

Abstract
Background and Purpose
Acamprosate is an anti‐craving drug used for the pharmacotherapy of alcohol use disorder (AUD). However, only some patients achieve optimal therapeutic outcomes. This study was designed to explore differences in metabolomic profiles between patients who maintained sobriety and those who relapsed, to determine whether those differences provide insight into variation in acamprosate treatment response phenotypes.
Experimental Approach
We previously conducted an acamprosate trial involving 442 AUD patients, and 267 of these subjects presented themselves for a 3‐month follow‐up. The primary outcome was abstinence. Clinical information, genomic data and metabolomics data were collected. Baseline plasma samples were assayed using targeted metabolomics.
Key Results
Baseline plasma arginine, threonine, α‐aminoadipic acid and ethanolamine concentrations were associated with acamprosate treatment outcomes and baseline craving intensity, a measure that has been associated with acamprosate treatment response. We next applied a pharmacometabolomics‐informed genome‐wide association study (GWAS) strategy to identify genetic variants that might contribute to variations in plasma metabolomic profiles that were associated with craving and/or acamprosate treatment outcome. Gene expression data for induced pluripotent stem cell‐derived forebrain astrocytes showed that a series of genes identified during the metabolomics‐informed GWAS were ethanol responsive. Furthermore, a large number of those genes could be regulated by acamprosate. Finally, we identified a series of single nucleotide polymorphisms that were associated with acamprosate treatment outcomes.
Conclusion and Implications
These results serve as an important step towards advancing our understanding of disease pathophysiology and drug action responsible for variation in acamprosate response and alcohol craving in AUD patients.
Keywords: acamprosate, alcohol use disorder, craving, metabolomics genome‐wide association study, multiple omics study
Abbreviations
- AUD
alcohol use disorder
- EBs
embryonic bodies
- FDA
US Food and Drug Administration
- FDR
false discovery rate
- GAD‐7
Generalized Anxiety Disorder 7‐item assessment
- GWAS
genome‐wide association study
- HRC
haplotype reference consortium
- iPSC
induced pluripotent stem cell
- MAF
minor allele frequency
- PACS
Penn Alcohol Craving Scale
- PHQ‐9
Patient Health Questionnaire 9
quantile–quantile
- SNP
single nucleotide polymorphism
- TLFB
Timeline Follow Back
- UPLC
ultraperformance liquid chromatography
What is already known
Metabolomics can be a useful approach to identify biomarkers that could potentially predict treatment response.
Craving is associated with alcohol use disorder relapse.
What does this study add
Baseline plasma metabolomic markers are associated with craving intensity.
Baseline plasma metabolomic markers are associated with alcohol relapse risk.
What is the clinical significance
Baseline craving intensity is associated with acamprosate treatment outcomes.
A pharmacometabolomics‐informed pharmacogenomics approach could identify genetic variants that are associated with acamprosate treatment outcomes.
1. INTRODUCTION
Alcohol use disorder (AUD) is the most common substance use disorder (Grant et al., 2004, 2015; Hunt et al., 2020). One third of US adults experience AUD during their lifetime (Grant et al., 2015). AUD is a disease that, like many other chronic diseases, typically requires long‐term treatment and care to prevent relapse (Witkiewitz et al., 2019). Acamprosate, naltrexone and disulfiram are the drugs that have received US Food and Drug Administration (FDA) approval for the treatment of AUD. However, only a small proportion of patients respond to treatment with these agents by achieving sustained abstinence (Anton et al., 2006). It would be a major achievement for precision medicine if we could develop ways to individualize the drug therapy of patients with alcohol addiction in order, to prevent alcohol relapse and to select the patients most likely to respond prior to the initiation of drug therapy (Ho et al., 2020; Litten et al., 2020). Acamprosate reduces the craving for alcohol (Hammarberg et al., 2009) and has also been reported to demonstrate antidepressant properties (Pałucha‐Poniewiera & Pilc, 2012). However, the mechanism(s) of action of acamprosate as an agent for the treatment of AUD remain unclear, and only a subset of patients with AUD achieve optimal therapeutic outcomes, which could be due, in part, to pharmacogenomic variation in response to this drug (Cheng et al., 2020). Therefore, it is important to understand mechanisms underlying variation in acamprosate efficacy and their interaction with the biological changes associated with alcohol use. That knowledge could potentially facilitate the development of individualized acamprosate treatment programmes and the design of new and better medications for the treatment of AUD.
Metabolomics can be a useful approach to identify biological markers that could potentially predict treatment response by identifying differential concentrations of metabolites associated with clinical variables such as treatment outcomes. The Mayo Clinic Center for the Individualized Treatment of Alcoholism Study recruited 442 patients with AUD to an open‐label study designed to investigate biomarkers associated with outcomes for patients treated for 3 months with acamprosate in community‐based programmes (Karpyak et al., 2014). We previously conducted a pilot study to identify baseline serum metabolomic biomarkers (i.e. glutamate, taurine, aspartate, ammonia, aminoadipic acid and threonine) that were associated with acamprosate treatment response in patients with AUD, using a small subset of these samples (relapse [n = 49] vs. non‐relapse [n = 71]) (Hinton et al., 2017). We have now collected a larger sample size (relapse [n = 110] vs. non‐relapse [n = 157]) in an attempt to expand and replicate our earlier findings. These 267 subjects presented themselves for a 3‐month follow‐up.
The present study was designed (1) to identify baseline plasma metabolomic biomarkers associated with alcohol relapse risk during 3 months of acamprosate treatment and (2) to apply our established pharmacometabolomics‐informed pharmacogenomics strategy to identify possible genetic variants that might contribute to the concentrations of these plasma metabolites. This study is similar in design to other studies that we have performed successfully with regard to selective serotonin reuptake inhibitor response in patients with major depressive disorder (Gupta et al., 2016; Ji et al., 2014; Liu et al., 2018). Specifically, metabolic profiles were associated with both clinical phenotypes and with genetic variants to identify novel genetic variants associated with drug response phenotypes. We hypothesized that metabolomic profiles might differ between patients who maintained sobriety and those who relapsed and that these differences might provide insight into mechanisms involved in variation in drug response phenotypes (Frye, Hinton, Karpyak, Biernacka, Gunderson, Feeder, et al., 2016; Frye, Hinton, Karpyak, Biernacka, Gunderson, Geske, et al., 2016; Hinton et al., 2017; Nam et al., 2015). We also explored differences in metabolomic profiles between men and women, because sex differences play a role in AUD pathophysiology (Karpyak et al., 2019; Mason & Lehert, 2012) and may also play a role in response to AUD pharmacotherapy. In addition, we identified a series of metabolites that were associated with craving intensity—an important clinical variable associated with alcohol relapse (Cavicchioli et al., 2020; Flannery et al., 2003; Subbaraman et al., 2013). Our results showed that baseline concentrations of metabolites associated with craving intensity might be associated with and contribute to acamprosate treatment response in patients with AUD. Finally, the utilization of a pharmacometabolomics‐informed pharmacogenomics approach allowed us to identify genetic variants that may contribute to variations in plasma metabolomics profiles associated with alcohol craving and/or acamprosate treatment outcomes. As a result, these findings could serve as an important step in advancing our understanding of both disease pathophysiology and drug mechanisms responsible for variation in acamprosate response and alcohol craving in patients with AUD.
2. METHODS
2.1. Study participants
The Mayo Clinic Center for the Individualized Treatment of Alcoholism Study previously recruited 442 participants with AUD (ClinicalTrials.gov Identifier: NCT00662571) (Biernacka et al., 2021; Karpyak et al., 2014, 2019). All participants in the study initially received acamprosate (one 333‐mg tablet three times a day) to determine tolerance to acamprosate treatment. A standard dose of acamprosate was then prescribed (two 333‐mg tablets three times a day). Confidentiality was maintained for all study participants. This study was conducted in accordance with protocols reviewed and approved by the Mayo Clinic Institutional Review Board (07‐007204 and 20‐000372).
2.2. Assessment
We collected clinical data that included the Patient Health Questionnaire 9 (PHQ‐9), Generalized Anxiety Disorder 7‐item (GAD‐7) assessment and Penn Alcohol Craving Scale (PACS). The study included two primary treatment outcomes: relapse and heavy relapse. Relapse was defined as return to alcohol use during 3 months of acamprosate treatment, whereas non‐relapse was defined as maintenance of abstinence from alcohol during those 3 months (Figure 1). Heavy relapse was defined as four or more standard drinks daily for a woman and five or more standard drinks daily for a man based on the Dietary Guidelines for Americans 2015–2020 recommendations (https://www.niaaa.nih.gov/alcohol-health/overview-alcohol-consumption/moderate-binge-drinking). Alcohol consumption during the 3 months of acamprosate treatment was determined by self‐report, using Timeline Follow Back (TLFB).
FIGURE 1.
