

SUMMARY
T follicular helper (TFH) cells promote expansion of germinal center (GC) B cells and plasma cell differentiation. Whether cognate peptide-MHCII (pMHCII) density instructs selection and cell fate decisions in a quantitative manner remains unclear. Using αDEC205-OVA to differentially deliver OVA peptides to GC B cells on the basis of DEC205 allelic copy number, we find DEC205+/+ B cells take up 2-fold more antigen than DEC205+/− cells, leading to proportional TFH cell help and B cell expansion. To validate these results, we establish a caged OVA peptide, which is readily detected by OVA-specific TFH cells after photo-uncaging. In situ uncaging of peptides leads to multiple serial B-T contacts and cell activation. Differential CD40 signaling, is both necessary and sufficient to mediate 2-fold differences in B cell expansion. While plasmablast numbers are increased, pMHCII density does not directly control the output or quality of plasma cells. Thus, we distinguish the roles TFH cells play in expansion versus differentiation.
Graphical Abstract
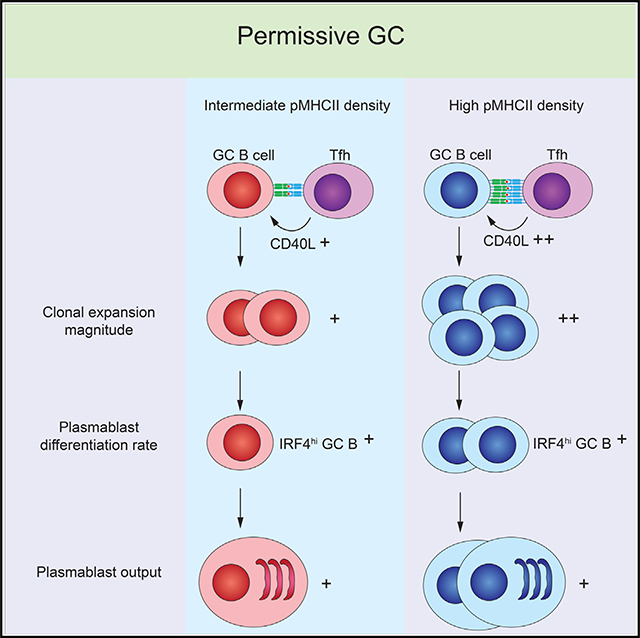
In brief
Jing et al. show that GC selection and plasmablast differentiation are permissive to lower-affinity clones even in competitive GCs, while maintaining proportional responses based on peptide-MHC density, via CD40L-CD40 strength. This egalitarian mechanism allows lower-affinity clones to participate in the humoral response but in a proportional manner.
INTRODUCTION
Germinal centers (GCs) are essential for generating somatically hypermutated and affinity-matured antibodies in T cell-dependent immune responses (Victora and Nussenzweig, 2012). GCs are divided micro-anatomically into the dark zone where antigen-activated B cells proliferate and mutate their BCR genes, and the light zone (LZ) where they bind antigens that are captured and presented by follicular dendritic cells (FDC) followed by selection by T follicular helper (TFH) cell recognition of cognate peptide-loaded major histocompatibility complex class II (pMHCII) (De Silva and Klein, 2015). Selected GC B cells can re-enter cell cycle for clonal expansion or differentiate into memory B cells or plasmablasts (PBs) (Shinnakasu and Kurosaki, 2017; Suan et al., 2017), whereas poorly selected GC B cells undergo a default apoptotic fate (Mayer et al., 2017). The fate of selected GC B cells in plasma cell (PC) differentiation is determined by cooperative signals from FDC and TFH cells in GCs, enabling high-affinity B cells to preferentially differentiate into PBs (Krautler et al., 2017; Phan et al., 2006; Smith et al., 2000). The molecular signature of PBs includes upregulation of Blimp1, IRF4, and XBP1, accompanied by the downregulation of Bcl6 and PAX5 (Shi et al., 2015). Naive B cells with high preimmune affinities of BCR have greater surface density of pMHCII than those with lower-affinity BCR (Batista and Neuberger, 1998), thus allowing better competencies for engaging TFH cells in the LZ through the TCR-pMHCII interaction (Schwickert et al., 2011). Consistent with that, a T cell-restricted scanning and selection model suggests a limited number of T cells controlling the positive selection of B cells based on their surface level of cognate pMHCII (Schwickert et al., 2011; Shulman et al., 2014).
TFH cells can provide help in a number of ways at different stages in GC reactions, including CD40L, LFA-1, and inducible costimulator (ICOS) expressed on the surface and TFH cell-derived cytokines interleukin-21 (IL-21) and IL-4, which are offered during the transient TFH cell-B cell contacts (Crotty, 2011; Liu et al., 2015; Reinhardt et al., 2009; Weinstein et al., 2016; Zaretsky et al., 2017). Complete and haploinsufficient deficiencies in any these factors can disrupt the overall GC response (Ise et al., 2018), but these results do not unravel the specific contribution of each factor during GC selection. Thus, it remains unknown whether these factors and other cell-extrinsic cues play a pre-requisite role in TFH cell-mediated selection versus an instructive role in directing proportional responses to GC B cells on the basis of differential pMHCII density. This is particularly relevant in vaccination efforts against highly variable strains of viruses, such as influenza, in which high-affinity clones that are specific to strain-dependent hemagglutinin epitopes outcompete broadly cross-reactive clones that tend to be lower affinity (Krammer and Palese, 2015; Schmidt et al., 2015).
Despite the essential role T cells play in the selection of B cells in GC, it remains unclear whether B cell fate in clonal expansion and PC differentiation are instructed by the surface density of pMHCII. Nussenzweig and colleagues have used αDEC205 antibody fused to OVA antigen (αDEC205-OVA) to deliver high doses of OVA peptide directly to GC B cells, driving selection by OVA-specific TFH cells in a synchronized manner (Victora et al., 2010). Prior to GC entry, it has been shown that monoclonal activated B cells targeted with αDEC205-OVA had higher pMHCII density and exhibited significantly more early proliferation and PB differentiation than in co-transferred B cells that did not receive αDEC205-OVA targeting (Schwickert et al., 2011). Similarly, in the GC, targeting αDEC205-OVA led to increased T cell help in the LZ in a dose-dependent manner, suggesting that T cell help was proportional to pMHCII (Gitlin et al., 2014). In these models, non-targeted B cells were not selected and diminished; however, the fate of B cells expressing intermediate levels of pMHCII were not addressed. In polyclonal GCs in mixed chimeric mice comprising a 50:50 mix of MHCII+/+ and MHCII+/− B cells, MHCII+/− B cells are less recruited to GC than MHCII+/+ B cells, but these cells persisted in the GC over time, even though pMHCII levels were reduced on MHCII+/− cells, suggesting a less instructive role for pMHCII density in GC selection (Yeh et al., 2018). However, in a polyclonal GC, it is difficult to tease out the role of BCR and T cell help in regulating the overall outcome.
Thus, it remains unclear if TFH cells can discriminate individual GC B cells on the basis of pMHCII density. In this study, our goal was to dynamically and differentially modulate antigen presentation on multiple GC B cells and monitor their fate using two different tools, one was using a caged peptide that could be photoactivated in vivo and the second was using multiple DEC205 genotypes to modulate uptake levels of surrogate antigens by αDEC205-OVA targeting.
RESULTS
Antigen uptake and presentation on MHCII is proportional to DEC205 expression
GC B cells highly express DEC205 (encoded by Ly75 gene), a cell surface lectin that can be targeted by αDEC205 Ab for uptake and processing of antigens and presentation of antigen-derived peptide on MHCII to TFH cells (Kamphorst et al., 2010; Victora et al., 2010). We hypothesized that, if DEC205 expression was proportional to allelic copy number, then we could deliver intermediate and low levels of cognate peptide to GC B cells in DEC205+/+, DEC205+/−, and DEC205−/− mice, respectively. Indeed, DEC205 surface expression on GC B cells of DEC205+/+ mice was 1.8-fold higher compared with cells from DEC205+/− mice and undetectable in DEC205−/− hosts, confirming our hypothesis (Figure 1A).
Figure 1. Differential pMHCII density can be controlled by αDEC-OVA targeting.
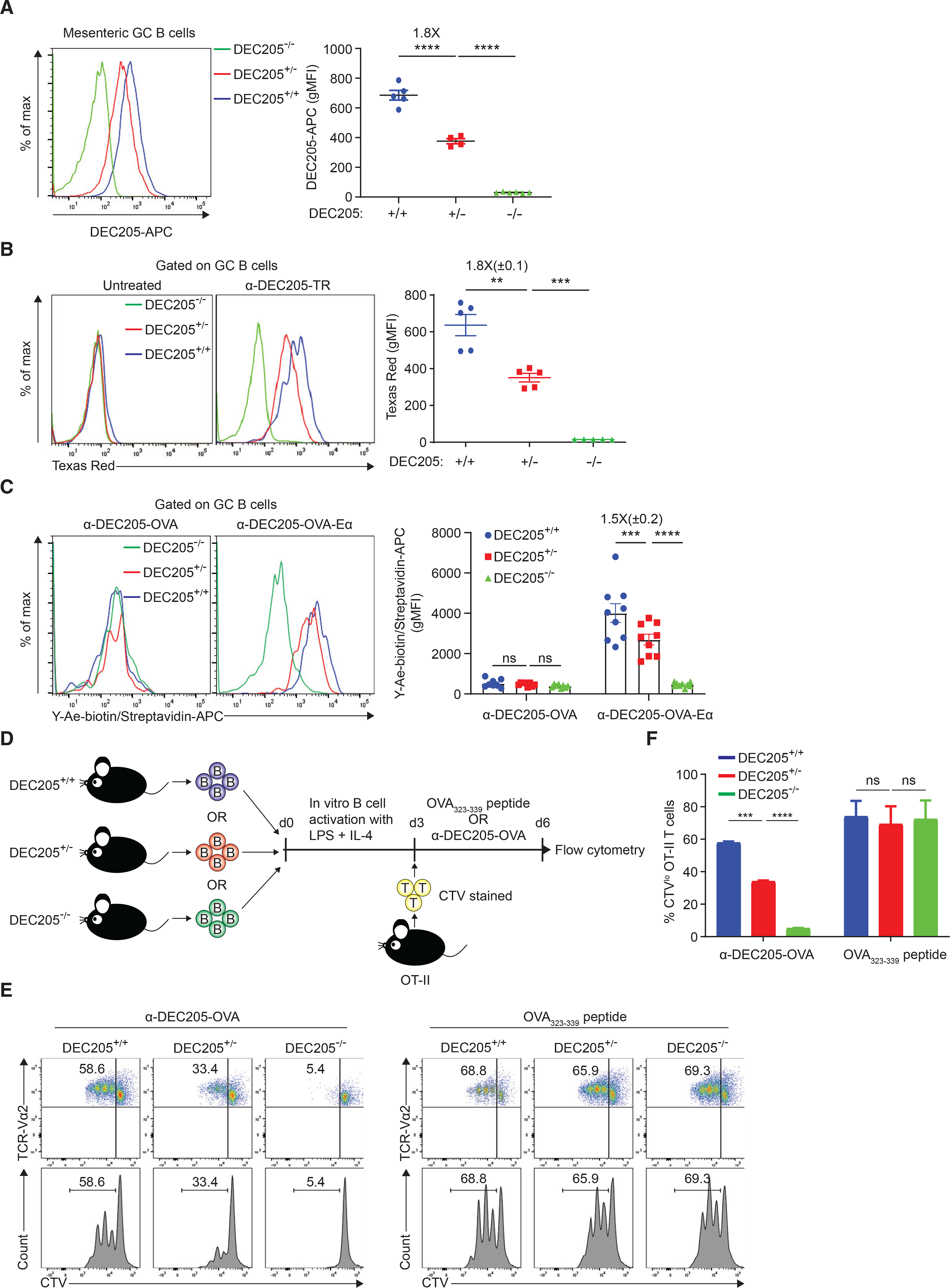
(A) DEC205 surface expression on GC B cells from mesenteric lymph nodes.
(B) αDEC205-Texas Red uptake level of DEC205+/+, DEC205+/−, and DEC205−/− GC B cells in the draining lymph node.
(C) Y-Ae staining of DEC205+/+, DEC205+/−, and DEC205−/− GC B cells in the draining lymph node.
(D) Experimental setup for (E) and (F).
(E) OT-II T cell proliferation is shown by CTV dilution, which is quantified in (F). Data are representative of two individual experiments. All bars show mean ± SEM. **p < 0.01; ***p < 0.001; ****p < 0.0001; ns, non-significant by unpaired Mann-Whitney U test (A and F) or paired Student’s t test (B and C). All graphs show pooled data from at least two independent experiments. (A) n = 5–6; (B) n = 5; (C) n = 8; (F) n = 2.
Next, we tested if in vivo uptake of αDEC205 cargo by GC B cells was proportional to differential expression of DEC205, based on allelic copy number in a cell-intrinsic manner. To do so, ovalbumin (OVA)-primed DEC205−/− hosts were co-transferred with naive B cells from DEC205+/+, DEC205+/−, and DEC205−/− mice that all expressed the B1–8 knock-in Ig heavy chain that, when paired with λ light chain, exhibits high affinity to the hapten 4-hydroxy-3-nitrophenylacetyl (NP) (Shih et al., 2002) and were congenically distinct based on CD45 isoforms. Subsequently, host mice were subcutaneously boosted in the hind footpad with OVA conjugated to NP (NP-OVA) to allow formation of GCs in the draining popliteal lymph node containing NP-specific B cells from all three transferred populations. On day 8 post boost, αDEC205 Ab conjugated to Texas Red, a pH-insensitive, non-degradable dye, was injected subcutaneously to directly assess in situ antigen uptake by GC B cells. Similarly, a 1.8-fold (±0.1) difference in Texas Red uptake level was seen between DEC205+/+ and DEC205+/− GC B cells with minimal uptake by DEC205−/− cells (Figures 1B and S1A). Thus, DEC205 can be targeted in the GC to deliver differential amount of antigen intracellularly based on the allelic genotype of an individual GC B cell.
