

Abstract
Aggregation of the multifunctional RNA‐binding protein TDP‐43 defines large subgroups of amyotrophic lateral sclerosis and frontotemporal dementia and correlates with neurodegeneration in both diseases. In disease, characteristic C‐terminal fragments of ~25 kDa ("TDP‐25") accumulate in cytoplasmic inclusions. Here, we analyze gain‐of‐function mechanisms of TDP‐25 combining cryo‐electron tomography, proteomics, and functional assays. In neurons, cytoplasmic TDP‐25 inclusions are amorphous, and photobleaching experiments reveal gel‐like biophysical properties that are less dynamic than nuclear TDP‐43. Compared with full‐length TDP‐43, the TDP‐25 interactome is depleted of low‐complexity domain proteins. TDP‐25 inclusions are enriched in 26S proteasomes adopting exclusively substrate‐processing conformations, suggesting that inclusions sequester proteasomes, which are largely stalled and no longer undergo the cyclic conformational changes required for proteolytic activity. Reporter assays confirm that TDP‐25 impairs proteostasis, and this inhibitory function is enhanced by ALS‐causing TDP‐43 mutations. These findings support a patho‐physiological relevance of proteasome dysfunction in ALS/FTD.
Keywords: ALS, neurodegeneration, phase separation, proteasome, TDP‐43
Subject Categories: Molecular Biology of Disease, Neuroscience
TDP‐25, a C‐terminal fragment of TDP‐43 found in ALS and FTD patients, forms cytoplasmic inclusions with gel‐like properties in primary neurons. Proteasome enrichment and impaired proteostasis support the relevance of proteasome dysfunction in ALS/FTD.

Introduction
TDP‐43 aggregation is the disease‐defining pathological hallmark in > 90% of patients with amyotrophic lateral sclerosis (ALS) and ~45% of patients with frontotemporal dementia (FTD) (Gao et al, 2017; Prasad et al, 2019; Tziortzouda et al, 2021). However, we still have a limited understanding of the native structure of TDP‐43 inclusions and their role in disease (Gao et al, 2017; Chien et al, 2021). Predominantly, cytoplasmic neuronal inclusions of TDP‐43 correlate strongly with regional neuron loss in spinal cord, motor cortex, or frontal‐temporal cortical regions (Mackenzie et al, 2013). These inclusions are enriched in 25‐35 kDa C‐terminal fragments of TDP‐43 that contain a glycine‐rich low‐complexity region (Neumann et al, 2006; Igaz et al, 2008). Rare autosomal dominant ALS‐causing mutations in TDP‐43 cluster in this region, although their pathomechanism is still poorly understood, and they have only modest effects in vitro and in knockin models (Prasad et al, 2019). However, overexpression of the inclusion‐forming 25 kDa C‐terminal fragment ("TDP‐25") triggers neurodegeneration in mice even without disease‐associated mutations (Walker et al, 2015).
Distinct biophysical mechanisms can drive inclusion formation in the context of neurodegenerative diseases: (i) formation of highly insoluble amyloid fibrils with cross β‐sheet conformation (Eisenberg & Jucker, 2012), (ii) liquid‐liquid phase separation (LLPS) into highly dynamic liquid‐like droplets (Gomes & Shorter, 2019), which may solidify and adopt amyloid‐like conformations under pathological conditions (Kato et al, 2012; Patel et al, 2015; Qamar et al, 2018). By adopting aberrant conformations, aggregated proteins may engage in toxic cellular interactions (Olzscha et al, 2011; Hipp et al, 2019). Rapid advances in cryo‐electron microscopy (cryo‐EM) elucidated the structure of amyloid fibrils formed by various disease‐associated aggregating proteins purified from patient tissue at near‐atomic resolution, most recently including TDP‐43 (Creekmore et al, 2021; Arseni et al, 2022). Cryo‐EM revealed a unique amyloid conformation of TDP‐43 fibrils purified from patients, with little resemblance to fibrillar structures derived from recombinant TDP‐43 C‐terminal region (at pH 4) and shorter synthetic peptide fragments (Guenther et al, 2018; Cao et al, 2019; Li et al, 2021; Arseni et al, 2022). Full‐length TDP‐43 has also been reported to form liquid droplets through LLPS in vitro (Conicella et al, 2016), and RNA‐free TDP‐43 can assemble into so‐called anisosomes consisting of a liquid TDP‐43 shell and a HSP70 core in the nucleus, suggesting that TDP‐43 condensates solidify into fibrils in disease (Yu et al, 2021; Arseni et al, 2022). ALS/FTD‐causing mutations in C9orf72, OPTN, SQSTM1, TBK1, UBQLN2, and VCP are linked to the ubiquitin‐proteasome system (UPS) and autophagy, which can clear aggregated and phase‐separated proteins, suggesting the proteostasis system is of particular importance at least in genetic ALS/FTD (Gitcho et al, 2009; Deng et al, 2011; Hipp et al, 2019). This view is reinforced by the observation that proteasome inhibition leads to the formation of TDP‐43 anisomes (Yu et al, 2021).
Cryo‐electron tomography (cryo‐ET) is a powerful method to visualize neurotoxic inclusions within their native cellular environment complementing the analysis of purified fibrils. Cryo‐ET studies have revealed a striking heterogeneity of ultrastructure and pathomechanisms for neuronal inclusion proteins (Bauerlein et al, 2020). In particular, we have used cryo‐ET to show that amyloid‐like poly‐GA inclusions linked to C9orf72 ALS/FTD disrupt neuronal proteostasis by sequestering proteasomes in a rare transition state, suggesting the cyclic conformational changes required for substrate processing (Collins & Goldberg, 2017) are altered by the association with the inclusions (Guo et al, 2018). Here, we aimed to elucidate gain‐of‐function mechanisms and the structure of cytoplasmic TDP‐43 aggregates found in sporadic and most genetic ALS/FTD cases. We focused on the aggregation‐prone TDP‐25 fragment (residues 220‐414 of full‐length human TDP‐43) (Neumann et al, 2006; Zhang et al, 2009) using a pipeline of cryo‐ET, proteomics, and functional assays. Neuronal TDP‐25 inclusions show amorphous morphology and gel‐like biophysical properties. The inclusions are enriched in stalled 26S proteasomes similar to poly‐GA inclusions, suggesting inhibition of the UPS is a common pathomechanism in ALS/FTD.
Results and Discussion
TDP‐25 inclusions are amorphous and enriched in 26S proteasomes
Expression of GFP‐tagged TDP‐25 in rat primary neurons resulted in abundant cytoplasmic inclusions phosphorylated at disease‐specific sites (Fig 1A) (Hasegawa et al, 2008). To explore the pathomechanisms enhanced by known pathogenic mutations, we additionally used a TDP‐25 variant containing eight mutations (G290A, G294V, G298S, A315T, M337V, G348C, N352S, and A382T) that individually cause ALS (Prasad et al, 2019). Wild‐type and mutant GFP‐TDP‐25 formed inclusions predominantly in the cell soma that were of similar appearance by light microscopy and showed a comparable degree of disease‐specific phosphorylation (TDP‐25 wild‐type: 95.8 ± 2.9% vs. TDP‐25 mutant: 96.2 ± 3.1%, mean ± SD from n = 3 biological replicates) (Fig 1A). While GFP‐TDP‐43 was almost completely soluble in RIPA buffer, a large fraction of wild‐type and mutant GFP‐TDP‐25 was only solubilized upon sequential extraction of the RIPA‐insoluble material with 2% SDS (Fig 1B) indicative of stronger intermolecular interactions in aggregating TDP‐25. Solubility of wild‐type and mutant TDP‐25 was not significantly different (Fig 1B).
Figure 1. TDP‐25 forms amorphous inclusions enriched in proteasomes.
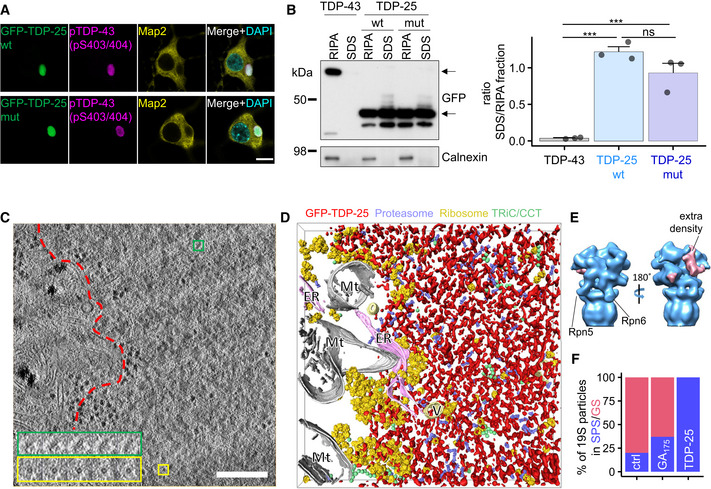
- Primary rat hippocampal neurons were transduced with constructs encoding for GFP‐tagged TDP‐25 (amino acids 220‐414 of TDP‐43) on day 5 in vitro and cultured for 8 additional days (DIV5+8). Immunofluorescence of GFP‐TDP‐25 wild‐type and variant containing eight ALS‐causing mutations shows disease‐related phosphorylation at serines 403/404. Counterstain to label the neuronal cytoskeleton (MAP2) and nuclei (DAPI). Scale bar = 10 µm.
- Hippocampal neurons transduced with GFP‐TDP‐25 constructs (DIV5+8) or GFP‐TDP‐43 (DIV5+4 due to higher toxicity) were sequentially extracted with RIPA buffer, followed by 2% SDS buffer and analyzed via immuoblotting. Left panel shows representative immunoblot for GFP and loading control calnexin. Upper arrow marks GFP‐TDP‐43 band, lower arrow marks GFP‐TDP‐25 band. Right panel shows quantification of the ratio of the respective GFP densiometric signal in SDS extract to RIPA extract. Barplots showing means ± SD from n = 3 independent experiments. TDP‐43 (0.039 ± 0.029, mean ± CI) vs. TDP‐25 wt (1.22 ± 0.296) vs. TDP‐25 mut (0.928 ± 0.564): F(2,6) = 51.4, ***P = 0.000168, η² = 0.94. TDP‐43 vs. TDP‐25 wt: ***P = 1.67*10−4, TDP‐43 vs. TDP‐25 mut: ***P = 7.983*10−4, TDP‐25 wt vs. TDP‐25 mut: P = 0.1182, One‐way ANOVA with Tukey's post‐hoc test.
