

Abstract
Mass spectrometry-based secretomics approaches frequently utilize serum-free culture conditions to circumvent serum-induced interference and to increase analytical depth. However, this can negatively affect a wide range of cellular functions and cell viability. These effects become particularly apparent when investigating transcriptionally regulated secretion events and feedback-loops in response to perturbations that require 48 h or more to fully manifest. We present an “interval-based” secretomics workflow, which determines protein secretion rates in short serum-free time windows. Relative quantification using tandem mass tags enables precise monitoring of time-dependent changes. We applied this approach to determine temporal profiles of protein secretion in the hepatocyte model cell lines HepG2 and HepaRG after stimulation of the acute-phase response (APR) by the cytokines IL1b and IL6. While the popular hepatocarcinoma cell line HepG2 showed an incomplete APR, secretion patterns derived from differentiated HepaRG cells recapitulated the expected APR more comprehensively. For several APR response proteins, substantial secretion was only observed after 72 h, a time window at which cell fitness is substantially impaired under serum-free cell culture conditions. The interval-based secretomics approach enabled the first comprehensive analysis of time-dependent secretion of liver cell models in response to these proinflammatory cytokines. The extended time range facilitated the observation of distinct chronological phases and cytokine-dependent secretion phenotypes of the APR. IL1b directed the APR toward pathogen defense over three distinct phases—chemotaxis, effector, clearance—while IL6 directed the APR toward regeneration. Protein shedding on the cell surface was pronounced upon IL1b stimulation, and small molecule inhibition of ADAM and matrix metalloproteases identified induced as well as constitutive shedding events. Inhibition of ADAM proteases with TAPI-0 resulted in reduced shedding of the sorting receptor SORT1, and an attenuated cytokine response suggesting a direct link between cell surface shedding and cytokine secretion rates.
Keywords: secretomics, acute-phase response, HepaRG cells, IL1b, IL6, cell surface shedding, inflammation
Abbreviations: AHA, azidohomoalanine; APR, acute-phase response; dHepaRG, differentiated HepaRG; DMSO, dimethyl sulfoxide; ECM, extracellular matrix; ER, endoplasmic reticulum; FDR, false discovery rate; GO, gene ontology; HCD, Higher energy collisional dissociation; LDH, lactate dehydrogenase; MMP, matrix metalloproteinase; TMT, tandem mass tag
Graphical Abstract
Highlights
-
•
Interval-based secretomics enables extended time course analysis.
-
•
Time-resolved acute phase response in liver model systems HepG2 and HepaRG.
-
•
IL1b response clusters in three phases.
-
•
Cell surface shedding is amplified during acute-phase response.
-
•
ADAM inhibition dampens secretion of inflammatory cytokines.
In Brief
Using short serum-free incubation intervals allows extended time course analysis downstream of perturbations and addresses limitations of classical serum-free secretomics workflows. The time-dependent secretion of liver cell models HepG2 and HepaRG upon stimulation of the acute-phase response with the proinflammatory cytokines IL1b and IL6 was studied and revealed time- and stimulus-dependent phases of protein secretion. Monitoring proteolysis at the cell surface linked ADAM inhibition to inflammatory cytokine secretion.
Hepatocytes are the key functional cells in the liver (1). Besides their essential roles in metabolism and detoxification, hepatocytes are true protein secretion factories with key immunological functions (1, 2, 3). In steady state, hepatocytes constitutively express and secrete a large variety of proteins into the bloodstream that perform pivotal body functions in transport, homeostasis, and innate immunity. Perturbations, such as inflammatory processes induce proinflammatory signals such as cytokines IL-1 and IL-6 that initialize the acute-phase response (APR) in hepatocytes and stimulate secretion of proteins (acute-phase proteins, APP) with immunomodulatory functions to support defense against pathogens and to restore homeostasis (4, 5, 6, 7, 8, 9).
The secretome of a cell or an organism consists of proteins secreted by the endoplasmic reticulum (ER)–Golgi secretory pathway and other direct or vesicle-based mechanisms (10, 11, 12). In addition, ectodomains of membrane proteins can be proteolytically released into the extracellular environment. The comprehensive and unbiased identification and quantification of proteins that constitute this subproteome is of particular interest as secreted proteins facilitate intercellular communication, function as cytokines, growth factors, hormones, or drug targets and can provide insights into disease states and physiological processes. However, the systematic investigation of secreted proteins by mass spectrometry–based proteomics faces several technical and biological challenges. The high dilution of secreted proteins in the cell culture medium often requires protein concentration steps prior to LC-MS analysis. The low abundance and high dynamic range of secreted proteins (13, 14) further complicates the analysis, especially when cells are maintained in serum-supplemented media. A variety of experimental strategies have been proposed to address these challenges. On one hand, metabolic labeling approaches by either the amino acid analog azidohomoalanine or azido sugars (15, 16, 17, 18, 19) enable enrichment of metabolically labeled proteins from the serum-supplemented cell culture supernatant and thus allow the focused and sensitive analysis of de-novo synthesized and secreted proteins. However, not all cell systems are compatible with these approaches and the required labeling reagents lead to metabolic stress and may impair protein function further narrowing down biological relevance of perturbation-induced secretomes. Direct proteomics analysis of cell supernatants using serum-free cell culture conditions is an alternative popular approach (20, 21, 22). However, serum-free cell culture conditions can affect cell viability and function, which limits the timeframe in which secretion may be probed without substantially impairing essential cell functions and response to stimuli. Thus, most published studies focused on monitoring protein secretion only for a few hours (20, 23, 24).
Hepatocytes interact with the immune system by expression and secretion of proteins with immunomodulatory functions. However, a detailed characterization of the secretome of hepatocytes in response to inflammatory stimuli is missing and could advance the understanding of liver inflammatory conditions. Different hepatocyte model systems have been established to facilitate studying liver biology (25). Among these, hepatoma cell lines like HepG2 cells are routinely used in in-vitro studies but their translational value is limited due to an abnormal hepatic phenotype which is reflected in perturbed signaling pathways, low expression levels of key metabolic enzymes, or impaired responses to cytokines and growth factors (25). HepaRG cells are a valuable alternative to hepatoma cell lines featuring major hepatocyte-specific functions that resemble those of primary human hepatocytes that are currently the gold standard for the study of liver biology (26, 27). HepaRG cells have gained growing interest especially for their use in in vitro hepatotoxicity studies (28, 29, 30, 31, 32). Established from a hepatocellular carcinoma of a patient with hepatitis C infection (27), undifferentiated HepaRG cells proliferate for approximately 7 days until complete confluency. From this point on, the monolayer develops into two distinct cell populations. After 14 days, the cell culture is continued in the presence of 2% dimethyl sulfoxide (DMSO) for further 14 days to induce a full differentiation into functional hepatocyte- and biliary-like cells. Differentiated cells exhibit a hepatocyte-like metabolism, secretory activity, and expression levels of structural constituents which is comparable to primary human hepatocytes and which render HepaRG cells a superior cell system (26). As such, we set out to study the effect of IL1b and IL6 on the secretion in differentiated HepaRG (dHepaRG) cells.
Here, we present an “interval-based” secretomics workflow that addresses some of the shortcomings of the common serum-free secretomics approach, by probing the secretion in short serum-free time windows. Minimizing the time in serum-free medium extents the time range within which stimulus-dependent secretion can be monitored and limits the impact of serum starvation on signaling and viability. We performed standard serum-free and interval-based secretomics to characterize the time-dependent secretion of proteins in the hepatocyte model systems HepG2 and HepaRG after stimulation of the APR. Relative quantification using tandem mass tags (TMTs) enabled the quantitative monitoring of dynamic changes in the secretome and revealed distinct cell type– and cytokine-dependent differences during the stimulation of the APR. The extension of the experimental time range uncovered distinct time-dependent phases of protein secretion during the APR and showed that IL1b and IL6 direct the APR toward different directions. Finally, inhibition of ADAM and matrix metalloproteinases (MMPs) with small molecule inhibitors allowed to identify induced as well as constitutive shedding events linking the ADAM-mediated shedding of the endosomal sorting receptor SORT1 to activation of the early cytokine response.
Experimental Procedures
Cytokines and Small Molecules
Recombinant human IL1b and recombinant human IL6 were purchased from PeproTech Inc. Broad-band MMP inhibitor Ilomastat and ADAM17 inhibitor TAPI-0 were purchased from Tocris Bioscience.
Cell Culture
Human hepatoma cell line HepG2 (obtained from ATCC) was maintained in minimal essential medium (Thermo Fisher) supplemented with 10% fetal calf serum, 2 mM glutamine, 1% nonessential amino acids, and 1 mM sodium pyruvate. Undifferentiated human HepaRG cells (HPR101056, Biopredic International) were cultured and differentiated according to the supplier’s instructions. Briefly, cryopreserved cells were thawed and maintained in William's E-medium (Thermo Fisher) supplemented with HepaRG Growth medium with antibiotics (ADD710, Biopredic International). Growth medium was exchanged twice a week. After 2 weeks in HepaRG growth medium, fully confluent HepaRG cells were trypsinized and passaged to maintain the cell line or seeded into 12-well plates with a density of 2.7 × 104 cells/cm2. After 2 weeks of proliferation in 12-well plates, HepaRG growth medium was replaced by differentiation medium with antibiotics (ADD720, Biopredic International), which causes the cells to differentiate to hepatocyte colonies and primitive biliary cells within 2 weeks. dHepaRG cells were maintained in differentiation medium until use for a maximum of four further weeks. All cell lines were maintained at 37 °C in a humidified atmosphere of 5% CO2.
Cell Treatments and Preparation of Secretome Samples
Secretomics experiments were conducted in 12-well plates. HepG2 cells were used at 90% confluency. All wash steps and treatments were performed with prewarmed media. For cumulative-based secretomics experiments, cell culture medium was removed, and the cells were carefully washed three times with serum- and phenol red-free medium. HepG2 cells were washed and treated in minimal essential medium. HepaRG cells were washed and treated in William's E-medium. APR was stimulated by addition of 1 ml prewarmed serum- and phenol red-free medium containing either 50 ng/ml recombinant human IL1b, 50 ng/ml recombinant human IL6, or treated with prewarmed serum- and phenol red-free medium without stimulant as the control. Cells were incubated for the indicated time points at 37 °C in a humidified atmosphere of 5% CO2.
For interval-based secretomics experiments, differentiation medium was exchanged with fresh differentiation medium containing 50 ng/ml recombinant human IL1b, IL6, or differentiation medium only. HepaRG cells were incubated for the indicated time points at 37 °C in an atmosphere of 5% CO2. Two hours before the end of a time point, the differentiation medium was removed, and the cells were washed carefully three times with serum- and phenol red-free William's E-medium. Cells were incubated for an additional 2 h in serum- and phenol red-free William's E-medium with 50 ng/ml recombinant human IL1b or IL6 or without stimulant. Cell supernatants from all samples were carefully collected, and 230 μl supernatant was transferred into a 0.45 μm 96-well filter-plate (Durapore, low protein-binding PVDF membrane, Merck Millipore) to remove detached cells and cell debris. Supernatants were cleared by centrifugation (2 min at 100g) and collected in a 96-well polypropylene plate. Two hundred microliter cleared supernatants were then transferred into a fresh 96-well polypropylene plate and subsequently sealed and stored at −80 °C.
For shedding experiments with protease inhibitors, the HepaRG differentiation medium was removed, and cells were carefully washed three times with William's E-medium. Samples were either mock treated (DMSO) as control, 50 ng/ml IL1b alone, inhibitor, or the combination of inhibitor and 50 ng/ml IL1b. The final DMSO concentration was set to 0.5%. All treatments were performed in phenol red- and serum-free William's E-medium, with 1 ml final volume per well. Protease inhibitors were used at the following concentrations: Ilomastat was used at 10 μM, and TAPI-0 was used of 50 μM. Cells were incubated for 8 h at 37 °C in a humidified atmosphere of 5% CO2. After 8 h, cell supernatants were carefully collected and processed as described above.
LDH-Assay
Cell viability was assessed by determination of lactate dehydrogenase (LDH) release into cell culture supernatants using the LDH-Glo cytotoxicity assay (Promega) following the manufacturer’s instructions. The maximal LDH activity was determined by the addition of 20 μl 10% Triton X-100 to the wells of the control cells.
