

Abstract
ADARs catalyze Adenosine-to-Inosine (A-to-I) editing of double-stranded RNA and regulate global gene expression output through interactions with RNA and other proteins. ADARs play important roles in development and disease, and previous work has shown that ADAR1 is oncogenic in a growing list of cancer types. Here we show that ADAR1 is a critical gene for triple-negative breast cancer cells, as ADAR1 loss results in reduced growth (viability and cell cycle progression), invasion, and mammosphere formation. Whole transcriptome sequencing analyses demonstrate that ADAR1 regulates both coding and non-coding targets by altering gene expression level, A-to-I editing, and splicing. We determine that a recoding edit in filamin B (FLNB chr3:58156064) reduces the tumor suppressive activities of the protein to promote growth and invasion. We also show that several tumor suppressor microRNAs are upregulated upon ADAR1 loss and suppress cell cycle progression and invasion. This work describes several novel mechanisms of ADAR1-mediated oncogenesis in triple-negative breast cancer, providing support to strategies targeting ADAR1 in this aggressive cancer type that has few treatment options.
Keywords: ADARs, RNA editing, microRNAs, breast cancer, cancer epigenetics
Introduction
Breast cancer (BC) is the most common malignancy in women, affecting 1 in 8 women, and encompasses multiple distinct subtypes with varying degrees of lethality. Triple-negative breast cancer (TNBC) is a more aggressive subtype defined by lack of hormone receptors (estrogen and progesterone) and human epidermal growth factor receptor 2, which therefore is unresponsive to therapies targeting those receptors. TNBC can be initially more responsive to chemotherapeutic agents, but nonresponsive tumors tend to be highly aggressive with few treatment options available.
The family of Adenosine Deaminases Acting on RNA (ADARs) are a group of enzymes that bind double-stranded RNA (dsRNA) and catalyze the hydrolytic deamination of adenosine to form inosine. Adenosine-to-inosine (A-to-I) editing occurs only at regions of dsRNA and alters the base pair preference from uracil to cytidine, thus functionally acting as an A-to-G mutation. There are three family members: ADAR1 (also called ADAR), ADAR2 (ADARB1), and ADAR3 (ADARB2). ADAR1 is ubiquitously expressed in nearly all tissues and consists of two isoforms, the nuclear p110 isoform and the interferon (IFN)-inducible p150 isoform which shuttles between the nucleus and cytoplasm. ADAR2 has more targeted expression to tissues like the brain, lungs, and arteries, while ADAR3 is brain-specific and has yet to demonstrate any editing activity1.
Dysregulation of ADAR expression and/or A-to-I editing has been associated with a variety of cancers, as well as several neurological and immunological diseases2,3. The oncogenic mechanisms of ADARs have been attributed to recoding events, altered microRNA (miRNA) targeting or processing, and even editing-independent effects4–7. Compelling new work suggests that ADAR1 modulates anti-tumor immunity, arising as a top hit for sensitizing murine cancers including TNBC to T-cell killing, and synergizing with anti-PD-1 treatment in melanoma8,9.
ADAR1 has been previously associated with breast cancer, with BC having higher ADAR1 expression and editing levels in tumors vs. normal tissue and higher levels for both correlating with poorer survival10–12. Previous work has shown that in breast cancer cell lines representing multiple subtypes, loss of ADAR1 reduces growth and increases apoptosis10,13,14. A recent study in TNBC demonstrated that ADAR1 loss yields growth and tumor-initiating defects, and confirmed in TNBC a known role for ADAR1 in suppressing the dsRNA sensor PKR and downstream IFN signaling14. Yet, there remains a lack of in-depth, unbiased investigation into the oncogenic mechanisms of ADAR1 in TNBC.
Given that the function of ADAR1 has proven to be highly context-dependent, we sought to understand how ADAR1 regulates the global transcriptome in TNBC to exert its oncogenic effect. Here, we demonstrate that multiple mechanisms contribute to the critical role for ADAR1 in TNBC cell growth and viability, cell cycle progression, invasion, and mammosphere formation. We performed mRNA and miRNA sequencing to identify ADAR1-regulated targets via editing and differential expression analysis. We investigated two miRNAs, miR-27a-5p and miR-448-3p, which play roles in growth and motility in TNBC. We also characterized a highly prevalent recoding edit site in FLNB, which acts to decrease the tumor suppressor’s activity on growth and invasion. Taken together, this work further solidifies the importance of ADAR1 in TNBC and reveals novel mechanisms contributing to ADAR1-mediated oncogenesis.
Materials & Methods
Cell lines & culture
BT20, MDA-MB-MB468 (MB468), and HEK293T cells were purchased from ATCC. HCC70 cells were a generous gift from Alex Toker. BT20 and MB468 cells were cultured in DMEM/F12 media with 10% FBS and penicillin/streptomycin (P/S) (Gibco). HEK293T cells were cultured in DMEM media with 10% FBS and P/S. HCC70 cells were cultured in RPMI media with 10% FBS and P/S. All cells were used within 20 passages and tested annually for mycoplasma contamination using MycoAlert (Lonza); STR authentication was not performed.
Transfections
All siRNA and miRNA experiments were performed via reverse transfection using RNAiMAX reagent (ThermoFisher). A final concentration of 5nM siRNA or 10nM miRNA was combined with 100X diluted RNAiMAX in Opti-MEM with GLUTAMAX, and let incubate for 15 minutes before adding to cells. The Silencer Select (ThermoFisher) siRNAs used were as follows: siControl (4390843), siADAR#1 (s1007), siADAR#2 (119581), siFLNB#1 (s5278), siFLNB#2 (s5279). mirVana miRNA mimics were used, along with Negative Control mimic #1 (ThermoFisher).
Lentivirus Transduction
pLentiCRISPRv2 constructs (Addgene) were transfected into HEK293T cells with pVSV-G and psPAX2 packaging vectors using TransIT-Lenti reagent according to manufacturer’s protocol (MirusBio). After 48hrs, media was collected, filtered, and concentrated to collect virus. BT20 cells were infected with 1:5 diluted virus in 10μg/ml polybrene media and moved to 0.8μg/ml puromycin media after 48hrs. Cells underwent selection for 5–7 days and then were maintained in 0.2μg/ml puromycin media.
For the ADAR1 rescue experiment, cells were transduced with HA-ADAR1p110, HA-ADAR1p150, or HA empty vector lentivirus (VectorBuilder) and transfected with siRNAs.
RT-qPCR
RNA samples were prepared in Trizol using the Direct-zol RNA Miniprep Plus kit (Zymo Research). For mRNA detection and RESS-qPCR, 1μg of total RNA was reverse transcribed using the SuperScript IV RT kit (ThermoFisher) and oligo(dT20) primers and diluted 10X. cDNA was quantified using LightCycler 480 SYBR Green I (Roche) and Cp values were normalized to GAPDH. For mature miRNA detection, 100ng of total RNA was reverse transcribed using the miRCURY LNA RT kit (Qiagen) and diluted 20X. cDNA was quantified using the miRCURY LNA SYBR Green PCR kit and miRCURY LNA miRNA PCR assays (Qiagen), and Cp values were normalized to miR-103a-3p.
For pre-miRNA and pri-miRNA detection, 1μg of total RNA was reverse transcribed using the miScript II RT kit (Qiagen) with HiFlex buffer and diluted 3X. For pre-miRNAs, quantification was performed using the miScript Universal Primer and custom pre-miRNA primers using Quantitect SYBR Green and Cp values were normalized to ACTB (Qiagen). For pri-miRNAs, quantification was performed using LightCycler 480 SYBR Green I (Roche), and Cp values were normalized to GAPDH. All qPCRs were performed on the Roche LightCycler 480 instrument according to the thermocycler conditions of the relevant SYBR master mix. qPCR primers are provided in Table S1.
Statistical analysis was performed using multiple unpaired two-tailed t tests for each treatment vs. control, not assuming consistent SD and using Holm-Sidak correction for multiple hypotheses, with alpha = 0.05.
Sanger sequencing
For FLNB editing detection, undiluted cDNA or genomic DNA (gDNA) was used as template to amplify the region surrounding the FLNB edit site using Q5 High-Fidelity Polymerase (ThermoFisher). PCR products were column purified using the Monarch PCR & DNA Cleanup Kit (NEB) and submitted for Sanger sequencing.
For detecting FLNB-ECS deletion, gDNA from FLNBdelECS cells was used to amplify the ECS and the PCR product was cloned into plasmid using the Zero Blunt TOPO PCR Cloning Kit (Thermo Fisher). Individual colonies were picked and submitted for sequencing, and deletions assessed by aligning to reference genome.
Western blot
Cells were lysed in RIPA buffer plus Halt protease & phosphatase inhibitors, spun down to clear cell debris, and quantified using BCA assay. Equal mass of each sample was denatured in sample lysis buffer with β-mercaptoethanol by boiling for 30 sec and loaded onto a NuPAGE Bis-Tris gel. Gels were transferred onto nitrocellulose membrane via wet transfer, blocked with 5% milk or BSA, and blotted using antibodies in Table S2. Membranes were probed with HRP-conjugated secondary antibody and imaged using SuperSignal West Pico PLUS substrate (ThermoFisher) on BioImager Chemlux.
MTT assay
Cells were plated in quintuplicate on four identical 96-well plates, including media-only control wells. BT20 and MB468 cells were plated at 3,000 cells/well, and HCC70s were plated at 1,000 cells/well. For each timepoint, cells were incubated with 25μl of 0.15% MTT reagent for 3 hours, protected from light. Plates were briefly spun down, media removed, and crystals resuspended in 100μl DMSO. Absorbance was measured at 560nm using GloMax Explorer (Promega). Blank-adjusted absorbance was determined by subtracting the average media-only reading, and samples were normalized as a percent of the D1 timepoint.
