

Abstract
By offering extremely long contiguous characterization of individual DNA molecules, rapidly emerging long‐read sequencing strategies offer comprehensive insights into the organization of genetic information in genomes and metagenomes. However, successful long‐read sequencing experiments demand high concentrations of highly purified DNA of high molecular weight (HMW), which limits the utility of established DNA extraction kits designed for short‐read sequencing. The challenges associated with input DNA quality intensify further when working with complex environmental samples of low microbial biomass, which requires new protocols that are tailored to study metagenomes with long‐read sequencing. Here, we use human tongue scrapings to benchmark six HMW DNA extraction strategies that are based on commercially available kits, phenol–chloroform (PC) extraction and agarose encasement followed by agarase digestion. A typical end goal of HMW DNA extractions is to obtain the longest possible reads during sequencing, which is often achieved by PC extractions, as demonstrated in sequencing of cultured cells. Yet our analyses that consider overall read‐size distribution, assembly performance and the number of circularized elements found in sequencing results suggest that column‐based kits with enzyme supplementation, rather than PC methods, may be more appropriate for long‐read sequencing of metagenomes.
Keywords: high‐molecular‐weight DNA, long‐read sequencing, metagenomics, nanopore
1. INTRODUCTION
High‐throughput sequencing of metagenomes offers unprecedented insights into the diversity and gene pool of naturally occurring microbes and viruses that occupy soils (Nesme et al., 2016), marine habitats (Gregory, Zayed, et al., 2019; Sunagawa et al., 2015) and host‐associated environments (Gregory, Zablocki, et al., 2020; Human Microbiome Project Consortium, 2012). The high accuracy and the high throughput of the modern sequencing platforms are afforded by read lengths that typically remain below 250 bases. These relatively short reads pose significant constraints on the data utility, especially in metagenomics (Wommack et al., 2008).
By stitching together the short reads that partially overlap, metagenomic assembly can reconstruct orders of magnitude longer contiguous segments of input DNA (Nurk et al., 2017) and enable the recovery of microbial genomes from metagenomes (Tyson et al., 2004). In recent years, this strategy has become a primary tool in microbiology to study the ecology and evolution of naturally occurring microbial populations (Al‐Shayeb et al., 2020; Delmont et al., 2018; Hug et al., 2016; Pasolli et al., 2019; Spang et al., 2015). However, metagenomic assembly is inherently challenging (Ayling et al., 2020) and the assembly of complex environments often leads to highly fragmented assemblies (Olson et al., 2019). These fragmented assemblies increase the likelihood of generating composite genomes that include contigs from multiple distinct populations (Chen et al., 2020), which risk erroneous insights into microbial ecology and evolution (Chen et al., 2020; Shaiber & Eren, 2019).
By circumventing the problems associated with short‐read assembly, long‐read sequencing offers a compelling solution to the ideal of reconstructing complete genomes from metagenomes (Driscoll et al., 2017; White et al., 2016). Nanopore sequencing, which resolves the identity and order of nucleotides based on changes in ionic current as a single‐stranded RNA or DNA molecule passes through a tiny pore (Kasianowicz et al., 1996), has rapidly gained popularity among researchers (Wang et al., 2014) thanks to its availability through affordable and easy to operate sequencing devices, such as MinION by Oxford Nanopore Technologies (“The Long View on Sequencing”, 2018). Despite the high error rates and relatively lower sequencing depth, long reads from nanopore sequencing of metagenomes led to key insights from challenging systems (Pessi et al., 2020; Reveillaud et al., 2019) and enabled the recovery of circular, complete genomes from metagenomes (Cusco et al., 2020; Moss et al., 2020; Nicholls et al., 2019; Sanderson et al., 2018; Singleton et al., 2021; Somerville et al., 2019).
The efficacy of long‐read sequencing depends heavily on the structural integrity of the input DNA (Schalamun et al., 2019), which poses a new and significant challenge. Commercial DNA extraction kits that emerged during the era of short‐read sequencing typically include steps that physically disrupt cells through mechanical lysis and generate highly fragmented DNA molecules. While these commercial kits improve short‐read sequencing outcomes as they ensure maximum yield and coverage of DNA in a sample, they set a critical limit to the outcomes of long‐read sequencing. Hence, establishing DNA extraction strategies that afford (i) preservation of high‐molecular‐weight (HMW) molecules, (ii) a high degree of sample purity and (iii) increased overall DNA yields have become critical considerations for the successful application of long‐read sequencing.
Phenol–chloroform DNA extractions, first popularized by Sambrook et al. (1989), have been making a resurgence as a “go‐to” method for extracting HMW DNA (Maghini et al., 2020; Quick & Loman, 2018). While recent studies have used this approach to recover ultralong DNA fragments (e.g., >100 kb) from cultured organisms (Cicha et al., 2020; Hosoe et al., 2020; Kinoshita et al., 2020; Takeshita et al., 2020; Tippelt et al., 2020), the utility of phenol–chloroform extractions for metagenomics is not yet clear. In parallel, long‐read sequencing surveys of metagenomes have largely focused on high‐microbial‐biomass samples including human stool (Moss et al., 2020) or activated sludge (Singleton et al., 2021), and best practices to study metagenomes of lower biomass samples are yet to emerge. Increasing the breadth of long‐read sequencing requires DNA extraction protocols that can both produce long reads and can scale to a range of systems, including those that are associated with low microbial biomass.
Here we designed six DNA extraction protocols and examined their relative effectiveness to extract HMW DNA in terms of total yield, concentration, purity, integrity and applicability for subsequent long‐read sequencing. Using an Oxford Nanopore MinION sequencer, we benchmarked our protocols based on the extent of host contamination, fragment size distribution, microbial taxonomy and metagenomic assembly outcomes across multiple sequencing runs, and compared taxonomic insights that emerged from long‐read sequencing to those from Illumina short‐read sequencing. We chose the human oral cavity as a model system, since it offers a challenging environment to benchmark DNA extraction strategies as it is home to complex microbial communities (Dewhirst et al., 2010) with relatively low biomass (Duran‐Pinedo & Frias‐Lopez, 2015) and is typically mixed with eukaryotic host DNA (Marotz et al., 2018).
2. MATERIALS AND METHODS
2.1. Tongue dorsum sample collection
A single healthy individual self‐collected scraping of their tongue dorsum on 13 separate occasions (one per day) over the course of 3 weeks. We used BreathRx Gentle Tongue Scrapers (Philips Sonicare) for sample collection, which was performed prior to eating, drinking or performing oral hygiene. Starting as far back as possible on the tongue, the scraper was passed forward over the entire surface three sequential times. We transferred the collected material to 520 µl of PBS (phosphate‐buffered saline) and immediately stored at −20°C until processing. The pooled 13 tongue dorsum samples were used for HMW DNA extraction comparisons, while another self‐collected tongue dorsum sample (named TD) from this same individual was obtained 2 weeks later for short‐read sequencing (amplicon and metagenomics).
2.2. DNA extraction methods
We compared six DNA extraction methods tailored for HMW DNA recovery. We included both commercially available kits and nonkit methods used in the published literature which incorporated different combinations of cell lysis mechanisms and DNA purification methodologies. To facilitate direct comparison between all extraction methods, we thawed and pooled the 13 samples immediately prior to DNA extraction. After homogenizing the pooled sample by vortexing for 15 s, we used a 500‐µl aliquot as the starting material for each extraction method, which was performed in duplicate. We resuspended the isolated genomic DNA from each method in a final 100 µl volume in a 1.5‐ml LoBind microfuge tube (Eppendorf). We sought to maximize read lengths by implementing best practices for handling HMW DNA throughout all the methods. We eliminated vortexing and mixing by pipetting, when possible, in favour of end‐over‐end tube rotation to minimize velocity gradients. We used wide‐bore pipette tips with gentle pipetting to reduce DNA breakage and avoided unnecessary freeze–thaw cycles by storing DNA at 4°C until sequence analysis.
2.3. DNeasy PowerSoil isolation kit with modified bead beating (PB)
DNeasy PowerSoil DNA isolation kits (Qiagen) are commonly used in metagenomics (Human Microbiome Project Consortium, 2012; Shaiber et al., 2020) to extract high‐quality DNA from environmental matrices. We sought to determine its compatibility with nanopore sequencing protocols by amending the PowerSoil DNA isolation protocol in two ways. In our first modification to the DNeasy PowerSoil DNA Isolation kit, we incorporated a modified bead beating step (Edwards et al., 2019), as a way to minimize velocity gradients and reduce DNA shearing, therefore improving fragment length. We transferred 500 µl of the pooled sample to the kit provided with a PowerBead tube, which we inserted flat into IKA Works MS2S8 Minishaker for Bioanalyzer DNA chips. Samples were agitated for 10 min (in 1 min pulse increments) at 2400 rpm (Edwards et al., 2019). We then followed the remainder of the manufacturer's instructions for DNA isolation and purification.
2.4. DNeasy PowerSoil Isolation kit supplemented with an enzymatic treatment (PE)
In the second modification to the DNeasy PowerSoil DNA Isolation kit, we fully replaced the use of mechanical cell lysis (or bead beating) with a heated, enzymatic treatment step (Yuan et al., 2012). We added 500 µl of the pooled sample to the kits’ PowerBeads tubes including the tube's solution (but lacking the beads). We added a lytic cocktail to facilitate cell lysis: 125 µl lysozyme (10 mg ml–1, Sigma‐Aldrich), 37.5 µl mutanolysin (10 KU ml–1, Sigma‐Aldrich), 7.5 µl lysostaphin (4000 U ml–1, Sigma‐Aldrich) and 5 µl RNase A (10 mg ml–1, Sigma‐Aldrich). We incubated for 1 h at 37°C. Then we added 50 µl of proteinase K (20 mg ml–1, Sigma‐Aldrich) alongside kit solution C1, and followed by incubation for 30 min at 56°C. After centrifugation of the tubes at 10,000 g for 30 s at room temperature (step 6), we continued the rest of the isolation protocol as described by the manufacturer.
2.5. DNeasy UltraClean Microbial Kit (UC)
We extracted genomic DNA using the DNeasy UltraClean Microbial Kit (Qiagen) and replaced its bead beating step with the manufacturer's alternative lysis procedure to reduce shearing of the DNA. This commercial kit was evaluated as a result of a direct recommendation by Oxford Nanopore. We added 500 µl of pooled sample to 300 µl of PowerBead solution. After the addition of 50 µl Solution SL, we incubated the sample for 10 min at 65°C. After centrifuging the tubes at 10,000 g for 30 s at room temperature (step 5), we continued the rest of the isolation protocol as described by the manufacturer.