Sample Preparation for Mass Spectrometry
Frozen secretome samples were dried in vacuo. Pellets were resuspended in 115 μl resuspension buffer (2% SDS, 0.5% IGEPAL CA-630) and incubated for 15 min at room temperature on an orbital shaker (Heidolph Titramax 1000) at 800 rpm. Hundred microliter resuspended samples were processed through a modified version of the single pot solid-phase sample preparation (SP3) protocol as described in ref (33). Briefly, proteins in resuspension buffer were bound to paramagnetic beads (SeraMag Speed beads, GE Healthcare, CAT#45152105050250, CAT#651521050502) by addition of 160 μl cleanup solution (130 μl ethanol, 27.5 μl 15% formic acid, 2.5 μl bead slurry) to a final ethanol concentration of 50%. Beads were washed four times with 200 μl 70% ethanol. Proteins were digested by resuspending in 40 μl 0.1 mM Hepes (pH 8.5) containing 1.25 mM TCEP, 5 mM chloroacetamide, 5 ng/μl trypsin, and 5 ng/μl LysC following overnight incubation. Peptides were labeled with isobaric mass tags (TMT-10/11, Thermo Fisher Scientific). The labeling reaction was performed in 100 mM Hepes (pH 8.5) 50% acetonitrile at 22 °C and quenched with 2.5% hydroxylamine. Labeled peptide extracts were pooled and purified using C18SCX stage-tips as described in ref (33).
LC-MS/MS Analysis
TMT-labeled samples were fractionated into eight fractions prior to LC-MS/MS analysis using the high pH reversed-phase peptide fractionation kit (Thermo Fisher Scientific) following the manufacturer’s instructions. Fractionated and lyophilized samples were resuspended in 0.05% TFA in water and 30% of each sample was injected into an Ultimate3000 nanoRLSC (Dionex) coupled to a Q Exactive (Thermo Fisher Scientific) or an Orbitrap Fusion Lumos mass spectrometer (Thermo Fisher Scientific). Peptides were separated on custom-made 50 cm × 100 μm (ID) reversed-phase columns (C18, 1.9 μm, Reprosil-Pur, Dr Maisch) at 55 °C. Gradient elution was performed from 2% acetonitrile to 40% acetonitrile in 0.1% formic acid and 3.5% DMSO over 65 min at a flow rate of 350 nl/min. Samples were online injected into the mass spectrometer. The Q Exactive Plus was operated in a data-dependent top ten acquisition method. MS spectra were acquired using 70,000 resolution and an ion target of 3 × 106 for MS1 scans. Higher energy collisional dissociation (HCD) scans were performed with 35% normalized collision energy at 35,000 resolution (at m/z 200), and ion target setting was set to 2 × 105 so as to avoid coalescence (34). The instruments were operated with Tune 2.4 and Xcalibur 3.0 build 63. The Orbitrap Fusion Lumos operated with a fixed cycle time of 3 s. MS1 spectra were acquired at a resolution of 60,000 and an ion target of 4 × 105. HCD fragmentation was performed at 38% normalized collision energy at a resolution of 30,000 and an ion target 1 × 105. The instrument was operated with Tune v.2.1.1565.23 and Xcalibur v.4.0.27.10.
Peptide and Protein Identification
Raw data were processed using an in-house pipeline based on the isobar quant package (35). Mascot 2.5 (Matrix Science) was used for protein identification. In a first search, 30 ppm peptide precursor mass and 30 mDa (HCD) mass tolerance for fragment ions was used for recalibration according to Cox et al. (36) followed by search using a 10 ppm mass tolerance for peptide precursors and 20 mDa (HCD) mass tolerance for fragment ions. Enzyme specificity was set to trypsin with up to three missed cleavages. The search database consisted of the SwissProt sequence database (SwissProt Human release December 2018, 42,423 sequences) combined with a decoy version of this database created using scripts supplied by Matrix Science. Carbamidomethylation of cysteine residues and TMT modification of lysine residues were set as fixed modification. Methionine oxidation, N-terminal acetylation of proteins, and TMT modification of peptide N-termini were set as variable modifications.
Unless stated otherwise, we accepted protein identifications as follows. (i) For single spectrum-to-sequence assignments, we required this assignment to be the best match and a minimum Mascot score of 31 and a 10× difference of this assignment over the next best assignment. Based on these criteria, the decoy search results indicated <1% false discovery rate (FDR). (ii) For multiple spectrum-to-sequence assignments and using the same parameters, the decoy search results indicate <0.1% FDR. All identified proteins were quantified; FDR for quantified proteins was below 1%.
Peptide and Protein Quantification
Reporter ion intensities were read from raw data and multiplied with ion accumulation times (the unit is milliseconds) so as to yield a measure proportional to the number of ions; this measure is referred to as ion area (37). Spectra matching to peptides were filtered according to the following criteria: mascot ion score >15, signal-to-background of the precursor ion >4, and signal-to-interference >0.5 (38). Fold changes were corrected for isotope purity as described and adjusted for interference caused by coeluting nearly isobaric peaks as estimated by the signal-to-interference measure (39). Protein quantification was derived from individual spectra matching to distinct peptides by using a sum-based bootstrap algorithm; 95% confidence intervals were calculated for all protein fold changes that were quantified with more than three spectra (37). Only proteins quantified with more than one quantified spectrum (qusm) and more than one unique peptide (qupm) were considered for downstream analysis.
Experimental Design and Statistical Rationale
Protein annotations were based on the UniProtKB database (November 14, 2019). Data analysis and visualizations were performed in R (version 4.0.2). All experiments were performed using a TMT-labeling strategy. Identified proteins were filtered for qusm >1 and qupm >1. For secretomics time course experiments, each biological replicate of cytokine treatment and the corresponding time-matched controls for each of the five time points were combined into one multiplexed TMT-10 experiment. All treatments were performed in biological triplicates resulting in three multiplexed analyses per cell system and cytokine. The triplicate MS experiments were merged for further analysis. Summed up ion areas of each TMT-channel and each replicate were log2 transformed and normalized to the median of the density maxima of all samples. The normalization factors of each TMT-channel and each replicate were used to calculate a median of normalization factors for every TMT channel across the replicate measurements. A TMT-channel was identified as an outlier, if (1) the normalization factor of this individual TMT-channel was greater than 2 and (2) the median normalization factor of the triplicates from this treatment was greater than 1.5 due to this individual TMT-channel. Statistical analysis was done separately for each time point by contrasting treatment to the time-matched control using LIMMA (40). Only proteins identified at least in two of the three replicates were considered for LIMMA analysis. Proteins were considered as significant when passing the following cut-offs: two times the median SD of ratios between replicates of each time point and treatment and by a p-value cut-off of 0.05 using Benjamini-Hochberg corrected p-values as described in (41). In the interval-based secretome analysis of IL6-treated dHepaRG cells, TMT channel 128L (IL6 treatment at 4 h) from replicate experiment 2 was excluded from the analysis as it did not meet the above described quality parameters for normalization factors. For the inhibition of extracellular proteases with the small molecule inhibitors Ilomastat or TAPI-0, two biological replicates of DMSO-treated control samples and three biological replicates of each treatment (IL1b only, inhibitor only or IL1b + inhibitor) were combined into one sample for mass spectrometric analysis per inhibitor using TMT-11 isobaric mass tags. K-means clustering across all time points was trained in a first step only with proteins annotated as secreted in their subcellular localization. The number of clusters was determined in an iterative process by manually testing different numbers of clusters. As starting point, we assigned eight clusters and reduced the number of clusters step by step, until no redundant clusters were left. Clusters were considered as redundant, when a calculated spearman correlation coefficient, based on the mean log2 fold changes of clusters, was greater than 0.90. In a second step, the clue R package (42) was used to assign all other proteins of the dataset to one of the defined clusters from step 1. Gene ontology enrichments were obtained using the topGO package in R (43). The enriched gene ontology (GO)-Terms were filtered for Benjamini-Hochberg corrected p-values of <0.05.
Results
Acute-phase proteins are mainly produced by hepatocytes in response to proinflammatory cytokines. To analyze the time-dependent release of proteins during the APR in hepatocyte model systems, we adapted a mass spectrometry–based secretomics approach using serum-free medium (20) to an isobaric mass tag-based chemical labeling strategy (TMT) to enable the precise quantification of time-dependent changes. Cells were treated with the proinflammatory cytokines IL1b or IL6 to stimulate the APR and serum-free cell culture supernatants were collected after 1, 2, 4, 8, and 12 h. One replicate of cytokine treatment and time-matched control for each of the five time points were labeled with TMT reagents and combined for multiplexed mass spectrometric analysis. All treatments were performed in triplicates resulting in three multiplexed mass spectrometric experiments per cell system and cytokine.
A total of 3882 proteins were identified from the supernatants of IL1b-treated HepG2 cells across the replicate experiments with 277 proteins being annotated as secreted or extracellular in UniProt (supplemental Table S1). While abundances of the majority of these proteins remained unaltered, 9, 6, 12, 13, and 13 proteins were significantly changing at the 1, 2, 4, 8 and 12 h time points, respectively (Fig. 1A and supplemental Table S1). As commonly observed in label-free secretomics analyses (20, 23), most identified proteins are predicted to be intracellular, likely resulting from cell death. These proteins mask the identification of secreted and shedded proteins. For example, upon cytokine stimulation. Hence, it is critical to keep cell death at the absolute minimum within the desirable experimental timeframe.
Fig. 1.

HepaRG cells secrete more acute-phase proteins upon IL1b treatment than HepG2 cells, but viability in serum-free medium limits the collection window to 12 h.A, secretomics analysis of IL1b-treated HepG2 cells. All identified proteins are depicted with their mean log2 fold change (n = 3) against the time-matched controls at the indicated time points. Proteins passing the significance thresholds (p(Benjamini-Hochberg) < 0.05 and log2 fold change > 2× SD of the individual treatment) are colored in orange, acute-phase response proteins are colored in purple, and acute-phase proteins passing the significance thresholds are indicated as triangles. IL1b is colored in blue and indicated with name. 2-fold increase in abundance is indicated by horizontal bars. B, heatmap of all proteins significantly secreted in HepG2 cells upon IL1b treatment for at least one time point. Displayed are mean log2 fold changes to the respective time-matched control. Statistically significant changes as described in (A) are denoted with asterisks (∗) in the respective cells. Bold names label known acute-phase proteins. C, same as (A) for differentiated HepaRG cells. D, same as (B) for proteins significantly secreted in HepaRG cells upon IL1b treatment. Due to the large number of significant proteins, only those proteins are displayed which show a log2 fold change >1.5. E, determination of LDH levels in the supernatant of HepaRG cells cultured in serum-free medium using a enzymatic assay at multiple time points (each time point in n = 3). Percentage of cell death is determined as percentage to the maximal LDH activity by lysing the cells with Triton X-100 at time point 0 h. F, light microscopic examination of differentiated HepaRG cells kept in serum-free medium for up to 72 h. White arrows denote areas with disruptions of the monolayer. LDH, lactate dehydrogenase.
IL1b induced the early release of cytokines and inflammation-related proteins from HepG2 cells, including the chemotactic factors CCL20, CX3CL1, CXCL8 (IL-8), CSF1, SDC4, the IL-1b receptor antagonist IL1RN, and the urokinase-type plasminogen activator PLAU (Fig. 1B). LCN2 was the only APP among the significantly changing proteins within the observed timeframe of 12 h (Fig. 1B), while the majority of known acute-phase proteins were either not detected or did not change in secretion rate (supplemental Fig. S1B). Additionally, we observed the release of the epidermal growth factor receptor ligand AREG, the growth/differentiation factor-15 (GDF-15), and the matricellular protein THBS1 that are related to inflammatory processes (44, 45, 46).
Upon treatment of HepG2 cells with the prototypic APR inducer IL6, we did not observe any changes in protein secretion (supplemental Fig. S1C and supplemental Table S1). Previous cell surface proteome mapping of these cells (47) did not identify the IL6 receptor, which could explain the lack of a biological response to IL6 treatment. In our data, we also did not detect the release of IL6 upon IL1b treatment, a biologically important reaction of hepatocytes to different stimuli, which is known to modulate the APR in an autocrine and endocrine fashion (48). The limited response of HepG2 cells toward the proinflammatory cytokines IL1b and IL6 raised the question whether this hepatocyte cell system reflects the true biology of the APR, hence, we chose dHepaRG cells as an alternative hepatic model cell line.
