

Abstract
The installation of gem-difluoromethylene groups into organic structures remains a daunting synthetic challenge despite their attractive structural, physical, and biochemical properties. A very efficient retrosynthetic approach would be the functionalization of a single C-F bond from a trifluoromethyl group. Recent advances in this line of attack have enabled the C-F activation of trifluoromethylarenes, but limit the accessible motifs to only benzylic gem-difluorinated scaffolds. In contrast, the C-F activation of trifluoroacetates would enable their use as a bifunctional gem-difluoromethylene synthon. Herein, we report a photochemically-mediated method for the defluorinative alkylation of a commodity feedstock: ethyl trifluoroacetate. A novel mechanistic approach was identified using our previously developed diaryl ketone HAT catalyst to enable the hydroalkylation of a diverse suite of alkenes. Furthermore, electrochemical studies revealed that more challenging radical precursors, namely trifluoroacetamides, could also be functionalized via synergistic Lewis acid/photochemical activation. Finally, this method enabled a concise synthetic approach to novel gem-difluoro analogs of FDA-approved pharmaceutical compounds
Keywords: Photochemistry, Carbon-Fluorine bond activation, Radical Chain
Graphical Abstract:
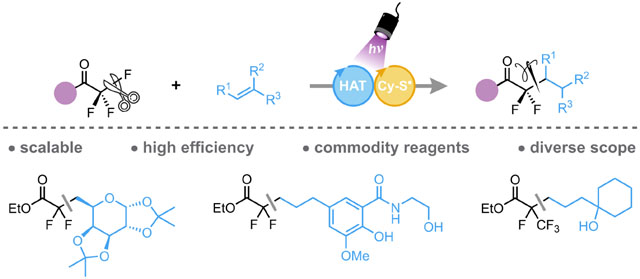
The emplacement of fluorine within organic substructures is among the more widely studied subsets of chemistry because of the numerous applications of fluorinated compounds. The gem-difluoromethylene unit is a particularly intriguing bioisosteric motif utilized in drug-discovery programs because of the exceptional pharmacokinetic (PK) properties, hydrogen bonding abilities, and enzymatic stability imparted by C-F bonds.1–6 However, incorporation of this unique fluorinated linkage, especially in a late-stage fashion, remains a significant synthetic challenge because of the dearth of protocols currently available for the construction of CF2 groups. A select range of methods for the incorporation of gem-difluoromethylene groups has been developed, most of which rely on specialized fluorinated functional groups and/or highly reactive fluorinating reagents (e.g., DAST or Selectfluor).7–21 The shortcomings of the aforementioned methods emphasize the urgent need for mild, general methods for gem-difluoromethylene installation from commodity starting materials.
The selective activation of a single C-F bond in a trifluoromethyl (CF3) group presents a significantly more attractive approach given the diverse array of readily available CF3-containing structures, but also poses a formidable synthetic challenge. Recent efforts have been made in this area, with most protocols focusing on the activation of a CF3-arene.22–30 Although novel, the benzylic gem-difluoromethylene products have minimal utility as synthetic intermediates, limiting the possibility for further elaboration to diverse chemical structures. In contrast, the C-F activation of α-trifluoromethyl carbonyl compounds, especially trifluoroacetates, would enable their use as bifunctional lynchpins in the synthesis of complex CF2-containing compounds (Figure 1). Following a defluorinative functionalization, the resulting gem-difluoro ester/carboxylates would offer several potential downstream transformations, allowing chemists to construct highly decorated fluorine-containing molecular scaffolds. The activation of a commodity, bench-stable starting material, such as ethyl trifluoroacetate ($0.06/mmol), has unquestionable advantages over niche materials such as bromo- or iododifluoroacetates ($2.58–7.15/mmol), which were previously the standard bifunctional feedstocks for such strategies. This concept is not without precedent, as a defluorinative alkylation (DFA) of α-trifluoromethyl carbonyl compounds via boryl radical activation was recently disclosed by Wang and Houk.31 The boryl radical addition triggers a spin-center shift (SCS),32 producing gem-difluoro α-carbonyl radicals (1b) that undergo Giese addition onto activated styrene derivatives. Although novel, the harsh and potentially dangerous conditions (super-stoichiometric tert-butyl peroxide at 120°C in MeCN) detract from the practicality of this method. Additionally, this report focused on trifluoroacetamides, including only a small subset of the more synthetically useful trifluoroacetates. Given these limitations, we sought to identify an alternate mode of catalysis for the DFA of trifluoroacetates via photochemical activation. The envisioned method would overcome the unresolved challenges in gem-difluoromethylene construction by transforming commodity starting materials under mild, scalable conditions.
Figure 1.
