

Abstract
The expression and turnover of Ag-specific peptide–MHC class II (pMHC-II) on the surface of dendritic cells (DCs) is essential for their ability to efficiently activate CD4 T cells. Ubiquitination of pMHC-II by the E3 ubiquitin ligase March-I regulates surface expression and survival of pMHC-II in DCs. We now show that despite their high levels of surface pMHC-II, MHC class II (MHC-II) ubiquitination–deficient mouse DCs are functionally defective; they are poor stimulators of naive CD4 T cells and secrete IL-12 in response to LPS stimulation poorly. MHC-II ubiquitination–mutant DC defects are cell intrinsic, and single-cell RNA sequencing demonstrates that these DCs have an altered gene expression signature as compared with wild-type DCs. Curiously, these functional and gene transcription defects are reversed by activating the DCs with LPS. These results show that dysregulation of MHC-II turnover suppresses DC development and function.
The expression of Ag-specific peptide–MHC class II (pMHC-II) on the surface of dendritic cells (DCs) is required for these cells to function as APCs that are capable of activating Ag-specific CD4 T cells. In their native (resting) state, DCs nonspecifically sample their microenvironrnent by a wide variety of endocytic processes, generate pMHC-II in endo-/lysosomal compartments, and ultimately express pMHC-II on the cell surface for recognition by Ag-specific CD4 T cells (reviewed in Ref. 1). In the absence of a DC activation signal, surface pMHC-II turns over rapidly (half-life ~4 h), thereby continually allowing for the generation and surface expression of novel pMHC-II (2). Upon encounter with an Ag containing either a TLR signal or an adjuvant, native DCs begin an activation process that leads to dramatic changes in DC biology, one of these being a complete termination of MHC class II (MHC-II) biosynthesis (3). Under these conditions the half-life of surface pMHC-II is dramatically increased (4, 5), thereby protecting pMHC-II from degradation and increasing the possibility that CD4 T cells can recognize the pMHC-II generated during exposure to the DC activating signal.
The turnover of pMHC-II in DCs and B cells is regulated by the E3 ubiquitin ligase March-I (2, 6–9). March-I is expressed in resting DCs and B cells, and the expression of this ubiquitin ligase is terminated upon activation of APCs (8). The target of March-I on MHC-II is a single lysine present in the β-chain cytosolic domain (K225), and in mice lacking March-I or in mice possessing a mutation of this lysine to arginine (MHC-II K225R), MHC-II molecules are not ubiquitinated (2, 10, 11). The net effect of preventing MHC-II ubiquitination in these mice is that pMHC-II molecules accumulate on the surface of resting DCs and B cells (2, 6). The accumulation of pMHC-II in these cells is likely due to the inability of nonubiquitinated MHC-II to interact with the endosomal complex required for transport (ESCRT) complex that normally delivers internalized ubiquitinated cargo proteins to late endosomes/lysosomes for degradation. As a result of this, in resting APCs in MHC-II ubiquitination–deficient mice, surface MHC-II has a prolonged half-life (2, 6).
A number of recent studies have analyzed APC function in MHC-II K225R and March-I knockout (KO) mice, and the overall conclusion from these studies is that pMHC-II turnover, regulated by ubiquitination by March-I, is an important regulator of APC function (12–14). In this study, we address the issue of DC "fitness" in MHC-II ubiquitination–mutant mice by analyzing in detail MHC-II K225R and March-I KO DCs isolated from mice of a variety of genetic backgrounds, in mixed bone marrow (BM) chimeras, and in F1 breeding of wild-type (WT) and MHC-II K225R mice. We find that MHC-II ubiquitination–deficient DCs are poor stimulators of naive CD4 T cells and secrete IL-12 in response to LPS stimulation poorly regardless of the genetic background of the mice. Furthermore, we find that these DC defects are cell autonomous, and single-cell RNA sequencing (scRNA-Seq) confirms that these DCs are phenotypically distinct from WT DCs. Curiously, activation of these DCs with LPS corrects their defects in T cell stimulatory capacity and IL-12 release and negates differences in gene expression between activated WT and MHC-II ubiquitination–mutant DCs. Thus, our data show that DC function in mice possessing ubiquitination-deficient MHC-II molecules is profoundly diminished because of cell-intrinsic alterations in gene expression.
Materials and Methods
Mice and cells
March-I KO mice and MHC-II K225R ubiquitination-mutant knock-in mice on the C57BL/6 background have been described (6, 15). Each strain was backcrossed at least 10 generations on the C57BL/6 background and was backcrossed an additional three generations on this background in our animal colony in Maryland. March-I KO mice on the H-2k background were generated by breeding March-I KO mice with congenic B10.BR mice and on the H-2d background were generated by breeding with congenic B10.D2 mice. MHC-II K225R mice were crossed with Rag1 KO mice to generate Rag1 KO MHC-II K225R mice. B6.SJL, OT-II, DO11.10, and 3A9 TCR-transgenic mice were from The Jackson Laboratory. CD45.1+ OT-II T cells were generated by crossing OT-II mice with congenic CD45.1+ B6.SJL mice. Naive CD4 T cells were isolated from the spleen of OT-II transgenic mice by using a CD4 T Cell Isolation Kit (Miltenyi Biotec). Mice were bred and maintained in-house at the National Cancer Institute at Frederick animal facility. All mice were cared for in accordance with National Institutes of Health guidelines with the approval of the National Cancer Institute Animal Care and Use Committee. DCs were isolated from mouse spleens by negative selection using an MACS mouse DC isolation kit (Miltenyi Biotec). Cells were isolated as quickly as possible and maintained on ice to limit spontaneous DC activation.
Abs and reagents
The following Abs were used in this study: anti-mouse MHC-II (clone M5/114 and clone 10.2.16; BD Biosciences), CD45.1 (eBioscience), CD45.2 (eBioscience), and anti-CD16/32 mAb 2.4G2 (BD Biosciences). I-Ak–hen egg lysozyme (HEL)(46–61) complexes were detected using mAb C4H3 (16). I-Ab–Eα(56–73) complexes were detected using mAb YAe (17). CellTracker Deep Red and CellTracker Deep Green were obtained from Thermo Fisher Scientific.
Flow cytometry analysis
For surface staining, cells were stained on ice using fluorochrome-conjugated primary Ab and appropriate mAb isotype-control Ab in FACS buffer (HBSS containing 2% FBS) and Fc block mAb. Stained cells were analyzed using an FACSCalibur or LSR II flow cytometer (BD Biosciences) and FlowJo software (Tree Star).
CD4 T cell activation
Where indicated, DCs were activated in vitro by incubation with 1 μg/ml LPS.
Spleen DCs (0.2 × 105 cells) were cocultured with CD4 T cells (1 × 105 cells) and various amounts of antigenic protein or preprocessed peptide in complete medium in a volume of 250 μl at 37°C. After 24 h, the amount of IL-2 secreted into the medium was determined using a Quantikine IL-2 ELISA kit (R&D Systems).
DC activation
To monitor IL-12 secretion from DCs, spleen DCs were cultured for 24 h in medium alone or medium containing 1 μ/ml LPS. The amount of IL-12 secreted into the medium was determined using an IL-12 ELISA (R&D Systems). In some experiments, after overnight culture, the DCs were washed and fresh prewarmed medium was added for 6 h. The amount of IL-12 released during this 6 h reculture was determined by ELISA.
BM chimeras
To generate mixed BM chimeric mice, 8–10-wk-old CD45.1+ B6.SJL mice were lethally irradiated with 1000 rad for 4 h before i.v. injection of 5 × 106 BM cells from either CD45.2+ B6 or CD45.2+ MHC-II K225R mice together with 5 × 106 BM cells from CD45.1+ B6.SJL mice. CD45.1/CD45.2 chimerism was assessed after at least 8 wk of reconstitution. Purified spleen DCs from chimeric mice were subjected to additional separation into CD45.2+ DCs (using anti-CD45.2 MicroBeads; Miltenyi Biotec) and CD45.1+ DCs (the flow through from the anti-CD45.2 MicroBead column). The purity of CD45.1+ control DCs and CD45.2+ test DCs was routinely >80% as assessed by FACS analysis.
Quantitation of T cell/DC conjugate formation by FACS
Conjugate formation between TCR-transgenic CD4 T cells and DCs was measured using a modification of our previously described protocol (18). Briefly, T cells were stained with 50 nM CellTracker Green in serum-free HBSS for 30 min at 37°C. DCs were incubated with the indicated amount of Ag for 4 h, washed, and stained with 150 nM CellTracker Deep Red in serum-free HBSS for 30 min at 37°C. After staining, the cells were washed three times in PBS containing 5% FCS. Stained CD4 T cells (1 × 106 cells) and DCs (1 × 106 cells) were combined, pelleted at 1200 rpm for 5 min, and incubated for 30 min at 37°C. The cells were then gently resuspended in ice-cold PBS/FCS and analyzed immediately in an FACSCalibur flow cytometer. Ag-specific conjugate formation was determined by assessing the percentage of cells in the CellTracker Deep Red+ CellTracker Green+ gate. The percentage of cells present this gate using Ag-free DCs was subtracted from each experimental value to obtain net Ag-specific conjugate formation.
Quantitation of T cell/DC conjugate formation by microscopy
Purified spleen DCs (1 × 106) were pulsed with the indicated Ag for 3 h and washed. CD4 T cells (1 × 106) were then combined with the DCs in 2 ml warm medium in 15-ml, round-bottom tubes (Falcon) and pelleted by centrifugation at 1200 rpm for 5 min at room temperature. The pellets were incubated at 37°C for 30 min, after which the medium was aspirated, and the cells were gently resuspended in 5 ml HBSS. Cells were fixed using 4% paraformaldehyde for 10 min at room temperature, washed twice for 10 min each with 50 mM NH4Cl in PBS and then once in HBSS before staining with the appropriate Alexa Fluor–conjugated Ab for 30 min at room temperature. Cells were washed three times with HBSS, incubated for 1 h on poly-l-lysine–coated coverslips, and mounted with Fluoromount-G (SouthemBiotech). Cells were imaged using a Zeiss LSM 880 confocal microscope using a 63× oil immersion objective lens. Ag-specific conjugate formation was scored by a blinded observer, and the percentage of cells with significant conjugate was expressed as a fraction of the total number of cells analyzed.
Immunization of mice with OVA
Purified naive CD4 T cells from CD45.1+ OT-II mice were labeled with CFSE (5 μM) in PBS for 15 min at 37°C and washed with complete medium. CFSE-labeled T cells (5 × 106 cells per mouse) were injected in the lateral tail vein of mice (day −1). On day 0, the mice received an i.p. injection of either 500 μg OVA in CFA or PBS-CFA as a control. After 3 d, spleens were collected, single-cell suspensions of spleen cells were stained with CD45.1 mAb, and the cells were analyzed by flow cytometry. CFSE dye dilution on CD45.1+ OT-II T cells was analyzed by FACS using an LSRFortessa flow cytometer. The extent of T cell proliferation was analyzed using FlowJo software and is shown as a division index.