The secretomics analysis of IL1b-treated dHepaRG cells identified a total of 4131 proteins across triplicates and five time points (Fig. 1C and supplemental Table S1); 306 proteins had a subcellular location annotation as secreted or being extracellular. Across the time course, 5, 8, 25, 40, and 56 proteins displayed significantly altered secretion rates after 1, 2, 4, 8, and 12 h, respectively (Fig. 1C). The secretome of IL1b-treated dHepaRG cells was characterized by an early release of chemokines and MMPs within the 12 h time window (Fig. 1D). In the first 2 h, we observed the release of CXCL8 (IL-8) and IL-6 in the supernatants, followed by further chemokines and pattern recognition molecules such as CCL20, CXCL1, CCL2 (MCP-1), CSF1, and LBP, within 4 h (Fig. 1D and supplemental Fig. S2C). The long pentraxin PTX3 was the first acute-phase protein showing significant abundance increase in cell culture supernatants within 4 h, followed by the acute-phase proteins CRP, LCN2, LBP, SERPINA3, CFB, and SERPINE1 starting at 8 h post stimulus (Fig. 1D and supplemental Fig. S2B).
The secretomics analysis of IL6-treated dHepaRG cells identified a total of 3859 proteins across triplicates and five time points (supplemental Fig. S2D and supplemental Table S1) with 283 proteins being annotated as secreted or extracellular. In the time course, strong stimulus-dependent secretion was observed after 4 to 8 h (supplemental Fig. S2G), indicating an earlier onset of APP release as compared to IL1b treatment. However, IL6-induced secretion was characterized by fibrinogens (FGA, FGB, FGG), CRP, LBP, and SERPINA3 but not an early release of chemokines (supplemental Fig. S2, F and G). This suggests that IL6 signaling is crucial for the secretion of APP in dHepaRG cells and further explains the delayed APP secretion upon IL1b stimulation.
In summary, the early phase of the IL1b-induced APR is characterized by the concerted secretion of chemokines and MMPs. In contrast to IL1b, IL6 treatment did not provoke the release of a chemotactic answer but stimulated the release of APP more directly. The direct comparison of the secretome data of IL1b- and IL6-treated HepG2 and dHepaRG cells featured a more diverse secretory response in dHepaRG cells than in HepG2 and comprised the biologically important secretion of IL-6 and the inflammation marker CRP (9). Based on these observations, we concluded that HepG2 cells are not a suitable model system to study the APR in-vitro.
However, even with the dHepaRG system, only seven acute-phase proteins were differentially secreted into the cell culture supernatant during this early inflammatory phase, while the majority of acute-phase proteins were either not secreted or did not change their secretion rate upon IL1b treatment. This made us hypothesize that these APP are either not responsive to the IL1b stimulus at all or the secretion of these proteins takes much longer than the 12 h timeframe that was monitored.
To evaluate whether an extension of the serum-free collection time would be feasible, we assessed the viability of dHepaRG cells in serum-free basal medium for up to 72 h by measurement of LDH (n = 3), a marker for plasma membrane damage, in cell culture supernatants (Fig. 1E). We observed a time-dependent increase of LDH by up to 15% within 72 h. Light microscopic examination revealed a marked disruption of the monolayer after 48 h in serum-free medium (Fig. 1F) consistent with increased levels of cell death that will hamper secretomics analysis. Furthermore, irrespective of effects on cell viability, prolonged incubation of cells in serum-free medium may result in changes of transcript and expression levels of proteins, altered protein phosphorylation, a reduction in the basal activity of signaling pathways, and can negatively impact protein secretion (15, 23, 49, 50, 51). To control for this, sampling times are preferred that are significantly shorter than the typical protein half-life.
Interval-Based Secretomics Enables Longer-Term Secretomics Experiments and Covers the Full Spectrum of the APR
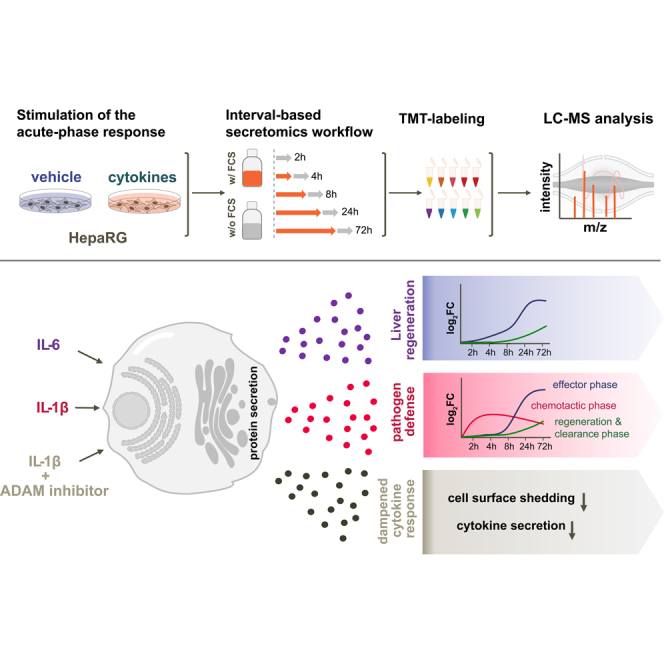
To address these challenges, we conceived an experimental design which allows secretome studies over multiple days (Fig. 2) by performing treatments in presence of serum, followed by 2-h incubation intervals in serum-free medium to collect secreted proteins. We reasoned that the interval-based incubation scheme would have two advantages: first, probing the secretion in intervals will no longer measure total protein amounts as this is the case with typical cumulative secretomics workflows but rather measures protein secretion rates and thus allows determining changes in protein secretion rates over time. Hence, basal, stimulus-independent secretion events are easier to detect as no change is expected. Second, minimizing the serum-free time window to 2 h should provide a good balance between achieving decent proteomic coverage and reducing the negative impact of nutrient and growth factor deficiency on protein expression.
Fig. 2.

Schematic representation of the secretomics workflow with both collection schemes: the standard serum-free ‘cumulative’ and the ‘interval-based’ approach. ①, cumulative experimental design: protein secretion is probed by collecting cell culture supernatants under serum-free conditions for a defined time range with the treatment. Interval experimental design: treatments are performed in presence of serum and secretion is probed in 2 h serum-free collection windows. ②, serum-free supernatants are cleared to remove potential cell debris by centrifugation through a 0.45 μm filter-plate. ③, proteins are digested, and peptides are chemically labeled with isobaric TMT reagents and are subsequently pooled. ④, pooled peptide samples are fractionated via high-pH spin column fractionation into eight fractions and analyzed by LC-MS/MS. TMT, tandem mass tag.
To cover early secretion events as well as late secretion events elicited by secondary, indirect mechanisms, we monitored secretion over 72 h. Upon stimulation of the APR in-vivo, changes in the plasma concentrations of acute-phase proteins, such as CRP or SAA, typically peak at 2 to 3 days (52). To cover and resolve the early response upon IL1b treatment, we included the time points 2, 4, and 8 h. For the four remaining channels of the TMT-10 plex experiment, we chose a mid-time point (24 h) to cover early transcription-dependent secretion events of proteins and a late time point (72 h) to also cover slow and indirect effects.
In total, we identified 4131 proteins across three replicates and five time points (2, 4, 8, 24 and 72 h) in the supernatants of IL1b-treated dHepaRG cells, of which 219 proteins were annotated to be secreted or extracellular (Fig. 3A and supplemental Table S3). Across the time course, 0, 0, 21, 80, and 162 proteins displayed significantly altered secretion rates after 2, 4, 8, 24, and 72 h, respectively.
Fig. 3.

Interval-based secretomics analysis of the acute-phase response triggered by IL1b or IL6 in differentiated HepaRG cells for up to 72 h.A, interval-based secretomics analysis of IL1b-treated HepaRG cells. All identified proteins are depicted with their mean log2 fold change (n = 3) against the time-matched controls at the indicated time points. Proteins passing the significance thresholds (p(Benjamini Hochberg) < 0.05 and log2 fold change > 2× standard deviation of the individual treatment) are colored in orange, acute-phase response proteins are colored in purple, and acute-phase proteins passing the significance thresholds are indicated as triangles. IL1b is colored in blue and indicated with name. 2-fold increase in abundance is indicated by horizontal bars. B, all acute-phase proteins identified in the secretomes of IL1b-treated differentiated HepaRG cells across different time points irrespective of their significance. Displayed are log2 fold changes to the respective time-matched control. Statistically significant changes are denoted with asterisks (∗). C, proteins released at 72 h of treatment with IL1b grouped by their subcellular location annotation (based on UniProt annotation). Gray: all proteins, blue: proteins with an extracellular domain, teal: proteins without a signal peptide (S. P.) and not annotated as secreted, purple: proteins with signal peptide and not annotated as secreted, yellow: proteins without signal peptide and annotated as secreted, pink: proteins with signal peptide and annotated as secreted. D–F, same as (A), (B), and (C) but for IL6-treated differentiated HepaRG cells.
We observed secretion of multiple acute-phase proteins starting at 8 h post stimulus (Fig. 3B). Protein release upon IL1b treatment was not limited to acute-phase proteins. Out of the 162 proteins that showed significantly changing secretion rates after 72 h, 106 proteins featured a significantly higher secretion rate. Out of these 106 proteins, 40 proteins were annotated as “secreted with signal peptide”, eight proteins were annotated as “secreted without signal peptide”, and 57 proteins were not annotated as secreted at all including nine transmembrane proteins with extracellular domains (Fig. 3C and supplemental Table S3). These observations suggest that a substantial fraction of proteins might be released via nonclassical secretion pathways, a phenomenon which was previously described for cancer cell lines (23). Of note, among the proteins that were annotated as “not secreted with signal peptide”, we found 18 ER-localized proteins such as the ER stress associated chaperones HSP90B1, HSPA13, HSPA5, and cochaperones DNAJB11, DNAJC3, the protein disulfide-isomerases PDIA3, PDIA4, PDIA6, TXNDC5, and the oxidoreductase ERO1A.
The interval-based secretome analysis of IL6-treated dHepaRG cells identified 4417 proteins across triplicates of which 215 proteins were annotated as secreted or extracellular (Fig. 3D and supplemental Table S3). The nonconventional protein secretion was less pronounced upon treatment with IL6 as compared to IL1b (Fig. 3E). We observed significantly different protein secretion rates starting at 24 h post stimulus including the acute phase proteins ORM1, CP, APCS, HP, SERPING1, CFB, SAA1 (Fig. 3F). The extended time range of the interval-based protocol not only allowed the characterization of positive secretion events, it now also enabled pinpointing the downregulation of protein secretion such as in case of the negative acute-phase proteins ALB, TF, and TTR (Fig. 3, B and F). Next, k-means clustering was used to characterize the time dependencies of the APR (Fig. 4 and supplemental Table S4). We first filtered the datasets for proteins annotated as secreted only to cluster the data and then matched the time-dependent secretion profiles of all remaining proteins to these clusters. The individual clusters mirror the different waves of the APR and help to pinpoint the major differences between the two inflammatory stimuli.
Fig. 4.

Time-dependent clustering of protein secretion discriminates early- and late-secretion events during the acute-phase response.A, time-resolved k-means cluster analysis of the interval-based secretomes of IL1b-treated HepaRG cells. Left panel, k-means clustering of all identified proteins (only clusters with time-dependent changes are shown here, for all clusters see supplemental Fig. S5). Clustering was performed by using proteins annotated to be secreted as training dataset. Colored ribbons indicate the mean log2 fold change ± SD in each cluster. Right panel, GO-term enrichment analysis showing the top ten biological processes for each cluster (brown bars indicate significant GO terms with p(Benjamini-Hochberg corrected) < 0.05). B, same as (A) for IL6 (only clusters with time-dependent changes are shown here, for all clusters see supplemental Fig. S6). C, heatmaps of time-dependent abundance change of all proteins grouped into cluster 1 and 2 from panel A of IL1b-treated HepaRG cells. Displayed are log2 fold changes to the respective time-matched control. Statistically significant changes are denoted with asterisks (∗). D, same as (C) for IL6-treated HepaRG cells. GO, gene ontology.
