, or downregulation of synaptic maintenance. For example, cluster 1 AD oligodendrocytes upregulate GO terms associated with axonogenesis and synapse organization (example associated DEGs include LRP4, TIAM1, CDH2). Decreases in synaptic cell adhesion protein N-cadherin (CDH2) have previously been reported in AD temporal cortex (Ando et al., 2011). Therefore, upregulation of CDH2 in this subpopulation of AD oligodendrocytes is an interesting discovery and may reflect a neuroprotective response to maintain contacts between axons and oligodendrocyte lineage cells (Schnadelbach et al., 2001). Cluster 2 AD oligodendrocytes upregulate pathways related to cholesterol metabolism. Cholesterol is an essential component of myelin (Saher et al., 2011), so identification of transcripts associated with cholesterol metabolism here (e.g. FM05, FDFT1) is not surprising. However, in the context of AD, upregulation of these transcripts in an oligodendrocyte subpopulation may suggest neurosupportive and/or neurotoxic effects (Guttenplan et al., 2021). For example, conditional knockdown of squalene synthase (FDFT1), a rate-limiting enzyme in cholesterol synthesis, significantly delays myelination in vivo (Saher et al., 2005). However, high cholesterol is also a risk factor for AD (Shepardson et al., 2011) and may exacerbate AD pathology by increasing Aβ production (as seen in APP/PS1 mice on a high cholesterol diet) (Refolo et al., 2000) or Aβ aggregation (Yip et al., 2001). Clusters 0 and 2 AD oligodendrocytes downregulate pathways involved in amino acid synthesis (Figure S4C). Fatty acid synthesis by oligodendrocyte lineage cells is critical for both myelination and remyelination, as seen in acute lesions in a focal spinal cord demyelination model (Dimas et al., 2019). Decreased expression of stearoyl-CoA desaturase (SCD), a rate-limiting enzyme in monounsaturated fatty acid synthesis (Paton and Ntambi, 2009), in these oligodendrocytes may reflect a loss of function regarding endogenous fatty acid synthesis and could imply limited myelination/remyelination capabilities. Cluster 0, which makes up almost 80% of all oligodendrocytes identified in our snRNA-seq dataset, downregulate GO terms associated with synapse transmission, synaptic vesicle regulation, and ion transmembrane transport (Figure S4D). In contrast to cluster 1 AD oligodendrocytes, those in cluster 0 downregulate a number of synaptic cell adhesion molecules like E-cadherin (CDH1) (Kilinc, 2018), liprin-α (PPFIA2) (Lie et al., 2018), and disrupted in schizophrenia 1 (DISC1) (Hattori et al., 2010), which suggests decreased contacts between oligodendrocytes and axons. Also, cluster 0 AD oligodendrocytes downregulate GO terms associated with metabolism (example DEGs include PDE8A, PDE10A, PDE1A, CNP, RORA). Inhibition of phosphodiesterases (PDEs), a group of enzymes that regulate cyclic nucleotide cAMP and cGMP levels, is used in the treatment of cardiovascular diseases, inflammatory airway diseases, and erectile dysfunction (Boswell-Smith et al., 2006) and have been evaluated as a therapy in AD (Prickaerts et al., 2017; Wu et al., 2018). In addition to the potential broader cognitive improvement by PDE inhibition, downregulation of PDEs (specifically PDE4) promotes oligodendrocyte lineage cell differentiation and remyelination in a focal demyelination model (Syed et al., 2013), suggesting these transcriptomic changes may serve a neuroprotective role in this specific oligodendrocyte subpopulation. We therefore hypothesize that some AD-associated transcriptional and functional changes in oligodendrocytes may also arise in other degenerative diseases like multiple sclerosis – even if the entire complement of transcriptional changes is not the same.
Integration of oligodendrocytes from multiple datasets recovers overlapping subtypes
Given the abundance of high quality, well-powered AD patient oligodendrocyte snRNAseq datasets in the literature (Del-Aguila et al., 2019; Gerrits et al., 2021; Grubman et al, 2019; Leng et al., 2021; Mathys et al., 2019; Zhou et al., 2020), we next sought to determine if we could resolve the same transcriptomic differences previously reported. We evaluated oligodendrocyte subtypes in each individual dataset and compared them to our own (Table S1, Figures S3I, S6–S8). By analyzing each dataset in isolation, we identified 5 oligodendrocyte clusters (M0-M4) in the Mathys dataset, 4 oligodendrocyte clusters (G0-G3) in the Grubman dataset, and 10 oligodendrocyte clusters (Z0-Z9) in the Zhou dataset (Figure 3A–B, Table S1). Using our oligodendrocyte subpopulation profiles as a reference (from now on referred to as oligodendrocyte clusters S0-S4), we identified subpopulations which were also recognizable in the individual datasets. This included S1-like oligodendrocytes (clusters M0, M3, G0, G3; defined by high expression of FTH1, CRYAB, CNP, and FRY) and S2-like oligodendrocytes (clusters M2, G1; defined by RBFOX1, RASGRF1, ACTN2, and SYNJ2). In the Grubman dataset, we also identified S4-like oligodendrocytes (cluster G2; which highly expresses S100A6, ITPKB, and NEAT1). The remaining oligodendrocyte clusters in the Mathys dataset were defined by either no highly unique transcripts (cluster M1; only enriched transcript is X-chromosome gene XIST, even though donors of both sex are represented in this cluster) or significantly fewer genes/UMI counts per nucleus compared to other clusters (cluster M4). In contrast, we identified twice as many oligodendrocyte subpopulations in the Zhou dataset compared to our own. However, although we detected more transcriptomically definable oligodendrocyte subpopulations, many of these clusters reflect gradations of transcript expression across multiple clusters. For example, oligodendrocyte clusters Z2, Z5, and Z9 express increasing levels of PLP1, CNP, CNTN1, FRY, and KCNIP4 (similar to S1 oligodendrocytes), and oligodendrocyte clusters Z4 and Z7 express ACTN2I, DTNA, RASGRF1, and RASGRF2 (similar to S2 oligodendrocytes). Overall, we found 3 out of 5 subpopulations were present in the Zhou dataset (spread over 7 clusters). The remaining three clusters are defined by either higher mitochondrial transcript expression and low gene/UMI counts per nucleus (cluster Z3) or have uniquely identified profiles that do not have a corresponding profile in our snRNA-seq dataset.
Integration of oligodendrocytes from multiple datasets reveals consistent identification of oligodendrocyte subtypes.
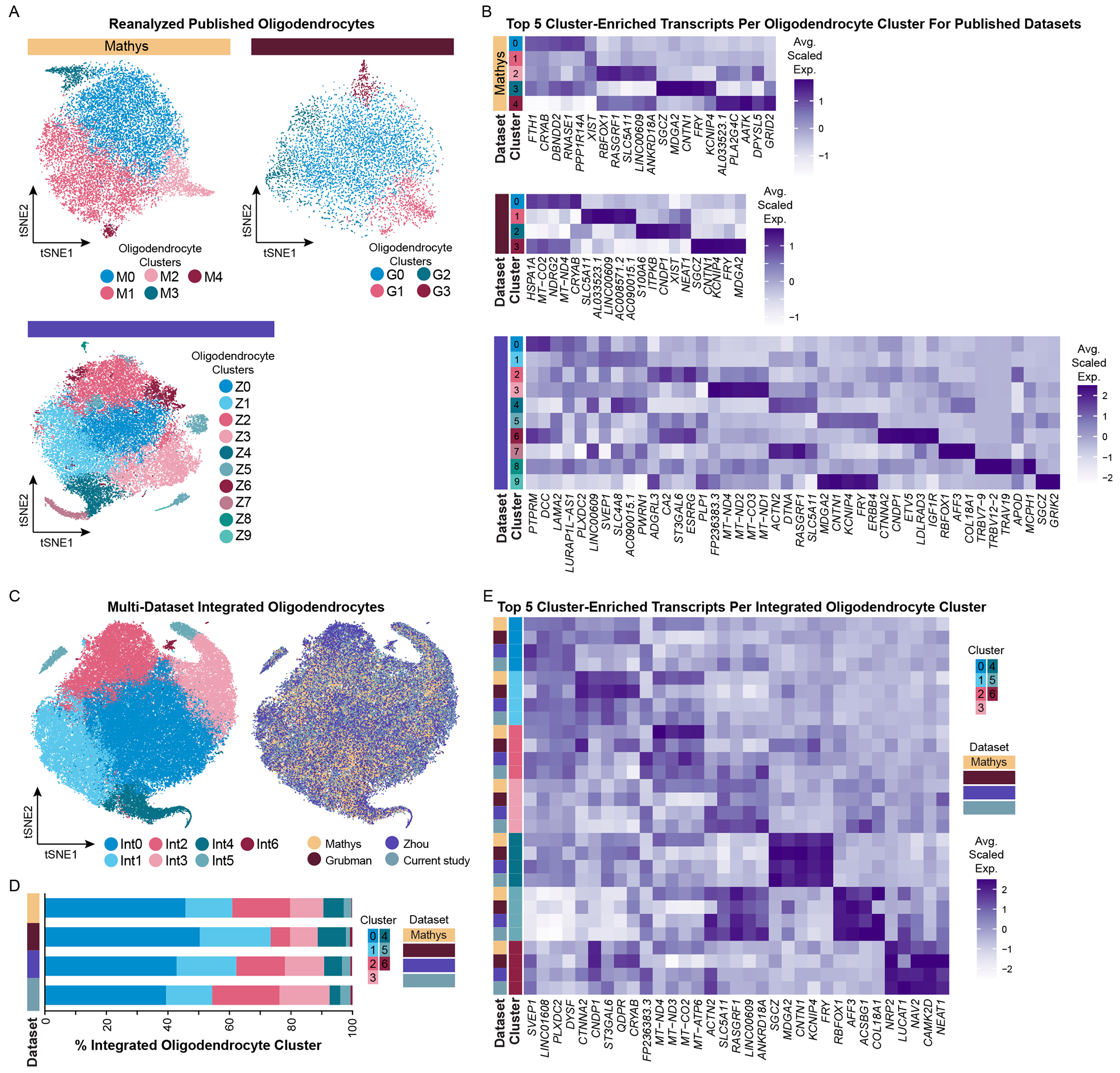
(A) tSNE plots of reanalyzed oligodendrocytes from published snRNA-seq datasets (Mathys, N = 18,229; Grubman, N = 7,604; Zhou, N = 34,949) and (B) their corresponding average scaled expression heatmap of the top 5 cluster-enriched/unique transcripts per cluster for each dataset. (C) tSNE plots of integrated oligodendrocytes (N = 84,622) visualized by cluster (left) and by dataset (right). (D) Proportion of integrated clusters split by dataset. (E) Average scaled expression heatmap of top 5 integrated oligodendrocyte cluster-enriched/unique transcripts by cluster and by dataset. See also Figures S6–S8, S13.
In addition to evaluating oligodendrocytes from published datasets in isolation, we also integrated these datasets with our own. By doing so, we defined 7 oligodendrocyte subpopulations (clusters Int0-Int6) and found highly consistent representation of clusters from each dataset (Figure 3C–D, Table S5). To determine if all datasets equally contributed to cluster-defining transcript expression, we evaluated the top five integrated oligodendrocyte cluster-enriched transcripts by cluster and dataset (Figure 3E). Overall, 5 out of 7 integrated oligodendrocyte clusters were well defined across all datasets (clusters Int1 and Int3-Int6). Oligodendrocyte cluster Int0 was unremarkably defined (i.e. lacked unique or highly enriched transcript features) across all datasets. Comparing transcriptomic profiles between integrated oligodendrocyte clusters with our own oligodendrocyte dataset, two clusters had a high degree of similarity. Oligodendrocyte cluster Int0 expresses SVEP1 and PLXDC2 (similar to S0 oligodendrocytes), and Int6 oligodendrocytes express NRP2, LUCAT1, and CAMK2D (similar to S4 oligodendrocytes). In contrast to these oligodendrocyte subpopulations, in the integrated dataset, we identified two pairs of clusters in which expression gradation of transcripts were now well definable across all datasets (and not just in the Zhou dataset). Specifically, clusters Int1 and Int4 share expression of transcripts FTH1, PLP1, APOD, and DBNDD2. In addition, Int4 oligodendrocytes highly expresses SGCZ, MDGA2, CNTN1, KCNIP4, and FRY (which we identified in S1 oligodendrocytes).
Transcriptionally distinct astrocyte subtypes are independent of disease state
Following our investigation into oligodendrocyte gene expression changes, we next investigated our captured astrocytes. We sought to determine if the increased numbers enabled us to detect novel subpopulations that had previously been missed. We identified 9 astrocyte subpopulations with unique transcriptomic signatures (Figure 4A–B) and also evaluated them by GO/pathway analysis to infer potential biological relevance (Table S6). For example, astrocyte clusters 0, 4, and 8 express unique sets of transcripts involved in synapse assembly, organization, and transmission (Cluster 0: EGFR, LRRC4C, EPHB1; Cluster 4: DCLK1, NTNG1, and several semaphorins; Cluster 8: EPHA4, AKAP12, NLGN4X). Astrocyte clusters 4 and 6 highly express transcripts involved in glutamate signaling (GRIA1, GRIK4, SHISA6). Clusters 2 and 5 express transcripts involved in extracellular matrix organization (Cluster 2: ADAMTSL3, L3MBTL4; Cluster 5: ADAMTSL3, FBN1), while cluster 5 also expresses transcripts involved in actin cytoskeletal organization (SORBS1, SPIRE1). The inclusion of ADAMTSL3 in clusters 2 and 5 may point to a protective role of (some) AD or aged astrocytes, as it has reported protective role in ischemia and cerebrovascular integrity in the APP/PS1 mouse model of AD (Cao et al., 2019). In contrast, cluster 3 astrocytes express transcripts involved in acute inflammatory responses (e.g. SERPINA3, C3, OSMR) that we have reported on previously in both mouse (Hasel et al., 2021; Liddelow et al., 2017) and human (Barbar et al., 2020). Astrocyte cluster 1 is highly enriched for transcripts involved in oxidative stress (PSAP, COX1, ND1/3) and associated with Aβ trafficking (e.g. APOE, CLU) and processing (e.g. ITM2B/2C). The inclusion of AD-risk genes, APOE and CLU, with integral membrane protein (ITM2B/2C) genes associated with cerebral amyloid angiopathy (Nelson et al., 2013; Vidal et al., 1999) in the same astrocyte cluster suggests a putative interaction. Astrocyte clusters 1 and 6 are both enriched in a number of metallothioneins and other transcripts involved in response to metal ions. Finally, cluster 7 expresses transcripts associated with apoptotic signaling and response to DNA damage. Most importantly, we did not identify any donor that singularly drove the identification of an astrocyte cluster (Figure S3A, F; see Figure S3A for rationale of why two donors were removed from final analyses). Like our oligodendrocyte populations, astrocyte heterogeneity was not driven by any definable underlying sample variable, including disease state, sex, RNA quality, age of donor, or PMI.
Figure 4. Astrocytes are heterogeneous and have both common and cluster-specific transcriptomic changes in Alzheimer’s disease.
