

Abstract
Aberrant smooth muscle cell (SMC) plasticity is etiological to vascular diseases. Cholesterol induces SMC phenotypic transition featuring high LGALS3 (galectin-3) expression. This proatherogenic process is poorly understood for its molecular underpinnings, in particular, the mechanistic role of sterol regulatory-element binding protein-1 (SREBP1), a master regulator of lipid metabolism. Herein we show that cholesterol loading stimulated SREBP1 expression in mouse, rat, and human SMCs. SREBP1 positively regulated LGALS3 expression (and vice versa), whereas Krüppel-like factor-15 (KLF15) acted as a negative regulator. Both bound to the Lgals3 promoter, yet at discrete sites, as revealed by chromatin immunoprecipitation-qPCR and electrophoretic mobility shift assays. SREBP1 and LGALS3 each abated KLF15 protein, and blocking the bromo/extraterminal domain-containing proteins (BETs) family of acetyl-histone readers abolished cholesterol-stimulated SREBP1/LGALS3 protein production. Furthermore, silencing bromodomain protein 2 (BRD2; but not other BETs) reduced SREBP1; endogenous BRD2 co-immunoprecipitated with SREBP1’s transcription-active domain, its own promoter DNA, and that of Lgals3. Thus, results identify a previously uncharacterized cholesterol-responsive dyad—SREBP1 and LGALS3, constituting a feedforward circuit that can be blocked by BETs inhibition. This study provides new insights into SMC phenotypic transition and potential interventional targets.
Keywords: vascular smooth muscle cell, cholesterol loading, phenotypic transition, SREBP1, LGALS3, feedforward circuit, BRD2, KLF15, MRTF-A
Graphical abstract
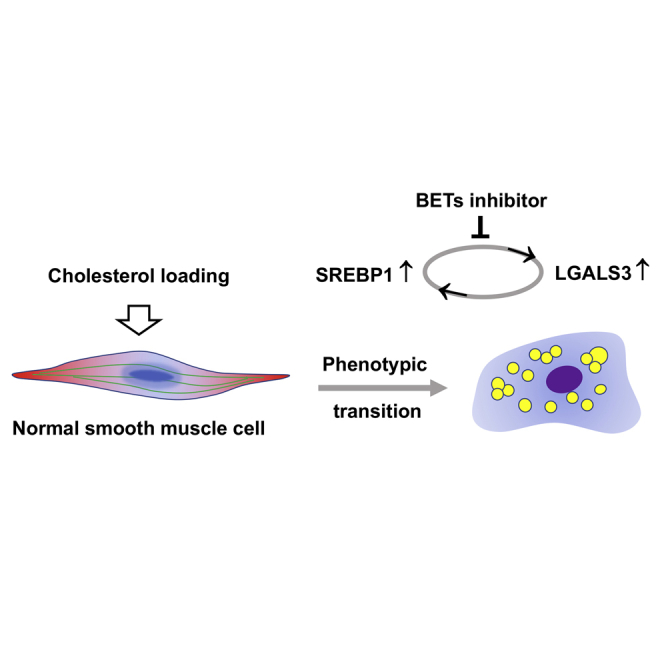
Smooth muscle cells in a cholesterol-rich environment transition to a LGALS3-expressing multi-potential state, a key event in atherogenesis. We identify a cholesterol-responsive feedforward dyad of SREBP1 and LGALS3 positively regulating each other’s expression in vitro, which can be abolished by inhibiting the BET family of histone code readers.
Introduction
Atherosclerosis is the principal cause of cardiovascular disease with widespread global prevalence. The extremely complex etiology of atherosclerosis involving vascular cell pathobiology is not well understood. Rong et al.1 showed that cholesterol loading of cultured vascular smooth muscle cells (SMCs) induced down-regulation of SMC markers and activation of Lgals3 (Mac2). Results were initially interpreted as evidence that SMCs were transitioning to a macrophage-like state. However, subsequent gene array studies by the Fisher group2 showed that cholesterol-loaded SMCs at best formed highly dysfunctional macrophages and suggested that the cholesterol loading may be a more appropriate model for foam cells, which is consistent with studies by Wang et al.,3 suggesting that SMCs are the major source of foam cells within lesions. Importantly, however, a recent study by Alencar et al.4 indicated that the identity of the LGALS3+ cells of SMC origin is far more complex. The study applied single-cell RNA sequencing (scRNA-seq) in both SMC lineage tracing ApoE−/− mice and in a novel myosin heavy chain 11 (Myh11)-tdTomato LGALS3 GFP ApoE−/− dual-recombinase mouse line that, after tamoxifen treatment, permits identification of Myh11+ SMCs that convert from being tdTomato+ to GFP+ on expression of LGALS3. Results indicated that Lgals3 activation in SMCs in late-stage brachiocephalic artery lesions of ApoE−/− mice did not represent their transition to a macrophage-like state.4 Rather, we showed that individual dedifferentiated medial SMCs that activate Lgals3 represent a multi-potential stem cell marker+ (e.g., stem cell antigen-1 [Sca1 or Ly6A]+; Klf4+) transition state that can give rise to multiple SMC-derived transcriptomic clusters or phenotypic states. Notably, SMCs that activate Lgals3 appear to preferentially give rise to osteochondrogenic and pro-inflammatory cells that are likely detrimental for plaque pathogenesis by contributing to advanced plaques and calcification.4 However, some of these cells gave rise to myofibroblast-like cells important for formation of the protective fibrous cap. Which phenotype they exhibit may be a function of lesion SMCs responding to spatially and temporally distinct environmental cues present in lesions. Alternatively, one cannot rule out the possibility that there are distinct subsets of SMCs that exhibit restricted plasticity, especially given the oligoclonal nature of SMC within lesions.5,6
In accordance with these recent studies indicating diverse cell types or states derived from LGALS3+ SMCs, LGALS3 is expressed by numerous cell types and has been implicated in the control of many cellular processes, including cancer cell and epithelial cell migration,7,8 stemness transformation of some cancer cell lines,9 proliferation of hepatic stellate cells,10 suppression of T cell apoptosis,11 and adipocyte and osteoblast differentiation.12,13 Moreover, LGALS3 is expressed in SMCs within pulmonary arteries during development of pulmonary hypertension and mediates proliferation, migration, and resistance to apoptosis of cultured SMCs.14 In addition, Lgals3 knockout rats are resistant to development of experimentally induced pulmonary hypertension.14 Although the preceding studies indicate that LGALS3 plays a key role in regulating SMC function, relatively little is known regarding mechanisms that activate its expression in this or other cell types.8,15
Sterol regulatory-element binding proteins (SREBPs) are master regulators of lipid metabolism.16 SREBP1 is key to lipogenesis and triglyceride deposition. In contrast, SREBP2 has been more closely associated with cholesterol synthesis and accumulation. Although SREBP2 is known to play an important role in cholesterol accumulation in rat aortic SMCs in vitro,17 SREBP1 is less understood for its role in SMCs,18 especially in cholesterol-induced SMC phenotypic transition. Whether SREBP1 plays a role in SMC expression of LGALS3 is not known. Activated in response to low cholesterol, SREBP2 is down-regulated in cholesterol-rich conditions.19 Interestingly, SREBP1 was reported to be upregulated by cholesterol loading of cultured SMCs,20 although the mechanistic details were not determined. This motivated us to determine whether SREBP1 is required for upregulation of LGALS3 in cholesterol-loaded SMCs.
Krüppel-like factor 15 (KLF15) is another cholesterol-responsive transcription factor that was initially shown to be crucial for gluconeogenesis.21 Of interest, recent studies unveiled its importance in lipid homeostasis22,23 and a role of inhibiting Srebp1 expression in hepatic lipogenesis.23 Moreover, it has been reported to impact migratory, proliferative, and inflammatory SMC states and associated disorders, such as neointimal hyperplasia and aneurysm.24, 25, 26 However, whether KLF15 is a transcriptional regulator of Lgals3 in cholesterol-loaded SMCs has not been reported.
Using the in vitro model of cholesterol-loaded SMCs,2 we found that SREBP1 and KLF15 positively and negatively regulated LGALS3 expression, respectively. This further led to the finding of a feedforward circuit formed by SREBP1 and LGALS3 positively regulating each other. Moreover, they each reduced KLF15 protein levels. In addition, we found that cholesterol-induced upregulation of the SREBP1/LGALS3 dyad involved cooperative actions of the transcription factor SREBP1 and bromodomain protein 2 (BRD2), a histone acetyl bookmark reader. Therefore, this study sheds new light on the mechanistic understanding of SMC phenotypic transition sensitive to cholesterol-rich environments.
