

Abstract
Colorectal carcinogenesis is a complex process, which is linked to dysregulation of human secretory phospholipases A2 (hsPLA2-G-IIA, hsPLA2-G-V, and hsPLA2-G-X), proteases (cathepsin-B, collagenase, thrombin, elastase, and trypsin), carbohydrate hydrolyzing enzymes (α-amylase and α-glucosidase), and free radical generating enzyme (xanthine oxidoreductase (XOR)). Therefore, some new quinazolinones were synthesized and evaluated as inhibitors against this array of enzymes as well as cytotoxic agents on LoVo and HCT-116 cells of colorectal cancer. Compounds 3g, 10, 8, 3c, and 1c exhibited promising cytotoxic effects with IC50 values ranging from 206.07 to 459.79 μM. Nine compounds showed promising enzymatic inhibitory effects, 3b, 3d, 3f, 5, 1a, and 12 (α-amylase), 8 (thrombin, elastase and trypsin), 10 (hsPLA2-G-IIA and hsPLA2-G-V), and 3f (α-glucosidase and XOR). Therefore, the most active inhibitors, were subjected to validated molecular docking studies to identify their affinities and binding modes. The expected physicochemical and pharmacokinetic features of the active candidates, 1a, 1c, 3b, 3c, 3d, 3f, 3g, 5, 8, 10, and 12 were predicted using bioavailability radar charts and boiled-egg graphical representations along with the Lipinski rule of five filter. Collectively, these studies showed the significance of derivatives 1c, 3b, 3c, 3d, 8, 10, and 12 as lead scaffolds for further optimization to develop enzymes inhibitors and anti-colorectal agents.
Introduction
Recently, colorectal cancer (CRC) has shown a more drastic increase in its incidence and mortality1 particularly in the younger population worldwide.2,3 Several enzymes have been proposed to contribute to colorectal carcinogenesis, in particular the human secretory phospholipases A2:4 hsPLA2-G-IIA, hsPLA2-G-V, and hsPLA2-G-X. The pro-tumorigenic roles5,6 of these isoforms are attributed to their potency to cleave the sn-2 ester bond of membrane glycerophospholipids, thus releasing the free fatty acid (mainly arachidonic acid, AA) and lysophospholipids. Further, AA is the precursor for the production of pro-inflammatory mediators termed eicosanoids,7 which comprise leukotrienes (LTs) and prostaglandins (PGs) including PGE2 under the catalytic effects of lipoxygenases (LOXs) and cyclooxygenase enzymes (COX-1 and COX-2), respectively. These mediators interfere with immunity,8 angiogenesis, apoptosis, and proliferation, leading to initiation and progression of CRC.9 Therefore, blocking the release of AA via inhibiting these enzymes is considered as a possible mechanism for prevention and treatment of CRC.
Likewise, proteases are responsible for breaking down proteins into polypeptides and amino acids. These important hydrolytic enzymes have been implicated in many signaling pathways and their deregulation is linked to cancer. Thus, overexpression of cathepsin B protein is observed in 60% of CRC patients. Moreover, previous studies demonstrated several casual roles of cathepsin B in tumor initiation, proliferation, angiogenesis, and invasion.10
Histological and bio-informatics analyses have shown that tumor progression and metastasis are accompanied by an abnormal remodeling of the matrix collagen (i.e., excessive deposition, altered proportions, and changed arrangement of collagen), which is induced by collagenase. Also, this enzyme enhances tumor growth via inducing platelet activation and angiogenesis.11 Consequently, inhibition of collagenase could inhibit tumor cell growth and invasiveness.
In addition, considerable evidence from studies on mice models have indicated that thrombin alters gene expression of tumor cells to promote oncogenesis. The increased thrombin production also augments tumor invasion via activation of platelets, which in association with fibrin aggregates around them stabilize and protect the cancerous cells from host immunity.12
Furthermore, high levels of other proteases, such as neutrophil elastase13 and trypsin,14 stimulate degradation of the extracellular matrix and promote evasion of immune system, invasion, metastasis, and resistance to apoptotic signals.15
Epidemiological and observational studies have indicated hyperglycemia, which is associated with increased levels of α-amylase and α-glucosidase, as a major risk factor in CRC.16−18 Other studies showed that glycemic control using low doses of the antidiabetic drug metformin has been associated with protective or better outcomes in cancer patients.19
Several studies suggest that reactive nitrogen species (RNS) and reactive oxygen species (ROS) play etiological roles in development of CRC via damaging vital macromolecules (lipids, proteins, RNA, and DNA), activating oncogenic signaling, turning off the expression of tumor suppressor genes, and stimulating angiogenesis (through activation of angiogenic factors and production of carcinogenic metabolites),20 proliferation, invasion, cell migration, and apoptosis.21 Despite their strictly modulated generation during cellular catabolism of purines by xanthine oxidoreductase (XOR), increased amounts of free radical are produced as a result of dysregulation of XOR. Moreover, elevated serum XOR activity was found to be associated with an increased risk of developing Type 2 diabetes mellitus (T2DM),22 which predisposes those patients to the risk of developing CRC.
Given these facts, management of hyperglycemia23 through inhibition of α-amylase and α-glucosidase enzymes, in addition to inhibition of XOR,24 phospholipases,25,26 and proteases,27 are expected to provide synergetic therapeutic opportunities for treatment of CRC.
In this regard, several studies have identified quinazolinone ring as an attractive pharmacophore (Figure 1), which can serve as a lead scaffold for designing new antitumor and anti-hyperglycemic agents as well as inhibitors for proteases and phospholipases.
Figure 1.

Structural features of some reported quinazolinone derivatives with antitumor, anti-hyperglycemic, antiproteases, and anti-phospholipases activities.28−31
Indeed, it has been documented that introducing a second chromophore at the 3-position of the quinazolinone ring enhanced the antiproliferative activities of compound I even more than 5-fluorouracil (5-FU) against various cancer cell lines, including M14 and SK-MEL-2 (melanoma), IGROVI (ovarian cancer), TK-10 (renal cancer), PC-3 (prostate cancer), MCF7 (breast cancer), and HT29 (CRC).28
Moreover, the enhanced anti-α-glucosidase activity of Schiff base derivative II has been rationalized using molecular docking analysis, which highlighted the role of quinazoline ring in the inhibition of the enzyme.29
Furthermore, in our previously published work,30 we have reported that Schiff bases III and IV, which are derived from 3-amino-6-bromo-2-methylquinazolin-4(3H)-one, demonstrated improved inhibitory efficiencies compared to oleanolic against hsPLA2-G-V and hsPLA2-G-X, respectively, whereas compound V exhibited strong anti-α-amylase, anti-α-glucosidase activities. In addition, thioacetamide derivative VII displayed more potent α-amylase inhibitory efficiency than quercetin and derivative VI showed improved cytotoxic activities compared to those of 5-FU against HT-29 and SW620 cells of CRC via downregulating the antiapoptotic proteins, Bcl2 and BclxL.
Besides, analogs VIII and IX are described as broad-spectrum antitumor agents with more potent antiproliferative efficiency (approximately 1.5–3.0-fold) than 5-FU.31
On the basis of the aforementioned structural characteristics of the reported bioactive quinazolinone derivatives, some new compounds, which comprise quinazolin-4(3H)-one Schiff base conjugates, 2-((6-chloro-2-methyl-4-oxoquinazolin-3(4H)-yl) amino)thiazol-4(5H)-one, N-(6-chloro-2-methyl-4-oxoquinazolin-3(4H)-yl)-2-((3,4,5-trimethoxyphenyl)amino) acetamide, and N-(2-methyl-4-oxoquinazolin-3(4H)-yl)-2-((4-oxo-3-phenyl-3,4-dihydroquinazolin-2-yl)thio)acetamide, were designed, synthesized, characterized, and examined in vitro as inhibitors for cathepsin B, collagenase, thrombin, neutrophil elastase, trypsin, hsPLA2-G-IIA, hsPLA2-G-V, hsPLA2-G-X, XOR, α-amylase, and α-glucosidase. Their anti-CRC activities were assessed against LoVo and HCT-116 cell lines. Finally, the most active candidates were subjected to molecular docking analyses against their target enzymes. In addition, their pharmacokinetic and drug-likeness properties were predicted using SwissADME server and Lipinski rule of five filter, respectively.
Results and Discussion
Synthesis and Characterization of the Target Quinazolinone Derivatives
3-Aminoquinazolin-4-ones (1a–d)32 were used as synthetic synthons to prepare 10 new quinazolinone derivatives, namely, 3a–f, 7b, 8, 10, and 12, in addition to the previously reported 3g, 7a, and 5 as outlined in Schemes 1 and 2 and described in detail in the Experimental Section.
Scheme 1.

Reagents and conditions: (i) EtOH, AcOH—catalytic amount, reflux 10 h; (ii) DMF, K2CO3, reflux 5 h.
Scheme 2.

Reagents and conditions: (i) Dry CHCl3, 0 °C, Et3N, compound 6, r.t., 1 h, reflux 8 h; (ii) EtOH, reflux, 6 h.; (iii) K2CO3, dry acetone, reflux 8 h; (iv) EtOH, Et3N, reflux 12 h.
The structures of these compounds were established on the basis of their spectroscopic data. Thus, the IR spectrum of Schiff base 3b indicated the absence of the stretching absorption bands at νmax (KBr)/cm–1 3419 and 3314 attributed to NH2 group (of its precursor 1a) and the presence of the stretching absorption bands at νmax 3427 and 1691 cm–1 due to the carboxylic -OH and -C=O groups, respectively.
The 1H NMR (300 MHz; DMSO-d6) spectrum of this Schiff base lacked the two protons singlet signal at δH = 4.89 ppm attributed to the NH2 group of 1a and characterized by the emergence of a new one proton singlet signal at chemical shift value δH = 9.10 ppm attributable to azomethine group. In addition, two new signals, were detected at δH = 8.12 and 8.07, each of them integrating to two protons with coupling constant values J = 8.0 Hz, attributable to the four protons of the p-COOH-C6H4 moiety.
The 13C NMR (150 MHz; DMSO-d6) spectrum revealed four characteristic signals at δC (ppm) = 168.55 and 166.96 (2 × C=O), 156.63 (Cq=N), 154.44 (HC=N).
Last, the mass spectrum showed the molecular ion peaks [M+] at m/z (%) = 387.22 (81Br) and 385.02 (79Br), with relative intensities of almost 1:1 ratio (8.94 and 8.77%, respectively), which is characteristic for the spectra of bromine containing compounds and corresponding to the molecular formula of C17H12BrN3O3. The base peak was detected at m/z = 75.13.
Likewise, the IR and 1H NMR (500 MHz; CDCl3) spectra of Schiff base 3e confirmed the disappearance of the absorption bands (at νmax/cm–1 = 3284 and 3112) and the signal (at δH = 5.65 ppm) due to the amino group of its precursor 1b. Moreover, the 1H NMR spectrum exhibited three new characteristic singlet signals at δH = 8.76, 5.23, and 3.95 ppm attributable to azomethine proton (CH=N-), benzylic methylene protons (CH2), and methoxy protons, respectively. Besides, the five aromatic protons of the benzyloxy moiety were displayed as a two protons doublet (J = 7.5 Hz), a two protons triplet (J = 7.5 Hz), and a one proton triplet (J = 7.5 Hz) at δH = 7.44, 7.37, 7.31 ppm, respectively.
In addition, the 13C NMR spectrum (125 MHz; CDCl3) revealed the presence of five new characteristic signals at δC = 153.13, 152.20, 150.09, 70.92, and 56.15 ppm due to 2 × Cq-O, CH=N, CH2-Ph and OCH3, respectively.
Last, its mass spectrum (EI) showed the anticipated molecular ion peaks [M+ + 1] and [M+] at m/z (%) = 414.33 (1.58), 413.29 (4.57), respectively for C25H23N3O3. The base peak was observed at m/z = 91.14.
Regarding the stereochemical assignment of the geometry of the azomethine double bond as E-configuration in compounds 3a–g and 5, it was deduced on the basis of the NMR data, which indicated the formation of a single isomer in each case. In addition, the azomethine-proton resonated at chemical shift values δH = 9.05, 9.10, 9.09, 9.04, 8.76, 9.01, and 8.70 ppm for the Schiff bases 3a–g, respectively, and at δH = 8.77 ppm for derivative 5, whereby these chemical shift values are consistent with the previously reported data for E-isomers (δH ranging from 9.09 to 8.683 ppm).33,34 Moreover, minimizing the energies of both Z and E isomers and calculation of their total energies, as shown in Supporting Information Table S1, showed that E-isomers possessed the lowered energies and consequently they are the more stabilized and preferable products.35
With respect to chloroacetamides 7a and 7b, their IR spectra revealed the absence of the stretching absorption bands due to the NH2 groups of their starting materials, which were previously observed at νmax (cm–1) = 3450, 3308 (1c), and 3311, 3211 (1d) and the presence of stretching absorption bands at νmax (cm–1) = 3227 and 1733 (7a) and 3251 and 1718 (7b) attributable to the NH and C=O groups of the chloroacetamide moiety, respectively.
Furthermore, their 1H NMR spectra elicited the conversion of the primary amino groups of the precursors 1c and 1d to the secondary amino (-NH) groups as a result of the substitution of one of the NH2 protons by the chloroacetamide moiety (-CO–CH2–Cl). Thus, each of these spectra exhibited a one proton singlet signal due to NH at δH = 11.52 (7a) and 8.99 (7b) ppm, in addition to a two protons AB quartet signal at δH = 4.50 and 4.43 (J = 13.8 Hz for 7a) and 4.37 and 4.30 ppm (J = 15.3 Hz for 7b).