Statistical significance was determined via two-way repeated measures ANOVA, assuming sphericity, performing multiple comparisons on treatments vs. control, with one family per row, using Holm-Sidak correction for multiple hypotheses, with alpha = 0.05.
Apoptosis assay
Cells were plated in quintuplicate in black-walled, flat clear bottom 96-well plates. BT20 and MB468 cells were plated at 10,000 cells/well. After 72hrs, media was aspirated and cells were incubated with 100μl of 2μM NucView 530 Caspase-3 Substrate (Biotium) and 20μM Hoechst (3.3μM Hoechst for HCC70) diluted in PBS for 30 mins. Plates were briefly spun down and cells were imaged using Celigo Image Cytometer (Nexcelom Bioscience). Hoechst staining was used to count cells, and cells that were NucView+/Hoechst+ were labeled as “Caspase-positive.” Percent caspase positive cells and total cell number were normalized to control treatment.
Cell cycle assay
Cells were transfected (and control wells plated) for 48/72hr in 6-well plates, and then pulsed with 10μM BrdU media for 2hrs. Cells were collected and fixed in ice-cold 70% ethanol for 2hrs. Cells were washed with 0.05% PBST and left in 2N HCl for 15min. Cells were washed in PBST and left in SuperBlock TBS blocking buffer (ThermoFisher) resuspended in 0.1M sodium tetraborate for 30min. Cells were resuspended in 100μl PBST with 20μl FITC-conjugated anti-BrdU antibody or IgG-FITC isotype control (BD Biosciences) and incubated, covered at 4C for 30min. Cells were washed with blocking buffer, then PBST, and resuspended in 500μl PBS with 50μg RNase and 2.5μg propidium iodide (PI). After 30min, cells were transferred to filter cap flow cytometry tubes and analyzed on CytoFLEX LX (Beckman Coulter).
Acquisition gains for FITC (BrdU) and ECD (PI) were set based on unstained, single-stain and IgG-FITC controls. Cells were gated to remove cell doublets and dead cells, and then at least 25,000 events were recorded for each sample. An ECD (linear) vs. FITC (log) plot was used to gate G1, S, and G2/M phase cells. Compensation and final analysis were performed on FlowJo software v10.7.
Invasion/migration assay
If transfected, cells were treated in 6-well plates for 48hrs prior to seeding. Cells were seeded at 2.5e5 cells per insert into BioCoat Matrigel Invasion Chambers and membrane-only Control Inserts (Corning). After 22hrs, the top compartment of each insert was scrubbed to remove noninvaded cells and matrigel, and cells remaining on the bottom of the insert were fixed and stained in a crystal violet formaldehyde solution. After washing in PBS and drying, membranes were imaged using a Primovert inverted microscope with the Axiocam 105 Color Camera (Zeiss). Five images were taken per well, and the area stained by invading/migrating cells was measured using ImageJ color threshold analysis. Average area measurements for the matrigel or control inserts were presented as average invasion or migration, respectively, and normalized invasion was calculated as 100%*(invasion/migration).
Mammosphere assay
After 24hr transfection in 6-well plates, BT20 cells were seeded at 500 cells/well into ultra-low-adherence 24-well plates and cultured in mammosphere media: DMEM/F12 with 2% B-27 supplement, 20ng/ml EGF, 4μg/ml insulin, and 100ng/ml SHH20. After five days, mammospheres of diameter at least 50μm were counted and representative images taken using EVOS microscope. Mammospheres were then spun down and collected for RNA isolation using the Direct-zol RNA Microprep kit (Zymo Research). Mammosphere formation efficiency (MFE) was calculated as 100%*(average # mammospheres)/(500 cells seeded). Mammosphere volume was approximated by measuring the diameter of each mammosphere twice, at its longest point (a) and approximately perpendicular to that (b), using ImageJ. Volume was calculated as .
Dual luciferase assay
The psiCHECK-2 vector (Promega) was used to create constructs with the WT or mutant binding site (BS) for each miRNA as controls, as well as the 3’UTR of each putative target tested with WT or mutant seed region for the miRNA BS(s). HEK293T cells were plated at 7.5e4 cells/well into 24-well plates, and after 24hrs cotransfected using Transit-X2 transfection reagent (Mirus Bio) along with 500ng plasmid and 40nM miRNA mimic. 24hrs after transfection, cells were lysed and plated in triplicate for luminescence reading using the Dual Luciferase Reporter Assay and GloMax Explorer (Promega). Renilla and Firefly luminescence readings were averaged, and Renilla normalized to Firefly (transfection efficiency control) to determine relative expression of each construct.
ELISA assay
Cells were transfected for 72hrs and supernatant collected and spun down to remove cell debris. IL6 concentration was detected using the Biolegend ELISA MAX Deluxe Set for Human IL6 according to manufacturer’s protocol. Log(Absorbance) vs. log(IL6 concentration) was plotted for IL6 standard curve values and fitted to a sigmoidal four-parameter logistics best-fit curve in Prism in order to interpolate IL6 concentration from samples.
Subcellular fractionation
Subcellular fractionation was performed using the NE-PER Nuclear and Cytoplasmic Extraction Reagents (ThermoFisher) according to manufacturer’s protocol. Final sample volumes were determined and 15% of each cytoplasmic and nuclear fraction was loaded, along with whole cell lysate controls. Blotting for nuclear (Lamin A) and cytoplasmic (alpha-tubulin) controls confirmed a clean fractionation. FLNB localization was determined by densitometry analysis to quantify band intensity, plotting the ratio of cytoplasmic/nuclear intensity normalized to the NT sample.
Immunofluorescence
Cells were plated in Lab-Tek II CC2 8-chamber slides at 10,000 cells per chamber. After 24hrs, cells were washed and fixed with 4% PFA for 15 mins, then washed and incubated in 5% BSA/0.3% Triton X-100 in PBS for 1hr. Cells were incubated in 1:500 diluted rabbit anti-FLNB antibody overnight at 4C. Cells were washed and incubated in 1:500 diluted anti-rabbit AlexaFluor 546 antibody and 1:300 diluted AlexaFluor 488 Phalloidin for two hours, washed, and mounted with ProLong Gold with DAPI (Thermo Fisher). Background staining was assessed with anti-rabbit only, phalloidin-only, and no stain conditions. Confocal images were acquired with a Carl Zeiss LSM 880 upright confocal microscope (Germany) using the Zen black software version SP2.3. The tile scan and z-stack images were acquired with a Plan-Apochromat 63x/NA 1.4 objective lens with 9–14 images per condition. Images were analyzed using Imaris 9.7 image analysis software (Oxford Instruments). Nuclear and cytoplasmic FLNB signals were isolated by creating regions of interest (ROIs). These ROIs were then analyzed for FLNB levels by intensity-based thresholding, and data was normalized to account for the difference in the cell volumes analyzed in each condition. Total FLNB intensity or percent nuclear/cytoplasmic FLNB were averaged across all images for a given condition. Statistical analysis was performed using ordinary one-way ANOVA.
RNA-seq sample collection & library preparation
Total RNA was isolated from cells transfected with siControl, siADAR#1, or siADAR#2 for 72hr using the miRvana miRNA isolation kit (ThermoFisher) and submitted to the Dana Farber Molecular Biology Core Facility. RNA quality was confirmed using Agilent Bioanalyzer. Library preparation was performed using the KAPA mRNA HyperPrep kit for poly(A+) RNA-seq and the Bioo Scientific NextFlex Small RNA-Seq kit with 4N adapters for small RNA-seq. Sequencing was performed on an Illumina NextSeq 500 using 75bp paired-end reads for poly(A+)-selected RNA-seq and 75bp single-end reads for small RNA-seq. Additional information can be found in Supplemental Methods.
Statistical analysis
With the exception of RNAseq and TCGA analyses (see Supplementary Methods), all statistical analyses were performed using Prism 9 software. Unless otherwise stated, statistical significance was determined by performing unpaired two-tailed t tests for each treatment vs. control, assuming consistent SD and using Holm-Sidak correction for multiple hypotheses, with alpha = 0.05. If multiple treatment conditions were compared simultaneously (ex. siADAR#1 and siADAR#2), an ordinary one-way ANOVA was performed assuming equal SDs, post-hoc comparisons of all treatments vs. control, using Holm-Sidak correction for multiple hypotheses, with alpha = 0.05. Unless otherwise stated, all experiments were done in triplicate, error bars indicate mean ± s.d., and p-values are: *p≤0.05, **p≤0.01, ***p≤0.001, ****p≤0.0001.
Results
Basal A cells are sensitive to ADAR1 loss
Analysis of ADAR1 expression in tumor bank samples from The Cancer Genome Atlas (TCGA) confirmed the observations from prior literature that ADAR1 has increased expression in breast cancer vs. normal tissues, as we confirmed in TNBC vs. normal (Fig. S1A). Comparing ADAR1 expression between the PAM50 breast cancer subtypes reveals distinctions among the subtypes, with luminal and basal samples having the highest ADAR1 expression (Fig. S1B). ADAR1 expression also has an increasing correlation trend with stage in breast cancer, with significant increase in Stage II over Stage I suggesting that ADAR1 activation may be an early event in cancer progression (Fig. S1C).
We next assessed ADAR1 sensitivity using the Broad Institute’s Cancer Dependency Map portal15. In the RNAi screening data, ADAR1 is considered “Strongly Selective,” meaning it is a selective dependency only in a specific subset of cancer cell lines. Of four cell lineages significantly enriched for ADAR1 sensitivity, Basal A breast cancer is second (p=2.8e-4), with the remaining three relating to Head & Neck Cancers (Fig. S1D). Basal A cell lines are the more epithelial triple-negative cells that best overlap with the Basal TNBC tumor subtype and feature enrichment of cytokeratins and integrins16. We thus selected three Basal A cell lines in which to study ADAR1: BT20, MDA-MB-468 (MB468), and HCC70 (Fig. S1E).