Results
SREBP1 positively regulates LGALS3 expression in cholesterol-loaded SMCs
Rong et al.1 discovered that cholesterol loading could activate Lgals3 expression in cultured SMCs. An important yet hitherto unaddressed question is whether the master regulator of lipid metabolism, SREBP1, plays a role in this activation. Herein we show that in mouse SMCs (MOVAS [mouse smooth muscle cell line]), LGALS3 increased in a cholesterol dose-dependent fashion and peaked at 80 μg/ml27 (Figure 1A). Cholesterol loading also increased the expression of ABCA1 and pro-inflammatory factors (VCAM-1 and MCP-1) (Figure S1), consistent with previous reports.2,27 CD68, which is often used together with LGALS3 as macrophage markers, was also elevated by cholesterol loading (Figure 1A). We thus determined two other bona fide macrophage markers, F4/80 and CD11b. However, their levels were not significantly altered by treatment with cholesterol (although CD11b was slightly increased in MOVAS cells) (Figure S2). This is consistent with the recent single-cell sequencing studies showing that LGALS3-expressing SMCs do not necessarily transition to macrophage-like cells.4,28 More interestingly, full-length SREBP1 and the N-terminal half molecule which is the transcription-active form (SREBP1Nterm), responded to cholesterol loading in the same fashion as LGALS3 (Figure 1A). This new information led us to explore a potential relationship between SREBP1 and LGALS3. We found that silencing SREBP1 reduced LGALS3 protein and mRNA expression in MOVAS cells (Figure 1B), as also observed in primary rat aortic SMCs (Figures 1C and S3). Conversely, SREBP1 overexpression significantly increased LGALS3 protein (Figure 1D). Furthermore, chromatin immunoprecipitation and quantitative PCR (ChIP-qPCR) experiments revealed strong SREBP1 association with the Lgals3 promoter at the S1 site (Figure 1E).
Figure 1.

SREBP1 positively regulates LGALS3 expression in mouse and rat SMCs
The SMCs used in experiments were mouse MOVAS cells unless otherwise specified (i.e., rat primary aortic SMCs in C). For loss or gain of function, SMCs were transduced with lentivirus for 24 h to express a specific shRNA or scrambled control, or to express a gene or empty vector (EVec) control. The culture was then changed to fresh medium and continued until subconfluence prior to cholesterol loading. Cells cultured in full medium were incubated with solvent control (0.25% BSA) or cholesterol (Chol; in a Chol-methyl-β-cyclodextrin soluble form) at indicated concentrations for 72 h prior to harvest for various assays. Data quantification for western blots: mean ± SEM, n ≥ 3 independent repeat experiments. Data quantification for quantitative real-time PCR and ChIP-qPCR: mean ± SD, n = 3 replicates; presented is one of two similar experiments. Statistics: one-way ANOVA followed by Tukey test; #p < 0.05, ##p < 0.01, ###p < 0.001 (between paired bars); ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, compared with the basal condition (the first bar in each plot); non-significant comparisons are not labeled. Note: Srebf1 produces two highly homologous mRNA variants and protein variants (SREBP1a and SREBP1c); the primers and antibody used here do not distinguish between these two variants. (A) Upregulation of LGALS3 and SREBP1 proteins (western blot) and mRNAs (quantitative real-time PCR) by increasing concentrations of cholesterol. (B) SREBP1 silencing in MOVAS cells reduces LGALS3 protein and mRNA expression. (C) SREBP1 silencing in primary rat aortic SMCs reduces LGALS3 protein. (D) SREBP1 overexpression in MOVAS cells increases LGALS3 protein. (E) ChIP-qPCR showing SREBP1 binding at a proximal site of the Lgals3 promoter. S1, S2, and S3 denote three software-predicted SREBP1-binding sequences at different Lgals3 promoter sites. ChIP was performed using an antibody specific for endogenous SREBP1, after MOVAS cells were treated for the indicated hours. r.u., relative unit; SREBP1Nterm, the N-terminal half molecule, which is the active form of the SREBP1 transcription factor.
KLF15 negatively regulates LGALS3 expression in cholesterol-loaded SMCs
KLF15 is another cholesterol-responsive transcription factor. Although KLF15 was recently found to regulate SMC pathophysiological states,24, 25, 26 it has not been reported to regulate LGALS3 expression. Our data show that KLF15 protein diminished in response to increasing cholesterol concentrations (Figure 2A), opposite to the response of SREBP1 (Figure 1A). More interestingly, KLF15 silencing (Figure 2B) and overexpression (Figure 2C) increased and decreased LGALS3, respectively. Furthermore, ChIP-qPCR data showed prominent binding of KLF15 at the S2 site of the Lgals3 promoter (Figure 2D). These results revealed KLF15-mediated negative regulation of LGALS3 expression in SMCs, contrary to SREBP1’s positive regulation described above.
Figure 2.

KLF15 negatively regulates LGALS3 expression in mouse SMCs
Mouse MOVAS cells were used. Cholesterol loading and lentiviral transduction were performed as described in detail for Figure 1. Data quantification for western blots: mean ± SEM, n ≥ 3 independent repeat experiments. Data quantification for quantitative real-time PCR and ChIP-qPCR: mean ± SD, n = 3 replicates; presented is one of two similar experiments. Statistics: ANOVA followed by Tukey test; #p < 0.05, ##p < 0.01, ###p < 0.001 (between paired bars); ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, compared with the basal condition (the first bar in each plot). (A) Down-regulation of KLF15 by increasing concentrations of cholesterol. (B) KLF15 silencing increases LGALS3 protein and mRNA. (C) KLF15 overexpression reduces LGALS3 protein. (D) ChIP-qPCR indicating KLF15 binding at the S2 site of the Lgals3 promoter. S1, S2, and S3 refer to the same sites in Figure 1E. ChIP was performed using an antibody specific for endogenous KLF15, after MOVAS cells were treated for indicated hours.
SREBP1 and KLF15 bind to the Lgals3 promoter DNA at different sites
We next pursued further evidence for transcription factor/promoter DNA interactions through electrophoretic mobility shift assay (EMSA), using biotin-labeled oligos that contain Lgals3 promoter DNA sequences (Figure 3). The EMSA data show that the oligos containing the KLF15-binding sequence (S2) or SREBP1-binding sequence (S1) ran at different locations on the gel (Figures 3A and 3B), suggestive of binding with different proteins. Multiple bands were detected for S1 most likely because of oligomerization of SREBP1 in non-denaturing native gel. Indicative of binding specificity, the EMSA signal was extinguished by increasing concentrations of a competitive unlabeled oligo (Figures 3C and 3D). In contrast, the oligo corresponding to the S3 site of the Lgals3 promoter, which did not show SREBP1 binding in the ChIP-qPCR experiment (Figure 1E), did not have an effect on the Biotin-S1/SREBP1 EMSA signal (Figure 3E). Further confirming the oligo sequence specificity, the EMSA signal of KLF15 or SREBP1 binding was attenuated by mutations in each respective oligo (Figures 3F and 3G). In addition, the biotin-S1/SREBP1 complex formation was not affected by KLF15 silencing (Figure 3H), consistent with the ChIP-qPCR result that SREBP1 and KLF15 bound at separate sites (S1 and S2, respectively) in the Lgals3 promoter.
Figure 3.

Electrophoretic mobility shift assays (EMSAs) showing transcription factor/promoter DNA interactions
MOVAS cells were cultured without or with 80 μg/mL cholesterol for 72 h. Nuclear proteins were then extracted and incubated with biotin-labeled oligonucleotides (A and B). Duplicate samples were loaded to the right six lanes in (A). S1, S2, and S3 denote the oligo sequences within the respective S1, S2, and S3 Lgals3 promoter sites (see Figures 1E and 2D). (C–E) A competitor oligo (without biotin label) in 2.5×, 5×, or 10× molar excess was added in nuclear extracts before adding the biotin-oligo of the same sequence. (F and G) A biotin-oligo without (WT control) or with a mutated sequence (mutant) was used. Shown in each blot is one of two to three similar experiments. (H) Experiments were performed in cells transduced with lentivirus expressing scrambled or KLF15-specific shRNA. (I and J) Schematics depict KLF15’s negative effect and SREBP1’s positive effect on Lgals3 transcription, respectively, as suggested by combined results presented in Figures 1 and 2 and this figure.
Aside from their differential binding to the Lgals3 promoter, SREBP1 and KLF15 exhibited opposite roles in regulating LGALS3’s expression, as presented above. We were thus inspired to investigate the relationship of these two transcription factors. As indicated in Figure 4A, while SREBP1 silencing and overexpression increased and decreased KLF15 protein, respectively, KLF15 silencing and overexpression increased and decreased SREBP1 protein. Of note, KLF15 protein did not increase significantly in response to SREBP1 silencing in the condition of cholesterol loading. This could be rationalized by the fact that high cholesterol (80 μg/mL) is a potent repression signal for KLF15 expression (Figure 2A), as also documented in the literature.23
Figure 4.

Reciprocal regulation of the transcription factors SREBP1 and KLF15
The SMCs used in experiments were mouse MOVAS cells unless otherwise specified (i.e., rat primary aortic SMCs in C). Cholesterol loading (80 μg/mL) and lentiviral transduction were performed as described in detail for Figure 1. TD139 was added at indicated concentrations 2 h prior to cholesterol loading. Quantification: mean ± SEM, n ≥ 3 independent repeat experiments. Statistics: ANOVA followed by Tukey test; #p < 0.05, ##p < 0.01, ###p < 0.001 (between paired bars); ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, compared with the basal condition (the first bar in each plot). (A) Reciprocal SREBP1 and KLF15 regulations in MOVAS cells. (B) Effect of LGALS3 inhibition with TD139 on SREBP1 and KLF15 protein levels in MOVAS cells. (C) Effect of LGALS3 inhibition with TD139 on SREBP1 and KLF15 protein levels in rat primary SMCs.
