

Abstract
The cellular energy and biomass demands of cancer drive a complex dynamic between uptake of extracellular FAs and their de novo synthesis. Given that oxidation of de novo synthesized FAs for energy would result in net-energy loss, there is an implication that FAs from these two sources must have distinct metabolic fates; however, hitherto, all FAs have been considered part of a common pool. To probe potential metabolic partitioning of cellular FAs, cancer cells were supplemented with stable isotope-labeled FAs. Structural analysis of the resulting glycerophospholipids revealed that labeled FAs from uptake were largely incorporated to canonical (sn-) positions on the glycerol backbone. Surprisingly, labeled FA uptake also disrupted canonical isomer patterns of the unlabeled lipidome and induced repartitioning of n-3 and n-6 PUFAs into glycerophospholipid classes. These structural changes support the existence of differences in the metabolic fates of FAs derived from uptake or de novo sources and demonstrate unique signaling and remodeling behaviors usually hidden from conventional lipidomics.
Supplementary key words: FA/transport, lipolysis and FA metabolism, lipase, phospholipid/metabolism, phospholipids/phosphatidylcholine, lipid isomers, stable-isotope tracing, imaging MS, ozone-induced dissociation
Abbreviations: AA, arachidonic acid; AT, activation time; CE, collision energy; CID, collision-induced dissociation; DB, double bond; dn-lipidome, de novo lipidome; ER, endoplasmic reticulum; FABP, fatty acyl binding protein; FDI, fractional distribution image; GPL, glycerophospholipid; LA, linoleic acid; LIT, linear ion trap; LPCAT, lysophosphatidylcholine acyltransferase; MSI, MS imaging; NCE, normalized collision energy; NL, neutral loss; NS, nil supplemented; OzID, ozone-induced dissociation; PA, palmitic acid; PC, phosphatidylcholine; PCa, prostate cancer; PE, phosphatidylethanolamine; PG, phosphatidylglycerol; PI, phosphatidylinositol; PLA2, phospholipase A2; PS, phosphatidylserine; ROS, reactive-oxidative species; SA, stearic acid; SCD-1, stearoyl-CoA desaturase 1; SFA, saturated FA; sPLA2-IIA, secretory-PLA2; TAG, triacylglycerol; up-lipidome, uptake lipidome
Graphical abstract
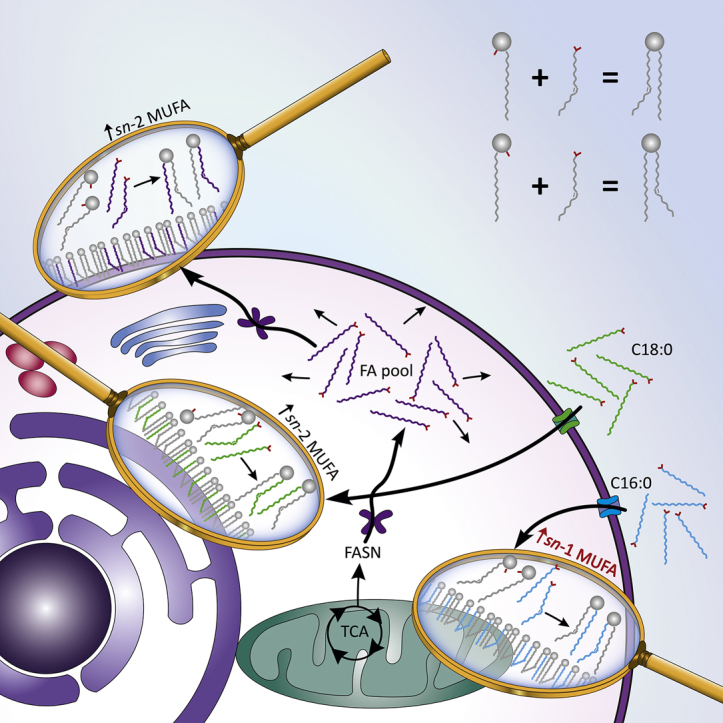
Although seemingly banal, the structure-function relationship of glycerophospholipid (GPL) is far more complex and insightful than first meets the eye. While the amphoteric nature of the molecule importantly allows for chemical partitioning, the individual functional groups serve numerous other specific purposes. The charged hydrophilic head group creates the possibility for lipids to be directed to particular subcellular compartments, whereby the lipophilic FAs can impart variable physicochemical properties to the molecule and initiate signaling (1). Arguably, two of the most important processes in dictating the modeling and remodeling of mammalian GPL structure are the Kennedy pathway and the Lands cycle. While the Kennedy pathway describes the de novo synthesis of phosphatidylcholine (PC) and phosphatidylethanolamine (PE) from cytidine-diphosphate diacylglycerol (2), the Lands cycle describes the process by which phospholipase and acyltransferase enzymes remodel GPL-FAs (3). While these processes logically have implications in cellular energy production, whether de novo generated FAs or extracellular FAs (from uptake) have identical or varied roles is not well understood. Given that cancer cells are known to modulate and oscillate between de novo and uptake FA metabolism to opportunistically source energy for growth (4, 5), understanding if subfractions of FAs serve discrete purposes is of importance when elucidating the molecular mechanisms of cancer. For example, a cancer cell actively increasing the production of FAs only to oxidize these to meet energy demands is counterproductive, and thus, the cell might instead be compelled to source extracellular FAs. For this to be possible however, there must be compartmentalization and discrete metabolic treatment of de novo or uptake FAs—a sentiment that is contrary to the current idea that the cell has a common “pool” of FAs, used to meet metabolic demand (4, 6).
One of the more important enzymes within the Lands cycle is phospholipase A2 (PLA2), a lipase that is responsible for the selective cleavage of the FA located at the sn-2 position within GPLs. Notably, PLA2 has been shown to exhibit a high-level of selectivity toward GPLs containing PUFAs, such as arachidonic acid (AA; 20:4 n-6) and DHA (22:6 n-3) (7, 8). Amongst other functions, the release of AA and DHA in turn initiates proinflammatory or anti-inflammatory responses through the cyclooxygenase and lipoxygenase pathways (8, 9, 10). Concurrent with the release of the sn-2 FA is the formation of a 1-acyl-2-lyso-GPL, or 2-lyso-GPL, with a hydroxyl group at the sn-2 position of the glycerol backbone. While these lysospecies are themselves signaling mediators (11), they also readily undergo re-esterification, reverting to diacyl-GPLs via acyltransferase enzymes, such as the lysophosphatidic acid acyltransferase or lysophosphatidylcholine acyltransferase (LPCAT) isoforms (3, 12, 13). The labile nature of lyso-GPLs also allows physicochemical properties such as pH and temperature to influence a behavior known as acyl chain migration, whereby a FA is able to migrate from one sn-position to a free hydroxyl group on the glycerol backbone. Unlike the sn-selectivity of PLA2, acyltransferase enzymes are rarely reported to exhibit selectivity or specificity for 1-lyso or 2-lyso acceptor lipids. Results from some studies however suggest that perhaps certain LPCAT isoforms (e.g., LPCAT1) are nonspecific to 1-lyso or 2-lyso lipids, whereas other isoforms (e.g., LPCAT3) and acyltransferase enzymes (e.g., LYCAT) exhibit a degree of selectivity depending on lysoacceptor and FA donor species (14). Generally, it is accepted that acyl chain migration occurring prior to esterification will be influential in lipid molecular structure. Because Kennedy and Lands metabolic pathways affect different structural motifs of the GPL molecule, they are able to dictate both the transport of lipids around the cell and the initiation of signaling cascades through the release of biologically active FAs. This allows for independent organelle membrane modifications as well as the initiation of signaling cascades through the release of biologically active FAs. Although the various mechanisms and outcomes of lipid remodeling have been studied extensively, little is known about the impact that sn-isomeric lipid species have on remodeling and transport, or conversely, the impact these mechanisms have on lipid isomer populations. While it is now known that the GPLs of both prokaryotic and eukaryotic cells exhibit a preference for unsaturated FAs to be at the sn-2 position, prior to 2003, there was little-to-no indication that sn-positional isomers were present in eukaryotic cells (15, 16). Ekroos et al. (15) displayed that not only was there a quantifiable population of sn-1 unsaturated GPLs, but the relative abundance of these regioisomer populations can vary between mammalian GPL samples. This was exemplified in more recent studies into mammalian GPL regioisomers, which revealed that bovine liver (17), egg yolk, and sheep kidney (18) exhibited a stronger preference for canonical sn-positions than synthetically prepared standards.
Although an independent lipase enzyme, phospholipase A1, is known to catalyze the cleavage of FAs in the sn-1 position of specific GPL classes (19), the preference for unsaturated FAs to exist in the sn-2 position is thought to be strongly linked with Lands cycle lipid remodeling and PLA2 functionality (7, 10, 20). This enhanced sn-positional specificity in biological systems implies that cellular intervention is required for maintenance. Studies into the peroxidation of unilamellar liposomes comprised of specific PC sn-isomers (containing palmitic acid [PA] and linoleic acid [LA]) revealed oxidation rates were sn-isomer dependent (21). Using radiolabeled FAs in mouse models, others showed that while the supplemented cis-FA (oleic acid) showed esterification specificity to the sn-2 position of GPLs, the trans-FA species (elaidic acid) was equally esterified to sn-1 and sn-2 positions. More recently, molecular dynamics simulations revealed that unsaturated FAs at the sn-1 position of GPLs create more ordered (and hence less fluid) membranes than their unsaturated sn-2 counterparts (22). These functional differences may help explain variations in sn-isomer distribution that are observed to exist in recent tissue models. In a pivotal study, Paine et al. (23) showed that lipid isomers were tightly regulated and highly organized within the gray and white matter structures of brain tissue and were subsequently disrupted in murine cancer tumors. Therefore, because lipid species containing saturated FAs (SFAs) at the sn-1 and unsaturated FAs at the sn-2 are the typical structures identified as the majority isomer in lipid canon, to aid with explanation throughout, these isomers will be referred to as “canonomers.” Conversely, lipids displaying atypical or apocryphal sn-isomeric structure (with the unsaturated FA contained at the sn-1 position) will be referred to “apocromers.”
Using stable isotope-labeled FAs and a prostate cancer (PCa) cell line (LNCaP), here we trace the metabolic fate of isotopically labeled FAs in cellular lipids, specifically tracking the sn-position at which they are incorporated into GPLs. Using a tandem mass spectrometric technique that combines collision-induced dissociation (CID) and ozone-induced dissociation (OzID; i.e., CID/OzID), we determine that lipids incorporating FAs from supplementation have isomer profiles that are distinct from lipids incorporating de novo synthesized (or previously stored) FAs. Monitoring the distribution of sn-isomers across resected LNCaP xenograft tumors (from mice) using isomer-resolved molecular imaging reveals that apocromeric species are spatially correlated to PUFA containing GPLs and lysolipids. Treating mice with a potent and selective PLA2 group IIA (group IIA secretory-PLA2 [sPLA2-IIA]) inhibitor (KH064) and conducting the same correlative analysis reveal that spatial correlation between apocromer, PUFA-GPLs, and lysolipids is weakened. Given that there are multiple known changes to the physicochemical environment of cancer tumors (24, 25) and lysolipid acyl chain migration is known to be affected by pH and temperature (26), we propose that lipid apocromer species are the product of lipid remodeling in a perturbed physicochemical environment and show potential as a biomarker for aberrant lipid metabolism in cancer tumors.
