

Abstract
Inflammasomes sense intracellular clues of infection, damage, or metabolic imbalances. Activated inflammasome sensors polymerize the adaptor ASC into micron‐sized “specks” to maximize caspase‐1 activation and the maturation of IL‐1 cytokines. Caspase‐1 also drives pyroptosis, a lytic cell death characterized by leakage of intracellular content to the extracellular space. ASC specks are released among cytosolic content, and accumulate in tissues of patients with chronic inflammation. However, if extracellular ASC specks contribute to disease, or are merely inert remnants of cell death remains unknown. Here, we show that camelid‐derived nanobodies against ASC (VHHASC) target and disassemble post‐pyroptotic inflammasomes, neutralizing their prionoid, and inflammatory functions. Notably, pyroptosis‐driven membrane perforation and exposure of ASC specks to the extracellular environment allowed VHHASC to target inflammasomes while preserving pre‐pyroptotic IL‐1β release, essential to host defense. Systemically administrated mouse‐specific VHHASC attenuated inflammation and clinical gout, and antigen‐induced arthritis disease. Hence, VHHASC neutralized post‐pyroptotic inflammasomes revealing a previously unappreciated role for these complexes in disease. VHHASC are the first biologicals that disassemble pre‐formed inflammasomes while preserving their functions in host defense.
Keywords: arthritis, extracellular inflammasomes, gout, nanobodies, pyroptosis
Subject Categories: Immunology, Musculoskeletal System
Inflammasome activation results in a lytic cell death named Pyroptosis. Following pyroptosis, inflammasome platforms called ASC specks are released into the extracellular space, where they spread inflammation through their pro‐inflammatory and prion‐like activities. However, if extracellular ASC specks have functions in the development of disease remains ill‐defined. The present study tackles this question using ASC‐targeting nanobodies.

Introduction
Inflammasomes are supramolecular organizing centers (SMOCs) that enable maturation of IL‐1‐family cytokines (e.g., IL‐1β) and trigger a pro‐inflammatory form of lytic cell death, termed pyroptosis. Inflammasomes are assembled following the activation of a sensor molecule which nucleates the oligomerization of the adaptor protein apoptosis‐associated speck‐like protein containing a CARD (ASC) (Martinon et al, 2002). ASC assembles into filamentous structures that are cross‐linked into star‐shaped complexes called specks, and which optimize signal transduction via proximity‐induced caspase‐1 activation. Caspase‐1 then drives the maturation of pro‐forms of IL‐1 cytokines and the proteolytic activation of gasdermin D (GSDMD). Active GSDMD oligomerizes and forms pores in the plasma membrane, which enable the release of IL‐1β and induce pyroptosis. At a later stage of pyroptosis, membrane damage is extensive andallows for the release of larger endogenous molecules including ASC specks (Baroja‐Mazo et al, 2014; Franklin et al, 2014; Volchuk et al, 2020).
Although IL‐1 cytokines are crucial to counter infection, their unbalanced production is responsible for a large spectrum of autoinflammatory disorders (Broderick et al, 2015; Spel & Martinon, 2020; Tartey & Kanneganti, 2020). Therapeutic strategies developed to prevent the negative effect of aberrant IL‐1 cytokines have mainly focused on targeting either cytokines (especially IL‐1β) or their receptors directly (Chauhan et al, 2020). However, the outputs of inflammasome activation are broader and include numerous danger signals (DAMPS), lipid mediators (von Moltke et al, 2012), and alarmins (Phulphagar et al, 2021). Given their essential roles in host defense, continuous inhibition of IL‐1 cytokines increases the risk of infections, as observed in patients receiving anakinra (Salliot et al, 2009). Hence, interest in targeting specific inflammasome sensors (e.g., NLRP3) has risen in the last years (Mangan et al, 2018; Chauhan et al, 2020). Preventing the activation of the upstream sensor would shortcut the cell death pathway triggered by a specific inflammasome and the ensuing release of DAMPs and ASC specks. However, this would also prevent beneficial roles of NLRP3 in host defense.
In addition to its central role as an inflammasome adaptor, inflammasome‐independent functions of ASC that amplify inflammation have been described (Ellebedy et al, 2011; Guo & Dhodapkar, 2012; Tsuchiya et al, 2014; Franklin et al, 2017; Venegas et al, 2017; Friker et al, 2020). We and others have shown that ASC specks are released from pyroptotic cells into the extracellular space (Baroja‐Mazo et al, 2014; Franklin et al, 2014). Once released, these post‐pyroptotic specks accumulate in tissues, where they sustain production of IL‐1β or are engulfed by surrounding phagocytes to propagate inflammation through seeding of further inflammasome structures within the recipient cells. Extracellular ASC specks are observed in mice after viral or bacterial infection (Sagoo et al, 2016; Tzeng et al, 2016), in the serum of patients with cryopyrin‐associated periodic syndrome (CAPS) (Baroja‐Mazo et al, 2014) or HIV infection (Ahmad et al, 2018), or in the bronchoalveolar lavage of chronic obstructive pulmonary disease (COPD) patients (Franklin et al, 2014). Recently, ASC specks were suggested as candidate biomarkers for medullary pyroptosis and ineffective hemopoiesis in patients with myelodysplastic syndromes (Basiorka et al, 2018). In fact, ASC‐targeting antibodies develop in patients with autoimmune disease (Franklin et al, 2014) indicating exposure of antigen‐presenting cells to circulating ASC. Furthermore, ASCPYD has prion‐like activities in vitro and in yeast (Cai et al, 2014; Lu et al, 2014) that are maintained in mammalian cells, where engulfed ASC specks nucleate the polymerization of soluble ASC (Franklin et al, 2014). In the brain, ASC specks from pyroptotic microglia are released into the parenchyma where they cross‐seed Amyloid‐β plaques and contribute to neuroinflammation in Alzheimer's Disease (AD; Venegas et al, 2017; Friker et al, 2020). Hence, there is high interest in targeting ASC specks to limit their extracellular pro‐inflammatory activity.
Our previous attempt to target ASC specks using conventional antibodies (Abs) resulted in increased inflammation in a silica‐induced model of peritonitis (Franklin et al, 2014). Anti‐ASC Abs promoted the uptake of extracellular ASC specks by phagocytes through Fc‐mediated opsonization leading to increased IL‐1β release from macrophages and immune cell infiltration into the peritoneal cavity. This common feature of conventional Abs used for therapy encouraged the development of alternative approaches, including single‐domain antibody fragments, such as nanobodies (VHHs), which are derived from larger heavy chain‐only Abs found in camelids. We recently generated a VHH against human ASC (VHHASC), which we over‐expressed in the cytosol of cells to study the molecular mechanisms involved in ASC oligomerization (Schmidt et al, 2016b). We showed that VHHASC binds the caspase‐recruitment domain (ASCCARD) of ASC, preventing formation of CARD/CARD interactions necessary to form full ASC specks.
In this study, we tested the therapeutic potential of VHHASC and a newly generated VHH against murine ASC (VHHmASC) to target ASC specks in vitro and in vivo. We show that pre‐incubation of extracellular ASC specks with VHHASC abrogated their inflammatory functions in vitro. Recombinant VHHASC rapidly disassembled pre‐formed ASC specks and thus inhibited their ability to seed the nucleation of soluble ASC. Notably, VHHASC required prior cytosolic access to prevent inflammasome activation within cells, but it was effective against extracellular ASC specks released following caspase‐1‐dependent loss of membrane integrity, and pyroptosis. Finally, systemic treatment with VHHmASC efficiently dampened the inflammation induced by intra‐articular challenges with either monosodium urate (MSU) crystals or mBSA in mouse models of acute gout or chronic rheumatoid arthritis (RA), respectively.
Together, our data provide the first evidence that extracellular ASC specks, or those exposed to the extracellular environment following pyroptosis, contribute to the development of chronic disease such as gout and rheumatoid arthritis. They also indicate that VHHASC can effectively disassemble pre‐formed inflammasomes, thus questioning for the first time the stabile nature of SMOCs. Hence, our study opens the possibility of targeting inflammasomes, and potentially other SMOCs, in established chronic inflammatory diseases in the clinic.
Results
VHHASC blocks pro‐inflammatory function of extracellular ASC specks
We first tested the effect of nanobody directed against human ASC (VHHASC) on the pro‐inflammatory function of ASC specks assembled in vitro from recombinant sources (Fernandes‐Alnemri et al, 2007; Fernandes‐Alnemri and Alnemri, 2008; Franklin et al, 2014). For this, recombinant human ASC specks were incubated with VHHASC prior to their addition to murine bone marrow‐derived macrophages (Fig 1A). To create a specific VHH control, we introduced a single structure‐guided amino acid mutation R50D into the complementarity determining regions (CDR2) of the VHHASC, generating a variant (mutVHHASC) unable to bind ASC, as shown by LUMIER assay (Fig EV1). We also tested several substitutions of single amino acids (T57, Y59, D62) present at the ASC/VHHASC interface (Schmidt et al, 2016) or their combinations (Fig EV1). The single R50D mutation showed the strongest loss of binding to ASC and was therefore used throughout this study (mutVHHASC). As expected (Baroja‐Mazo et al, 2014; Franklin et al, 2014; de Almeida et al, 2015), macrophages incubated with ASC specks released IL‐1β into the culture supernatants. Notably, the pre‐incubation of ASC specks with VHHASC, but not with mutVHHASC or a conventional polyclonal anti‐ASC Ab (anti‐ASC pAb), blunted the ability of ASC specks to trigger IL‐1β secretion from macrophages (Fig 1A). Treatment of macrophages with VHHASC alone did not induce IL‐1β release (Fig 1A). These results indicate that VHHASC interferes with the capacity of extracellular ASC specks to stimulate further intracellular processing of IL‐1β after their phagocytosis by surrounding macrophages.
Figure 1. VHHASC blocks the extracellular pro‐inflammatory function of ASC specks.
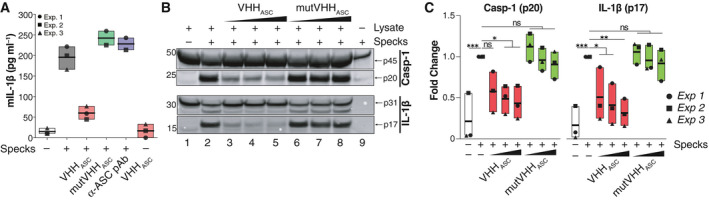
- Mouse IL‐1β (mIL‐1β) concentrations in cell‐free supernatants of LPS‐primed BMDMs (200 ng ml−1, for 2 h), that were stimulated with in vitro‐generated human ASC specks (200 µg ml−1, O/N). ASC specks were pre‐incubated with 200 µg ml−1 of an anti‐human ASC nanobody (VHHASC), its mutated variant, mutVHHASC or 50 µg ml−1 of polyclonal ASC antibody (a‐ASC pAb, AL177) for 15 min at RT. Data is pooled from at least two independent experiments, each represented by a different symbol, and is displayed as floating bars with the max/min values and mean (thicker band).
- Immunoblot analysis of pro‐ (p31) and cleaved (p17) IL‐1β, and pro‐ (p45) and cleaved (p20) caspase‐1 in cytosolic fractions of ASC deficient (Pycard −/−) immortalized murine macrophages that were primed with LPS (200 ng ml−1, for 3 h). Fractions were incubated with recombinant ASC specks in the presence of 2, 20 or 200 µg ml−1 of VHHASC or mutVHHASC. One representative of three independent experiments is shown.
- Quantitative analysis of band densitometry of three independent experiments as shown in B. Data represents the fold change from all conditions (lanes 1–9) vs. Pycard −/− lysates incubated with ASC specks alone (lane 2). ns P > 0.05; *P < 0.05; **P < 0.005; ***P < 0.0002, One‐way ANOVA, multiple comparison (Dunnet test). Data is displayed as floating bars with the max/min values and mean (thicker band).
Source data are available online for this figure.
Figure EV1. Binding activity and species specificity of ASC VHHs.
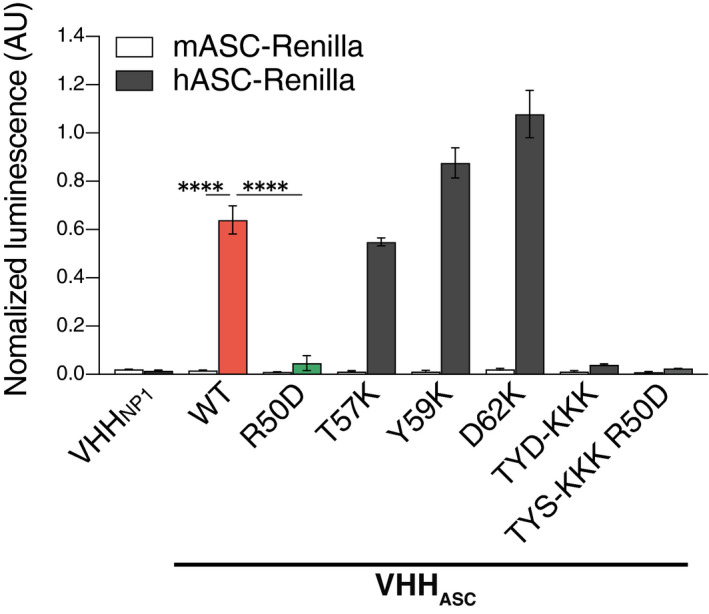
Lysates of HEK 293T cells transiently expressing HA‐tagged VHHASC (wild‐type or point mutants), an unrelated control (VHHNP1) and the indicated bait proteins fused to Renilla luciferase were used to immunoprecipitate VHHs with immobilized anti‐HA antibody. Renilla luciferase activity of the co‐immunoprecipitated proteins was measured and normalized to the input luciferase. Data represents mean values ± SEM from three independent experiments. ****P < 0.0001, One‐way ANOVA, multiple comparison (Tukey test). 'TYD‐KKK' indicates the triple mutant T57K, Y59K, and D62K.
Because extracellular ASC specks can also induce the maturation of caspase‐1 and IL‐1β in the extracellular space (Baroja‐Mazo et al, 2014; Franklin et al, 2014), we assessed the proteolytic processing of pro‐caspase‐1 and pro‐IL‐1β in the lysates of LPS‐primed ASC‐deficient (Pycard −/−) immortalized murine macrophages incubated with ASC specks in the presence of increasing concentrations of VHHASC or mutVHHASC. As previously reported (Baroja‐Mazo et al, 2014; Franklin et al, 2014; de Almeida et al, 2015), exogenous ASC specks induced the activation of pro‐caspase‐1 (p45), and pro‐IL‐1β (p31) contained in Pycard −/− lysates into their mature forms (p20 and p17, respectively, Fig 1B and C). VHHASC dose‐dependently inhibited the ASC specks‐induced activation of pro‐caspase‐1 and pro‐IL‐1β, while mutVHHASC had no effect. Consistent with our previous observations (Franklin et al, 2014), ASC specks contained little caspase‐1, but no detectable IL‐1β (Fig 1B), supporting that the detected proteins originate from the cleavage of their pro‐forms in the lysates. Together these data demonstrate that VHHASC blocks the pro‐inflammatory activity of extracellular ASC specks.