Thus, up to this point, our data have demonstrated that the two prominent cholesterol-responsive transcription factors, SREBP1 and KLF15, positively and negatively regulate Lgals3 expression, respectively, likely via binding at discrete sites on the Lgals3 promoter (see schemes in Figures 3I and 3J).
LGALS3 positively regulates SREBP1, but negatively regulates KLF15
Although poorly understood, interaction of LGALS3 with nuclear components has been recently reported.29 We thus surmised that LGALS3 might influence SREBP1 and/or KLF15 levels. We first explored this possibility with a commonly used LGALS3 inhibitor, TD13930 (Figures 4B and 4C). The data show that TD139 dose-dependently reduced SREBP1 protein, in cholesterol-loaded MOVAS cells and rat primary SMCs. In contrast, TD139 increased KLF15 in non-loaded cells, at a concentration as low as 50 nM. In the cholesterol-loading condition, however, TD139 increased KLF15 protein only in rat primary SMCs. This dampened potency of TD139 was likely due to the aforementioned strong inhibitory effect of cholesterol on KLF15 levels. We then sought to confirm LGALS3’s functional specificity using genetic approaches. As shown in Figure 5A, LGALS3-specific silencing diminished SREBP1 protein and mRNA, in either cholesterol-loaded or non-loaded MOVAS cells; the opposite occurred to KLF15 protein. LGALS3 overexpression increased SREBP1 protein (full-length and N-terminal half) (Figure S4) and decreased KLF15, and the LGALS3 inhibitor TD139 dampened these changes (Figure 5B). The experiments with rat primary SMCs led to essentially the same results, albeit with varied statistical outcomes in some conditions (Figures 5C and 5D).
Figure 5.

LGALS3 positively regulates SREBP1 and negatively regulates KLF15
The SMCs used in experiments were mouse MOVAS cells unless otherwise specified (i.e., rat primary aortic SMCs in C and D). Cholesterol loading (80 μg/mL) and lentiviral transduction were performed as described in detail for Figure 1. TD139 was added (final 1 μM) 2 h prior to cholesterol loading. Data quantification for western blots: mean ± SEM, n ≥ 3 independent repeat experiments. Data quantification for quantitative real-time PCR: mean ± SD, n = 3 replicates; presented is one of two similar experiments. Statistics: ANOVA followed by Tukey test; #p < 0.05, ##p < 0.01, ###p < 0.001 (between paired bars); ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, compared with the basal condition (the first bar in each plot). (A) Effect of LGALS3 silencing on SREBP1 and KLF15 levels in MOVAS cells. SREBP1 (primer1) refers to a sequence in the C-terminal domain; SREBP1 (primer2) refers to a sequence in the N-terminal domain. (B) Effect of LGALS3 overexpression on SREBP1 and KLF15 protein levels in MOVAS cells. (C) Effect of LGALS3 silencing on SREBP1 and KLF15 protein levels in rat primary SMCs. (D) Effect of LGALS3 overexpression on SREBP1 and KLF15 protein levels in rat primary SMCs.
Thus, SREBP1 and LGALS3 appeared to form a feedforward signaling circuit that is augmented by cholesterol loading. We therefore focused on this dyad in the following experiments.
Upregulation of SREBP1 and LGALS3 can be blocked by BETs inhibition
Epigenetic factors play critical roles in transmitting extra- or intra-cellular perturbations to transcriptional reprogramming and subsequent cell-state changes. A prominent example is the BETs (bromo/extraterminal domain-containing proteins), including BRD2, BRD3, BRD4, and BRDt (testis restricted and hence herein irrelevant), which have been recently implicated in lipid metabolism.31,32 Given our results showing that SREBP1 is a powerful regulator of the response of SMCs to cholesterol loading, we next asked whether in this context BETs served as upstream determinants controlling SREBP1 expression. As shown in Figure 6A (and Figure S5), treating SMCs with JQ1, the first-in-class BETs-selective inhibitor,33 abolished cholesterol-induced upregulation of SREBP1, LGALS3, and CD68. Taken together, these results suggest that blocking BETs may provide an effective means to suppress cholesterol-induced activation of the SREBP1/LGALS3 dyad.
Figure 6.

BETs inhibition abrogates cholesterol-induced upregulation of SREBP1 and LGALS3 proteins
Mouse MOVAS cells were used unless otherwise specified. Cholesterol loading (80 μg/mL) and lentiviral transduction were performed as described in detail for Figure 1. Data quantification for western blots: mean ± SEM, n ≥ 3 independent repeat experiments. Data quantification for qPCR: mean ± SD, n = 3 replicates; presented is one of two similar experiments. Statistics: ANOVA followed by Tukey test; #p < 0.05, ##p < 0.01, ###p < 0.001 (between paired bars); ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, compared with the basal condition (the first bar in each plot). (A) Treatment of MOVAS cells with BET-selective inhibitor JQ1 abolishes cholesterol-induced protein upregulation of SREBP1, LGALS3, and CD68. JQ1 was added (final 0.5 μM) 2 h prior to cholesterol loading (final 80 μg/mL). (B) Effect of silencing individual BETs on SREBP1 protein levels in MOVAS cells. (C) Co-immunoprecipitation (coIP) of endogenous BRD2 with SREBP1Nterm. The vector for expressing FLAG-GFP (EVec control) or FLAG-SREBP1Nterm was transfected into HEK293 cells. An anti-FLAG antibody was used for IP, and coIPed endogenous BRD2 was detected via immunoblotting (IB). Mean ± SEM was calculated from n = 4 independent repeat experiments. Pre-stained marker bands are labeled with molecular weights. (D) IB of IPed FLAG-SREBP1Nterm. The same samples from (C) were used. (E) Lack of coIP of LGALS3 with SREBP2Nterm. Experiments were performed as in (C) except for the IB detecting endogenous LGALS3. (F) Chromatin immunoprecipitation (ChIP) followed by qPCR. MOVAS cells were cultured without or with cholesterol (80 μg/mL) for 1 h prior to harvest for ChIP-qPCR. The value from IgG control was used for data normalization. ChIP was performed using an antibody specific for endogenous BRD2 or H3K27ac. Primer 2 was used to detect Srebf1 promoter; primer S1 (within the sequence of S1 site) was used to detect Lgals3 promoter (see Table S6). (G) A schematic depicts co-occupancy of BRD2 and SREBP1 at the Srebf1 or Lgals3 promoter, as suggested by the results presented in Figure 6 (A–F). (H) A schematic depicts the feedforward interplay of the SREBP1/LGALS3 dyad, as suggested by the results presented in Figures 1, 2, 3, 4, and 5 and this figure. SREBP1 positively regulates LGALS3 expression and vice versa. They each suppress KLF15 protein levels, and KLF15 negatively regulates their expression. Pre-treatment with BET-selective inhibitor JQ1 abrogates cholesterol-induced SREBP1/LGALS3 protein upregulation.
BRD2 co-immunoprecipitates with the SREBP1 N-terminal domain and the promoters of Srebf1 and Lgals3
We then performed knockdown experiments to distinguish the role of individual BET family proteins. The data indicate that BRD2 silencing, but not BRD3 or BRD4 silencing, reduced SREBP1 protein in cholesterol-loaded MOVAS cells (Figure 6B). Recent evidence suggests that BETs collaborate with transcription factors to assume their functional specificity in cell-type/state transitions.19,31,34 In particular, master transcription factors that are key to cell identity often activate their own gene transcription.33 We therefore next investigated possible BRD2/SREBP1 functional cooperation. Interestingly, we first observed their physical association (Figure 6C), as evidenced by the co-immunoprecipitation (coIP) of endogenous BRD2 with SREBP1Nterm.16 The specificity of the coIP was supported by equal BRD2 protein levels in SREBP1Nterm-expressing cells and empty vector control cells (see input in Figure 6C) and also by efficient expression and immunoprecipitation (IP) of SREBP1Nterm (Figure 6D). Furthermore, as an additional negative control for the positive coIP of BRD2 with SREBP1Nterm, there was a lack of coIP of LGALS3 with SREBP1Nterm (Figure 6E). To assess the functional significance of the BRD2/SREBP1Nterm association, we performed ChIP-qPCR experiments in MOVAS cells using an antibody against endogenous BRD2 (Figure 6F). The data show that although the Srebf1 promoter DNA coIPed with BRD2 (∼6-fold over the IgG background) under basal conditions, the coIP was further enhanced by cholesterol loading (∼17-fold above the IgG background). A similar result occurred to the Lgals3 promoter DNA, consistent with Lgals3 being a target gene of SREBP1 (Figure 1E). Moreover, in line with BETs being histone-3 acetyl bookmark readers,35 Srebf1 and Lgals3 promoter DNA fragments also coIPed with the H3K27ac antibody in ChIP experiments (Figure 6F). In aggregate, these results suggest that BRD2 and SREBP1Nterm form a protein complex while co-localizing on the promoter of Srebf1, and they also co-localize on the promoter of Lgals3 (see schematic in Figure 6G), consistent with the observed inhibitory effect of JQ1 on the SREBP1/LGALS3 feedforward dyad (see Figure 6A and the schematic in Figure 6H).
