

Abstract
Robinow syndrome is characterized by a triad of craniofacial dysmorphisms, disproportionate-limb short stature and genital hypoplasia. A significant degree of phenotypic variability seems to correlate with different genes/loci. Disturbances of the non-canonical WNT-pathway have been identified as the main cause of the syndrome. Biallelic variants in ROR2 cause an autosomal recessive form of the syndrome with distinctive skeletal findings. Twenty-two patients with a clinical diagnosis of autosomal recessive Robinow syndrome were screened for variants in ROR2 using multiple molecular approaches. We identified 25 putatively pathogenic ROR2 variants, 16 novel, including single nucleotide variants and exonic deletions. Detailed phenotypic analyses revealed that all subjects presented with a prominent forehead, hypertelorism, short nose, abnormality of the nasal tip, brachydactyly, mesomelic limb shortening, short stature and genital hypoplasia in male patients. A total of 19 clinical features were present in more than 75% of the subjects, thus pointing to an overall uniformity of the phenotype. Disease-causing variants in ROR2, contribute to a clinically recognizable AR trait phenotype with multiple skeletal defects. A comprehensive quantitative clinical evaluation this cohort delineated the phenotypic spectrum of ROR2-related Robinow syndrome. The identification of exonic deletion variant alleles further supports the contention of a loss-of-function mechanism in the etiology of the syndrome.
Keywords: Skeletal dysplasia, craniofacial morphology, WNT pathway, next generation sequencing, chromosome microarray analysis, exonic deletion, HPO terms, quantitative phenotyping cluster heatmap
Graphical Abstract
Introduction
Robinow syndrome (RS) is characterized by a triad of craniofacial dysmorphism, disproportionate-limb short stature and genital hypoplasia, with an extensive degree of clinical variability (Robinow et al. 1969; Mazzeu et al. 2007). More pronounced skeletal involvement and marked short stature are observed in the autosomal recessive (AR) form of RS (AR-RS), initially described as COVESDEM syndrome (COstoVErtebral Segmentation DEfects with Mesomelia; MIM# 268310) (Wadia et al. 1978). AR-RS is mostly caused by biallelic variants in the tyrosine kinase-like orphan receptor gene ROR2 (Afzal et al. 2000; Van Bokhoven et al. 2000). Autosomal recessive inheritance has also been described in a rare form of RS with biallelic variants in NXN (MIM# 618529) (White et al. 2018; Zhang et al. 2020). Gain-of-function (GoF) ROR2 variants have been associated with autosomal dominant brachydactyly type B (MIM 113000) including nonsense/frameshift variants resulting in premature termination codons (PTC) mapping to the last coding exon, or −55 bp of the penultimate exon, that escape nonsense mediated decay (NMD) (Schwabe et al. 2000; Ben-Shachar et al. 2009).
The autosomal dominant forms of RS (AD-RS) are associated with a milder skeletal phenotype and are usually caused by heterozygous pathogenic variants in WNT5A (DRS1; MIM# 180700) (Person et al. 2010), DVL1 (DRS2; MIM# 616331) (Bunn et al. 2015; White et al. 2015), DVL3 (DRS3; MIM# 616894) (White et al. 2016), FZD2 (White et al. 2018; Zhang et al. 2020) or DVL2 (Zhang et al., 2022). Nevertheless, there is also one report of biallelic WNT5A variants in RS inherited from unaffected heterozygous parents (Birgmeier et al. 2018).
All genes associated with RS, play a role in the β-catenin-independent WNT/planar cell polarity (PCP) pathway. Therefore, despite the genetic heterogeneity, the genes implicated in causing RS to date converge on the WNT signaling pathway, resulting in a recognizable clinical syndrome (White et al. 2018, Zhang et al., 2022).
The receptor tyrosine kinase-like orphan receptors (RORs) are involved in the regulation of multiple biological processes during embryonic development, including development of axial and paraxial mesoderm, nervous system and neural crest, axial and appendicular skeleton, and kidneys. The characteristic skeletal phenotype of AR-RS includes vertebral malformations, which were observed in Ror2-null mouse model and are caused by the reduced size of presomitic mesoderm (Schwabe et al., 2004). Animal model studies have also identified several WNT pathway components in the mechanisms of craniofacial and limb formation (Nohno et al. 1999; Geetha-Loganathan et al. 2009; Sisson and Topczewski 2009).
RS-associated genes not only encode components in a common pathway, but the individual protein component directly interact with each other in signal transduction. WNT5A acts as a soluble extracellular ligand of ROR2; and together with FZD2 transmembrane receptor they trigger the DVL homologs to transduce the β-catenin independent pathway. The WNT5A-ROR2 pathway is a proposed additional branch of the non-canonical WNT-signaling network. Unlike the canonical WNT pathway, other branches of this signaling pathway are not well-defined, resulting in a paucity of information regarding constituent components (Stricker et al. 2017).
Facing the challenges in the clinical diagnosis of RS and in a first attempt to clinically differentiate the AR-RS and AD-RS forms, Mazzeu et al. (2007) investigated the frequency of clinical signs and symptoms in 88 patients with RS, considering rib fusions as indicative and potentially pathognomonic of the AR-RS form. However, despite the more severe bone involvement in AR-RS, rib fusion is not universally present in AR-RS, evident by its absence in a small proportion of molecularly-confirmed cases. (Mehawej et al. 2012; Aglan et al. 2015).
The identification of the causative genes in RS further illuminated the underlying patho-mechanism of disease and enhanced the understanding of potentially how the molecular lesions lead to the phenotypic expression. Molecular diagnosis together with quantitative deep phenotyping using Human Phenotype Ontology (HPO) terms and similarity analysis, have recently become a powerful tools for delineating disease contributing molecular pathways, the biology of disease, and the definition of the etiology of many syndromes, including RS (Zhang et al., 2022). A detailed phenotypic characterization of patients with an identified disease-causing variant allows more precise genotype-phenotype correlation, delineation of allele-specific phenotypic differences and increases the accuracy of clinical diagnosis and management.
There are few reports of AR-RS patients with confirmed molecular diagnosis. Thirty-two different ROR2 pathogenic variants have been identified in patients of different ethnicities (Suppl. Table S1). Most variants were located in exons 5, 6 and 9. While truncating variant mRNAs are degraded by nonsense-mediated decay (Ben-Shachar et al. 2009), mutant protein caused by missense variants are retained in the endoplasmic reticulum and ultimately lead to the absence of the ROR2 receptor (Chen et al., 205, Ali et al. 2007).
We report the genotype and detailed HPO-term based quantitative phenotypic analyses of 22 patients with biallelic ROR2 variants, aiming to further characterize and expand the phenotypic and genotypic spectrum of ROR2 related AR-RS.
Subject and Methods
Clinical data
Twenty-two patients with a clinical diagnosis of RS or RS-like phenotype were referred from different medical genetic clinics worldwide for identification of the causative variants and/or clarification of the definitive diagnosis for conditions with phenotypic overlap (Suppl. Figure S1, S2). Seventeen patients were referred by clinical geneticists. Five patients were evaluated by a genetic counselor specialized in the clinical phenotyping of RS, during the family conventions organized by the Robinow Syndrome Foundation (www.robinow.org). Informed consent/assent was obtained and pretest counselling was provided to all patients and/or their legal guardians. This study was approved by the institutional ethics committee of the Faculdade de Medicina, Universidade de Brasília (CEP FM: 079/2009; 25/11/2009), and the Institutional Review Board at Baylor College of Medicine (IRB protocols no. H-43246 and no. H-29697).
The clinical information of three patients was partially included in previous publications: A16 (Beiraghi et al. 2011); A6 and A21 (Abu-Ghname et al. 2020; Conlon et al. 2021; Gerber et al. 2020; Schwartz et al. 2020; Zhang et al. 2020; Shayota et al. 2020). Patients A2, A5, A6, A8, A9 and A16 were included in the clinical review by Mazzeu et al. (2007).
Clinical data were collected using a standardized table including all clinical signs present in more than 25% of the patients with AR-RS, according to Mazzeu et al. (2007). Detailed family history, anthropometric data, radiographic images and other investigations and results were obtained during the consultation or from patient’s clinical records.
Quantitative phenotypic analyses based on Human Phenotype Ontology (HPO) terms
Phenotypes were annotated with HPO terms for each affected individual (N=22). All diseases (n = 8,114, including number symbol, plus sign, percent sign, and no symbol in OMIM) and genes (n = 4,216, asterisk symbol in OMIM) that have been annotated with HPO terms by OMIM were downloaded from the Human Phenotype Ontology resource page (https://hpo.jax.org/app/download/annotation). Individual similarity matrices were generated with the OntologyX suite of R packages using the Lin’s semantic similarity score and the average method (Lin, 1998; Liu et al., 2019). Similarity matrices were then used to generate distance matrices of individual similarity. Hierarchical agglomerative clustering (HAC) was performed on distance matrices with the Ward’s method (Ward, 1963) with the number of clusters set based on visualization of the gap statistic curve. Individual similarity scores were visualized using the ComplexHeatmap package in R, and statistical analysis of individual groups was done using the OntologyX suite. Annotation grids were generated with the OntologyX suite of packages, and then edited to exclude ancestral terms and to order columns by phenotype frequency. A cohort-to-gene and cohort-to-disease HPO analysis was performed.
These 22 individuals, 21 unrelated research subjects with RS, were separately assessed for phenotypic similarity to all 1) genes and 2) diseases with OMIM HPO annotation. HPO-annotated phenotypes for the 22 individuals were queried against all disease-associated genes (n=4,216) or all diseases (n=8,114) annotated with HPO terms by OMIM for phenotypic similarity. Lin semantic similarity scores between all pairs of the 22 individuals and all genes or diseases annotated with HPO terms were calculated. The top 10 phenotypically similar gene-associated or disease HPO term sets to each disease in the group of 31 diseases described above was parsed and duplicates removed. Every combination of 2 that includes 1 member from the group of 22 individuals and 1 from the top phenotypically similar gene associated phenotype matches was taken, and p-value calculated via comparison of the phenotypic similarity score between that group of 2 and 100,000 randomly selected groups of 2 from all OMIM HPO annotated genes or diseases, respectively (p-value cutoff < 0.001). The gap statistic was calculated for cluster number k = 1 to 11 (gene analysis) or 8 (disease analysis), and the resultant curve was visualized to select optimal number of clusters to use. HAC analysis and visualization of phenotypic similarity and clustering was then performed as described above for RS proband phenotypes.
Analysis of variant type-associated phenotypes
The 22 ROR2 probands were categorized into two groups based on variant type. The missense group (N=7), composed of all individuals carrying bi-allelic missense variants, and the loss of function (LoF) group, composed of all individuals carrying bi-allelic LoF (nonsense and frameshifting) variants. Individuals with other variant types were not included in this analysis because of limited numbers (N<3). Prevalence of each phenotype in each group was calculated. Prevalence of each phenotype across all 22 individuals and prevalence of each phenotype across all probands published in Mazzeu et al. 2007 (Mazzeu et al., 2007) were also included. Patient prevalence of each phenotype in each group were visualized by using the ComplexHeatmap package in R language.
Molecular analysis
DNA was extracted from peripheral blood or saliva lymphocytes, according to standard laboratory procedures. Screening approaches for ROR2 variants (Table 1) included targeted Sanger sequencing (patients A1, A2, A4, A6, A7, A8, A9, A16, A17 and A21), next generation sequencing (NGS) panels (patients A3, A5, A10, A11, A14 and A19), exome sequencing (ES) (patients A12, A13, A20 and A22), Multiplex Ligation-dependent Probe Amplification (MLPA) (patients A4 and A11) and chromosome microarray (A4 and A11).
Table.I -.