VHHASC disassemble ASC oligomers and block the prionoid activities of ASC specks
In addition to its central function in assembling inflammasomes, ASC specks have prion‐like activities (Cai et al, 2014; Franklin et al, 2014; Lu et al, 2014) which propagate inflammation by nucleating the oligomerization of soluble ASC in vitro, and in the cytosol of macrophages that phagocytose ASC specks. We therefore assessed the effect of VHHASC or mutVHHASC on the nucleation of soluble ASC‐mTurquoise by ASC‐GFP specks. We imaged the recruitment of soluble ASC‐mTurquoise to ASC‐GFP specks that had been pre‐incubated with VHHs (Fig 2A and B). In line with our previous observations (Franklin et al, 2014), ASC‐GFP specks rapidly seeded the polymerization of soluble ASC‐mTurquoise. However, pre‐incubation with VHHASC prevented ASC‐GFP specks from seeding the nucleation of soluble ASC‐mTurquoise, while mutVHHASC had no effect (Fig 2A and B). To test whether VHHASC affected the integrity of specks, we assessed the stability of ASC oligomers using DSS cross‐link and immunoblotting (Fernandes‐Alnemri et al, 2007; Fernandes‐Alnemri and Alnemri, 2008; Franklin et al, 2014; Hoss et al, 2018; Fig 2C). VHHASC dose‐dependently disrupted the oligomeric conformation of ASC specks, demonstrated by the decreased proportion of ASC oligomers to monomers (Fig 2C), confirming previous findings that VHHASC shifts GFP‐ASCCARD from an oligomeric to a monomeric conformation (Schmidt et al, 2016b). In contrast, mutVHHASC had no effect on the conformation of ASC specks, nor did the incubation with an ASC‐specific pAb (Fig 2C). Hence, VHHASC masks and prevents critical ASCCARD‐ASCCARD interactions within specks disassembling ASC oligomers, and blocking their prion‐like functions.
Figure 2. VHHASC disrupts the “prionoid” activities and disassembles ASC specks.
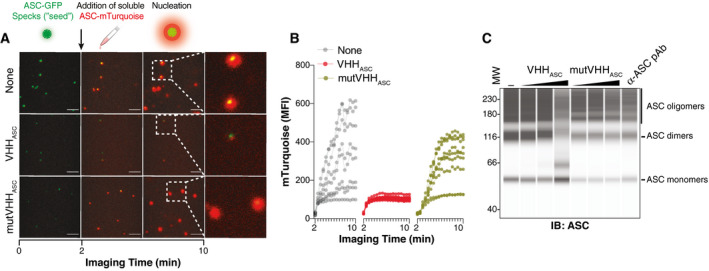
- Time‐lapse confocal imaging of the in vitro nucleation of soluble ASC‐mTurquoise (red) by ASC‐TagGFP (ASC‐GFP) specks (green), that were left untreated (None), or pre‐incubated with VHHASC or mutVHHASC (200 µg ml−1 for 15 min). Scale bars: 10 μm.
- Median fluorescence intensity (MFI) of mTurquoise graphed over time showing its polymerization seeded by ASC‐TagGFP specks. Each line shows the mTurquoise MFI around a seeding ASC‐TagGFP speck. Data is one representative of three independent experiments.
- WES capillary electrophoresis and immunoblotting of DSS cross‐linked in‐vitro‐generated human ASC‐mTurquoise (ASC) specks that were incubated for 1 h (RT) with 2, 20 or 200 µg ml−1 of VHHASC, mutVHHASC or with 20 µg ml−1 of a polyclonal anti‐ASC Ab (a‐ASC pAb). Data is from one representative of four independent experiments.
Source data are available online for this figure.
VHHASC requires access to cytosolic ASC to prevent inflammasome activation in intact cells
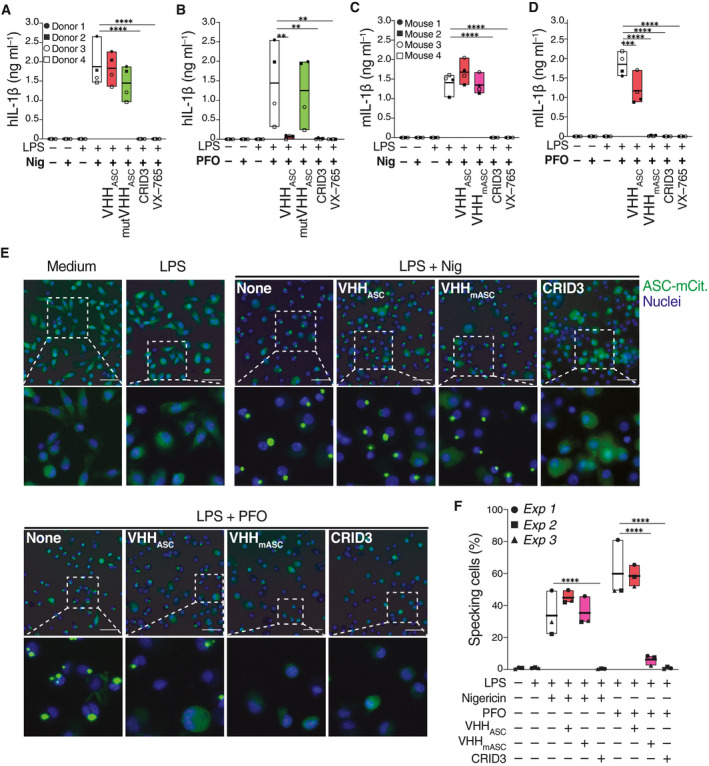
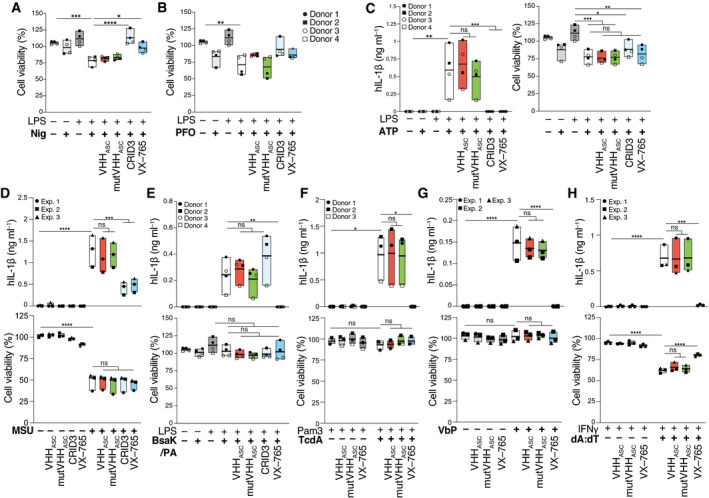
Next, we investigated whether the addition of VHHASC directly into culture supernatants of inflammasome‐activated cells affects the intracellular processing of caspase‐1 and IL‐1β. We pre‐incubated LPS‐primed primary human macrophages (hMDMs) with VHHASC or mutVHHASC. As controls, we pre‐treated hMDMs with CRID3 (MCC950) (Coll et al, 2015) or VX‐765, two well‐characterized inhibitors of the NRLP3 and caspase‐1, respectively. Cells were then treated with nigericin, for the activation of NLRP3 (Figs 3A and EV2A). As expected, both CRID3 and VX‐765 abrogated the IL‐1β release (Fig 3A) and prevented cell death (Fig EV2A) induced by stimulation with nigericin. Notably, neither VHHASC nor mutVHHASC had any effect on IL‐1β secretion or the viability of NLRP3‐activated cells (Figs 3A and EV2A). Similar results were observed in cells stimulated with ATP (NLRP3 in, Fig EV2C), MSU (NLRP3 in PMA‐treated THP‐1, Fig EV2D), a combination of LFn‐BsaK and PA (NLRC4 in hMDMs, Fig EV2E), TcdA (Pyrin in CD14+ monocytes, Fig EV2F), Val‐boroPro (NLRP1 in N/TERT‐1 keratinocytes, Fig EV2G), or poly(dA:dT) (AIM2 in PMA‐ and IFNγ‐treated THP‐1, Fig EV2H), indicating that our finding can be generalized to other NLRP3 activators, and other inflammasomes. Based on these findings, we speculated that VHHASC was unable to cross the intact plasma membrane early enough to prevent inflammasome activation in intact cells. To address this hypothesis, we stimulated LPS‐primed hMDMs with the pore‐forming toxin perfringolysin O (PFO), which forms membrane pores of ~40 nm reminiscent of GSDMD pores (Czajkowsky et al, 2004; Mulvihill et al, 2018), and was previously shown to induce the potassium efflux‐dependent activation of NLRP3 (Yamamura et al, 2019). Supporting our hypothesis, the presence of VHHASC in the supernatants completely abrogated the release of IL‐1β from PFO‐treated macrophages, similarly to CRID3 and VX‐765 (Fig 3B). The mutant nanobody, mutVHHASC, failed to block IL‐1β release. While IL‐1β release was efficiently blunted in VHHASC‐treated cells, VHHASC could not restore cell viability induced by PFO (Fig EV2B), likely due to the loss of membrane integrity caused by PFO pores. Supporting this view, neither CRID3 nor VX‐765 could rescue cell viability in PFO‐activated cells (Fig EV2B).
Figure 3. VHHASC and VHHmASC prevent IL‐1β release and ASC speck formation induced by PFO, but not nigericin in primary human and mouse cells.
-
A–D(A, B) Human IL‐1β (hIL‐1β) concentrations in cell‐free supernatants of LPS‐primed (10 ng ml−1, 150 min) primary human macrophages that were left untreated, or pre‐incubated with VHHASC or mutVHHASC (100 µg ml−1), CRID3 (50 µM) or VX‐765 (50 µM) for 30 min before stimulation with (A) nigericin (10 µM), or (B) PFO (30 ng ml−1) for 2 h. (C‐D) Mouse IL‐1β (mIL‐1β) concentrations in cell‐free supernatants of LPS‐primed mouse BMDMs (200 ng ml−1, 150 min), incubated with VHHs, CRID3 or VX765, before activation with nigericin (10 µM), or PFO (250 ng ml−1). Data is combined from two independent experiments, each performed with two donors (A, B) or mice (C, D), represented with individual symbols (4 donors or mice in total). Data is displayed as floating bars with the max/min values and mean (thicker band).
-
E, F(E) Epifluorescence microscopy imaging and (F) quantification of ASC speck formation in BMDMs from ASC‐mCitrine (Green) transgenic mice. Cells were primed with LPS (200 ng ml−1, 150 min), pre‐treated with VX‐765 (50 µM, 30 min), then treated with VHHASC, VHHmASC (100 µg ml−1) or CRID3 (50 µM) for another 30 min before stimulation with nigericin (top), or PFO (bottom) for 2 h and finally fixed with 4% PFA. Nuclei was stained with DRAQ5 (Blue). Scale bars: 100 μm. Images in (E) are from one representative out of three independent experiments that were quantified in F. Data in F is displayed as floating bars with the max/min values and mean (thicker band).
Data information: **P < 0.005; ***P < 0.0002; ****P < 0.0001, One‐way ANOVA, multiple comparison (Tukey test).
Figure EV2. VHHASC preserves endogenous inflammasome activation in human living cells.
-
A, BCell viability (CTB) assay on LPS‐primed primary human macrophages that were left untreated, or pre‐incubated with VHHASC or mutVHHASC (100 µg ml−1), CRID3 (50 µM) or VX‐765 (50 µM) for 30 min before being activated with (A) nigericin (10 µM), or (B) PFO (30 ng ml−1) for 2 h. Data is from the experiments displayed in Fig 3A and B.
-
CHuman IL‐1β (hIL‐1β) concentrations in cell‐free supernatants (left), and cell viability assay (right) of LPS‐primed primary human macrophages that were incubated with VHHASC or mutVHHASC (100 µg ml−1), CRID3 (50 µM) or VX‐765 (50 µM) for 30 min before being stimulated with 2.5 mM ATP.
-
DhIL‐1β concentrations in cell‐free supernatants (top), and cell viability assay (bottom) of PMA‐differentiated THP‐1 cells treated with VHHASC or mutVHHASC (100 µg ml−1), CRID3 (10 µM) or VX‐765 (50 µM) for 30 min before 4.5 h stimulation with 250 µg ml−1 MSU crystals.
-
EhIL‐1β concentrations in cell‐free supernatants (top), and cell viability assay (bottom) of LPS‐primed primary human macrophages that were incubated with VHHASC or mutVHHASC (100 µg ml−1), CRID3 (50 µM) or VX‐765 (50 µM) for 30 min before being stimulated with 0.1 µg ml−1/0.5 µg ml−1 mixture of LFn‐BsaK and PA for 2 h.
-
FhIL‐1β concentrations in cell‐free supernatants (top), and cell viability assay (bottom) of Pam3CysK4‐primed (1 µg ml−1) primary human CD14+ monocytes that were incubated with VHHASC or mutVHHASC (100 µg ml−1), or VX‐765 (50 µM) for 30 min before being stimulated with 1 µg ml−1 TcdA.
-
GhIL‐1β concentrations in cell‐free supernatants (top), and cell viability assay (bottom) of keratinocyte cells (N‐TERT) that were treated with VHHASC or mutVHHASC (100 µg ml−1), or VX‐765 (50 µM), then directly stimulated with 30 µM Val‐boroPro (VbP) for 22 h.
-
HhIL‐1β concentrations in cell‐free supernatants (top), and cell viability assay (bottom) of PMA‐differentiated THP‐1 cells treated with IFNγ (500 U ml−1) for 16 h and that were incubated with VHHASC or mutVHHASC (100 µg ml−1), CRID3 (10 µM) or VX‐765 (50 µM) for 30 min before 2 h stimulation with 1 µg ml−1 poly(dA:dT) in complex with Lipofectamine 2000.
Data information: Data is representative of either two independent experiments, each run with one to two donors (A–C, E, F, 3 or 4 donors in total) or at least 3 independent experiments (D, G, H). Each symbol represents one donor or independent experiment. ns P > 0.05; *P < 0.05; **P < 0.005; ***P < 0.0002; ****P < 0.0001, One‐way ANOVA, multiple comparison (Tukey test). Data is displayed as floating bars with the max/min values and mean (thicker band).
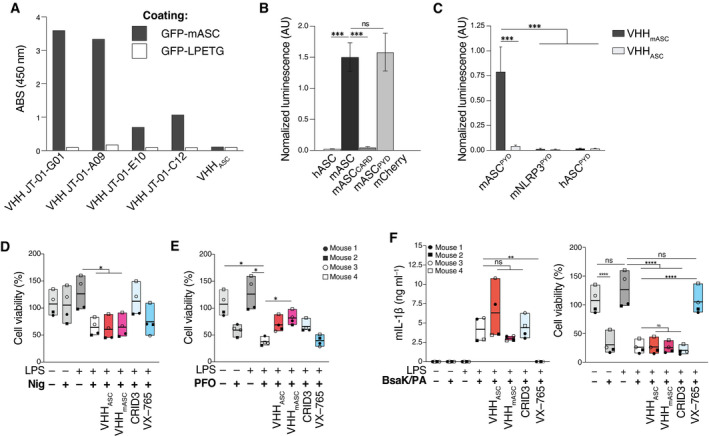
As shown by the stringent LUMIER assay (Fig EV1), VHHASC does not efficiently bind murine ASC. We therefore generated mouse‐specific anti‐ASC VHHs (VHHmASC) by immunizing an alpaca with murine ASC, and screening for nanobodies able to target mouse ASC. We identified several VHHs specific for murine ASC by phage display and confirmed binding by ELISA assay with recombinant proteins (Fig EV3A). VHH JT‐01‐A09 was selected for further use in this manuscript (VHHmASC). We further characterized binding of VHHmASC to mouse ASC with LUMIER assays, which evaluate the cytosolic interaction of HA‐tagged VHHmASC with bait proteins fused to Renilla luciferase. These experiments revealed that VHHmASC functions in the reducing environment of the cytosol, and that VHHmASC binds to the pyrin domain (ASCPYD) of mouse ASC, but neither the CARD (ASCCARD), nor the human ASC (Fig EV3B). To demonstrate the specificity of VHHmASC to mouse ASCPYD, we also tested for binding of VHHmASC to mouse NLRP3PYD or human ASCPYD (Fig EV3C). As expected, VHHmASC exclusively bound mouse ASCPYD, but no other PYDs (Fig EV3C). We then tested whether VHHmASC is also capable of inhibiting the formation of ASC specks in mouse bone marrow‐derived macrophages (BMDMs). Similar to the human‐specific VHHASC, VHHmASC had no effect on IL‐1β release or survival of LPS‐primed BMDMs stimulated with nigericin (Figs 3C and EV3D) or BsaK (Fig EV3F). In line with the activity of VHHASC in human macrophages (Fig 3B), pre‐treatment of murine BMDMs with VHHmASC fully abrogated the PFO‐induced release of IL‐1β to a similar degree to CRID3 or VX‐765 (Fig 3D), and partially rescued cell viability (Fig EV3B). Interestingly, when tested in mouse BMDMs, the anti‐human ASC nanobody (VHHASC) partially impaired the PFO‐induced release of IL‐1β, and rescued cell viability (Figs 3D and EV3B), indicating some limited cross‐reactivity.