2.6. Qiagen Genomic Tip 20/G supplemented with an enzymatic treatment (GT)
We extracted DNA using the Qiagen Genomic Tip 20/G (Qiagen) and followed the manufacturer's protocol for “Preparation Gram‐negative and some Gram‐positive Bacterial Samples.” We centrifuged 500 µl of pooled sample for 10 min at 10,000 g and resuspended the pellet in Buffer B1 supplemented with 20 µl DNAse‐free RNAse (10 mg ml–1, Sigma‐Aldrich), 45 µl Proteinase K (20 mg ml–1, Sigma‐Aldrich) and 20 µl lysozyme (100 mg ml–1, Sigma‐Aldrich), as outlined. We modified the lytic cocktail to include 9 µl lysostaphin (4000 U ml–1, Sigma‐Aldrich) and 45 µl mutanolysin (10 KU ml–1 Sigma‐Aldrich) in order to improve the lysis potential in Gram‐positive bacteria. After incubation for 2 h at 37°C, we added Buffer B2 and extended the incubation at 50°C to 90 min. As the lysate had not cleared after this initial period, we extended the incubation for an additional 2 h. We removed any remaining particulate matter by centrifugation at 5000 g for 10 min as recommended by the manufacturer. Finally, we used the columns in the kit according to the described isolation protocol.
2.7. Phenol/chloroform extraction (PC)
We extracted DNA using a phenol/chloroform extraction protocol modified from Chapter 6, protocol 1 of Sambrook et al. (1989). For SDS cell lysis, we added 500 µl of pooled sample to 10 ml of TLB (10 mm Tris‐HCL pH 8.0, 25 mm EDTA pH 8.0, 100 mm NaCl, 0.5% [w/v] SDS, 20 µg ml–1 RNase A) and vortexed at full speed for 5 s followed by an incubation at 37°C for 1 h. Proteinase K (Qiagen) was added to a final concentration of 200 µg ml–1 and we mixed the sample by slow inversion three times, followed by 2 h at 50°C with gentle mixing every 30 min. We purified the lysate with 10 ml buffer saturated phenol using phase‐lock gel falcon tubes, followed by phenol/chloroform‐isoamyl alcohol (1:1). We precipitated the DNA by adding 4 ml of 5 m ammonium acetate and 30 ml ice‐cold ethanol. We recovered DNA by one of the following methods: a glass hook followed by washing twice in 70% ethanol (if a DNA mass was visible) or centrifugation at 4500 g for 10 min followed by washing twice in 70% ethanol (if no DNA mass was visible). After spinning down at 10,000 g, we removed ethanol by drying at ambient temperature for 10 min. We added 100 µl EB (elution buffer, 10 mm Tris‐HCl, pH 8.5) to the DNA and left it at 4°C overnight to resuspend the pellet.
2.8. Agarose plug encasement and extraction (AE)
From the 500‐µl pooled sample aliquot, we pelleted the cells by centrifugation at 10,000 g for 10 min and removed the supernatant. We performed DNA extraction on the pellet using a pulsed‐field gel electrophoresis (PFGE)‐based agarose encasement extraction protocol (Matushek et al., 1996). The pelleted cells were resuspended in 300 µl 2× lysis solution (12.5 mm Tris‐HCl pH 7.6, 2 m NaCl, 20 mm EDTA pH 8.0, 1% [w/v] Brij 58, 1% [w/v] deoxycholate, 1% [w/v] sodium lauroyl sarcosine) to which we added lysozyme (1 mg ml–1) and DNAse‐free RNAse (30 µg ml–1) on the day of the experiment. We combined the entire suspension with 300 µl of molten 1.6% low‐melting point (LMP) agarose (TopVision, Thermo Fisher Scientific) which we pipetted into a plug mould and allowed to solidify. We placed each plug into 3 ml of 1× lysis solution containing: lysozyme, 0.5 mg ml–1; DNAse‐free RNAse, 100 µg ml–1; lysostaphin, 50 U ml–1; and mutanolysin, 0.3 KU ml–1. We added the enzymes on the day of the experiment and incubated the plugs overnight at 37°C with gentle shaking. After incubation, we replaced the lysis solution sequentially with ESP (10 mm Tris‐HCl pH 7.6, 1 mm EDTA) to which we added proteinase K (Sigma) at a final concentration of 100 µg ml–1 and 1% sodium dodecyl sulfate at 50°C for 1 h; and then two rounds of washing in sterile, dilute TE (10 mm Tris‐HCl pH 7.6, 0.1 mm EDTA) first at 50°C for 1 h and then at 35°C for 30 min with gentle shaking. We transferred the plug to 5 ml of fresh dilute TE in a clean tube for storage until we performed a β‐agarase I digestion of the agarose and isopropanol DNA precipitation according to the manufacturer's instructions (New England Biolabs). We resuspended the precipitated DNA in 100 µl EB (10 mm Tris‐HCl, pH 8.5).
2.9. Determination of DNA yield, purity metrics and fragment size distribution
For each extraction, we quantified the DNA yield on a Qubit 1.0 Fluorometer (Thermo Scientific), using the dsDNA HS (high sensitivity, 0.2–100 ng) Assay kit according to the manufacturer's protocols; a sample volume of 1 µl was added to 199 µl of a Qubit working solution. We assessed the purity of the extracted nucleic acids with the A260/280 and A260/230 absorbance ratios obtained using a NanoDrop spectrophotometer (NanoDrop Technologies). We assessed the DNA fragment size distribution by electrophoresis (90 V for 1.5 h) of genomic DNA (22 or 44 ng, as available) on a 0.8% (w/v) agarose gel followed by staining with ethidium bromide and UV light visualization. We used λ‐HindIII DNA size standards to estimate fragment sizes.
2.10. MinION library preparation and sequencing
Upon receipt, and again immediately prior to sequencing, we measured the flowcell pore counts using the Platform QC script (minknow). We stored the flowcells in their original packaging, which we resealed with parafilm and tape, at 4°C until use. Unless stated otherwise, we omitted size selection and the optional shearing step to allow us to evaluate the full distribution of fragment sizes produced by each method. We performed the library preparation using the Ligation Sequencing Kit (SQK‐LSK108) and Native Barcoding Kit (EXP‐NBD103) for genomic DNA, according to the standard 1D Native barcoding protocol provided by the manufacturer (Oxford Nanopore) (version: NBE_9006_v103_rev2_21DEC16) unless indicated. We carried out all reactions at room temperature. We followed the input DNA mass recommendation of 1.0 µg genomic DNA (gDNA) and performed DNA repair (NEBNext FFPE DNA Repair Mix, NEB M6630) to maximize read length. For the 1.0× AMPure clean‐up step, we used gentle rotation and an extended elution time (minimum 20 min) to assist in the release of long DNA fragments from the beads. We quantified 1‐µl aliquots by fluorometry (Qubit) after each phase of the library preparation (i.e., damage repair, end preparation, barcoding, pooling and adaptor ligation) to quantify DNA recovery, and identified substantial DNA loss after each step (Table 1).
TABLE 1.
Summary of DNA concentration and quality metrics. Technical replicates are denoted as “XX_1” and “XX_2”
| Extraction methodology | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PB | PE | UC | GT | PC | AE | ||||||||
| Method | Metric | PB_1 | PB_2 | PE_1 | PE_2 | UC_1 | UC_2 | GT_1 | GT_2 | PC_1 | PC_2 | AE_1 | AE_2 |
| Qubit | DNA concentration (µg ml–1) | 2.34 | 1.71 | 4.08 | 4.21 | 30.7 | 36.2 | 110 | 110 | 75.8 | 77.9 | 90.6 | 95.5 |
| Nanodrop | A260/A280 a | 6.01 | 5.53 | 1.75 | 2.27 | 1.95 | 1.74 | 1.74 | 1.82 | 1.97 | 1.95 | 1.8 | 1.76 |
| Nanodrop | A260/A230 b | 0.26 | 0.16 | 0.33 | 0.08 | 1.98 | 2.02 | 1.86 | 2.27 | 2.07 | 2.07 | 1.56 | 1.36 |
Primary measure of nucleic acid purity—expected value for “pure” DNA: ~1.8 while >2 indicates RNA contamination.
Secondary measure of nucleic acid purity—evaluates residual chemical contamination (phenol, guanidine HCl, carbohydrate carryover). Expected values are in the range of 1.8–2.2. Values significantly lower indicate chemical contamination and are undesired.
We used two sequencing runs (three samples multiplexed on each flowcell) for the sequencing. We loaded the completed libraries onto R9.4 flowcells as per instructions from ONT and scheduled sequencing runs for 48 h. Sequencing continued until time expired or until pore exhaustion (defined by <10 functional pores).
2.11. BluePippin size selection
To quantify the magnitude of its impact, if incorporated into a long‐read sequencing workflow, we performed size selection using DNA from GT_1 and GT_2 with the BluePippin (Sage Science) system with 0.75% dye‐free agarose cassettes and marker S1. We selected fragments >6 kb in high‐pass collection mode (an approach enabling the collection of DNA fragments above a user‐defined size). Our 3.3‐µg sample DNA input was less than the recommended 5 µg; however, this input was maximized given sample DNA concentrations and loading volume constraints.
2.12. Long‐read sequence analysis
We uploaded the raw MinION FAST5 files produced with the minknow software (versions 1.15.4 to 3.1.19) to our cluster to perform the base‐calling and demultiplexing with guppy version 2.3.1. FastQ files were generated only for reads meeting a minimum quality threshold (quality score of 7). We used minimap2 version 2.14 (Li, 2018) and samtools version 1.9 (Li et al., 2009) to remove sequences that mapped to the human genome build 38 (GRCh38) from NCBI and estimate the amount of host contamination. We used anvi’o version 6.2 (2021) and the contig snakemake workflow to compute the sequence metrics (Köster & Rahmann, 2012). Briefly, the workflow created a contigs database with “anvi‐gen‐contigs‐database,” which used prodigal version 2.6.3 (Hyatt et al., 2010) with the metagenome mode to identify open reading frames. It used “anvi‐run‐hmm” to detect the single‐copy core genes from bacteria (n = 71, modified from Lee, 2019), archaea (n = 76, Lee, 2019), eukarya (n = 83, http://merenlab.org/delmont‐euk‐scgs) and ribosomal RNAs (rRNAs) (n = 12, modified from https://github.com/tseemann/barrnap). We used “anvi‐display‐contigs‐stats” to obtain the number of sequences, total length, N50, longest sequence, number of genes, number of single‐copy core genes and ribosomal genes. We used blast against the NCBI’s nr/nt database to get the best taxonomy, percentage identity and query alignment for each longest read per extraction method. We used “anvi‐get‐sequences‐for‐hmm‐hits” to recover 16S rRNA genes for each condition and used the Human Oral Microbiome Database (HOMD) online blast tool for taxonomic assignment. We assembled the long reads with flye and the metagenomic option (Kolmogorov et al., 2020), which takes into account the uneven coverage nature of metagenomes. We used short‐reads generated using HMW DNA extraction sample PB_2 (see method below) to polish the assemblies using pilon (Walker et al., 2014). We created anvi’o contigs databases, as described above, with the polished flye’s contigs to summarize the assembly metrics.
