

Abstract
Immunization or microbial infection can establish long-term B cell memory not only systemically but also locally. Evidence has suggested that local B cell memory contributes to early local plasmacytic responses after secondary challenge, however; it is unclear whether locality plays any role in participation of memory B cells in recall germinal centers (GCs) which is essential for updating their B cell antigen receptors (BCRs). Using single B cell culture and fate-mapping, we have characterized BCR repertoires in recall GCs following boost immunizations at sites local or distal to the priming. Local boosts with homologous antigen recruit the progeny of primary GC B cells to recall GCs more efficiently than do distal boosts. Recall GCs elicited by local boosts contain significantly more B cells with elevated levels of Immunoglobulin (Ig) mutation and higher avidity BCRs. This local preference is unaffected by blocking CD40:CD154 interaction to terminate active, GC responses. Local boosts with heterologous antigens elicit secondary GCs with B cell populations enriched for cross-reactivity to the prime and boost antigens; in contrast, cross-reactive GC B cells are rare following distal boosts. Our results suggest that local B cell memory is retained in the form of memory B cells, GC B cells and GC-phenotype B cells that are independent of organized GC structures and that these persistent “primed B cells” contribute to recall GC responses at local sites. Our findings indicate the importance of locality in humoral immunity and inform serial vaccination strategies for evolving viruses.
One Sentence Summary:
The participation of memory B cells in recall germinal centers depends on whether the boost is local or distal to the priming site.
INTRODUCTION
Vaccinations or microbial infections activate the adaptive immune system and establish, predominantly through germinal center (GC) responses, long-lasting protective immunity (1). This durable immunity comprises long-lived plasma cells and memory B (Bmem) cells. The former maintain circulating antibody (Ab) to provide a first line of defense against reinfection. The latter exhibit multiple fates on re-encountering antigen (2, 3). Bmem cells either proliferate and differentiate into short-lived plasmablasts/cytes (PBs/PCs) or (re)enter GCs in which they undergo new rounds of antigen-driven selection and immunoglobulin (Ig) somatic hypermutation (SHM) to “update” their B cell receptors (BCRs). Some fraction of re-activated Bmem cells may also return to their resting state (4). Rapid Bmem differentiation into PBs/PCs contributes to prompt, high affinity Ab responses and protective activity, the Bmem cells that re-enter GCs and emerge with updated BCRs play critical roles in combatting evolving viruses (e.g., HIV-1, influenza, SARS-CoV-2) (5–7). Viruses that accumulate fitness-enhancing mutations in response to immune pressure (in individuals or populations) can generate escape variants to which pre-existing Bmem BCRs and circulating Abs bind weakly or not at all (8).
Studies in mice and humans have provided substantial evidence for the recruitment of Bmem cells into recall GCs (9–13) and for the updating of Bmem BCRs (12, 14–16). Nonetheless, questions have arisen concerning participation of Bmem cells in recall GC responses (17–20), and recent studies have suggested that recruitment of Bmem cells to secondary GCs is not an efficient process (21). Fate-mapping experiments have shown that the marked progeny of primary GC B cells account for a small subset (≤5%) of secondary GC B cells and that newly-activated, mature B cells dominate the recall GC responses (21, 22). In addition to this overall inefficiency, recruitment of high affinity Bmem cells into recall GCs may be further reduced as higher affinity Bmem cells appear to be biased for PBs/PCs differentiation rather than GC re-entry (23–28).
That immunization or microbial infection can establish humoral memory that is local as well as systemic is now recognized. For example, pulmonary infection of mice with influenza viruses establishes resident Bmem cells in the lung, and these local Bmem cells exhibit distinct BCR specificities and contribute to early local plasmacytic responses that provide stronger protection against influenza challenge than do circulating, systemic Bmem cells (29, 30). This property of local humoral memory may be general. Early studies indicate that antigen-retention in local LNs has significant roles in the migration and retention of specific Bmem cells and that LNs linked to primary immunization sites can produce more Ab-secreting cells than do distal LNs after secondary challenge (31–34).
Here, we aimed to determine whether a distinct population of local Bmem cells in peripheral LNs support secondary GC responses that are distinct from those at distal sites, and whether the location in humoral immunity is relevant only to non-lymphoid tissues, e.g., lung, or if it extends to secondary lymphoid tissues. To address these questions, we compared secondary GC responses to boost immunizations given in the same (ipsilateral) or opposite (contralateral) leg of primed mice. The magnitude of secondary GC and serum Ab responses to homologous boosts were similar for both ipsilateral and contralateral boosts. We found, however, that the quality of these responses differed. Ipsilateral boosts elicited GCs with higher numbers of cells that were the progeny of the primary GC response than did contralateral boosts. Consequently, secondary GCs elicited by ipsilateral boosting contained higher numbers of B cells with elevated Ig mutation frequencies and higher avidity for antigen. Disruption of active, primary GC responses by injecting anti-CD154 Ab did not reduce this local recall bias for previously mutated, higher affinity cells. In response to boosts with heterologous antigens, ipsilateral boosts were more effective in producing secondary GC B cells that bound both the primary and boost antigens than contralateral boosts. Our findings suggest that local B cell memory is retained in the form of Bmem cells, GC B cells and GC-phenotype B cells. The results have implications for strategies of vaccination against evolving pathogens, for which updating BCRs is essential for durable protection.
RESULTS
Boost immunizations at local sites and at distal sites elicit comparable levels of recall serum IgG
To compare Ab responses following boost immunizations at local and distal sites, we immunized B6 mice with influenza hemagglutinin (HA) H1 SI-06 in the right footpad, and then boosted these animals 1-3 month(s) later with homologous HAs either in the right hock (ipsilateral boosts) or in the left hock (contralateral boosts). Eight days after boosting, we quantified HA-specific IgG Abs in sera by a Luminex multiplex bead assay, and enumerated PBs/PCs in the draining LNs by flow cytometry (Figure. 1A). Boost immunizations raised the concentrations of HA-specific serum IgG Abs by ~12-fold (p < 0.001; ipsilateral boosts, 14-fold; contralateral boosts, 10-fold; Figure. 1B–C), but there were no significant differences in H1 HA-specific serum IgGs between ipsilateral and contralateral boosts (p > 0.99; Figure. 1B–C).
Figure. 1. Boost immunizations at local and distal sites elicit comparable levels of recall serum IgG.
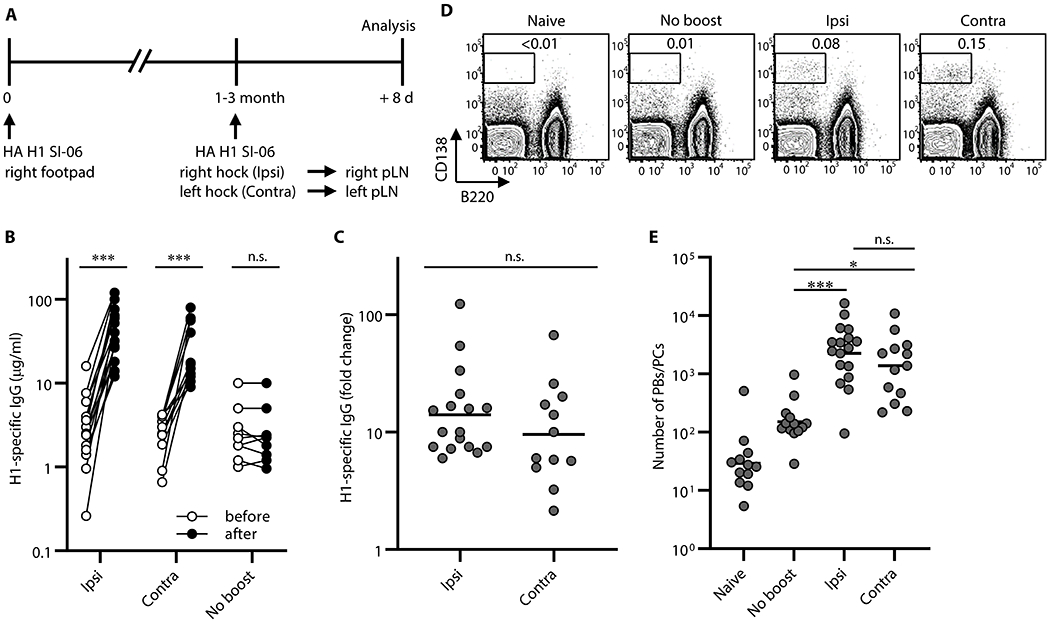
Plasmacytic responses following boosts with homologous antigens. (A) Graphic representation of the immunization strategy used in experiments in Figs. 1–4. (B) Concentrations of serum IgGs specific to H1 SI-06 before and after boosts ipsilaterally (n = 17) or contralaterally (n = 12). No-boost group (n = 9) received primary immunizations but not boosts. Each symbol represents an individual mouse, and serum samples from the same animals are connected with lines. ***, p < 0.001; n.s., p > 0.05 by Wilcoxon matched-pairs signed rank test. (C) Fold changes in concentrations of H1 HA-specific IgGs in serum samples in (B) after boosts. n.s., p > 0.05 by Mann-Whitney’s U test. (D and E) Representative flow diagrams (D) and number (E) of B220loCD138hi PBs/PCs in the draining LNs from naïve (n = 12), No-boost (n = 13), and boosted mice (ipsilateral boost, n = 17; contralateral boost, n = 13). Numbers near boxes in (D) represent frequency of B220loCD138hi cells among live lymphocyte population. ***, p < 0.001; *, p < 0.05; n.s., p > 0.05 by Kruskal-Walis test with Dunn’s multiple comparisons. Combined data from 9 independent experiments are shown.
Consistent with robust serum IgG responses, the number of B220loCD138hi PBs/PCs in the draining LNs were higher after boosting, ~15-fold (p = 0.001) and ~8-fold (p = 0.038) following ipsilateral and contralateral boosts, respectively than in no-boost controls (Figure. 1D–E). Between boost regimens there were no significant differences in the number of PBs/PCs in the draining LNs (p > 0.99; Figure. 1E). We sorted these samples into three groups by intervals between the priming and boosting (i.e., 4-5 week, 8-10 week, and 12-14 week intervals, respectively) and found across all intervals that ipsilateral boosts and contralateral boosts elicited comparably robust serum IgG responses and plasmacytic responses in the draining LNs (Figure. S1).
Secondary GCs following local boosts contain high avidity, antigen-specific B cells at elevated frequency
We characterized secondary GC responses in the draining LNs following ipsilateral or contralateral boosts by enumerating the number of B220+CD138−GL-7+CD38loIgD− GC B cells and by determining their BCR reactivity and avidity. Flow cytometric analysis showed that GC responses elicited by the primary immunization had greatly waned, but not completely, by the time of boosting (Figure. 2A–B). From the draining LNs of no-boost controls we recovered ~6 times as many GC phenotype B cells than from naïve controls. Although this difference was not statistically significant (p = 0.72), ~85% (11 out of 13) of LN samples from no-boost controls contained more than 1,000 GC B cells, while only one third (4 out of 12) of LN samples from naïve mice did (Figure. 2B).
