

Summary
Gasdermins (GSDMs) are a family of pore-forming effectors that permeabilize the cell membrane during the cell death program of pyroptosis1. GSDMs are activated by proteolytic removal of autoinhibitory C-terminal domains, typically by caspase regulators1–9. However, no activator is known for one member of this family, GSDMA. Here we show that the major human pathogen group A Streptococcus (GAS) secretes a protease virulence factor, SpeB, that induces GSDMA-dependent pyroptosis. SpeB cleavage of GSDMA releases an active N-terminal fragment that can insert into membranes to form lytic pores. GSDMA is primarily expressed in the skin10, and keratinocytes infected with SpeB-expressing GAS die of GSDMA-dependent pyroptosis. Mice have three homologs of human GSDMA, and triple knockout mice are more susceptible to invasive infection by a pandemic hypervirulent M1T1 clone of GAS. These results indicate GSDMA is critical in the immune defence against invasive skin infections by GAS. Furthermore, this shows GSDMs can act independently of host regulators as direct sensors of exogenous proteases. Since SpeB is essential for tissue invasion and survival within skin cells, these results suggest that GSDMA can act akin to a guard protein that directly detects concerning virulence activities of microbes that present a severe infectious threat.
Keywords: pyroptosis, inflammasome, group A Streptococcus, gasdermin A, keratinocytes, invasion, virulence factors, skin infection
Introduction
Group A Streptococcus (GAS, Streptococcus pyogenes) lives in close association with cells of the human nasopharynx and epidermis. Infection is typically self-limiting, but hypervirulent strains are associated with the resurgence of invasive infections in recent decades11. While GAS are often extracellular, they can bind the receptor CD46 (membrane cofactor protein) to specifically adhere to and invade human keratinocytes12. These intracellular GAS (iGAS) escape into the cytosol and are targeted for destruction by autophagosomes13 like other intracellular pathogens14. Hypervirulent M1T1 serotype clones escape killing by autophagy/xenophagy by cleaving essential regulators with the secreted protease SpeB15. SpeB targets several other immune effectors, broadly promoting pharyngitis, skin infection, and severe invasive infections of the soft-tissue16,17.
We recently demonstrated that SpeB cleaves the proinflammatory cytokine IL-1β, removing an N-terminal autoinhibitor domain17,18. IL-1β cleavage is conventionally regulated by endogenous caspase-family proteases, but IL-1β detection of SpeB adds to a rapid burst of inflammation that limits invasive infection18. Caspases also regulate the cell death program pyroptosis19 by cleaving inert GSDM-family death effector proteins to release a C-terminal autoinhibitory domain from a lytic pore-forming subunit1–9. Our work with GAS also observed SpeB-dependent cell death during infection18. Thus, we hypothesised SpeB also cleaves a GSDM to initiate death. Here we show SpeB directly cleaves GSDMA, an orphan member of this family expressed in keratinocytes, which initiate GSDMA-dependent pyroptosis. This counters SpeB-expressing iGAS and GSDMA-deficient mice are susceptible to GAS in a necrotising fasciitis model. These findings define a new function for GSDMA in the cell-autonomous defence against invasive microbes.
GAS cleaves and activates gasdermins
We hypothesised that since SpeB can cleave IL-1β then perhaps it may also cleaves a GSDM protein18. To examine SpeB activation of GSDMs, we first expressed GSDMA, GSDMB, GSDMC, GSDMD, and GSDME individually in HEK293T cells, which express low levels of endogenous GSDMs and caspases20. Low cytotoxicity was observed, and lysis was significantly elevated in every instance by SpeB co-transduction (Fig. 1a). Expression of a truncated N-terminal domain of GSDMD was sufficient for lysis in the absence of protease (Extended Data Fig. S1a), as previously shown5. Caspase-1, which can cleave other GSDMs, did not lead to GSDMA-dependent cell lysis, nor did the catalytic mutant SpeBC192S15 (Extended Data Fig. S1a). To confirm GSDM cleavage we next expressed Flag-tagged GSDMA, GSDMB, GSDMC, GSDMD, and GSDME individually in HEK293T cells and incubated cell lysates in the presence or absence of purified SpeB. SpeB generated distinct N-terminal cleavage products of GSDMA, GSDMC, and GSDMD (Fig. 1b).
Fig. 1 |. SpeB cleaves GSDMA.
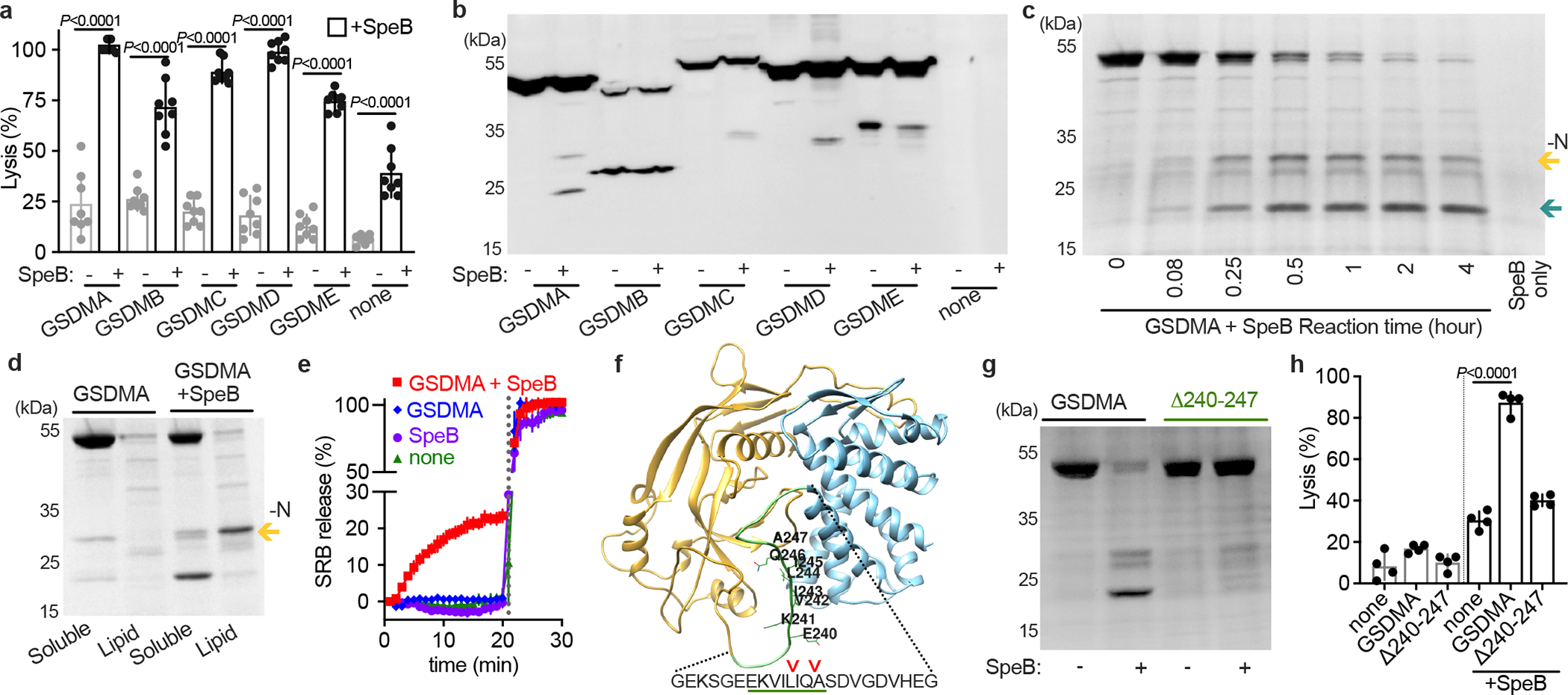