Moreover, their 13C NMR spectra indicated the presence of two new signals at δC = 165.95 and 40.73 (7a) and 166.44 and 41.13 ppm (7b) attributable to the carbonyl group and the methylene groups of chloroacetamide substituent, respectively.
With regard to derivative 8, its IR spectrum revealed a shift in the frequency of the stretching absorption band due to the C=O to a higher value as compared to its precursor 7b. Similarly, the 1H and 13C NMR spectra exhibited different chemical shift values due to the protons and the carbons, respectively, which confirmed the transformation to a new product via substitution of the chloride anion by thiocyanate anion and the subsequent intramolecular cyclization. Moreover, a new signal due to the S–C=N group emerged at δC = 153.90 ppm. The MS (DART-ToF) spectrum showed the molecular ion peak at m/z [M+ + 1] 309.020 corresponding to the molecular formula of C12H935ClN4O2S.
For compound 10, the IR and the 1H NMR spectra indicated the absence of the stretching absorption band due to the C=S group (at vmax = 1195 cm–1) and the signal of the thiol proton (at δH = 13.05 ppm) of its precursor 9. Furthermore, the 13C NMR spectrum was characterized by the absence of the signal at δC = 176.05 ppm (C=S of 9) and the presence of three carbonyl groups/and two C=N groups, which exhibited five signals at chemical shift values δC = 169.26, 161.26, 159.71, 158.08, and 155.11 ppm, respectively. The mass spectrum (DART-ToF) displayed the molecular ion peak [M+ + 1] at m/z = 470.12 for the molecular formula C25H19N5O3S.
Considering compound 12, the 3,4,5-trimethoxyphenylamine moiety displayed a stretching absorption band at vmax = 3379 cm–1 (for NH) in the IR spectrum and four characteristic signals in the 1H NMR spectrum (850 MHz; CDCl3) as follows: a two protons singlet, a six protons singlet, a three protons singlet, and a one proton singlet at δH = 6.03, 3.82, 3.73, and 3.48 ppm attributable to two aromatic methine, two methoxy, one methoxy, and NH groups, respectively. Further, this moiety displayed in the 13C NMR (213 MHz; CDCl3) spectrum three distinctive signals at chemical shift values δC = 91.43, 61.08, and 56.11 ppm, attributable to 2 × CH-Ph, 2 × OCH3, and the third OCH3, respectively. Last, the mass spectrum (DART-ToF) displayed the molecular ion peak [M+ – C9H11O3] at m/z 267.00 for C20H2137ClN4O5 (Scheme 3).
Scheme 3. Possible Fragmentation Pattern of Compound 12.

Enzymatic Inhibitory Activities
Antiproteases Activities
The results of antiproteases assays (Table S2) of the synthesized compounds disclosed that they exhibited higher IC50 (μg/mL) values ranging from 9.00 ± 1.41 (compound 7b) to 55.77 ± 1.85 (compound 3f) against cathepsin-B and from 2.50 ± 0.71 (compound 1c) to 57.83 ± 2.69 (compound 3d) against collagenase in comparison to 0.175 ± 0.04 and 0.15 ± 0.07, which were exerted by cocktail, the reference proteases’ inhibitor. Moreover, these derivatives exerted higher IC50 (μg/mL) values spanning from 100.36 ± 5.51 (3c) to 0.20 ± 0.00 (8) against elastase and ranging from 109.85 ± 1.77 (3c) to 0.55 ± 0.07 (8) against trypsin in comparison to 0.25 ± 0.07 and 0.125 ± 0.04, respectively, which were displayed by cocktail.
With regard to thrombin, the highest recorded IC50 value of 83.65 ± 4.78 μg/mL was displayed by derivative 3d. However, the lowest IC50 value of 0.225 ± 0.04 μg/mL was exerted by compound 8, which was also lowered than that of cocktail (0.25 ± 0.07). Therefore, compound 8 could be considered as a potent antithrombin agent. This finding is of great importance due to cancer-associated thrombosis is a common sign of malignancy, and it is currently the second leading cause of mortality in cancer patients.36 In addition, thrombin supports early events linked to inflammation-driven tumorigenesis in colitis-associated colon cancer37 as well as it plays crucial roles in tumor proliferation, stroma formation, angiogenesis, and metastasis.38
Anti-phospholipases Activities
Likewise, the results of sPLA2 assays (Table S3) revealed that hsPLA2-G-IIA, was the least sensible isoform as all of the studied compounds expressed higher IC50 values ranging from 13.84 ± 2.12 (compound 10) to 127.42 ± 2.12 μM (compound 7b) as compared to 11.50 μM, which was exerted by oleanolic acid, the used reference phospholipases inhibitor.
Contrarily, hsPLA2-G-V was the most sensible isoenzyme, whereby it was inhibited efficiently with lowered IC50 (μM) of 16.14 ± 0.85 (1a) and 14.70 ± 0.42 (10), in comparison to 16.42 ± 0.71 μM produced by oleanolic acid. The remaining compounds exhibited IC50 values ranging from 22.28 ± 0.92 (compound 3g) to 151.39 ± 2.12 μM (compound 9).
With regard to the hsPLA2-G-X isoform, it was potently inhibited by derivative 3g, which exhibited a lowered IC50 value of 14.55 ± 0.92 μM compared to that of oleanolic acid (16.53 ± 0.64 μM). The rest of the compounds demonstrated IC50 values spanning from 21.45 ± 2.31 (compound 3d) to 109.5 ± 5.01 μM (compound 3a).
On the basis of these results, the new quinazolinone derivatives 3g and 10 might be utilized to develop new anti-colorectal agents whose mode of actions depend upon inhibiting the hsPLA2-G-V and hsPLA2-G-X. It is noteworthy, to indicate that previous reports elicited the roles of these isoforms in generation of arachidonic acid (AA), which serves as a substrate for intracellular biochemical pathways generating the bioactive eicosanoids including prostaglandins, thromboxanes, leukotrienes, and lipoxins.39 Eicosanoids act through numerous signaling pathways to modulate tumor occurrence, angiogenesis, invasion, metastasis, immunity, and cell apoptosis processes.40 Thus, inhibiting this cascade of biochemical and signaling pathways would provide antiproliferative effects.
Antiglycemic and Anti-free-radical-generating Activities
Finally, the results of in vitro assessment of the studied compounds, against α-amylase, α-glucosidase, and XOR, are summarized in Table S4. Compound 3f displayed potent α-glucosidase inhibitory efficiency with lowered IC50 value (μM) of 12.43 ± 0.83 in comparison to 12.57 ± 0.28 by the reference inhibitor, quercetin. The remaining compounds demonstrated values ranging from 13.65 ± 1.41 (compound 5) to 93.36 ± 4.39 μM (compound 3c).
None of the studied compounds was capable of inhibiting XOR by a lowered IC50 (μM) than the value of 4.78 ± 0.07 μM, which was displayed by allopurinol, whereas they displayed values ranging from 7.34 ± 0.33 (compound 3f) to 339.63 ± 4.95 (compound 1c).
Contrarily, improved inhibitory efficiencies against α-amylase with lowered IC50 (μM) values of 264.11 ± 10.98, 310.22 ± 10.04, 357.85 ± 4.95, 374.64 ± 11.51, 399.48 ± 4.95, and 400.82 ± 6.36 were exhibited by compounds 3b, 3f, 5, 3d, 1a, and 12, respectively, relative to 406.97 ± 2.83 by quercetin.
Overall, compounds 3b, 3d, 3f, 5, 1a, and 12 would be beneficial for developing anti-hyperglycemic agents, which could also contribute to modulation of CRC due to the role of hyperglycemia in chemoresistance as well as in induction of VEGF gene transcription, leading to angiogenesis and tumor invasion.41
Cytotoxicity Assays on HCT-116 and LoVo Cells of CRC
Moreover, the studied compounds were further assessed in vitro for their antiproliferative activities against LoVo and HCT-116 cell lines of CRC by determination of the percentage of residual viable cells after being treated with each compound at six concentrations (25, 50, 75, 100, 200, and 400 μg/mL); then, the half-maximal inhibitory concentrations (μM) were deduced from the standard curves (Table S5). The obtained results are compared to the percentage of viable untreated cells (negative control) and cells treated by 0.1% Triton X-100 in the assay medium (positive control).
On the basis of our previous work, which was carried out on similar compounds, the viability assays were performed at a concentration of 200 μg/mL of each studied compound. The data indicated that compounds 1a, 3a, 3b, 3d, 3e, 3f, 5, 7b, 9, and 12 were incapable of suppressing the growth of cancerous cells effectively as they exhibited percentages of viable cells ranging from 90.5 ± 3.54 (3d) to 58.0 ± 4.24% (3f) against LoVo type and spanning from 93.50 ± 2.12 (5) to 48.25 ± 1.06% (1a) against HCT-116 cells.
Contrarily, the viabilities of LoVo cells were suppressed effectively to 13.50 ± 0.71 (8), 16.75 ± 0.35 (3g), 22.00 ± 1.41 (10), and 28.0 ± 4.24% (1c). Considering HCT-116 cells, their viabilities were greatly reduced to 4.75 ± 0.35 (10), 12.75 ± 1.06 (3g), 28.00 ± 1.41 (8), 36.0 ± 4.24% (3c), and to 46.50 ± 2.12 (1c).
The calculated IC50 (μM) values against LoVo cells were ranging from 206.07 ± 7.28 to 1052.80 ± 64.01 μM, with compounds 3g, 10, 8, 3c, and 1c, displaying the lowest half-maximal inhibitory concentrations of 206.07 ± 7.29, 272.62 ± 9.04, 319.04 ± 11.45, 320.53 ± 13.20, and 339.63 ± 12.10 μM, respectively, whereas the half-maximal concentrations against HCT-116 cells were spanning from 1450.99 ± 22.24 to 230.02 ± 9.04, with compounds 10, 3g, 8, 3c, and 1c being the most active with 230.02 ± 9.04, 284.63 ± 9.11, 448.60 ± 11.46, 459.79 ± 12.11, and 530.85 ± 16.15 μM, respectively.
Collectively, the obtained results revealed that compounds 3g (anti-hsPLA2-G-X), 8 (antithrombin), and 10 (anti-hsPLA2-G-V) also possessed cytotoxic potential; therefore, they would be considered as lead compounds with multifunctional profiles against CRC.
Molecular Docking Studies
Molecular docking is a modern approach to recognizing structural features, which decide the biological profile of a molecule, especially when the 3D structure of a target enzyme is known. In the present work, the results of the enzymatic assays were further cross-investigated through molecular docking analysis of the most active compounds against the respective target enzymes. In addition, their binding energies and molecular interactions were studied in comparison to their cocrystallized ligands and reference inhibitors (Supporting Information, Table S6).
Initially, self-redocking of the cocrystallized ligands in the vicinity of the binding site of each target enzyme was performed (Figure 2) and the root-mean-square deviation (RMSD) values (Å) between the original crystal ligand and the conformation of the redocked ligand were calculated, which were found to be less than the permissible cutoff value (2 Å) suggesting the accuracy and reliability of the performed procedures.42 Moreover, the docking scores (S) for the interactions were predicted as shown in Table S7. The more negative score indicates the better affinity of the ligand to the specified molecular target and its tendency to form stronger interactions with the amino acid residues in the active pocket.
Figure 2.

3D representations of the superimposition of the cocrystallized ligands (purple) and the re-docked poses (gray) of (a) thrombin, (b) elastase, (c) trypsin, (d) sPLA2-hG-IIA, (e) sPLA2-hG-X, (f) α-amylase, (g) α-glucosidase, and (h) xanthine oxidoreductase.
Docking against Thrombin
The cocrystal ligand amino{[(4S)-5-[(2R,4R)-2-carboxy-4-methylpiperidin-1-yl]-4-({[(3R)-3-methyl-1,2,3,4-tetrahydroquinolin-8-yl]sulfonyl}amino)-5-oxopentyl]amino}methaniminium (MIT) displayed binding energy of −15.0807 kcal/mol in the active site of thrombin.43 It formed strong hydrogen bond interactions with Ser195, Gly216, and Gly219 residues in addition to ionic interactions with Glu192 and Asp189 (Figure S1).
Studying the most favorable conformation of compound 8 with thrombin (binding score = −10.4683 kcal/mol) indicated that it fitted well inside the active pocket through formation of a conventional hydrogen bond with a distance of 3.05 Å between its carbonyl oxygen and Ser-H195, which is supported by other amide−π stacked and π–alkyl interactions. Thus, the docking results (Table S8 and Figure S2) are consistent with the experimental inhibitory assay.
Docking against Elastase
The cocrystal elastase inhibitor, 2-[5-methanesulfonylamino-2-(4-aminophenyl)-6-oxo-1,6-dihydro-1-pyrimidinyl]-n-(3,3,3-trifluoro-1-isopropyl-2-oxopropyl)acetamide (TFI)44,45 was better fitted within the active pocket with binding energy of −11.4465 kcal/mol as compared to compound 8, which is predicted to form a complex with the elastase with binding score of −9.8673 kcal/mol. The most prominent interactions of the cocrystallized ligand were in the form of hydrogen bonds with His57, Asp194, Ser214, Val216, and Arg217 amino acids (Figure S3).