2.13. 16S rRNA gene amplicon DNA extraction, library preparation, sequencing and analysis
For the 16S rRNA gene amplicon sequencing, we used a sample (TD), which was collected from the same individual 2 weeks after the pooled samples that were used for HMW DNA extraction. We performed sample DNA extraction using the DNeasy Powersoil kit (Qiagen) following the manufacturer's protocol. We amplified the V4–V5 hypervariable regions of the bacterial small subunit (SSU) rRNA gene using degenerate primers: 518F (CCAGCAGCYGCGGTAAN) and 926R (CCGTCAATTCNTTTRAGT, CCGTCAATTTCTTTGAGT, and CCGTCTATTCCTTTGANT). Amplification was done with fusion primers containing the 16S‐only sequences fused to Illumina adapters. The forward primers included a 5‐nt multiplexing barcode and the reverse primers a 6‐nt index. We generated PCR (polymerase chain reaction) amplicons in triplicate 33‐µl reaction volumes with an amplification cocktail containing 0.67 U SuperFi Taq Polymerase (Invitrogen), 1× enzyme buffer (includes MgCl2), 200 µm dNTP mix (ThermoFisher), and 0.3 µm of each primer. We added ~10–25 ng template DNA to each PCR and ran a no‐template control for each primer pair. Amplification conditions were: initial 94°C, 3‐min denaturation step; 30 cycles of 94°C for 30 s, 57°C for 45 s, and 72°C for 60 s; final 2‐min extension at 72°C. The triplicate PCRs were pooled after amplification, visualized with the negative controls on a Caliper LabChipGX, and purified using Ampure followed by PicoGreen quantification and Ampure size selection. Libraries were sequenced on an Illumina Miseq 250‐cycle paired‐end run. We used illumina‐utils version 2.7 (Eren et al., 2015) for the quality filtering, following the recommendations of Minoche et al. (2011), and the merging of the paired‐end reads. We used vsearch to remove chimeric sequences (Rognes et al., 2016) and Minimum Entropy Decomposition (MED) (Eren et al., 2015) to cluster the merged reads into oligotypes. We assigned taxonomy using dada2 (Callahan et al., 2016) and the Silva version 132 nonredundant database (Quast et al., 2013).
2.14. Short‐read metagenomic library preparation, sequencing and analysis
For the short‐read metagenomic sequencing, we used both the sample TD collected 2 weeks after the initial sampling, and the HMW DNA extraction sample PB_2 (the closest methodology to gold standard short‐read sequencing extraction methodology). Sample DNA concentrations, determined via a PicoGreen assay, were 67 ng µl–1 (TD) and 0.75 ng µl–1 (PB_2). We used 100 and 28 ng, respectively, for library construction. DNA was sheared to ~400 bp using the Covaris S2 acoustic platform and libraries were constructed using the Nugen Ovation Ultralow kit. Each required an amplification step: seven cycles (TD) or 11 cycles (PB_2). The products were visualized on an Agilent Tapestation 4200 and size‐selected to an average of 482 bp using BluePippin (Sage Biosciences). The final library pool was quantified with the Kapa Biosystems qPCR protocol and sequenced on the Illumina NextSeq500 in a 2 × 150 paired‐end sequencing run using dedicated read indexing. We used anvi’o version 6.2 and the metagenomics snakemake workflow for the assembly and analysis of the short reads. Briefly, the workflow uses illumina‐utils for quality filtering followed by a metagenomics assembly with idba‐ud version 1.1.3 (Peng et al., 2012) and generates contig databases as described in the long‐read sequence analysis section. The workflow also uses bowtie2 version 2.3.5.1 (Langmead & Salzberg, 2012) to map short reads on the assembled contigs and samtools (Li et al., 2009) to sort, index and convert sam files into bam files used by anvi’o to generate profiles databases. To compute taxonomic profiles of metagenomes we used the anvi’o program “anvi‐estimate‐scg‐taxonomy,” which aligns ribosomal proteins found in a metagenomic assembly to those that are found in reference genomes from the Genome Taxonomy Database (GTDB) (Parks et al., 2020).
2.15. Visualization
We generated the figures in R with ggplot2 (Wickham, 2009) and modified them with inkscape.
3. RESULTS
Our benchmarks here include four DNA extraction protocols based on three commercially available Qiagen DNA extraction kits (each incorporated modifications to their cell lysis procedures): the Qiagen DNeasy PowerSoil with a modified bead beating step (PB) or with bead beating replaced by enzymatic cell lysis (PE); the Qiagen DNeasy UltraClean Microbial Kit (UC) using the manufacturer's alternative lysis procedure to reduce DNA shearing; and the Qiagen Genomic Tip 20/G extraction kit (GT) augmented with additional enzymatic cell lysis. The remaining two protocols included in our study are a phenol–chloroform protocol (PC), and a pulsed‐field gel electrophoresis (PFGE)‐based agarose encasement extraction protocol (AE) followed by agarase digestion. Throughout the text we refer to these protocols as PB, PE, UC, GT, PC and AE, and we denote technical replicates as “XX_1” or “XX_2.” To benchmark the protocols that are detailed in the Methods section, we used a tongue dorsum sample pooled together from 13 samples collected from the same individual. An additional sample (TD) collected from the same individual has been used to compare taxonomic composition between short and long‐read sequencing.
3.1. Yield and quality metrics of isolated DNA vary between protocols
To ascertain their suitability for long‐read sequence analysis, we first analysed the quantity and quality of DNA isolated from each extraction protocol using both fluorometric (Qubit) and spectrophotometric (Nanodrop) methods (Table 1). The fluorescent dye used by Qubit binds specifically to its target molecule, dsDNA, and provides the most accurate DNA quantification. DNA concentrations were comparable between technical replicates for all methods, and three extraction protocols (GT, PC and AE) were distinguished from the others with concentrations >75 µg ml–1. These concentrations were comparable to previously published HMW DNA extractions from oral samples (Yahara et al., 2021). GT exhibited the highest sample concentrations with a mean of 110 µg ml–1. In comparison, the mean concentrations from PB, PE and UC were far less at 2.03, 4.15 and 33.45 µg ml–1, respectively. The MinION sequencing protocol for the Ligation Sequencing Kit (SQK‐LSK108) advises using 1–1.5 µg DNA. As we resuspended DNA into a final volume of 100 µl, only UC, GT, PC and AE had sufficient DNA yield to meet the recommended input.
We then considered the absorption spectra to assess sample purity and identify potential non‐nucleic acid contamination. The A260/A280 ratio indicates DNA purity with expected values around 1.8 for pure DNA. All extraction methods fell within the desired range (1.74–1.97), except for both PB replicates and one replicate of PE; however, samples with concentrations approaching the lower limit of 2 µg ml–1 may result in unacceptable A260/A280 ratios. The A260/A230 ratio signifies possible residual chemical contamination such as EDTA, phenol, guanidine salts (often used in column‐based kits) or carbohydrate carryover. UC, GT and PC had ratios close to the expected value of 2. Overall, GT had the best combined yield and purity metrics; PC and AE were deemed suitable alternatives. The purity metrics and congruence of Qubit and Nanodrop concentrations in UC were also desirable, yet UC’s ~2–3‐fold lower DNA yield proved less appealing.
We ran an agarose gel electrophoresis to visually assess the crude DNA fragment size distribution (Figure 1). All DNA samples migrated predominantly as a single HMW DNA band that aligned with (or was larger) than the reference 23.1‐kbp fragment of HindIII‐digested lambda DNA. A light smear, visible to 2.0 kb, was observed in GT, indicating the presence of smaller fragments. Recovery of larger DNA fragments by PC was denoted by the predominant band running higher than the 23.1‐kbp fragment.
FIGURE 1.
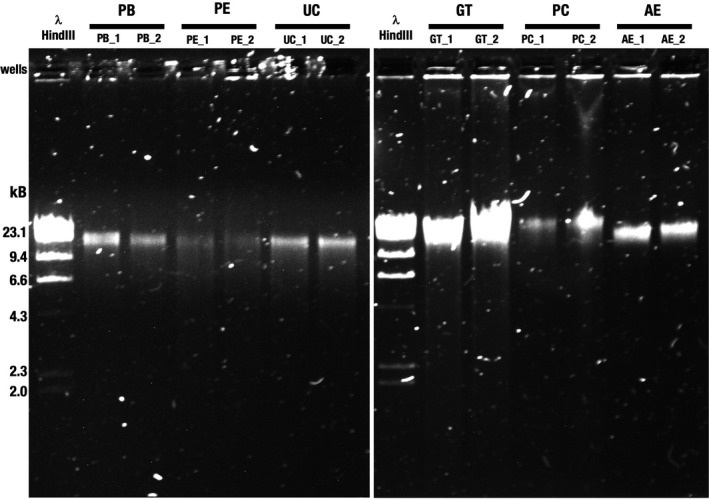
Agarose gel electrophoresis of genomic DNA isolated from a pool of tongue dorsum samples. Genomic DNA was electrophoresed on a 0.8% (w/v) agarose gel. PB, PE and UC with replicates (22 ng input, left panel) and GT, PC and AE replicates (44 ng input, right panel) are shown. Different DNA inputs were used based on overall sample availability. λ‐HindIII, Lambda DNA, digested with the restriction endonuclease HindIII, was used to assess fragment size distribution. PB, DNeasy PowerSoil with modified bead beating; PE, DNeasy PowerSoil with enzymatic treatment; UC, DNeasy UltraClean Microbial Kit; GT, Qiagen Genomic Tip 20/G with enzymatic treatment; PC, phenol–chloroform; AE, agarose encasement. The designations “_1” and “_2” indicate replicate 1 and replicate 2, respectively
Low DNA yield can result in sample loss during library preparation or low pore occupancy on MinION during sequencing. While working with samples of low DNA yield is inevitable, spiking in known DNA (such as lambda DNA) to “pad” samples with low DNA yields may be used to start sequencing, as we demonstrated previously (Reveillaud et al., 2019). However, to minimize the need for additional DNA to “pad” samples with low DNA yield due to the extraction protocol, we eliminated protocols PB, PE and UC from any further evaluation as they consistently resulted in mediocre DNA yield.
Next, we sequenced the DNA from GT, PC and AE using two MinION flow cells to ensure the replicates of the same protocol were sequenced on different runs (Run 1: GT_1, PC_1 and AE_1; Run 2: GT_2, PC_2 and AE_2). The sequencing runs generated 4.84 and 7.79 Gbp, respectively, which were within the expected range of MinION sequencing output (Cusco et al., 2020; Moss et al., 2020). The increase in the output in the second run could be attributed to less sample loss during library preparation and subsequent input of three times more DNA (142.8 vs. 466.2 ng) into the flow cell. After performing a quality filtering step (using a minimum Q‐score of 7), the percentage of reads passing the quality check (Pass_Reads) was similar between extraction methods within each flow cell (96% and 93%–94% respectively, Table S1a). However, a comparison between runs demonstrated that a higher percentage of sequences were removed (Fail_Reads) in the second run (Table S1a). The total number of nucleotides was comparable between GT and PC within sequencing runs (1,699,213,259 and 1,641,389,967 on average, respectively), and was much smaller for AE (949,615,901 on average) in both runs.