a, Cytotoxicity of each human GSDMs transfected ±SpeB in HEK293Ts. Data are the mean±s.d. of 8 technical replicates. b, Lysates of GSDM-transfected HEK293Ts incubated with SpeB and analysed by immunoblot. c, SDS–PAGE of recombinant human GSDMA cleavage by SpeB over time. GSDMA-N (gold arrow), GSDMA-C (teal arrow). d, PC:PE:cardiolipin liposome binding of GSDMA ±SpeB analysed by SDS–PAGE. GSDMA-N (gold arrow). e, Liposome leakage monitored by Sulforhodamine B (SRB) fluorescence on incubation with GSDMA ±SpeB. Detergent was added after 21 min (dotted line). f, hGSDMA model (Alphafold:Q96QA5) with SpeB cleavage sites identified by Edman sequencing indicated by arrows; GSDMAΔ240-247 deletion underlined in green. g, Cleavage of recombinant GSDMA or GSDMAΔ240-247 ±SpeB was analysed by SDS-PAGE. h, Cytotoxicity of HEK293Ts transfected with GSDMA or GSDMAΔ240-247 ±SpeB. Data are the mean±s.d. of 4 technical replicates. P values were calculated by two-tailed unpaired Student’s t-test (a,h) Data (a-h) are representative of three independent experiments. For gel source data, see Supplementary Figure 1.
We decided to further focus on GSDMA since it is cleaved by SpeB, little was known about this family member, and its expression profile closely matched the body site tropism of GAS10. To confirm whether SpeB cleaved GSDMA is stable, we co-incubated recombinant forms of each protein over time (Fig. 1c). SpeB cleaved GSDMA to yield two large, stable fragments. Some GSDMs are known to bind cardiolipin and other acidic phospholipids, which mediates their targeting to the inner leaflet of the plasma membrane4,5,8. GSDMA had increased binding of these lipids after SpeB cleavage (Extended Data Fig. S1b). On incubation with liposomes consisting of a mix of the base uncharged lipids phosphatidylcholine and phosphatidylethanolamine with 20% cardiolipin, GSDMA-N generated by SpeB cleavage, but neither unprocessed GSDMA nor the C-terminal fragment (GSDMA-C), segregated into fractions containing charged lipids (Fig. 1d). We next measured whether GSDMA-N form pores when inserting into liposomes (Fig. 1e). In liposomes packed with a high-concentration of sulforhodamine B (SRB), fluorescence is quenched until release upon membrane disruption by detergent or pore. GSDMA cleaved by SpeB, but neither GSDMA nor SpeB alone, disrupted liposomes (Fig. 1e).
The N-terminal fragment (GSDMA-N), amino acids 1–239, contain the structural features required for membrane insertion4 and fully removes the C-terminal autoinhibitor domain (amino acids 248–445). Edman sequencing of the C-terminal fragment detected multiple SpeB cleavage sites in GSDMA, all within a disordered solvent-accessible loop present in other GSDMs and targeted by their cognate protease activators loop that mediates the activation of other GSDMs (Fig. 1f). Deletion of residues 240–247 of GSDMA encompassing the predicted cleavage sites generated a stable protein (Extended Data Fig. S1c) that abolished SpeB cleavage (Fig. 1g) and prevented SpeB induced cell death (Fig. 1h). Collectively, these data demonstrate that SpeB cleaves and activates GSDMA.
GSDMA defends against GAS skin infection
To examine whether GSDMA is important in the immune defence against GAS we turned to the established murine model of infection, where intradermal inoculation of wild-type GAS induces a necrotic lesion with deep dermal injury and bacterial proliferation as observed in human infection18,21,22. Mice express three tandem alleles (mGSDMA1–3) on chromosome 11 (Fig. 2a). Like human GSDMA they are expressed in the skin and upper gastrointestinal tract, but with different abundance in each tissue; mGSDMA3 is highest in the skin, mGSDMA2 in the stomach, and mGSDMA1 abundant in both10. Only mGSDMA1 encodes identical SpeB cleavage sites identified in human GSDMA (Extended Data Fig. S2), but SpeB cleaved all three mGSDMAs to similar sizes as human N-GSDMA (Fig. 2b). Since this suggested the possibility of functional redundancy between alleles during GAS infection, we generated mGSDMA123-triple-knockout mice to phenocopy the single GSDMA present in humans. GAS-infected mGSDMA123-knockout mice developed larger lesions (Fig. 2c) carrying a significantly greater GAS burden (Fig. 2d). GSDMA knockout did not restore the virulence capacity of ΔspeB GAS, consistent with its known, dominant and independent virulence contributions through the cleavage of antibodies, extracellular matrix, cytokines, chemokines, and complement proteins16. Together these results demonstrate that GSDMA plays a significant role in controlling GAS skin infection.