Figure EV3. VHHmASC is specific to ASCPYD but retains the same activity as VHHASC .
-
ASpecificity of different single‐domain antibodies (VHHs) probed by ELISA. Recombinant murine ASC as an eGFP fusion or eGFP alone (GFP‐LPETG) as control, were coated onto ELISA plates at 1 µg ml−1/well. Wells were incubated with HA‐tagged VHHs (100 nM), anti‐HA‐tag mouse mAb coupled to HRP, and the HRP substrate TMB. Binding was quantified by measuring the absorbance at 450 nm.
-
B, CLysates of HEK 293T cells transiently expressing HA‐tagged VHHmASC or VHHASC and the indicated bait proteins fused to Renilla luciferase were used to immunoprecipitate VHHs with immobilized anti‐HA antibody. Renilla luciferase activity of the co‐immunoprecipitated proteins was measured and normalized to the input luciferase. Data represents mean values ± SEM from three independent experiments.
-
D, ECell viability (CTB) assay on LPS‐primed (200 ng ml−1) primary mouse BMDMs that were left untreated, or pre‐incubated with VHHASC or VHHmASC (100 µg ml−1), CRID3 (50 µM) or VX‐765 (50 µM) for 30 min before being activated with (D) nigericin (10 µM), or (E) PFO (250 ng ml−1) for 2 h. Data is from the experiments displayed in Fig 3C and D. Data is displayed as floating bars with the max/min values and mean (thicker band).
-
FMouse IL‐1β (mIL‐1β) concentrations in cell‐free supernatants (left), and cell viability assay (right) of LPS‐primed mouse BMDMs incubated with VHHs, CRID3 or VX–765, before stimulation with 0.1 µg ml−1/0.5 µg ml−1 mixture of LFn‐BsaK and PA for 2 h. Each symbol represents one mouse. Data is displayed as floating bars with the max/min values and mean (thicker band).
Data information: ns P > 0.05; ***P < 0.001; ****P < 0.0001, Two‐way (A, C) or One‐way (B, D–F) ANOVA, multiple comparison (Tukey test).
Next, we tested the activity of VHHASC and VHHmASC on BMDMs derived from transgenic mice expressing mouse ASC in fusion with mCitrine (Tzeng et al, 2016). This allowed us to study the activity of VHHASC or VHHmASC on the intracellular formation of ASC‐mCitrine specks (Fig 3E and F). To prevent the formation of GSDMD pores and restrict nanobody access to the cytosol, we treated cells with VX‐765. As expected, primed macrophages treated with nigericin or PFO displayed the typical fluorescent ASC puncta (Fig 3E). Pre‐treatment with CRID3 entirely abrogated the formation of specks in either nigericin‐ or PFO‐treated cells. In cells stimulated with nigericin, pre‐treatment with VHHASC or VHHmASC had no effect (Fig 3E and F). In contrast, in PFO‐stimulated cells, VHHmASC strongly prevented ASC specks formation, while VHHASC had no effect, confirming the limited cross‐reactivity between VHHASC and mouse ASC.
Together, our data suggest that membrane pores are required for VHHs to access the cytosol of inflammasome‐activated cells. To consolidate these findings, we tested the contribution of GSDMD in the effect of VHHASC. For this, we used THP‐1 cells expressing a doxycycline (Dox)‐inducible CRISPR‐Cas9 cassette as well as a sgRNA targeting GSDMD (Budden et al, 2021; Fig 4). Doxycycline treatment initiated GSDMD knockout and efficiently abrogated GSDMD expression (Fig 4A). As expected, GSDMD deficiency strongly attenuated the release of IL‐1β and LDH from nigericin‐activated cells (Fig 4B, left panels), but had minor impact on the release of IL‐1β from PFO‐treated cells (Fig 4B, upper right panel). Irrespective of GSDMD expression, VHHASC blunted the release of IL‐1β induced by PFO (Fig 4B, upper right panel), while it had no effect on nigericin‐activated cells (Fig 4B, upper left panel). As expected, CRID3 treatment completely blocked IL‐1β and LDH release in response to both nigericin and PFO (Fig 4B, upper panels). PFO‐treated GSDMD‐KO cells released lower amounts of LDH compared with GSDMD competent cells, indicating that LDH release induced by PFO partly depends on GSDMD (Fig 4B, lower right panel). The presence of VHHASC further decreased the release of LDH in both GSDMD competent and knockout cells (Fig 4B, lower right panel). To study the kinetic of VHHASC uptake by inflammasome‐activated cells, we performed live imaging on PMA‐primed and nigericin‐stimulated ASC‐GFP expressing THP‐1 cells in the presence of low dose (10 µg ml−1) of fluorescently‐labeled VHHASC (VHHASC‐AF647) directly applied into the culture supernatants (Fig 4C). Inflammasome activation allowed the entry of VHHASC‐AF647 which co‐localized to endogenous ASC‐GFP specks. VHHASC‐AF647 staining of GFP specks was prevented by the caspase‐1 inhibition with VX‐765 (Fig 4C). Confirming the requirement for GSDMD for VHHASC entry into cells, deletion of GSDMD (+Dox) prevented both cell death (propidium iodide staining) and VHHASC‐AF647 internalization (Fig EV4A and Movie EV1). Notably, upon stimulation with PFO, VHHASC‐AF647 rapidly entered the cells (~ 5 min) in a manner that was independent of GSDMD expression (Fig EV4B and Movie EV1). Together, these findings confirm that early access to the cell cytosol correlates with the ability of VHHASC to interfere with ASC specks formed from inflammasome activation inside cells. Hence, in our previous experiments, PFO pores and not GSDMD allowed VHHASC access to the cytosol to target endogenous ASC and prevent inflammasome activation and IL‐1β release. These experiments also demonstrate that upon stimulation with conventional inflammasome activators (e.g., nigericin), VHHASC predominantly targets ASC specks in the extracellular space, or exposed in pyroptotic cells, and that GSDMD‐dependent membrane permeabilization is required for targeting intracellular ASC. We could also demonstrate the species specificity of VHHASC, and discovered a new mouse‐specific nanobody targeting ASCPYD with inhibitory properties similar to VHHASC.
Figure 4. Contribution of GSDMD in the effect of VHHASC on the release of IL‐1β and cell death induced by PFO or nigericin.
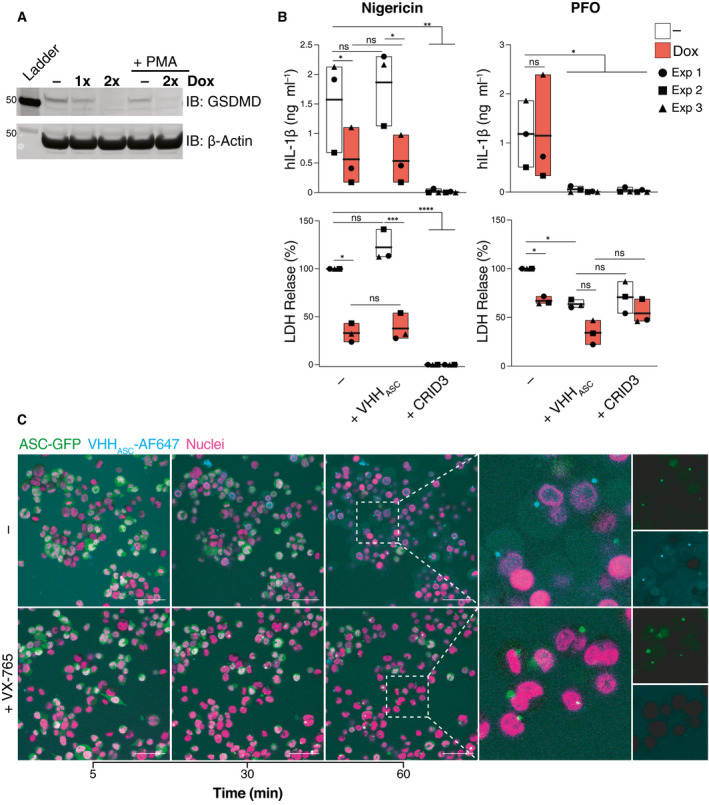
- Immunoblot analysis of GSDMD expression following the indicated course of Dox treatment and PMA‐differentiation, as indicated. Data is from one representative of two independent experiments.
- IL‐1β concentration or percentage of LDH released into cell‐free supernatants of PMA‐differentiated THP‐1 cells that were treated with VHHASC (200 µg ml−1) or CRID3 (25 µM) for 30 min prior to stimulation with nigericin (10 µM, left panels) or PFO (30 ng ml−1, right panels) for 2 h. Data is average of experimental duplicates from three independent experiments, each represented by a different symbol. ns P > 0.05; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001, Two‐way ANOVA, multiple comparison (Tukey test). Data is displayed as floating bars with the max/min values and mean (thicker band).
- Live confocal imaging of PMA‐differentiated and nigericin‐treated (10 µM) THP‐1 cells expressing human ASC‐GFP (green) in the presence of AlexaFluor647‐labeled VHHASC (VHHASC‐AF647, 10 µg ml−1, cyan) in the medium. Cells were either left untreated (–) or incubated with VX‐765 (50 µM) for 1 h prior to nigericin stimulation. Nuclei were stained with Hoechst 34580 (magenta). Scale bar: 50 µm. Data is from one representative out of two independent experiments.
Source data are available online for this figure.
Figure EV4. Contribution of GSDMD in the effect of VHHASC on the internalization of VHHASC in cells stimulated with PFO or nigericin.
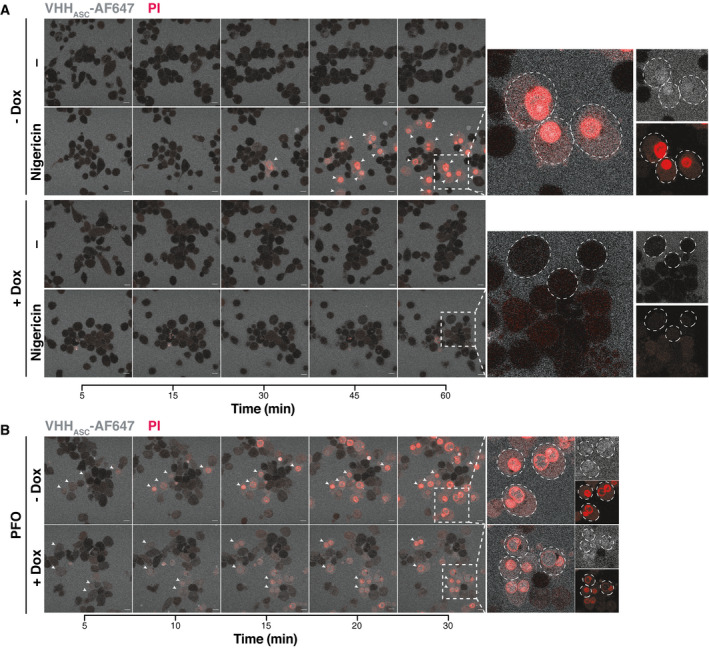
-
A, BConfocal microscopy images of GSDMD competent (–Dox) or GSDMD‐KO (+Dox) cells primed with PMA and that were either left untreated or treated with (A) nigericin (10 µM) or (B) PFO (30 ng ml−1) for the indicated periods of time and in the presence of AlexaFluor647‐labeled VHHASC (VHHASC‐AF647, 10 µg ml−1, blue) and propidium iodide (PI, 3.33 µg ml−1, red). White arrows indicate of cells with intracellular VHHASC‐AF647 signal. Dashed circles define boundaries of representative cells. Data is from one representative out of three independent experiments. Scale bar: 10 µm. Also see Movie EV1.
VHHASC targets ASC specks in inflammasome‐activated cells undergoing pyroptosis
Having shown that ectopic treatment with VHHASC does not prevent the initial assembly of ASC specks in cells treated with nigericin, we wondered whether VHHASC could engage with their intracellular targets after the onset of pyroptosis. We therefore performed live cell imaging on PMA‐differentiated THP‐1 macrophages expressing fluorescent ASC‐GFP and followed the dynamics of ASC speck formation upon treatment with nigericin or PFO (Fig 5A and B and Movie EV2). As expected, nigericin and PFO induced a rapid increase in the proportion of cells assembling ASC specks that plateaued after 100 or 40 min, respectively (Fig 5A and B and Movie EV2). The presence of VHHASC in PFO‐treated cells almost entirely abrogated the formation of ASC specks, confirming that the rapid access of VHHASC to the cytosol (Fig EV4B) is allowed by PFO pores (Fig 5B, right panel and Movie EV2). In contrast, the proportion of nigericin‐treated cells containing ASC‐GFP specks in the presence of VHHASC initially increased, peaking at 50 min (Fig 5B, left panel and Movie EV2), but decreased to levels close to untreated cells at later time points (Fig 5B, left panel and Movie EV2). These findings indicate that the GSDMD‐dependent membrane perforation granting VHHASC access to the cytosol (Figs 4C and EV4A) enabled interference with ASC specks inside pyroptotic cells. Further supporting this conclusion and previous data (Fig 4C), caspase‐1 inhibition with VX‐765 prevented VHHASC from targeting and disassembling cytosolic ASC‐GFP specks in nigericin‐treated cells (Fig 5B, left panel and Movie EV2). Hence, GSDMD‐dependent plasma membrane perforation allows VHHASC to target exposed ASC specks in pyroptotic cells without affecting pre‐pyroptotic IL‐1β maturation (Zhou & Abbott, 2021). To exclude that the decreased number of specks detected in the presence of VHHASC was not due to the release of specks into the extracellular space (Baroja‐Mazo et al, 2014; Franklin et al, 2014), we quantified ASC specks in the culture supernatants (Fig 5A and B) by flow cytometry, or the formation of ASC oligomers by DSS‐cross‐linking and immunoblotting (Fig 5C and D and Appendix Fig S1). Consistent with the ability of VHHASC to disassemble ASC oligomers, supernatants of cells treated with nigericin or PFO in the presence of VHHASC had decreased amounts of GFP‐positive specks (Fig 5C and D and Appendix Fig S1B). These findings were reproducible in cells stimulated with poly(dA:dT) (Fig EV5A and B and Movie EV3), MxiH (Fig EV5C and D and Movie EV4), or MSU (Fig EV5E and Movie EV5) confirming the broad effects of the VHHASC against ASC specks assembled by AIM2, NLRC4, and different NLRP3 triggers. In cells treated with nigericin (NLRP3), poly(dA:dT) (AIM2), and MxiH (NLRC4), the inhibitory effect of VHHASC was lost when caspase‐1 (and thus GSDMD pore formation) was inhibited with VX‐765. A notable exception were MSU‐treated cells, in which prolonged incubation with VHHASC decreased the number of ASC‐GFP speck even in the presence of VX‐765, (Fig EV5E and Movie EV5). This suggested that prolonged exposure to MSU crystals enabled the entry of VHHASC into cells, likely due to crystal‐mediated disruption of cellular membranes (Hornung et al, 2008; Duewell et al, 2010).