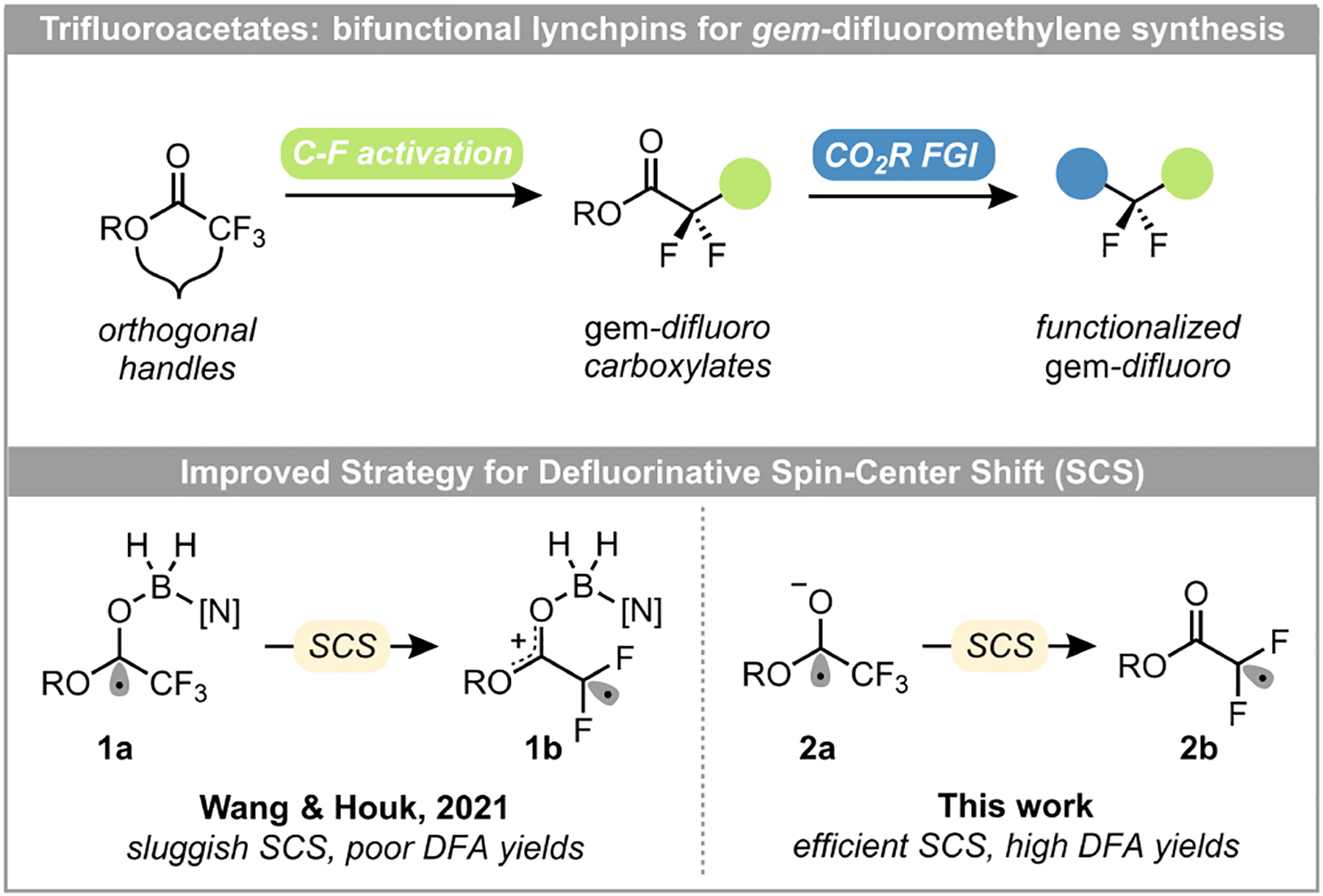
Defluorinative functionalization strategies ([N] = N,N-dimethyl-4-aminopyridine)
According to calculations by Wang and Houk, the defluorinative SCS barrier for trifluoroacetates (1a → 1b) was significantly high, contributing to the diminished DFA yields. Preliminary studies by Janata and Giese demonstrated that SCS fragmentation is generally much faster for charged species than their neutral counterparts.33, 34 Therefore, we reasoned that the corresponding trifluoroacetate radical anion (2a), generated via single electron reduction, would undergo a faster defluorinative SCS compared to the neutral intermediate 1a, which was generated via boryl radical addition. Given the challenging reduction potential of alkyl trifluoroacetates (−2.0 V vs SCE), we turned to a recently discovered method utilizing CO2•− as a potent single electron reductant (−2.2 V vs SCE).35, 36 We began our optimization studies (see SI for full details) by identifying ethyl trifluoroacetate (3) as a readily available commodity CF3 radical precursor. Attempts to use photoredox catalysts to access the CO2•− reductant as previously described35, 36 only furnished the desired DFA product in poor yields with a complex mixture of byproducts (see SI). We therefore envisioned an alternative mechanism relying on a photoactive hydrogen atom transfer (HAT) catalyst to activate the formate salt. Gratify-ingly, utilization of our previously developed diaryl ketone HAT catalyst (5)37 accomplished the desired defluorinative alkylation of ethyl trifluoroacetate in quantitative yield. Examination of a suite of related diaryl ketone catalysts revealed that ketones substituted with one electron-withdrawing and one electron-donating aryl group performed the best. Such diaryl ketones exhibit low excited state energies because of captodative stabilization of the triplet ketyl diradical, making them particularly effective for photochemical HAT.37 In addition to improved reactivity, this HAT catalyst tolerated a more diverse set of functional groups compared to the typical photocatalysts previously employed. Furthermore, we discovered that the structure of the thiol had a significant impact on the DFA yield. We reasoned that the thiol was critical to the mechanism as it engages in HAT with the radical Giese adduct as well as the formate, sustaining the proposed radical chain. Therefore, the optimal bond dissociation enthalpy should, in theory, fall between that of the newly formed C-H bond in the product (97–99 kcal/mol)38 and the formyl C-H bond (88 kcal/mol).38 A significant decrease in yield was observed when using aryl- (S-H BDE = ~80 kcal/mol)38 rather than alkyl thiols (S-H BDE = 87–89 kcal/mol),38 likely because the aryl S-H bond dissociation enthalpies are below that of the formyl C-H bond, resulting in inefficient HAT. Lower loadings (1–4 equivalents) of ethyl trifluoroacetate resulted in incomplete consumption of the olefin. Sodium outperformed any other alkaline earth metal or organic counterions of the formate salt. Consistent with literature precedent,35,36 this reaction functioned best in strongly polar, aprotic solvents. Control studies confirmed the necessity of diaryl ketone catalyst 5, thiol, and light for efficient reactivity (see SI). Additional experiments revealed that the loading of benzophenone 5 could be dropped to as low as 1 mol % with only a minimal decrease in yield given the same irradiation time, enabling practical DFA on multi-gram scale (see SI). Ultimately, these studies demonstrate that this novel strategy of diaryl ketone-mediated HAT is uniquely suited to facilitate the reduction of trifluoroacetates via CO2•− in the desired DFA.