- Tomographic slice of an aggregate within a GFP‐TDP‐25 wild‐type transduced neuron (DIV5+8). Colored boxes show a series of higher magnification tomographic slices of representative protein complexes detected in the tomogram. Green boxes show side views of single‐capped, yellow boxes show ring‐like cross‐sections of 26S proteasomes. Red dotted line segments aggregate area. Scale bar = 200 nm.
- 3D rendering of the aggregate shown in (C). Amorphous aggregate material is labeled in red, proteasomes in violet, ribosomes in yellow, TRiC/CCT chaperones in green, mitochondria (Mt) in white, endoplasmic reticulum (ER) in pink and other vesicles (V) in light yellow. The irregular aggregate structures were approximately segmented using a threshold‐based approach for visualization purposes. Average structures of proteasomes, ribosomes, and TRiC/CCT were pasted at the locations and orientations determined by template matching. See also Movie EV1.
- Subtomogram averaging of macromolecules in GFP‐TDP‐25 inclusions reveals the proteasome structure at ~20 Å resolution (see Fig EV1). The positions of Rpn5/PSMD11 and Rpn6/PSDM12 are indicated. Prominent extra densities in the substrate binding region are colored in pink in the 3D rendering.
- Classification based on the conformation of the regulatory particle in TDP‐25 inclusions compared with non‐transduced control neurons (Asano et al, 2015) and poly‐GA inclusions (Guo et al, 2018). GS: Ground State, SPS: Substrate Processing State. Due to the uncertainties inherent to the classification procedure, it is possible that a small fraction of particles adopted other conformations.
To investigate the downstream consequences of TDP‐25 aggregation, we analyzed neuronal GFP‐TDP‐25 inclusions in situ using cryo‐ET (Bauerlein et al, 2020). To that end, neurons were cultured, transduced with GFP‐tagged TDP‐25 constructs (wild‐type and mutant) and vitrified on EM grids. Vitrified grids were imaged by cryo‐light microscopy to determine the subcellular location of TDP‐25 inclusions. Correlative light‐electron microscopy (CLEM) was performed to locate back the inclusions in a dual‐beam cryo‐focused ion beam/scanning electron microscope (cryo‐FIB/SEM). Thin (100–200 nm) lamellae were prepared at those positions. Finally, lamellae were transferred to a cryo‐transmission electron microscope, where they were imaged at high magnification. Wild‐type TDP‐25 inclusions appeared amorphous and seemingly lacked fibrillar structure, although they were clearly demarcated within the cytoplasm (Fig 1C and D, Movie EV1). These findings are in stark contrast to previously investigated poly‐Q, poly‐GA, and α‐synuclein neuronal inclusions, which show amyloid‐like conformation in neurons in our cryo‐ET pipeline (Bauerlein et al, 2017; Guo et al, 2018; Trinkaus et al, 2021). Introducing the eight ALS‐causing mutations had no obvious effect on the morphology of TDP‐25 inclusion (Fig EV1A).
Figure EV1. Proteasomes are enriched in TDP‐25 inclusions.
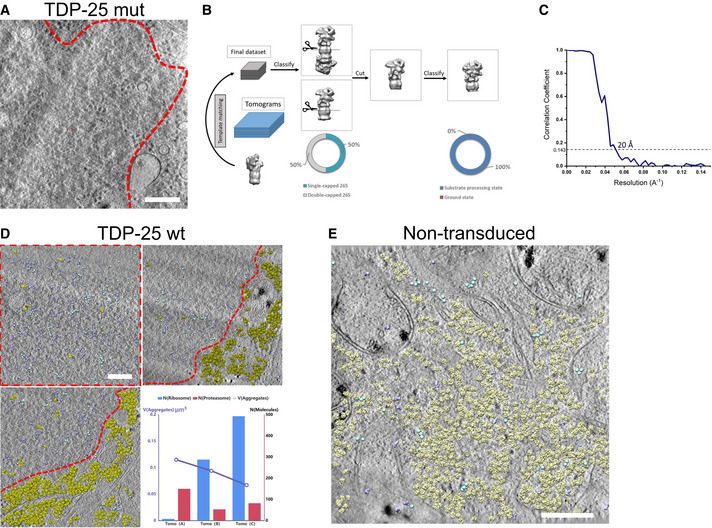
- Tomographic slice of an aggregate within a neuron transduced with GFP‐TDP‐25 mutant construct (DIV5+8). Red dotted line segments aggregate area. Scale bar = 200 nm.
- Workflow of subtomogram averaging and classification. Subtomograms were identified using a low‐resolution single‐capped proteasome as template. All proteasomes were firstly classified into single‐capped or double‐capped. To further analyze the conformation of the regulatory particles, all proteasomes were cut in silico between the β‐rings of the core particle, resulting in two independent particles for double‐capped ones. 50% of the particles were assigned to the single and double‐capped classes, respectively. Cut out regulatory particles were merged and subjected to a further round of classification. All regulatory particles were assigned to substrate processing conformations. However, due to the uncertainties inherent to the classification procedure, it is possible that a small fraction of particles adopted other conformations.
- Gold‐standard Fourier shell correlation curve of the proteasome structure showing a resolution of 20 Å.
- Molecular mapping in three tomograms of GFP‐TDP‐25 wild‐type inclusions in transduced neurons (DIV5+8). Regions containing GFP‐TDP‐25 are outlined in red. For the whole tomogram, proteasomes (purple), TRiC (cyan) and ribosomes (yellow) are mapped to their original positions and orientations using the information from template matching and subtomogram averaging. The numbers of proteasomes and ribosomes detected in the tomograms are plotted versus the volume of the tomogram. Scale bar = 200 nm.
- Molecular mapping in a tomogram recorded on a non‐transduced control neuron, with proteasomes (purple), ribosomes (yellow) and TRiC (cyan) plotted as in Fig EV1D. This tomogram contains 24 proteasomes, 1,222 ribosomes and 73 TRiC molecules. Scale bar = 200 nm.
Similar to our findings in C9orf72 poly‐GA inclusions (Guo et al, 2018), we detected ring‐like structures (Fig 1C) accumulating within TDP‐25 inclusions. Sub‐tomogram averaging of these objects (Fig EV1B) converged to a proteasome structure at ~20 Å resolution (Fig 1E, Fig EV1C). Interestingly, an extra density that was not accounted for by proteasomal subunits was present on the proteasome regulatory particle, possibly reflecting substrates or adaptor proteins (Fig 1E).
Whereas ribosomes were largely excluded from TDP‐25 inclusions, we found an approximately eightfold enrichment of proteasomes compared with proteasome concentration in control neurons (Asano et al, 2015; Guo et al, 2018; Figs 1D and EV1D and E). Strikingly, classification based on the conformation of 19S regulatory particles revealed that virtually all proteasome particles within the inclusions were in substrate processing states (Fig 1E and F). In comparison, only 20 and 37% of proteasomes were in substrate processing states in control neurons and poly‐GA inclusions, respectively (Asano et al, 2015; Guo et al, 2018; Fig 1F). It should be noted that, due to the uncertainties inherent to the classification procedure, we cannot rule out that a small fraction of particles adopted other conformations. The little to no detectable amount of ground state proteasomes suggests that proteasomes inside TDP‐25 inclusions are stalled, as proteasome function requires cyclic transition through activated and ground states (Collins & Goldberg, 2017).
Amorphous TDP‐25 inclusions have gel‐like properties
Given the amorphous appearance of TDP‐25 inclusions, we asked whether they may represent phase‐separated liquid droplets. Thus, we analyzed the mobility of GFP‐TDP‐25 in neuronal inclusions using fluorescence recovery after photobleaching (FRAP) in comparison with known liquid and solid reference proteins, that is, nucleolar NPM1 and inclusion‐forming poly‐Q, respectively (Bauerlein et al, 2017; Frottin et al, 2019). In contrast to TDP‐25, GFP‐NPM1 fluorescence recovered within seconds after bleaching, consistent with high mobility of the protein within the liquid‐like nucleolus (Fig 2A and B). This clearly argues against a liquid droplet character of TDP‐25 inclusions. Some neurons showed diffuse cytoplasmic TDP‐25 expression without inclusion formation. Photobleaching of such diffuse TDP‐25 resulted in very quick recovery suggesting that inclusion formation greatly reduces mobility (Fig EV2). Moreover, nuclear full‐length TDP‐43 showed a much higher mobile fraction than TDP‐25 inclusions (Fig EV2), in line with its good solubility (Fig 1B). However, compared with fibrillar poly‐Q (huntingtin exon 1 containing 97 glutamines fused to GFP; Htt97Q‐GFP), a classical amyloid previously shown to interact with ER membranes (Bauerlein et al, 2020), TDP‐25 mobility was much higher (~10% vs. ~25% recovery at 22 min, Fig 2C and D).
Figure 2. TDP‐25 inclusions are neither liquid‐like nor solid‐like.