Fig. 2 |. GSDMA protects mice against severe GAS skin infection.
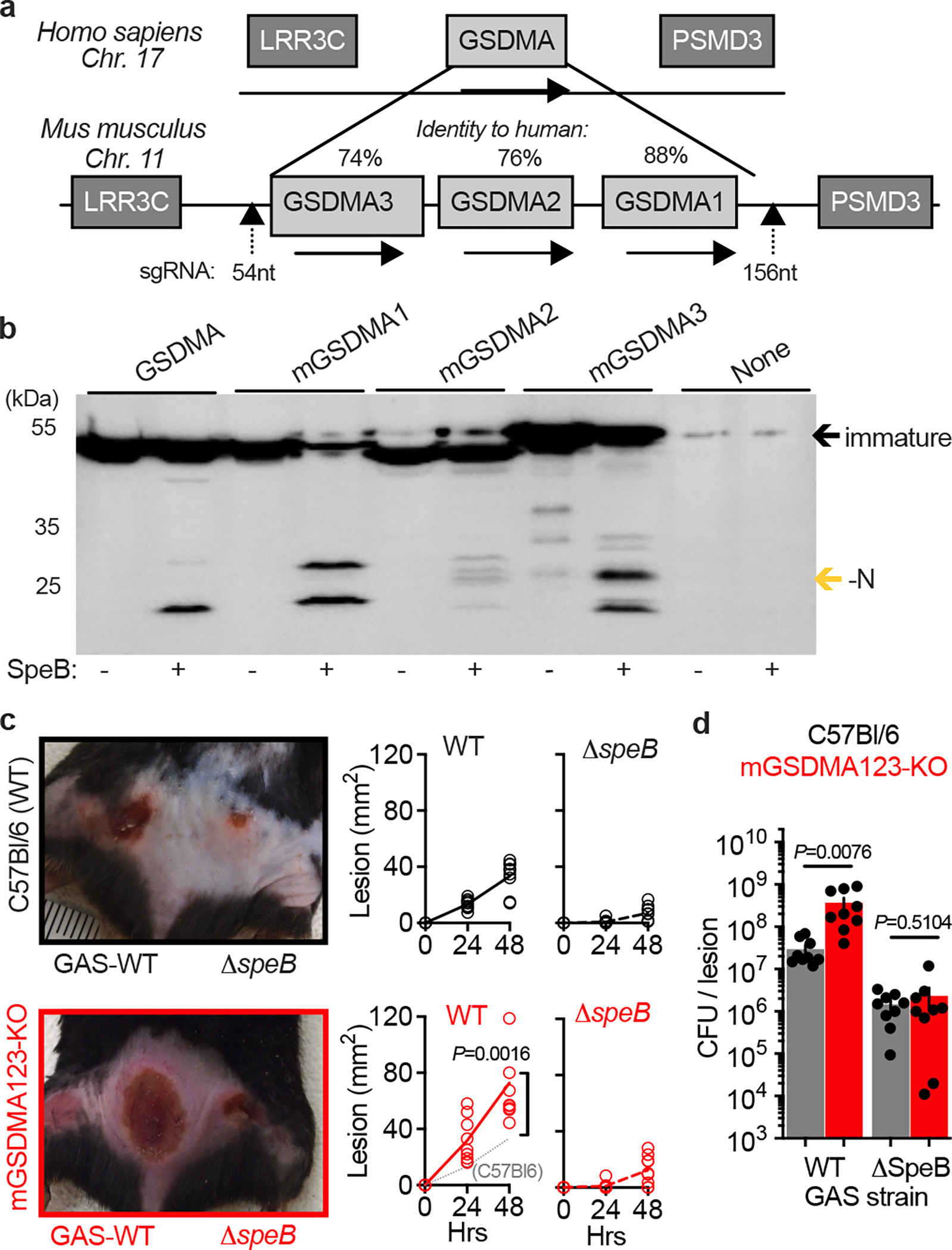
a, GSDMA gene arrangement in mice, with identity to human GSDMA and site of CRISPR knockout indicated. b, Lysates of HEK293Ts transfected with indicated plasmids were incubated with purified SpeB and analysed by immunoblot for Flag-tagged GSDMs (GSDM-N indicated by gold arrow). One of three repeats is shown. For gel source data, see Supplementary Figure 1. c,d, Wild-type (C57Bl/6) and GSDMA-deficient (mGSDMA123-KO) mice were inoculated intradermally with 108 CFU GAS 5448 or ΔspeB and (c) lesion area measured and compared to GAS-WT in C57Bl/6 mice (dotted line). Representative images 48 h post-infection are shown. (d) GAS CFU enumeration in the same mice 48 h post-infection. Data (c-d) are pooled from two independent experiments and are the mean±s.d. (n=9). P values were calculated by two-tailed unpaired Student’s t-test.
SpeB induces pyroptosis in keratinocytes
Keratinocytes highly express GSDMA10 and in the skin are a primary cell type that GAS is in contact with15,23,24. We examined this host-pathogen interaction by infecting primary keratinocytes from human donors with GAS 5448, a highly-virulent isolate from the globally disseminated M1T1 serotype responsible for the invasive infection epidemic of recent decades25. GAS 5448 can specifically bind cells of the epithelium and establish an intracellular replicative niche15,23,24. Accordingly, cleavage of GSDMA in GAS-infected keratinocytes was detected (Fig. 3a) and was proportional to amount of active SpeB during infection (Fig. 3a). SpeB complementation under control of an anhydrotetracycline (ATc) promoter allows overexpression during infection, which increased GSDMA cleavage (Fig. 3a). To determine whether this GSDMA cleavage resulted in cell death, keratinocytes infected with SpeB-expressing GAS were examined microscopically for permeabilization to propidium iodide (PI) (Fig. 3b). Cells infected with SpeB-expressing GAS stained PI+ and released lactate dehydrogenase into the supernatant, the hallmark measure for cell lysis (Fig. 3c). During infection, substantial activity by SpeB could be detected (Fig. 3d). Activity by Caspase-1, a protease that can activate other GSDM-family members, was not detected during keratinocyte infection. Primary keratinocytes from mGSDMA123-knockout mice were resistant to SpeB-mediated pyroptosis (Fig. 3e), as were human primary keratinocytes treated with GSDMA-targeting CRISPR vectors (Fig. 3f). Together, these results show GSDMA induces cell death in keratinocytes infected with SpeB-expressing GAS.