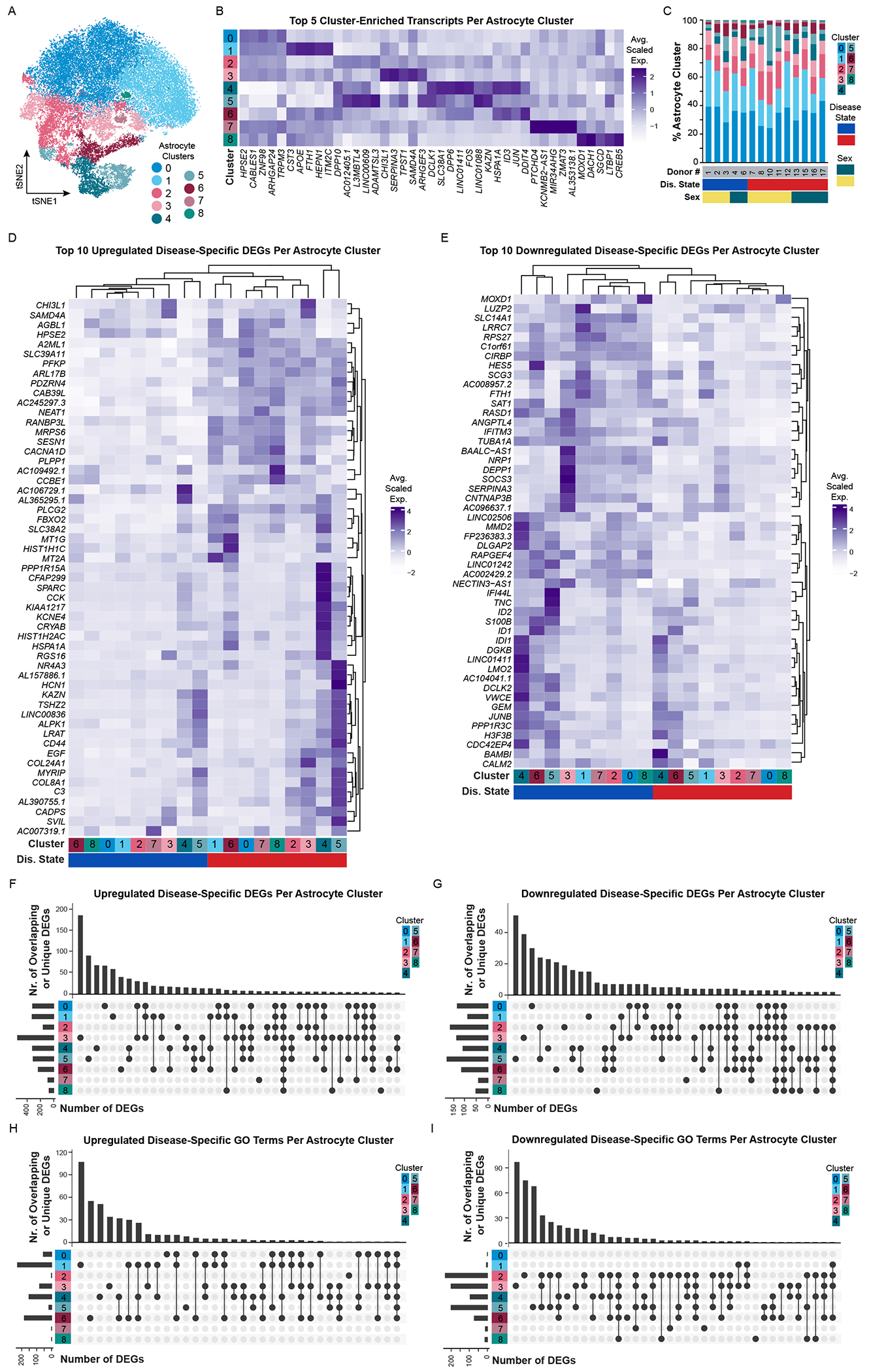
(A) tSNE plot of astrocyte nuclei (N = 41,071) and (B) corresponding average scaled expression heatmap of top 5 enriched/unique transcripts per cluster. (C) Proportion of astrocyte clusters identified in each donor. Additional donor metavariables highlighted include disease state (blue, NS donors; red, AD donors) and sex (green, female; yellow, male). Average scaled expression of the top 10 (D) upregulated and (E) downregulated disease-specific differentially expressed genes (DEGs) split by cluster. (F-I) UpSetR plots highlighting upregulated and downregulated DEGs or GO terms that are unique to or shared between clusters. Abbreviations: AD, Alzheimer’s disease; D#, donor number; DEG, differentially expressed gene; Dis., disease; F, female; GO, gene ontology; M, male; NS, non-symptomatic. See also Figure S11.
Astrocytes have both common and cluster-specific transcriptomic changes in Alzheimer’s disease
We next analyzed samples for AD-associated differential gene expression and identified both common and cluster-specific transcriptomic changes between AD and NS astrocytes. In total, we identified 1,084 unique upregulated DEGs and 450 unique downregulated DEGs between astrocyte clusters that were presumably driven by AD (Table S6). When comparing the top 10 up-/down-regulated DEGs by cluster and disease state through hierarchical clustering, all AD and NS astrocyte subpopulations clustered separately (Figure 4D–E). Across all clusters, AD astrocytes upregulate transcripts including HPSE2, SLC39A11, PFKP, NEAT1, RANBP3L, PLPP1, and PLCG2. HPSE2, a heparanase homolog, antagonizes heparanase activity (e.g., degradation of heparan sulfate proteoglycans in the extracellular matrix – important for removal of Aβ deposits that can aggregate with heparan sulfates (Lorente-Gea et al., 2017; O’Callaghan et al., 2008)). Given that HPSE2 acts as a competitive inhibitor of HPSE, increased HPSE2 release by astrocytes may enable expansion of Aβ deposits (Lorente-Gea et al., 2017), and therefore inhibition of astrocyte-produced HPSE2 may provide therapeutic benefit. NEAT1 (nuclear enriched abundant transcript 1) is also an enticing target for future investigation as it is upregulated in several mouse models of AD, including APP/PS1 mice, and is putatively associated with regulation of PINK1 degradation and impaired mitophagy (Huang et al., 2020). Additionally, across all clusters, AD astrocytes downregulate transcripts including SLC14A1, C1orf61, CIRBP, and SAT1. Some of these transcripts have important putative protective roles in neurodegenerative disease, so their decreased expression levels could be problematic. For instance, catabolic polyamine enzyme spermidine/spermine N1-acetyltransferase 1 (SAT1) levels are reduced by treatment with the dimanazene aceturate drug Berenil, which leads to worsened pathology in patients with Parkinson’s disease (Lewandowski et al., 2010). In contrast to these pan-astrocytic DEGs, we also identified DEGs that were unique to single or combinations of astrocyte clusters (Figure 4F–G) and evaluated these gene sets by GO/pathway analysis (Figure 4H–I). By doing so, we were able to contextualize transcriptomic expression differences between AD and NS astrocytes in a cluster-specific manner and ultimately infer the potential biological role of astrocyte subpopulations in AD.
Astrocytes have both putative gain and loss of function in Alzheimer’s disease
We next evaluated astrocyte cluster-specific ‘GO descriptions’ associated with either upregulated or downregulated pathways (Figures 5, S9, S10, Table S6). Cluster 1 AD astrocytes upregulate GO terms associated with cell death and oxidative stress (Figure 5A, e.g. RGCC, PRDX1, DDIT4). The Regulator of Cell Cycle protein (RGCC), previously reported as upregulated in AD patients (Counts and Mufson, 2017), may be important for the re-entry of post-mitotic astrocytes to the cell cycle to enable proliferation around regions of pathology/degeneration. In comparison, peroxiredoxin (PRDX1), which is increased in AD patient hippocampi at the protein level (Chang et al., 2014), may have important antioxidant protection functions and suggests a supportive role of these astrocytes in AD. Cluster 5 AD astrocytes upregulate pathways related to lipid storage and fatty acid oxidation (Figure 5B, e.g. C3, ABCA1, PPARGC1, ACACB). We, and others, have reported upregulation of complement component 3 (C3) in a specific sub-state of reactive astrocytes that respond to inflammation in a range of neurodegenerative diseases (Diaz-Castro et al., 2019; Guttenplan et al., 2020; Liddelow et al., 2017; Shi et al., 2017) including AD (Liddelow et al., 2017) and AD mouse models (Lian et al., 2016; Wu et al., 2019). C3+ astrocytes are associated with a neurotoxic function and are only found in regions of neurodegeneration. As such, their inclusion here is not surprising given we completed pathological analysis and snRNA-seq on regions with high pathology load. We performed immunofluorescent quantification of C3+ astrocytes in NS and AD patient samples and report no difference at the protein level (Figure S11) – though this may be a result of a reported increase in C3+ astrocytes with normal aging (Boisvert et al., 2018; Clarke et al., 2018), broader transcription versus translation differences between gene expression and protein levels, or the fact that these DEGs are specific to only a few subtypes of astrocytes in our dataset and therefore quantifying samples using single markers poses difficulties as it is unclear if these astrocytes belong to the subtypes under investigation. We also quantified another cluster 5 DEG, SPARC, and report no difference in the number of SPARC+GFAP+ cells or overall fluorescence intensity between NS and AD patient cortex (Figure S11) – which given our sequencing of astrocyte nuclei from prefrontal cortex may also be a result of reported upregulation of SPARC in cortical astrocytes with normal aging (Clarke et al., 2018). Clusters 0, 1, 4, and 6 AD astrocytes share upregulation of pathways involved in response to metal ions (Figure S9A). DEGs associated with these GO terms include features that are cluster-specific (e.g. PRKN in cluster 0) as well as those that are shared by multiple clusters (e.g., DUSP1 in clusters 4 and 6; MT1G in clusters 0, 1, 4, and 6). Upregulation of DUSP1 (dual-specificity phosphatase, also known as mitogen-activated protein kinase, MKP1) has previously been reported in models of Parkinson’s disease (Collins et al., 2013) and Huntington’s disease (Taylor et al., 2013) as well as following ischemic stroke (Boutros et al., 2008; Ramsay et al., 2019) and seizures (Kedmi and Orr-Urtreger, 2007). DUSP1 upregulation is associated with repression of pro-apoptosis and neuronal cell death pathways in neuroblastoma (Nunes-Xavier et al., 2019), suggesting another putatively important protective response in astrocyte subpopulations in AD. Cluster 4 and 6 AD astrocytes also share upregulation of protein folding/unfolding pathways (example DEGs include HSPA1B, DNAJB1, ATF3I; Figure S9A) as well as downregulation of signaling receptor activity and axonal guidance pathways (e.g. GRIA1, NLGN1; Figure S9B). Increased expression of activating transcription factor 3 (ATF3) suggests a response to endoplasmic reticulum stress, a pathway that propagates through the induction of eukaryotic initiation factor 2 (eIF2) kinase-associated genes (Jiang et al., 2004). In addition, since these astrocyte clusters downregulate transcripts critical for both synaptogenesis and astrocyte morphogenesis (like neuroligin-1, NLGN1), this may implicate that putative decreased synaptic maintenance functions could be due to stunted/altered astrocyte territories and limited infiltration of surrounding neuropil (Stogsdill et al., 2017). The intersection of unfolding protein response (UPR) and synaptic maintenance mechanisms was recently evaluated by Smith and colleagues who found that chronic PERK-eIFα signaling in astrocytes induced an UPR-associated reactivity state in which astrocytes lose synaptic support functions and ultimately induce neuron death in vitro and in prion infection in mice (Smith et al., 2020). Cluster 3 AD astrocytes downregulate GO terms associated with angiogenesis regulation and BBB maintenance (Figure 5C). As a key modulator of vascular permeability and angiogenesis, downregulation of vascular endothelial growth factor A (VEGFA) may be a protective astrocyte response to limit BBB breakdown. Administration of human recombinant VEGF165 1 hour post-ischemic stroke exacerbated BBB leakage in a middle cerebral artery occlusion rat model (Zhang et al., 2000), and inhibition of VEGFA improved BBB integrity around active lesions in an experimental autoimmune encephalomyelitis mouse model, decreasing immune cell infiltration and reducing overall demyelination (Argaw et al., 2012).
Figure 5. Astrocyte transcriptomic profiles suggest cluster-specific gain and loss of functional changes in Alzheimer’s disease.
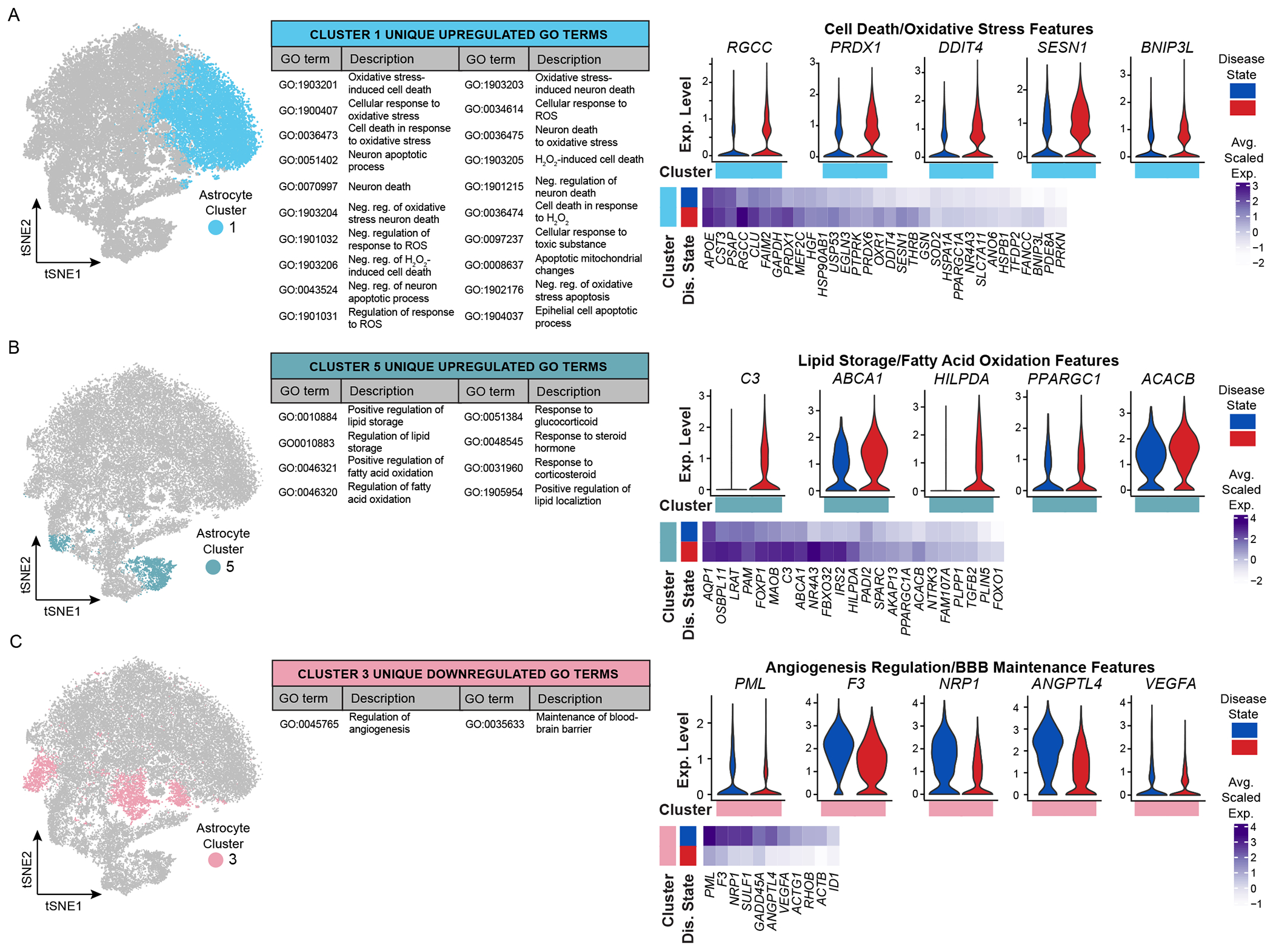
tSNE plots highlighting several clusters of interest, unique/shared GO terms, and differentially expressed genes (DEGs) associated with GO terms. GO-associated DEGs are presented as average scaled expression heatmaps by cluster of interest and split by disease state (blue, NS donors; red, AD donors). DEGs are highlighted on violin plots to resolve the range of expression (log normalized UMI counts) across all astrocytes in single or multiple clusters. (A) Upregulated cell death and oxidative stress features unique to cluster 1. (B) Upregulated lipid storage and fatty acid oxidation features unique to cluster 5. (C) Downregulation of angiogenesis regulation and blood brain barrier maintenance features unique to cluster 3. Abbreviations: AD, Alzheimer’s disease; BBB, blood-brain barrier; DEG, differentially expressed genes; Dep., dependent; Dis., disease; GO, gene ontology; H2O2, hydrogen peroxide; Neg., negative; NS, non-symptomatic; Reg., regulation; ROS, reactive oxygen species; UMI, unique molecular identifier. See also Figures S9–S11.