With regard to compound 8, it formed five strong hydrogen bonding interactions (Figure S4) using its two carbonyl groups and sulfur atom as H-bond acceptors with Gln192 (2.63 Å), Gly193 (2.77 Å), Asp194 (2.92 Å), Ser195 (2.15 Å), and Val216 (3.01 Å). The results summarized in Table S8 indicated that compound 8 formed only two conventional hydrogen bonds similar to those of the cocrystallized ligand, which may account for its lowered binding affinity.
Docking against Trypsin
The cocrystallized ligand 3,3′-[ethane-1,2-diylbis(nitrilomethylylidene)]bis(4-hydroxybenzenecarboximidamide) (A2C) bound to trypsin enzyme with binding energy of −14.3302 kcal/mol. It showed hydrogen bonding interactions with Phe24, His40, Ser172, Ser195, and Gly196 amino acids besides ionic interaction46 with Asp171 as shown in Figure S5.
Docking results of compound 8 indicated its high affinity to trypsin (the binding score −10.1979 kcal/mol) as well as the importance of the chlorine atom, which was involved in the formation of two hydrogen bonds with Ser195 (2.89 Å) and Lys202 (3.25 Å) residues. Moreover, 8 formed an amide-π stacked interaction with Trp193 amino acid. The results are depicted in Table S8 and Figure S6.
Docking against Secretory Phospholipases
Searching in PDB indicated that the crystal structure coordinates are available only for hsPLA2-G-IIA and hsPLA2-G-X isozymes, while the representation for hsPLA2-G-V is missed.47
Docking against hsPLA2-G-IIA
The cocrystallized inhibitor 6-phenyl-4(R)-(7-phenyl-heptanoylamino)hexanoic acid (BR4)48 displayed binding energy of −13.9194 kcal/mol in the active pocket of hsPLA2-G-IIA. It interacted through three hydrogen bonds with Gly29, Gly31, and Asp48 residues. Also, it formed ionic interaction with Lys62 amino acid (Figure S7).
With regard to oleanolic acid, it showed slightly lowered binding affinity (with higher binding energy value of −12.4222 kcal/mol) than that of the cocrystallized ligand toward the protein and it exhibited only two hydrogen bonding interactions with Lys62 and Thr61 (Figure S8). However, the bisquinazolinone derivative 10 fitted within the enzyme active pocket with a binding affinity of −12.3560 kcal/mol and it was stabilized through three conventional hydrogen bonds with Gly29 (2.68 Å), Gly31 (2.14 Å), and Lys62 (2.88 Å) amino acids (Figure S9). These interactions were supported by several π and alkyl interactions (Table S8).
Docking against hsPLA2-G-X
Investigation of the binding modes of the complex 5G3M,49 which was formed as a result of fitting of the cocrystallized ligand 4-benzylbenzamide (9JH) in the active pocket of hsPLA2-G-X with binding energy of −13.9194 kcal/mol indicated that the amide group of the ligand coordinated the sPLA2-hG-X’s Gly28, His46, and Asp47 residues through conventional hydrogen bonds. π-Sulfur interaction was formed with Met21. In addition, carbon–hydrogen interaction with Cys27 and several hydrophobic forces were observed (Figure S10).
Although 3g exhibited improved inhibitory efficiency (lowered IC50 value) as compared to oleanolic acid; the latter displayed better affinity with lowered binding energy −13.188 kcal/mol as compared to compound 3g (−11.7267 kcal/mol). This could be attributed to the capability of oleanolic acid to establish two conventional hydrogen bonds with the same amino acids (Gly28 and His46) as the cocrystal ligand but with a smaller number of electrostatic interactions. However, compound 3g bound to sPLA2-hG-X through nonclassical carbon hydrogen bond using its nitrogen and oxygen atoms with Leu29 (2.23 Å) and His46 (2.79 Å) residues. Moreover, 3g formed other interactions with Lys61 through π-cation force (2.91 Å to phenyl and 2.17 Å to pyrimidine) and with Asp47 through π-anion force (3.79 Å to phenyl and 3.54 Å to pyrimidine). These results are summarized in Table S8 and Figures S11 and S12.
Docking against α-Glucosidase
Castanospermine (CTS), the potent α-glucosidase cocrystallized inhibitor and the surrounding active site residues superposed perfectly (binding score = −15.3919 kcal/mol). It formed conventional hydrogen bonding interactions using its four hydroxyl groups as H-bond donors with three glutamate residues; Glu439, Glu391, and Glu532. In addition, these hydroxyl groups interacted as H-bond acceptors with Trp331, His437, Lys467, and His507 as reported previously.50 Furthermore, the ring nitrogen atom of CTS was involved in ionic interactions with the Glu391, Glu439, Glu508, and Glu532 amino acids (Figure S13).
Quercetin, the used polyhydroxy flavonoid α-glucosidase inhibitor, showed comparable binding affinity to the cocrystallized ligand (binding score = −16.1296 kcal/mol). It formed four hydrogen bonds, two as an acceptor with Lys467 (using its C=O) and His507 (suing one of its OH groups) and two as donor with Glu194 and Glu532 (Figure S14). In addition, it showed π–anion interactions with glutamate residues Glu439 and Glu532 (2.20 Å).
Although, compound 3f fitted well within the active pocket, it exhibited lowered binding affinity than quercetin (−14.5846 kcal/mol), which can be attributed to the absence of hydrogen bonding interactions with the amino acids. Instead, this ligand showed π–anion interactions with glutamate residue Glu439 (3.49 and 3.51 Å). These interactions were supported by other several π–π stacked and π–alkyl interactions. The results are summarized in Table S8 and Figure S15.
Docking against Xanthine Oxidoreductase
The cocrystallized ligand lumazine (LUZ)51 was optimized within the active pocket of xanthine oxidoreductase with binding energy of −15.3919 kcal/mol. LUZ demonstrated hydrogen bonding interactions as H-bond donor through its NH group (at position 3) with Thr1010 and ionic interactions with Arg880 (Figure S16).
Compound 3f laid deep in the active pocket, and it exhibited better binding affinity to the protein as indicated by its more negative binding score (−14.9338 kcal/mol) in comparison to that of allopurinol, the reference XO inhibitor (binding score of −9.1790 kcal/mol). Indeed, 3f formed two conventional hydrogen bonding interactions (Figure S18) through its carbonyl group and nitrogen atom with Ser1080 (1.77 Å) and Arg912 (2.87 Å), respectively. In addition, π–π stacking and π–alkyl interactions were monitored, whereas allopurinol demonstrated three hydrogen bonding interactions, two as a H-bond acceptor through its carbonyl with Thr1010 and Arg880 and one as a H-bond donor through its NH group with Glu802. Also, it formed a π–cation interaction with Met1038 and other π–alkyl and van der Waal’s interactions (Figure S17). All results are summarized in Table S8.
It is noteworthy to indicate that allopurinol is oxidized to oxypurinol by xanthine oxidoreductase enzyme at the molybdenum cofactor center, where oxypurinol covalently inhibits the enzyme activity,52,53 which may account for the improved experimental activity of allopurinol as compared to 3f.
Docking against α-Amylase
The redocked cocrystallized acarbose (AC1) ligand was involved in salt bridge interactions with Asp197, Glu233, and Asp300, in addition to extensive hydrogen bonding interactions with Trp59, Gln63, Thr163, Arg195, Lys200, His201, Glu240, His299, and His305 residues (Figure S19) as described previously (PDB code: 1B2Y).54
Compound 3b showed a better binding score (−11.4264 kcal/mol) than quercetin (−10.2531 kcal/mol) due to it formed two extra stronger conventional hydrogen bonding interactions with Arg195 (3.29 and 3.32 Å) through the two oxygen atoms of the carboxylic acid substituent, which may account for the improved experimental inhibitory efficiency of 3b as compared to quercetin (Figures S20 and S21). Other interactions of compound 3b and quercetin are summarized in Table S8.
Overall, these docking simulations provide some information about the expected binding modes of the tested compounds with the respective target enzymes, which need to be investigated at the cellular level.
ADME Profiling
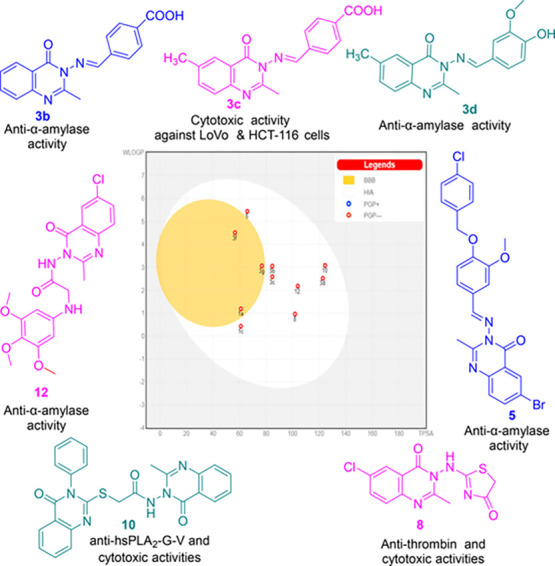
The physicochemical properties and the pharmacokinetic (absorption, distribution, metabolism, and excretion, ADME) parameters of the newly synthesized bioactive compounds 1a, 1c, 3b, 3c, 3d, 3f, 3g, 5, 8, 10, and 12 were studied using the freely accessible in silico SwissADME web tool (http://www.swissadme.ch), which provides a number of proficient methods such as the bioavailability radar and boiled-egg charts.
The bioavailability radar chart is used to investigate the physicochemical and drug-likeness properties of a molecule through prediction of six physicochemical descriptors: lipophilicity (LIPO, the partition coefficient between n-octanol and water log Po/w value should be between −0.7 and +5.0), size (the acceptable molecular weight lies between 150 and 500 Da), polarity (POLAR), topological polar surface area (TPSA between 20 and 130 Å2), insolubility (INSOL, the decimal logarithm of the molar solubility in water log S should not exceed 6), unsaturation (UNSAT, fraction of carbons in the sp3 hybridization ≥0.25), and flexibility (FLEX, the number of rotatable bonds should not be greater than 9). Ideally, the optimal physicochemical range on each axis is depicted as a pink area in which the radar plot of the molecule has to fall entirely to be considered drug-like.55
As shown in the bioavailability radar chart (Figure 3), compound 12 did not violate any of the studied parameters, whereas compounds 1a, 1c, 3b, 3c, 3d, 3f, 3g, 8, and 10 violated the unsaturation parameter as they have a low percentage of sp3 carbons (0.11 for 1a, 1c, and 3c; 0.06% for 3b; 0.17 for 3d; 0.10 for 3f; 0.12% for 3g; 0.17 for 8; and 0.08% for 10). Considering compound 5, it violated the unsaturation (0.12% of sp3 carbons) and the insolubility (it is poorly soluble in water with log S of −6.45) parameters. Concerning the predicted logP values, the studied compounds showed intermediate values of 1.58 (1a), 0.92 (1c), 3.05 (3b), 2.83 (3c), 2.11 (3d), 4.32 (3f), 2.94 (3g), 1.92 (8), 3.39 (10), and 2.35 (12), except for compound 5, which showed a slightly higher value of 5.09.
Figure 3.

Bioavailability radar chart for the bioactive compounds (the colored zone is the suitable physicochemical space for oral bioavailability).
Moreover, the SwissADME server provides a boiled-egg chart, which is used to evaluate the ADME behavior of each molecule under investigation individually. This model provides information about the passive human gastrointestinal absorption (HIA) and blood–brain barrier (BBB) permeation, in addition to it allowing the prediction for the probability of the studied molecule to be a substrate or nonsubstrate of the permeability glycoprotein (P-gp) using the most important members among ATP-binding cassette transporters (ABC-transporters). The latter parameter is essential to predict whether or not the drug will suffer from drug resistance (DR) due to restricting the entry of the drug to the target cells through efflux pump.56 Thus, being a P-gp inhibitor is a beneficial property for the anticancer candidate to maintain its minimum therapeutic concentration at the target site for better therapeutic efficacy.
In the boiled-egg model, the yolk (i.e., the physicochemical space for highly probable passive BBB permeation) and the inside of the white (i.e., the physicochemical space for highly probable HIA absorption), besides the outside gray compartment (outside the egg) indicates that the tested molecule is not passively absorbed through gastrointestinal wall and BBB. In addition, the active efflux from the CNS or to the gastrointestinal tract is indicated by color-coding: blue dots for P-gp substrates (PGP+) and red dots for P-gp nonsubstrate (PGP−),55 respectively.
Thus, as shown in the graphical output of the boiled-egg chart for this set of the compounds (Figure 4), it can be concluded that all of them would have good intestinal absorption. While only compounds 1a, 3f, and 3g are predicted to be BBB penetrant (inside the yolk), implying that they would cause serious side effects to CNS. On the other hand, derivatives 1c, 3b, 3c, 3d, 5, 8, 10, and 12 are not predicted to penetrate the BBB.
Figure 4.

Boiled-egg chart for the studied bioactive compounds
Considering the permeability glycoprotein (P-gp) property, all of the studied derivatives are not predicted to be subjected to the active efflux P-gP mechanism (as indicated by their red colors). This prediction would highlight the importance of the cytotoxic candidates, 10, 8, 3c and 1c as they are not expected to suffer from DR
Finally, computation of the parameters of the Lipinski rule of five57,58 was used to predict the expected drug-likeness characteristics of the biologically active candidates. The results of this filter presented in Table S9 revealed that none of the investigated compounds violated the rule except for compound 5, which violated the solubility (logP value = 5.09) and the molecular weight (512.78 g/mol) parameters.