Figure 5. Pyroptosis allows VHHASC to access the cytosol and target ASC specks.
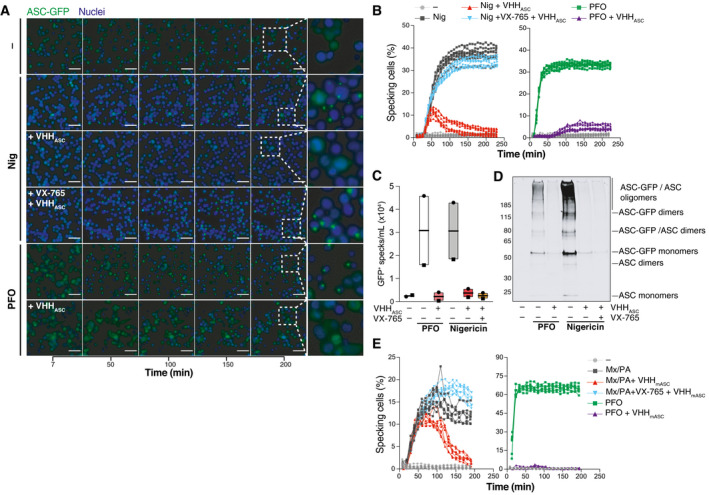
-
AWide‐field fluorescence microscopy imaging of live PMA‐differentiated and nigericin‐ (10 µM, top) or PFO‐activated (25 ng ml−1, bottom) THP‐1 expressing human ASC‐GFP. Cells were incubated with VHHASC (200 µg ml−1) alone or in the presence of the caspase‐1 inhibitor VX‐765 (50 µM). Nuclei were stained with DRAQ5 (blue). Cells were imaged live with a CellDiscoverer 7 microscope. For each condition, a total of 8 positions within 2 wells (2 × 4 images/well) were imaged. Data is maximal intensity projections from Z‐stacks. Scale bars: 50 µm. See also Movie EV2.
-
BGraphic representation of the percent of specking cells over time. Maximal intensity projections from Z‐stacks were generated for each image set before the number of cells and specks per field were calculated using CellProfiler. (A) represents images from one experiment out of three independent experiments of which percentage of specking cells data is represented in (B) with each line representing one field‐of‐view.
-
C, D(C) Flow cytometry quantification, and (D) immunoblotting analysis of ASC‐GFP oligomers (DSS cross‐linked) in extracellular specks recovered from supernatants of cells treated as in A. Data in (C) is from two independent experiments (each represented by a different symbol). And in (D) is from one out of at least two independent experiments. Data is displayed as floating bars with the max/min values and mean (thicker band).
-
EMouse BMDMs expressing ASC‐mCitrin and primed with LPS (200 ng ml−1) for 3 h were pre‐treated with VHHmASC (200 µg ml−1) alone or in the presence of the caspase‐1 inhibitor VX‐765 (50 µM) for 30 min. Cells were then treated with a mixture of LFn‐MxiH and PA (Mx/PA, 100 ng ml−1/1 µg ml−1) or PFO (250 ng ml−1) and imaged as in (A) for 3 h. Data is a graphic representation of specking cells over time calculated as in (B). Data is from one experiment out of three independent experiments. See also Movie EV6.
Source data are available online for this figure.
Figure EV5. VHHASC disrupts post‐pyroptotic ASC specks.
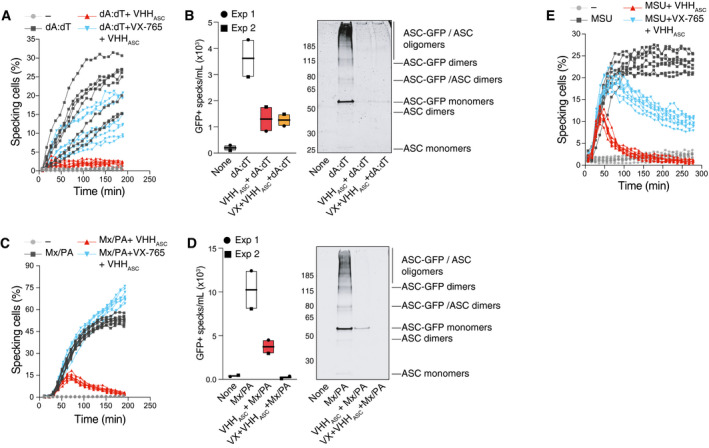
- PMA‐differentiated (50 ng ml−1) THP‐1 ASC‐GFP cells were treated with IFNγ (500 U ml−1) for 16 h. Then, cells were pre‐incubated with VHHASC (100 µg ml−1), or VX‐765 (50 µM) for 30 min before being activated with poly(dA:dT) (dA:dT, 1 µg ml−1) and imaged live with a CellDiscoverer 7 microscope as described in Fig 4 over 3 h. Data is a graphic representation of the percent of specking cells over time. Maximal intensity projections from Z‐stacks were generated for each image set (8 images per condition) before the number of cells and specks per field were calculated using CellProfiler. Also see Movie EV3.
- Flow cytometric quantification (left), or western‐blot (right, following DSS cross‐linking) assessement of GFP+ specks in the supernatants of cells pre‐treated with VHHASC or VX‐765 and then activated with poly(dA:dT) for 3.5 h. Data in left panel is displayed as floating bars with the max/min values and mean (thicker band).
- Flow cytometric quantification or western‐blotting of GFP+ specks released by PMA‐differentiated THP‐1 ASC‐GFP cells pre‐incubated with VHHASC (100 µg ml−1), or VX‐765 (50 µM) for 30 min before stimulation with LFn‐MxiH/PA (100 ng ml−1/1 µg ml−1) for 3 h and imaged live with a CellDiscoverer 7 microscope. Also see Movie EV4.
- GFP+ specks detected by flow cytometry (left) or western‐blot (right, following DSS cross‐linking) in the supernatants of cells pre‐treated with VHHASC or VX‐765 and then activated with LFn‐MxiH/PA for 3.5 h. Data in left panel is displayed as floating bars with the max/min values and mean (thicker band).
- PMA‐differentiated THP‐1 ASC‐GFP cells were pre‐incubated with VHHASC (100 µg ml−1), or VX‐765 (50 µM) for 30 min before stimulation with MSU crystals (250 µg ml−1) for 4.5 h and imaged live with a CellDiscoverer 7 microscope. Imaging and quantifications were done as in (A). Also see Movie EV5.
Source data are available online for this figure.
As VHHASC can access ASC specks in the cytosol of nigericin‐treated cells at later time points, we wondered whether delayed addition of VHHASC to culture supernatants could affect the IL‐1β output of cells stimulated with nigericin for a longer period. We treated LPS‐primed human primary macrophages with nigericin for 90 min, then added either VHHASC or CRID3 for another 7 h (Appendix Fig S2). Notably, neither VHHASC nor CRID3 had any effect on the output of IL‐1β from these cells. Indeed, cells treated for extended times released similar amounts of IL‐1β as in the first 90 min of nigericin‐activation (Appendix Fig S2), indicating that the maximal IL‐1β production is reached early during inflammasome activation in primary human macrophages. This would explain why pre‐treatment with VHHASC had no effect on IL‐1β release in nigericin‐treated macrophages (Fig 3), while it still disassembled post‐pyroptotic ASC speck (Fig 5).
Finally, we tested whether VHHmASC can target post‐pyroptotic specks upon stimulation of NLRC4 (MxiH) or NLRP3 (PFO) in mouse macrophages (Fig 5E and Movie EV6). Similar to the activity of VHHASC in nigericin‐treated human macrophages (Fig 5B), the presence of VHHmASC initially allowed for the initial formation of ASC‐mCitrine specks inside stimulated cells (Fig 5E, left panel). However, VHHmASC induced a decrease in the proportion of cells containing ASC‐mCitrine specks at later time points. This effect was abolished by VX‐765, confirming the requirement for pyroptosis to allow VHHmASC access to the cytosol (Fig 4E, left panel). In contrast, in cells stimulated with PFO, no assembly of ASC‐GFP specks was observed in the presence VHHmASC (Fig 4E, right panel), as seen in human cells (Fig 4B, right panel). Taken together, these data demonstrate that both VHHASC and VHHmASC can target and disassemble ASC specks in pyroptotic cells, while preserving initial IL‐1β secretion. They further demonstrate that VHHASC and VHHmASC can target inflammasomes that recruit ASC either via its CARD or via its PYD, indicating that the activity of VHHASC and VHHmASC resides at the interface between ASC molecules rather than between ASC and the inflammasome sensor.
VHHmASC ameliorates inflammation and clinical disease in gout
Next, we tested the inhibitory activity of a mouse‐specific VHHmASC in vivo in the inflammasome‐dependent inflammatory model of gouty arthritis, induced by the intra‐articular injection of monosodium urate (MSU) crystals (Martinon et al, 2006). We first used a prophylactic approach where mice were given an intra‐peritoneal injection of VHHmASC or PBS (as vehicle) 1 h prior to challenge with MSU crystals (Appendix Fig S3A). We measured the clinical and immunological parameters of gout, such as joint swelling, pain sensitivity, leukocyte infiltration, and tissue levels of pro‐inflammatory markers (Martinon et al, 2006). As expected, mice treated with MSU had a marked decrease in mechanical withdrawal threshold, indicating an increased sensitivity to painful stimuli when compared with vehicle‐treated or unchallenged VHHmASC‐treated animals (Appendix Fig S3B). Notably, the systemic administration of VHHmASC significantly reduced the nociceptive behavior (Appendix Fig S3B) and joint swelling (Appendix Fig S3C) in MSU‐challenged animals. The MSU challenge also resulted in the infiltration of granulocytes (CD45+/Ly6G+) and inflammatory monocytes (CD45+/Ly6G−/Ly6C+) into the synovial space, which were markedly reduced in animals pre‐treated with VHHmASC (Appendix Fig S3D and Appendix Fig S4D). In line with an anti‐inflammatory effect of VHHmASC, pre‐treatment with VHHmASC strongly decreased the MSU‐induced tissue concentrations of IL‐1β and IL‐6 while it only partially decreased TNFα levels (ANOVA, P = 0.29) (Appendix Fig S3E).
Together with our observations that ASC‐specific VHHs target ASC following pyroptosis (Figs 3 and 4), these findings suggest that VHHmASC administered systemically can reach the inflamed tissue to target ASC specks exposed to the extracellular space. VHHmASC may thus inhibit the amplification of the inflammation induced by ASC specks exposed to the extracellular space. To validate these findings, we also tested the efficacy of local injection of VHHmASC together with MSU crystals into the joints. Recapitulating their systemic effect, local injection of VHHmASC reversed the MSU‐induced nociception and edema (Appendix Fig S4A and B). Cell infiltration, especially of granulocytes, were reduced in animals treated with VHHmASC together with MSU, when compared with those treated with MSU alone (Appendix Fig S4C).
We next tested whether VHHmASC treatment could ameliorate gout after MSU inflammation is established. We thus induced disease by i.a. injection of MSU crystals and treated the mice with systemic injection of VHHmASC (i.p.) after 3 h (Fig 6A). As controls, we employed both a vehicle treatment and VHH NP‐1 (Ashour et al, 2015), an unrelated nanobody against the nucleoprotein of influenza A virus. Strikingly, VHHmASC treatment rapidly ameliorated pain sensitivity and joint swelling (6 h), when compared with the vehicle‐ or VHH NP‐1‐treated groups (Fig 6B). VHHmASC treatment additionally decreased total infiltrated leukocytes and granulocytes (Fig 6C, ANOVA, P = 0.059 and P = 0.18, respectively) and the tissue concentrations of TNFα, IL‐6 and IL‐1β (Fig 6D, ANOVA, P = 0.056). Hence, both preventive and delayed administration of VHHmASC have a strong therapeutic benefit in gout, demonstrating that a substantial fraction of MSU inflammation is driven by ASC specks exposed to the extracellular space. These findings provide the first evidence for the in vivo relevance of ASC specks, and support their potential as target for treatment of inflammasome‐dependent diseases. The inhibitory effect measured following systemic exposure suggests that VHHmASC penetrates tissues in time to prevent the amplification of the inflammation induced by MSU crystals.
Figure 6. VHHmASC ameliorates MSU gouty inflammation.
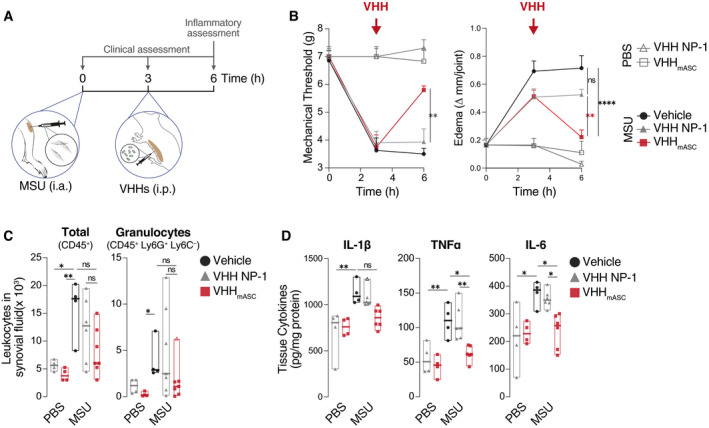
- Schematic representation of the experimental setting used for the MSU‐gout model. Mice were injected i.a with 100 µg of monosodium urate (MSU) crystals into the knee. After 3 h, mice were treated intraperitonially (i.p.) with VHHmASC, VHH NP‐1 as unrelated nanobody (both 5 mg kg−1), or vehicle (PBS).
- Mechanical allodynia threshold and edema were evaluated at 3 and 6 h post‐MSU challenge. Error bars represent SEM from biological replicates: t0, n = 3 for all groups; t3 and t6: PBS + VHH NP‐1 and PBS + VHHmASC, n = 3 (Mechanical threshold) or n = 4 (Edema); MSU + Vehicle, n = 4; MSU + VHH NP‐1, n = 7; MSU + VHHmASC, n = 7.
- Flow cytometric assessment of infiltrating total leukocytes (CD45+) and granulocytes (CD45+ Ly6G+ Ly6C−) recovered in the synovial fluid of the knee joints of mice treated as in (A). Data in (C) is displayed as floating bars with the max/min values and mean (thicker band). Biological replicates are: PBS + VHH NP‐1, n = 4; PBS + VHHmASC, n = 4; MSU + Vehicle, n = 4; MSU + VHH NP‐1, n = 7; MSU + VHHmASC, n = 7.
- ELISA of IL‐1β, TNFα and IL‐6 in tissue homogenates of knee joints of mice treated as in (A). Data in (C) is displayed as floating bars with the max/min values and mean (thicker band). Biological replicates are PBS + VHH NP‐1, n = 4; PBS + VHHmASC, n = 4; MSU + Vehicle, n = 4; MSU + VHH NP‐1, n = 7; MSU + VHHmASC, n = 7.
Data information: ns P > 0.05; *P < 0.05; **P < 0.005, One‐way ANOVA, multiple comparison (Tukey test). Data with non‐normal distribution were tested with Krustal‐Wallis test and multiple comparison using Dunn’s test. Outlier in (D, IL‐1β) was determined by the ROUT method and are represented with Δ.