The early phase of the IL1b-induced inflammatory response was characterized by a fast release of chemotactic factors (Fig. 4A Cluster 1, Fig. 4C and supplemental Table S4), MMPs, and antimicrobial proteins showing a steep increase in secretion rates within the first 4 h. This data is in agreement with the cumulative IL1b dataset (supplemental Fig. S3 and supplemental Table S2) that identified CXCL8, CXCL1, CCL20, and CCL2 as well as MMP1, MMP3, MMP10, and MMP2 and the cytokine IL6 for the cytokine and MMP cluster. However, lower proteomic coverage was achieved likely due to compromises made to balance early and late time points and shorter collection times for secreted proteins.
The early chemotactic response was followed by the secretion of effector molecules (Fig. 4A, cluster 2) with strongly increased secretion rates from 8 h to 72 h. Several proteins of this effector cluster such as HP, LCN2, ORM1, ORM2, SERPINA3, and SERPINB3 are mapping to the neutrophil degranulation pathway or have further immunomodulatory effects like, for example, ORM1 that can enhance the secretion of several cytokines (53). Of note, the three nonacute-phase proteins SERPINB3/B4 and CHI3L2 were among the APP in Cluster 2 and are associated with inflammatory processes (54, 55). In summary, our data characterize SERPINB3/B4 and CHI3L2 as additional acute-phase proteins in the context of an IL1b response.
Cluster 3 (late response) contained proteins with moderately increased secretion rates including complement factors (C1R, C1S, C3, C4BPA, CFH), fibrinogens (FGA, FGB, FGG), and the proteins S100A8/A9 as well as the chaperones HSP90B1, HSPA13, HSPA5, the cochaperones DNAJB11, DNAJC3, the protein disulfide-isomerases PDIA3, PDIA4, PDIA6, TXNDC5, and the oxidoreductase ERO1A. Among the members with the strongest changes in this cluster, we found several proteins with anti-inflammatory or protease inhibitory properties, such as PLA2G2A or SLPI. With GO terms “extracellular matrix organization” and “blood coagulation, fibrin clot formation” being enriched in cluster 3, the composition and time profile of this cluster suggest it to represent the regeneration and clearance phase of inflammation that involves enzymes such as B4GALT1, BMP1, and COLGALT1 and constituents of the extracellular matrix (ECM) such as COL5A1, FBLN1, and FN1. Additionally, after 72 h, the secretion rate of MMPs (MM3, MMP7, (cluster 1)) was equal to the time-matched controls and as such reached the basal secretion rate again (Fig. 4C). We also found CHI3L1 to be secreted 72 h post stimulus which is known to influence tissue remodeling by inhibition of IL1-induced MMP secretion and regulation of MMP activity (56, 57). Of note, TNFAIP2 was among the proteins with the highest abundance relative to the time-matched control within this cluster and which is known to modulate the inflammatory response by inhibition of NFkB activity.
Approximately, 30% of the proteins in this group were neither annotated as secreted nor contained a signal peptide (supplemental Table S4). However, the secretion profiles of these proteins suggest a release via a nonclassical secretion pathway rather than cell death, since they are clearly separated from the background cluster 5 (supplemental Fig. S5) that contained high abundant intracellular proteins like LDHA. Consistent with this, the majority of these proteins matched with the vesiclepedia database (58) including TNFAIP8, B4GALT1, and NFKB2 that were previously found to be unconventionally released via extracellular vesicles (59, 60, 61). Additionally, ER-localized chaperones such as HSP90B1, HSPA5, DNAJC3 have been reported to be transported to the extracellular space during inflammatory conditions (62, 63, 64).
Cluster 4 consisted of proteins for which secretion is downregulated upon IL1b treatment, such as the known negative acute-phase proteins ALB, TF, TTR, SERPINC1, RBP4. Of note, cluster 4 also contained proteins that were observed to be downregulated in plasma of patients with liver cirrhosis (65), (e.g., F2, SERPINC1, SERPIND1, CPB2, C6, RBP4, and TTR) the final stage of hepatic disarrangement in, for example, response to chronic inflammatory conditions. We also observed a downregulation of proteins mapping to triglyceride metabolism and transport, such as APOA1, APOB, APOC3, reflecting a change in lipoprotein particles and consistent with a change in serum lipid profiles upon inflammation. It was shown that the APR is associated with changes in lipoproteins, for example, APOA1 plasma levels are reduced and APOA1 is displaced in high density lipoprotein particles by SAA1 (66, 67, 68). Of note, we observed increased secretion rates of SAA1.
Cluster 5 with 3374 protein members reflected the background proteome and included secreted proteins that were IL1b independent (supplemental Fig. S5). As reflected by the GO enrichment, this cluster contained mainly high abundant intracellular proteins such as ribosomal proteins, splicing factors, and structural proteins, for example, ACTB, TUBB, and the LDH subunit A.
K-means clustering of the IL6 interval-secretome data revealed a fundamentally different response compared to the IL1b dataset (Fig. 4B and supplemental Table S4). Within the first 24 h, dHepaRG cells mainly responded to IL6 stimulation with the secretion of fibrinogens (FGA, FGB, FGG), HP, SERPINA3, CP, and LBP (Fig. 4B, Cluster 1) and the GO term “blood coagulation, fibrin clot formation” was strongly enriched. The late response (Fig. 4B, cluster 2) resembles the proposed regeneration and clearance phase of cluster 3 of the IL1b treatment (Fig. 4A). Among the highest abundant proteins in this group, we found ORM1/2 to be secreted after 24 h. Increased levels of ORM1 were observed after hepatectomy in humans and a ORM1 KO in mice was reported to impair liver regeneration (69). ORM1/2 are also mapping to the GO-term “platelet degranulation”, which was under the top three enriched GO terms of cluster 2. Furthermore, and consistent with previous reports, IL6 reduced the secretion of the negative acute-phase proteins ALB, TF, TTR, RBP4 (Fig. 4B cluster 3) (70, 71).
IL1b Stimulation Induced Ectodomain Shedding in HepaRG Cells
Closer inspection of proteins released by dHepaRG cells upon IL1b stimulation revealed several transmembrane proteins with extracellular domains (Fig. 5A). As all identified peptides mapped to the extracellular domains (supplemental Fig. S7A) and, in addition, the IL1b treatment induced the release of several MMPs, we hypothesized that those proteins are released into the secretome via proteolytic cleavage of the ectodomains. Ectodomain shedding is an important posttranslational modification with implications in a variety of cellular processes, such as cell adhesion, proliferation, differentiation, migration, apoptosis, necroptosis, and inflammation (72, 73, 74, 75, 76). To further investigate induced ectodomain shedding during the APR in dHepaRG cells, we studied the effect of inhibition of extracellular proteases of the ’a disintegrin and metalloprotease’ (ADAM)- family and MMPs by two small molecule inhibitors. Cells were either treated with 10 μM of the broad spectrum MMP inhibitor Ilomastat (GM6001) or with 50 μM of the ADAM17 and MMP inhibitor TAPI-0. The nonmembrane permeable inhibitor TAPI-0 was used at 50 μM to ensure effective inhibition of a wide range of known targets within the ADAM and MMP families. Samples were treated with a combination of IL1b and TAPI-0 or Ilomastat to specifically inhibit IL1b-induced shedding events. Moreover, the treatment with TAPI-0 or Ilomastat alone allowed for identification of potential constitutive shedding events. As controls, we included time-matched mock-treated samples (DMSO) as well as samples that were only treated with IL1b (Fig. 5B). Supernatants were harvested after 8 h, and the samples for each inhibitor were encoded with isobaric mass tags and combined for mass spectrometric analysis(Fig. 5B). In the cumulative secretome data of IL1b-treated HepaRG cells, high levels of the proteases MMP-1, MMP-3, MMP-7, MMP-10, MMP-12, and MMP-13 were detected already at 8 h post stimulus, suggesting that this time point would provide good sensitivity and coverage of the relevant biology.
Fig. 5.

Constitutive and IL1b-induced ectodomain shedding in HepaRG can be modulated by metalloprotease inhibitors and leads to secondary effects on cytokine and metalloprotease secretion.A, proteins released by IL1b-treated differentiated HepaRG cells compared to time-matched controls at indicated time points (same dataset as in Fig. 3A). Transmembrane proteins containing an extracellular domain are indicated in blue, matrix metalloproteinases are indicated in pink. 2-fold increase in abundance is indicated by horizontal bars. B, schematic illustration of the shedding experiments. Differentiated HepaRG cells in 12-well plates were treated with IL1b, inhibitor, the combination or DMSO control. Tested inhibitors were Ilomastat (10 μM) and TAPI-0 (50 μM). Supernatants were harvested after 8 h, and the samples were combined into one sample for mass spectrometric analysis individually per inhibitor using TMT-11 isobaric mass tags. C, changes in protein secretion patterns upon addition of the broadband MMP inhibitor Ilomastat after 8 h treatment. Displayed proteins are filtered for transmembrane proteins containing an extracellular domain with a significant change (p(Benjamini Hochberg) < 0.05 and log2 fold change > 2× SD of the individual treatment) upon inhibitor treatment in at least one of the indicated treatments. Asterisks indicate significance for the respective contrast. Bold names indicate known ADAM targets. D, same as (C) for the ADAM17 inhibitor TAPI-0. E, abundance change of SORT1 in the supernatant in the TAPI-0 experiment. Displayed is log2 protein fold change of the indicated contrasts. F and G, inhibition of ectodomain shedding by TAPI-0 reduces the IL1B-induced secretion of (F) cytokines and (G) MMPs into the cell culture supernatant. Asterisks indicate a significant difference in the comparison of IL1b versus control to IL1b stimulation in presence of TAPI-0 versus control (n = 3): ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001. MMP, matrix metalloproteinase; TMT, tandem mass tag.
First, we investigated whether cell surface shedding contributes to the constitutive secretome of dHepaRG cells. We compared the secretomes of cells treated with inhibitor alone to the secretomes of the untreated cells. Upon treatment with the broad spectrum MMP inhibitor Ilomastat which has also been described to inhibit the activity of ADAM10 and ADAM17 (77), 24 proteins were significantly reduced in abundance in these cell culture supernatants (supplemental Table S5). Among these, we found 14 annotated transmembrane proteins with extracellular domain, such as the membrane associated chemokine CX3CL1, for which constitutive and induced shedding by ADAMs and MMPs has been previously described (78). Treatment with the ADAM17 and MMP inhibitor TAPI-0 significantly affected the release of 36 proteins compared to the untreated controls, with 21 annotated transmembrane proteins with extracellular domains being in this group. Of these, ten transmembrane proteins with extracellular domain were overlapping as hits in experiments with Ilomastat and TAPI-0 (supplemental Fig. S7B).
Second, we investigated the effect of MMP/ADAM inhibition on surface shedding upon stimulation of the APR with IL1b. To identify shedding events upon stimulation of the APR, the secretomes of combined IL1b and inhibitor-treated cells were compared to the treatment with IL1b only. Upon inhibition with the broad spectrum MMP inhibitor Ilomastat, the abundances of 37 proteins in the supernatant were significantly reduced of which 25 are transmembrane proteins with an extracellular domain. Treatment with the ADAM17 and MMP inhibitor TAPI-0 led to a significant reduction of protein abundance in the supernatant of 80 proteins compared to IL1b only. Of those proteins, 41 were annotated as transmembrane proteins with extracellular domain, 21 proteins were affected by both inhibitors, while 20 proteins were exclusively dependent on TAPI-0 (supplemental Fig. S7C).
Upon inhibitor treatment with Ilomastat (Fig. 5C) and TAPI0 (Fig. 5D), several known ADAM substrates (76, 78, 79, 80, 81) were reduced in abundance in the supernatants such as CX3CL1, ALCAM, SDC4, NEO1, APP, APLP2, and CD44 consistent with shedding of these proteins upon IL1B stimulation. Shedding was also observed constitutively in dHepaRG cells, as transmembrane proteins like, for example, CX3CL1, ALCAM, NEO1, APP, or SDC4 showed a reduced abundance upon treatment with Ilomastat (Fig. 5C) or TAPI-0 (Fig. 5D) in the absence of IL1b.