SMCs with Lgals3 activation do not transition to an adipocyte phenotype
Thus far, our data have shown transcriptional control of LGALS3 by SREBP1, likely in combination with BRD2. Of note, LGALS3 was initially used as a macrophage marker reciprocally regulating SREBP1 levels (Figure 5). In fact, increasing evidence in the literature supports the notion that LGALS3 is not merely a marker, but it actively participates in various cellular events while distributed broadly in intra- or extra-cellular locations of different cell types.30 We were thus prompted to examine its possible influence on cholesterol-induced SMC phenotype. The data in Figure 7A show that LGALS3 silencing and overexpression potently mitigated and promoted an SMC migratory behavior, respectively, and LGALS3 knockdown without cholesterol loading appeared to be pro-apoptotic (Figure S6). These results agree with previous reports.30,36 Consistent with SREBP1 regulation of LGALS3 (Figure 1), silencing SREBP1 reduced SMC migration as well (Figure 7B). Because cholesterol loading resulted in remarkable lipid accumulation inside SMCs (Figure 7C), we were curious as to whether this treatment had turned SMCs into an adipocyte-like phenotype, a question not previously addressed. We found that although three lipid-storage factors, SREBP1 (Figure 1), FABP4, and ACC1, were elevated after cholesterol loading, other adipogenic factors including CEBPA, PPARγ, adiponectin, and ACAT2, decreased (Figure 7D). This result was confirmed by experiments with rat primary SMCs (Figure S7). Indicative of a specific role for LGALS3, its silencing significantly inhibited cholesterol-induced upregulation of SREBP1 (Figure 5), FABP4, and ACC1, albeit without a significant effect on other markers (Figure 7E). Accordingly, the effects of SREBP1 silencing on these markers largely followed the pattern that resulted from LGALS3 silencing (Figure 7F). This differential LGALS3 regulation of adipogenic markers in cholesterol-loaded SMCs was not previously reported and may deserve future investigation, including genomic analysis at different time points.
Figure 7.

LGALS3 mediates SMC migration and activates expression of a subset of adipogenic marker genes
Mouse MOVAS cells were used unless otherwise specified (i.e., rat primary aortic SMCs in (B) and (F). Cholesterol loading (80 μg/mL) and lentiviral transduction were performed as described in detail for Figure 1. Data quantification for western blots: mean ± SEM, n ≥ 3 independent repeat experiments. Data quantification for quantitative real-time PCR: mean ± SD, n = 3 replicates; presented is one of two similar experiments. Statistics: ANOVA followed by Tukey test; #p < 0.05, ##p < 0.01, ###p < 0.001 (between paired bars); ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, compared with the basal condition (the first bar in each plot). (A) Effect of LGALS3 silencing or overexpression on MOVAS cell migration. Cells were illuminated with calcein fluorescence. Larger non-occupied scratch wound area indicates less migration. (B) Effect of SREBP1 silencing on rat primary SMC migration. Cells were illuminated with calcein fluorescence. (C) Lipid storage in MOVAS cells. Shown are representative images of oil red O staining. Unstained circles are nuclei. Scale bar: 100 μm. (D) Effect of cholesterol loading (to MOVAS cells) on adipogenic marker expression (protein and mRNA). (E) Effect of LGALS3 silencing on protein levels of adipogenic markers. MOVAS cells were transduced with lentivirus expressing scrambled or LGALS3-specific shRNA. (F) Effect of SREBP1 silencing on protein levels of adipogenic markers. Rat primary SMCs were transfected with scrambled or SREBP1-specific siRNA.
LGALS3 represses protein levels of the SMC master differentiation control factor myocardin-related transcription factor A (MRTF-A)
Loss of smooth muscle α-actin (αSMA), a common etiology of various vascular pathologies, is another conspicuous SMC phenotypic change induced by cholesterol loading.2,27 Evidence exists for an influence of LGALS3 on αSMA expression in SMCs,36 and we found that SMCs that activate Lgals3 during development of atherosclerosis show reductions in expression of SMC differentiation marker genes, including αSMA and Myh11.4 However, the underlying mechanisms remain unclear. As expected, cholesterol loading dose-dependently reduced αSMA protein (Figures 8A and 8B) and Myh11 mRNA (Figure S1A) in MOVAS cells. Interestingly, this inhibitory effect of cholesterol loading also occurred on MRTF-A, the transcription co-activator for the expression of αSMA. MRTF-A protein was significantly elevated by inhibiting LGALS3 with TD139 at a higher concentration (1 μM) in non-loaded MOVAS cells, but not in cholesterol-loaded cells (Figure 8B), consistent with the strong inhibitory effect of cholesterol on MRTF-A and αSMA observed in Figure 8A. Nevertheless, genetic manipulations of LGALS3, via LGALS3 silencing or overexpression, effectively increased and decreased αSMA and MRTF-A proteins, respectively (Figure 8C). More detailed analysis indicated that elevating LGALS3 protein reduced MRTF-A protein in both the nucleus and the cytosol (Figure 8D). In accordance, silencing SREBP1, which positively regulates LGALS3 (Figure 1), increased MRTF-A protein levels (Figure 8F). Taken together, these results provide novel information that LGALS3 and SREBP1 negatively regulate SMC MRTF-A protein levels.
Figure 8.

LGALS3 negatively regulates MRTF-A protein levels
Mouse MOVAS cells were used. Cholesterol loading and lentiviral transduction were performed as described in detail for Figure 1. Data quantification for western blots: mean ± SEM, n ≥ 3 independent repeat experiments. Data quantification for quantitative real-time PCR: mean ± SD, n = 3 replicates; presented is one of two similar experiments. Statistics: ANOVA followed by Tukey test; #p < 0.05, ##p < 0.01, ###p < 0.001 (between paired bars); ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, compared with the basal condition (the first bar in each plot). (A) Down-regulation of αSMA and MRTF-A expression by cholesterol loading to MOVAS cells. (B) Effect of LGALS3 inhibitor TD139 on MRTF-A protein levels. (C) Negative LGALS3 regulation of αSMA and MRTF-A protein levels. (D) Effect of LGALS3 overexpression on nuclear and cytosolic MRTF-A protein levels. (E) Schematic of negative regulation of MRTF-A protein by LGALS3, as suggested by the results in (A)–(D). (F) Effect of SREBP1 silencing on MRTF-A protein levels.
The SREBP1/LGALS3 dyad occurs in cholesterol-loaded human primary SMCs
Following the above experiments performed with both the mouse MOVAS cell line and rat primary SMCs, we determined whether LGALS3 regulation by SREBP1 also occurs in human primary aortic SMCs. As shown in Figure 9A, whereas SREBP1, LGALS3, and CD68 proteins were upregulated by cholesterol loading, KLF15, MRTF-A, and αSMA were down-regulated, largely recapitulating the results obtained from mouse and rat SMCs. Indeed, SREBP1 silencing diminished LGALS3 and increased KLF15 protein levels (Figure 9B). Interestingly, pre-treating human SMCs with JQ1 prevented cholesterol-induced upregulation of SREBP1, LGALS3, and CD68 and partially restored the protein production of KLF15, MRTF-A, and αSMA that was inhibited by cholesterol loading (Figure 9C). In addition, LGALS3 silencing reduced CD68 protein in the cholesterol-loaded condition and elevated MRTF-A and αSMA protein levels in human SMCs without cholesterol loading (Figure 9D). In sum, these results of SREBP1/LGALS3 regulations in human primary aortic SMCs largely reproduced that from rodent SMCs, although in the cholesterol-loaded condition, LGALS3 silencing in human SMCs did not significantly increase MRTF-A and αSMA proteins as it did in rodent SMCs.
Figure 9.

SREBP1 positively regulates LGALS3 protein levels in human primary SMCs
Human primary aortic SMCs in full medium were transfected with a specific siRNA or scrambled control. The culture was then changed to fresh medium and continued until subconfluence. The cells were incubated with solvent control (0.25% BSA) or cholesterol at indicated concentrations for 72 h prior to harvest for assays. Because LGALS3 (and full-length SREBP1) protein levels peaked at 40 μg/mL cholesterol (see A), this concentration was used in the following experiments (see B–D). Data quantification (A–D): mean ± SEM, n ≥ 3 independent repeat experiments. Statistics: one-way ANOVA followed by Tukey test; #p < 0.05, ##p < 0.01, ###p < 0.001 between paired bars; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 compared with the basal condition (the first bar in each plot); non-significant comparisons are not labeled. (A) Upregulation of SREBP1 and LGALS3 proteins in cholesterol-loaded human SMCs. The colored bands are molecular weight markers. (B) SREBP1 silencing in human SMCs down-regulates LGALS3 protein. For KLF15, p = 0.031 and 0.057 when compared pairwise using Student’s t test. (C) Pre-treatment of human SMCs with JQ1 (0.1 or 0.5 μM) inhibits cholesterol-induced SREBP1 and LGALS3 protein production. Cells were incubated with JQ1 (0.1 or 0.5 μM) or vehicle (DMSO) for 1 h prior to cholesterol loading. (D) LGALS3 negatively regulates MRTF-A protein levels in human SMCs.