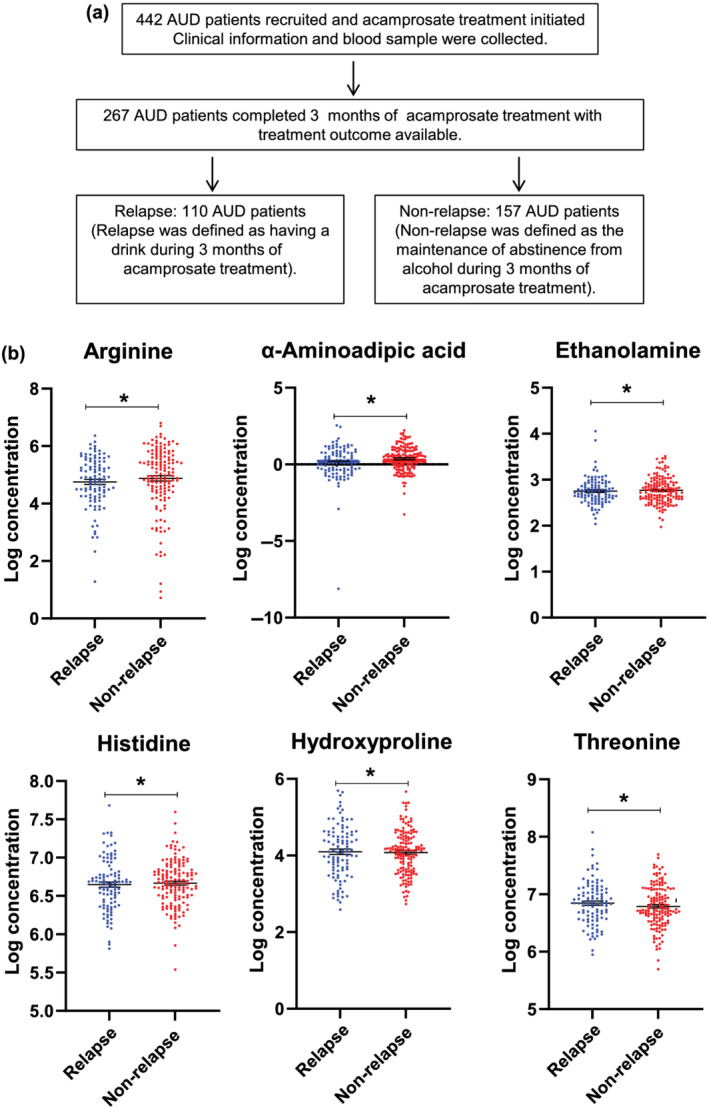
(a) Schematic outline of study design and sample numbers. (b) Plasma concentrations of metabolites in relapse and non‐relapse groups for metabolites that displayed significant differences between those two groups. Data distribution was tested using a Shapiro–Wilk test. Arginine, ethanolamine and α‐aminoadipic acid concentrations were not normally distributed; as a result, non‐parametric tests were performed. *P < 0.05. AUD indicates alcohol use disorder
2.3. Targeted metabolomics using ultraperformance liquid chromatography (UPLC)–tandem mass spectrometry
Baseline plasma samples from patients with AUD were assayed for the amino acid panel and the neuromodulator panel in the Mayo Clinic Metabolomics Core Facility (see Table S1 for a list of the metabolites assayed). Briefly, blood samples for the isolation of plasma were collected in EDTA tubes. Blood samples were centrifuged at 2900 g for 15 min at 4°C for biobanking. Thawed plasma samples (10 μl) were spiked with an internal standard and were then derivatized. The amino acid derivatizing reagent used was 6‐aminoquinolyl‐N‐hydroxysuccinimidyl carbamate. Concentrations of metabolites were determined based on standard curves. A 10‐point standard concentration curve was made from the calibration standard solution to calculate amino acid concentrations in plasma samples. High‐resolution separation was performed using an Acquity UPLC system and injection of 1 ml of derivatized solution using a UPLC BEH C18 column (Waters Corp). Mass detection was performed using a TSQ Ultra Quantum running in ESI positive mode (Thermo Fisher Scientific) (Hinton et al., 2017; Nam et al., 2015). Analyses reported subsequently included metabolites with fewer than 25% of samples below the limit of detection.
2.4. Data and statistical analysis
Statistical analysis was performed using R Statistical Software (Version 4.0.2; R Foundation for Statistical Computing). GraphPad Prism Software v7 (GraphPad Prism, RRID:SCR_002798) was used to generate the figures. The data and statistical analyses comply with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology (Curtis et al., 2018). Experiments were performed using randomization and blinding analysis where technically feasible and appropriate. Statistical analysis was conducted only when the experimental group where n ≥ 5 (n refers to independent values, not replicates). The exact sample size for each experiment is listed in the figure legends. Log‐transformed metabolomics and gene expression data were analysed. Data distribution was tested using a Shapiro–Wilk test. Continuous variables were compared using unpaired t test or Mann–Whitney U test (when the datasets were not normally distributed). Categorical variables were tested for correlation using χ 2 test or Fisher's exact test. P < 0.05 was considered statistically significant. We also applied p.adjust function in R (Version 4.0.2) to estimate false discovery rate (FDR). In addition, logistic regression models of treatment outcomes (i.e., 3‐month relapse to any drinking or heavy drinking) were used to examine each metabolite as a predictor of relapse individually. Models examined included unadjusted (univariate) models, with relapse or heavy relapse (yes/no) as the outcome and the metabolite as the predictor. Two types of multivariable logistic regression models were then examined. The first was a model adjusted for sex, still including the metabolite as a predictor. The second model examined was a multivariable model including sex, using the metabolites as main effects (predictors), as well as a sex–metabolite interaction term.
2.5. Genotyping and single nucleotide polymorphism (SNP) data analysis
DNA samples were first genotyped using Illumina Human Core arrays with only ~300,000 markers at the Mayo Clinic Medical Genome Facility. The National Institute of Alcohol Abuse and Alcoholism then genotyped these samples using Infinium OmniExpressExome‐8 BeadChips, a high‐density SNP genotyping array with ~958,497 markers. Data from the two arrays were quality‐controlled, combined and checked for concordance, and additional quality control was performed using the combined dataset. Samples were excluded from analysis if they had a low call rate, extreme heterozygosity or disagreement between reported sex and genetically determined sex. Sample relatedness was checked by pairwise identical‐by‐descent estimation. Imputation was conducted using the Michigan Imputation Server with the haplotype reference consortium (HRC) reference panel (Version HRC.r1‐1.GRCh37.wgs.mac5.sites). We excluded SNPs with a call rate < 95%, SNPs not in Hardy–Weinberg equilibrium, and SNPs with minor allele frequency (MAF) < 0.01 among the AUD patients. Of the 6,654,675 SNPs, 6,621,773 passed the initial QC and were used in the genome‐wide association studies (GWAS). Concentration values for each metabolite were transformed using van der Waerden's method (VDW score). Associations between variants and metabolites were tested using linear regression, adjusted for sex, age, site and baseline PACS score. These analyses were done in R Version 3.6.3 and PLINK 1.9 (PLINK, RRID:SCR_001757).
Genetic variants within the PTPRD gene (GRCh37/hg19: chromosome 9: 8314246–10612723) were tested for association with a series of drug response phenotypes. Participants who had at least 1 week of follow‐up were included in the analysis. Single SNPs were tested individually as predictors of time until alcohol use, or time until heavy drinking after initiating acamprosate therapy, using multivariable Cox proportional hazard models. Heavy drinking was defined as four or more standard drinks daily for a woman and five or more standard drinks daily for a man. SNP associations with binary outcomes, that is, relapse to alcohol use or relapse to heaving drinking during 3 months of acamprosate therapy, were determined using multivariable logistic regression models. Models were adjusted for the number of days sober prior to treatment, baseline PACS and study sites. Results of the Cox proportional hazard analyses are displayed in Figure 5 using Kaplan–Meier curves and have not been adjusted for multiple testing. The odds ratios and hazard ratios are represented as OR and HR (95% confidence interval), with a value > 1 indicating worse outcome.
FIGURE 5.
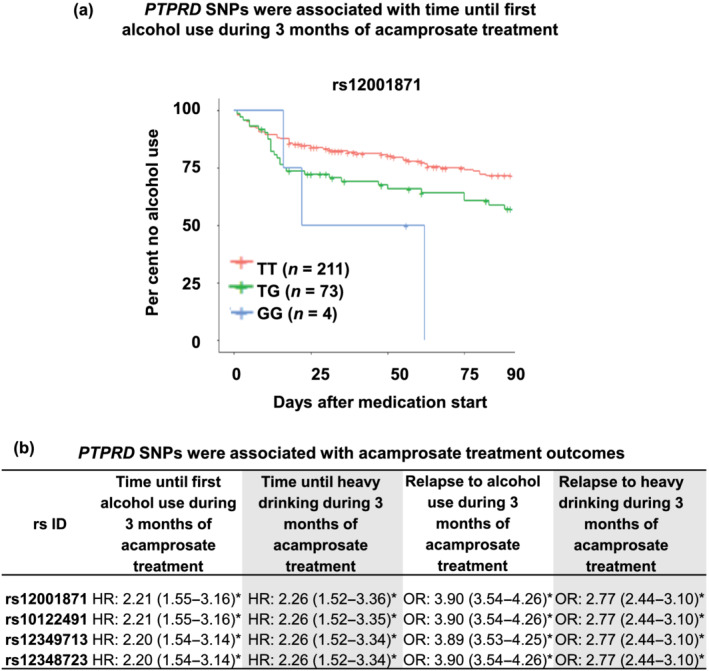
PTPRD SNPs were associated with acamprosate treatment response. (a) Kaplan–Meier curves for the time until first alcohol use during 3 months of acamprosate therapy. Participants who had at least 1 week of follow‐up were included in the analysis. The wild‐type genotype was associated with better outcomes, that is, longer abstinence length until first drink during 3 months of acamprosate treatment, P: 1.35E‐05. (b) PTPRD SNPs were associated with acamprosate treatment response phenotypes. SNPs within the PTPRD gene (GRCh37/hg19: chromosome 9: 8314246–10612723) were tested for association with a series of phenotypes. Single SNPs were tested individually as predictors of time until alcohol use or time until heavy drinking after initiating acamprosate therapy using multivariable Cox proportional hazard models. Heavy drinking was defined as four or more standard drinks daily for a woman and five or more standard drinks daily for a man. SNP associations with binary outcomes, that is, relapse to alcohol use or relapse to heaving drinking during 3 months of acamprosate therapy, were determined using multivariable logistic regression models. Models were adjusted for the number of days sober prior to treatment, baseline PACS and study site. Results of the Cox proportional hazard analyses were not adjusted for multiple testing. The odds ratios or hazard ratios are represented as OR and HR (95% confidence interval), with a value > 1 indicating worse outcome. *P < 0.05
2.6. Generation of iPSC‐derived forebrain‐specific astrocytes and organoids
Peripheral blood mononuclear cells (n = 6) were used to perform induced pluripotent stem cell (iPSC) reprogramming using the CytoTune‐iPS 2.0 Sendai Reprogramming Kit (A16517, Thermo Fisher Scientific), as previously described (Vadodaria, Ji, Skime, Paquola, Nelson, Hall‐Flavin, Fredlender, et al., 2019; Vadodaria, Ji, Skime, Paquola, Nelson, Hall‐Flavin, Heard, et al., 2019). All iPSCs had normal karyotypes and expressed pluripotency markers. All of these cell lines were regularly verified to be free from mycoplasma infection. iPSCs were differentiated into forebrain‐specific astrocytes (1801; ScienCell Research Laboratories). Forebrain‐specific astrocytes derived from iPSCs were treated with 25 mM of ethanol (EtOH), a concentration that is considered physiologically relevant for EtOH use, with 25 mM of EtOH being slightly higher than the 0.08% blood alcohol concentration often used as a measure of intoxication (Lira et al., 2020). The concentrations of acamprosate (5 μM) and naltrexone (30 nM) used to perform these experiments were selected to fall within the range of blood drug concentrations in patients taking standard clinical doses of these two drugs (Mason et al., 2002). Drug treatment was conducted at 50–57 days of astrocyte differentiation with a daily medium change. We also generated iPSC‐derived 3D forebrain organoids. Briefly, pre‐patterned floating embryonic bodies (EBs) formed from intact iPSC colonies were embedded in Matrigel and cultured with small molecules and proteins including 1× N2 (Life Technologies, Grand Island, NY, USA), 1× NEAA and 1× Glutamax (Invitrogen, Grand Island, NY, USA), 1‐μM SB‐431542 and 1‐μM CHIR99021 (Selleckchem, Carlsbad, CA, USA) for 7 days. On Day 14, organoids were mechanically dissociated from the Matrigel and were cultured in a 12‐well plate using a bioreactor (Qian et al., 2018). Culture medium from Days 14 to 70 consisted of DMEM/F12 medium supplemented with 1× N2, 1× B27, 1× NEAA and 1× Glutamax, 1× 2‐metabptoethanol, 100× pen/strp and 2.5 μg·ml−1 insulin (Sigma‐Aldrich, St Louis, MO, USA). To improve maturation during prolonged culture, medium was replaced every other day. From Day 70 onwards, supplementing media with 20 ng·ml−1 BDNF, 20 ng·ml−1 GDNF (Peprotech, Rocky Hill, NJ, USA), 0.2‐mM l‐ascorbic acid (Sigma‐Aldrich, St Louis, MO, USA) and 0.5‐mM cAMP (Sigma‐Aldrich, St Louis, MO, USA) were used. Drug treatment was conducted at 83–90 days of forebrain organoid differentiation with a daily medium change. Cultured cells were used for RNA isolation.