To ensure that differential αDEC205-OVA targeting and uptake to GC B cells in vivo leads to proportional changes in pMHCII presentation, we generated a chimeric αDEC205 antibody (αDEC205-OVA-Eα) that co-expressed a fragment of α-chain of I-E class II molecules (Eα52–68), which, when presented in I-Ab molecules, is detectable by the monoclonal Ab, Y-Ae (Rudensky et al., 1991) (Figure S1B). Using this system, we found a 1.5-fold (±0.2) increase in Y-Ae gMFI on DEC205+/+ GC B cells compared with DEC205+/− GC B cells (Figure 1C), indicating αDEC205 targeting correlated directly with subsequent pMHCII surface density on GC B cells.
Next, we tested if targeting different amounts of antigen, based on DEC205 receptor expression, leads to proportional presentation of OVA peptides-loaded pMHCII molecules and T cell recognition in vitro. To do so, OT-II transgenic T cells, which recognize OVA323–339:MHCII in the context of I-Ab, were purified and co-cultured with (I-Ab) B cell blasts from DEC205+/+, DEC205+/−, or DEC205−/− mice, in the presence of chimeric αDEC205 antibodies that were fused to OVA full protein (αDEC205-OVA) (Bonifaz et al., 2002) or OVA323–339 free peptide (Figure 1D). Prior to co-culturing, OT-II T cells were labeled with CellTrace Violet (CTV) dye, and CTV dilution on day 3 was used to measure antigen presentation. When B cells were exogenously loaded with OVA323–339 peptide, all three populations induced equally robust (~70% CTVlow) T cell proliferation, excluding any underlying differences in antigen presentation on the basis of DEC205 genotype (Figures 1E and 1F). In contrast, when treated with αDEC205-OVA, DEC205+/+ B cells produced a 1.8-fold higher level of OT-II proliferation than DEC205+/− B cells (58.6% versus 33.4%; Figures 1E and 1F), whereas DEC205−/− B cells were unable to promote T cell proliferation (5.4% CTVlow; Figures 1E and 1F), in line with their uptake of antigen. Taken together, we find that surrogate antigens can be differentially delivered to GC B cells in vivo based on DEC205 allelic genotype, on a per cell basis, leading to proportional differences in antigen uptake, antigen presentation, and T cell recognition on a per cell basis in vitro.
GC B clonal expansion magnitude is proportional to pMHCII density
We hypothesized that T cell help may vary on the basis of pMHCII densities on GC B cells, leading to alternative cell fates and outcomes. In particular, we wondered whether GC B cells expressing intermediate levels of pMHCII (e.g., low-affinity clones) could be selected, when in direct competition with cells expressing higher amounts (e.g., high-affinity clones), as could occur in polyclonal GCs with either intra-clonal or inter-clonal competition or both. To reduce clonal complexity, we studied the competition between GC B cells bearing the same high-affinity NP-specific BCR, B1–8hi, but presenting differential levels of pMHCII, we adoptively transfer a mixture of naive monoclonal B cells from B1–8hi CD45.1 DEC205+/+ mice, B1–8hi CD45.2 DEC205+/− mice, and B1–8hi CD45.1/2 DEC205−/− mice at equal ratio (33%:33%:33%) into OVA-primed CD45.1/2 DEC205−/− hosts (Figures 2A and 2B). Recipient mice were boosted with NP-OVA so that all three populations enter the GC and receive help from OVA-specific TFH cells. To synchronize the TFH cell help, DEC205 Ab fused to cognate antigen OVA (αDEC205-OVA) were given to one footpad to target the DEC205+/+ and DEC205+/− donor B cells only, robustly forcing the GC selection of these cells by TFH cells (Victora et al., 2010). To assess the kinetics of selection, popliteal draining lymph nodes were analyzed at 24, 48, and 72 h after αDEC205-OVA injection in the left footpad, in comparison with the popliteal lymph node from the untreated right leg. We found that, at 72 h, DEC205+/+ GC B cell numbers had an average 2-fold increase relative to DEC205+/− GC B cells 72 h after treatment (8.6- versus 4.4-fold), whereas DEC205−/− GC B cells (comprising both transferred B1–8hi and endogenous polyclonal cells) did not increase after αDEC205-OVA injection (Figure 2C). While there was a broad variance in fold increases of DEC205+/+ GC B cells (ranging from 2- to 20-fold increases) in any given mouse, the 2-fold proportional difference over DEC205+/− B cells in the same host was maintained (Figure 2D), indicating that discrimination of pMHCII density on a per cell basis occurs at broad ranges of signal strength. To measure changes in selection on the basis of pMHCII density, we plotted the ratio of DEC205+/+/DEC205+/− GC B cells at 72 h post treatment and found a 2-fold increase in αDEC205-OVA-treated legs over untreated legs (Figure 2E). We did not observe increase of DEC205+/+ and DEC205+/− GC B numbers in the contralateral popliteal lymph node (in the untreated leg), which had similar ratios to mice that were not boosted with αDEC205-OVA at all, indicating that no substantial amounts of αDEC205-OVA was draining into the untreated leg (Figures S1C and S1D). The increase in DEC205+/+/DEC205+/− ratio was not observed after treatment with unconjugated control αDEC205, excluding any possible effect caused by targeting DEC205 alone (Figure 2E). While it is known that some rejection of DEC205+ cells can occur in DEC205−/− hosts (Pasqual et al., 2015), which might confound some results, we obtained similar increases in DEC205+/+/DEC205+/− ratio when using wild-type (WT) (DEC205+/+) hosts (Figure 2E). This 2-fold increase in DEC205+/+ GC B cells was manifested by an increased proliferation rate over DEC205+/− cells at 48 h post treatment based on EdU incorporation (Figures 2F and S2A). We asked if this advantage in clonal expansion of DEC205+/+ GC B cells could eventually lead to the loss of DEC205+/− cells, by multiple rounds of αDEC205-OVA treatment every 3 days (Figure S2B). However, the frequency and numbers of both DEC205+/+ and DEC205+/− GC B cells gradually dropped after the second and third doses of αDEC205-OVA treatment, suggesting that DEC205+/− GC B cells could not be outcompeted by DEC205+/+ cells or due to technical limitations with the model system, such as GC decay (Figures S2C and S2D).
Figure 2. GC B cell clonal expansion magnitude is proportional to pMHCII density.
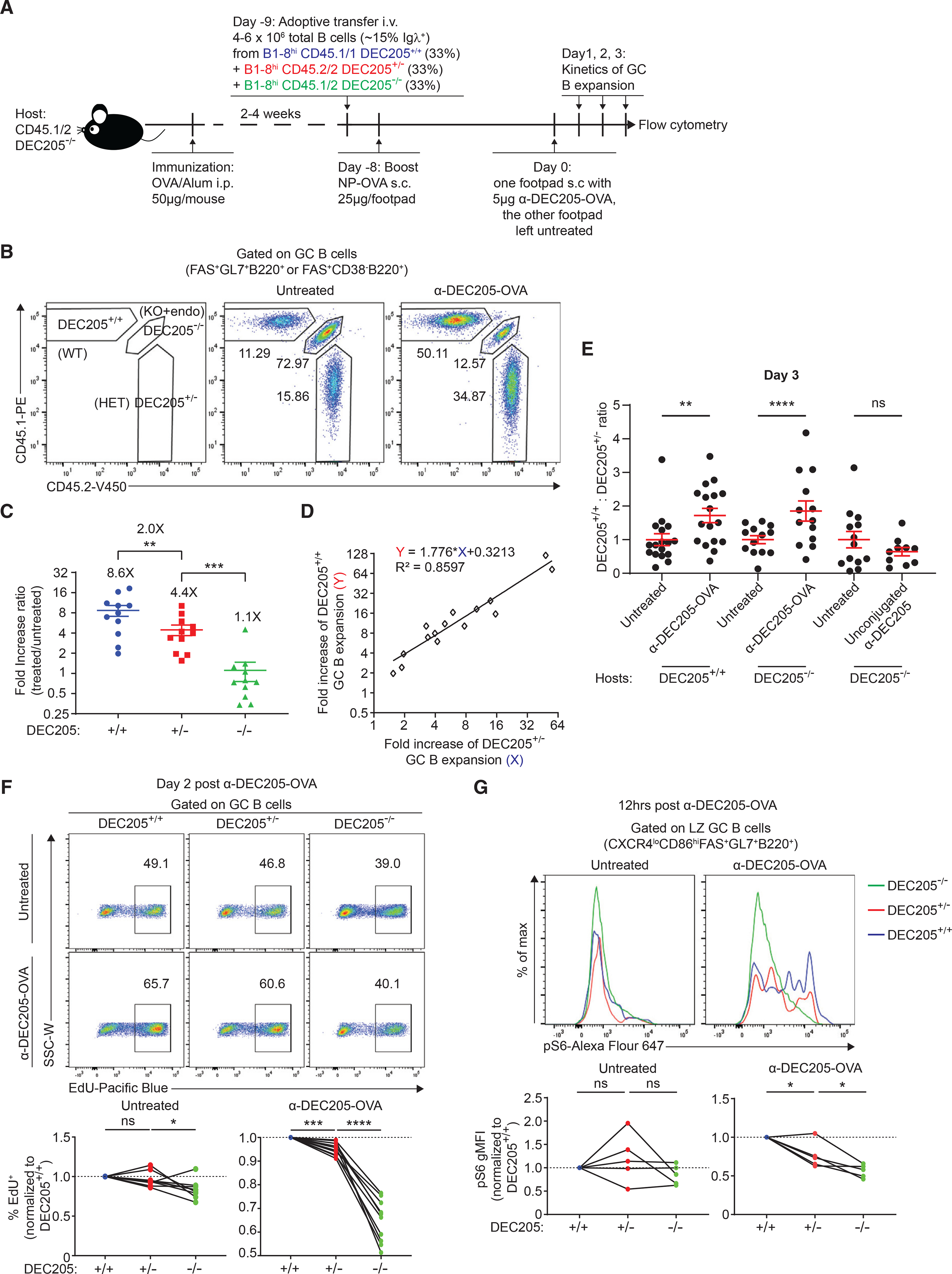
(A) Experimental setup for (B–E).
(B) Representative plot of FACS staining for differentiating DEC205+/+, DEC205+/−, and DEC205−/− GC B cells, a key of gating each population is shown on the left.
C) αDEC-OVA/untreated fold increase ratio of GC B cell absolute numbers.
(D) Linear association between the fold change of DEC205+/+ and DEC205+/− GC B cell expansion.
(E) The DEC205+/+/DEC205+/− ratio by absolute numbers of GC B cells 72 h after αDEC-OVA or unconjugated αDEC205 treatment compared with untreated contralateral footpad in DEC205+/+ or DEC205−/− hosts.
(F) Normalized EdU incorporation of GC B cell subsets 2 days post αDEC-OVA treatment compared with untreated footpad on the top, which is quantified at the bottom.
(G) Normalized phospho-S6 staining of DEC205+/+, DEC205+/−, and DEC205−/− GC B cells 12 h post αDEC-OVA treatment compared with untreated footpad on the top, which is quantified at the bottom. All bars show mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; ns, non-significant by paired Student’s t test. All graphs show pooled data from at least two independent experiments. (C) n = 11; (E) n = 13–17; (F) n = 8; (G) n = 5.
Positively selected GC B cells activate mTORC1 and its downstream target S6 kinase, and the extent of phosphorylation of S6 can be used as a readout for mTORC1 activation as well as T cell help via CD40 stimulation (Ersching et al., 2017). To determine if DEC205+/+ GC B cells acquired more T cell help than DEC205+/− counterparts after αDEC205-OVA injection, we stained GC B cells for phospho-S6 12 h post treatment. Indeed, LZ DEC205+/+, DEC205+/−, and DEC205−/− GC B cells showed the highest, intermediate, and lowest amounts of phospho-S6, respectively, whereas all three populations had similar phosphorylation of S6 in the absence of treatment (Figure 2G). Therefore, we conclude that, in mixed GCs, both DEC205+/+ and DEC205+/− GC B cells were selected and capable of clonal expansion, although at different rate in line with distinct amounts of T cell help, in proportion to their surface cognate pMHCII.
CD40-CD40L signaling strength controls the magnitude of pMHCII-density-dependent clonal expansion
Having found that pMHCII density proportionally controls the GC B cell clonal expansion, we next sought to address which T cell signal(s) regulates this process. While B-T interactions in the GC involve many factors, both soluble and membrane bound, we hypothesized that the strength of CD40-CD40L signaling governs clonal expansion and acute interference would disrupt pMHCII-dependence of this process.
To identify the timing and duration of T cell help after αDEC205-OVA treatment, we imaged fluorescently labeled OT-II TFH cells and GC B cells in vivo using two-photon laser scanning microscopy (Figures S3A and S3B). We monitored T cell motility in the GC, as a proxy for T cell activation and B-T interactions, over time following αDEC205-OVA administration. TFH cell velocity kinetics were analyzed at 6, 12, 24, 48, and 72 h after treatment (Figure S3C). Before treatment, TFH cells were highly motile (9.6 μm/min); however, their velocities decreased by 41% and 37% at 6 and 12 h (5.7 and 6.0 μm/min, respectively) post treatment, suggesting that GC selection was probably ongoing during this time window, which was consistent with previous findings (Shulman et al., 2014). By 24 h, and through 72 h, TFH cell velocities had approached levels similar to untreated conditions, suggesting that TFH cell and GC B cell interactions had returned to baseline level (Figure S3C). These findings indicated selection interactions were short-lived, likely occurring during a 6–12 h window, and were completed before bursts in B cell proliferation and differentiation 48–72 h later.
To see if CD40 signal strength mediated pMHCII-dependent difference in proliferation, we evaluated how neutralizing CD40L signaling with anti-CD40L antibody (MR-1) (Han et al., 1995) augments αDEC205-OVA treatment (Figure 3A). As expected, i.v. treatment with anti-CD40L reduced the GC size, leading to a ~10-fold decrease in total GC B cell numbers as compared with isotype-treated control mice (Figure S3D). However in the presence of anti-CD40L, s.c. administration of αDEC205-OVA still provided an increase in DEC205+/+ and DEC205+/− GC B cell numbers in the treated leg in comparison with the other leg (Figures 3B and 3C). Interestingly, while in isotype-treated mice αDEC-OVA led to a 2-fold increase in DEC205+/+/DEC205+/− ratio (Figure 3D), as seen in controls (Figures 2D and 2E), in anti-CD40L-treated mice, DEC205+/+/DEC205+/− ratio did not increase after αDEC205-OVA targeting (Figure 3D), indicating that selection on the basis of pMHCII density is CD40L dependent. Delayed treatment with anti-CD40L at 24 h similarly reduced GC numbers (Figure S3D), but did not disrupt the αDEC-OVA-mediated increase in DEC205+/+/DEC205+/− ratio. This result indicates that αDEC205-OVA-mediated T cell help was temporal rather than chronic and help had already been provided by 24 h, consistent with kinetics of B-T cell contacts over time (Figure S3C).