Materials and methods
Materials
Methanol (LC-MS grade), acetonitrile (Optima®), water (Optima®), and isopropanol (Optima® LC-MS grade) for lipid LC-MS were obtained from Fisher Scientific, Scorseby, Victoria, Australia. Ammonium acetate for lipid LC-MS was obtained from Sigma-Aldrich, North Ryde, New South Wales, Australia. For MALDI-MS-related sample preparation and analysis, sodium acetate (≥99%), 2,5-dihydroxyacetophenone (≥99.5%, Ultrapure), methanol (LC-MS grade), and chloroform (HPLC grade) were obtained from Sigma-Aldrich, Zwijndrecht, The Netherlands.
Biological sample preparation
Ethics and murine model
Animal ethics approval was granted by the Office of Research Ethics, University of Queensland and Queensland University of Technology, and assigned AEC approval number QUT/260/18. All testings were undertaken in accordance with accepted standards of humane animal care outlined by the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes. Cell line use was approved by Queensland University of Technology Human Research Ethics. Six-week-old male nonobese diabetic/severe combined immunodeficieny mice were purchased from the Animal Resource Centre (Australia) and maintained under temperature-controlled conditions and a 12 h light-dark cycle. Mice were allowed to acclimate for 2 weeks prior to experiment and were provided chow and water ad libitum for the duration of the experiment. Mice were injected subcutaneously in the right flank with LNCaP cells (1 M cells) in a 1:1 ratio of cells and Matrigel® (100 μl; Corning®). Bloods were collected once weekly, and prostate-specific antigen was measured by ELISA to monitor PCa progression. Mice were weighed, and tumor volume was measured using digital callipers thrice weekly. Tumors were allowed to grow until prostate-specific antigen reached 25–50 ng/ml before mice were castrated. One week following castration, mice were randomized to receive an sPLA2-IIa inhibitor (KH064; 5 mg−1 kg−1 day−1 by oral gavage) or vehicle control (0.5% carboxymethylcellulose). Mice were culled when the ethical end point was reached, defined as maximum tumor volume (1,000 mm3), or when an Animal Ethics Committee-approved welfare score required euthanasia. Xenografts and tissues were collected and weighed. Tumors were H&E stained for morphological assessment, whereas tissues were halved and either: i) placed in liquid nitrogen until moved to storage at −80°C or ii) stored in neutral-buffered formalin fixation and paraffin embedded.
Tissue sectioning and mounting
Frozen resected tissues were freeze mounted to the microtome block using water and sectioned at 10 μM thickness for lipid analyses using a CM 1950 Cryostat (Leica Biosystems, Nussloch, Germany) and using a blade that was free from OCT compound. Tissue sections were thaw mounted on standard glass slides (SuperFrost Plus, Menzel-Gläser, Braunschweig, Germany) before MALDI-MS imaging (MSI)-OzID protocol (see the “MSI of lipid isomers” section) and the procedure used for H&E staining (see the “H&E staining” section).
H&E staining
A standard H&E protocol was used for staining of xenograft tissues (70% ethanol 2× 3 min, water 1× 3 min, hematoxylin 1× 3 min, water 1× 3 min, eosin 1× 30 s, water 1× 3 min, ethanol 1× 1 min, and xylene 1× 30 s). High-resolution optical images of stained tissues were generated using an Aperio CS2 digital pathology slide scanner (Leica Biosystems, Wetzlar, Germany). Hematoxylin and Entellan® were purchased from Merck (Darmstadt, Germany) and eosin Y from J.T. Baker (Center Valley).
Cell culturing
LNCaP (Research Resource Identifier: CVCL_0395) cells were obtained from the ATCC (Manassas, VA) and were cultured in RPMI medium (Thermo Fisher, Waltham, MA) supplemented with 5% fetal bovine serum (Invitrogen, Waltham, MA) and incubated at 37°C in 5% CO2. Medium was changed every 3 days, and cells were passaged at approximately 80% confluency by trypsinization. Cell lines were authenticated by short tandem repeat profiling in March 2018 and December 2020 by Genomics Research Centre (Brisbane, Australia) and routinely tested to exclude mycoplasma infection (Lonza MycoAlert). Cell number and viability was determined by trypan blue staining and using a TC20 Automated Cell Counter (Bio-Rad).
13Carbon tracing
Cells were seeded and cultured under the conditions mentioned previously (see the “Cell culturing” section). After 48 h of seeding, cells were switched to fresh RPMI media supplemented with 10% charcoal-stripped bovine serum (Invitrogen, Waltham, MA) and incubated at 37°C for 30 min to remove FAs from the binding sites of BSA. FA-depleted cells were then supplemented with either unlabeled (i.e., 12C16-PA) or labeled (i.e., 13C16-PA or 13C18-stearic acid [SA]) FAs at a final concentration of 20 μM. All FAs were purchased from Sigma-Aldrich, Castle Hill, Australia. Cells were grown for a further 72 h and washed twice with ice-cold PBS before lipid extraction. Cell number and viability were determined by trypan blue exclusion with a TC20 Automated Cell Counter.
Lipid extraction
Cell lipids were extracted using methods similar to those described by Matyash et al. (27) and quantified with deuterated lipid internal standards (SPLASH Lipid-o-mix®; Avanti Polar Lipids, Alabaster, AL). To reduce pipetting error, a bulk stock solution for the internal standards was made (720 μl methyl tert-butyl ether [0.01% butylated hydroxytoluene], 40 μl SPLASH Lipid-o-mix®, and 20 μl nonadecanoic acid in methyl tert-butyl ether [3.35 mM] per 2 M cells). Cell pellet clear glass vials (∼2 M cells) were washed with PBS solution twice before adding 220 μl of methanol and 780 μl of bulk internal standard solution. Vials were vortexed for 20 s prior to 1.5 h bench-top agitation. About 200 μl of aqueous ammonium acetate (150 mM) was added to each sample to induce phase separation before samples were vortexed for 20 s and centrifuged for 5 min at 2,000 g. Supernatants were collected in a clean labeled glass vial via pipette and stored at −20°C before analyses.
Analytical methods
MSI of lipid isomers
Samples for MALDI-MSI were placed into microcentrifuge tubes, purged with nitrogen gas, and stored on dry ice for interlaboratory shipping. Samples were stored at −80°C upon arrival. Immediately prior to analysis, tissues were first sectioned as per the “Tissue sectioning and mounting” section and thinly coated with 12 passes (45 mm spray height, 30°C, 10 psi, 2 mm track spacing) of 100 mM sodium acetate dissolved in 2:1 methanol/chloroform using an HTX TM-Sprayer (HTXImaging, Chapel Hill, NC). 2,5-Dihydroxyacetophenone was then sublimated (40 mg, 160°C, 4 min) to the sample slides using a sublimator (HTXImaging). Two separate MALDI-MSI experiments were undertaken on 10 μm tissue sections that were adjacent to H&E-stained tissues. The first was the identification and spatial distribution of sum composition lipid species via high mass resolution (120 k at m/z 400) and mass analysis (≤3 Δppm), and the second was the identification and spatial distribution of PC 34:1 sn-isomers (using MALDI-MSI-CID/OzID). All MSI experiments were performed on an Orbitrap Elite (Thermo Fisher Scientific, Bremen, Germany) mass spectrometer with MALDI-MSI pixel size of ∼50 μm. The commercial ion source and stacked ring ion guide were replaced with an elevated-pressure MALDI ion source incorporating a dual-ion funnel interface (Spectroglyph LLC, Kennewick, WA) as has been described previously (28). The MALDI laser was operated at a repetition rate of 100 Hz and pulse energy of ∼1.0 μJ. The laser was focused to a spot size of ∼15 × 12 μm as determined by the size of ablation craters in desorbed matrix. Pressure within the ion source was set to 10 mbar in the first ion funnel and 2 mbar in the second ion funnel. The mass spectrometer was modified to allow for ozonolysis, as previously described (23). Briefly, using oxygen as a feed gas (99.999% purity; Linde Gas Benelux BV, The Netherlands), ozone was generated via a high-concentration generator (TG-40; Ozone Solutions, Hull, IA) and was introduced into the helium buffer gas flow before conduction through to the high-pressure region of the linear ion trap (LIT). Ozone concentration was measured online using a UV absorption-based ozone monitor (106-H; 2B Technologies, Boulder, CO). For the determination of FA sn-position, precursor ions were mass selected using an isolation width of m/z 1. Operating in positive polarity mode, CID/OzID and full MS scans were alternated between for each pixel. MS3 CID/OzID of m/z 782 (using normalized collision energy [NCE] of 42 for MS2 and an activation time (AT) of 250 ms for MS3) was conducted and detected in the LIT, and full MS detection was undertaken by the Fourier Transform-orbitrap detector operating with a mass resolving power of ∼60,000.
Direct infusion ESI-OzID of lipid double bonds
The double bonds (DBs) of intact GPLs were determined via MS using an Orbitrap Elite high-resolution mass spectrometer (Thermo Fisher Scientific, Bremen, Germany) modified with an ozone generator (Titan-30UHC Absolute Ozone, Edmonton, Canada), as described in “MSI of lipid isomers” section. In this instance, a diverter valve was placed on the nitrogen gas inlet to the higher energy collisional dissociation cell, and nitrogen was replaced with generated ozone gas (29). Operating in positive-ion mode for PC lipid OzID, cell line lipid extract samples were mixed 1:1 (v/v) with 1 mM methanolic sodium acetate solution and introduced to the mass spectrometer via a chip-based nanoelectrospray source (TriVersa Nanomate, Advion, Ithaca, NY) using 1.35 kV/0.35 psi spray parameters. Using the Thermo Xcalibur software package, a data-independent acquisition sequence was created to perform sequential OzID (AT = 5 s [higher energy collisional dissociation] and collision energy [CE] = 1 V) and CID/OzID (MS2: AT = 5 ms [LIT], NCE = 33; MS3: AT = 500 ms, NCE = 0) for six labeled and unlabeled sodiated PC precursor ion masses. Maximum injection time was 100 ms, and isolation window was ± 0.5 Da across a 175–1,000 Da scan range. Included sodiated precursor ion m/z values were 754.6, 782.6, 770.6, 772.6, 798.6, and 800.6. The product ions from all fragmentation experiments were detected using the orbitrap mass analyzer for high resolution and accurate mass, allowing for unambiguous assignment of characteristic fragments to specific lipids and not isobaric lipids or isotopes. Intensity values obtained from OzID and CID/OzID mass spectrometric experiments were the average of 11 and 28 scans, respectively.
Adapting the aforementioned positive-ion mode experiments, MS4 experiments were created for simultaneous full structure elucidation, including DB location and sn-position. In brief, sodiated PC precursor ions were mass selected to undergo OzID in the LIT (MS2: NCE = 0 and AT = 10 s). The OzID-aldehyde product ion of one DB position was then mass selected to undergo CID in the LIT (MS3: NCE = 33 and AT = 5 ms) to form a product ion corresponding to the loss of phosphocholine, [M-183]+. This product ion was then mass selected for additional OzID in the LIT (MS4: NCE = 0 and AT = 500 ms) to yield fourth-generation product ions that are characteristic to FA chain lengths at either sn-1 or sn-2 positions for the DB location that was selected for MS3 activation. Because of the multiple levels of MS for species confirmation, detection was achieved by the low-resolution detector of the LIT. This experiment was then repeated for the remaining OzID-aldehyde product ions from each DB location.