2.7. Immunofluorescence staining and confocal imaging analysis
The immunofluorescence staining procedures comply with the recommendations of the British Journal of Pharmacology on immunoblotting and immunochemistry (Alexander et al., 2018). Specifically, cells were fixed in 4% paraformaldehyde at room temperature for 15 min. The cells were washed in cold PBS and permeabilized with 0.2% Triton X‐100 in PBS. After blocking for 30 min with 3% normal donkey serum in PBS, cells were incubated with primary antibody in 5% BSA overnight. The secondary antibody was used at a 1:2000 dilution. Antifade mounting media with DAPI (VECTOR laboratory, Burlingame, CA, USA) was used to stain the cell nuclei. Slides were visualized using fluorescence microscopy (Olympus, FV1200).
2.8. Real‐time PCR
The PCR reactions contained 100 ng of total RNA, 5 μl of 2X SYBR green qPCR master mix (Life Technologies, CA, USA), 1 μl of gene‐specific primer and distilled water up to 10 μl of final volume per reaction. Real‐time PCR reactions were performed in duplicate using the Applied Biosystems ViiA 7™ Real‐Time PCR System (Life Technologies, Carlsbad, CA, USA). The 2−ΔΔCt method was employed for statistical data analysis.
2.9. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in the IUPHAR/BPS Guide to PHARMACOLOGY, http://www.guidetopharmacology.org, and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20 (Alexander et al., 2019).
3. RESULTS
3.1. Characteristics of study participants
The Mayo Clinic Center for the Individualized Treatment of Alcoholism Study is an acamprosate clinical trial that recruited 442 participants with AUD, all of whom were treated with acamprosate. Two hundred and sixty‐seven of these AUD subjects presented themselves for the 3‐month follow‐up. As mentioned previously, acamprosate has received FDA approval for the pharmacotherapy of AUD. It should be emphasized that our trial was not designed to determine the efficacy of acamprosate, but rather to study biomarkers associated with individual variation in treatment response to acamprosate. Clinical information obtained included rating scales for depression, anxiety and craving. Blood samples for DNA genotyping were also collected at baseline. However, as pointed out above, only 267 of the 442 participants were evaluated after 3 months of acamprosate treatment and, as a result, had treatment outcome data available (Karpyak et al., 2014, 2019). Alcohol consumption was measured using the TLFB every month for 3 months, which permitted the calculation of various treatment outcome measures including relapse to drinking or to heavy drinking (Karpyak et al., 2019). Relapse (n = 110) was defined as return to any alcohol consumption during the 3 months of acamprosate treatment, whereas non‐relapse (n = 157) was defined as abstinence from alcohol (no alcohol use) during 3 months of acamprosate treatment (see Figure 1a). Baseline characteristics of the AUD participants in both groups are listed in Table 1. Among the AUD participants, two thirds were men, consistent with most studies of AUD—a disorder that displays a striking difference in incidence between the sexes (Blendberg et al., 2020; Erol & Karpyak, 2015). Psychiatric and medical comorbidities are common among patients with AUD and may influence the efficacy of AUD pharmacotherapy. Therefore, those comorbidities are potentially important clinical variables for studies of drug response biomarkers. Our data showed that the frequency of psychiatric comorbidities such as depression, anxiety and panic disorder did not display significant differences between the relapse and non‐relapse groups (Table 1). Nevertheless, the relapse group displayed significantly higher baseline PHQ‐9 scores—an assessment of depressive symptoms. Baseline alcohol craving scores as determined by the PACS were also significantly higher in the relapse group (Table 1). These clinical variables may be associated with outcomes among patients with AUD. However, even if that is the case, the underlying molecular biology remains to be determined. Therefore, we set out to identify and study baseline plasma metabolomic markers that were associated with alcohol craving and alcohol relapse risk during 3 months of acamprosate treatment.
TABLE 1.
Clinical and demographic characteristics of study participants
| Characteristic | All participants (N = 442) | Relapse (n = 110) | Non‐relapse (n = 157) | Relapse versus non‐relapse |
|---|---|---|---|---|
| Mean ± SD or no. (%) | Mean ± SD or no. (%) | Mean ± SD or no. (%) | P value | |
| Age (years) | 42 ± 11.8 | 41.59 ± 12.04 | 42.39 ± 11.58 | NS |
| Male sex | 286 (65) | 67 (60.9) | 112 (71.3) | NS |
| White race | 412 (93) | 100 (90.9) | 147 (93.6) | NS |
| Baseline PHQ‐9 score | 9.4 ± 6.1 | 10.24 ± 6.14 | 8.79 ± 6.04 | * |
| Baseline PACS score | 13.38 ± 8.0 | 15.44 ± 8.47 | 11.91 ± 7.37 | * |
| Baseline GAD‐7 score | 9.0 ± 5.9 | 9.35 ± 5.88 | 8.66 ± 5.80 | NS |
| Depression | 102 (23) | 30 (27.2) | 33 (21.0) | NS |
| Anxiety | 141 (31.9) | 33 (30) | 45 (28.6) | NS |
| OCD | 22 (4.9) | 6 (5.4) | 8 (5.0) | NS |
| Social phobia | 54 (12.2) | 14 (12.7) | 16 (10.1) | NS |
| PTSD | 79 (17.8) | 19 (17.2) | 21 (13.3) | NS |
| Panic disorder | 31 (7) | 10 (9.0) | 13 (8.2) | NS |
Note: Relapse was defined as having a standard drink during 3 months of acamprosate treatment, whereas non‐relapse was defined as the maintenance of abstinence from alcohol during 3 months of acamprosate treatment.
Abbreviations: GAD‐7, General Anxiety Disorder 7; NS, not significant; OCD, obsessive–compulsive disorder; PACS, Penn Alcohol Craving Scale; PHQ‐9, Patient Health Questionnaire 9; PTSD, post‐traumatic stress disorder.
P < 0.05.
3.2. Plasma metabolomic markers associated with alcohol relapse risk
Targeted metabolomic assays were performed using the baseline plasma samples (see Figure 1a for an outline of the study design). To identify metabolomic biomarkers associated with alcohol relapse risk during 3 months of acamprosate treatment, we studied baseline plasma samples from the 267 AUD participants in whom we know the treatment outcome. Four metabolites (histidine, hydroxyproline, arginine and α‐aminoadipic acid) were significantly elevated in the non‐relapse group as compared with the relapse group. Two metabolites (threonine and ethanolamine) were significantly lower in the non‐relapse than in the relapse group (see Figure 1b and, for complete results for all metabolites assayed, see Table S2). We also studied relapse to heavy drinking as an additional treatment outcome and found that concentrations of α‐aminoadipic acid, threonine, hydroxyproline, arginine, hydroxyproline and ethanolamine also differed between the heavy relapse and non‐heavy relapse groups (Table S3).
3.3. Sex‐specific differences in metabolite concentrations
It has been reported that men and women display distinct metabolomic profiles (Mittelstrass et al., 2011), and we also observed a series of plasma metabolites that displayed sex‐related differences in our AUD patients (see Table S4). As stated previously, AUD is more highly prevalent in men than in women. However, sex did not substantially influence acamprosate treatment response (Table 1), a result that is consistent with observations made by the COMBINE study (Anton et al., 2006). We also performed analyses using logistic models that can accommodate covariates (i.e., sex), because sex differences in concentrations of metabolites were observed (Table S4). Specifically, we set out to evaluate evidence for a sex‐by‐metabolite interaction effect on the outcomes (i.e., relapse and heavy relapse). Those analyses tested whether associations between the metabolites and outcomes differed significantly between men and women. We observed no significant interactions (Tables S2 and S3).
3.4. Correlation of plasma metabolomics with alcohol craving intensity
Acamprosate is an anti‐craving drug (Boothby & Doering, 2005; Shen, 2018), and elevated craving increases the likelihood of alcohol relapse (Cavicchioli et al., 2020; McHugh et al., 2016; Schneekloth et al., 2012; Stohs et al., 2019). In line with previous reports, baseline alcohol craving scores in our relapse group were significantly higher than in the non‐relapse group (Table 1). Because no objective biochemical measures have been associated with alcohol craving intensity, we set out to explore possible correlations between craving intensity and plasma metabolite concentrations. We found that concentrations for 18 of the metabolites that we had assayed were significantly associated with baseline PACS scores, as listed in Table 2. Among metabolites that were correlated with baseline craving intensity, arginine, α‐aminoadipic acid, threonine and ethanolamine concentrations also differed significantly between the relapse and non‐relapse groups, as is shown in Figure 1b. This series of observations demonstrates that craving intensity, at least in this study, was associated with plasma metabolite concentrations, thus raising the question of the underlying biology that might drive the differences in metabolite concentrations that we observed, a question that we began to address in the studies described subsequently.
TABLE 2.
Correlation of plasma metabolomics with baseline Penn Alcohol Craving Scale
| Metabolite | Pearson's r | FDR |
|---|---|---|
| Arginine | −0.220 | 0.00029 |
| α‐Aminoadipic acid | −0.211 | 0.00014 |
| Tyrosine | −0.168 | 0.00614 |
| Ethanolamine | 0.155 | 0.00998 |
| Valine | −0.152 | 0.01125 |
| α‐Amino‐N‐butyric acid | 0.150 | 0.01125 |
| Taurine | 0.145 | 0.01439 |
| Tryptophan | −0.145 | 0.01439 |
| Hydroxylysine 2 | −0.132 | 0.02409 |
| Cysteine | −0.129 | 0.03331 |
| Lysine | −0.124 | 0.03889 |
| Serine | 0.123 | 0.03889 |
| γ‐Amino‐N‐butyric acid | 0.118 | 0.01125 |
| Isoleucine | −0.118 | 0.04739 |
| Threonine | 0.108 | 0.07349 |
| Leucine | −0.105 | 0.07470 |
| Glycine | 0.104 | 0.07556 |
| Asparagine | 0.101 | 0.08154 |
| Citrulline | 0.086 | 0.14279 |
| Alanine | −0.085 | 0.14377 |
| Hydroxyproline | −0.074 | 0.21020 |
| Phenylalanine | −0.067 | 0.26264 |
| β‐Aminoisobutyric acid | 0.058 | 0.30557 |
| Ornithine | 0.057 | 0.33495 |
| Histidine | −0.043 | 0.47689 |
| Aspartic acid | 0.041 | 0.47689 |
| Proline | −0.025 | 0.70599 |
| Sarcosine | −0.024 | 0.70599 |
| Phosphoethanolamine | 0.022 | 0.69447 |
| Glutamine | −0.019 | 0.76604 |
| Glutamic acid | 0.013 | 0.82345 |
| Methionine | −0.010 | 0.85729 |
Abbreviation: FDR, false discovery rate.