Astrocyte subtypes are regionally heterogeneous in human and mouse
Given recent discoveries highlighting astrocytes as increasingly variable across the CNS (Bayraktar et al., 2020), we next sought to explore whether our heterogeneous astrocyte subtypes reside in different cortical locations. However, examining regional differences in these astrocyte subtypes or disease-associated reactive sub-states is a challenging prospect given many clusters are defined by slight differences in expression of dozens or hundreds of genes rather than expression of individual DEGs specific to a single cluster, making it difficult to evaluate these gene signatures using traditional in situ methods. To overcome these challenges, we leveraged published spatial transcriptomics datasets to explore regional differences in our astrocyte subtypes in the NS human brain (Maynard et al., 2021) and compared this with the healthy and inflamed mouse brain (Hasel et al., 2021).
To determine the likely location of each astrocyte population, we created modules of marker genes from each cluster (Hasel et al., 2021; Tirosh et al., 2016) and examined the expression of the modules across the human and mouse spatial transcriptomics data (Figure S12A). We found that all astrocyte cluster modules exhibited significant differences in expression in at least one cortical layer (Kruskal-Wallis test, p < 0.05); however, there were large differences in the degree to which cluster gene signatures were region-specific (Figure S12F). Some cluster modules were strongly enriched in select regions. For example, the Cluster 6 gene signature is significantly enriched in layer 1 and the white matter (WM) in both human and mouse brain (Figure 6A). Several Cluster 6 marker genes, like ID1, ID3, and AGT, have been previously reported in WM and L1 astrocytes in the mouse (Bayraktar et al., 2020), supporting this localization. Cluster 8, in contrast, was enriched in upper cortical layers L1-L3 in both species (Figure 6B), fitting with the recent description of several genes in this set having elevated expression in upper cortical astrocytes, such as GRM3, SLCO1C1, and EPHB1 (Bayraktar et al., 2020). In aggregate, we found significant heterogeneity in the cortical regions most enriched for each astrocyte cluster (Figure 6C; Figure S12G). Regional cluster enrichment was similar between the human and mouse datasets, supporting the robustness of the gene module approach and suggesting the astrocyte subtypes we identified may be conserved between species (Figure 6D).
Figure 6. Astrocyte subtypes are regionally heterogeneous.
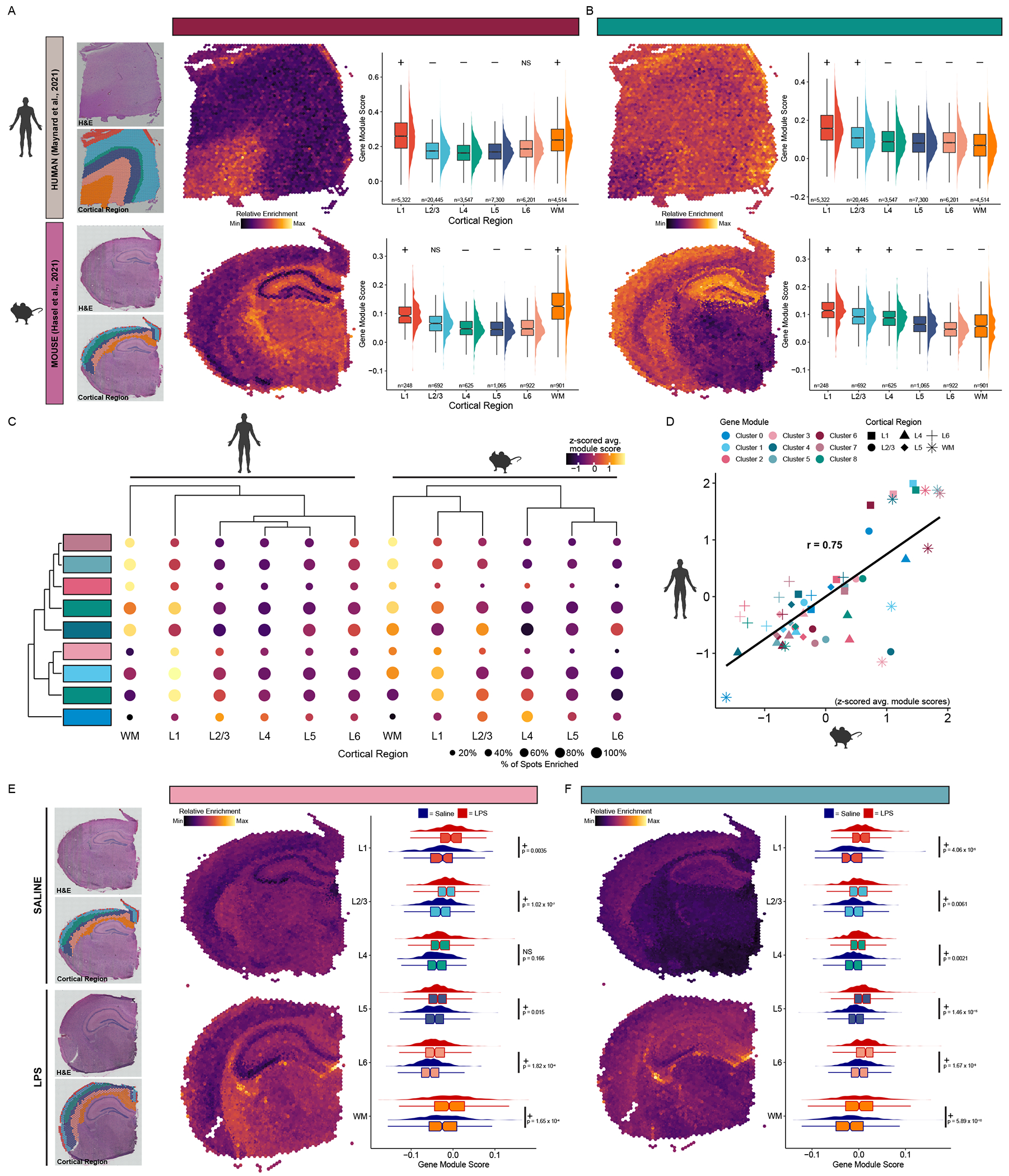
Visualization and differential enrichment results for Cluster 6 marker genes enriched in Layer 1 and white matter (A), and Cluster 8 genes enriched in the upper layers of the cortex (B). For both: Upper: human spatial transcriptomics data from Maynard et al. (2021). Lower: mouse spatial transcriptomics data from Hasel et al. (2021). (leftmost) H&E staining and regional annotation of spots from the representative Visium section. Relative enrichment of cluster gene module section, and box and density plots of gene module scores across all spots and all sections grouped by cortical region for Clusters 6 and 8. Cluster gene modules were significantly enriched (+) or de-enriched (−) in spots from the indicated region compared to the rest of the cortex (Wilcoxon rank sum test with Bonferroni correction). (C) Summary dot plot of astrocyte cluster gene modules across human (left) and mouse (right) cortical regions. Dots colored by z-scored average gene module score. Dot sizes correspond to the percentage of spots with a gene module score greater than zero, indicating elevated expression of the geneset compared to control genesets (see Methods). (D) Scatter plot comparing z-scored average gene module scores across region and clusters between human and mouse showing cluster module enrichment is similar. A linear regression line is shown (r refers to Pearson’s r correlation coefficient). (E) Relative enrichment of Cluster 3 module overlaid on saline (upper) and LPS (lower) sections. Box & density plot comparing expression of Cluster 3 module across spots in LPS-versus saline-injected mice (right). (F) Relative enrichment of genes upregulated in Cluster 8 in AD overlaid on saline (top) and LPS (bottom) sections. Box & density plot comparing expression of Cluster 8 AD module across cortical regions in LPS-versus saline-injected mice (right). For (E-F): +/− symbol represents whether the Cluster 3 module is significantly upregulated or downregulated in spots from the indicated region in LPS versus saline-injected mice (Wilcoxon rank sum test with Bonferroni correction). See Table S7 for test statistics and p-values. Abbreviations: AD, Alzheimer’s disease; NS, non-significant; H&E, hematoxylin & eosin; LPS, lipopolysaccharide. See also Figure S12.
Cluster 3 astrocytes were denoted by several genes previously described as upregulated in response to acute inflammation. Correspondingly, it was less well defined in the NS human and healthy mouse brain (Figure S12G) as compared to the inflamed mouse brain where this gene signature was upregulated across nearly all cortical regions (Figure 6E). This may indicate that Cluster 3 astrocytes are not region-specific but rather generally associated with inflammation. To explore whether some of the AD-associated gene expression changes in each astrocyte subtype may also be associated with inflammation, we next compared the expression of AD gene modules for each cluster between the healthy and inflamed mouse brain. We found that the Cluster 5 AD module was upregulated across all cortical layers in the inflamed mouse brain (Figure 6F), suggesting these AD-associated gene expression changes may be attributable to inflammatory mechanisms. Notably, AD gene signatures for nearly all other astrocyte clusters were not significantly enriched in the inflamed mouse brain relative to the healthy control brain (Figure S12H), indicating non-inflammatory mechanisms may be responsible for these changes.
Data integration increases astrocyte numbers and enables consistent identification of unique subpopulations
While we defined 9 transcriptomically distinct astrocyte clusters in our snRNA-seq dataset (from now on referred to as astrocyte clusters S0-S8), when reanalyzing published astrocyte datasets in isolation, we identified 5 clusters (M0-M4) in the Mathys dataset, 5 clusters (G0-4) in the Grubman dataset, and 7 clusters (Z0-Z6) in the Zhou dataset (Figure 7A–B, Table S1). With our dataset serving as a reference, we identified one or two previously defined astrocyte subpopulations in the Mathys and Grubman datasets: S1-like astrocytes (clusters M0, G1; which are defined by high expression of CST3, FTH1, APOE, and ITM2C) and S0-like astrocytes (cluster G0; which highly expresses CACNB2, GPC5, and RORA). In contrast to the Mathys and Grubman datasets, we identified 6 out of 9 astrocyte subpopulations in the Zhou dataset, which were similar to S0-S5 astrocytes. Like with our own astrocyte snRNA-seq dataset, we did not identify any singular sample variable that was exclusively associated with a single astrocyte cluster across all three datasets (Figure S7). This is in contrast with original findings presented in Grubman et al. (2019), which reported differences in the proportion of astrocyte clusters between AD and NS patients. However, we believe these detected differences may stem from lack of sample integration.
Figure 7. Integrating astrocyte snRNA-seq datasets allows for improved resolution of unique astrocyte subpopulations.
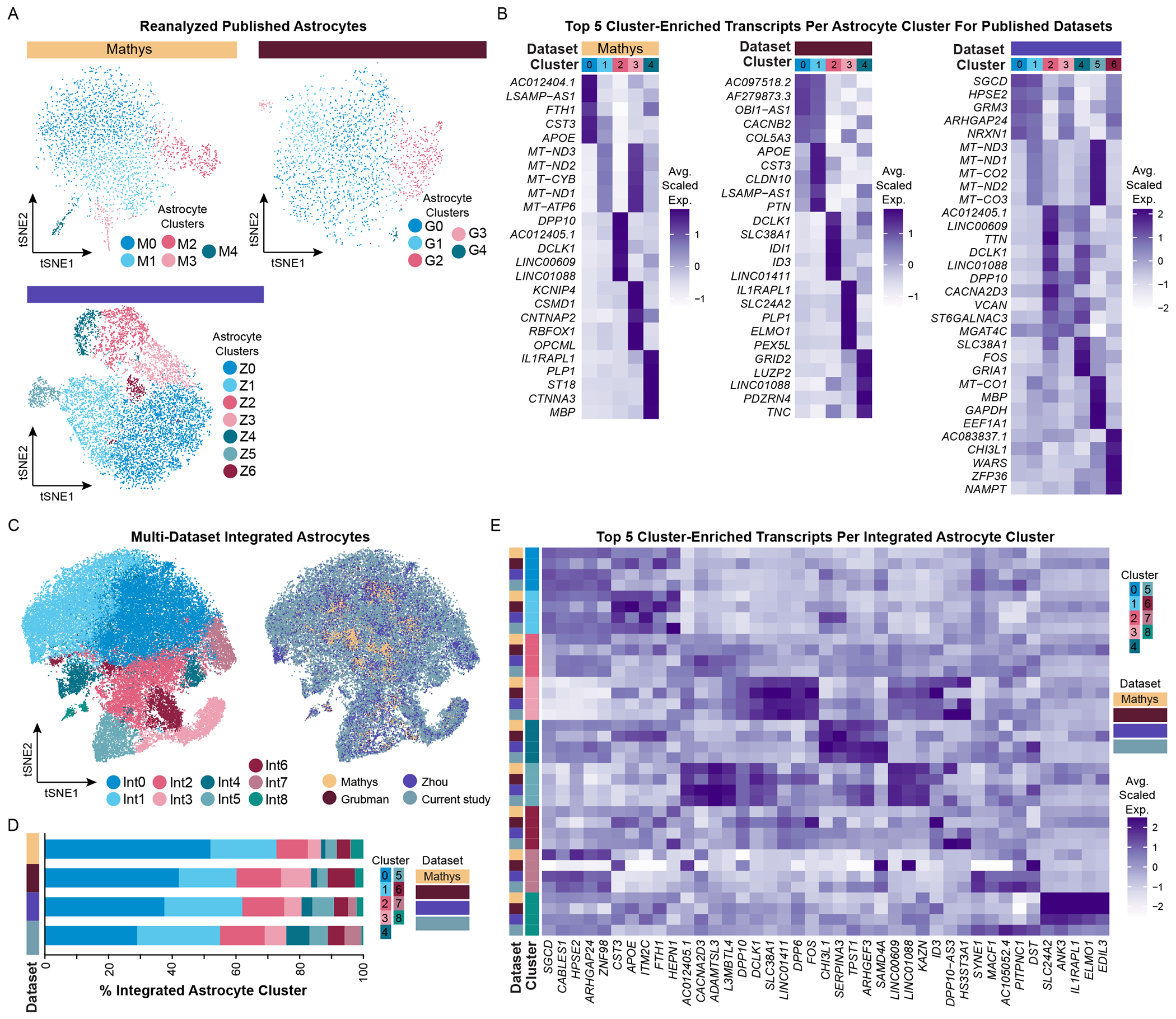
(A) tSNE plots of reanalyzed astrocytes from published snRNA-seq AD datasets (Mathys, N = 3,079; Grubman, N = 2,330; Zhou, N = 10,538) and (B) their corresponding average scaled expression heatmap of the top 5 cluster-enriched/unique transcripts. (C) tSNE plots of integrated astrocytes (N = 57,018) as visualized by cluster (left) and by dataset (right). Mathys data are in yellow, Grubman data are in dark red, Zhou data are in violet, and the current study’s data are in steel blue. (D) Proportion of integrated astrocyte clusters identified in the integrated dataset. (E) Average scaled expression heatmap of top 5 integrated astrocyte cluster-enriched/unique transcripts by cluster and by dataset. See also Figures S1, S3, S6–S8, S13.
Cross-comparing astrocyte subpopulation profiles between datasets is a useful exercise to evaluate what potential similarities and differences exist. In doing so, we defined 9 astrocyte subpopulations (referred to as astrocyte clusters Int0-Int8) (Figure 7C) and found that these astrocyte subpopulations had markedly similar proportions in each dataset (Figure 7C–D, Table S5). This is exciting as it highlights the feasibility of improving subpopulation identification post-hoc via data integration, thereby overcoming limitations in total number of astrocytes captured and depth of sequencing levels in individual datasets (Figure S8A). To determine if all datasets equally contributed to cluster-defining transcript expression, we evaluated the top five integrated astrocyte cluster-enriched transcripts by cluster and dataset (Figure 7E). Remarkably, 7 out of 9 integrated clusters were well-defined across all datasets (clusters Int0-Int6), and their corresponding transcriptomic profiles were similar to S0-S6 astrocytes. In contrast, cluster Int7 defining features were primarily present in the Zhou dataset and our dataset, and its profile most resembles astrocyte cluster Int0. However, this cluster had lower total genes and UMIs identified per nucleus, which is likely why it was identified as unique. Additionally, cluster Int8 was primarily identified in Mathys, Grubman, and Zhou datasets, and its profile is defined by higher expression of oligodendrocyte-associated and mitochondrial transcripts.
This integration method enabled identification of unique astrocyte subpopulations not previously resolvable in published astrocyte datasets. Next we explored whether AD-associated astrocyte transcriptional changes originally reported in each study were resolvable when integrating these datasets with our own (Figure S13A–D). Only the DEGs highlights in the Zhou et al. dataset were detectable in the integrated dataset (upregulated – Z5/Int2; downregulated – Z2/Int5; Figure S13D), likely due to increased numbers of sequenced astrocytes. Other reported disease-associated astrocyte DEGs were not specific to individual integrated clusters. Conversely, reverse-probing for previously described disease-associated oligodendrocyte cluster-specific DEGs was more successful (Figure S13E–H).