3.2. Eukaryotic contamination is enriched in the pool of shorter fragments
Samples from the human oral cavity, prepared for metagenomic sequencing, are typically associated with extensive eukaryotic contamination, which can account for up to 45% of the short‐read sequencing product (Shaiber et al., 2020). Therefore, we assessed the amount of host contamination in each DNA sample by mapping reads to a reference human genome from the NCBI (GRCh38). Host DNA contamination was high for all protocols. AE had the least amount of human DNA (on average, 63%) compared to GT (on average, 75%) and PC (on average, 81%) (Table 2). This trend persisted when comparing their cumulative sequence lengths, although our analysis of read length distribution showed that the human reads were predominantly composed of shorter fragments (Figure 2, insets). The increased representation of human DNA in PC compared to other extraction methods is probably due to the use of detergents vs. enzymes. Lytic detergents exert their effect on both bacterial and eukaryotic cells, while lytic enzymes target bacterial cells only. PC lacked lytic enzymes and included SDS, a stronger lytic detergent than those used in GT (Tween 20, Triton X‐100) and AE (SLS, Brij, deoxycholate).
TABLE 2.
The impact of HMW DNA extraction protocol on proportional read numbers and sequence lengths according to read type (microbial vs. human)
| Qiagen Genomic Tip (GT) | Phenol–chloroform (PC) | Agarose encasement (AE) | ||||
|---|---|---|---|---|---|---|
| GT_1 | GT_2 | PC_1 | PC_2 | AE_1 | AE_2 | |
| All reads | ||||||
| Number of reads | 2,052,300 | 3,681,083 | 2,008,795 | 4,338,575 | 739,186 | 962,065 |
| Sequencing yield (Gbp) | 1.440 | 1.959 | 1.311 | 1.972 | 0.767 | 1.133 |
| Human reads | ||||||
| Number of reads | 1,584,229 | 2,674,013 | 1,664,447 | 3,433,184 | 490,377 | 585,733 |
| % | 77.19 | 72.64 | 82.86 | 79.13 | 66.34 | 60.88 |
| Sequencing yield (Gbp) | 0.908 | 1.199 | 1.026 | 1.536 | 0.405 | 0.463 |
| % | 63.07 | 61.23 | 78.26 | 77.88 | 52.79 | 40.91 |
| Microbial reads | ||||||
| Number of reads | 468,071 | 1,007,070 | 344,348 | 905,391 | 248,809 | 376,332 |
| % | 22.81 | 27.36 | 17.14 | 20.87 | 33.66 | 39.12 |
| Sequencing yield (Gbp) | 0.532 | 0.759 | 0.285 | 0.436 | 0.362 | 0.669 |
| % | 36.93 | 38.77 | 21.74 | 22.12 | 47.21 | 59.09 |
FIGURE 2.
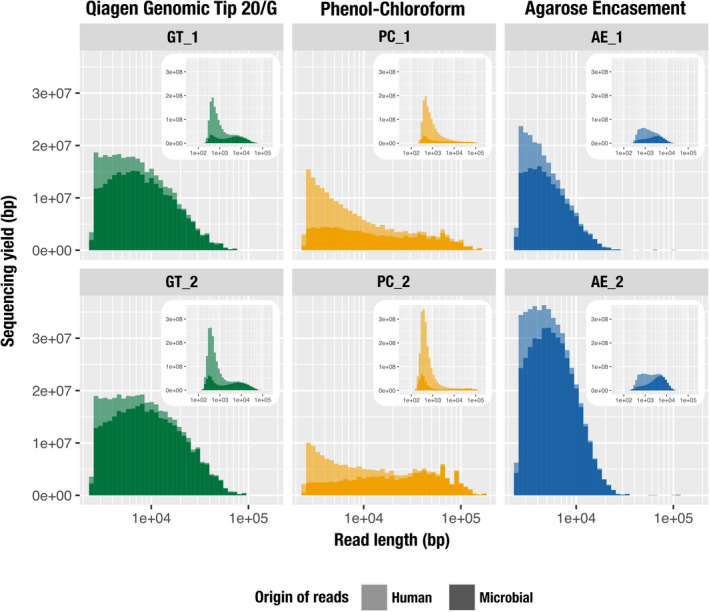
The impact of DNA extraction protocol on the distribution of human (lighter colour) and microbial (darker colour) read lengths from MinION sequencing. These histograms visualize the total accumulative length (total number of nucleotides sequenced) per range of individual read lengths. The x‐axis represents the read length on a log scale and the y‐axis represents the cumulative length for a given size bin (bar width). The main panel shows the size distribution of reads >2500 bp for GT (green), PC (yellow) and AE (blue) while the inset panel shows the size distribution of all reads, using the same data. Results are outlined vertically by extraction method (replicate 1, top panel; replicate 2, lower panel)
3.3. Read size distribution is not uniform across extraction methods
After the removal of sequences that match the human genome, we assumed that the vast majority of the sequences were of microbial origin. We further focused on sequences that were longer than 2500 nucleotides, and quantified the number of reads and their length distribution across GT, PC and AE (Table 3). Overall, we observed a size distribution that was coherent with other long‐read metagenomics studies, with reads reaching 100,000 bp (Moss et al., 2020; Somerville et al., 2019).
TABLE 3.
Microbial read size distribution. All percentages are relative to the total reads (or sequencing yield) of the quality filtered reads, prior to removal of human reads
| Qiagen Genomic Tip (GT) | Phenol–chloroform (PC) | Agarose encasement (AE) | ||||
|---|---|---|---|---|---|---|
| GT_1 | GT_2 | PC_1 | PC_2 | AE_1 | AE_2 | |
| All reads | ||||||
| Number of reads | 2,052,300 | 3,681,083 | 2,008,795 | 4,338,575 | 739,186 | 962,065 |
| Sequencing yield (Gbp) | 1.440 | 1.959 | 1.311 | 1.972 | 0.767 | 1.133 |
| All microbial reads | ||||||
| Number of reads | 468,071 | 1,007,070 | 344,348 | 905,391 | 248,809 | 376,332 |
| % | 22.81 | 27.36 | 17.14 | 20.87 | 33.66 | 39.12 |
| Sequencing yield (Gbp) | 0.532 | 0.759 | 0.285 | 0.436 | 0.362 | 0.669 |
| % | 36.93 | 38.77 | 21.74 | 22.12 | 47.21 | 59.09 |
| N50 | 2810 | 1929 | 1116 | 449 | 2524 | 3649 |
| L50 | 38,513 | 62,126 | 34,874 | 155,142 | 38,768 | 52,182 |
| Median length (bp) | 468 | 326 | 427 | 307 | 782 | 837 |
| Longest microbial reads (bp) | 73,029 | 90,424 a | 163,320 | 180,460 | 68,189 | 116,730 |
| The top hit for the longest microbial read on NCBI’s nr database (identity/alignment) | Streptococcus salivarius (93.6%/99%) | Streptococcus salivarius (92%/98%) a | Streptococcus sp. (89.9%/81%) | Veillonella dispar (88.7%/69%) | Veillonella nakazawae (90.2%/89%) | Prevotella histicola (92.5%/96%) |
| Microbial reads >2.5 kb | ||||||
| Number of reads | 43,173 | 49,572 | 13,752 | 10,182 | 39,270 | 82,221 |
| % | 2.10 | 1.35 | 0.68 | 0.23 | 5.31 | 8.55 |
| Sequencing yield (Gbp) | 0.278 | 0.352 | 0.109 | 0.117 | 0.182 | 0.426 |
| % | 19.32 | 17.98 | 8.29 | 5.92 | 23.77 | 37.58 |
Showing the second longest read as the first longest read (92,515 bp) had no hits on NCBI.
Our comparison of the five longest reads per replicate revealed that PC produced the longest fragments (130,355–180,460 bp) while GT ranged from 68,275 to 92,515 bp and AE had a single 116,730‐bp read followed by significantly shorter reads (28,218–68,189 bp) (Table 3). The replicates of PC yielded 24 and 38 reads that were over 100,000 bp (Table S1b), but this extraction method was also associated with the smallest N50 score due to the large fraction of shorter reads (Figure 2, insets). The contribution of reads above 2500 bp to the sequencing yield (total number of nucleotides) was greater in GT (mean, 18.65%) and AE (mean, 30.68%), while dropping to 7.11% for PC (Table 3, Figure 2). AE had a few short reads contributing to the sequencing yield, but also lacked long reads as there were only 40 and 186 reads above 20 kbp in the two runs (Table S1b). We find it surprising that AE, an extraction strategy that can yield over a million base pair DNA fragments (Anand, 1986), produced only four reads that were longer than 50 kbp when we used identical DNA mass inputs for all extractions. We speculate that, in the absence of a fragmentation step, the very long fragments AE might have produced may have been lost during library preparation steps or become stuck in the pores during sequencing.
3.4. Size selection has limited utility and leads to substantial loss of biomass
Despite adding the nanopore recommended 1× volume of solid‐phase reversible immobilization (SPRI) beads to reaction mixtures to remove small fragments during library preparation steps, there was a large contribution of shorter reads (<2500 bp) in our initial sequencing effort. Thus, we evaluated additional options to reduce their numbers and increase the representation of longer fragments. We sought to determine the effect of using the BluePippin system on fragment size metrics by utilizing the “high‐pass collection mode,” which enables the collection of DNA molecules above a certain user‐defined size. To maximize sequences that might contain rRNA genes, which are particularly useful for taxonomic assignments (Camanocha & Dewhirst, 2014), we chose 6 kbp as our minimum threshold value. Due to its ample material availability, we used replicates of GT to compare the size‐selected sequencing metrics with data from the previous untreated sequencing runs. High sample loss is a known drawback of BluePippin high‐pass size filtering as the manufacturer warns to expect a loss between 20% and 50% of the sample input. However, we were able to recover only 16%–18% of the 3300 ng of input DNA for each replicate after the size selection step, in agreement with low recovery rates (25%–35%) also reported by others (Schalamun et al., 2019). Even though we started the size selection step with more than three times the amount of DNA than was used for the untreated workflow, our recovery post‐size selection was 550–600 ng, meaning that our sample input into the start of library preparation was half of the DNA input that went into the untreated sequencing run. Strikingly, the number of reads and sequencing yields were reduced by 80%–90% compared to the untreated sample (Table 4; Table S1c), probably as a consequence of the reduced sample input. Other notable shifts included a reduced proportion of human contamination and an increased N50 (Table 4). The number of microbial reads above 2.5 kbp and their cumulative length were comparable for both approaches, which is quite impressive given the low amount of input DNA in the size‐selected samples. However, this parallel did not persist when evaluating longer DNA fragments as the cumulative length of microbial reads above 20 kbp was greater for nonsize‐selected samples. Propelled by vastly reduced read numbers (and despite the superior N50), the size selection step did not result in the substantial improvements we hypothesized in overall read lengths and the cumulative nucleotide sequences. Considering the additional demands on (i) sample requirements, (ii) reagent/personnel costs, and (ii) sample handling and processing times, we consider size selection of this type to have limited utility in this context.
TABLE 4.