Figure 2. Secondary GCs following local boosts contain a greater proportion of high avidity, antigen-specific B cells than do those following distal boosts.
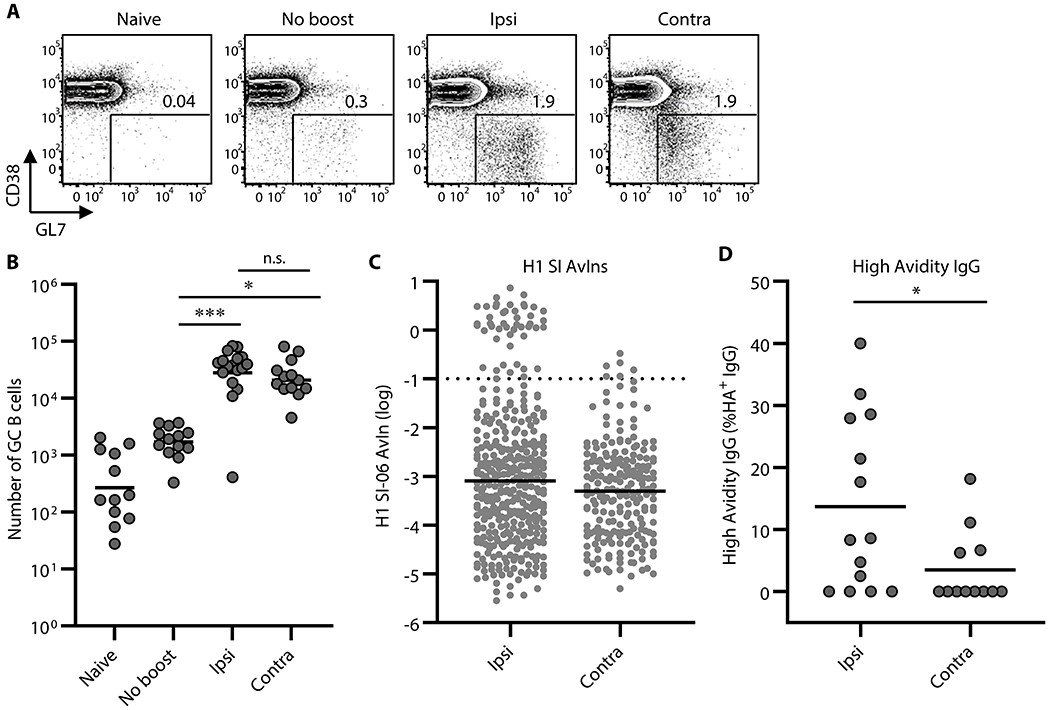
GC responses following boosts with homologous antigens. (A and B) Representative flow diagrams of GL-7 and CD38 expressions on B220+CD138− cells (A) and number of B220+CD138−GL-7+CD38loIgD− GC B cells in the draining LNs of naïve (n = 12), No-boost (n = 13), and boosted mice (ipsilateral boosts, n = 17; contralateral boosts, n = 13). Numbers near boxes in (A) represent frequency of GL-7+CD38loIgD− cells among B220hiCD138−/lo cells. Horizontal bars in (B) represent geometric mean. ***, p < 0.001; *, p < 0.05; n.s., p > 0.05 by Kruskal-Walis test with Dunn’s multiple comparisons. Combined data from 9 independent experiments are shown. (C) Distributions of avidity index (AvIn) values for H1 HA-specific IgGs from single-cell cultures of GC B cells are shown (n = 368 and 214 for ipsilateral and contralateral boosts, respectively). Each dot represents individual culture supernatant IgG. Horizontal bars represent geometric mean. Dotted line represents AvIn = 0.1, which we considered high avidity. Combined data from 6–8 independent experiments are shown. (D) Frequency of high avidity IgGs (AvIn > 0.1) among HA H1 SI-06 specific IgGs. Each dot represents individual mouse (n = 14, and 12 for ipsilateral and contralateral boosts, respectively). *, p < 0.05 by Mann-Whitney’s U test.
Both ipsilateral and contralateral boosts elicited strong secondary GC responses; numbers of GC B cells were ~17-fold (p < 0.001) and ~13-fold (p = 0.011) higher following ipsilateral and contralateral boosts, respectively, than in no-boost controls (Figure. 2A–B). Ipsilateral and contralateral boosts elicited secondary GC responses of comparable magnitudes (p > 0.99; Figure. 2A–B). This relationship held when we boosted mice at 4-5 weeks, 8-10 weeks or 12-14 weeks after priming (Figure. S2A–C).
To determine the specificity and affinity of B cells in secondary GCs, we sorted individual GC B cells from the draining LNs of boosted animals into single-cell Nojima cultures (35). After culture, we screened clonal IgG Abs in culture supernatants by a Luminex multiplex assay for binding to HA H1 SI-06 (Figure. 2C). We obtained 5,113 clonal IgG examples from secondary GC B cells following ipsilateral (n = 2,255) or contralateral boosts (n = 2,858) (Table S1). In contrast to the similar magnitudes of GC responses induced by ipsilateral or contralateral boosting, the frequency of antigen-specific clonal IgGs from secondary GC B cells elicited by ipsilateral boosts was significantly (p = 0.0011) higher [16% (±5.9%)] than that of GC B cells following contralateral boosts ([7.7% (±4.7%)]; Figure. S2D, Table S1).
To compare the distribution of BCR affinities for HA H1 SI-06, we determined the avidity index (AvIn) for each clonal IgG Ab (35). The median AvIn for the GC B cells following ipsilateral boosts was 8 × 10−4, which was moderately but significantly different from median AvIn value for their contralateral boost counterparts (median AvIn = 5 × 10−4; p = 0.019; Figure. 2C). In addition, ipsilateral secondary GCs contained more high avidity (AvIn > 0.1) B cells. High avidity B cells constituted 14% (±14%) of HA-specific GC B cells elicited by ipsilateral boosts, about 4-fold above that of contralateral boost GC B cells (p = 0.035; Figure. 2D). Secondary GCs after local boosts were similar in size to those induced at distal sites but contained more high affinity B cell clones.
Secondary GCs following local boosts contain highly mutated B cells at elevated frequency
To examine somatic hypermutation in secondary GC B cells, we determined VDJ gene sequences for subsets of Nojima culture samples (ipsilateral boosts, n = 837; contralateral boosts, n = 375). We amplified VDJ rearrangements from cDNA prepared and sequenced from the cell pellets of individual IgG+ cultures (35). Secondary GC B cells following ipsilateral boosts and contralateral boosts carried on average 2.7 and 2.0 mutations, respectively, in the heavy chain variable region (VH) (Figure. 3A), corresponding to VH mutation frequencies (number of nucleotide substitutions per base pairs sequenced) of 1.0% and 0.7% (p = 0.73; Figure. 3A). These mutation frequencies were significantly higher than those in day 8 primary GC B cells (p < 0.001, average 0.5%, Figure. 3A and (35)). Despite similar VH mutation frequencies, following ipsilateral boosts secondary GC B cells had a broader range of VH mutations (0-29) than their contralateral-boost counterparts (0-14, Figure. 3A). 7.3% of secondary GC B cells following ipsilateral boosts carried 8 or more VH mutations, compared to <1% seen in their contralateral-boost counterparts (p < 0.001, Figure. 3B).
Figure 3. Somatic hypermutation in secondary GC B cells.
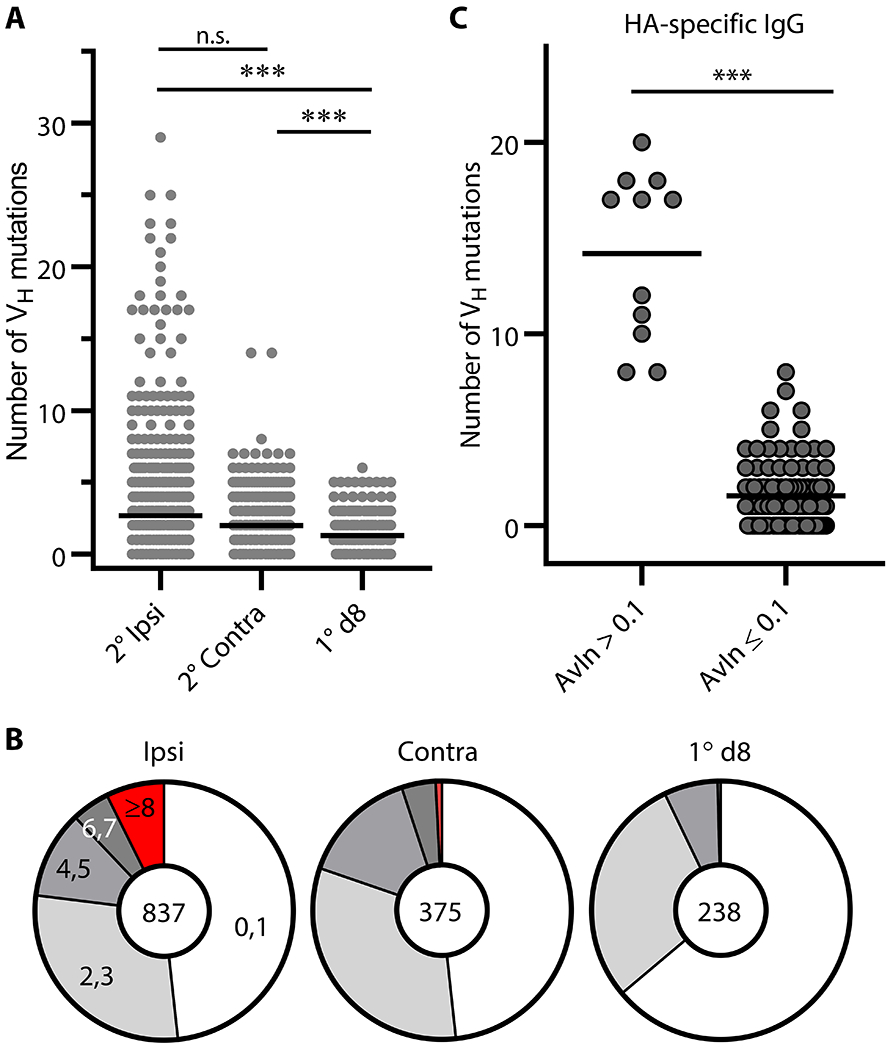
(A) Distribution of the number of VH point mutations recovered from secondary (2°) GC B cells following ipsilateral boosts (n = 837) and distal boosts (n = 375). Data for primary (1°) d8 GC B cells (n = 238) are re-plotted from published work of our own (35). Each dot represents an individual IgG+ sample. Horizontal bars represent mean. ***, p < 0.001; n.s., p > 0.05 by Kruskal-Walis test with Dunn’s multiple comparisons. (B) Pie charts depict proportion of IgG examples that carry indicated number of VH point mutations. (C) VH gene sequences for HA H1 SI-06 reactive IgGs were split by their AvIn values into high avidity (AvIn > 0.1; n = 11) and the rest (AvIn ≤ 0.1; n = 102). ***, p < 0.001 by Mann-Whitney’s U test.