Discussion
In a cholesterol-rich environment, arterial wall resident SMCs activate Lgals3, giving rise to a multi-potential stem cell-like transition state that contributes to distinct phenotypic states that can be either beneficial or detrimental for atherosclerotic lesion pathogenesis.4,28 However, these previous studies did not determine mechanisms that activate Lgals3. To this end, we used the in vitro model of cholesterol-induced SMC phenotypic transition27 to investigate cholesterol-responsive regulators and the interplay thereof. Major findings are the following: (1) the master regulator of lipid metabolism, SREBP1 positively regulates LGALS3 expression and vice versa; (2) KLF15 inhibits the expression of LGALS3 and SREBP1; (3) BETs inhibition abrogates cholesterol-induced upregulation of the SREBP1/LGALS3 dyad; and (4) LGALS3 negatively regulates MRTF-A protein levels. Taken together, our findings provide new insights regarding mechanisms by which cholesterol loading regulates SMC plasticity.
SREBP1 has been implicated in atherosclerosis.37,38 However, these studies mainly focused on leukocytes and endothelial cells, cell types that respond to environmental perturbations differently than SMCs.39 Moreover, a role for SREBP1 in governing Lgals3 transcription was not previously reported. Our data revealed that SREBP1 bound to the proximal side of the Lgals3 promoter, and this binding was enhanced by cholesterol loading into SMCs. The SREBP1 specificity was further demonstrated by the contrasting result that KLF15 negatively regulated Lgals3 expression. The two transcription factors bound to the Lgals3 promoter with spatially and temporally distinct patterns. This raises an interesting question as to how KLF15 represses Lgals3 transcription. Apparently, it cannot be fully accounted for by an effect of KLF15 reducing SREBP1 protein, given our data showing direct KLF15 binding to the Lgals3 promoter DNA. Another scenario is that SREBP1 binding with the Lgals3 promoter sterically blocks KLF15 from activating Lgals3 transcription. However, counter to this possibility, the KLF15 ChIP-qPCR signal had already ebbed before a surge of SREBP1 ChIP-qPCR signal induced by cholesterol. Alternatively, by analogy with a recent study using hepatocytes,23 a plausible speculation is that KLF15 may recruit a yet-to-be-identified repressor or repressor protein complex that inhibits Lgals3 transcription.
Recent research discovered other transcription factors key to SMC phenotypic transition on cholesterol loading. For instance, in vivo and in vitro studies indicated that KLF4 not only promotes SMC dedifferentiation via inhibiting SMC contractile gene expression, it also mediates expression of LGALS3 and CD68, as well as pro-inflammatory cytokines.27 Similar to MRTF-A, myocardin is a master regulator of SMC contractile gene expression, which was previously reported to negatively regulate cholesterol-induced SMC phenotypic transitions.2 However, whether KLF4 or myocardin regulates Lgals3 expression by directly binding to its promoter remains unclear. Herein we show that cholesterol loading enhanced SREBP1 binding to a proximal Lgals3 promoter site. Furthermore, our data revealed a negative regulation of MRTF-A protein levels by SREBP1, as well as LGALS3, in cholesterol-loaded SMCs, an outcome in favor of SMC dedifferentiation and hence in accordance with these previous reports. A new question is whether SREBP1 cross-talks with KLF4 and myocardin pathways, which may deserve future studies to explore.
Transcription factors rarely act alone. Rather, along with other transcription factors and co-regulatory proteins, they nearly always regulate interactions of cis-regulatory elements to activate or repress gene expression in concert with epigenetic controls, including histone modifications.33,40 BETs are powerful epigenetic effectors that recognize histone acetyl marks as anchoring sites to render cell-state transitions.35 Indeed, our data show that inhibition of the BET family effectively contained SREBP1 and LGALS3 protein levels that were otherwise amplified because of cholesterol loading. Of particular interest, BRD2 appeared to be primarily responsible for the BET family function in this context. This result underscores differential functions of individual BETs, especially in view of a reported prominent role of BRD4 in other conditions, including proliferative and inflammatory cell states34,35,41,42 and, more recently, in adipogenesis31,32 and mitochondrial dysfunction.43 Furthermore, our data uncovered novel mechanistic details, namely, a BRD2 physical association with SREBP1 at SREBP1’s own gene and its target gene, Lgals3. This concurs with SREBP1 being a master transcription factor, a term used for a limited number of transcription factors key to cell identity that often bind at their own promoters.33 Echoing this SREBP1 story, SREBP2 was reported to also collaborate with BRD2 in activating gene expression, however, in cholesterol-deprived conditions instead in retinal pigment epithelial cells.19 It is also worth noting that in terms of restraining SREBP1 and LGALS3 from upregulation, blocking the BET family with JQ1 appeared to be more effective than silencing BRD2. This is rationalizable, because it may not be mechanistically equivalent to eliminate a BET protein via genetic silencing or to use a small molecule to block the binding of BET bromodomains to the histone acetyl-lysine marks. These differential outcomes hence lend new knowledge that may prove useful for identifying future therapeutic interventions for diseases such as atherosclerosis where SMC phenotypic transitions play a critical role.44
LGALS3 used to be deemed a macrophage marker, but more recent studies have unveiled its broad signaling and regulatory functions.30 Of note, genetic and pharmacological studies suggested its importance in atherogenesis,30,45,46 and in vitro evidence showed its positive role in promoting SMC migration and pro-inflammatory cytokine expression.36 Beyond this knowledge, our data contributed new information showing that LGALS3 decreased expression of MRTF-A protein, a well-established transcription co-activator of SMC contractile genes. Collectively, our results and existing reports suggest that rather than merely a cell-type marker, LGALS3 is a key effector that promotes cholesterol-induced SMC phenotypic states, including migration, inflammation, dedifferentiation, lipid storage, and resistance to apoptosis (Figure S3). Moreover, we identified it as an atypical target gene of both SREBP1 and KLF15. In addition, we observed that LGALS3 promotes SREBP1 expression but represses KLF15 production. Our coIP experiments did not show an obvious LGALS3/SREBP1 physical association, although it has been reported that LGALS3 participates in nuclear function, such as forming an early splicing machinery.29 However, it will be important that future studies determine how LGALS3 regulates SREBP1 and KLF15 expression.
In view of the SREBP1→LGALS3→SREBP1 feedforward circuit identified herein, and previous evidence that SMCs with activated Lgals3 preferentially give rise to atherosclerotic lesion cells,4 it is interesting to consider LGALS3 as a potential interventional target for breaking this vicious cycle. However, the effectiveness of using an LGALS3 inhibitor to attenuate SREBP1 upregulation varied in different conditions, and conflicting reports exist with regard to its role in atherosclerosis in animal and human studies.45, 46, 47 In contrast, BETs inhibition with JQ1 abrogated cholesterol-induced increases of LGALS3 and SREBP1 protein levels, in rodent SMCs and also in human primary SMCs. This potent effect implicates an alternative strategy to inhibit this SREBP1/LGALS3 pathway. BETs inhibitors have shown anti-atherogenic efficacy in preclinical models.35,40 In a phase II clinical trial, the pan-BETs inhibitor RVX208, which is known to increase apolipoprotein A-I,48 has exhibited a favorable profile of safety and efficacy in amelioration of major adverse cardiovascular events.49 In contrast, the molecular mechanisms concerning BETs in atherosclerosis, especially in the perspective of the SREBP1/LGALS3 dyad in SMC phenotypic transition, remain to be better understood.
Conclusions
In this study, we focused on unraveling molecular mechanisms that underlie the SMC pathobiology elicited by cholesterol overload. We found a SREBP1/LGALS3 feedforward circuit that was antagonized by KLF15. We further found that pharmacological blockade of the BET epigenetic reader family prevented this SREBP1/LGALS3 dyad from surging in a cholesterol-rich environment. It will be important that future studies examine these regulatory pathways in vivo in an atherogenic disease background. Nevertheless, our findings shed new light on molecular determinants of SMC plasticity and potential interventional strategies.
Materials and methods
Materials
Various resources, including kits and reagents, are presented in Table S1 or included in the corresponding texts below.
Cell culture and cholesterol loading
MOVAS, which is a commonly used mouse aortic SMC line, and human primary aortic SMCs were purchased from ATCC (Manassas, VA, USA). The cells were cultured in DMEM high-glucose full medium (catalog no. [cat.] 11965092; ThermoFisher) supplemented with 10% FBS and 50 μg/mL G418 (antibiotic, cat. 4727878001; Sigma-Aldrich) at 5% CO2 and 37°C. Primary rat aortic SMCs were isolated as described previously42 with minor modifications based on a recent report.50 The cells were cultured in smooth muscle complete (full) medium (cat. M2268; Cell Biologics) and used at passage 5. To load cholesterol into SMCs, we followed a previously reported method using a soluble form of cholesterol, i.e., cholesterol-methyl-β-cyclodextrin (cat. C4951; Sigma-Aldrich).1,2 Cholesterol-methyl-β-cyclodextrin (concentration indicated in the figure legends) in 0.25% (w/v) BSA was added to subconfluent SMCs and incubated for 72 h prior to harvest for various assays.2,27 No obvious apoptosis was observed after cholesterol loading (Figure S1B). The cells incubated with 0.25% BSA for 72 h without cholesterol-methyl-β-cyclodextrin served as solvent controls.2
Lenti-vector construction
For loss-of-function experiments using MOVAS cells, mouse short hairpin RNAs (shRNAs) designed with web tools (https://portals.broadinstitute.org/gpp/public/) were expressed by lentivirus. Their sequences are listed in Table S2. The shRNA-expressing constructs were generated using the vector pLKO.1 puro (cat. 8453; Addgene). For gain-of-function experiments, stable cell lines were generated using lentiviral vectors. First, mouse and rat mRNAs were extracted from MOVAS cells and rat primary aortic SMCs, respectively, using TRIzol reagents (cat. 15596018; Thermo Fisher Scientific). Second, reverse transcription was performed with the extracted mRNA, and a full-length cDNA was cloned. Coding sequences were Mouse KLF15 (GenBank: NM_023184.4), rat KLF15 (GenBank: NM_053536.2), mouse LGALS3 (GenBank: NM_001145953.1), and rat LGALS3 (GenBank: NM_031832.1). Mouse SREBP1 (GenBank: XM_006532716.3) cDNA was cloned from the pLKO-puro FLAG-SREBP1 vector (cat. 32017; Addgene). KLF15 and SREBP1 cDNAs were each subcloned into the pLenti-puro vector (cat. 39481; Addgene) with a FLAG tag at the N terminus. Mouse and rat LGALS3 cDNAs were also subcloned into this vector but with a V5 tag at the C terminus. The primers used for cloning are listed in Table S3.