Comparison of sequencing run read metrics between untreated and BluePippin size‐selected samples. All percentages are relative to the total reads (or sequencing yield) of the quality filtered reads
| Untreated | BluePippin high‐pass size selection | |||
|---|---|---|---|---|
| GT_1 | GT_2 | GT_1 SS | GT_2 SS | |
| All reads | ||||
| Total reads | 2,052,300 | 3,681,083 | 221,344 | 430,986 |
| Sequencing yield (Gbp) | 1.440 | 1.959 | 0.410 | 0.450 |
| Human reads | ||||
| Number of reads | 1,584,229 | 2,674,013 | 117,071 | 282,113 |
| % | 77.19 | 72.64 | 52.89 | 65.46 |
| Sequencing yield (Gbp) | 0.908 | 1.199 | 0.104 | 0.162 |
| % | 63.07 | 61.23 | 25.31 | 35.92 |
| Microbial reads | ||||
| Number of reads | 468,071 | 1,007,070 | 104,273 | 148,873 |
| % | 22.81 | 27.36 | 47.11 | 34.54 |
| Sequencing yield (Gbp) | 0.532 | 0.759 | 0.306 | 0.289 |
| % | 36.93 | 38.77 | 74.69 | 64.08 |
| N50 | 2810 | 1929 | 6106 | 5594 |
| Microbial reads >2.5 kb | ||||
| Number of reads | 43,173 | 49,572 | 40,248 | 34,949 |
| % | 2.10 | 1.35 | 18.18 | 8.11 |
| Sequencing yield (Gbp) | 0.278 | 0.352 | 0.252 | 0.215 |
| % | 19.32 | 17.98 | 61.26 | 47.74 |
| Microbial reads >20 kb | ||||
| Number of reads | 1269 | 2294 | 300 | 407 |
| % | 0.06 | 0.06 | 0.14 | 0.09 |
| Sequencing yield (Gbp) | 0.035 | 0.067 | 0.008 | 0.011 |
| % | 2.42 | 3.41 | 1.86 | 2.49 |
GT, Qiagen Genomic Tip 20/G with enzymatic treatment; SS size‐selection.
3.5. Extraction method and read length distribution alter taxonomic profiles
Extraction methods may have distinctive biases that can impact the determination of microbial community from metagenomic data, such as a differential ability to lyse Gram‐negative vs. Gram‐positive organisms. To investigate the microbial community composition of our long‐read metagenomes we used bacterial, archaeal and eukaryotic single‐copy core genes (SCGs) and rRNAs. Given the high rate of insertion or deletion errors in nanopore sequencing, predicting genes accurately is a significant challenge in uncorrected reads. While we assumed that these biases would similarly impact all extraction methods and elected to not correct our raw reads, we suggest the use of appropriate bioinformatics tools to correct frame‐shift errors in long‐read sequencing results (Arumugam et al., 2019; Huang et al., 2020).
In agreement with the sequencing yield of bacterial reads across extraction strategies, GT had the most and PC had the fewest number of genes predicted (Table 5). PC also yielded the lowest number of 16S rRNA genes with an average of 123 genes, as compared to GT and AE, which yielded an average of 418 and 398 genes, respectively. Read size distribution played an important role in the recovery of SCGs and rRNA genes: for example, PC_2 and AE_1 had a comparable sequencing yield of microbial reads (436 and 361 Mbp respectively, Table 3) but the numbers of SCGs and rRNA genes were two times larger in AE_1, despite a smaller number of predicted genes. These two extraction strategies differed drastically in their read size distribution (Figure 2) with AE_1 having fewer very long reads (>50,000 bp, Table S1b) but more medium‐sized ones (>2.5 kbp, Table 3). To complement this observation, AE_2 had the most SCGs and rRNA genes out of all extractions and also the most reads above 2.5 kbp (Table 3), despite having very few long reads (only three >50 kbp, Table S1b) and being the second smallest sample in terms of number of reads and sequencing yield before the removal of human reads (Table 3).
TABLE 5.
Results of prodigal gene calling and HMM hits for single‐copy core genes (SCGs) and ribosomal RNAs (rRNAs). Analysis was performed after removal of human‐reads
| Qiagen Genomic Tip (GT) | Phenol–chloroform (PC) | Agarose encasement (AE) | ||||
|---|---|---|---|---|---|---|
| GT_1 | GT_2 | PC_1 | PC_2 | AE_1 | AE_2 | |
| Number of genes | 676,577 | 994,050 | 391,675 | 665,986 | 440,845 | 771,653 |
| Bacterial SCGs | 2893 | 3131 | 1309 | 1210 | 2136 | 3559 |
| rRNAs (per 1000 genes) | 901 (1.33) | 1107 (1.11) | 295 (0.75) | 307 (0.46) | 655 (1.49) | 1355 (1.76) |
| Bacterial 16S rRNA | 373 | 462 | 120 | 125 | 249 | 547 |
We used the HOMD to assign taxonomy to the 16S rRNA genes found in our long reads. The top genera included Prevotella, Rothia, Streptococcus and Veillonella, which are commonly found in oral samples (Mark Welch et al., 2016; Zaura et al., 2009). We observed a comparable taxonomic profile between GT and AE, with Streptococcus, Veillonella, Prevotella and Actinomyces identified as the most abundant genera (Figure 3). PC differed with relatively more Prevotella and Haemophilus (both Gram‐negative bacteria) and less Streptococcus (a Gram‐positive bacterium).
FIGURE 3.
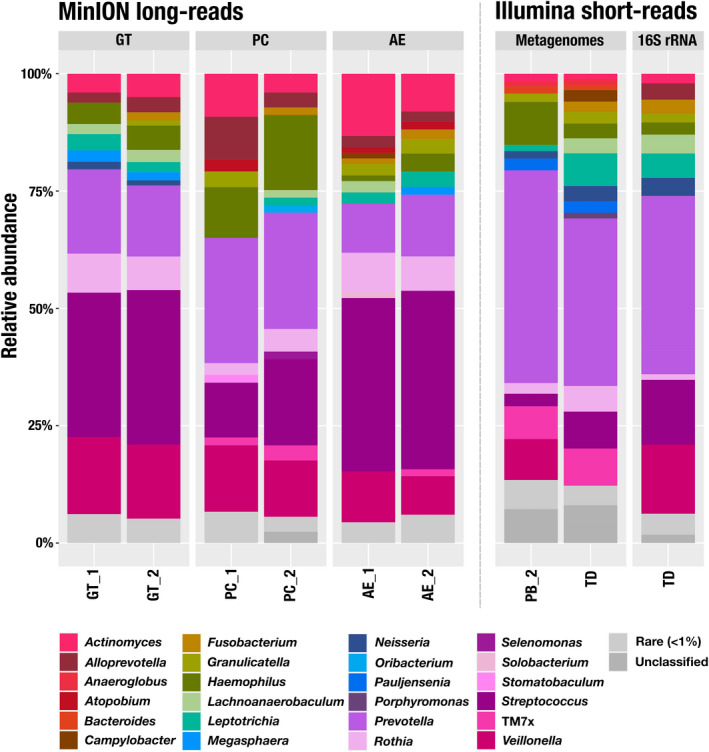
Relative abundance of 16S rRNA at the genus level. We used the Human Oral Microbiome Database (HOMD) to assign taxonomy to the 16S rRNA from the MinION reads. For the short‐read metagenomes, we used the taxonomy of the ribosomal gene S7 with the Genome Taxonomy Database (GTDB). We processed the 16S rRNA amplicons with the Minimum Entropy Decomposition (MED) algorithm and used Silva version 132 to assign taxonomy. Genera representing less than 1% of a sample were pooled as rare (in light grey). Samples noted as TD correspond to an additional sampling performed 2 weeks after the initial pool of samples used for the long‐read extractions. PB, DNeasy PowerSoil with modified bead beating; GT, Qiagen Genomic Tip 20/G with enzymatic treatment; PC, phenol–chloroform; AE, agarose encasement. The designations “_1” and “_2” indicate replicate 1 and replicate 2, respectively
We then compared these profiles to the short‐read sequencing of 16S rRNA gene amplicons found in a sample (TD) collected for the same individual 2 weeks after the initial sampling. While long‐read and amplicon sequencing results were similar to each other qualitatively, the relative abundance estimates between these approaches differed (Figure 3; Table S2). Multiple sources of bias may have contributed to these differences, including the variable detection of rare taxa due to differences in depth of sequencing, differences in sampling time, the use of a single time point for TD vs. the pooling of samples from all other sites, and the use of primers. For instance, our 16S rRNA gene primers did not match to TM7, a prevalent taxon in the human oral cavity, which explains their absence in the amplicon sequencing.
Finally, we compared taxonomic profiles across protocols to the short‐read sequencing of two metagenomes from the same individual generated from (i) the sample used for PB_2 and (ii) the sample used for 16S rRNA gene amplicon sequencing (sample TD). To estimate the relative abundance of taxa in metagenomes, we used short read coverage of single‐copy core genes matching to ribosomal protein S7, which was the most frequently found ribosomal protein in the assemblies of short‐read metagenomes (Table S3). While the genus composition was very comparable with long‐read metagenomes, we observed a lower relative abundance of Streptococcus and higher relative abundance of TM7, which is probably due to copy number of the rRNA operons in these genera (Stoddard et al., 2015) that skew the relative abundance estimates based on short‐read amplicons and long‐read metagenomes. We also investigated the distribution and taxonomic assignment of ribosomal proteins in long‐read metagenomes (Table S3, Figure S1), but the relatively low sequencing depth of these data and challenges associated with gene calling in uncorrected MinION sequences prevented reliable insights.
Overall, these results highlight (i) the differences observed between extraction methods (i.e., Gram‐negative/positive biases), but also (ii) the critical role that the read size distribution has on the recovery of SCGs, rRNAs and, consequently, its impact on taxonomic profiling.
3.6. Read size distribution dramatically influences assembly outcomes
We finally compared different extraction methods by assembling microbial reads using a long‐read assembler, flye (Kolmogorov et al., 2020). We also used short reads generated with PB_2 to polish the assemblies using pilon (Walker et al., 2014). GT and AE resulted in larger assemblies with an average size of 31,518,599 and 28,354,223 bp respectively, while the average assembly size of PC was much smaller at 11,269,952 bp (Table 6). The extraction method GT stood out for its high N50, but also by having the longest contig assembled (1,176,789 bp) and the greatest number of contigs above 100 kbp. The number of predicted genes was directly related to the assembly size. For that reason, GT had on average more genes predicted and more SCGs and rRNAs, which led to more bacterial genomes expected in the assembly (based on the most frequently occurring count of SCG hits). The assembly step did not result in any circular bacterial chromosomes but there were a few circular plasmids and phage genomes. PC had the fewest circular contigs (Table 6), and while AE had more, they were shorter. GT stood out again with the most circular contigs.