Single-cell capture, library preparation, and sequencing
CD11c MicroBead–purified spleen DCs were either incubated for 16 h in ice-cold medium (immature DCs) or in medium containing 1 μg/ml LPS at 37°C (mature DCs). Single-cell suspensions were washed twice with ice-cold PBS containing 0.04% BSA. Cell counts and viability were determined using a fluorescent cell counter and propidium iodide and acridine orange dyes (LUNA-FL; Logos Biosystem). Cells were loaded onto the 10× Genomics Chromium platform using the 3′ v2 gene expression chemistry targeting 6000 cells when sample amounts and viabilities allowed. Preparation of libraries were performed according to manufacturer recommendations. Sequencing was performed at the Center for Cancer Research Sequencing Facility on a NextSeq 500/550 instrument using High Output v2.5 kits with a 28-bp read to identify cell barcodes and unique molecular indices, an 8-bp read for sample indices, and a 98-bp read to identify cDNA inserts. Samples were multiplexed for sequencing and reads were combined from multiple sequencing runs. Data were processed using the 10× Genomics Cell Ranger pipeline (v2.2.0) to demultiplex reads and then align reads to an mm10 reference (refdata-cellranger-mm10–2.1.0) and to generate a single-cell barcode and gene expression matrix that was used for downstream analysis.
scRNA-Seq analysis
Initial scRNA-Seq data were analyzed using Partek Flow, following the standard recommended processing pipeline. Additional analysis was performed using the R statistical program (v3.6.0, Vienna, Austria). Quality control was performed for each sample individually. Cell filtering for mitochondrial fraction and gene and read distribution was performed using the Routliers package (19). Read count normalization was performed using the SCTransform tool, as provided in the Seurat package (20, 21). Cell types for each sample were determined using the Single R package, using Immunological Genome Project as a reference (22,23). The DoubletFinder package was used to identify and remove multiplet cell droplets (24). After quality control was completed, the samples were merged using the standard Seurat workflow. The top 30 principal components were used to create the Uniform Manifold Approximation and Projection (25). Differentially expressed genes were identified as having a q value of ≤0.05 via the model-based analysis of single-cell transcriptomics algorithm (26).
Statistical analysis
Data in all figures ate expressed as the mean ± SD. Differences between pairs of samples were calculated using an unpaired, two-tailed Student t test. In all figures, statistical significance is represented as *p < 0.05 and **p < 0.005.
Results
MHC-II K225R ubiquitination–mutant DCs make large amounts of pMHC-II but are poor stimulators of CD4 T cells
MHC-II ubiquitination–deficient APCs (isolated from either March-I KO mice or MHC-II K225R mice) have been reported to behave in vitro as both superior APCs (12, 27) and inferior APCs as compared with WT DCs (13, 15). To address this question in vivo, we have immunized MHC-II K225R mice with OVA protein and examined the ability of adoptively transferred, CFSE-labeled OT-II CD4 T cells to proliferate. (OT-II T cells recognize OVA323–339 bound to I-Ab.) Unlike OT-II T cells in WT mice, OT-II T cells transferred into MHC-II K225R mice failed to proliferate after immunization (Fig. 1), strongly suggesting that in vivo Ag presentation to OT-II T cells by spleen DCs was defective in these mice.
FIGURE 1.
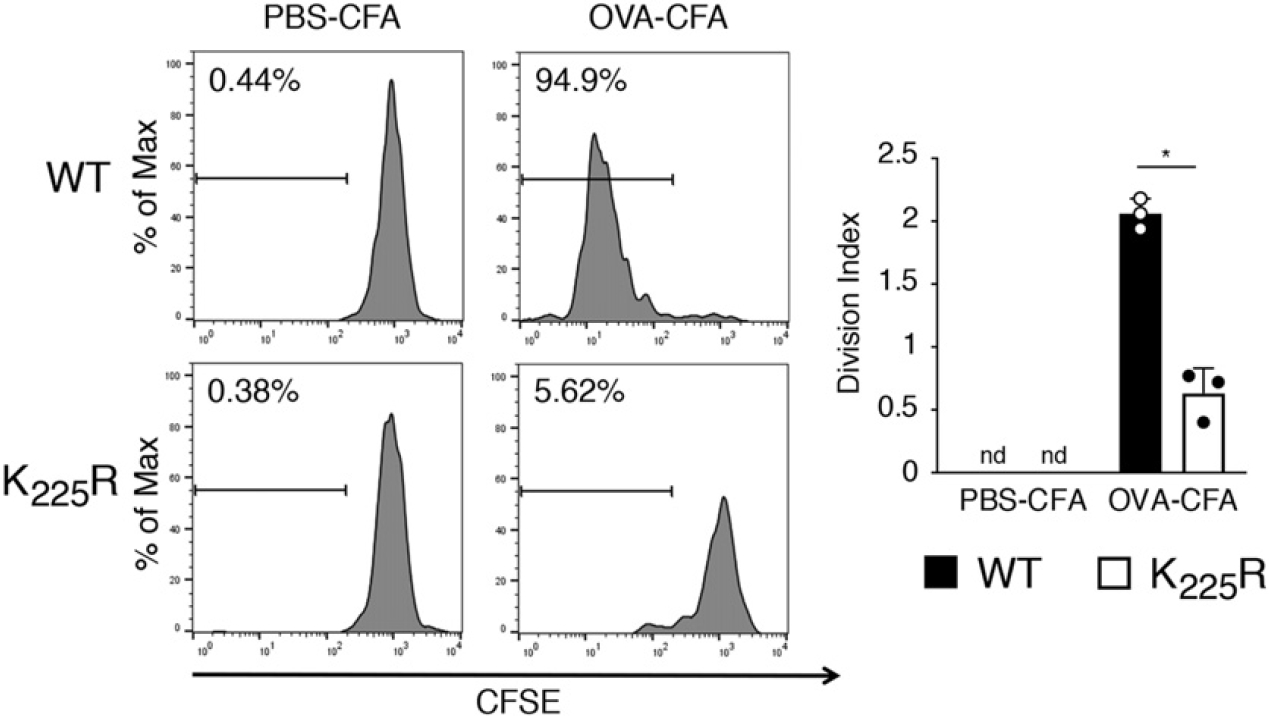
In vivo Ag presentation to OT-II T cells by spleen DCs are defective in MHC-II K225R ubiquitination–mutant mice. CFSE-labeled 5 × 106 CD4+ OT-II T cells were injected i.v. into mice. One day after, mice were immunized i.p. with OVA-CFA or PBS-CFA. After 3 d, spleen cells were harvested and stained with anti-CD45.1, CD4 mAb. CFSE profiles of OT-II cells from mice immunized with PBS-CFA are shown as control. In each experiment the division index was calculated by using FlowJo software. The data shown are the mean ± SD from three independent experiments. *p < 0.05.
There could be many different causes for the inability of the transferred CD4 T cells to proliferate in the above assay, so we therefore directly assessed DC APC function by removing spleen DCs from WT and MHC-II K225R mice and monitoring the ability of the cells to generate specific pMHC-II and by measuring ex vivo Ag presentation. WT and MHC-II K225R spleen DCs were incubated with an OVA–Eα(56–73) fusion protein as a model Ag, and surface expression of I-Ab–Eα(56–73) complexes was detected by flow cytometry using the pMHC-II complex–specific mAb YAe (17). Spleen DCs from MHC-II K225R mice expressed more I-Ab–Eα(56–73) complexes as compared with WT DCs (Fig. 2A), confirming that processing of the fusion protein and loading of Eα(56–73) onto MHC-II proceeded normally in these DCs. Nevertheless, OVA protein–pulsed MHC-II K225R DCs were poor stimulators of naive OT-II CD4 T cells, and these DCs were even poor APCs when pulsed with preprocessed OVA(323–339) peptide (Fig. 2B), demonstrating that the observed T cell activation defect was not simply a consequence of altered OVA processing.
FIGURE 2.
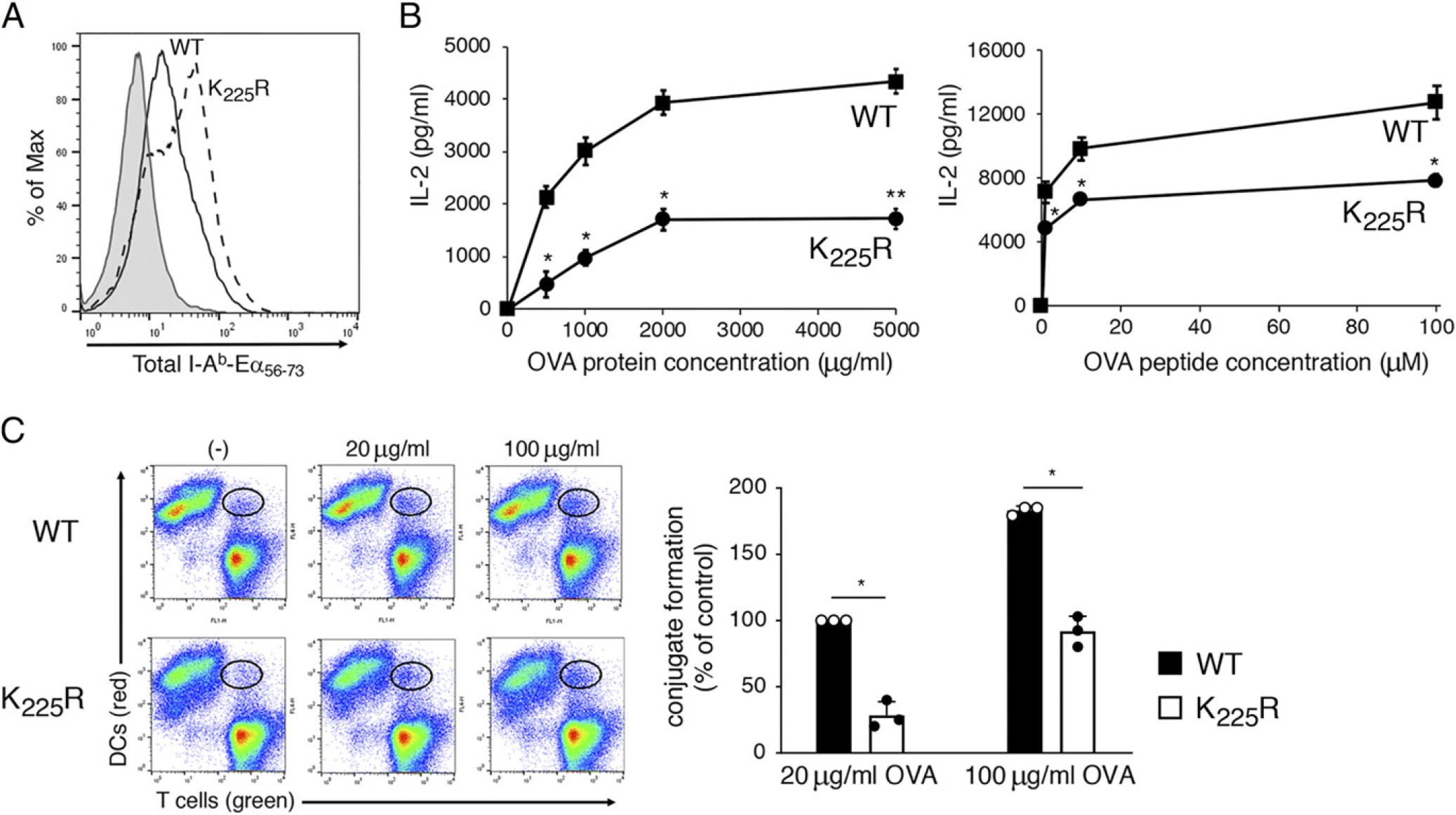
MHC-H K225R ubiquitination–mutant spleen DCs are poor stimulators of Ag-specific CD4 T cells. DCs were isolated by magnetic bead purification from spleen of WT C57BL/6 mice or MHC-H K225R ubiquitination–mutant mice. (A) Purified DCs were incubated with 50 μg/ml of Eα–OVA(56–73) fusion protein for 3 h, and surface expression of I-Ab–Eα(56–73) complexes was determined by flow cytometry using the pMHC-H complex-specific mAb YAe. A representative histogram showing staining of WT DCs incubated with no Ag (gray fill) and either WT DCs (black line) or MHC-H K225R DCs (dash line) is shown. (B) Spleen DCs from either WT mice (squares) or MHC-H K225R ubiquitination–mutant mice (circles) were cocultured with naive OT-H CD4+ T cells and various amounts of OVA protein or OVA(323–339) peptide. The amount of IL-2 secreted after 24 h of culture was determined by ELISA. (C) CellTracker Deep Red–stained DCs were incubated in the absence of presence of the indicated amount of OVA for 3 h. DCs were then combined with CellTracker Green–stained OT-H CD4+ T cells for 30 min, and DC/T cell conjugate formation was analyzed by flow cytometry. A representative contour plot is shown, and the percentage of T ceUs present in DC/T cell conjugates (relative to that using WT DCs incubated with 20 μg/ml OVA) was determined. The data shown are the mean ± SD obtained from three independent experiments. *p < 0.05, **p < 0.005.