Conventional lipidomics for phospholipid profiles
Lipid extracts from cell lines were run through an automated lipidomics workflow using an LC-20A HPLC (Shimadzu, Kyoto, Japan) set to deliver 100 μl sample loop injections into a mobile phase of 5 mM methanolic ammonium acetate flowing at 15 μl/min. The sample column and column oven were bypassed with Viper PEEKsil (50 mm; Thermo Fisher Scientific, Waltham, MA) to maintain instrument back pressure limits. Sample lipids were then directly infused through the electrospray ionization source of a QTRAP 6500 hybrid triple quadrupole/LIT mass spectrometer (SCIEX; Concord, ON, Canada) using a spray voltage of 5 kV, a source temperature of 150°C, and both source gasses set to 15 (arb.). Various precursor ion and neutral loss scans were employed to confirm lipid head group, with the detected m/z being indicative of summed-fatty-acyl composition (PC: precursor ion scan m/z 184.2, CE: 39 V; PE: neutral loss [NL] m/z 141.1, CE: 29 V; phosphatidylserine [PS]: NL m/z 185.1, CE: 29 V; phosphatidylglycerol [PG]: NL m/z 189.1, CE: 29 V; phosphatidylinositol [PI]: NL m/z 277.1, CE: 29 V; and cholesteryl esters: precursor ion scan m/z 369.4, CE: 29 V). Instrument blanks were run throughout to ensure no sample carryover, and pooled batch quality controls were used to gauge instrument performance over the duration of the experiment.
Quantification and statistical analysis
MALDI-MSI OzID for lipid DB imaging
MS spectral data files were first stitched together with MALDI imaging co-ordinate positions and collated as a MATLAB data table. Total ion current normalization of the data was achieved by dividing each spectral peak by the total ion abundance in each MSI pixel and comparing this to all other pixels. Lipid abundance and distribution data were then visualized through MATLAB scripts developed by Eijkel et al. (23, 28). Fractional distribution images (FDIs) were calculated by plotting the intensity of one monitored ion divided by the combined intensity of all monitored ions (i.e., Ia/[Ia + Ib]).
Conventional lipidomics for phospholipid profiles
LipidView (version 1.3 beta; SCIEX) was used for data processing of SCIEX data files obtained from the QTRAP 6500. Lipid assignments were based on the software lipid tables and shortlisted to include even-chain lipids with 0–6 s. Odd-chain/ether-lipid data were obtained but were not included in this study because of the ambiguity in assigning isobars in low-resolution MS. Isotope correction factors were applied, and MS peaks were ratioed to the isotope-corrected internal standard included in each scan type. The inclusion of deuterated and odd-chain FAs within the internal standard lipids sufficiently mass shifted internal standards away from any biological lipids, therefore allowing accurate and reliable peak intensity measurements to be discerned. Data tables were extracted from LipidView and imported to Microsoft Excel for cell count normalization, internal standard concentration factoring, statistical analysis, and graphing.
Statistics and error analysis
The mean with 95% confidence intervals was used for error analysis on column charts throughout and was calculated using Microsoft Excel and conventional equations. For Fig. 1, the mean and variance were established using Microsoft Excel to calculate a paired t-test. Subsequent t-distribution values were translated to statistical significance (P values) via relevant degrees of freedom and a critical t-value table. While statistical analyses for isotope tracing studies were conducted on small biological replicate populations (n = 2), the inclusion of statistical significance values remains valid as reporting followed the recommendations of de Winter (30) for t-testing of extremely small sample sizes. In Fig. 4, heatmap was calculated using R x64 3.6.1 packages and built-in functions (i.e., PerformanceAnalytics, Hmisc(), and corrplot()). The images in Fig. 5 refer to precursor ions from the same tissue section, except for the sn-isomer FDI and the H&E, which were conducted on two separate serial sections. Equivalent analyses were conducted on a replicate resected tumor and an sPLA2-IIa inhibited (KH064) resected tumor, both of which can be found in the supplemental data. Specific statistical details for each of the figures (including number of replicates, P value representation, and error model) can be found within the respective figure captions.
Fig. 1.

The abundance of GPL classes and PC sum compositions (including label-incorporated species) arising from 12PA, 13PA, and 13SA supplementation. Left: GPL sum compositions summed by class and separated by dn-lipidome (blue) and up-lipidome (yellow) abundances; represented in (A) absolute abundance (nmol/1 M cells) and (B) relative abundance (mol%). Right: The effects of supplementation on the top 15 most abundant PC lipids; represented in (C) absolute abundance (nmol/1 M cells), and (D) relative abundance (mol%). Biological replicates: n = 2 (NS: n = 3) and technical replicates: n = 7, mean ± 95% confidence interval displayed. Paired t-test P values: ∗0.05, ∗∗0.01, and ∗∗∗0.001. 12C16-PA supplement (12PA), 13C16-PA supplement (13PA), 13C18-SA supplement (13SA).
Fig. 4.

The effects of FA supplementation on the degree of unsaturation of GPL species. Left: Bar charts showing the degree of unsaturation in PSs in either absolute (A) or relative (B) abundance scales. Degree of unsaturation has been separated into SFA (blue), MUFA (orange), polyunsaturated with two or three DBs (PUFA ≤3, green), or polyunsaturated with four or five DBs (PUFA ≥4, yellow). C: Heatmap showing the influence of 13PA or 13SA FA supplementation on either the labeled/up-lipidomes or unlabeled/dn-lipidomes relative to the lipidome arising from 12PA supplementation. Sum composition species are indicated across five GPL classes. Biological replicates: n = 2 and technical replicates: n = 7, mean value displayed for absolute abundance or mean value ± 95% confidence interval displayed for relative abundance.
Fig. 5.

MALDI-MSI and MALDI-MSI-OzID molecular imaging of lipids and the H&E stain of resected LNCaP xenograft tissues. A–F: The abundance and spatial distribution of (A) PC 32:0, (B) PC 34:1, (C) PC 36:1, (D) lyso-PC (LPC) 18:0, (E) PC 38:4, and (F) PC 40:6. G–I: Tissue FDIs expressing the relative abundance of the indicated lipid(s) as a function of both lipids for (G) (PC 38:4 + PC 40:6)/PC 34:1, (H) (PC 18:1/16:0)/PC 16:0/18:1, and (I) (TAG [48:0, 48:1, 48:2, 50:0, 50:1, 50:2, 50:3, 50:4, 52:0, 52:1, 52:2, 52:3, 52:4, 52:5, 52:6, 54:1, 54:2, 54:3, 54:4, 54:5, and 54:6])/PC 34:1. J: The H&E-stained tissue showing the tumor cells (red) and host adipocyte cells (black) in a representative treatment-naïve (vehicle) LNCaP xenograft.
Results
FA origin and its influence on isomeric lipid remodeling
To investigate the impact of FA supplementation, LNCaP cells were either nil supplemented (NS) or supplemented with 12C16-16:0 (12PA), 13C16-16:0 (13PA), or 13C18-18:0 (13SA). Using conventional lipidomics methods, the GPL sum composition for each supplement was determined, giving total GPL class abundance (Fig. 1A, B). Given the mass spectrometric approach to analysis, unlabeled lipids generated by the cell (now referred to as the de novo [dn]-lipidome [Fig. 1A, B, blues]) and lipids incorporating the isotope labels from FA uptake (now referred to as the uptake [up]-lipidome [Fig. 1A, B, yellows]) were easily distinguishable by their mass (example structures in Fig. 1). Comparison of relative GPL abundance (Fig. 1B) is preferred over absolute abundance (Fig. 1A) as this minimized uncertainty when comparing between experiments. This comparison revealed that SFA supplementation had no apparent impact on GPL class populations (Fig. 1B) although slight differences in contributions of de novo (blue) and uptake (yellow) fractions from either 13PA or 13SA cells could be observed. In contrast, much greater variation was observed when exploring abundance shifts of the top 15 PC sum composition lipids (Fig. 1C, D), including specific differences between the dn-lipidomes and up-lipidomes. For example, statistically significant increases in unlabeled PC 34:0 within the dn-lipidome were apparent after the labeled 13PA (P ≤ 0.01) and 13SA (P ≤ 0.05) supplements were introduced (Fig. 1D, blues). Concurrently, within the up-lipidome (Fig. 1D, yellows), 13PA or 13SA caused significant increases in PUFA PC lipids, PC 34:2 (P ≤ 0.01 & P ≤ 0.001), PC 34:3 (P ≤ 0.001 & P ≤ 0.05), and PC 36:3 (P ≤ 0.05 & P ≤ 0.05).
Considering the variation between the dn- and up-lipidome(s) at the sum composition level, sn-isomer analysis populations were established using CID/OzID. Observing the sn-isomer compositions of PC 32:1 within the dn-lipidome (Fig. 2A), LNCaP cells grown without FA supplementation (NS) were seen to favor canonical sn-2 unsaturated lipids, showing a combined apocromer contribution of 13% the total dn-PC 32:1 (9% PC 16:1/16:0 [light orange] and 4% PC 18:1/14:0 [light gray]). Supplementing LNCaP cells with 12PA, 13PA, or 13SA decreased the overall contribution of canonomers PC 14:0/18:1 (3–5%) and PC 16:0/16:1 (3–5%), resulting in a concomitant increase of the apocromer PC 16:1/16:0 (8–10%). Figure 2B shows the individual percentage contribution of canonomer (white bars and lipid structure) and apocromer (black bars and lipid structure) for the two FA compositions comprising dn-PC 32:1. Notably, this showed that the uptake (but not incorporation) of extracellular FA increased the relative abundance of apocromer species in the dn-lipidome.
Fig. 2.

The distribution of DB and sn-isomers within the dn-lipidomes and up-lipidomes after FA supplementation. A: Bar charts showing the FA sn-isomer distribution of two de novo PC 32:1 FA compositional isomers, namely, PC 16:0_16:1 (oranges) and PC 14:0_18:1 (grays) and the pie charts showing the distribution of DB isomers within these two lipid species. B: Proportions of PC 32:1 apocromer (black) and canonomer (cream) for each cell supplement—representations of the chemical structure for each are found topmid. C: Bar charts showing the sn-isomer distribution of two uptake PC 32:1 lipid species, namely, PC 16:0_16:1 (oranges) and PC 14:0_18:1 (grays). D: Labeled PC 32:1 lipid displayed in C can either have the labeled FA unmodified or modified by desaturation and/or β-oxidation (chemical structures represented bottom-mid). The proportions of apocromer (black) and canonomer (white) are compared between these unique isomers and isotopomers with the labeled FA chain being indicated with an asterisk. Pie charts in (D) are representative of the DB isomer distribution from the unsaturated FA chain. The values displayed throughout are the relative abundance mean for biological replicates: n = 2 (NS: n = 3) and technical replicates: n = 7. FA supplements are indicated below each chart (+), and isotope labeling is indicated in blue with a double dagger (ⱡ).
As with changes to FA sn-position, variation in the distribution of DB locational isomers within each supplement experiment was observed (Fig. 2A, pie charts). Comparative to NS cells, FA supplemented cells (12PA, 13PA, and 13SA) exhibited slight decreases of n-8 (≤1%), n-9 (1–3%), and n-10 (1–2%) and a concomitant increase of n-7 (3–5%). Together, this combination of increased apocromer contribution (Fig. 2A, bar charts) and increased n-7 DB isomers (Fig. 2A, pie charts) suggested an association between the n-7 DB and the sn-1 position of PC 32:1. A similar pattern was evident in the PC 34:1 analysis (supplemental Fig. S1D, pie charts), where n-7 and n-10 appeared to be positively correlated with sn-1 isomers, whereas n-9 correlated with sn-2.