This highlights we can leverage large astrocyte datasets to better resolve astrocyte subpopulations in smaller datasets. For example, we were originally unable to detect a C3+ astrocyte subpopulation in both Mathys and Grubman datasets (Figure S11H). In comparison, we identified C3+ astrocytes in both our dataset (astrocyte clusters S3 and S5) and the Zhou dataset (astrocyte cluster Z2; however, this identification was driven by a single donor) (Tables S1 and S3). Once all datasets were evaluated together, this C3+ astrocyte subpopulation was uniquely ascribed to astrocyte clusters Int4 and Int5 (Figure S11G, Table S5), with all datasets contributing to C3 expression in astrocyte cluster Int4 and our dataset primarily contributing to C3 expression in astrocyte cluster Int5. This difference may be due to differences in pathological loads of the same samples being sequenced – as we previously reported C3+ astrocytes are only present in high pathology brain regions (Liddelow et al., 2017). Given the cluster-specific transcriptomic changes we identified between AD and NS patients, we believe this underscores the importance of resolving these unique astrocyte subpopulations in the context of health and disease.
DISCUSSION
Here we present a snRNA-seq dataset and paired pathology assessment resource for both astrocytes and oligodendrocytes from a well-defined human AD and age-matched NS patient cohort. This approach enabled us to identify putative biologically important astrocyte subpopulations. To localize these transcriptomically distinct populations of astrocytes we profiled 10X Visium spatial transcriptomics datasets and localized astrocyte subgroups in both the human and mouse brain. In addition, by leveraging our astrocyte snRNA-seq dataset with published astrocyte snRNA-seq datasets, we identified unique and previously undefinable astrocyte subpopulations in all datasets.
Integral for the success of this resource, we limited donor genetic variance and characterized the pathology of donor tissue from the same sample as sequenced material. Given the spatial heterogeneity that can occur due to differences in disease pathology and progression in adjacent brain regions (Komarova and Thalhauser, 2011; Murray et al., 2011), we are enormous proponents of being self-critical about what is driving results throughout analyses – being particularly wary of outlier donors and/or donor features. For example, after the first round of analyzing our astrocyte snRNA-seq data, we identified one cluster that was entirely representative of a single donor (D5). When evaluating this cluster, we found that it was highly enriched for transcripts associated with neuroinflammation and interferon gamma signaling (e.g., IFIT-1/2/3/6, IFI-44/44L/H1; see Figure S3A–E). Classified as a NS control, we double-checked our pathology characterization, which corroborated this donor as seemingly NS due to very low pathology load. However, upon further investigation of clinical evaluations associated with this donor, we discovered D5 had vascular dementia. This unique astrocyte subpopulation shares remarkable similarity to an interferon responsive subpopulation of neuroinflammatory reactive astrocytes following acute inflammation in mice and several neurodegenerative disease models, including AD, that we published recently (Hasel et al., 2021).
While our dataset and integration with published datasets highlight novel and putative functional populations of glia in AD, future functional studies are required to evaluate these subpopulations and their potential for modulation by therapies. How heterogeneity of astrocyte and oligodendrocyte responses might differ with disease progression or other patient cohort characteristics, like AD-associated mutations, secondary disease contraindications, or ethnic backgrounds, is an open question. Future human stem cell organoid and novel AD mouse models and isolated rodent cells used in in vitro functional testing will be able to address these questions. We also hope that continued integration of our data with future snRNA-seq and spatial datasets will add greater insight to these and many new questions.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
For further information and resource/reagent requests, please direct all inquiries to the Lead Contact, Shane Liddelow (shane.liddelow@nvulanqone.org).
Materials availability
This study did not generate any unique reagents.
Data and code availability
Raw snRNA-seq data generated in this study (including both FASTQ and Cell Ranger-generated matrix files) are available at GEO (GSE167494). Microscopy data reported in this paper are available through the Cell Image Library (www.cellimagelibrary.org, CIL group#: 54423).
All code for analysis of original and previously published snRNA-seq, scRNA-seq, and spatial transcriptomic datasets are available on the Liddelow Lab GitHub page: https://github.com/liddelowlab/Sadick_et_al._2022.
Analyzed snRNA-seq and pseudobulk snRNA-seq data is available on an open-access, interactive website: www.qliaseq.com. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Postmortem human cohorts
Pilot cohort.
Five de-identified human donor prefrontal cortex samples (non-symptomatic patients (NS), N = 3; AD patients, N = 2) were provided by Rhode Island Hospital’s Brain Tissue Resource Center (Title 45 CRF Part 46.102(f)) and New York University’s AD Research Center (ADRC). All tissues were donated with pre-mortem informed consent as regulated by Institute Review Boards at each respective Institution. In total, this donor cohort was comprised of one female and four male donors with ages ranging between 60-90 years of age and APOE genotypes of ε2/3, ε3/3, and ε3/4. Post-mortem intervals ranged between 12-24 hours for all tissues.
Final cohort.
Sixteen de-identified human donor prefrontal cortex samples (NS patients, N = 6; AD patients, N = 10; with an additional N = 6 NS and AD patient samples for immunostaining validation studies) were provided by NYU Grossman School of Medicine’s Alzheimer’s Disease Research Center and University of California San Diego Shiley-Marcos ADRC. All tissues were donated with pre-mortem informed consent as regulated by Institute Review Boards at each respective Institution. In total, this donor cohort was comprised of 7 female and 9 male donors with ages ranging between 56-100 years of age. All donors had APOE genotypes of ε2/3. Postmortem intervals ranged between 5-100 hours across all tissues. All samples had RNA Integrity Number (RIN) scores greater than 8. Based on sequencing analysis, two donors (Donors 5 and 9) were removed from final analyses, resulting in a total of 5 NS and 9 AD patients. Additional donor information is listed in Table S2 (any patient-specific information that is not included in these de-identified tables can be obtained from the original brain bank – donor IDs provided for coordination).
METHOD DETAILS
Tissue RNA quality verification
In order to ensure high quality outputs from snRNA-seq experiments, all donor tissue samples were first evaluated for bulk RNA quality, as quantified by RIN scores. Bulk RNA was extracted from each donor (~15-30 mg of postmortem frozen human prefrontal cortex tissue) using QIAshredder (QIAGEN, 79656) and RNeasy Plus Mini kits (QIAGEN, 74136), following manufacturer’s instructions. In brief, after each tissue was homogenized in lysis buffer using a Wheaton Dounce tissue grinder (DWK Life Sciences, 357538), samples were transferred to QIAshredder columns, flowthroughs were processed using RNeasy spin columns, and RNA from each sample was eluted in 30 μL DNase- and RNase-free water (Invitrogen, 10977015). RIN scores were then generated using an Agilent 2100 Bioanalyzer (Table S2).
APOE genotype verification
To confirm APOE genotype identification provided by NYU and UCSD ADRCs, all final human cohort samples were validated in-house by PCR using single nucleotide polymorphism-specific primers for each allele (i.e., ε2, ε3, ε4) and loading positive control β-actin, as designed by Zhong and colleagues (Zhong et al., 2016). The HotSHOT method was followed to isolate genomic DNA (gDNA). In brief, 10-30 mg of postmortem frozen human prefrontal cortex tissue per sample was digested in 75 μL of alkaline lysis reagent (25 mM NaOH, 0.2 mM EDTA in DNase- and RNase-free water) at 98°C in a thermomixer for 1 hour. To stop the reaction, 75 μL of neutralizing reagent (40 mM Tris-HCl, pH 5.5 in DNase- and RNase-free water) was added to each sample, and the solution was centrifuged at 4,000 rpm for 3 minutes. The supernatant, now containing gDNA, was then used in all following PCR reactions. Twenty-five μL PCR reactions were prepared for each sample and primer combination using GoTaq Green Master Mix (Promega, M7123) and respective primers (working dilution of 0.25 μM), following manufacturer’s instructions. PCR products were then run on 1.5% agarose gels (Thermo Fisher Scientific, 16-500-100) with ethidium bromide (VWR, 470024-556) for 40 minutes and were imaged using a Bio-Rad Gel Doc system (Figure S1D).
Immunohistochemistry and imaging of postmortem frozen human prefrontal cortex tissue
To evaluate pathological load in the final donor cohort, ~100 mg of postmortem frozen human prefrontal cortex tissue per donor was sent to the Neuropathology Brain Bank and Research CoRE at the Icahn School of Medicine at Mount Sinai for tissue fixation/embedding, sectioning, and staining. In brief, flash frozen tissues were fixed in 10% neutral buffered formalin and were then embedded in paraffin. Five μm sections were cut of each sample block for subsequent immunohistochemistry (IHC) assessments (N = 2-5 sections per stain). Primary antibodies used for IHC stains included: Anti-amyloid-β (4G8) (BioLegends, 800701, 1:8000), anti-phosphorylated tau (AT8) (Thermo Fisher Scientific, MN1020, Ser202/Thr205 monoclonal, 1:1000), and anti-glial fibrillary acidic protein (GFAP) (Ventana, 760-4345, 1:10). Primary antibodies were detected using DISCOVERY universal secondary biotinylated antibody cocktail (Roche, 760-4205). Roche ultraView reagents were used in the preparation of all IHCs, and IHC stains were performed on a Ventana Benchmark XT following manufacturer’s instructions. All IHC slides were counterstained with Hematoxylin prior to visualization. In addition to IHC stains, donor samples were prepared with a modified Bielschowksy’s silver stain in order to visualize diffuse plaques, neuritic plaques, and neurofibrillary tangles. Slides were scanned using a Leica SCN400 F whole-slide scanner through NYU Langone’s Experimental Pathology Core. All slides were blinded for imaging and subsequent evaluation/quantification using QuPath (v0.2.3) software (Bankhead et al., 2017). Raw images are available through the Cell Image Library (CIL group#: 54423).
Quantification of4G8 andAT8 IHC.
For 4G8 and AT8 quantification, hematoxylin and DAB stains were separated using the default H-DAB color deconvolution settings for downstream quantifications. To quantify the area of each tissue section, the region of the section was defined using a custom pixel classifier, classifying pixels at a resolution of 2 μm, with a hematoxylin channel threshold of 0.01 after applying a gaussian filter (sigma = 2). To quantify amyloid-β plaques, 4G8 staining within each section’s region of interest (ROI) was quantified using a second custom pixel classifier, which classified pixels at a resolution of 0.25 μm, with a DAB stain value greater than 0.2 as 4G8-positive and excluded any positive signal that was < 5 μm2 in area. To quantify neurofibrillary tangles, AT8 staining within each section’s ROI was quantified using a second custom pixel classifier, which classified pixels at a resolution of 0.25 μm, with a DAB stain value greater than 0.5 as AT8-positive. Additionally, to count AT8+ cell bodies, the same custom pixel classifier was applied with an additional parameter to remove any AT8+ signal that was smaller than 60 μm2 or larger than 1200 μm2 (completed post-hoc in R). For both 4G8 and AT8 staining, normalized pixel density (4G8 or AT8 staining/total tissue area) was calculated in R (v4.0.3), and total counts and areas of each feature (i.e., 4G8+ plaques and AT8+ cell bodies) were also tallied. All raw quantifications for 4G8 and AT8 are provided in Table S2.
Quantification of GFAP IHC.
For GFAP quantification, hematoxylin and DAB stains were first separated using the default H-DAB color deconvolution settings to separate stains for downstream quantification. To quantify the area of each tissue section, the region of the section was defined using a custom pixel classifier, classifying pixels at a resolution of 2 μm, with a value greater than or equal to 215 after applying a gaussian filter (sigma = 1.5). Continuous regions of pixels exceeding the threshold were classified as belonging to the tissue section, after excluding small regions less than 10,000 μm2 in area which were detached from the majority of the section. Densely stained regions of artifact on the edges of the sections, if present, were manually selected using the brush tool and excluded from the final ROI defining the section. Next, the total area of GFAP staining within each section was quantified. The area of GFAP staining within each section’s ROI was then quantified using a second custom pixel classifier, which classified pixels at a resolution of 0.25 μm, with a DAB stain value greater than 0.25 as GFAP-positive. To normalize across sections of different areas, GFAP staining was reported as the proportion of the pixels in the section’s ROI that were GFAP+. The average proportion of pixels that were GFAP+ for the sections from each donor was then calculated using R.
In addition to normalized pixel density, the average area of the astrocytes in each section was quantified using QuPath, Fiji (Schindelin et al., 2012), and R. First, quantifiable astrocytes in each section were identified manually as GFAP+ cells with an identifiable cell body and several processes that could be distinguished from the processes of adjacent astrocytes. All quantifiable astrocytes from a given section were numbered, and a maximum of 8 astrocytes from each section were randomly selected for quantification using R. In sections from two (out of 16) donors (specifically D2 and D6), no quantifiable astrocytes could be identified, and thus these donors were excluded from further analysis. Each randomly chosen astrocyte was then analyzed using Fiji. ROIs were manually drawn around each astrocyte using the freehand tool such that the GFAP staining contained within each ROI appeared to correspond to only the chosen astrocyte. The region outside the ROI was then excluded, and the included astrocyte was then segmented via manual thresholding using the default method. Then, the area of the segmented astrocyte was measured. The average area of the astrocytes from each donor was then calculated using R. All raw quantifications for GFAP are provided in Table S2.
Semi-quantification of Bielschowsky’s silver stain.
At 15X magnification, 5 ROIs were drawn randomly in each tissue section for manual assessment. Diffuse plaques, neuritic plaques, and neurofibrillary tangles were manually counted in each ROI. Based on total pathology feature counts, samples were given broad descriptors indicating none, low, moderate, or severe pathology. Raw pathological feature counts and overall descriptors are listed in Table S2.
Validation of transcriptomic DEGs using antibody staining.
Formalin-Fixed Paraffin-Embedded (FFPE) human brain tissue was sectioned to 5 μm and mounted on microscope slides. The sections were dewaxed at 60 °C for 30 min and then transferred into HistoChoice (Sigma) for 2 washes for 5 min each. Sections were then moved into 100%, 95% and 70% ethanol for rehydration followed by three washes in PBS. For C3 staining, sections underwent antigen retrieval in M6 buffer (2.1% citric acid monohydrate, 2.94% tri-sodium citrate in dH2O, pH 6) at 95 °C for 10 min.
Sections were then blocked in blocking buffer containing 10% normal goat serum (NGS), 0.4% Triton X-100 (Sigma) in PBS for 1h at room temperature. The following primary antibodies were used: C3d (Dako A0063, rabbit 1:600), GFAP (Dako Z0034, rabbit, 1:500), GFAP (Sigma G3893, mouse, 1:400) and SPARC (R&D MAB941, mouse, 25 ug/mL). Sections were incubated in the primary antibodies over night at 4 °C in blocking buffer followed by three washes in PBS. Sections were then incubated in the following secondary antibodies: goat anti-rabbit Alexa 594 (Invitrogen) and goat anti-mouse Alexa 488 (Abeam) at room temperature for 1h. Sections were then washed in PBS, incubated in TrueBlack (biotium) for 1 min, counter-stained with DAPI and mounted using Fluoromount-G (SouthernBiotech). All images were acquired on a Keyence BZ-X710 using a 20x objective and processed in Fiji.
Isolation of nuclei from postmortem frozen human prefrontal cortex tissue
The protocol followed to isolate nuclei from postmortem frozen human brain tissue was based off of a previously published study (Hodge et al., 2019). Processing of tissue was completed on ice or at 4°C for the entirety of the protocol. In brief, ~100 mg of postmortem frozen human prefrontal cortex per donor was homogenized in 2 mL homogenization buffer (10 mM Tris pH 8 (Invitrogen, Am9010), 250 mM sucrose (Invitrogen, Am9010), 25 mM KCl (Invitrogen, Am9010), 5 mM MgCl2 (Invitrogen, Am9010), 0.1% Triton X100 (Sigma, T8787), 1%RNasin Plus (Promega, N2615), 1X Protease inhibitor (Promega, G6521), and 0.1 mM DTT (Sigma, D9779) in DNase- and RNase-free water) using a Wheaton Dounce tissue grinder (10 strokes with loose pestle, followed by 10 strokes with the tight pestle). Then, the homogenized tissue was filtered through 30 μm pre-separation filters (Miltenyi, 130-041-407) to remove major debris. Samples were then centrifuged at 900 g for 10 minutes at 4°C, and resulting nuclei pellet was resuspended in blocking buffer in preparation for immunolabel-based sorting.