As we set out to examine the scope of the DFA of ethyl trifluoroacetate, we found that a wide variety of alkenes bearing complex functional groups could be incorporated (Table 1). Unactivated terminal alkenes, which are unreactive under typical photoredox conditions,39 produced the desired product in good to excellent yield. Remarkably, alkenes bearing alcohols, amines, amides, anhydrides, boronate esters, epoxides, and electron-rich heterocycles offered minimal to no off-target reactivity, demonstrating the excellent functional group tolerance of this reaction. Products 9, 10, and 14 were furnished from alkenes bearing alpha-heteroatomic, allylic C-H bonds without undesired activation at these homolytically labile positions. A suite of alkene-containing natural products was also incorporated, including limonene oxide to afford 19, demonstrating the applicability of this method to the synthesis of complex molecular architectures and sensitive functional groups. Compound 21, derived from (−)-carvone, demonstrates the chemoselectivity for radical addition to an electron-rich alkene over an electron-poor enone. A range of internal alkenes also underwent DFA with excellent regioselectivity (25–27), remarkably including access to anhydride 26. The fragmentation of the bridged-bicyclobutane observed in production of 27, derived from (−)-nopol, confirms the radical nature of this process. In addition to unactivated alkenes, the scope was extended to electron-rich substrates including enamines, enamides, and enols (28–37). An enol ether derived from (D)-galactose underwent the DFA with excellent diastereoselectivity. Surprisingly, product 34 was isolated without any cleavage of the silyl ether despite the generation of stoichiometric fluoride byproducts. Unfortunately, styrene derivatives were unable to be incorporated into this DFA protocol.
Table 1.
Scope of Alkenes in the DFA of Perfluorinated Esters
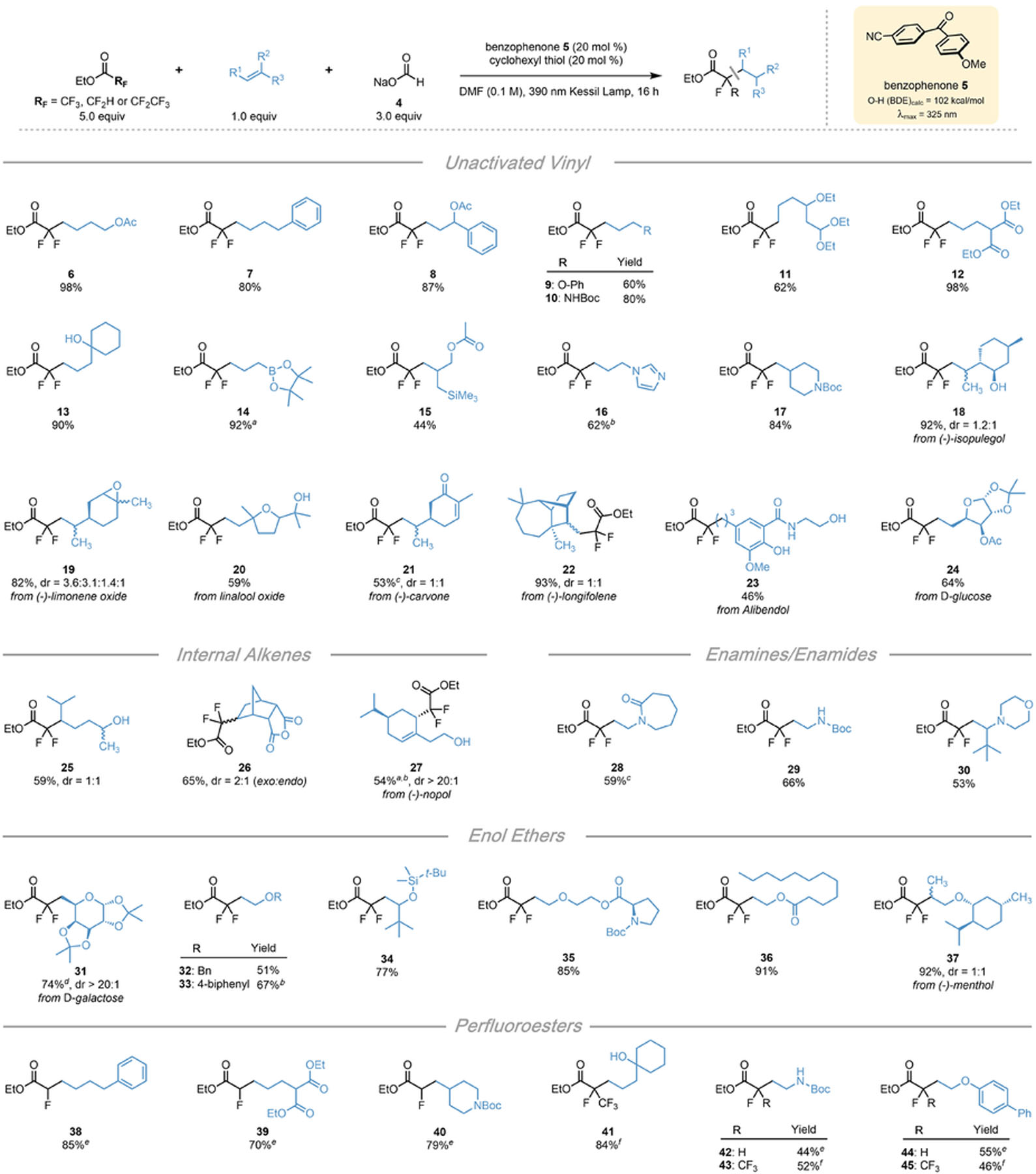
|
All values indicate the yield of the isolated product. Unless otherwise noted: alkene (1 equiv, 0.5 mmol), ethyl trifluoroacetate (5 equiv, 2.5 mmol), sodium formate (3 equiv, 1.5 mmol) benzophenone 5 (20 mol %, 0.10 mmol), cyclohexanethiol (20 mol %, 0.10 mmol), DMF (0.1 M), 16 h, irradiating with a 390 nm PR160 Kessil lamp.
H NMR yield of crude reaction.
Using 5 equiv of sodium formate and 10 equiv of ethyl trifluoroacetate.
Using 10 equiv of ethyl trifluoroacetate.
Isolated as the corresponding carboxylic acid.
Using ethyl difluoroacetate (10 equiv, 5.0 mmol) in place of ethyl trifluoroacetate with 48 h reaction time.
Using ethyl pentafluoropropionate (5 equiv, 2.5 mmol) in place of ethyl trifluoroacetate.
We then sought to expand the protocol to other α-fluoroesters, enabling the construction of unique fluoroalkyl groups. A diverse range of alkenes were engaged in hydroalkylation with ethyl difluoroacetate, producing α-monofluoro esters. Ethyl difluoroacetate exhibited sluggish reactivity compared to ethyl trifluoroacetate, likely because of a more challenging reduction potential (−2.9 V vs SCE). However, at increased loadings and extended reaction times compounds 38–40, 42 and 44 were obtained in good to excellent yield. Ethyl pentafluoropropionate (−2.6 V vs SCE) underwent a single, regioselective defluorinative alkylation with a similar range of alkenes (41, 43, 45), requiring no alteration of our standard reaction conditions. To our knowledge, such perfluoroesters have yet to be successfully engaged in defluorinative alkylation protocols.31 These results demonstrate the excellent chemoselectivity and broad applicability of this method for the facile preparation of complex, synthetically challenging fluoroalkyl groups.