Together, it can be suggested that compounds 1c, 3b, 3c, 3d, 8, 10, and 12 are expected to possess molecular features that are compatible with acceptable pharmacokinetics properties, which make them interesting candidates for further optimization to develop anti-colorectal drugs.
Conclusions
Ten new quinazolinone derivatives have been designed, synthesized, characterized, and evaluated in vitro as inhibitors for selected proteases, phospholipases, and glycolytic enzymes as well as antiproliferative agents against LoVo and HCT-116 cell lines of CRC. Some compounds showed promising and more potent enzymatic inhibitory efficiency as compared to the respective reference inhibitors: 8 (anti-thrombin); 10 (anti-hsPLA2-G-V); 3g (anti-hsPLA2-G-X); 1a, 3b, 3d, 3f, 5, and 12 (anti-α-amylase); and 3f (anti-α-glucosidase). Contrarily, none of the studied compounds demonstrated improved efficiencies as compared to cocktail against cathepsin-B, collagenase, elastase and trypsin, or oleanolic acid against hsPLA2-G-IIA or allopurinol against XO with cathepsin-B being the least sensible enzyme. In spite of this, compounds 1c/8/10 and 3f exhibited the highest inhibitory efficiency among the studied compounds against collagenase/(elastase and trypsin)/sPLA2-hG-IIA and XO, respectively. In view of these results, the in silico molecular docking simulations were performed to identify the important interactions between the cocrystallized ligands, the used reference inhibitors, and the active compounds with their target enzymes. Moreover, the antiproliferative assays showed that quinazolinones, 3g, 8, and 10, reduced the viability of the cancerous cells in the micromolar range with IC50 values of 206.07 ± 2.83, 319.04 ± 3.54, and 272.62 ± 4.24 against LoVo cells and 284.63 ± 3.54, 448.60 ± 3.54, and 230.02 ± 4.24 against HCT-116 cells, respectively. The compliance with the Lipinski rule of five, in addition to the expected pharmacokinetic profiles of the active candidates, were predicted using the bioavailability radar charts and boiled-egg model. Collectively, these predictions revealed that the anti-hyperglycemic (3b, 3d, and 12) and the cytotoxic (1c, 3c, 8, and 10) candidates are expected to possess acceptable molecular, drug-likeness, and pharmacokinetic characteristics, particularly they are not expected to cause CNS toxicity nor drug resistance, which suggest the suitability of these derivatives to serve as potential leads for optimization as anti-hyperglycemic and anti-colorectal agents.
Experimental Section
Chemistry
General Information
All of the melting points were determined with a Gallenkamp melting point apparatus (°C) and uncorrected. The IR spectra were recorded on a PerkinElmer FTIR spectrophotometer, Spectrum BX 1000 in wavenumber (cm–1) with potassium bromide (KBr) discs. Nuclear magnetic resonance spectra were recorded using Bruker NMR spectrometer: on (1) Ascend 850 MHz for 1H and 213 MHZ for 13C or on (2) Avance 600 MHz for 1H and 150 MHZ for 13C (Nuclear Magnetic Resonance Center, KAU, Jeddah, KSA) or on (3) a Bruker Avance 500 spectrometer operating at 500 MHZ for 1H and 125 MHZ for 13C at 25 °C (Research Unit, College of Pharmacy, Prince Sattam Bin Abdulaziz University, AlKharj, KSA) or on (4) an Eclipse 300 FT NMR Spectrometer operating at 300 MHz for 1H and 75 MHZ for 13C (at KSU, Riyadh, KSA). The chemical shifts are expressed in ppm downfield from tetramethylsilane (TMS) as internal standard; coupling constants (J) are expressed in Hz. Deuterated chloroform (CDCl3) and deuterated dimethyl sulfoxide (DMSO-d6) were used as solvents; the splitting patterns (multiplicities) in 1H NMR were designated as s (singlet), br s (broad singlet), app s (apparent singlet), d (doublet), dd (doublet of doublet), t (triplet), q (quartet), app dd (apparent doublet of doublet), and m (multiplet). The mass spectra were obtained on a Shimadzu Qp-2010 Plus mass spectrometer that works by using ionization mode (EI; Micro Analytical Center, Cairo University, Egypt) or on a AccuToF LC-plus JMS-T100LP (Joel). Time-of-flight mass spectrometer (DART-ToFMS) works by accelerating an ionized sample and calculating mass per charge on the basis of how long each “object” is in flight for (Acc-TOF LC-Plus; KSU, Riyadh, KSA).
General Procedures for the Syntheses of Schiff Bases 3a–g
An equimolar mixture (0.002 mol) of 3-amino-6-substituted-2-methyl-3H-quinazolin-4-one derivative (1a,b), and the appropriate aromatic aldehyde (2a–f), namely, 5-ethylthiophene-2-carbaldehyde, 4-formylbenzoic acid, 5-nitrovanillin, 3-(benzyloxy)-3-methoxybenzaldehyde, 6-methoxy-2-naphthaldehyde, or vanillin in absolute ethanol (20 mL) containing a catalytic amount of glacial acetic acid (3 mL), was refluxed for 10 h. The separated solid was collected by filtration, washed with water, air-dried, and recrystallized from the appropriate solvent to give the corresponding Schiff base derivative (3a–g).
(E)-3-[((5-Ethylthiophen-2-yl)methylene)amino]-2,6-dimethylquinazolin-4(3H)-one (3a)
Yellow crystals (hexane); yield, 56%; mp 129–130 °C. νmax (KBr)/cm–1: 3050 (CH-aromatic), 2924 and 2967 (CH-aliphatic), 1681 (C=O), 1603 (C=N), 1473 and 1427 (C=C), 1370, 1332, 1287, 1239, 1205, 1120, 1060, 1025, 941, 809, 701, 648, 597, 540, 478, 443. 1H NMR (500 MHZ; CDCl3): δH 9.05 (1H, s, CH=N), 8.04 (1H, s, CH5-quinazolin-4(3H)-one), 7.53 (2H, d, J = 8.5 Hz, CH7, CH8-quinazolin-4(3H)-one), 7.34 (1H, d, J = 3.5 Hz, CH-thiophene), 6.84 (1H, app d, J = 3.0 Hz, CH-thiophene), 2.90 (2H, q, J = 7.5 Hz, CH2-CH3), 2.61 (3H, s, CH3), 2.47 (3H, s, CH3), 1.35 (3H, t, J = 7.5 Hz, CH2–CH3). 13C NMR (150 MHZ; CDCl3): δC 159.76 (C=O), 158.84 (Cq=N), 154.96 (HC=N), 153.23, 144.38, 136.33, 135.63, 134.83, 134.77, 126.62, 126.46, 124.64, 121.13 (5 × CH-aromatic and 5 × Cq-aromatic), 24.02 (CH2–CH3), 22.70 (CH3), 21.26 (CH3), 15.57 (CH2–CH3). MS (EI; m/z; %): [M+ + 2] 313.13 (12.82); [M+ + 1] 312.29 (41.47); [M+] 311.28 (100.00) for C17H17N3OS, 310.36 (11.78), 281.55 (3.30), 174.37 (7.19), 173.86 (17.47), 157.30 (2.65), 132.17 (6.81), 124.12 (5.75), 91.20 (15.57), 90.16 (19.99), 89.14 (48.96), 78.15 (22.91), 77.17 (59.61), 76.14 (22.12), 65.10 (59.49), 64.17 (50.13), 63.15 (83.42), 53.16 (12.82), 51.17 (15.51), 45.05 (11.42), 41.13 (8.49).
(E)-4-[(6-Bromo-2-methyl-4-oxo-4H-quinazolin-3-ylimino)methyl]benzoic acid (3b)
Beige powder (MeOH/CHCl3); yield, 66%; mp 298–299 °C. νmax (KBr)/cm–1: 3427 (OH), 3073 and 3000 (CH-aromatic), 2935 (CH-aliphatic), 1691 (2 × C=O), 1599 (C=N), 1512, 1469, and 1424 (C=C) 1375, 1318, 1292, 1235, 1157, 1123, 1037, 1014, 954,880, 832, 799, 768, 742, 696, 674, 547, 506, 465. 1H NMR (300 MHZ; DMSO-d6): δH 9.10 (1H, s, CH=N), 8.20 (1H, d, J = 2.1 Hz, CH5-quinazolin-4(3H)-one), 8.12 (2H, d, J = 8.0 Hz, 2 × CH-Ar), 8.07 (2H, d, J = 8.0 Hz, 2 × CH-Ar), 7.96 (1H, dd, J = 8.7, 2.1 Hz, CH7-quinazolin-4(3H)-one), 7.60 (1H, d, J = 8.7 Hz, CH8-quinazolin-4(3H)-one), 2.52 (3H, s, CH3). 13C NMR (150 MHZ; DMSO-d6): δC 168.55 (C=O), 166.96 (C=O), 156.63 (Cq=N), 154.44 (HC=N), 145.50, 137.58, 136.28, 134.49, 130.21, 129.52, 129.17, 128.91, 122.92, 119.07 (7 × CH-aromatic and 5 × Cq-aromatic), 22.59 (CH3). MS (EI; m/z; %): [M+, 81Br] 387.22 (8.94), [M+, 79Br] 385.02 (8.77) for C17H12BrN3O3, 343.23 (16.81), 342.20 (14.13), 326.00 (12.06), 324.81 (9.35), 312.65 (5.22), 284.93 (5.58), 265.80 (5.64), 252.47 (5.17), 237.96 (94.16), 208.41 (3.98), 196.54 (19.27), 162.45 (10.75), 155.88 (5.64), 153.38 (4.93), 128.44 (10.30), 119.41 (3.40), 101.16 (26.26), 89.26 (56.51), 75.13 (100.00), 63.28 (25.55), 50.19 (35.31), 42.19 (12.46).
(E)-4-[((2,6-Dimethyl-4-oxoquiazolin-3(4H)-yl)imino)methyl]benzoic acid (3c)
Beige powder (MeOH/CHCl3); yield, 81%; mp 275–276 °C. νmax (KBr)/cm–1: 3430 (OH), 3055 and 3013 (CH-aromatic), 2971 and 2927 (CH-aliphatic), 1690 (C=O), 1616 (C=O), 1588 (C=N), 1490 and 1424 (C=C), 1370, 1309, 1288, 1219, 1170, 1118, 1013, 950, 855, 827, 772, 696, 657, 596, 557, 596, 545, 450. 1H NMR (300 MHZ; DMSO-d6): δH 9.09 (1H, s, CH=N), 8.11 (2H, d, J = 8.1 Hz, 2 × CH-Ar), 8.04 (2H, d, J = 8.1 Hz, 2 × CH-Ar), 7.92 (1H, s, CH5-quinazolin-4(3H)-one), 7.64 (1H, d, J = 8.4 Hz, CH7-quinazolin-4(3H)-one), 7.54 (1H, d, J = 8.1 Hz, CH8-quinazolin-4(3H)-one), 2.51 (3H, s, CH3), 2.44 (3H, s, CH3). 13C NMR (75 MHZ; DMSO-d6): δC 167.51 (C=O), 166.68 (C=O), 157.35 (Cq=N), 152.53 (HC=N), 144.24, 136.22, 136.10, 135.79, 134.044, 129.88, 128,76, 126.65, 125.95, 120.72 (7 × CH-aromatic and 5 × Cq-aromatic), 22.14 (CH3), 20.78 (CH3). MS (EI; m/z; %): [M+ + 2] 322.18 (47.24), [M+ + 1] 322.18 (47.24), [M+] 321.20 (100.00), and [M+ – 1] 320.38 (25.80) for C18H15N3O3, 174.12 (31.27), 145.25 (4.25), 131.13 (20.42), 116.09 (3.87), 105.10 (9.25), 89.21 (50.82), 77.17 (48.86), 63.22 (49.33), 51.10 (9.36), 42.23 (2.68).
(E)-3-[(4-Hydroxy-3-methoxy-5-nitrobenzylidene)amino]-2,6-dimethylquinazolin-4(3H)-one (3d)
Yellow crystals (MeOH/CHCl3); yield, 76%; mp 219–220 °C. νmax (KBr)/cm–1: 3277 (OH), 3091 and 3013 (CH-aromatic) 2971 and 2927 (CH-aliphatic), 1673 (C=O), 1604 (C=N), 1544 and 1488 (C=C), 1421, 1373, 1336, 1271, 1236, 1189, 1105, 1054, 987, 960, 915, 875, 831, 780, 706, 675, 637, 608, 536, 462. 1H NMR (500 MHZ; CDCl3): δH 11.12 (1H, s, OH), 9.04 (1H, s, CH=N), 8.10–8.02 (2H, m, CH7 and CH8-quinazolin-4(3H)-one), 7.78 (1H, d, J = 1.5 Hz, CH5-quinazolin-4(3H)-one), 7.56 (2H, s, 2 × CH-Ar), 4.03 (3H, s, OCH3), 2.64 (3H, s, CH3), 2.48 (3H, s, CH3). 13C NMR (150 MHZ; CDCl3): δC 163.15 (C=O), 158.77 (Cq=N), 152.82 (HC=N), 150.72 (Cq-O), 149.55 (Cq—O), 144.32, 136.79, 136.03, 133.65, 126.76, 126.64, 124.44, 121.07, 118.87, 114.00 (5 × CH-aromatic and 5 × Cq-aromatic), 56.88 (OCH3), 22.82 (CH3), 21.30 (CH3). MS (EI; m/z; %): [M+ + 1] 369.65 (42.76) and [M+] 368.04 (66.09) for C18H16N4O5, 363.96 (71.43), 358.61 (27.38), 339.34 (21.81), 322.55 (48.49), 311.11 (30.89), 307.36 (30.63), 286.34 (19.75), 277.99 (20.32), 264.81 (26.35), 246.17 (57.44), 245.62 (37.55), 229.27 (17.30), 214.58 (48.39), 201.83 (49.93), 200.16 (38.48), 197.90 (60.82), 180.76 (44.15), 177.82 (57.18), 154.75 (33.05), 139.95 (26.02), 122.36 (38.75), 108.82 (100.00), 91.43 (39.31), 90.15 (51.08), 80.03 (33.44), 66.23 (33.15), 58.82 (26.18), 45.74 (32.91).