Figure 3. CD40-CD40L signaling strength controls the magnitude of pMHCII-density-dependent clonal expansion.
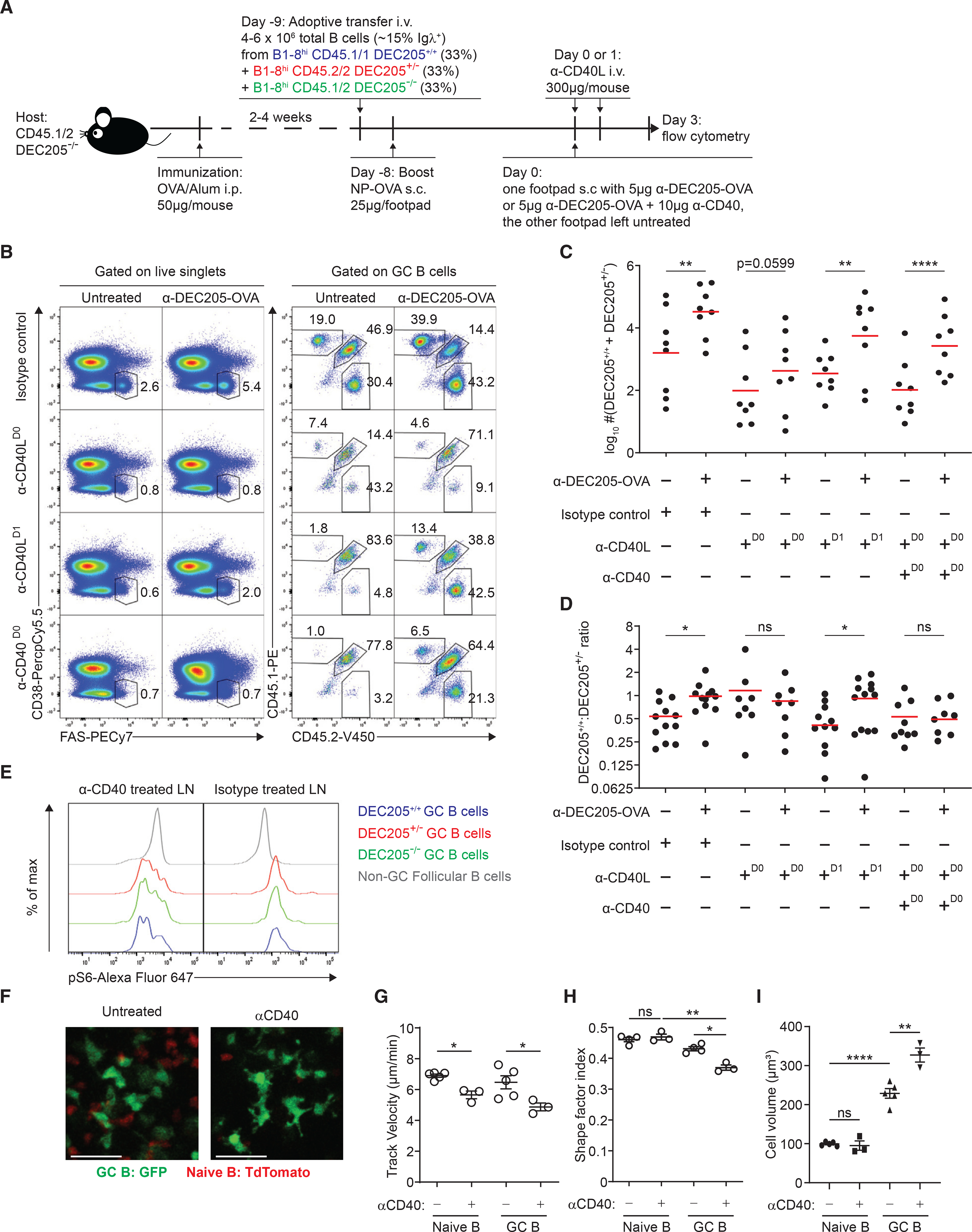
(A) Experimental setup for direct competition of GC B cells with differential pMHCII density in the presence of CD40-CD40L signaling stimulus or blockade by using agonist αCD40 or antagonist αCD40L.
(B) Representative plot of total GC B cell frequency and frequency of DEC205+/+, DEC205+/−, and DEC205−/− GC. B cells within the GC in the presence of agonist αCD40, antagonist αCD40L, or isotype control with or without αDEC-OVA.
(C) Log-transformed sum of DEC205+/+ and DEC205+/− GC B cell absolute numbers in the presence of agonist αCD40, antagonist αCD40L, or isotype control 3 days after αDEC-OVA treatment.
(D) DEC205+/+/DEC205+/− ratio by GC B cell absolute numbers in the presence of agonist αCD40, antagonist αCD40L, or isotype control 3 days after αDEC-OVA treatment.
(E) Phospho-S6 staining of DEC205+/+, DEC205+/−, and DEC205−/− GC B cells in mice treated with stimulating αCD40 compared with isotype-treated controls.
(F) Example two-photon images of GFP+ GC B cells (green) and tdTomato+ naive B cells in the pLN, with or without agonist anti-CD40. Scale bars, 27 μm.
(G–I) (G) Track velocity, (H) shape factor, and (I) cell volume of transferred GFP+ GC B cells (green) and endogenous tdTomato+ naive B cells (red) with or without agonist anti-CD40 in the GC. All bars show mean (C and D) or mean ± SEM (G–I). *p < 0.05; **p < 0.01; ****p < 0.0001; ns, non-significant or exact p values shown by paired (C and D, G–I) or unpaired Student’s t test (H–I). All graphs show pooled data from at least two independent experiments. (C and D) n = 7–9; (G) n = 3–5; (H) n = 3–4; (I) n = 3–5.
These results prompted us to investigate whether pMHCII-dependent B cell expansion rate is instructed on the basis of differential CD40-CD40L signal strength. Alternatively, CD40 signaling could be a pre-requisite costimulatory factor that acts in concert with other T cell signals to control differential B cell selection. To resolve these two possibilities, we tested if delivering equivalent CD40 signaling to GC B cells, during αDEC205-OVA-driven T cell selection, would also eliminate differential expansion on the basis of pMHCII. First, we tried to saturate GC B cells with increasing concentrations of agonist anti-CD40 (1C10) in the presence of αDEC205-OVA treatment but found the effects on GC B expansion were always additive even at the highest levels of antibody (data not shown). Therefore, to deliver equal levels of CD40 signaling, we combined both anti-CD40L (i.v.) blockade, with a dual s.c. injection of agonist anti-CD40 and αDEC205-OVA to one footpad only (Figure 3A). Sufficient and equal CD40 signaling throughout all GC B cells was confirmed by a similar amount of ribosomal protein phospho-S6 in DEC205+/+, DEC205+/−, and DEC205−/− GC B cells (Figure 3E). Agonist anti-CD40 increased the absolute number of DEC205+/+ and DEC205+/− GC B cells compared with mice treated with αDEC205-OVA alone in the presence of blocking anti-CD40L, suggesting a synergistic effect on clonal expansion (Figure 3C). Interestingly, in stimulating anti-CD40 treated mice, the DEC205+/+/DEC205+/− ratio was not increased after αDEC205-OVA targeting, consistent with effects of blocking signaling with anti-CD40L alone (Figure 3D). Together, we conclude that CD40-CD40L signaling strength is both necessary and sufficient for controlling magnitude of pMHCII-density-dependent clonal expansion.
GC B cell dynamics and morphology are intimately linked with the efficiency of T cell recognition in the GC. GC B and T cells have reduced motility compared with naive counterparts, also exhibit a deformed, elongated cell body with a characteristically long uropod, possibly facilitating cell contacts (Allen et al., 2007a; Schwickert et al., 2007). Previous work demonstrated that αDEC205-OVA-triggered selection decreased B and T cell speeds during and/or following selection and increased deformation of the cell shape (Shulman et al., 2013). To see if this was CD40-mediated, we imaged GC B cells 24 h after agonist anti-CD40 stimulation and observed decreased B cell track velocity and increased GC B cell size and deformation of the cell shape (Figures 3F–3I). While systemic CD40 stimulation may have off-target effects in vivo, B cell blasts stimulated with anti-CD40 in vitro exhibited similar changes in speed and morphology when imaged on ICAM-1-coated substrates (Figure S3E–S3G), suggesting that CD40 signaling could directly alter GC B cell dynamics and shape, similar to T cell help. Thus, in addition to promoting expansion, CD40 signaling directly promotes the “GC-like” B cell morphology and motility pattern, which may support signal integration.
Generation and characterization of a caged OVA peptide
Previous intravital imaging has shown that, in the DEC model, the increased duration of B-T interactions happened several hours following DEC205-OVA, concomitant with decreases in B and T cell motility (Shulman et al., 2014). Since antigen processing and presentation occurs over hours, it is unclear if these observed longer contacts represent the initial recognition of increased pMHCII density or an integration of multiple rounds of interactions and CD40 signaling, thus leading to the reduced cell motility. Thus, to directly test if T cells can rapidly and directly detect changes in pMHCII density on a per cell basis, in the GC, we developed a second approach to acutely and dynamically manipulate pMHCII in situ in an ongoing GC. We developed a caged version of the model OVA peptide, recognized by OT-II T cells, which can regulate GC B cell selection following OVA-based immunogens. Based on previous studies (Robertson et al., 2000), OT-II TCR recognizes OVA323–339 peptide in the context of I-Ab MHC class II molecules, with E333 and H331 as the key peptide residues that are required for TCR recognition but not for MHC II binding. We generated a variant of the OVA323–339 peptide, replacing glutamate at position 333 with a caged glutamate containing a photo-labile nitroindoline blocking group on the side chain (Sigma G3291). We posited that this caged OVA peptide (cOVA) should still be able to bind into the I-Ab pocket, but would not be recognized by OT-II TCR unless the blocking group were removed, by UV irradiation.
To validate this system, (I-Ab) B cell blasts pre-loaded with limiting dilutions of peptide were co-cultured with CFSE-labeled tdTomato+ OT-II T cell blasts. On the basis of CFSE dilution, OT-II T cells recognized the conventional OVA peptide but not the cOVA peptide, indicating that the caging group was robustly conjugated to the peptide (Figures 4A and 4B). However, when cOVA peptide was pre-treated by UV light in vitro to induce uncaging, the uncaged cOVA (uOVA) peptide was now able to induce comparable CFSE dilution of OT-II T cells to conventional OVA peptide. Similarly, injection of the uOVA peptide was capable of expanding OT-II cells in vivo, but not the cOVA peptide, indicating that the photo-labile blocking group was stable in vivo as well (Figures 4C and 4D). To test if antigen recognition could be modulated based on the level of photouncaging, peptide-loaded B blasts were exposed to varying amounts of UV light and then co-cultured with tdTomato+ OT-II blasts to assess B-T conjugate formation. UV illumination had no effect on conjugate formation in the absence of peptide or in the presence of conventional OVA peptide (Figures 4E and 4F). However, cOVA peptide-loaded B cell blasts formed B-T conjugates with increasing UV light and, importantly, no conjugates were formed in the absence of any UV irradiation (Figures 4E and 4F). To determine if photo-cleavage had a cell-intrinsic effect on antigen recognition, we plated peptide-loaded photoactivatable GFP (PA-GFP)-expressing B cell blasts and tdTomato+ OT-II blasts on ICAM-1-coated substrates, which allows for cell migration and interactions. A central region of interest was photoactivated with UV laser scanning microscopy, resulting in photoconverting (PA-GFP) GFP− B cells to GFP+ (green) and concomitantly uncaging cOVA peptides loaded only on these cells as well (Figure 4G), leading to increased contact durations with OT-II T cells (Figure 4H) immediately after contact and accumulating over time compared with conditions no peptide present (Figure 4I, Videos S1 and S2). As B-T cell conjugates formed rapidly upon irradiation, it is likely that these cOVA peptides must have already been loaded on surface MHCII molecules prior to uncaging. Taken together, cOVA peptides can be readily recognized by OT-II T cells in vitro upon UV-mediated photo-cleavage.
Figure 4. Generation and characterization of a caged OVA peptide.