Furthermore, we observed a significantly reduced abundance of SORT1 (sortilin) by inhibition with TAPI-0 (Fig. 5E). SORT1 is a VPS10 domain sorting receptor which is predominantly involved in the secretory pathway binding to various ligands. SORT1 functions as a clearance receptor at the cell surface where it can be shed by ADAM proteases (82, 83, 84). Interestingly, recent studies have demonstrated that SORT1 is involved in the regulation of lipoprotein metabolism and cytokine secretion (85, 86). Loss of SORT1 impairs the secretion of IL-6 and INF-γ in macrophages, leading to reduced levels of these cytokines in cell culture supernatants, and a SORT1 KO study in mice previously observed significantly attenuated expression levels of cholangiocyte-derived cytokines like CXCL1, CXCL2, CCL2, and IL6 (87). A direct interaction of SORT1 with IL6 and other cytokines has been demonstrated by SPR analysis (86, 88) further underscoring its involvement in cytokine trafficking and secretion. Treatment with TAPI-0 not only inhibited the shedding of SORT1 (Fig. 5E), it also impaired the secretion of the cytokines IL6, CCL2, CCL20, CXCL1, CXCL8 (Fig. 5F), and the matrix metalloproteinases MMP-3, MMP-10, MMP-12 (Fig. 5G). However, the release of other secreted proteins that are induced upon IL1b treatment were not affected by TAPI-0 (supplemental Fig. S5D). In summary, ADAM inhibition during the early phase of the IL1b-induced APR leads to a dampening of the early cytokine response.
Discussion
The majority of in-vitro secretome studies are performed under serum-free cell culture conditions which strongly limits the observable time window to only a few hours as prolonged incubation times in serum-free medium can compromise cellular integrity, narrowing down the biological relevance of such secretome data. We addressed this shortcoming with a modified experimental design that allows secretomics analysis from cells treated in the presence of serum by probing the protein secretion rate into the supernatant in 2-h intervals of serum-free medium. The limited time window in which cells are maintained in serum-free medium extends the experimentally covered time range to multiple days without a negative impact on cell viability. The interval-based secretomics approach enables the study of protein secretion in cell systems (i) which are highly susceptible to serum-free cell culture conditions, (ii) in which cell culture relies on specific media compositions, for example, differentiation, or (iii) for which the use of metabolic labeling strategies for selective enrichment of secreted proteins, such as azidohomoalanine (AHA) or azido sugars (15, 16, 19) is challenging due to limited cell numbers, incompatibilities with the labeling reagents, general toxic effects, or undesirable variations in cell growth. Although elegant, metabolic strategies have inherent weaknesses which can be addressed by the interval-based protocol. Azidosugar-based enrichment methods work via protein glycosylations leading to potentially false-positives if the applied perturbations affect the cellular glycosylation machinery; and in addition, unconventional secretion events of nonglycosylated proteins are missed. Pulsed AHA labeling approaches are vulnerable to perturbations of the cellular translation machinery; and proteins which were synthesized before the AHA-pulse, for example, pre-existing secretory granules cannot be specifically enriched. Additionally, AHA labeling in combination with the necessary methionine starvation can potentially disturb cellular signaling pathways.
Protein quantification is another crucial design element for secretomics experiments. We employed a TMT-based isobaric labeling strategy in which up to 11 samples were pooled into one single MS run. We combined either multiple time points or multiple treatments and treatment combinations into one TMT experiment. Especially for time course experiments, TMT labeling ensures complete quantitative data for all identified proteins at all time points. This data completeness, which cannot be guaranteed with a label-free quantitative approach, enables the identification of time-dependent phases during a cellular treatment in addition to the high precision of relative quantification typically achieved with isobaric mass tags (89).
We demonstrated the utility of the interval-based secretomics approach by studying the APR, an indispensable reaction of hepatocytes to inflammatory processes that is characterized through cytokine-regulated secretion changes of proteins that elicit pivotal functions to restore body homeostasis. A hallmark of the hepatic APR is the control on the transcriptional level (6, 90). Monitoring such regulation on secretome level requires observation times of several days which is incompatible with serum-free cell treatment due to impaired cell viability. Consistent with this, short-term treatments of HepG2 and HepaRG cells for up to 12 h led to incomplete APR with marked differences between these two hepatocyte cell lines, questioning the biological relevance of HepG2 cells for the study of liver inflammatory conditions. The interval-based secretomics approach enabled the characterization of protein secretion in dHepaRG cells over a time period of 72 h while circumventing the detrimental effects of prolonged serum starvation. This, for the first time allowed studying direct early secretion events as well as indirect effects resulting from downstream transcriptional activation providing detailed insights in the orchestration of the APR in hepatocyte cell models upon stimulation with IL1b and IL6.
IL1b induced extensive remodeling of protein secretion which can be classified into three consecutive phases (Fig. 4A). First, the early release of chemokines including CXCL8, CXCL1, CCL20, CCL2, and IL6 as well as MMPs such as MMP1, MMP3, MMP10, and MMP2 with only few APP (LCN2, PTX3, LBP, SERPINA3) being secreted (Figs. 1D and 3E). This first response to IL1b triggers the attraction and activation of the cellular component of the innate immune system. CC chemokines such as CCL20 and CCL2 promote a migration of monocytes and lymphocytes from the bloodstream into the tissue (91), while CXC chemokines, such as CXCL8, mobilize neutrophils to enter the inflamed or damaged tissue (92, 93, 94). Neutrophils are among the first cells that arrive at the site of inflammation. The early release of CXCL8 not only functions as cell attractant, signaling in neutrophils but also promotes the augmentation of a respiratory burst that generates oxygen radicals and nitric oxide and induces a discharge of neutrophilic granules (92). The early chemokine release was accompanied by the secretion of MMPs for the modification of the extracellular matrix and diverse humoral components such as CRP, PTX3, and CFB that bind to pathogens and are capable of modulating inflammation through the complement system. The concerted secretion of chemokines and ECM modifying proteases, such as MMPs, stimulates trafficking of immune cells through tissue barriers or basement membranes which is only possible via the modification or degradation of the ECM. Furthermore, MMP biology is not limited to ECM degradation: MMPs are modulators of inflammation and innate immunity, adding an additional regulatory level during acute and chronic inflammatory processes by modification of cytokines and chemokines (95, 96).
The early chemokine response is followed by an effector phase, which is characterized by the secretion of proteins, that are capable of stimulating further downstream effects on the attracted immune cells (HP, LCN2, ORM1, ORM2, SERPINA3, SERPINB3, and CHI3L2), such as the degranulation of neutrophils or the enhanced release of TNFα and other cytokines. Of note, SERPINB3 and SERPINB4 are usually undetectable in the liver under steady state conditions, whereas expression is associated with chronic inflammatory conditions and hepatic fibrosis (54, 55). The non-acute phase protein CHI3L2 has been described to be secreted in osteoarthritis and exerts angiogenic and monocyte chemotactic properties (97, 98, 99, 100). The APP ORM1 for example can promote proinflammatory and anti-inflammatory effects on immune cells, dependent on its local concentration. On mononuclear cells, ORM1 induces the secretion of cytokines, while it simultaneously inhibits the chemotaxis of neutrophils and modulates their function (53). The orosomucoid family member ORM2 conversely possesses anti-inflammatory and immunomodulatory properties through, for example, the restriction of neutrophil migration (101), hence indicating the beginning resolution of the inflammatory response. This process is characterized by the local and temporal production of proresolving mediators that aid to clear immune cells from the site of inflammation and restore the ECM. In our secretome data, we found that the secretion rates of such mediators, for example, PLA2G2A, SLPI, FN1, COL5A1, FBLN1, SEMA4B, were continuously increased, starting at 24 h after stimulation with IL1b. This shows that proinflammatory and anti-inflammatory events coincide at this stage.
The regeneration and clearance phase was characterized by proteins with anti-inflammatory, protease inhibitory, or growth-promoting properties such as phospholipase PL2G2A, SLPI, or FGL1. Interestingly, we also found CHI3L1 to fall into the regeneration and clearance cluster. CHI3L1 is regarded as potential biomarker for liver fibrosis (102) and is known to promote a transition of macrophages to the anti-inflammatory M2 phenotype (103) that are associated with wound healing and tissue repair. Moreover, the significant release of ER-localized proteins upon IL1b but not upon IL6 such as the chaperones HSP90B1, HSPA13, HSPA5, the cochaperones DNAJB11, DNAJC3, and the protein disulfide-isomerases PDIA3, PDIA4 and PDIA6 point toward a crosstalk between IL1b-dependent inflammation and ER stress in the liver. It was shown that inflammatory cytokines can initiate the activation of an ER stress response and that the activation of the NFkB pathway plays a central role within this ER stress and inflammatory network under different pathological conditions (63, 64). Upon ER stress conditions, chaperones such as HSPA5 are upregulated and can be released into the extracellular space where they exert specific immune modulatory functions. Secretion of HSPA5 for example has been described as a component of resolution-associated molecular patterns (64).
The IL6 secretome data point toward a protective or regenerative role in dHepaRG cells which is marked by an early release of fibrinogens, HP, SERPINA3, CP, and LBP. A previous study has shown that intrahepatic fibrinogen deposition and platelet accumulation is a driving force for liver regeneration (104). Moreover, IL6 induced the release of ORM1/2, two proteins that activate platelets and participate in the exocytosis of platelet granules. A growing body of evidence points toward a central role of platelets in liver homeostasis and as regulators of liver regeneration (105, 106, 107, 108). Platelets have been shown to accumulate in the liver after a resection, releasing secretory granules (106, 109) with mitogenic proteins that are able to stimulate a regenerative process (110). Moreover, ORM1 was shown to be secreted after partial hepatectomy exerting growth-promoting activities on hepatocytes (69). Consistently, besides its role as proinflammatory cytokine and inducer of the APR, a growing body of evidence connects IL6 with a protective and regenerative role in the liver (111, 112) as IL6 KO mice show impaired liver regeneration (112) and a inhibition of IL6 signaling exacerbates liver injury (113).
The early release of IL6 upon IL1b observed in the cumulative secretome data suggests a central role for IL6 in the development of the APR. Different studies have shown that IL6 can be regarded as a key mediator of the hepatic APR (48), which induces gene expression via the transcription factor STAT3 (5), leading to transcriptional activation of the CRP gene (114). The critical involvement of STAT3 in the synthesis and secretion of APP was further demonstrated in mice with a specific deletion of the gp130 signal-transducing receptor subunit (115) that led to impaired STAT3 signaling and abrogation of the APP expression. There is a growing body of evidence that suggests that IL6 is the main inducer of the APR whereas IL1-like cytokines seem to play a modulating role by inhibiting or enhancing the expression of various proteins (6, 8, 116, 117, 118), most likely through interaction between NF-kB and STAT3 signaling. The fact that IL6 stimulated a different response in dHepaRG cells compared to IL1b suggests that both cytokines direct the APR in different directions. IL1b-treated dHepaRG cells displayed an early release of cytokines, including IL6, while only a few APP were secreted during this timeframe. This IL1b characteristic cytokine response was not present upon IL6 treatment, which suggests that the secretion of cytokines in dHepaRG cells is mediated via NFkB activation. As such, our data propose that IL1b directs the APR toward defense against pathogens, whereas the exclusive stimulation with IL6 directs the APR toward tissue repair or regeneration processes. Moreover, our secretome data show that the secretion of APP is (i) dependent on the nature of the stimulus and (ii) that the pattern of coacting cytokines influences the secretion phenotype of the APR.
Finally, inhibition of ADAM proteases by TAPI-0 resulted in reduced constitutive as well as stimulus-dependent shedding of transmembrane proteins. This included reduced shedding of the endosomal sorting receptor SORT1 which was accompanied by an attenuated cytokine response suggesting a direct link between cell surface shedding and cytokine secretion rates. Of note, it has been demonstrated that SORT1 is involved in the exocytic trafficking of cytokines, such as IL-6 and IL-12 (88). As such, our data suggest that the cytokines and MMPs released by dHepaRG cells upon IL1b treatment are SORT1 ligands and ADAM-mediated shedding of SORT1 is necessary for the full secretion of those proteins. The modulation of liver inflammatory conditions through ADAM inhibition thus may have therapeutic potential, and oligonucleotide-based inhibition of ADAM biosynthesis offers the opportunity to achieve tissue selectivity, thus limiting off target tissue–based toxicities (119).
In summary, this study provides a deep analysis of the APR in models of hepatocyte cells. Our results highlight the complexity of the inflammatory secretome and provide a comprehensive view on proteins released by hepatocytes during inflammatory processes. Moreover, our data provide evidence that inflammatory signaling–pathways and liver-specific functions are functional in dHepaRG cells, rendering this cell line an interesting surrogate to primary hepatocytes for the study of liver biology such as liver inflammatory conditions or regenerative processes. We highlight different secretion phenotypes that are stimulated by the cytokines IL1b and IL6 in dHepaRG cells and suggest ADAM inhibition as a potential therapeutic strategy for liver inflammatory conditions.