TABLE 6.
flye assembly statistics. Assemblies were polished using pilon and short‐reads from the extraction PB_2
| Qiagen Genomic Tip (GT) | Phenol–chloroform (PC) | Agarose encasement (AE) | ||||
|---|---|---|---|---|---|---|
| GT_1 | GT_2 | PC_1 | PC_2 | AE_1 | AE_2 | |
| Total length (bp) | 28,002,213 | 35,034,984 | 10,630,822 | 11,909,082 | 17,557,138 | 39,151,308 |
| Number of contigs | 401 | 466 | 137 | 106 | 483 | 855 |
| No.of contigs >5 kb | 369 | 412 | 130 | 101 | 467 | 775 |
| No. of contigs >10 kb | 347 | 383 | 124 | 100 | 440 | 714 |
| No. of contigs >20 kb | 299 | 336 | 107 | 97 | 354 | 564 |
| No. of contigs >50 kb | 159 | 194 | 73 | 67 | 84 | 203 |
| No. of contigs >100 kb | 63 | 84 | 33 | 35 | 21 | 69 |
| Longest contig (bp) | 1,025,627 | 1,176,789 | 504,505 | 676,998 | 304,763 | 875,613 |
| Shortest contig (bp) | 512 | 528 | 2103 | 1025 | 913 | 511 |
| N50 (bp) | 129,677 | 155,366 | 122,828 | 166,496 | 44,461 | 71,755 |
| Number of genes | 32,540 | 41,818 | 11,628 | 13,025 | 21,392 | 48,784 |
| Single‐copy core genes | ||||||
| Bacteria_71 | 845 | 1072 | 298 | 339 | 551 | 1163 |
| Archaea_76 | 428 | 554 | 139 | 167 | 283 | 614 |
| Protista_83 | 34 | 48 | 11 | 14 | 26 | 54 |
| Ribosomal RNAs | 75 | 116 | 30 | 43 | 44 | 100 |
| No. of expected bacterial genome | 12 | 12 | 5 | 5 | 7 | 15 |
| Circular contigs | ||||||
| Number of circular contigs | 20 | 32 | 8 | 5 | 9 | 24 |
| Max. length | 86,329 | 155,366 | 155,422 | 155,411 | 24,494 | 88,098 |
4. DISCUSSION
Prior to undertaking this evaluation, we established two key conditions that a successful HMW DNA extraction method to study host‐associated environments with low microbial biomass would need to fulfil: (i) optimizing a read length distribution profile to improve downstream sequence analyses and (ii) minimizing the proportion of host‐associated DNA contamination. While the utility of minimizing nontarget DNA in sequencing libraries is relatively obvious from a resource conservancy point of view, the impact of the read length distribution on studies of metagenomes may escape attention when a sequencing strategy focuses on obtaining the longest possible reads. Based on the assembly of 2267 genomes and the average length of rDNA operons, Koren et al. (2013) had suggested 7 kbp as the ideal read length that could span through most repeats for dramatic improvements in genome assembly. The modified column‐based extraction method (GT) consistently yielded the greatest number of reads over 10 kbp and led to most successful downstream assemblies, as indicated by the total size, the length of longest contigs and the number of circularized elements.
The representation of host DNA contamination varied between the extraction methods, and reached its maximum in the phenol–chloroform extraction (PC). As a consequence, the phenol–chloroform extraction resulted in fewer microbial reads for an equal amount of DNA input (Table S2). While host DNA was enriched in the pool of shorter sequences and thus could be eliminated through size selection, our analyses revealed a high cost for this step as size selection removed many of the very long reads (>60 kbp) and required three times more DNA as input. Size selection can also be performed using SPRI, which holds great potential for improvements on read‐size distributions. Indeed, a recent analysis of diverse and customized SPRI formulations demonstrated significant increases in the sizing threshold of SPRI beads from 150–800 bp to 1.5–7 kbp (Stortchevoi et al., 2020). The modified PowerSoil kit methods resulted in low DNA yield, which was probably due to our modifications of the manufacturer protocols regarding the bead beating step. Extraction methods that rely on mechanical sample lysis will probably result in much lower yields when modified to diverge from the manufacturer's guidelines to maximize the recovery of HMW DNA. A unique advantage of nanopore sequencing platforms is the real‐time access to the product that is being sequenced, which promotes new methods for reference‐based identification of nontarget DNA molecules and their real‐time rejection from an active pore to minimize their impact on sequencing results (Kovaka et al., 2020). Maximizing the input DNA quality, and the use of novel assembly algorithms (Kolmogorov et al., 2020; Koren et al., 2017; Li, 2016; Ruan & Li, 2020) and effective polishing strategies (Loman et al., 2015; Morisse et al., 2021; Vaser et al., 2017; Walker et al., 2014), will lead to new insights in metagenomics especially with complete circular MAGs.
AUTHOR CONTRIBUTIONS
F.T., K.L. and A.M.E. conceived the study. K.L. developed and implemented long‐read sequencing protocols. F.T. performed analyses of short‐ and long‐read sequencing data. K.L., E.F. and A.S. helped with data analysis. H.G.M. performed short‐read sequencing. H.G.M., L.B., B.J. and A.M.E. supervised research. F.T., K.L. and A.M.E. wrote the paper with critical input from all authors.
OPEN RESEARCH BADGES
This article has earned an Open Data Badge for making publicly available the digitally‐shareable data necessary to reproduce the reported results. The reproducible workflow and the data can found at the URL https://merenlab.org/data/hmw‐dna‐extraction‐strategies/.
Supporting information
Fig S1
Table S1
Table S2
Table S3
ACKNOWLEDGEMENTS
We thank Nicole Robichaud for technical help with short‐read sequencing. This research was supported by the Gastro‐Intestinal Research Foundation, the Mutchnik Family Fund, and the Simons Foundation (no. 687269, A.M.E.). In addition, a Helmsley Foundation grant to L.B., B.J. and A.M.E. supported F.T., and an NIH NIDDK grant (RC2 DK122394) supported K.L. and A.M.E.
Trigodet, F. , Lolans, K. , Fogarty, E. , Shaiber, A. , Morrison, H. G. , Barreiro, L. , Jabri, B. , & Eren, A. M. (2022). High molecular weight DNA extraction strategies for long‐read sequencing of complex metagenomes. Molecular Ecology Resources, 22, 1786–1802. 10.1111/1755-0998.13588
Trigodet and Lolans equal contribution.
DATA AVAILABILITY STATEMENT
Raw sequences for long‐ and short‐read sequencing data for shotgun metagenomes and 16S rRNA gene amplicons are available under the BioProject PRJNA703035. We also made available FASTA files and anvi’o contigs databases for (i) assembled and polished long‐read sequences at https://doi.org/10.6084/m9.figshare.14141228, (ii) unassembled long‐read sequences at https://doi.org/10.6084/m9.figshare.14141414, and (iii) assembled shotgun metagenomes at https://doi.org/10.6084/m9.figshare.14141819. Supporting Tables and Figures are also available via https://doi.org/10.6084/m9.figshare.14141918. A reproducible bioinformatics workflow is available at https://merenlab.org/data/hmw‐dna‐extraction‐strategies/, where https://github.com/merenlab/web/tree/master/data/hmw‐dna‐extraction‐strategies and the commit hash 557f6e37381d212a22a17d67c7b0e774e5abee82 gives access to its final version at the time of publication.
REFERENCES
- Al‐Shayeb, B. , Sachdeva, R. , Chen, L.‐X. , Ward, F. , Munk, P. , Devoto, A. , Castelle, C. J. , Olm, M. R. , Bouma‐Gregson, K. , Amano, Y. , He, C. , Méheust, R. , Brooks, B. , Thomas, A. , Lavy, A. , Matheus‐Carnevali, P. , Sun, C. , Goltsman, D. S. A. , Borton, M. A. , … Banfield, J. F. (2020). Clades of huge phages from across Earth’s ecosystems. Nature, 578(7795), 425–431. 10.1038/s41586-020-2007-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand, R. (1986). Pulsed field gel electrophoresis: a technique for fractionating large DNA molecules. Trends in Genetics: TIG, 2, 278–283. 10.1016/0168-9525(86)90269-6 [DOI] [Google Scholar]
- Arumugam, K. , Bağcı, C. , Bessarab, I. , Beier, S. , Buchfink, B. , Górska, A. , Qiu, G. , Huson, D. H. , & Williams, R. B. H. (2019). Annotated bacterial chromosomes from frame‐shift‐corrected long‐read metagenomic data. Microbiome, 7(1), 61. 10.1186/s40168-019-0665-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayling, M. , Clark, M. D. , & Leggett, R. M. (2020). New approaches for metagenome assembly with short reads. Briefings in Bioinformatics, 21(2), 584–594. 10.1093/bib/bbz020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan, B. J. , McMurdie, P. J. , Rosen, M. J. , Han, A. W. , Johnson, A. J. A. , & Holmes, S. P. (2016). DADA2: High‐resolution sample inference from Illumina amplicon data. Nature Methods, 13(7), 581–583. 10.1038/nmeth.3869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camanocha, A. , & Dewhirst, F. E. (2014). Host‐associated bacterial taxa from Chlorobi, Chloroflexi, GN02, Synergistetes, SR1, TM7, and WPS‐2 Phyla/candidate divisions. Journal of Oral Microbiology, 6, 10.3402/jom.v6.25468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, L. X. , Anantharaman, K. , Shaiber, A. , Murat Eren, A. , & Banfield, J. F. (2020). Accurate and complete genomes from metagenomes. Genome Research, 30(3), 315–333. 10.1101/gr.258640.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cicha, C. , Hedges, J. , Novak, I. , Snyder, D. , Jutila, M. , & Wiedenheft, B. (2020). Complete genome sequence of Brucella abortus Phage EF4, determined using long‐read sequencing. Microbiology Resource Announcements, 9(18), e00212‐20. 10.1128/MRA.00212-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cusco, A. , Perez, D. , Viñes, J. , & Francino, O. (2020). Long‐read metagenomics to retrieve high‐quality metagenome‐assembled genomes from canine feces. Research Square, 22(1), 330. 10.21203/rs.3.rs-60068/v1 [DOI] [Google Scholar]
- Delmont, T. O. , Quince, C. , Shaiber, A. , Esen, Ö. C. , Lee, S. T. , Rappé, M. S. , McLellan, S. L. , Lücker, S. , & Eren, A. M. (2018). Nitrogen‐fixing populations of Planctomycetes and Proteobacteria are abundant in surface ocean metagenomes. Nature Microbiology, 3(7), 804–813. 10.1038/s41564-018-0176-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewhirst, F. E. , Chen, T. , Izard, J. , Paster, B. J. , Tanner, A. C. R. , Yu, W.‐H. , Lakshmanan, A. , & Wade, W. G. (2010). The human oral microbiome. Journal of Bacteriology, 192(19), 5002–5017. 10.1128/JB.00542-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driscoll, C. B. , Otten, T. G. , Brown, N. M. , & Dreher, T. W. (2017). Towards long‐read metagenomics: complete assembly of three novel genomes from bacteria dependent on a diazotrophic cyanobacterium in a freshwater lake co‐culture. Standards in Genomic Sciences, 12, 9. 10.1186/s40793-017-0224-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duran‐Pinedo, A. E. , & Frias‐Lopez, J. (2015). Beyond microbial community composition: functional activities of the oral microbiome in health and disease. Microbes and Infection, 17(7), 505–516. 10.1016/j.micinf.2015.03.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards, A. , Debbonaire, A. R. , Nicholls, S. M. , Rassner, S. M. E. , Sattler, B. , Cook, J. M. , Davy, T. , Soares, A. , Mur, L. A. J. , & Hodson, A. J. (2019). In‐field metagenome and 16S rRNA gene amplicon nanopore sequencing robustly characterize glacier microbiota. bioRxiv, 73965. 10.1101/073965 [DOI] [Google Scholar]