3.5. Pharmacometabolomics informs pharmacogenomics
We next performed GWAS using baseline metabolomic profiles as quantitative biological traits to identify genetic variants associated with variations in concentrations of metabolites that were associated with drug response phenotypes (Figure S1). Specifically, we applied the pharmacometabolomics‐informed pharmacogenomic approach outlined schematically in Figure 2a. Among the metabolites that were associated with acamprosate treatment outcomes, as shown in Figure 1b, we observed a signal that was near genome‐wide significant in the baseline ethanolamine GWAS (Figure 2b with the corresponding quantile–quantile [QQ] plot shown in Figure S2). Among the SNP loci with suggestive evidence for association (P ≤ 5E‐06), the majority of SNPs mapped to protein coding genes that were ethanol responsive, as determined by real‐time PCR using iPSC‐derived forebrain‐specific astrocytes (see Figure 2c,d). We subsequently differentiated iPSC‐derived forebrain organoids, which are 3D self‐assembled structures composed of multiple brain cell types (Figure 2e). We then observed that the expression of GRM7, PTPRD and AUTS2 could be significantly induced by ethanol in iPSC‐derived forebrain organoids (Figure 2f).
FIGURE 2.
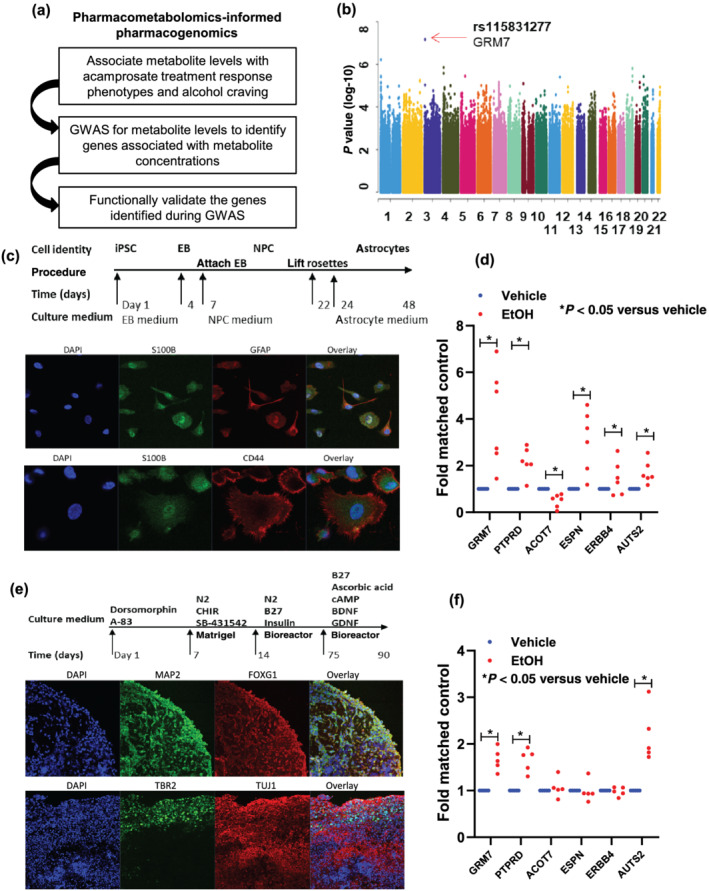
(a) Schematic outline of pharmacometabolomics‐informed pharmacogenomics research strategy. (b) Manhattan plot for GWAS of plasma concentrations of ethanolamine that were associated with acamprosate treatment outcomes. (c) A schematic outline of procedures used during the differentiation of iPSC‐derived forebrain astrocytes. The panel below the schematic displays representative examples of staining for astrocyte markers (S100β and GFAP). iPSC indicates induced pluripotent stem cell. EB indicates embryonic body. NPC indicates neural progenitor cell. (d) Effect of ethanol (EtOH: 25 mM) on mRNA expression in iPSC‐derived forebrain astrocytes for SNP loci identified during the ethanolamine GWAS with P value < 10−6. Real‐time PCR was performed using iPSC‐derived forebrain astrocytes from six AUD subjects. *P < 0.05, significantly different as indicated; Mann–Whitney U test. (e) A schematic outline of procedures used during the differentiation of iPSC‐derived forebrain organoids. The panel below the schematic displays representative examples of staining for neuronal markers (MAP 2 [Sigma‐Aldrich Cat# M2320, RRID:AB_609904], FOXG1 [Abcam Cat# ab18259, RRID:AB_732415] and TUJ1 [Covance Cat# MMS‐435P, RRID:AB_2313773]) and for a cortical layer marker (TBR2 [Abcam Cat# ab75720, RRID:AB_1310743]). (f) Effects of ethanol (EtOH: 25 mM) on gene expression in iPSC‐derived forebrain organoids for SNP loci identified during the ethanolamine GWAS with P value < 10−6. Real‐time PCR was performed using iPSC‐derived forebrain organoids (n = 5). Thedot plot displays mRNA expression levels (EtOH treatment vs. vehicle treatment). Real‐time PCR results were analysed using 2−ΔΔct method. As a result, the control mean is 1, and there is no variance in the control. Real‐time PCR data were then analysed using non‐parametric statistical tests (Mann–Whitney U test). *P < 0.05, significantly different as indicated
We next performed metabolomics‐informed GWAS for the metabolites that were associated with alcohol craving intensity (Table 2), an important variable that has been associated with acamprosate treatment response. We observed genome‐wide or near genome‐wide significant SNPs in the GWAS for α‐amino‐N‐butyric acid, asparagine, ethanolamine, lysine, taurine and tyrosine (Figure 3). Among the SNP loci with suggestive evidence of association (P ≤ 5E‐06) with concentrations of the six metabolites (Figure 3), a large number of SNPs were located in or near protein coding genes. Of particular interest, a large number of those genes were ethanol responsive in iPSC‐derived forebrain‐specific astrocytes (n = 6) (see Figure 4a and Table S5). In addition, a large number of those genes displayed ‘inverted’ gene expression patterns in response to acamprosate treatment, that is, the directionality after EtOH exposure was opposite to that after acamprosate exposure (Figure 4b). Even more striking, very similar results were observed when the cells were exposed to naltrexone, one of the two US FDA approved anti‐craving drugs for the treatment of AUD (Figure 4c). In summary, our multiple omics studies have identified a series of genes that might contribute to treatment response and/or disease pathophysiology for further studies designed to understand underlying mechanisms for the effects of those genes. However, it would be impractical to functionally study all of the genes that we had identified. The subsequent paragraph will demonstrate one of the strategies for prioritizing genes or SNPs for future functional genomic study.
FIGURE 3.
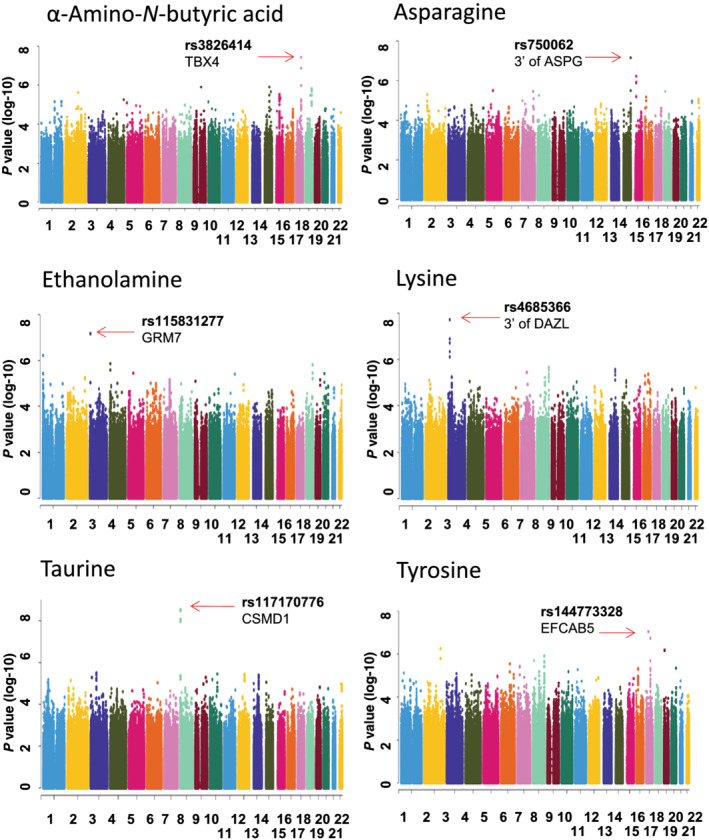
Manhattan plots for GWAS for plasma concentrations of metabolites that were associated with alcohol craving
FIGURE 4.