High avidity (AvIn > 0.1), HA-reactive IgG Abs from secondary GC B cells carried increased numbers of VH mutations (range 8-20) (Figure. 3C), indicating that these cells were the progeny of affinity-matured, primary GC B cells rather than from newly-activated, naïve B cells (21). The elevated frequencies of B cells with higher number of VH mutations in secondary GCs following ipsilateral boosts suggest the efficient engagement of the progeny of primary GC B cells at local sites.
Local boosts more efficiently recruit the progeny of the primary GC B cells to secondary GCs than distal boosts
Secondary GCs contain both the re-activated progeny of primary GC B cells (e.g., persistent GC B and recalled Bmem cells), and newly-activated naïve B cells. Although the latter B cell type dominates the response, at least in mice (21), elevated frequencies of higher affinity B cells and of highly-mutated B cells in secondary GCs following ipsilateral boosts (Figures. 2 and 3), suggest that persistent GC B and Bmem cells contribute most efficiently to secondary GCs elicited by local boosting. To directly compare the efficiency of ipsilateral and contralateral boosts in activating/recruiting the progeny of primary GC B cells into secondary GC responses, we used the AID-Cre-EYFP mouse model (9, 21). Here, we labeled GC B cells during primary responses and traced the fates of the labeled B cells following boost immunizations. We immunized AID-Cre-EYFP mice in the footpad with HA H1 SI-06 and subsequently induced YFP expression in AID-expressing GC B cells by i.p. injections of tamoxifen (days 8-12). Beginning tamoxifen injections at day 8 after priming mitigates any labeling of AID+ early activated precursors that may generate GC-independent Bmem cells (36, 37) as most have exited cell cycle and become quiescent by this time (37). Eight- to ten weeks after primary immunization, we boosted these animals in the ipsilateral or contralateral hock with homologous HA and enumerated YFP+ cells in PBs/PCs and GC B cell compartments in the draining LNs eight days later.
Tamoxifen injections can induce YFP expression not only in AID-expressing cells that are elicited by primary immunization but also in those irrelevant to the priming event (e.g., Peyer’s patch GC B cells constitutively active in the gut). To ensure that YFP+ cells that engage in secondary responses originated from cells specifically generated by the priming, we compared frequencies of YFP+ cells following “boost” immunizations with or without prior immunization. In the absence of priming, “boost” immunizations following tamoxifen injections recruited little or no YFP+ cells into PBs/PCs and GC B cell compartments, while YFP+ cells were readily detectable in these B cell compartments in mice that had been primed previously (Figure. S3A). We conclude that most, if not all, YFP+ PBs/PCs and GC B cells in draining LNs following boost immunizations are the progeny of primary GC B cells elicited by earlier footpad immunization.
Both boost regimens elicited nearly identical frequencies and numbers of YFP+ PBs/PCs in draining LNs (Figure. S3), consistent with comparable levels of recall serum IgG responses following ipsilateral and contralateral boosts (Figure. 1). Frequencies of YFP+ cells among PBs/PCs were 19% (±12%) and 18% (±9.6%) following ipsilateral and contralateral boosts, respectively (p > 0.99; Figure. S3B–C). Similarly, there were no significant differences in the number of YFP+ PBs/PCs in the draining LNs following ipsilateral and contralateral boosts (2,600 ±2,900 and 2,000 ±2,700, respectively; p > 0.99; Figure. S3D).
The levels of YFP+ GC B cells in the draining LNs following ipsilateral boosts were higher than those following contralateral boosts (Figure. 4A–C), in contrast to plasmacytic responses but consistent with over-representation of both high affinity B cells and highly-mutated B cells in secondary GCs (Figures. 2 and 3). The frequency and number of YFP+ GC B cells after ipsilateral boosts were significantly higher than after contralateral boosts (Figure. 4A–C). We obtained similar results with S1pr2-ERT2cre-tdTomato mice, in which labeling efficiency of primary GC B cells is higher than that of AID-Cre-EYFP mice (85% vs. 25%; Figure. S4A; (9, 27, 38)). With S1pr2-ERT2cre-tdTomato mice, the frequency and number of labeled, secondary GC B cells was significantly higher in ipsilateral boosts (Figure. S4B–C). Therefore, local boosts activate and recruit the progeny of primary GC B cells into secondary GC responses more efficiently than do distal boosts.
Figure 4. Progeny of the primary GC B cells engage in secondary GCs more efficiently after local boosts than after distal boosts.
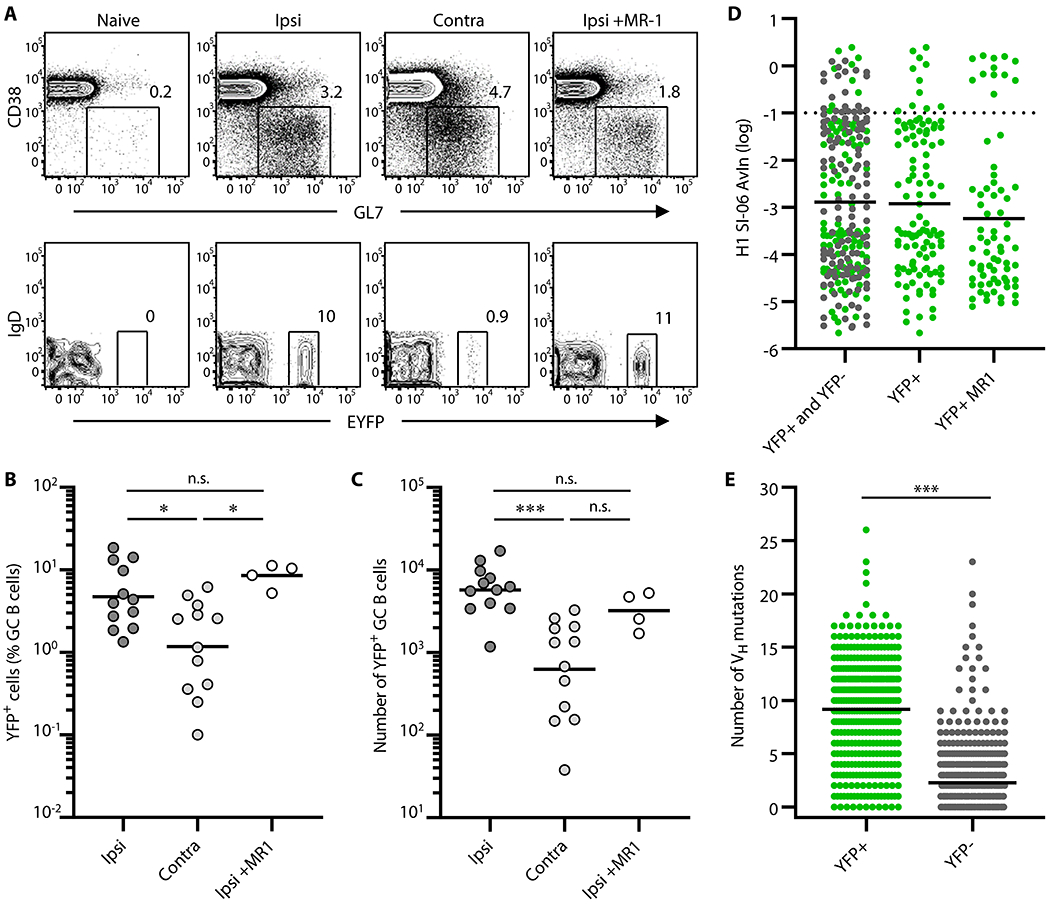
AID-Cre-EYFP mice were primed and boosted with H1 SI-06 as shown in Figure. 1A. Primed animals were given tamoxifen i.p. daily through days 8–12 after priming. In some experiments, primed animals received MR-1 Abs or control IgGs 4 weeks after the priming. Representative flow diagrams (A), and frequency (B) and number (C) of YFP+ GC B cells in the draining LNs are shown. (B and C) Each dot represents an individual mouse. Horizontal bars represent geometric mean. ***, p < 0.001; *, p < 0.05; n.s., p > 0.05 by Kruskal-Walis test with Dunn’s multiple comparisons. (D) Distributions of AvIn values for clonal IgGs from GC B cells following ipsilateral boosts are shown (left, YFP+ and YFP− combined, n = 227; middle, YFP+, n = 109; right, YFP+ receiving MR-1, n = 71). Horizontal bars represent geometric mean. Dotted line represents AvIn = 0.1, our cutoff for “high avidity”. Combined data from 4 independent experiments are shown. No statistical significance was seen among groups by Kruskal-Walis test with Dunn’s multiple comparisons. (E) Distributions of the number of VH point mutations in YFP+ (n = 399) and YFP− (n = 618) secondary GC B cells following ipsilateral boosts. Each dot represents an individual IgG+ sample. Horizontal bars represent mean. ***, p < 0.001 by Mann-Whitney’s U test.
Late disruption of primary GCs does not impair recruitment of their progeny to local boost GCs
Although GC responses are generally thought to be transient, persistent GCs following immunization have been reported (9, 12, 39, 40). Indeed, we observed small but detectable levels of GC B cells by flow cytometry for ~2-3 months after primary immunizations (Figures. 2A–B and Figures. S2A–C). Therefore, in all preceding experiments, ipsilateral boosts may have (re)activated residual, primary GC B cells in addition to Bmem cells. To determine whether disruption of ongoing primary GC responses would reduce the engagement of YFP+ cells in secondary GCs, we injected a cohort of H1-primed AID-Cre-EYFP mice with CD154-specific MR-1 Ab or control hamster IgG 4 weeks after the priming (41–43). These mice were boosted with H1 SI-06 4-6 weeks later (i.e., 8-10 weeks after priming). Eight days after boosts, we enumerated the frequency and number of YFP+ GC B cells.
Flow cytometric analysis performed at 5 weeks after MR-1 Ab injections (i.e., 9 weeks after priming, without boost) showed the number of YFP+ GC B cells was reduced by ~70% compared to mice given control IgG (580 vs. 1,900; Figure. S5A). Consistent with flow cytometric analysis, we observed clusters of YFP+IgD− cells in association with the CD21/CD35bright follicular dendritic cell (FDC) network in the draining LNs of control mice (Figure. S5B). In the MR-1 treated mouse LNs, FDC areas were filled with IgD+ B cells and only rarely were YFP+IgD− cells detected (Figure. S5C), confirming that MR-1 administration effectively disrupted the primary GC response. Despite absence of organized GC structures in MR-1-treated mice, the majority (≥70%) of YFP+B220+CD138− cells were GL-7+CD38lo, a surface phenotype indistinguishable from that of GC B cells (Figure. S5D). In contrast, in the contralateral LNs, virtually all (99% ±2.5%) YFP+B220+CD138− cells had a GL-7−CD38+ classical Bmem phenotype (Figure. S5D). YFP+IgD− cells present within GC-like structures were CD38− (Figure. S5E–F) while those that were present within B cell follicles contained both CD38− and CD38+ subsets (Figure. S5G–J).