Fig. 3 |. GAS induce SpeB-dependent keratinocyte pyroptosis.
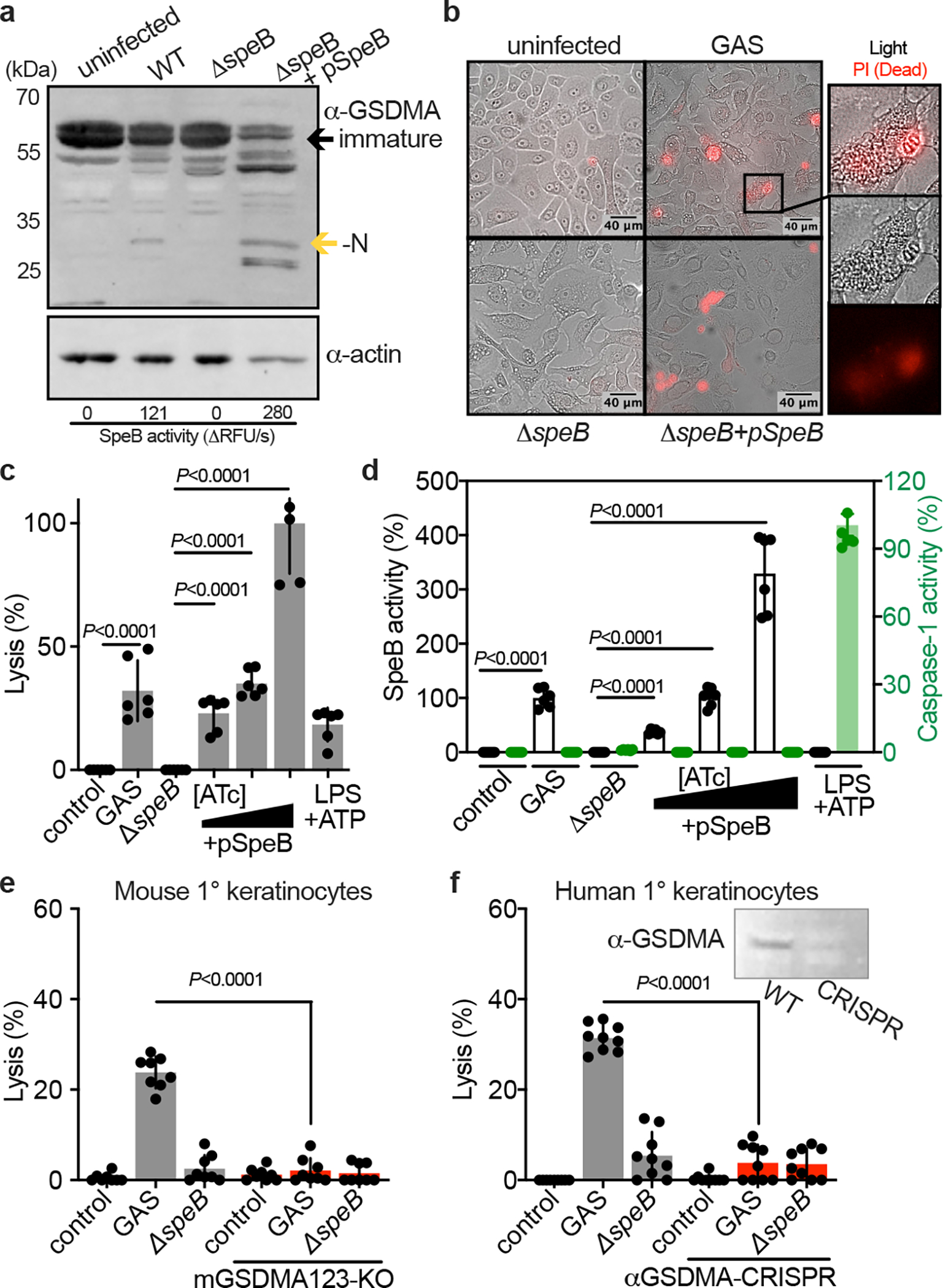
a, Keratinocytes were infected with GAS (MOI=100) for 24 h and GSDMA cleavage was detected by immunoblot. SpeB activity reported as initial kinetic velocity was measured using specific substrate Mca-IFFDTWDNE-Lys-Dnp. b, Immunofluorescent microscopy images of keratinocyte permeabilization to propidium iodide (PI; red) during GAS infection (MOI=100), 2 h. (a,b) Data are representative of three independent experiments. c,d, Human keratinocytes infected 4 h with GAS (MOI=100), ΔspeB, and pSpeB complemented ΔspeB, inducible with 0.1, 1, or 10 ng/ml Anhydrotetracycline (ATc). (c) Lysis measured by LDH release and (d) SpeB and Caspase activity measured using specific substrates Mca-IFFDTWDNE-Lys-Dnp and YVAD-pNA. Cells were treated 18 h with LPS 100 ng/ml and 2 h ATP 5 mM as a caspase-1 control. Data are the mean±s.d. of 6 technical replicates. e,f, Keratinocytes were infected for 4 h with GAS or ΔspeB (MOI=100), treated with gentamycin at 1 h, and lysis measured by LDH release. Data are the mean±s.d. of 9 technical replicates. (e) Keratinocytes were isolated from C57Bl/6 or mGSDMA123-KO mice. (f) Human keratinocytes transfected with GSDMA-targeting sgRNA:Cas9 complex or mock-treated 72 h before infection. Efficacy was evaluated by GSDMA immunoblot. (c,d,e,f) P values were calculated by two-tailed unpaired Student’s t-test and are representative of three independent experiments. For gel source data, see Supplementary Figure 1.