To demonstrate this method as a general protocol for DFA of trifluoromethyl carbonyl species, we sought to expand the scope of radical precursors to trifluoroacetamides, but initially only trace consumption of starting materials was observed. Cyclic voltammetry studies of trifluoroacetamide 46 (−2.2 V vs SCE) rationalized the inability of CO2•− to effect DFA under our original conditions. However, examination of a suite of Lewis acids revealed that a catalytic loading of Zn(OTf)2 restored reactivity for N-aryl trifluoroacetamides by lowering the reduction potential by 0.5 V. Under these conditions, a set of unactivated, activated, and internal alkenes underwent DFA with super-stoichiometric loading of trifluoroacetamide 46 (Table 2). The reaction displayed minimal degradation of the excess trifluoroacetamide, and typically >90% of the remaining starting material was recovered by crystallization prior to chromatography.
Table 2.
Scope of Alkenes in the DFA of N-Aryl Trifluoroacetamides
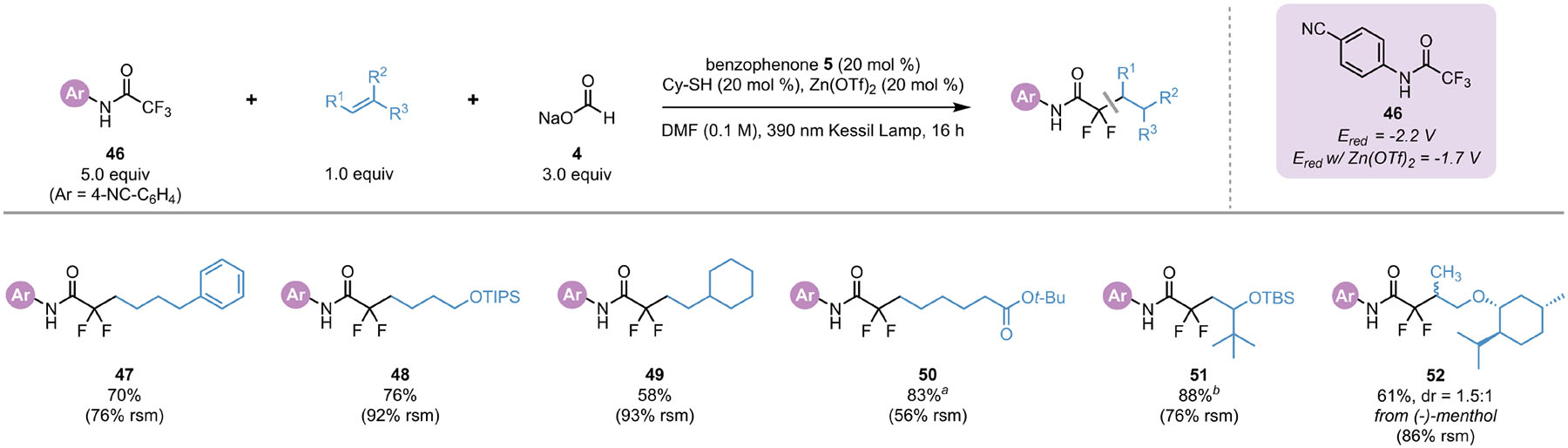
|
All values indicate the yield of the isolated product. Unless otherwise noted: alkene (1 equiv, 1.0 mmol), N-(4-cyanophenyl)-trifluoroacetamide (5 equiv, 5.0 mmol), sodium formate (3 equiv, 3.0 mmol) benzophenone 5 (20 mol %, 0.20 mmol), cyclohexanethiol (20 mol %, 0.20 mmol), Zn(OTf)2 (20 mol %, 0.20 mmol), DMF (0.1 M), 16 h, irradiating with a 390 nm PR160 Kessil lamp. RSM = percent recovered N-(4-cyanophenyl)-trifluoroacetamide (46) based on 4 equiv.
Isolated as the corresponding carboxylic acid.
Using “recycled” 46 recovered by precipitation protocol from previous DFA reactions.
Given the efficiency of the developed protocol, we probed its scalability (Scheme 1). Using 4-phenyl-1-butene as a model alkene, the reaction scale was increased 20- and 100-fold in batch with only a minimal decrease in yield. Importantly, no specialized glassware, equipment, or irradiation source was required, and the reaction was completed within the same time frame as in the small-scale reactions. We also demonstrated how increased scale allowed more efficient purification methods to be used, such as vacuum distillation, dramatically decreasing the process mass intensity (PMI)40 for this transformation. In particular, pinacol boronate 14 could not be isolated on 0.5 mmol because it decomposed when exposed to SiO2 chromatography, but was successfully isolated by distillation on 25 mmol scale. The ease of scalability and cost-effectiveness of this photochemical transformation make it a remarkably practical method for the multi-gram preparation of gem-difluoromethylene compounds.
Scheme 1.