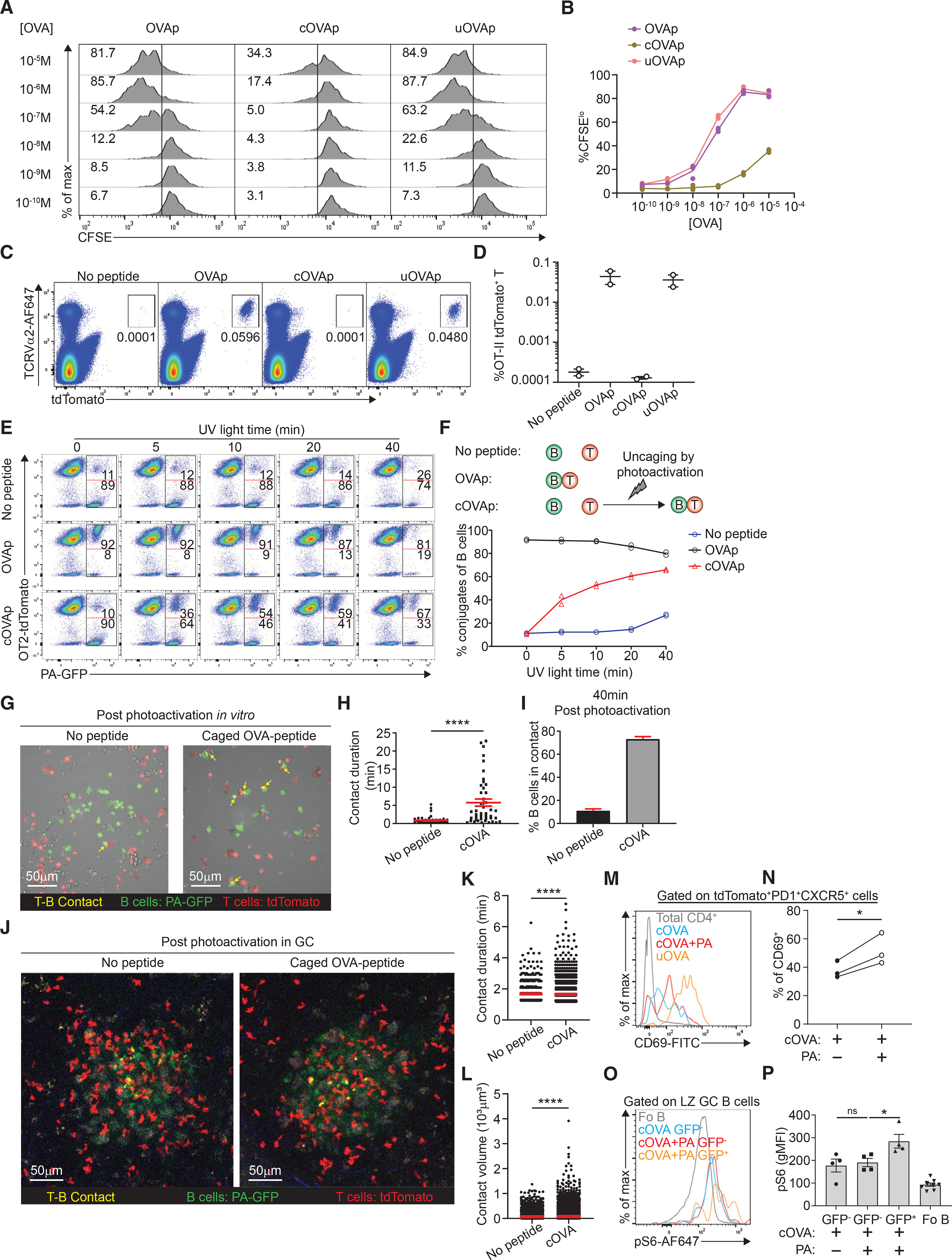
(A and B) (A) In vitro OT-II T cell proliferation measured by CFSE dilution co-cultured with B cell blasts incubated with varying concentration of OVA peptide, caged OVA peptide (cOVAp), or uncaged OVA peptide (uOVAp). Numbers in the gating show the frequency of OT-II T cells that are CFSElo, which is quantified in (B).
(C and D) (C) Expansion of transferred OT-II tdTomato+ T cells following treatment with no peptide, OVA peptide, caged OVA peptide (cOVAp), or uncaged OVA peptide (uOVAp), Numbers in the gating show the frequency of OT-II T cells of live singlets, which is quantified in (D).
(E) Representative plot of B-T cell conjugate formation in the presence of no peptide, OVA peptide, or caged OVA peptide (cOVAp) after photoactivation with increasing exposure time of UV light in vitro. Numbers in the gating show the frequency of PA-GFP+ B cells that formed conjugates with OT-II tdTomato+ T cells on the top versus B cells that did not form conjugates at the bottom.
(F) Schematic for B-T conjugate formation assay in the various conditions (upper panel), with the quantification of frequency of B cells that formed conjugates (lower panel).
(G) Example of single time point snapshot of dynamics of TFH cell (red) and B cell (green) interactions in the presence of no peptide or caged OVA peptide (cOVAp) post photoactivation by two-photon laser scanning microscopy in vitro.
(H) T-B contact duration in the presence of no peptide or caged OVA peptide (cOVAp) post photoactivation.
(I), Frequency of B cells in contact in the presence of no peptide or caged OVA peptide (cOVAp) 40 min post photoactivation. </p/>(J) Example of single-time point snapshot of dynamics of TFH cell (red) and B cell (green) interactions in the presence of no peptide or caged OVA peptide (cOVAp) post photoactivation by two-photon laser scanning microscopy in the GC. </p/>(K and L) (K) T-B contact duration and (L) T-B contact volume in the presence of no peptide or caged OVA peptide (cOVAp) post photoactivation in the GC. </p/>(M and N) (M) A representative histogram of CD69 surface expression on tdTomato+ OT-II TFH cells in the presence of caged OVA peptide (cOVAp), uncaged OVA peptide (uOVAp) in vitro with UV light, or uncaged OVA peptide (uOVAp) in the GC by two-photon excitation, which is quantified in (N).
(O and P) (O) A representative histogram of phospho-S6 level in non-GC follicular B cells, GFP− (not photoactivated) GC B cells in the presence of caged OVA peptide (cOVAp), GFP− (not photoactivated) GC B cells in the presence of caged OVA peptide (cOVAp) after photo-uncaging, and GFP+ (photoactivated) GC B cells in the presence of caged OVA peptide (cOVAp) after photo-uncaging, which is quantified in (P).
All bars show mean (K and L) or mean ± SEM (D, H, I, and P). *p < 0.05; ****p < 0.0001; ns, non-significant by Mann-Whitney U test (H, K, and L), unpaired Student’s t test (H–I), or one-way ANOVA with Dunnett’s test (P). All data are representative of two independent experiments (B, D, F, H, I, M, and O) or pooled data from two (K, L, N, and P) independent experiments. (B) n = 2; (D) n = 2; (F) n = 2; (H and I) n = 1; (K and L) n = 2; (N) n = 3; (P) n = 4–8.
To see if we could trigger uncaging in the GC, we transferred OT-II T cells and NP-specific B1–8hi PA-GFP+ B cells, and immunized with NP-OVA s.c. in the footpad. After GC formation, cOVA peptide was injected s.c. in the same footpad and photoactivated in a sub-region (~20%) of the GC using two-photon laser scanning microscopy to simulate the energy of a single UV photon. Using this approach, we were able to uncage cOVA molecules and photo-convert PA-GFP into GFP in the same volume, simultaneously, thus marking GC B cells that had been in situ peptide loaded. Following photoactivation, we analyzed GFP+ redistributed around the GC once (Figure 4J). Photoactivated (GFP+), peptide-loaded uncaged GC B cells exhibited longer and larger contacts with T cells compared with control photoactivated GCs with no peptide uncaging or no peptide loading (Figures 4K and 4L, Videos S3 and S4). We also noted that GFP+ peptide-loaded B cells engaged in serial interactions with multiple T cells (Video S4), rather than long monogamous interactions reported with DEC targeting. These results indicate that TFH cells can rapidly sense and respond to cell-intrinsic changes in pMHCII in the GC.
To verify that these uOVA peptides were functionally relevant, we photoactivated all GCs and analyzed OT-II TFH cell activation 6 h later. We found CD69 upregulation on OT-II TFH cells after photo-uncaging in the GCs compared with control LNs treated with cOVA without photoactivation (Figures 4M and 4N), indicating that OVA-specific T cell activation was increased. Analogously, we found that photoactivated GFP+ GC B cells had higher pS6 levels compared with GFP− GC B cells in the same LN (Figures 4O and 4P), indicating that photo-cleavage of cOVA led to a cell-intrinsic enhancement of T cell help. As photo-converted GFP molecules decay rapidly in GC B cells (Victora et al., 2010), we were unable to follow GFP+ cells any further, longitudinally.
pMHCII density contributes to PB expansion but not differentiation rate or maturation
One important fate of positively selected GC B cells is differentiation into PBs (IRF4hi Blimp1hi CD138hi), which requires the signal from BCR itself and signals provided by TFH cells (Krautler et al., 2017). Since T cell help, which is based on cognate pMHCII interaction, is the limiting factor for positive selection of GC B cells (Victora et al., 2010), we next sought to investigate whether differential levels of pMHCII density can instruct PB commitment of GC B cells. The transcription factor IRF4 is essential and highly expressed in GC B cells during PB differentiation, while the transcription factor IRF8 acts antagonistically, inhibiting PB differentiation (Sciammas et al., 2006; Xu et al., 2015). Thus, we tested if GC B cells’ commitment to PB precursor fate might be regulated by surface pMHCII density using the same competitive transfer model (as shown in Figure 2A). We monitored PB differentiation commitment based on the frequency of IRF8loIRF4hi in the GC B cell compartment, which are also Bcl6lo (Figure S4A), consistent with other reports (Ise et al., 2018). Treatment with αDEC205-OVA had no effect on the frequency of PB precursors per subset, compared with untreated, which averaged at ~1% of GC B cells (Figures 5B and 5C). Next, we assessed if PB proliferation rates were dependent on pMHCII density using EdU incorporation by both DEC205+/+ and DEC205+/− IRF8loIRF4hi GC B cells 48 h post treatment. However, the frequency of EdU+ IRF8loIRF4hi GC B cells was indistinguishable between DEC205+/+ and DEC205+/− populations from 24 to 72 h post treatment (Figures 5D and 5E), suggesting that pMHCII density does not drive proportional PB expansion as in the GC pool.
Figure 5. Plasmablast and plasmablast precursor differentiation rate and output efficiency are independent of pMHCII density.
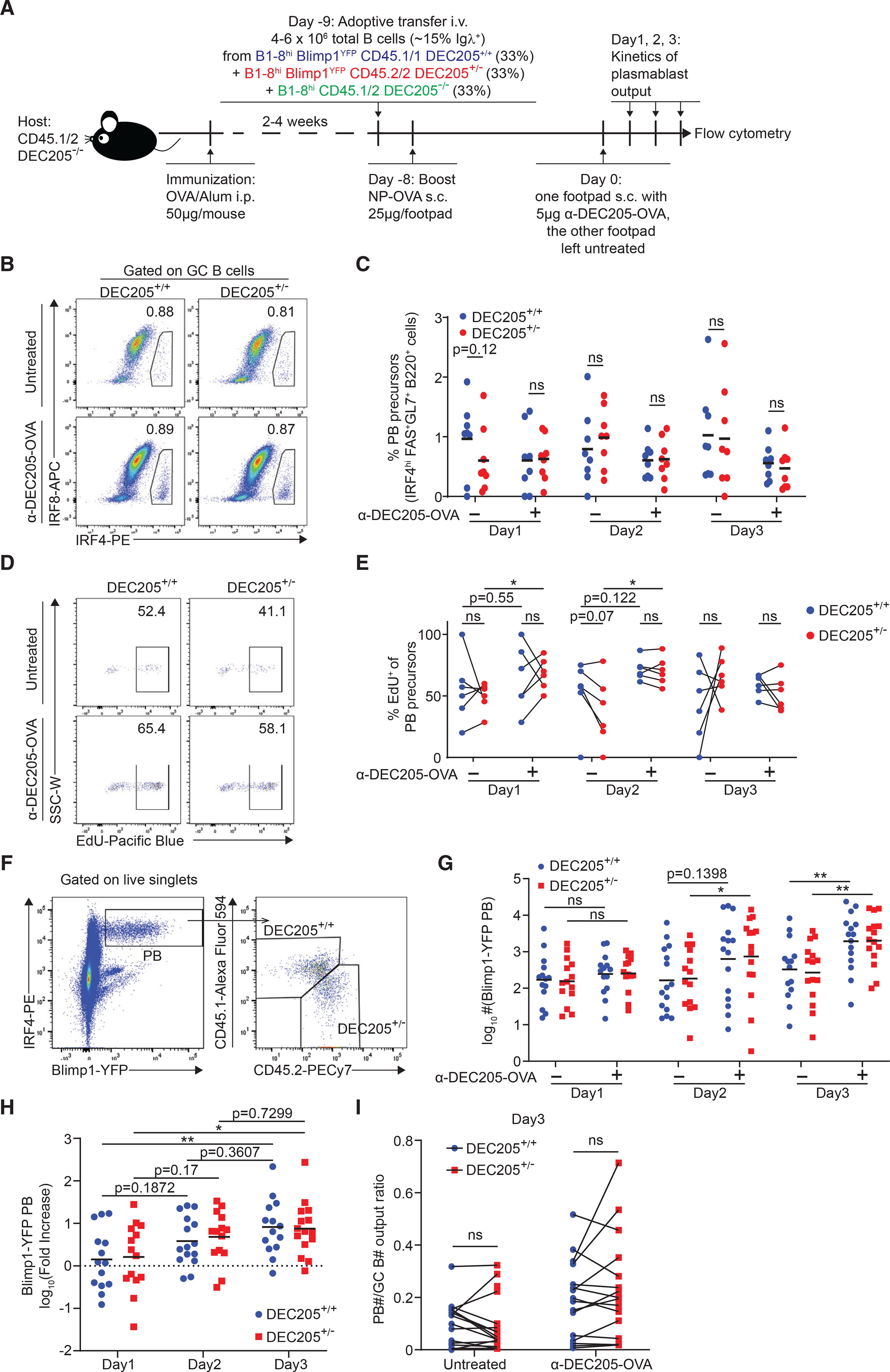
(A) Experimental setup used in (B–H).
(B) Representative plot of IRF4hiIRF8lo plasmablast precursor frequency of DEC205+/+ and DEC205+/− GC B cells in αDEC-OVA-treated footpad compared with untreated contralateral footpad.
(C) Kinetics of plasmablast precursor frequency of GC B cells after αDEC-OVA treatment compared with untreated control footpad.
(D) Representative plot of percentage of EdU incorporation of DEC205+/+ and DEC205+/− GC B cell plasmablast precursor.
(E) Kinetics of EdU incorporation frequency of DEC205+/+ and DEC205+/− GC B cell plasmablast precursor after αDEC-OVA treatment compared with untreated control footpad.
(F) Representative plot of gating on Blimp1-YFP+IRF4hi plasmablasts generated from DEC205+/+ and DEC205+/− GC B cells.
(G) Kinetics of log-transformed plasmablast absolute numbers after αDEC-OVA treatment compared with untreated control footpad.
(H) Kinetics of αDEC-OVA/untreated fold increase ratio of plasmablast after αDEC-OVA treatment.
(I) Plasmablast/GC B cell output ratio 3 days post αDEC-OVA treatment compared with untreated control footpad.
All bars show mean (C, G, and H). *p < 0.05; **p < 0.01; ns, non-significant or exact p values by paired Student’s t test (C, E, G, and I) or unpaired Student’s t test (H). All graphs show pooled data from at least two independent experiments. (C) n = 8; (E) n = 6; (G and H) n = 14–15; (I) n = 14.