In order to assess the importance of ADAR1 in TNBC cells, we compared the phenotypes of cells transfected with control and ADAR1-targeting siRNAs. We first validated that ADAR1 loss results in decreased A-to-I editing using qPCR primer pairs against the unedited, or WT, and edited versions of two transcripts using a previously described assay called RNA Editing Site-Specific (RESS)-qPCR (Fig. 1A, S2A–B)17. AZIN1 and FLNB contain previously identified recoding edit sites that are over-edited in HCC18. AZIN1 editing has been shown to promote tumorigenesis in HCC, but the effect of FLNB editing at this site has not been studied4. However, in the course of our studies we identified and functionally characterized FLNB M2293V editing in TNBC (see below). ADAR1-KD cells had demonstrable growth defects, as determined by MTT assay and PCNA expression (Fig. 1B–C). This growth defect was mediated in part by a higher rate of apoptosis, as measured by PARP cleavage and caspase activation (Fig. 1C–D). Activation of PKR, a hallmark of ADAR1 loss known to trigger apoptosis via translational shutdown and activation of the IFN response, was also detected, in agreement with previous work14,19 (Fig. 1C). In addition, siADAR cells displayed altered cell cycle profiles as measured by BrdU/PI flow cytometry, with a higher proportion of cells in G1 phase and fewer in S phase, suggesting a block in the G1-S transition (Fig. 1E). Western blotting confirmed the dysregulation, with increased expression of G1-S inhibitors p27/p21 and decreased CDK1 phosphorylation (Fig. 1F). A third Basal A cell line, HCC70, also yielded growth defects in response to ADAR1 KD (Fig. S2C). Overall, ADAR1 appears critical for the proliferation and viability of several Basal A TNBC lines.
Figure 1. Basal A cells are sensitive to loss of ADAR1.
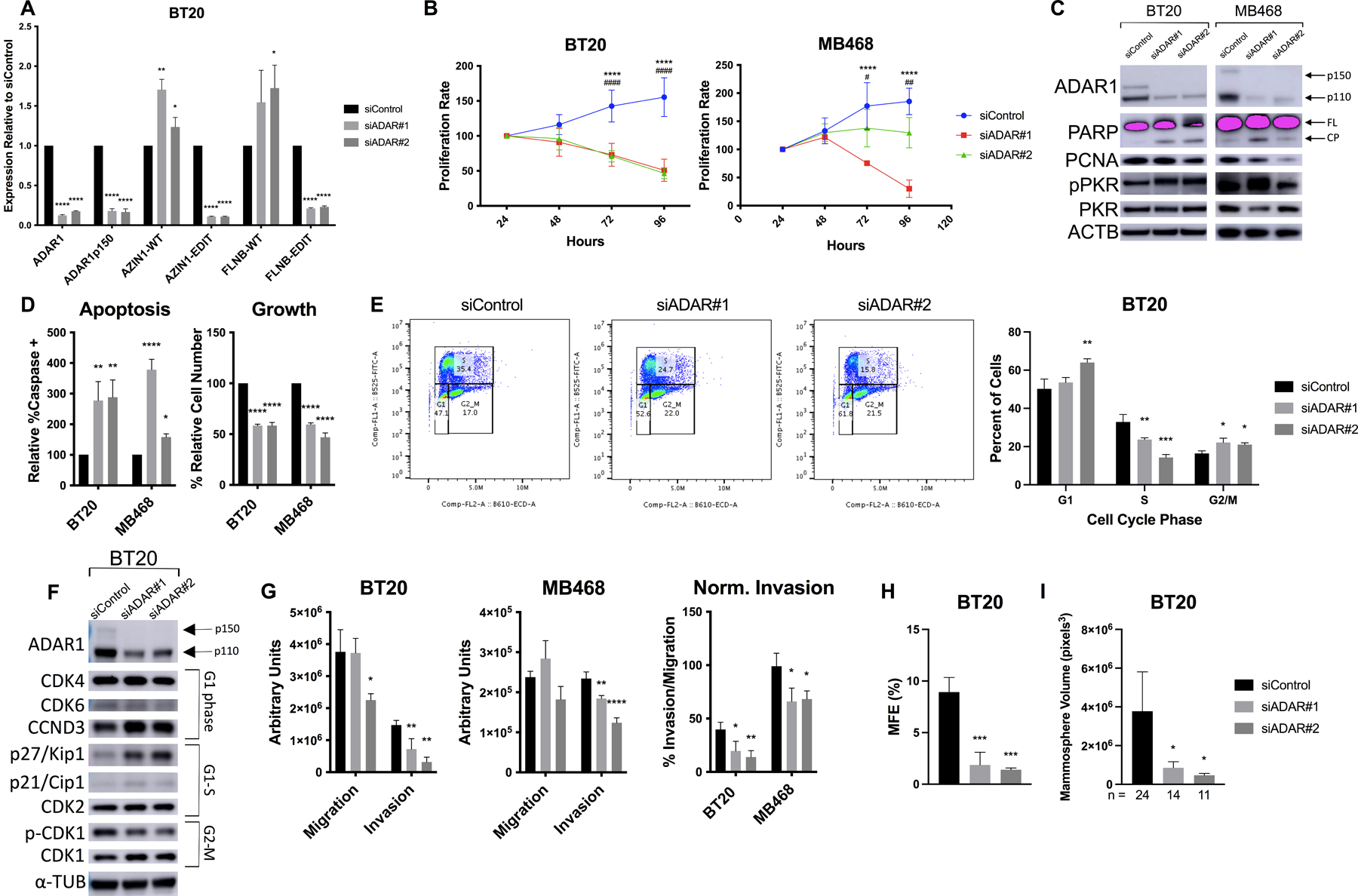
BT20 and MB468 cells were transfected with siRNAs against ADAR1. A) qPCR after 72hr KD for total ADAR1 or the p150-specific transcript, and the wildtype (WT) or edited (EDIT) form of two recoding edit sites, AZIN1 c.1099A>G (S367G) and FLNB c.6970A>G (M2293V). B) MTT assay at indicated timepoints, normalized to D1. p-value symbols are * = siADAR#1 and # = siADAR#2. C) Western blot in 48hr ADAR1-KD cell lysates. Two isoforms of ADAR1 (p110 and p150) are indicated. FL = full length, CP = cleavage product. D) Apoptosis assay, measuring both caspase activation (left) and cell count (right). E) Cell cycle assay measuring PI and BrdU-FITC staining by flow cytometry at 48hr. (Left) Representative dot plot. (Right) Average of three replicates. F) Western blotting for cell cycle markers, with relevant phase indicated. G) Invasion and migration assay using transwell inserts. ImageJ was used to quantify cell area in five images per insert. H) Mammosphere Formation Efficiency (MFE) assay, counting spheres ≥50μm diameter at day 5 after seeding. I) Mammospheres were imaged at day 5 and approximate volume determined from two diameter measurements per sphere; n indicates # spheres per condition.
We next assessed the role of ADAR1 in cell motility via transwell inserts and found that ADAR1-KD cells have reduced invasion, even when normalizing to account for effects on migration (Fig. 1G). We also tested ADAR1 loss in a mammosphere formation efficiency (MFE) assay, which quantifies the ability of individual cells to propagate in 3D suspension culture20. ADAR1-KD BT20s had significantly reduced MFE compared to control and yielded smaller mammospheres overall (Fig. 1H–I, S3A). Mammospheres at day 5 still maintained reduced ADAR1 and editing levels (Fig. S3B). Taken together, these results demonstrate that ADAR1 is a critical regulator of multiple cancer hallmarks, including growth and viability, motility, and sphere formation in TNBC cells.
ADAR1-regulated miRNAs control cell cycle & invasion
After demonstrating that ADAR1 is important for growth & motility in TNBC cells, we examined to what extent ADAR1 regulates global gene expression and thus mediates its effects on cancer cell aggressiveness. To do so, we performed RNA-seq on both poly(A)+ and small RNAs in ADAR1-KD BT20 cells and identified differentially expressed genes and A-to-I edited transcripts. We compared all four siADAR replicates against siControl to call differentially expressed (DE) genes, which yielded over 1,700 poly(A)+ genes (padj < 0.05), with over 800 having |log2fc| > 0.5 (Table S3). Importantly, ADAR1 was the most significant downregulated gene, confirming the efficacy of silencing (Fig. 2A).
Figure 2. RNA-seq analysis identifies ADAR1-regulated pathways.

A) Volcano plot for high-confidence DE poly(A)+ RNAs, with ADAR1 noted. B) Enrichment results for siADAR-upregulated genes using GSEA on KEGG terms. Normalized enrichment score (NES) and −log10(FDR) are shown in blue and red, respectively. FDR cutoffs are indicated. C) qPCR for IL6/TNF in 72hr ADAR1-KD BT20s. D) ELISA for IL6 in 72hr ADAR1-KD BT20s. E) Enrichment results for siADAR-downregulated genes using GSEA on KEGG terms. Normalized enrichment score (NES) and −log10(FDR) are shown in blue and red, respectively. FDR cutoffs are indicated. F) Volcano plot for high-confidence DE miRNAs, with miRNAs 27a-5p and 4485-3p indicated and miRNAs with previous connection to ADAR1 highlighted in red (see Table S5). G) Enrichment results for TargetScan predicted targets of all siADAR-upregulated miRNAs using GSEA on Hallmark terms. Normalized enrichment score (NES) and −log10(FDR) are shown in blue and red, respectively. FDR cutoffs are indicated.