Following ipsilateral boosts, the frequency and number of YFP+ GC B cells were no different between mice that had received control IgGs and MR-1 Abs (Figure. 4A–C). Therefore, disruption of primary GC responses by CD154 blockade did not impair local participation of YFP+ cells in secondary GCs. These imply that the majority of YFP+ B cells in secondary GCs after ipsilateral boosting are generated by the primary GC response but become resistant to CD40:CD154 signaling blockade.
Progeny of the primary GC B cells with a broad range of BCR affinities and of VH SHM engage in secondary GCs
To examine the BCR repertoire of YFP+ secondary GC B cells following ipsilateral boosts, we FACS sorted GC B cells into single-cell Nojima cultures, and determined binding specificity and avidity of the clonal IgG Abs by Luminex™ assay. Of the 514 clonal IgG Abs we recovered from YFP+ secondary GC B cells, 109 (21%) bound to HA H1 SI-06 (Table S1). These HA-reactive clonal IgG Abs had a broad range of BCR affinities for the HA immunogen. This avidity distribution was no different from that of overall secondary GC B cells (compare Figure. 2C, Figure. 4D left column, and Figure. 4D middle column). Suppression of primary GC responses by MR-1 treatment had no significant effect on BCR avidity distributions among secondary YFP+ GC B cells (Figure. 4D right column).
YFP+ secondary GC B cells had a broad range of VH SHM (0-26; Figure. 4E), like all secondary GC B cells (Figure. 3A). Consistent with a primary GC B cell origin, YFP+ secondary GC B cells had higher VH SHM (median number = 10; Figure. 4E) than did primary newly activated, d8 GC B cells (median = 1; Figure. 3A). In contrast to YFP+ cells, YFP− secondary GC B cells included both recently-activated naïve B cells and progeny of primary GC B cells that were not labeled by tamoxifen ((9, 38) and Figure. S4). Highly-mutated clones (VH mutations ≥8) constituted ~5% (31/618) of the YFP− secondary GC B cells (Figure. 4E), unlike primary d8 GC B cells (Figure. 3A). These uncommon, highly-mutated clones are likely the unlabeled progeny of primary GC B cells. In contrast, the comparable frequencies of overall VH SHM among all YFP− secondary GC B cells and primary d8 GC B cells (median = 1, Figures. 3A and 4E) indicate that most YFP− secondary GC B cells originated from recently-activated, naïve B cells. Both HA-binding and non-binding YFP+ secondary GC B cells had higher VH SHM than did their respective YFP− counterparts (Figure. S6A–B). Although high avidity (AvIn > 0.1), HA-binding YFP+ cells had elevated VH SHM (range: 9-15), neither the number of VH mutations nor of amino acid substitutions was correlated with AvIn values (Figure. S6C–D). Thus, progeny of primary GC B cells with a broad range of BCR affinities and VH SHM, including high affinity clones and highly-mutated clones, are reactivated to secondary GC responses by ipsilateral boosts with homologous antigen.
Recall of H1-specific serum IgG following local boosts with H3 HAs in H1-primed mice
To determine whether locality had any impact on recall humoral responses following boosts with heterologous antigens, we immunized B6 mice with HA H1 SI-06 in the right footpad, and then boosted these animals 8-12 weeks later in the hock ipsilaterally or contralaterally with a heterologous HA (HA H3 X31) or an irrelevant antigen (recombinant protective antigen of Bacillus anthracis, rPA). Eight days after boosting, we collected serum samples for quantifying HA-specific IgG Ab and draining LNs for characterizing GC B cells (Figure. 5A).
Figure 5. Recall of H1 HA-binding IgG Abs following local boosts with H3 HAs in H1-primed mice.
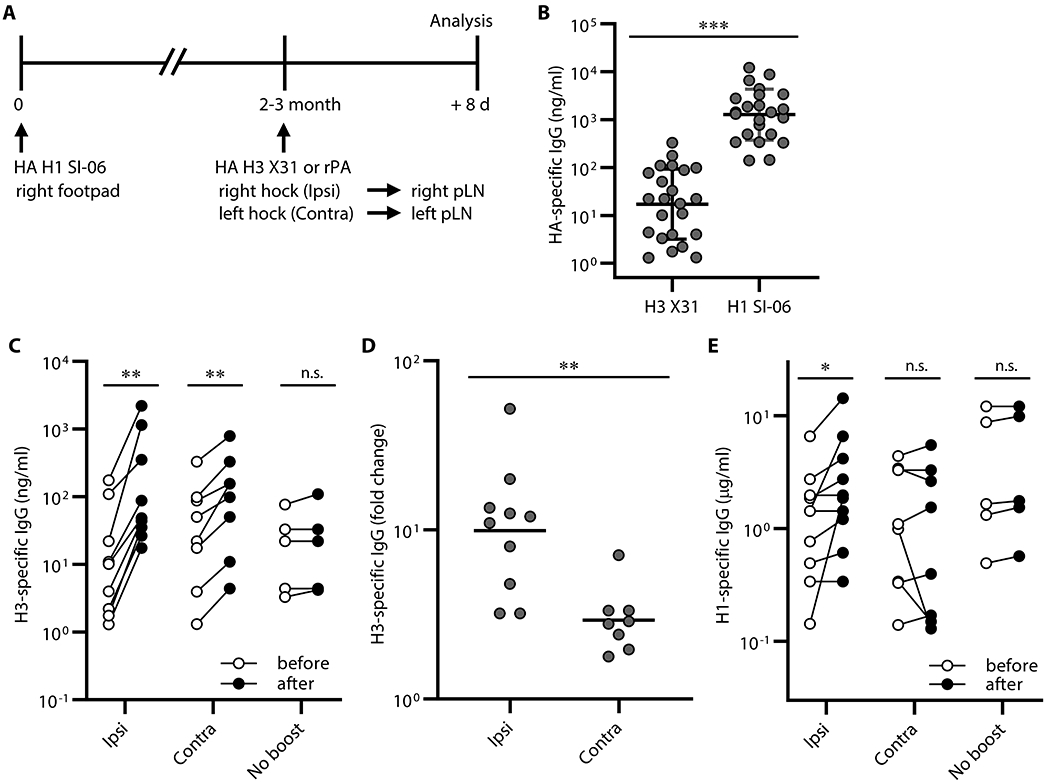
Serum IgG responses following boosts with heterologous antigens. (A) Graphic representation of the immunization strategy used in experiments described in Figures. 5 and 6. (B) Concentrations of serum IgGs binding to HA H1 SI-06 and HA H3 X31 before boosts (n = 23). ***, p < 0.001 by Mann-Whitney’s U test. (C-D) Mice were randomly selected for ipsilateral (n = 10) or contralateral (n = 8) or no boosts (n = 5). (C) Concentrations of serum IgGs specific to HA H3 X31 before and after boosts ipsilaterally or contralaterally. No-boost group received primary immunizations but not boosts. **, p < 0.01; n.s., p > 0.05 by Wilcoxon matched-pairs signed rank test. (D) Fold changes in concentrations of H3 HA-specific IgGs in serum samples in (C) after boosts. **, p < 0.01; by Mann-Whitney’s U test. (E) Concentrations of serum IgGs specific to HA H1 SI-06 in serum samples in (C). Each symbol represents an individual mouse (B-E), and serum samples from the same animals are connected with lines (C and E). *, p < 0.05; n.s., p > 0.05 by Wilcoxon matched-pairs signed rank test. Combined data from 4 independent experiments are shown.
Before boosting, sera from H1-primed animals contained H3-binding serum IgG Ab at 17 ng/ml (geometric mean), ~70-fold lower than the concentration of H1-binding IgG Ab levels (1,300 ng/ml; Figure. 5B). Following boosts, concentrations of H3-specific serum IgG Ab increased by 10-fold (from 13 ng/ml to 130 ng/ml, ipsilateral boosts) and 3-fold (from 27 ng/ml to 79 ng/ml, contralateral boosts; Figure. 5C–5D). In contrast to H3-binding IgG responses, ipsilateral, but not contralateral, boosts with H3 HA increased H1-specific serum IgG Ab levels 2-fold (from 1.1 μg/ml to 2.1 μg/ml, p < 0.05; Figure. 5D), implying that ipsilateral boosts with HA H3 X31 re-activated H1/H3 cross-reactive cells generated by priming with HA H1 SI-06. Contralateral boosts with H3 HA had no significant effect on H1 HA serum Ab (from 0.98 μg/ml to 0.75 μg/ml, p < 0.99; Figure. 5D).
More H1/H3 cross-reactive secondary GC B cells after local boosts with H3 HA in H1-primed mice
We determined the number and frequency of secondary GC B cells by flow cytometry. Boosting with heterologous HAs elicited robust GC responses in the draining LNs after both ipsilateral and contralateral boosts (Figure. 6A–B), similar to secondary GC responses to homologous HA boosts (Figure. 2A). To determine the reactivity profile of secondary GC B cells after boosting with heterologous HA antigens, we isolated GC B cells from the draining LNs of boosted animals and established single-cell Nojima cultures (35). On average, antigen-specific B cells (i.e., clonal IgGs that bound HAs of H1 SI-06, H3 X31 or both) accounted for 16% (±9.4%) and 18% (±8.2%) of clonal IgGs recovered from secondary GC B cells following ipsilateral and contralateral boosts, respectively (Figure. S7A). For both boost regimens, most HA reactive B cells bound the boosting antigen, HA H3 X31 (90% and 97% for ipsilateral and contralateral boosts, respectively) (Figure. 6C). Despite these similarities, the distribution of HA reactivity of secondary GC B cells was different between boost regimens. The frequency of H1/H3 cross-reactive cells among HA-specific secondary GC B cells was significantly higher after ipsilateral boosts than after contralateral boosts (Figure. 6C–D). On average, H1/H3 cross-reactive B cells constituted 12% (±11%) of HA-reactive GC B cells after ipsilateral boosts, but only 0.9% (±1.5%) following contralateral boosts. We recovered H1/H3 cross-reactive B cells from 8 of 10 mice given an ipsilateral boost, but only 2 of 6 mice after a contralateral boost (Figure. 6D).
Figure 6. Participation of H1/H3 cross-reactive B cells in secondary GCs following local boosts with H3 HAs in H1 HA-primed mice.