IL-2 release is a relatively late event in CD4 T cell activation by APCs, so to examine an early event in T cell activation, we monitored the ability of OVA-pulsed DCs to form stable conjugates with naive OT-II T cells after incubation for only 30 min. Both flow cytometric analysis (Fig. 2C) and visualization by immunofluorescence microscopy (Supplemental Fig. 1) revealed a profound defect in the ability of OVA-pulsed MHC-II K225R spleen DCs to form stable conjugates with OT-II T cells, suggesting that defective T cell activation (as monitored by IL-2 release) is due to inefficient conjugate formation between OVA-pulsed MHC-II K225R spleen DCs and OT-II T cells.
The March-I KO DC Ag presentation defect is MHC haplotype and Ag independent
March-I is the E3 ubiquitin ligase responsible for MHC-II K225R ubiquitination (6, 15). Ubiquitination of MHC-II controls MHC-II recycling and lysosomal degradation (6, 8), and we have found that ubiquitination-dependent alterations in these processes are identical in March-I KO and MHC-II K225R APCs (8, 28). Like spleen DCs from MHC-II K225R mice, OVA–Eα(56–73)-pulsed March-I KO spleen DCs expressed more I-Ab–Eα(56–73) complexes as compared with WT DCs (Fig. 3A) and were also defective stimulators of OT-IIT cells when pulsed with OVA protein or OVA peptide (Fig. 3B). OVA-pulsed March-I KO spleen DCs were also defective in their ability to form conjugates with OT-II T cells (Fig. 3C); however, the extent of the defect seemed to be more modest than that observed with MHC-II K225R spleen DCs.
FIGURE 3.
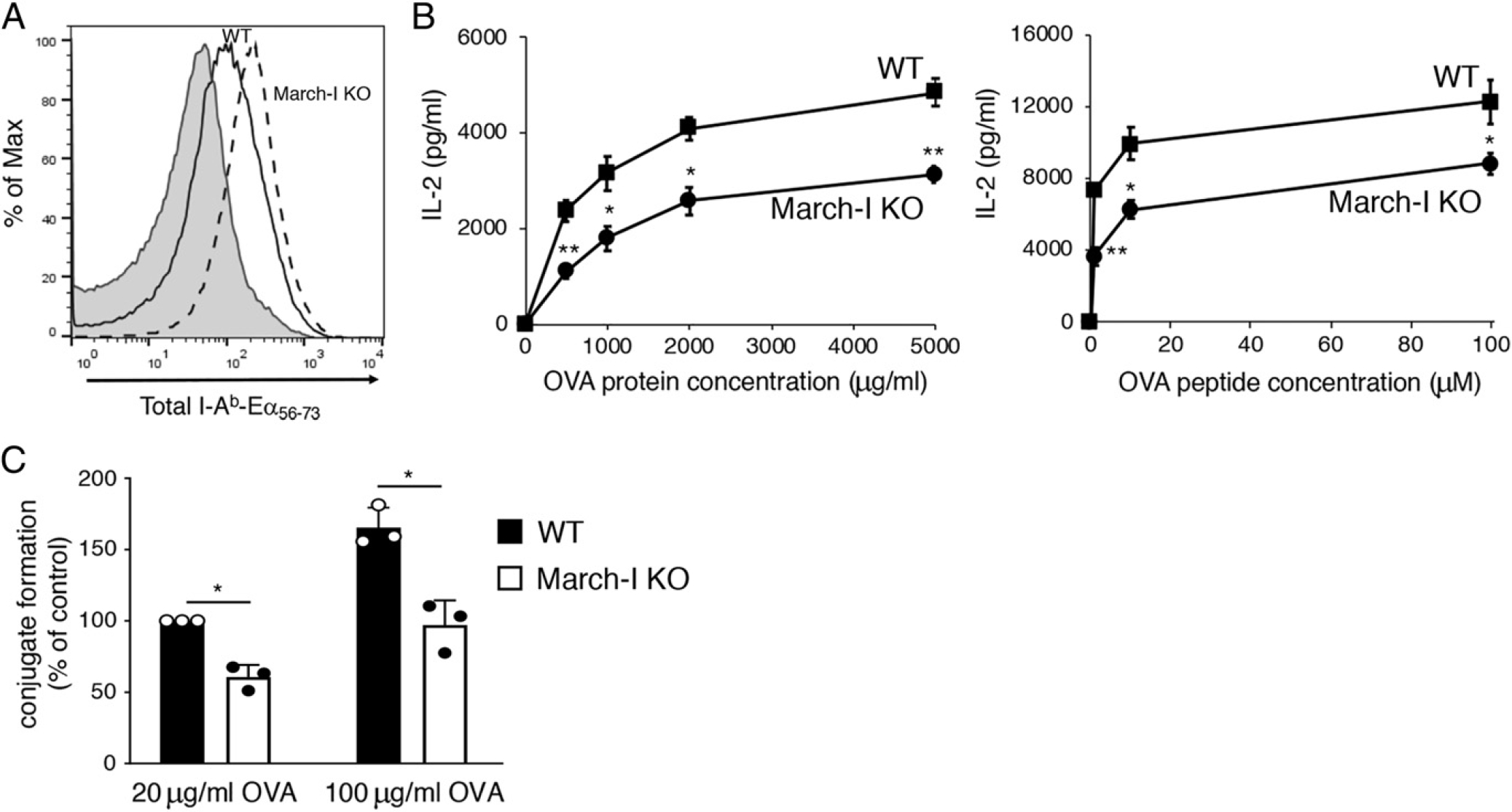
March-I KO spleen DCs make large amounts of pMHC-II but are poor stimulators of Ag-specific CD4 T cells. DCs were isolated by magnetic bead purification from spleen of WT C57BL/6 mice or March-I KO mice on a C57BL/6 background. (A) Purified DCs were incubated with 50 μg/ml of Eα-OVA(56–73) fusion protein for 3 h and surface expression of I-Ab–Eα(56–73) complexes was determined by flow cytometry using the pMHC-II complex–specific mAb YAe. A representative histogram showing staining of WT DCs incubated with no Ag (gray fill) and either WT DCs (black line) or March-I KO DCs (dash line) is shown. (B) Spleen DCs from either WT mice (filled bars) or March-I KO mice (open bars) were cocultured with naive OT-II CD4+ T cells and various amounts of OVA protein or OVA(323–339) peptide. The amount of IL-2 secreted after 24 h of culture was determined by ELISA. (C) CellTracker Deep Red–stained DCs were incubated in the absence of presence of the indicated amount of OVA for 3 h. DCs were then combined with CellTracker Green–stained OT-II CD4+ T cells for 30 min and DC/T cell conjugate formation was analyzed by flow cytometry. The percentage of T cells present in DC/T cell conjugates (relative to that using WT DCs incubated with 20 μg/ml OVA) was determined. The data shown are the mean ± SD obtained from three independent experiments. *p < 0.05, **p < 0.005.
The Ag presenting defects described above were obtained using C57BL/6 (H-2b) mice and OVA as a model Ag. To ask whether the MHC-H ubiquitination defect was MHC haplotype or Ag specific, we crossed March-I KO mice onto different genetic backgrounds and monitored processing and presentation of the model protein Ag HEL. Like spleen DCs from March-I KO mice on a C57BL/6 background, spleen DCs from March-I KO mice on the H-2k background expressed large amounts of MHC-H on their surface (Fig. 4A), and by using the I-Ak–HEL(46–61)-specific pMHC-H mAb C4H3, we determined that incubation of these DCs with HEL resulted in the expression of more I-Ak–HEL(46–61) complexes as compared with WT DCs (Fig. 4B). In agreement with our studies using OVA, incubation of these March-I KO DCs with HEL resulted in poor activation of I- Ak–restricted, HEL48–62-specific naive 3A9 CD4 T cells (Fig. 4C), demonstrating that the observed defect in Ag presentation by March-I KO spleen DCs was not MHC-II allele or Ag specific.
FIGURE 4.