In order to understand the global effects of ADAR1 gene dysregulation, we performed Gene Set Enrichment Analysis (GSEA)21. Analysis of all DE poly(A)+ genes (padj<0.05) yielded significant enrichment (FDR<0.05) for Cytokine and Cytokine Receptor Interaction genes in the upregulated genes (Fig. 2B). Upregulation of these genes, especially the top two hits, IL6 and TNF, is associated with an inflammatory immune response, which can play dual roles in cancer progression and antitumor immunity22,23. We confirmed that ADAR1 loss generally induces IL6/TNF upregulation in Basal A cells, with some exception or nonsignificant trends in HCC70s and mammospheres (Fig. 2C, S2D–E, S3C). We also assessed cytokine secretion by ELISA for IL6 in ADAR1-KD cells, which demonstrated significant upregulation, although much less than that observed for mRNA (Fig. 2D). Additionally, TNF consistently fell below the minimum detectable concentration of our kit (2pg/ml), preventing quantitative analysis. These observations are in line with previous work demonstrating activation of IFN response upon loss of ADAR1 and reinforces the connection between ADAR1 and antitumor immunity in TNBC8,19,24.
Additional pathways meeting a less stringent significance cutoff (FDR<0.25) included enrichment for cancer signaling (Pathways in Cancer, MAPK Signaling), cell motility (Focal Adhesion), and innate immune response genes (Toll-like & Nod-like Receptor), confirming that ADAR1 is a key regulator of cancer and immune pathways (Fig. 2B). Although below the FDR<0.25 cutoff, we also saw an enrichment for Cell Cycle in ADAR1-KD downregulated genes, supporting the observed cell cycle defects in siADAR cells and previous RNA-seq pathway analysis in TNBC (Fig. 2E)13.
Surprisingly, there were far fewer hits in the analysis of miRNAs from the smRNAseq data, compared to previous studies25. Only 12 DE miRNAs were called at padj < 0.05, and all had |log2fc| > 0.5 (Fig. 2F, Table S4). This may be due to the lower number of sequencing replicates, and thus our data appears to reflect only the mostly robustly altered miRNAs. Of the DE miRNAs called, we found that many had previous evidence of ADAR1 regulation, either by direct binding or editing (Table S5). We used TargetScan to call all predicted targets for the eight upregulated miRNAs and observed that the targets downregulated in siADAR were enriched for cell cycle (E2F Targets, G2M Checkpoint) and cell-cell contact (Apical Junction) pathways (Fig. 2G). Thus, these miRNAs are likely to function in pathways relevant to ADAR1 and could contribute to its effects on cell cycle & invasion.
We chose to study miR-27a-5p and miR-4485-3p in detail, as these miRNAs had strong evidence for ADAR1 regulation (Table S5) and had demonstrated relevance in breast cancer. miR-27a-5p has been shown to suppress growth and invasion in cancer cells, and we found that its expression is significantly decreased in TNBC vs. normal tissue (p=1.1E-5) in TCGA samples, in addition to having negative correlation with ADAR1 (R=−0.073, p=0.035) (Fig. 3A, Fig. S4A)26,27. Notably, there is previous evidence for A-to-I editing of miR-27a, but we were unable to detect this in our cells. miR-4485-3p was shown to suppress BC xenograft growth and suppress cyclins, though we were unable to compare expression in tumors as miR-4485-3p was not available in TCGA samples28,29. We used qPCR first to validate that the mature miRNAs are upregulated in siADAR cells, and confirmed for miR-27a that the opposite strand miR-27a-3p (with known oncogenic roles) was unchanged (Fig. 3B)30. We then determined that the pre-miRNA and pri-miRNA transcripts were unchanged or decreased, suggesting that ADAR1 regulates these miRNAs post-transcriptionally by altering the processing efficiency, as has been described in several studies (Fig. 3B)25,31. In order to further test the relationship between ADAR1 and these miRNAs, we rescued expression of the ADAR1 p110 or p150 isoform in ADAR1-KD cells. We used qPCR to confirm a significant decrease in miR-27a-5p expression in p150-expressing cells, suggesting a direct role in suppressing miRNA expression, but did not observe a significant change in miR-4485-3p expression (Fig. S4B).
Figure 3. ADAR1-regulated miRNAs alter growth and invasion.
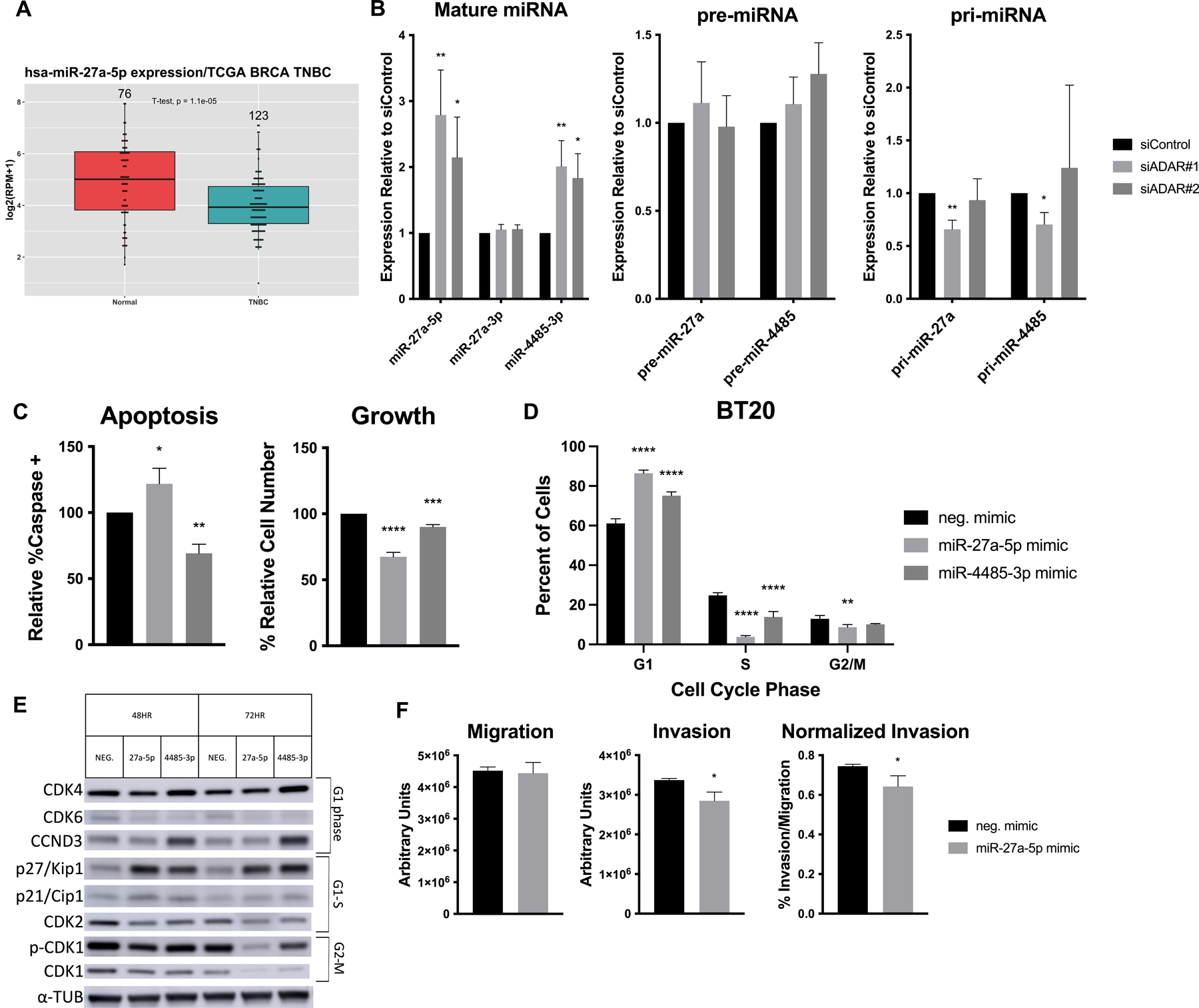
A) miR-27a-5p expression in TNBC vs. normal tissue. Number of samples indicated above, p = 1.1E-5. (B-F) BT20 cells were transfected with siRNAs or miRNA mimics. B) qPCR on mature, precursor, and primary miRNA transcripts in siADAR cells. C) Apoptosis assay, measuring caspase cleavage (left) and cell count (right). D) BrdU/PI cell cycle assay at 72hr, showing dot plot averages. E) Western blotting for cell cycle markers, with relevant phase indicated. F) Invasion/migration assay in miR-27a-5p transfected cells.
To determine the function of each individual miRNA, we transfected BT20 cells with a control or miRNA mimic (Fig. S4C–D). Overexpression of either miRNA led to reduced growth, but only miR-27a-5p could explain this phenotype at least in part by increasing apoptosis (Fig. 3C, S4E). Given that ADAR1-KD cells also showed cell cycle defects, we examined the effect of miRNA overexpression on cell cycle progression. Both miRNAs promoted a G1 arrest, with the most notable reduction of cells in S phase, similar to the phenotype in ADAR1-KD cells (Fig. 3D, S4F). Western blotting for key cell cycle regulators revealed an increase in CDK2 inhibitor p27/Kip1 in both mimic conditions, as well as decreased CDK2 and, especially at 72hr, CDK1 (Fig. 3E). Overexpression of miR-27a-5p led to a significant decrease in invasion, even when normalized to migration (Fig. 3F). Lastly, we assessed the effect of these miRNAs on IL6 and TNF and observed significant mRNA upregulation of both by miR-27a-5p overexpression, as well as increased IL6 secretion (Fig. S4G–H). These results demonstrate that both miR-27a-5p and miR-4485-3p contribute to the suppression of cancer phenotypes, and suggests that ADAR1 acts to inhibit the production of these tumor suppressor miRNAs.