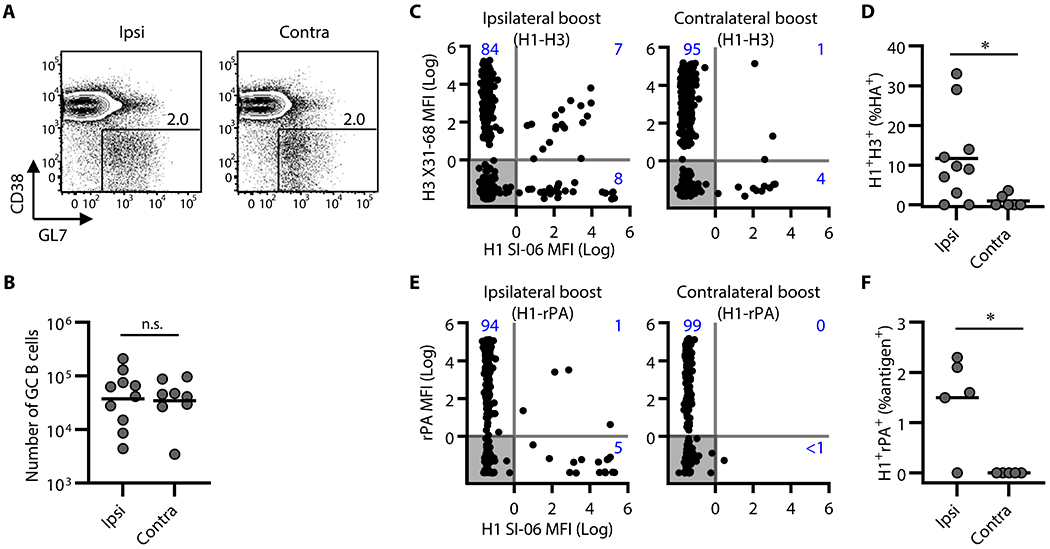
GC responses following boosts with heterologous antigens. (A and B) Representative flow diagrams of GL-7 and CD38 expression on B220+CD138− cells (A) and number of B220+CD138−GL-7+CD38loIgD− GC B cells in the draining LNs of boosted mice (ipsilateral boosts, n = 10; contralateral boosts, n = 8). Numbers near boxes in (A) represent frequency of GL-7+CD38loIgD− cells among B220hiCD138−/lo cells. Horizontal bars in (B) represent geometric mean. n.s., p > 0.05 by two-tailed Student’s t test. Combined data from 4 independent experiments are shown. (C-F) Individual GC B cells were placed in Nojima cultures for determination of BCR reactivity. H3 X31 ipsilateral boosts (n = 1,825), H3 X31 contralateral boosts (n = 1,696), rPA ipsilateral boosts (n = 1,130), and rPA contralateral boosts (n = 1,082). (C and E) Background-subtracted, normalized MFI values (Log10) for H1 SI-06 (x-axes) and H3 X31 (C, y-axes) or rPA (E, y-axes). Numbers in each quadrant represent frequencies of H1 or H3 or H1/H3 (C) reactive IgGs and H1 or rPA or H1/rPA reactive IgGs among antigen-specific clonal IgGs. Each dot represents an individual clonal IgG. (D and F) Frequencies of H1/H3 cross-reactive IgGs (D) and of H1/rPA cross-reactive IgGs (F) among all antigen-specific IgG. Each dot represents an individual mouse. *, p < 0.05 by Mann-Whitney’s U test. Combined data from 2 (rPA boosts) and 4 (H3 X31 boosts) independent experiments are shown.
Selection for cross-reactive, secondary GC B cells was much lower when we boosted H1 SI-06 primed mice with an unrelated protein antigen, rPA. Antigen-specific B cells (i.e., clonal IgGs that bound H1 SI-06, rPA or both) accounted for 28% (±10%) and 18% (±3.3%) of clonal IgGs recovered from secondary GC B cells following ipsilateral boosts and contralateral boosts, respectively (Figure. S7B). Ipsilateral boosts with rPA yielded rare H1/rPA cross-reactive GC B cells, representing 1% of antigen-specific B cells (Figure. 6E–F); contralateral boosts with rPA produced no H1/rPA binders (Figure. 6E–F). Structural similarity of epitopes between the priming antigens and boosting antigens is a key determinant for cross-reactive B cells to engage in secondary GC responses.
DISCUSSION
In this study, we have shown an important role for locality in participation of Bmem cells in recall GC responses. Using a combined approach of single B cell cultures with a fate-mapping mouse model, we found that the progeny of primary GC B cells with a broad range of BCR affinities and VH mutations, including high affinity cells and highly-mutated cells, participated in secondary GC responses significantly more efficiently after local boosts compared to boosts at distal sites (Figures. 2–4). In response to heterologous HA antigens, local boosts recruited to secondary GCs cross-reactive B cells that bound both the prime and boost HAs more efficiently than did distal boosts (Figure. 6). Advanced engagement of HA cross-reactive B cells in secondary GCs was accompanied by an increase of serum IgGs that bound the priming HAs along with those that bound boosting HAs (Figure. 5).
The factors which contribute to an efficient engagement of the progeny of primary GC B cells in secondary GCs following ipsilateral boosts are not well defined. As immunizations substantially change local microenvironments in the draining LNs, numerous factors (e.g., local antigen retention, focused cytokine and chemokine milieu, local populations of T follicular helper cells (44, 45) and other cell types (46)) might cooperatively contribute to the locality. Antigens retained on FDCs (47) have significant roles in retaining “primed B cells” within the draining LNs without further differentiation into PBs/PCs and thereby contribute to long-lasting, local B cell memory (32–34). We propose that local B cell memory is retained in the primed LNs in the form of Bmem cells that may include both GC-dependent and GC-independent subsets, persistent GC B cells, and GC-phenotype B cells that are independent of organized GC structures (Figure. S5) and that these persistent “primed B cells” contribute to recall GC responses at local sites. For example, “re-fueling” of persistent GCs with cognate antigens (9, 12, 39, 40) (Figure. 2, Figures. S2 and S5) may expand the persistent GC B cells following ipsilateral boosts (21). In addition to this re-fueling model, our observations show significant contributions of the MR-1 resistant B cells in secondary GC responses following local boosts, as the disruption of active GC structures by the MR-1 Ab treatment did not impair participation of the fate-mapped, high affinity B cells in secondary GCs (Figure. 4 and Figure. S5). The majority (≥70%) of the MR-1 resistant, fate-mapped cells found in the primed LNs had a surface phenotype (GL-7+CD38lo) shared by active GC B cells despite the absence of organized GC structures (Figure. S5).
Our observation that secondary GCs elicited by ipsilateral boosts recruit the progeny of primary GC B cells more efficiently than contralateral boosts provides an explanation for an apparent discrepancy between recent observations concerning Bmem recruitment to recall GCs. Mesin et al. (21) have suggested that recall of Bmem cells may be an inefficient process based on observations in fate-mapping mice. They find that fate-mapped cells (i.e., progeny of the primary GC B cells) are less frequent in secondary GCs following contralateral boosts. In contrast, Turner et al. (13) have analyzed the fine needle aspirates of the LN samples from recent influenza vaccinees and shown that a substantial fraction of antigen-binding GC B cells have elevated Ig SHM and that they are broadly cross-reactive, consistent with Bmem origin. Although speculative, it is possible that the human donors studied by Turner et al. had established local B cell memory from previous vaccinations and that ipsilateral boosts with an influenza vaccine activate local B cell memory populations (described above), resulting in substantial engagement of Bmem cells in recall GCs.
Recall GCs following local boosts contain B cells of heterogeneous origin, including recently-activated, naïve B cells and recalled “primed B cells” – a category that comprises both GC-dependent and GC-independent Bmem cells, persistent GC B cells and MR-1 resistant, GC-phenotype B cells (Figure. S5). The question remains at to what would be the outcome of recall GC responses, that is, which population(s) wins the Darwinian selection that would be operated in recall GCs? While activated, naïve B cells that are overrepresented in early recall GCs (21, 22) (Figure. 4) may eventually outnumber the rarer, recalled “primed B cells” over the course of the GC reactions, a relative population size in early GCs does not necessarily predict an outcome of the B cell selection. For example, a subset of recalled Bmem cells may have a “head start” in selection fitness and outcompete the naïve counterparts as they express high avidity BCRs that are infrequent in the naïve counterparts (Figures. 2–4). It is also possible that secondary GCs, like primary GCs, are permissive environments and overall BCR affinity maturation could operate without losing BCR diversity (35). In this model, both the rarer recalled “primed B cells” and more frequent, activated naïve B cells could co-exist throughout the recall GC responses and produce their respective progenies.
HA-binding IgGs accounted for only ~20% of all clonal IgGs recovered from secondary GC B cells (Table S1). This frequency did not change when we limited our analysis to fate-mapped, YFP+ cells (Table S1). In other words, most (80%) of the progeny of primary GC B cells participating in secondary GC responses express BCRs with unmeasurable affinity to the eliciting immunogen. One possible explanation would be that these HA non-binding B cells lost affinity to the boost HA by new rounds of SHM during secondary GC responses. Indeed, without strong selection, any stochastic mutational process has a much greater likelihood of lowering BCR affinity than of increasing it. It is also possible that these HA non-binding YFP+ cells represent progeny of the antigen non-binding B cells found among primary GC B cell populations (35, 48) and/or among the contemporaneously generated Bmem pool (22). Recovery of HA non-binding, YFP+ secondary GC B cells that carried no IgH SHM or only silent (no amino acid change) IgH SHM (Figure. S6) strongly suggests that they were activated by both the primary and secondary immunizations without measurable BCR affinities (as IgG) and yet participated in secondary GC responses. Although the nature of these antigen non-binding secondary GC B cells still needs to be elucidated, secondary GCs, like primary GCs, are permissive environments, at least for entry and the early phase of the reaction, and support B cell clones with a broad range of BCR affinities, including those with undetectable affinities for the native form of the immunogen by standard binding assays.
H1/H3 cross-reactive B cells participated in secondary GCs following ipsilateral boosts with H3 HAs, and to a lesser extent, following contralateral boosts in H1-primed mice (Figure. 6). This advanced engagement of H1/H3 cross-reactive B cells in local, secondary GCs accompanied an increase, following ipsilateral boosts, in binding of serum IgG Abs to H1 HAs as well as of those binding to H3 HAs (Figure. 5). Our observations suggest that local boosts with H3 HAs effectively activate H1/H3 cross-reactive B cells that remain within the persistent pool of “primed B cells” we described above. Participation of H1/H3 cross-reactive B cells in secondary GCs would give them an opportunity to update their BCRs to fit with newly-introduced antigens (i.e., H3 HA). A further question is how these cross-reactive B cells compete with other B cells (e.g., H1-specific or H3-specific) and how they co-evolve in recall GCs.
This study has several limitations. We used a fate-mapping mouse model and showed that progeny of the primary GC B cells enter secondary GCs more efficiently after local rather than distal boosts. As AID expression is not restricted to GC B cells, we cannot formally exclude the possibility that we labeled GC-independent Bmem cells descended from early “activated precursors” (36, 37) although we started tamoxifen injections when a majority of these cells had already become quiescent (37). We also note that we likely underestimated frequency of Bmem cells that participated in secondary GCs. First, AID-Cre reporter system used in this study is known to be inefficient (9, 38). Indeed, we labeled only ~25% of primary GC B cells. Second, we were unable to label B cells that left GCs and downregulated AID expressions prior to tamoxifen or B cells that were recruited to GCs after tamoxifen lost its activity. Other limitations include incapability to assess 1) precursor/progeny relationships between Bmem cells and secondary GC B cells and 2) clonal relationships between secondary GC B cells in the ipsilateral and contralateral LNs. Further experiments that involve different fate-mapping strategies, serial samplings of LN biopsies, and bilateral boost immunizations in the same animals will be required to address these questions.