Since previous work had shown αDEC205-OVA treatment could increase CD138high PBs (Victora et al., 2010), we tested if PB output depended strictly on pMHCII density. We performed adoptive transfer experiments, as before, but using donor DEC205+/+ and DEC205+/− that also co-expressed Blimp1-YFP, which express YFP after PB differentiation (Fooksman et al., 2010) (Figures 5A and 5F). The absolute number of DEC205+/+ and DEC205+/− Blimp1-YFP+ PBs started to expand 2 days post treatment, and maintained similar fold increase 3 days post treatment, compared with the untreated footpad (Figures 5G and 5H), suggesting increased PB output after αDEC205-OVA treatment. Although GC B cells with higher pMHCII density would receive more T cell help, both DEC205+/+ and DEC205+/− PB exhibited similar expansion as the size of their GC B cell pools with or without αDEC205-OVA treatment, indicating a similar efficiency of PB output irrespective of pMHCII level (Figure 5I).
GC-derived PBs preferentially migrate to bone marrow and can mature into long-lived PCs providing lifelong production of antibodies (Weisel et al., 2016). To assess if pMHCII density, and thus T cell help, alters the quality of PBs produced, we assessed their migration to spleen and bone marrow. Both DEC205+/+ and DEC205+/− YFP+ PBs were present in the draining lymph nodes, spleen, and bone marrow by flow cytometry 7 days after PBS or αDEC205-OVA treatment (Figures S5A–S5C). The PB migration efficiency to bone marrow or spleen was quantified by the migration index, as determined by the ratio of total numbers of bone marrow or spleen YFP+ PBs to that of draining lymph node YFP+ PBs. There was no difference between the migration index (spleen or bone marrow) of DEC205+/+ and DEC205+/− YFP+ PB in mice treated with PBS or αDEC205-OVA (Figures S5D and S5E). Taken together, these results indicate that, while T cell help supports PB production, pMHCII density on GC B cells is not directly regulating the efficiency of PB differentiation, expansion, or PB capacity to migrate to survival niches.
We reasoned that similar rate of PB differentiation in our model could come about as a result of all the B cells expressing the same high-affinity BCR, B1–8hi, which has a 10-fold higher affinity for NP than unmutated B1–8 BCR (Krautler et al., 2017; Schwickert et al., 2011). To explore the possible synergy between pMHCII complex density and BCR affinity, we immunized WT (polyclonal) mice with OVA/Alum for 4 weeks and boosted the draining lymph nodes with NP-OVA. Eight days after boost, mouse footpads were treated with either PBS or αDEC205-OVA for 3 days and IgG1-switched IRF8loIRF4hi GC B PB precursors were subdivided into high- (NPhi) and low-affinity (NPlo) NP-specific B cells on the basis of NP-PE staining (Figures 6A and 6B). More NPhi GC B cells differentiated into IRF8loIRF4hi PB precursors than NPlo GC B cells at day 8 post NP-OVA boost (Figures 6B and 6C), which is consistent with previous reports showing that high-affinity GC B cells preferentially differentiate into early PBs (Krautler et al., 2017; Phan et al., 2006). Treatment of WT mice with αDEC205-OVA should target all GC B cells (and other cells, such as DCs) and, thus, both NPhi and NPlo GC B cells expanded after induced T cell selection by αDEC205-OVA treatment compared with PBS treated mice (Figure 6D) but there was similar fold increase after αDEC205-OVA treatment based on ratio of GC B cell numbers after αDEC205-OVA treatment to that after PBS treatment (Figure 6E), suggesting that αDEC205-OVA targeting indeed promoted similar selection and expansion of both NPhi and NPlo GC B cells. We next asked whether equal T cell help conferred by DEC205 targeting promotes similar PB precursor differentiation of NPhi and NPlo GC B cells. As expected, both NPhi and NPlo GC B cells exhibited significantly increased differentiation into IRF8loIRF4hi PB precursors after αDEC205-OVA treatment compared with PBS-treated mice (Figure 6F). However, there was no significant difference in magnitude of fold increase of differentiation into IRF8loIRF4hi PB precursors between NPhi and NPlo GC B cells (Figure 6G), suggesting that equalized pMHCII density equally promotes PB differentiation of high- and low-affinity NP-specific GC B cells.
Figure 6. pMHCII density promotes plasmablast precursor differentiation of both high- and low-affinity NP-specific GC B cells in polyclonal GC responses.
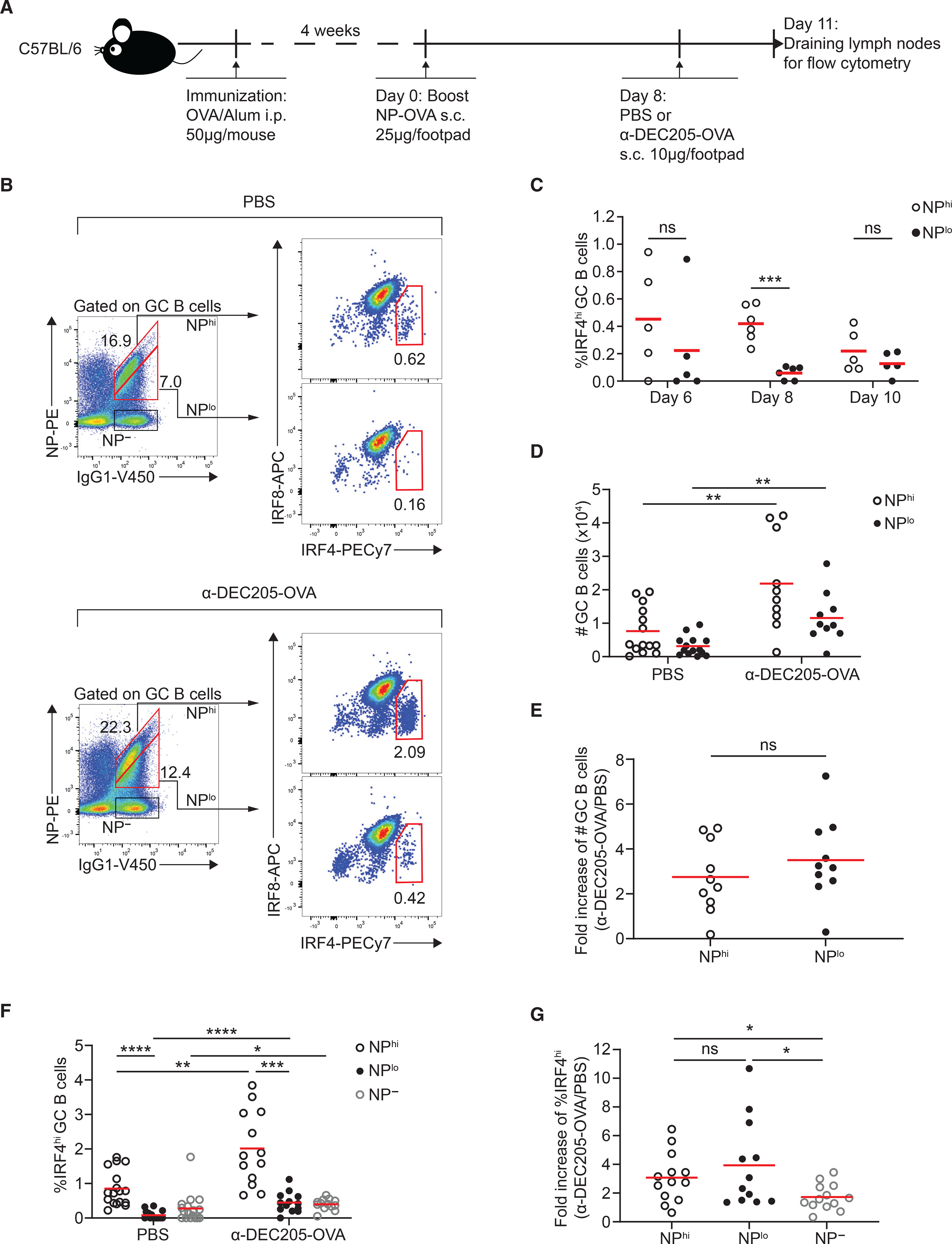
(A) Experimental setup for (B–G).
(B) Representative plot of high- and low-affinity NP-specific IgG1-switched GC B cells and further gated for IRF4hi plasmablast precursors in the presence of PBS (upper panel) or αDEC-OVA (lower panel). Numbers show frequency.
(C) Kinetics of IRF4hi plasmablast precursors’ frequency of high- and low-affinity NP-specific IgG1-switched GC B cells 6–10 days after boost.
(D) Absolute numbers of high- and low-affinity NP-specific IgG1-switched GC B cells in PBS or αDEC-OVA.
(E) Fold increase of high- and low-affinity NP-specific IgG1-switched GC B cell numbers after αDEC205-OVA treatment is determined by the ratio of GC B cell numbers after αDEC205-OVA treatment to that after PBS treatment.
(F) Frequency of IRF4hi plasmablast precursors of high- and low-affinity NP-specific IgG1-switched GC B cells as well as non-NP-binding IgG1-switched GC B cells in the presence of PBS or αDEC-OVA treatment.
(G) Fold increase of IRF4hi plasmablast precursors’ frequency of high- and low-affinity NP-specific IgG1-switched GC B cells as well as non-NP-binding IgG1-switched GC B cells is determined by the ratio of IRF4hi plasmablast precursors’ frequency after αDEC205-OVA treatment to that after PBS treatment.
All bars show mean. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; ns, non-significant by Mann-Whitney U test (C, D, F, and G) or paired Student’s t test (E and F). All graphs show pooled data from at least two independent experiments. (C) n = 5–6; (D) n = 10–14; (E) n = 10; (F) n = 13–17; (G) n = 12–13.
DISCUSSION
GC selection for B cells with high affinity for a diversity of epitopes is essential for an efficient and cross-protective immune protection against mutating pathogens. At the center of this selection process are B-T interactions and bi-directional signaling that regulate clonal competition while providing sufficient slack in the system to allow intermediate clones to evolve over time.
Our results on the role of pMHCII density may help resolve a disconnect between two previous lines of inquiry on its role. Previous work from Gitlin et al. (2014) showed, using a DEC-205 targeting model, that T cell help and B cell proliferation correlated with antigen dose when compared with DEC205−/− B cells that received no antigen targeting at all. However, they did not resolve level of T cell sensitivity to varying levels of pMHCII in a calibrated manner. In contrast, Yeh et al. (2018) showed that MHCII+/+ outnumber MHCII+/− B cells for GC entry by 2-fold, presenting 2-fold more cognate antigen at 16 h post immunization but, nevertheless, MHC+/− were not lost in GC over time or defective in affinity maturation, suggesting that TFH cells in the GC are not sensitive to 2-fold differences in pMHCII levels. In our study, we find that, by unequal targeting of antigen to subsets of B cells in situ in the GC, we are able to modulate a 2-fold difference in pMHCII in the GC leading to proportional changes in T cell help and proliferation. One explanation that may explain the difference we obtain may be due to the levels of antigen. At early hours after immunization, antigen levels are peaking, possibly even saturating antigen processing, leading to differences in pMHCII and T cell selection on the basis of MHC genotype. However, at the GC stage, where antigen levels are substantially lower, MHC molecules may no longer be a limiting factor for efficient peptide loading and thus permissive to presenting similar amounts of pMHCII complexes in WT versus MHC+/− GC B cells. In our model, we can artificially modulate pMHCII levels directly in the GC, thus showing that T cells can indeed discern a 2-fold difference in the peptide at this stage. We would posit that, if Yeh et al. could measure pMHCII in the GC in their model, they would find similar levels on B cells from both MHC genotypes, consistent with their results. While antigen in the GC may be limiting following protein immunizations, the same may not be true for GCs with replicating pathogens or in autoimmune models, where self-antigen is abundant. In such cases, T cell help is likely driving selection on the basis of pMHCII.
Signaling through CD40L-CD40 is essential for GC reaction and disrupting it quickly ablates the GC, affinity maturation, and PC differentiation (Han et al., 1995). CD40L may engage CD40 in the extensive junctions between TFH cells and GC B cells, and our results suggest that both TCR-pMHCII and CD40L-CD40 contribute to expansion of the T-B synapsis. While CD40 signaling stimulates upregulation of adhesion molecules like ICAM-1 on B cells and DC, its ability to mediate inside-out activation of LFA-1 has only been hinted at previously (Mayumi et al., 1995). CD40L may also be transferred to B cells (Gardell and Parker, 2017) in extracellular vesicles (Saliba et al., 2019). While CD40 signaling clearly is necessary for the GC, correlates with selection (Victora et al., 2010) and is sufficient to drive PB differentiation (Noelle et al., 1992), it was one of many signals transmitted between B and T cells in the GC, including cytokines, and other costimulatory molecules. However, we find that CD40 signal strength is the rheostat of pMHCII density, necessary and sufficient to instruct levels of B cell proliferation differences, via mTOR-driven MYC production (Finkin et al., 2019). Blocking CD40L or equalizing CD40 signaling was able to block any B cell proliferation increases on the basis of pMHCII density.
Nevertheless, despite more efficient proliferation, WT B cells were not able to fully diminish DEC205+/− B cell clones from the GC with repeated αDEC205, demonstrating that T cell help is not limited to high-affinity clones only, but to any pMHCII-bearing B cells. This is consistent with studies demonstrating a permissive nature of GC selection as reported in response to complex antigen immunization (Kuraoka et al., 2016). This permissive nature undoubtedly contributes to the development and persistence of a small fraction of broadly neutralizing antibodies (bNAbs) in patients infected with HIV or influenza (Johnston and Fauci, 2011; Pappas et al., 2014). The B cell clones expressing bNAbs are usually directed against subdominant epitopes and appeared in a significant delay in the course of infection (Mascola and Haynes, 2013; Pappas et al., 2014).