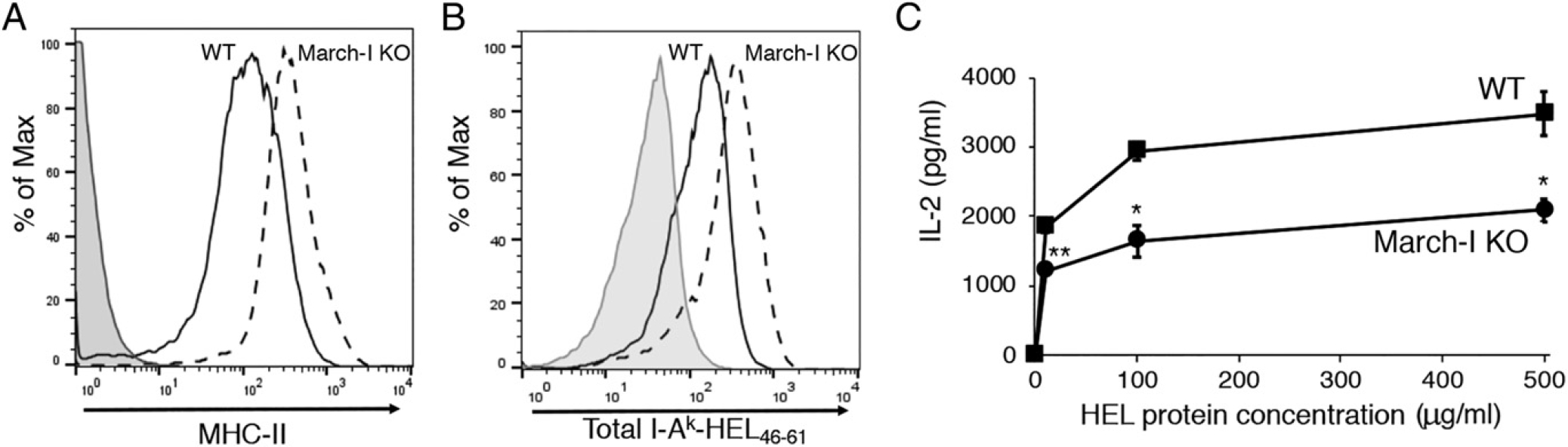
The March-I KO DC Ag present defect is not haplotype or Ag specific. DCs were isolated by magnetic bead purification from spleen of WT B10.BR mice or March-I KO mice on a B10.BR background. (A and B) Purified DCs were incubated with 50 (μg/ml of HEL for 3 h. Total surface I-Ak expression was determined using mAb 10.2.16 (A), and surface expression of I-Ak–HEL(46–61) complexes was determined by flow cytometry using the pMHC-n complex–specific mAb C4H3 (B). A representative histogram showing staining of WT DCs incubated with no Ag (gray fill) and either WT DCs (black line) or March-I KO DCs (dash line) is shown. (C) Spleen DCs from either WT mice (squares) or March-I KO mice (circles) were cocultured with naive 3A9 CD4+ T cells and various amounts of HEL protein. The amount of IL-2 secreted after 24 h of culture was determined by ELISA. The data shown are the mean ± SD obtained from three independent experiments. *p < 0.05, **p < 0.005.
MHC-n K225R and March-I KO spleen DCs on the C57BL/6 background are defective in LPS-mediated IL-12 and TNF-α secretion (15). We therefore examined IL-12 release from March-I KO spleen DCs crossed onto the H-2k and H-2d background to ask whether the defects observed previously are MHC haplotype specific. We observed significant defects in LPS-dependent IL-12 release from spleen DCs isolated from MHC-II K225R mice or March-I KO mice on the C57BL/6 (H-2b), B10.BR (H-2k), or B10.D2 (H-2d) genetic backgrounds (Fig. 5), demonstrating that the defect in IL-12 secretion from March-I KO DCs is independent of MHC haplotype. Taken together with our Ag presentation results, these data suggest that the observed Ag presentation defect in MHC-II ubiquitination–deficient DCs is not due to a specific defect in Ag presentation per se but is instead a consequence of an alteration in DC function that is independent of Ag presentation.
FIGURE 5.
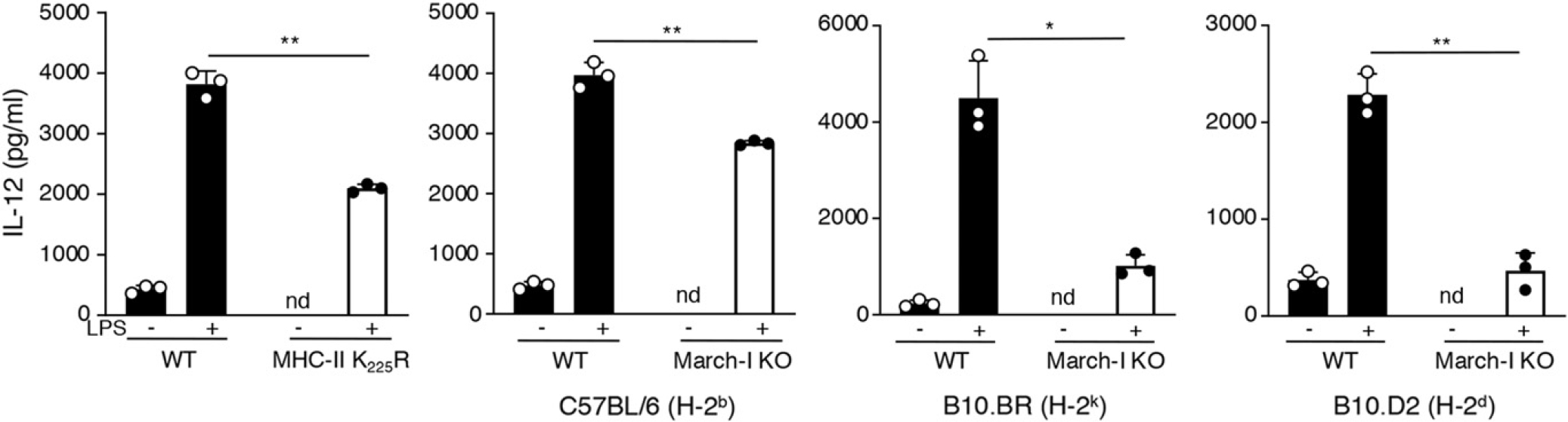
LPS-stimulated MHC-II ubiquitination–mutant DCs secrete IL-12 poorly. Spleen DCs were isolated from WT or MHC-II K225R ubiquitination-mutant mice or WT or March-I KO mice crossed onto a C57BL/6 background (H-2b), B10.BR background (H-2k), or B10.D2 (H-2d) background. The cells were incubated for 24 h in medium alone (−) or medium containing LPS (+). The amount of IL-12 secreted from the cells was determined by ELISA and the data shown are the mean ± SD obtained from three independent experiments. *p < 0.05, **p < 0.005.
The DC defect in MHC-II K225R mice is cell intrinsic
To examine whether the defect in MHC-II K225R mice is cell intrinsic, we generated mixed BM chimeric mice. Lethally irradiated mice were reconstituted with equal parts of B6.SJL (CD45.1) control BM and either C57BL/6 or MHC-II K225R (CD45.2) test BM (Fig. 6A). Spleen DCs were generated with equally efficiency regardless of whether C57BL/6 or MHC-II K225R BM was used to generate the chimera (Supplemental Fig. 2). OVA-pulsed CD45.1+ DCs isolated from control chimera mice or test chimera mice stimulated OT-II CD4 T cells equally well, demonstrating that the presence of CD45.2+ MHC-II K225R DCs did not alter the function of CD45.1+ B6 BM-derived DCs in this same mouse (Fig. 6B). By contrast, and unlike CD45.2+ C57BL/6 DCs, OVA-pulsed CD45.2+ MHC-II K225R DCs were defective stimulators of OT-II T cells. Furthermore, analysis of LPS-induced IL-12 release revealed that CD45.2+ MHC-II K225R DCs were defective in secreting IL-12 as compared with CD45.2+ C57BL/6 DCs (Fig. 6C). These data reveal that the Ag presentation and IL-12 secretion defects observed in MHC-II K225R DCs are cell intrinsic.
FIGURE 6.
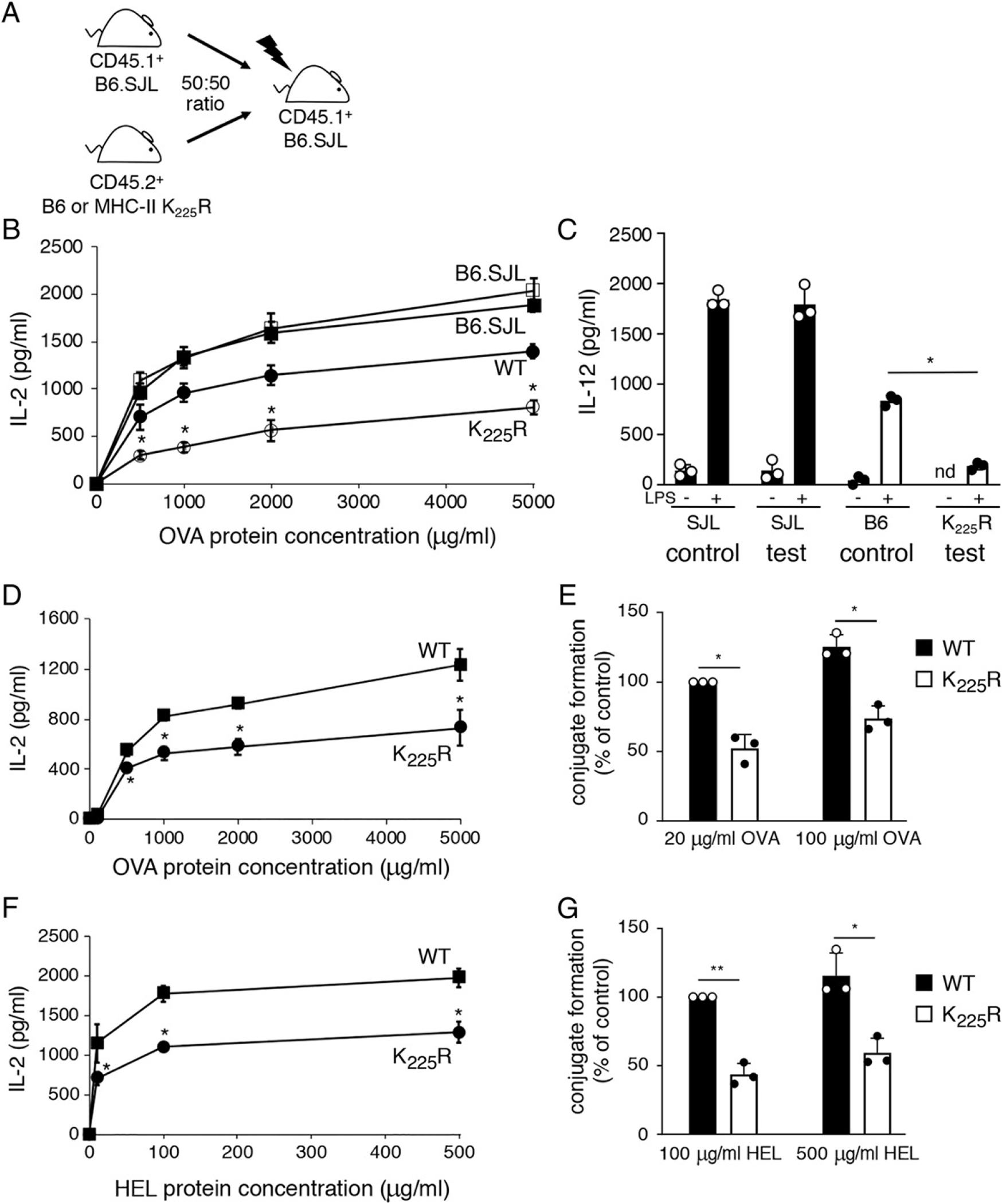
The functional defects in MHC-II K225R DCs are cell intrinsic. (A and B) Spleen DCs from either control chimeras (SJL, filled squares; B6, filled circles) or test chimeras (SJL, open squares; MHC-II K225R, open circles) were cocultured with OT-II CD4+ T cells in the presence of increasing concentrations of OVA protein. After 24 h, the amount of IL-2 secreted was examined by ELISA. (C) Spleen DCs from either control chimeras or test chimeras were cultured in the absence or presence of LPS for 24 h and the amount of IL-12 secreted was determined by ELISA. The data shown are the mean ± SD obtained from three independent experiments. *p < 0.05. (D and E) Spleen DCs were purified from C57BL/6 × B10.BR FI mice (control FI, squares) or MHC-II K225R × B10.BR FI mice (test FI, circles). (D) Spleen DCs were cocultured with OT-II CD4+ T cells in the presence of increasing amounts of OVA, and after 24 h, the amount of IL-2 secreted was examined by ELISA. (E) CellTracker Deep Red–stained spleen DCs were cultured in the absence or presence of the indicated amount of OVA protein for 3 h. The DCs were incubated with CellTracker Green–stained OT-II CD4+ T cells for 30 min, and DC/T cell conjugate formation was analyzed by flow cytometry. The percentage of OT-II T cells present in DC/T cell conjugates (relative to that using WT DCs incubated with 20 μg/ml OVA) was calculated. The data shown are the mean ± SD obtained from three independent experiments. *p < 0.05. (F) Spleen DCs were cultured with 3A9 T cells in the presence of increasing amounts of HEL, and the amount of IL-2 secreted was determined as described above. (G) The percentage of 3A9 T cells present in DC/T cell conjugates (relative to that using WT DCs incubated with 100 μg/ml HEL) was calculated. The data shown are the mean ± SD obtained from three independent experiments. *p < 0.05, **p < 0.005.