- Eren, A. M. , Kiefl, E. , Shaiber, A. , Veseli, I. , Miller, S. E. , Schechter, M. S. , Fink, I. , Pan, J. N. , Yousef, M. , Fogarty, E. C. , Trigodet, F. , Watson, A. R. , Esen, Ö. C. , Moore, R. M. , Clayssen, Q. , Lee, M. D. , Kivenson, V. , Graham, E. D. , Merrill, B. D. , Karkman, A. , … Willis, A. D. (2021). Community‐led, integrated, reproducible multi‐omics with anvi'o. Nature microbiology, 6(1), 3–6. 10.1038/s41564-020-00834-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eren, A. M. , Morrison, H. G. , Lescault, P. J. , Reveillaud, J. , Vineis, J. H. , & Sogin, M. L. (2015). Minimum entropy decomposition: Unsupervised oligotyping for sensitive partitioning of high‐throughput marker gene sequences. The ISME Journal, 9(4), 968–979. 10.1038/ismej.2014.195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory, A. C. , Zablocki, O. , Zayed, A. A. , Howell, A. , Bolduc, B. , & Sullivan, M. B. (2020). The gut virome database reveals age‐dependent patterns of virome diversity in the human gut. Cell Host & Microbe, 28(5), 724–740. 10.1101/655910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory, A. C. , Zayed, A. A. , Conceição‐Neto, N. , Temperton, B. , Bolduc, B. , Alberti, A. , Ardyna, M. , Arkhipova, K. , Carmichael, M. , Cruaud, C. , Dimier, C. , Domínguez‐Huerta, G. , Ferland, J. , Kandels, S. , Liu, Y. , Marec, C. , Pesant, S. , Picheral, M. , Pisarev, S. , … Wincker, P. (2019). Marine DNA viral macro‐ and microdiversity from pole to pole. Cell, 177(5), 1109–1123. 10.1016/j.cell.2019.03.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosoe, A. , Suenaga, T. , Sugi, T. , Iizumi, T. , Nagai, N. , & Terada, A. (2020). Complete Genome Sequence of Pseudomonas putida Strain TS312, Harboring an HdtS‐Type N‐Acyl‐Homoserine Lactone Synthase, Isolated from a Paper Mill. Microbiology Resource Announcements, 9(13), e00055‐20. 10.1128/MRA.00055-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, Y.‐T. , Liu, P.‐Y. , & Shih, P.‐W. (2020). High‐quality genomes of nanopore sequencing by homologous polishing. Cold Spring Harbor Laboratory; (p. 2020.09.19.304949). 10.1101/2020.09.19.304949 [DOI] [Google Scholar]
- Hug, L. A. , Baker, B. J. , Anantharaman, K. , Brown, C. T. , Probst, A. J. , Castelle, C. J. , Butterfield, C. N. , Hernsdorf, A. W. , Amano, Y. , Ise, K. , Suzuki, Y. , Dudek, N. , Relman, D. A. , Finstad, K. M. , Amundson, R. , Thomas, B. C. , & Banfield, J. F. (2016). A new view of the tree of life. Nature Microbiology, 1, 16048. 10.1038/nmicrobiol.2016.48 [DOI] [PubMed] [Google Scholar]
- Human Microbiome Project Consortium (2012). Structure, function and diversity of the healthy human microbiome. Nature, 486(7402), 207–214. 10.1038/nature11234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyatt, D. , Chen, G.‐L. , Locascio, P. F. , Land, M. L. , Larimer, F. W. , & Hauser, L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics, 11, 119. 10.1186/1471-2105-11-119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasianowicz, J. J. , Brandin, E. , Branton, D. , & Deamer, D. W. (1996). Characterization of individual polynucleotide molecules using a membrane channel. Proceedings of the National Academy of Sciences of the United States of America, 93(24), 13770–13773. 10.1073/pnas.93.24.13770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita, Y. , Niwa, H. , Uchida‐Fujii, E. , & Nukada, T. (2020). Complete genome sequence of mycoplasma felis Strain Myco‐2, isolated from an equine tracheal wash sample in Japan. Microbiology Resource Announcements, 9(9), e00057‐20. 10.1128/MRA.00057-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolmogorov, M. , Bickhart, D. M. , Behsaz, B. , Gurevich, A. , Rayko, M. , Shin, S. B. , Kuhn, K. , Yuan, J. , Polevikov, E. , Smith, T. P. L. , & Pevzner, P. A. (2020). metaFlye: scalable long‐read metagenome assembly using repeat graphs. Nature Methods, 17(11), 1103–1110. 10.1038/s41592-020-00971-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolmogorov, M. , Bickhart, D. M. , Behsaz, B. , Gurevich, A. , Rayko, M. , Shin, S. B. , Kuhn, K. , Yuan, J. , Polevikov, E. , Smith, T. P. L. , & Pevzner, P. A. (2020). metaFlye: scalable long‐read metagenome assembly using repeat graphs. Nature Methods, 17(11), 1103–1110. 10.1101/637637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren, S. , Harhay, G. P. , Smith, T. P. L. , Bono, J. L. , Harhay, D. M. , Mcvey, S. D. , Radune, D. , Bergman, N. H. , & Phillippy, A. M. (2013). Reducing assembly complexity of microbial genomes with single‐molecule sequencing. Genome Biology, 14(9), R101. 10.1186/gb-2013-14-9-r101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren, S. , Walenz, B. P. , Berlin, K. , Miller, J. R. , Bergman, N. H. , & Phillippy, A. M. (2017). Canu: scalable and accurate long‐read assembly via adaptive k‐mer weighting and repeat separation. Genome Research, 27(5), 722–736. 10.1101/gr.215087.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köster, J. , & Rahmann, S. (2012). Snakemake–a scalable bioinformatics workflow engine. Bioinformatics, 28(19), 2520–2522. 10.1093/bioinformatics/bts480 [DOI] [PubMed] [Google Scholar]
- Kovaka, S. , Fan, Y. , Ni, B. , Timp, W. , & Schatz, M. C. (2020). Targeted nanopore sequencing by real‐time mapping of raw electrical signal with UNCALLED. Nature Biotechnology, 39(4), 431–441. 10.1038/s41587-020-0731-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead, B. , & Salzberg, S. L. (2012). Fast gapped‐read alignment with Bowtie 2. Nature Methods, 9(4), 357–359. 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, M. D. (2019). GToTree: a user‐friendly workflow for phylogenomics. Bioinformatics, 35(20), 4162–4164. 10.1093/bioinformatics/btz188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. (2016). Minimap and miniasm: fast mapping and de novo assembly for noisy long sequences. Bioinformatics, 32(14), 2103–2110. 10.1093/bioinformatics/btw152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. (2018). Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics, 34(18), 3094–3100. 10.1093/bioinformatics/bty191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , Handsaker, B. , Wysoker, A. , Fennell, T. , Ruan, J. , Homer, N. , Marth, G. , Abecasis, G. , & Durbin, R. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics, 25(16), 2078–2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loman, N. J. , Quick, J. , & Simpson, J. T. (2015). A complete bacterial genome assembled de novo using only nanopore sequencing data. Nature Methods, 12(8), 733–735. 10.1038/nmeth.3444 [DOI] [PubMed] [Google Scholar]
- Maghini, D. G. , Moss, E. L. , Vance, S. E. , & Bhatt, A. S. (2020). Improved high‐molecular‐weight DNA extraction, nanopore sequencing and metagenomic assembly from the human gut microbiome. Nature Protocols, 16(1), 458–471. 10.1038/s41596-020-00424-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mark Welch, J. L. , Rossetti, B. J. , Rieken, C. W. , Dewhirst, F. E. , & Borisy, G. G. (2016). Biogeography of a human oral microbiome at the micron scale. Proceedings of the National Academy of Sciences of the United States of America, 113(6), E791–E800. 10.1073/pnas.1522149113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marotz, C. A. , Sanders, J. G. , Zuniga, C. , Zaramela, L. S. , Knight, R. , & Zengler, K. (2018). Improving saliva shotgun metagenomics by chemical host DNA depletion. Microbiome, 6(1), 42. 10.1186/s40168-018-0426-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matushek, M. G. , Bonten, M. J. , & Hayden, M. K. (1996). Rapid preparation of bacterial DNA for pulsed‐field gel electrophoresis. Journal of Clinical Microbiology, 34(10), 2598–2600. https://www.ncbi.nlm.nih.gov/pubmed/8880529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minoche, A. E. , Dohm, J. C. , & Himmelbauer, H. (2011). Evaluation of genomic high‐throughput sequencing data generated on Illumina HiSeq and Genome Analyzer systems. Genome Biology, 12(11), R112. 10.1186/gb-2011-12-11-r112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morisse, P. , Marchet, C. , Limasset, A. , Lecroq, T. , & Lefebvre, A. (2021). Scalable long read self‐correction and assembly polishing with multiple sequence alignment. Scientific Reports, 11(1), 761. 10.1038/s41598-020-80757-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss, E. L. , Maghini, D. G. , & Bhatt, A. S. (2020). Complete, closed bacterial genomes from microbiomes using nanopore sequencing. Nature Biotechnology, 38(6), 701–707. 10.1038/s41587-020-0422-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesme, J. , Achouak, W. , Agathos, S. N. , Bailey, M. , Baldrian, P. , Brunel, D. , Frostegård, Å. , Heulin, T. , Jansson, J. K. , Jurkevitch, E. , Kruus, K. L. , Kowalchuk, G. A. , Lagares, A. , Lappin‐Scott, H. M. , Lemanceau, P. , Le Paslier, D. , Mandic‐Mulec, I. , Murrell, J. C. , Myrold, D. D. , … Simonet, P. (2016). Back to the future of soil metagenomics. Frontiers in Microbiology, 7, 73. 10.3389/fmicb.2016.00073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls, S. M. , Quick, J. C. , Tang, S. , & Loman, N. J. (2019). Ultra‐deep, long‐read nanopore sequencing of mock microbial community standards. GigaScience, 8(5), giz043. 10.1093/gigascience/giz043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurk, S. , Meleshko, D. , Korobeynikov, A. , & Pevzner, P. A. (2017). metaSPAdes: a new versatile metagenomic assembler. Genome Research, 27(5), 824–834. 10.1101/gr.213959.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson, N. D. , Treangen, T. J. , Hill, C. M. , Cepeda‐Espinoza, V. , Ghurye, J. , Koren, S. , & Pop, M. (2019). Metagenomic assembly through the lens of validation: recent advances in assessing and improving the quality of genomes assembled from metagenomes. Briefings in Bioinformatics, 20(4), 1140–1150. 10.1093/bib/bbx098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks, D. H. , Chuvochina, M. , Chaumeil, P.‐A. , Rinke, C. , Mussig, A. J. , & Hugenholtz, P. (2020). A complete domain‐to‐species taxonomy for Bacteria and Archaea. Nature Biotechnology, 38(9), 1079–1086. 10.1038/s41587-020-0501-8 [DOI] [PubMed] [Google Scholar]