GSDMA detects GAS virulence traits
Patterns of disease are variable between GAS isolates and are correlated with M serotype21–23,25. To examine whether induction of keratinocyte pyroptosis was specific to the hypervirulent M1T1 clone, or potentially broadly relevant, we gathered a panel of contemporary GAS isolates. SpeB activity was variable between isolates, as previously observed26, and proportional to the degree to which they induced keratinocyte lysis (Fig. 4a, Extended Data Fig. S3a). Strains with mutations in covRS can arise naturally during infection and no longer express SpeB18,22 and are hyperencapsulated, which further blocks keratinocyte internalization and can redirect GAS toward paracellular transcytosis27. These mutants did not produce SpeB (Extended Data Fig. S3b) and poorly bound human keratinocytes (Extended Data Fig. S3c). Notably, several SpeB-producting isolates induced less cytotoxicity (Fig. 4a). These also poorly bound human keratinocytes (Extended Data Fig. S3c), further suggesting bacterial cell contact was important for SpeB action, and that variable bacterial virulence determinants contributed to whether a particular GAS isolate activates GSDMA.
Fig. 4 |. GSDMA activation requires cell contact and restricts iGAS.
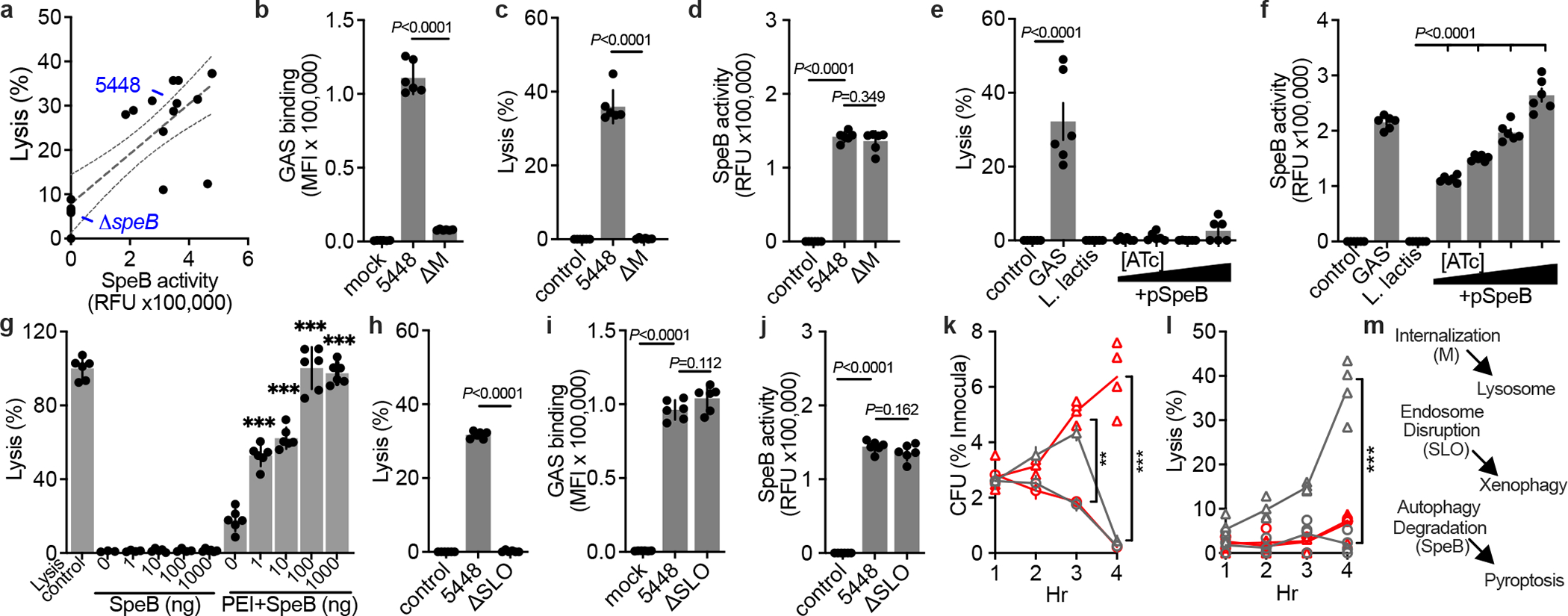
a-f,h-j. Human keratinocytes were infected MOI=100, treated with gentamycin at 1 h, then (a,c,e,h), lysis measured by LDH release after 4 h, (a,d,f,j) SpeB activity measured using specific substrate Mca-IFFDTWDNE-Lys-Dnp after 4 h, and (b,i) Bocillin-labeled GAS adherence after PBS wash was measured by fluorescence after 1 h incubation. (f) 0, 0.1, 1, or 10 ng/mL ATc was included for SpeB induction. g, Keratinocyte lysis measured by LDH release 2 h after treatment with SpeB dilutions (P=0.307; 0.082; 0.330; 0.0243) or packaged within PEI liposomes (all P<0.0001). (a-j) Data are representative of three independent experiments and presented as mean±s.d. of 6 technical replicates. k,l, Human normal (gray) or GSDMA-KO (red) keratinocytes were infected (MOI=100) with JRS4 (circles) or JRS4+pSpeB (triangles) and (k) intracellular CFU and (l) lysis evaluated by LDH release. JRS4+pSpeB has a growth advantage at 3 h (P=0.0004) lost at 4 h, concurrent with cell lysis (P<0.0001). The growth advantage is sustained in lysis-defective GSDMA-KO keratinocytes(P<0.0001). (k,l) Data are representative of three independent experiments and presented as mean±s.d. of 4 technical replicates. (b-l) P values were calculated by two-tailed unpaired Student’s t-test; **P<0.001,***P<0.0001. m, Proposed model of iGAS virulence factor activities and their detection by host cells.
Strains with high attachment to skin cells are efficiently internalised, and are epidemiologically linked to skin infection28. The M protein adhesin mediates cell binding, and tight cell-cell interaction is required for cytotoxicity toward HaCat epithelial-like cells12,29. ΔM 5448 failed to adhere (Fig. 4b) and also lost the ability to induce pyroptosis in keratinocytes (Fig. 4c), despite expressing SpeB (Fig. 4d). Furthermore, keratinocyte pyroptosis was not induced by the non-invasive related species Lactococcus lactis15,18 (Fig. 4e), made to express SpeB (Fig. 4f). Since a requirement for bacterial binding suggested that extracellular SpeB may not translocate across the plasma membrane to access GSDMA, we added SpeB to keratinocyte cultures and examined pyroptosis (Fig. 4g). Purified SpeB only induced lysis when packaged in liposomes that allow transfection directly into cells (Fig. 4g). Together these data suggest extracellular SpeB does not efficiently activate GSDMA and cytosolic translocation of SpeB requires bacteria association with the cell, a process that can involve bacterial adhesins and capsule.