We next examined DCs possessing both WT and MHC-II K225R MHC-H molecules to determine if the observed APC defect was MHC-H molecule specific or cell specific. Hybrid mice were generated by crossing B10.BR (I-Ak) mice with either C57BL/6 (I-Ab) mice (control [B10.BR × B6] FI hybrid) or MHC-II K225R (I-Ab) mice (test [B10.BR × K225R] FI hybrid). As expected, stimulation of OT-II T cells by OVA-pulsed test fl DCs (containing K225R I-Ab) was reduced when compared with OT-II stimulation by control OVA-pulsed control hybrid DCs (containing WT I-Ab; Fig. 6D) and test fl DCs were also defective in DC/OT-II T cells conjugate formation when directly compared with OVA-pulsed control fl DCs (Fig. 6E). Surprisingly, despite the fact that each hybrid mouse contained WT I-Ak molecules, HEL-pulsed test hybrid DCs were also poor stimulators of 3A9 T cells when compared with stimulation by control HEL-pulsed control hybrid DCs (Fig. 6F), and these cells were also defective in DC/3A9 T cell conjugate formation (Fig. 6G). These data demonstrate that the presence of MHC-H K225R molecules alters the ability of DCs to function as APCs even by unrelated MHC-II molecules present on the same cell.
Decreasing MHC-II reduces APC function in WT mice but enhances APC function in MHC-II K225R mice
MHC-n K225R and March-I KO APCs express much more surface MHC-H than WT APCs because of diminished lysosomal degradation of MHC-II ubiquitination–mutant molecules (6, 28). We therefore asked whether the total amount of MHC-II on the surface of MHC-II K225R DCs was responsible for their altered function. We addressed this hy creating MHC-II hemizygous mice by breeding MHC-II KO mice with either WT or MHC-II K225R mice. As expected, MHC-H expression on hemizygous WT and MHC-H K225R spleen DCs was approximately half that observed in their homozygous counterpart (Fig. 7A). Although OVA-pulsed homozygous MHC-II K225R spleen DCs were poor stimulators of OT-II T cells, and hemizygous MHC-H K225R spleen DCs were not defective and stimulated OT-II T cells as well as hemizygous WT DCs (Fig. 7B). Similarly, although homozygous MHC-H K225R spleen DCs were clearly defective in LPS-induced IL-12 secretion, hemizygous MHC-H K225R spleen DCs were not (Fig. 7C). These data show that reducing the amount of MHC-H on the surface of MHC-II K225R spleen DCs restores the normal function of these cells.
FIGURE 7.
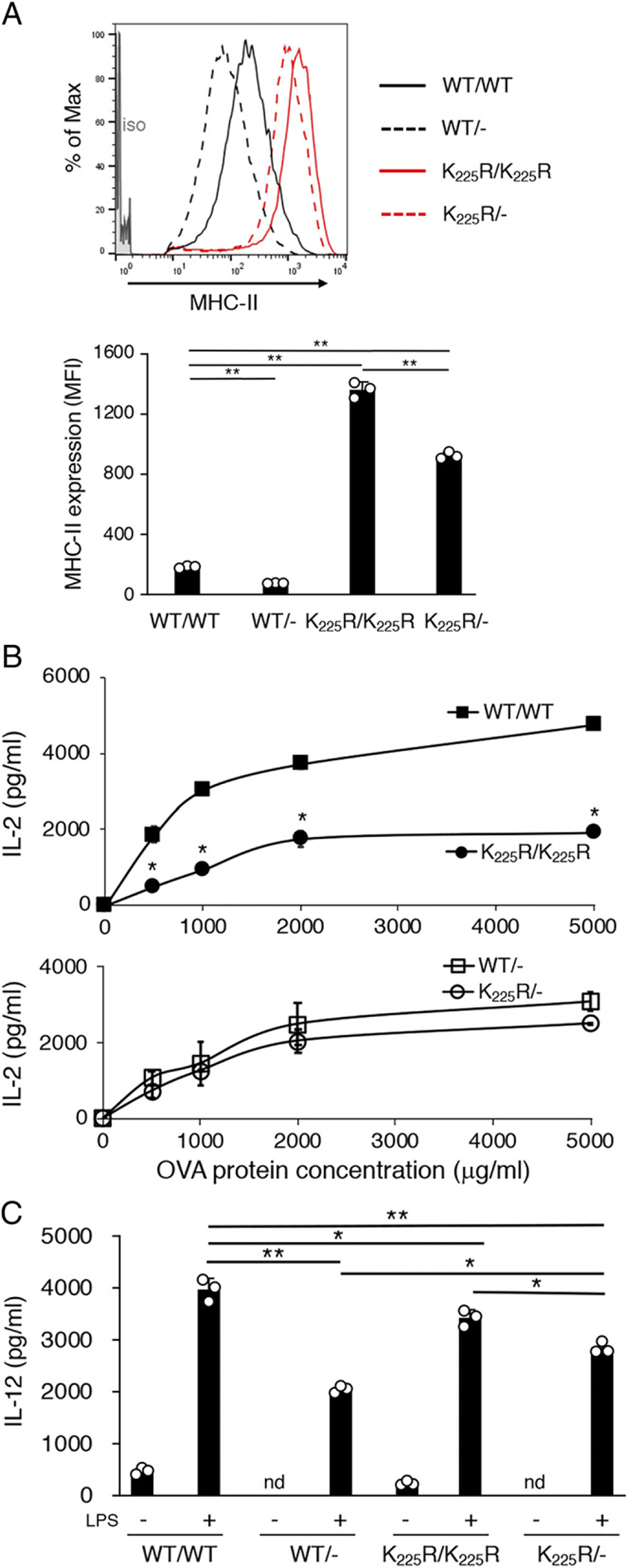
Reducing the total amount of MHC-II suppresses defects in MHC-D K225R DCs. Spleen DCs were isolated from C57BL/6 (WT) mice, C57BL/6 hemizygous mice (WT Hemi), MHC-II K225R (K > R) mice, or MHC-H K225R hemizygous (K > R Hemi) mice. (A) Surface MHC-II expression on spleen DCs isolated from WT (black line), WT Hemi (black dash line), K > R (red line), or K > R Hemi (red dash line) mice was determined DCs by FACS analysis. The staining of an isotype-control mAh is shown in gray fill. Net mean fluorescence intensity (MFT) of MHC-II expression was determined, and the data shown are the mean ± SD obtained from three independent experiments. **p < 0.005. (B) WT (filled squares) and WT Hemi (open squares) spleen DCs (left panel) or K > R (filled circles) and K > R Hemi (open circles) spleen DCs were cocultured with OT-II CD4+ T cells in the presence of increasing amounts of OVA protein. After 24 h, the amount of secreted IL-2 was examined by ELISA. The data shown are the mean ± SD obtained from three independent experiments. *p < 0.05. (C) The indicated preparation of spleen DCs was cultured in the absence or presence of LPS for 24 h, and the amount of IL-12 secreted was determined by ELISA. The data shown are the mean ± SD obtained from three independent experiments. *p < 0.05, **p < 0.005.
MHC-II K225R DCs are defective in mice lacking T cells and B cells
In an attempt to understand how enhanced MHC-II expression could suppress DC function, we considered the possibility that increased amounts of MHC-II K225R on DCs could interact nonspeciflcally with the TCR on T cells in lymphoid organs, thereby functionally tolerizing these DCs. To address this, we generated MHC-E K225R mice lacking all T cells (and B cells) by crossing these mice onto an RAG1 KO background. As was observed in WT (Ragl+) mice, OVA-pulsed MHC-II K225R/RAG1 KO DCs were still defective stimulators of OT-II T cells as compared with WT/RAG1 KO DCs (Fig. 8A) and MHC-II K225R/RAG1 KO DCs still exhibited diminished LPS-induced IL-12 secretion as compared with WT/RAG1 KO DCs (Fig. 8B). These data show that DCs that develop in MHC-II K225R mice lacking T and B cells are defective, thereby ruling out contacts with CD4 T cells as the cause of the observed DC defect in these mice.
FIGURE 8.