Whereas most secretome studies solely cover early secretory responses to a stimulus, the here described interval approach extents the experimental time range and enables long-term secretome studies. This allows the analysis of the full breath of the transcriptional regulation including feedback-loops to study the effects of cytokines but also enables the study of secondary compound effects, for example, as a result of compound metabolization.
Data Availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (120) partner repository with the dataset identifier PXD027821 (https://www.ebi.ac.uk/pride).
Supplemental data
This article contains supplemental data.
Conflict of interest
All authors are employees of GSK and M. B. and H. C. E. are shareholders of GSK. The authors declare that they have no conflicts of interest with the contents of this article.
Acknowledgments
We thank J. Stuhlfauth, N. Garcia-Altrieth, K. Beß, and B. Dlugosch for supporting the cell culture; M. Klös-Hudak, K. Kammerer, P. Sauer, and M. Steidel for technical assistance with mass spectrometry; and M. Kalxdorf, C. Boecker and T. Mathieson for statistical as well as IT and computational support. This work was funded by Cellzome GmbH/GlaxoSmithKline.
Author contributions
M. B. and H. C. E. conceptualization; M. B. and H. C. E. supervision; S. K. investigation; S. K., H. C. E., and M. B. formal analysis; S. K., H. C. E., and M. B. writing–original draft.
Contributor Information
H. Christian Eberl, Email: hans-christian.h.eberl@gsk.com.
Marcus Bantscheff, Email: marcus.x.bantscheff@gsk.com.
Supplemental Data
References
- 1.Zhou Z., Xu M.J., Gao B. Hepatocytes: a key cell type for innate immunity. Cell. Mol. Immunol. 2016;13:301–315. doi: 10.1038/cmi.2015.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schulze R.J., Schott M.B., Casey C.A., Tuma P.L., McNiven M.A. The cell biology of the hepatocyte: a membrane trafficking machine. J. Cell Biol. 2019;218:2096–2112. doi: 10.1083/jcb.201903090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Robinson M.W., Harmon C., O'Farrelly C. Liver immunology and its role in inflammation and homeostasis. Cell. Mol. Immunol. 2016;13:267–276. doi: 10.1038/cmi.2016.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moshage H. Cytokines and the hepatic acute phase response. J. Pathol. 1997;181:257–266. doi: 10.1002/(SICI)1096-9896(199703)181:3<257::AID-PATH756>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 5.Heinrich P.C., Behrmann I., Haan S., Hermanns H.M., Muller-Newen G., Schaper F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem. J. 2003;374:1–20. doi: 10.1042/BJ20030407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bode J.G., Albrecht U., Haussinger D., Heinrich P.C., Schaper F. Hepatic acute phase proteins--regulation by IL-6- and IL-1-type cytokines involving STAT3 and its crosstalk with NF-kappaB-dependent signaling. Eur. J. Cell Biol. 2012;91:496–505. doi: 10.1016/j.ejcb.2011.09.008. [DOI] [PubMed] [Google Scholar]
- 7.Schmidt-Arras D., Rose-John S. IL-6 pathway in the liver: from physiopathology to therapy. J. Hepatol. 2016;64:1403–1415. doi: 10.1016/j.jhep.2016.02.004. [DOI] [PubMed] [Google Scholar]
- 8.Andus T., Geiger T., Hirano T., Kishimoto T., Tran-Thi T.A., Decker K., et al. Regulation of synthesis and secretion of major rat acute-phase proteins by recombinant human interleukin-6 (BSF-2/IL-6) in hepatocyte primary cultures. Eur. J. Biochem. 1988;173:287–293. doi: 10.1111/j.1432-1033.1988.tb13997.x. [DOI] [PubMed] [Google Scholar]
- 9.Kramer F., Torzewski J., Kamenz J., Veit K., Hombach V., Dedio J., et al. Interleukin-1beta stimulates acute phase response and C-reactive protein synthesis by inducing an NFkappaB- and C/EBPbeta-dependent autocrine interleukin-6 loop. Mol. Immunol. 2008;45:2678–2689. doi: 10.1016/j.molimm.2007.12.017. [DOI] [PubMed] [Google Scholar]
- 10.Stuhler K. The secrets of protein secretion: what are the key features of comparative secretomics? Expert Rev. Proteomics. 2020;17:785–787. doi: 10.1080/14789450.2020.1881890. [DOI] [PubMed] [Google Scholar]
- 11.Ramazanov B.R., Tran M.L., von Blume J. Sending out molecules from the TGN. Curr. Opin. Cell Biol. 2021;71:55–62. doi: 10.1016/j.ceb.2021.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dimou E., Nickel W. Unconventional mechanisms of eukaryotic protein secretion. Curr. Biol. 2018;28:R406–R410. doi: 10.1016/j.cub.2017.11.074. [DOI] [PubMed] [Google Scholar]
- 13.Mukherjee P., Mani S. Methodologies to decipher the cell secretome. Biochim. Biophys. Acta. 2013;1834:2226–2232. doi: 10.1016/j.bbapap.2013.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chevallet M., Diemer H., Van Dorssealer A., Villiers C., Rabilloud T. Toward a better analysis of secreted proteins: the example of the myeloid cells secretome. Proteomics. 2007;7:1757–1770. doi: 10.1002/pmic.200601024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eichelbaum K., Winter M., Berriel Diaz M., Herzig S., Krijgsveld J. Selective enrichment of newly synthesized proteins for quantitative secretome analysis. Nat. Biotechnol. 2012;30:984–990. doi: 10.1038/nbt.2356. [DOI] [PubMed] [Google Scholar]
- 16.Laughlin S.T., Agard N.J., Baskin J.M., Carrico I.S., Chang P.V., Ganguli A.S., et al. Metabolic labeling of glycans with azido sugars for visualization and glycoproteomics. Methods Enzymol. 2006;415:230–250. doi: 10.1016/S0076-6879(06)15015-6. [DOI] [PubMed] [Google Scholar]
- 17.Kuhn P.H., Koroniak K., Hogl S., Colombo A., Zeitschel U., Willem M., et al. Secretome protein enrichment identifies physiological BACE1 protease substrates in neurons. EMBO J. 2012;31:3157–3168. doi: 10.1038/emboj.2012.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Serdaroglu A., Muller S.A., Schepers U., Brase S., Weichert W., Lichtenthaler S.F., et al. An optimised version of the secretome protein enrichment with click sugars (SPECS) method leads to enhanced coverage of the secretome. Proteomics. 2017;17 doi: 10.1002/pmic.201600423. [DOI] [PubMed] [Google Scholar]
- 19.Tushaus J., Muller S.A., Kataka E.S., Zaucha J., Sebastian Monasor L., Su M., et al. An optimized quantitative proteomics method establishes the cell type-resolved mouse brain secretome. EMBO J. 2020;39 doi: 10.15252/embj.2020105693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meissner F., Scheltema R.A., Mollenkopf H.J., Mann M. Direct proteomic quantification of the secretome of activated immune cells. Science. 2013;340:475–478. doi: 10.1126/science.1232578. [DOI] [PubMed] [Google Scholar]
- 21.Frauenstein A., Meissner F. Quantitative proteomics of secreted proteins. Methods Mol. Biol. 2018;1714:215–227. doi: 10.1007/978-1-4939-7519-8_14. [DOI] [PubMed] [Google Scholar]
- 22.Tanzer M.C., Frauenstein A., Stafford C.A., Phulphagar K., Mann M., Meissner F. Quantitative and dynamic catalogs of proteins released during apoptotic and necroptotic cell death. Cell Rep. 2020;30:1260–1270.e5. doi: 10.1016/j.celrep.2019.12.079. [DOI] [PubMed] [Google Scholar]
- 23.Villarreal L., Mendez O., Salvans C., Gregori J., Baselga J., Villanueva J. Unconventional secretion is a major contributor of cancer cell line secretomes. Mol. Cell. Proteomics. 2013;12:1046–1060. doi: 10.1074/mcp.M112.021618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Franko A., Hartwig S., Kotzka J., Ruoss M., Nussler A.K., Konigsrainer A., et al. Identification of the secreted proteins originated from primary human hepatocytes and HepG2 cells. Nutrients. 2019;11:1795. doi: 10.3390/nu11081795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guguen-Guillouzo C., Guillouzo A. General review on in vitro hepatocyte models and their applications. Methods Mol. Biol. 2010;640:1–40. doi: 10.1007/978-1-60761-688-7_1. [DOI] [PubMed] [Google Scholar]
- 26.Tascher G., Burban A., Camus S., Plumel M., Chanon S., Le Guevel R., et al. In-depth proteome analysis highlights HepaRG cells as a versatile cell system surrogate for primary human hepatocytes. Cells. 2019;8:192. doi: 10.3390/cells8020192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gripon P., Rumin S., Urban S., Le Seyec J., Glaise D., Cannie I., et al. Infection of a human hepatoma cell line by hepatitis B virus. Proc. Natl. Acad. Sci. U. S. A. 2002;99:15655–15660. doi: 10.1073/pnas.232137699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rasmussen T.P., Guillouzo A., Guguen-Guillouzo C. Stem Cells in Birth Defects Research and Developmental Toxicology. John Wiley & Sons, Inc.; New York, NY: 2018. HepaRG cells as a model for hepatotoxicity studies; pp. 309–339. [Google Scholar]
- 29.Guillouzo A., Corlu A., Aninat C., Glaise D., Morel F., Guguen-Guillouzo C. The human hepatoma HepaRG cells: a highly differentiated model for studies of liver metabolism and toxicity of xenobiotics. Chem. Biol. Interact. 2007;168:66–73. doi: 10.1016/j.cbi.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 30.Rubin K., Janefeldt A., Andersson L., Berke Z., Grime K., Andersson T.B. HepaRG cells as human-relevant in vitro model to study the effects of inflammatory stimuli on cytochrome P450 isoenzymes. Drug Metab. Dispos. 2015;43:119–125. doi: 10.1124/dmd.114.059246. [DOI] [PubMed] [Google Scholar]
- 31.Lubberstedt M., Muller-Vieira U., Mayer M., Biemel K.M., Knospel F., Knobeloch D., et al. HepaRG human hepatic cell line utility as a surrogate for primary human hepatocytes in drug metabolism assessment in vitro. J. Pharmacol. Toxicol. Methods. 2011;63:59–68. doi: 10.1016/j.vascn.2010.04.013. [DOI] [PubMed] [Google Scholar]
- 32.Andersson T.B., Kanebratt K.P., Kenna J.G. The HepaRG cell line: a unique in vitro tool for understanding drug metabolism and toxicology in human. Expert Opin. Drug Metab. Toxicol. 2012;8:909–920. doi: 10.1517/17425255.2012.685159. [DOI] [PubMed] [Google Scholar]
- 33.Werner T., Steidel M., Eberl H.C., Bantscheff M. Affinity enrichment chemoproteomics for target deconvolution and selectivity profiling. Methods Mol. Biol. 2021;2228:237–252. doi: 10.1007/978-1-0716-1024-4_17. [DOI] [PubMed] [Google Scholar]
- 34.Werner T., Sweetman G., Savitski M.F., Mathieson T., Bantscheff M., Savitski M.M. Ion coalescence of neutron encoded TMT 10-plex reporter ions. Anal. Chem. 2014;86:3594–3601. doi: 10.1021/ac500140s. [DOI] [PubMed] [Google Scholar]
- 35.Franken H., Mathieson T., Childs D., Sweetman G.M., Werner T., Togel I., et al. Thermal proteome profiling for unbiased identification of direct and indirect drug targets using multiplexed quantitative mass spectrometry. Nat. Protoc. 2015;10:1567–1593. doi: 10.1038/nprot.2015.101. [DOI] [PubMed] [Google Scholar]