Lentivirus production and transduction
Lentivirus was produced using HEK293FT cells (cat. A14527; Invitrogen) with the second-generation packaging system pSPAX2 (cat.12260; Addgene) and pMD2.G (cat. 12259; Addgene). Lentiviral titer was determined using Lenti-X GoStix Plus (cat. 631280; TakaRa) by measuring the viral RNA content in the supernatant. MOVAS cells or rat primary SMCs were cultured in their respective full medium until 80–90% confluence and then transduced with >1 × 108 lentivirus for 48 h while selected with 5 μg/mL puromycin. Prior to experiments such as cholesterol loading, selected cells were replated and cultured to subconfluence in fresh full medium still under the selection pressure of 5 μg/mL puromycin.
Gene silencing
Gene silencing in MOVAS cells was performed using shRNA-expressing lentivirus. Lentiviral transduction was done as described above. Gene silencing in rat primary SMCs was achieved via small interfering RNAs (siRNAs) transfection for 24 h using the Lipofectamine RNAiMAX Transfection Reagent (Cat. 13778075; ThermoFisher Scientific), and cholesterol was then added and cultured for another 72 h before cell harvest. Sequences of siRNAs are listed in Table S2.
Quantitative real-time PCR
We followed our previous report.19 Briefly, total RNA was isolated from cultured cells using the TRIzol reagent (cat. 15596018; ThermoFisher Scientific). Potential contaminating genomic DNA was removed by using gDNA Eliminator columns provided in the kit. RNA was quantified with a NanoDrop NP-1000 spectrometer, and 1 μg was used for the first-strand cDNA synthesis. Quantitative real-time PCR was then performed using Quant Studio 3 (Applied Biosystems, Carlsbad, CA, USA). The housekeeping gene GAPDH was used for normalization using the ΔΔCt method. Each cDNA template was amplified in triplicate reactions using PerfeCTa SYBR Green SuperMix (cat. 95054; Quantabio) with gene-specific primers listed in Table S4.
Western blotting analysis
At the end of each treatment, cells were harvested and lysed on ice in the RIPA buffer (cat. 89900; ThermoFisher Scientific) that includes a Halt protease and phosphatase inhibitor cocktail (cat. 78440; ThermoFisher Scientific). Cell lysates were quantified for protein concentrations using the Bio-Rad DC Protein Assay kit (cat. 5000112; Bio-Rad). Proteins were separated on a 5–12% SDS-PAGE gel, transferred to a polyvinylidene fluoride (PVDF) membrane, and then incubated with a primary antibody (see list in Table S5). The signal from a specific protein was amplified by Goat Anti-mouse IgG (H + L)-HRP Conjugate (cat. 1706516; Bio-Rad) or Goat Anti-rabbit IgG (H + L)-HRP Conjugate (cat. 1706515; Bio-Rad) and illuminated with Clarity ECL Western Blotting Substrates (cat. 1705060; Bio-Rad). Western blot images were immediately recorded with Azure C600 Imager (Azure Biosystems). Protein band densitometry was quantified using National Institutes of Health (NIH) ImageJ and normalized to loading control for statistical analysis.
Cell migration assay
The assay was performed using the scratch method as we previously described.51 Briefly, MOVAS cells were cultured to 90% confluence in six-well plates. A sterile pipette tip was used to scrape out an ∼1-mm cell-free gap. Dislodged cells were washed away with PBS followed by refilling fresh basal medium supplemented with 10% FBS. For gene silencing using shRNA, cells were transduced with lentivirus and selected with puromycin prior to cholesterol loading, as described above. TD139 (LGALS3 inhibitor, 1 μM) or vehicle control (DMSO) was added 2 h prior to cholesterol loading. For illumination of the cells, Calcein-AM (cat. C1430; Thermo Scientific) was added (final 2 μM) and incubated for 10 min at the end of each treatment. Images were taken after at least four times of gentle rinse with PBS. Cell migration was quantified with ImageJ (NIH) based on the area of the cell-free gap.
ChIP-qPCR assay
We followed the manufacturer’ instruction and our recently reported protocol.34 Briefly, MOVAS cells cultured under various conditions (indicated in the figure legends) were cross-linked with 1% formaldehyde for 10 min at room temperature. The reaction was terminated for 5 min with 0.125 M glycine. The cells were washed with ice-cold PBS and lysed in the buffer containing 10 mM Tris-HCl (pH 7.9), 0.25% Triton X-100, 10 mM EDTA, and protease inhibitors. The lysate was centrifuged at 4,000 rpm for 5 min. The pellet was suspended in the nuclei extraction buffer (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 0.1% SDS, 1% Triton X-100, 1 mM EDTA, and protease inhibitors) and sonicated (forty 30-s pulses at 20 W per 1×107 cells) to disrupt the nuclear membrane. Chromatin extracts containing DNA fragments (each with ∼100–1,000 bp) were IPed by incubating with a specific antibody (or nonspecific IgG) overnight at 4°C. ChIP-grade Protein A/G Magnetic beads were added and incubated for 4 h at 4°C. RNase A (5 U) and Proteinase K (2 μg/μL) were used to digest RNA and proteins. The purified DNA was used for qPCR with primers listed in Table S6.
EMSA
Assays were performed using 5′-biotinylated double-stranded DNA oligonucleotide probes and LightShift Chemiluminescent EMSA Kit (cat. 20148; ThermoFisher Scientific), with a procedure described in our recent publication.52 In brief, equimolar complementary strands were mixed and heated to 95°C followed by gradual cooling to ambient temperature for at least 5 h to anneal the probes. Double-stranded DNA probes (40 fmol) were mixed with 10 μg of nuclear extracts from MOVAS cells in 20 μL reaction buffer containing 10 mM Tris (pH 7.5), 50 mM KCl, 1 mM DTT, 50 ng/μL poly(dI:dC), 5% glycerol, 0.05% Nonidet P-40 (NP-40), 5 mM MgCl2, and 1 mM EDTA. After 30-min reaction at room temperature, the protein:DNA complexes were separated at 4°C on a pre-run 6% native polyacrylamide gel in 0.5× Tris-borate-EDTA (TBE) and transferred to a positively charged nylon membrane. The mobility of biotinylated DNA probe was illuminated using Chemiluminescent Nucleic Acid Detection Module (cat. 89880; ThermoFisher Scientific). For competition experiments, an unlabeled oligonucleotide in 2.5×, 5×, or 10× molar excess was incubated with the respective biotinylated probe. Wild-type (WT) and mutant probes with a 5′-biotin label were synthesized by Invitrogen (Table S7).
CoIP
We followed the method used in our recent report19 with minor modifications. In brief, cells (HEK293) were transfected with pEGFP-N1-FLAG (empty vector) (cat. 60360; Addgene) or pFLAG-SREBP1Nterm (N-terminal transcription-active domain, amino acids 1–490) (cat. 26801; Addgene) and cultured for 24 h. The cells were harvested and lysed on ice for 30 min in Pierce IP Lysis Buffer (cat. 87788; ThermoFisher Scientific) containing Halt Protease Inhibitor Cocktail (cat. 87785; ThermoFisher Scientific) and then centrifuged at 13,200 × g for 15 min at 4°C. The supernatant was incubated overnight at 4°C with 50 μL of Pierce Anti-FLAG (DYKDDDDK) Magnetic Agarose beads (cat. A36797; ThermoFisher Scientific). To elute the immunoprecipitates, we washed the beads three times with cold PBS and then incubated in 100 mM glycine (pH 2.8) for 10 min at room temperature while frequently vortexed. The eluate was neutralized with 1 M Tris-HCl (pH 8.5; 15 μL per 100 μL eluate), briefly heated at 95°C in SDS sample buffer, and then loaded for SDS-PAGE and western blot analysis. An antibody specific for endogenous BRD2 or LGALS3 was used for immunoblotting (IB) to detect their coIP with SREBP1Nterm. Antibodies are listed in Table S5.