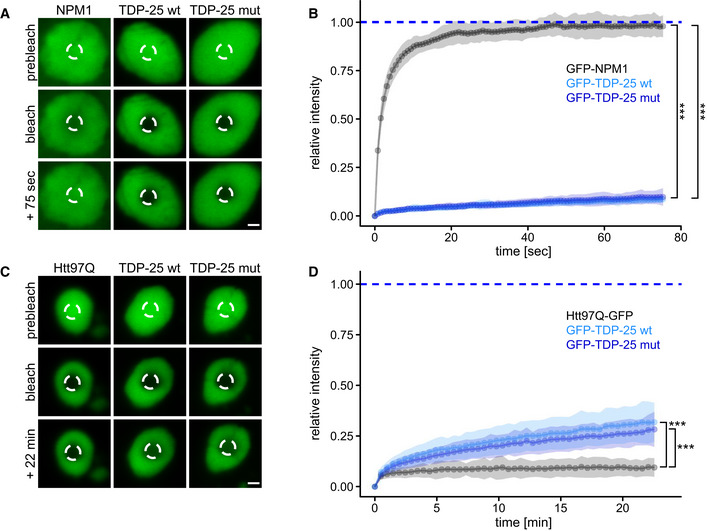
-
A–DPrimary rat hippocampal neurons were transduced with GFP‐TDP‐25 variants, GFP‐NPM1 or Htt97Q‐GFP and analyzed by fluorescence recovery after photobleaching (FRAP) at DIV5+8, except Htt97Q‐GFP which was analyzed at DIV5+6 to avoid excessive toxicity. Example images with indicated bleach regions (dashed circles) are shown (A, C). Scale bar = 1.5 µm. Resulting normalized FRAP curves (relative fluorescence intensity over time) with values representing means ± SD are shown in (B, D). (B) Comparison of the averaged recovery fractions of the last six timepoints obtained from three independent experiments. GFP‐NPM1 (0.982 ± 0.019, mean ± CI, n = 37 cells) vs. GFP‐TDP‐25 wt (0.097 ± 0.013, n = 35 cells) vs. GFP‐TDP‐25 mut (0.085 ± 0.006, n = 38 cells): H(1) = 74.134, df = 2, ***P < 2.2*10−16, η²[H] = 0.674, Kruskal‐Wallis Test. GFP‐NPM1 vs. GFP‐TDP‐25 wt: ***P < 2*10−16, GFP‐NPM1 vs. GFP‐TDP‐25 mut: ***P < 2*10−16, GFP‐TDP‐25 wt vs. GFP‐TDP‐25 mut: P = 0.11, Pairwise Wilcoxon Rank Sum Tests with Benjamini‐Hochberg correction. In (D), the recovery fractions averaged at >20 min timepoints were compared from four independent experiments. Htt97Q‐GFP (0.094 ± 0.017, n = 25 cells) vs. GFP‐TDP‐25 wt (0.312 ± 0.041, n = 23 cells) vs. GFP‐TDP‐25 mut (0.271 ± 0.029, n = 24 cells): H(1) = 46.376, df = 2, ***P = 8.501*10−11, η²[H] = 0.643, Kruskal‐Wallis Test. Htt97Q‐GFP vs. GFP‐TDP‐25 wt: ***P = 1.9*10−11, Htt97Q‐GFP vs. GFP‐TDP‐25 mut: ***P = 4.3*10−12, GFP‐TDP‐25 wt vs. GFP‐TDP‐25 mut: P = 0.12, Pairwise Wilcoxon Rank Sum Tests with Benjamini‐Hochberg correction.
Figure EV2. TDP‐25 inclusions are less mobile than TDP‐43.
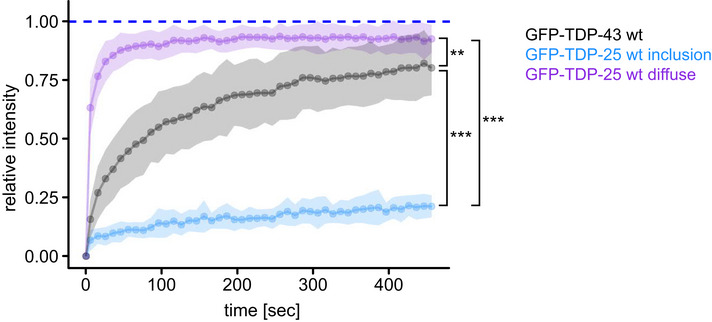
Rat primary hippocampal neurons were transduced with GFP‐TDP‐25 wild‐type (DIV5+8) or GFP‐TDP‐43 wild‐type (DIV9+4) and analyzed by FRAP. The resulting normalized FRAP curves (relative fluorescence intensity over time) with values representing means ± SD are shown. Comparison of the averaged recovery fractions of the last six timepoints obtained from three independent experiments: GFP‐TDP‐43 (0.802 ± 0.06, mean ± CI, n = 20 cells) vs. GFP‐TDP‐25 inclusion (0.209 ± 0.019, n = 18 cells) vs. GFP‐TDP‐25 diffuse (0.926 ± 0.045, n = 10 cells): H(1) = 36.295, df = 2, P = 1.314*10−8, η²[H] = 0.762, Kruskal‐Wallis Test. GFP‐TDP‐43 vs. GFP‐TDP‐25 inclusion: ***P = 1.8*10−10, GFP‐TDP‐43 vs. GFP‐TDP‐25 diffuse: **P = 0.0033, GFP‐TDP‐25 inclusion vs. GFP‐TDP‐25 diffuse: ***P = 2.3*10−7, Pairwise Wilcoxon Rank Sum Tests with Benjamini‐Hochberg correction.
Overall, these findings suggest that non‐fibrillar TDP‐25 adopts a specific conformation within the inclusions that is best described by a gel‐like state. Solidification into hydrogels has been reported for phase‐separated droplets of FUS, involving the formation of amyloid‐like fibrils (Murray et al, 2017; Qamar et al, 2018). A similar process has been postulated for TDP‐43, and very recently, amyloid‐like fibrils of TDP‐43 have been obtained from ALS/FTD patient brains (Guenther et al, 2018; Babinchak et al, 2019; Cao et al, 2019; Gasset‐Rosa et al, 2019; Mann et al, 2019; Li et al, 2021; Arseni et al, 2022). Taken together, GFP‐TDP‐25 inclusions are clearly less dynamic than classical phase‐separated compartments, but more dynamic than fibrillar aggregates.
TDP‐25 sequesters and impairs the proteasome
Phase‐separated inclusions may form through weak multivalent interactions with other cellular components (Martin & Mittag, 2018; Gomes & Shorter, 2019). To further investigate this gel‐like state and the molecular consequences of the TDP‐25 inclusions, we analyzed the interactomes of TDP‐25 and full‐length TDP‐43 using mass‐spectrometry‐based proteomics. We detected several hundred specific interactors for full‐length TDP‐43 and TDP‐25 in primary neurons, in comparison with the GFP‐only control (Figs 3A and B, and EV3A, Dataset EV1). TDP‐43‐specific interactors were strongly enriched in splicing‐associated proteins consistent with the prominent involvement of TDP‐43 in RNA splicing and in line with previous interactome studies for TDP‐43 (Figs 3A and D, and EV3A; Prasad et al, 2019).
Figure 3. TDP‐25 sequesters and impairs the proteasome.
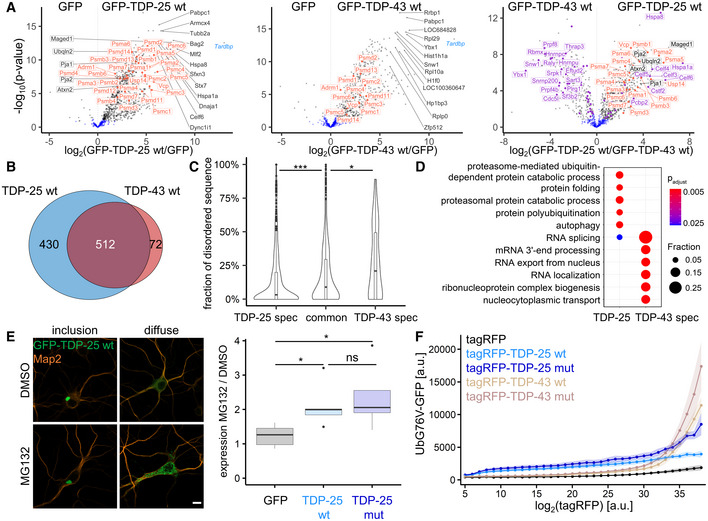
- Rat primary cortical neurons were transduced with GFP‐TDP‐25 wild‐type, GFP‐TDP‐43 wild‐type or GFP at DIV5. Due to high toxicity, GFP‐TDP‐43 (n = 6) and GFP control samples (n = 6) were harvested at DIV5+4, while GFP‐TDP‐25 (n = 5) and additional GFP control samples (n = 6) were harvested at DIV5+8. Immunoprecipitates were analyzed by LC‐MS/MS. Volcano plots indicate enrichment and statistical significance (gray dots: FDR‐corrected P < 0.05). Proteins associated with GO term “proteasome complex” (GO:0000502) are labeled in orange and proteins associated with GO term “RNA splicing” (GO:0008380) in purple. Full data are listed in Dataset EV1. A comparison with previous proteomic studies is shown in Fig EV3A.
- Overlap of GFP‐TDP‐25 and GFP‐TDP‐43 interactomes. Significant interactors in FDR‐based approach (see Methods) were considered for comparison.
- Combined violin and box plot (box shows lower quartile, median and upper quartile, whiskers extend to the most extreme data point within 1.5 the interquartile range of the box) of the proportion of low‐complexity regions (D2P2) of wild‐type GFP‐TDP‐25‐specific, GFP‐TDP‐43‐specific or shared/common interactors. TDP‐25‐specific (0.144 ± 0.021, n = 432 Uniprot IDs) vs. TDP‐43‐specific (0.266 ± 0.061, n = 78) vs. common interactors (0.184 ± 0.02, n = 481): H(1) = 22.276, df = 2, P = 1.455*10−5, η²[H] = 0.0205, Kruskal‐Wallis test. TDP‐25‐specific vs. TDP‐43‐specific: ***P = 0.00054, TDP‐25‐specific vs. common: ***P = 0.00035, TDP‐43‐specific vs. common interactors: *P = 0.04893, Pairwise Wilcoxon Rank Sum Tests with Benjamini‐Hochberg correction.
- Gene ontology (GO) enrichment analysis (biological process) in interactors specific for either wild‐type GFP‐TDP‐25 or GFP‐TDP‐43. Full list of GO terms is found in Dataset EV2.
- Immunofluorescence of hippocampal neurons transduced with GFP, GFP‐TDP‐25 wild‐type or mutant and treated with MG132 (10 µM, 14 h, DIV5+8). Representative images of GFP‐TDP‐25 inclusions and diffuse cytoplasmic staining. Automated quantification of GFP intensity normalized to the number of DAPI‐stained nuclei (omitted here for clarity) in MG132‐treated cells compared to DMSO control. Counterstain to label the neuronal cytoskeleton (MAP2). Scale bar = 10 µm. Box plot as in Fig 3C. GFP (1.23 ± 0.393, n = 5 independent experiments) vs. GFP‐TDP‐25 wt (2.11 ± 0.804, n = 5) vs. GFP‐TDP‐25 mut (2.36 ± 1.16, n = 5): H(1) = 7.34, df = 2, P = 0.02548, η²[H] = 0.445, Kruskal‐Wallis test. GFP vs. GFP‐TDP‐25 wt: *P = 0.048, GFP vs. GFP‐TDP‐25 mut: *P = 0.048, GFP‐TDP‐25 wt vs. GFP‐TDP‐25 mut: P = 0.690, Pairwise Wilcoxon Rank Sum Tests with Benjamini‐Hochberg correction.