In summary, our observations suggest that re-activation of local B cell memory populations by repeated immunizations at the same sites might be required to ensure efficient participation of “primed B cells” in recall GC responses in which they undergo new rounds of SHM, clonal selection and affinity maturation to newly-introduced antigens. This participation is necessary for all serial vaccine strategies against rapidly-mutating viruses (e.g., SARS-CoV-2, influenza, HIV-1), for which updating BCRs is essential for durable protection, including conventional prime and boost as well as so-called B cell lineage design vaccine strategies (49).
MATERIALS AND METHODS
Study Design
The purpose of this study was to determine roles of relative locations of the prime and boost on the recall of Bmem cells, with specific focus on the participation of Bmem cells in secondary GC responses. We used a combined approach of single B cell cultures with a fate-mapping mouse model and determined the frequency and number of fate-mapped cells as well as BCR reactivity (specificity and avidity) of >12,000 clonal IgG Abs produced by the progeny of single, GC B cells. We compared secondary GC responses to boost immunizations given in the same (ipsilateral) or opposite (contralateral) leg of primed mice.
Mice and Immunizations
Female, C57BL/6 mice were obtained from the Jackson Laboratory. AicdaCre-ERT2 x Rosa26loxP-EYFP (AID-Cre-EYFP) mice (9, 38) and S1pr2-ERT2cre-tdTomato mice (27) were provided by Drs. Claude-Agnes Reynaud and Jean-Claud Weill and by Dr. Tomohiro Kurosaki, respectively. All mice were maintained under specific pathogen-free conditions at the Duke University Animal Care Facility. Eight to 12-week-old female mice were immunized with 20 μg of HA H1 SI-06 (see below) in the footpad of the right hind leg. One to three months later, cohorts of mice were boosted with 20 μg of HA SI-06 or HA H3 X31 or rPA (protective antigen from Bacillus Anthracis, BEI Resources) in the hock ipsilaterally (right hind leg) or contralaterally (left hind leg). Mice were analyzed 8 days after the boosts. All antigens were mixed with Alhydrogel® adjuvant 2% (final 1%) before immunizations. In some experiments, we injected total 900 μg of MR-1 Abs or control hamster IgGs (300 μg daily for three consecutive days) i.v. 4 weeks after primary immunizations to disrupt ongoing GC reactions. LNs draining the site of the most recent immunizations (right popliteal LNs for no boosts and ipsilateral boosts, and left popliteal LNs for contralateral boosts) were analyzed. Sera were collected before boosts (one day before or same day as boosts) and after boosts (8 days after boosts). All experiments involving animals were approved by the Duke University Institutional Animal Care and Use Committee.
Expression and purification of HAs
Hemagglutinin (HA) proteins for the full-length, soluble ectodomains (FLsE) of A/Solomon Islands/03/2006 (H1 SI-06) and A/Aichi/2/1968 (H3 X31) were cloned, expressed, and purified as previously described (35, 50–54). Briefly, codon-optimized cDNA of the recombinant FLsEs containing N-terminal gp67 secretion signal and C-terminal T4 fibritin (foldon) trimerization and a 6xHis tag was subcloned into a pFastBac vector between NheI and KasI restriction sites using standard restriction endonuclease cloning protocols. Recombinant baculoviruses were generated by transfection of Spodoptera frugiperda (Sf9) cells and sequential passage of the viral stock to generate “high titer P3” viral stocks. Trichoplusia ni (Hi5) cells were infected with the recombinant baculoviruses, and at 72 h post-infection, the supernatants were harvested and clarified by centrifugation at 3,000xg. The HAs were purified using Co2+-NTA metal affinity resin (Takara). The bound protein was washed with 20 column volumes of TBS (10 mM Tris-HCL and 150 mM NaCl, pH 7.5) before elution with TBS supplemented with 500 mM imidazole. The eluted protein was concentrated and further purified on a Superdex S200 size-exclusion column. The eluted protein was then incubated overnight with PreScission protease (Thermo Fisher Scientific) at a 1:100 molar mass ratio to remove the foldon and 6xHis purification tags. HAs was further purified by orthogonal Co2+-NTA agarose chromatography to remove the tags and the un-cleaved protein. HAs was not activated by the addition of trypsin, and the resulting protein was uncleaved HA0. The purified protein was concentrated to >2 mg/mL and stored at 4 °C until use.
Flow Cytometry
GC B cells (GL-7+B220hiCD38loIgD−CD138−) and plasmablasts/-cytes (B220loCD138hi) in the draining LNs were identified as described (35, 55, 56). Labeled cells were analyzed/sorted in a FACS Canto (BD Bioscience) or a FACS LSRII (BD Biosciences) or a FACS Vantage with DIVA option (BD Bioscience). Flow cytometric data were analyzed with FlowJo software (Treestar Inc.). Doublets were excluded by FSC-A/FSC-H gating strategy. Cells that take up propidium iodide were excluded from our analyses. mAbs used in this study: FITC- or PE-conjugated anti-GL-7 (GL7, BD Bioscience or BioLegend, respectively), BV421-conjugated anti-B220 (RA3-6B2, Biolegend), PE-Cy7-conjugated anti-CD38 (90, BioLegend), BV510-conjugated anti-IgD (11-26c.2a, BioLegend), PE- or APC-conjugated anti-CD138 (281-2, BD Bioscience or BioLegend, respectively).
Single-cell Nojima culture
GC B cells were expanded in single cell cultures (35). Briefly, NB-21.2D9 cells feeder cells were seeded into 96-well plates at 2,000 cells/well in B cell media (BCM); RPMI-1640 (Invitrogen) supplemented with 10% HyClone FBS (Thermo scientific), 5.5 × 10−5 M 2-mercaptoethanol, 10 mM HEPES, 1 mM sodium pyruvate, 100 units/ml penicillin, 100 μg/ml streptomycin, and MEM nonessential amino acid (all Invitrogen). Next day, recombinant mouse IL-4 (Peprotech; 2 ng/ml) was added to the cultures, and then single B cells were directly sorted into each well of 96-well plates using a FACS Vantage. Two days after culture, 50% (vol.) of culture media were removed from cultures and 100% (vol.) of fresh BCM were added to the cultures. Two-thirds of the culture media were replaced with fresh BCM every day from day 4 to day 8. On day 10, culture supernatants were harvested for ELISA determinations and culture plates were stored at −80°C for V(D)J amplifications.
ELISA and Luminex assays
Presence of total and antigen-specific IgG in serum samples and in culture supernatants were determined by ELISA and Luminex multiplex assay (35). Diluted culture supernatants (1:10 in PBS containing 0.5% BSA and 0.1% Tween-20) were screened for the presence of IgGs by standard ELISA (35). IgG+ samples were then screened for binding to immunogen antigens (HA H1 SI-06, HA H3 X31, and rPA) by Luminex assay (35). Briefly, serum samples or culture supernatants were diluted (initial dilutions of 1: 100, and then 3-fold, 11 serial dilutions for serum samples; 1: 10 or 1: 100 for culture supernatants) in 1×PBS containing 1% BSA, 0.05% NaN3 and 0.05% Tween20 (assay buffer) with 1% milk and incubated for 2 hours with the mixture of antigen-coupled microsphere beads in 96-well filter bottom plates (Millipore). After washing with assay buffer, these beads were incubated for 1 hour with PE-conjugated goat anti-mouse IgG Abs (Southern Biotech). After three washes, the beads were re-suspended in assay buffer and the plates were read on a Bio-Plex 3D Suspension Array System (Bio-Rad). The following antigens were coupled with carboxylated beads (Luminex Corp): BSA (Affymetrix), goat anti-mouse Igκ, goat anti-mouse Igλ, goat anti-mouse IgG (all Southern Biotech), HA H1 SI-06, HA H3 X31, and rPA. For each IgG+ culture supernatant sample and serum sample, concentrations of antigen-binding IgG were determined in reference to monoclonal standards; “musinized” CH67 (35) and HC19 (57, 58) for H1-specific IgG and H3-specific IgG, respectively. Measured KD of CH67 and HC19 IgG are 2.4-24 nM and 28 ±8 nM (58), respectively. AvIn values were obtained for HA H1 SI-06 binding IgG samples in reference to monoclonal standard, “musinized” CH67 (35).
Amplification of V(D)J rearrangements
V(D)J rearrangements of cultured B cells were amplified by a semi-nested PCR. Total RNA was extracted from selected samples using Quick-RNA 96 kit (Zymo Research). cDNA was synthesized from DNase I-treated RNA using SMARTScribe™ Reverse Transcriptase (Clontech) with 0.2 μM each of gene-specific reverse primers (mIgGHGC-RT, mIgKC-RT, mIgLC23-RT, mIgLC1-RT, and mIgLC4-RT, Table S2) and 1 μM of 5’ SMART template-switching oligo that contained plate-associated barcodes (Table S2) at 50°C for 50 min followed by 85°C for 5 min. cDNA was then subjected to two rounds of PCR using Herculase II fusion DNA polymerase (Agilent Technologies) with combinations of forward primers and reverse primers that contained well-associated barcodes (Table S2, Set-2). Primary PCR: 95°C for 4 min, followed by 2 cycles of 95°C for 30 sec, 64°C for 20 sec, 72°C for 30 sec; 3 cycles of 95°C for 30 sec, 62°C for 20 sec, 72°C for 30 sec; 25 cycles of 95°C for 30 sec, 55°C for 20 sec, 72°C for 30 sec; and 72°C for 10 min. Secondary PCR: 95°C for 4 min, followed by 40 cycles of 95°C for 30 sec, 45°C for 20 sec, 72°C for 30 sec; and 72°C for 10 min.
In some experiments, cDNA was synthesized using Superscript III (Invitrogen) with 0.2 μM each of gene-specific reverse primers (mIgGHGC-RT, mIgKC-RT, mIgLC23-RT, mIgLC1-RT, and mIgLC4-RT) at 50°C for 50 min followed by 85°C for 5 min. cDNA samples were then subjected to two rounds of PCR using Herculase II fusion DNA polymerase with combinations of forward primers and reverse primers that contained well-associated barcodes (Table S2, Set-1). Primary PCR: 95°C for 4 min, followed by 5 cycles of 95°C for 30 sec, 62°C-54°C (−2°C per cycle) for 30 sec, 72°C for 30 sec; 25 cycles of 95°C for 30 sec, 52°C for 30 sec, 72°C for 30 sec; and 72°C for 10 min. Secondary PCR: 95°C for 4 min, followed by 2 cycles of 95°C for 30 sec, 64°C for 20 sec, 72°C for 30 sec; 3 cycles of 95°C for 30 sec, 62°C for 20 sec, 72°C for 30 sec; 25 cycles of 95°C for 30 sec, 55°C for 20 sec, 72°C for 30 sec; and 72°C for 10 min.