ROR2 variants identified in 22 patients with a clinical diagnosis of the recessive form of Robinow syndrome
| Patient | Disease causing variants | Protein effect | Zygosity | Novel? (Y/N) | CADD SNV PHRED | ACMG/AMP classification | Inheritance | Method |
|---|---|---|---|---|---|---|---|---|
| A1 | c.323G>A | p.(Arg108Gln) | Hom | N | 25.7 | Likely Pathogenic PM2/PP4/PP5 | Parents not tested | Sanger sequencing |
| A2 | c.323G>A | p.(Arg108Gln) | Hom | N | 25.7 | Likely Pathogenic PM2/PP4/PP5 | Parents not tested | Sanger sequencing |
| A3 | c.355C>T | p.(Arg119*) | Hom | N | 35.0 | Pathogenic PVS1/PM2/PP4/PP5 | Parents not tested | NGS panel |
| A4 | Seq[GRCh37]/hg19]del(9)(q22.31) chr9:g.94498192_94519323del |
p.? | Hom | Y | - | Uncertain +0.45 | Heterozygous parents | Sanger sequencing /MLPA/CMA |
| A5 | c.613C>T c.1189C>T |
p.(Arg205*) p.(Arg397*) |
Comp Het | N, N | 36.0 40.0 |
Pathogenic PVS1/PM2/ PP4/PP5 Pathogenic PVS1/PM2/PP4/PP5 |
Parents not tested | NGS panel |
| A6 | c.899G>T c.990delC |
p.(Cys300Phe) p.(Thr331Profs*114) |
Comp Het | N, N | 27.4 - |
Likely Pathogenic PM2/PM3/PP3/PP4/PP5 Pathogenic PVS1/PM2/PP4/PP5 |
Mother heterozygous for c.899G>T. Father not tested | Sanger sequencing |
| A7 | c.717C>A | p.(Cys239*) | Hom | Y | 37.0 | Pathogenic PVS1/PM2/PP/4PP5 | Parents not tested | Sanger sequencing |
| A8 | c.675delG | p.(Gln225Hisfs*220) | Hom | Y | - | Pathogenic PVS1/PM2/PP4/PP5 | Heterozygous parents | Sanger sequencing |
| A9 | c.2074C>A | p.(Pro692Thr) | Hom | Y | 25.7 | Uncertain Significance PM2/PP3/PP4/PP5 | Heterozygous parents | Sanger sequencing |
| A10 | c.1516_1520delinsT | p.(Ile506*) | Hom | Y | - | Pathogenic PVS1/PM2/PP3/PP4 | Heterozygous parents | NGS panel |
| A11 | c.1970G>A Seq[GRCh37]/hg19]del(9)(q22.31) chr9:g.94371974_94844329del |
p.(Arg657His) - |
Comp Het | Y, Y | 31.0 - |
Uncertain significance PM2/PM3/PP4 Pathogenic +1.00 |
Parents not tested | NGS panel/MLPA/CMA |
| A12 | c.494+4_494+7del | - | Hom | Y | - | Uncertain significance PM2/PP3/PP4 | Parents not tested | Exome sequencing |
| A13 | c.2T>G | p.? | Hom | Y | 20.4 | Likely Pathogenic PVS1/PM2//PP4/PP5 | Heterozygous parents | Exome sequencing |
| A14 | c.79_80delTC | p.(Ser29Profs*5) | Hom | Y | - | Pathogenic PVS1/PM2/PP4 | Parents not tested | NGS panel |
| A15 | c.1855C>A c.2215T>C |
p.(Arg619Ser) p.(Phe739Leu) |
Comp Het | Y, Y | 26.4 25.7 |
Uncertain significance PM2/PP3/PP4/PP5 Uncertain significance PM2/PP3/PP4/PP5 |
Parents not tested | Sanger sequencing |
| A16 | c.1096C>T c.2207G>A |
p.(Arg366Trp) p.(Arg736Gln) |
Comp Het | N, Y | 29.1 28.1 |
Likely Pathogenic PM1/PM2/PM3/PP3/PP4/PP5 Likely Pathogenic PM2/ PM3/PP3/PP4/PP5 |
Father: c.1096C>T Mother: c.2207G>A |
Sanger sequencing |
| A17/A18 | c.623–11G>A | -- | Hom | Y | - | Uncertain significance PM2/PP4/PP5/BP7 | Heterozygous parents | Sanger sequencing |
| A19 | c.2074C>A | p.(Pro692Thr) | Hom | Y | 25.7 | Uncertain Significance PM2/PP3/PP4/PP5 | Heterozygous parents | NGS panel |
| A20 | c.1083delG c.1324C>T |
p.(His362Thrfs*83) p.(Arg442*) |
Comp Het | Y, N | - 36.0 |
Pathogenic PVS1/PM2/PM3/PP3/PP4/PP5 Pathogenic PVS1/PM2/PM3/PP4/PP5 |
Father:c.1324C>T Mother:c.1083delG |
Exome sequencing |
| A21 | c.904C>T c.1970G>A |
p.(Arg302Cys) p.(Arg657His) |
Comp Het | N, Y | 27.4 31.0 |
Uncertain Significance PM2/ PP4 Uncertain significance PM2/PP3/PP4 |
Father: c.904C>T Mother: c.1970G>A |
Sanger sequencing |
| A22 | c.1100A>T c.1189C>T |
p.(Asn367Ile) p.(Arg397*) |
Comp Het | Y, N | 26.5 40.0 |
Likely Pathogenic PM1/PM2/PM3/PP3/PP4/PP5 Pathogenic PVS1/PM2/PM3/PP4/PP5 |
Father: c.1189C>T Mother: c.1100A>T |
Exome sequencing |
Comp Het: Compound Heterozygous; Hom: Homozygous
All variants are represented in NM_004560.4. CADD model GRCh37-v1.6. ACMG/AMP classification automated using Franklin (Genoox) acessed on June 15th 2021.
Sanger sequencing of all ROR2 coding regions and intron-exon boundaries was performed as a first screening method for 10 patients, and for confirmation of the causative variants identified through next generation sequencing for the remaining subjects.
For four patients, screening was performed using the ION PGM™ Inherited Disease Panel, as described by the manufacturer. Library construction was carried out using the Inherited Disease Panel (IDP) and the Ion Ampliseq™ Library Kit 2.0 (Thermo Fisher Scientific, Carlsbad, USA), using 30ng/primer pool of genomic DNA, following the manufacturer’s recommendation. The amplicons were enriched with the Ion PGM™ Template OT2 200 Kit on the Ion OneTouch™ 2 instrument (OT2) and Ion OneTouch™ ES (Thermo Fisher Scientific, Carlsbad, USA). Sequencing was performed using the Ion PGM™ Sequencing 200 kit v2 on Ion Torrent PGM™ System using the Ion 318™ Chip v2 with two samples per microchip. Enrichment and sequencing were performed following the manufacturer’s recommendation. The data were processed using the Ion Torrent Suite™ Server using hg19 as the reference genome for alignment, and the Ion Reporter™ Software v5.2, for variant analysis (Thermo Fisher Scientific, Carlsbad, USA).
Samples from four patients were submitted to (ES) at the Human Genome Sequencing Center (HGSC) at Baylor College of Medicine, through the Baylor-Hopkins Center for Mendelian Genomics Initiative (Posey et al. 2019). ES was performed using 0.5ug of DNA in an Illumina (Illumina, San Diego, USA) paired-end pre-captured library constructed according to the manufacturer’s protocol with modifications. Six to ten pre-captured libraries were pooled and then hybridized in solution to the HGSC in-house developed VCRome 2.1 design with custom spike-in according to the manufacturer’s protocol NimbleGen SeqCap EZ Exome Library SR User’s Guide with minor revisions. Illumina sequencing was performed with a sequencing yield averaging 11Gb, samples achieved 97.5% of the targeted exome bases covered to a depth of 20´ or greater with an average depth of coverage of 118.6´. In parallel to the exome workflow, a cSNP array was generated for a final quality assessment. This included orthogonal confirmation of sample identity and purity using the Error Rate In Sequencing (ERIS) pipeline developed at the HGSC. Using an “e-GenoTyping” approach, ERIS screens all sequence reads for exact matches to probe sequences defined by the variant and position of interest. A successfully sequenced sample must meet quality control metrics of ERIS SNP array concordance (>90%) and ERIS average contamination rate (<5%). Two variant discovery methods were used in parallel in order to prevent bias in the filtering and parsing of variants starting with the HGSC Mercury analysis pipeline (Reid et al. 2014), which moves data from the initial sequence generation on the instrument to annotated variant call files (VCF) via various analysis tools including xAtlas for variant calling. In addition, we used the Genome Analysis Toolkit (GATK) HaplotypeCaller to produce joint called files with indel realignment and base recalibration in all patients that underwent ES. Candidate variants were filtered against exome data in publicly available databases, including the Genome Aggregation Database (gnomAD), the NHLBI Exome Sequencing Project (ESP) Exome Variant Server, the Atherosclerosis Risk in Communities Study (ARIC) database, and the internal Baylor-Hopkins Centers for Mendelian Genomics variant analyzer database (~13,000). In parallel, webtools were applied to parsed rare variant data that can predict functional effects of candidate variants into consideration, such as Polymorphism Phenotyping v2 (PolyPhen-2), Sorting Intolerant From Tolerant (SIFT), and Combined Annotation Dependent Depletion (CADD) (Adzhubei et al. 2010; Sim et al. 2012; Kircher et al. 2014). A B-allele frequency was calculated from ES data using BafCalculator (Eldomery et al. 2017) to delineate genomic intervals showing absence of heterozygosity (AOH) as a surrogate measure for Runs of Homozygosity (ROH) and consistent with identity by descent.
Pathogenicity was ascertained automatically using Franklin (Genoox) according to ACMG/AMP guidelines (Richards et al. 2015). Variants were scored for a likelihood of damaging effect, or deleteriousness, by combined annotation dependent depletion (CADD).
Screening for deletion and duplication variant alleles affecting ROR2 was performed by MLPA using kit P179 (MRC-Holland, Amsterdam, The Netherlands) in cases with no variant or only a single variant allele was identified by Sanger sequencing or NGS. Reactions were performed according to manufacturer’s protocol and analyses were performed using Coffalyser software.
Deletion in patient A11 was independently confirmed by chromosome microarray analysis, using the Cytoscan 750K platform (Thermo Scientific, Carlsbad, USA). The procedures for sample purification, hybridization and washing were those described by the manufacturer and analysis was performed using CHAS software (Thermo Scientific, Carlsbad, USA).
Customized aCGH in 4x180K format (AMADID# 086154, Agilent Technologies, Santa Clara, USA), which covers RS related genes, genes related to conditions within the differential diagnosis of RS, and genes in WNT signaling (837 mean probe space), was performed on genomic DNA isolated from blood obtained from subject A4 and A11. Experimental steps of aCGH, including DNA fragmentation, DNA labeling and clean-up, array hybridization, array washing, and scanning were performed as previously described (Beck et al. 2019). Junctions of deletions were then confirmed by conventional PCR and Sanger dideoxy capillary sequencing. The primers used were: 1) A4 ROR2 F: TGAAACCGTTCCCTAGGGCC; 2) A4 ROR2 R: GGACAATCTTGTGCCCTGGA; 3) A11 ROR2 F: CACCTCTTATGAGCCAGGCA; 4) A11 SPTLC1 R: CGAGACCAGCCTCAGCATG.
Patients A15, A17 and A19 were screened for ROR2 variants using NGS panels in different certified clinical laboratories. Patient 18 was screened by Sanger sequencing for the variant identified in her sister.