The pore-forming toxin SLO is conserved among GAS and is required for the bacteria to gain access to the cytosol13,24,30. Pyroptosis was not observed during infection by ΔSLO 5448 (Fig. 4h), which could still bind cells (Fig. 4i) and express SpeB (Fig. 4j). The GAS clone JRS4 has been commonly used to study mechanisms of killing of iGAS since it has mutations that prevent SpeB expression15. Accordingly, JRS4 made to express SpeB have an initial growth advantage over the WT JRS4 clone in primary human keratinocytes (Fig. 4k), but now induce pyroptosis (Fig. 4l), which eliminates SpeB-expressing iGAS within GSDMA-competent cells (Fig. 4k).
DISCUSSION
Pyroptosis is conventionally regulated by caspase-119 though cleavage of GSDMD, though endogenous proteases that activate GSDMD and other GSDM-family members have been identified1–9. The exception is GSDMA, which was known to generate a lytic effector when experimentally truncated31, but had no known activator. We show GSDMs including GSDMA are directly cleaved by SpeB, a protease of the important bacterial pathogen GAS. This activates pyroptosis, a cell death mechanism previously only known to be initiated by host proteases. Cell death can be important in the defence against microbial infection. Mice deficient in GSDMA are susceptible to GAS infection, in a SpeB dependent manner, indicating an important contribution in the defence against this major skin pathogen.
The skin is both the primary site for GSDMA expression and of GAS colonization and infection10. Keratinocytes form the top layer of the skin and provide the first resistance to infectious, chemical, and physical insults. SpeB is important for allowing GAS to penetrate this layer, survive within cells, and during invasive infection, for penetrating deeper into tissue16,18. Internalised GAS are normally targeted for lysosomal degradation, but escape by puncturing the pathogen-containing vacuole with SLO13. Disruption of this compartment is detected by autophagy regulators that target the iGAS for xenophagic killing13, but GAS counter this using SpeB15. GSDMA detection of SpeB provides a mechanism to eliminate persistent iGAS, by directing cell suicide by pyroptosis (Fig. 4m). GSDMs are protected from indiscriminate activation due to its intracellular localization, suggesting that can be sentinels of exogenous proteases in the cytosol, such as those delivered by pathogens as virulence factors. SpeB activation of GSDMA requires GAS also use virulence factors for keratinocyte attachment, internalisation, and endosome disruption13,15. A pathogen with this combination of activities is a proven threat, suggesting a model where GSDMA allows keratinocytes to rapidly respond to serious threats, but ignore the ubiquitous extracellular proteases from microbiota and endogenous sources.
In summary, our results highlight a crucial role of GSDMA in the keratinocyte defence against invasion by GAS. Gasdermins abundant in other tissues may have similar function in host defense as sensors of pathogen proteases.
Online-Only Methods
Bacterial strains.
The invasive GAS isolate M1T1 5448, its isogenic ΔspeB, Δemm1, Δslo, ΔcovRS, JRS4, L. lactis, and complemented pSpeB and pM strains have been previously described15,18,32,33. pM is expressed from the native promoter; pSpeB was controlled with titrations of anhydrotetracycline (Cayman Chemical) as previously15. Additional clinical isolates (16357, 16357, 61109, 76029, 835777, 16752, 83676, 87132, 87279, 83825, 87277, 78463, 78136, 74264, and 87401) were provided through the Emory University Investigational Clinical Microbiology Core. GAS strains were routinely propagated statically at 37°C in Todd Hewitt broth (Difco) supplemented with 1% yeast (THY), washed two times with phosphate-buffered saline (PBS), and diluted to a multiplicity of infection (MOI) of 100 for in vitro infections.
Reagents.
Mouse anti-FlagM2 (Sigma, F3165). Polyclonal rabbit anti-GSDMA (Invitrogen, PA-598753 or Abcam, ab237615). Anti-Rabbit Dylight 800 (Novus, NBP1–72970). Anti-Rabbit IgG Starbright Blue 700 (12004162), anti-mouse IgG Starbright Blue 700 (12004158), and hFAB Rhodamine anti-actin (12004164) from BioRad. OneBlock Western-FL Blocking buffer (Prometheus, 20–314).
Plasmids.
Plasmids for transfection of each human GSDMs were generated by InvivoGen as pUNO1-hGSDMA (Genbank: NM_178171.4), pUNO1-hGSDMB (Genbank: NM_001165958.1), pUNO1-hGSDMC (Genbank: NM_031415.2), pUNO-hGSDMD (Genbank: NM_024736.6), and pUNO-hGSDME (Genbank: NM_004403.2). pET-SUMO-hGSDMD was a gift from Hongbo Luo (Addgene #111559). cDNA containing the murine ORFs for mGSDMA1 (Genbank: NM_021347.4), mGSDMA2 (Genbank: NM_029727.3), and mGSDMA3 (Genbank: NM_001007461.2) were obtained from GenScript. Full length human GSDMA was cloned from pUNO1-hGSDMA into pET-SUMO with a cleavable His-SUMO tag using Polymerase Incomplete Primer Extension (PIPE) cloning technique34. pCMV-Flag-hGSDMA, pCMV-Flag-hGSDMB, pCMV-Flag-hGSDMC, pCMV-Flag-hGSDMD, pCMV-Flag-hGSDME, pCMV-Flag-mGSDMA1, pCMV-Flag-mGSDMA2, pCMV-Flag-mGSDMA3 were created for this study by PIPE cloning from these ORFs. GSDMAΔ240-247 was created by PIPE cloning. pSpeB and pSpeBC192A are previously described15. pCaspase-1 was a gift from Michael Karin18. mGSDMD-N and mGSDMD-N-4A were gifts from Judy Lieberman (Addgene #80951 and 80952). All plasmids were verified by sequencing. All cloning, primer design, and sequence analysis was performed with DNASTAR v17. Refer to supplemental table 1 for list of primers used.