Gel-purified, pooled V(D)J amplicands were submitted to DNA Link, Inc. to obtain DNA sequences using PacBio sequencing platform. Obtained circular consensus sequences (CCS) were analyzed to build a consensus sequence for each sample. Obtained CCS were first sorted according to the barcode. Consensus sequences for individual samples were then built from 20 randomly-selected CCS (or from a minimum of 5 CCS) using established alignment programs MUSCLE v3.8.31 (59) and Kalign (60) with default settings. The rearranged V, D, and J gene segments were first identified using IMGT/V-QUEST (http://www.imgt.org/) or Cloanalyst (61), and then numbers and kinds of point mutations were determined.
Immunofluorescence
Popliteal LNs from immunized AID-Cre/EYFP mice were fixed with 1% paraformaldehyde in PBS for overnight at 4°C, and then placed in PBS containing 10-30% sucrose at 4 °C (10% for 2 hours, 20% for 2 hours, and then 30% for overnight). Tissues were then embedded in Tissue-Tek O.C.T. compound (Sakura Finetek) and snap-frozen in 2-methylbutane cooled with liquid nitrogen. Frozen tissues were stored at −80 °C until use. Serial 5-μm-thick cryosections were cut on a CM1850 cryostat (Leica) and thaw-mounted onto glass slides and stored at −80°C until use. After air drying, sections were rehydrated in a washing buffer (PBS with 0.5% BSA and 0.1% Tween-20) for 30 minutes at room temperature, and then blocked with rat anti-mouse CD16/32 (2.4G2) and rat IgG (Sigma-Aldrich) for 15 minutes at room temperature. Sections were labeled with combinations of mAbs in a humidified chamber for 3 hours at room temperature in the dark. After washing, labeled sections were mounted in Fluoremount-G (SouthernBiotech) and imaged with SP8 upright confocal microscope (Leica) or LSM 780 upright confocal microscope (Zeiss). The images were processed with ImageJ software (Fiji package, NIH) or Adobe Photoshop CS6 version 13.0. mAbs used for immunofluorescence: AlexaFluor488-conjugated anti-GFP/YFP (FM264G, BioLegend), PE or BV510-conjugated anti-IgD (11-26c.2a, BioLegend), BV421-conjugated anti-CD3 (KT3.1.1, BioLegend), APC-conjugated anti-CD38 (90, BioLegend) and AlexaFluor647-conjugated anti-CD21/35 (7E9, BioLegend).
Statistics
Statistical significance (p < 0.05) was determined by Wilcoxon matched-pairs signed rank test, Kruskal-Walis test with Dunn’s multiple comparisons, and Mann-Whitney’s U test using GraphPad Prism software (version 9.2.0, GraphPad Software). Statistic test is indicated within each figure legend.
Supplementary Material
Table S1. Summary of single B cell cultures
Table S2. Primer sequences
Table S3. Raw data excel file
Figure. S1. Robust recall antibody responses following boost immunizations
Figure. S2. Robust GC responses following boosts at local sites or distal sites
Figure. S3. Tracing the fates of the progeny of primary GC B cells in AID-Cre-EYFP mice
Figure. S4. Tracing the fates of the progeny of primary GC B cells in S1pr2-ERT2cre-tdTomato mice
Figure. S5. Tracing the fates of the progeny of primary GC B cells after late ablation of GC structures
Figure. S6. SHM of YFP+ secondary GC B cells in AID-Cre-EYFP mice
Figure. S7. Frequency of antigen-specific IgGs among recall GC B cells
Acknowledgments:
We thank W. Zhang, X. Liang, D. Liao, A. Watanabe, and X. Nie (Duke University) for assistance. We thank Drs. C.A. Reynaud and J.C. Weill (Universite Paris-Descartes) for their gift of AID-Cre-EYFP mice. We thank Dr. T. Kurosaki (RIKEN) for S1pr2-ERT2cre-tdTomato mice. We thank Drs. R.W. Rountree, R. Spreng, and Y. Wang (Duke Human Vaccine Institutes) for advice on statistical analysis.
Funding:
This study was supported by National Institutes of Health grant AI100645 (G.K.) and National Institutes of Health grant AI089618 (S.C.H.)
Footnotes
Competing interests: Authors declare that they have no competing interests.
Data and materials availability:
All data needed to evaluate the conclusions in the paper are available in the main text or the supplementary materials. AID-Cre-ERT2 mice are available under an MTA with INSERM laboratory, S1pr2-ERT2cre mice are available under an MTA with RIKEN. VDJ sequence data are available at GenBank (www.ncbi.nlm.nih.gov/genbank, accession numbers: OM732509-OM734737).
References and Notes
- 1.Dhenni R, Phan TG, The geography of memory B cell reactivation in vaccine-induced immunity and in autoimmune disease relapses. Immunol Rev 296, 62–86 (2020). [DOI] [PubMed] [Google Scholar]
- 2.Kurosaki T, Kometani K, Ise W, Memory B cells. Nat Rev Immunol 15, 149–159 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Weisel F, Shlomchik M, Memory B Cells of Mice and Humans. Annu Rev Immunol 35, 255–284 (2017). [DOI] [PubMed] [Google Scholar]
- 4.Cancro MP, Tomayko MM, Memory B cells and plasma cells: The differentiative continuum of humoral immunity. Immunol Rev 303, 72–82 (2021). [DOI] [PubMed] [Google Scholar]
- 5.Haynes BF, Shaw GM, Korber B, Kelsoe G, Sodroski J, Hahn BH, Borrow P, McMichael AJ, HIV-Host Interactions: Implications for Vaccine Design. Cell Host Microbe 19, 292–303 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Quast I, Tarlinton D, B cell memory: understanding COVID-19. Immunity 54, 205–210 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Inoue T, Shinnakasu R, Kurosaki T, Generation of High Quality Memory B Cells. Frontiers in immunology 12, 825813 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cobey S, Hensley SE, Immune history and influenza virus susceptibility. Curr Opin Virol 22, 105–111 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dogan I, Bertocci B, Vilmont V, Delbos F, Megret J, Storck S, Reynaud CA, Weill JC, Multiple layers of B cell memory with different effector functions. Nat Immunol 10, 1292–1299 (2009). [DOI] [PubMed] [Google Scholar]
- 10.Pape KA, Taylor JJ, Maul RW, Gearhart PJ, Jenkins MK, Different B Cell Populations Mediate Early and Late Memory During an Endogenous Immune Response. Science, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zuccarino-Catania GV, Sadanand S, Weisel FJ, Tomayko MM, Meng H, Kleinstein SH, Good-Jacobson KL, Shlomchik MJ, CD80 and PD-L2 define functionally distinct memory B cell subsets that are independent of antibody isotype. Nat Immunol 15, 631–637 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McHeyzer-Williams LJ, Milpied PJ, Okitsu SL, McHeyzer-Williams MG, Class-switched memory B cells remodel BCRs within secondary germinal centers. Nat Immunol 16, 296–305 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Turner JS, Zhou JQ, Han J, Schmitz AJ, Rizk AA, Alsoussi WB, Lei T, Amor M, McIntire KM, Meade P, Strohmeier S, Brent RI, Richey ST, Haile A, Yang YR, Klebert MK, Suessen T, Teefey S, Presti RM, Krammer F, Kleinstein SH, Ward AB, Ellebedy AH, Human germinal centres engage memory and naive B cells after influenza vaccination. Nature 586, 127–132 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wrammert J, Smith K, Miller J, Langley WA, Kokko K, Larsen C, Zheng NY, Mays I, Garman L, Helms C, James J, Air GM, Capra JD, Ahmed R, Wilson PC, Rapid cloning of high-affinity human monoclonal antibodies against influenza virus. Nature 453, 667–671 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kaji T, Furukawa K, Ishige A, Toyokura I, Nomura M, Okada M, Takahashi Y, Shimoda M, Takemori T, Both mutated and unmutated memory B cells accumulate mutations in the course of the secondary response and develop a new antibody repertoire optimally adapted to the secondary stimulus. Int Immunol 25, 683–695 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schmidt AG, Do KT, McCarthy KR, Kepler TB, Liao HX, Moody MA, Haynes BF, Harrison SC, Immunogenic Stimulus for Germline Precursors of Antibodies that Engage the Influenza Hemagglutinin Receptor-Binding Site. Cell Rep 13, 2842–2850 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shlomchik MJ, Do Memory B Cells Form Secondary Germinal Centers? Yes and No. Cold Spring Harb Perspect Biol 10, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McHeyzer-Williams LJ, Dufaud C, McHeyzer-Williams MG, Do Memory B Cells Form Secondary Germinal Centers? Impact of Antibody Class and Quality of Memory T-Cell Help at Recall. Cold Spring Harb Perspect Biol 10, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pape KA, Jenkins MK, Do Memory B Cells Form Secondary Germinal Centers? It Depends. Cold Spring Harb Perspect Biol 10, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wong R, Belk JA, Govero J, Uhrlaub JL, Reinartz D, Zhao H, Errico JM, D’Souza L, Ripperger TJ, Nikolich-Zugich J, Shlomchik MJ, Satpathy AT, Fremont DH, Diamond MS, Bhattacharya D, Affinity-Restricted Memory B Cells Dominate Recall Responses to Heterologous Flaviviruses. Immunity 53, 1078–1094 e1077 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mesin L, Schiepers A, Ersching J, Barbulescu A, Cavazzoni CB, Angelini A, Okada T, Kurosaki T, Victora GD, Restricted Clonality and Limited Germinal Center Reentry Characterize Memory B Cell Reactivation by Boosting. Cell 180, 92–106 e111 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Viant C, Weymar GHJ, Escolano A, Chen S, Hartweger H, Cipolla M, Gazumyan A, Nussenzweig MC, Antibody Affinity Shapes the Choice between Memory and Germinal Center B Cell Fates. Cell 183, 1298–1311 e1211 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smith KG, Light A, Nossal GJ, Tarlinton DM, The extent of affinity maturation differs between the memory and antibody-forming cell compartments in the primary immune response. Embo J 16, 2996–3006 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Paus D, Phan TG, Chan TD, Gardam S, Basten A, Brink R, Antigen recognition strength regulates the choice between extrafollicular plasma cell and germinal center B cell differentiation. J Exp Med 203, 1081–1091 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Phan TG, Paus D, Chan TD, Turner ML, Nutt SL, Basten A, Brink R, High affinity germinal center B cells are actively selected into the plasma cell compartment. J Exp Med 203, 2419–2424 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Taylor JJ, Pape KA, Steach HR, Jenkins MK, Humoral immunity. Apoptosis and antigen affinity limit effector cell differentiation of a single naive B cell. Science 347, 784–787 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shinnakasu R, Inoue T, Kometani K, Moriyama S, Adachi Y, Nakayama M, Takahashi Y, Fukuyama H, Okada T, Kurosaki T, Regulated selection of germinal-center cells into the memory B cell compartment. Nat Immunol 17, 861–869 (2016). [DOI] [PubMed] [Google Scholar]