Construction of ROR2 mutants
The plasmid pcDNA3-Ror2WT-HA (Ali et al, 2007) served as a template to generate ROR2 mutants R108Q, R366W, P693T and R736Q. QuickChange™ Mutagenesis (Stratagene) was performed according to the manufacturer’s instructions using the following primers R108Q: Ror2-R108Q-F GTGCAAGAGCCACGACAGGTCGTCATCCGGAAG and Ror2-R108Q-R CTTCCGGATGACGACCTGTCGTGGCTCTTGCAC; R366W: Ror2-R366W-F -GGCCATGCCTACTGCTGGAACCCCGGGGGC and Ror2-R366W-R-GCCCCCGGGGTTCCAGCAGTAGGCATGGCC CTTCCGGATGACGACCTGTCGTGGCTCTTGCAC; P693T: Ror2-P693T-F CTTTAGCTACGGCCTGCAAACCTACTGTGGGTACTCC and Ror2-P693T-R GGAGTACCCACAGTAGGTTTGCAGGCCGTAGCTAAAG; R736Q: Ror2-R736Q-F GAGTTCCCAAGCCGGCAGCCCCGCTTTAAGGAC and Ror2-R736Q-R GTCCTTAAAGCGGGGCTGCCGGCTTGGGAACTC. The generated mutants were confirmed by Sanger sequencing.
Cell culture, transfection, and fluorescence microscopy
The protocols used have been described previously (Ali et al, 2007). HeLa cells were cultured in Dulbecco’s modified Eagle’s medium (Invitrogen) supplemented with 10% fetal calf serum, 2 mM L-glutamine, and 100 U/ml penicillin/streptomycin at 37°C with 10% CO2. For immunofluorescence, HeLa cells were grown on coverslips in a 24-well plate for 24 h and transiently transfected using the liposomal transfection reagent FuGENE 6 (Roche Biochemicals) according to the manufacturer’s instructions. In co-transfection, a mixture of 0.5 mg of EGFP-hRas, 1 mg of mRor2 wildtype or mutant DNA and 5 ml of FuGENE 6 in 94 ml of OPTIMEM I medium (Invitrogen) was applied to each well of the HeLa cells at about 60% confluence. The cells were then fixed and processed for microscopy 24 h later. For immunofluorescence, coverslip-grown HeLa cells were washed with phosphate-buffered saline (PBS), fixed in cold methanol at −20°C for 4 min, washed in PBS three times and incubated in 1X PBS containing 0.5% BSA for 15 min. The fixed cells were then incubated at room temperature for 1 h with either mouse monoclonal anti-HA antibody alone, or co-stained with both mouse monoclonal anti-HA antibody and rabbit polyclonal anti-calnexin antibody. After washing with PBS, the cells were incubated with the appropriate secondary antibodies for 1 h at room temperature, washed several times with PBS and mounted in Immuno Fluor medium (ICN Biomedicals), and visualized under a Leica DM-IRBE confocal microscope. Images were acquired using Leica TCS-NT software associated with the microscope and processed with Adobe Photoshop® (Adobe Inc.).
Immuno-localization of Ror2 mutant alleles
Mouse anti-HA-Tag monoclonal antibody was obtained from Cell Signaling Technology (USA) and used at 1:200 dilution for immunofluorescence, rabbit anti-calnexin polyclonal antibody from StressGen Biotechnologies and used at 1:500 dilution, Alexa Fluor 568-goat anti-mouse IgG and Alexa Fluor 488-goat anti-rabbit IgGs were from Molecular Probes and used at 1:200 dilution.
Results
We analyzed the genotype and phenotype of 22 patients (12 males and 10 females) from 21 unrelated families and from different ethnic backgrounds. Twelve patients have presumed consanguinity by clinical history. In four families the index cases had affected siblings (Suppl. Figure S1). The only sibling pair described in detail is the pair A17/ A18 for whom we had comprehensive clinical data on each affected family member.
ROR2 variant screening
Using different molecular approaches, biallelic causative variants were identified in all 22 patients (Table 1, Fig. 1A). All families with historical report of consanguinity presented homozygous alleles. BAF calculator provided further evidence for identity-by-descent and thus confirmed the consanguinity in the two homozygous cases where such data were available (A12 and A13) (Fig. 1B). In total, twenty-five different putatively pathogenic variants were found in 21 patients: including 10 missense, 5 nonsense, 5 small indels and two large deletions. Two patients had a splicing variant, and one patient had a variant affecting the initiation codon. Sixteen of them (64%) are novel, i.e. not yet reported, variants.
Figure 1.
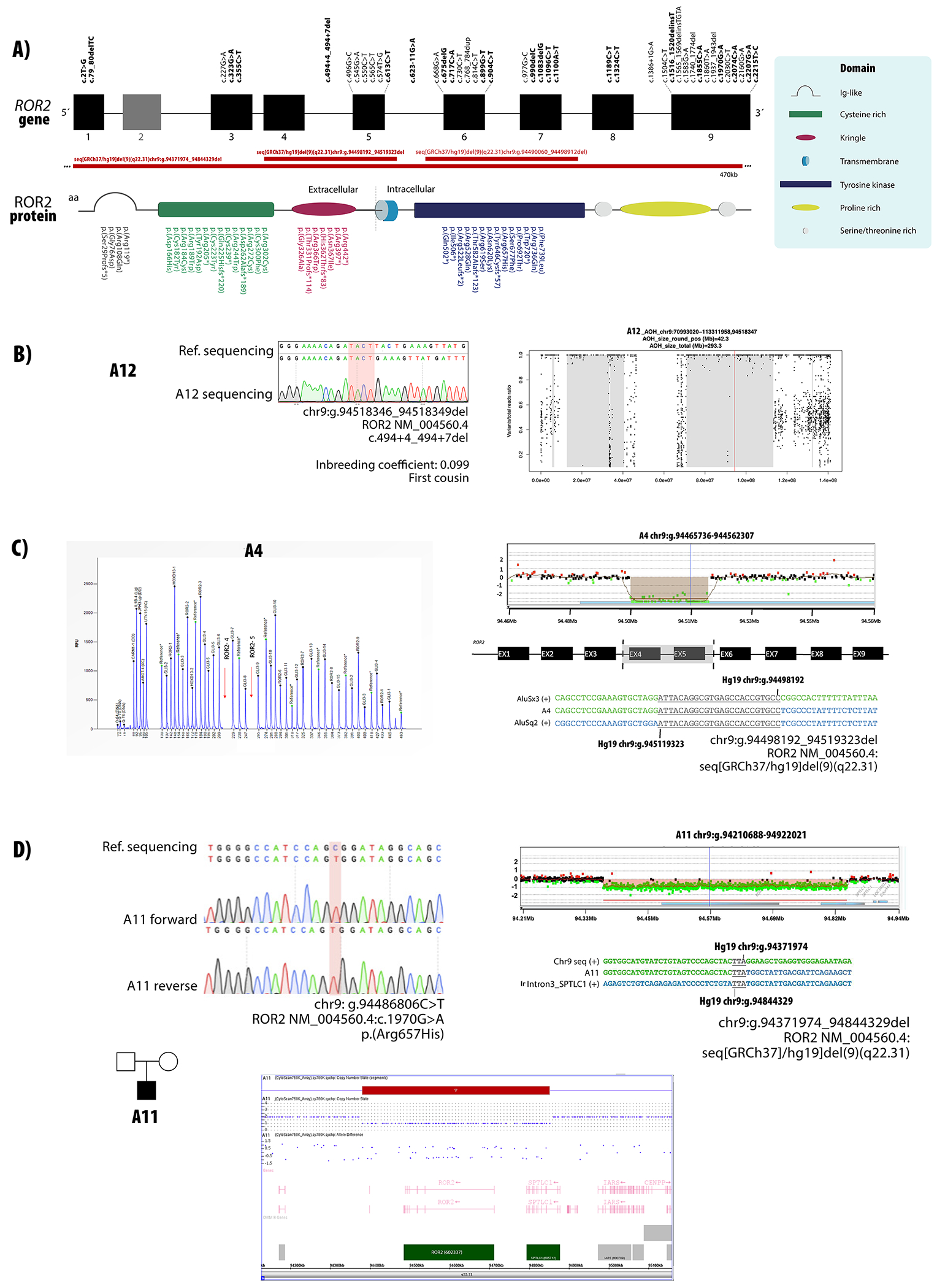
ROR2 variants causative of AR-RS. A) Localization of variants described in the present study and in previous reports in relation to their position in the exons and protein domains. Novel variants are highlighted in bold. B) Sanger sequencing chromatogram for patient A12 carrying a homozygous variant in ROR2. AOH plot shows that this variant is within an AOH region of 42.3 Mb. C) Electropherogram of P179 MLPA reaction showing a homozygous deletion of probes corresponding to exons 4 and 5 of ROR2. Customized array CGH plots confirms the 21 kb deletion. Breakpoints determined by Sanger sequencing are represented below. D) Sanger sequencing chromatogram for patient A11 carrying a hemizygous variant in ROR2. Chromosome microarray analysis showing a 470 kb deletion including whole ROR2 and partial SPTLC1 genes. Breakpoints determined by Sanger sequencing are represented below.
In patient A4, exons 4 and 5 of ROR2 could not be amplified by PCR, which suggest an exonic deletion. MLPA was then performed and confirmed that these two exons were deleted in both alleles (c.(431_494+60)_(574_672)del). Customized array CGH followed by Sanger sequencing mapped the breakpoints within introns 3 and 5 Seq[GRCh37]del(9)(q22.31) NC_000009.11:g.94498192_94519323del) (Fig. 1C).
In Patient A11 (Fig. 1D), a single variant (c.1970G>A, p.Arg657His), was called as homozygous by NGS analysis software. Since there was no reported history of consanguinity, and only one allele was detected for other SNPs in ROR2, copy-number investigation by MLPA and chromosome microarray were performed, and a 470 kb deletion encompassing the ROR2 and SPTLC1 genes was identified - arr[hg19] 9q22.31(94,381,136-94,851,388)x1. Customized array CGH followed by Sanger sequencing confirmed the proximal breakpoint upstream of ROR2 and the distal breakpoint at intron 3 of SPTLC1 (Seq[GRCh37] del(9)(q22.31) NC_000009.11:g.94371974_94844329del).
In eight unrelated patients, compound heterozygous variants were identified. In four families (A16, A20, A21 and A22) Sanger sequencing of both parents confirmed that the variants were in trans. For A6, TA cloning of exons 6 and 7 amplified as a single PCR product confirmed that the two variants were present in trans. Patient A11 had compound heterozygosity for a large deletion and a missense variant. Parental samples of the patients A5 and A15 were not available (Table 1).
Characterization of DNA rearrangement alleles at the ROR2 locus
Detailed characterization of the large deletions allowed precise identification of the breakpoints. In patient A11, the proximal breakpoint maps to an intergenic region upstream of ROR2 and distal breakpoint maps to SPTLC1 intron 3. A TTA microhomology at the breakpoint junction (Fig. 1D) suggests that the deletion arose by microhomology-mediated break induced replication (MMBIR) or by non-homologous end-joining (Carvalho and Lupski 2016).
Patient A4 has an exonic deletion of exons 4 and 5 of ROR2. Breakpoint mapping confirmed the breakpoints as intronic, revealing a 21 kb genomic deletion, thus confirming deletion of both exons. A 24 bp region within Alu elements was identical in both introns (Fig. 1C), suggesting that this deletion was mediated by AAMR (Alu-Alu mediated rearrangement). This rearrangement is similar to Alu-mediated CNVs reported at other disease loci, due to breakpoint microhomology, small size and rearrangement between different Alu family members (Song et al. 2018). The genomic instability score for ROR2 as calculated from AluAluCNVpredictor, (http://alualucnvpredictor.research.bcm.edu:3838/), 0.506 for RefSeq genes, suggests the possibility for encountering other exonic deletion alleles at this locus.