In order to better understand how these miRNAs were involved in suppressing growth and invasion, we examined potential targets. We identified predicted targets for each miRNA using TargetScan and overlapped the results with mRNAs that decreased in the siADAR RNA-seq analysis (where miRNAs increased)32. Prioritizing genes with previous literature linking them to cancer, we first tested the expression of candidates upon miRNA mimic transfection (Fig. 4A). Candidates that showed promising anticorrelation with the miRNA were also tested in siADAR cells to confirm downregulation (Fig. 4B). From this work, the top candidates for each miRNA were tested for direct miRNA targeting at the 3’UTR using the psicheck dual luciferase assay, whereby the 3’UTR of a gene is inserted downstream of Renilla luciferase and luminescence reading used as a proxy for expression. We also cloned a miRNA binding site (BS) mutant form of each construct. We then compared the mimic over control luminescence ratio for WT and mutant constructs to identify miRNA BS-dependent regulation. Using this assay, we confirmed direct miR-4485 targeting of GALNT14 and PARK7 but were unable to validate any miR-27a-5p targets (Fig. 4C). GALNT14 is a glycosyltransferase that promotes lung-specific metastasis in TNBC cells via suppression of inhibitory BMP signaling33. PARK7, also called DJ-1, is known to promote PI3K/Akt signaling and cell growth under stress34. We confirmed that miR-4485-3p overexpression resulted in decreased protein expression for both genes (Fig. 4D). Confirming the tumor relevance of both targets, we observed increased expression for PARK7 and GALNT14 in TNBC vs. normal samples (p=0.022 and 4.9E-10, respectively, Fig. 4E–F). Examining all predicted targets of each miRNA, we observed enrichment of invasion (ECM Receptor Interaction), Cytokine-Cytokine Receptor Interaction, and Cell Cycle pathways for miR-27a-5p and Pathways in Cancer for miR-4485-3p (Fig. 4G–H). Thus, upregulation of these genes via suppression of miR-27a-5p and miR-4485-3p targeting could help explain the oncogenic phenotypes promoted by ADAR1 in Basal A cells.
Figure 4. Validation of miRNA targets.
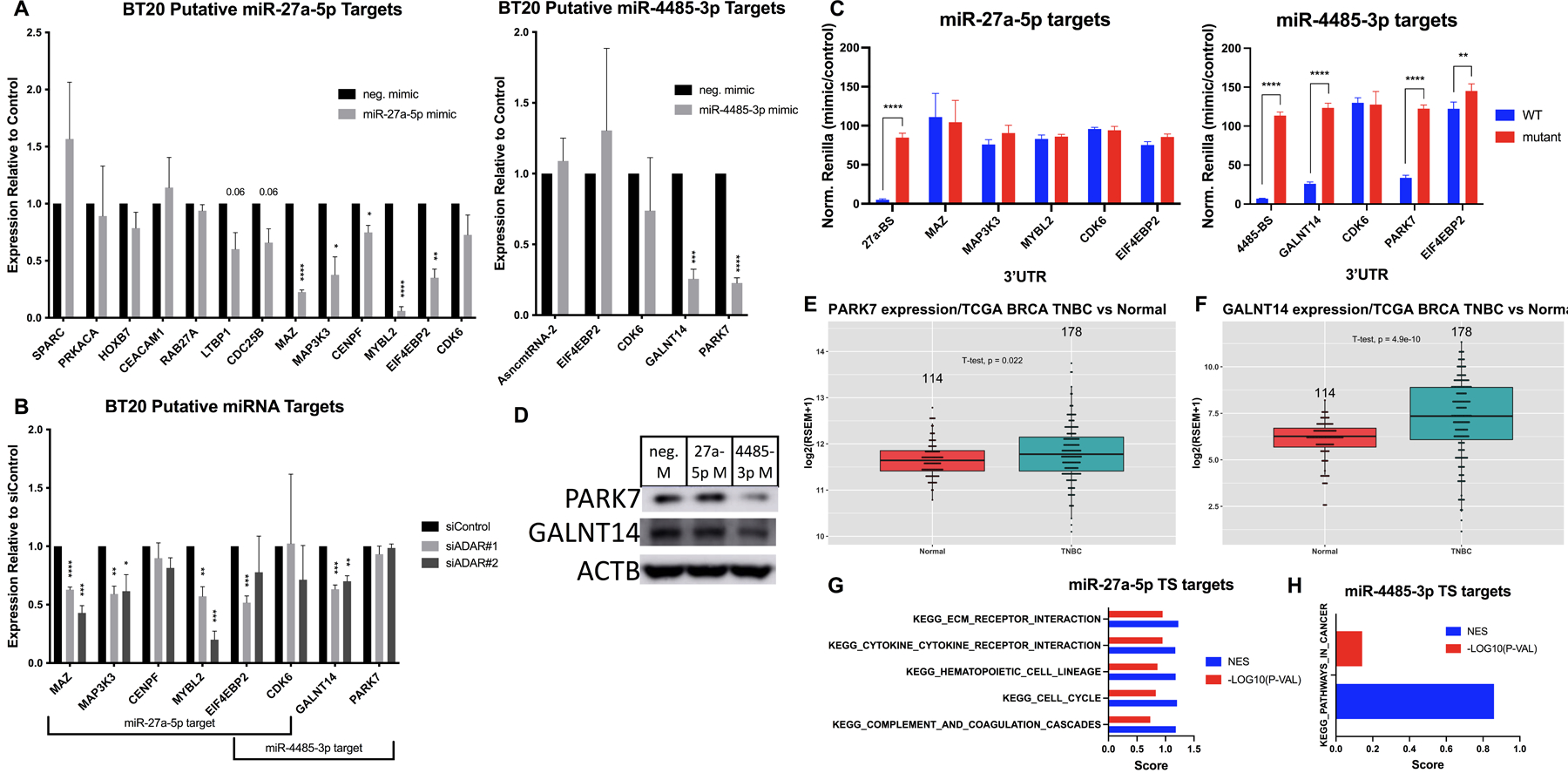
A) Mimic-transfected cells were analyzed by qPCR on putative targets for miR-27a-5p (left) and miR-4485-3p (right). B) siADAR cells were analyzed by qPCR on putative miRNA targets. C) HEK293T cells were cotransfected with miRNA mimics and psicheck constructs for each putative target mRNA 3’UTR containing WT or mutant miRNA binding sites (BS). Normalized Renilla/Firefly luminescence in miRNA vs. control mimic conditions is plotted for each plasmid construct. D) Western blot for PARK7 and GALNT14 in mimic-transfected cells. E) PARK7 expression in TNBC vs. normal tissue. Number of samples indicated above, p=0.022. F) GALNT14 expression in TNBC vs. normal tissue. Number of samples indicated above, p=4.9E-10. G) Enrichment results for miR-27a-5p TargetScan predicted targets using GSEA on KEGG terms. Normalized enrichment score (NES) and −log10(p-value) are shown in blue and red, respectively. H) Enrichment results for miR-4485-3p TargetScan predicted targets using GSEA on KEGG terms. Normalized enrichment score (NES) and −log10(p-value) are shown in blue and red, respectively.
FLNB editing regulates growth & invasion
We also utilized our RNAseq data to profile the A-to-I editome in Basal A cells. GATK variant calling in siControl poly(A)+ samples yielded over 2,500 A-to-I edit sites after filtering out known SNPs. As expected, the majority (84%) of called variants were A-to-I edits (A>G and T>C are considered A-to-I as sequencing was not strand-specific), while another 6% are likely C-to-U edits, mediated by APOBECs (Fig. 5A, Table S6)1. By taking into account the change in edited transcript allele frequency (AF) in siADAR samples (which we expect to decrease), we identified 500 high-confidence edit sites (FDR<0.1), most of which were found in the 3’UTR (Fig. 5B). The high-confidence edit sites have a median edit AF of 0.37, and the average log2FC in edit AF is −0.35, confirming that ADAR1-KD cells have globally decreased editing (Fig. 5C, Table S6). GSEA analysis of edited genes ranked by editing prevalence revealed enrichment for cell cycle (Neg. Regulation of Cell Cycle Process, Regulation of Cell Cycle Phase Transition) and cytokine (Cytokine Mediated Signaling Pathway, Response to Cytokine) pathways (Fig. 5D).
Figure 5. Identification of A-to-I edited genes.