Fluorescence-activated cell sorting for astrocytic nuclei enrichment
All procedures were completed on ice or at 4°C, and centrifugations were completed at 400 g for 5 minutes at 4°C unless otherwise specified. Post-isolation, nuclei suspensions were incubated in blocking buffer (0.8% reagent-grade bovine serum albumin (BSA; Proliant Biologicals, 68700), 0.5% RNasin Plus, and 10% goat serum (MP Biomedicals, 191356) in 1X phosphate buffer saline (PBS; VWR, 16750-102)) for 20 minutes. Each sample was split into aliquots in order to prepare all respective controls for sorting, including unstained, secondary antibody-only, and single antibody-labeled controls. All antibody dilutions were prepared in fluorescence-activated cell sorting (FACS) buffer (0.8% reagent-grade BSA and 0.5% RNasin Plus in 1X PBS). Samples were then pelleted, resuspended in either FACS buffer or diluted primary antibodies, and incubated on ice for 20 minutes in the dark. Post-incubation, samples were pelleted and washed with FACS buffer prior to resuspension in FACS buffer or diluted secondary antibodies. Finally, samples were pelleted and washed with FACS buffer prior to resuspension in FACS buffer for sorting. Immediately prior to sorting, samples were spiked with 4′,6 Diamidino 2 Phenylindole, Dihydrochloride (DAPI; Thermo Fisher Scientific, D1306, 1:10,000) in order to better visualize nuclei. All samples were sorted using a MoFlo XDP sorter with a 100 μm nozzle at 4°C. Unstained, isotype control, and single antibody-labeled controls were used to established gating scheme for each donor (see Figure S2 for representative gating schemes). All sorting data was visualized and quantified using FlowJo (v10.7.1).
S0X9 sorts.
Primary antibodies used for SOX9-based sorts included: Anti-SOX9 (Abeam, ab185966, 1:100) and rabbit IgG isotype control (Abeam, ab172730, 1:200). Primary antibodies were detected with goat anti-rabbit IgG (H&L) secondary antibody conjugated to Alexa Fluor 594 (Thermo Fisher Scientific, R37117, 2 drops/mL). Gates were set to collect DAPI+ and SOX9+ singlet events, and on average, ~75,000 DAPP/SOX9+ nuclei were captured per pilot donor (see Table S3 for all SOX9 sorting outputs).
LHX2+/NeuN− sorts.
Primary antibodies used for LHX2+/NeuN−-based sorts included: Anti-LIM Homeobox 2 LHX2 (EMD Millipore, AB5756, 1:500), anti-NeuN (Millipore Sigma, MAB377, 1:2500), rabbit IgG isotype control (Abcam, ab172730, 1:1000), mouse IgG1 isotype control (Millipore Sigma, MABC002, 1:1250). Primary antibodies were detected with goat anti-rabbit IgG (H&L) secondary antibody conjugated to Alexa Fluor 488 (Invitrogen, A11034, 1:4,000) or goat anti-mouse IgG (H&L) secondary antibody conjugated to Alexa Fluor 647 (Invitrogen, A21235, 1:7,000), respectively. Gates were set to collect DAPI+, LHX2+, and NeuN− singlet events, and on average, ~88,000 DAPI+/LHX2+/NeuN− nuclei were captured per donor (see Table S3 for all LHX2+/NeuN−sorting outputs).
Single-nuclei RNA sequencing pipeline
After sorting, collected nuclei were pelleted at 900 g for 10 minutes at 4°C, were resuspended in ~50 μL of 0.04% BSA in PBS, and counted using a hemocytometer. Based on nuclei counts, samples were then resuspended in additional 0.04% BSA in PBS buffer in order that nuclei concentrations were ideal for 10x Chromium loading (between 100-1,700 nuclei/μL). Nuclei were processed using the Single Cell 3’ Gene Expression kit v3 (10x Chromium, 1000076) according to manufacturer’s instructions. In brief, 4,800-16,000 nuclei per sample were loaded onto Single Cell Chips B in order to recover as many nuclei as possible (targeting 3,000-10,000 nuclei per sample), while limiting potential for doublets. Using a Chromium Controller, Gel Bead-In Emulsions were generated, and samples were subsequently processed to isolate and amplify cDNA and ultimately construct libraries. Quality and concentration of cDNA was evaluated on an Agilent 2100 Bioanalyzer. Quality and concentration of libraries were evaluated by qPCR and on an Agilent 2200 TapeStation, and libraries were sequenced an Illumina NovaSeq 6000 through NYU Langone’s Genomic Technology Core. Basecalling was completed using Illumina NovaSeq 6000 RTA v3.4.4 software, and BCL base call files were converted to FASTQ files using bcl2fastq Conversion software (v2.20). Using Cell Ranger software suite (v4.0.0) (1 OX Genomics), FASTQ files were aligned to a premRNA-modified GRCh38 human reference genome (modification steps provided by 10x Genomics), and gene-barcode count matrices were generated for all demultiplexed samples.
Single-nuclei RNA sequencing data analysis
The majority of code used to evaluate snRNA-seq data is based off of analysis completed in the original muscat R package vignette (Crowell et al., 2019). This process was repeated separately for the SOX9-sorted pilot donor cohort as well as for iterations of LHX2+/NeuN−-sorted donor cohort. Exact code used to analyze each dataset analysis is provided on the Liddelow Lab GitHub page: hhttps://github.com/liddelowlab/Sadick_et_al.2022.
Quality control.
All sample raw gene-barcode count matrices were converted into a SingleCellExperiment (SCE) object in R (v3.6.1 - 4.0.3) for initial quality control filtering (Amezquita et al., 2020). Undetected genes were removed based on the total summed counts per gene. Doublets were removed using scds, in which a threshold was applied assuming that 1% of every 1,000 nuclei captured was a doublet. Sample-specific outliers were identified using scater::isOutlier, and nuclei were removed if total counts, total features, and/or percentage of mitochondrial genes was greater than 2.5 median absolute deviations away from the sample median. Finally, genes were only kept if they had a count of at least one in more than 20 nuclei. After filtering SOX9-sorted donor pilot data, a total of 18,991 nuclei remained across 2 NS and 3 AD patients, with a median of 8,885 counts and 3,406 features per nucleus. After filtering LHX2+/NeuN−-sorted donor data (final donor cohort), a total of 80,247 nuclei remained across 5 NS and 9 AD patients, with a median of 6,714 counts and 2,929 features per nucleus.
Normalization, integration, and dimension reduction.
Functions in the Seurat (v3.2.2) package were used for the following analyses (Stuart et al., 2019). Data were log normalized, and the top 2,000 variable features were identified on a per sample basis. Samples were then anchored and integrated using Canonical Correlation Analysis (dims = 30). After scaling the data, linear and non-linear dimension reduction was performed by Principle Component Analysis of variable features and t-Distributed Stochastic Neighbor Embedding (tSNE) analysis, respectively, using the top 30 principle components. For each dataset, the number of dimensions used for dimensional reduction analyses was determined based on the inflection point on an Elbow plot.
Clustering, annotation, and marker identification.
Clustering was calculated using the functions FindNeighbors and FindClusters, with a range in resolution between 0.1-1. Ultimately, for each dataset, a resolution of 0.1 was used for initial clustering. To identify major cell types present, the FindAllMarkers function (log2 fold change > 0.25, using Wilcoxon Rank Sum test, adjusted p-value < 0.05 using the Bonferroni correction) was used to determine unique and/or highly enriched differentially expressed genes (DEGs) in one cluster compared to all other clusters. These cluster-specific features were then queried against a set of canonical cell type-specific markers from the literature. Data were visualized using Seurat package functions, including DimPlot, FeaturePlot, DotPlot, VlnPlot, and DoHeatmap. For SOX9-positive sorted data, only 6.6% of nuclei were astrocytes, while the vast majority of nuclei (72.2%) were neurons. For LHX2+/NeuN−-based sorted data, astrocytes made up the largest captured nuclei population (51.5%), with the second largest captured nuclei population as oligodendrocytes (29.7%). Please refer to Table S3 for cell type captures and DEGs identified for each sorting strategy.
Cell type-specific sub-clustering.
Using the function Seurat::subset, astrocyte and oligodendrocyte nuclei were reanalyzed in isolation. For SOX9-based sorts, 1,832 astrocytes were analyzed. For LHX2+/NeuN−-based sorts (final cohort), 41,340 astrocytes and 23,840 oligodendrocytes were analyzed, respectively. Astrocyte- and oligodendrocyte-specific analyses were completed as described above (from identifying a new set of top 2,000 variable features through clustering and marker identification). Please note that two rounds of subsetting and analysis were required for LHX2+/NeuN−-sorted astrocytes in order to remove contaminating, non-astrocytic nuclei. Please see to Table S3 for the number of principle components and resolutions used in each analysis as well as astrocyte- and oligodendrocyte-specific DEGs. Data were visualized using Seurat package functions (as listed above). Additionally, DEGs for LHX2+/NeuN−-sorted astrocytes and oligodendrocytes were evaluated by pathway analysis. In brief, cluster-specific astrocyte or oligodendrocyte DEG gene IDs were converted to ENSEMBL IDs (using AnnotationDbi package org.Hs.eg.db::mapIDs) and then to Entrez IDs (using biomaRt::getBM). For each individual cluster in each cell type, Entrez IDs were analyzed using clusterProfiler::enrichGO, and gene ontology (GO) terms were identified (adjusted p-values < 0.05 using the Benjamini-Hochberg method, false discovery rate < 0.1) (Table S3).
Cluster-specific differential gene expression and pathway analysis
Differential gene expression and pathway analysis was completed for LHX2+/NeuN−-sorted astrocyte and oligodendrocyte clusters in parallel. For each cluster in each cell type, these analyses were completed based on disease state (i.e., comparing all NS donors with AD donors). Data was read in using the Seurat::subset function. The subsetted Seurat object was then converted into a SCE object for additional threshold filtering. Lowly expressed genes were removed, as identified by having fewer than 5 transcripts counted in less than 5 cells. Then, the top 2000 variable genes were identified using scater::modelGeneVar and getTopHVG functions, and the zinbwave function was run using observational weights generated for each gene (K = 0, epsilon = 1e12) (Risso et al., 2018). The remaining DEG analysis was completed using the edgeR package (Robinson et al., 2010) by calculating normalized factors, estimating dispersion, model fitting using glmFit, comparing disease state (i.e., NS versus AD), and passing zinbwave-generated observational weights to the glmWeightedF function. Genes were identified as DEGs if they had an adjusted p-value < 0.05 using the Benjamini-Hochberg method and had a log2 fold change > ±0.25. For pathway analysis, all DEGs were converted to their ENSEMBL IDs and subsequently their Entrez IDs (as described above) prior to being separated into upregulated and downregulated lists with their accompanying log2 fold changes. Each list was then analyzed separately to determine upregulated and downregulated GO terms (as described above). DEGs were visualized using ComplexHeatmap::Heatmap function. Upregulated and downregulated DEGs as well as GO terms were compared across clusters and were visualized using UpSetR package. Lists of DEGs and pathways are provided in Tables S4 and S5.
Reanalysis of previously published snRNA-seq and scRNA-seq datasets
We obtained FASTQ files from previously published snRNA-seq (Grubman et al., 2019; Mathys et al., 2019; Zhou et al., 2020) datasets in order that all datasets were analyzed under identical protocols to our own generated data, as described above beginning at alignment using the Cell Ranger software suite (v4.0.0). All snRNA-seq datasets were aligned to the premRNA-modified GRCh38 human reference genome. As described above, each dataset was processed for quality control, normalization, anchoring, integration, dimension reduction, clustering, annotation, and marker identification (Figure S6). For each dataset, astrocyte and oligodendrocyte nuclei were subsetted as unique Seurat objects for reanalysis in isolation (Figures 3 and 7). For Mathys astrocyte and oligodendrocyte subsetted analyses, multiple donors were removed because their nuclei yields were lower than the number of principle components used to evaluate the data (donors removed are listed in Table S1). Please note that reference-based integration was used for all objects in Mathys and Zhou datasets due to memory constraints (parameters listed in Table S1). Additionally, please note that two rounds of subsetting and analysis were required for Zhou astrocytes in order to remove contaminating, non-astrocytic nuclei/cells. Principle components/resolution used for analyses, cell type captures, and DEGs identified per cluster per dataset are listed in Table S1.
Cell type-specific multi-dataset analyses
Once only astrocyte and oligodendrocyte Seurat objects were created for each dataset, we merged either all astrocyte-or oligodendrocyte-specific objects to create two multi-dataset objects: (1) astrocyte nuclei from Mathys, Grubman, Zhou, and our LHX2+/NeuN−-sorted snRNA-seq datasets (Figure 7) and (2) oligodendrocyte nuclei from Mathys, Grubman, Zhou, and our LHX2+/NeuN−-sorted snRNA-seq datasets (Figure 3). Once individual dataset cell type-specific Seurat objects were merged using the function Seurat::merge, multi-dataset objects were processed as described above (from identifying a new set of top 2,000 variable features through clustering and marker identification). Reference-based integration was used for astrocyte and oligodendrocyte nuclei snRNAseq merged objects due to memory constraints (parameters listed in Table S5). Please refer to Table S5 for multi-dataset cell type-specific cluster yields and DEGs identified.
Analysis of previously published spatial transcriptomic data
Code used to analyze the spatial localization of astrocyte gene signatures is available on the Liddelow Lab GitHub page: https://github.com/liddelowlab/Sadick_et_al.2022. All statistics, plots, and analysis for this portion of the manuscript were created with R (v4.1.1).
Datasets.
Previously published 10X Visium spatial transcriptomics data from twelve pathology-free human dorsolateral prefrontal cortex (DLPFC)88 samples (3 donors) was obtained from the Globus repository (available at http://research.libd.org/globus/jhpce_HumanPilot10x/index.html) in the form of filtered feature-barcode matrices, corresponding TIFF images, and spot coordinate TSV files. We also utilized our lab’s recently published 10X Visium data from whole hemisphere saline or LPS-injected mice (n = 3 per condition), referred to in this manuscript as “healthy”/“control” and “inflamed” (data available at the Gene Expression Omnibus repository: Series GSE165098).25 These mice were injected intraperitoneally with saline or 5 mg/kg lipopolysaccharide (LPS), and tissue was collected 24 hours post-injection.25
Creating astrocyte cluster gene modules.
We defined gene signatures for each astrocyte cluster using positive marker genes (log2-fold change > 0.3, adjusted p-value < 0.05) from our earlier differential expression testing (see Cluster-specific differential gene expression and pathway analysis and Table S3). To explore expression of these gene signatures in the mouse data as well, we created mouse gene modules comprised of one-to-one orthologs of the human module genes, which we identified using the biomaRt R package (v2.48.3) (Durinck et al., 2009). These mouse gene modules were then filtered to remove genes not expressed in the spatial transcriptomics data. The resultant modules of genes for each astrocyte cluster are available in Table S7. We also created mouse modules using these same methods for genes upregulated in each astrocyte cluster in AD (log2-fold change > 0.5, adjusted p-value < 0.05) using our differential expression testing results (see Cluster-specific differential gene expression and pathway analysis and Table S6). The genes comprising these AD gene modules are also available in Table S7.
Regional annotation.