Investigating the sn-isomer distributions of the isotope-labeled GPLs that constituted the up-lipidome of 13PA and 13SA cells, the labeled FAs from uptake were observed to either be directly incorporated into the lipid or were incorporated in combination with cellular modification (e.g., desaturation, elongation, or β-oxidation). It has previously been shown that the ordering of these metabolic events determines the species of the FA (31). The strength of supplementing with isotopically labeled FAs combined with mass spectrometric fragmentation strategies for isomer elucidation is that the shift in m/z caused by the inclusion of isotopes provides additional confirmation of FA modification processes. For example, using CID and CID/OzID strategies, the 13SA supplement (i.e., FA 18:0) displayed four distinct metabolic fates prior to (or post) incorporation into PC 32:1 (Fig. 2C, D, pie charts). These included i) direct desaturation to form FA 18:1 n-9, ii) β-oxidation to form FA 16:0, iii) sequential β-oxidation and desaturation to form FA 16:1 n-7 and FA 16:1 n-10, and iv) the inverse sequence desaturation and β-oxidation to form FA 16:1 n-9. In addition, FA 16:1 n-12 was formed, which given literature on desaturase Δ-position specificity, could potentially arise from either sequence of β-oxidation and desaturation or direct Δ4-desaturation of FA 16:0. Notably, the modification sequence that the labeled FA underwent was observed to influence the sn-position at which it was incorporated (Fig. 2C, D, bar charts). For example, compared with the sn-positional isomer distribution of NS LNCaP (Fig. 2B), in Fig. 2D, the β-oxidation of 13SA to FA 16:0 increased favorability of the canonomer fraction, whereas combined β-oxidation and desaturation of 13SA to FA 16:1 increased favorability of the apocromer fraction.
As was expected, 13PA cells exhibited an increase within the relative abundance of the PC 16:0_16:1 composition (Fig. 2C). Unexpectedly however, 13SA cells exhibited a greater increase of PC 16:0_16:1 (β-oxidation) over PC 14:0_18:1 (direct desaturation). For comparison back to the unlabeled lipidome (Fig. 2B), the labeled (ⱡ) PC 32:1ⱡ data were investigated as a percentage contribution of canonomer or apocromer species (Fig. 2D). As was observed for the 13PA cells in the left panels and mid panels of Fig. 2D, the 13PA supplement and its modified progeny (FA 16:1ⱡ) favor apocromer distribution (PC 16:1/16:0) 11–12%—equivalent to the isomer distribution of NS cells (Fig. 2B, left; 11%). Within 13SA cells, for the 13SA supplement (FA 18:0ⱡ) to incorporate into PC 16:0_16:1, it required cellular modification by β-oxidation (to form FA 16:0ⱡ) or by combined β-oxidation and desaturation (to form FA 16:1ⱡ). When FA 16:0ⱡ was formed from 13SA, it showed an 8% distribution to the sn-2 position (PC 16:1_16:0ⱡ; Fig. 2D, left)—∼3% less favored than NS cells (Fig. 2B, left; 11%). In contrast, when 13SA cells formed FA 16:1ⱡ (Fig. 2D, mid), this instead occupied the sn-1 position (PC 16:1ⱡ/16:0) in ∼22% of the species, which was similar to the unlabeled/dn-lipidome of cells having received FA supplements (Fig. 2B, left; 20–22%). Shifting focus to the variation observed within the PC 14:0_18:1 FA composition, when the 13PA supplement from 13PA cells underwent two cellular modification events (i.e., desaturation and elongation; Fig. 2D, right), FA 13C16-18:1ⱡ displayed an isomer distribution of ∼19% to the sn-1 position (PC 18:1ⱡ/14:0). This result is near identical to the sn-isomer distribution of PC 14:0_18:1 within NS cells (PC 18:1/14:0; 20%; Fig. 2B, right). 13SA cells again showed contrast with this result, with modification of the 13SA supplement by direct desaturation showing a 4% distribution of the FA 18:1ⱡ to the sn-1 position (PC 18:1ⱡ/14:0; Fig. 2D, right). Together, these results present that within 13PA cells the incorporation of 13PA and its progeny to labeled PC 32:1ⱡ showed close correlation with the sn-isomeric distribution of the unlabeled PC 32:1 within NS cells. In contrast, within 13SA cells, the incorporation of the cell-modified 13SA supplement to specific sn-positions of labeled PC 32:1ⱡ was significantly influenced by the types and number of modifications it underwent.
Considering that the fatty acyl composition (and sn-isomer distribution) of labeled PC 32:1ⱡ have been established (Fig. 2D), DB isomers can be associated with specific lipid species, as is seen within the pie charts of Fig. 2D. Exploring the position of DBs in labeled PC 32:1ⱡ from cell samples using OzID (32), the site of unsaturation was seen to exist in either the unlabeled/dn-FA or the labeled/extracellular FA. If the cells were supplemented with 13PA and no further cellular modification occurs (i.e., FA 16:1 is the unlabeled acyl chain), n-10 fractions remained the same, whereas n-9 decreased and n-7 increased. In contrast, if 13PA instead underwent cellular desaturation, n-7 and n-10 fractions increased, whereas n-9 fractions decreased. From supplementation with 13SA cells, it could be observed that n-9 was the only detectable DB position in the label when being incorporated to PC 14:0_18:1ⱡ. Interestingly, if 13SA underwent both β-oxidation and desaturation (FA 16:1ⱡ), an additional FA 16:1 n-12 DB isomer arose alongside the increase in FA 16:1 n-9—perhaps indicating the origin of the species from a specific subcellular compartment. Alongside the sn-isomer distributions (Fig. 2D, bar chart), these DB isomer fractions (Fig. 2D, pie charts) suggested that FA 18:1 n-9 is distributed more commonly to the sn-2 position and n-7 FAs are more likely associated with the sn-1 position (attribution of this effect cannot be specified to FA 16:1 n-7 or FA 18:1 n-7 in this analysis; for specification of FA, vide infra Fig. 3).
Fig. 3.

Simultaneous elucidation of fatty acyl composition, sn-isomeric position, and DB locations for the full structural elucidation of PC 32:1 within LNCaP cells. Bar charts showing the sn-isomeric distribution (apocromeric in light and canonomeric in dark colors), for each of three DB positions (n-7: purple, n-9: red, and n-10: green) across the two fatty acyl compositional isomers (PC 16:0_16:1, PC 14:0_18:1) within the PC 32:1 species. The 12 molecular structures for each of the measured species are located either side of the chart. n = 3, mean value ± SEM (95% confidence interval).
The relationship between sn- and DB position
To investigate an explicit relationship between DB and sn-positional isomers, mass spectral dimensionality was increased (MS4) to combine both OzID and CID/OzID experiments (i.e., ordered as OzID/CID/OzID). As such, the distribution of canonomer and apocromer for each FA DB location is quantitative; however, absolute mole quantitation of each DB FA sn-isomer would still require establishing OzID reaction rates or developing calibration curves. This technique is not without limitations, however; to achieve the analysis requires i) the lipid to be detectable in positive polarity MS, ii) the ability of precursor ions to form sodiated adducts, and iii) high concentrations of ozone to be generated and passivated into the high-pressure trapping regions of the MS instrument. As such, applications of this technique are currently limited to PC lipids with high cellular abundance such as PC 32:1 and PC 34:1.
Applying this methodology to PC 32:1 from NS LNCaP (Fig. 3) revealed the presence of two fatty acyl composition isomers (PC 16:0_16:1 and PC 14:0_18:1) with the DB in each existing in one of three possible locations (n-7, n-9, or n-10), and with the monosaturated FA being in either canonomeric (sn-2) or apocromeric (sn-1) positions on the glycerol backbone. In total, 12 discrete isomers were identified within the PC 32:1 sum composition from NS LNCaP. From a quantitative perspective, within the PC 16:0_16:1 composition (Fig. 3, left), the n-7 (purples), n-9 (reds), and n-10 (greens) DBs appear in the apocromer PC 16:1/16:0 (light colors) with a frequency of 29% (±4), 46% (±9), and 54% (±3), respectively. Within the PC 14:0_18:1 composition (Fig. 3, right), the n-7, n-9, and n-10 DB isomers displayed an sn-isomer distribution that was distinct from PC 16:0_16:1, with apocromer PC 18:1/14:0 contributing 63% (±11), 27% (±8), and 68% (±5), respectively. These results supported the findings from the aforementioned analyses and confirmed that the 18:1 n-7 isomer indeed had a higher probability of existing in the sn-1 position of the PC 14:0_18:1 species (i.e., PC 18:1 n-7/14:0). Similarly, n-10 DBs display a similar preference for the sn-1 positions of the PC 16:0_16:1 and PC 14:0_18:1 species (i.e., PC 16:1 n-10/16:0 and PC 18:1 n-10/14:0), whereas n-9 shows mixed sn-position occupancy that appeared dependant on fatty acyl composition (i.e., increased relative abundance of both PC 16:1 n-9/16:0 and PC 14:0/18:1 n-9 isomers). An equivalent full structure analysis for PC 34:1 can be found in supplemental Fig. S1E and shows that within the PC 16:0_18:1 species, n-7 and n-10 DBs were associated with sn-1 isomers, whereas n-9 showed strong preference for the sn-2 position.
Influence of SFA supplements on unsaturated membrane lipids
Figure 4 displays the widespread effects FA uptake has on GPL lipid remodeling and FA degree of unsaturation. Complex lipidomic analysis across five GPL subclasses was conducted using the 12PA, 13PA, and 13SA cell samples. Within all GPL classes, the uptake of SFAs impacted the degree of unsaturation in both the unlabeled/dn-lipidomes and labeled/up-lipidomes (for full lipid profiles, see supplemental Fig. S2). Using the PS class as an example (Fig. 4B), lipids were separated by the degree of unsaturation to reflect the total contribution of SFA (blue), MUFA (orange), and PUFAs to PS profiles. PUFA PS was further separated into lipids with 2 or 3 DBs (as a proxy for mainly de novo desaturation; green) and lipids with 4, 5, or 6 DBs (to approximately represent lipids containing “dietary” FAs likely obtained from uptake; yellow). In Fig. 4B and relative to the 12PA cells, PS within the unlabeled dn-lipidome of 13PA cells appeared to display similar fractions of SFA, MUFA, and PUFA. Notably, the ordering of these mole fractions was maintained between 12PA and dn-13PA—with MUFA being the most abundant, sequentially followed by PUFA ≤3, PUFA ≥4, and SFA. In contrast and again relative to the 12PA PS profile, the PS within the labeled up-lipidome of 13PA cells displayed an increase of SFAs along with decreases in MUFA and PUFA ≥4. Interestingly, the degree of unsaturation ordering these lipids within up-13PA is perturbed and instead showed PUFA ≤3 being almost equally abundant as MUFA, and SFA now being more abundant than PUFA ≥4. When comparing the PS lipids of up-13SA, slight increases within the PUFA ≥4 fraction (5%) were observed; however, the ordering of degree of unsaturation fractions appeared equivalent to the PS profile from 12PA. When instead comparing the dn-13SA PS profile, large-scale change could be observed, with MUFA, PUFA ≤3, and PUFA ≥4 displaying approximately equal abundance. What is most intriguing about this perturbance in the PS lipids of dn-13SA is that the 13SA supplement itself did not contribute to the observed variation as this lipidome fraction did not carry any isotopic label. Instead, this result implies that SA from uptake induced lipid remodeling to include twice as much PUFA ≥4 to the unlabeled dn-PS fractions, whereas this effect is only minor in any newly formed PS that incorporated the 13SA label in the labeled up-lipidome. This effect was apparent within all monitored GPLs (cf., supplemental Fig. S2).