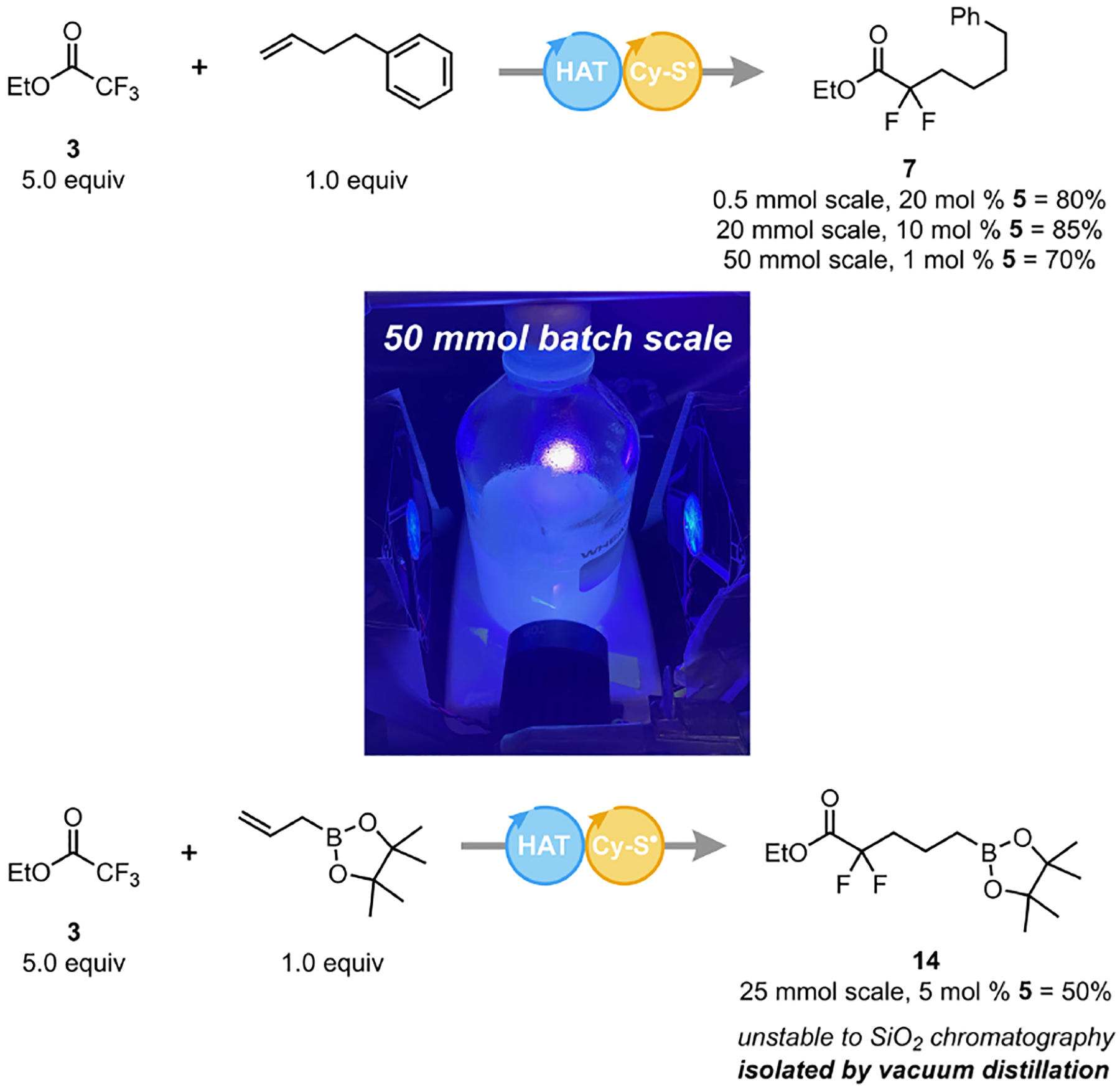
Multi-gram DFA of Ethyl Trifluoroacetate
Standard DFA conditions as in Table 1 using either 10, 5 or 1 mol % benzophenone 5. See SI for further details
To showcase the applicability of this DFA method toward medicinal chemistry, we prepared three novel gem-difluoro precursor derivatives of FDA-approved small molecule APIs via concise syntheses (Scheme 2). The advantages of this photochemical DFA of unactivated alkenes (53, 55 and 57) are evident when juxtaposed with classical approaches using bromo- or iododifluoroacetates, which necessitate unsustainable noble metal catalysis.41 In addition to accessing the illustrated targets, incorporation of the gem-difluoroester motif renders these DFA products as excellent synthetic intermediates, enabling rapid exploration of chemical space for structure-activity relationship (SAR) studies. We anticipate that this method will enable the preparation of many novel gem-difluoro-containing scaffolds in medicinal chemistry that would have previously been infeasible.
Scheme 2.
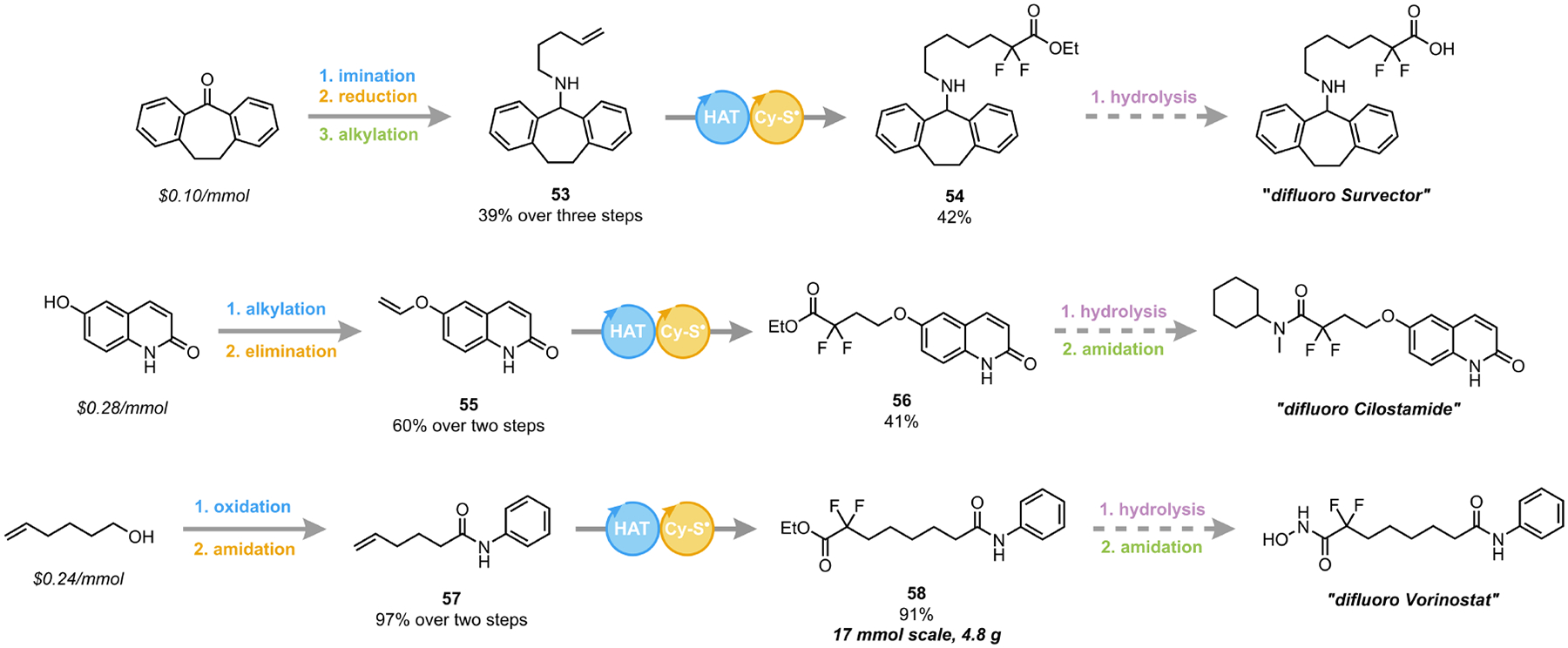
Synthesis of gem-difluoro analogs of select APIs
See SI for alkene preparation details. Standard DFA conditions as in Table 1
Given previous literature precedent,35 we propose that the developed DFA mechanism operates via a highly efficient radical chain process, mediated by both diaryl ketone and thiol HAT events (Figure 2). Photoexcitation of the diaryl ketone catalyst (I) accesses the triplet state diradical (II) that undergoes HAT from IV, generating V. Intermediate V accomplishes a single electron reduction of the α-trifluorocarbonyl substrate VI, producing the corresponding radical anion (VII) and CO2 as a byproduct. Intermediate VII then undergoes an efficient defluorinative spin-center shift to produce the desired gem-difluoro radical (VIII), which engages in a Giese addition with an alkene. The