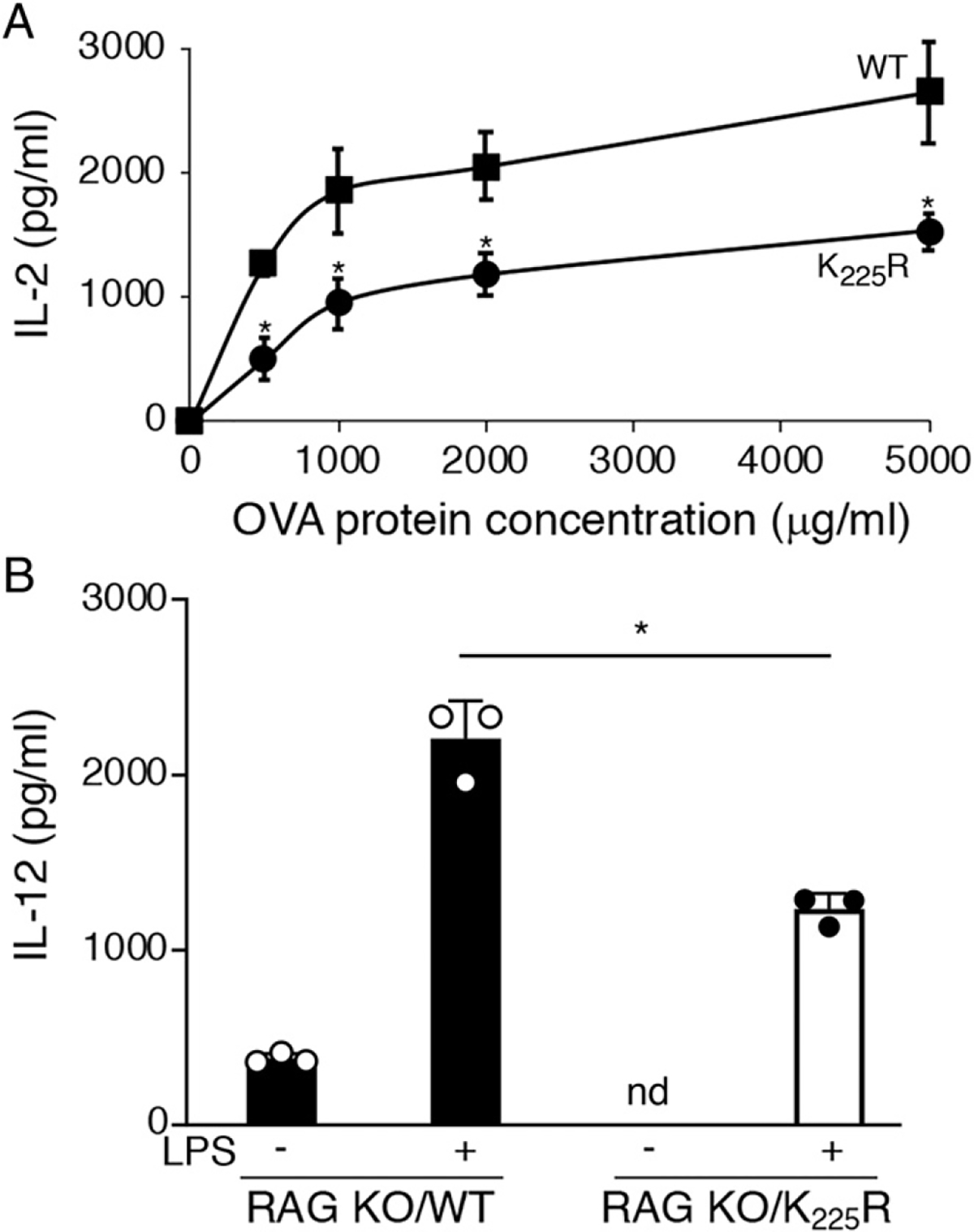
The MHC-II K225R DC defect is present in mice lacking T cells. Spleen DCs were isolated from RAG 1-deficient mice or MHC-II K225R mice crossed onto an RAGl-deficient background. (A) RAGl-deficient DCs (squares) or MHC-II K225R/RAGl-deficient DCs (circles) were cocultured with OT-II CD4+ T cells in the presence of increasing amounts of OVA protein. After 24 h, the amount of IL-2 secreted was determined by ELISA. The data shown are the mean ± SD obtained from three independent experiments. *p < 0.05. (B) RAGl-deficient DCs (filled bars) or MHC-II K225R/RAGl-deficient DCs (open bars) were cultured in the absence or presence LPS for 24 h, and the amount of IL-12 secreted was determined by ELISA. The data shown are the mean ± SD obtained from three independent experiments. *p < 0.05.
DCs from MHC-II K225R mice have an altered gene expression signature as compared with WT DCs
To analyze in detail the DCs used in our functional assays, we performed scRNA-Seq of our isolated DC preparations. Initial dimensionality reduction through t-distributed stochastic neighbor embedding (tSNE) visualization of the DC populations isolated from WT and K225R mice revealed that, overall, the cell populations isolated from these mice were very similar (Fig. 9A). Furthermore, analysis of the cell populations present in the DC samples through the use of the Single R tool using the Immunological Genome Project database as reference showed that spleen DCs isolated by negative selection from each mouse had similar amounts of CD8− DCs, CD8+ DCs, and plasmacytoid DCs and were similarly contaminated by small amounts of non-DCs (Fig. 9B). Nevertheless, volcano plots examining gene expression in CD8− and CD8+ cDCs revealed significant differences in expression in many genes in WT and K225R mice (Fig. 9C), as determined through differential expression using the model-based analysis of single-cell transcriptomics algorithm. The identity of these genes in cDCs present in the samples is highlighted in heatmaps (Fig. 9D), and pathway analysis using protein analysis through evolutionary relationships classification system analysis revealed that many of these genes are present in cell activation pathways that are downregulated in K225R DCs as compared with WT DCs (Supplemental Table I). These data demonstrate that although K225R and WT DCs are similar overall, there are significant differences in expression of certain genes in K225R DCs that likely lead to the diminished function of these cells.
FIGURE 9.
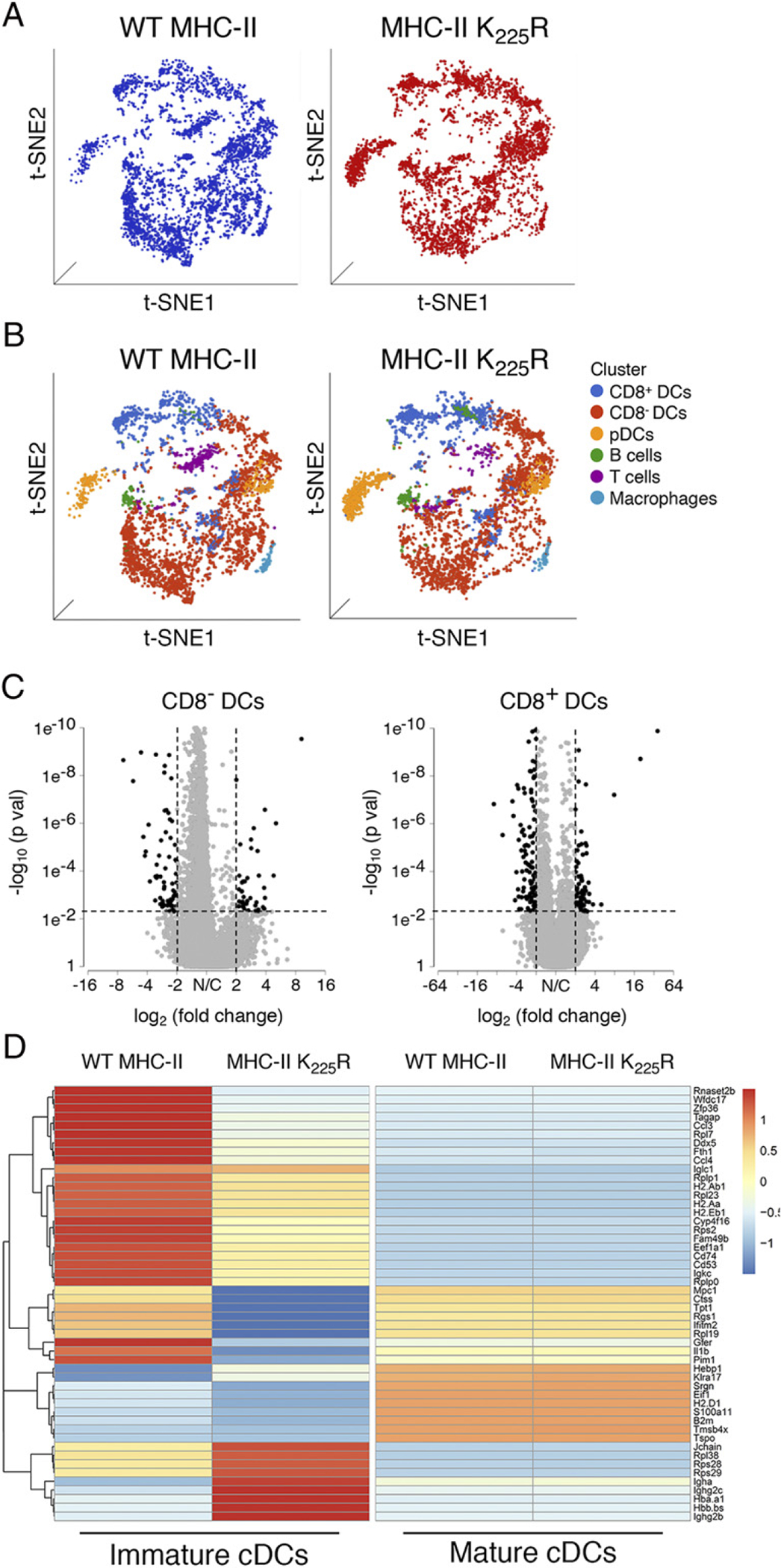
The MHC-II K225R mice DC have an altered gene expression signature as compared with WT DCs. (A) tSNE visualization of whole-spleen DCs samples from WT and MHC-II K225R mutant mice. (B) Identification of cell populations present in the spleen DC samples from WT and MHC-II K225R mutant mice was visualized by tSNE plots. Each dot represents one cell, and colors represent cell clusters as indicated. (C) Volcano plot showing the differentially expressed genes in between WT and MHC-II K225R mutant CD8− cDCs and CD8+ cDCs. Black dots indicate downregulated genes or upregulated genes, and gray dots indicate nonsignificant genes. The x- and y-axes show log2 fold change and – log10 p value, respectively. Thresholds are shown as dashed lines. (D) Heatmaps of scRNA-Seq expression data from cDCs (both CD8− and CD8+ cDCs) isolated from spleens of WT and MHC-H K225R mutant mice of the 50 most dysregulated genes in immature cDCs. The display is row scaled and indicates the relative average expression in the groups by number of standard deviations from the mean.
In addition to performing scRNA-Seq on freshly isolated spleen DCs, we also performed scRNA-Seq on these same DCs after they were cultured with LPS for 16 h. Surprisingly, after DC activation, there were no statistically significant differences in gene expression between WT and MHC-II K225R mutant DCs (Fig. 9D). The identity of these genes in cDCs present in the samples is highlighted in heatmaps. Volcano plots examining gene expression in LPS-activated spleen DCs failed to reveal any significant differences in gene expression in WT and K225R DCs (Supplemental Fig. 3). These data confirmed that the DCs isolated from MHC-II K225R mutant mice were fully capable of becoming identical to WT DCs after activation and that the spleen DCs isolated from these mice were indeed conventional DCs.
Activation corrects the functional defects in MHC-II K225R DCs
Given that scRNA-Seq failed to reveal significant changes in gene expression between LPS-activated WT and MHC-II K225R DCs, we hypothesized that activated MHC-II K225R DCs would be fully functional APCs. Indeed, incubation with LPS for 4 h prior to incubation with OVA and OT-II T cells failed to reveal any defect in T cell activation by MHC-II K225R DCs (Fig. 10A). Similarly, whereas freshly isolated MHC-II K225R spleen DCs exhibited defective LPS-induced IL-12 secretion, IL-12 release from LPS-activated MHC-II K225R DCs was essentially identical from that observed from activated WT DCs (Fig. 10B). Taken together with the results of scRNA-Seq and the functional assays described in this study, these results demonstrate that the presence of ubiquitination-deficient MHC-II molecules alters DCs development in mice and that this perturbation can be overcome by activating the cells.
FIGURE 10.