Cell culture and transfection.
HEK293T cells (ATCC) were routinely cultured in RPMI supplemented with 10% fetal bovine serum. Transient transfection of HEK293 cells was performed with Lipofectamine 2000 (Invitrogen) according to manufacturer instructions. Transfection of proteins was performed with polyethylenimine (PEI; VWR) as detailed previously35. Pooled neonatal normal human epidermal keratinocytes from Lonza or PromoCell were cultured in supplemented KGM2 (Promocell). GSDMA knockout keratinocytes were generated by multi-guide sgRNA editing CRISPR-Cas9 Gene Knockout Kit (Synthego; GKO_HS1_GSDMA). After expansion, only clonal populations with levels of expression undetectable by western blot were used for experiments. 100 U/mL penicillin, 100 μg/mL streptomycin, and 100 μg/mL Normocin was supplemented during routine culture, and omitted during experimental infections, unless otherwise noted. Mouse keratinocytes were isolated from adult tails as detailed previously36, and cultured in KGM2. All cells were maintained at 37 ℃ and 5% CO2.
Protein expression and purification.
Protein expression in E. coli BL21 (DE3) was induced at 18 °C overnight with 0.2 mM isopropyl-β-d-thiogalactopyranoside (IPTG) when OD600 reached 0.6. Cells were collected and resuspended in lysis buffer containing 20 mM Tris-HCl (pH 8.0), 150 mM NaCl, 1 mM DTT, and lysates were homogenized by ultrasonication. The His6-SUMO-tagged proteins were purified by affinity chromatography using HisPur Cobalt Resin (Thermo Scientific) and the His-SUMO tag removed by overnight ULP1 protease digestion at 4°C. His-SUMO was removed using HisPur Cobalt Resin and GSDM was found in the flow through. SpeB was purified by previously described methods18. Recombinant Human Caspase-1 protein was purchased from NovusBio (NBP1-99608).
GSDM thermal shift assay.
Protein unfolding was monitored by fluorescence of Sypro orange (Invitrogen) as temperature was increased at a ramp rate of 1°C per 30 seconds using a BioRad CFX Connect Real-Time PCR detection system.
GSDM cleavage.
Purified recombinant GSDMA (~6 μM) was incubated with SpeB (1 ng/ul) in assay buffer (PBS, 2 mM DTT) for indicated times at 37°C. Cleavage of GSDMs was examined by AcquaStain (Bulldog Bio) staining of proteins separated by SDS–PAGE. For protein sequencing, 12 μg GSDMA was incubated with 40 ng SpeB for 30 minutes at 37°C in assay buffer. Proteins were run on SDS-PAGE, transferred to PVDF membrane, and stained with Coomassie blue. The C-terminal band was cut out and sequenced by Edman degradation by Tufts University Core Facility. For cleavage of transiently transfected Flag-GSDMs, HEK293 cells were transfected overnight and lysed by sonication in assay buffer. Cell lysates were treated with 50 ng SpeB for 1 h at 37 °C and processed for immunoblot. Cleavage sites are diagramed schematically on the human GSDMA structure for Q96QA5 previously created from the AlphaFold Monomer v2.0 pipeline37.
Protein–lipid binding assay.
Lipid strips (Echelon Biosciences) were preincubated with blocking buffer for 1 h at room temperature. Strips were incubated with protein (2 μg/ml) diluted in blocking buffer for 1 h at room temperature and then washed with wash buffer (0.1% Tween-20 in PBS). Lipid strips were incubated with anti-GSDMA for 1 h at room temperature, washed and followed by incubation for 1 h with anti-rabbit Dylight 800. After washing, bound GSDMA was imaged using a BioRad ChemiDoc MP.
Liposome preparation.
Liposomes were prepared by hydration of lipids in PBS or 50mM Sulforhodamine B (SRB; Sigma, S1402) in PBS, and extrusion through a 100-nm polycarbonate membrane (~21 passages; Avanti mini-extruder). Liposomes were comprised of a 4:1 ratio of 1,2-Dioleoyl-sn-glycero-3-phosphocholine (DOPC, L-1182) and 1,2-Dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE, L-2182), to which we added 5% (leakage assay) or 20% (binding assay) Cardiolipin (Avanti, 710333P). Unincorporated Sulforhodamine B was removed by dialysis into PBS.
Liposome binding.
Reaction mixtures from GSDMA cleavage assays were incubated with the indicated liposome (500 μM) at room temperature for 30 min in a total volume of 500 μl. Samples were centrifuged at 4 °C for 20 min at 100,000 g. The supernatant was collected to examine unbound proteins. The liposome pellets were washed twice with 500 μl PBS. Supernatant and pellet fractions were analysed by SDS–PAGE and staining with AcquaStain (Bulldog Bio).
Liposome leakage.
SRB-loaded liposomes were mixed with buffer containing reaction mixture from GSDM cleavage assays described above. Leakage of liposomes encapsulating SRB was determined by an increase in fluorescence intensity (Fn) at 560/600 nm (ex/em) for 20 min at 1 min intervals using a Nivo plate reader at 37°C. At the end of the reactions, 0.4% Triton-X 100 was added to determine maximum fluorescence (F100). Percentage of SRB released is defined as % = ((Fn-F0)/(F100-F0))*100
Cell infection assays.
Adherent cells were infected with GAS 2 h, unless otherwise noted. Cell death was quantified by release of lactate dehydrogenase (CytoTox 96 kit; Promega). Caspase activity was monitored with YVAD-pNA (Enzo) and SpeB activity monitored with Mca-IFFDTWDNE-Lys-Dnp (CPC Scientific) as previously described38. Activity was normalized to positive controls, wt GAS for SpeB, and LPS+ATP for caspase-1. Survival of intracellular GAS was examined by incubating cells with 100 μg/mL gentamicin (VWR) after 1 h, then washing and replacing with antibiotic-free media before the addition of 0.05 % triton X-100 (Sigma) to release intracellular bacteria for enumeration by CFU.
Immunoblot.
Cells were lysed in RIPA lysis buffer (EMD Millipore) containing protease inhibitors (Sigma, P2714) unless otherwise indicated. Cell lysates were heated to 37°C for 10 min with sample buffer, resolved on SDS-PAGE gel, and transferred to a polyvinylidene difluoride (PVDF) membrane (Invitrogen). PVDF membrane was probed with indicated antibodies according to manufacturer instructions. Bands were visualized using a BioRad ChemiDoc MP.