- 36.Cox J., Michalski A., Mann M. Software lock mass by two-dimensional minimization of peptide mass errors. J. Am. Soc. Mass Spectrom. 2011;22:1373–1380. doi: 10.1007/s13361-011-0142-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Savitski M.M., Sweetman G., Askenazi M., Marto J.A., Lang M., Zinn N., et al. Delayed fragmentation and optimized isolation width settings for improvement of protein identification and accuracy of isobaric mass tag quantification on Orbitrap-type mass spectrometers. Anal. Chem. 2011;83:8959–8967. doi: 10.1021/ac201760x. [DOI] [PubMed] [Google Scholar]
- 38.Savitski M.M., Fischer F., Mathieson T., Sweetman G., Lang M., Bantscheff M. Targeted data acquisition for improved reproducibility and robustness of proteomic mass spectrometry assays. J. Am. Soc. Mass Spectrom. 2010;21:1668–1679. doi: 10.1016/j.jasms.2010.01.012. [DOI] [PubMed] [Google Scholar]
- 39.Savitski M.M., Mathieson T., Zinn N., Sweetman G., Doce C., Becher I., et al. Measuring and managing ratio compression for accurate iTRAQ/TMT quantification. J. Proteome Res. 2013;12:3586–3598. doi: 10.1021/pr400098r. [DOI] [PubMed] [Google Scholar]
- 40.Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43 doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kalxdorf M., Gunthner I., Becher I., Kurzawa N., Knecht S., Savitski M.M., et al. Cell surface thermal proteome profiling tracks perturbations and drug targets on the plasma membrane. Nat. Methods. 2021;18:84–91. doi: 10.1038/s41592-020-01022-1. [DOI] [PubMed] [Google Scholar]
- 42.Hornik K. A CLUE for CLUster ensembles. J. Stat. Softw. 2005;14:1–25. [Google Scholar]
- 43.Alexa A., Rahnenfuhrer J. Bioconductor; 2020. topGO: Enrichment Analysis for Gene Ontology. R Package Version 2.40.0. [Google Scholar]
- 44.Pardo-Saganta A., Latasa M.U., Castillo J., Alvarez-Asiain L., Perugorria M.J., Sarobe P., et al. The epidermal growth factor receptor ligand amphiregulin is a negative regulator of hepatic acute-phase gene expression. J. Hepatol. 2009;51:1010–1020. doi: 10.1016/j.jhep.2009.06.030. [DOI] [PubMed] [Google Scholar]
- 45.Wischhusen J., Melero I., Fridman W.H. Growth/differentiation factor-15 (GDF-15): from biomarker to novel targetable immune checkpoint. Front. Immunol. 2020;11:951. doi: 10.3389/fimmu.2020.00951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lopez-Dee Z., Pidcock K., Gutierrez L.S. Thrombospondin-1: multiple paths to inflammation. Mediators Inflamm. 2011;2011:296069. doi: 10.1155/2011/296069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kalxdorf M., Gade S., Eberl H.C., Bantscheff M. Monitoring cell-surface N-glycoproteome dynamics by quantitative proteomics reveals mechanistic insights into macrophage differentiation. Mol. Cell. Proteomics. 2017;16:770–785. doi: 10.1074/mcp.M116.063859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Norris C.A., He M., Kang L.I., Ding M.Q., Radder J.E., Haynes M.M., et al. Synthesis of IL-6 by hepatocytes is a normal response to common hepatic stimuli. PLoS One. 2014;9 doi: 10.1371/journal.pone.0096053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pirkmajer S., Chibalin A.V. Serum starvation: caveat emptor. Am. J. Physiol. Cell Physiol. 2011;301:C272–C279. doi: 10.1152/ajpcell.00091.2011. [DOI] [PubMed] [Google Scholar]
- 50.Cooper S. Reappraisal of serum starvation, the restriction point, G0, and G1 phase arrest points. FASEB J. 2003;17:333–340. doi: 10.1096/fj.02-0352rev. [DOI] [PubMed] [Google Scholar]
- 51.Hasan N.M., Adams G.E., Joiner M.C. Effect of serum starvation on expression and phosphorylation of PKC-alpha and p53 in V79 cells: implications for cell death. Int. J. Cancer. 1999;80:400–405. doi: 10.1002/(sici)1097-0215(19990129)80:3<400::aid-ijc11>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 52.Gabay C., Kushner I. Acute-phase proteins and other systemic responses to inflammation. N. Engl. J. Med. 1999;340:448–454. doi: 10.1056/NEJM199902113400607. [DOI] [PubMed] [Google Scholar]
- 53.Hochepied T., Berger F.G., Baumann H., Libert C. Alpha(1)-acid glycoprotein: an acute phase protein with inflammatory and immunomodulating properties. Cytokine Growth Factor Rev. 2003;14:25–34. doi: 10.1016/s1359-6101(02)00054-0. [DOI] [PubMed] [Google Scholar]
- 54.Novo E., Villano G., Turato C., Cannito S., Paternostro C., Busletta C., et al. SerpinB3 promotes pro-fibrogenic responses in activated hepatic stellate cells. Sci. Rep. 2017;7:3420. doi: 10.1038/s41598-017-03744-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Turato C., Calabrese F., Biasiolo A., Quarta S., Ruvoletto M., Tono N., et al. SERPINB3 modulates TGF-beta expression in chronic liver disease. Lab. Invest. 2010;90:1016–1023. doi: 10.1038/labinvest.2010.55. [DOI] [PubMed] [Google Scholar]
- 56.Zhao T., Su Z., Li Y., Zhang X., You Q. Chitinase-3 like-protein-1 function and its role in diseases. Signal. Transduct. Target. Ther. 2020;5:201. doi: 10.1038/s41392-020-00303-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ling H., Recklies A.D. The chitinase 3-like protein human cartilage glycoprotein 39 inhibits cellular responses to the inflammatory cytokines interleukin-1 and tumour necrosis factor-alpha. Biochem. J. 2004;380:651–659. doi: 10.1042/BJ20040099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pathan M., Fonseka P., Chitti S.V., Kang T., Sanwlani R., Van Deun J., et al. Vesiclepedia 2019: a compendium of RNA, proteins, lipids and metabolites in extracellular vesicles. Nucleic Acids Res. 2019;47:D516–D519. doi: 10.1093/nar/gky1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liang B., Peng P., Chen S., Li L., Zhang M., Cao D., et al. Characterization and proteomic analysis of ovarian cancer-derived exosomes. J. Proteomics. 2013;80:171–182. doi: 10.1016/j.jprot.2012.12.029. [DOI] [PubMed] [Google Scholar]
- 60.Dozio V., Sanchez J.C. Characterisation of extracellular vesicle-subsets derived from brain endothelial cells and analysis of their protein cargo modulation after TNF exposure. J. Extracell Vesicles. 2017;6:1302705. doi: 10.1080/20013078.2017.1302705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xiao J., Zhang H., Yang F., Xiao M., Zhou L., Yu R., et al. Proteomic analysis of plasma sEVs reveals that TNFAIP8 is a new biomarker of cell proliferation in diabetic retinopathy. J. Proteome Res. 2021;20:1770–1782. doi: 10.1021/acs.jproteome.0c01048. [DOI] [PubMed] [Google Scholar]
- 62.de Seny D., Bianchi E., Baiwir D., Cobraiville G., Collin C., Deliege M., et al. Proteins involved in the endoplasmic reticulum stress are modulated in synovitis of osteoarthritis, chronic pyrophosphate arthropathy and rheumatoid arthritis, and correlate with the histological inflammatory score. Sci. Rep. 2020;10:14159. doi: 10.1038/s41598-020-70803-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rahmati M., Moosavi M.A., McDermott M.F. ER stress: a therapeutic target in rheumatoid arthritis? Trends Pharmacol. Sci. 2018;39:610–623. doi: 10.1016/j.tips.2018.03.010. [DOI] [PubMed] [Google Scholar]
- 64.Shields A.M., Thompson S.J., Panayi G.S., Corrigall V.M. Pro-resolution immunological networks: binding immunoglobulin protein and other resolution-associated molecular patterns. Rheumatology (Oxford) 2012;51:780–788. doi: 10.1093/rheumatology/ker412. [DOI] [PubMed] [Google Scholar]
- 65.Niu L., Geyer P.E., Wewer Albrechtsen N.J., Gluud L.L., Santos A., Doll S., et al. Plasma proteome profiling discovers novel proteins associated with non-alcoholic fatty liver disease. Mol. Syst. Biol. 2019;15 doi: 10.15252/msb.20188793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Khovidhunkit W., Memon R.A., Feingold K.R., Grunfeld C. Infection and inflammation-induced proatherogenic changes of lipoproteins. J. Infect. Dis. 2000;181 Suppl 3:S462–S472. doi: 10.1086/315611. [DOI] [PubMed] [Google Scholar]
- 67.Carpintero R., Pineiro M., Andres M., Iturralde M., Alava M.A., Heegaard P.M., et al. The concentration of apolipoprotein A-I decreases during experimentally induced acute-phase processes in pigs. Infect. Immun. 2005;73:3184–3187. doi: 10.1128/IAI.73.5.3184-3187.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cabana V.G., Siegel J.N., Sabesin S.M. Effects of the acute phase response on the concentration and density distribution of plasma lipids and apolipoproteins. J. Lipid Res. 1989;30:39–49. [PubMed] [Google Scholar]
- 69.Qin X.Y., Hara M., Arner E., Kawaguchi Y., Inoue I., Tatsukawa H., et al. Transcriptome analysis uncovers a growth-promoting activity of orosomucoid-1 on hepatocytes. EBioMedicine. 2017;24:257–266. doi: 10.1016/j.ebiom.2017.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gruys E., Toussaint M.J., Niewold T.A., Koopmans S.J. Acute phase reaction and acute phase proteins. J. Zhejiang Univ. Sci. B. 2005;6:1045–1056. doi: 10.1631/jzus.2005.B1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mohd M.A., Ahmad Norudin N.A., Muhammad T.S.T. Transcriptional regulation of retinol binding protein 4 by interleukin-6 via peroxisome proliferator-activated receptor alpha and CCAAT/enhancer binding proteins. Mol. Cell. Endocrinol. 2020;505:110702. doi: 10.1016/j.mce.2020.110702. [DOI] [PubMed] [Google Scholar]
- 72.Weber S., Saftig P. Ectodomain shedding and ADAMs in development. Development. 2012;139:3693–3709. doi: 10.1242/dev.076398. [DOI] [PubMed] [Google Scholar]
- 73.Garton K.J., Gough P.J., Raines E.W. Emerging roles for ectodomain shedding in the regulation of inflammatory responses. J. Leukoc. Biol. 2006;79:1105–1116. doi: 10.1189/jlb.0106038. [DOI] [PubMed] [Google Scholar]
- 74.Cai Z., Zhang A., Choksi S., Li W., Li T., Zhang X.M., et al. Activation of cell-surface proteases promotes necroptosis, inflammation and cell migration. Cell Res. 2016;26:886–900. doi: 10.1038/cr.2016.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Morsing S.K.H., Rademakers T., Brouns S.L.N., Stalborch A.D.V., Donners M., van Buul J.D. ADAM10-mediated cleavage of ICAM-1 is involved in neutrophil transendothelial migration. Cells. 2021;10:232. doi: 10.3390/cells10020232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Qian M., Shen X., Wang H. The distinct role of ADAM17 in APP proteolysis and microglial activation related to Alzheimer's disease. Cell Mol. Neurobiol. 2016;36:471–482. doi: 10.1007/s10571-015-0232-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Oh M., Im I., Lee Y.J., Kim Y.H., Yoon J.H., Park H.G., et al. Structure-based virtual screening and biological evaluation of potent and selective ADAM12 inhibitors. Bioorg. Med. Chem. Lett. 2004;14:6071–6074. doi: 10.1016/j.bmcl.2004.09.082. [DOI] [PubMed] [Google Scholar]
- 78.Bourd-Boittin K., Basset L., Bonnier D., L'Helgoualc'h A., Samson M., Theret N. CX3CL1/fractalkine shedding by human hepatic stellate cells: contribution to chronic inflammation in the liver. J. Cell Mol. Med. 2009;13:1526–1535. doi: 10.1111/j.1582-4934.2009.00787.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kuhn P.H., Colombo A.V., Schusser B., Dreymueller D., Wetzel S., Schepers U., et al. Systematic substrate identification indicates a central role for the metalloprotease ADAM10 in axon targeting and synapse function. Elife. 2016;5 doi: 10.7554/eLife.12748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Okamura Y., Kohmura E., Yamashita T. TACE cleaves neogenin to desensitize cortical neurons to the repulsive guidance molecule. Neurosci. Res. 2011;71:63–70. doi: 10.1016/j.neures.2011.05.012. [DOI] [PubMed] [Google Scholar]