Immuno localization of Ror2 mutant alleles
We generated mutant Ror2 alleles for four different missense variants identified in our cohort (R108Q, and R366W, located at the extracellular part of Ror2 receptor and P692T and R736Q located at the intracellular part of Ror2 receptor). Co-expression of Ror2 and EGFP-hRas or calnexin in Hela-cells showed that wild-type Ror2 localizes predominantly to the plasma membrane, while mutant Ror2 proteins do not migrate to the plasma membrane and are retained to the endoplasmic reticulum (Fig. S3, S4). The results did not differ for mutations localized at the intracellular or extracellular domains (data not shown).
Phenotype analysis
Phenotypes of the 22 patients with biallelic variants in ROR2 are summarized in Table 2 (Detailed phenotype described in Suppl. Table S2) and the photographs from available patients are shown in Fig. 2. All patients manifested at least 16/61 clinical features; 11 clinical features were present in more than 90% of the cohort, and 19 features, in more than 75%, pointing to an overall consistent phenotype.
Table.II -.
Clinical signs and symptoms in patients with biallelic ROR2 variants grouped according to their frequency.
| Frequency % | Clinical signs |
|---|---|
| 75-100 | Prominent forehead (100%) |
| Hypertelorism (100%) | |
| Wide nasal bridge (95.45%) | |
| Short nose (100%) | |
| Abnormality of the nasal tip (100%) | |
| Anteverted nares (95.45%) | |
| Midface retrusion (95.45%) | |
| Downturned corners of mouth (77.27%) | |
| Gengival overgrowth (77.27%) | |
| Bifid tongue (77.27%) | |
| Abnormalities of the dentition (75%) | |
| Short stature (100%) | |
| Mesomelia (100%) | |
| Short palms (86.36%) | |
| Clinodactyly (80,95%) | |
| Brachydactyly (100%) | |
| Rib fusion (86.36%) | |
| Hemivertebrae (86.36%) | |
| Micropenis (100%) | |
| 50-74 | Proptosis (63.63%) |
| Long eyelashes (72.72%) | |
| Long palpebral fissures (68.18%) | |
| Upslanted palpebral fissures (50%) | |
| Depressed nasal bridge (68.18%) | |
| Long philtrum (50.00%) | |
| Triangular mouth (72.72%) | |
| Thin upper lip vermilion (59.09%) | |
| Micrognathia (68.18%) | |
| Retrognathia (68.18%) | |
| Low-set ears (66.66%) | |
| Short neck (63.63%) | |
| Scoliosis (68.18%) | |
| Limited pronation/ supination of forearm (65%) | |
| Broad thumbs (59.09%) | |
| Cryptorchidism (50.00%) | |
| 25-49 | Dowslanted palpebral fissures (31.81%) |
| Epicanthus (31.81%) | |
| Short philtrum (27.27%) | |
| Hypoplasia of the tongue (35.29%) | |
| Highly narrow palate (41,17%) | |
| Tooth agenesis (30.00%) | |
| Pectus excavatum (47.61%) | |
| Nail dysplasia (38.09%) | |
| Syndactyly (28.57%) | |
| Single transversal palmar crease (26.31%) | |
| Broad halux (47.05%) | |
| Hypoplastic labia minora (36.36%) | |
| 5-24 | Melanocytic nevus (20.00%) |
| Ptosis (13,63%) | |
| Strabismus (19.04%) | |
| Oral cleft (22.72%) | |
| Microtia (9,52%) | |
| Camptodactyly (19.04%) | |
| Hip dislocation (15.78%) | |
| Hypospadias (20.00%) | |
| Hypoplastic labia majora (16.67%) | |
| Sacral dimple (5,26%) | |
| Inguinal hernia (15.78%) | |
| Abnormal heart morphology (18.18%) | |
| Abnormality of the kidney (14.28%) | |
| Hearing impairment (13.63%) |
Figure 2.

Facial and whole-body photographs of eight individuals from our cohort showing the spectrum of ROR2-related Robinow syndrome. All individuals exhibit typical dysmorphic features that characterize the syndrome. Patient A6 is shown at different ages to document the evolving facial gestalt.
Phenotypic features were classified in four groups, according to frequency: more than 75%, between 50-75%, between 25-50%, and below 25%. Prominent forehead, hypertelorism, short nose, abnormality of the nasal tip, brachydactyly, mesomelic limb shortening, short stature and micropenis were present in all patients. Midface retrusion, wide nasal bridge, anteverted nares, downslanted mouth corners, bifid tongue, gum hyperplasia, abnormalities of the dentition, short palms, clinodactyly, hemivertebrae and rib fusion were present in more than 75% of subjects. Therefore, these features should be considered as major defining phenotypic criteria in the clinical diagnosis of ROR2-related Robinow syndrome. Three patients did not present rib fusions, a sign formerly considered pathognomonic for AR-RS (Mazzeu et al. 2007). Intraoral manifestations were also prevalent (above 75%), including bifid tongue, gingival overgrowth and abnormalities of the dentition. Genital hypoplasia was present in all male patients, but in less than 50% of the females. Major congenital anomalies, such as abnormal heart and kidneys were present in less than 25% of the patients. Hypoplasia of the tongue was present in 35% of the patients and considered a novel phenotypic feature, not previously associated with AR-RS.
Quantitative assessment of RS clinical phenotypes
To quantify and visualize genotype-phenotype correlations, semantic similarity scores were calculated using an HPO-based analysis. Phenotypic similarity scores between each AR-RS proband and OMIM annotated gene phenotypes were calculated and visualized in a cluster heatmap.
Subjects with ROR2 variants in our cohort were clustered with DVL1, WNT5A, ROR2, DVL3 and NXN gene phenotypes (Fig. 3). Three different subclusters were observed: one cluster included patients A1, A2 and A21, all carrying missense variants only. The second subcluster included patients with at least one LoF allele (A3, A13, A6, A8, A14, A11, A20) and a third cluster was composed of two siblings (A17 and A18), who both carry biallelic splicing variants. No difference was observed between patients with compound heterozygous variants and patients with homozygous variants. Variants affecting extracellular or intracellular domains did not cluster as shown in Fig 3., suggesting that domain localization does not contribute to clinical phenotypic variability in this cohort.
Figure 3.
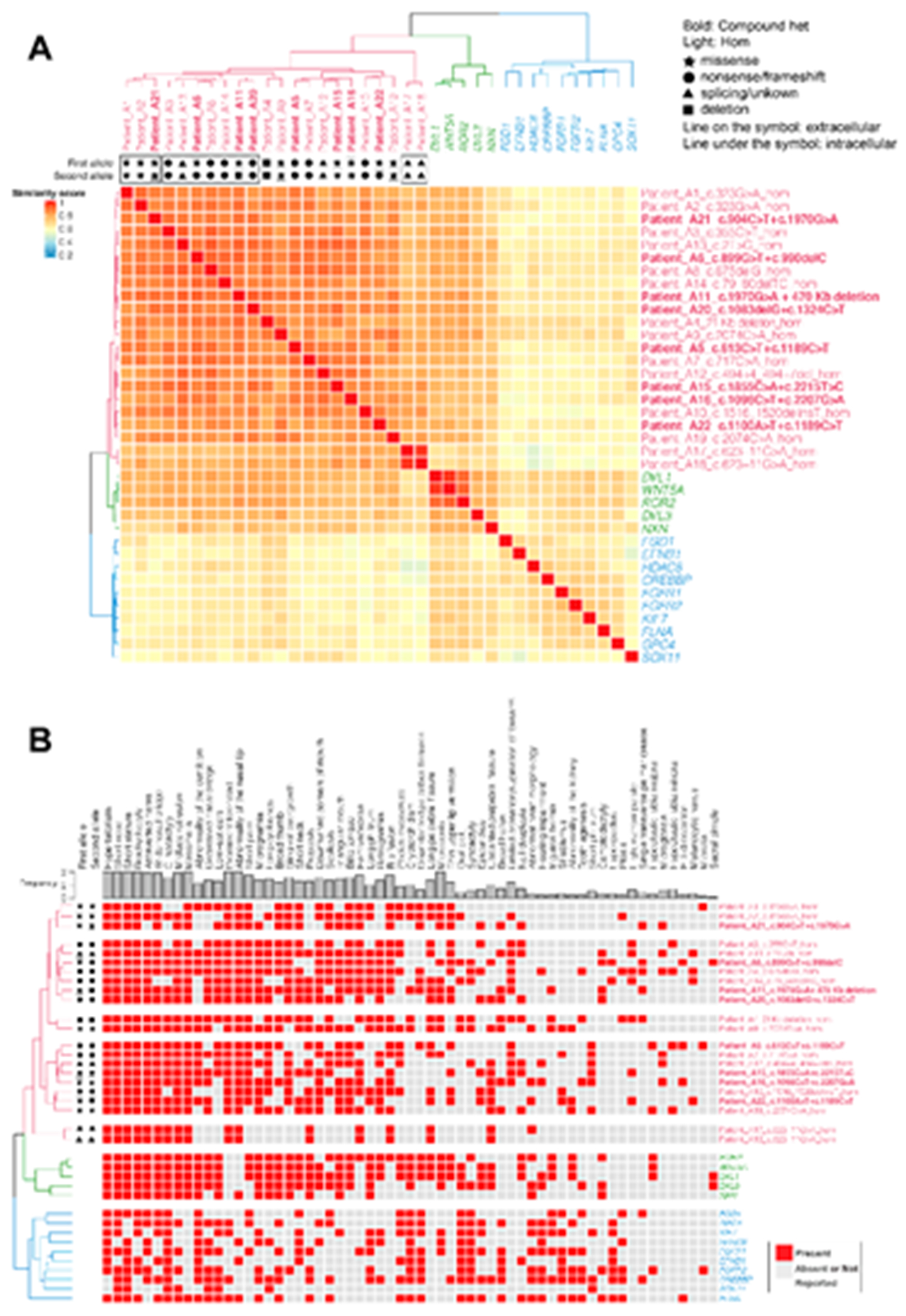
Semantic similarity heatmap and phenotypic annotation grid results between research subjects with biallelic ROR2 variants and significantly similar OMIM annotated known disease gene phenotypes
A. *Hierarchical agglomerative clustering (HAC) and visualization of quantitative phenotypic similarity. The dendrogram shown at the top and to the left of the heatmap is based on HAC analysis of the dissimilarity matrix produced from Lin semantic similarity scores and with k set to 3. Unique clusters are represented by different colors, individual probands and significantly similar known disease genes are labeled on top of and to the right of the heatmap. Within the heatmap, dark red indicates a higher similarity while dark blue indicates lower similarity. A key is provided on the left. Bold: subjects who have compound heterozygous variant alleles. Light font type: subjects who have homozygous variant alleles. Star: missense variants. Circle: loss of function (LoF) variants including nonsense variants and frameshifting variant alleles. Triangle: splicing variants or variants with unknown consequence. Rectangle: large exonic deletion (> 50bp) variant alleles. Line on the symbol: variants in the extracellular region. Line under the symbol (i.e. underlined font): variants in the intracellular region. B. Phenotypic annotation grid. Phenotypic annotation grid of phenotypes of all subjects and significantly similar known disease genes. To interpret and understand biology of phenotypes driving semantic similarity in these analyses, HPO terms associated with all subjects and significantly similar known disease genes were annotated and visualized in a gridded array format. Red indicates presence of a phenotype while gray represents absence or not reported. Probands and significantly similar known disease genes are labeled to the right (italicized gene symbols) and are ordered by HAC. The frequency of each phenotype in probands from this cohort is shown on top of the grid.
To investigate whether there are mutation-type specific phenotypes as suggested by the initial heatmap analysis, we sorted the cohort for bi-allelic missense variants (N=7) and bi-allelic LoF variants (N=7) (Fig. 5, Suppl. Figure S5). Such analysis revealed that none of the patients in the missense group had camptodactyly, hypospadia or melanocytic nevus and long palpebral fissures, whereas low-set ears, micrognathia and retrognathia were less-frequent in this group. In contrast, patients with bi-allelic LoF variants do not present cryptorchidism, abnormal heart morphology, inguinal hernia and abnormality of the kidney whereas a few patients (N=3/7) had a broad thumb.