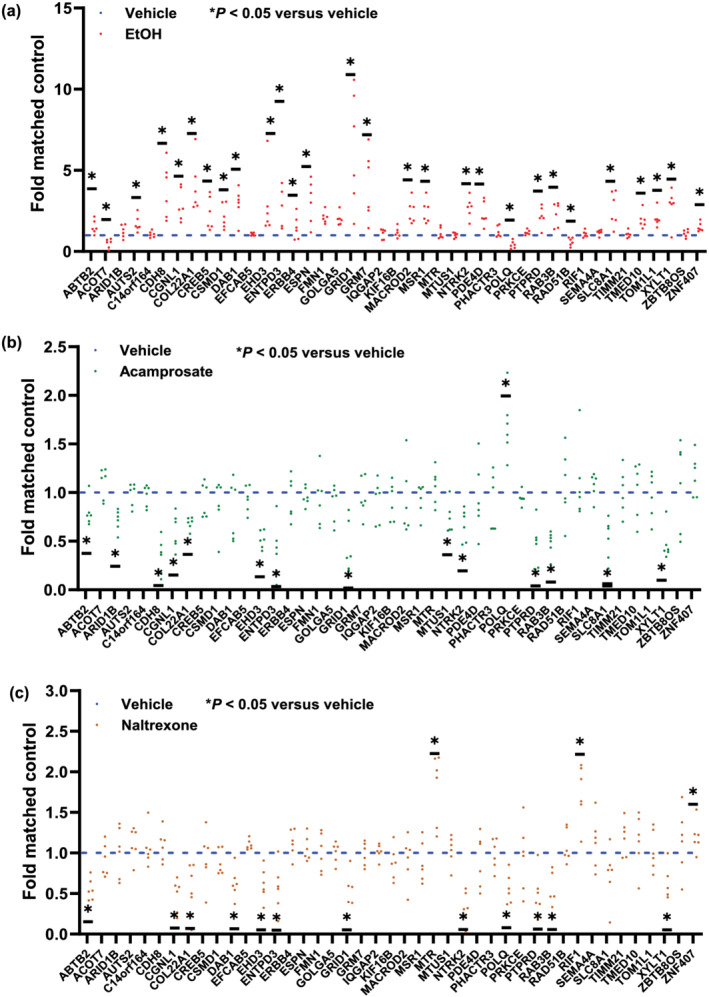
Effects of ethanol (a) or the anti‐craving drugs, acamprosate (b) or naltrexone (c), on gene expression in iPSC‐derived forebrain astrocytes for genes identified by the pharmacometabolomics‐informed GWAS shown in Figure 3. Real‐time PCR (n = 6) was used to determine mRNA expression before and after drug exposure. Specifically, forebrain‐specific astrocytes derived from iPSCs were treated with 25 mM of ethanol (EtOH), a concentration that is considered physiologically relevant for EtOH use, with 25 mM of EtOH being slightly higher than the 0.08% blood alcohol concentration often used as a measure of intoxication (Lira et al., 2020). The concentrations of acamprosate (5 μM) and naltrexone (30 nM) used to perform these experiments were selected to fall within the range of blood drug concentrations in patients taking standard clinical doses of these two drugs (Mason et al., 2002). Real‐time PCR results were analysed using 2−ΔΔct method. As a result, the control mean is 1, and there is no variance in the control. Real‐time PCR data were then analysed using non‐parametric statistical tests (Mann–Whitney U test). *P < 0.05, significantly different as indicated
As a first step, we used the STRING database to identify a protein–protein integration network using the genes listed in Figure 4, all of which were identified in the course of our metabolomics‐informed GWAS. Of interest is the fact that we found that the PTPRD gene might be functionally important because it appeared to be the ‘hub’ protein in the network with the highest degree of connectivity with other proteins in the module (Figure S3). Furthermore, a series of genes shown in Figure 2d (PTPRD, AUTS2, GRM7 , ERBB4 and ESPN) that were identified in the ethanolamine GWAS also appeared to have either direct or indirect protein–protein interactions. As a result, we set out to determine whether SNPs within the PTPRD gene might be associated with acamprosate treatment outcomes (Karpyak et al., 2014, 2019). We found that the four most significant PTPRD SNPs (rs12001871, rs10122491, rs12349713 and rs12348723), all with MAF ~ 14%, were in tight linkage disequilibrium (R 2 ≥ 0.98) and were associated with time until first alcohol use during 3 months of acamprosate treatment (see Figure 5a and Table S6). The wild‐type genotypes for these SNPs were associated with longer abstinence length until first drink during 3 months of acamprosate treatment. Those same four SNPs were also associated with a series of drug response phenotypes, that is, time until heavy drinking, return to alcohol use (binary variable) or return to heavy alcohol use (binary variable) during 3 months of acamprosate treatment (Figure 5b). Taken together, this series of studies illustrated that the experimental approach that we had taken might represent a potentially important step towards generating functional hypotheses that could be tested to gain insight into molecular mechanisms underlying acamprosate treatment response phenotypes.
4. DISCUSSION
AUD is the most prevalent substance use disorder globally, and, like many other chronic diseases, it typically requires long‐term treatment and care with a goal of preventing relapse (Carvalho et al., 2019). Among common primary outcome measures for AUD treatment are per cent of days abstinent from alcohol and time to first heavy drinking (Anton et al., 2006; Karpyak et al., 2014; Mann et al., 2018). Like most psychiatric disorders, AUD is diagnosed primarily on the basis of symptoms included in a list of diagnostic criteria (i.e., Diagnostic and Statistical Manual for Mental Disorders, 5th edition), rather than through the use of biologically based biomarkers. The present study was designed to use metabolomics data as a quantitative biological trait as a step towards identifying biomarkers that might be associated with acamprosate treatment outcomes. Specifically, we performed targeted metabolomic assays to identify genes/signals that might contribute to individual variation in metabolomic markers that were associated with acamprosate treatment response and/or alcohol craving in AUD patients. We then performed GWAS for metabolomic markers associated with acamprosate treatment outcomes and/or alcohol craving intensity, followed by initial functional studies of the genes identified (Figure 2).
We identified a series of baseline plasma metabolite concentrations, including those for histidine, threonine, ethanolamine, hydroxyproline, arginine and α‐aminoadipic acid, that were associated with alcohol relapse risk, although those observations require replication. Our acamprosate study is the only existing AUD study cohort that includes genomics, metabolomics, transcriptomics, clinical data and acamprosate treatment outcome data. Little is known about the possible role of these metabolites in the pathophysiology of AUD or acamprosate treatment response. In the present study, we replicated our previous findings for serum threonine and α‐aminoadipic acid, in this case using plasma rather than serum, and were able to identify additional metabolites that were associated with acamprosate treatment outcomes. However, to our knowledge, no prior studies have reported associations of elevated plasma histidine, ethanolamine, hydroxyproline or arginine with acamprosate treatment response. It should be pointed out that, in addition to acamprosate treatment outcome (Figure 1b), baseline arginine, α‐aminoadipic acid, threonine and ethanolamine concentrations were also correlated with baseline alcohol craving scores (Table 2). Among these metabolites, only arginine has been reported to be associated with alcohol withdrawal symptoms in a rodent model (Uzbay & Erden, 2003). Specifically, Uzbay and Erden (2003) reported that l‐arginine, an NO precursor, had beneficial effects on ethanol withdrawal symptoms in rats. However, the biological role of arginine in AUD disease risk and/or acamprosate treatment response has not been systematically addressed. Our data showed that elevated arginine was found in the non‐relapse group (Figure 1b), and those values were negatively associated with craving scores (Table 2). We did not observe that any SNPs achieved genome‐wide significance in our GWAS for arginine concentrations. That may be due, at least in part, to the fact that the effect size is too small to detect in this relatively small sample. However, we should once again point out that our study is currently the only study with both genomic and metabolomic data from patients with AUD and acamprosate treatment outcome information. Future studies will be required to investigate the potential role of these metabolites in molecular mechanisms underlying AUD pathophysiology and individual variation in drug treatment response.
Craving intensity has been associated with AUD treatment response (Schneekloth et al., 2012; Subbaraman et al., 2013; Verheul et al., 2005). In line with those previous reports, the present study showed that baseline craving intensity was associated with acamprosate treatment outcomes (Table 1). However, the mechanism of action of acamprosate relative to anti‐craving remains unclear. In addition to arginine, α‐aminoadipic acid, threonine and ethanolamine—all of which were associated with acamprosate treatment response—together with concentrations of several other metabolites, as shown in Table 2, were correlated with baseline craving intensity as determined by PACS scores. We recognized that the effect sizes appear to be minimal. Therefore, caution is necessary when interpreting results of the correlation analyses. Obviously, future studies will be required to explore additional metabolites within these pathways. Once again, there are, to our knowledge, no prior studies addressing the biological roles of these metabolites in AUD disease risk and/or acamprosate treatment response. As a result, we have identified genetic variants associated with the concentrations of these metabolites that may contribute to or be associated with variation in acamprosate response. These findings may also help to stimulate the study of molecular mechanisms involved in variation in AUD drug response phenotypes, in this case, response to acamprosate therapy. Specifically, we observed that several SNPs reached or were near genome‐wide significance, with P values ≤ 5 × 10−8 in the GWAS for metabolites associated with either acamprosate treatment response or alcohol craving intensity (Figures 2 and 3)—although these observations must be replicated. We observed that several proteins identified in our GWAS for baseline ethanolamine concentration might display protein–protein interactions based on the STRING database. It should also be pointed out that several genes involved in the protein interactive network have been implicated in substance use disorders (Narita et al., 2016; Uhl et al., 2008; Yeung et al., 2017). For example, the autism susceptibility candidate 2 (AUTS2) gene has implications for alcohol consumption (Narita et al., 2016). Several genetic variants (rs10085696, rs13229395 and rs6943555) in AUTS2 have been associated with alcohol consumption (P ≤ 5 × 10−8, n = 1,039,210) (Liu et al., 2019). The protein tyrosine phosphatase receptor type D (PTPRD) has been implicated in alcohol dependence (Yeung et al., 2017) and cocaine reward (Uhl et al., 2018). Both ethanol and acamprosate can regulate the expression of PTPRD in iPSC‐derived forebrain astrocytes generated from AUD participants (see Figure 4). Finally, we identify PTPRD SNPs that were associated with acamprosate treatment outcomes. Taken as a whole, these results help to demonstrate that the application of a pharmacometabolomics‐informed pharmacogenomics strategy could help to prioritize genes for subsequent functional mechanistic studies—as has already been done successfully during our previous studies of selective serotonin reuptake inhibitors in the treatment of major depressive disorder (Gupta et al., 2016; Liu et al., 2018).
Our study also has limitations. We should point out that our acamprosate clinical trial was not originally designed to study metabolomic biomarkers, but rather genomic biomarkers associated with acamprosate treatment response (Karpyak et al., 2014). Therefore, blood samples were collected as the source of DNA, and plasma samples were stored in the Mayo Clinic Biobank for future studies such as those reported here. The present study was designed as an extension of our previous preliminary serum metabolomics study, with a focus on amino acid and neuromodulator metabolomics panels (Hinton et al., 2017; Nam et al., 2015). We know that concentrations of amino acids and their derivatives could be influenced by diet, a variable that we found difficult to control. It should also be emphasized that plasma metabolites may not directly reflect metabolite levels in the brain. However, some of these metabolites may be predictors for acamprosate treatment response in patients with AUD and, if so, relevant pathways require further exploration with regard to their possible role in disease pathophysiology. Future studies should also be performed to explore the value of the assay of additional metabolites. A larger prospective study that includes functional genomic experiments should be conducted to validate and replicate our findings. However, the application of a pharmacometabolomics‐informed pharmacogenomic research strategy might make it possible to identify genetic variants contributing to variation in acamprosate response (Gupta et al., 2016). Finally, we used human iPSC‐derived forebrain astrocytes to perform our functional genomic studies. Those cell lines, like any cell lines, also have limitations; that is, iPSC‐derived brain cells are ‘region specific’. The present study utilized forebrain‐specific astrocytes, cells that have been implicated in the pathophysiology of AUD (Bradshaw et al., 2017; Ho et al., 2020; Swift & Aston, 2015). Future studies that include different brain cell types and different brain regions will be required to pursue the results reported here.