- 81.Pruessmeyer J., Martin C., Hess F.M., Schwarz N., Schmidt S., Kogel T., et al. A disintegrin and metalloproteinase 17 (ADAM17) mediates inflammation-induced shedding of syndecan-1 and -4 by lung epithelial cells. J. Biol. Chem. 2010;285:555–564. doi: 10.1074/jbc.M109.059394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Navarro V., Vincent J.P., Mazella J. Shedding of the luminal domain of the neurotensin receptor-3/sortilin in the HT29 cell line. Biochem. Biophys. Res. Commun. 2002;298:760–764. doi: 10.1016/s0006-291x(02)02564-0. [DOI] [PubMed] [Google Scholar]
- 83.Hermey G., Sjogaard S.S., Petersen C.M., Nykjaer A., Gliemann J. Tumour necrosis factor alpha-converting enzyme mediates ectodomain shedding of Vps10p-domain receptor family members. Biochem. J. 2006;395:285–293. doi: 10.1042/BJ20051364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Evans S.F., Irmady K., Ostrow K., Kim T., Nykjaer A., Saftig P., et al. Neuronal brain-derived neurotrophic factor is synthesized in excess, with levels regulated by sortilin-mediated trafficking and lysosomal degradation. J. Biol. Chem. 2011;286:29556–29567. doi: 10.1074/jbc.M111.219675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Strong A., Patel K., Rader D.J. Sortilin and lipoprotein metabolism: making sense out of complexity. Curr. Opin. Lipidol. 2014;25:350–357. doi: 10.1097/MOL.0000000000000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mortensen M.B., Kjolby M., Gunnersen S., Larsen J.V., Palmfeldt J., Falk E., et al. Targeting sortilin in immune cells reduces proinflammatory cytokines and atherosclerosis. J. Clin. Invest. 2014;124:5317–5322. doi: 10.1172/JCI76002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hubel E., Saroha A., Park W.J., Pewzner-Jung Y., Lavoie E.G., Futerman A.H., et al. Sortilin deficiency reduces ductular reaction, hepatocyte apoptosis, and liver fibrosis in cholestatic-induced liver injury. Am. J. Pathol. 2017;187:122–133. doi: 10.1016/j.ajpath.2016.09.005. [DOI] [PubMed] [Google Scholar]
- 88.Yabe-Wada T., Matsuba S., Takeda K., Sato T., Suyama M., Ohkawa Y., et al. TLR signals posttranscriptionally regulate the cytokine trafficking mediator sortilin. Sci. Rep. 2016;6:26566. doi: 10.1038/srep26566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Dayon L., Affolter M. Progress and pitfalls of using isobaric mass tags for proteome profiling. Expert Rev. Proteomics. 2020;17:149–161. doi: 10.1080/14789450.2020.1731309. [DOI] [PubMed] [Google Scholar]
- 90.Kushner I., Rzewnicki D.L. The acute phase response: general aspects. Baillieres Clin. Rheumatol. 1994;8:513–530. doi: 10.1016/s0950-3579(05)80113-x. [DOI] [PubMed] [Google Scholar]
- 91.Cao S., Liu M., Sehrawat T.S., Shah V.H. Regulation and functional roles of chemokines in liver diseases. Nat. Rev. Gastroenterol. Hepatol. 2021;18:630–647. doi: 10.1038/s41575-021-00444-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.El-Benna J., Hurtado-Nedelec M., Marzaioli V., Marie J.C., Gougerot-Pocidalo M.A., Dang P.M. Priming of the neutrophil respiratory burst: role in host defense and inflammation. Immunol. Rev. 2016;273:180–193. doi: 10.1111/imr.12447. [DOI] [PubMed] [Google Scholar]
- 93.Russo R.C., Garcia C.C., Teixeira M.M., Amaral F.A. The CXCL8/IL-8 chemokine family and its receptors in inflammatory diseases. Expert Rev. Clin. Immunol. 2014;10:593–619. doi: 10.1586/1744666X.2014.894886. [DOI] [PubMed] [Google Scholar]
- 94.Bickel M. The role of interleukin-8 in inflammation and mechanisms of regulation. J. Periodontol. 1993;64:456–460. [PubMed] [Google Scholar]
- 95.Hu J., Van den Steen P.E., Sang Q.X., Opdenakker G. Matrix metalloproteinase inhibitors as therapy for inflammatory and vascular diseases. Nat. Rev. Drug Discov. 2007;6:480–498. doi: 10.1038/nrd2308. [DOI] [PubMed] [Google Scholar]
- 96.Parks W.C., Wilson C.L., Lopez-Boado Y.S. Matrix metalloproteinases as modulators of inflammation and innate immunity. Nat. Rev. Immunol. 2004;4:617–629. doi: 10.1038/nri1418. [DOI] [PubMed] [Google Scholar]
- 97.Steck E., Breit S., Breusch S.J., Axt M., Richter W. Enhanced expression of the human chitinase 3-like 2 gene (YKL-39) but not chitinase 3-like 1 gene (YKL-40) in osteoarthritic cartilage. Biochem. Biophys. Res. Commun. 2002;299:109–115. doi: 10.1016/s0006-291x(02)02585-8. [DOI] [PubMed] [Google Scholar]
- 98.Knorr T., Obermayr F., Bartnik E., Zien A., Aigner T. YKL-39 (chitinase 3-like protein 2), but not YKL-40 (chitinase 3-like protein 1), is up regulated in osteoarthritic chondrocytes. Ann. Rheum. Dis. 2003;62:995–998. doi: 10.1136/ard.62.10.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Shao R., Hamel K., Petersen L., Cao Q.J., Arenas R.B., Bigelow C., et al. YKL-40, a secreted glycoprotein, promotes tumor angiogenesis. Oncogene. 2009;28:4456–4468. doi: 10.1038/onc.2009.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Liu T., Larionova I., Litviakov N., Riabov V., Zavyalova M., Tsyganov M., et al. Tumor-associated macrophages in human breast cancer produce new monocyte attracting and pro-angiogenic factor YKL-39 indicative for increased metastasis after neoadjuvant chemotherapy. Oncoimmunology. 2018;7 doi: 10.1080/2162402X.2018.1436922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Fang T., Cui M., Sun J., Ge C., Zhao F., Zhang L., et al. Orosomucoid 2 inhibits tumor metastasis and is upregulated by CCAAT/enhancer binding protein beta in hepatocellular carcinomas. Oncotarget. 2015;6:16106–16119. doi: 10.18632/oncotarget.3867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Huang H., Wu T., Mao J., Fang Y., Zhang J., Wu L., et al. CHI3L1 is a liver-enriched, noninvasive biomarker that can be used to stage and diagnose substantial hepatic fibrosis. OMICS. 2015;19:339–345. doi: 10.1089/omi.2015.0037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Xu N., Bo Q., Shao R., Liang J., Zhai Y., Yang S., et al. Chitinase-3-like-1 promotes M2 macrophage differentiation and induces choroidal neovascularization in neovascular age-related macular degeneration. Invest. Ophthalmol. Vis. Sci. 2019;60:4596–4605. doi: 10.1167/iovs.19-27493. [DOI] [PubMed] [Google Scholar]
- 104.Groeneveld D., Pereyra D., Veldhuis Z., Adelmeijer J., Ottens P., Kopec A.K., et al. Intrahepatic fibrin(ogen) deposition drives liver regeneration after partial hepatectomy in mice and humans. Blood. 2019;133:1245–1256. doi: 10.1182/blood-2018-08-869057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lesurtel M., Graf R., Aleil B., Walther D.J., Tian Y., Jochum W., et al. Platelet-derived serotonin mediates liver regeneration. Science. 2006;312:104–107. doi: 10.1126/science.1123842. [DOI] [PubMed] [Google Scholar]
- 106.Murata S., Ohkohchi N., Matsuo R., Ikeda O., Myronovych A., Hoshi R. Platelets promote liver regeneration in early period after hepatectomy in mice. World J. Surg. 2007;31:808–816. doi: 10.1007/s00268-006-0772-3. [DOI] [PubMed] [Google Scholar]
- 107.Myronovych A., Murata S., Chiba M., Matsuo R., Ikeda O., Watanabe M., et al. Role of platelets on liver regeneration after 90% hepatectomy in mice. J. Hepatol. 2008;49:363–372. doi: 10.1016/j.jhep.2008.04.019. [DOI] [PubMed] [Google Scholar]
- 108.Meyer J., Balaphas A., Fontana P., Morel P., Robson S.C., Sadoul K., et al. Platelet interactions with liver sinusoidal endothelial cells and hepatic stellate cells lead to hepatocyte proliferation. Cells. 2020;9:1243. doi: 10.3390/cells9051243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Starlinger P., Haegele S., Offensperger F., Oehlberger L., Pereyra D., Kral J.B., et al. The profile of platelet alpha-granule released molecules affects postoperative liver regeneration. Hepatology. 2016;63:1675–1688. doi: 10.1002/hep.28331. [DOI] [PubMed] [Google Scholar]
- 110.Matsuo R., Ohkohchi N., Murata S., Ikeda O., Nakano Y., Watanabe M., et al. Platelets strongly induce hepatocyte proliferation with IGF-1 and HGF in vitro. J. Surg. Res. 2008;145:279–286. doi: 10.1016/j.jss.2007.02.035. [DOI] [PubMed] [Google Scholar]
- 111.Streetz K.L., Luedde T., Manns M.P., Trautwein C. Interleukin 6 and liver regeneration. Gut. 2000;47:309–312. doi: 10.1136/gut.47.2.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Cressman D.E., Greenbaum L.E., DeAngelis R.A., Ciliberto G., Furth E.E., Poli V., et al. Liver failure and defective hepatocyte regeneration in interleukin-6-deficient mice. Science. 1996;274:1379–1383. doi: 10.1126/science.274.5291.1379. [DOI] [PubMed] [Google Scholar]
- 113.Yamaguchi K., Itoh Y., Yokomizo C., Nishimura T., Niimi T., Umemura A., et al. Blockade of IL-6 signaling exacerbates liver injury and suppresses antiapoptotic gene expression in methionine choline-deficient diet-fed db/db mice. Lab. Invest. 2011;91:609–618. doi: 10.1038/labinvest.2011.2. [DOI] [PubMed] [Google Scholar]
- 114.Zhang D., Sun M., Samols D., Kushner I. STAT3 participates in transcriptional activation of the C-reactive protein gene by interleukin-6. J. Biol. Chem. 1996;271:9503–9509. doi: 10.1074/jbc.271.16.9503. [DOI] [PubMed] [Google Scholar]
- 115.Streetz K.L., Wustefeld T., Klein C., Kallen K.J., Tronche F., Betz U.A., et al. Lack of gp130 expression in hepatocytes promotes liver injury. Gastroenterology. 2003;125:532–543. doi: 10.1016/s0016-5085(03)00901-6. [DOI] [PubMed] [Google Scholar]
- 116.Albrecht U., Yang X., Asselta R., Keitel V., Tenchini M.L., Ludwig S., et al. Activation of NF-kappaB by IL-1beta blocks IL-6-induced sustained STAT3 activation and STAT3-dependent gene expression of the human gamma-fibrinogen gene. Cell Signal. 2007;19:1866–1878. doi: 10.1016/j.cellsig.2007.04.007. [DOI] [PubMed] [Google Scholar]
- 117.Zhang Z., Fuller G.M. Interleukin 1beta inhibits interleukin 6-mediated rat gamma fibrinogen gene expression. Blood. 2000;96:3466–3472. [PubMed] [Google Scholar]
- 118.Zhang Z., Fuller G.M. The competitive binding of STAT3 and NF-kappaB on an overlapping DNA binding site. Biochem. Biophys. Res. Commun. 1997;237:90–94. doi: 10.1006/bbrc.1997.7082. [DOI] [PubMed] [Google Scholar]
- 119.Sehgal A., Vaishnaw A., Fitzgerald K. Liver as a target for oligonucleotide therapeutics. J. Hepatol. 2013;59:1354–1359. doi: 10.1016/j.jhep.2013.05.045. [DOI] [PubMed] [Google Scholar]
- 120.Perez-Riverol Y., Csordas A., Bai J., Bernal-Llinares M., Hewapathirana S., Kundu D.J., et al. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 2019;47:D442–D450. doi: 10.1093/nar/gky1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (120) partner repository with the dataset identifier PXD027821 (https://www.ebi.ac.uk/pride).