Human: The spots in the human spatial transcriptomics dataset were previously manually annotated with their cortical layer locations by Maynard et al.88, and these annotations were retrieved using the spatialLIBD R package (v1.4.0) (Pardo et al., 2021). Layers 2 and 3 were combined and labeled layer 2/3 to aide in comparison to the mouse dataset. Mouse: To first identify spots as white matter or gray matter, we assigned white matter scores to individual spots on the basis of expression of white matter/myelin genes (Mbp, Mobp, Plp1, Mag, Mog, and Mal) using the AddModuleScore function in Seurat (v4.0.5) (Hao et al., 2021) (Figure S12C, see Differential Enrichment Testing for detailed explanation of module score calculations). Initial classification of spots as white matter or gray matter was then determined by thresholding the white matter module score; spots with a module score greater than the 78.75th percentile were classified as white matter, and spots below this threshold were classified as gray matter. This threshold was chosen based on agreement with anatomy apparent from the H&E staining. Individual spot classification was then manually adjusted to correct mistaken assignments (e.g. a spot clearly in the corpus callosum but assigned as gray matter). Gray matter spots were then labeled with their cortical layer location based on gene expression. To this end, spot-level gene expression data was log-normalized. Then top 3,000 highly variable genes most common across the six samples were identified using the FindIntegrationFeatures function. Technical variables (the number of genes detected per spot, the number of UMIs per spot, and the percentage of UMIs mapping to the mitochondrial genome per spot) were regressed against each gene in the normalized feature-barcode matrix, and the residuals were scaled and centered. PCA was then performed on the scaled data, using the previously identified highly variable genes. The resulting PCA embeddings were then iteratively corrected to remove the effects of sample identity and condition (LPS or saline) using the Harmony R package (v0.1.0) (Korsunsky et al., 2019). UMAP dimensionality reduction was then performed using the first 15 principal components from the integrated, corrected PCA embeddings. A shared nearest-neighbor graph was constructed using the FindNeighbors function, and unbiased Louvain clustering was performed using the FindClusters function with a resolution parameter of 0.6. Several resolution parameters were examined, and 0.6 was selected because the resultant clusters appeared to map most closely to the layers of the cortex. Four clusters appearing largely in the retrosplenial and somatomotor areas were subsetted, re-integrated, and sub-clustered using identical methods to capture the cortical layers in those regions (with 2,000 variable features, 20 principal components for UMAP dimensionality reduction and clustering, and a clustering resolution parameter of 0.5). In aggregate, clustering and sub-clustering the cortical mouse Visium spots resulted in 11 clusters, which were each largely contained within one cortical layer. Each cluster was then assigned to a cortical layer – L1, L2/3, L4, L5, or L6 – based on its position in the Visium sections and its average expression of canonical markers. Finally, the individual layer labels were adjusted for individual outlier spots to match their true anatomical locations. Final cortical layer groups were generally well separated on the basis of gene expression (Figure S12D), validating their annotations. To verify that our mouse cortical layers were well labeled and comparable to the previously annotated human Visium data, we next sought to compare the cross-species similarity of each cortical layer. To this end, we z-scored the average expression of all one-to-one orthologous genes that were highly variable (top 3,000 genes) in both the human and mouse Visium datasets across the cortical layers in each dataset (in total, 707 genes). We then calculated the Spearman correlation coefficients for all layer combinations between the datasets. This revealed each cortical layer in a given species was most highly correlated with the same cortical layer in the corresponding species (Figure S12E), supporting the validity of our mouse cortical layer annotations and cross-species comparison of the spatial transcriptomics datasets.
Differential Enrichment Testing.
To quantify enrichment of astrocyte subtype and reactive sub-state gene signatures across cortical regions, we first calculated expression scores for each gene module within each spatial transcriptomics spot using Seurat and the AddModuleScore function89,25. In summary, module scores reflect the average expression of the gene set subtracted by expression of a control gene set 100 times larger than the gene module, where each gene in the module is matched with 100 genes in the control gene set with similar expression levels 89. Subtraction of the control gene set allows for standardization of module scores across spots and Visium sections despite technical variation. Most astrocyte cluster gene modules contained dozens or even hundreds of genes, making these module scores robust to gene drop out and technical variation. To test whether each cluster gene module differed in enrichment across cortical regions, we pooled all sections of a given species together and compared gene module scores between spots of each cortical region (L1-L6 and WM) using a Kruskal-Wallis test with a Bonferroni correction for multiple comparisons. All cluster gene modules exhibited statistically significant differences (adjusted p-value < 0.05; see Table S7 for test statistics and p-values). To compare how strongly different cluster modules varied in score across the cortex in Figure S12F, Kruskal-Wallis H test statistics were z-scored within each species and a heatmap was created using the ComplexHeatmap R package (v2.8.0) (Gu et al., 2016). To test whether each gene module was enriched (more highly expressed) or de-enriched (less highly expressed) in a given cortical region compared to the rest of the cortex, we next performed Wilcoxon rank sum tests with Bonferroni multiple comparisons corrections comparing the spots from a given region to all other spots (pooling all other cortical regions; see Table S7 for p-values and test statistics). For Figure 6A–B & Figure S12G, + or − symbol indicates the gene module is significantly enriched or de-enriched, respectively, in a given cortical region compared to the rest of the cortex (adjusted p-value < 0.05). The sign of the symbol corresponds to the sign (positive or negative) of the median of the estimated difference between a spot from the cortex region in question compared to spots from the rest of the cortex (i.e. the median location shift). We next tested whether cluster gene modules were upregulated between spots from the inflamed (LPS-injected) mouse brain compared to the control (saline-injected) brain using Wilcoxon rank sum tests with Bonferonni corrections (Table S7). Lastly, we examined whether AD gene modules for each astrocyte subtype were upregulated in spots from the inflamed mouse brain compared to controls using Wilcoxon rank sum tests with Bonferonni corrections (Table S7). For Figure 6E–F and Figure S12H, + or − symbols indicate the gene module is significantly enriched or de-enriched, respectively, in spatial transcriptomics spots from the LPS-injected samples compared to spots from the saline-injected samples within the indicated cortical region (adjusted p-value < 0.05). For all box and density plots in Figure 6 and Figure S12: transecting line denotes the median; notches denote the 95% confidence interval surrounding the median; and whiskers denote 1.5x the interquartile range. All box and density plots were created with the ggplot2 (v3.3.5) and ggdist (v3.0.1) (Kay, 2021) packages in R.
Visualization.
Visualization of astrocyte cluster gene signatures across representative Visium sections is challenging given sparse expression, the prevalence of gene drop out in spatial transcriptomics data, and the size of Visium spots (which often capture several cells within each spot). To overcome these difficulties, we utilized a recent Bayesian method of spatial gene expression resolution enhancement 93. Individual samples were first normalized with Seurat (Hafemeister and Satija, 2019),, then preprocessing, PCA, and spatial clustering were performed using the BayesSpace R package (v1.2.1)93. For the human samples, seven clusters (chosen based on the elbow in a log-likelihood plot) and 15 principal components (chosen based on the elbow in a plot of the standard deviation of each principal component from the PCA results) were used for spatial clustering. For the mouse samples, 20 principal components and 17-18 clusters were used. Spatial clustering was then enhanced to sub-spot resolution using the spatialEnhance function with default parameters. Spatial gene expression for all genes was then enhanced to sub-spot resolution using the enhanceFeatures function, which fits a linear model for each gene with the top principal components from each spot and predicts sub-spot expression using this model.93 We then calculated enrichment scores for each sub-spot using the above module score method (see Differential Enrichment Testing) with the same gene modules (see Creating astrocyte cluster gene modules) using the resolution-enhanced expression data. Enrichment scores were then plotted with a minimum-maximum scale on representative sections. We found calculating enrichment scores using these resolution enhancement methods aided in recognition of spatial patterns and were generally representative of spot-level module score patterns within each cortical region (Figure S12B). All differential enrichment testing and statistical tests were performed using original, spot-level gene expression and module scores; Bayesian resolution-enhanced gene expression and enrichment scores were only used for visualization.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistics
Throughout this manuscript, quantified cell/nuclei outputs are displayed as the arithmetic mean (± s.d., if applicable), and plots were generated using the ggplot2 and ggpubr packages in R and/or using GraphPad Prism (v9.0.0) unless otherwise noted. Please note that all quantified raw values for sorting yields, IHC measurements, snRNA-seq and spatial transcriptomics analyses are also available in Tables S1–7.
Wilcox test.
For Figure 1A, statistical tests used were two-tailed Wilcoxon signed-rank tests (α = 0.05). All quantification and analysis were performed blinded to the condition of the donors.
Randomization.
No randomization was used in the analysis of snRNA-seq or IHC imaging data.
Sample size estimation.
No methods were used to predetermine the sample size used in this study. However, our donor cohort is similar in size to published works (Grubman et al., 2019; Leng et al., 2021).
ADDITIONAL RESOURCES
The following datasets were produced during this study and are available for download and further analysis.
Cell Image Library:
http://cellimagelibrarv.org/groups/54423
All pathology imaging data are available through the Cell Image Library repository.
Raw sequencing data:
https://www.ncbi.nlm.nih.gov/geo/guery/acc.cgi?acc=GSE167494
Raw single nuclei RNA sequencing (snRNA-seq) data generated in this study (including both FASTQ and Cell Ranger-generated matrix files) are available at GEO (Gene Expression Omnibus) under accession number GSE167494.
Analyzed snRNA-seq data (pseudobulk):
We created an open-access, interactive website as a resource to share and easily navigate our analyzed snRNA-seq data for astrocytes and oligodendrocytes.
GitHub:
https://github.com/liddelowlab/Sadick_et_al._2022
All code for analysis of original and previously published snRNA-seq and spatial transcriptomic datasets are available on the Liddelow Lab GitHub page.
Supplementary Material
Table S3 (Related to Figures S1, S2). Sorting outputs and cluster-specific DEG and GO analyses.
Table S5 (Related to Figures 3, 7, S7, S8, S13). Astrocyte and oligodendrocyte multi-dataset integrations.
KEY RESOURCE TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-amyloid-beta 17-24 | BioLegends | Cat#800701; RRID:AB_2564633 |
| Mouse anti-phospho-tau, Ser202/Thr205 monoclonal | Thermo Fisher Scientific | Cat#MN1020; RRID:AB_223647 |
| Rabbit anti-glial fibrillary acidic protein (EP672Y) | Ventana | Cat#760-4345 |
| DISCOVERY universal secondary biotinylated antibody cocktail | Roche | Cat#760-4205; RRID:AB_10805231 |
| Rabbit anti-SOX9 (EPR14335-78) | Abcam | Cat#ab185966; RRID:AB_2728660 |
| Rabbit IgG isotype control (EPR25A) | Abcam | Cat#ab172730; RRID:AB_2687931 |
| Mouse anti-Human SPARC (Clone: 122511) | R&D Systems | Cat# MAB941, RRID:AB_2195073 |
| Mouse anti-GFAP | Sigma-Aldrich | Cat# G3893, RRID:AB_477010 |
| Rabbit anti-GFAP | DAKO | Cat# Z0034 |
| Rabbit anti-Human C3d | DAKO | Cat# A0063 |
| Goat anti-Rabbit IgG (H+L) Cross-Adsorbed ReadyProbes Secondary Antibody, Alexa Fluor 594 | Thermo Fisher Scientific | Cat#R37117; RRID:AB_2556545 |
| Rabbit anti-LIM Homeobox 2 LHX2 | EMD Millipore | Cat#AB5756; RRID:AB_92012 |
| Mouse anti-NeuN (Clone A60) | Millipore Sigma | Cat#MAB377; RRID:AB_2298772 |
| Mouse IgG1 isotype control (Clone Ci4) | Millipore Sigma | Cat#MABC002; RRID:AB_97846 |
| Goat anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | Invitrogen | Cat#A11034; RRID:AB_2576217 |
| Goat anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 594 | Invitrogen | Cat# A11012, RRID:AB_2534079 |
| Goat anti-Mouse IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 647 | Invitrogen | Cat#A21235; RRID:AB_2535804 |
| Bacterial and Virus Strains | ||
| Biological Samples | ||
| De-identified human, post-mortem prefrontal cortex samples from Alzheimer’s disease and age-matched non-symptomatic patients | Rhode Island Hospital’s Brain Tissue Resource Center Table S2 | https://www.brown.edu/research/facilities/brain-tissue-resource-center/ |
| De-identified human, post-mortem prefrontal cortex samples from Alzheimer’s disease and age-matched non-symptomatic patients | Alzheimer’s disease Research Center at NYU Langone Table S2 | https://med.nyu.edu/departments-institutes/neurology/divisions-centers/center-cognitive-neurology/alzheimers-disease-center |
| De-identified human, post-mortem prefrontal cortex samples from Alzheimer’s disease and age-matched non-symptomatic patients | Shiley-Marcos Alzheimer’s disease Research Center at UCSD Table S2 | https://medschool.ucsd.edu/som/neurosciences/centers/adrc/Pages/default.aspx |
| Chemicals, Peptides, and Recombinant Proteins | ||
| RNase-free buffer kit | Invitrogen | Cat#Am9010 |
| Triton X100 | Sigma | Cat#T8787 |
| RNasin Plus | Promega | Cat#N2615 |
| Protease inhibitor cocktail | Promega | Cat#G6521 |
| DL-Dithiothreitol | Sigma | Cat#D9779 |
| DNase- and RNase-free water | Invitrogen | Cat#10977015 |
| Reagent-grade bovine serum albumin | Proliant Biologicals | Cat#68700 |
| Goat serum | MP Biomedicals | Cat#191356 |
| 4′,6 Diamidino 2 Phenylindole, Dihydrochloride | Thermo Fisher Scientific | Cat#D1306 |
| Phosphate buffer saline | VWR | Cat#16750-102 |
| Critical Commercial Assays | ||
| QIAshredder | QIAGEN | Cat#79656 |
| RNeasy Plus Mini kits | QIAGEN | Cat#74136 |
| GoTaq Green Master Mix | Promega | Cat#M7123 |
| Single Cell 3’ Gene Expression kit v3 | 10x Chromium | Cat#1000076 |
| Deposited Data | ||
| Pathology imaging data | This paper | Cell Image Library: http://cellimagelibrary.org/groups/54423 |
| Single nuclei RNA sequencing data (FASTQ and Cell Ranger-generated matrix files) | This paper | GEO: GSE167494; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE167494 |
| Single nuclei RNA sequencing data (FASTQ files) | Mathys et al., 2019 | Synapse: syn18485175; https://www.synapse.org/#!Synapse:syn18485175 |
| Single nuclei RNA sequencing data (FASTQ files) | Grubman et al., 2019 | GEO: GSE138852; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE138852 |
| Single nuclei RNA sequencing data (FASTQ files) | Zhou et al., 2020 | Synapse: syn21670836; https://www.synapse.org/#!Synapse:syn21670836 |
| Experimental Models: Cell Lines | ||
| Experimental Models: Organisms/Strains | ||
| Oligonucleotides | ||
| APOE_e2_FWD: 5′-GCGGACATGGAGGACGTGT-3′ | Zhong et al., 2016 | N/A |
| APOE_e2_REV: 5′-CCTGGTACACTGCCAGGCA-3′ | Zhong et al., 2016 | N/A |
| APOE_e3_FWD: 5′-CGGACATGGAGGACGTGT-3′ | Zhong et al., 2016 | N/A |
| APOE_e3_REV: 5′-CTGGTACACTGCCAGGCG-3′ | Zhong et al., 2016 | N/A |
| APOE_e4_FWD: 5′-CGGACATGGAGGACGTGC-3′ | Zhong et al., 2016 | N/A |
| APOE_e4_REV: 5′-CTGGTACACTGCCAGGCG-3′ | Zhong et al., 2016 | N/A |
| B-actin_FWD: 5′-GACGTGGACATCCGCAAAGAC-3′ | Zhong et al., 2016 | N/A |
| B-actin_REV: 5′-CAGGTCAGCTCAGGCAGGAA-3′ | Zhong et al., 2016 | N/A |
| Recombinant DNA | ||
| Software and Algorithms | ||
| QuPath (version 0.2.3) | Bankhead et al., 2017 | https://qupath.github.io/; RRID:SCR_018257 |
| Fiji-ImageJ (version 2.1.0) | Schindelin et al., 2012 | https://imagej.net/Fiji; RRID:SCR_003070 |
| Prism (version 9.0.0) | GraphPad | https://www.graphpad.com/scientific-software/prism/; RRID:SCR_002798 |
| Flowjo (version 10.7.1) | BD | https://www.flowjo.com/; RRID:SCR_008520 |
| NovaSeq 6000 RTA (version 3.4.4) | Illumina | https://www.illumina.com/systems/sequencing-platforms/novaseq.html; RRID:SCR_014332 |
| bcl2fastq conversion software (version 2.20) | Illumina | https://support.illumina.com/downloads/bcl2fastq-conversion-software-v2-20.html; RRID:SCR_015058 |
| Cell Ranger software suite (version 4.0.0) | 10x Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/installation; RRID:SCR_017344 |
| R (versions 3.6.2 - 4.0.3) | The R Foundation | https://www.r-project.org/foundation/; RRID:SCR_001905 |
| SingleCellExperiment (version 1.12.0) | Amezquita et al., 2020 | https://bioconductor.org/packages/release/bioc/html/SingleCellExperiment.html |
| Seurat (version 3.2.2) | Stuart et al., 2019 | https://satijalab.org/seurat/index.html; RRID:SCR_016341 |
| zinbwave (version 1.12.0) | Risso et al., 2018 | https://bioconductor.org/packages/release/bioc/html/zinbwave.html |
| edgeR (version 3.32.0) | Robinson et al., 2010 | https://bioconductor.org/packages/release/bioc/html/edgeR.html; RRID:SCR_012802 |
| All Rscripts used to analyze single nuclei RNA sequencing data | This paper | https://github.com/liddelowlab/Sadick_et_al. |
| Other | ||
| Open-access, interactive website highlighting all analyzed single nuclei RNA sequencing data | This paper | www.gliaseq.com |
Highlights.