Giese adduct (IX) is then quenched via HAT from a thiol (XII), generating a thiyl radical (XI) and the hydroalkylated product X. Thiyl radical XI then takes part in a single electron transfer with III, returning the diaryl ketone to its neutral ground state (I). Alternatively, this thiyl radical (XI) could perform HAT from IV, initiating a radical chain. Quantum yield experiments (ϕ = 4.9) are in good agreement with a radical chain process as is suggested by previous mechanistic studies by Jui.35 Examination of bond dissociation enthalpies (O-H in III = 102 kcal/mol;37 C-H in IV = 87 kcal/mol35) and relative radical philicities suggest that the rate of HAT between II and formate IV is much faster than between II and XII. Therefore, we believe the diaryl ketone catalyst is not only critical in the initiation of this mechanism, but likely sustains the radical chain throughout the course of the reaction.
Figure 2.
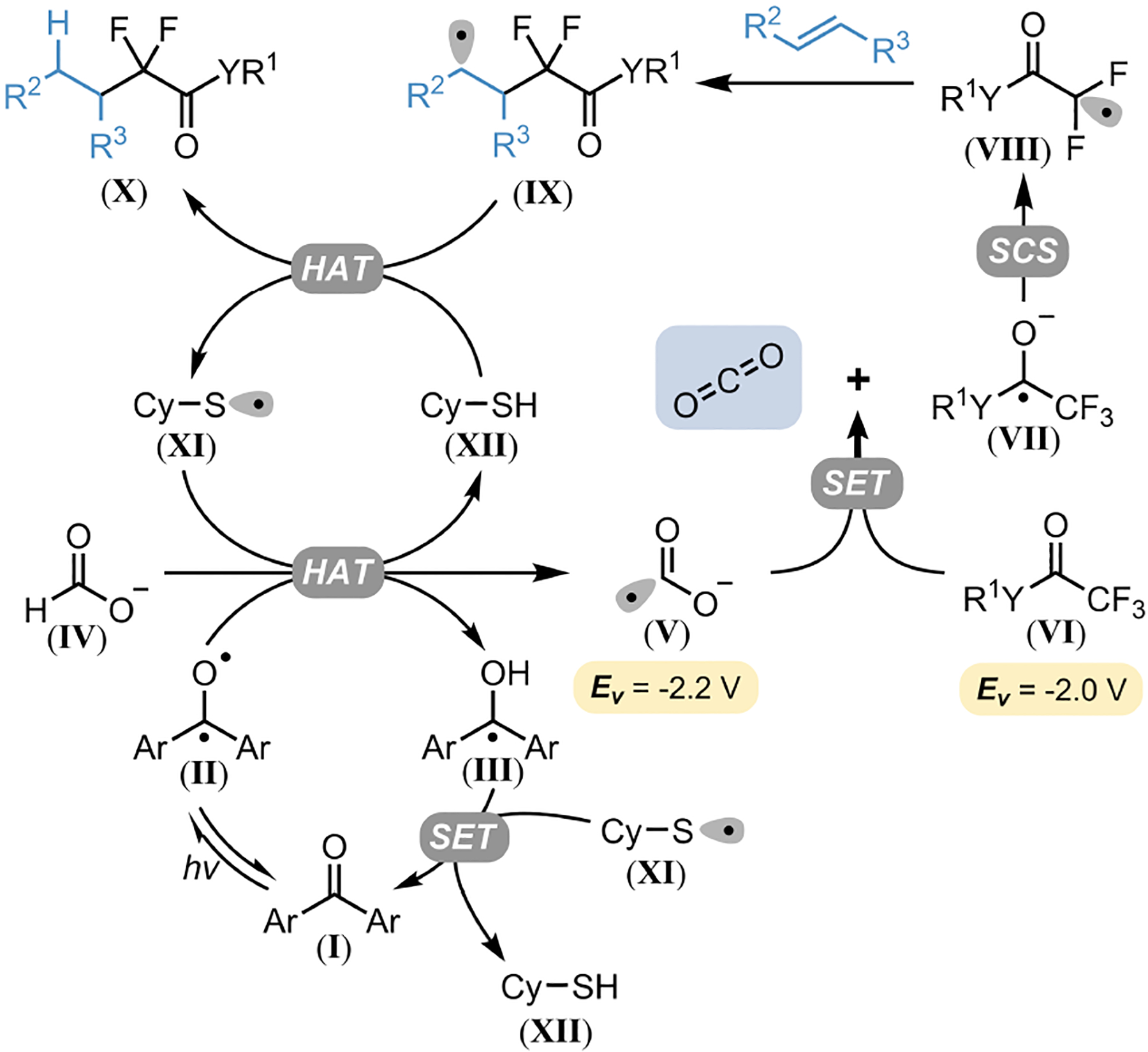
Proposed Mechanism for Photochemical DFA of α-Trifluorocarbonyl Compounds
The method developed addresses a pressing need in the synthesis of fluorinated small molecules. The incorporation of gemdifluoromethylene groups, a previously daunting synthetic challenge, is now readily accomplished by photochemical DFA. A novel HAT-initiated approach to radical chain reduction via CO2•− has been identified, facilitating the hydroalkylation of unactivated and electronically rich alkenes. This protocol serves as an exceptionally efficient and functionally tolerant strategy to prepare complex α,α-difluorocarbonyl derivatives, making it a prime candidate for the late-stage functionalization of complex molecules. The cost effectiveness of all reagents and starting materials, combined with the simple reaction set up and short irradiation time, make this reaction applicable to multi-gram scale preparation of fluorinated motifs in organic synthesis.