- Pasolli, E. , Asnicar, F. , Manara, S. , Zolfo, M. , Karcher, N. , Armanini, F. , Beghini, F. , Manghi, P. , Tett, A. , Ghensi, P. , Collado, M. C. , Rice, B. L. , DuLong, C. , Morgan, X. C. , Golden, C. D. , Quince, C. , Huttenhower, C. , & Segata, N. (2019). Extensive unexplored human microbiome diversity revealed by over 150,000 genomes from metagenomes spanning age, geography, and lifestyle. Cell, 176(3), 649–662. 10.1016/j.cell.2019.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng, Y. , Leung, H. C. M. , Yiu, S. M. , & Chin, F. Y. L. (2012). IDBA‐UD: a de novo assembler for single‐cell and metagenomic sequencing data with highly uneven depth. Bioinformatics, 28(11), 1420–1428. 10.1093/bioinformatics/bts174 [DOI] [PubMed] [Google Scholar]
- Pessi, I. S. , Viitamäki, S. , Eronen‐Rasimus, E. , Delmont, T. O. , Luoto, M. , & Hultman, J. (2020). Truncated denitrifiers dominate the denitrification pathway in tundra soil metagenomes (p. 2020.12.21.419267). Cold Spring Harbor Laboratory. 10.1101/2020.12.21.419267 [DOI] [Google Scholar]
- Quast, C. , Pruesse, E. , Yilmaz, P. , Gerken, J. , Schweer, T. , Yarza, P. , Peplies, J. , & Glöckner, F. O. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web‐based tools. Nucleic Acids Research, 41(D1), D590–D596. 10.1093/nar/gks1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quick, J. , & Loman, N. J. (2018). DNA extraction strategies for nanopore sequencing. In Nanopore Sequencing (pp. 91–105). World Scientific. 10.1142/9789813270619_0007 [DOI] [Google Scholar]
- Reveillaud, J. , Bordenstein, S. R. , Cruaud, C. , Shaiber, A. , Esen, Ö. C. , Weill, M. , Makoundou, P. , Lolans, K. , Watson, A. R. , Rakotoarivony, I. , Bordenstein, S. R. , & Eren, A. M. (2019). The Wolbachia mobilome in Culex pipiens includes a putative plasmid. Nature Communications, 10(1), 1051. 10.1038/s41467-019-08973-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rognes, T. , Flouri, T. , Nichols, B. , Quince, C. , & Mahé, F. (2016). VSEARCH: a versatile open source tool for metagenomics. PeerJ, 4, e2584. 10.7717/peerj.2584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan, J. , & Li, H. (2020). Fast and accurate long‐read assembly with wtdbg2. Nature Methods, 17(2), 155–158. 10.1038/s41592-019-0669-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook, J. , Fritsch, E. F. , & Maniatis, T. (1989). Molecular cloning: a laboratory manual. In Molecular cloning: A laboratory manual. Cold Spring Harbor Laboratory Press. https://www.cabdirect.org/cabdirect/abstract/19901616061 [Google Scholar]
- Sanderson, N. D. , Street, T. L. , Foster, D. , Swann, J. , Atkins, B. L. , Brent, A. J. , McNally, M. A. , Oakley, S. , Taylor, A. , Peto, T. E. A. , Crook, D. W. , & Eyre, D. W. (2018). Real‐time analysis of nanopore‐based metagenomic sequencing from infected orthopaedic devices. BMC Genomics, 19(1), 714. 10.1186/s12864-018-5094-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schalamun, M. , Nagar, R. , Kainer, D. , Beavan, E. , Eccles, D. , Rathjen, J. P. , Lanfear, R. , & Schwessinger, B. (2019). Harnessing the MinION: An example of how to establish long‐read sequencing in a laboratory using challenging plant tissue from Eucalyptus pauciflora. Molecular Ecology Resources, 19(1), 77–89. 10.1111/1755-0998.12938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaiber, A. , & Eren, A. M. (2019). Composite metagenome‐assembled genomes reduce the quality of public genome repositories. MBio, 10(3), e00725‐19. 10.1128/mBio.00725-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaiber, A. , Willis, A. D. , Delmont, T. O. , Roux, S. , Chen, L.‐X. , Schmid, A. C. , Yousef, M. , Watson, A. R. , Lolans, K. , Esen, Ö. C. , Lee, S. T. M. , Downey, N. , Morrison, H. G. , Dewhirst, F. E. , Mark Welch, J. L. , & Eren, A. M. (2020). Functional and genetic markers of niche partitioning among enigmatic members of the human oral microbiome. Genome Biology, 21(1), 292. 10.1186/s13059-020-02195-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton, C. M. , Petriglieri, F. , Kristensen, J. M. , Kirkegaard, R. H. , Michaelsen, T. Y. , Andersen, M. H. , Kondrotaite, Z. , Karst, S. M. , Dueholm, M. S. , Nielsen, P. H. , & Albertsen, M. (2021). Connecting structure to function with the recovery of over 1000 high‐quality metagenome‐assembled genomes from activated sludge using long‐read sequencing. Nature Communications, 12(1), 2009. 10.1038/s41467-021-22203-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somerville, V. , Lutz, S. , Schmid, M. , Frei, D. , Moser, A. , Irmler, S. , Frey, J. E. , & Ahrens, C. H. (2019). Long‐read based de novo assembly of low‐complexity metagenome samples results in finished genomes and reveals insights into strain diversity and an active phage system. BMC Microbiology, 19(1), 143. 10.1186/s12866-019-1500-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spang, A. , Saw, J. H. , Jørgensen, S. L. , Zaremba‐Niedzwiedzka, K. , Martijn, J. , Lind, A. E. , van Eijk, R. , Schleper, C. , Guy, L. , & Ettema, T. J. G. (2015). Complex archaea that bridge the gap between prokaryotes and eukaryotes. Nature, 521(7551), 173–179. 10.1038/nature14447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoddard, S. F. , Smith, B. J. , Hein, R. , Roller, B. R. K. , & Schmidt, T. M. (2015). rrnDB: improved tools for interpreting rRNA gene abundance in bacteria and archaea and a new foundation for future development. Nucleic Acids Research, 43(D1), D593–D598. 10.1093/nar/gku1201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stortchevoi, A. , Kamelamela, N. , & Levine, S. S. (2020). SPRI Beads‐based Size Selection in the Range of 2–10kb. Journal of Biomolecular Techniques, 31(1), 7–10. 10.7171/jbt.20-3101-002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunagawa, S. , Coelho, L. P. , Chaffron, S. , Kultima, J. R. , Labadie, K. , Salazar, G. , Djahanschiri, B. , Zeller, G. , Mende, D. R. , Alberti, A. , Cornejo‐Castillo, F. M. , Costea, P. I. , Cruaud, C. , d'Ovidio, F. , Engelen, S. , Ferrera, I. , Gasol, J. M. , Guidi, L. , Hildebrand, F. , … Velayoudon, D. (2015). Ocean plankton. Structure and function of the global ocean microbiome. Science, 348(6237), 1261359. 10.1126/science.1261359 [DOI] [PubMed] [Google Scholar]
- Takeshita, K. , Jang, S. , & Kikuchi, Y. (2020). Complete genome sequence of Burkholderia sp. strain THE68, a bacterial symbiont isolated from midgut crypts of the seed bug togo hemipterus. Microbiology Resource Announcements, 9(10), e00041‐20. 10.1128/MRA.00041-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The long view on sequencing (2018). Nature Biotechnology, 36(4), 287. 10.1038/nbt.4125 [DOI] [PubMed] [Google Scholar]
- Tippelt, A. , Busche, T. , Rückert, C. , & Nett, M. (2020). Complete Genome Sequence of the Cryptophycin‐Producing Cyanobacterium Nostoc sp. Strain ATCC 53789. Microbiology Resource Announcements, 9(14), e00040‐20. 10.1128/MRA.00040-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyson, G. W. , Chapman, J. , Hugenholtz, P. , Allen, E. E. , Ram, R. J. , Richardson, P. M. , Solovyev, V. V. , Rubin, E. M. , Rokhsar, D. S. , & Banfield, J. F. (2004). Community structure and metabolism through reconstruction of microbial genomes from the environment. Nature, 428(6978), 37–43. 10.1038/nature02340 [DOI] [PubMed] [Google Scholar]
- Vaser, R. , Sović, I. , Nagarajan, N. , & Šikić, M. (2017). Fast and accurate de novo genome assembly from long uncorrected reads. Genome Research, 27(5), 737–746. 10.1101/gr.214270.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker, B. J. , Abeel, T. , Shea, T. , Priest, M. , Abouelliel, A. , Sakthikumar, S. , Cuomo, C. A. , Zeng, Q. , Wortman, J. , Young, S. K. , & Earl, A. M. (2014). Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One, 9(11), e112963. 10.1371/journal.pone.0112963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y. , Yang, Q. , & Wang, Z. (2014). The evolution of nanopore sequencing. Frontiers in Genetics, 5, 449. 10.3389/fgene.2014.00449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White, R. A. , Bottos, E. M. , Roy Chowdhury, T. , Zucker, J. D. , Brislawn, C. J. , Nicora, C. D. , Fansler, S. J. , Glaesemann, K. R. , Glass, K. , & Jansson, J. K. (2016). Moleculo long‐read sequencing facilitates assembly and genomic binning from complex soil metagenomes. mSystems, 1(3), e00045‐16. 10.1128/mSystems.00045-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham, H. (2009). ggplot2. Springer. 10.1007/978-0-387-98141-3 [DOI] [Google Scholar]
- Wommack, K. E. , Bhavsar, J. , & Ravel, J. (2008). Metagenomics: read length matters. Applied and Environmental Microbiology, 74(5), 1453–1463. 10.1128/AEM.02181-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yahara, K. , Suzuki, M. , Hirabayashi, A. , Suda, W. , Hattori, M. , Suzuki, Y. , & Okazaki, Y. (2021). Long‐read metagenomics using PromethION uncovers oral bacteriophages and their interaction with host bacteria. Nature Communications, 12(1), 27. 10.1038/s41467-020-20199-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan, S. , Cohen, D. B. , Ravel, J. , Abdo, Z. , & Forney, L. J. (2012). Evaluation of methods for the extraction and purification of DNA from the human microbiome. PLoS One, 7(3), e33865. 10.1371/journal.pone.0033865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaura, E. , Keijser, B. J. F. , Huse, S. M. , & Crielaard, W. (2009). Defining the healthy “core microbiome” of oral microbial communities. BMC Microbiology, 9(1), 259. 10.1186/1471-2180-9-259 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Table S1
Table S2
Table S3
Data Availability Statement
Raw sequences for long‐ and short‐read sequencing data for shotgun metagenomes and 16S rRNA gene amplicons are available under the BioProject PRJNA703035. We also made available FASTA files and anvi’o contigs databases for (i) assembled and polished long‐read sequences at https://doi.org/10.6084/m9.figshare.14141228, (ii) unassembled long‐read sequences at https://doi.org/10.6084/m9.figshare.14141414, and (iii) assembled shotgun metagenomes at https://doi.org/10.6084/m9.figshare.14141819. Supporting Tables and Figures are also available via https://doi.org/10.6084/m9.figshare.14141918. A reproducible bioinformatics workflow is available at https://merenlab.org/data/hmw‐dna‐extraction‐strategies/, where https://github.com/merenlab/web/tree/master/data/hmw‐dna‐extraction‐strategies and the commit hash 557f6e37381d212a22a17d67c7b0e774e5abee82 gives access to its final version at the time of publication.