Considering the independent changes to the degree of unsaturation within the dn-lipidomes and up-lipidomes after FA supplementation, it was decided that individual lipid speciation should be explored for any variation. To achieve this, a heatmap was created where the mol% of FA sum compositions across five GPLs from the dn-lipidomes and up-lipidomes of 13PA and 13SA cells were compared against 12PA cells as a fold change (Fig. 4C). Overall, this effect of increased PUFA for dn-13SA could be observed across all GPL classes and was exemplified by increases in PI, PS, PC, and PE 36:5 and 36:6 and also PI, PS, PC, PG, and PE 38:6. Although affecting fewer GPL classes, the up-13SA profiles also displayed this effect but in addition showed decreases in PE 38:4 and PE 40:4 and increases in PC, PE, and PI 34:4. The change within GPLs with five or six degrees of unsaturation (i.e., EPA or DHA) was less significant in both dn- and up-13PA profiles; however, the impact 13PA supplementation had on GPLs with four degrees of unsaturation (i.e., AA) was more widespread than 13SA.
As mentioned previously, PUFA ≥4 is broadly representative of “dietary” FAs, and its increase amongst all monitored dn-GPLs after PA or SA supplementation suggests that these PUFAs are being sourced and remodeled from another lipid class, such as other GPLs or more likely, being sequestered from triacylglycerols (TAGs) (33, 34). Interestingly, within the dn-13SA profiles, an increase within the PC 40:5, PC 40:6, and PE, PS, PI 40:6 could be observed. This is in contrast to the up-13SA profiles, where PS, and PE 40:5 and 40:6 were seen to decrease. Together, these increases in 40:5 and 40:6 from the dn-lipidome and decreases in PUFA PS and PE from the up-lipidome suggest that the remodeling of PUFA from other lipid classes after FA uptake was GPL class specific. Overall, these results display that the uptake of FAs not only impacted the preexisting (de novo) lipidome but was also influential in the remodeling of PUFAs between the GPLs (and other unmonitored classes) in a class-specific manner.
Implications of FA signaling and lipid remodeling in tissue
We have shown that FA supplementation influences the position and degree of unsaturation as well as the sn-positions of FA in PCa cells. These nutrient-driven changes are highly relevant to in vivo type experiments, such as xenograft tissue models, where extant cancer cells are frequently subject to variations in the physicochemical environment and nutrient supply. Indeed, these environmental variations also better represent the usual conditions that cancer cells regularly must endure and thus improve applicability of findings to human cancers. To search LNCaP xenograft tumors for the signatures of metabolic shifts discussed earlier, we deployed MSI technologies capable of lipid-isomer resolution (MALDI-MSI-OzID).
Figure 5 shows the MS signal intensity for lipid species as a function of position to display the relative abundance of the indicated lipid across the resected LNCaP xenograft tumors. Sum composition lipids (Fig. 5A–F) were characterized with high mass resolution and subsequently identified by comparisons to theoretical exact mass (≤3 ppm), and GPL sn-isomers were resolved using CID/OzID. To compare and correlate the distributions of these lipids (Fig. 5G–I), FDIs were created (23, 35, 36). All images were compared against the structural features of the adjacent H&E-stained tissue (Fig. 5J). The H&E image reveals two main features; a round tumor bolus that is dense with cancer cells, and host adipocyte cells surrounding the bolus. Comparing the sum composition lipid images of the top row to the tissue H&E stain, particular lipids showed high-levels of spatial correlation with either of the tissue features. For example, PC 32:0 (Fig. 5A) appeared in higher abundance at the perimeter of the fatty tissue, whereas in contrast, PC 36:1 (Fig. 5C) was confined to the center. It has previously been shown that the acyl chain composition of these two species is dominated by the 16:0_16:0 and 18:0_18:1 fatty acyl compositions (37), which represent two unique pathways for formation. It is interesting to see that these two unique synthesis pathways are also spatially distinct, which alongside the differences observed in the labeled supplement study, might suggest that the production and release of PAs and SAs differs between the tumor cells and host adipocyte cells.
One of the more striking observations is the large variation in the abundance of PC 34:1, in either the tumor or host adipocyte cells. While this particular lipid appears to be more abundant in the bolus and less so in the fatty tissue, other lipids such as lyso-PC 18:0, PC 38:4, and PC 40:6 display the complete inverse. This was best observed within the FDI image (Fig. 5G), which revealed that PUFA-GPLs, PC 38:4 and PC 40:6, were indeed spatially distinct to high-abundance PC 34:1. If PC 34:1 content was divided into its sn-positional isomer distributions (i.e., PC 16:0/18:1 and PC 18:1/16:0; Fig. 5H) and compared against the spatial distribution of PUFA-GPLs (Fig. 5G, H), the apocromer showed high positive correlation with the presence of these species. Given that PUFA-GPLs (e.g., PC 18:0_20:4 and PC 18:0_22:6) are the primary substrate for PLA2 (7), and the intermediary products of this activity are lysolipids, the presence of a lyso-PC 18:0 (Fig. 5D) in this same region is suggestive of PLA2 activity. With the altered pH environments that are commonplace in tumors (24), acyl chain migration (26) occurring in these lysolipids prior to lipid remodeling and the reformation of diacyl lipids would lead to the occurrence of sn-apocromer lipid species. Indeed, the overexpression of sPLA2-IIA is observed in LNCaP cells and is correlated with poor clinical prognosis in PCas (38). Within murine models that were treated with an sPLA2-IIA inhibitor (KH064), correlations between PUFA GPLs and apocromeric lipids were less apparent, suggesting that this migration activity can be partially attenuated by limiting lipase activity (cf., supplemental Fig. S3).
An unexpected result (Fig. 5I) is the correlation between TAG species and the region of extensive lipid remodeling in the host adipocyte cells. Recent studies investigating how external stimuli can affect cancer cell lipid homeostasis found that the degree of FA unsaturation in TAG and GPLs was disrupted by nutrient deprivation and hypoxia (33, 39). Given that the physical conditions in the study attenuate cellular FA desaturation, researchers were able to show that in order to maintain appropriate ratios of membrane-bound saturated and unsaturated FAs, cells sequestered unsaturated FAs from TAG fractions and transferred to membrane GPLs. Therefore, because of the colocalization of TAG and PUFA-GPLs, the increased abundance of PUFA species in GPLs is assumed to come from the repartitioning of TAG stores. Similar to the TAG localization, the fatty tissue appears to house an increased abundance of PUFA ether-lipid species (PC O-32:0 and PC O-34:1) and the tentatively assigned TAG ethers (cf., supplemental Fig. S4). These have been previously identified in mammalian lipid droplets and play a central role in ether lipid metabolism and intracellular lipid traffic (40). Speculatively, given that the main route for DHA synthesis requires peroxisomal β-oxidation (41) and the peroxisomes are fundamental in the production of ether lipids (42), these data suggest differences in peroxisome metabolism between the host adipocyte cells and tumor cells. Indeed, it was recently found that tumor cells can evade ferroptosis by downregulating the peroxisomal synthesis of PUFA ether lipids (43).
Discussion
With exception to mitochondria, FA modification is predominantly undertaken in the endoplasmic reticulum (ER) and thus trafficking of FAs and lipids is necessary to reach the plasma and organelle membranes. Although there are exceptions, lipids mainly require active transport mechanisms to shuttle around the cell. The three conventionally accepted trafficking mechanisms involve membrane transport (e.g., budding from a donor membrane), carrier proteins (e.g., fatty acyl binding proteins [FABPs]), or transfer across membrane contact sites (e.g., ER/mitochondria lipid exchange) (44). Alongside de novo lipogenesis, cells can also obtain extracellular FAs from uptake and incorporate these into lipid structures. From our work within, these differential trafficking mechanisms can be inferred from the variations in the PC (and other GPL) lipid species profile(s) (vide supra Fig. 1D; other GPLs, cf., supplemental Fig. S2). For example, tracing incorporation and modification(s) of labeled PAs and SAs reveals that each FA is either incorporated into more highly unsaturated GPLs or are themselves desaturated more frequently (Fig. 1D; up 13PA/up 13SA; e.g., PC 32:2, PC 34:2, PC 34:3, and PC 36:3). In contrast, GPLs solely incorporating FAs synthesized by cellular machinery are influenced by the supplemented FA such that unlabeled PC 34:0 and PC 36:6 species are discretely increased (Fig. 1D; dn 13PA/dn 13SA). These differences suggest that the cellular management of FAs is dependent on both FA source origin and FA speciation. Thus, we propose that there are distinct mechanisms for the trafficking of FAs originating from de novo lipogenesis and those the cell has acquired from uptake. Previous research has also revealed FA-dependent trafficking differences. One such example can be found in FABP5, a FA binding protein that delivers FAs from the cytosol to nuclear PPARβ/δ. Researchers were able to show that while FAs from uptake all complexed with FABP5, a SFA (PA) inhibited FABP5 activation of the PPARβ/δ pathway, whereas a PUFA (LA; FA 18:2 n-6) instead activated PPARβ/δ (45). This FA-driven modulation of PPARβ/δ has downstream effects in the activation of lipometabolic-centered genes such as pyruvate dehydrogenase kinase 4 (PDK4), angiopoietin 4 (ANGPTL4), perilipin 2 (PLIN2), and cluster of differentiation/FA translocase (CD36). Considering the changes that occur to FA metabolism in cancer, especially including FA uptake and energy production from FAs, the translation of these genes into proteins has widespread implications for oncometabolism (46, 47, 48, 49, 50, 51). Furthermore, this FABP5/PPARβ/δ mechanism highlights how trafficking mechanisms can be entangled with signaling events, while this work provides the foundation for the discovery of the proteins involved in partitioning of dn- and up-FAs.
Given the widespread impact FA trafficking can have on signaling for gene expression and protein translation, it is logical to assume that the cell has fine control over FA uptake, FA trafficking, and regulation of lipid remodeling. This is exemplified in our work by the DB or sn-position differences observed between labeled or unlabeled FAs and possibly relates to the known compartmentalization of desaturase enzymes. Within the pie charts of Fig. 2D, the percentage of n-10 (requiring FADS2 desaturation) is observed to be higher when the labeled PA is modified by desaturation or by combined desaturation and elongation, whereas n-7 and n-9 (requiring stearoyl-CoA desaturase 1 [SCD-1] desaturation) are seen to be higher within labeled lipids where the unlabeled FA is the unsaturated chain. This would suggest that a higher proportion of FAs from uptake are transported to the mitochondria (where FADS2 desaturation mainly occurs) (52), whereas de novo synthesized FAs are more readily transported to the ER (where SCD-1 desaturation occurs) (53). Furthermore, this would indicate that FA speciation and origin (i.e., uptake or de novo) both determine subcellular destination as opposed to the conventional theory that all FAs form a common “pool.” One explanation for this compartmentalization of FA fractions would be for net-positive energy production. Catabolizing FAs that have been actively de novo synthesized, although sometimes required, is counterproductive to energy production. Instead, trafficking FAs from uptake to β-oxidation sites would provide externally produced energy (and carbon) to the cell. Interestingly, FA uptake influences PPARβ/δ activity and subsequent activation of PDK4, which regulates the conversion of pyruvate and glucose to acetyl-CoA and suppresses ferroptosis (46). These functions directly implicate compartmentalized FA metabolism as mitochondria are responsible for cellular energy production via β-oxidation and FA synthesis, whereas the peroxisomes mediate an alternate FA β-oxidation pathway and are largely responsible for the formation of reactive oxidative species and ferroptosis.