VHHmASC ameliorates antigen‐induced arthritis in vivo
Finally, to test the activity of VHHmASC in a chronic model of inflammatory disease, we used the well‐established mBSA‐induced arthritis model (Fig 7A; Pinto et al, 2010, 2015). We immunized mice with a mixture of methylated bovine serum albumin (mBSA) and Freund's adjuvant (M. tuberculosis extract) injected sub‐cutaneously on days 0 and 7. Joint inflammation was induced by intra‐articular injection (i.a.) of mBSA on days 21 and 26. Mice were treated daily with either VHHmASC, IL‐1 receptor antagonist (IL‐1RA) or vehicle between challenges. On day 27, we measured clinical and inflammatory parameters (Fig 7A). As expected, i.a. treatment with mBSA induced an increased sensitivity to painful stimuli (Fig 7B), joint swelling (Fig 7C), increased infiltration of leukocytes such as monocytes and granulocytes (Fig 7D), as well as an increase concentration of cytokines (Fig 7E), when compared with vehicle or VHHmASC‐only conditions. In line with its activity in MSU‐induced gouty arthritis (Fig 6), VHHmASC rescued the nociceptive sensitization as well as the joint swelling phenotypes induced by challenge with i.a. mBSA (Fig 7B and C). VHHmASC treatment also abrogated the infiltration of pro‐inflammatory cells into the joint (Fig 7D) and strongly reduced the concentration of pro‐inflammatory cytokines in the tissue (Fig 7E). Strikingly, in all readouts, VHHmASC showed a similar, or better, activity to the benchmark treatment for arthritis, anakinra (IL‐1RA, Fig 7B and E). Together, these data conclusively demonstrate the efficacy of VHHmASC for the treatment of chronic arthritis in vivo and further suggest an important post‐pyroptotic role for ASC specks in the development of RA.
Figure 7. VHHmASC ameliorates antigen‐induced arthritis.
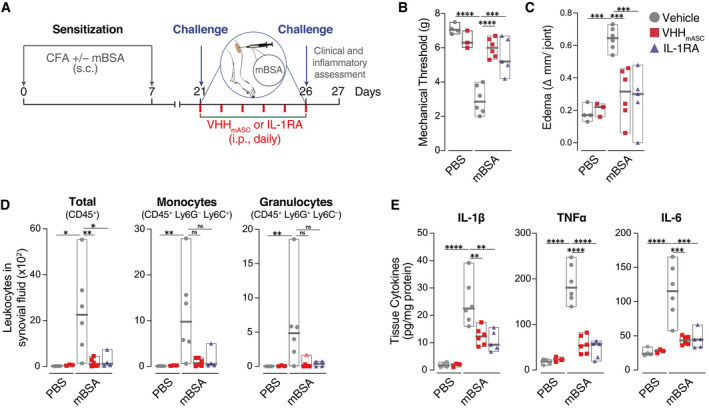
-
ASchematic representation of the experimental setting used for the mBSA‐induced arthritis model. Mice were injected sub‐cutaneously (s.c.) with mBSA (500 µg/animal) in an emulsion containing 1 mg ml−1 Freund’s adjuvant on day 0 and day 7. Control mice received injections lacking mBSA. Mice were then immunized with an intra‐articular injection of mBSA (100 µg, right knee) on days 21 and 26. From day 21 until day 26, mice were treated daily with an intra‐peritoneal (i.p.) injection of VHHmASC (5 mg kg−1), IL‐1RA (50 µg kg−1), or vehicle (PBS).
-
B, CMechanical allodynia threshold (B) and edema (C) were evaluated on day 27. Data is displayed as floating bars with the max/min values and mean (thicker band). Biological replicates are: PBS + Vehicle, n = 4; PBS + VHHmASC, n = 3; mBSA + Vehicle, n = 6; mBSA + VHHmASC, n = 6; mBSA + IL‐1RA, n = 5.
-
DFlow cytometric assessment of infiltrating total leukocytes (CD45+), granulocytes (CD45+ Ly6G+ Ly6C−), and inflammatory monocytes (CD45+ Ly6G− Ly6C+) recovered in the synoival fluid of the knee joints of mice treated as in (A). Data is displayed as floating bars with the max/min values and mean (thicker band). Biological replicates are: PBS + Vehicle, n = 4; PBS + VHHmASC, n = 3; mBSA + Vehicle, n = 6; mBSA + VHHmASC, n = 6; mBSA + IL‐1RA, n = 5.
-
EELISA of IL‐1β, TNFα and IL‐6 in tissue homogenates of knee joints of mice treated as in (A). Data is displayed as floating bars with the max/min values and mean (thicker band). Biological replicates are: PBS + Vehicle, n = 4; PBS + VHHmASC, n = 3; mBSA + Vehicle, n = 6; mBSA + VHHmASC, n = 6; mBSA + IL‐1RA, n = 5.
Data information: ns P > 0.05; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001, One‐way ANOVA, multiple comparison (Tukey test). Data with non‐normal distribution were tested with Krustal–Wallis test and multiple comparison using Dunn’s test. Outliers were determined by the ROUT method and are represented with Δ.
Discussion
Research on autoimmune and inflammatory diseases has been advancing quickly, and the last years have seen major investments in novel approaches to target inflammasomes and IL‐1 cytokines. In particular, several NLRP3 inhibitors are now in clinical trials for a wide array of diseases (Carroll, 2018; Ben, 2019; Jonson & Jonson, 2021). These events follow the successful clinical application of small molecules, decoy receptors, and humanized Abs targeting IL‐1β or its signaling receptor, which became standard treatments against autoinflammatory syndromes (Kuijk et al, 2007; Calligaris et al, 2008; van der Hilst et al, 2016), and have also shown clinical benefits against conditions such as atherosclerosis (Ridker et al, 2017a), lung cancer (Ridker et al, 2017b), gout (Janssen et al, 2019), and RA (Ruscitti et al, 2019; Chauhan et al, 2020). However, a major concern surrounding blocking therapies targeting innate immunity molecules with such essential functions in host defense is that they considerably increase the risk of infections (Ottaviani et al, 2013; Ridker et al, 2017a). For example, the risk of serious opportunistic infections and cancer is increased in patients with rheumatologic diseases treated with interleukin inhibitors (Bilal et al, 2019). Moreover, recent off target effects of the small molecule inhibitor of NLRP3, MCC950 (or CRID3), have been reported, which may jeopardize its chances for clinical application (Kennedy et al, 2021).
In this study, we present camelid‐derived nanobodies as viable alternatives to target inflammasome complexes (ASC specks) that outlive pyroptotic cells and propagate inflammation in the extracellular space (Baroja‐Mazo et al, 2014; Franklin et al, 2014). Nanobodies directed against human (VHHASC) and mouse ASC (VHHmASC) supplied to the culture medium efficiently blocked the inflammatory and prion‐like activities of ASC specks after pore formation in pyroptotic cells, while preserving initial IL‐1β release. Importantly, mouse‐specific VHHmASC largely abolished the clinical manifestations of MSU‐induced gout, and AIA, providing the first evidence for the in vivo relevance of extracellular inflammasomes, and revealing the potential of VHHASC against acute and chronic inflammatory diseases.
ASC is an interesting therapeutic target as it plays a central role in inflammasome activation, but also displays inflammasome‐independent functions (Ellebedy et al, 2011; Guo & Dhodapkar, 2012; Tsuchiya et al, 2014; Franklin et al, 2017; Venegas et al, 2017; Friker et al, 2020). Recent reports have shown that extracellular ASC specks can seed amyloid aggregates such as amyloid‐β plaques at the root of Alzheimer's disease (Venegas et al, 2017; Friker et al, 2020). Hence, targeting ASC specks inside the brain using VHHASC may have clinical relevance in AD. In a mouse model of Alzheimer's disease, intra‐cranial delivery of anti‐ASC Abs prevented the ASC speck‐induced aggregation of Aβ in the hippocampus of APP/PS1 mice injected with Aβ seeds from APP/PS1 brain lysates (Venegas et al, 2017). Of note, no inflammatory readouts were assessed in this model, and the effect of anti‐ASC Abs in neuroinflammation remains to be determined. Other attempts to target ASC in disease have generated conflicting results. While partial benefits in brain and spinal cord injury models have been reported (de Rivero Vaccari et al, 2008; Desu et al, 2020), our previous attempt to target ASC specks using conventional Abs worsened inflammation in silica‐induced peritonitis, likely due to increased Fc‐mediated opsonization of extracellular specks (Franklin et al, 2014). As VHHs lack the Fc domains that interact with immune cells, they offer an alternative to target inflammasomes without engaging immune cells.
Furthermore, VHHASC are the first biologicals able to disassemble pre‐formed supramolecular organizing centers (SMOCs), the signaling organelles of the innate immune system. This feature highlight their potential as tools to investigate the structures and composition of other SMOCs, such as the Myddosome, and the poorly defined Triffosome that assemble downstream of TLR activation, or MAVS filaments formed after activation of RIG‐I‐like receptors (Kagan et al, 2014).
In our study, we observed that VHHASC requires cytosolic access to target intracellular inflammasomes, which is prevented by caspase‐1 inhibition or genetic ablation of GSDMD. Recent studies have placed the formation of GSDMD membrane pores upstream of some NLPR3 triggers. Indeed, human caspase‐4/5 (or mouse caspase‐11) and caspase‐8 were shown to activate GSDMD, leading to potassium efflux and downstream NLRP3 activation (Orning et al, 2018; Demarco et al, 2020; Santos et al, 2020). In these settings, the presence of VHHASC would most likely prevent full oligomerization of ASC specks and therefore inhibit the release of IL‐1, downstream of caspase‐1 activation. Further experiments are required to define the impact of VHHASC (or VHHmASC) on caspase‐8‐ or caspase‐4/11‐dependent anti‐microbial defenses.
The similarity with which VHHASC and VHHmASC affect active ASC specks is informative for the contribution of ASCCARD or ASCPYD in the assembly of ASC specks. In fact, both CARD‐ and PYD‐mediated interactions between ASC molecules contribute to the overall structure of inflammasomes (Cai et al, 2014; Dick et al, 2016; Schmidt et al, 2016b). The destabilization we observed validates a role of ASCCARD in the maintenance of the quaternary structure of ASC specks. Furthermore, the disruption of existing ASC specks by targeting either ASCCARD or ASCPYD with VHHASC or VHHmASC, respectively, suggests some reversibility as well as the vulnerability of both CARD‐CARD and PYD‐PYD interactions to interference, enabling both VHHASC or VHHmASC to bind ASC and counteract inflammation. Our experiments targeting inflammasomes formed downstream of the NLRP3 or NLRC4 activation using either VHHASC or VHHmASC demonstrate that it is sufficient to interfere with the interaction between ASC molecules rather than between ASC and the sensor. Indeed, while NLRP3 recruits ASC via PYD‐PYD interactions, NLRC4 recruits ASC via its CARD. In either case, both CARD‐ or PYD‐targeting VHHASC or VHHmASC, respectively, could disassemble ASC specks seeded by NLRP3 or NLRC4, indicating that ASC‐ASC interactions are critical for inflammation. In our previous work (Schmidt et al, 2016b), we found that VHHASC can stabilize the formation of PYD filaments when present prior to ASC oligomerization. Here, we could not detect such filaments when targeting already assembled ASC oligomers with VHHASC. This could be explained by a different stoichiometry between ASC and VHHASC in our experiments. Smaller filaments would also be hard to detect with the kinetic and imaging resolution used in our experiments.
It is tempting to speculate that the use of VHHASC could also destabilize the structure of ASC oligomers in existing Aβ plaques in AD, as it does for fully functional inflammasomes. Interference with ASC oligomerization may also break the observed circle of inflammatory exacerbation fed by VHH‐accessible ASC specks. A major hurdle to target the CNS is the low penetrance of protein drugs through the blood‐brain barrier (BBB). It remains unknown whether VHHASC is able to cross the BBB and penetrate the brain. However, recently established single‐domain antibodies that cross the BBB by engaging cargo molecules of transcytosis may overcome this barrier (Gao et al, 2020; Wouters et al, 2020; Stocki et al, 2021). Once established, these could be fused to VHHASC to promote transport of bivalent nanobodies across the BBB. The ever‐growing toolbox of nanobody modifications such as multivalent nanobodies, Fc fusions, or fusion with albumin‐binding domains or albumin nanobodies could also be applied to VHHASC to enhance stability and half‐life in the circulation (Kontermann, 2016). Similar strategies could direct VHHs to specific cells or tissues. Whether the inhibitory function of VHHASC shown in this study remains within such a construct would need to be tested. Additionally, the in vivo activity of the human‐specific VHH (VHHASC) would benefit from additional experiments using, for example, transgenic mice expressing human ASC.
To achieve a prolonged exposure with VHHASC, the use of nanobody‐secreting bacteria strains or gene therapy may become interesting avenues for delivery in patients (Gurbatri et al, 2020). Nevertheless, it is important to note that nanobodies are already used in the clinic to treat thrombotic thrombocytopenic purpura and thrombosis (e.g., caplacizumab, HERCULES trial; Scully et al, 2019; Volker et al, 2020).
Gout and RA are painful and debilitating inflammatory diseases affecting the joints and other organs (Dalbeth et al, 2019). Current therapies include managing acute attacks using nonsteroidal anti‐inflammatory drugs (NSAIDs), corticosteroids, colchicine (gout), or disease‐modifying immunosuppressant drugs (DMARDs, RA). The growing appreciation of the NLRP3 inflammasome activation and bioactive IL‐1β release in acute gouty inflammation or in the more chronic rheumatoid arthritis has prompted studies of agents blocking the IL‐1β receptor or soluble IL‐1β (canakinumab, rilonacept, and anakinra). An NLRP3 inhibitors (Dapansutrile) has shown promising results against gout flares in early clinical trials (Kluck et al, 2020). While these therapies are improving acute and chronic symptoms in the clinic, our study raises the possibility of targeting post‐pyroptotic inflammasomes directly with nanobodies. Our findings suggest that the inflammatory response initiated by MSU crystals or intra‐articular antigen challenge include several rounds of amplification involving accessible ASC specks. Interestingly, VHHmASC rescued the phenotype of AIA mice to the same extent than the benchmark drug anakinra. This is of significant importance because, although VHHmASC and anakinra have a similar therapeutic effect in this model, their targets and mechanisms of action are different. Indeed, anakinra targets the IL‐1R and thus blocks IL‐1β‐induced downstream signaling and prevents recruitment of immune cells such as neutrophils, thus decreasing inflammation. VHHmASC not only decreased swelling and pain sensitivity in AIA mice, but it also inhibited production of IL‐1β and infiltration of immune cells. This suggests that post‐pyroptotic ASC specks play an important role in the development of AIA by promoting the production of IL‐1β, probably through several rounds of amplification. Importantly, inhibiting the amplification of inflammation induced by extracellular ASC specks will still permit the initial intracellular activation of inflammasomes, essential to the immune defense against microbes. We thus describe a clinical scenario that is highly desirable for therapeutic control of inflammasome activation. Hence, nanobodies against human ASC may be a radically new alternative to other therapies that broadly target inflammasome downstream signals leaving patients with increased risks of infections.
Materials and Methods
Study design
In this study, we evaluated the anti‐inflammatory potency of VHHASC or VHHmASC against un‐regulated inflammasomes, more specifically against extracellular inflammasomes. We first conducted in vitro studies using immortalized cell lines and when necessary, using primary cells. Human PBMCs were isolated from buffy coats obtained from the blood bank of the University hospital of Bonn, with consent of healthy donors and according to protocols accepted by the institutional review board of the University of Bonn (local ethics votes Lfd. Nr. 075/14). To study the activity of VHHmASC in vivo, we used a well‐established model of MSU‐induced gout disease. These experiments were performed on male C57BL/6 wild‐type mice weighing between 18 and 22 g. They were housed in temperature‐controlled rooms (22–25°C) and given water and food ad libitum at the animal facility in the Department of Pharmacology of Ribeirao Preto Medical School, University of Sao Paulo, Brazil. Animal husbandry and procedures were in accordance with the guidelines of the Animal Ethics Committee of the Ribeirao Preto Medical School (241/2018). Group size were determined using the 3R principle.