- A HEK293 cell line expressing the proteostasis reporter UbG76V‐GFP was transiently transfected with tagRFP, tagRFP‐TDP‐25 wild‐type, tagRFP‐TDP‐25 mutant, tagRFP‐TDP‐43 wild‐type or tagRFP‐TDP‐43 mutant. UbG76V‐GFP levels were analyzed by flow cytometry 72 h after transfection. The relationship of binned tagRFP fluorescence and UbG76V‐GFP fluorescence is plotted. Values shown are means ± SD obtained from n = 4 independent experiments. For clarity, adjusted P‐values of pairwise comparisons within each tagRFP intensity bin are shown in Fig EV3C. Note that at lower expression levels (bin < 30) both TDP‐25 variants induce higher UbG76V‐GFP levels than their respective full‐length counterparts, indicative of stronger impairment of proteostasis. The full‐length variants show meaningful accumulation of the reporter levels only at the high‐expression levels. The mutant versions of TDP‐25 and TDP‐43 lead to an increased reporter signal at higher expression levels compared with wild‐type variants.
Figure EV3. Additional interactome analyses.
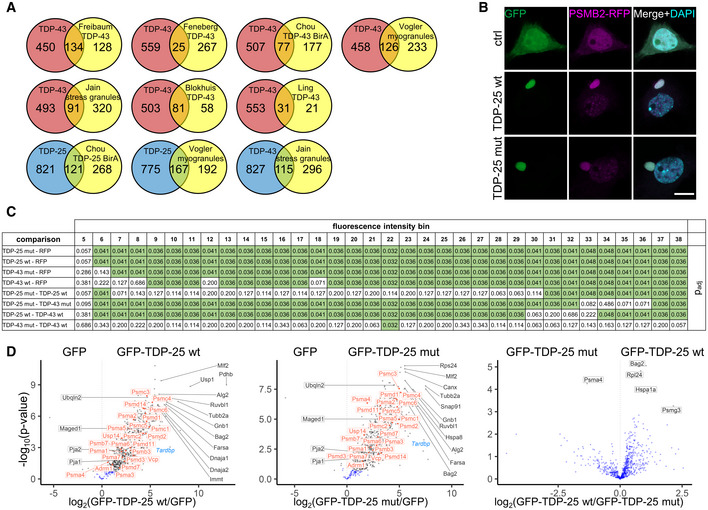
- Proteins identified in the GFP‐TDP‐43 interactome from Fig 3A (uniquely or in a cluster) were compared to published datasets (Freibaum et al, 2010; Ling et al, 2010; Blokhuis et al, 2016; Chou et al, 2018; Feneberg et al, 2020). LOC and RGD gene names that usually represent minor components of the identified clusters were removed, because such entries could not be mapped based on gene names between species. A core set of 32 proteins from our TDP‐43 interactome was found in four (DDX3X, DHX9, ELAVL1, HNRNPA0, HNRNPD, HNRNPH1, HNRNPL, HNRNPM, HNRNPR, ILF2, MATR3, PABPC4, RALY, YBX1) or three (DDX5, DDX6, EFTUD2, EIF3A, HNRNPA3, HNRNPAB, HNRNPC, HNRNPK, HNRNPU, HNRNPUL2, ILF3, NONO, NOP56, NOP58, PABPC1, SNRNP200, SNRPA1, SSB) interactome datasets (Freibaum et al, 2010; Ling et al, 2010; Blokhuis et al, 2016; Feneberg et al, 2020). There was also substantial overlap of our TDP‐25 interactome with a BirA proximity labeling dataset for TDP‐25 (Chou et al, 2018), the stress granule proteome (Jain et al, 2016) and TDP‐43 myogranules in regenerating muscle (Vogler et al, 2018).
- Colocalization of proteasomes with TDP‐25 inclusions. Primary rat hippocampal neurons were co‐transduced with lentivirus encoding for GFP‐tagged TDP‐25 variants or GFP and PSMB2‐tagRFP lentivirus on day 5. Immunofluorescence images were taken 8 days after transduction (DIV5+8). Counterstain to label nuclei (DAPI). Scale bar = 10 µm.
- Statistical analysis for Fig 3F. Pairwise comparisons using Wilcoxon Rank Sum Tests with Benjamini‐Hochberg correction were performed for each tagRFP intensity bin respectively. Comparisons with P < 0.05 are highlighted in green.
- Interactome analysis of TDP‐25 mutant. Rat primary cortical neurons were transduced with GFP‐TDP‐25 wild‐type (n = 5), GFP‐TDP‐25 mutant (n = 5) or GFP (n = 4) at DIV5 and harvested at DIV5+8. Immunoprecipitates were analyzed by LC‐MS/MS. Volcano plots indicate enrichment and statistical significance (gray dots: FDR‐corrected P < 0.05). Proteins associated with GO term “proteasome complex” (GO:0000502) are labeled in orange. Full data are listed in Dataset EV3. This data was obtained from a second experiment independent from Fig 2A.
The loss of the N‐terminus in TDP‐25 led to the loss of 72 interactors compared with full‐length TDP‐43 but resulted in over 400 new interactions, which may shape biophysical properties and drive gain‐of‐function toxicity in ALS/FTD (Fig 3B). This gain of interactors may be explained by misfolding of the protein fragment (Gottlieb et al, 2019) or co‐partitioning of additional proteins in the gel‐phase and is consistent with previous findings using proximity labeling (Chou et al, 2018). To investigate the role of LLPS in inclusion formation in our neuronal model, we analyzed the content of low‐complexity domains or intrinsically disordered regions in interacting proteins, as such sequences drive phase separation through multivalent homo‐ or heterotypic interactions (Martin & Mittag, 2018). Interestingly, the proportion of disordered regions/low‐complexity regions was lower in TDP‐25‐specific interactors than in TDP‐43‐specific and shared interactors (Fig 3C), in line with the higher dynamics of nuclear TDP‐43 (Fig EV2). The relative absence of proteins with low‐complexity domains within TDP‐25 inclusions argues against their formation through LLPS or may reflect solidification driven by loss of LLPS‐promoting low‐complexity interactors.
Consistent with our cryo‐ET data, proteomics showed that a large number of proteasome subunits were highly enriched in the TDP‐25 interactome (Fig 3A and D and Dataset EV2). Furthermore, immunofluorescence confirmed partial sequestration of proteasomes into GFP‐TDP‐25 inclusions (Fig EV3B), similar to poly‐GA inclusions in C9orf72 ALS/FTD (Guo et al, 2018), although proteasomal sequestration was less prominent for TDP‐25. Our mass spectrometry data also confirmed the previously reported selective interaction of TDP‐25 with other UPS components, such as the ALS‐linked Ubiquilin 2 (Deng et al, 2011; Cassel & Reitz, 2013) and the E3 ubiquitin ligases Pja1 (Watabe et al, 2020), which could indicate ongoing degradation attempts. These or other UBL domain proteins may contribute to the extra density found on the proteasome regulatory particle (Fig 1E).
Finally, we investigated the functional consequences of proteasome recruitment to TDP‐25 inclusions and asked whether TDP‐25 is primarily a substrate or an inhibitor of the proteasome. Treating GFP‐TDP‐25 expressing neurons with the proteasome inhibitor MG132 increased the level of punctate GFP‐TDP‐25, without obvious effects on the larger inclusions (Fig 3E), in line with a previous report (Scotter et al, 2014). Quantification showed a similar twofold accumulation of GFP‐TDP‐25 wild‐type and mutant upon proteasome inhibition, suggesting that the proteasome is actively degrading largely soluble GFP‐TDP‐25 under basal conditions.
Our cryo‐ET data suggested alterations in the functional cycle of proteasomes recruited to TDP‐25 inclusions. To interrogate whether TDP‐25 affects the cellular proteasome capacity, we expressed RFP‐TDP‐25 in an established UbG76V‐GFP reporter HEK293 cell line (Dantuma et al, 2000). Already starting at low expression levels, we noticed a robust dose‐dependent accumulation of the UPS reporter in cells expressing wild‐type and mutant RFP‐TDP‐25 compared with the RFP‐only control and full‐length RFP‐TDP‐43, indicative of competitive inhibition (Fig 3F). Compared with RFP‐only control, RFP‐TDP‐43 induced strong upregulation of the reporter only at higher expression levels, which is consistent with an overall less pronounced interaction of TDP‐43 with the proteasome subunits compared with TDP‐25 (Fig 3A). Introducing the eight ALS‐causing mutations into TDP‐25 and TDP‐43 further impaired overall protein degradation at high‐expression levels, although this only reached statistical significance for TDP‐25 (Figs 3F and EV3C). Nevertheless, the interactomes of wild‐type and mutant TDP‐25 appeared very similar (Fig EV3D). These findings corroborate our structural data, indicating that proteasomes enriched in TDP‐25 inclusions are functionally impaired similar to proteasomes sequestered in poly‐GA inclusions (Guo et al, 2018).
In summary, we used cryo‐ET, proteomics and functional assays in primary neurons to show that TDP‐43 C‐terminal fragments adopt a gel‐like conformation and impair proteostasis by sequestering stalled proteasomes in situ. This gel‐like conformation could be a transition state from a liquid compartment (Conicella et al, 2016; Yu et al, 2021) to mature amyloid fibrils (Arseni et al, 2022) or an alternative state like the SDS‐resistant cytoplasmic myo‐granules of full‐length TDP‐43 found in regenerating muscle that lack classical amyloid conformation (Vogler et al, 2018). So far, cryo‐ET has only detected proteasome accumulation and impairment within inclusions related to ALS/FTD (Guo et al, 2018 and this article), but not in other protein aggregates such as poly‐Q, artificial β‐sheet proteins or α‐synuclein (Bauerlein et al, 2017; Riera‐Tur et al, 2021; Trinkaus et al, 2021). Additionally, we show that ALS‐causing mutations may further impair proteasome function. These data resonate with previous reports on the crucial role of the UPS for motor neuron function and survival (Bett et al, 2009; Tashiro et al, 2012; Bax et al, 2019), pointing to proteasome dysfunction as a major specific hallmark of ALS/FTD pathogenesis.