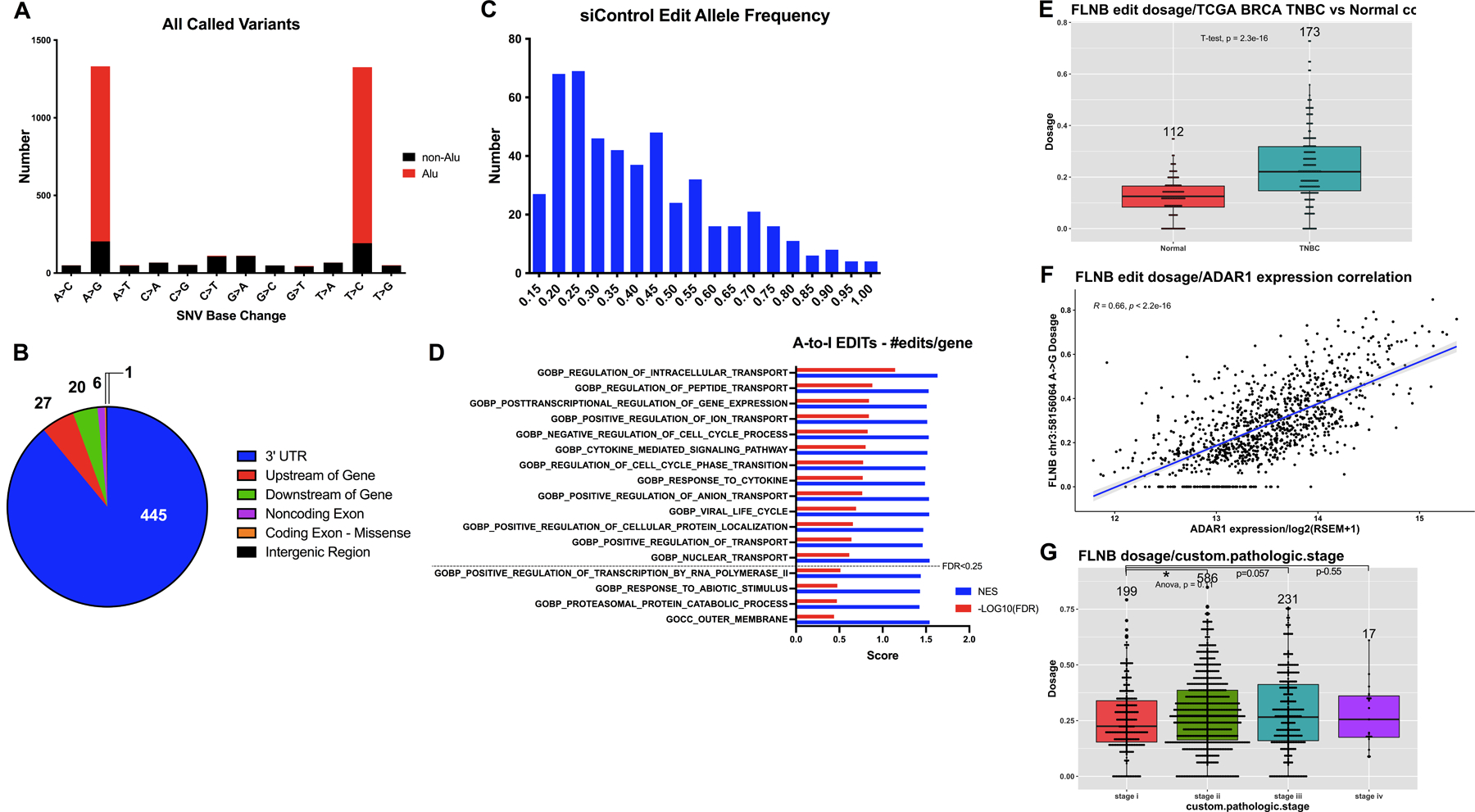
A) Frequency of each base change for all non-SNP variants, with those overlapping Alu repeats in red. A-to-I edits are represented by A>G and T>C variants, as sequencing was not strand-specific. B) Pie chart depicting relative abundance of high-confidence (FDR<0.1) edit site localization. C) Histogram of the average edit allele frequency for high-confidence (FDR<0.1) edit sites. Median AF was 0.37, mean AF was 0.41. D) Enrichment results for the most prevalently edited genes using GSEA on GO terms. Normalized enrichment score (NES) and −log10(FDR) are shown in blue and red, respectively. FDR cutoffs are indicated. E) FLNB chr3:58156064 editing dosage (AF) in TNBC vs. normal tissue. Number of samples indicated above, p = 2.3E-16. F) FLNB edit dosage correlation with ADAR1 expression in BC samples. R = 0.66, p < 2.2E-16. G) FLNB edit dosage correlation with pathologic stage in all BC samples. Individual t-tests with Stage I are as follows: Stage II p=0.01, III p=0.057, IV p=0.055.
Although rare, edit sites within the coding regions of mRNAs are particularly intriguing due to their clear potential for impacting the sequence, and thus function, of a protein. We investigated the most significant recoding edit site in our dataset, a missense mutation in filamin B (FLNB) that leads to an M2293V substitution (chr3:58156064). FLNB is a cytoskeleton component that binds actin and scaffolds a diverse array of proteins, thus regulating motility and growth signaling. Mutations in filamins are associated with musculoskeletal abnormalities, but their dysregulation has also been implicated in tumorigenesis35–37. FLNB has been reported as a human BC tumor-suppressive susceptibility gene with survival prognosis value, and it plays a localization-dependent role in suppressing EMT signatures in BC38,39. FLNB editing at chr3:58156064 has been identified in HCC and glioblastoma cells and ADAR1 binding to FLNB detected in Galipon et al. 2017 via RIP-seq, but any functional effect of this substitution has yet to be described18,40,41. Confirming its relevance in tumors, we determined that FLNB editing is higher in TNBC tumors vs. normal tissue (p=2.3E-16), strongly correlated with ADAR1 expression (R=0.66, p<2.2E-16), and increased with higher stage in BC (Fig. 5E–G).
In our RNA-seq data, the FLNB edit site has about 20% edit AF, and we validated this by Sanger sequencing of BT20 cDNA (Fig. S5A). Importantly, the ‘G’ peak is lost in siADAR cDNA, and absent in genomic DNA, confirming that that it is an RNA- and ADAR-specific variant. The edit site is located in IgG-like repeat 22, within the C-terminal region involved in many protein-protein interactions, and is predicted “Pathogenic” (score 0.80) by FATHMM-MKL35,42,43. To test the function of FLNB in Basal A cells, we first manipulated its expression. While knockdown of FLNB did not significantly affect proliferation, the cells had reduced PARP cleavage and PCNA expression (Fig. S5B–D). FLNB-KD cells also had increased invasion and migration (Fig. S5E). Given that FLNB has been associated with EMT, we assessed expression of the pro-EMT transcription factor Slug and observed concomitant increase and decrease in siFLNB and siADAR cells, respectively (Fig. S5F–G). Thus, we hypothesized that ADAR1 editing of FLNB decreases the activity of this tumor suppressor to promote invasion, but direct testing of the functional consequence of the edit was needed.
Poor plasmid transfection efficiency and a gene too large for viral constructs (FLNB cDNA ~8kb) prevented a straightforward overexpression experiment, so we used CRISPR-Cas9 editing to specifically alter endogenous FLNB editing. With similar reasoning to previous work, we took advantage of the requirement for ADAR1 to bind dsRNA and deleted the intronic sequence predicted to hybridize with the FLNB edit site, termed the edit complementary site (ECS) (Fig. 6A, Fig. S6A)42. Cells were transduced with plentiCRISPR constructs expressing both an upstream and downstream guideRNA (gRNA) targeting the ECS or single nontargeting control (NT) and underwent puromycin selection (Fig. S6B). We first confirmed that total FLNB and ADAR1 expression were not altered compared to NT cells, but that FLNB editing was specifically abrogated in FLNBdelECS cells (Fig. 6B, S6C–D). We validated robust ECS deletion by amplification of the gDNA and TOPO cloning individual amplicons for sequencing, which confirmed ~85% deletion efficiency (Fig. S6E). FLNBdelECS cells had significantly reduced growth rate and invasive capability, as well as concomitant decrease in Slug expression (Fig. 6C–E). These experiments validate that FLNB editing, even at moderate levels, has a biologically meaningful effect on FLNB protein to promote oncogenic phenotypes in TNBC cells.
Figure 6. Loss of FLNB editing suppresses growth & invasion.
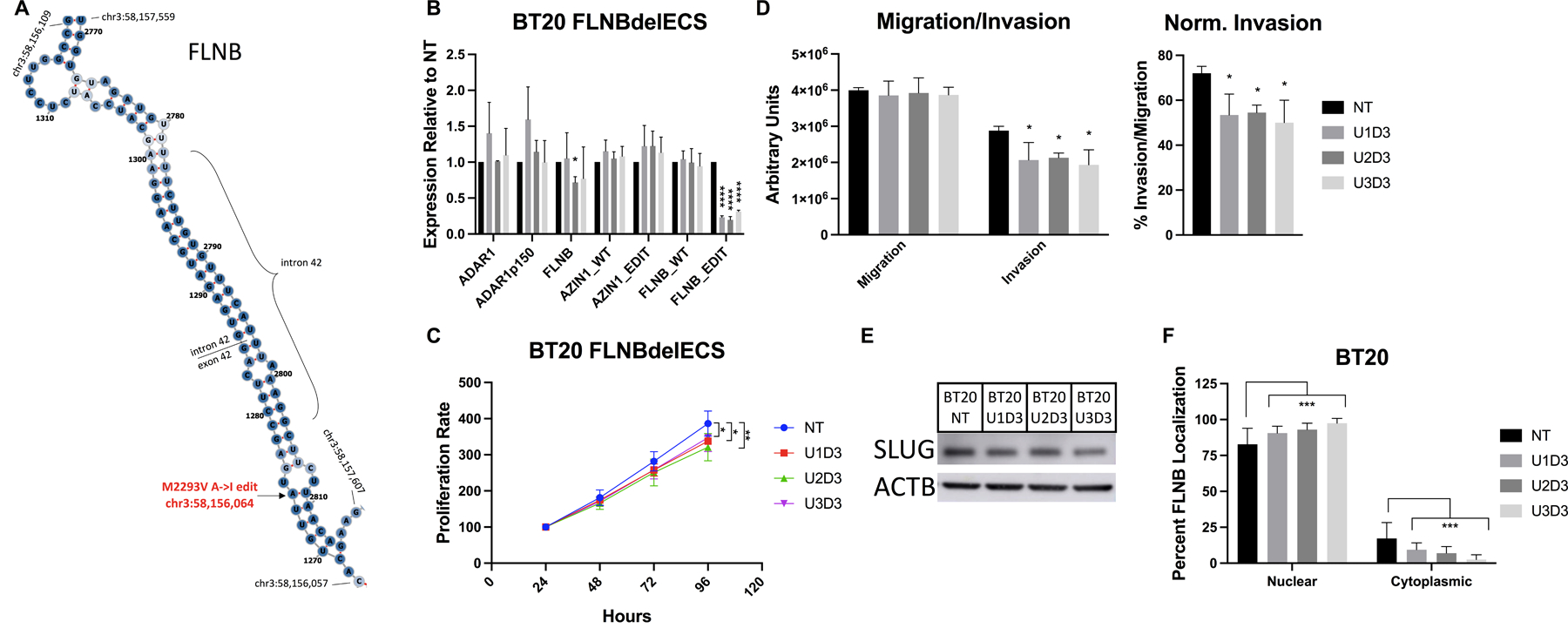
A) Schematic of FLNB mRNA predicted structure, generated using RNAfold (http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi). FLNB edit site, intron/exon boundaries, and boundary coordinates are indicated. (B-F) Cells were stably transduced with plentiCRISPR-nontargeting control (NT) or dual FLNBdelECS gRNAs (U1-3D3). B) qPCR for ADAR and FLNB, and AZIN1/FLNB edit sites. C) MTT assay, normalized to D1. D) Invasion/migration assay using transwell inserts. E) Western blot for Slug. F) Quantification of FLNB localization from immunofluorescence images. Nuclear localization called by overlap with DAPI signal.