Reagents
Cell culture reagents (e.g., PBS, DMEM, RPMI, Trypsin‐EDTA) were from Gibco, Thermo Fisher Scientific. Stimuli were as follow: LPS (UltraPure, Invivogen), nigericin (Thermo Fisher Scientific), PFO (Biotrend), LFn‐BsaK, LFn‐MxiH and PA were produced in‐house as described previously (19), and uric acid used for in‐house production of MSU crystals (Sigma‐Aldrich). Inhibitors used were CRID3 (Tocris) and VX‐765 (Selleckchem). Antibodies were as follow: anti‐ASC polyclonal antibody (AL177, Adipogen, 50 µg ml−1) or anti‐ASC monoclonal antibody (N‐15, Santa Cruz, 1:500), anti‐Caspase‐1 p20 antibody (Casper‐1, Adipogen, 1:1,000), anti‐IL‐1β antibody (BAF401, R&D Systems, 1:1,000), and anti‐GSDMD (L60, Cell Signaling Technology, 1:1,000). DRAQ5 was from Thermo Fisher Scientific. For FACS, Abs used were anti‐CD45‐BV421, Ly6G‐FITC and Ly6C‐PerCP (BD Biosciences, 1:100). HTRF for human or mouse IL‐1β were from CisBio. ELISA kits were used to detect IL‐1β, TNFα, IL‐6 or CXCL1 (DuoSet, R&D Systems). Cell viability was measured with the Cell Titer Blue kit (CTB, Promega).
Cell lines
THP‐1 cells (ATCC TIB‐202) expressing human ASC‐GFP and Pycard −/− immortalized macrophages (iMøs) expressing human ASC‐mTurquoise were generated via retroviral transduction and fluorescence‐activated cell sorting of TagGFP+ or mTurquoise+ cells, respectively. Pycard −/− iMøs were described previously (Franklin et al, 2014). Cell lines are routinely tested for contamination with Mycoplasma.
Alpaca immunization and panning for novel VHHmASC
Heavy chain‐only antibodies against murine ASC (mASC) were elicited by immunizing one alpaca five times with a mixture of proteins including GFP‐mASC as a 1:1 (v/v) mixture with GERBU Adjuvant Fama (GERBU Biotechnik GmbH). GFP‐mASC was kindly provided by Liman Zhang and Hao Wu (Harvard Medical School, Boston, MA, USA). Alpaca immunizations were conducted at the research farm of preclinics GmbH (Bruchhausen‐Vilsen, Germany), according to animal procedures approved by the state of Lower Saxony. A VHH plasmid library in the M13 phagemid vector pJSC was generated as described before (Maass et al, 2007; Koenig et al, 2021). In brief, RNA from peripheral blood lymphocytes was extracted and used as a template to generate cDNA using three sets of primers (random hexamers, oligo(dT), and primers specific for the constant region of the alpaca heavy chain gene). VHH coding sequences were amplified by PCR using VHH‐specific primers, cut with AscI and NotI, and ligated into pJSC linearized with the same restriction enzymes. E. coli TG1 (Agilent) cells were electroporated with the ligation reaction and the obtained ampicillin‐resistant colonies harvested, pooled, and stored as glycerol stocks.
Mouse ASC‐specific VHHs were obtained by phage display and panning with a protocol modified from (Schmidt et al, 2016b). Escherichia coli TG1 cells containing the VHH library were infected with helper phage VCSM13 to produce phages displaying the encoded VHHs as pIII fusion proteins. Phages in the supernatant were purified and concentrated by precipitation; phages displaying GFP‐specific VHHs were removed in a depletion step with GFP immobilized to the bottom of tissue culture flasks. Phages presenting mASC‐specific VHHs were enriched using biotinylated GFP‐mASC immobilized to MyOne Streptavidin T1 Dynabeads (ThermoFisher Scientific). The obtained phages were used to infect E. coli ER2837 and subjected to a second round of panning. 96 E. coli ER2837 colonies yielded in the second panning were grown in 96‐well plates and VHH expression induced with IPTG. VHHs released into the supernatant were tested for specificity using ELISA plates coated with GFP or GFP‐mASC; bound VHHs were detected with HRP‐coupled rabbit anti‐E‐Tag antibodies (Bethyl) and the chromogenic substrate TMB. The amino acid sequence of VHHmASC (JT‐01‐A09) is: QVQLVETGGG MVHPGGSLRL SCAASGFTFS EYGMTWVRQA PGKGPEWVSR INSSGGYTVY RASVKGRFTV SRDNAKNTLY LQMNSLKPED TALYYCARTT NWETRLSQGT QVTVSS.
LUMIER assay
Protein interactions in transfected HEK 293T cells were quantified using the LUMIER assay according to a protocol modified from Schmidt et al (2016a). HEK 293T cells in 24‐wells were transfected with 0.5 μg bait expression vectors (pCAGGS VHH‐HA) and 0.5 μg prey expression vectors (pEXPR hASC‐Renilla, pEXPR mASC‐Renilla, pEXPR mASCCARD‐Renilla, or pEXPR hASCPYD‐Renilla, all derived from pcDNA3‐ccdB‐Renilla, a kind gift of Mikko Taipale, Susan Lindquist lab, Whitehead Institute) using Lipofectamine 2000 (Thermo Fisher Scientific). 2 days post‐transfection, cells were lysed in 120 μl LUMIER IP buffer (50 mM Hepes pH 7.9, 150 mM NaCl, 2 mM EDTA pH 8.0, 0.5% Triton X‐100, 5% glycerol, protease inhibitor cocktail (Roche)). 90 μl of the lysates were transferred to blocked LUMITRAC™ 600 plates (Greiner) coated with mouse anti‐HA.11 and incubated at 4°C for 3 h. After extensive washing steps with IP buffer, incubated wells (or 10 μl lysate) were incubated with Coelenterazine‐containing Renilla luciferase substrate mix (BioLux Gaussia Luciferase Assay Kit, New England BioLabs) and light emission quantified using a SpectraMax M3 microplate reader (Molecular Devices). Renilla luciferase activity in the immunoprecipitation samples was normalized by Renilla luciferase activity in the lysates.
Expression and purification of recombinant VHH proteins
Recombinant VHH proteins were produced as previously described (Schmidt et al, 2016b; Koenig et al, 2021). In short, proteins were expressed in E. coli WK6 strain transformed with pHEN6‐based expression vectors. Cultures in terrific broth (TB) complemented with 100 µg ml−1 ampicillin were grown at 37°C until OD of 0.6. Protein expression was induced by addition of 1 mM IPTG and cultures grown at 30°C for 16 h. Periplasmic extracts were generated from cell pellets by osmotic shock involving incubation with TES buffer (200 mM Tris pH 8.0, 0.65 mM EDTA, 0.5 M sucrose) for 1 h, followed by dilution in 0.25× TES buffer and further incubation overnight at 4°C. Periplasmic extracts were cleared by centrifugation at 10.000 x g. Proteins were isolated using Ni‐NTA agarose beads (Qiagen), followed by size‐exclusion chromatography with a HiLoad 16/600 Superdex 75 pg column (Cytiva).
In vitro generation of ASC specks
ASC specks were prepared in vitro as previously described (Fernandes‐Alnemri et al, 2007; Franklin et al, 2014). In short, iMøs expressing human ASC‐mTurquoise were lysed in CHAPS buffer (20 mM HEPES‐KOH, pH 7.5, 5 mM MgCl2, 0.5 mM EGTA, 0.1 mM PMSF and 0.1% CHAPS) by passing cells 25 times through a 20G needle. “S100” lysates were prepared at 4°C by centrifugation of the cell lysate at 20,000 g for 8 min followed by centrifugation at 100,000 g for 30 min. Lysates were finally filtered through a 0.22 µm PVDF filter spin column (Millipore). Cleared lysates were then incubated at 37°C for approximately 45 min. Specks were isolated by centrifugation at 660 g for 8 min, then washed with CHAPS buffer.
Generation of bone marrow‐derived macrophages and stimulation
Bone marrow of C57BL/6 WT or ASC‐mCitrine‐expressing transgenic mice was flushed from isolated tibia and femur. Cells were then incubated for 6 days in the presence of L929 culture supernatants generated in‐house. Adherent differentiated macrophages were detached from their culture vessel using PBS supplemented with 5 mM EDTA and 2% FBS. Cells were seeded in 384‐well plates at 4 × 104 cells per well. Cells were finally incubated at 37°C, 5% CO2 overnight, prior to stimulation. Cells were primed with 200 ng ml−1 LPS, then stimulated overnight with in vitro‐generated ASC specks (200 µg ml−1) that had been pre‐incubated 15 min at room temperature with either VHHASC or mutVHHASC (200 µg ml−1), or anti‐ASC pAb (50 µg ml−1). Prior to stimulation with nigericin (10 µM), PFO (250 ng ml−1), or the combination of the rod protein from Burkholderia pseudomallei (Bsak, fused to lethal factor N‐terminus) and Bacillus anthracis protective antigen (PA, 0.1 µg/ml/0.5 µg/ml) (Figs 3 C and D and Appendix Fig S1A and B), cells were primed with 200 ng/ml LPS for 2:30 h followed by 30‐min incubation with VHHASC or VHHmASC (100 µg ml−1), CRID3 (50 µM) or VX‐765 (50 µM). For microscopy experiments (Fig 3E and F), cells were primed with LPS (200 ng ml‐1) for 2 h and then treated with the caspase‐1 inhibitor VX‐765 (50 µM) for 30 min. VHHASC or mutVHHASC (200 µg ml−1), or CRID3 (25 µM) were added to the cells 30 min prior to addition of PFO (250 ng ml−1) or nigericin (10 µM). After 2 h stimulation, cells were fixed with 4% PFA (in PBS) and nuclei stained with DRAQ5.
Differentiation and stimulation of human M‐CSF‐differentiated macrophages
Human macrophages were generated as previously described (Rolfes et al, 2020). In short, PBMCs were isolated from buffy coats using a density gradient (Ficoll‐Paque Plus, GE Healthcare) followed by positive selection of monocytes using a CD14 MACS bead kit (Miltenyi Biotech). Cells were then differentiated into macrophages with 500 U ml−1 rhM‐CSF (Immunotools) for 3 days at 37°C, 5% CO2. Adherent cells were harvested using PBS complemented with 2% FBS and 2 mM EDTA and seeded into a 384‐well plate at 4 × 104 cells per well, covered with a Breath‐Easy plate seal (Sigma‐Aldrich) and incubated at 37°C, 5% CO2 overnight. Cells were primed with 10 ng ml−1 LPS for 2.5 h before addition of VHHASC (100 µg ml−1), mutVHHASC (100 µg ml−1), CRID3 (50 µM) or VX‐765 (50 µM) and further incubation for 30 min. Cells were stimulated either with PFO (30 ng ml−1), nigericin (10 µM), ATP (2.5 mM, with pH equilibration with a final concentration of 3.75 mM NaOH), or the combination of LFn‐BsaK and PA (0.1 µg ml−1/0.5 µg ml−1) for 2 h at 37°C. For nigericin stimulation at 7 h (Appendix Fig S2), LPS‐primed cells were first stimulated with nigericin for 90 min. VHHASC (100 µg ml−1), mutVHHASC (100 µg ml−1), or CRID3 (50 µM) were then added and cells were further incubated for a total of 7 h at 37°C, 5% CO2. As control for the 90‐min time point, cell‐free supernatants were harvested directly after 90 min. Cell‐free supernatants were collected and IL‐1β concentration was measured by HTRF. CTB assay was performed on the remaining living cells following the manufacturer's recommendations. For activation of NLRP3 with MSU crystals, PMA‐differentiated (20 nM, 16 h) THP‐1 macrophages were pre‐treated with VHHASC and mutVHHASC (100 µg ml−1), CRID3 (10 µM) or VX‐765 (50 µM) 30 min prior to addition of MSU crystals (250 µg ml−1).
Stimulation of other inflammasomes and challenge with ASC‐targeting VHHs
The pyrin inflammasome was stimulated for 90 min with TcdA (1 µg ml−1) in CD14+ monocytes isolated with MACS beads (Miltenyi Biotech) following the manufacturer’s instructions and primed with Pam3CysK4 for 3 h (1 µg ml−1). AIM2 was stimulated for 2 h with poly(dA:dT) (1 µg ml−1) transfected using Lipofectamine 2000 (Thermo Fisher Scientific) in PMA‐differentiated (50 ng ml−1, 16 h) and IFNγ‐treated (500 U ml−1, 16 h) THP‐1 (ASC‐GFP) cells. As previously described, in monocytes and THP‐1, VHHASC and mutVHHASC (100 µg ml−1) or VX‐765 (50 µM) were added 30 min prior to inflammasome stimulation. NLRP1 was stimulated for 22 h with Val‐boroPro (VbP, 30 µM) in N‐TERT keratinocytes. Here, VHHs and VX‐765 were added just before Val‐boroPro.
Stimulation of GSDMD knockout THP‐1 cells
Doxycycline (Dox)‐inducible CRISPR‐Cas9 GSDMD knockout (GSDMD‐KO) THP‐1 cells were generously provided Seth Masters, Melbourne University. Knockout was performed as previously described (Budden et al, 2021). GSDMD‐KO was confirmed using SDS‐PAGE and immunoblotting. THP‐1 cells were differentiated overnight with 20 nM PMA at 37°C, 5% CO2. Cells were then harvested using trypsin and seeded into a 96‐well microscopy plate at a density of 5 × 104 cells per well. PMA‐differentiated THP‐1 cells were allowed to adhere at least 30 min. Cells were treated with VHHASC (200 µg ml−1) or CRID3 (25 µM) and incubated for 30 min. Cells were then stimulated either with PFO (30 ng ml−1) or nigericin (10 µM) for 2 h at 37°C. Cell‐free supernatants were collected and IL‐1β concentration was measured by HTRF and LDH release was measured using a CyQUANT LDH cytotoxicity assay (ThermoFisher Scientific) following the manufacturer's recommendations. Data are represented as percentage of LDH release normalized to the maximal release (100%) from cells treated with Triton‐X100 solution (0.5% v/v in PBS) and background subtraction of signal from untreated cells (0%).
For analysis of the entry of VHHASC into the cell cytosol, CRISPR‐Cas9 GSDMD THP‐1 cells treated with doxycycline as described above were primed overnight with PMA (20 nM). Medium was replaced with imaging medium (RPMI free of phenol‐red, 10% FBS and 30 mM HEPES) before cells were treated with fluorescently‐labeled VHHASC (VHHASC‐AlexaFluor647, 10 µg ml−1). Cells were then either left unstimulated, or stimulated with nigericin (10 µM) or PFO (25 ng ml−1) and imaged live on a SP8 confocal microscope (Leica) for 60 min, one time frame every 2.5 min. To detect cell death, propidium iodide (3.33 µg ml−1) was added to the cells before stimulation.
Live imaging of ASC‐GFP specks
THP‐1 ASC‐GFP cells were differentiated overnight with 20 nM PMA at 37°C, 5% CO2. Cells were then harvested using trypsin and seeded into a 96‐well microscopy plate at a density of 5 × 104 cells per well. Differentiated THP‐1 cells were allowed to adhere for 2 h. Nuclei were stained using DRAQ5 (1:1,500 dil.) and VHHASC (200 µg ml−1) or a combination of VHHASC and VX‐765 (50 µM) were added to the cells followed by 30 min incubation at 37°C, 5% CO2. PFO (25 ng ml−1) or nigericin (10 µM) were added to the cells followed by centrifugation at 340 g for 1 min. The plate was then transferred into a CellDiscoverer 7 microscope (Zeiss) already set for imaging and the inner chamber equilibrated at 37°C and 5% CO2. Imaging was started 7 min post‐stimulation and each frame was taken every 7 min for a total of 3h30. For each condition, a total of 8 positions within 2 wells (2 × 4 images/well) were imaged using 6 Z‐slices per image. Maximal intensity projections were generated for each image set before the number of cells and specks per field were calculated using Cell Profiler software.