How are T cells able to reconcile and respond proportionally to 2-fold differences in pMHCII? Do they count? Our photoactivatable peptide imaging suggests that multiple cognate TFH cells can contact pMHCII-loaded B cells, in addition to increased durations observed after DEC205 targeting. These serial encounters would be compatible with B cells presenting a mix of different pMHCII detectable by various cognate TFH cells, thus leading to integration of multiple T cell help signals. In addition, CD40 signaling was accompanied by the many morphological and dynamic features that distinguish GC B cells. These changes may further facilitate subsequent contacts by additional polyclonal TFH cells migrating and scanning for cognate pMHCII complexes in a dynamic and heterogenous landscape, leading to more signal integration through cMYC (Finkin et al., 2019). Since, potentially, multiple T cell interactions are integrating into CD40 signal strength, which is proportional to pMHCII, it is essential that these detected pMHCII molecules are either removed by the T cells via trogocytosis (Zhou et al., 2011) as blebs (Allen et al., 2007b), or internally degraded in B cell lysosomes following recognition (Bannard et al., 2016) to avoid double counting pMHC complexes. Interestingly, these essential B-T interactions were early (occurring only within the first few hours after DEC205 or seconds after peptide activation) and temporally restricted, yet the effects on proliferation and differentiation took days to accumulate and complete. These long delays may provide opportunities for other, intermediate B cell clones to also gain access to T cell help.
Finally, despite increased CD40 signaling, we did not observe any pMHCII-density-dependent increase in the propensity of GC B cells to differentiate into PBs. While we cannot conclude that pMHCII density is instructive in PB differentiation, it is clearly a pre-requisite, and enhances overall PB numbers following DEC205 targeting. Potentially, significant variance in the data could be obscuring a pMHCII-dependent role in PB differentiation. However, our data are in line with previous findings that showed that BCR signaling is important for PB differentiation (Krautler et al., 2017). Indeed, in GCs with polyclonal NP-specific B cells, we found high-affinity NP-specific GC B cells preferentially differentiated into IRF8loIRF4hi PB precursors compared with low-affinity NP-specific GC B cells. Other signals during TFH cell-GC B cell interaction including ICOS-ICOSL signaling and IL21-pSTAT3 signaling have also been shown to regulate PB differentiation (Ise et al., 2018; Liu et al., 2015; Weinstein et al., 2016). Alternatively, stochastic processes, such as asymmetric cell division (Lin et al., 2015), may underlie differentiation, while higher proliferation on the basis of pMHCII may ultimately lead to more PBs indirectly through expansion of high-affinity clones in the GC.
Nevertheless, we did not see any qualitative differences in PC differentiation or homing, suggesting that all of these clones may have equal potential to contribute to long-lived immune protection regardless of pMHCII density. These lower-affinity clones are often cross-reactive and broadly neutralizing in the case of influenza, and thus may be important for protection in secondary infections.
Limitations of the study
This study of immune responses following vaccination was conducted in mice only, using a single NP-OVA model antigen with alum adjuvant only to track antigen-specific responses. Thus, it is unclear how generalizable these immune regulatory mechanisms are for other types of vaccinations, infections, or in humans. Higher levels of pMHCII density in the GC through DEC205 targeting or caged peptide injections used here may not be physiologically relevant to normal vaccinations. Thus, additional studies using titrating levels of peptide would be needed to see if these findings hold at other concentrations. While we investigated the role of CD40 and its implication in reading out pMHCII density, this may not be unique to CD40 signaling. We did not explore cytokines (IL-21, IL-4, etc.) or costimulatory surface molecules (B7 family members, integrins) that are implicated in GC reaction. Potentially CD40 signaling may not be unique.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, David R. Fooksman (david.fooksman@einsteinmed.edu).
Materials availability
[DEC-OVA plasmids, DEC-OVA-Ealpha plasmids, and caged OVA-peptide] generated in this study will be made available on request, but we may require a payment and/or a completed Materials Transfer Agreement if there is potential for commercial application.
Data and code availability
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
HEK293T cells were revived from liquid nitrogen by defrosting in the 37°C water bath and added dropwise to DMEM+5%FBS (Gibco) prewarmed at 37°C followed by spin down, resuspension, and transferred to 100 mm × 20 mm plate. Cells were incubated in 37°C at 5% CO2 for 2–3 days until it reached over 90% confluency. The cells were splitted at 1:2 ratio when it reached 70% confluency before transfection.
Mice
B1–8hi mice (Shih et al., 2002), DEC205−/− mice (Guo et al., 2000), and Blimp1-YFP mice (Fooksman et al., 2010) were generated previously and bred in-house, which can also be obtained from the Jackson Laboratory. To generate DEC205+/− mice, C57BL/6 (DEC205+/+) mice were bred to DEC205−/− mice. ECFP mice, tdTomato mice, OT-II mice, and PA-GFP mice were purchased from The Jackson Laboratory. C57BL/6 (CD45.2) and B6-Ly5.1/Cr (CD45.1) mice were purchased from Charles River. All mice were housed in groups of 2–5 animals per cage in specific pathogen-free facilities at Albert Einstein College of Medicine. The animal protocol in this study was approved by Albert Einstein College of Medicine Institutional Animal Care Use Committee. For adoptive transfer experiments, host mice including both males and females that are 6–10 weeks old were used, and donor mice including both males and females that are 8–16 weeks old were used. For other experiments, age- and sex-matched males and females, 6–8-week-old mice were used.
METHOD DETAILS
Construction and production of chimeric antibodies, peptides
The cDNA encoding Eα (52–68) peptide flanked by overlap sequences was synthesized (IDT), cloned in frame with the carboxyl terminus of the heavy chain of the chimeric antibody αDEC-OVA (Boscardin et al., 2006) by NEBuilder HiFi DNA Assembly Cloning Kit (New England Biolabs), and confirmed by sequencing (GeneWiz). Chimeric antbodies αDEC-OVA-Eα and αDEC-OVA were transiently expressed in 293T cells after transfection using calcium-phosphate. Cells were grown in serum-free DMEM supplemented with Nutridoma SP (Roche) at 37°C, 5% CO2 in a humidified incubator for 5 days. Supernatant were collected for antibody purification using Protein G Sepharose Fast Flow (GE Healthcare Life Sciences) followed by buffer exchange in pH 7.4 PBS using Amicon Ultra-15 centrifugal filter unit (50 KDa, MilliporeSigma). The concentrations of purified antibodies were determined by NanoDrop.
For Figure 4 experiments, OVA caged peptide, and control OVA peptide was synthesized by Dr. Henry Zebroski in the proteomics core at Rockefeller University using standard FMOC peptide synthesis to generate GSGEEFAKFISQAVHAAHAXINEAGR peptide using a caged glutamate (Sigma G3291) substituted at (Q) position.
Adoptive transfers, immunizations, and treatments
For adoptive transfer experiments, spleen and lymph nodes harvested from donor mice were pressed through a 70 μm nylon filter in PBS containing 0.5% BSA and 1 mM EDTA followed by red blood cell lysis using ACK buffer (Lonza). Naïve B cells were then purified by magnetic cell separation (MACS) using anti-CD43 beads (Miltenyi Biotec) according to manufacturer’s protocol. Prior to adoptive transfer, ratio of B1–8hi cells in the mixture of DEC205+/+, DEC205+/−, and DEC205−/− cells were determined by flow cytometry after staining an aliquot of mixture with NP(19)-PE (Biosearch Technologies) and antibody for surface markers including B220, IgMa, CD45.1, and CD45.2. Recipient mice were immunized intraperitoneally (i.p.) with 50 μg of OVA (Biosearch Technologies) emulsified in alum (Imject Alum; Thermo Fisher Scientific) at 2:1 v:v ratio in 150 μL volume for 2–4 weeks, followed by boost immunization with 25 μg of NP(16)-OVA (Biosearch Technologies) in PBS in 30 μL volume that are delivered subcutaneously (s.c.) into one hind footpad and the other left untreated (for most experiments) or into all footpads (for long-term αDEC-OVA effect experiments and polyclonal GC experiments). Boosted footpads were treated s.c. with 5 μg αDEC-OVA (Pasqual et al., 2015) in PBS in 30 μL volume 8 days after immunization with NP-OVA. DEC205−/− recipient mice were used to exclude host endogenous DEC-205 expression. For pMHCII density quantification and validation experiments, boosted footpads were treated s.c. with either 5 μg αDEC-OVA, αDEC-OVA0-Eα, or unconjugated αDEC205 in PBS in 30 μL volume 8 days after immunization with NP-OVA. For CD40L blockade experiments, the mice were first treated s.c. with 5 μg of αDEC-OVA in one footpad in parallel with the other footpad untreated day 8 post boost, and were treated i.v. immediately after or 24 h later with 300 μg of anti-CD40L blocking antibody (MR-1; Bio X Cell) in PBS in 100 μL volume or 300 μg of Armenian Hamster IgG isotype control (Bio X Cell) in 100 μL volume. For CD40 saturation experiments, the mice CD40L were first blocked i.v. by anti-CD40L treatment the same way described before, and then were treated s.c. with 10 μg of anti-CD40 stimulating antibody (1C10; eBioscience) together with 5 μg of αDEC-OVA in one footpad in parallel with the other footpad untreated day 8 post boost. For validating stimulating anti-CD40 effect by phospho-S6 staining, 10 μg isotype control (eBR2a; eBioscience) was used.
For testing caged peptide stability in vivo (Figures 4C and 4D), naïve purified CFSE-labeled OT-II tdTomato cells (50,000) were adoptively transferred into B6 recipients, i.p. immunized with 0.5 nmoles of control or caged OVA peptides were emulsified with 50 μL alum (Pierce).
For in vivo GC imaging of peptide uncaging experiments, 50,000–100,000 tdTomato+ OT-II naïve T cells were transferred to B6 mice, and i.p. immunized with Ovalbumin (50 μg) emulsified in alum (150 μL) to generate memory T cells that would dominate GCs (Shulman et al., 2013). Two weeks later, 3–5 million B1–8hi PA-GFP naïve B cells were adoptively transferred and boosted in the hind footpads with NP-OVA (25 μg per footpad) to generate GCs. On day 7–9, mice were treated with 5 μg caged OVA-peptide or PBS in the footpad 1hr prior to initiating surgery and GC imaging in the popliteal lymph node, as previously described (Fooksman et al., 2010). Proper GC dynamics, based on GC B cells and TFH motility, were monitored as proxies for proper oxygen, temperature, and low inflammation, and used to exclude any non-physiological imaging sessions from analysis. To photoactivate PA-GFP cells, a region of interest was selected about 20% of the GC volume, (100 × 100 × 50 micron XYZ, 0.5micron slices, 15microsec/micron, 15–25% laser power at 2.5 W 830 nm Ti-Sapphire pulsed laser). To uncage cOVA peptide in conjunction with PA-GFP photoactivation, an additional irradiation cycle was added at 750 nm of the same volume of interest for imaging for Figures 4J–4L and Videos S3 and S4. Alternatively, high-power irradiation at 800 nm was also capable of triggering both photo-uncaging cOVA peptide and GFP photoactivating simultaneously (Figures 4M–4P). Successful photoactivation was empirically optimized based on activation of GFP+ cells and TFH cells were still properly motile and non-apoptotic from excessive UV-induced laser damage. Time-lapse 3D images were analyzed for contacts using measurements feature on Volocity to identify GFP+ and Tomato+ cell-cell contacts, track their duration (above 5 timepoints or 1.5 min) and volume of contact. Custom scripts were applied to all conditions, pooled data from multiple mice were analyzed and compared on Prism.
Flow cytometry
Single cell suspensions of popliteal draining lymph nodes (for most experiments) or of axial, brachial, and popliteal draining lymph nodes (for long-term αDEC-OVA effect experiments and polyclonal GC experiments in MD4 mice) were filtered through a 70 μm nylon mesh and prepared in PBS containing 0.5% BSA and 1 mM EDTA. Cells were incubated with anti-CD16/32 (2.4G2, Bio X Cell) for Fc receptor blockade, surface-stained with antibodies on ice for 30 min, and analyzed on a FACS Aria III (BD) or LSR-II (BD). Fluorescent counting beads (Spherotech) were added to the cells for calculating absolute number of cells in each lymph node. Anti-B220 (RA3–6B2), CD4 (GK1.5), CD45.2 (104), CD138 (281–2), CD205 (NLDC-145), GL7 (GL7), IgMa (MA-69), TCR-Va2 (B20.1), CD86 (GL-1) were purchased from BioLegend. Anti-CD38 (90), CD45.1 (A20), CD45.2 (104), CD95 (Jo2), GL7 (GL7), IgMa (DS-1), CD184 (2B11) were purchased from BD Biosciences. Anti-CD45.1 (A20), CD69 (H1.2F3), and Ea52–68 peptide bound to I-Ab Monoclonal Antibody (Y-Ae) was purchased from eBiosciences. NP-PE was purchased from Biosearch Technologies. Phospho-S6 staining was done exactly as described previously (Ersching et al., 2017). For intracellular staining, Foxp3 staining kit (eBiosciences) were used to fix and permeabilize the cells followed by staining using anti-IRF4 (3E4; eBiosciences) and anti-IRF8 (V3GYWCH; eBiosciences). For EdU incorporation assays, mice were injected i.p. with 1 mg EdU (Thermo Fisher Scientific) in DMSO in 50 μL volume 5 h before sacrifice. Popliteal draining lymph node cells were Fc-blocked, surface-stained, and then EdU-incorporation was detected using a Click-it Plus EdU Flow Cytometry Assay kit (Life Technologies) following the manufacturer’s instructions. For detection of EdU incorporation by IRF4hiIRF8lo GC plasmablast precursors, lymph node cells were Fc-blocked, surface-stained, pre-fixed in 2% paraformaldehyde for 10 min at room temperature, and then fixed and permeabilized and stained for intracellular IRF4 and IRF8 using Foxp3 staining kit as described above followed by EdU detection at last. Data were analyzed in Flowjo v10.4.2 (BD). For validating TFH and GC B cell activation status after photoactivation and photouncaging in the GC, mice were sacrificed 4 h after imaging, and popliteal lymph nodes were taken for flow cytometry.
Intravital imaging and analysis
Surgical preparation of popliteal lymph node intravital imaging has been described previously (Fooksman et al., 2010). Mice were kept under anesthesia using isoflurane gas during imaging process. All imaging was conducted on an Olympus FVE-1200 upright microscope, using 25×1.04 NA objective, and Deepsee MaiTai Ti-Sapphire pulsed laser (Spectra-Physics) tuned to 905 nm. For photoactivation, the laser was tuned to 830 nm and then imaged at 920 nm. To maintain temperature and limit room light, the microscope was fitted with custom-built incubator chamber and heated 37°C platform. Time lapses were conducted every 30 s as 50–90 μm deep Z-stacks (5 μm steps) with 1x-1.5× zoom and with 512 × 512 X-Y resolution.