(E)-3-[(4-(Benzyloxy)-3-methoxybenzylidene)amino]-2,6-dimethylquinazolin-4(3H)-one (3e)
Beige powder (EtOH/CHCl3); yield, 78%; mp 198–199 °C. νmax (KBr)/cm–1: 3033 and 3012 (CH-aromatic), 2961; 2929 and 2864 (CH-aliphatic), 1675 (C=O), 1596 (C=N), 1512; 1460 and 1421 (C=C), 1374, 1338, 1309, 1271, 1231, 1192, 1142, 1085, 1036, 996, 921, 862, 830, 806, 748, 700, 623, 537, 499, 458, 429. 1H NMR (500 MHZ; CDCl3): δH 8.76 (1H, s, CH=N), 8.04 (1H, s, CH-aromatic), 7.57 (1H, d, J = 1.5 Hz, CH-aromatic), 7.56–2.75 (2H, m, 2 × CH-aromatic), 7.44 (2H, d, J = 7.5 Hz, 2 × CH-aromatic), 7.37 (2H, t, J = 7.5 Hz, 2 × CH-aromatic), 7.31 (1H, t, J = 7.5 Hz, CH-aromatic), 7.24 (1H, d, J = 1.5 Hz, CH-aromatic), 6.94 (1H, d, J = 8.5 Hz, CH-aromatic), 5.23 (2H, s, OCH2), 3.95 (3H, s, OCH3), 2.60 (3H, s, CH3), 2.46 (3H, s, CH3). 13C NMR (125 MHZ; CDCl3): δC 167.01 (C=O), 158.76 (Cq=N), 153.13 (HC=N), 152.20 and 150.09 (2 × Cq–O), 144.63, 136.54, 136.36, 135.82, 128.78, 128.21, 127.30, 126.74, 126.63, 125.84, 124.74, 121.28, 112.98, 109.54 (11 × CH-aromatic and 5 × Cq-aromatic), 70.92 (OCH2), 56.15 (OCH3), 22.80 (CH3), 21.38 (CH3). MS (EI; m/z; %): [M+ + 1] 414.33 (1.58) and [M+] 413.29 (4.57) for C25H23N3O3, 174.23 (2.34), 91.14 (100.00), 77.14 (4.85), 65.15 (14.14), 51.41 (2.64), 42.18 (1.66).
(E)-6-Bromo-3-[((6-methoxynaphthalen-2-yl)methylene)amino]-2-methylquinazolin-4(3H)-one (3f)
Maroon powder (EtOH/CHCl3); yield, 59%; mp 234–235 °C. νmax (KBr)/cm–1: 3086; 3004 and 3055 (CH-aromatic), 2965 and 2930 (CH-aliphatic), 1673 (C=O), 1597 (C=N), 1469 (C=C), 1372, 1337, 1313, 1274, 1173, 1031, 960, 891, 865, 833, 746, 697, 672, 640, 566, 539, 474, 423. 1H NMR (500 MHZ; DMSO-d6): δH 9.01 (1H, s, CH=N), 8.34 (1H, s, CH-aromatic), 8.23 (1H, d, J = 2.0 Hz, CH-aromatic), 8.10 (1H, d, J = 8.5 Hz, CH-aromatic), 7.98 (2H, d, J = 8.5 Hz, 2 × CH-aromatic), 7.62 (2H, d, J = 8.5 Hz, 2 × CH-aromatic), 7.46 (1H, s, CH-aromatic), 7.28 (1H, app dd, J = 9.0, 2.5 Hz, CH-aromatic), 3.93 (3H, s, OCH3), 2.49 (3H, s, CH3). 13C NMR (150 MHZ; CDCl3): δC 167.09 (C=O), 159.59 (Cq=N), 157.60 (HC=N and Cq–O), 145.31, 137.33, 137.13, 132.22, 130.51, 129.63, 128.72, 128.22, 127.95, 127.71, 123.59, 119.70, 106.17 (9 × CH-aromatic and 6 × Cq-aromatic), 55.40 (OCH3), 22.88 (CH3). MS (EI; m/z; %): [M+ + 2, 81Br] 4.25.09 (3.81), [M+ + 1, 81Br] 424.16 (23.23), [M+, 81Br] 423.13 (81.92), [M+ + 1, 79Br] 422.20 (36.74), and [M+, 79Br] 421.06 (100.00) for C21H1681BrN3O2, 380.12 (1.36), 369.23 (16.50), 368.23 (59.79), 367.25 (14.19), 342.23 (2.01), 341.22 (7.11), 340.30 (4.10), 183.09 (3.220), 140.10 (1.27), 75.14 (5.06).
(E)-6-Bromo-3-[(4-hydroxy-3-methoxybenzylidene)amino]-2-methyl-3H-quinazolin-4-one (3g)
White powder (EtOH); yield, 77%; mp 233–235 °C. vmax (KBr)/cm–1: 3370, 2929, 2340, 1657, 1587, 1520, 1465, 1378, 1296, 1201, 1037, 823, 674, 604, 455. 1H NMR (600 MHz; DMSO-d6): δH 10.09 (1H, br. s, OH), 8.70 (1H, s, CH=N), 8.20 (1H, d, J = 2.4 Hz, CH5-quinazolin-4(3H)-one), 7.96 (1H, dd, J = 8.4, 2.4 Hz, CH7-quinazolin-4(3H)-one), 7.60 (1H, d, J = 8.4 Hz, CH8-quinazolin-4(3H)-one), 7.54 (1H, d, J = 1.8 Hz, one of CH of p-hydroxy-m-methoxyphenyl group), 7.35 (1H, app dd, J = 8.4, 1.8 Hz, one of CH of p-hydroxy-m-methoxyphenyl group), 6.95 (1H, d, J = 7.8 Hz, one of CH of p-hydroxy-m-methoxyphenyl group), 3.86 (3H, s, OCH3), 2.49 (3H, s, CH3). 13C NMR (150 MHz; DMSO-d6): δC 169.86 (C=O), 156.46 (Cq=N), 154.24 (HC=N), 151.59, 148.16, 145.38, 137.07, 129.22, 128.50, 124.77, 123.42, 122.66, 118.57, 115.61, 110.47 (6 × CH-aromatic and 6 × Cq-aromatic), 55.64 (OCH3), 22.30 (CH3). MS (EI; m/z; %) [M+] 389.00 (4.94) for C17H1481BrN3O3, 388.05 (1.40), 387.00 (4.92), 240.95 (21.80), 239.95 (100.00), 238.95 (25.05), 237.95 (98.74), 197.90 (11.11), 149.10 (45.03), 134.05 (16.83), 75.00 (23.19), 65.00 (10.72), 59.05 (14.66).
Synthesis of (E)-6-Bromo-3-{[4-(4-chlorobenzyloxy)-3-methoxybenzylidene]amino}-2-methyl-3H-quinazolin-4-one (5)
An equimolar mixture (0.00129 mol) of compound 3g (0.5 g) and 4-chlorobenzyl chloride 4 (0.2 g) in DMF (20 mL) containing potassium carbonate (3 equiv, 0.0039 mol, 0.53 g) was heated under reflux for 5 h.59 The reaction mixture was poured onto cold water and the precipitated solid was filtered off, air-dried, and recrystallized from ethyl acetate to afford compound 5 as a greenish white powder: yield, 76%; mp 218–220 °C. νmax (KBr)/cm–1: 3087, 2927, 1665, 1600, 1511, 1467, 1418, 1374, 1316, 1274, 1207, 1172, 1133, 1097, 1038, 988, 999, 941, 897, 862, 832, 803, 728, 700, 674, 610, 534, 505, 460. 1H NMR (850 MHz; CDCl3): δH 8.77 (1H, s, CH=N), 8.40 (1H, d, J = 2.6 Hz, CH5-quinazolin-4(3H)-one), 7.81 (1H, app dd, J = 9.4, 2.6 Hz, CH7-quinazolin-4(3H)-one), 7.58 (1H, d, J = 1.7 Hz, H2′), 7.54 (1H, d, J = 9.4 Hz, CH8-quinazolin-4(3H)-one), 7.39 (2H, d, J = 8.5 Hz, 2 × CH of p-chlorophenyl moiety), 7.36 (2H, d, J = 8.5 Hz, 2 × CH of p-chlorophenyl moiety), 7.28 (1H, app dd, J = 8.5, 1.7 Hz, H6′), 6.93 (1H, d, J = 7.7 Hz, H5′), 5.21 (2H, s, benzylic-CH2), 3.97 (3H, s, OCH3), 2.63 (3H, s, CH3). 13C NMR (213 MHz; CDCl3): δC 167.21 (C=O), 157.54 (HC=N), 154.44 (C=N), 152.03, 150.07, 137.44, 134.73, 134.03, 129.66, 128.93, 128.73, 128.61, 125.76, 124.78, 122.87, 119.84, 112.94, 109.53 (10 × CH-aromatic and 8 × Cq-aromatic), 70.13 (CH2), 56.09 (OCH3), 22.82 (CH3). MS (DART-ToF; m/z): [M+ + 1] at m/z 514 for C24H1981Br35ClN3O3 or C24H1979Br37ClN3O3.
General Procedures for Synthesis of 2-Chloro-N-(6-unsubsituted/6-chloro-2-methyl-4-oxo-4H-quinazolin-3-yl)acetamides (7a,b)
α-Chloroacetyl chloride 6 (2 equiv, 0.019 mol, 2.15 g, 1.5 mL) was added slowly over a period of 5 min to a cooled mixture (0 °C) of 3-amino-2-methyl-3H-quinazolin-4-one derivatives 1c,d (0.0095 mol) and triethylamine (2 equiv, 0.019 mol, 1.93 g, 2.7 mL) in dry chloroform (30 mL).60 Thereafter, the resulting reaction mixture in each case was stirred at room temperature for 1 h, then it was further heated under reflux for an additional 8 h. Evaporation of chloroform under reduced pressure gave the crude products, which were washed with water, air-dried, and recrystallized from benzene to yield the pure chloroacetamide derivatives 7a,b.
2-Chloro-N-(2-methyl-4-oxo-4H-quinazolin-3-yl)acetamide (7a)
Beige powder; yield, 65%; mp 183–185 °C. νmax (KBr) /cm–1: 3227, 3000, 2924, 1733, 1662, 1608, 1518, 1469, 1425, 1378, 1322, 1265, 1226, 1148, 1029, 974, 950, 871, 777, 692, 659, 630, 590, 521, 477. 1H NMR (500 MHz; CDCl3): δH 11.52 (1H, s, NH), 8.12 (1H, d, J = 7.8 Hz, CH-quinazolin-4(3H)-one), 7.85 (1H, t, J = 8.1 Hz, CH-quinazolin-4(3H)-one), 7.65 (1H, d, J = 8.1 Hz, CH-quinazolin-4(3H)-one), 7.54 (1H, t, J = 7.8 Hz, CH-quinazolin-4(3H)-one), 4.50 and 4.43 (2H, ABq, J = 13.8 Hz, CH2), 2.42 (3H, s, CH3). 13C NMR (125 MHz; CDCl3): δC 165.95 (C=O), 158.63 (C=O), 155.70 (C=N), 146.44, 135.05, 128.27, 126.88, 126.80, 126.39, 120.49 (4 × CH-quinazolin-4(3H)-one, 2 × Cq-quinazolin-4(3H)-one), 40.73 (CH2), 20.86 (CH3). MS (EI; m/z; %) [M+, 37Cl] 253.05 (17.87), [M+, 35Cl] 251.05 (52.15) for C11H10ClN3O2, 203.00 (12.81), 202.00 (100.00), 175.05 (54.64), 160.10 (11.43), 146.10 (63.60), 118.10 (10.62), 117.10 (35.40), 90.05 (17.09), 77.00 (39.79), 76.00 (38.02), 50.00 (15.59).