Supplementary Material
ACKNOWLEDGMENT
The authors thank Dr. Charles W. Ross, III (University of Pennsylvania) for obtaining HRMS data. We acknowledge Kessil Lighting for lights used in this study.
Funding Sources
The authors are grateful for the financial support (RG2020) provided by Merck KGaA, Darmstadt, Germany, and NIGMS (R35 GM 131680 to G.A.M.). M.W.C. is grateful for the financial support provided by an NSF Graduate Research Fellowship. The NIH supplement awards 3R01GM118510-03S1 and 3R01GM087605-06S1, as well as the Vagelos Institute for Energy Science and Technology supported the purchase of NMR spectrometers used in this study
Footnotes
The Supporting Information including all experimental details, reaction optimization data, spectral characterization of novel compounds and cyclic voltammetry data is available free of charge on the ACS Publications website.
REFERENCES
- 1.Bohm HJ; Banner D; Bendels S; Kansy M; Kuhn B; Muller K; Obst-Sander U; Stahl M, Fluorine in medicinal chemistry. Chembiochem, 2004, 5, 637–643. [DOI] [PubMed] [Google Scholar]
- 2.Erickson JA; McLoughlin JI, Hydrogen-bond Donor Properties of the Difluoromethyl Group. J. Org. Chem, 1995, 60, 1626–1631. [Google Scholar]
- 3.Gillis EP; Eastman KJ; Hill MD; Donnelly DJ; Meanwell NA, Applications of Fluorine in Medicinal Chemistry. J. Med. Chem, 2015, 58, 8315–8359. [DOI] [PubMed] [Google Scholar]
- 4.Meanwell NA, Fluorine and Fluorinated Motifs in the Design and Application of Bioisosteres for Drug Design. J. Med. Chem, 2018, 61, 5822–5880. [DOI] [PubMed] [Google Scholar]
- 5.Muller K; Faeh C; Diederich F, Fluorine in pharmaceuticals: Looking beyond intuition. Science, 2007, 317, 1881–1886. [DOI] [PubMed] [Google Scholar]
- 6.Sessler CD; Rahm M; Becker S; Goldberg JM; Wang F; Lippard SJ, CF2H, a Hydrogen Bond Donor. J. Am. Chem. Soc, 2017, 139, 9325–9332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Feng Z; Xiao YL; Zhang XG, Transition-Metal (Cu, Pd, Ni)-Catalyzed Difluoroalkylation via Cross-Coupling with Difluoroalkyl Halides. Acc. Chem. Res, 2018, 51, 2264–2278. [DOI] [PubMed] [Google Scholar]
- 8.Fier PS; Hartwig JF, Copper-Mediated Difluoromethylation of Aryl and Vinyl Iodides. J. Am. Chem. Soc, 2012, 134, 5524–5527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fujikawa K; Fujioka Y; Kobayashi A; Amii H, A New Method for Aromatic Difluoromethylation: Copper-Catalyzed Cross-Coupling and Decarboxylation Sequence from Aryl Iodides. Org. Lett, 2011, 13, 5560–5563. [DOI] [PubMed] [Google Scholar]
- 10.Haycock-Lewandowski SJ; Wilder A; Ahman J, Development of a Bulk Enabling Route to Maraviroc (UK-427,857), a CCR-5 Receptor Antagonist. Org. Process Res. Dev, 2008, 12, 1094–1103. [Google Scholar]
- 11.Hu JB; Zhang W; Wang F, Selective difluoromethylation and monofluoromethylation reactions. Chem. Commun, 2009, 48, 7465–7478. [DOI] [PubMed] [Google Scholar]
- 12.Krishnamoorthy S; Kar S; Kothandaraman J; Prakash GKS, Nucleophilic difluoromethylation of aromatic aldehydes using trimethyl (trifluoromethyl)silane (TMSCF3). J. Fluor. Chem, 2018, 208, 10–14. [Google Scholar]
- 13.Kucera R; Goetzke FW; Fletcher SP, An Asymmetric Suzuki-Miyaura Approach to Prostaglandins: Synthesis of Tafluprost. Org. Lett, 2020, 22, 2991–2994. [DOI] [PubMed] [Google Scholar]
- 14.Lafrance D; Caron S, New Synthetic Route to a Dipeptidyl Peptidase-4 Inhibitor. Org. Process Res. Dev, 2012, 16, 409–414. [Google Scholar]
- 15.Li X; He ST; Song QL, Diethylzinc-Mediated Radical 1,2-Addition of Alkenes and Alkynes. Org. Lett, 2021, 23, 2994–2999. [DOI] [PubMed] [Google Scholar]
- 16.Middleton WJ, New Fluorinating Reagents - Dial-kylaminosulfur Fluorides. J. Org. Chem, 1975, 40, 574–578. [Google Scholar]
- 17.Miele M; Pace V, (Difluoromethyl)trimethylsilane (TMSCHF2): A Useful Difluoromethylating Nucleophilic Source. Aust. J. Chem, 2021, 74, 623–625. [Google Scholar]
- 18.Min QQ; Yin ZS; Feng Z; Guo WH; Zhang XG, Highly Selective gem-Difluoroallylation of Organoborons with Bromodifluoromethylated Alkenes Catalyzed by Palladium. J. Am. Chem. Soc, 2014, 136, 1230–1233. [DOI] [PubMed] [Google Scholar]
- 19.Shishimi T; Hara S, BrF3-KHF2: An air-stable fluorinating reagent. J. Fluor. Chem, 2014, 168, 55–60. [Google Scholar]
- 20.Bychek RM; Hutskalova V; Bas YP; Zaporozhets OA; Zozulya S; Levterov VV; Mykhailiuk PK, Difluoro-Substituted Bicyclo[1.1.1]pentanes for Medicinal Chemistry: Design, Synthesis, and Characterization. J. Org. Chem, 2019, 84, 15106–15117. [DOI] [PubMed] [Google Scholar]