All image analysis was conducted using Imaris software 9.2 (Bitplane) or Volocity 6.3 (Quorum Technologies) to detect and track tdTomato+ T cells and CFP+ or PA-GFP+ B cells and to correct drift. For T-B conjugate detection and tracking after photoactivation, colocalization tool in Imaris Software was used.
In vitro culture
For in vitro B cell blasts and T cells co-culture experiments, naïve B cells from the spleen of DEC205+/+, DEC205+/−, and DEC205−/− mouse (8–16-week-old, both males and females were used) were purified using anti-CD43 beads (Miltenyi Biotec) by MACS as described above, and then were cultured in RPMI media supplemented with 10% FBS, Pen/Strep, 0.05 mM b-mercaptoethanol, 20 μg/mL LPS (Sigma), and 4 ng/mL recombinant mouse IL-4 (PeproTech) for 3 days at 37°C. Naïve CD4+ T cells were isolated from the spleen of OT-II mouse (8–16 weeks old males and females) using CD4+ T cell isolation kit (Miltenyi Biotec) before B cells blasts harvest. Single cell suspension of naïve CD4+ T cells were then labeled by CellTrace Violet dye (CTV; Life Technologies) following manufacturer’s instructions. The B cell blasts were harvested, washed, and cultured with 3 times more numbers of CTV-labeled naïve CD4+ T cells in LPS/IL-4-free RPMI media containing either 10 μg/mL αDEC-OVA or 5 μg/mL OVA323–339 (InvivoGen) for another 3 days at 37°C. Finally, Co-culture was harvested and CTV dilution in CD4+ T cells was measured by flow cytometry on a FACS Aria III (BD Biosciences).
For CD40 in vitro imaging (Figures S3E–S3G), GFP+ naïve B cell were purified and activated with LPS and used on day 2 post activation, and treated with or without anti-CD40 (5 μg/mL), and imaged the next day on ICAM-1 coated substrates using interference reflection microscopy or confocal fluorescence microscopy using a 20× oil dipping objective on Zeiss 710 inverted microscope as previously described (Fooksman et al., 2010), collecting images every 15 s for 20 min. Images were analyzed on Volocity 6.3 (Quorum Technologies).
Mixed co-culture with caged and uncaged peptide utilized a similar setup, except OT-II tdTomato+ T cells were preactivated, with OVA-pulsed splenocytes and maintained in IL-2 for 1–2 weeks. OT-II T cells were labeled with CFSE and analyzed 3-days post incubation. For uncaged experiments (Figures 4E and 4F), peptide loaded B cells in glass bottom flat wells, were exposed to (filtered 440/60) UV light from Excite Arc lamp, on a Zeiss 710 magnified by 10× objective for the specified times. Then cells were incubated with OT-II tdTomato T cells for 30 min in RPMI+10%FBS, and analyzed by flow cytometry to identify doublets. To maintain conjugates, all media contained calcium and EDTA-free.
For in vitro imaging conjugate formation (Figures 4G–4I), PA-GFP+ LPS-induced B cell blasts were generated and used 2 days after activation, with tdTomato+ OT-II T cell blasts. B cells were peptide loaded as above, washed extensively, and plated on ICAM-1-fc coated substrates as described for CD40 in vitro imaging above. Photoactivation was achieved using 405 nm laser scanning (25% laser power, 4 microns2/μsec) confined to a region of interest (approximately 100 × 100 micron field). Images were analyzed on Imaris.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were performed using a two-tailed unpaired T-Test to compare two groups or paired T-Test to compare two cell populations in the same mouse. A p value of less than or equal to 0.05 was considered statistically significant. Data are presented as the mean ± SD or mean ± SEM. Statistical tests were performed using GraphPad Prism (versions 7 and 8). Specific test used are listed in the figure legends with p values.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Armenian Hamster monoclonal anti-CD40L (clone MR-1) | Bio X Cell | Cat# BE0017-1; RRID:AB_1107601 |
| Armenian Hamster monoclonal anti-CD69 FITC (clone H1.2F3) | eBiosciences | Cat# 11-0691-82; RRID:AB_465119 |
| Armenian Hamster monoclonal anti-CD95 BV510 (clone Jo2) | BD Biosciences | Cat# 563646; RRID:AB_2738345 |
| Hamster monoclonal anti-CD95 PE-Cy7 (clone Jo2) | BD Biosciences | Cat# 557653; RRID:AB_396768 |
| Armenian Hamster polyclonal IgG | Bio X Cell | Cat# BE0091; RRID:AB_1107773 |
| Eα52-68 peptide bound to I-Ab Monoclonal Antibody Biotin (Y-Ae) | eBiosciences | Cat# 13-5741-82; RRID:AB_657821 |
| Rat monoclonal anti-B220 APC-Cy7 (clone RA3-6B2) | BioLegend | Cat# 103224; RRID:AB_313007 |
| Rat monoclonal anti-CD4 BV421 (clone GK1.5) | BioLegend | Cat# 100438; RRID:AB_11203718 |
| Rat monoclonal anti-CD16/CD32 (clone 2.4G2) | Bio X Cell | Cat# CUS-HB-197; RRID:AB_2687830 |
| Rat monoclonal anti-CD38 PerCP-Cy5.5 (clone 90) | BD Biosciences | Cat# 562770; RRID:AB_2737782 |
| Rat monoclonal anti-CD40 (clone 1C10) | eBiosciences | Cat# 16-0401-82; RRID:AB_468941 |
| Mouse monoclonal anti-CD45.1 AF700 (clone A20) | BD Biosciences | Cat# 561235; RRID:AB_10611577 |
| Mouse monoclonal anti-CD45.1 eFluor 450 (clone A20) | eBiosciences | Cat# 48-0453-82; RRID:AB_1272189 |
| Mouse monoclonal anti-CD45.1 PE (clone A20) | eBiosciences | Cat# 12-0453-83; RRID:AB_465676 |
| Mouse monoclonal anti-CD45.2 PerCP- Cy5.5 (clone 104) | BioLegend | Cat# 109828; RRID:AB_893350 |
| Mouse monoclonal anti-CD45.2 PerCP- Cy5.5 (clone 104) | BD Biosciences | Cat# 552950; RRID:AB_394528 |
| Mouse monoclonal anti-CD45.2 V450 (clone 104) | BD Biosciences | Cat# 560697; RRID:AB_1727495 |
| Rat monoclonal anti-CD86 BV421 (clone GL1) | BD Biosciences | Cat# 564198; RRID:AB_2738663 |
| Rat monoclonal anti-CD138 APC (clone 281-2) | BD Biosciences | Cat# 558626; RRID:AB_1645216 |
| Rat monoclonal anti-CD138 PE (clone 281-2) | BD Biosciences | Cat# 553714; RRID:AB_395000 |
| Rat monoclonal anti-CD138 PerCP-Cy5.5 (clone 281-2) | BioLegend | Cat# 142510; RRID:AB_2561601 |
| Rat monoclonal anti-CD184 PE (clone 2B11) | BD Biosciences | Cat# 551966; RRID:AB_394305 |
| Rat monoclonal anti-CD205 APC (clone NLDC-145) | BioLegend | Cat# 138206; RRID:AB_10613641 |
| Rat monoclonal anti-CD205 (clone NLDC-145) | BioLegend | Cat# 138202; RRID:AB_2281398 |
| Rat monoclonal anti-GL7 AF647 (clone GL7) | BD Biosciences | Cat# 561529; RRID:AB_10716056 |
| Rat monoclonal anti-GL7 PerCP-Cy5.5 (clone GL7) | BioLegend | Cat# 144610; RRID:AB_2562979 |
| Mouse monoclonal anti-IgMa FITC (clone DS-1) | BD Biosciences | Cat# 553516; RRID:AB_394897 |
| Mouse monoclonal anti-IgMa PE (clone MA-69) | BioLegend | Cat# 408608; RRID:AB_940545 |
| Rat IgG2a kappa Isotype Control (eBR2a) | eBiosciences | Cat# 16-4321-82; RRID:AB_470156 |
| Rat monoclonal anti-IRF4 PE (clone 3E4) | eBiosciences | Cat# 12-9858-82; RRID:AB_10852721 |
| Mouse monoclonal anti-IRF8 APC (clone V3GYWCH) | eBiosciences | Cat# 17-9852-82; RRID:AB_2573318 |
| Rabbit monoclonal anti-PhosphoS6 (Ser235/236) AF647 (clone D57.2.2E) | CST | Cat# 4851S; RRID:AB_10695457 |
| Rat monoclonal anti-TCR Vα2 AF647 (clone B20.1) | BioLegend | Cat# 127812; RRID:AB_1186115 |
| DEC-OVA | This paper | N/A |
| DEC-OVA-Eα | This paper | N/A |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Advanced DMEM | Gibco | Cat# 12491-023 |
| Bovine Serum Albumin, Fraction V (Modified Cohn) | Sigma-Aldrich | Cat# 12-660-100GM |
| 5(6)-Carboxyfluorescein diacetate N-succinimidyl ester (CFSE) | Sigma-Aldrich | Cat# 21888-25MG-F |
| Calcium phosphate | Thermo Fisher Scientific | Cat# AC446390010 |
| EDTA (0.5 M), pH 8.0, RNase-free | Thermo Fisher Scientific | Cat# AM9261 |
| EdU (5-ethynyl-2’-deoxyuridine) | Thermo Fisher Scientific | Cat# A10044 |
| Gibco Fetal Bovine Serum | Thermo Fisher Scientific | Cat# 10-437-028 |
| Glutamate, Caged hydrate | Sigma-Aldrich | Cat# G3291 |
| Imject Alum Adjuvant | Thermo Fisher Scientific | Cat# 77161 |
| Isothesia (Isoflurane) Solution | Covetrus | Cat# 029405 |
| Lactated Ringer’s Solution | Henry Schein | Cat# 14792 |
| Lonza ACK Lysing Buffer | Thermo Fisher Scientific | Cat# BW10548E |
| Lipopolysaccharides from Escherichia coli O111:B4 | Sigma-Aldrich | Cat# L-2630 |
| 2-Mercaptoethanol | Thermo Fisher Scientific | Cat# 21985023 |
| NP(16)-OVA | Biosearch Technologies | Cat# N-5051-100 |
| NP(19)-PE | Biosearch Technologies | Cat# N-5070-1 |
| Nutridoma-SP | Sigma-Aldrich | Cat# 11011375001 |
| Ovalbumin | Biosearch Technologies | Cat# O-1000-100 |
| OVA323-339 | InvivoGen | Cat# vac-isq |
| PBS, 10X Powder, pH 7.4 | Thermo Fisher Scientific | Cat# BP665-1 |
| Penicillin-Streptomycin (10, 000U/ml) | Gibco | Cat# 15140-122 |
| Penicillin-Streptomycin-Glutamine (100X) | Gibco | Cat# 10378016 |
| Protein G Sepharose 4 Fast Flow | Cytiva | Cat# 17061802 |
| 16% Paraformaldehyde (formaldehyde) aqueous solution | Electron Microscopy Company | Cat# 15710 |
| Recombinant Murine IL-4 | PeproTech | Cat# 214-14 |
| Recombinant Mouse ICAM-1/CD54 Fc Chimera Protein | R&D Systems | Cat# 796-IC-050 |
| RPMI 1640 Medium | Thermo Fisher Scientific | Cat# SH3009601 |
| Streptavidin APC | BioLegend | Cat# 405207 |
|
| ||
| Critical commercial assays | ||
|
| ||
| BD Cytofix/Cytoperm Fixation/Permeabilization Kit | BD Biosciences | Cat# 554714 |
| CellTrace Violet Cell Proliferation Kit | Life Technologies | Cat# C34571 |
| Click-iT Plus EdU Pacific Blue Flow Cytometry Assay Kit | Life Technologies | Cat# C10636 |
| CD43 microbeads | Miltenyi Biotec | Cat#130-049-801 |
| CD4+ T Cell Isolation Kit | Miltenyi Biotec | Cat#130-104-454 |
| Foxp3/Transcription Factor Fixation/Permeabilization Concentrate and Diluent | eBioscience | Cat# 00-5521-00 |
| Live/Dead Fixable Aqua Dead Cell Stain Kit | Thermo Fisher Scientific | Cat# L34957 |
| NEBuilder HiFi DNA Assembly Cloning Kit | New England Biolabs | Cat# E5520S |
| Texas Red-X, Succinimidyl Ester, single isomer | Life Technologies | Cat# T20175 |
| UltraComp eBeads Compensation Beads | Thermo Fisher Scientific | Cat# 01-2222-41 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| HEK293T | ATCC | Cat# CRL-3216; RRID:CVCL_0063 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mouse: Blimp1-YFP | The Jackson Laboratory | JAX stock# 008828 |
| Mouse: B1-8hi | The Jackson Laboratory | JAX stock# 007775 |
| Mouse: B6-Ly5.1/Cr | Charles River Laboratories | NCI stock# 564 |
| Mouse: C57BL/6 | Charles River Laboratories | NCI stock# 556 |
| Mouse: CAG-ECFP | The Jackson Laboratory | JAX stock# 003773 |
| Mouse: CMV-cre | The Jackson Laboratory | JAX stock# 006054 |
| Mouse: DEC205−/− | The Jackson Laboratory | JAX stock# 005528 |
| Mouse: Rosa26Stop-tdTomato (AI14) | The Jackson Laboratory | JAX stock# 007914 |
| Mouse: OT-II | The Jackson Laboratory | JAX stock# 004194 |
| Mouse: Rosa26Stop–CAG-PA-GFP | The Jackson Laboratory | JAX stock# 035687 |
|
| ||
| Oligonucleotides | ||
|
| ||
| Synthesized duplex ultramer cDNA containing Eα (52–68) peptide and flanking overlap sequences (sense strand): 5′-CTTCTTTGGCAGATGTGTTTCCCC TGCCAGCTTTGAGGCTCAGGGTGCA CTGGCTAATATAGCTGTGGACAAAGC TCTAGAGGGCCCGCGGTTCGAAC AAAAACT-3′ |
Integrated DNA Technologies | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| Adobe After Effects | Adobe | N/A |
| Adobe Illustrator | Adobe | RRID: SCR_010279 |
| Excel | Microsoft | RRID: SCR_016137 |
| FlowJo | BD Biosciences | RRID: SCR_008520 |
| Imaris | Bitplane | RRID: SCR_007370 |
| Prism | Graphpad | RRID: SCR_002798 |
| Volocity | Quorum Technologies | RRID: SCR_002668 |
Highlights.