Statistical analysis
Western blot analysis was repeated in at least three independent experiments performed in different days, and the averaged data values are presented as mean ± standard error of the mean (SEM). For PCR-based analysis, the same result was repeated in two independent experiments performed in different days, and one of two sets of data is presented as mean ± standard deviation (SD) of triplicates, as specified in the figure legends. Comparison between two groups was analyzed using Student’s t test. In the case of multi-group analysis, one-way ANOVA with post hoc Tukey test was performed. p < 0.05 was regarded as statistically significant. Note that because of small sample sizes, normality and variance could not be tested to determine whether the applied parametric tests were appropriate. Statistics and graphical data plots were generated using GraphPad Prism (Version 8.0).
Acknowledgments
This work was supported by NIH R01 grant HL133665 (to L.-W. G.) and NIH Grant R01 HL136314 (to G.K.O.). We would like to thank Dr. Xiujie Xie and Dr. Qingwei Wang for their assistance in methodology. We thank Dr. Avril Somlyo for important discussions and Dr. Vlad Serbulea for suggestions on statistics.
Author contributions
J.L. and H.S. performed experiments; G.K.O. criticized the data and critically reviewed and revised the manuscript; J.L. and L.-W. G. analyzed data and wrote the manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2022.05.028.
Supplemental information
References
- 1.Rong J.X., Shapiro M., Trogan E., Fisher E.A. Transdifferentiation of mouse aortic smooth muscle cells to a macrophage-like state after cholesterol loading. Proc. Natl. Acad. Sci. U S A. 2003;100:13531–13536. doi: 10.1073/pnas.1735526100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vengrenyuk Y., Nishi H., Long X., Ouimet M., Savji N., Martinez F.O., Cassella C.P., Moore K.J., Ramsey S.A., Miano J.M., Fisher E.A. Cholesterol loading reprograms the microRNA-143/145-myocardin axis to convert aortic smooth muscle cells to a dysfunctional macrophage-like phenotype. Arterioscler. Thromb. Vasc. Biol. 2015;35:535–546. doi: 10.1161/atvbaha.114.304029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang Y., Dubland J.A., Allahverdian S., Asonye E., Sahin B., Jaw J.E., Sin D.D., Seidman M.A., Leeper N.J., Francis G.A. Smooth muscle cells contribute the majority of foam cells in ApoE (apolipoprotein E)-Deficient mouse atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2019;39:876–887. doi: 10.1161/atvbaha.119.312434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alencar G.F., Owsiany K.M., Karnewar S., Sukhavasi K., Mocci G., Nguyen A.T., Williams C.M., Shamsuzzaman S., Mokry M., Henderson C.A., et al. Stem cell pluripotency genes Klf4 and Oct4 regulate complex SMC phenotypic changes critical in late-stage atherosclerotic lesion pathogenesis. Circulation. 2020;142:2045–2059. doi: 10.1161/circulationaha.120.046672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chappell J., Harman J.L., Narasimhan V.M., Yu H., Foote K., Simons B.D., Bennett M.R., Jorgensen H.F. Extensive proliferation of a subset of differentiated, yet plastic, medial vascular smooth muscle cells contributes to neointimal formation in mouse injury and atherosclerosis models. Circ. Res. 2016;119:1313–1323. doi: 10.1161/circresaha.116.309799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang Y., Nanda V., Direnzo D., Ye J., Xiao S., Kojima Y., Howe K.L., Jarr K.U., Flores A.M., Tsantilas P., et al. Clonally expanding smooth muscle cells promote atherosclerosis by escaping efferocytosis and activating the complement cascade. Proc. Natl. Acad. Sci. U S A. 2020;117:15818–15826. doi: 10.1073/pnas.2006348117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang D., Chen Z.G., Liu S.H., Dong Z.Q., Dalin M., Bao S.S., Hu Y., Wei F. Galectin-3 gene silencing inhibits migration and invasion of human tongue cancer cells in vitro via downregulating beta-catenin. Acta Pharmacol. Sin. 2013;34:176–184. doi: 10.1038/aps.2012.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Margadant C., van den Bout I., van Boxtel A.L., Thijssen V.L., Sonnenberg A. Epigenetic regulation of galectin-3 expression by β1 integrins promotes cell adhesion and migration. J. Biol. Chem. 2012;287:44684–44693. doi: 10.1074/jbc.m112.426445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nangia-Makker P., Hogan V., Raz A. Galectin-3 and cancer stemness. Glycobiology. 2018;28:172–181. doi: 10.1093/glycob/cwy001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maeda N., Kawada N., Seki S., Arakawa T., Ikeda K., Iwao H., Okuyama H., Hirabayashi J., Kasai K., Yoshizato K. Stimulation of proliferation of rat hepatic stellate cells by galectin-1 and galectin-3 through different intracellular signaling pathways. J. Biol. Chem. 2003;278:18938–18944. doi: 10.1074/jbc.m209673200. [DOI] [PubMed] [Google Scholar]
- 11.Yang R.Y., Hsu D.K., Liu F.T. Expression of galectin-3 modulates T-cell growth and apoptosis. Proc. Natl. Acad. Sci. U S A. 1996;93:6737–6742. doi: 10.1073/pnas.93.13.6737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blasetti Fantauzzi C., Iacobini C., Menini S., Vitale M., Sorice G.P., Mezza T., Cinti S., Giaccari A., Pugliese G. Galectin-3 gene deletion results in defective adipose tissue maturation and impaired insulin sensitivity and glucose homeostasis. Sci. Rep. 2020;10:20070. doi: 10.1038/s41598-020-76952-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nakajima K., Kho D.H., Yanagawa T., Harazono Y., Gao X., Hogan V., Raz A. Galectin-3 inhibits osteoblast differentiation through notch signaling. Neoplasia. 2014;16:939–949. doi: 10.1016/j.neo.2014.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barman S.A., Li X., Haigh S., Kondrikov D., Mahboubi K., Bordan Z., Stepp D.W., Zhou J., Wang Y., Weintraub D.S., et al. Galectin-3 is expressed in vascular smooth muscle cells and promotes pulmonary hypertension through changes in proliferation, apoptosis, and fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2019;316:L784–L797. doi: 10.1152/ajplung.00186.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Knights A.J., Yik J.J., Mat Jusoh H., Norton L.J., Funnell A.P., Pearson R.C., Bell-Anderson K.S., Crossley M., Quinlan K.G. Kruppel-like factor 3 (KLF3/BKLF) is required for widespread repression of the inflammatory modulator galectin-3 (Lgals3) J. Biol. Chem. 2016;291:16048–16058. doi: 10.1074/jbc.m116.715748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shimano H., Sato R. SREBP-regulated lipid metabolism: convergent physiology - divergent pathophysiology. Nat. Rev. Endocrinol. 2017;13:710–730. doi: 10.1038/nrendo.2017.91. [DOI] [PubMed] [Google Scholar]
- 17.Ma K.L., Liu J., Wang C.X., Ni J., Zhang Y., Wu Y., Lv L.L., Ruan X.Z., Liu B.C. Activation of mTOR modulates SREBP-2 to induce foam cell formation through increased retinoblastoma protein phosphorylation. Cardiovasc. Res. 2013;100:450–460. doi: 10.1093/cvr/cvt203. [DOI] [PubMed] [Google Scholar]
- 18.Zhou R.H., Pesant S., Cohn H.I., Eckhart A.D. Enhanced sterol response element-binding protein in postintervention restenotic blood vessels plays an important role in vascular smooth muscle proliferation. Life Sci. 2008;82:174–181. doi: 10.1016/j.lfs.2007.10.025. [DOI] [PubMed] [Google Scholar]
- 19.Shen H., Li J., Xie X., Yang H., Zhang M., Wang B., Kent K.C., Plutzky J., Guo L.W. BRD2 regulation of sigma-2 receptor upon cholesterol deprivation. Life Sci. Alliance. 2021;4 doi: 10.26508/lsa.201900540. e201900540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qin L., Yang Y.B., Yang Y.X., Zhu N., Gong Y.Z., Zhang C.P., Li S., Liao D. Ezetimibe suppresses cholesterol accumulation in lipid-loaded vascular smooth muscle cells in vitro via MAPK signaling. Acta Pharmacol. Sin. 2014;35:1129–1136. doi: 10.1038/aps.2014.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gray S., Wang B., Orihuela Y., Hong E.G., Fisch S., Haldar S., Cline G.W., Kim J.K., Peroni O.D., Kahn B.B., Jain M.K. Regulation of gluconeogenesis by Kruppel-like factor 15. Cell Metab. 2007;5:305–312. doi: 10.1016/j.cmet.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fan L., Sweet D.R., Prosdocimo D.A., Vinayachandran V., Chan E.R., Zhang R., Ilkayeva O., Lu Y., Keerthy K.S., Booth C.E., et al. Muscle Kruppel-like factor 15 regulates lipid flux and systemic metabolic homeostasis. J. Clin. Invest. 2021;131:e139496. doi: 10.1172/jci139496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takeuchi Y., Yahagi N., Aita Y., Murayama Y., Sawada Y., Piao X., Toya N., Oya Y., Shikama A., Takarada A., et al. KLF15 enables rapid switching between lipogenesis and gluconeogenesis during fasting. Cell Rep. 2016;16:2373–2386. doi: 10.1016/j.celrep.2016.07.