Astrocytes and oligodendrocytes have altered and heterogeneous transcriptomes in AD
Dataset integration improves glia clustering and suggests putative altered function
‘reactive’ sub-states of glia are likely spatially restricted
Astrocyte inflammation responses mimic some AD-associated gene expression changes
ACKNOWLEDGEMENTS
We thank members of the Liddelow lab for review of this manuscript. We also thank the Genome Technology Center at NYU Langone Medical Center for sequence alignment and data preprocessing, the Cytometry and Cell Sorting Laboratory at NYU Langone Medical Center for assistance sorting samples, the Neuropathology Brain Bank & Research CoRE at the Icahn School of Medicine at Mount Sinai for preparing and staining all pathology samples, and the Experimental Pathology Research Laboratory for scanning all pathology samples. Donor samples were provided by the Brain Tissue Resource Center at Rhode Island Hospital, UCSD Alzheimer’s Disease Center, and New York University Alzheimer’s Disease Center (which is sponsored in part by PHS Grant P30 AG08051). SAL was supported by NYU School of Medicine, the Bias Frangione Foundation, the Neurodegenerative Diseases Consortium from MD Anderson, Alzheimer’s Research UK, the Gifford Family Neuroimmune Consortium as part of the Cure Alzheimer’s Fund, The Alzheimer’s Association, and generous anonymous donors. JSS was supported by the National Institutes of Health (T32 via T32AG052909 (Wisniewski, Scharfman)). Additionally, SAL and JSS were supported by the Alzheimer’s Disease Resource Center at NYU Langone Medical Center. AF was supported by the Alzheimer’s Disease Resource Center at NYU Langone Medical Center (P30 AG066512 (Wisniewski)).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATIONS OF INTEREST
SAL is a founder of AstronauTx Ltd. and a member of its scientific advisory board. All other authors declare no competing interests.
INCLUSION AND DIVERSITY
We worked to ensure sex balance in the selection of human samples. One or more of the authors of this paper self-identifies as a member of the LGBTQ+ community. One or more of the members of this paper self-identifies as an underrepresented ethnic minority in science. While citing references scientifically relevant for this work, we also actively worked to promote gender balance in our reference list.
REFERENCES
- Amezquita RA, Lun ATL, Becht E, Carey VJ, Carpp LN, Geistlinger L, Marini F, Rue-Albrecht K, Risso D, Soneson C, et al. (2020). Orchestrating single-cell analysis with Bioconductor. Nat Methods 17, 137–145. 10.1038/s41592-019-0654-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ando K, Uemura K, Kuzuya A, Maesako M, Asada-Utsugi M, Kubota M, Aoyagi N, Yoshioka K, Okawa K, Inoue H, et al. (2011). N-cadherin regulates p38 MAPK signaling via association with JNK-associated leucine zipper protein: implications for neurodegeneration in Alzheimer disease. J Biol Chem 286, 7619–7628. 10.1074/jbc.M110.158477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argaw AT, Asp L, Zhang J, Navrazhina K, Pham T, Mariani JN, Mahase S, Dutta DJ, Seto J, Kramer EG, et al. (2012). Astrocyte-derived VEGF-A drives blood-brain barrier disruption in CNS inflammatory disease. J Clin Invest 122, 2454–2468. 10.1172/JCI60842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alzheimer’s Association (2021). 2021 Alzheimer’s disease facts and figures. Alzheimers Dement 17, 327–406. 10.1002/alz.12328. [DOI] [PubMed] [Google Scholar]
- Bankhead P, Loughrey MB, Fernandez JA, Dombrowski Y, McArt DG, Dunne PD, McQuaid S, Gray RT, Murray LJ, Coleman HG, et al. (2017). QuPath: Open source software for digital pathology image analysis. Sci Rep 7, 16878. 10.1038/s41598-017-17204-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbar L, Jain T, Zimmer M, Kruglikov I, Sadick JS, Wang M, Kalpana K, Rose IVL, Burstein SR, Rusielewicz T, et al. (2020). CD49f Is a Novel Marker of Functional and Reactive Human iPSC-Derived Astrocytes. Neuron 107, 436–453. 10.1016/j.neuron.2020.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayraktar OA, Bartels T, Holmqvist S, Kleshchevnikov V, Martirosyan A, Polioudakis D, Ben Haim L, Young AMH, Batiuk MY, Prakash K, et al. (2020). Astrocyte layers in the mammalian cerebral cortex revealed by a single-cell in situ transcriptomic map. Nature Neuroscience 23, 500–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boisvert MM, Erikson GA, Shokhirev MN, and Allen NJ (2018). The Aging Astrocyte Transcriptome from Multiple Regions of the Mouse Brain. Cell Rep 22, 269–285. 10.1016/j.celrep.2017.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boswell-Smith V, Spina D, and Page CP (2006). Phosphodiesterase inhibitors. Br J Pharmacol 147 Suppl 1, S252–257. 10.1038/sj.bjp.0706495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutros T, Chevet E, and Metrakos P (2008). Mitogen-activated protein (MAP) kinase/MAP kinase phosphatase regulation: roles in cell growth, death, and cancer. Pharmacol Rev 60, 261310. 10.1124/pr.107.00106. [DOI] [PubMed] [Google Scholar]
- Cao Y, Xu H, Zhu Y, Shi MJ, Wei L, Zhang J, Cheng S, Shi Y, Tong H, Kang L, et al. (2019). ADAMTS13 maintains cerebrovascular integrity to ameliorate Alzheimer-like pathology. PLoS Biol 17, e3000313. 10.1371/journal.pbio.3000313. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Carter SF, Scholl M, Almkvist O, Wall A, Engler H, Langstrom B, and Nordberg A (2012). Evidence for astrocytosis in prodromal Alzheimer disease provided by 11C-deuterium-L-deprenyl: a multitracer PET paradigm combining 11C-Pittsburgh compound B and 18F-FDG. Journal of Nuclear Medicine 53, 37–46. 10.2967/jnumed.110.087031. [DOI] [PubMed] [Google Scholar]
- Chang RY, Etheridge N, Dodd PR, and Nouwens AS (2014). Targeted quantitative analysis of synaptic proteins in Alzheimer’s disease brain. Neurochem Int 75, 66–75. 10.1016/j.neuint.2014.05.011. [DOI] [PubMed] [Google Scholar]
- Clarke LE, Liddelow SA, Chakraborty C, Munch AE, Heiman M, and Barres BA (2018). Normal aging induces A1-like astrocyte reactivity. Proc Natl Acad Sci U S A 115, E1896–E1905. 10.1073/pnas.1800165115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins LM, O’Keeffe GW, Long-Smith CM, Wyatt SL, Sullivan AM, Toulouse A, and Nolan YM (2013). Mitogen-activated protein kinase phosphatase (MKP)-1 as a neuroprotective agent: promotion of the morphological development of midbrain dopaminergic neurons. Neuromolecular Med 15, 435–446. 10.1007/s12017-013-8230-5. [DOI] [PubMed] [Google Scholar]
- Counts SE, and Mufson EJ (2017). Regulator of Cell Cycle (RGCC) Expression During the Progression of Alzheimer’s Disease. Cell Transplant 26, 693–702. 10.3727/096368916X694184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowell HL, Soneson C, Germain P-L, Calini D, Collin L, Raposo C, Malhotra D, and Robinson MD (2019). muscat detects subpopulation-specific state transitions from multisample multi-condition single-cell transcriptomics data. Nat Commun 10, 6077. 10.1038/S41467-020-19894-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Torre JC (2004). Is Alzheimer’s disease a neurodegenerative or a vascular disorder? Data, dogma, and dialectics. The Lancet Neurology 3, 184–190. 10.1016/s1474-4422(04)00683-0. [DOI] [PubMed] [Google Scholar]
- De Strooper B, and Karran E (2016). The cellular phase of Alzheimer’s disease. Cell 164, 603–615. 10.1016/j.cell.2015.12.056. [DOI] [PubMed] [Google Scholar]
- Del-Aguila JL, Li Z, Dube U, Mihindukulasuriya KA, Budde JP, Fernandez MV, Ibanez L, Bradley J, Wang F, Bergmann K, et al. (2019). A single-nuclei RNA sequencing study of Mendelian and sporadic AD in the human brain. Alzheimers Res Ther 11, 71. 10.1186/s13195-019-0524-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Castro B, Gangwani MR, Yu X, Coppola G, and Khakh BS (2019). Astrocyte molecular signatures in Huntington’s disease. Science Translational Medicine 11. [DOI] [PubMed] [Google Scholar]
- Dimas P, Montani L, Pereira JA, Moreno D, Trotzmuller M, Gerber J, Semenkovich CF, Kofeler HC, and Suter U (2019). CNS myelination and remyelination depend on fatty acid synthesis by oligodendrocytes. Elife 8. 10.7554/eLife.44702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durinck S, Spellman PT, Birney E, and Huber W (2009). Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat Protoc 4, 1184–1191. 10.1038/nprot.2009.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farkas E, and Luiten PGM (2001). Cerebral microvascular pathology in aging and Alzheimer’s disease. Progress in Neurobiology 64, 575–611. [DOI] [PubMed] [Google Scholar]
- Funfschilling U, Supplie LM, Mahad D, Boretius S, Saab AS, Edgar J, Brinkmann BG, Kassmann CM, Tzvetanova ID, Mobius W, et al. (2012). Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature 485, 517–521. 10.1038/nature11007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerrits E, Brouwer N, Kooistra SM, Woodbury ME, Vermeiren Y, Lambourne M, Mulder J, Kummer M, Moller T, Biber K, et al. (2021). Distinct amyloid-beta and tau-associated microglia profiles in Alzheimer’s disease. Acta Neuropathol. 10.1007/s00401-021-02263-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginsberg SD, Che S, Counts SE, and Mufson EJ (2006). Single cell gene expression profiling Alzheimer’s disease. NeuroRx 3, 302–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg TE, Huey ED, and Devanand DP (2020). Association of APOE e2 genotype with Alzheimer’s and non-Alzheimer’s neurodegenerative pathologies. Nat Commun 11, 4727. 10.1038/S41467-020-18198-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Reyes RE, Nava-Mesa MO, Vargas-Sanchez K, Ariza-Salamanca D, and Mora-Munoz L (2017). Involvement of astrocytes in Alzheimer’s disease from a neuroinflammatory and oxidative stress perspective. Frontiers in Molecular Neuroscience 10 (427). 10.3389/fnmol.2017.00427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubman A, Chew G, Ouyang JF, Sun G, Choo XY, McLean C, Simmons RK, Buckberry S, Vargas-Landin DB, Poppe D, et al. (2019). A single-cell atlas of entorhinal cortex from individuals with Alzheimer’s disease reveals cell-type-specific gene expression regulation. Nat Neurosci 22, 2087–2097. 10.1038/s41593-019-0539-4. [DOI] [PubMed] [Google Scholar]
- Gu Z, Eils R, and Schlesner M (2016). Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849. 10.1093/bioinformatics/btw313. [DOI] [PubMed] [Google Scholar]
- Guttenplan KA, Weigel MK, Adler DI, Couthouis J, Liddelow SA, Gitler AD, and Barres BA (2020). Knockout of reactive astrocyte activating factors slows disease progression in an ALS mouse model. Nat Commun 11, 3753. 10.1038/s41467-020-17514-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttenplan KA, Weigel MK, Prakash P, Wijewardhane PR, Hasel P, Rufen-Blanchette U, Munch AE, Blum JA, Fine J, Neal MC, et al. (2021). Neurotoxic reactive astrocytes induce cell death via saturated lipids. Nature. 10.1038/s41586-021-03960-y. [DOI] [PubMed] [Google Scholar]
- Hafemeister C, and Satija R (2019). Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol 20, 296. 10.1186/s13059-019-1874-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao Y, Hao S, Andersen-Nissen E, Mauck WM 3rd, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager M, et al. (2021). Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587 e3529. 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasel P, Rose IVL, Sadick JS, Kim RD, and Liddelow SA (2021). Neuroinflammatory astrocyte subtypes in the mouse brain. Nat Neurosci 24, 1475–1487. 10.1038/s41593-021-00905-6. [DOI] [PubMed] [Google Scholar]
- Hattori T, Shimizu S, Koyama Y, Yamada K, Kuwahara R, Kumamoto N, Matsuzaki S, Ito A, Katayama T, and Tohyama M (2010). DISC1 regulates N-cadherin and β1-integrin expression in neurons. Molecular Psychiatry 15, 778–778. 10.1038/mp.2010.83. [DOI] [PubMed] [Google Scholar]
- Hodge RD, Bakken TE, Miller JA, Smith KA, Barkan ER, Graybuck LT, Close JL, Long B, Johansen N, Penn O, et al. (2019). Conserved cell types with divergent features in human versus mouse cortex. Nature 573, 61–68. 10.1038/s41586-019-1506-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, Merry KM, Shi Q, Rosenthal A, Barres BA, et al. (2016). Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352, 712–716. 10.1126/science.aad8373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z, Zhao J, Wang W, Zhou J, and Zhang J (2020). Depletion of LncRNA NEAT1 Rescues Mitochondrial Dysfunction Through NEDD4L-Dependent PINK1 Degradation in Animal Models of Alzheimer’s Disease. Front Cell Neurosci 14, 28. 10.3389/fncel.2020.00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang HY, Wek SA, McGrath BC, Lu D, Hai T, Harding HP, Wang X, Ron D, Cavener DR, and Wek RC (2004). Activating transcription factor 3 is integral to the eukaryotic initiation factor 2 kinase stress response. Mol Cell Biol 24, 1365–1377. 10.1128/mcb.24.3.1365-1377.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay M (2021). ggdist: Visualizations of Distributions and Uncertainty. DOI: 10.5281/zenodo.3879620. [DOI] [PubMed] [Google Scholar]
- Kedmi M, and Orr-Urtreger A (2007). Expression changes in mouse brains following nicotine-induced seizures: the modulation of transcription factor networks. Physiol Genomics 30, 242–252. 10.1152/physiolgenomics.00288.2006. [DOI] [PubMed] [Google Scholar]