2-Chloro-N-(6-chloro-2-methyl-4-oxo-4H-quinazolin-3-yl)acetamide (7b)
Beige powder; yield, 55%; mp 164–165 °C. νmax (KBr) /cm–1: 3251, 3077, 3011, 2931, 1718, 1677, 1606, 1517, 1467, 1432, 1373, 1354, 1319, 1266, 1127, 1072, 1032, 974, 903, 850, 801, 775, 689, 652, 575, 537, 462. 1H NMR (850 MHz; CDCl3): δH 8.99 (1H, s, NH), 8.13 (1H, app s, CH5-quinazolin-4(3H)-one), 7. 69 (1H, app dd, J = 8.5, 2.2 Hz, CH7-quinazolin-4(3H)-one), 7.60 (1H, d, J = 8.5 Hz, CH8-quinazolin-4(3H)-one), 4.37 and 4.30 (2H, ABq, J = 15.3 Hz, CH2), 2.54 (3H, s, CH3). 13C NMR (125 MHz; CDCl3): δC 166.44 (C=O), 158.85 (C=O), 155.07 (C=N), 145.39, 135.61, 132.81, 129.02, 126.18, 121.58 (3 × CH-quinazolin-4(3H)-one, 3 × Cq-quinazolin-4(3H)-one), 41.13 (CH2), 21.27 (CH3). MS (EI; m/z; %): [M+, 37Cl2] 289.00 (11.63), [M+ – 1, 37Cl2] 288.00 (9.73), [M+, 37Cl and 35Cl ] 287.00 (63.97), [M+ + 1, 35Cl2] 286.00 (15.13), [M+, 35Cl2] 285.00 (100.00) for C11H9Cl2N3O2, 238.00 (32.55), 237.00 (12.67), 236.00 (95.96), 211.00 (17.05), 209.00 (48.08), 180.00 (30.06), 153.05 (10.16), 151.05 (17.68), 111.00 (15.31), 110.00 (26.59), 76.95 (23.20), 75.00 (41.42), 74.00 (14.79), 57.10 (9.46), 50.95 (10.73).
Synthesis of 6-Chloro-2-methyl-3-(4-oxo-4,5-dihydrothiazol-2-ylamino)-3H-quinazolin-4-one (8)
A mixture of compound 7b (0.0015 mol, 0.45 g), potassium thiocyanate (2 equiv, 0.003 mol, 0.3 g) in absolute ethanol (20 mL) was refluxed for 6 h.61 Concentration of the solvent under reduced pressure and triturating the residue with ice/water gave a solid, which was collected by filtration, air-dried, and recrystallized from ethanol to get the title compound as brown crystals; yield, 83%; mp 213-215 °C. νmax (KBr)/cm–1: 3290, 3063, 2972, 2927, 2366, 1748, 1702, 1614, 1464, 1430, 1377, 1355, 1231, 1182, 1113, 1080, 1035, 992, 898, 845, 783, 688, 663, 623, 579, 539, 507, 468. 1H NMR (850 MHz; CDCl3): δH 8.18 (1H, d, J = 1.7 Hz, CH5-quinazolin-4(3H)-one), 8.14 (1H, br s, NH), 7.72 (1H, dd, J = 8.5, 1.7 Hz, CH7-quinazolin-4(3H)-one), 7.63 (1H, d, J = 8.5 Hz, CH8-quinazolin-4(3H)-one), 4.22 and 4.09 (2H, ABq, J = 17.0 Hz, CH2-thiazol-4-one), 2.45 (3H, s, CH3). 13C NMR (213 MHz; CDCl3): δC 166.83 (C=O), 156.68 (C=O), 154.47 (C=N), 153.90 (C=N), 145.45, 135.72, 132.99, 129.18, 126.67, 121.95 (3 × CH-quinazolin-4(3H)-one, 3 × Cq-quinazolin-4(3H)-one), 31.52 (CH2), 20.74 (CH3). MS (DART-ToF; m/z): [M+ + 1] 309.020 for C12H935ClN4O2S.
2-Mercapto-3-phenyl-3H-quinazolin-4-one (9)
(62) White crystals; yield, 99%; mp 288–290 °C. νmax (KBr)/cm–1: 3220, 3132, 3064, 2962, 2366, 1953, 1846, 1661, 1621, 1529, 1485, 1403, 1339, 1267, 1226, 1195, 1069, 1025, 986, 911, 880, 835, 800, 756, 687, 640, 558, 526. 1H NMR (850 MHz; DMSO-d6): δH 13.05 (1H, s, SH), 7.96 (1H, d, J = 7.7 Hz, CH-aromatic), 7.78 (1H, t, J = 7.7 Hz, CH-aromatic), 7.49 (2H, t, J = 7.7 Hz, 2 × CH-aromatic), 7.46 (1H, d, J = 8.5 Hz, CH-aromatic), 7.41 (1H, t, J = 7.7 Hz, CH-aromatic), 7.35 (1H, t, J = 7.7 Hz, CH-aromatic), 7.29 (2H, d, J = 7.7 Hz, 2 × CH-aromatic). 13C NMR (213 MHz; DMSO-d6): δC 176.05 (C=S), 159.79 (C=O), 139.57, 139.29, 135.57, 128.99, 128.91, 128.10, 127.39, 124.33, 116.16, 115.68 (9 × CH-aromatic and 3 × Cq-aromatic). MS (EI; m/z; %): [M+ + 1] 255.00 (14.03) for C14H10N2OS, [M+] 254.05 (64.28), 253.05 (100.00), 119.05 (16.38), 92.05 (12.64), 77.00 (11.93), 76.00 (16.94).
Synthesis of N-(2-Methyl-4-oxo-4H-quinazolin-3-yl)-2-(4-oxo-3-phenyl-3,4-dihydroquinazolin-2-ylsulfanyl)acetamide (10)
A mixture of compound 7a (0.0012 mol), 2-mercapto-3-phenyl-3H-quinazolin-4-one 9 (0.0012 mol) and potassium carbonate (2.5 equiv, 0.003 mol) in dry acetone (20 mL) was heated under reflux for 8 h. The hot reaction mixture was filtered off, and the filtrate was concentrated under reduced pressure. The precipitated solid was washed with water, air-dried, and recrystallized from acetone to afford the title compound as beige powder: yield, 54%; mp 209–210 °C. νmax (KBr)/cm–1: 3436, 3110, 2959, 1675, 1606, 1546, 1466, 1334, 1300, 1260, 1202, 1025, 964, 874, 766, 693, 639, 561, 501. 1H NMR (850 MHz; CDCl3): δH 10.29 (1H, br. s, NH), 8.25–7.26 (13H, m, 13 × CH-aromatic), 4.03 and 3.99 (2H, ABq, J = 12.8 Hz, CH2–S), 2.50 (3H, s, CH3). 13C NMR (213 MHz; CDCl3): δC 169.26, 161.26, 159.71 (3 × C=O), 158.08 (Cq=N), 155.11 (Cq=N), 146.91, 146.88, 135.29, 135.18, 134.88, 130.59, 130.18, 129.93, 129.33, 128.88, 127.66, 127.13, 126.98, 126.86, 126.66, 125.60, 120.77, 119.95 (13 × CH-aromatic and 5 × Cq-aromatic), 34.06 (CH2), 21.34 (CH3). MS (DART-ToF; m/z): [M+ + 1] 470.12 for C25H19N5O3S.
Synthesis of N-(6-Chloro-2-methyl-4-oxo-4H-quinazolin-3-yl)-2-(3,4,5-trimethoxyphenylamino)acetamide (12)
An equimolar mixture (0.0017 mol) of compound 7b (0.5 g), 3,4,5-trimethoxyaniline (11; 0.3 g), and triethylamine (0.17 g, 0.24 mL) in ethanol (20 mL) was refluxed for 12 h. The reaction mixture was poured onto an ice/H2O mixture; the precipitated product was filtered off, air-dried, and recrystallized from benzene to afford the title compound as a beige powder: yield, 50%; mp 210–215 °C. νmax (KBr)/cm–1: 3379, 3259, 3079, 2932, 2837, 2340, 2366, 1714, 1684, 1604, 1508, 1466, 1406, 1331, 1263, 1230, 1128, 1039, 1003, 931, 836, 806, 776, 687, 613, 582, 534, 486, 449. 1H NMR (850 MHz; CDCl3): δH 8.10 (1H, app. s, CH5-quinazolin-4(3H)-one), 7.68 (1H, dd, J = 8.5, 2.6 Hz, CH7-quinazolin-4(3H)-one), 7.59 (1H, d, J = 8.5 Hz, CH8-quinazolin-4-one), 7.34 (1H, s, NH), 6.03 (2H, s, 2 × CH-trimethoxyphenyl moiety), 4.12 (2H, s, CH2), 3.82 (6H, s, 2 × O CH3), 3.73 (3H, s, O CH3), 3.48 (1H, s, NH), 2.48 (3H, s, CH3). 13C NMR (213 MHz; CDCl3): δC 171.19 (C=O), 158.65 (C=O), 155.65 (C=N), 154.17, 153.77, 145.20, 143.19, 135.45, 132.72, 128.81, 128.34, 126.20, 121.67, 91.43 (5 × CH-aromatic, 7 × Cq-aromatic), 61.08, 56.11 (3 × OCH3), 48.78 (CH2), 21.39 (CH3). MS (DART-ToF; m/z): [M+ – C9H11O3] 267.00 for C20H2137ClN4O5.
Biological Evaluation
Anti-phospholipases Assays
The in vitro anti-phospholipases activities assays of the studied compounds were performed against three isoforms: hsPLA2-G-II, hsPLA2-G-V, and hsPLA2-G-X as reported by de Araújo and Radvanyi.63 Briefly, 10 μL of each compound at different concentrations (5–50 μg/mL) was mixed with 10 μL of each hsPLA2 solution (20 μg/mL); then the resulting mixture was incubated for 20 min at room temperature. Thereafter, 1 mL of the sPLA2 substrate, which consisted of 3.5 mM lecithin suspended in 10 mM CaCl2, 3 mM sodium taurodeoxycholate (NaTDC), and 100 mM NaCl; in addition, a 0.055 mM red phenol as the colorimetric indicator was added. The pH of the resulting mixture was adjusted to 7.6. The kinetics of the hydrolysis was followed spectrophotometrically (BIBBY, Anadéo RS232.UV–vis spectrophotometer) by recording the optical density at the wavelength of 558 nm for 5 min. The results were reported as the inhibition percentage that was calculated by comparison with a negative control experiment (absence of the test compound), and the half-maximal inhibitory concentrations (IC50) values were deduced from the standard calibration curves.
Anti-proteases Assays
The inhibitory activities of the selected compounds on five available therapeutically important proteases, including cathepsin-B, collagenase, thrombin, elastase, and trypsin, were determined calorimetrically according to the method described previously by Kunitz.64 Briefly, the inhibitions of the enzymes were assayed by adding different concentrations of each compound (5–75 μg/mL) to the respective reaction mixture and pre-incubation for 15 min at 37 °C. Then the remaining enzyme activity was followed by the addition of 2 mL of 1% casein (enzyme substrate), and the resulting mixture was allowed to stand for 30 min at 37 °C. Thereafter, 2.5 mL of 5% trichloroacetic acid (TCA) solution was added to stop the reaction. Centrifugation of the reaction mixture (12,000 rpm, 15 min) was done, and the absorbance of the filtrate was measured at 280 nm. The proteases inhibitory activities were expressed as percent inhibitions, which were compared to control experiment. Moreover, the IC50 values were calculated from the standard curve. The standard protease inhibitor cocktail (Sigma) was used as the positive control.
In Vitro Assessment of α-Amylase Inhibitory Activity
The α-amylase inhibitory activity of the tested compounds was evaluated according to the reported method.65 Briefly, 10 μL of α-amylase enzyme (3,3 U, EC 3.2.1.1, Sigma Chemical Co., St. Louis, MO, USA) was mixed with 10 μL of each compound at different concentrations ranging from 20 to 200 μg/mL, appropriate solvent, or quercetin (positive control) at 37 °C for 5 min. Afterward 180 μL of the amylase substrate (Labtest) was added and the samples were incubated for 8 min, then the first reaction was measured at 620 nm. Thereafter, the reaction mixture was incubated for an additional 5 min at 37 °C; then, the second reaction was measured to obtain the final reading. Labtest was diluted in distilled water (1:1) before being added to the microplate (Bio Tek ELX-800, USA). Quercetin was used at the same compound concentrations. The α-amylase inhibition was calculated as follows: % inhibition = 100 – (X2 sample – X1 sample/X2 control – X1 control) × 100, where X1 is the absorbance of the initial reading and X2 is the absorbance of the final reading. The results were expressed in terms of IC50, which were deduced from the standard curve.
In Vitro Assessment of α-Glucosidase Inhibitory Activity
α-Glucosidase inhibitory efficiency of the specified compounds was determined on the basis of measuring the release of 4-nitrophenol (NP) from 4-nitrophenyl α-d-glucopyranoside (4-NPGP) as described by Andrade-Cetto and his collaborators.66 Thus, 20 μL of the test compound at different concentrations ranging from 5 to 50 μg/mL, appropriate solvent, or quercetin (positive control) was mixed with 180 μL of the α-glucosidase enzyme (2 U, EC 3.2.1.20, Sigma) from Saccharomyces cereviseae, and the obtained mixture was incubated at 37 °C for 2 min. Then, 150 μL of the color reagent 4-NPGP was added and the samples were further incubated for 15 min at 37 °C. The colorimetric assay included 2U of α-glucosidase, 5 mM of 4-NPGP and 10 mM potassium phosphate buffer at pH 6.9. Reading of the assay was carried out by using a microplate reader (Bio-Tek ELX-800, USA) at 405 nm. The α-glucosidase inhibition was calculated as follows: % inhibition = 100 – (X2 sample – X1 sample/X2 control – X1 control) × 100, where X1 is the absorbance of the initial reading and X2 is the absorbance of the final reading, control is the absorbance of the assay with the appropriate solvent. The results were expressed in terms of IC50 values, which were obtained from the calibration curve.