Figure 5.
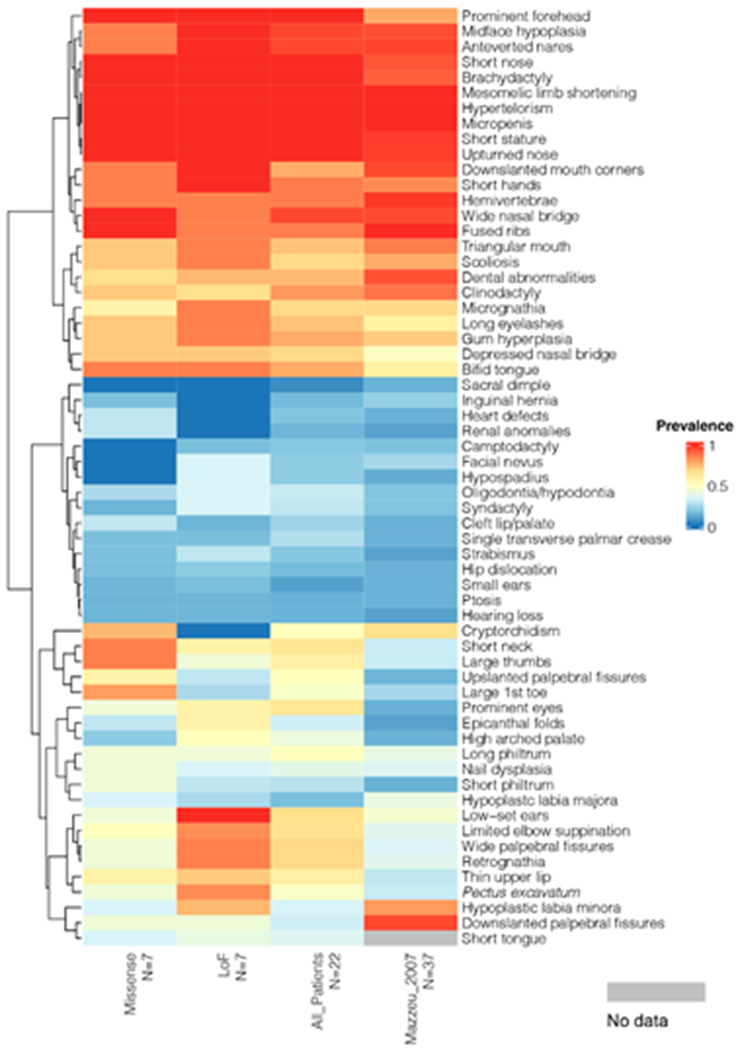
Phenotypic analysis of subjects with bi-allelic missense variants and LoF variants. Prevalence (0-1.0) of phenotypes in subjects with bi-allelic missense variants (A1, A2, A9, A15, A16, A19, A21), bi-allelic LoF variants (A3, A5, A7, A10, A14, A20), all subjects (N=22), and subjects published in Mazzeu et al. 2007 (N=37) is displayed by heatmap. Probands with other mutation types were not included in this analysis because of their limited numbers (N<3). Within the heatmap, red indicates a higher prevalence while blue indicates lower prevalence; light grey indicates these specific data are not available. The phenotypes are ordered by dendrogram shown on the left based on hierarchical agglomerative clustering (HAC) analysis. A prevalence key is provided on the right.
The overall clinical phenotype was consistent in all patients as the majority of clinical signs were present in all patients independently of the type of variant (Fig 5). However, some signs were more prevalent (difference value >28%) in the missense or LoF groups allowing discrimination between them (Fig 5).
Cryptorchidism, broad halux, broad thumbs, abnormal heart morphology, abnormalities of the kidney, short neck and upslanted palpebral fissures were more prevalent in patients with bi-allelic missense variants. Low-set ears, retrognathia, long palpebral fissures, hypoplastic labia minora, pectus excavatum, limited pronation/ supination of forearm, hypospadia, melanocytic nevus, highly narrow palate, micrognathia and epicanthus were more prevalent in the LoF group.
A similarity analysis between ROR2 subjects and OMIM annotated disease phenotypes showed that ROR2 subjects strongly clustered with other forms of RS caused by variants in WNT5A, DVL1, DVL3 and NXN. FZD2_OMOD2 grouped into a distinct cluster. Diseases that have phenotypic overlap with RS are matched using a less stringent p-value cutoff (p=0.005). This aided in viewing the similar sets of diseases to ROR2 patient phenotypes, however subclusters were more poorly resolved due to the increased number of phenotype sets to cluster.
Discussion
Here we report a cohort of 22 individuals with AR-RS caused by biallelic ROR2 variants. Twenty-five disease-causing variants in ROR2 were identified, and 16 of these were novel. Although most of the variants were missense, further description of frameshift, initiation site, splice-site variants and large exonic deletion adds to the evidence that the syndrome is caused by biallelic loss-of-function variants and to the mutational and allelic complexity for this rare disease trait.
Six of the detected variants have been previously described (Suppl. Table S1). The majority of the previous reports of AR-RS were from Turkey potentially due to the high frequency of consanguineous marriages. However, 13 different variants have been described in Turkish patients, which is inconsistent with a founder effect and more suggestive of Clan Genomics hypothesis proposing recently arisen biallelic rare alleles are more likely to be unmasked due to identity-by-descent homozygosity. (Lupski et al. 2011; Lupski 2021)
The ROR2 gene comprises nine exons. Disease-causing variants were more frequent in exons 5, 6 and 9, usually affecting the extracellular domains, though variants affecting the tyrosine kinase domain were also identified (Fig. 1A, Table 1). None of the variants modified interdomain regions which is consistent with previous studies showing that variants affecting interdomain regions can act as gain-of-function (GoF) alleles and cause brachydactyly type B (Schwabe et al. 2000). Considering all single nucleotide variants in ROR2 described in patients with AR-RS (Suppl. Table S1) most of them (22/36) occurred at CpG nucleotides. Cytosine residues in CpG dinucleotides might undergo modifications such as methylation, deamination, and halogenation that can contribute to the formation of mutational hotspots (Sassa et al. 2016). The preponderance of alleles involving CpG is also consistent with Clan Genomics and the derivation of the allele as a new mutation in antecedent generations of the clan that is then brought to homozygosity by identity-by-decent (IBD) (Lupski et al. 2011, Lupski 2021).
Patient A11 had a possible homozygous variant detected by NGS however the parents were not consanguineous. Further studies showed a large deletion of the other allele encompassing the entire ROR2 and part of SPTLC1 genes. Interestingly, heterozygous SPTLC1 variant alleles are associated with Hereditary Sensory and Autonomic Neuropathy type 1A (HSANIA; MIM# 162400). Whether this specific SPTLC1 exonic deletion allele behaves as a LoF or Gof mutation remains to be explored.
According to ACMG/AMP (Richards et al. 2015), 11 variants were classified as pathogenic, six as likely pathogenic and ten as uncertain significance. The variants classified as uncertain failed PM1 criteria for being out of mutational hotspots. In our cohort, disease-causing variants were identified throughout the gene except for exons 2 and 4, the smaller ROR2 exons. Therefore, we did not find evidence of mutational hotspots in ROR2 and so it seems that this PM1 classification criteria is not useful for ROR2 variant classification.
Phenotypic analysis comparing missense variants with LOF variants showed minor differences as depicted in Fig.5. These results are discordant with functional studies that demonstrate retention of ROR2 mutant proteins to the endoplasmic reticulum (Suppl. Figure S3 and S4, Ali et al., 2007) as well as loss-of-function alleles from absent mRNA due to degradation by NMD surveillance mechanism. (Ben-Shachar et al., 2009). This partial genotype-phenotype correlation might indicate a residual function of ROR2 in patients with missense variants.
Patients carrying variants that affect the extracellular or intracellular ROR2 domains do not cluster according to this feature as shown in Fig. 3, indicating that location of the variant in the protein may not be relevant for the overall clinical phenotype. This is consistent with the retention of all missense variants affecting intracellular or extracellular domains in the endoplasmic reticulum (Suppl. Figure S3 and S4, Ali et al., 2007).
We also showed that subjects with ROR2 variants clustered with phenotypes associated with other non-ROR2 gene forms of the syndrome confirming the identity of Robinow syndrome as a single syndrome with genetic heterogeneity and confirming that disruption of this pathway leads to a specific group of phenotypes (Fig.4). This is in accordance with our recent phenotypic analysis of dominant RS showing that ROR2-RS was closely clustered with other gene forms of the syndrome (Zhang et al., 2021).
Figure 4.
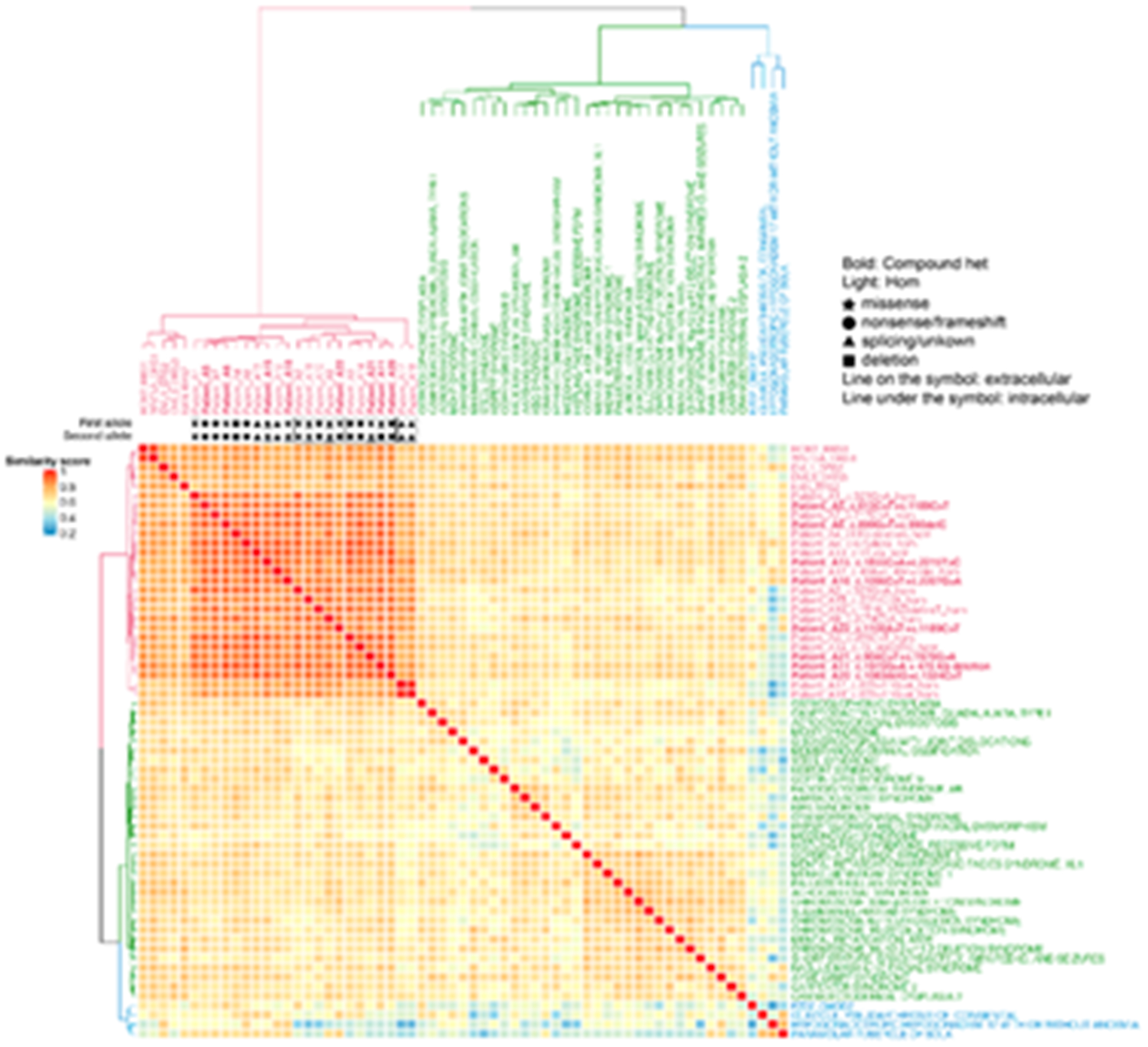
Semantic similarity heatmap between ROR2 subjects and significantly similar OMIM annotated known disease phenotypes (p<0.005).