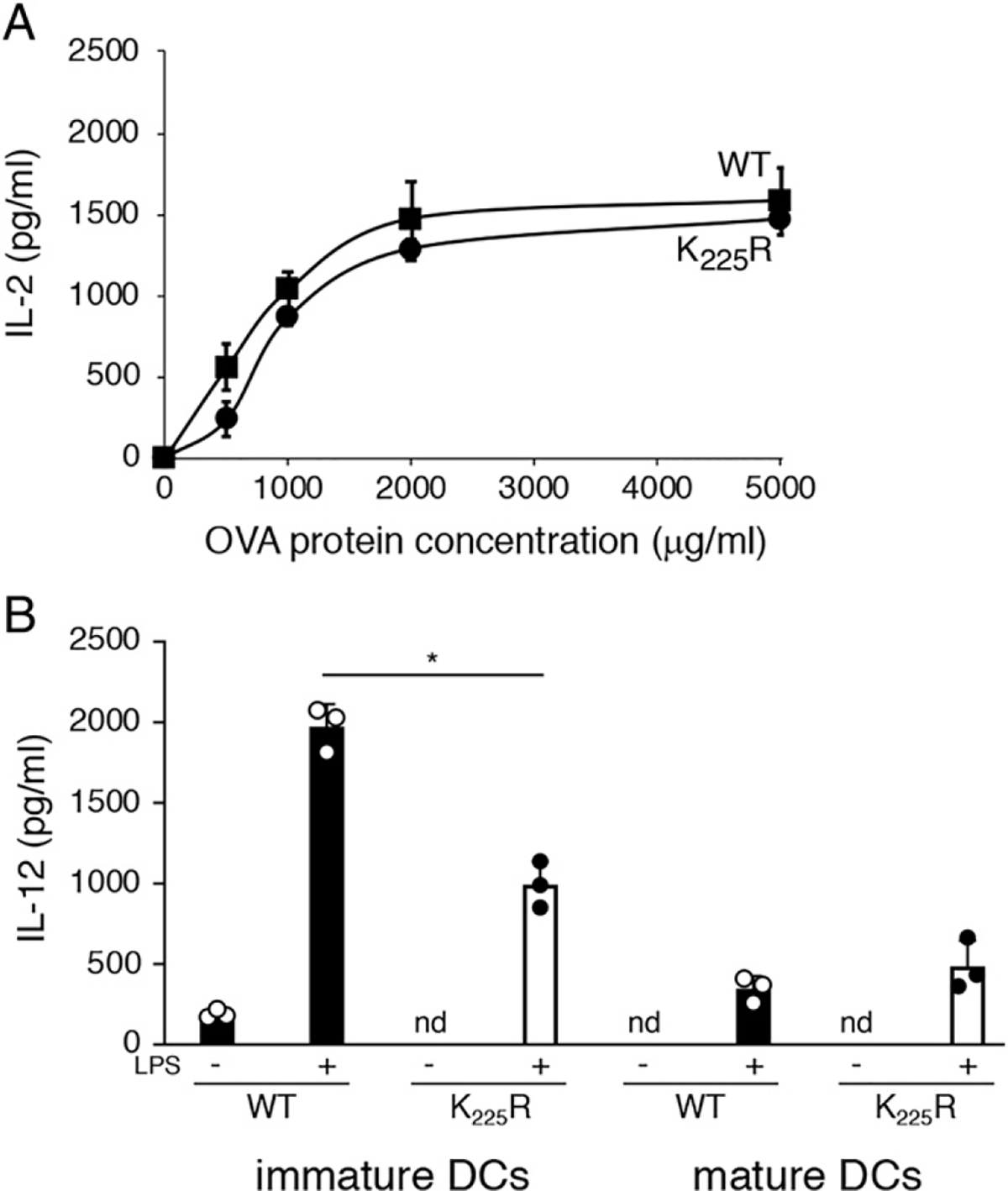
Maturation reverses the functional defects observed in MHC-H K225R DCs. (A) Spleen DCs purified from WT mice (squares) or MHC-n K225R mice (circles) were stimulated with LPS for 4 h, washed, and cultured with OT-II CD4+ T cells in the presence of increasing amounts of OVA protein. After 24 h, the amount of secreted IL-2 was examined by ELISA. The data shown are the mean ± SD obtained from three independent experiments, ns, not significant. (B) Spleen DCs from WT mice (filled bars) or MHC-II K225R mice (open bars) were stimulated with LPS overnight, and an aliquot of the supernatant was harvested for analysis of IL-12 content. The cells were then washed and cultured in fresh medium. After 6 h, another aliquot of the supernatant was harvested, and the amount of IL-12 present in each sample was determined by ELISA. The data shown are the mean ± SD obtained from three independent experiments. *p < 0.05.
Discussion
Ubiquitination of MHC-II by March-I reroutes internalized MHC-II from a pathway of early endosomal recycling to the pathway of lysosomal degradation in APCs. The net effect of preventing MHC-II ubiquitination in either March-I KO or MHC-II K225R mutant mice is a dramatic increase in MHC-II expression on the plasma membrane (6, 15). Unlike in WT immature DCs, MHC-II does not appear to reside in late endosomal Ag processing compartments in these mutant DCs but instead accumulates at the plasma membrane (2, 10). This is not due to a failure of nascent MHC-II trafficking to these compartments [because MHC-II expression on MVB-derived exosomes is normal in these cells (29, 30)] but instead reflects diminished lysosomal degradation of MHC-II in immature DCs from MHC-II ubiquitination–mutant mice.
Despite this unremarkable role for ubiquitination in regulating the cell biology of MHC-II expression, studies have attributed MHC-H turnover to a wide variety of immunological phenomena in MHC-H ubiquitination–deficient mice, including modulation of regulatory T cell generation (12, 13), affinity maturation in germinal centers (9), DnaK-induced alloimmunity (31), MHC class I expression and function on DCs (32), and migration of CD206+ DCs to skin-draining lymph nodes (14). Incubation of MHC-H ubiquitination–deficient DCs with specific Ags ex vivo enhances expression of Ag-specific pMHC-II (2, 12), a phenomenon that has been proposed to reflect enhanced Ag presentation by these cells in vivo (12). We now show that although it is true that expression of Ag-specific pMHC-H is enhanced in DCs isolated from MHC-II ubiquitination–mutant mice, the DCs are actually very poor stimulators of Ag-specific CD4 T cells. This was shown by examining DCs obtained from March-I KO DCs on the H-2b, H-2k, and H-2d backgrounds, using either OVA or HEL as model Ags and using different TCR-transgenic naive CD4 T cells to measure APC function. Mixed BM chimeras revealed that the defects observed are cell autonomous. Furthermore, DCs possessing both WT I-Ak and mutant I-Ab MHC-II molecules are poor stimulators of both I-Ak– and I-Ab–restricted T cells, revealing that the defects observed are cell specific and not molecule specific.
In addition to exhibiting poor T cell stimulatory activity, DCs isolated from all MHC-II ubiquitination–mutant mice used in this study had profound defects in LPS-induced IL-12 secretion as compared with WT DCs. Although our study, to our knowledge, is the first to characterize in detail defects in Ag presentation and LPS-induced IL-12 secretion in primary spleen DCs from MHC-II ubiquitination–mutant mice, previous work demonstrated that BM-derived DCs generated from these mice are defective stimulators of Ag-specific T cells (13, 33). Additionally, although BM cells from March-I KO mice cultured with Flt-3L developed into functionally competent DCs in vitro, injection of these cells into C57BL/6 mice revealed defects in IL-12 production (15), suggesting the spleen microenvironment is necessary for the APC defects to be manifest. Curiously, it is not the presence of T cells (or B cells) in the spleen that is the cause of this APC defect because DCs isolated from MHC-II ubiquitination–mutant mice on an RAG KO background remained defective in both T cell activation and LPS-stimulated IL-12 release. The results of our study therefore demonstrate that the immunological defects observed in MHC-II ubiquitination–mutant mice cannot solely be attributed to changes in MHC-II turnover or enhanced Ag presentation but are instead a consequence of altered DC phenotype and genotype.
By creating MHC-II hermzygous mice, we round that hemizygous MHC-H ubiquitination–mutant DCs, which express half as much MHC-H as their homozygous counterparts, were affected much less than were homozygous MHC-H ubiquitination–mutant DCs, suggesting that the absolute amount of mutant MHC-II played an important role in the observed defects. Recently, it has been shown that defects in regulatory T cell generation in MHC-II K225R hemizygous mice were not as severe as that observed in MHC-II K225R homozygous mice (13), a finding that is consistent with our findings of APC functional defects in these mice. We considered the possibility that enhanced MHC-II expression on MHC-H ubiquitination–mutant spleen DCs results in nonspecific interaction of DCs with spleen T cells that could lead to suppression of DC function; however, this is not likely to be the case because March-I KO spleen DCs are defective even when isolated from T cell–deficient (Ragl KO) mice.
What then is the reason for decreased APC function and diminished IL-12 secretion from MHC-II ubiquitination–mutant DCs? Clearly this defect is dependent on the absolute amount of ubiquitination-deficient MHC-II on the DC surface because MHC-II K225R hemizygous mice, which express half as much mutant MHC-n, functioned normally. It is interesting to note that transgenic overexpression of MHC-II in mouse APCs also results in immune defects, including severe reductions in immature B cell numbers (34), and overexpression of MHC-II in pancreatic β cells leads to β cell death and diabetes (35, 36). It is important to note that despite high levels of pMHC-II on the surface of MHC-II ubiquitination–deficient DCs, the rate of pMHC-II biosynthesis in these cells is identical to that obtained using DCs isolated from WT mice (8). Oh et al. (13) recently reported that BM-derived DCs from MHC-II K225R mice have diminished ability to activate TCR-transgenic mouse thymocytes. The authors attributed this defect to diminished expression of lipid rafts on the surface of the cells because MHC-H association with lipid rafts is important for efficient T cell activation by APCs (37). However, we have extensively analyzed MHC-H K225R and March-I KO spleen DCs and found no anomaly in lipid rafts expression on the cell surface (based on staining with cholera toxin B subunit), no alteration in expression of lipid raft/tetraspanin protein on the DC surface, and no alteration in the fraction of MHC-H associated with lipid raft microdomains in these DCs (H.J. Kim and P.A. Roche, unpublished observations).
In an attempt to understand why DCs isolated from MHC-II K225R mice were abnormal, we performed scRNA-Seq analysis comparing WT and MHC-II ubiquitination–mutant DCs hoping to find a small number of genes to investigate in detail. Instead, we found significant differences in expression of hundreds of genes in both CD8+ and CD8− subsets of spleen DCs from MHC-II K225R mice; however, it is unclear whether these changes are the cause or the consequence of the altered DC function. This take-home point is clear from the scRNA-Seq data: DCs isolated from MHC-II ubiquitination–mutant mice are not normal DCs, and thus, it is not surprising that immunological phenomena that rely on DC function are abnormal. We also have preliminary evidence that Ag presentation by spleen B cells to CD4 T cells is also defective in these mice, potentially explaining the observed defects in germinal center reactions in March-I KO mice (9). Analysis of March-If1/f1 mice should assist in attributing changes in germinal center reactions to MHC-II on B cells or on DCs.