GC clonal expansion magnitude is directly proportional to cognate pMHCII
CD40 signaling strength is necessary and sufficient to control GC B cell expansion
Plasmablast differentiation is not directly instructed based on pMHCII density
Caged OVA peptide can be used to optically trigger rapid selection in vivo
ACKNOWLEDGMENTS
This work was supported by NIH R01AI72529 (D.R.F. and M.L.D.), HL141491 (D.R.F., Z.J., and M.J.M.), S10 RR023704 (M.L.D.), the Kennedy Trust for Rheumatology Research (M.L.D.), and the Albert Einstein NCI Cancer Center P30CA013330. We would like to thank Michel Nussenzweig.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
INCLUSION AND DIVERSITY
We worked to ensure sex balance in the selection of non-human subjects. One or more of the authors of this paper self-identifies as a member of the LGBTQ+ community. One or more of the authors of this paper received support from a program designed to increase minority representation in science. While citing references scientifically relevant for this work, we also actively worked to promote gender balance in our reference list.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.110763.
REFERENCES
- Allen CD, Okada T, and Cyster JG (2007a). Germinal-center organization and cellular dynamics. Immunity 27, 190–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen CD, Okada T, Tang HL, and Cyster JG (2007b). Imaging of germinal center selection events during affinity maturation. Science 315, 528–531. [DOI] [PubMed] [Google Scholar]
- Bannard O, McGowan SJ, Ersching J, Ishido S, Victora GD, Shin JS, and Cyster JG (2016). Ubiquitin-mediated fluctuations in MHC class II facilitate efficient germinal center B cell responses. J. Exp. Med. 213, 993–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batista FD, and Neuberger MS (1998). Affinity dependence of the B cell response to antigen: a threshold, a ceiling, and the importance of off-rate. Immunity 8, 751–759. [DOI] [PubMed] [Google Scholar]
- Bonifaz L, Bonnyay D, Mahnke K, Rivera M, Nussenzweig MC, and Steinman RM (2002). Efficient targeting of protein antigen to the dendritic cell receptor DEC-205 in the steady state leads to antigen presentation on major histocompatibility complex class I products and peripheral CD8+ T cell tolerance. J. Exp. Med. 196, 1627–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boscardin SB, Hafalla JC, Masilamani RF, Kamphorst AO, Zebroski HA, Rai U, Morrot A, Zavala F, Steinman RM, Nussenzweig RS, and Nussenzweig MC (2006). Antigen targeting to dendritic cells elicits long-lived T cell help for antibody responses. J. Exp. Med. 203, 599–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crotty S (2011). Follicular helper CD4 T cells (TFH). Annu. Rev. Immunol. 29, 621–663. [DOI] [PubMed] [Google Scholar]
- De Silva NS, and Klein U (2015). Dynamics of B cells in germinal centres. Nat. Rev. Immunol. 15, 137–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ersching J, Efeyan A, Mesin L, Jacobsen JT, Pasqual G, Grabiner BC, Dominguez-Sola D, Sabatini DM, and Victora GD (2017). Germinal center selection and affinity maturation require dynamic regulation of mTORC1 kinase. Immunity 46, 1045–1058.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkin S, Hartweger H, Oliveira TY, Kara EE, and Nussenzweig MC (2019). Protein amounts of the MYC transcription factor determine germinal center B cell division capacity. Immunity 51, 324–336.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fooksman DR, Schwickert TA, Victora GD, Dustin ML, Nussenzweig MC, and Skokos D (2010). Development and migration of plasma cells in the mouse lymph node. Immunity 33, 118–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardell JL, and Parker DC (2017). CD40L is transferred to antigen-presenting B cells during delivery of T-cell help. Eur. J. Immunol. 47, 41–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitlin AD, Shulman Z, and Nussenzweig MC (2014). Clonal selection in the germinal centre by regulated proliferation and hypermutation. Nature 509, 637–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo M, Gong S, Maric S, Misulovin Z, Pack M, Mahnke K, Nussenzweig MC, and Steinman RM (2000). A monoclonal antibody to the DEC-205 endocytosis receptor on human dendritic cells. Hum. Immunol. 61, 729–738. [DOI] [PubMed] [Google Scholar]
- Han S, Hathcock K, Zheng B, Kepler TB, Hodes R, and Kelsoe G (1995). Cellular interaction in germinal centers. Roles of CD40 ligand and B7–2 in established germinal centers. J. Immunol. 155, 556–567. [PubMed] [Google Scholar]
- Ise W, Fujii K, Shiroguchi K, Ito A, Kometani K, Takeda K, Kawakami E, Yamashita K, Suzuki K, Okada T, and Kurosaki T (2018). T follicular helper cell-germinal center B cell interaction strength regulates entry into plasma cell or recycling germinal center cell fate. Immunity 48, 702–715.e4. [DOI] [PubMed] [Google Scholar]
- Johnston MI, and Fauci AS (2011). HIV vaccine development–improving on natural immunity. N. Engl. J. Med. 365, 873–875. [DOI] [PubMed] [Google Scholar]
- Kamphorst AO, Guermonprez P, Dudziak D, and Nussenzweig MC (2010). Route of antigen uptake differentially impacts presentation by dendritic cells and activated monocytes. J. Immunol. 185, 3426–3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krammer F, and Palese P (2015). Advances in the development of influenza virus vaccines. Nat. Rev. Drug Discov. 14, 167–182. [DOI] [PubMed] [Google Scholar]
- Krautler NJ, Suan D, Butt D, Bourne K, Hermes JR, Chan TD, Sundling C, Kaplan W, Schofield P, Jackson J, et al. (2017). Differentiation of germinal center B cells into plasma cells is initiated by high-affinity antigen and completed by Tfh cells. J. Exp. Med. 214, 1259–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuraoka M, Schmidt AG, Nojima T, Feng F, Watanabe A, Kitamura D, Harrison SC, Kepler TB, and Kelsoe G (2016). Complex antigens drive permissive clonal selection in germinal centers. Immunity 44, 542–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin WH, Adams WC, Nish SA, Chen YH, Yen B, Rothman NJ, Kratchmarov R, Okada T, Klein U, and Reiner SL (2015). Asymmetric PI3K signaling driving developmental and regenerative cell fate bifurcation. Cell Rep. 13, 2203–2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Xu H, Shih C, Wan Z, Ma X, Ma W, Luo D, and Qi H (2015). T-B-cell entanglement and ICOSL-driven feed-forward regulation of germinal centre reaction. Nature 517, 214–218. [DOI] [PubMed] [Google Scholar]
- Mascola JR, and Haynes BF (2013). HIV-1 neutralizing antibodies: understanding nature’s pathways. Immunol. Rev. 254, 225–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer CT, Gazumyan A, Kara EE, Gitlin AD, Golijanin J, Viant C, Pai J, Oliveira TY, Wang Q, Escolano A, et al. (2017). The microanatomic segregation of selection by apoptosis in the germinal center. Science 358, eaao2602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayumi M, Sumimoto S, Ohshima Y, Katamura K, Heike T, Hata D, Kanazashi S, and Furusho K (1995). Role of LFA-1/ICAM-1-dependent cell adhesion in CD40-mediated inhibition of anti-IgM antibody-induced B-cell death. J. Allergy Clin. Immunol. 96, 1136–1144. [DOI] [PubMed] [Google Scholar]
- Noelle RJ, Ledbetter JA, and Aruffo A (1992). CD40 and its ligand, an essential ligand-receptor pair for thymus-dependent B-cell activation. Immunol. Today 13, 431–433. [DOI] [PubMed] [Google Scholar]
- Pappas L, Foglierini M, Piccoli L, Kallewaard NL, Turrini F, Silacci C, Fernandez-Rodriguez B, Agatic G, Giacchetto-Sasselli I, Pellicciotta G, et al. (2014). Rapid development of broadly influenza neutralizing antibodies through redundant mutations. Nature 516, 418–422. [DOI] [PubMed] [Google Scholar]
- Pasqual G, Angelini A, and Victora GD (2015). Triggering positive selection of germinal center B cells by antigen targeting to DEC-205. Methods Mol. Biol. 1291, 125–134. [DOI] [PubMed] [Google Scholar]
- Phan TG, Paus D, Chan TD, Turner ML, Nutt SL, Basten A, and Brink R (2006). High affinity germinal center B cells are actively selected into the plasma cell compartment. J. Exp. Med. 203, 2419–2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhardt RL, Liang HE, and Locksley RM (2009). Cytokine-secreting follicular T cells shape the antibody repertoire. Nat. Immunol. 10, 385–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson JM, Jensen PE, and Evavold BD (2000). DO11.10 and OT-II T cells recognize a C-terminal ovalbumin 323–339 epitope. J. Immunol. 164, 4706–4712. [DOI] [PubMed] [Google Scholar]
- Rudensky A, Rath S, Preston-Hurlburt P, Murphy DB, and Janeway CA Jr. (1991). On the complexity of self. Nature 353, 660–662. [DOI] [PubMed] [Google Scholar]
- Saliba DG, Cespedes-Donoso PF, Balint S, Compeer EB, Korobchev-skaya K, Valvo S, Mayya V, Kvalvaag A, Peng Y, Dong T, et al. (2019). Composition and structure of synaptic ectosomes exporting antigen receptor linked to functional CD40 ligand from helper T cells. eLife 8, e47528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt AG, Therkelsen MD, Stewart S, Kepler TB, Liao HX, Moody MA, Haynes BF, and Harrison SC (2015). Viral receptor-binding site antibodies with diverse germline origins. Cell 161, 1026–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwickert TA, Lindquist RL, Shakhar G, Livshits G, Skokos D, Kosco-Vilbois MH, Dustin ML, and Nussenzweig MC (2007). In vivo imaging of germinal centres reveals a dynamic open structure. Nature 446, 83–87. [DOI] [PubMed] [Google Scholar]
- Schwickert TA, Victora GD, Fooksman DR, Kamphorst AO, Mugnier MR, Gitlin AD, Dustin ML, and Nussenzweig MC (2011). A dynamic T cell-limited checkpoint regulates affinity-dependent B cell entry into the germinal center. J. Exp. Med. 208, 1243–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciammas R, Shaffer AL, Schatz JH, Zhao H, Staudt LM, and Singh H (2006). Graded expression of interferon regulatory factor-4 coordinates isotype switching with plasma cell differentiation. Immunity 25, 225–236. [DOI] [PubMed] [Google Scholar]
- Shi W, Liao Y, Willis SN, Taubenheim N, Inouye M, Tarlinton DM, Smyth GK, Hodgkin PD, Nutt SL, and Corcoran LM (2015). Transcriptional profiling of mouse B cell terminal differentiation defines a signature for antibody-secreting plasma cells. Nat. Immunol. 16, 663–673. [DOI] [PubMed] [Google Scholar]
- Shih TA, Roederer M, and Nussenzweig MC (2002). Role of antigen receptor affinity in T cell-independent antibody responses in vivo. Nat. Immunol. 3, 399–406. [DOI] [PubMed] [Google Scholar]
- Shinnakasu R, and Kurosaki T (2017). Regulation of memory B and plasma cell differentiation. Curr. Opin. Immunol. 45, 126–131. [DOI] [PubMed] [Google Scholar]
- Shulman Z, Gitlin AD, Targ S, Jankovic M, Pasqual G, Nussenzweig MC, and Victora GD (2013). T follicular helper cell dynamics in germinal centers. Science 341, 673–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulman Z, Gitlin AD, Weinstein JS, Lainez B, Esplugues E, Flavell RA, Craft JE, and Nussenzweig MC (2014). Dynamic signaling by T follicular helper cells during germinal center B cell selection. Science 345, 1058–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KG, Light A, O’Reilly LA, Ang SM, Strasser A, and Tarlinton D (2000). bcl-2 transgene expression inhibits apoptosis in the germinal center and reveals differences in the selection of memory B cells and bone marrow antibody-forming cells. J. Exp. Med. 191, 475–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suan D, Sundling C, and Brink R (2017). Plasma cell and memory B cell differentiation from the germinal center. Curr. Opin. Immunol. 45, 97–102. [DOI] [PubMed] [Google Scholar]
- Victora GD, and Nussenzweig MC (2012). Germinal centers. Annu. Rev. Immunol. 30, 429–457. [DOI] [PubMed] [Google Scholar]
- Victora GD, Schwickert TA, Fooksman DR, Kamphorst AO, Meyer-Hermann M, Dustin ML, and Nussenzweig MC (2010). Germinal center dynamics revealed by multiphoton microscopy with a photoactivatable fluorescent reporter. Cell 143, 592–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein JS, Herman EI, Lainez B, Licona-Limon P, Esplugues E, Flavell R, and Craft J (2016). TFH cells progressively differentiate to regulate the germinal center response. Nat. Immunol. 17, 1197–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisel FJ, Zuccarino-Catania GV, Chikina M, and Shlomchik MJ (2016). A temporal switch in the germinal center determines differential output of memory B and plasma cells. Immunity 44, 116–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Chaudhri VK, Wu Z, Biliouris K, Dienger-Stambaugh K, Rochman Y, and Singh H (2015). Regulation of bifurcating B cell trajectories by mutual antagonism between transcription factors IRF4 and IRF8. Nat. Immunol. 16, 1274–1281. [DOI] [PubMed] [Google Scholar]
- Yeh CH, Nojima T, Kuraoka M, and Kelsoe G (2018). Germinal center entry not selection of B cells is controlled by peptide-MHCII complex density. Nat. Commun. 9, 928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaretsky I, Atrakchi O, Mazor RD, Stoler-Barak L, Biram A, Feigelson SW, Gitlin AD, Engelhardt B, and Shulman Z (2017). ICAMs support B cell interactions with T follicular helper cells and promote clonal selection. J. Exp. Med. 214, 3435–3448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou G, Ding ZC, Fu J, and Levitsky HI (2011). Presentation of acquired peptide-MHC class II ligands by CD4+ regulatory T cells or helper cells differentially regulates antigen-specific CD4+ T cell response. J. Immunol. 186, 2148–2155. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.