Materials and Methods
Primary rat neurons
Hippocampi and neocortices from E19 rat embryos were dissected in ice‐cold dissection media (HBBS, 1% penicillin/streptomycin, 10 mM HEPES pH 7.3, all Thermo Fisher Scientific), followed by enzymatic dissociation in dissection media (15 min in 0.15% trypsin for hippocampi, 20 min in 0.25% trypsin, 0.7 mg/ml DNase I for cortices). Hippocampal neurons were plated in Neurobasal media supplemented with 2% B27, 1% penicillin‐streptomycin, 0.5 mM L‐glutamine and 12.5 µM L‐glutamate (all Thermo Fisher Scientific) at 85,000 cells/ml on gold grids for cryo‐ET experiments, on poly‐D‐lysine‐coated glass coverslips (VWR) in 12‐well plates for immunofluorescence, in 35 mm petri dishes with a 14 mm glass bottom inlay (MatTek Corporation) for FRAP analysis and in 6‐well plates for biochemical analysis. For mass spectrometry, cortical neurons were plated in 10 cm petri dishes at a density of 400,000 cells/ml in the above media without L‐glutamate supplementation.
DNA constructs
cDNA encoding for TDP‐25 (amino acids 220‐414), TDP‐43 (amino acids 1‐414) and NPM1 were PCR amplified from HEK293FT (Thermo Fisher Scientific) cDNA and subsequently cloned into the FhSynW backbone (human synapsin promoter) with an N‐terminal EGFP tag (May et al, 2014). TDP‐43 variants with N‐terminal tagRFP were cloned in a lentiviral backbone with UbC promoter (FUW3), while human PSMB2 was expressed with a C‐terminal tagRFP. TDP‐43 point‐mutations were introduced using standard PCR methods. The Htt97Q‐EGFP construct was generated by subcloning the open reading frame from Addgene #1186 into FhSynW.
Lentiviral packaging and transduction
HEK293FT cells of low passage number were seeded into three 10 cm dishes (5 × 106 cells/dish) using DMEM (Thermo Fisher Scientific) supplemented with 10% fetal bovine serum (Sigma‐Aldrich), 1% penicillin‐streptomycin and 1% Non‐Essential Amino Acids (both Thermo Fisher Scientific) as described previously (Guo et al, 2018). On the next day, cells were co‐transfected with 18.6 µg transfer vector, 11 µg pSPAX2 and 6.4 µg pVSVG using Lipofectamine 2000 (Thermo Fisher Scientific). The transfection media was replaced by plating media supplemented with 13 mg/ml bovine serum albumin (Sigma Aldrich) on the following day. Lentivirus from the cell supernatant was collected 24 h later by ultracentrifugation (87,000 g, 2 h, 4°C). Finally, lentiviral particles were resuspended in Neurobasal media and stored at −80°C in aliquots.
Immunofluorescence, image acquisition and quantitative analysis
Cells grown on coverslips were briefly washed with PBS once before fixing for 10 min at room temperature using 4% paraformaldehyde (Sigma‐Aldrich) and 4% sucrose (Sigma‐Aldrich) in PBS. Primary antibodies (anti‐phospho TDP‐43 [pS403/404], Cat# TIP‐PTD‐P05, Cosmo Bio Co., Ltd., 1:1,000; anti‐Map2, Cat# M1406, clone AP‐20, Sigma‐Aldrich, 1:250) as well as secondary antibodies (Goat anti‐rabbit Alexa 555 or Goat anti‐mouse Alexa 647, Thermo Fisher Scientific, 1:400) were diluted in GDB buffer containing 0.1% gelatin (Sigma‐Aldrich), 0.3% Triton X‐100 (Merck), 450 mM NaCl, and 16 mM sodium phosphate pH 7.4. Coverslips were incubated with primary antibodies overnight at 4°C and secondary antibodies for 1 h at room temperature, each followed by three washes with PBS. Coverslips were mounted using Vectashield Vibrance with DAPI (Cat# VEC‐H‐1800, Biozol) to counterstain nuclei.
For colocalization studies, confocal microscopy was performed using an inverted Zeiss LSM800 Axio Observer.Z1 / 7 confocal laser scanning microscope (Carl Zeiss) with a Plan‐Apochromat 63×/1.40 Oil DIC M27 objective and equipped with the ZEN 2.5 software package (blue edition, Carl Zeiss). All images were acquired at 2048 × 2048 pixel resolution with two‐times averaging using two GaAsP PMT detectors at 8‐bit depth.
To quantify effects of proteasomal inhibition (MG132 vs. DMSO), tile scan images (> 100 tiles per condition) were taken on a Leica DMi8 fluorescence microscope using a HC PL APO 40×/0,95 CORR objective, a DFC9000 camera and LAS X Software (Leica Microsystems). Images were quantified via ImageJ/Fiji software (version 1.53c) to determine differences in GFP signal between conditions using a custom script. Integrated Density of GFP and GFP‐TDP‐25 variants were measured using a fixed threshold, and the resulting medians were normalized to the number of detected nuclei. DAPI‐stained nuclei were counted as particles with circularity factor 0.5–1.0 and size between 60 and 150 µm2 after thresholding and binary water shedding.
Cellular fractionation and immunoblotting
Transduced neurons were washed with PBS and lysed in RIPA buffer (137 mM NaCl, 20 mM Tris‐HCl pH 7.5, 0.1% SDS, 10% glycerol, 1% Triton X‐100, 0.5% deoxycholate, 2 mM EDTA) freshly supplemented with 67 U/ml Benzonase Nuclease (Sigma‐Aldrich), protease inhibitor cocktail (Sigma‐Aldrich) and phosphatase inhibitor cocktail (Sigma‐Aldrich) on ice for 30 min. After centrifugation (18,000 g, 30 min, 4°C), the supernatant was collected as RIPA‐soluble fraction. Pellets were resuspended in 2% SDS buffer (2% SDS, 100 mM Tris‐HCl pH 7.0) and incubated for 2 h at room temperature. Upon centrifugation (18,000 g, 30 min, 4°C), the supernatant was collected as SDS‐soluble fraction. Samples were denatured for 10 min at 95°C after adding 3× loading buffer (200 mM Tris‐HCl pH 6.8, 6% SDS, 20% glycerol, 0.1 g/ml DTT, 0.1 mg Bromophenol Blue). Protein concentrations were determined for the RIPA‐soluble fraction by BCA assay (Interchim) and equivalent fractions of the total cell population were loaded on 10% SDS–PAGE gels, followed by transfer to Immobilon®‐P PVDF membranes (Merck). Membranes were blocked in 0.2% iBlock (Thermo Fisher Scientific). The following primary antibodies were used: anti‐GFP (Cat# 75‐131, clone N86/8, UC Davis/NIH Neuromab Facility) and anti‐Calnexin (Cat# ADI‐SPA‐860, Enzo Life Sciences).
CLEM, cryo‐FIB and cryo‐ET
CLEM, cryo‐FIB/SEM and cryo‐ET tomographic data collection was performed as described in detail before (Guo et al, 2018). In brief, EM grids were mounted onto modified Autogrids sample carriers (Rigort et al, 2012) and then transferred into the cryo‐stage of an FEI CorrSight microscope. Images of grid and GFP signal were, respectively, acquired with FEI MAPS software in transmitted light and widefield mode using 5× and 20× lenses. The samples were then transferred into a FIB/SEM dual‐beam microscope (Quanta 3D FEG, FEI) using a cryo‐transfer system (PP3000T, Quorum). Cryo‐light microscope and SEM images were correlated in 2D with MAPS 2.1 software (Thermo Fisher Scientific). Lamellas were prepared using Ga2+ ion beam at 30 kV in the regions of GFP signal with final thickness of 100–200 nm.
The grids were transferred to an FEI Titan Krios transmission electron microscope for tomographic data collection. For the whole procedure, samples were kept at liquid N2 temperature. Tomographic tilt series were recorded with a Gatan K2 Summit direct detector in counting mode. A GIF‐quantum energy filter was used with a slit width of 20 eV to remove inelastically scattered electrons. Tilt series were collected dose‐symmetrically between −51° and +69° starting from +12° with an increment of 3° and total dose of 110 e−/Å2 using SerialEM software (Mastronarde, 2005) at a pixel size of 3.52 Å.
cET Image processing
Image frames were aligned using Motioncor2 (Zheng et al, 2017). IMOD software package (Kremer et al, 1996) was used for tomogram reconstruction: The tilt series were firstly aligned using fiducial‐less patch tracking, and tomograms were then reconstructed by weighted back projection of the resulting aligned images. Contrast was enhanced by filtering the tomograms using MATLAB script tom_decov (https://github.com/dtegunov/tom_deconv).
MATLAB with TOM toolbox (Nickell et al, 2005) was used as general platform for image processing. For segmentation, tomograms were rescaled with a binning factor of four. The membranes were firstly segmented automatically with TomoSegMemTV (Martinez‐Sanchez et al, 2014) using a tensor voting method, and then manually optimized with Amira (Thermo Fisher Scientific). Proteasome, ribosome, and TRiC were detected using an template matching procedure with PyTOM software (Hrabe et al, 2012), templates were generated by filtering the corresponding structures from previous work (Guo et al, 2018) to 40 Å. The resulting coordinates were used to crop the full size subtomograms from the original tomograms, which were then CTF corrected, classified and refined using RELION (Bharat & Scheres, 2016). In total, 1,170 proteasome subtomograms were picked from 11 tomograms with dominant TDP‐25 aggregates for further analysis.
FRAP assay
FRAP (fluorescence recovery after photobleaching) experiments were performed on transduced hippocampal neurons on glass bottom dishes at 37°C in HBSS buffer (Thermo Fisher Scientific) supplemented with 20 mM HEPES (Thermo Fisher Scientific) and 4.5 g/l glucose using an inverted LSM710 Axio Observer.Z1 confocal laser scanning system (Carl Zeiss) equipped with a Plan‐Apochromat 63×/1.40 Oil DIC M27 objective, a PMT detector and the ZEN 2011 software (black edition, Carl Zeiss). Images were acquired with two‐line averages. Three prebleach images were taken. Within the investigated structure (inclusion or nucleolus), a circular region of interest (ROI) of approx. 1.3 µM diameter was photobleached at 100% laser power and fluorescent recovery was monitored over 75 s (image acquisition in 0.8 s intervals), 22 min (30 s intervals), or 450 s (10 s intervals). For analysis, recorded movies were aligned using the StackReg plugin of the ImageJ/Fiji software. Afterwards, the mean intensity values of each region of interest (ROI) were monitored over time with ROI1 being the bleached region, ROI2 the whole nucleolus/inclusion/nucleus/surrounding cytoplasm and ROI3 the background noise. Values were then analyzed using easyFRAP (version 9.0.1) by applying the “full scale” normalization method (Rapsomaniki et al, 2012).