A related result that supports the hypothesis of specific FA species compartmentalization can be found in the unusual lipid DB profile displaying 16:1 n-12 and increased 16:1 n-9 (Fig. 2D) after a labeled SA has undergone partial β-oxidation and desaturation to FA 16:1ⱡ. As stated, within the cell, both the mitochondria and peroxisomes have the capacity for β-oxidation; however, peroxisome β-oxidation and stimulation has been shown to have a complex relationship with the genetic expression of FADS1 and FADS2 and hence FADS1 and FADS2 enzymes (54, 55). These enzymes catalyze Δ4, Δ5, Δ6, and Δ8 desaturation (37, 52) and in conjunction with β-oxidation would lead to the formation of the unusual 16:1 n-12 species observed within Fig. 2D (mid). This may suggest that the SA supplement is being split between two trafficking pathways: i) to the ER for SCD-1 desaturation and then membrane-contact transport to the peroxisomes for β-oxidation to yield 16:1 n-9 and stimulate the peroxisomal influence on FADS2 and ii) to the mitochondria for FADS2 desaturation and β-oxidation to yield 16:1 n-12.
Another example of remodeling compartmentalization can be observed in Fig. 3 (left), where PC 16:0_16:1 n-9 and PC 16:0_16:1 n-10 from nonsupplemented LNCaP showed equal apocromer/canonomer distribution, whereas PC 16:0_16:1 n-7 markedly favored canonomeric incorporation. This is a remarkable result as both β-oxidation and FADS2 desaturation (leading to FA 16:1 n-9 and n-10, respectively) are mitochondrial processes (52, 56), whereas the SCD-1 desaturation forming FA 16:1 n-7 would occur at the ER. It should also be noted that the desaturation of FA 16:0 by FADS2 is performed at a rate determined by the relative abundance of FA components in the substrate mixture (56). Similarly, the PC 14:0_18:1 in Fig. 3 (right) shows that sn-isomer distribution of FA 18:1 n-7 and n-10 is quite similar, whereas n-9 is distinctly different. To form PC 14:0_18:1, both FA 16:1 n-7 and FA 16:1 n-10 would require elongation, whereas FA 18:1 n-9 would be directly desaturated from FA 18:0. Interestingly, subcellular organelles are observed to have discrete pH ranges with mitochondrial pH being 7.58 for the matrix and 6.88 for the intermembrane space (57), ER pH being 7.1 at a resting state (58) and peroxisome pH varying between 7.4 and 8.1 depending on metabolic activity and cell-type properties (59), Considering acyl chain migration (while lipids exist as lysospecies) is a pH-dependant equilibrium, the remodeling of lipids under the different pH conditions of subcellular compartments could indeed lead to differences in the sn-positional isomer distributions. Therefore, the sn-positional isomers may be a further indication of compartmentalized FA modification events. Because FA 16:1 n-9 has been recently identified as a mediator for an anti-inflammatory response (60, 61), FA 16:1 n-7 is known as a lipokine that regulates SCD-1 expression (62) and FA 16:1 n-10 has antimicrobial properties (63), it is paramount to observe and measure any changes to the sn-isomeric composition in order to better understand the enzymes responsible for their catalytic release in metabolic diseases such as cancer.
FAs in their free form have the potential for signaling cellular processes and mechanisms (20, 45, 64, 65, 66). Along with previous examples (i.e., FA 16:1 n-7, FA 16:1 n-9, and FA 16:1 n-10), the FAs comprising the bulk of research surrounding signaling behaviors are those from dietary (extracellular) sources. These include FA 16:1 n-7(trans) and FA 18:1 n-7(trans) from milk products, which stimulate endocannabinoid and noncannabinoid production to in turn impact PPARα, lipolysis, inflammation, and ANGPTL4 (67, 68); LA and AA mainly from plant sources, which are precursory to eicosanoid formation and the proinflammation response (10); and, alpha-linolenic acid, EPA, and DHA from mainly marine sources, which mediate an anti-inflammatory response through resolvins, protectins, maresins, and the disruption of lipid rafts (9, 69). Furthermore, the presence or the absence of specific FA species within particular GPL classes and the degree of unsaturation in membrane lipid FAs has been shown to be influential factors in cell signaling (70, 71). As seen in Fig. 4C, notably our work shows that the uptake of SA drives a sharp increase of DHA into GPLs. Given that these GPL-DHA species are prime targets for PLA2 activity, the increase in these substrates would likely lead to the release of DHA from the lipid and mediate anti-inflammatory signaling cascades. This signal by uptake FAs presents a promising target for inducing proinflammatory or anti-inflammatory responses via nontoxic interventions. For example, by pre-exposing the cells to DHA substrates for a period and then supplementing with SA. This should repartition DHA into the GPLs, which upon lipase activity would initiate a strong anti-inflammatory effect and provide a nontoxic alternative to conventional anti-inflammatory drug therapies.
Given that FAs from uptake are shown to signal for the remodeling of PUFAs into the GPL lipid fraction (cf., Fig. 4), one explanation for this distinction between de novo and uptake FAs might lie within homeostasis. Within Fig. 2, it is observed that FA uptake causes a large variation to the abundance of lipid apocromers in the dn-lipidome fraction comparative to the nil-supplemented control. In contrast, the supplementation and incorporation of PA (and its metabolic progeny) into the up-lipidome is seen to influence the apocromer abundance such that it replicates the nil-supplemented control. In the more biologically relevant instance, where the cell is constantly taking up extracellular FAs that are causing the signaling for remodeling events, it is logical to assume that the incorporation of these new FAs into lipids would follow a homeostatic cellular template. Accordingly, the more that FAs are taken up, the larger this uptake-lipid fraction becomes until it exists in majority and thus dilutes the lipid fraction that was perturbed by remodeling—thereby restoring homeostasis. Disruption of these mechanisms, such as changes to environmental physicochemical properties, would therefore lead to the incorrect formation of lipid sn-isomers and have dire implications on homeostatic metabolism. The spatial correlations observed in Fig. 5 between PUFA lipids, lipid apocromers, lysolipids, and TGs indicate that there was dynamic lipid remodeling occurring within the fatty tissue of the xenograft model; however, this same remodeling is not observed within the bulk of the tumor (round bolus). Here, it is speculated that the hallmark switch to FA metabolism by tumor cells led to the production and secretion of PAs and SAs, while simultaneously creating a more acidic environment for the surrounding tissues and an increased expression of sPLA2-IIa in an attempt to decrease inflammation. Subsequently, the secreted PAs and SAs were taken up by the host adipocyte cells, which caused the repartitioning of PUFAs from TGs to GPLs, especially PIs that are mainly located on the outer leaflet of the plasma membrane. These outwardly exposed PUFA PIs are a prime target for the increased presence of sPLA2-IIa, which would in turn lead to the formation of lysolipid and free FA species. The hypothetical change in the pH caused by the tumor would then lead to lysoacyl chain migration and the formation of apocromeric lipid species. Consequently, lipids normally containing signaling FAs at the sn-2 position, which would be released by PLA2 under normal circumstances, would now release and excrete the sn-2 SFA—which could potentially go on to initiate the same cataclysmic remodeling response in surrounding cells. Considering the changes that are occurring within the TAG, ether-lipid (TAG O- and PC O-) and GPL sum compositional fractions (cf., Figs. 5 and S4) and their roles and biosynthetic origins, it is tempting to speculate that the excretion of these SFAs would also lead to the earlier mentioned FABP5 inhibition/activation of PPARβ/δ and its influence on the expression of ANGPTL4, Plin2, and CD36. Each of these enzymes can be inferred within the observed changes. Changes to ANGPTL4 (known for its roles in reactive-oxidative species [ROS] regulation, hypoxia response, and anoikis) (47, 48) expression would be assistive in the prevention of ferroptosis brought on by ROS coming from increased high-levels of GPL unsaturation and lipid peroxidation. Interestingly, the formation of ROS is also controlled by the peroxisomes, which along with previous implications, peroxisomal function/dysfunction can also be implied through the codistribution of ether-lipid species (peroxisomal lipid class) to the PUFA-GPL tissue regions. TAG speciation and spatial distribution (cf., supplemental Fig. S4) also alludes to changes in lipid droplet formation in PUFA-GPL tissue regions, whereas Plin2 is known to interact with lipid droplets to indirectly regulate droplet structure and function (49). Finally, changes to FA/lipid uptake and trafficking are readily observed throughout this work, which are known functions of CD36 (50), and together, the relationship between FA metabolism and these downstream effects of FA activated PPARβ/δ activity suggests that the trafficking of FAs and lipids is tightly regulated and highly influential in cell homeostasis.
Although previous research has provided a wealth of knowledge with regard to lipid remodeling, trafficking, and signaling, the recent progress made in lipid isomer research calls for an update on how these processes are impacted by or impact upon isomeric lipid species. Within, we present an array of isomeric changes that occur after FA supplementation and how these mechanisms are manifesting in tumor tissues. Importantly, these metabolic differences are heavily related to known oncogenic enzymes that are influential in cancer pathogenesis. Together, the indication that there exists distinct subcellular fractions of FA that have different metabolic fates, functions, and flow-on effects suggests that lipid isomers could be potential biomarkers for lipid remodeling and disease progression in tumor tissues. Thus, the work presented within provides the foundation for the discovery of proteins that are responsible for the partitioning of FAs derived from de novo synthesis or uptake and the mechanisms responsible for the influx of PUFAs to GPLs upon SFA uptake.
Data availability
The datasets generated during and/or analyzed during the current study are available as a data archive from QUT Research Data Finder using the following DOI: https://doi.org/10.25912/RDF_1638495738287.
Supplemental data
This article contains supplemental data.
Conflict of interest
S. J. B. holds patents on ozone-induced dissociation technology (A method for the determination of the position of unsaturation in a compound, US8242439 and US7771943). All other authors declare that they have no conflicts of interest with the contents of this article.
Acknowledgments
The project team acknowledge and thank the Central Analytical Research Facility operated by the Queensland University of Technology for provision of instrumentation and training in support of this project; the Translational Research Institute, including the Biological Resources Facility, Preclinical Imaging, and Histology facilities (and supporting grants from the Australian and Queensland Governments); and David L. Marshall and Gert B. Eijkel for helpful discussions and software developments.
Author contributions
R. S. E. Y., M. C. S., and S. J. B. conceptualization; R. S. E. Y., B. L. J. P., S. R. E., M. C. S., and S. J. B. methodology; R. S. E. Y. software; R. S. E. Y., A. P. B., K. D. T., and M. C. S. validation; R. S. E. Y. formal analysis; R. S. E. Y., A. P. B., K. D. T., and M. C. S. investigation; J. H. G., L. K. P., B. L. J. P., C. C. N., S. R. E., R. M. A. H., and S. J. B. resources; R. S. E. Y. data curation; R. S. E. Y. writing–original draft; R. S. E. Y., B. L. J. P., S. R. E., M. C. S., and S. J. B. writing–review & editing; R. S. E. Y. visualization; B. L. J. P., M. C. S., and S. J. B. supervision; R. S. E. Y., M. C. S., and S. J. B. project administration; B. L. J. P., C. C. N., S. R. E., R. M. A. H., and S. J. B. funding acquisition.