- 28.Krautler NJ, Suan D, Butt D, Bourne K, Hermes JR, Chan TD, Sundling C, Kaplan W, Schofield P, Jackson J, Basten A, Christ D, Brink R, Differentiation of germinal center B cells into plasma cells is initiated by high-affinity antigen and completed by Tfh cells. J Exp Med 214, 1259–1267 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Adachi Y, Onodera T, Yamada Y, Daio R, Tsuiji M, Inoue T, Kobayashi K, Kurosaki T, Ato M, Takahashi Y, Distinct germinal center selection at local sites shapes memory B cell response to viral escape. J Exp Med 212, 1709–1723 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Allie SR, Bradley JE, Mudunuru U, Schultz MD, Graf BA, Lund FE, Randall TD, The establishment of resident memory B cells in the lung requires local antigen encounter. Nat Immunol 20, 97–108 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ponzio NM, Chapman-Alexander JM, Thorbecke GJ, Transfer of memory cells into antigen-pretreated hosts. I. Functional detection of migration sites for antigen-specific B cells. Cell Immunol 34, 79–92 (1977). [DOI] [PubMed] [Google Scholar]
- 32.Baine Y, Ponzio NM, Thorbecke GJ, Transfer of memory cells into antigen-pretreated hosts. II. Influence of localized antigen on the migration of specific memory B cells. Eur J Immunol 11, 990–996 (1981). [DOI] [PubMed] [Google Scholar]
- 33.Baine Y, Ponzio NM, Thorbecke GJ, Unilateral localization of hapten-specific B memory cells in lymph node draining a footpad injection of antigen. Adv Exp Med Biol 149, 167–178 (1982). [DOI] [PubMed] [Google Scholar]
- 34.Baine Y, Thorbecke GJ, Induction and persistence of local B cell memory in mice. J Immunol 128, 639–643 (1982). [PubMed] [Google Scholar]
- 35.Kuraoka M, Schmidt AG, Nojima T, Feng F, Watanabe A, Kitamura D, Harrison SC, Kepler TB, Kelsoe G, Complex Antigens Drive Permissive Clonal Selection in Germinal Centers. Immunity 44, 542–552 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roco JA, Mesin L, Binder SC, Nefzger C, Gonzalez-Figueroa P, Canete PF, Ellyard J, Shen Q, Robert PA, Cappello J, Vohra H, Zhang Y, Nowosad CR, Schiepers A, Corcoran LM, Toellner KM, Polo JM, Meyer-Hermann M, Victora GD, Vinuesa CG, Class-Switch Recombination Occurs Infrequently in Germinal Centers. Immunity 51, 337–350 e337 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Glaros V, Rauschmeier R, Artemov AV, Reinhardt A, Ols S, Emmanouilidi A, Gustafsson C, You Y, Mirabello C, Bjorklund AK, Perez L, King NP, Mansson R, Angeletti D, Lore K, Adameyko I, Busslinger M, Kreslavsky T, Limited access to antigen drives generation of early B cell memory while restraining the plasmablast response. Immunity 54, 2005–2023 e2010 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Le Gallou S, Nojima T, Kitamura D, Weill JC, Reynaud CA, The AID-Cre-ERT2 Model: A Tool for Monitoring B Cell Immune Responses and Generating Selective Hybridomas. Methods Mol Biol 1623, 243–251 (2017). [DOI] [PubMed] [Google Scholar]
- 39.Kasturi SP, Skountzou I, Albrecht RA, Koutsonanos D, Hua T, Nakaya HI, Ravindran R, Stewart S, Alam M, Kwissa M, Villinger F, Murthy N, Steel J, Jacob J, Hogan RJ, Garcia-Sastre A, Compans R, Pulendran B, Programming the magnitude and persistence of antibody responses with innate immunity. Nature 470, 543–547 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aiba Y, Kometani K, Hamadate M, Moriyama S, Sakaue-Sawano A, Tomura M, Luche H, Fehling HJ, Casellas R, Kanagawa O, Miyawaki A, Kurosaki T, Preferential localization of IgG memory B cells adjacent to contracted germinal centers. Proc Natl Acad Sci U S A 107, 12192–12197 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Han S, Hathcock K, Zheng B, Kepler TB, Hodes R, Kelsoe G, Cellular interaction in germinal centers. Roles of CD40 ligand and B7–2 in established germinal centers. J Immunol 155, 556–567 (1995). [PubMed] [Google Scholar]
- 42.Han S, Zheng B, Dal Porto J, Kelsoe G, In situ studies of the primary immune response to (4-hydroxy-3-nitrophenyl)acetyl. IV. Affinity-dependent, antigen-driven B cell apoptosis in germinal centers as a mechanism for maintaining self-tolerance. J Exp Med 182, 1635–1644 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Takahashi Y, Dutta PR, Cerasoli DM, Kelsoe G, In situ studies of the primary immune response to (4-hydroxy-3-nitrophenyl)acetyl. V. Affinity maturation develops in two stages of clonal selection. Journal of Experimental Medicine 187, 885–895 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Suan D, Nguyen A, Moran I, Bourne K, Hermes JR, Arshi M, Hampton HR, Tomura M, Miwa Y, Kelleher AD, Kaplan W, Deenick EK, Tangye SG, Brink R, Chtanova T, Phan TG, T follicular helper cells have distinct modes of migration and molecular signatures in naive and memory immune responses. Immunity 42, 704–718 (2015). [DOI] [PubMed] [Google Scholar]
- 45.Yeh CH, Finney J, Okada T, Kurosaki T, Kelsoe G, Primary germinal center-resident T follicular helper cells are a physiologically distinct subset of CXCR5(hi)PD-1(hi) T follicular helper cells. Immunity 55, 272–289 e277 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Netea MG, Dominguez-Andres J, Barreiro LB, Chavakis T, Divangahi M, Fuchs E, Joosten LAB, van der Meer JWM, Mhlanga MM, Mulder WJM, Riksen NP, Schlitzer A, Schultze JL, Stabell Benn C, Sun JC, Xavier RJ, Latz E, Defining trained immunity and its role in health and disease. Nat Rev Immunol 20, 375–388 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Heesters BA, Myers RC, Carroll MC, Follicular dendritic cells: dynamic antigen libraries. Nat Rev Immunol 14, 495–504 (2014). [DOI] [PubMed] [Google Scholar]
- 48.Tas JM, Mesin L, Pasqual G, Targ S, Jacobsen JT, Mano YM, Chen CS, Weill JC, Reynaud CA, Browne EP, Meyer-Hermann M, Victora GD, Visualizing antibody affinity maturation in germinal centers. Science 351, 1048–1054 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Haynes BF, Kelsoe G, Harrison SC, Kepler TB, B-cell-lineage immunogen design in vaccine development with HIV-1 as a case study. Nat Biotechnol 30, 423–433 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Whittle JR, Zhang R, Khurana S, King LR, Manischewitz J, Golding H, Dormitzer PR, Haynes BF, Walter EB, Moody MA, Kepler TB, Liao HX, Harrison SC, Broadly neutralizing human antibody that recognizes the receptor-binding pocket of influenza virus hemagglutinin. Proc Natl Acad Sci U S A 108, 14216–14221 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schmidt AG, Therkelsen MD, Stewart S, Kepler TB, Liao HX, Moody MA, Haynes BF, Harrison SC, Viral receptor-binding site antibodies with diverse germline origins. Cell 161, 1026–1034 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McCarthy KR, Watanabe A, Kuraoka M, Do KT, McGee CE, Sempowski GD, Kepler TB, Schmidt AG, Kelsoe G, Harrison SC, Memory B Cells that Cross-React with Group 1 and Group 2 Influenza A Viruses Are Abundant in Adult Human Repertoires. Immunity 48, 174–184 e179 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Raymond DD, Bajic G, Ferdman J, Suphaphiphat P, Settembre EC, Moody MA, Schmidt AG, Harrison SC, Conserved epitope on influenza-virus hemagglutinin head defined by a vaccine-induced antibody. Proc Natl Acad Sci U S A 115, 168–173 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bajic G, Maron MJ, Adachi Y, Onodera T, McCarthy KR, McGee CE, Sempowski GD, Takahashi Y, Kelsoe G, Kuraoka M, Schmidt AG, Influenza Antigen Engineering Focuses Immune Responses to a Subdominant but Broadly Protective Viral Epitope. Cell Host Microbe, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kuraoka M, Holl TM, Liao D, Womble M, Cain DW, Reynolds AE, Kelsoe G, Activation-induced cytidine deaminase mediates central tolerance in B cells. Proc Natl Acad Sci U S A, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Reynolds AE, Kuraoka M, Kelsoe G, Natural IgM is produced by CD5- plasma cells that occupy a distinct survival niche in bone marrow. J Immunol 194, 231–242 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bizebard T, Gigant B, Rigolet P, Rasmussen B, Diat O, Bosecke P, Wharton SA, Skehel JJ, Knossow M, Structure of influenza virus haemagglutinin complexed with a neutralizing antibody. Nature 376, 92–94 (1995). [DOI] [PubMed] [Google Scholar]
- 58.Fleury D, Barrere B, Bizebard T, Daniels RS, Skehel JJ, Knossow M, A complex of influenza hemagglutinin with a neutralizing antibody that binds outside the virus receptor binding site. Nat Struct Biol 6, 530–534 (1999). [DOI] [PubMed] [Google Scholar]
- 59.Edgar RC, MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32, 1792–1797 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lassmann T, Sonnhammer EL, Kalign--an accurate and fast multiple sequence alignment algorithm. BMC Bioinformatics 6, 298 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kepler TB, Reconstructing a B-cell clonal lineage. I. Statistical inference of unobserved ancestors. F1000Res 2, 103 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Summary of single B cell cultures
Table S2. Primer sequences
Table S3. Raw data excel file
Figure. S1. Robust recall antibody responses following boost immunizations
Figure. S2. Robust GC responses following boosts at local sites or distal sites
Figure. S3. Tracing the fates of the progeny of primary GC B cells in AID-Cre-EYFP mice
Figure. S4. Tracing the fates of the progeny of primary GC B cells in S1pr2-ERT2cre-tdTomato mice
Figure. S5. Tracing the fates of the progeny of primary GC B cells after late ablation of GC structures
Figure. S6. SHM of YFP+ secondary GC B cells in AID-Cre-EYFP mice
Figure. S7. Frequency of antigen-specific IgGs among recall GC B cells
Data Availability Statement
All data needed to evaluate the conclusions in the paper are available in the main text or the supplementary materials. AID-Cre-ERT2 mice are available under an MTA with INSERM laboratory, S1pr2-ERT2cre mice are available under an MTA with RIKEN. VDJ sequence data are available at GenBank (www.ncbi.nlm.nih.gov/genbank, accession numbers: OM732509-OM734737).