Immunoprecipitation
Transduced neurons were lysed in 2% Triton X‐100, 750 mM NaCl, 1 mM KH2PO4, freshly supplemented with 67 U/ml Benzonase Nuclease, 1× protease inhibitor cocktail and 1× phosphatase inhibitor cocktail under constant rotation for 45 min at 4°C and then centrifuged at low speed (1,000 g, 5 min, 4°C) to remove cellular debris but retain inclusion proteins (Hartmann et al, 2018). Supernatants were incubated with anti‐GFP antibody (Cat# N86/38, clone N86/38, UC Davis/NIH NeuroMab) pre‐bound to Protein G Dynabeads (Cat# 10004D, Life Technologies) on a shaking rotator for 3 h at 4°C. Beads were subsequently washed three times (150 mM NaCl, 50 mM Tris‐HCl pH 7.5, 5% glycerol) and analyzed by mass spectrometry.
Sample preparation for LC‐MS/MS
Beads were resuspended in 155 µl denaturing buffer (8 M urea, 50 mM Tris‐HCl pH 7.5, 1 mM dithiothreitol). Proteins were digested off the beads with LysC (220 ng/sample) for 1 h at room temperature while shaking at 1,200 rpm. The suspension was diluted fourfold to lower urea concentration permitting trypsin digestion. Iodoacetamide (final concentration 5 mM) to alkylate cysteines and trypsin (220 ng/sample) was added to digest for another hour before the supernatant was transferred to another tube and digested overnight. The digest was stopped by addition of trifluoroacetic acid to 1% v/v and half of the peptide solution was further processed by desalting chromatography on three disks of C18 material using the STAGE‐tip format (Kulak et al, 2014). Briefly, STAGE‐tips were washed with 100 µl buffer B (50% v/v acetonitrile, 0.5% v/v acetic acid), conditioned with 100 µl methanol, washed twice with 100 µl buffer A (2% v/v acetonitrile, 0.5% v/v acetic acid), loaded with sample peptides, washed twice with 100 µl buffer A, and subjected to peptide elution by 60 µl of buffer B. The eluate was evaporated to dryness in a vacuum concentrator. Peptides were resuspended in 10 µl 2% v/v acetonitrile, 0.5% v/v acetic acid, 0.1% v/v trifluoroacetic acid and stored at −20°C. Peptide concentration was measured spectrophotometrically at 280 nm and 500 ng of peptide/sample were subjected to LC‐MS/MS analysis.
MS data acquisition
Peptides were separated on an EASY‐nanoLC 1200 HPLC system (Thermo Fisher Scientific) via in‐house packed columns (75‐μm inner diameter, 50‐cm length, and 1.9‐μm C18 particles [Dr. Maisch GmbH]) in a gradient of buffer A (0.5% formic acid) to buffer B (80% acetonitrile, 0.5% formic acid). The gradient started at 5% B, increasing to 30% B in 40 min, further to 60% B in 4 min, to 95% B in 4 min, staying at 95% B for 4 min, decreasing to 5% B in 4 min and staying at 5% B for 4 min at a flow rate of 300 nl/min and a temperature of 60°C. A Quadrupole Orbitrap mass spectrometer (Q Exactive HF‐X for first dataset [wt TDP‐25 and TDP‐43], Exploris 480 for second dataset [wt and mut TDP‐25]; both Thermo Fisher Scientific) was directly coupled to the LC via a nano‐electrospray source. The mass spectrometers were operated in a data‐dependent mode. The survey scan range was set from 300 to 1,650 m/z, with a resolution of 60,000 at m/z 200. The most abundant isotope patterns (up to 12 or 10 on Q Exactive HF‐X and Exploris 480, respectively) with a charge of two to five were isolated and subjected to collision‐induced dissociation fragmentation (normalized collision energy of 27 or 30 on Q Exactive HF‐X and Exploris 480, respectively), an isolation window of 1.4 Th, and a MS/MS resolution of 15,000 at m/z 200. Dynamic exclusion to minimize re‐sequencing was set to 30 s.
MS raw data processing
To process MS raw files, we employed the MaxQuant software (version 1.6.0.15 and 1.6.17.0 for the first and second dataset, respectively) (Cox & Mann, 2008), searching against the UniProtKB rat FASTA database using canonical and isoform protein sequences downloaded in March 2018. Default search parameters were utilized unless stated differently. In brief, tryptic peptides with a minimum length of 7 amino acids, a maximum mass of 4,600 Da, and two miscleavages at maximum were searched. Carbamidomethlyation was set as a fixed modification and methionine oxidation and protein N‐terminal acetylation as variable modifications. A maximum of five modifications per peptide was permitted. A false discovery rate (FDR) cutoff of 1% was applied at the peptide and protein level. The search feature “Match between runs,” which allows the transfer of peptide identifications in the absence of MS/MS‐based identification after nonlinear retention time alignment, was enabled with a maximum retention time window of 0.7 min. Protein abundances were normalized with the MaxLFQ label‐free normalization algorithm built into MaxQuant (Cox et al, 2014).
Bioinformatic data analysis
The datasets were further processed and separately analyzed in the Perseus environment version 1.6.1.3 (Tyanova et al, 2016). Reverse (decoy) hits, proteins only identified by site, potential contaminants, and proteins not quantified in at least 65% of samples of at least one condition (construct and DIV) were removed. Protein abundances were log2‐transformed. Missing values for protein abundances were imputed from a normal distribution around the detection limit with a standard deviation of 0.3 times that of the quantified protein abundance distribution combining all samples and a downshift of 1.8 standard deviations. Volcano plot data were generated with the built‐in Perseus tool using a SAM statistic with an s0‐paramater of 0.1 to integrate the effect size and a permutation‐based FDR control set to 5% (Tusher et al, 2001). Proteins significantly (q‐value < 5%) associated with TDP‐25 or TDP‐43 were termed interactors and included in the further analysis.
To analyze the content of low‐complexity regions of neuronal TDP‐25/TDP‐43 interactors, we automatically queried the D2P2 database based on MaxQuant reported UniProt identifiers, using the first entry if multiple identifiers were reported in protein groups. Low complexity regions were defined as regions with a consensus of at least 6 of 9 analysis pipelines within D2P2 (Oates et al, 2013). Gene ontology (GO) analysis was performed focusing on the “biological process” categories of differentially immunoprecipitated proteins using clusterProfiler version 3.12 (Yu et al, 2012). A manual selection is shown to best represent the key pathways. The full list of significant GO terms is shown in Dataset EV2. The MS proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifiers PXD024358 and PXD031083.
Flow cytometry
Flow cytometry experiments were performed as described before (Guo et al, 2018). UbG76V‐GFP proteasome reporter cells were dissociated 72 h after transfection and analyzed with a Thermo Fisher Attune NxT Flow Cytometer (Thermo Fisher Scientific). To compensate for cross talk between GFP and tagRFP, HEK293 were transfected with tagRFP constructs only. Raw flow cytometry data were analyzed using FlowJo software (version 9.9, Treestar). The tagRFP channel was subdivided into different gates, corresponding to the log of fluorescence intensity of the transfected protein.
Data visualization and statistical analysis
Data were visualized and statistically analyzed using R (version 3.6.1). Assumptions for parametric tests were analyzed with Shapiro‐Wilk test and Levene’s test. One‐way ANOVA with post‐hoc Tukey's HSD test was used as parametric test to compare more than two groups. Kruskal‐Wallis test and subsequent Pairwise Wilcoxon Rank Sum Tests with Benjamini‐Hochberg correction were used as non‐parametric tests. Data are plotted as mean ± standard deviation (SD) or box plots/violin plots. In the legends mean ± 95% CI is noted. P‐values within graphs are reported as follows: * denotes P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, P ≥ 0.05, ns.
Author contribution
Henrick Riemenschneider: Conceptualization; Formal analysis; Investigation; Visualization; Methodology; Writing – original draft; Writing – review & editing. Qiang Guo: Conceptualization; Data curation; Formal analysis; Investigation; Visualization; Methodology; Writing – original draft; Writing – review & editing. Jakob Bader: Conceptualization; Data curation; Formal analysis; Investigation; Methodology. Frédéric Frottin: Formal analysis; Methodology. Daniel Farny: Methodology. Gernot Kleinberger: Methodology. Christian Haass: Supervision; Writing – review & editing. Matthias Mann: Funding acquisition. F Ulrich Hartl: Supervision; Funding acquisition; Writing – review & editing. Wolfgang Baumeister: Funding acquisition. Mark S Hipp: Conceptualization; Formal analysis; Supervision; Funding acquisition; Investigation; Writing – review & editing. Felix Meissner: Supervision. Rubén Fernández‐Busnadiego: Conceptualization; Formal analysis; Supervision; Funding acquisition; Investigation; Writing – original draft; Writing – review & editing. Dieter Edbauer: Conceptualization; Formal analysis; Supervision; Funding acquisition; Investigation; Visualization; Writing – original draft; Writing – review & editing.