Isolation and analysis of released ASC specks from stimulated cells
THP‐1 cells expressing ASC‐GFP were differentiated overnight in the presence of 20 nM PMA. Cells were then detached from their culture dish using trypsin and transferred into 6‐well plates (3 × 106 cells per well). Once cells adhered to the well, they were stimulated as indicated for 3.5 h. Cell‐free supernatants containing specks were harvested by centrifugation of the culture supernatants twice at 340 g for 5 min. An aliquot of the supernatant was used for FACS analysis using the gating strategy presented in Appendix Fig S1A. Specks contained in the remaining supernatants were sedimented at 5,000 g for 10 min and washed in HEPES buffer (20 mM Hepes pH 7.4, 150 mM NaCl, 10% glycerol). The oligomeric state of specks was analyzed by DSS cross‐linking, SDS‐PAGE and immunoblotting.
DSS cross‐linking of ASC specks
Specks were incubated with the indicated dose of VHHASC, mutVHHASC or anti‐ASC pAb for 1 h at RT. DSS was added to a final concentration of 2 mM and further incubated at RT. Cross‐linking reaction was halted by mixing with electrophoresis samples buffer (either LDS‐sample buffer complemented with reducing buffer – ThermoFisher Scientific; or provided for WES readout by ProteinSimple) and heating at 95°C for 5 min. Samples were then run on WES following manufacturer's instruction or run for Western blot on a 4–12% SDS‐PAGE (ThermoFisher Scientific – using MOPS buffer, ThermoFisher Scientific) and transferred onto a PVDF membrane (Immobilon FL, Merck‐Millipore) prior to blocking with 3% BSA (w/v) in TBS buffer and detection of ASC distribution (monomeric to oligomeric state) using a monoclonal anti‐ASC antibody (N‐15).
MSU‐induced gout model
The experiments were performed on male C57BL/6 wild‐type animals. Animal allocation to groups was randomized. For therapeutical dosage, mice were treated with knee intra‐articular (i.a.) injection of 100 µg monosodium urate (MSU) crystal in 10 µl endotoxin‐free PBS 3 h prior to intra‐peritoneal (i.p.) injection with VHHmASC (5 mg kg−1) or VHH52 (targeting the nucleocapsid of influenza virus, 5 mg kg−1) as unrelated nanobody control. For preventive dosage, mice were injected with vehicle or VHHmASC (5 mg kg−1, i.p.) 1 h prior to knee i.a. injection of 100 µg monosodium urate (MSU) crystal in 10 µl endotoxin‐free PBS. Alternatively, VHHmASC (2 mg kg−1) was injected i.a. together with MSU crystals. Mechanical allodynia threshold was measured using a von Frey testing device at 3 h and 6 h post‐MSU injection. Additionally, edema was evaluated by measuring the size of the joint before and 6 h after MSU injection using a digital caliper. Phenotypical readouts were conducted blinded. (Δ) represents the difference between measurement at 6 h and t‐0 h. Leukocyte infiltration was analyzed by flow cytometry using surface markers for granulocytes (CD45+/Ly6G+) and inflammatory monocytes (CD45+/Ly6G−/Ly6C+). Harvesting of synovial fluid was performed by four injections of 2.5 µl PBS in each joint. Lavages were then collected in 50 µl PBS placed in a 96‐well plate. Cells were then pelleted and resuspended in staining mix. Joint tissue was frozen in liquid nitrogen, crushed with pestle and homogenized in 500 µl PBS supplemented with protease inhibitors. A panel of pro‐inflammatory cytokines (IL‐1β, TNFα, and IL‐6) in the tissue homogenate were determined by ELISA (R&D Systems).
Antigen‐induced arthritis (AIA) model
The experiment was performed as described by Pinto et al (2010, 2015) using male C57BL/6 wild‐type animals. Animal allocation to groups was randomized. Briefly, mice were sensitized with subcutaneous injection of 500 µg of methylated bovine serum albumin (mBSA) mixed with 100 µg Freund's adjuvant (Mycobacterium tuberculosis) on days 0 and 7. Control mice received injections of Freund's adjuvant without mBSA in saline (vehicle). Arthritis was induced by intra‐articular injection of mBSA (100 µg/cavity) into the right knee joint on days 21 and 26. During arthritis induction (day 21–26), mice received daily intra‐peritoneal injections of VHHmASC (5 mg/kg), IL1RA (50 µg/kg), or saline (vehicle). After 24 h of challenge (on day 27), we assessed nociceptive mechanical threshold using electronic von Frey, and measured knee joint thickness using a caliper, as described (Pinto et al, 2010). Phenotypical readouts were conducted blinded. (Δ) represents the difference between the basal thickness (day 0) and the thickness on day 27. Immune cell infiltration and tissue cytokine concentrations were measured as described for the gout model.
Statistical analysis
Experiments were run with the indicated numbers of repeats (please see figure legends). For in vivo experiments, mice were attributed to each group randomly at the beginning of the experiment. Wherever possible, readouts were carried out blindly by a different experimentator, especially for phenotypical readouts. For statistical analysis, data were first tested for normal (Gaussian) distribution. Normal distributed data were analyzed using one‐way ANOVA with multiple comparison (Tukey test). Non‐gaussian distributed data were first tested for the presence of outliers using the ROUT method (in figures, represented with Δ). Gaussian distribution was then again evaluated. Data with non‐normal distribution were tested with Krustal‐Wallis test and multiple comparison using Dunn's test. Statistical significance is showed as follow: ns P > 0.05; *P < 0.05; **P < 0.005; ***P < 0.0002; ****P < 0.0001.
Author contributions
Damien Bertheloot: Formal analysis; Supervision; Investigation; Visualization; Methodology; Writing—original draft; Writing—review and editing. Carlos WS Wanderley: Investigation. Ayda H Schneider: Investigation. Lisa DJ Schiffelers: Investigation. Jennifer D Wuerth: Investigation. Jan MP Tödtmann: Investigation. Salie Maasewerd: Investigation. Ibrahim Hawwari: Investigation. Fraser Duthie: Investigation. Cornelia Rohland: Investigation. Lucas S Ribeiro: Investigation. Lea‐Marie Jenster: Investigation. Nathalia Rosero: Investigation. Yonas M Tesfamariam: Investigation. Fernando Q Cunha: Resources; Investigation; Writing ‐ review and editing. Florian I Schmidt: Conceptualization; Resources; Investigation; Methodology; Writing—review and editing. Bernardo S Franklin: Conceptualization; Resources; Data curation; Software; Formal analysis; Supervision; Funding acquisition; Validation; Investigation; Visualization; Methodology; Writing—original draft; Project administration; Writing—review and editing.
In addition to the CRediT author contributions listed above, the contributions in detail are:
Conceptualization: FIS and BSF. Methodology: DB, CWSW, JDW, FD, FQC, FIS, and BSF. Formal Analysis: DB, FIS, FQC, and BSF. Investigation: DB, CWSW, AHS, JDW, SM, JT, LS, IH, FD, CR, LSR, LJ, NR, YMT, FIS, and BSF. Visualization: DB and BSF. Writing – original draft: DB and BSF. Writing – review & editing: DB, CWSW, FQC, FIS, and BSF. Funding acquisition: BSF. Project administration: BSF. Supervision: DB, FQC, FIS, and BSF.
Disclosure and competing interests statement
DB is a past employee and shareholder of IFM Therapeutics (unrelated to this work). FIS is a cofounder and shareholder of Dioscure Therapeutics SE as well as a consultant and shareholder of IFM Therapeutics (unrelated to this work). All other authors declare that they have no conflict of interest.
The paper explained.
Problem
Inflammasomes are central signaling hubs of the innate immune system. These cytosolic sensors patrol the intracellular environment of a cell for signs of pathogen invasion of other stressors. Emerging evidence indicate that inflammasomes also have roles in the extracellular space. Activation of inflammasomes in different immune cells results in a lytic form of cell death called pyroptosis. Upon lysis, pyroptotic cells spill their intracellular content, including these multimeric inflammasome platforms, into the extracellular space. Extracellular ASC specks have been reported in the circulation and in inflamed tissues in different disease states. However, little is known about their contribution to the development of inflammation and disease.
Results
Using camelid‐derived single‐domain antibodies (nanobodies, VHHs), we demonstrate that extracellular ASC specks that remain after the demise of pyroptotic cells mediate a substantial part of the inflammation in two models of arthritis. VHH directed against ASC (VHHASC) target and disrupt inflammasome complexes (ASC specks) in pyroptotic cells preventing their inflammatory and prion‐like extracellular functions. Furthermore, we show that VHHASC can only access cytosolic ASC specks through membrane pores formed after the demise of inflammasome‐activated cells, thus, without compromising early cytokine production (IL‐1β), important for host defense.
Impact
Biotherapies aimed against pro‐inflammatory cytokines, such as IL‐1β, are currently used against Rheumatoid arthritis (RA) and other inflammasome‐related diseases. Given its essential role in host defense, IL‐1‐blockade increases the risk of serious infections in patients. Hence, the use of nanobodies against the inflammasome adaptor ASC is an attractive therapeutic alternative to overcome immunosuppression as it preserves inflammasome‐activation and IL‐1β production, while blocking the extracellular functions of inflammasomes that persist in inflamed tissues. Nanobodies targeting ASC are thus tailored to modulate the host response to inflammation and limit chronic disease.
Supporting information
Appendix
Expanded View Figures PDF
Movie EV1
Movie EV2
Movie EV3
Movie EV4
Movie EV5
Movie EV6
Source Data for Expanded View
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 4
Source Data for Figure 5
Acknowledgments
We thank Matthew Mangan, and Tomasz Prochnicki for scientific discussions, input and sharing of reagents. We also acknowledge Gudrun Engels and Maximilian Rothe for technical help, and organization and maintenance of mouse lines, respectively. We are also grateful to Rainer Stahl for help and input with cloning, and Alvin Lu for advice to generate VHHASC mutants. We also tank Gabor Horvath and the Microscopy Core Facility of the Medical Faculty at the University of Bonn for providing help, services, and devices funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation, project number 388158066). We thank Paul‐Albert Koenig and the Core Facility Nanobodies of the Medical Faculty at the University of Bonn for supporting nanobody generation and characterization. We also thank Peter Wurst and the FACS Core Facility of the Medical Faculty at the University of Bonn for providing help, services, and devices funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation, project numbers 216372545 and 388159768). Finally, we thank Pia Langhoff for laboratory management and Andrea Schlichting and Leonie Verwohlt for administrative support. BSF is supported by grants from the European Research Council (EC | H2020 | H2020 Priority Excellent Science | H2020 European Research Council (ERC) PLAT‐IL‐1 714175); and the Germany's Excellence Strategy (EXC 2151 – 390873048) from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation). FIS is funded by the Emmy Noether Programme 322568668 and TRR237‐369799452, and EXC 2151 – 390873048 of the DFG. This study was fully supported by intramural funding from the Medical Faculty of the University of Bonn to BSF. Open access funding enabled and organized by Projekt DEAL.
EMBO Mol Med (2022) 14: e15415.
See also: S Adriouch & P Pelegrin (June 2022)
Contributor Information
Damien Bertheloot, Email: dbertheloot@uni-bonn.de.
Florian I Schmidt, Email: fschmidt@uni-bonn.de.
Bernardo S Franklin, Email: franklin@uni-bonn.de.
Data availability
This study includes no dataset requiring the deposition of primary data into a public database.