To further examine how FLNB editing may alter protein function, we tested the effect of editing on localization by immunofluorescence. FLNB localization is tightly regulated, as shown by previous work detailing an exon skipping event that reduces nuclear FLNB to promote EMT (a mechanism not prevalent in our cells)39. Upon quantification of confocal images we observed that the cytoplasmic localization of FLNB is significantly decreased in FLNBdelECS cells (Fig. 6F, S7). We confirmed that total FLNB intensity was not significantly altered upon ECS deletion (Fig. S6F). The specificity of the antibody was confirmed by Western blot in siFLNB cells, and the fluorescent signal showed no background staining (Fig. S6F–G). To test whether FLNB localization is altered in ADAR1-KD cells, we performed subcellular fractionation, but only observed a nonsignificant trend of decreased cytoplasmic/nuclear ratio in ADAR1-KD cells (Fig. S6H).
Discussion
This work demonstrates the sensitivity of triple-negative, Basal A breast cancer cells to ADAR1 loss and identifies novel mechanisms of ADAR1-mediated oncogenesis. While recent work has evaluated ADAR1 sensitivity in triple-negative breast cancer with a focus on cell/tumor growth and apoptosis, our work shows that ADAR1 loss induces additional defects in cell cycling and invasion. To our knowledge, our work represents the first effort to investigate downstream mechanisms of ADAR1 in Basal A BC based on unbiased RNA-seq screening. We have identified genes that are differentially expressed and/or edited upon ADAR1 perturbation, which are enriched for pathways involving cytokine signaling, cell cycle, motility, and broader cancer signaling. We also identified several thousand alternately spliced genes, of which the majority were exon skipping events (Fig. S8A). The most differentially spliced genes were also enriched for cell cycle, cytokine signaling, and apoptosis pathways, suggesting that the global effects of ADAR1 on the global transcriptome converge upon common pathways important in cancer (Fig. S8B).
We were particularly interested in Basal A TNBC as it is enriched for cells sensitive to ADAR1 loss. Previous work in breast and other cancer contexts has attempted to identify predictors for ADAR1 sensitivity, and some connection has been made to baseline expression of IFN-stimulated genes, like PKR14,24. However, more work is needed, especially towards predicting clinical responders of potential anti-ADAR1 therapeutics.
We have demonstrated that ADAR1-downregulated miRNAs 27a-5p and 4485-3p inhibit cell cycle progression, and that miR-27a-5p also suppresses invasion and promotes IL6/TNF expression in TNBC cells. While there is conflicting literature on the role of miR-27a, both 5p and 3p, in regulating inflammatory cytokines like IL6/TNF, it appears that this miRNA contributes to the altered expression of cytokines observed in ADAR1-KD cells44,45. Inflammatory cytokines can play dual roles in cancer cells, promoting stemness and metastasis or death and growth suppression, and can also recruit or suppress immune cells in the tumor microenvironment22,23. However, our results indicate that ADAR1 loss suppresses growth and invasion even with upregulated inflammatory cytokines, and others have observed these phenotypes translate to in vivo mouse studies in diverse cancer contexts6,14,46.
Target prediction for all siADAR-upregulated miRNAs yielded enrichment for genes involved in cell cycle and motility. We validated direct binding of miR-4485-3p to oncogenic GALNT14 and PARK7, which likely contribute to the tumor suppressive role of this miRNA. We were unable to validate direct targets of miR-27a-5p, although predicted targets were enriched for pathways involved in invasion, cytokine signaling, and cell cycle.
While we focused on dysregulated miRNAs with connection to ADAR1, we also observed upregulation of miR-4488 for which recent literature has suggested a role in breast cancer (Table S5). Exclusion of miR-4488 from extracellular vesicles promoted angiogenesis in TNBC cells and was decreased in vesicles from TNBC patient serum47. While we did not assess angiogenesis phenotypes in our study, this work complements ours in suggesting an additional miRNA-mediated mechanism by which ADAR1 may promote metastasis in TNBC.
Given previous evidence for ADAR1 binding to precursors of both miRNAs and that only the mature forms are significantly dysregulated upon ADAR1 KD, it is likely that ADAR1 binding affects the processing of these miRNAs. Previous work has shown that ADAR1 binding can alter miRNA processing in either direction, and can be editing-independent, possibly even via association with DICER25,31,48. We were unable to detect evidence of A-to-I editing of either pri-miRNA by Sanger sequencing. As ADAR1 regulates the global transcriptome, the dysregulated miRNA processing could also be due to an indirect effect. Rescue of ADAR1 expression after knockdown yielded decreased miR-27a-5p, but not miR-4485-3p, expression, suggesting the latter may be regulated indirectly. Interestingly, DICER1 is decreased in ADAR1-KD cells (log2FC −0.62) which would be expected to decrease mature miRNA expression, so perhaps the activity of ADAR1 is able to more than compensate for this effect.
Given that the majority of edit sites fall within the 3’UTR, we looked for edit sites that overlap with a miRNA seed sequence, considering seeds that match either the unedited or edited version of the transcript. We identified de novo miRNA BSs where the seed matches the edited transcript and the mRNA has log2fc > 0.20 in siADAR samples. We also found disrupted miRNA BSs that match the unedited transcript, and where the mRNA has log2fc < −0.20 in siADAR samples. However, this analysis yielded only 19 genes with de novo sites and 9 genes with disrupted sites, although two de novo sites were identified in the cell cycle regulator RAD1 (Table S7, S8). Editing analysis failed to confidently call any edited miRNAs, which are rare but have been documented49. Most likely, the sequencing was not deep enough and/or relied on too few replicates to accurately call edited miRNAs.
We provided novel characterization of the highly prevalent FLNB chr3:58156064 edit site, demonstrating a role in tuning down the growth- and invasion-suppressing activities of the protein. Additionally, we demonstrated that FLNB editing suppresses the nuclear localization of the protein, which may impact invasion by reducing the EMT-suppressive role of nuclear FLNB, as previously described39. The mechanism for how FLNB editing modifies localization remains unclear, although the edit site falls within a region important for protein interactions and may alter key binding affinities35. A nearby edit site found in both FLNA and FLNB (Q2341R/Q2296R) was shown in mice to have dozens of distinct protein interactions for edited vs. unedited FLNA (no FLNB or FLNA editing outside of FLNB-M2293V was detected in our cells)42. Thus, we hypothesize that the M2293V edit reduces FLNB activity via disruption of binding partners and consequently of localization.
Here we have expanded the role for ADAR1 in TNBC by demonstrating novel cell cycle, invasion, and mammosphere phenotypes, further solidifying its importance especially in Basal A cells. We have also provided a broad investigation into the multiple parallel pathways by which ADAR1 modulates the global transcriptome. These results highlight the diverse roles that ADAR1 performs within cells, suggesting that ADAR1 could be an important upstream regulator for diverse processes, fine-tuning many genes to create a large, cumulative impact on the cancer cell transcriptome. Additionally, we have characterized several novel mechanisms of ADAR1-mediated oncogenesis, involving miRNA dysregulation and FLNB editing. This work contributes to a growing body of evidence that supports investigation of ADAR1 as a drug target in TNBC and diverse additional cancer contexts.
Supplementary Material
Implications:
Targeting ADAR1 and thus downstream FLNB editing and miRNA regulation represents a possible novel therapeutic strategy in triple-negative breast cancer.
Acknowledgements
The authors would like to thank Lorena Pantano of the Harvard Chan Bioinformatics Core, Harvard T.H. Chan School of Public Health, Boston, MA for assistance with differential expression and editing analysis of the RNAseq data. We also thank Lay-Hong Ang and Aniket Gad of the Confocal Imaging Core Facility at BIDMC for assistance with image collection and analysis for immunofluorescence experiments.
Financial support:
We acknowledge the NCI Outstanding Investigator Award (R35CA232105) and funding from the Ludwig Center at Harvard to FJS.