In summary, we have compared metabolomic profiles between patients who maintained sobriety and those who relapsed based on any alcohol use during 3 months of acamprosate treatment and identified novel genes or genetic variants that may be associated with variation in metabolite concentrations, thus contributing to variation in acamprosate treatment outcomes. These data suggest that the application of metabolomics may be a useful approach for identifying biological markers that could potentially predict drug treatment response as well as clinical variables such as craving. Finally, this union of genomics, metabolomics and transcriptomic, that is, of ‘multiple omics’, could help us to move beyond ‘biomarkers’ to novel hypotheses with regard to biological mechanisms underlying alcohol craving and drug response phenotypes.
AUTHOR CONTRIBUTIONS
M. Ho and R. Weinshilboum wrote the manuscript; all authors designed the research; M. Ho, C. Zhang and I. Moon performed the research; M. Ho, C. Zhang, L. Zhang, L. Wei, J. Geske, J. Biernacka and H. Li analysed the data; C. Zhang, L. Wei, J. Geske, J. Biernacka and H. Li contributed analytical tools. All authors have given final approval of the version to be published.
CONFLICT OF INTEREST
Dr Weinshilboum is a cofounder of and stockholder in OneOme LLC, a pharmacogenomics decision‐support company. Dr Choi is a scientific advisory board member for Peptron Inc. Dr Frye reports grant support from Assurex Health, Mayo Foundation, Medibio Consultant (Mayo)—Actify Neurotherapies, Allergan, Intra‐Cellular Therapies Inc., Janssen, Myriad, Neuralstem Inc., Takeda and Teva Pharmaceuticals. He also reports CME/travel/honoraria from the American Physician Institute, CME Outfitters and Global Academy for Medical Education. All other authors have no conflicts to declare.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for Design and Analysis, and Immunoblotting and Immunochemistry, and as recommended by funding agencies, publishers and other organizations engaged with supporting research.
Supporting information
Figure S1. Manhattan plots for the 6 GWAS for baseline plasma concentrations of metabolites that were associated with relapse during 3 months of acamprosate treatment. GWAS indicates genome‐wide association study.
Figure S2. Quantile‐quantile plots for the GWAS for plasma concentrations of metabolites that were associated with alcohol craving. GWAS indicates genome‐wide association study.
Figure S3. A protein‐protein interaction network of genes identified in the metabolomics‐informed GWAS shown in Figure 4 was constructed by using the STRING database (https://string-db.org/). The number of lines indicates the strength of predicted functional interactions between the proteins using a Markov Cluster Algorithm. The color of the lines connecting the nodes indicates the particular lines of evidence used to establish a functional association. There is no particular meaning of the node color itself because they are used only as a visual aid. The distance between the nodes is a measure of the confidence of the interaction as determined by a Bayesian scoring system.
Table S1. A list of metabolites included in the current study.
Table S2. Plasma concentrations of metabolites in relapse and non‐relapse groups. Logistic regression models of 3‐month relapse to any drinking were used to examine each metabolite individually as a predictor of relapse. Models examined included unadjusted (univariate) models, with relapse (yes/no) as the outcome and the metabolite as the predictor. Two types of multivariable logistic regression models were examined, both with relapse (yes/no) as the outcome. The first was a model adjusted for gender, still including metabolites as predictors. The second model type was a multivariable model that included gender and the metabolite as main effects (predictors), as well as a gender‐metabolite interaction term. There were no significant gender‐metabolite interactions in the prediction of relapse at an uncorrected for multiple testing p‐value level of P = 0.05.
Table S3. Plasma concentrations of metabolites in the heavy relapse and non‐heavy relapse groups. Logistic regression models of 3‐month relapse to heavy drinking were used to examine each metabolite individually as a predictor of relapse. Models examined included unadjusted (univariate) models, with heavy relapse (yes/no) as the outcome and the metabolite as the predictor. Subsequently two types of multivariable logistic regression models were examined, both with heavy relapse (yes/no) as the outcome. The first was a model adjusted for gender, still including the metabolite as a predictor. The second model type examined was a multivariable model including gender, with the metabolites as main effects (predictors), as well as a gender‐metabolite interaction term. No significant gender‐metabolite interactions with the exception of Hydroxylysine 2 was observed (uncorrected for multiple testing p‐value = 0.0278).
Table S4. Sex‐specific differences in metabolite concentrations. P < 0.05 was considered statistically significant.
Table S5. Effects of ethanol (EtOH) or the anti‐craving drugs acamprosate or naltrexone on gene expression in iPSC‐derived forebrain astrocytes for genes identified in pharmacometabolomics‐informed GWAS shown in Figure 3. iPSC‐derived astrocytes (n = 6) were used to determine mRNA expression before and after drug exposure. Real‐time PCR results were analyzed using the 2−ddct method. As a result, the control mean is 1, and there is no variance in the control. Real‐time PCR data were then analyzed using non‐parametric statistical tests (Mann–Whitney U‐test). *P < 0.05, significantly different as indicated in bold.
Table S6. PTPRD SNPs associated with acamprosate treatment outcomes.
ACKNOWLEDGEMENTS
This work was supported in part by the National Institute of General Medical Sciences (R01GM28157, U19 GM61388), National Institute on Alcohol Abuse and Alcoholism (P20 AA17830, R21 AA25214, R01 AA27486, U01 AA27487, K01 AA28050), National Center for Advancing Translational Sciences (UL1TR000135), Mayo Clinic Center for Individualized Medicine, Terrance and Bette Noble Foundation and Mayo Clinic SC Johnson Genomics of Addiction Program.
Ho, M.‐F. , Zhang, C. , Wei, L. , Zhang, L. , Moon, I. , Geske, J. R. , Skime, M. K. , Choi, D.‐S. , Biernacka, J. M. , Oesterle, T. S. , Frye, M. A. , Seppala, M. D. , Karpyak, V. M. , Li, H. , & Weinshilboum, R. M. (2022). Genetic variants associated with acamprosate treatment response in alcohol use disorder patients: A multiple omics study. British Journal of Pharmacology, 179(13), 3330–3345. 10.1111/bph.15795
Acamprosate: Gene associated with response. ClinicalTrials.gov Identifier: NCT00662571.
Funding information National Institute of General Medical Sciences, Grant/Award Numbers: R01GM28157, U19 GM61388; National Institute on Alcohol Abuse and Alcoholism, Grant/Award Numbers: P20 AA17830, R21 AA25214, R01 AA27486, U01 AA27487, K01 AA28050; National Center for Advancing Translational Sciences, Grant/Award Number: UL1TR000135; Mayo Clinic Center for Individualized Medicine Terrance and Bette Noble Foundation Mayo Clinic SC Johnson Genomics of Addiction Program
Contributor Information
Ming‐Fen Ho, Email: ho.mingfen@mayo.edu.
Richard M. Weinshilboum, Email: weinshilboum.richard@mayo.edu.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request. Individual researchers may request the data for specific projects on a collaborative basis. Some data may not be made available because of privacy or ethical restrictions.
REFERENCES
- Alexander, S. P. H. , Christopoulos, A. , Davenport, A. P. , Kelly, E. , Mathie, A. , Peters, J. A. , Veale, E. L. , Armstrong, J. F. , Faccenda, E. , Harding, S. D. , Pawson, A. J. , Sharman, J. L. , Southan, C. , Davies, J. A. , & CGTP Collaborators . (2019). The Concise Guide to PHARMACOLOGY 2019/20: G protein‐coupled receptors. British Journal of Pharmacology, 176, S21–S141. 10.1111/bph.14748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H., Roberts, R. E., Broughton, B. R. S., Sobey, C. G., George, C. H., Stanford, S. C., Cirino, G., Docherty, J. R., Giembycz, M. A., Hoyer, D., Insel, P. A., Izzo, A. A., Ji, Y., MacEwan, D. J., Mangum, J., Wonnacott, S., & Ahluwalia, A. (2018). Goals and practicalities of immunoblotting and immunohistochemistry: A guide for submission to the British Journal of Pharmacology. British Journal of Pharmacology, 175(3), 407–411. 10.1111/bph.14112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anton, R. F. , O'Malley, S. S. , Ciraulo, D. A. , Cisler, R. A. , Couper, D. , Donovan, D. M. , Gastfriend, D. R. , Hosking, J. D. , Johnson, B. A. , LoCastro, J. S. , Longabaugh, R. , Mason, B. J. , Mattson, M. E. , Miller, W. R. , Pettinati, H. M. , Randall, C. L. , Swift, R. , Weiss, R. D. , Williams, L. D. , & Zweben, A. (2006). Combined pharmacotherapies and behavioral interventions for alcohol dependence: The combine study: A randomized controlled trial. Jama, 295, 2003–2017. 10.1001/jama.295.17.2003 [DOI] [PubMed] [Google Scholar]
- Biernacka, J. M., Coombes, B. J., Batzler, A., Ho, A.‐C., Geske, J. R., Frank, J., Hodgkinson, C., Skime, M., Colby, C., Zillich, L., Pozsonyiova, S., Ho, M.‐F., Kiefer, F., Rietschel, M., Weinshilboum, R., O’Malley, S. S., Mann, K., Anton, R., Goldman, D., & Karpyak, V. M. (2021). Genetic contributions to alcohol use disorder treatment outcomes: A genome‐wide pharmacogenomics study. Neuropsychopharmacology, 46(12), 2132–2139. 10.1038/s41386-021-01097-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blendberg, J. M. , Àrnadottir, S. , Tarp, K. , & Bilberg, R. (2020). Gender differences in alcohol treatment. Alcohol and Alcoholism. 10.1093/alcalc/agaa071 [DOI] [PubMed] [Google Scholar]
- Boothby, L. A. , & Doering, P. L. (2005). Acamprosate for the treatment of alcohol dependence. Clinical Therapeutics, 27, 695–714. 10.1016/j.clinthera.2005.06.015 [DOI] [PubMed] [Google Scholar]