In addition to the CRediT author contributions listed above, the contributions in detail are:
Conceptualisation, HR, QG, JB, MSH, RFB, and DE; Formal analysis, HR, QG, JB, FF, MSH, RFB, and DE; Methodology, HR, QG, JB, FF, DF, and GK; Investigation, HR, QG, JB, MSH, RFB, and DE; Data curation, HR, QG, and JB; Visualisation, HR, QG, and DE; Writing—original draft, HR, QG, RFB, and DE; Review & editing, HR, QG, CH, FUH, MSH, RFB, and DE; Supervision, CH, FUH, MSH, FM, RFB, and DE; Funding acquisition, MM, FUH, WB, MSH, RFB, and DE.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Dataset EV3
Movie EV1
Acknowledgements
We thank Philipp Erdmann, Günter Pfeifer, Jürgen Plitzko and Miroslava Schaffer for electron microscopy support. We thank Ina Wendland and the Imaging Facility of the Max Planck Institute of Biochemistry for technical support. We thank Christian Behrends, Mareike Czuppa, Dorothee Dormann, Bettina Schmid, Ali Rezaei and Qihui Zhou for critical comments to the manuscript. This work was supported by NOMIS foundation (D.E.). M.S.H., F.U.H., Q.G., W.B., and R.F.‐B. have received funding from the European Commission (FP7 GA ERC‐2012‐SyG_318987‐ToPAG). D.E., C.H., M.S.H., F.U.H., and R.F.‐B. acknowledge funding from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) through Germany’s Excellence Strategy ‐ EXC 2067/1‐ 390729940 (R.F.‐B.) and EXC 2145 – 390857198 (C.H., F.U.H., M.S.H., and D.E.). The synopsis image has been created with BioRender (adapted from “Ubiquitin Proteasome System,” by BioRender.com (2021), retrieved from https://app.biorender.com/biorender‐templates). Open Access funding enabled and organized by Projekt DEAL.
EMBO reports (2022) 23: e53890.
Contributor Information
Rubén Fernández‐Busnadiego, Email: ruben.fernandezbusnadiego@med.uni-goettingen.de.
Dieter Edbauer, Email: dieter.edbauer@dzne.de.
Data availability
The datasets produced in this study are available in the following databases: Protein interaction AP‐MS data: PRIDE PXD024358 (http://www.ebi.ac.uk/pride/archive/projects/PXD024358), PRIDE PXD031083 (http://www.ebi.ac.uk/pride/archive/projects/PXD031083). cryoET Imaging datasets: EMDB EMD‐32216 (http://www.ebi.ac.uk/pdbe/entry/EMD‐32216; Subtomogram averaging of 26S proteasome from the cell with TDP‐25 inclusion) and EMDB EMD‐32217 (http://www.ebi.ac.uk/pdbe/entry/EMD‐32217; Electron tomogram of a rat primary neuron harboring TDP‐25 inclusion).
References
- Arseni D, Hasegawa M, Murzin AG, Kametani F, Arai M, Yoshida M, Ryskeldi‐Falcon B (2022) Structure of pathological TDP‐43 filaments from ALS with FTLD. Nature 601: 139–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asano S, Fukuda Y, Beck F, Aufderheide A, Förster F, Danev R, Baumeister W (2015) Proteasomes. A molecular census of 26S proteasomes in intact neurons. Science 347: 439–442 [DOI] [PubMed] [Google Scholar]
- Babinchak WM, Haider R, Dumm BK, Sarkar P, Surewicz K, Choi JK, Surewicz WK (2019) The role of liquid‐liquid phase separation in aggregation of the TDP‐43 low‐complexity domain. J Biol Chem 294: 6306–6317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauerlein FJB, Fernandez‐Busnadiego R, Baumeister W (2020) Investigating the structure of neurotoxic protein aggregates inside cells. Trends Cell Biol 30: 951–966 [DOI] [PubMed] [Google Scholar]
- Bäuerlein FJB, Saha I, Mishra A, Kalemanov M, Martínez‐Sánchez A, Klein R, Dudanova I, Hipp MS, Hartl FU, Baumeister W et al (2017) In situ architecture and cellular interactions of PolyQ inclusions. Cell 171: 179–187.e10 [DOI] [PubMed] [Google Scholar]
- Bax M, McKenna J, Do‐Ha D, Stevens CH, Higginbottom S, Balez R, Cabral‐da‐Silva MEC, Farrawell NE, Engel M, Poronnik P et al (2019) The ubiquitin proteasome system is a key regulator of pluripotent stem cell survival and motor neuron differentiation. Cells 8: 581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bett JS, Cook C, Petrucelli L, Bates GP (2009) The ubiquitin‐proteasome reporter GFPu does not accumulate in neurons of the R6/2 transgenic mouse model of Huntington's disease. PLoS One 4: e5128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bharat TA, Scheres SH (2016) Resolving macromolecular structures from electron cryo‐tomography data using subtomogram averaging in RELION. Nat Protoc 11: 2054–2065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blokhuis AM, Koppers M, Groen EJN, van den Heuvel DMA, Dini Modigliani S, Anink JJ, Fumoto K, van Diggelen F, Snelting A, Sodaar P et al (2016) Comparative interactomics analysis of different ALS‐associated proteins identifies converging molecular pathways. Acta Neuropathol 132: 175–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Q, Boyer DR, Sawaya MR, Ge P, Eisenberg DS (2019) Cryo‐EM structures of four polymorphic TDP‐43 amyloid cores. Nat Struct Mol Biol 26: 619–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassel JA, Reitz AB (2013) Ubiquilin‐2 (UBQLN2) binds with high affinity to the C‐terminal region of TDP‐43 and modulates TDP‐43 levels in H4 cells: characterization of inhibition by nucleic acids and 4‐aminoquinolines. Biochim Biophys Acta 1834: 964–971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien HM, Lee CC, Huang JJ (2021) The different faces of the TDP‐43 low‐complexity domain: the formation of liquid droplets and amyloid fibrils. Int J Mol Sci 22: 8213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou C‐C, Zhang YI, Umoh ME, Vaughan SW, Lorenzini I, Liu F, Sayegh M, Donlin‐Asp PG, Chen YH, Duong DM et al (2018) TDP‐43 pathology disrupts nuclear pore complexes and nucleocytoplasmic transport in ALS/FTD. Nat Neurosci 21: 228–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins GA, Goldberg AL (2017) The logic of the 26S proteasome. Cell 169: 792–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conicella AE, Zerze GH, Mittal J, Fawzi NL (2016) ALS mutations disrupt phase separation mediated by alpha‐helical structure in the TDP‐43 low‐complexity C‐terminal domain. Structure 24: 1537–1549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, Hein MY, Luber CA, Paron I, Nagaraj N, Mann M (2014) Accurate proteome‐wide label‐free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol Cell Proteomics 13: 2513–2526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, Mann M (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.‐range mass accuracies and proteome‐wide protein quantification. Nat Biotechnol 26: 1367–1372 [DOI] [PubMed] [Google Scholar]
- Creekmore BC, Chang YW, Lee EB (2021) The Cryo‐EM effect: structural biology of neurodegenerative disease aggregates. J Neuropathol Exp Neurol 80: 514–529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dantuma NP, Lindsten K, Glas R, Jellne M, Masucci MG (2000) Short‐lived green fluorescent proteins for quantifying ubiquitin/proteasome‐dependent proteolysis in living cells. Nat Biotechnol 18: 538–543 [DOI] [PubMed] [Google Scholar]
- Deng H‐X, Chen W, Hong S‐T, Boycott KM, Gorrie GH, Siddique N, Yang YI, Fecto F, Shi Y, Zhai H et al (2011) Mutations in UBQLN2 cause dominant X‐linked juvenile and adult‐onset ALS and ALS/dementia. Nature 477: 211–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg D, Jucker M (2012) The amyloid state of proteins in human diseases. Cell 148: 1188–1203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feneberg E, Gordon D, Thompson AG, Finelli MJ, Dafinca R, Candalija A, Charles PD, Mäger I, Wood MJ, Fischer R et al (2020) An ALS‐linked mutation in TDP‐43 disrupts normal protein interactions in the motor neuron response to oxidative stress. Neurobiol Dis 144: 105050 [DOI] [PubMed] [Google Scholar]
- Freibaum BD, Chitta RK, High AA, Taylor JP (2010) Global analysis of TDP‐43 interacting proteins reveals strong association with RNA splicing and translation machinery. J Proteome Res 9: 1104–1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frottin F, Schueder F, Tiwary S, Gupta R, Korner R, Schlichthaerle T, Cox J, Jungmann R, Hartl FU, Hipp MS (2019) The nucleolus functions as a phase‐separated protein quality control compartment. Science 365: 342–347 [DOI] [PubMed] [Google Scholar]
- Gao FB, Almeida S, Lopez‐Gonzalez R (2017) Dysregulated molecular pathways in amyotrophic lateral sclerosis‐frontotemporal dementia spectrum disorder. EMBO J 36: 2931–2950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasset‐Rosa F, Lu S, Yu H, Chen C, Melamed Z, Guo L, Shorter J, Da Cruz S, Cleveland DW (2019) Cytoplasmic TDP‐43 de‐mixing independent of stress granules drives inhibition of nuclear import, loss of nuclear TDP‐43, and cell death. Neuron 102: 339–357.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitcho MA, Strider J, Carter D, Taylor‐Reinwald L, Forman MS, Goate AM, Cairns NJ (2009) VCP mutations causing frontotemporal lobar degeneration disrupt localization of TDP‐43 and induce cell death. J Biol Chem 284: 12384–12398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes E, Shorter J (2019) The molecular language of membraneless organelles. J Biol Chem 294: 7115–7127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb CD, Thompson ACS, Ordureau A, Harper JW, Kopito RR (2019) Acute unfolding of a single protein immediately stimulates recruitment of ubiquitin protein ligase E3C (UBE3C) to 26S proteasomes. J Biol Chem 294: 16511–16524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenther EL, Cao Q, Trinh H, Lu J, Sawaya MR, Cascio D, Boyer DR, Rodriguez JA, Hughes MP, Eisenberg DS (2018) Atomic structures of TDP‐43 LCD segments and insights into reversible or pathogenic aggregation. Nat Struct Mol Biol 25: 463–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Q, Lehmer C, Martínez‐Sánchez A, Rudack T, Beck F, Hartmann H, Pérez‐Berlanga M, Frottin F, Hipp MS, Hartl FU et al (2018) In situ structure of neuronal C9orf72 Poly‐GA aggregates reveals proteasome recruitment. Cell 172: 696–705.e12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann H, Hornburg D, Czuppa M, Bader J, Michaelsen M, Farny D, Arzberger T, Mann M, Meissner F, Edbauer D (2018) Proteomics and C9orf72 neuropathology identify ribosomes as poly‐GR/PR interactors driving toxicity. Life Sci Alliance 1: e201800070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa M, Arai T, Nonaka T, Kametani F, Yoshida M, Hashizume Y, Beach TG, Buratti E, Baralle F, Morita M et al (2008) Phosphorylated TDP‐43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Ann Neurol 64: 60–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hipp MS, Kasturi P, Hartl FU (2019) The proteostasis network and its decline in ageing. Nat Rev Mol Cell Biol 20: 421–435 [DOI] [PubMed] [Google Scholar]