List of abbreviations
- ADAR
adenosine deaminase acting on RNA
- AF
allele frequency
- A-to-I
adenosine-to-inosine
- BC
breast cancer
- BS
binding site
- DE
differentially expressed
- dsRNA
double-stranded RNA
- ECS
edit complementary site
- FLNB
filamin B
- gDNA
genomic DNA
- gRNA
guideRNA
- GSEA
Gene Set Enrichment Analysis
- HCC
hepatocellular carcinoma
- IFN
interferon
- MB468
MDA-MB-468
- MFE
mammosphere formation efficiency
- miRNA
microRNA
- NES
normalized enrichment score
- NT
nontargeting control
- PI
propidium iodide
- P/S
penicillin/streptomycin
- RESS-qPCR
RNA editing site-specific qPCR
- TCGA
The Cancer Genome Atlas
- TNBC
triple-negative breast cancer
Footnotes
Conflict of Interest: The authors declare no competing interests.
Availability of data and materials
RNA sequencing data is available at GEO under accession number GSE177056. Remaining data generated and/or analyzed during the current study are included in this published article (and its supplementary files) or available from the corresponding author upon reasonable request.
References
- 1.Nishikura K A-to-I editing of coding and non-coding RNAs by ADARs. Nat. Rev. Mol. Cell Biol 17, 83–96 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xu L-D & Öhman M ADAR1 Editing and its Role in Cancer. Genes 10, 12 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Slotkin W & Nishikura K Adenosine-to-inosine RNA editing and human disease. Genome Med 5, 105 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen L et al. Recoding RNA editing of AZIN1 predisposes to hepatocellular carcinoma. Nat. Med 19, 209–216 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zipeto M et al. ADAR1 Activation Drives Leukemia Stem Cell Self-Renewal by Impairing Let-7 Biogenesis. Cell Stem Cell 19, 177–191 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu X et al. ADAR1 promotes the epithelial-to-mesenchymal transition and stem-like cell phenotype of oral cancer by facilitating oncogenic microRNA maturation. J. Exp. Clin. Cancer Res 38, 315 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nemlich Y et al. MicroRNA-mediated loss of ADAR1 in metastatic melanoma promotes tumor growth. J. Clin. Invest 123, 2703–2718 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ishizuka JJ et al. Loss of ADAR1 in tumours overcomes resistance to immune checkpoint blockade. Nature (2018) doi: 10.1038/s41586-018-0768-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lawson KA et al. Functional genomic landscape of cancer-intrinsic evasion of killing by T cells. Nature 586, 120–126 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fumagalli D et al. Principles Governing A-to-I RNA Editing in the Breast Cancer Transcriptome. Cell Rep 13, 277–289 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Paz-Yaacov N et al. Elevated RNA Editing Activity Is a Major Contributor to Transcriptomic Diversity in Tumors. Cell Rep 13, 267–276 (2015). [DOI] [PubMed] [Google Scholar]
- 12.Sagredo EA et al. ADAR1-mediated RNA-editing of 3′UTRs in breast cancer. Biol. Res 51, 36 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sagredo EA et al. ADAR1 Transcriptome editing promotes breast cancer progression through the regulation of cell cycle and DNA damage response. Biochim. Biophys. Acta BBA - Mol. Cell Res 118716 (2020) doi: 10.1016/j.bbamcr.2020.118716. [DOI] [PubMed] [Google Scholar]
- 14.Kung C-P et al. Evaluating the therapeutic potential of ADAR1 inhibition for triple-negative breast cancer. Oncogene 40, 189–202 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tsherniak A et al. Defining a Cancer Dependency Map. Cell 170, 564–576.e16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dai X, Cheng H, Bai Z & Li J Breast Cancer Cell Line Classification and Its Relevance with Breast Tumor Subtyping. J. Cancer 8, 3131–3141 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Crews LA et al. An RNA editing fingerprint of cancer stem cell reprogramming. J. Transl. Med 13, 52 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chan THM et al. A disrupted RNA editing balance mediated by ADARs (Adenosine DeAminases that act on RNA) in human hepatocellular carcinoma. Gut 63, 832–843 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Samuel CE Adenosine deaminase acting on RNA (ADAR1), a suppressor of double-stranded RNA–triggered innate immune responses. J. Biol. Chem 294, 1710–1720 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sirkisoon SR et al. TGLI1 transcription factor mediates breast cancer brain metastasis via activating metastasis-initiating cancer stem cells and astrocytes in the tumor microenvironment. Oncogene 39, 64–78 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Subramanian A et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Montfort A et al. The TNF Paradox in Cancer Progression and Immunotherapy. Front. Immunol 10, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fisher DT, Appenheimer MM & Evans SS The Two Faces of IL-6 in the Tumor Microenvironment. Semin. Immunol 26, 38–47 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gannon HS et al. Identification of ADAR1 adenosine deaminase dependency in a subset of cancer cells. Nat. Commun 9, 5450 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bahn JH et al. Genomic analysis of ADAR1 binding and its involvement in multiple RNA processing pathways. Nat. Commun 6, ncomms7355 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu X et al. Coordinated Targeting of the EGFR Signaling Axis by MicroRNA-27a*. Oncotarget 4, 1388–1398 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mizuno K et al. The microRNA expression signature of small cell lung cancer: tumor suppressors of miR-27a-5p and miR-34b-3p and their targeted oncogenes. J. Hum. Genet 62, 671–678 (2017). [DOI] [PubMed] [Google Scholar]
- 28.Fitzpatrick C et al. Mitochondrial ncRNA targeting induces cell cycle arrest and tumor growth inhibition of MDA-MB-231 breast cancer cells through reduction of key cell cycle progression factors. Cell Death Dis 10, 1–12 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sripada L et al. hsa-miR-4485 regulates mitochondrial functions and inhibits the tumorigenicity of breast cancer cells. J. Mol. Med 95, 641–651 (2017). [DOI] [PubMed] [Google Scholar]
- 30.Zhang J et al. MicroRNA-27a (miR-27a) in Solid Tumors: A Review Based on Mechanisms and Clinical Observations. Front. Oncol 9, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Qi L et al. An RNA editing/dsRNA binding-independent gene regulatory mechanism of ADARs and its clinical implication in cancer. Nucleic Acids Res 45, 10436–10451 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Agarwal V, Bell GW, Nam J-W & Bartel DP Predicting effective microRNA target sites in mammalian mRNAs. eLife 4, e05005 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Song K-H et al. GALNT14 promotes lung-specific breast cancer metastasis by modulating self-renewal and interaction with the lung microenvironment. Nat. Commun 7, 13796 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vasseur S et al. DJ-1/PARK7 is an important mediator of hypoxia-induced cellular responses. Proc. Natl. Acad. Sci 106, 1111–1116 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stossel TP et al. Filamins as integrators of cell mechanics and signalling. Nat. Rev. Mol. Cell Biol 2, 138–145 (2001). [DOI] [PubMed] [Google Scholar]
- 36.Iguchi Y et al. Filamin B Enhances the Invasiveness of Cancer Cells into 3D Collagen Matrices. Cell Struct. Funct 40, 61–67 (2015). [DOI] [PubMed] [Google Scholar]
- 37.Bandaru S et al. Targeting filamin B induces tumor growth and metastasis via enhanced activity of matrix metalloproteinase-9 and secretion of VEGF-A. Oncogenesis 3, e119 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen L et al. Transposon insertional mutagenesis in mice identifies human breast cancer susceptibility genes and signatures for stratification. Proc. Natl. Acad. Sci 114, E2215–E2224 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li J et al. An alternative splicing switch in FLNB promotes the mesenchymal cell state in human breast cancer. eLife 7, e37184 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kang L et al. Genome-wide identification of RNA editing in hepatocellular carcinoma. Genomics 105, 76–82 (2015). [DOI] [PubMed] [Google Scholar]
- 41.Galipon J, Ishii R, Suzuki Y, Tomita M & Ui-Tei K Differential Binding of Three Major Human ADAR Isoforms to Coding and Long Non-Coding Transcripts. Genes 8, 68 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jain M et al. RNA editing of Filamin A pre-mRNA regulates vascular contraction and diastolic blood pressure. EMBO J e94813 (2018) doi: 10.15252/embj.201694813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shihab HA et al. An integrative approach to predicting the functional effects of non-coding and coding sequence variation. Bioinformatics 31, 1536–1543 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Boucher A et al. The miR-23a~27a~24–2 microRNA Cluster Promotes Inflammatory Polarization of Macrophages. J. Immunol (2020) doi: 10.4049/jimmunol.1901277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kong L et al. Effect of microRNA-27a-5p on apoptosis and inflammatory response of pancreatic acinar cells in acute pancreatitis by targeting PTEN. J. Cell. Biochem 120, 15844–15850 (2019). [DOI] [PubMed] [Google Scholar]
- 46.Anadón C et al. Gene amplification-associated overexpression of the RNA editing enzyme ADAR1 enhances human lung tumorigenesis. Oncogene 35, 4407–4413 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zheng X et al. MCU-dependent negative sorting of miR-4488 to extracellular vesicles enhances angiogenesis and promotes breast cancer metastatic colonization. Oncogene 39, 6975–6989 (2020). [DOI] [PubMed] [Google Scholar]
- 48.Kawahara Y, Zinshteyn B, Chendrimada TP, Shiekhattar R & Nishikura K RNA editing of the microRNA-151 precursor blocks cleavage by the Dicer–TRBP complex. EMBO Rep 8, 763–769 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nigita G et al. microRNA editing in seed region aligns with cellular changes in hypoxic conditions. Nucleic Acids Res 44, 6298–6308 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McFarland JM et al. Improved estimation of cancer dependencies from large-scale RNAi screens using model-based normalization and data integration. Nat. Commun 9, 4610 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA sequencing data is available at GEO under accession number GSE177056. Remaining data generated and/or analyzed during the current study are included in this published article (and its supplementary files) or available from the corresponding author upon reasonable request.