In Vitro Assessment of Xanthine Oxidoreductase Inhibitory Activity
Xanthine oxidoreductase inhibitory activity of the same set of compounds was determined by following the formation of uric acid from xanthine using a modified version of the reported procedures by Morgan and co-workers.67 A 40 μL aliquot of xanthine oxidoreductase enzyme (EC 1.17.3.2, Sigma) and 15 μL of each compound (5–150 μg/mL), allopurinol (positive control), or appropriate solvent (negative control) were added to each microplate well and incubated for 5 min at 37 °C. Then, 95 μL from reagent 1 (mixture of hydroxylamine (0.2 mM), EDTA (0.1 mM), and xanthine (667 mM) all in 50 mM phosphate buffer solution at pH 7.5) were added to the reaction mixture and incubated at the same temperature for 30 min. After that, the absorbance was measured at 295 nm using a microplate reader (Bio-Tek ELX-800, USA). Finally, 150 μL of uric acid reagent was added and the absorbance was measured again. Allopurinol (positive control) was used at the same concentration of the tested compounds. The xanthine oxidoreductase inhibition was calculated as follows: % inhibition = 100 – (X2 sample – X1 sample/X2 control – X1 control) × 100, where X1 is the absorbance of the initial reading and X2 is the absorbance of the final reading. The results were expressed in terms of IC50 values, which were calculated from the standard curve.
Cytotoxicity Assay
Cytotoxic potency was examined on human colon cancer cell lines HCT-116 and LoVo (American Type Culture Collection; USA) using various amounts of tested compounds to obtain final concentrations of 25, 50, 75, 100, 200, and 400 μg/mL. Samples were diluted in Dulbecco’s modified Eagles medium, consisting of 10% fetal bovine serum, added to cells grown and cultured for 24 h in a 5% CO2-humidified incubator at 37 °C. Then, the activity of lactate dehydrogenase released from the damaged cells was determined in the collected supernatant aliquots using an ELISA end-point assay (Benchmark Plus, Bio-Rad, Hercules, CA, USA). 0.1% Triton X-100 in the assay medium and the assay medium only were used as positive and negative controls, respectively. Cell viability, expressed as a relative percentage of the OD values (at 550 nm) for compound-treated cells (final concentration of 200 μg/mL) and the control, is shown as mean ± SD (n = 2). The plot of the cell viability (%) versus the compound concentration was also performed to determine the compound concentration providing 50% inhibition (IC50).
Molecular Docking
All of the molecular modeling studies were carried out using Molecular Operating Environment (MOE, 2019.0102) software. All minimizations were performed with MOE until an RMSD gradient of 0.1 kcal mol–1 Å–1 with MMFF94x force field and the partial charges were automatically calculated.
The X-ray crystallographic structure of the target protein complexed with its cocrystallized ligand, Table S5, was downloaded from Protein Data Bank68 (pdb) accessed on Oct. 20–23, 2021. The pdb file was first prepared before the docking procedure through assessing the quality of the data using temperature factors, protein geometry checks, and electron density. Then the software replaced the missing protein sections using homology modeling and rotamer exploring. Moreover, it considered whether fixing bonding patterns in cofactors and ligands and deleting unbound water molecules are necessary. Hydrogens were added and optimized at their positions. Energy minimization of the structure was performed. The amino acid interactions were visualized by Discovery Studio Visualizer v17.2.0.16349.
ADME Study
SwissADME is a free web tool to evaluate pharmacokinetics, drug-likeness, and medicinal chemistry friendliness of small molecules. It was accessed on Oct. 28, 2021 and Mar. 23, 2022 to predict the ADME properties69 of compounds 1a, 1c, 3b, 3c, 3d, 3f, 3g, 5, 8, 10, and 12.
Statistical Analysis
Microsoft Excel software was used for statistical analyses. The values are presented as the arithmetical mean value ± standard deviation (SD) of two replicates for each sample. P values ≤ 0.01 were considered to be statistically significant.
Acknowledgments
We extend our appreciation to the Deanship of Scientific Research at King Saud University for funding this work through Research Group No. RGP-070.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c00812.
Minimized total energies of E and Z isomers of 3a−g and 5, mean IC50 values against studied enzymes, cytotoxic effects, the PDB codes and cocrystallized ligands of target proteins, binding scores, docking results, the drug-likeness properties (Tables S1–S9); interaction representations within active sites of target enzymes (Figures S1–S21); 1H and 13C NMR of some newly synthesized derivatives (PDF)
Author Contributions
N.N.E.E. designed the work, interpreted all of the biological results, supervised the student, and wrote the manuscript. N.M.A. and Z.M.A. conducted the syntheses and characterizations. N.N.E.E. and Z.M.A. provided all chemicals. M.K.E. and N.N.E.E. performed and contributed to interpretation of docking and ADMET studies. A.B. performed biological experiments. M.I.A. provided editing and formatting.
The authors declare no competing financial interest.
Supplementary Material
References
- Wong M. C.; Huang J. J.; Lok V.; Wang J. X.; Fung F.; Ding H. Y.; Zheng Z. J. Differences in incidence and mortality trends of colorectal cancer worldwide based on sex, age, and anatomic location. Clin. Gastroenterol. Hepatol. 2021, 19, 955. 10.1016/j.cgh.2020.02.026. [DOI] [PubMed] [Google Scholar]
- Siegel R. L.; Miller K. D.; Goding Sauer A.; Fedewa S. A.; Butterly L. F.; Anderson J. C.; Cercek A.; Smith R. A.; Jemal A. Colorectal cancer statistics. CA: Cancer J. Clin. 2020, 70, 145. 10.3322/caac.21601. [DOI] [PubMed] [Google Scholar]
- Vuik F. E.; Nieuwenburg S. A. V.; Bardou M.; Lansdorp-Vogelaar I.; Dinis-Ribeiro M.; Bento M. J.; Zadnik V.; Pellise M.; Esteban L.; Kaminski M. F.; et al. Increasing incidence of colorectal cancer in young adults in Europe over the last 25 years. Gut 2019, 68, 1820. 10.1136/gutjnl-2018-317592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vecchi L.; Araújo T. G.; Azevedo F.V.P.D.V.; Mota S. T. S.; Ávila V. D. M. R.; Ribeiro M. A.; Goulart L. R. Phospholipase A2 Drives Tumorigenesis and Cancer Aggressiveness through Its Interaction with Annexin A1. Cells 2021, 10, 1472. 10.3390/cells10061472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leiguez E.; Motta P.; Maia Marques R.; Lomonte B.; Sampaio S. V.; Teixeira C. A Representative GIIA phospholipase A2 activates preadipocytes to produce inflammatory mediators implicated in obesity development. Biomolecules 2020, 10, 1593. 10.3390/biom10121593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap W. H.; Phang S. W.; Ahmed N.; Lim Y. M. Differential effects of sPLA 2-GV and GX on cellular proliferation and lipid accumulation in HT29 colon cancer cells. Mol. Cell. Biochem. 2018, 447, 93. 10.1007/s11010-018-3295-y. [DOI] [PubMed] [Google Scholar]
- Sheng J.; Sun H.; Yu F. B.; Li B.; Zhang Y.; Zhu Y. T. The role of cyclooxygenase-2 in colorectal cancer. Int. J. Med. Sci. 2020, 17, 1095. 10.7150/ijms.44439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno R.; Kawada K.; Sakai Y. Prostaglandin E2/EP signaling in the tumor microenvironment of colorectal cancer. Int. J. Mol. Sci. 2019, 20, 6254. 10.3390/ijms20246254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surrel F.; Jemel I.; Boilard E.; Bollinger J. G.; Payré C.; Mounier C. M.; Talvinen K. A.; Laine V. J. O.; Nevalainen T. J.; Gelb M. H.; Lambeau G. Group X phospholipase A2 stimulates the proliferation of colon cancer cells by producing various lipid mediators. Mol. Pharmacol. 2009, 76, 778. 10.1124/mol.108.053371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudzińska M.; Parodi A.; Soond S. M.; Vinarov A. Z.; Korolev D. O.; Morozov A. O.; Daglioglu C.; Tutar Y.; Zamyatnin A. A. The role of cysteine cathepsins in cancer progression and drug resistance. Int. J. Mol. Sci. 2019, 20, 3602. 10.3390/ijms20143602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y.; Lv Z.; Huang G.; Qin J.; Li H.; Nong F.; Wen B. Prognostic significance of abnormal matrix collagen remodeling in colorectal cancer based on histologic and bioinformatics analysis. Oncol. Rep. 2020, 1671. 10.3892/or.2020.7729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddel C. J.; Tan C. W.; Chen V. M. Thrombin generation and cancer: Contributors and consequences. Cancers 2019, 11, 100. 10.3390/cancers11010100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno R.; Kawada K.; Itatani Y.; Ogawa R.; Kiyasu Y.; Sakai Y. The role of tumor-associated neutrophils in colorectal cancer. Int. J. Mol. Sci. 2019, 20, 529. 10.3390/ijms20030529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soreide K.; Janssen E. A.; Körner H.; Baak J. P. A. Trypsin in colorectal cancer: molecular biological mechanisms of proliferation, invasion, and metastasis. J. Pathol. 2006, 209, 147. 10.1002/path.1999. [DOI] [PubMed] [Google Scholar]
- Vizovisek M.; Ristanovic D.; Menghini S.; Christiansen M. G.; Schuerle S. The tumor proteolytic landscape: A challenging frontier in cancer diagnosis and therapy. Int. J. Mol. Sci. 2021, 22, 2514. 10.3390/ijms22052514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez N.; Prieto I.; del Puerto-Nevado L.; Portal-Nunez S.; Ardura J. A.; Corton M.; Fernandez-Fernandez B.; Aguilera O.; Gomez-Guerrero C.; Mas S.; et al. 2017 update on the relationship between diabetes and colorectal cancer: epidemiology, potential molecular mechanisms and therapeutic implications. Oncotarget 2017, 8, 18456. 10.18632/oncotarget.14472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Kort S.; Masclee A. A. M.; Sanduleanu S.; Weijenberg M. P.; van Herk-Sukel M. P. P.; Oldenhof N. J. J.; van den Bergh J. P. W.; Haak H. R.; Janssen-Heijnen M. L. Higher risk of colorectal cancer in patients with newly diagnosed diabetes mellitus before the age of colorectal cancer screening initiation. Sci. Rep. 2017, 7, 46527. 10.1038/srep46527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guraya S. Y. Association of type 2 diabetes mellitus and the risk of colorectal cancer: a meta-analysis and systematic review. World J. Gastroenterol. 2015, 21, 6026. 10.3748/wjg.v21.i19.6026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higurashi T.; Nakajima A. Metformin and colorectal cancer. Front. Endocrinol. 2018, 9, 622. 10.3389/fendo.2018.00622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phipps O.; Al-Hassi H. O.; Quraishi M. N.; Kumar A.; Brookes M. J. Influence of iron on the gut microbiota in colorectal cancer. Nutrients 2020, 12, 2512. 10.3390/nu12092512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh S. H.; Choi S. Y.; Choi H. J.; Ryu H. M.; Kim Y. J.; Jung H. Y.; Cho J. H.; Kim C. D.; Park S. H.; Kwon T. H.; Kim Y. L. The emerging role of xanthine oxidase inhibition for suppression of breast cancer cell migration and metastasis associated with hypercholesterolemia. FASEB J. 2019, 33, 7301. 10.1096/fj.201802415RR. [DOI] [PubMed] [Google Scholar]
- Li X.; Meng X.; Gao X.; Pang X.; Wang Y.; Wu X.; Deng X.; Zhang Q.; Sun C.; Li Y. Elevated serum xanthine oxidase activity is associated with the development of type 2 diabetes: a prospective cohort study. Diabetes Care 2018, 41, 884. 10.2337/dc17-1434. [DOI] [PubMed] [Google Scholar]