- Bradshaw, S. D. , Shumway, S. T. , Dsauza, C. M. , Morris, N. , & Hayes, N. D. (2017). Hope, coping skills, and the prefrontal cortex in alcohol use disorder recovery. The American Journal of Drug and Alcohol Abuse, 43, 591–601. 10.1080/00952990.2017.1286500 [DOI] [PubMed] [Google Scholar]
- Carvalho, A. F. , Heilig, M. , Perez, A. , Probst, C. , & Rehm, J. (2019). Alcohol use disorders. The Lancet, 394, 781–792. 10.1016/S0140-6736(19)31775-1 [DOI] [PubMed] [Google Scholar]
- Cavicchioli, M. , Vassena, G. , Movalli, M. , & Maffei, C. (2020). Is craving a risk factor for substance use among treatment‐seeking individuals with alcohol and other drugs use disorders? A meta‐analytic review. Drug and Alcohol Dependence, 212, 108002. 10.1016/j.drugalcdep.2020.108002 [DOI] [PubMed] [Google Scholar]
- Cheng, H.‐Y. , McGuinness, L. A. , Elbers, R. G. , MacArthur, G. J. , Taylor, A. , McAleenan, A. , McGuinness, L. A. , Elbers, R. G. , MacArthur, G. J. , Taylor, A. , McAleenan, A. , Dawson, S. , López‐López, J. A. , Higgins, J. P. T. , Cowlishaw, S. , Lingford‐Hughes, A. , Hickman, M. , & Kessler, D. (2020). Treatment interventions to maintain abstinence from alcohol in primary care: Systematic review and network meta‐analysis. BMJ (Clinical Research Ed), 371, m3934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis, M. J. , Alexander, S. , Cirino, G. , Docherty, J. R. , George, C. H. , Giembycz, M. A. , Hoyer, D. , Insel, P. A. , Izzo, A. A. , Ji, Y. , MacEwan, D. J. , Sobey, C. G. , Stanford, S. C. , Teixeira, M. M. , Wonnacott, S. , & Ahluwalia, A. (2018). Experimental design and analysis and their reporting II: Updated and simplified guidance for authors and peer reviewers. British Journal of Pharmacology, 175, 987–993. 10.1111/bph.14153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erol, A. , & Karpyak, V. M. (2015). Sex and gender‐related differences in alcohol use and its consequences: Contemporary knowledge and future research considerations. Drug and Alcohol Dependence, 156, 1–13. 10.1016/j.drugalcdep.2015.08.023 [DOI] [PubMed] [Google Scholar]
- Flannery, B. A. , Poole, S. A. , Gallop, R. J. , & Volpicelli, J. R. (2003). Alcohol craving predicts drinking during treatment: An analysis of three assessment instruments. Journal of Studies on Alcohol, 64, 120–126. 10.15288/jsa.2003.64.120 [DOI] [PubMed] [Google Scholar]
- Frye, M. A. , Hinton, D. J. , Karpyak, V. M. , Biernacka, J. M. , Gunderson, L. J. , Feeder, S. E. , Choi, D. S. , & Port, J. D. (2016). Anterior cingulate glutamate is reduced by acamprosate treatment in patients with alcohol dependence. Journal of Clinical Psychopharmacology, 36, 669–674. 10.1097/JCP.0000000000000590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frye, M. A. , Hinton, D. J. , Karpyak, V. M. , Biernacka, J. M. , Gunderson, L. J. , Geske, J. , Feeder, S. E. , Choi, D. S. , & Port, J. D. (2016). Elevated glutamate levels in the left dorsolateral prefrontal cortex are associated with higher cravings for alcohol. Alcoholism: Clinical and Experimental Research, 40, 1609–1616. 10.1111/acer.13131 [DOI] [PubMed] [Google Scholar]
- Grant, B. F. , Goldstein, R. B. , Saha, T. D. , Chou, S. P. , Jung, J. , Zhang, H. , Pickering, R. P. , Ruan, W. J. , Smith, S. M. , Huang, B. , & Hasin, D. S. (2015). Epidemiology of DSM‐5 alcohol use disorder: Results from the National Epidemiologic Survey on Alcohol and Related Conditions III. JAMA Psychiatry, 72, 757–766. 10.1001/jamapsychiatry.2015.0584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant, B. F. , Stinson, F. S. , Dawson, D. A. , Chou, S. P. , Dufour, M. C. , Compton, W. , Pickering, R. P. , & Kaplan, K. (2004). Prevalence and co‐occurrence of substance use disorders and independent mood and anxiety disorders: Results from the National Epidemiologic Survey on Alcohol and Related Conditions. Archives of General Psychiatry, 61, 807–816. 10.1001/archpsyc.61.8.807 [DOI] [PubMed] [Google Scholar]
- Gupta, M. , Neavin, D. , Liu, D. , Biernacka, J. , Hall‐Flavin, D. , Bobo, W. V. , Frye, M. A. , Skime, M. , Jenkins, G. D. , Batzler, A. , Kalari, K. , Matson, W. , Bhasin, S. S. , Zhu, H. , Mushiroda, T. , Nakamura, Y. , Kubo, M. , Wang, L. , Kaddurah‐Daouk, R. , & Weinshilboum, R. M. (2016). TSPAN5, ERICH3 and selective serotonin reuptake inhibitors in major depressive disorder: Pharmacometabolomics‐informed pharmacogenomics. Molecular Psychiatry, 21, 1717–1725. 10.1038/mp.2016.6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammarberg, A. , Jayaram‐Lindström, N. , Beck, O. , Franck, J. , & Reid, M. S. (2009). The effects of acamprosate on alcohol‐cue reactivity and alcohol priming in dependent patients: A randomized controlled trial. Psychopharmacology, 205, 53–62. 10.1007/s00213-009-1515-6 [DOI] [PubMed] [Google Scholar]
- Hinton, D. J. , Vázquez, M. S. , Geske, J. R. , Hitschfeld, M. J. , Ho, A. M. C. , Karpyak, V. M. , Biernacka, J. M. , & Choi, D. S. (2017). Metabolomics biomarkers to predict acamprosate treatment response in alcohol‐dependent subjects. Scientific Reports, 7, 2496–2496. 10.1038/s41598-017-02442-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho, M.‐F. , Zhang, C. , Zhang, L. , Wei, L. , Zhou, Y. , Moon, I. , Geske, J. R. , Choi, D. S. , Biernacka, J. , Frye, M. , Wen, Z. , Karpyak, V. M. , Li, H. , & Weinshilboum, R. (2020). TSPAN5 influences serotonin and kynurenine: Pharmacogenomic mechanisms related to alcohol use disorder and acamprosate treatment response. Molecular Psychiatry, 26, 3122–3133. 10.1038/s41380-020-0855-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt, G. E. , Malhi, G. S. , Lai, H. M. X. , & Cleary, M. (2020). Prevalence of comorbid substance use in major depressive disorder in community and clinical settings, 1990–2019: Systematic review and meta‐analysis. Journal of Affective Disorders, 266, 288–304. 10.1016/j.jad.2020.01.141 [DOI] [PubMed] [Google Scholar]
- Ji, Y. , Schaid, D. J. , Desta, Z. , Kubo, M. , Batzler, A. J. , Snyder, K. , Mushiroda, T. , Kamatani, N. , Ogburn, E. , Hall‐Flavin, D. , Flockhart, D. , Nakamura, Y. , Mrazek, D. A. , & Weinshilboum, R. M. (2014). Citalopram and escitalopram plasma drug and metabolite concentrations: Genome‐wide associations. British Journal of Clinical Pharmacology, 78, 373–383. 10.1111/bcp.12348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpyak, V. M. , Biernacka, J. M. , Geske, J. R. , Jenkins, G. D. , Cunningham, J. M. , Rüegg, J. , Kononenko, O. , Leontovich, A. A. , Abulseoud, O. A. , Hall‐Flavin, D. K. , Loukianova, L. L. , Schneekloth, T. D. , Skime, M. K. , Frank, J. , Nöthen, M. M. , Rietschel, M. , Kiefer, F. , Mann, K. F. , Weinshilboum, R. M. , … Choi, D. S. (2014). Genetic markers associated with abstinence length in alcohol‐dependent subjects treated with acamprosate. Translational Psychiatry, 4, e453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpyak, V. M. , Geske, J. R. , Hall‐Flavin, D. K. , Loukianova, L. L. , Schneekloth, T. D. , Skime, M. K. , Seppala, M. , Dawson, G. , Frye, M. A. , Choi, D. S. , & Biernacka, J. M. (2019). Sex‐specific association of depressive disorder and transient emotional states with alcohol consumption in male and female alcoholics. Drug and Alcohol Dependence, 196, 31–39. [DOI] [PubMed] [Google Scholar]
- Lira, M. C. , Sarda, V. , Heeren, T. C. , Miller, M. , & Naimi, T. S. (2020). Alcohol policies and motor vehicle crash deaths involving blood alcohol concentrations below 0.08. American Journal of Preventive Medicine, 58, 622–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litten, R. Z. , Falk, D. E. , Ryan, M. L. , Fertig, J. , & Leggio, L. (2020). Five priority areas for improving medications development for alcohol use disorder and promoting their routine use in clinical practice. Alcoholism: Clinical and Experimental Research, 44, 23–35. [DOI] [PubMed] [Google Scholar]
- Liu, D. , Ray, B. , Neavin, D. R. , Zhang, J. , Athreya, A. P. , Biernacka, J. M. , Bobo, W. V. , Hall‐Flavin, D. K. , Skime, M. K. , Zhu, H. , Jenkins, G. D. , Batzler, A. , Kalari, K. R. , Boakye‐Agyeman, F. , Matson, W. R. , Bhasin, S. S. , Mushiroda, T. , Nakamura, Y. , Kubo, M. , … Weinshilboum, R. M. (2018). Beta‐defensin 1, aryl hydrocarbon receptor and plasma kynurenine in major depressive disorder: Metabolomics‐informed genomics. Translational Psychiatry, 8, 10–23. 10.1038/s41398-017-0056-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, M. , Jiang, Y. , Wedow, R. , Li, Y. , Brazel, D. M. , Chen, F. , Datta, G. , Davila‐Velderrain, J. , McGuire, D. , Tian, C. , Zhan, X. , 23andMe Research Team; HUNT All‐In Psychiatry , Choquet, H. , Docherty, A. R. , Faul, J. D. , Foerster, J. R. , Fritsche, L. G. , Gabrielsen, M. E. , Gordon, S. D. , … Vrieze, S. (2019). Association studies of up to 1.2 million individuals yield new insights into the genetic etiology of tobacco and alcohol use. Nature Genetics, 51, 237–244. 10.1038/s41588-018-0307-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann, K. , Roos, C. R. , Hoffmann, S. , Nakovics, H. , Leménager, T. , Heinz, A. , & Witkiewitz, K. (2018). Precision medicine in alcohol dependence: A controlled trial testing pharmacotherapy response among reward and relief drinking phenotypes. Neuropsychopharmacology: Official Publication of the American College of Neuropsychopharmacology, 43, 891–899. 10.1038/npp.2017.282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason, B. J. , Goodman, A. M. , Dixon, R. M. , Hameed, M. H. A. , Hulot, T. , Wesnes, K. , Hunter, J. A. , & Boyeson, M. G. (2002). A pharmacokinetic and pharmacodynamic drug interaction study of acamprosate and naltrexone. Neuropsychopharmacology, 27, 596–606. 10.1016/S0893-133X(02)00368-8 [DOI] [PubMed] [Google Scholar]