- Hrabe T, Chen Y, Pfeffer S, Cuellar LK, Mangold AV, Forster F (2012) PyTom: a python‐based toolbox for localization of macromolecules in cryo‐electron tomograms and subtomogram analysis. J Struct Biol 178: 177–188 [DOI] [PubMed] [Google Scholar]
- Igaz LM, Kwong LK, Xu Y, Truax AC, Uryu K, Neumann M, Clark CM, Elman LB, Miller BL, Grossman M et al (2008) Enrichment of C‐terminal fragments in TAR DNA‐binding protein‐43 cytoplasmic inclusions in brain but not in spinal cord of frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Am J Pathol 173: 182–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain S, Wheeler JR, Walters RW, Agrawal A, Barsic A, Parker R (2016) ATPase‐modulated stress granules contain a diverse proteome and substructure. Cell 164: 487–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato M, Han T, Xie S, Shi K, Du X, Wu L, Mirzaei H, Goldsmith E, Longgood J, Pei J et al (2012) Cell‐free formation of RNA granules: low complexity sequence domains form dynamic fibers within hydrogels. Cell 149: 753–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremer JR, Mastronarde DN, McIntosh JR (1996) Computer visualization of three‐dimensional image data using IMOD. J Struct Biol 116: 71–76 [DOI] [PubMed] [Google Scholar]
- Kulak NA, Pichler G, Paron I, Nagaraj N, Mann M (2014) Minimal, encapsulated proteomic‐sample processing applied to copy‐number estimation in eukaryotic cells. Nat Methods 11: 319–324 [DOI] [PubMed] [Google Scholar]
- Li Q, Babinchak WM, Surewicz WK (2021) Cryo‐EM structure of amyloid fibrils formed by the entire low complexity domain of TDP‐43. Nat Commun 12: 1620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling SC, Albuquerque CP, Han JS, Lagier‐Tourenne C, Tokunaga S, Zhou H, Cleveland DW (2010) ALS‐associated mutations in TDP‐43 increase its stability and promote TDP‐43 complexes with FUS/TLS. Proc Natl Acad Sci U S A 107: 13318–13323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie IR, Arzberger T, Kremmer E, Troost D, Lorenzl S, Mori K, Weng S‐M, Haass C, Kretzschmar HA, Edbauer D et al (2013) Dipeptide repeat protein pathology in C9ORF72 mutation cases: clinico‐pathological correlations. Acta Neuropathol 126: 859–879 [DOI] [PubMed] [Google Scholar]
- Mann JR, Gleixner AM, Mauna JC, Gomes E, DeChellis‐Marks MR, Needham PG, Copley KE, Hurtle B, Portz B, Pyles NJ et al (2019) RNA binding antagonizes neurotoxic phase transitions of TDP‐43. Neuron 102: 321–338.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin EW, Mittag T (2018) Relationship of sequence and phase separation in protein low‐complexity regions. Biochemistry 57: 2478–2487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez‐Sanchez A, Garcia I, Asano S, Lucic V, Fernandez JJ (2014) Robust membrane detection based on tensor voting for electron tomography. J Struct Biol 186: 49–61 [DOI] [PubMed] [Google Scholar]
- Mastronarde DN (2005) Automated electron microscope tomography using robust prediction of specimen movements. J Struct Biol 152: 36–51 [DOI] [PubMed] [Google Scholar]
- May S, Hornburg D, Schludi MH, Arzberger T, Rentzsch K, Schwenk BM, Grässer FA, Mori K, Kremmer E, Banzhaf‐Strathmann J et al (2014) C9orf72 FTLD/ALS‐associated Gly‐Ala dipeptide repeat proteins cause neuronal toxicity and Unc119 sequestration. Acta Neuropathol 128: 485–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray DT, Kato M, Lin Y, Thurber KR, Hung I, McKnight SL, Tycko R (2017) Structure of FUS protein fibrils and its relevance to self‐assembly and phase separation of low‐complexity domains. Cell 171: 615–627.e16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM et al (2006) Ubiquitinated TDP‐43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314: 130–133 [DOI] [PubMed] [Google Scholar]
- Nickell S, Forster F, Linaroudis A, Net WD, Beck F, Hegerl R, Baumeister W, Plitzko JM (2005) TOM software toolbox: acquisition and analysis for electron tomography. J Struct Biol 149: 227–234 [DOI] [PubMed] [Google Scholar]
- Oates ME, Romero P, Ishida T, Ghalwash M, Mizianty MJ, Xue B, Dosztanyi Z, Uversky VN, Obradovic Z, Kurgan L et al (2013) D(2)P(2): database of disordered protein predictions. Nucleic Acids Res 41: D508–D516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olzscha H, Schermann SM, Woerner AC, Pinkert S, Hecht MH, Tartaglia GG, Vendruscolo M, Hayer‐Hartl M, Hartl FU, Vabulas RM (2011) Amyloid‐like aggregates sequester numerous metastable proteins with essential cellular functions. Cell 144: 67–78 [DOI] [PubMed] [Google Scholar]
- Patel A, Lee H, Jawerth L, Maharana S, Jahnel M, Hein M, Stoynov S, Mahamid J, Saha S, Franzmann T et al (2015) A liquid‐to‐solid phase transition of the ALS protein FUS accelerated by disease mutation. Cell 162: 1066–1077 [DOI] [PubMed] [Google Scholar]
- Prasad A, Bharathi V, Sivalingam V, Girdhar A, Patel BK (2019) Molecular mechanisms of TDP‐43 misfolding and pathology in amyotrophic lateral sclerosis. Front Mol Neurosci 12: 25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qamar S, Wang G, Randle SJ, Ruggeri FS, Varela JA, Lin JQ, Phillips EC, Miyashita A, Williams D, Strohl F et al (2018) FUS phase separation is modulated by a molecular chaperone and methylation of arginine cation‐pi interactions. Cell 173: 720–734.e715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapsomaniki MA, Kotsantis P, Symeonidou IE, Giakoumakis NN, Taraviras S, Lygerou Z (2012) easyFRAP: an interactive, easy‐to‐use tool for qualitative and quantitative analysis of FRAP data. Bioinformatics 28: 1800–1801 [DOI] [PubMed] [Google Scholar]
- Riera‐Tur I, Schäfer T, Hornburg D, Mishra A, Fernández‐Mosquera L, Feigenbutz D, Auer P, Mann M, Baumeister W, Klein R et al (2021) Amyloid‐like aggregates cause lysosomal defects in neurons via gain‐of‐function toxicity. bioRxiv. 10.1101/2019.12.16.877431 [PREPRINT] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigort A, Bauerlein FJ, Villa E, Eibauer M, Laugks T, Baumeister W, Plitzko JM (2012) Focused ion beam micromachining of eukaryotic cells for cryoelectron tomography. Proc Natl Acad Sci U S A 109: 4449–4454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scotter EL, Vance C, Nishimura AL, Lee YB, Chen HJ, Urwin H, Sardone V, Mitchell JC, Rogelj B, Rubinsztein DC et al (2014) Differential roles of the ubiquitin proteasome system and autophagy in the clearance of soluble and aggregated TDP‐43 species. J Cell Sci 127: 1263–1278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tashiro Y, Urushitani M, Inoue H, Koike M, Uchiyama Y, Komatsu M, Tanaka K, Yamazaki M, Abe M, Misawa H et al (2012) Motor neuron‐specific disruption of proteasomes, but not autophagy, replicates amyotrophic lateral sclerosis. J Biol Chem 287: 42984–42994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinkaus VA, Riera‐Tur I, Martinez‐Sanchez A, Bauerlein FJB, Guo Q, Arzberger T, Baumeister W, Dudanova I, Hipp MS, Hartl FU et al (2021) In situ architecture of neuronal alpha‐Synuclein inclusions. Nat Commun 12: 2110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tusher VG, Tibshirani R, Chu G (2001) Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A 98: 5116–5121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, Mann M, Cox J (2016) The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat Methods 13: 731–740 [DOI] [PubMed] [Google Scholar]
- Tziortzouda P, Van Den Bosch L, Hirth F (2021) Triad of TDP43 control in neurodegeneration: autoregulation, localization and aggregation. Nat Rev Neurosci 22: 197–208 [DOI] [PubMed] [Google Scholar]
- Vogler TO, Wheeler JR, Nguyen ED, Hughes MP, Britson KA, Lester E, Rao B, Betta ND, Whitney ON, Ewachiw TE et al (2018) TDP‐43 and RNA form amyloid‐like myo‐granules in regenerating muscle. Nature 563: 508–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker AK, Tripathy K, Restrepo CR, Ge G, Xu Y, Kwong LK, Trojanowski JQ, Lee VM (2015) An insoluble frontotemporal lobar degeneration‐associated TDP‐43 C‐terminal fragment causes neurodegeneration and hippocampus pathology in transgenic mice. Hum Mol Genet 24: 7241–7254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watabe K, Kato Y, Sakuma M, Murata M, Niida‐Kawaguchi M, Takemura T, Hanagata N, Tada M, Kakita A, Shibata N (2020) Praja1 RING‐finger E3 ubiquitin ligase suppresses neuronal cytoplasmic TDP‐43 aggregate formation. Neuropathology 40: 570–586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Wang LG, Han Y, He QY (2012) clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16: 284–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Lu S, Gasior K, Singh D, Vazquez‐Sanchez S, Tapia O, Toprani D, Beccari MS, Yates JR, Da Cruz S et al (2021) HSP70 chaperones RNA‐free TDP‐43 into anisotropic intranuclear liquid spherical shells. Science 371: eabb4309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y‐J, Xu Y‐F, Cook C, Gendron TF, Roettges P, Link CD, Lin W‐L, Tong J, Castanedes‐Casey M, Ash P et al (2009) Aberrant cleavage of TDP‐43 enhances aggregation and cellular toxicity. Proc Natl Acad Sci U S A 106: 7607–7612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng SQ, Palovcak E, Armache JP, Verba KA, Cheng Y, Agard DA (2017) MotionCor2: anisotropic correction of beam‐induced motion for improved cryo‐electron microscopy. Nat Methods 14: 331–332 [DOI] [PMC free article] [PubMed] [Google Scholar]