The dendrogram shown at the top and to the left of the heatmap is based on HAC analysis of the dissimilarity matrix produced from Lin semantic similarity scores and with k set to 3. Unique clusters are represented by different colors, individual probands and significantly similar known diseases are labeled on top of and to the right of the heatmap. Within the heatmap, dark red indicates a higher similarity while dark blue indicates lower similarity. A key is provided on the left. Bold: subjects who have compound heterozygous variant alleles. Light font type: subjects who have homozygous variant alleles. Star: missense variants. Circle: loss of function (LoF) variants including nonsense variants and frameshifting variant alleles. Triangle: splicing variants or variants with unknown consequence. Rectangle: large exonic deletion (> 50bp) variant alleles. Line on the symbol: variants in the extracellular region. Line under the symbol (i.e. underlined font): variants in the intracellular region.
The overall phenotype of the patients reported herein is in accordance with the previous clinical characterization of AR-RS. Though some discrepancies were observed in relation to the report of Mazzeu et al. (2007), most clinical signs had similar frequencies in both studies (Table 2, Suppl. Table S2, Fig 5). Minor discrepancies appeared more evident in clinical signs with mild clinical impact that might have been unreported or overlooked but could still be present. Clinical signs present in all patients (prominent forehead, hypertelorism, short nose, abnormality of the nasal tip, brachydactyly, mesomelic limb shortening, short stature and micropenis) as well as those present in more than 75% of subjects (midface retrusion, wide nasal bridge, anteverted nares, downslanted mouth corners, bifid tongue, abnormalities of the dentition, short palms, clinodactyly, hemivertebrae and rib fusion) should be considered when evaluating variants of uncertain significance in ROR2.
All skeletal changes (short stature, brachydactyly, clinodactyly, mesomelia, rib fusion and hemivertebrae) had frequencies above 75%. Craniofacial characteristics were also consistent between different patients, including prominent forehead, hypertelorism, midface retrusion, wide nasal bridge, short nose, abnormality of the nasal tip, anteverted nares, and downturned corners of mouth likely providing the recognizable pattern allowing clinical diagnosis (Table 2).
As with many craniofacial disorders, facial characteristics become attenuated with age in RS patients. We have followed up five patients through adulthood. The typical facial characteristics are very prominent in early childhood, but become less pronounced in adulthood (Fig. 2B). An important consideration in the diagnosis of AR-RS is the characterization of the skeletal defects, considering their high prevalence. Therefore, a thorough radiological documentation is essential for clinical diagnosis and management. As a diagnostic tool, the most important findings are mesomelia, brachydactyly, rib fusions and hemivertebrae, as depicted in Fig. 6. The variable severity of the vertebral defects is remarkable, some patients having a single hemivertebrae while others have all vertebrae involved with a major impact on prognosis (Fig. 6). The presence of rib fusions is highly suggestive of AR-RS diagnosis. Despite the previous report of two patients with AR-RS without rib fusions (Mehawej et al. 2012; Aglan et al. 2015), also absent in Patients A12, A20 and A22, in the absence of rib fusions other diagnoses should also be considered, including other forms of the syndrome. Scoliosis is also a common finding that progresses with age and is the result of multiple vertebral anomalies. In our cohort it has been described in 69% of the patients. Patients without scoliosis were usually evaluated at a very young age except for patient A5, an adult woman.
Figure 6.
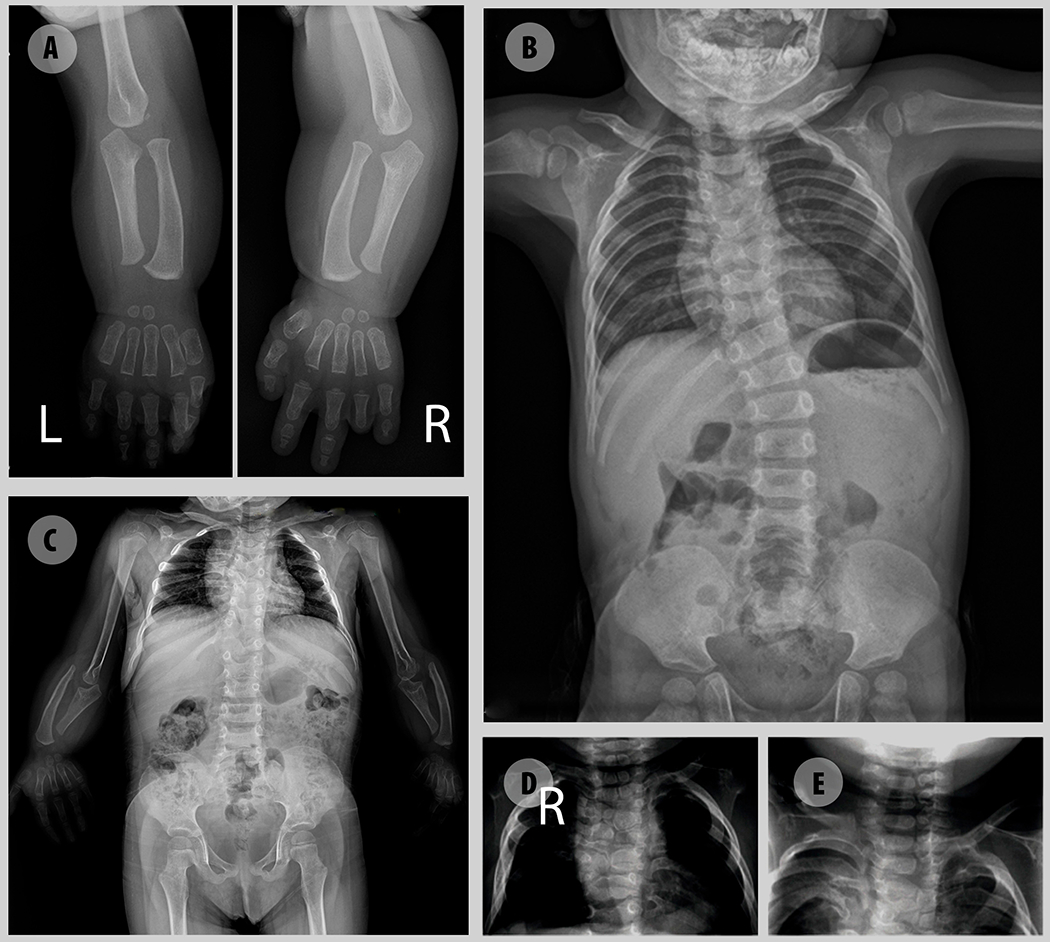
Radiographic images illustrative of major skeletal defects in AR-RS. A – Upper limb showing mesomelia, brachydactyly with pronounced shortening of distal phalanges and absence of medial and distal phalanges of 4th finger. B –Thoracic scoliosis due to multiple hemivertebra C- Multiple hemivertebra, butterfly vertebrae, rib fusions and mesomelia with malformation of the olecranon and coronoid process. Absence of the humero-radial joint. D, E – Hemivertebra.
Brachydactyly was also described in all patients, although it might consist of a minimal shortening of distal phalanges or even absence of distal and medial phalanges, as shown in Fig. 6. Clinodactyly is also frequent (81,8%) and some patients might also present syndactyly (28,6%).
Individuals of all ages had short stature and the final height of five adults, both females and males, ranged between 128 to 145 cm (<3rd centile) in our cohort.
Genital anomalies do not have a major impact on female patients but are a major concern for males. At birth, the penis can be extremely small and buried, often accompanied by cryptorchidism (50%), requiring chromosomal confirmation of the genetic sex (Gerber et al. 2021). Psychological follow-up is recommended. The few male adults followed have reported normal erection and significant growth of the penis after puberty.
This study provides an in-depth quantitative clinical delineation of the ROR2-related recessive Robinow syndrome in a large cohort of patients from diverse ethnic background originating from multiple continents and with confirmed molecular diagnoses. Sixteen novel variants were detected that mapped throughout the coding regions of ROR2, with no evident hot-spots. Both a total gene deletion allele and an exonic deletion CNV allele were characterized; the latter formed by Alu-Alu mediated rearrangement (AAMR). Despite consistency of the overall phenotype, minor phenotypic differences were observed in missense and LoF cases. Functional data and the identification of large deletions further supports the LoF mechanism in the etiology of the ROR2-related Robinow syndrome.
Supplementary Material
Supplementary Table 1. - ROR2 pathogenic variants identified to date related to AR-RS, including the present study
Supplementary Table 2. - The clinical phenotype of patients with ROR2-related Robinow syndrome.
Supplementary Figure 1 – Pedigrees of the families included in the study.
Supplementary Figure 2 – Country origin of patients with ROR2-related Robinow syndrome.
Supplementary Figure 3 – ROR2 missense variants cause the Ror2 protein to be retained intracellularly. HA-tagged Ror2 wild type and mutants were co-transfected with EGFP-hRas DNA into HeLa cells. Cells were fixed and stained using mouse anti-HA monoclonal antibody which was detected using Alexa 568-conjugated goat anti-mouse secondary antibodies (red, left hand panels). Expression of EGFP-hRas protein was detected by intrinsic GFP fluorescence (middle panels). Merged fluorescence shows that Ror2 wild-type protein co-localizes with the EGFP-hRas protein at the plasma membrane of the cells (Panel A), while Ror2 mutant proteins do not but are retained intracellularly in a reticular pattern typical of the ER region of the cell (panels B-E).
Supplementary Figure 4 - Ror2 mutant proteins are co-localized with the ER marker protein Calnexin. HeLa cells were transfected with HA-tagged Ror2 wild type and mutant constructs. Cells were co-stained using mouse anti-HA monoclonal antibody (left panels) and rabbit polyclonal antibody reactive to calnexin, (middle panels). Monoclonal antibody was detected using Alexa 568- conjugated goat anti-mouse secondary antibody (red) and polyclonal antibody was detected using Alexa 488-conjugated goat-anti rabbit secondary antibody (green). The merged images show that all of the Ror2 mutant proteins co-localized with the ER marker (panels B-E) in contrast to the wild type which was located on the plasma membrane (panel A).
Supplementary Figure 5- Phenotypic annotation grid of research subjects with biallelic ROR2 variants and significantly similar OMIM annotated known disease gene phenotypes ordered by mutation type
Phenotypic annotation grid of phenotypes of all subjects and significantly similar known disease genes. To interpret and understand biology of the phenotypes driving semantic similarity in this analysis, HPO terms associated with all subjects and significantly similar known disease genes were annotated and visualized in a gridded format. Red indicates presence of a phenotype while gray represents absence or not reported. Probands and significantly similar known disease genes are labeled to the left (italicized gene symbols) and probands are ordered by mutation type. Bold: subjects who have compound heterozygous variant alleles. Light font type: subjects who have homozygous variant alleles. Star: missense variants. Circle: loss of function (LoF) variants including nonsense variants and frameshifting variants. Triangle: splicing variants or variant with unknown consequence. Rectangle: large deletion (> 50 bp) variants. Line on the symbol: variants in the extracellular region. Line under the symbol (i.e. underlined font): variants in the intracellular region. Six (6) groups of subjects and mutation types are separated by white horizontal space (described as numbered groups from top to bottom): First group: probands who have bi-allelic missense variants. Second group: probands who have compound heterozygous missense and LoF variants. Third group: proband who has a large homozygous deletion. Fourth group: probands who have bi-allelic LoF variants. Fifth group: proband who has a homozygous variant affecting the start codon (c.2T>G). Sixth group: probands who have bi-allelic splicing variants. The frequency of each phenotype in probands from this cohort is shown on top of the grid.