- Mason, B. J. , & Lehert, P. (2012). Acamprosate for alcohol dependence: A sex‐specific meta‐analysis based on individual patient data. Alcoholism, Clinical and Experimental Research, 36, 497–508. 10.1111/j.1530-0277.2011.01616.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHugh, R. K. , Fitzmaurice, G. M. , Griffin, M. L. , Anton, R. F. , & Weiss, R. D. (2016). Association between a brief alcohol craving measure and drinking in the following week. Addiction (Abingdon, England), 111, 1004–1010. 10.1111/add.13311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittelstrass, K. , Ried, J. S. , Yu, Z. , Krumsiek, J. , Gieger, C. , Prehn, C. , Roemisch‐Margl, W. , Polonikov, A. , Peters, A. , Theis, F. J. , Meitinger, T. , Kronenberg, F. , Weidinger, S. , Wichmann, H. E. , Suhre, K. , Wang‐Sattler, R. , Adamski, J. , & Illig, T. (2011). Discovery of sexual dimorphisms in metabolic and genetic biomarkers. PLoS Genetics, 7, e1002215. 10.1371/journal.pgen.1002215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam, H. W. , Karpyak, V. M. , Hinton, D. J. , Geske, J. R. , Ho, A. M. C. , Prieto, M. L. , Biernacka, J. M. , Frye, M. A. , Weinshilboum, R. M. , & Choi, D. S. (2015). Elevated baseline serum glutamate as a pharmacometabolomic biomarker for acamprosate treatment outcome in alcohol‐dependent subjects. Translational Psychiatry, 5, e621. 10.1038/tp.2015.120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narita, S. , Nagahori, K. , Nishizawa, D. , Yoshihara, E. , Kawai, A. , Ikeda, K. , & Iwahashi, K. (2016). Association between AUTS2 haplotypes and alcohol dependence in a Japanese population. Acta Neuropsychiatrica, 28, 214–220. 10.1017/neu.2015.70 [DOI] [PubMed] [Google Scholar]
- Pałucha‐Poniewiera, A. , & Pilc, A. (2012). Involvement of mGlu5 and NMDA receptors in the antidepressant‐like effect of acamprosate in the tail suspension test. Progress in Neuro‐Psychopharmacology and Biological Psychiatry, 39, 102–106. 10.1016/j.pnpbp.2012.05.015 [DOI] [PubMed] [Google Scholar]
- Qian, X. , Jacob, F. , Song, M. M. , Nguyen, H. N. , Song, H. , & Ming, G.‐l. (2018). Generation of human brain region–specific organoids using a miniaturized spinning bioreactor. Nature Protocols, 13, 565–580. 10.1038/nprot.2017.152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneekloth, T. D. , Biernacka, J. M. , Hall‐Flavin, D. K. , Karpyak, V. M. , Frye, M. A. , Loukianova, L. L. , Stevens, S. R. , Drews, M. S. , Geske, J. R. , & Mrazek, D. A. (2012). Alcohol craving as a predictor of relapse. The American Journal on Addictions, 21, S20–S26. 10.1111/j.1521-0391.2012.00297.x [DOI] [PubMed] [Google Scholar]
- Shen, W. W. (2018). Anticraving therapy for alcohol use disorder: A clinical review. Neuropsychopharmacology Reports, 38, 105–116. 10.1002/npr2.12028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stohs, M. E. , Schneekloth, T. D. , Geske, J. R. , Biernacka, J. M. , & Karpyak, V. M. (2019). Alcohol craving predicts relapse after residential addiction treatment. Alcohol and Alcoholism, 54, 167–172. 10.1093/alcalc/agy093 [DOI] [PubMed] [Google Scholar]
- Subbaraman, M. S. , Lendle, S. , van der Laan, M. , Kaskutas, L. A. , & Ahern, J. (2013). Cravings as a mediator and moderator of drinking outcomes in the COMBINE study. Addiction, 108, 1737–1744. 10.1111/add.12238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swift, R. M. , & Aston, E. R. (2015). Pharmacotherapy for alcohol use disorder: Current and emerging therapies. Harvard Review of Psychiatry, 23, 122–133. 10.1097/HRP.0000000000000079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhl, G. R. , Drgon, T. , Liu, Q.‐R. , Johnson, C. , Walther, D. , Komiyama, T. , Harano, M. , Sekine, Y. , Inada, T. , Ozaki, N. , Iyo, M. , Iwata, N. , Yamada, M. , Sora, I. , Chen, C. K. , Liu, H. C. , Ujike, H. , & Lin, S. K. (2008). Genome‐wide association for methamphetamine dependence: Convergent results from 2 samples. Archives of General Psychiatry, 65, 345–355. 10.1001/archpsyc.65.3.345 [DOI] [PubMed] [Google Scholar]
- Uhl, G. R. , Martinez, M. J. , Paik, P. , Sulima, A. , Bi, G.‐H. , Iyer, M. R. , Gardner, E. , Rice, K. C. , & Xi, Z. X. (2018). Cocaine reward is reduced by decreased expression of receptor‐type protein tyrosine phosphatase D (PTPRD) and by a novel PTPRD antagonist. Proceedings of the National Academy of Sciences, 115, 11597–11602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uzbay, I. T. , & Erden, B. F. (2003). Attenuation of ethanol withdrawal signs by high doses of L‐arginine in rats. Alcohol and Alcoholism, 38, 213–218. 10.1093/alcalc/agg075 [DOI] [PubMed] [Google Scholar]
- Vadodaria, K. C. , Ji, Y. , Skime, M. , Paquola, A. , Nelson, T. , Hall‐Flavin, D. , Fredlender, C. , Heard, K. J. , Deng, Y. , Le AT, D. S. , Fung, L. , Li, X. , Marchetto, M. C. , Weinshilboum, R. , & Gage, F. H. (2019). Serotonin‐induced hyperactivity in SSRI‐resistant major depressive disorder patient‐derived neurons. Molecular Psychiatry, 24, 795–807. 10.1038/s41380-019-0363-y [DOI] [PubMed] [Google Scholar]
- Vadodaria, K. C. , Ji, Y. , Skime, M. , Paquola, A. C. , Nelson, T. , Hall‐Flavin, D. , Heard, K. J. , Fredlender, C. , Deng, Y. , Elkins, J. , Dani, K. , Le AT, M. M. C. , Weinshilboum, R. , & Gage, F. H. (2019). Altered serotonergic circuitry in SSRI‐resistant major depressive disorder patient‐derived neurons. Molecular Psychiatry, 24, 808–818. 10.1038/s41380-019-0377-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verheul, R. , Lehert, P. , Geerlings, P. J. , Koeter, M. W. , & van den Brink, W. (2005). Predictors of acamprosate efficacy: Results from a pooled analysis of seven European trials including 1485 alcohol‐dependent patients. Psychopharmacology, 178, 167–173. 10.1007/s00213-004-1991-7 [DOI] [PubMed] [Google Scholar]
- Witkiewitz, K. , Litten, R. Z. , & Leggio, L. (2019). Advances in the science and treatment of alcohol use disorder. Science Advances, 5, eaax4043. 10.1126/sciadv.aax4043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung, E. W. , Craggs, J. G. , & Gizer, I. R. (2017). Comorbidity of alcohol use disorder and chronic pain: Genetic influences on brain reward and stress systems. Alcoholism, Clinical and Experimental Research, 41, 1831–1848. 10.1111/acer.13491 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Manhattan plots for the 6 GWAS for baseline plasma concentrations of metabolites that were associated with relapse during 3 months of acamprosate treatment. GWAS indicates genome‐wide association study.
Figure S2. Quantile‐quantile plots for the GWAS for plasma concentrations of metabolites that were associated with alcohol craving. GWAS indicates genome‐wide association study.
Figure S3. A protein‐protein interaction network of genes identified in the metabolomics‐informed GWAS shown in Figure 4 was constructed by using the STRING database (https://string-db.org/). The number of lines indicates the strength of predicted functional interactions between the proteins using a Markov Cluster Algorithm. The color of the lines connecting the nodes indicates the particular lines of evidence used to establish a functional association. There is no particular meaning of the node color itself because they are used only as a visual aid. The distance between the nodes is a measure of the confidence of the interaction as determined by a Bayesian scoring system.
Table S1. A list of metabolites included in the current study.
Table S2. Plasma concentrations of metabolites in relapse and non‐relapse groups. Logistic regression models of 3‐month relapse to any drinking were used to examine each metabolite individually as a predictor of relapse. Models examined included unadjusted (univariate) models, with relapse (yes/no) as the outcome and the metabolite as the predictor. Two types of multivariable logistic regression models were examined, both with relapse (yes/no) as the outcome. The first was a model adjusted for gender, still including metabolites as predictors. The second model type was a multivariable model that included gender and the metabolite as main effects (predictors), as well as a gender‐metabolite interaction term. There were no significant gender‐metabolite interactions in the prediction of relapse at an uncorrected for multiple testing p‐value level of P = 0.05.
Table S3. Plasma concentrations of metabolites in the heavy relapse and non‐heavy relapse groups. Logistic regression models of 3‐month relapse to heavy drinking were used to examine each metabolite individually as a predictor of relapse. Models examined included unadjusted (univariate) models, with heavy relapse (yes/no) as the outcome and the metabolite as the predictor. Subsequently two types of multivariable logistic regression models were examined, both with heavy relapse (yes/no) as the outcome. The first was a model adjusted for gender, still including the metabolite as a predictor. The second model type examined was a multivariable model including gender, with the metabolites as main effects (predictors), as well as a gender‐metabolite interaction term. No significant gender‐metabolite interactions with the exception of Hydroxylysine 2 was observed (uncorrected for multiple testing p‐value = 0.0278).
Table S4. Sex‐specific differences in metabolite concentrations. P < 0.05 was considered statistically significant.
Table S5. Effects of ethanol (EtOH) or the anti‐craving drugs acamprosate or naltrexone on gene expression in iPSC‐derived forebrain astrocytes for genes identified in pharmacometabolomics‐informed GWAS shown in Figure 3. iPSC‐derived astrocytes (n = 6) were used to determine mRNA expression before and after drug exposure. Real‐time PCR results were analyzed using the 2−ddct method. As a result, the control mean is 1, and there is no variance in the control. Real‐time PCR data were then analyzed using non‐parametric statistical tests (Mann–Whitney U‐test). *P < 0.05, significantly different as indicated in bold.
Table S6. PTPRD SNPs associated with acamprosate treatment outcomes.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request. Individual researchers may request the data for specific projects on a collaborative basis. Some data may not be made available because of privacy or ethical restrictions.