- Vasconcelos-Dos-Santos A.; Loponte H. F. B. R.; Mantuano N. R.; Oliveira I. A.; De Paula I. F.; Teixeira L. K.; De-Freitas-Junior J. C. M.; Gondim K. C.; Heise N.; Mohana-Borges R.; et al. Hyperglycemia exacerbates colon cancer malignancy through hexosamine biosynthetic pathway. Oncogenesis 2017, 6, e306. 10.1038/oncsis.2017.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battelli M. G.; Polito L.; Bortolotti M.; Bolognesi A. Xanthine oxidoreductase in cancer: more than a differentiation marker. Cancer Med. 2016, 5, 546. 10.1002/cam4.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim R. R.; Chen Z.; Mann T. J.; Bastard K.; Scott K. F.; Church W. B. Structural and Functional Aspects of Targeting the Secreted Human Group IIA Phospholipase A2. Molecules 2020, 25, 4459. 10.3390/molecules25194459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarla N. S.; Satyakumar K.; Srinivasu D.; DSVGK K.; Aliev G.; Dharmapuri G.; Putta G. R. SP. S.; Jagarlapoodi S.; Bheeram V.; Sadu S. P.; Duddukuri G. R. Phospholipase A2: A potential therapeutic target in inflammation and cancer iIn silico, in vitro, in vivo and clinical approach). J. Cancer Sci. Ther. 2015, 7, 249. 10.4172/1948-5956.1000357. [DOI] [Google Scholar]
- Herszényi L.; Barabás L.; Hritz I.; István G.; Tulassay Z. Impact of proteolytic enzymes in colorectal cancer development and progression. World J. Gastroenterol. 2014, 20, 13246. 10.3748/wjg.v20.i37.13246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Badry Y. A.; El-Hashash M. A.; Al-Ali K. Synthesis of bioactive quinazolin-4 (3H)-one derivatives via microwave activation tailored by phase-transfer catalysis. Acta Pharm. 2020, 70, 161. 10.2478/acph-2020-0001. [DOI] [PubMed] [Google Scholar]
- Tokalı F. S.; Taslimi P.; Demircioglu I. H.; Karaman M.; Gultekin M. S.; Gulcin I.; Sendil K. Design, synthesis, molecular docking, and some metabolic enzyme inhibition properties of novel quinazolinone derivatives. Arch. Pharm. (Weinheim, Ger.) 2021, 354, 2000455. 10.1002/ardp.202000455. [DOI] [PubMed] [Google Scholar]
- El-Sayed N. N.; Almaneai N. M.; Ben Bacha A.; Al-Obeed O.; Ahmad R.; Abdulla M.; Alafeefy A. M. Synthesis and evaluation of anticancer, antiphospholipases, antiproteases, and antimetabolic syndrome activities of some 3H-quinazolin-4-one derivatives. J. Enzyme Inhib. Med. Chem. 2019, 34, 672. 10.1080/14756366.2019.1574780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Suwaidan I. A.; Abdel-Aziz A.A.-M.; Shawer T. Z.; Ayyad R. R.; Alanazi A. M.; El-Morsy A. M.; Mohamed M. A.; Abdel-Aziz N. I.; El-Sayed M. A.-A.; El-Azab A. S. Synthesis, antitumor activity and molecular docking study of some novel 3-benzyl-4(3H) quinazolinone analogues. J. Enzyme Inhib. Med. Chem. 2016, 31, 78. 10.3109/14756366.2015.1004059. [DOI] [PubMed] [Google Scholar]
- AL-maneai N. M.Synthesis and Pharmacological Evaluation of Some Novel 4-(3H)-Quinazolinone Derivatives. M.S. Dissertation, King Saud University, Riyadh, Saudi Arabia, 2017. [Google Scholar]
- Hricovíniová Z.; Hricovíni M.; Kozics K. New series of quinazolinone derived Schiff’s bases: synthesis, spectroscopic properties and evaluation of their antioxidant and cytotoxic activity. Chem. Pap. 2018, 72, 1041. 10.1007/s11696-017-0345-y. [DOI] [Google Scholar]
- Tariq S.; Avecilla F.; Sharma G. P.; Mondal N.; Azam A. Design, synthesis and biological evaluation of Quinazolin-4 (3H)-one Schiff base conjugates as potential antiamoebic agents. J. Saudi Chem. Soc. 2018, 22, 306. 10.1016/j.jscs.2016.05.006. [DOI] [Google Scholar]
- Mansour M. A.; AboulMagd A. M.; Abdel-Rahman H. M. Quinazoline-Schiff base conjugates: in silico study and ADMET predictions as multi-target inhibitors of coronavirus (SARS-CoV-2) proteins. RSC Adv. 2020, 10, 34033. 10.1039/D0RA06424F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitrugno A.; Tassi Yunga S.; Sylman J. L.; Zilberman-Rudenko J.; Shirai T.; Hebert J. F.; Kayton R.; Zhang Y.; Nan X.; Shatzel J. J.; et al. The role of coagulation and platelets in colon cancer-associated thrombosis. Am. J. Physiol. Cell Physiol. 2019, 316, C264. 10.1152/ajpcell.00367.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turpin B.; Miller W.; Rosenfeldt L.; Kombrinck K.; Flick M. J.; Steinbrecher K. A.; Harmel-Laws E.; Mullins E. S.; Shaw M.; Witte D. P.; et al. Thrombin drives tumorigenesis in colitis-associated colon cancer. Cancer Res. 2014, 74, 3020. 10.1158/0008-5472.CAN-13-3276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojtukiewicz M. Z.; Mysliwiec M.; Sierko E.; Sobierska M.; Kruszewska J.; Lipska A.; Radziwon P.; Tucker S. C.; Honn K. V. Elevated microparticles, thrombin-antithrombin and VEGF levels in colorectal cancer patients undergoing chemotherapy. Pathol. Oncol. Res. 2020, 26, 2499. 10.1007/s12253-020-00854-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyanovsky B. B.; Webb N. R. Biology of secretory phospholipase A 2. Cardiovasc. Drugs Ther. 2009, 23, 61. 10.1007/s10557-008-6134-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X.; Li Q. Prostaglandin EP2 receptor: novel therapeutic target for human cancers. Int. J. Mol. Med. 2018, 42, 1203. 10.3892/ijmm.2018.3744. [DOI] [PubMed] [Google Scholar]
- Cheng H. C.; Chang T. K.; Su W. C.; Tsai H. L.; Wang J. Y. Narrative review of the influence of diabetes mellitus and hyperglycemia on colorectal cancer risk and oncological outcomes. Transl. Oncol. 2021, 14, 101089. 10.1016/j.tranon.2021.101089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizutani M. Y.; Takamatsu Y.; Ichinose T.; Nakamura K.; Itai A. Effective handling of induced-fit motion in flexible docking. Proteins: Struct., Funct., Bioinf. 2006, 63, 878. 10.1002/prot.20931. [DOI] [PubMed] [Google Scholar]
- Mena-Ulecia K.; Tiznado W.; Caballero J. Study of the differential activity of thrombin inhibitors using docking, QSAR, molecular dynamics, and MM-GBSA. PLoS One 2015, 10, e0142774. 10.1371/journal.pone.0142774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veale C. A.; Bernstein P. R.; Bryant C.; Ceccarelli C.; Damewood J. R.; Earley R.; Feeney S. W.; Gomes B.; Kosmider B. J.; Steelman G. B.; et al. Nonpeptidic inhibitors of human leukocyte elastase. 5. Design, synthesis, and X-ray crystallography of a series of orally active 5-aminopyrimidin-6-one-containing trifluoromethyl ketones. J. Med. Chem. 1995, 38, 98. 10.1021/jm00001a015. [DOI] [PubMed] [Google Scholar]
- Edwards P. D.; Andisik D. W.; Strimpler A. M.; Gomes B.; Tuthill P. A. Nonpeptidic inhibitors of human neutrophil elastase. 7. Design, synthesis, and in vitro activity of a series of pyridopyrimidine trifluoromethyl ketones. J. Med. Chem. 1996, 39, 1112. 10.1021/jm950684z. [DOI] [PubMed] [Google Scholar]
- Iyaguchi D.; Kawano S.; Takada K.; Toyota E. Structural basis for the design of novel Schiff base metal chelate inhibitors of trypsin. Bioorg. Med. Chem. 2010, 18, 2076. 10.1016/j.bmc.2010.02.016. [DOI] [PubMed] [Google Scholar]
- Abhithaj J.; Arun K. G.; Sharanya C. S.; Haridas M.; Jayadevi Variyar E. Isozymes inhibited by active site blocking: versatility of calcium indifferent hesperidin binding to phospholipase A2 and its significance. J. Recept. Signal Transduction 2019, 39, 60. 10.1080/10799893.2019.1606239. [DOI] [PubMed] [Google Scholar]
- Hansford K. A.; Reid R. C.; Clark C. I.; Tyndall J. D. A.; Whitehouse M. W.; Guthrie T.; McGeary R. P.; Schafer K.; Martin J. L.; Fairlie D. P. D-Tyrosine as a chiral precursor to potent inhibitors of human nonpancreatic secretory phospholipase A2 (IIa) with antiinflammatory activity. ChemBioChem 2003, 4, 181. 10.1002/cbic.200390029. [DOI] [PubMed] [Google Scholar]
- Giordanetto F.; Pettersen D.; Starke I.; Nordberg P.; Dahlström M.; Knerr L.; Selmi N.; Rosengren B.; Larsson L.; Sandmark J.; et al. Discovery of AZD2716: a novel secreted phospholipase A2 (sPLA2) inhibitor for the treatment of coronary artery disease. ACS Med. Chem. Lett. 2016, 7, 884. 10.1021/acsmedchemlett.6b00188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gloster T. M.; Turkenburg J. P.; Potts J. R.; Henrissat B.; Davies G. J. Divergence of catalytic mechanism within a glycosidase family provides insight into evolution of carbohydrate metabolism by human gut flora. Chem. Biol. 2008, 15, 1058. 10.1016/j.chembiol.2008.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauff J. M.; Cao H.; Hille R. Substrate orientation and catalysis at the Molybdenum site in xanthine oxidase. J. Biol. Chem. 2009, 284, 8760. 10.1074/jbc.M804517200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley E. E.; Batthyany C. I.; Hundley N. J.; Woodcock S. R.; Bonacci G.; Del Rio J. M.; Schopfer F. J.; Lancaster J. R.; Freeman B. A.; Tarpey M. M. Nitro-oleic acid, a novel and irreversible inhibitor of xanthine oxidoreductase. J. Biol. Chem. 2008, 283, 36176. 10.1074/jbc.M802402200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong C.; Montes M.; Al-Sawai W. M. Xanthine oxidoreductase inhibition–A review of computational aspect. J. Theor. Comput. Chem. 2020, 19, 2040008. 10.1142/S0219633620400088. [DOI] [Google Scholar]
- Nahoum V.; Roux G.; Anton V.; Rougé P.; Puigserver A.; Bischoff H.; Henrissat B.; Payan F. Crystal structures of human pancreatic α-amylase in complex with carbohydrate and proteinaceous inhibitors. Biochem. J. 2000, 346, 201. 10.1042/bj3460201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daina A.; Michielin O.; Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharom F. J. The P-glycoprotein efflux pump: how does it transport drugs?. J. Membr. Biol. 1997, 160, 161. 10.1007/s002329900305. [DOI] [PubMed] [Google Scholar]
- Lipinski C. A.; Lombardo F.; Dominy B. W.; Feeney P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Delivery Rev. 1997, 23, 3. 10.1016/S0169-409X(96)00423-1. [DOI] [PubMed] [Google Scholar]
- Giménez B. G.; Santos M. S.; Ferrarini M.; Fernandes J. P. S. Evaluation of blockbuster drugs under the rule-of-five. Pharmazie 2010, 65, 148. [PubMed] [Google Scholar]
- Chen W.; Yang X. D.; Li Y.; Yang L. J.; Wang X. Q.; Zhang G. L.; Zhang H. B. Design, synthesis and cytotoxic activities of novel hybrid compounds between dihydrobenzofuran and imidazole. Org. Biomol. Chem. 2011, 9, 4250. 10.1039/c1ob05116d. [DOI] [PubMed] [Google Scholar]
- AL-Balawi N. A. M.Synthesis of novel 2-aminothiazole derivatives of anticipated biological activities. Master’s thesis, King Saud University, Riyadh, Saudi Arabia, April 29, 2015. [Google Scholar]
- Liu H. L.; Lieberzeit Z.; Anthonsen T. Synthesis and fungicidal activity of 2-imino-3-(4-arylthiazol-2-yl)-thiazolidin-4-ones and their 5-arylidene derivatives. Molecules 2000, 5, 1055. 10.3390/50901055. [DOI] [Google Scholar]
- Kim H.; Lee H. J.; Kim D. P. Integrated One-Flow Synthesis of Heterocyclic Thioquinazolinones through Serial Microreactions with Two Organolithium Intermediates. Angew. Chem., Int. Ed. 2015, 54, 1877. 10.1002/anie.201410062. [DOI] [PubMed] [Google Scholar]
- de Araújo A. L.; Radvanyi F. Determination of phospholipase A2 activity by a colorimetric assay using a pH indicator. Toxicon 1987, 25, 1181. 10.1016/0041-0101(87)90136-X. [DOI] [PubMed] [Google Scholar]
- Kunitz M. Crystalline soybean trypsin inhibitor II. General properties. J. Gen. Physiol. 1947, 30, 291. 10.1085/jgp.30.4.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian R.; Asmawi M. Z.; Sadikun A. In vitro alpha-glucosidase and alpha-amylase enzyme inhibitory effects of Andrographis paniculata extract and andrographolide. Acta Biochim. Polym. 2008, 55, 391. 10.18388/abp.2008_3087. [DOI] [PubMed] [Google Scholar]
- Andrade-Cetto A.; Becerra-Jiménez J.; Cárdenas-Vázquez R. Alfa-glucosidase-inhibiting activity of some Mexican plants used in the treatment of type 2 diabetes. J. Ethnopharmacol. 2008, 116, 27. 10.1016/j.jep.2007.10.031. [DOI] [PubMed] [Google Scholar]
- Dew T. P.; Day A. J.; Morgan M. R. Xanthine oxidase activity in vitro: effects of food extracts and components. J. Agric. Food Chem. 2005, 53, 6510. 10.1021/jf050716j. [DOI] [PubMed] [Google Scholar]
- RSCB Protein Data Bank (PDB); https://www.rcsb.org (last accessed 2021-10-23).
- SwissADME, Swiss Institute of Bioinformatics; http://www.swissadme.ch/index.php (last accessed 2022-03-23).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.