Acknowledgements
We thank all the patients and their families for participating in the study.
Funding
This study was partially supported by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP, Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Coordenação de Aperfeiçoamento de Pessoal de Ensino Superior (CAPES), United States National Human Genome Research Institute (NHGRI)/ National Heart Lung and Blood Institute (NHLBI) grant number UM1HG006542 to the Baylor-Hopkins Center for Mendelian Genomics (BHCMG), the National Institute of Neurological Disorders and Stroke (NINDS) R35 NS105078 (J.R.L.), the National Institute of General Medical Sciences (NIGMS): R01 GM132589 (C.M.B.C), the Eunice Kennedy Shriver National Institute of Child Health & Human Development (NICHD) R03HD092569 (C.M.B.C and V.R.S.).
Footnotes
Ethics declaration: Patients or their legal guardians signed an informed consent declaration, including consent for publication and the study was approved by the institutional ethics committee of the Faculdade de Medicina, Universidade de Brasília (CEP FM: 079/2009; 25/11/2009) and the Institutional Review Board at Baylor College of Medicine (IRB protocols no. H-43246 and no. H-29697). Informed consent was obtained from all patients for the use of photos.
Conflict of interests:
Baylor College of Medicine (BCM) and Miraca Holdings have formed a joint venture with shared ownership and governance of the Baylor Genetics (BG), which performs clinical microarray analysis and clinical exome sequencing and whole genome sequencing. J.R.L. serves on the Scientific Advisory Board of the BG. J.R.L. has stock ownership in 23andMe, is a paid consultant for Regeneron Genetics Center, and is a coinventor on multiple United States and European patents related to molecular diagnostics for inherited neuropathies, eye diseases, genomic disorders and bacterial genomic fingerprinting. The Department of Molecular and Human Genetics at Baylor College of Medicine derives revenue from molecular genetics and clinical genomics testing offered at BG. The other authors declare no competing financial interests.
Data availability
The accession numbers for identified variants were deposited in ClinVar (http://www.https://www.ncbi.nlm.nih.gov/clinvar/) with the following identifiers: SCV001441486 - SCV001441509. The dbGAP accession number for exome sequences, for which informed consent for data sharing in controlled-access databases has been provided, is dbGAP:phs000711.v5.p1 (https://www.ncbi.nlm.nih.gov/gap/).
References
- Abu-Ghname A, Trost J, Davis MJ, Sutton VR, Zhang C, Guillen DE, Carvalho C, & Maricevich RS (2021). Extremity anomalies associated with Robinow syndrome. American journal of medical genetics. Part A, 185(12), 3584–3592. 10.1002/ajmg.a.61884 [DOI] [PubMed] [Google Scholar]
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, & Sunyaev SR (2010). A method and server for predicting damaging missense mutations. Nature methods, 7(4), 248–249. 10.1038/nmeth0410-248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afzal AR, Rajab A, Fenske CD, Oldridge M, Elanko N, Ternes-Pereira E, Tüysüz B, Murday VA, Patton MA, Wilkie AO, & Jeffery S (2000). Recessive Robinow syndrome, allelic to dominant brachydactyly type B, is caused by mutation of ROR2. Nature genetics, 25(4), 419–422. 10.1038/78107 [DOI] [PubMed] [Google Scholar]
- Aglan M, Amr K, Ismail S, Ashour A, Otaify GA, Mehrez MA, Aboul-Ezz EH, El-Ruby M, Mazen I, Abdel-Hamid MS, & Temtamy SA (2015). Clinical and molecular characterization of seven Egyptian families with autosomal recessive robinow syndrome: Identification of four novel ROR2 gene mutations. American journal of medical genetics. Part A, 167A(12), 3054–3061. 10.1002/ajmg.a.37287 [DOI] [PubMed] [Google Scholar]
- Ali BR, Jeffery S, Patel N, Tinworth LE, Meguid N, Patton MA, & Afzal AR (2007). Novel Robinow syndrome causing mutations in the proximal region of the frizzled-like domain of ROR2 are retained in the endoplasmic reticulum. Human genetics, 122(3-4), 389–395. 10.1007/s00439-007-0409-0 [DOI] [PubMed] [Google Scholar]
- Beck CR, Carvalho C, Akdemir ZC, Sedlazeck FJ, Song X, Meng Q, Hu J, Doddapaneni H, Chong Z, Chen ES, Thornton PC, Liu P, Yuan B, Withers M, Jhangiani SN, Kalra D, Walker K, English AC, Han Y, Chen K, … Lupski JR (2019). Megabase Length Hypermutation Accompanies Human Structural Variation at 17p11.2. Cell, 176(6), 1310–1324.e10. 10.1016/j.cell.2019.01.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beiraghi S, Leon-Salazar V, Larson BE, John MT, Cunningham ML, Petryk A, & Lohr JL (2011). Craniofacial and intraoral phenotype of Robinow syndrome forms. Clinical genetics, 80(1), 15–24. 10.1111/j.1399-0004.2011.01683.x [DOI] [PubMed] [Google Scholar]
- Ben-Shachar S, Khajavi M, Withers MA, Shaw CA, van Bokhoven H, Brunner HG, & Lupski JR (2009). Dominant versus recessive traits conveyed by allelic mutations - to what extent is nonsense-mediated decay involved?. Clinical genetics, 75(4), 394–400. 10.1111/j.1399-0004.2008.01114.x [DOI] [PubMed] [Google Scholar]
- Birgmeier J, Esplin ED, Jagadeesh KA, Guturu H, Wenger AM, Chaib H, Buckingham JA, Bejerano G, & Bernstein JA (2018). Biallelic loss-of-function WNT5A mutations in an infant with severe and atypical manifestations of Robinow syndrome. American journal of medical genetics. Part A, 176(4), 1030–1036. 10.1002/ajmg.a.38636 [DOI] [PubMed] [Google Scholar]
- Bunn KJ, Daniel P, Rösken HS, O’Neill AC, Cameron-Christie SR, Morgan T, Brunner HG, Lai A, Kunst HP, Markie DM, & Robertson SP (2015). Mutations in DVL1 cause an osteosclerotic form of Robinow syndrome. American journal of human genetics, 96(4), 623–630. 10.1016/j.ajhg.2015.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho CM, & Lupski JR (2016). Mechanisms underlying structural variant formation in genomic disorders. Nature reviews. Genetics, 17(4), 224–238. 10.1038/nrg.2015.25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Bellamy WP, Seabra MC, Field MC, & Ali BR (2005). ER-associated protein degradation is a common mechanism underpinning numerous monogenic diseases including Robinow syndrome. Human molecular genetics, 14(17), 2559–2569. 10.1093/hmg/ddi259 [DOI] [PubMed] [Google Scholar]
- Conlon CJ, Abu-Ghname A, Raghuram AC, Davis MJ, Guillen DE, Sutton VR, Carvalho C, & Maricevich RS (2021). Craniofacial phenotypes associated with Robinow syndrome. American journal of medical genetics. Part A, 185(12), 3606–3612. 10.1002/ajmg.a.61986 [DOI] [PubMed] [Google Scholar]
- Eldomery MK, Coban-Akdemir Z, Harel T, Rosenfeld JA, Gambin T, Stray-Pedersen A, Küry S, Mercier S, Lessel D, Denecke J, Wiszniewski W, Penney S, Liu P, Bi W, Lalani SR, Schaaf CP, Wangler MF, Bacino CA, Lewis RA, Potocki L, … Lupski JR (2017). Lessons learned from additional research analyses of unsolved clinical exome cases. Genome medicine, 9(1), 26. 10.1186/s13073-017-0412-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geetha-Loganathan P, Nimmagadda S, Antoni L, Fu K, Whiting CJ, Francis-West P, & Richman JM (2009). Expression of WNT signalling pathway genes during chicken craniofacial development. Developmental dynamics : an official publication of the American Association of Anatomists, 238(5), 1150–1165. 10.1002/dvdy.21934 [DOI] [PubMed] [Google Scholar]
- Gerber JA, Sheth KR, & Austin PF (2021). Robinow syndrome: Genital analysis, genetic heterogeneity, and associated psychological impact. American journal of medical genetics. Part A, 185(12), 3601–3605. 10.1002/ajmg.a.61981 [DOI] [PubMed] [Google Scholar]
- Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, & Shendure J (2014). A general framework for estimating the relative pathogenicity of human genetic variants. Nature genetics, 46(3), 310–315. 10.1038/ng.2892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin D (1998). An Information-Theoretic Definition of Similarity. Icml 98, 296–304. 10.5555/645527.657297 [DOI] [Google Scholar]
- Liu P, Meng L, Normand EA, Xia F, Song X, Ghazi A, Rosenfeld J, Magoulas PL, Braxton A, Ward P, Dai H, Yuan B, Bi W, Xiao R, Wang X, Chiang T, Vetrini F, He W, Cheng H, Dong J, … Yang Y (2019). Reanalysis of Clinical Exome Sequencing Data. The New England journal of medicine, 380(25), 2478–2480. 10.1056/NEJMc1812033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupski JR, Belmont JW, Boerwinkle E, & Gibbs RA (2011). Clan genomics and the complex architecture of human disease. Cell, 147(1), 32–43. 10.1016/j.cell.2011.09.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupski JR (2021). Clan genomics: From OMIM phenotypic traits to genes and biology. American journal of medical genetics. Part A, 185(11), 3294–3313. 10.1002/ajmg.a.62434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzeu JF, Pardono E, Vianna-Morgante AM, Richieri-Costa A, Ae Kim C, Brunoni D, Martelli L, de Andrade CE, Colin G, & Otto PA (2007). Clinical characterization of autosomal dominant and recessive variants of Robinow syndrome. American journal of medical genetics. Part A, 143(4), 320–325. 10.1002/ajmg.a.31592 [DOI] [PubMed] [Google Scholar]
- Mehawej C, Chouery E, Maalouf D, Baujat G, Le Merrer M, Cormier-Daire V, & Mégarbané A (2012). Identification of a novel causative mutation in the ROR2 gene in a Lebanese family with a mild form of recessive Robinow syndrome. European journal of medical genetics, 55(2), 103–108. 10.1016/j.ejmg.2011.11.003 [DOI] [PubMed] [Google Scholar]
- Nagasaki K, Nishimura G, Kikuchi T, Nyuzuki H, Sasaki S, Ogawa Y, & Saitoh A (2018). Nonsense mutations in FZD2 cause autosomal-dominant omodysplasia: Robinow syndrome-like phenotypes. American journal of medical genetics. Part A, 176(3), 739–742. 10.1002/ajmg.a.38623 [DOI] [PubMed] [Google Scholar]
- Nohno T, Kawakami Y, Wada N, Komaguchi C, & Nishimatsu S (1999). Differential expression of the frizzled family involved in Wnt signaling during chick limb development. Cellular and molecular biology (Noisy-le-Grand, France), 45(5), 653–659. [PubMed] [Google Scholar]