- Kilinc D (2018). The Emerging Role of Mechanics in Synapse Formation and Plasticity. Front Cell Neurosci 12, 483. 10.3389/fncel.2018.00483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komarova NL, and Thalhauser CJ (2011). High degree of heterogeneity in Alzheimer’s disease progression patterns. PLoS Comput Biol 7, e1002251. 10.1371/journal.pcbi.1002251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korsunsky I, Millard N, Fan J, Slowikowski K, Zhang F, Wei K, Baglaenko Y, Brenner M, Loh PR, and Raychaudhuri S (2019). Fast, sensitive and accurate integration of single-cell data with Harmony. Nat Methods 16, 1289–1296. 10.1038/s41592-019-0619-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau SF, Cao H, Fu AKY, and Ip NY (2020). Single-nucleus transcriptome analysis reveals dysregulation of angiogenic endothelial cells and neuroprotective glia in Alzheimer’s disease. Proc Natl Acad Sci U S A 117, 25800–25809. 10.1073/pnas.2008762117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Viqar F, Zimmerman ME, Narkhede A, Tosto G, Benzinger TL, Marcus DS, Fagan AM, Goate A, Fox NC, et al. (2016). White matter hyperintensities are a core feature of Alzheimer’s disease: Evidence from the dominantly inherited Alzheimer network. Ann Neurol 79, 929–939. 10.1002/ana.24647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leng K, Li E, Eser R, Piergies A, Sit R, Tan M, Neff N, Li SH, Rodriguez RD, Suemoto CK, et al. (2021). Molecular characterization of selectively vulnerable neurons in Alzheimer’s disease. Nat Neurosci. 10.1038/s41593-020-00764-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewandowski NM, Ju S, Verbitsky M, Ross B, Geddie ML, Rockenstein E, Adame A, Muhammad A, Vonsattel JP, Ringe D, et al. (2010). Polyamine pathway contributes to the pathogenesis of Parkinson disease. Proc Natl Acad Sci U S A 107, 16970–16975. 10.1073/pnas.1011751107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian H, Litvinchuk A, Chiang AC, Aithmitti N, Jankowsky JL, and Zheng H (2016). Astrocyte-Microglia Cross Talk through Complement Activation Modulates Amyloid Pathology in Mouse Models of Alzheimer’s Disease. J Neurosci 36, 577–589. 10.1523/JNeUrOSCI.2117-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Munch AE, Chung WS, Peterson TC, et al. (2017). Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487. 10.1038/nature21029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lie E, Li Y, Kim R, and Kim E (2018). SALM/Lrfn Family Synaptic Adhesion Molecules. Front Mol Neurosci 11, 105. 10.3389/fnmol.2018.00105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorente-Gea L, Garcia B, Martin C, Quiros L, and Fernandez-Vega I (2017). Heparan sulfate proteoglycans and heparanases in Alzheimer’s disease: current outlook and potential therapeutic targets. Neural Regeneration Research 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathys H, Davila-Velderrain J, Peng Z, Gao F, Mohammadi S, Young JZ, Menon M, He L, Abdurrob F, Jiang X, et al. (2019). Single-cell transcriptomic analysis of Alzheimer’s disease. Nature. 10.1038/s41586-019-1195-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattsson N, Schott JM, Hardy J, Turner MR, and Zetterberg H (2016). Selective vulnerability in neurodegeneration: insights from clinical variants of Alzheimer’s disease. J Neurol Neurosurg Psychiatry 87, 1000–1004. 10.1136/jnnp-2015-311321. [DOI] [PubMed] [Google Scholar]
- Maynard KR, Collado-Torres L, Weber LM, Uytingco C, Barry BK, Williams SR, Catallini JL 2nd, Tran MN, Besich Z, Tippani M, et al. (2021). Transcriptome-scale spatial gene expression in the human dorsolateral prefrontal cortex. Nat Neurosci 24, 425–436. 10.1038/S41593-020-00787-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohamed LA, Keller JN, and Kaddoumi A (2016). Role of P-glycoprotein in mediating rivastigmine effect on amyloid-beta brain load and related pathology in Alzheimer’s disease mouse model. Biochimica et Biophysica Acta 1862, 778–787. 10.1016/j.bbadis.2016.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray ME, Graff-Radford NR, Ross OA, Petersen RC, Duara R, and Dickson DW (2011). Neuropathologically defined subtypes of Alzheimer’s disease with distinct clinical characteristics: a retrospective study. The Lancet Neurology 10, 785–796. 10.1016/s1474-4422(11)70156-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedergaard M, Ransom B, and Goldman SA (2003). New roles for astrocytes: redefining the functional architecture of the brain. Trends Neurosci 26, 523–530. 10.1016/j.tins.2003.08.008. [DOI] [PubMed] [Google Scholar]
- Nelson PT, Pious NM, Jicha GA, Wilcock DM, Fardo DW, Estus S, and Rebeck GW (2013). APOE-e2 and APOE-e4 Correlate With Increased Amyloid Accumulation in Cerebral Vasculature. J Neuropathol Exp Neurol 72, 708–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nott A, Holtman IR, Coufal NG, Schlachetzki JCM, Yu M, Hu R, Han CZ, Pena M, Xiao J, Wu Y, et al. (2019). Brain cell type-specific enhancer-promoter interactome maps and disease-risk association. Science 366, 1134–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunes-Xavier CE, Zaldumbide L, Aurtenetxe O, Lopez-Almaraz R, Lopez JI, and Pulido R (2019). Dual-Specificity Phosphatases in Neuroblastoma Cell Growth and Differentiation. Int J Mol Sci 20. 10.3390/ijms20051170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Callaghan P, Sandwall E, Li JP, Yu H, Ravid R, Guan ZZ, van Kuppevelt TH, Nilsson LN, Ingelsson M, Hyman BT, et al. (2008). Heparan sulfate accumulation with Abeta deposits in Alzheimer’s disease and Tg2576 mice is contributed by glial cells. Brain Pathol 18, 548–561. 10.1111/j.1750-3639.2008.00152.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen JB, Di Domenico F, Sultana R, Perluigi M, Cini C, Pierce WM, and Butterfield DA (2009). Proteomics-determined differences in the concanavalin-A-fractionated proteome of hippocampus and inferior parietal lobule in subjects with Alzheimer’s disease and mild cognitive impairment: implications for progression of AD. Journal of Proteome Research 8, 471–482. 10.1021/pr800667a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardo B, Spangler A, Weber LM, Hicks SC, Jaffe AE, Martinowich K, Maynard KR, and Collado-Torres L (2021). spatialLIBD: an R/Bioconductor package to visualize spatially-resolved transcriptomics data. bioRxiv, 2021.2004.2029.440149. 10.1101/2021.04.29.440149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paton CM, and Ntambi JM (2009). Biochemical and physiological function of stearoyl-CoA desaturase. Am J Physiol Endocrinol Metab 297, E28–37. 10.1152/ajpendo.90897.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prickaerts J, Heckman PRA, and Blokland A (2017). Investigational phosphodiesterase inhibitors in phase I and phase II clinical trials for Alzheimer’s disease. Expert Opin Investig Drugs 26, 1033–1048. 10.1080/13543784.2017.1364360. [DOI] [PubMed] [Google Scholar]
- Radanovic M, Ramos F, Pereira S, Stella F, Aprahamian I, Ferreira LK, Forlenza OV, and Busatto GF (2013). White matter abnormalities associated with Alzheimer’s disease and mild cognitive impairment: a critical review of MRI studies. Expert Review in Neurotherapeutics 13, 483–493. [DOI] [PubMed] [Google Scholar]
- Ramsay L, Quille ML, Orset C, de la Grange P, Rousselet E, Ferec C, Le Gac G, Genin E, and Timsit S (2019). Blood transcriptomic biomarker as a surrogate of ischemic brain gene expression. Ann Clin Transl Neurol 6, 1681–1695. 10.1002/acn3.50861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Refolo LM, Malester B, LaFrancois J, Bryant-Thomas T, Wang R, Tint GS, Sambamurti K, Duff K, and Pappolla MA (2000). Hypercholesterolemia accelerates the Alzheimer’s amyloid pathology in a transgenic mouse model. Neurobiol Dis 7, 321–331. 10.1006/nbdi.2000.0304. [DOI] [PubMed] [Google Scholar]
- Reiman EM, Arboleda-Velasquez JF, Quiroz YT, Huentelman MJ, Beach TG, Caselli RJ, Chen Y, Su Y, Myers AJ, Hardy J, et al. (2020). Exceptionally low likelihood of Alzheimer’s dementia in APOE2 homozygotes from a 5,000-person neuropathological study. Nat Commun 11, 667. 10.1038/s41467-019-14279-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risso D, Perraudeau F, Gribkova S, Dudoit S, and Vert JP (2018). A general and flexible method for signal extraction from single-cell RNA-seq data. Nat Commun 9, 284. 10.1038/S41467-017-02554-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saab AS, Tzvetavona ID, Trevisiol A, Baltan S, Dibaj P, Kusch K, Mobius W, Goetze B, Jahn HM, Huang W, et al. (2016). Oligodendroglial NMDA Receptors Regulate Glucose Import and Axonal Energy Metabolism. Neuron 91, 119–132. 10.1016/j.neuron.2016.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadick JS, and Liddelow SA (2019). Don’t forget astrocytes when targeting Alzheimer’s disease. British Journal of Pharmacology. 10.1111/bph.14568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saher G, Brugger B, Lappe-Siefke C, Mobius W, Tozawa R, Wehr MC, Wieland F, Ishibashi S, and Nave KA (2005). High cholesterol level is essential for myelin membrane growth. Nat Neurosci 8, 468–475. 10.1038/nn1426. [DOI] [PubMed] [Google Scholar]
- Saher G, Quintes S, and Nave KA (2011). Cholesterol: a novel regulatory role in myelin formation. Neuroscientist 17, 79–93. 10.1177/1073858410373835. [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods 9, 676–682. 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schipper HM, Bennett DA, Liberman A, Bienias JL, Schneider JA, Kelly J, and Arvanitakis Z (2006). Glial heme oxygenase-1 expression in Alzheimer disease and mild cognitive impairment. Neurobiology of Aging 27, 252–261. 10.1016/j.neurobiolaging.2005.01.016. [DOI] [PubMed] [Google Scholar]
- Schnadelbach O, Ozen I, Blaschuk OW, Meyer RL, and Fawcett JW (2001). N-cadherin is involved in axon-oligodendrocyte contact and myelination. Mol Cell Neurosci 17, 1084–1093. 10.1006/mcne.2001.0961. [DOI] [PubMed] [Google Scholar]
- Shepardson NE, Shankar GM, and Selkoe DJ (2011). Cholesterol level and statin use in Alzheimer disease: I. Review of epidemiological and preclinical studies. Arch Neurol 68, 1239–1244. 10.1001/archneurol.2011.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Yamada K, Liddelow SA, Smith ST, Zhao L, Luo W, Tsai RM, Spina S, Grinberg LT, Rojas JC, et al. (2017). ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 549, 523–527. 10.1038/nature24016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith HL, Freeman OJ, Butcher AJ, Holmqvist S, Humoud I, Schatzl T, Hughes DT, Verity NC, Swinden DP, Hayes J, et al. (2020). Astrocyte Unfolded Protein Response Induces a Specific Reactivity State that Causes Non-Cell-Autonomous Neuronal Degeneration. Neuron 105, 855–866 e855. 10.1016/j.neuron.2019.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stogsdill JA, Ramirez J, Liu D, Kim YH, Baldwin KT, Enustun E, Ejikeme T, Ji RR, and Eroglu C (2017). Astrocytic neuroligins control astrocyte morphogenesis and synaptogenesis. Nature 551, 192–197. 10.1038/nature24638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902 e1821. 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syed YA, Baer A, Hofer MP, Gonzalez GA, Rundle J, Myrta S, Huang JK, Zhao C, Rossner MJ, Trotter MW, et al. (2013). Inhibition of phosphodiesterase-4 promotes oligodendrocyte precursor cell differentiation and enhances CNS remyelination. EMBO Mol Med 5, 1918–1934. 10.1002/emmm.201303123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor DM, Moser R, Regulier E, Breuillaud L, Dixon M, Beesen AA, Elliston L, Silva Santos Mde F, Kim J, Jones L, et al. (2013). MAP kinase phosphatase 1 (MKP-1/DUSP1) is neuroprotective in Huntington’s disease via additive effects of JNK and p38 inhibition. J Neurosci 33, 2313–2325. 10.1523/JNEUROSCI.4965-11.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teaktong T, Graham A, Court J, Perry R, Jaros E, Johnson M, Hall R, and Perry E (2003). Alzheimer’s disease is associated with a selective increase in alpha7 nicotinic acetylcholine receptor immunoreactivity in astrocytes. Glia 41, 207–211. 10.1002/glia.10132. [DOI] [PubMed] [Google Scholar]
- Tirosh I, Izar B, Prakadan SM, Wadsworth MH 2nd, Treacy D, Trombetta JJ, Rotem A, Rodman C, Lian C, Murphy G, et al. (2016). Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196. 10.1126/science.aad0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal R, Frangione B, Rostagno A, Mead S, Revesz T, Plant G, and Ghiso J (1999). A stop-codon mutation in the BRI gene associated with familial British dementia. Nature 399, 776–781. [DOI] [PubMed] [Google Scholar]
- Viswanathan A, and Greenberg SM (2011). Cerebral amyloid angiopathy in the elderly. Annals of Neurology 70, 871–880. 10.1002/ana.22516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Bartheld CS, Bahney J, and Herculano-Houzel S (2016). The search for true numbers of neurons and glial cells in the human brain: A review of 150 years of cell counting. J Comp Neurol 524, 3865–3895. 10.1002/cne.24040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu T, Dejanovic B, Gandham VD, Gogineni A, Edmonds R, Schauer S, Srinivasan K, Huntley MA, Wang Y, Wang TM, et al. (2019). Complement C3 Is Activated in Human AD Brain and Is Required for Neurodegeneration in Mouse Models of Amyloidosis and Tauopathy. Cell Rep 28, 2111–2123 e2116. 10.1016/j.celrep.2019.07.060. [DOI] [PubMed] [Google Scholar]
- Wu Y, Li Z, Huang YY, Wu D, and Luo HB (2018). Novel Phosphodiesterase Inhibitors for Cognitive Improvement in Alzheimer’s Disease. J Med Chem 61, 5467–5483. 10.1021/acs.jmedchem.7b01370. [DOI] [PubMed] [Google Scholar]
- Wu Z, Zhao L, Chen X, Cheng X, and Zhang Y (2015). Galantamine attenuates amyloid-beta deposition and astrocyte activation in APP/PS1 transgenic mice. Experimental Gerontology 72, 244–250. 10.1016/j.exger.2015.10.015. [DOI] [PubMed] [Google Scholar]
- Yip CM, Elton EA, Darabie AA, Morrison MR, and McLaurin J (2001). Cholesterol, a modulator of membrane-associated Abeta-fibrillogenesis and neurotoxicity. J Mol Biol 311, 723–734. 10.1006/jmbi.2001.4881. [DOI] [PubMed] [Google Scholar]
- Yue C, and Jing N (2015). The promise of stem cells in the therapy of Alzheimer’s disease. Transl Neurogegener 4, 8. 10.1186/s40035-015-0029-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamanian JL, Xu L, Foo LC, Nouri N, Zhou L, Giffard RG, and Barres BA (2012). Genomic analysis of reactive astrogliosis. J Neurosci 32, 6391–6410. 10.1523/JNEUROSCI.6221-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Sloan SA, Clarke LE, Caneda C, Plaza CA, Blumenthal PD, Vogel H, Steinberg GK, Edwards MS, Li G, et al. (2016). Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron 89, 37–53. 10.1016/j.neuron.2015.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang ZG, Zhang L, Jiang Q, Zhang R, Davies K, Powers C, van Bruggen N, and Chopp M (2000). VEGF enhances angiogenesis and promotes blood-brain barrier leakage in the ischemic brain. Journal of Clinical Investigation 106, 829–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong L, Xie YZ, Cao TT, Wang Z, Wang T, Li X, Shen RC, Xu H, Bu G, and Chen XF (2016). A rapid and cost-effective method for genotyping apolipoprotein E gene polymorphism. Mol Neurodegener 11, 2. 10.1186/s13024-016-0069-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Song WM, Andhey PS, Swain A, Levy T, Miller KR, Poliani PL, Cominelli M, Grover S, Gilfillan S, et al. (2020). Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nat Med 26, 131–142. 10.1038/s41591-019-0695-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S3 (Related to Figures S1, S2). Sorting outputs and cluster-specific DEG and GO analyses.
Table S5 (Related to Figures 3, 7, S7, S8, S13). Astrocyte and oligodendrocyte multi-dataset integrations.
Data Availability Statement
Raw snRNA-seq data generated in this study (including both FASTQ and Cell Ranger-generated matrix files) are available at GEO (GSE167494). Microscopy data reported in this paper are available through the Cell Image Library (www.cellimagelibrary.org, CIL group#: 54423).
All code for analysis of original and previously published snRNA-seq, scRNA-seq, and spatial transcriptomic datasets are available on the Liddelow Lab GitHub page: https://github.com/liddelowlab/Sadick_et_al._2022.
Analyzed snRNA-seq and pseudobulk snRNA-seq data is available on an open-access, interactive website: www.qliaseq.com. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.