Funding and additional information
This work was financially supported by the Australian Research Council, Australia through the Discovery Program (grant no.: DP190101486) and the Linkage Program (grant no.: LP180100238, partnered with Waters Corporation for the development of MALDI-MSI OzID technology); the Prostate Cancer Foundation of Australia, Australia and the Australian Government Department of Health, Australia through a Movember Revolutionary Team Award; 2019 Cancer Program Initiative for Metabolism (Institute of Health and Biomedical Innovation, Queensland University of Technology, Australia); and, the Dutch Province of Limburg as part of the “LINK” program. R.S.E.Y acknowledges financial support from the Australian Government through award of an RTP scholarship; S. R. E., A. P. B., and R. M. A. H. acknowledge funding from Interreg V EMR and the Netherlands Ministry of Economic Affairs, Netherlands within the “EURLIPIDS” project (project no.: EMR23); and S. R. E. acknowledges funding from the Australian Research Council Future Fellowship Scheme (grant no.: FT190100082).
Contributor Information
Martin C. Sadowski, Email: martin.sadowski@pathology.unibe.ch.
Stephen J. Blanksby, Email: stephen.blanksby@qut.edu.au.
Supplemental data
References
- 1.Jackson Catherine L., Walch L., Verbavatz J.-M. Lipids and their trafficking: an integral part of cellular organization. Dev. Cell. 2016;39:139–153. doi: 10.1016/j.devcel.2016.09.030. [DOI] [PubMed] [Google Scholar]
- 2.Kennedy E.P., Weiss S.B. The function of cytidine coenzymes in the biosynthesis of phospholipides. J. Biol. Chem. 1956;222:193–214. [PubMed] [Google Scholar]
- 3.Lands W.E.M. Stories about acylchains. BBA-Mol. Cell Biol. L. 2000;1483:1–14. doi: 10.1016/s1388-1981(99)00177-8. [DOI] [PubMed] [Google Scholar]
- 4.Koundouros N., Poulogiannis G. Reprogramming of fatty acid metabolism in cancer. Br. J. Cancer. 2020;122:4–22. doi: 10.1038/s41416-019-0650-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hosios Aaron M., Hecht Vivian C., Danai Laura V., Johnson Marc O., Rathmell Jeffrey C., Steinhauser Matthew L., et al. Amino acids rather than glucose account for the majority of cell mass in proliferating mammalian cells. Dev. Cell. 2016;36:540–549. doi: 10.1016/j.devcel.2016.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nagarajan S.R., Butler L.M., Hoy A.J. The diversity and breadth of cancer cell fatty acid metabolism. Cancer Metab. 2021;9:2. doi: 10.1186/s40170-020-00237-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Diez E., Chilton F.H., Stroup G., Mayer R.J., Winkler J.D., Fonteh A.N. Fatty acid and phospholipid selectivity of different phospholipase A2 enzymes studied by using a mammalian membrane as substrate. Biochem. J. 1994;301:721–726. doi: 10.1042/bj3010721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lands B. Lipid nutrition: “In silico” studies and undeveloped experiments. Prog. Lipid Res. 2022;2021:101142. doi: 10.1016/j.plipres.2021.101142. [DOI] [PubMed] [Google Scholar]
- 9.Calder P.C. Omega-3 fatty acids and inflammatory processes. Nutrients. 2010;2:355–374. doi: 10.3390/nu2030355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Piomelli D. Arachidonic acid in cell signaling. Curr. Opin. Cell Biol. 1993;5:274. doi: 10.1016/0955-0674(93)90116-8. [DOI] [PubMed] [Google Scholar]
- 11.Meyer Zu Heringdorf D., Jakobs K.H. Lysophospholipid receptors: signalling, pharmacology and regulation by lysophospholipid metabolism. Biochim. Biophys. Acta Biomem. 2007;1768:923–940. doi: 10.1016/j.bbamem.2006.09.026. [DOI] [PubMed] [Google Scholar]
- 12.Hishikawa D., Shindou H., Kobayashi S., Nakanishi H., Taguchi R., Shimizu T. Discovery of a lysophospholipid acyltransferase family essential for membrane asymmetry and diversity. Proc. Natl. Acad. Sci. U. S. A. 2008;105:2830. doi: 10.1073/pnas.0712245105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hishikawa D., Hashidate T., Shimizu T., Shindou H. Diversity and function of membrane glycerophospholipids generated by the remodeling pathway in mammalian cells. J. Lipid Res. 2014;55:799–807. doi: 10.1194/jlr.R046094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kawana H., Kano K., Shindou H., Inoue A., Shimizu T., Aoki J. An accurate and versatile method for determining the acyl group-introducing position of lysophospholipid acyltransferases. BBA-Mol. Cell Biol. L. 2019;1864:1053–1060. doi: 10.1016/j.bbalip.2019.02.008. [DOI] [PubMed] [Google Scholar]
- 15.Ekroos K., Ejsing C.S., Bahr U., Karas M., Simons K., Shevchenko A. Charting molecular composition of phosphatidylcholines by fatty acid scanning and ion trap MS3 fragmentation. J. Lipid Res. 2003;44:2181–2192. doi: 10.1194/jlr.D300020-JLR200. [DOI] [PubMed] [Google Scholar]
- 16.Renooij W., Van Golde L.M., Zwaal R.F., Van Deenen L.L. Topological asymmetry of phospholipid metabolism in rat erythrocyte membranes. Evidence for flip-flop of lecithin. Eur. J. Biochem. 1976;61:53–58. doi: 10.1111/j.1432-1033.1976.tb09996.x. [DOI] [PubMed] [Google Scholar]
- 17.Wozny K., Lehmann W.D., Wozny M., Akbulut B.S., Brügger B. A method for the quantitative determination of glycerophospholipid regioisomers by UPLC-ESI-MS/MS. Anal. Bioanal. Chem. 2018;411:915–924. doi: 10.1007/s00216-018-1517-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kozlowski R.L., Mitchell T.W., Blanksby S.J. A rapid ambient ionization-mass spectrometry approach to monitoring the relative abundance of isomeric glycerophospholipids. Sci. Rep. 2015;5:9243. doi: 10.1038/srep09243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aoki J., Inoue A., Makide K., Saiki N., Arai H. Structure and function of extracellular phospholipase A1 belonging to the pancreatic lipase gene family. Biochimie. 2007;89:197–204. doi: 10.1016/j.biochi.2006.09.021. [DOI] [PubMed] [Google Scholar]
- 20.Hughes-Fulford M., Tjandrawinata R.R., Li C.-F., Sayyah S. Arachidonic acid, an omega-6 fatty acid, induces cytoplasmic phospholipase Ainf2/inf in prostate carcinoma cells. Carcinogenesis. 2005;26:1520–1526. doi: 10.1093/carcin/bgi112. [DOI] [PubMed] [Google Scholar]
- 21.Mazari A., Iwamoto S., Yamauchi R. Effects of linoleic acid position in phosphatidylcholines and cholesterol addition on their rates of peroxidation in unilamellar liposomes. Biosci. Biotechnol. Biochem. 2010;74:1013–1017. doi: 10.1271/bbb.90896. [DOI] [PubMed] [Google Scholar]
- 22.Martinez-Seara H., Róg T., Karttunen M., Vattulainen I., Reigada R. Why is the sn-2 chain of monounsaturated glycerophospholipids usually unsaturated whereas the sn-1 chain is saturated? studies of 1-Stearoyl-2-oleoyl-sn-glycero-3-phosphatidylcholine (SOPC) and 1-Oleoyl-2-stearoyl-sn-glycero-3-phosphatidylcholine (OSPC) membranes with and without cholesterol. J. Phys. Chem. B. 2009;113:8347–8356. doi: 10.1021/jp902131b. [DOI] [PubMed] [Google Scholar]
- 23.Paine M.R.L., Poad B.L.J., Eijkel G.B., Marshall D.L., Blanksby S.J., Heeren R.M.A., et al. Mass spectrometry imaging with isomeric resolution enabled by ozone-induced dissociation. Angew. Chem. Int. Ed. 2018;57:10530. doi: 10.1002/anie.201802937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Swietach P., Vaughan-Jones R.D., Harris A.L., Hulikova A. The chemistry, physiology and pathology of pH in cancer. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014;369:20130099. doi: 10.1098/rstb.2013.0099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Korenchan D.E., Flavell R.R. Spatiotemporal pH heterogeneity as a promoter of cancer progression and therapeutic resistance. Cancers. 2019;11:1026. doi: 10.3390/cancers11071026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Croset M., Brossard N., Polette A., Lagarde M. Characterization of plasma unsaturated lysophosphatidylcholines in human and rat. Biochem. J. 2000;345:61–67. [PMC free article] [PubMed] [Google Scholar]
- 27.Matyash V., Liebisch G., Kurzchalia T.V., Shevchenko A., Schwudke D. Lipid extraction by methyl-tert-butyl ether for high-throughput lipidomics. J. Lipid Res. 2008;49:1137–1146. doi: 10.1194/jlr.D700041-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Belov M.E., Ellis S.R., Dilillo M., Paine M.R.L., Danielson W.F., Anderson G.A., et al. design and performance of a novel interface for combined matrix-assisted laser desorption ionization at elevated pressure and electrospray ionization with orbitrap mass spectrometry. Anal. Chem. 2017;89:7493–7501. doi: 10.1021/acs.analchem.7b01168. [DOI] [PubMed] [Google Scholar]
- 29.Marshall D.L., Criscuolo A., Young R.S.E., Poad B.L.J., Zeller M., Reid G.E., et al. Mapping unsaturation in human plasma lipids by data-independent ozone-induced dissociation. J. Am. Soc. Mass Spectrom. 2019;30:1621–1630. doi: 10.1007/s13361-019-02261-z. In press. [DOI] [PubMed] [Google Scholar]
- 30.de Winter J.C.F. Using the Student's t-test with extremely small sample sizes. Pract. Assess. Res. Eval. 2013;18 doi: 10.7275/e4r6-dj05. [DOI] [Google Scholar]
- 31.Guillou H., Zadravec D., Martin P.G.P., Jacobsson A. The key roles of elongases and desaturases in mammalian fatty acid metabolism: insights from transgenic mice. Prog. Lipid Res. 2010;49:186–199. doi: 10.1016/j.plipres.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 32.Thomas M.C., Mitchell T.W., Harman D.G., Deeley J.M., Nealon J.R., Blanksby S.J. Ozone-induced dissociation: elucidation of double bond position within mass-selected lipid ions. Anal. Chem. 2008;80:303–311. doi: 10.1021/ac7017684. [DOI] [PubMed] [Google Scholar]