- Person AD, Beiraghi S, Sieben CM, Hermanson S, Neumann AN, Robu ME, Schleiffarth JR, Billington CJ Jr, van Bokhoven H, Hoogeboom JM, Mazzeu JF, Petryk A, Schimmenti LA, Brunner HG, Ekker SC, & Lohr JL (2010). WNT5A mutations in patients with autosomal dominant Robinow syndrome. Developmental dynamics : an official publication of the American Association of Anatomists, 239(1), 327–337. 10.1002/dvdy.22156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posey JE, O’Donnell-Luria AH, Chong JX, Harel T, Jhangiani SN, Coban Akdemir ZH, Buyske S, Pehlivan D, Carvalho C, Baxter S, Sobreira N, Liu P, Wu N, Rosenfeld JA, Kumar S, Avramopoulos D, White JJ, Doheny KF, Witmer PD, Boehm C, … Centers for Mendelian Genomics (2019). Insights into genetics, human biology and disease gleaned from family based genomic studies. Genetics in medicine : official journal of the American College of Medical Genetics, 21(4), 798–812. 10.1038/s41436-018-0408-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid JG, Carroll A, Veeraraghavan N, Dahdouli M, Sundquist A, English A, Bainbridge M, White S, Salerno W, Buhay C, Yu F, Muzny D, Daly R, Duyk G, Gibbs RA, & Boerwinkle E (2014). Launching genomics into the cloud: deployment of Mercury, a next generation sequence analysis pipeline. BMC bioinformatics, 15, 30. 10.1186/1471-2105-15-30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, & ACMG Laboratory Quality Assurance Committee (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in medicine : official journal of the American College of Medical Genetics, 17(5), 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinow M, Silverman FN, & Smith HD (1969). A newly recognized dwarfing syndrome. American journal of diseases of children (1960), 117(6), 645–651. 10.1001/archpedi.1969.02100030647005 [DOI] [PubMed] [Google Scholar]
- Sassa A, Kanemaru Y, Kamoshita N, Honma M, & Yasui M (2016). Mutagenic consequences of cytosine alterations site-specifically embedded in the human genome. Genes and environment : the official journal of the Japanese Environmental Mutagen Society, 38(1), 17. 10.1186/s41021-016-0045-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwabe GC, Tinschert S, Buschow C, Meinecke P, Wolff G, Gillessen-Kaesbach G, Oldridge M, Wilkie AO, Kömec R, & Mundlos S (2000). Distinct mutations in the receptor tyrosine kinase gene ROR2 cause brachydactyly type B. American journal of human genetics, 67(4), 822–831. 10.1086/303084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwabe GC, Trepczik B, Süring K, Brieske N, Tucker AS, Sharpe PT, Minami Y, & Mundlos S (2004). Ror2 knockout mouse as a model for the developmental pathology of autosomal recessive Robinow syndrome. Developmental dynamics : an official publication of the American Association of Anatomists, 229(2), 400–410. 10.1002/dvdy.10466 [DOI] [PubMed] [Google Scholar]
- Schwartz DD, Fein RH, Carvalho C, Sutton VR, Mazzeu JF, & Axelrad ME (2021). Neurocognitive, adaptive, and psychosocial functioning in individuals with Robinow syndrome. American journal of medical genetics. Part A, 185(12), 3576–3583. 10.1002/ajmg.a.61854 [DOI] [PubMed] [Google Scholar]
- Shayota BJ, Zhang C, Shypailo RJ, Mazzeu JF, Carvalho C, & Sutton VR (2020). Characterization of the Robinow syndrome skeletal phenotype, bone micro-architecture, and genotype-phenotype correlations with the osteosclerotic form. American journal of medical genetics. Part A, 182(11), 2632–2640. 10.1002/ajmg.a.61843 [DOI] [PubMed] [Google Scholar]
- Sim NL, Kumar P, Hu J, Henikoff S, Schneider G, & Ng PC (2012). SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic acids research, 40(Web Server issue), W452–W457. 10.1093/nar/gks539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sisson BE, & Topczewski J (2009). Expression of five frizzleds during zebrafish craniofacial development. Gene expression patterns : GEP, 9(7), 520–527. 10.1016/j.gep.2009.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X, Beck CR, Du R, Campbell IM, Coban-Akdemir Z, Gu S, Breman AM, Stankiewicz P, Ira G, Shaw CA, & Lupski JR (2018). Predicting human genes susceptible to genomic instability associated with Alu/Alu-mediated rearrangements. Genome research, 28(8), 1228–1242. 10.1101/gr.229401.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stricker S, Rauschenberger V, & Schambony A (2017). ROR-Family Receptor Tyrosine Kinases. Current topics in developmental biology, 123, 105–142. 10.1016/bs.ctdb.2016.09.003 [DOI] [PubMed] [Google Scholar]
- van Bokhoven H, Celli J, Kayserili H, van Beusekom E, Balci S, Brussel W, Skovby F, Kerr B, Percin EF, Akarsu N, & Brunner HG (2000). Mutation of the gene encoding the ROR2 tyrosine kinase causes autosomal recessive Robinow syndrome. Nature genetics, 25(4), 423–426. 10.1038/78113 [DOI] [PubMed] [Google Scholar]
- Wadia RS, Shirole DB, & Dikshit MS (1978). Recessively inherited costovertebral segmentation defect with mesomelia and peculiar facies (Covesdem syndrome): A new genetic entity? Journal of medical genetics, 15(2), 123–127. 10.1136/jmg.15.2.123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward JH (1963). Hierarchical grouping to optimize an objective function. J Am Stat Assoc 58, 236–244. [Google Scholar]
- White J, Mazzeu JF, Hoischen A, Jhangiani SN, Gambin T, Alcino MC, Penney S, Saraiva JM, Hove H, Skovby F, Kayserili H, Estrella E, Vulto-van Silfhout AT, Steehouwer M, Muzny DM, Sutton VR, Gibbs RA, Baylor-Hopkins Center for Mendelian Genomics, Lupski JR, Brunner HG, … Carvalho CM (2015). DVL1 frameshift mutations clustering in the penultimate exon cause autosomal-dominant Robinow syndrome. American journal of human genetics, 96(4), 612–622. 10.1016/j.ajhg.2015.02.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JJ, Mazzeu JF, Coban-Akdemir Z, Bayram Y, Bahrambeigi V, Hoischen A, van Bon B, Gezdirici A, Gulec EY, Ramond F, Touraine R, Thevenon J, Shinawi M, Beaver E, Heeley J, Hoover-Fong J, Durmaz CD, Karabulut HG, Marzioglu-Ozdemir E, Cayir A, … Carvalho C (2018). WNT Signaling Perturbations Underlie the Genetic Heterogeneity of Robinow Syndrome. American journal of human genetics, 102(1), 27–43. 10.1016/j.ajhg.2017.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JJ, Mazzeu JF, Hoischen A, Bayram Y, Withers M, Gezdirici A, Kimonis V, Steehouwer M, Jhangiani SN, Muzny DM, Gibbs RA, Baylor-Hopkins Center for Mendelian Genomics, van Bon B, Sutton VR, Lupski JR, Brunner HG, & Carvalho C (2016). DVL3 Alleles Resulting in a −1 Frameshift of the Last Exon Mediate Autosomal-Dominant Robinow Syndrome. American journal of human genetics, 98(3), 553–561. 10.1016/j.ajhg.2016.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Mazzeu JF, Eisfeldt J, Grochowski CM, White J, Akdemir ZC, Jhangiani SN, Muzny DM, Gibbs RA, Lindstrand A, Lupski JR, Sutton VR, & Carvalho C (2021). Novel pathogenic genomic variants leading to autosomal dominant and recessive Robinow syndrome. American journal of medical genetics. Part A, 185(12), 3593–3600. 10.1002/ajmg.a.61908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Jolly A, Shayota BJ, Mazzeu JF, Du H, Dawood M, Soper PC, Ramalho de Lima A, Ferreira BM, Coban-Akdemir Z, White J, Shears D, Thomson FR, Douglas SL, Wainwright A, Bailey K, Wordsworth P, Oldridge M, Lester T, Calder AD, … Carvalho C (2021). Novel pathogenic variants and quantitative phenotypic analyses of Robinow syndrome: WNT signaling perturbation and phenotypic variability. HGG advances, 3(1), 100074. 10.1016/j.xhgg.2021.100074 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1. - ROR2 pathogenic variants identified to date related to AR-RS, including the present study
Supplementary Table 2. - The clinical phenotype of patients with ROR2-related Robinow syndrome.
Supplementary Figure 1 – Pedigrees of the families included in the study.
Supplementary Figure 2 – Country origin of patients with ROR2-related Robinow syndrome.
Supplementary Figure 3 – ROR2 missense variants cause the Ror2 protein to be retained intracellularly. HA-tagged Ror2 wild type and mutants were co-transfected with EGFP-hRas DNA into HeLa cells. Cells were fixed and stained using mouse anti-HA monoclonal antibody which was detected using Alexa 568-conjugated goat anti-mouse secondary antibodies (red, left hand panels). Expression of EGFP-hRas protein was detected by intrinsic GFP fluorescence (middle panels). Merged fluorescence shows that Ror2 wild-type protein co-localizes with the EGFP-hRas protein at the plasma membrane of the cells (Panel A), while Ror2 mutant proteins do not but are retained intracellularly in a reticular pattern typical of the ER region of the cell (panels B-E).
Supplementary Figure 4 - Ror2 mutant proteins are co-localized with the ER marker protein Calnexin. HeLa cells were transfected with HA-tagged Ror2 wild type and mutant constructs. Cells were co-stained using mouse anti-HA monoclonal antibody (left panels) and rabbit polyclonal antibody reactive to calnexin, (middle panels). Monoclonal antibody was detected using Alexa 568- conjugated goat anti-mouse secondary antibody (red) and polyclonal antibody was detected using Alexa 488-conjugated goat-anti rabbit secondary antibody (green). The merged images show that all of the Ror2 mutant proteins co-localized with the ER marker (panels B-E) in contrast to the wild type which was located on the plasma membrane (panel A).
Supplementary Figure 5- Phenotypic annotation grid of research subjects with biallelic ROR2 variants and significantly similar OMIM annotated known disease gene phenotypes ordered by mutation type
Phenotypic annotation grid of phenotypes of all subjects and significantly similar known disease genes. To interpret and understand biology of the phenotypes driving semantic similarity in this analysis, HPO terms associated with all subjects and significantly similar known disease genes were annotated and visualized in a gridded format. Red indicates presence of a phenotype while gray represents absence or not reported. Probands and significantly similar known disease genes are labeled to the left (italicized gene symbols) and probands are ordered by mutation type. Bold: subjects who have compound heterozygous variant alleles. Light font type: subjects who have homozygous variant alleles. Star: missense variants. Circle: loss of function (LoF) variants including nonsense variants and frameshifting variants. Triangle: splicing variants or variant with unknown consequence. Rectangle: large deletion (> 50 bp) variants. Line on the symbol: variants in the extracellular region. Line under the symbol (i.e. underlined font): variants in the intracellular region. Six (6) groups of subjects and mutation types are separated by white horizontal space (described as numbered groups from top to bottom): First group: probands who have bi-allelic missense variants. Second group: probands who have compound heterozygous missense and LoF variants. Third group: proband who has a large homozygous deletion. Fourth group: probands who have bi-allelic LoF variants. Fifth group: proband who has a homozygous variant affecting the start codon (c.2T>G). Sixth group: probands who have bi-allelic splicing variants. The frequency of each phenotype in probands from this cohort is shown on top of the grid.
Data Availability Statement
The accession numbers for identified variants were deposited in ClinVar (http://www.https://www.ncbi.nlm.nih.gov/clinvar/) with the following identifiers: SCV001441486 - SCV001441509. The dbGAP accession number for exome sequences, for which informed consent for data sharing in controlled-access databases has been provided, is dbGAP:phs000711.v5.p1 (https://www.ncbi.nlm.nih.gov/gap/).