- 21.Tang XJ; Zhang ZX; Dolbier WR, Direct Photoredox-Catalyzed Reductive Difluoromethylation of Electron-Deficient Alkenes. Chem. Eur. J, 2015, 21, 18961–18965. [DOI] [PubMed] [Google Scholar]
- 22.Chen K; Berg N; Gschwind R; Konig B, Selective Single C(sp(3))-F Bond Cleavage in Trifluoromethylarenes: Merging Visible-Light Catalysis with Lewis Acid Activation. J. Am. Chem. Soc, 2017, 139, 18444–18447. [DOI] [PubMed] [Google Scholar]
- 23.Vogt DB; Seath CP; Wang HB; Jui NT, Selective C-F Functionalization of Unactivated Trifluoromethylarenes. J. Am. Chem. Soc, 2019, 141, 13203–13211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang HB; Jui NT, Catalytic Defluoroalkylation of Trifluoromethylaromatics with Unactivated Alkenes. J. Am. Chem. Soc, 2018, 140, 163–166. [DOI] [PubMed] [Google Scholar]
- 25.Yamauchi Y; Fukuhara T; Hara S; Senboku H, Electrochemical carboxylation of alpha,alpha-difluorotoluene derivatives and its application to the synthesis of alpha-fluorinated nonsteroidal anti-inflammatory drugs. Synlett, 2008, 3, 438–442. [Google Scholar]
- 26.Fuchibe K; Ohshima Y; Mitomi K; Akiyama T, Low-valent niobium-catalyzed reduction of alpha,alpha,alpha-trifluorotoluenes. Org. Lett, 2007, 9, 1497–1499. [DOI] [PubMed] [Google Scholar]
- 27.Stahl T; Klare HFT; Oestreich M, Main-Group Lewis Acids for C-F Bond Activation. ACS Catal, 2013, 3, 1578–1587. [Google Scholar]
- 28.Sugihara N; Suzuki K; Nishimoto Y; Yasuda M, Photoredox-Catalyzed C−F Bond Allylation of Perfluoroalkylarenes at the Benzylic Position. J. Am. Chem. Soc, 2021, 143, 9308–9313 [DOI] [PubMed] [Google Scholar]
- 29.Luo C; Bandar J, Selective Defluoroallylation of Trifluoromethylarenes. J. Am. Chem. Soc, 2019, 141, 14120–14125. [DOI] [PubMed] [Google Scholar]
- 30.Yan S; Liu S; Chen L; Lan Y; Luo S; Yu D, Visible-light photoredox-catalyzed selective carboxylation of C(sp3)−F bonds with CO2. Chem, 2021, 7, 1–15. [Google Scholar]
- 31.Yu YJ; Zhang FL; Peng TY; Wang CL; Cheng J; Chen C; Houk KN; Wang YF, Sequential C-F bond functionalizations of trifluoroacetamides and acetates via spin-center shifts. Science, 2021, 371, 1232–1240. [DOI] [PubMed] [Google Scholar]
- 32.Wessig P; Muehling O, Spin-center shift (SCS) - A versatile concept in biological and synthetic chemistry. Eur. J. Org. Chem, 2007, 14, 2219–2232. [Google Scholar]
- 33.Bansal KM; Gratzel M; Henglein A; Janata E, Polarographic and optical absorption studies of radicals produced in the pulse radiolysis of aqueous solutions of ethylene glycol. J. Phys. Chem, 1973, 77, 16–19. [Google Scholar]
- 34.Beckwith ALJ; Crich D; Duggan PJ; Yao Q, Chemistry of β-(Acyloxy)alkyl and β-(Phosphatoxy)alkyl Radicals and Related Species: Radical and Radical Ionic Migrations and Fragmentations of Carbon−Oxygen Bonds. Chem. Rev, 1997, 97, 3273–3312 and references therein. [DOI] [PubMed] [Google Scholar]
- 35.Hendy CM; Smith GC; Xu ZH; Lian TQ; Jui NT, Radical Chain Reduction via Carbon Dioxide Radical Anion (CO2•−). J. Am. Chem. Soc, 2021, 143, 8987–8992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chmiel AF; Williams OP; Chernowsky CP; Yeung CS; Wickens ZK, Non-innocent Radical Ion Intermediates in Photoredox Catalysis: Parallel Reduction Modes Enable Coupling of Diverse Aryl Chlorides. J. Am. Chem. Soc, 2021, 143, 10882–10889. [DOI] [PubMed] [Google Scholar]
- 37.Campbell MW; Yuan M; Polites VC; Gutierrez O; Molander GA, Photochemical C–H Activation Enables Nickel-Catalyzed Olefin Dicarbofunctionalization. J. Am. Chem. Soc, 2021, 143, 3901–3910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Luo Y-R, Handbook of Bond Dissociation Energies in Organic Compounds. CRC Press: New York, USA, 2003, 5–299. [Google Scholar]
- 39.Campbell MW; Compton JS; Kelly CB; Molander GA, Three-Component Olefin Dicarbofunctionalization Enabled by Nickel/Photoredox Dual Catalysis, J. Am. Chem. Soc, 2019, 141, 20069–20078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Edward R; Monteith E; Mampuys P; Summerton L; Clark J; Maes BU; McElroy CR, Why we might be misusing process mass intensity (PMI) and a methodology to apply it effectively as a discovery level metric, Green Chem, 2020, 22, 123–135. [Google Scholar]
- 41.Sumino S; Uno M; Ryu I; Matsuura M; Kishikawa Y, Photoredox-Catalyzed Hydrodifluoroalkylation of Alkenes Using Difluorohaloalkyl Compounds and a Hantzsch Ester, J. Org. Chem, 2017, 82, 5469–5474. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.