- 33.Ackerman D., Tumanov S., Qiu B., Michalopoulou E., Spata M., Azzam A., et al. Triglycerides promote lipid homeostasis during hypoxic stress by balancing fatty acid saturation. Cell Rep. 2018;24:2596–2605.e5. doi: 10.1016/j.celrep.2018.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tousignant K.D., Rockstroh A., Poad B.L.J., Talebi A., Young R.S.E., Taherian Fard A., et al. Therapy-induced lipid uptake and remodeling underpin ferroptosis hypersensitivity in prostate cancer. Cancer Metab. 2020;8:11. doi: 10.1186/s40170-020-00217-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Claes B.S.R., Bowman A.P., Poad B.L.J., Young R.S.E., Heeren R.M.A., Blanksby S.J., et al. Mass spectrometry imaging of lipids with isomer resolution using high-pressure ozone-induced dissociation. Anal. Chem. 2021;93:9826–9834. doi: 10.1021/acs.analchem.1c01377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Young R.S.E., Claes B.S.R., Bowman A.P., Williams E.D., Shepherd B., Perren A., et al. Isomer-resolved imaging of prostate cancer tissues reveals specific lipid unsaturation profiles associated with lymphocytes and abnormal prostate epithelia. Front. Endocrinol. 2021;12:689600. doi: 10.3389/fendo.2021.689600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Young R.S.E., Bowman A.P., Williams E.D., Tousignant K.D., Bidgood C.L., Narreddula V.R., et al. Apocryphal FADS2 activity promotes fatty acid diversification in cancer. Cell Rep. 2021;34:108738. doi: 10.1016/j.celrep.2021.108738. [DOI] [PubMed] [Google Scholar]
- 38.Graff J.R., Konicek B.W., Deddens J.A., Chedid M., Hurst B.M., Colligan B., et al. Expression of group IIa secretory phospholipase A2 increases with prostate tumor grade. Clin. Cancer Res. 2001;7:3857. [PubMed] [Google Scholar]
- 39.Lisec J., Jaeger C., Rashid R., Munir R., Zaidi N. Cancer cell lipid class homeostasis is altered under nutrient-deprivation but stable under hypoxia. BMC Cancer. 2019;19:501. doi: 10.1186/s12885-019-5733-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bartz R., Li W.-H., Venables B., Zehmer J.K., Roth M.R., Welti R., et al. Lipidomics reveals that adiposomes store ether lipids and mediate phospholipid traffic. J. Lipid Res. 2007;48:837–847. doi: 10.1194/jlr.M600413-JLR200. [DOI] [PubMed] [Google Scholar]
- 41.Voss A., Reinhart M., Sankarappa S., Sprecher H. The metabolism of 7,10,13,16,19-docosapentaenoic acid to 4,7,10,13,16,19-docosahexaenoic acid in rat liver is independent of a 4-desaturase. J. Biol. Chem. 1991;266:19995–20000. [PubMed] [Google Scholar]
- 42.van den Bosch H., Schrakamp G., Hardeman D., Zomer A.W.M., Wanders R.J.A., Schutgens R.B.H. Ether lipid synthesis and its deficiency in peroxisomal disorders. Biochimie. 1993;75:183–189. doi: 10.1016/0300-9084(93)90076-5. [DOI] [PubMed] [Google Scholar]
- 43.Zou Y., Henry W.S., Ricq E.L., Graham E.T., Phadnis V.V., Maretich P., et al. Plasticity of ether lipids promotes ferroptosis susceptibility and evasion. Nature. 2020;585:603–608. doi: 10.1038/s41586-020-2732-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Blom T., Somerharju P., Ikonen E. Synthesis and biosynthetic trafficking of membrane lipids. Cold Spring Harb. Perspect. Biol. 2011;3 doi: 10.1101/cshperspect.a004713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Levi L., Wang Z., Doud M.K., Hazen S.L., Noy N. Saturated fatty acids regulate retinoic acid signalling and suppress tumorigenesis by targeting fatty acid-binding protein 5. Nat. Commun. 2015;6:8794. doi: 10.1038/ncomms9794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Song X., Liu J., Kuang F., Chen X., Zeh H.J., III, Kang R., et al. PDK4 dictates metabolic resistance to ferroptosis by suppressing pyruvate oxidation and fatty acid synthesis. Cell Rep. 2021;34:108767. doi: 10.1016/j.celrep.2021.108767. [DOI] [PubMed] [Google Scholar]
- 47.Baba K., Kitajima Y., Miyake S., Nakamura J., Wakiyama K., Sato H., et al. Hypoxia-induced ANGPTL4 sustains tumour growth and anoikis resistance through different mechanisms in scirrhous gastric cancer cell lines. Sci. Rep. 2017;7:11127. doi: 10.1038/s41598-017-11769-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhu P., Tan Ming J., Huang R.-L., Tan Chek K., Chong Han C., Pal M., et al. Angiopoietin-like 4 protein elevates the prosurvival intracellular O2−:H2O2 ratio and confers Anoikis resistance to tumors. Cancer Cell. 2011;19:401–415. doi: 10.1016/j.ccr.2011.01.018. [DOI] [PubMed] [Google Scholar]
- 49.McIntosh A.L., Senthivinayagam S., Moon K.C., Gupta S., Lwande J.S., Murphy C.C., et al. Direct interaction of Plin2 with lipids on the surface of lipid droplets: a live cell FRET analysis. Am. J. Physiol. Cell Physiol. 2012;303:C728–C742. doi: 10.1152/ajpcell.00448.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hao J.-W., Wang J., Guo H., Zhao Y.-Y., Sun H.-H., Li Y.-F., et al. CD36 facilitates fatty acid uptake by dynamic palmitoylation-regulated endocytosis. Nat. Commun. 2020;11:4765. doi: 10.1038/s41467-020-18565-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jay A.G., Hamilton J.A. The enigmatic membrane fatty acid transporter CD36: new insights into fatty acid binding and their effects on uptake of oxidized LDL. Prostag. Leukotr. Ess. 2018;138:64–70. doi: 10.1016/j.plefa.2016.05.005. [DOI] [PubMed] [Google Scholar]
- 52.Park H.G., Park W.J., Kothapalli K.S.D., Brenna J.T. The fatty acid desaturase 2 (FADS2) gene product catalyzes Δ4 desaturation to yield n-3 docosahexaenoic acid and n-6 docosapentaenoic acid in human cells. FASEB J. 2015;29:3911–3919. doi: 10.1096/fj.15-271783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Miyazaki M., Ntambi J.M. Role of stearoyl-coenzyme A desaturase in lipid metabolism. Prostag. Leukotr. Ess. 2003;68:113–121. doi: 10.1016/s0952-3278(02)00261-2. [DOI] [PubMed] [Google Scholar]
- 54.Hall D., Poussin C., Velagapudi V.R., Empsen C., Joffraud M., Beckmann J.S., et al. Peroxisomal and microsomal lipid pathways associated with resistance to hepatic steatosis and reduced pro-inflammatory state. J. Biol. Chem. 2010;285:31011–31023. doi: 10.1074/jbc.M110.127159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Song He W., Nara T.Y., Nakamura M.T. Delayed induction of Δ-6 and Δ-5 desaturases by a peroxisome proliferator. Biochem. Biophys. Res. Commun. 2002;299:832–838. doi: 10.1016/s0006-291x(02)02743-2. [DOI] [PubMed] [Google Scholar]
- 56.Park H.G., Kothapalli K.S.D., Park W.J., Deallie C., Liu L., Liang A., et al. Palmitic acid (16:0) competes with omega-6 linoleic and omega-3 ɑ-linolenic acids for FADS2 mediated Δ6-desaturation. BBA-Mol. Cell Biol. L. 2016;1861:91–97. doi: 10.1016/j.bbalip.2015.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Porcelli A.M., Ghelli A., Zanna C., Pinton P., Rizzuto R., Rugolo M. pH difference across the outer mitochondrial membrane measured with a green fluorescent protein mutant. Biochem. Biophys. Res. Commun. 2005;326:799–804. doi: 10.1016/j.bbrc.2004.11.105. [DOI] [PubMed] [Google Scholar]
- 58.Kim J.H., Johannes L., Goud B., Antony C., Lingwood C.A., Daneman R., et al. Noninvasive measurement of the pH of the endoplasmic reticulum at rest and during calcium release. Proc. Natl. Acad. Sci. U. S. A. 1998;95:2997. doi: 10.1073/pnas.95.6.2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Godinho L.F., Schrader M. In: Peroxisomes: Methods and Protocols. Schrader M., editor. Springer; New York, NY: 2017. Determination of peroxisomal pH in living mammalian cells using pHRed; pp. 181–189. [DOI] [PubMed] [Google Scholar]
- 60.Astudillo A.M., Meana C., Guijas C., Pereira L., Lebrero P., Balboa M.A., et al. Occurrence and biological activity of palmitoleic acid isomers in phagocytic cells. J. Lipid Res. 2018;59:237. doi: 10.1194/jlr.M079145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Guijas C., Meana C., Astudillo Alma M., Balboa María A., Balsinde J. Foamy monocytes are enriched in cis-7-Hexadecenoic fatty acid (16:1n-9), a possible biomarker for early detection of cardiovascular disease. Cell Chem. Biol. 2016;23:689–699. doi: 10.1016/j.chembiol.2016.04.012. [DOI] [PubMed] [Google Scholar]
- 62.Cao H., Gerhold K., Mayers J.R., Wiest M.M., Watkins S.M., Hotamisligil G.S. Identification of a lipokine, a lipid hormone linking adipose tissue to systemic metabolism. Cell. 2008;134:933–944. doi: 10.1016/j.cell.2008.07.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fischer C.L., Blanchette D.R., Brogden K.A., Dawson D.V., Drake D.R., Hill J.R., et al. The roles of cutaneous lipids in host defense. BBA-Mol. Cell Biol. L. 2014;1841:319–322. doi: 10.1016/j.bbalip.2013.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhou H., Urso C.J., Jadeja V. Saturated fatty acids in obesity-associated inflammation. J. Inflamm. Res. 2020;13:1–14. doi: 10.2147/JIR.S229691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Greene E.L., Lu G., Zhang D., Egan B.M. Signaling events mediating the additive effects of oleic acid and angiotensin II on vascular smooth muscle cell migration. Hypertension. 2001;37:308–312. doi: 10.1161/01.hyp.37.2.308. [DOI] [PubMed] [Google Scholar]
- 66.Carta G., Murru E., Banni S., Manca C. Palmitic acid: physiological role, metabolism and nutritional implications. Front. Physiol. 2017;8:902. doi: 10.3389/fphys.2017.00902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jacome-Sosa M., Vacca C., Mangat R., Diane A., Nelson R.C., Reaney M.J., et al. Vaccenic acid suppresses intestinal inflammation by increasing anandamide and related N-acylethanolamines in the JCR:LA-cp rat. J. Lipid Res. 2016;57:638–649. doi: 10.1194/jlr.M066308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nielsen S.D., Young J.F., Mortensen G., Petersen R.K., Kristiansen K., Dalsgaard T.K. Activation of the angiopoietin-like 4 (ANGPLT4) gene by milk fat and casein. Int. Dairy J. 2014;36:136–142. [Google Scholar]
- 69.Calder P.C. Omega-3 fatty acids and inflammatory processes: from molecules to man. Biochem. Soc. Trans. 2017;45:1105–1115. doi: 10.1042/BST20160474. [DOI] [PubMed] [Google Scholar]
- 70.Sabogal-Guáqueta A.M., Villamil-Ortiz J.G., Arias-Londoño J.D., Cardona-Gómez G.P. Inverse phosphatidylcholine/phosphatidylinositol levels as peripheral biomarkers and phosphatidylcholine/lysophosphatidylethanolamine-phosphatidylserine as hippocampal indicator of postischemic cognitive impairment in rats. Front. Neurosci. 2018;12:989. doi: 10.3389/fnins.2018.00989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schoeniger A., Fuhrmann H., Schumann J. LPS- or Pseudomonas aeruginosa-mediated activation of the macrophage TLR4 signaling cascade depends on membrane lipid composition. PeerJ. 2016;4:e1663. doi: 10.7717/peerj.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during and/or analyzed during the current study are available as a data archive from QUT Research Data Finder using the following DOI: https://doi.org/10.25912/RDF_1638495738287.