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lu Y., Zhang L., Liao X., Sangwung P., Prosdocimo D.A., Zhou G., Votruba A.R., Brian L., Han Y.J., Gao H., et al. Kruppel-like factor 15 is critical for vascular inflammation. J. Clin. Invest. 2013;123:4232–4241. doi: 10.1172/JCI68552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lu Y., Haldar S., Croce K., Wang Y., Sakuma M., Morooka T., Wang B., Jeyaraj D., Gray S.J., Simon D.I., Jain M.K. Kruppel-like factor 15 regulates smooth muscle response to vascular injury--brief report. Arterioscler. Thromb. Vasc. Biol. 2010;30:1550–1552. doi: 10.1161/atvbaha.110.207050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haldar S.M., Lu Y., Jeyaraj D., Kawanami D., Cui Y., Eapen S.J., Hao C., Li Y., Doughman Y.Q., Watanabe M., et al. Klf15 deficiency is a molecular link between heart failure and aortic aneurysm formation. Sci. Transl. Med. 2010;2:26ra26. doi: 10.1126/scitranslmed.3000502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shankman L.S., Gomez D., Cherepanova O.A., Salmon M., Alencar G.F., Haskins R.M., Swiatlowska P., Newman A.A.C., Greene E.S., Straub A.C., et al. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat. Med. 2015;21:628–637. doi: 10.1038/nm.3866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pan H., Xue C., Auerbach B.J., Fan J., Bashore A.C., Cui J., Yang D.Y., Trignano S.B., Liu W., Shi J., et al. Single-cell genomics reveals a novel cell state during smooth muscle cell phenotypic switching and potential therapeutic targets for atherosclerosis in mouse and human. Circulation. 2020;142:2060–2075. doi: 10.1161/circulationaha.120.048378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fritsch K., Mernberger M., Nist A., Stiewe T., Brehm A., Jacob R. Galectin-3 interacts with components of the nuclear ribonucleoprotein complex. BMC Cancer. 2016;16:502. doi: 10.1186/s12885-016-2546-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gao Z., Liu Z., Wang R., Zheng Y., Li H., Yang L. Galectin-3 is a potential mediator for atherosclerosis. J. Immunol. Res. 2020;2020:5284728. doi: 10.1155/2020/5284728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brown J.D., Feldman Z.B., Doherty S.P., Reyes J.M., Rahl P.B., Lin C.Y., Sheng Q., Duan Q., Federation A.J., Kung A.L., et al. BET bromodomain proteins regulate enhancer function during adipogenesis. Proc. Natl. Acad. Sci. U S A. 2018;115:2144–2149. doi: 10.1073/pnas.1711155115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee J.E., Park Y.K., Park S., Jang Y., Waring N., Dey A., Ozato K., Lai B., Peng W., Ge K. Brd4 binds to active enhancers to control cell identity gene induction in adipogenesis and myogenesis. Nat. Commun. 2017;8:2217. doi: 10.1038/s41467-017-02403-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bradner J.E., Hnisz D., Young R.A. Transcriptional addiction in cancer. Cell. 2017;168:629–643. doi: 10.1016/j.cell.2016.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang Q., Ozer H.G., Wang B., Zhang M., Urabe G., Huang Y., Kent K.C., Guo L.W. A hierarchical and collaborative BRD4/CEBPD partnership governs vascular smooth muscle cell inflammation. Mol. Ther. Methods Clin. Dev. 2021;21:54–66. doi: 10.1016/j.omtm.2021.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Borck P.C., Guo L.W., Plutzky J. BET epigenetic reader proteins in cardiovascular transcriptional programs. Circ. Res. 2020;126:1190–1208. doi: 10.1161/circresaha.120.315929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tian L., Chen K., Cao J., Han Z., Gao L., Wang Y., Fan Y., Wang C. Galectin-3-induced oxidized low-density lipoprotein promotes the phenotypic transformation of vascular smooth muscle cells. Mol. Med. Rep. 2015;12:4995–5002. doi: 10.3892/mmr.2015.4075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Karasawa T., Takahashi A., Saito R., Sekiya M., Igarashi M., Iwasaki H., Miyahara S., Koyasu S., Nakagawa Y., Ishii K., et al. Sterol regulatory element-binding protein-1 determines plasma remnant lipoproteins and accelerates atherosclerosis in low-density lipoprotein receptor-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2011;31:1788–1795. doi: 10.1161/atvbaha.110.219659. [DOI] [PubMed] [Google Scholar]
- 38.Liu Y., Major A.S., Zienkiewicz J., Gabriel C.L., Veach R.A., Moore D.J., Collins R.D., Hawiger J. Nuclear transport modulation reduces hypercholesterolemia, atherosclerosis, and fatty liver. J. Am. Heart Assoc. 2013;2:e000093. doi: 10.1161/jaha.113.000093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pan X., Wang B., Yuan T., Zhang M., Kent K.C., Guo L.W. Analysis of combined transcriptomes identifies gene modules that differentially respond to pathogenic stimulation of vascular smooth muscle and endothelial cells. Sci. Rep. 2018;8:395. doi: 10.1038/s41598-017-18675-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brown J.D., Lin C.Y., Duan Q., Griffin G., Federation A.J., Paranal R.M., Bair S., Newton G., Lichtman A.H., Kung A.L., et al. NF-κB directs dynamic super enhancer formation in inflammation and atherogenesis. Mol. Cell. 2014;56:219–231. doi: 10.1016/j.molcel.2014.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang M., Wang B., Urabe G., Huang Y., Plutzky J., Kent K.C., Guo L.W. The BD2 domain of BRD4 is a determinant in EndoMT and vein graft neointima formation. Cell. Signal. 2019;61:20–29. doi: 10.1016/j.cellsig.2019.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang B., Zhang M., Takayama T., Shi X., Roenneburg D.A., Craig Kent K., Guo L.W. BET bromodomain blockade mitigates intimal hyperplasia in rat carotid arteries. EBioMedicine. 2015;2:1650–1661. doi: 10.1016/j.ebiom.2015.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim S.Y., Zhang X., Schiattarella G.G., Altamirano F., Ramos T.A.R., French K.M., Jiang N., Szweda P.A., Evers B.M., May H.I., et al. Epigenetic reader BRD4 (Bromodomain-Containing protein 4) governs nucleus-encoded mitochondrial transcriptome to regulate cardiac function. Circulation. 2020;142:2356–2370. doi: 10.1161/circulationaha.120.047239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bennett M.R., Sinha S., Owens G.K. Vascular smooth muscle cells in atherosclerosis. Circ. Res. 2016;118:692–702. doi: 10.1161/circresaha.115.306361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mortensen M.B., Fuster V., Muntendam P., Mehran R., Baber U., Sartori S., Falk E. Negative risk markers for cardiovascular events in the elderly. J. Am. Coll. Cardiol. 2019;74:1–11. doi: 10.1016/j.jacc.2019.04.049. [DOI] [PubMed] [Google Scholar]
- 46.Ding N., Yang C., Ballew S.H., Kalbaugh C.A., McEvoy J.W., Salameh M., Aguilar D., Hoogeveen R.C., Nambi V., Selvin E., et al. Fibrosis and inflammatory markers and long-term risk of peripheral artery disease: the ARIC study. Arterioscler. Thromb. Vasc. Biol. 2020;40:2322–2331. doi: 10.1161/atvbaha.120.314824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Di Gregoli K., Somerville M., Bianco R., Thomas A.C., Frankow A., Newby A.C., George S.J., Jackson C.L., Johnson J.L. Galectin-3 identifies a subset of macrophages with a potential beneficial role in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2020;40:1491–1509. doi: 10.1161/atvbaha.120.314252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bailey D., Jahagirdar R., Gordon A., Hafiane A., Campbell S., Chatur S., Wagner G.S., Hansen H.C., Chiacchia F.S., Johansson J., et al. RVX-208: a small molecule that increases apolipoprotein A-I and high-density lipoprotein cholesterol in vitro and in vivo. J. Am. Coll. Cardiol. 2010;55:2580–2589. doi: 10.1016/j.jacc.2010.02.035. [DOI] [PubMed] [Google Scholar]
- 49.Ghosh G.C., Bhadra R., Ghosh R.K., Banerjee K., Gupta A. RVX 208: a novel BET protein inhibitor, role as an inducer of apo A-I/HDL and beyond. Cardiovasc. Ther. 2017;35:e12265. doi: 10.1111/1755-5922.12265. [DOI] [PubMed] [Google Scholar]
- 50.Chi J., Meng L., Pan S., Lin H., Zhai X., Liu L., Zhou C., Jiang C., Guo H. Primary culture of rat aortic vascular smooth muscle cells: a new method. Med. Sci. Monit. 2017;23:4014–4020. doi: 10.12659/msm.902816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li J., Urabe G., Huang Y., Zhang M., Wang B., Marcho L., Shen H., Kent K.C., Guo L.W. A role for polo-like kinase 4 in vascular fibroblast cell-type transition. JACC Basic Transl. Sci. 2021;6:257–283. doi: 10.1016/j.jacbts.2020.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xie X., Urabe G., Marcho L., Stratton M., Guo L.W., Kent C.K. ALDH1A3 regulations of matricellular proteins promote vascular smooth muscle cell proliferation. iScience. 2019;19:872–882. doi: 10.1016/j.isci.2019.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.