Curiously, the observed defect in Ag presentation, IL-12 secretion, and gene expression profile in MHC-II ubiquitination–mutant DCs can be reversed by activating the cells with LPS ex vivo. This finding confirms that the CDllc+ cells isolated were in fact DCs, albeit of altered function, and suggests that the freshly isolated DCs from these mice are immature resting DCs. Although identifying the molecular events leading to altered development of these MHC-II ubiquitination defective DCs is beyond the scope of this study, our results clearly demonstrate that despite the fact that they can be loaded with even larger amounts of Ag-specific pMHC-II as compared with WT DCs, MHC-II ubiquitination-mutant DCs are in fact functionally deficient.
Supplementary Material
Acknowledgments
We thank Dr. Michael Kelly for single-cell RNA analysis. This work used the computational resources of the National Institutes of Health High Performance Computing Biowulf cluster (http://hpc.nih.gov).
This work was supported by the Intramural Research Program of the National Institutes of Health (to P.A.R.). Support for the National Cancer Institute Center for Cancer Research Single Cell Analysis Facility and Center for Cancer Research Sequencing Facility was funded by Frederick National Laboratory for Cancer Research Contract HHSN261200800001E.
Abbreviations used in this article:
- BM
bone marrow
- DC
dendritic cell
- HEL
hen egg lysozyrne
- KO
knockout
- MHC-II
MHC class II
- pMHC-II
peptide–MHC class II complex
- scRNA-Seq
single-cell RNA sequencing
- tSNE
t-distributed stochastic neighbor embedding
- WT
wild-type
Footnotes
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Roche PA, and Furuta K 2015. The ins and outs of MHC class II-mediated antigen processing and presentation. Nat. Rev. Immunol. 15: 203–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Walseng E, Furuta K, Bosch B, Weih KA, Matsuki Y, Bakke O, Ishido S, and Roche PA 2010. Ubiquitination regulates MHC class II-peptide complex retention and degradation in dendritic cells. Proc. Natl. Acad. Sci. USA 107: 20465–20470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wilson NS, El-Sukkari D, and Villadangos JA 2004. Dendritic cells constitutively present self antigens in their immature state in vivo and regulate antigen presentation by controlling the rates of MHC class II synthesis and endocytosis. Blood 103: 2187–2195. [DOI] [PubMed] [Google Scholar]
- 4.Cella M, Engering A, Pinet V, Pieters J, and Lanzavecchia A 1997. Inflammatory stimuli induce accumulation of MHC class II complexes on dendritic cells. Nature 388: 782–787. [DOI] [PubMed] [Google Scholar]
- 5.Pierre P, Turley SJ, Gatti E, Hull M, Meltzer J, Mirza A, Inaba K, Steinman RM, and Mellman I 1997. Developmental regulation of MHC class II transport in mouse dendritic cells. Nature 388: 787–792. [DOI] [PubMed] [Google Scholar]
- 6.Matsuki Y, Ohmura-Hoshino M, Goto E, Aoki M, Mito-Yoshida M, Uematsu M, Hasegawa T, Koseki H, Ohara O, Nakayama M, et al. 2007. Novel regulation of MHC class II function in B cells. EMBO J. 26: 846–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.De Gassart A, Camosseto V, Thibodeau J, Ceppi M, Catalan N, Pierre P, and Gatti E 2008. MHC class II stabilization at the surface of human dendritic cells is the result of maturation-dependent MARCH I down-regulation. Proc. Natl. Acad. Sci. USA 105: 3491–3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cho KJ, Walseng E, Ishido S, and Roche PA 2015. Ubiquitination by March-I prevents MHC class II recycling and promotes MHC class II turnover in antigen-presenting cells. Proc. Natl. Acad. Sci. USA 112: 10449–10454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bannard O, McGowan SJ, Ersching J, Ishido S, Victora GD, Shin JS, and Cyster JG 2016. Ubiquitin-mediated fluctuations in MHC class II facilitate efficient germinal center B cell responses. J. Exp. Med. 213: 993–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shin JS, Ebersold M, Pypaert M, Delamarre L, Hartley A, and Mellman I 2006. Surface expression of MHC class II in dendritic cells is controlled by regulated ubiquitination. Nature 444: 115–118. [DOI] [PubMed] [Google Scholar]
- 11.van Niel G, Wubbolts R, Ten Broeke T, Buschow SI, Ossendorp FA, Melief CJ, Raposo G, van Balkom BW, and Stoorvogel W 2006. Dendritic cells regulate exposure of MHC class II at their plasma membrane by oligoubiquitination. Immunity 25: 885–894. [DOI] [PubMed] [Google Scholar]
- 12.Oh J, Wu N, Baravalle G, Cohn B, Ma J, Lo B, Mellman I, Ishido S, Anderson M, and Shin JS 2013. MARCH 1-mediated MHCII ubiquitination promotes dendritic cell selection of natural regulatory T cells. J. Exp. Med. 210: 1069–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oh J, Perry JSA, Pua H, Irgens-Möller N, Ishido S, Hsieh CS, and Shin JS 2018. MARCH 1 protects the lipid raft and tetraspanin web from MHCE proteotoxicity in dendritic cells. J. Cell Biol. 217: 1395–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Majdoubi A, Lee JS, Balood M, Sabourin A, DeMontigny A, Kishta OA, Moulefera MA, Galbas T, Yun TJ, Talbot S, et al. 2019. Downregulation of MHC class II by ubiquitination is required for the migration of CD206+ dendritic cells to skin-draining lymph nodes. J. Immunol. 203: 2887–2898. [DOI] [PubMed] [Google Scholar]
- 15.Ohmura-Hoshino M, Matsuki Y, Mito-Yoshida M, Goto E, Aoki-Kawasumi M, Nakayama M, Ohara O, and Ishido S 2009. Cutting edge: requirement of MARCH-I-mediated MHC II ubiquitination for the maintenance of conventional dendritic cells. J. Immunol. 183: 6893–6897. [DOI] [PubMed] [Google Scholar]
- 16.Zhong G, Reis e Sousa C, and Germain RN 1997. Production, specificity, and functionality of monoclonal antibodies to specific peptide-major histocompatibility complex class II complexes formed by processing of exogenous protein. Proc. Natl. Acad. Sci. USA 94: 13856–13861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.AYu R, Rath S, Preston-Hurlburt P, Murphy DB, and Janeway CA Jr. 1991. On the complexity of self. Nature 353: 660–662. [DOI] [PubMed] [Google Scholar]
- 18.Hiltbold EM, Poloso NJ, and Roche PA 2003. MHC class Il-peptide complexes and APC lipid rafts accumulate at the immunological synapse. J. Immunol. 170: 1329–1338. [DOI] [PubMed] [Google Scholar]
- 19.Leys C, Ley C, Klein O, Bernard P, and Licata L 2013. Detecting outliers: do not use standard deviation around the mean, use absolute deviation around the median. J. Exp. Soc. Psychol. 49: 764–766. [Google Scholar]
- 20.Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM III, Hao Y, Stoeckius M, Smibert P, and Satija R 2019. Comprehensive integration of single-cell data. Cell 177: 1888–1902.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hafemeister C, and Satija R 2019. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 20: 296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aran D, Looney AP, Liu L, Wu E, Fong V, Hsu A, Chak S, Naikawadi RP, Wolters PJ, Abate AR, et al. 2019. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol. 20: 163–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Robinette ML, Fuchs A, Cortez VS, Lee JS, Wang Y, Durum SK, Gilfillan S, and Colonna M, Immunological Genome Consortium. 2015. Transcriptional programs define molecular characteristics of innate lymphoid cell classes and subsets. Nat. Immunol. 16: 306–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McGinnis CS, Murrow LM, and Gartner ZJ 2019. DoubletFinder: doublet detection in single-cell RNA sequencing data using artificial nearest neighbors. Cell Syst. 8: 329–337.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Becht E, Mclnnes L, Healy J, Dutertre CA, Kwok IWH, Ng LG, Ginhoux F, and Newell EW 2018. Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol DOI: 10.1038/nbt4314. [DOI] [PubMed] [Google Scholar]
- 26.Finak G, McDavid A, Yajima M, Deng J, Gersuk V, Shalek AK, Slichter CK, Miller HW, McElrath MJ, Prlic M, et al. 2015. MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol. 16: 278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tze LE, Horikawa K, Domaschenz H, Howard DR, Roots CM, Rigby RJ, Way DA, Ohmura-Hoshino M, Ishido S, Andoniou CE, et al. 2011. CD83 increases MHC II and CD86 on dendritic cells by opposing IL-10-driven MARCH1-mediated ubiquitination and degradation. J. Exp. Med. 208: 149–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Furuta K, Walseng E, and Roche PA 2013. Internalizing MHC class II-peptide complexes are ubiquitinated in early endosomes and targeted for lysosomal degradation. Proc. Natl. Acad. Set. USA 110: 20188–20193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gauvreau ME, Côté MH, Bourgeois-Daigneault MC, Rivard LD, Xiu F, Brunet A, Shaw A, Steimle V, and Thibodeau J 2009. Sorting of MHC class II molecules into exosomes through a ubiquitin-independent pathway. Traffic 10: 1518–1527. [DOI] [PubMed] [Google Scholar]
- 30.Buschow SI, Nolte-’t Hoen EN, van Niel G, Pols MS, ten Broeke T, Lauwen M, Ossendorp F, Melief CJ, Raposo G, Wubbolts R, et al. 2009. MHC II in dendritic cells is targeted to lysosomes or T cell-induced exosomes via distinct multivesicular body pathways. Traffic 10: 1528–1542. [DOI] [PubMed] [Google Scholar]
- 31.Borges TJ, Murakami N, Machado FD, Murshid A, Lang BJ, Lopes RL, Bellan LM, Uehara M, Antunes KH, Pérez-Saéz MJ, et al. 2018. March1-dependent modulation of donor MHC II on CD103+ dendritic cells mitigates alloimmunity. Nat. Commun. 9: 3482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wilson KR, Liu H, Healey G, Vuong V, Ishido S, Herold MJ, Villadangos JA, and Mintem JD 2018. MARCH1-mediated ubiquitination of MHC II impacts the MHC I antigen presentation pathway. PLoS One 13: e0200540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ishikawa R, Kajikawa M, and Ishido S 2014. Loss of MHC II ubiquitination inhibits the activation and differentiation of CD4 T cells. Int. Immunol. 26: 283–289. [DOI] [PubMed] [Google Scholar]
- 34.Gilfillan S„, Aiso S, Michie SA, and McDevitt HO 1990. Immune deficiency due to high copy numbers of an Ak beta transgene. Proc. Natl. Acad Set. USA 87: 7319–7323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sarvetnick N, Liggitt D, Pitts SL, Hansen SE, and Stewart TA 1988. Insulin-dependent diabetes mellitus induced in transgenic mice by ectopic expression of class II MHC and interferon-gamma. Cell 52: 773–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lo D, Burkly LC, Widera G, Cowing C, Flavell RA, Palmiter RD, and Brinster RL 1988. Diabetes and tolerance in transgenic mice expressing class II MHC molecules in pancreatic beta cells. Cell 53: 159–168. [DOI] [PubMed] [Google Scholar]
- 37.Anderson HA, Hiltbold EM, and Roche PA 2000. Concentration of MHC class II molecules in lipid rafts facilitates antigen presentation. Nat. Immunol. 1: 156–162. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.