References
- Ahmad F, Mishra N, Ahrenstorf G, Franklin BS, Latz E, Schmidt RE, Bossaller L (2018) Evidence of inflammasome activation and formation of monocyte‐derived ASC specks in HIV‐1 positive patients. AIDS 32: 299–307 [DOI] [PubMed] [Google Scholar]
- Ashour J, Schmidt FI, Hanke L, Cragnolini J, Cavallari M, Altenburg A, Brewer R, Ingram J, Shoemaker C, Ploegh HL (2015) Intracellular expression of camelid single‐domain antibodies specific for influenza virus nucleoprotein uncovers distinct features of its nuclear localization. J Virol 89: 2792–2800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baroja‐Mazo A, Martín‐Sánchez F, Gomez AI, Martínez CM, Amores‐Iniesta J, Compan V, Barberà‐Cremades M, Yagüe J, Ruiz‐Ortiz E, Antón J et al (2014) The NLRP3 inflammasome is released as a particulate danger signal that amplifies the inflammatory response. Nat Immunol 15: 738–748 [DOI] [PubMed] [Google Scholar]
- Basiorka AA, McGraw KL, Abbas‐Aghababazadeh F, McLemore AF, Vincelette ND, Ward GA, Eksioglu EA, Sallman DA, Ali NA, Padron E et al (2018) Assessment of ASC specks as a putative biomarker of pyroptosis in myelodysplastic syndromes: an observational cohort study. Lancet Haematol 5: e393–e402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben H (2019) Novartis bets $1.6bn on inflammasome‐focused biotech. Outsourcing‐Pharma https://www.outsourcing‐pharma.com/Article/2019/04/01/Novartis‐acquires‐IFM‐Tre‐for‐1.6bn
- Bilal J, Berlinberg A, Riaz IB, Faridi W, Bhattacharjee S, Ortega G, Murad MH, Wang Z, Prokop LJ, Alhifany AA et al (2019) Risk of infections and cancer in patients with rheumatologic diseases receiving interleukin inhibitors: a systematic review and meta‐analysis. JAMA Netw Open 2: e1913102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broderick L, De Nardo D, Franklin BS, Hoffman HM, Latz E (2015) The inflammasomes and autoinflammatory syndromes. Annu Rev Pathol 10: 395–424 [DOI] [PubMed] [Google Scholar]
- Budden CF, Gearing LJ, Kaiser R, Standke L, Hertzog PJ, Latz E (2021) Inflammasome‐induced extracellular vesicles harbour distinct RNA signatures and alter bystander macrophage responses. J Extracell Vesicles 10: e12127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai X, Chen J, Xu H, Liu S, Jiang Q‐X, Halfmann R, Chen ZJ (2014) Prion‐like polymerization underlies signal transduction in antiviral immune defense and inflammasome activation. Cell 156: 1207–1222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calligaris L, Marchetti F, Tommasini A, Ventura A (2008) The efficacy of anakinra in an adolescent with colchicine‐resistant familial Mediterranean fever. Eur J Pediatr 167: 695–696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll J (2018) Giant Roche wades into the preclinical NLRP3 pool, bagging Jecure for their anti‐inflammatory team at Genentech. Endpoints News https://endpts.com/giant‐roche‐wades‐into‐the‐preclinical‐nlrp3‐pool‐bagging‐jecure‐for‐their‐anti‐inflammatory‐team‐at‐genentech/
- Chauhan D, Vande Walle L, Lamkanfi M (2020) Therapeutic modulation of inflammasome pathways. Immunol Rev 297: 123–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coll RC, Robertson AAB, Chae JJ, Higgins SC, Muñoz‐Planillo R, Inserra MC, Vetter I, Dungan LS, Monks BG, Stutz A et al (2015) A small‐molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med 21: 248–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czajkowsky DM, Hotze EM, Shao Z, Tweten RK (2004) Vertical collapse of a cytolysin prepore moves its transmembrane beta‐hairpins to the membrane. EMBO J 23: 3206–3215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalbeth N, Choi HK, Joosten LAB, Khanna PP, Matsuo H, Perez‐Ruiz F, Stamp LK (2019) Gout. Nat Rev Dis Primers 5: 69 [DOI] [PubMed] [Google Scholar]
- de Almeida L, Khare S, Misharin A, Patel R, Ratsimandresy R, Wallin M, Perlman H, Greaves D, Hoffman H, Dorfleutner A et al (2015) The PYRIN domain‐only protein POP1 inhibits inflammasome assembly and ameliorates inflammatory disease. Immunity 43: 264–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demarco B, Grayczyk JP, Bjanes E, Le Roy D, Tonnus W, Assenmacher C‐A, Radaelli E, Fettrelet T, Mack V, Linkermann A et al (2020) Caspase‐8‐dependent gasdermin D cleavage promotes antimicrobial defense but confers susceptibility to TNF‐induced lethality. Sci Adv 6: eabc3465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desu HL, Plastini M, Illiano P, Bramlett HM, Dietrich WD, de Rivero Vaccari JP, Brambilla R, Keane RW (2020) IC100: a novel anti‐ASC monoclonal antibody improves functional outcomes in an animal model of multiple sclerosis. J Neuroinflammation 17: 143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick MS, Sborgi L, Ruhl S, Hiller S, Broz P (2016) ASC filament formation serves as a signal amplification mechanism for inflammasomes. Nat Commun 7: 11929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, Abela GS, Franchi L, Nuñez G, Schnurr M et al (2010) NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 464: 1357–1361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellebedy AH, Lupfer C, Ghoneim HE, DeBeauchamp J, Kanneganti TD, Webby RJ (2011) Inflammasome‐independent role of the apoptosis‐associated speck‐like protein containing CARD (ASC) in the adjuvant effect of MF59. Proc Natl Acad Sci 108: 2927–2932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes‐Alnemri T, Alnemri ES (2008) Chapter thirteen assembly, purification, and assay of the activity of the ASC pyroptosome. In Programmed Cell Death,General Principles for Studying Cell Death, Part A, Khosravi‐Far, R , Zakeri, Z , Lockshin, RA , Piacentini, M (eds), Vol. 442, pp. 251–270. Methods in Enzymology. Academic Press Inc; [DOI] [PubMed] [Google Scholar]
- Fernandes‐Alnemri T, Wu J, Yu J‐W, Datta P, Miller B, Jankowski W, Rosenberg S, Zhang J, Alnemri ES (2007) The pyroptosome: a supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase‐1 activation. Cell Death Differ 14: 1590–1604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin BS, Bossaller L, De Nardo D, Ratter JM, Stutz A, Engels G, Brenker C, Nordhoff M, Mirandola SR, Al‐Amoudi A et al (2014) The adaptor ASC has extracellular and ‘prionoid’ activities that propagate inflammation. Nat Immunol 15: 727–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin BS, Latz E, Schmidt FI (2017) The intra‐ and extracellular functions of ASC specks. Immunol Rev 281: 74–87 [DOI] [PubMed] [Google Scholar]
- Friker LL, Scheiblich H, Hochheiser IV, Brinkschulte R, Riedel D, Latz E, Geyer M, Heneka MT (2020) beta‐Amyloid Clustering around ASC Fibrils Boosts Its Toxicity in Microglia. Cell Rep 30: 3743–3754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Zhu J, Lu H (2020) Single domain antibody‐based vectors in the delivery of biologics across the blood–brain barrier: a review. Drug Deliv Transl Res 11: 1818–1828 [DOI] [PubMed] [Google Scholar]
- Guo X, Dhodapkar KM (2012) Central and overlapping role of Cathepsin B and inflammasome adaptor ASC in antigen presenting function of human dendritic cells. Hum Immunol 73: 871–878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurbatri CR, Lia I, Vincent R, Coker C, Castro S, Treuting PM, Hinchliffe TE, Arpaia N, Danino T (2020) Engineered probiotics for local tumor delivery of checkpoint blockade nanobodies. Sci Transl Med 12: eaax0876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Hilst J, Moutschen M, Messiaen PE, Lauwerys BR, Vanderschueren S (2016) Efficacy of anti‐IL‐1 treatment in familial Mediterranean fever: a systematic review of the literature. Biologics 10: 75–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, Fitzgerald KA, Latz E (2008) Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol 9: 847–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoss F, Rolfes V, Davanso MR, Braga TT, Franklin BS (2018) Detection of ASC speck formation by flow cytometry and chemical cross‐linking. In Innate Immune Activation, De Nardo, D , De Nardo, C (eds), Vol. 1714, pp. 149–165. Methods in Molecular Biology. New York, NY: Humana Press. 10.1007/978-1-4939-7519-8_10 [DOI] [PubMed] [Google Scholar]
- Janssen CA, Oude Voshaar MAH, Vonkeman HE, Jansen T, Janssen M, Kok MR, Radovits B, van Durme C, Baan H, van de Laar M (2019) Anakinra for the treatment of acute gout flares: a randomized, double‐blind, placebo‐controlled, active‐comparator, non‐inferiority trial. Rheumatology 58(8): 1344–1352 [DOI] [PubMed] [Google Scholar]
- Jonson and Jonson (2021) Janssen to Acquire Investigational Bermekimab from XBiotech, INNOVATION. Johnson & Johnson https://www.jnj.com/janssen‐to‐acquire‐investigational‐bermekimab‐from‐xbiotech
- Kagan JC, Magupalli VG, Wu H (2014) SMOCs: supramolecular organizing centres that control innate immunity. Nat Rev Immunol 14: 821–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy CR, Goya Grocin A, Kovacic T, Singh R, Ward JA, Shenoy AR, Tate EW (2021) A probe for NLRP3 inflammasome inhibitor MCC950 identifies carbonic anhydrase 2 as a novel target. ACS Chem Biol 16: 982–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klück V, Jansen TLTA, Janssen M, Comarniceanu A, Efdé M, Tengesdal IW, Schraa K, Cleophas MCP, Scribner CL, Skouras DB et al (2020) Dapansutrile, an oral selective NLRP3 inflammasome inhibitor, for treatment of gout flares: an open‐label, dose‐adaptive, proof‐of‐concept, phase 2a trial. Lancet Rheumatol 2: e270–e280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenig P‐A, Das H, Liu H, Kümmerer BM, Gohr FN, Jenster L‐M, Schiffelers LDJ, Tesfamariam YM, Uchima M, Wuerth JD et al (2021) Structure‐guided multivalent nanobodies block SARS‐CoV‐2 infection and suppress mutational escape. Science 371: eabe6230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kontermann RE (2016) Half‐life extended biotherapeutics. Expert Opin Biol Ther 16: 903–915 [DOI] [PubMed] [Google Scholar]
- Kuijk LM, Govers AM, Frenkel J, Hofhuis WJ (2007) Effective treatment of a colchicine‐resistant familial Mediterranean fever patient with anakinra. Ann Rheum Dis 66: 1545–1546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu A, Magupalli VG, Ruan J, Yin Q, Atianand MK, Vos MR, Schröder GF, Fitzgerald KA, Wu H, Egelman EH (2014) Unified polymerization mechanism for the assembly of ASC‐dependent inflammasomes. Cell 156: 1193–1206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maass DR, Sepulveda J, Pernthaner A, Shoemaker CB (2007) Alpaca (Lama pacos) as a convenient source of recombinant camelid heavy chain antibodies (VHHs). J Immunol Methods 324: 13–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangan MSJ, Olhava EJ, Roush WR, Seidel HM, Glick GD, Latz E (2018) Targeting the NLRP3 inflammasome in inflammatory diseases. Nat Rev Drug Discov 17: 688 [DOI] [PubMed] [Google Scholar]
- Martinon F, Burns K, Tschopp J (2002) The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL‐beta. Mol Cell 10: 417–426 [DOI] [PubMed] [Google Scholar]
- Martinon F, Pétrilli V, Mayor A, Tardivel A, Tschopp J (2006) Gout‐associated uric acid crystals activate the NALP3 inflammasome. Nature 440: 237–241 [DOI] [PubMed] [Google Scholar]
- Mulvihill E, Sborgi L, Mari SA, Pfreundschuh M, Hiller S, Muller DJ (2018) Mechanism of membrane pore formation by human gasdermin‐D. EMBO J 37: e98321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orning P, Weng D, Starheim K, Ratner D, Best Z, Lee B, Brooks A, Xia S, Wu H, Kelliher MA et al (2018) Pathogen blockade of TAK1 triggers caspase‐8‐dependent cleavage of gasdermin D and cell death. Science 362: 1064–1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottaviani S, Moltó A, Ea H‐K, Neveu S, Gill G, Brunier L, Palazzo E, Meyer O, Richette P, Bardin T et al (2013) Efficacy of anakinra in gouty arthritis: a retrospective study of 40 cases. Arthritis Res Ther 15: R123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phulphagar K, Kuhn LI, Ebner S, Frauenstein A, Swietlik JJ, Rieckmann J, Meissner F (2021) Proteomics reveals distinct mechanisms regulating the release of cytokines and alarmins during pyroptosis. Cell Rep 34: 108826 [DOI] [PubMed] [Google Scholar]
- Pinto LG, Cunha TM, Vieira SM, Lemos HP, Verri WA Jr, Cunha FQ, Ferreira SH (2010) IL‐17 mediates articular hypernociception in antigen‐induced arthritis in mice. Pain 148: 247–256 [DOI] [PubMed] [Google Scholar]
- Pinto LG, Talbot J, Peres RS, Franca RF, Ferreira SH, Ryffel B, Aves‐Filho JC, Figueiredo F, Cunha TM, Cunha FQ (2015) Joint production of IL‐22 participates in the initial phase of antigen‐induced arthritis through IL‐1beta production. Arthritis Res Ther 17: 235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD et al (2017a) Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med 377: 1119–1131 [DOI] [PubMed] [Google Scholar]
- Ridker PM, MacFadyen JG, Everett BM, Ridker P, Lorenzatti A, Krum H, Varigos J, Siostrzonek P, Sinnaeve P, Fonseca F et al (2017b) Effect of interleukin‐1β inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: exploratory results from a randomised, double‐blind, placebo‐controlled trial. The Lancet 390: 1833–1842 [DOI] [PubMed] [Google Scholar]
- de Rivero Vaccari JP, Lotocki G, Marcillo AE, Dietrich WD, Keane RW (2008) A molecular platform in neurons regulates inflammation after spinal cord injury. J Neurosci 28: 3404–3414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolfes V, Ribeiro LS, Hawwari I, Böttcher L, Rosero N, Maasewerd S, Santos MLS, Próchnicki T, Silva CMDS, Wanderley CWDS et al (2020) Platelets fuel the inflammasome activation of innate immune cells. Cell Rep 31: 107615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruscitti P, Masedu F, Alvaro S, Airò P, Battafarano N, Cantarini L, Cantatore FP, Carlino G, D'Abrosca V, Frassi M et al (2019) Anti‐interleukin‐1 treatment in patients with rheumatoid arthritis and type 2 diabetes (TRACK): A multicentre, open‐label, randomised controlled trial. PLoS Med 16: e1002901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagoo P, Garcia Z, Breart B, Lemaître F, Michonneau D, Albert ML, Levy Y, Bousso P (2016) In vivo imaging of inflammasome activation reveals a subcapsular macrophage burst response that mobilizes innate and adaptive immunity. Nat Med 22: 64–71 [DOI] [PubMed] [Google Scholar]
- Salliot C, Dougados M, Gossec L (2009) Risk of serious infections during rituximab, abatacept and anakinra treatments for rheumatoid arthritis: meta‐analyses of randomised placebo‐controlled trials. Ann Rheum Dis 68: 25–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos JC, Boucher D, Schneider LK, Demarco B, Dilucca M, Shkarina K, Heilig R, Chen KW, Lim RYH, Broz P (2020) Human GBP1 binds LPS to initiate assembly of a caspase‐4 activating platform on cytosolic bacteria. Nat Commun 11: 3276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt FI, Hanke L, Morin B, Brewer R, Brusic V, Whelan SP, Ploegh HL (2016a) Phenotypic lentivirus screens to identify functional single domain antibodies. Nat Microbiol 1: 16080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt FI, Lu A, Chen JW, Ruan J, Tang C, Wu H, Ploegh HL (2016b) A single domain antibody fragment that recognizes the adaptor ASC defines the role of ASC domains in inflammasome assembly. J Exp Med 213: 771–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scully M, Cataland SR, Peyvandi F, Coppo P, Knöbl P, Kremer Hovinga JA, Metjian A, de la Rubia J, Pavenski K, Callewaert F et al (2019) Caplacizumab treatment for acquired thrombotic thrombocytopenic purpura. N Engl J Med 380: 335–346 [DOI] [PubMed] [Google Scholar]
- Spel L, Martinon F (2020) Inflammasomes contributing to inflammation in arthritis. Immunol Rev 294: 48–62 [DOI] [PubMed] [Google Scholar]
- Stocki P, Szary J, Rasmussen CLM, Demydchuk M, Northall L, Logan DB, Gauhar A, Thei L, Moos T, Walsh FS et al (2021) Blood‐brain barrier transport using a high affinity, brain‐selective VNAR antibody targeting transferrin receptor 1. FASEB J 35: e21172 [DOI] [PubMed] [Google Scholar]
- Tartey S, Kanneganti TD (2020) Inflammasomes in the pathophysiology of autoinflammatory syndromes. J Leukoc Biol 107: 379–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchiya K, Hara H, Fang R, Hernandez‐Cuellar E, Sakai S, Daim S, Chen X, Dewamitta SR, Qu H, Mitsuyama M et al (2014) The adaptor ASC exacerbates lethal Listeria monocytogenesinfection by mediating IL‐18 production in an inflammasome‐dependent and ‐independent manner. Eur J Immunol 44: 3696–3707 [DOI] [PubMed] [Google Scholar]
- Tzeng T‐C, Schattgen S, Monks B, Wang D, Cerny A, Latz E, Fitzgerald K, Golenbock DT (2016) A fluorescent reporter mouse for inflammasome assembly demonstrates an important role for cell‐bound and free ASC specks during in vivo infection. Cell Rep 16: 571–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venegas C, Kumar S, Franklin BS, Dierkes T, Brinkschulte R, Tejera D, Vieira‐Saecker A, Schwartz S, Santarelli F, Kummer MP et al (2017) Microglia‐derived ASC specks cross‐seed amyloid‐β in Alzheimer’s disease. Nature 552: 355–361 [DOI] [PubMed] [Google Scholar]
- Volchuk A, Ye A, Chi L, Steinberg BE, Goldenberg NM (2020) Indirect regulation of HMGB1 release by gasdermin D. Nat Commun 11: 4561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Völker LA, Kaufeld J, Miesbach W, Brähler S, Reinhardt M, Kühne L, Mühlfeld A, Schreiber A, Gaedeke J, Tölle M et al (2020) Real‐world data confirm the effectiveness of caplacizumab in acquired thrombotic thrombocytopenic purpura. Blood Adv 4: 3085–3092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Moltke J, Trinidad NJ, Moayeri M, Kintzer AF, Wang SB, van Rooijen N, Brown CR, Krantz BA, Leppla SH, Gronert K et al (2012) Rapid induction of inflammatory lipid mediators by the inflammasome in vivo . Nature 490: 107–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wouters Y, Jaspers T, De Strooper B, Dewilde M (2020) Identification and in vivo characterization of a brain‐penetrating nanobody. Fluids Barriers CNS 17: 62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamura K, Ashida H, Okano T, Kinoshita‐Daitoku R, Suzuki S, Ohtani K, Hamagaki M, Ikeda T, Suzuki T (2019) Inflammasome activation induced by Perfringolysin O of clostridium perfringens and its involvement in the progression of gas gangrene. Front Microbiol 10: 2406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B, Abbott DW (2021) Gasdermin E permits interleukin‐1 beta release in distinct sublytic and pyroptotic phases. Cell Rep 35: 108998 [DOI] [PMC free article] [PubMed] [Google Scholar]