SpeB activity.
Internally quenched peptide, IFFDTWDNE, corresponding to amino acids 103 to 111 of the reference human pro–IL-1β sequence (UniProt: P01584), was labeled on the N terminus with Mca and on the C terminus with Lys-Dnp (CPC Scientific). In quadruplicate, 6 μM peptides were incubated in assay buffer with 10 μL supernatants from keratinocytes infected with GAS. The reaction was continuously monitored using a Victor plate reader (PerkinElmer) with fluorophore excitation at 323 nm and emission at 398 nm.
GAS binding.
GAS cultures were incubated 5 minutes with 10 μg/mL Bocillin-FL (ThermoFisher) for surface staining. Upon washing twice with PBS, labeled bacteria were added to confluent monolayers of human keratinocytes and incubated 1 h. After washing twice with PBS, fluorescence signal was measured on a Victor plate reader (PerkinElmer) with excitation at 485 and emission at 520 nm, with a 500 nm bandpass filter. Signal from uninfected keratinocytes was subtracted as background, and inoculum was separated measured to monitor labeling efficiency; no differences were observed between strains.
Immunofluorescent microscopy.
Keratinocytes were seeded directly onto microscopy slides and allowed to adhere overnight. 30 min before imaging, infected cells were stained with propidium iodide (10 mg/ml; ImmunoChemistry Technologies), the washed once in PBS and transmitted light and epifluorescence using a far-red filter imaged on a Zeiss AxioOberver Z1 microscope. Imaging and processing in ImageJ v1.53 was kept consistent between experimental samples.
Animal model.
Generation of knockout mice by CRISPR/Cas9 technology was performed by the Emory Mouse Transgenic and Gene Targeting Core. Fertilized zygotes were microinjected with Cas9/sgRNA RNP and implanted into pseudopregnant females by standard techniques. Knockouts were maintained on the C57Bl/6 line. mGSDMA123 was targeted with the 5’ gRNA 135 rev (TTTATGCATCCATCAAGGCT), which cut the DNA 54 bp upstream of Gsdma3 Exon 1, and the 3’ gRNA 160 fw (TCGGAGTCATTCATCGGCCA), which cut the DNA 156 bp downstream of Gsdma Exon 12, for an effective knockout region of ~52kb that eliminates the coding sequence of all 3 Gsdma genes and include no overlapping genes on either DNA strand. Knockout was confirmed by sequencing, and primers Gsdma-F (GCCTGAGGTGAGGCACTTCTAACAG) and Gsdma-R (GCACGAGGCACACAAGTGGTGCAC) used for genotyping.
Animal experiments
GAS cultures were grown statically overnight at 37°C in Todd Hewitt broth and washed twice in PBS, and 1 × 108 colony-forming units (CFU) suspended in 100 μl of PBS. Mice were injected intradermally into six- to twelve- week-old mice of both genders as previously described18,32. Each injection site is prepared by shaved and exfoliation for visualization, and ethanol swabbed immediately before inoculation. Mice were housed in specific pathogen-free conditions with a 14-hour light/10-hour dark cycle in standard ambient environment (~ 20C and ~50% humidity) in ABSL-2 conditions and lesions were imaged daily. At 48 h, mice were euthanised by CO2 asphyxiation, lesions were excised, homogenized, and dilutions plated onto THY agar plates for enumeration of bacterial CFU. Animal use and procedures were approved by the Emory Institutional Animal Care and Use Committee.
Statistics.
Graphpad Prism 9 was used to evaluate statistical significance. Student’s t-test (two-tailed) was used for the statistical analysis of experiments. P values <0.05 were considered significant.
Extended Data
Extended Data Fig. 1 |. SpeB cleaves GSDMA.
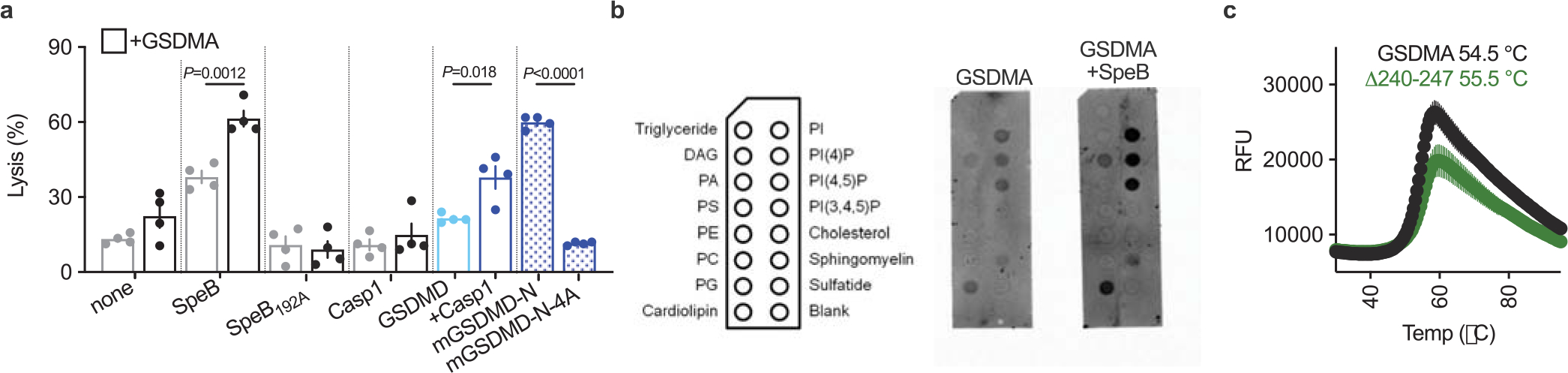
a, Cytotoxicity of HEK293Ts transfected with human GSDMs ± SpeB. Data are the mean±s.d. of 4 technical replicates. P values were calculated by two-tailed Student’s t-test. b, Membranes spotted with lipids were incubated with indicated proteins and binding was assessed by immunoblot for GSDMA. c, Thermal melt analysis of GSDMA and GSDMAΔ240-247 with melting temperatures indicated. (a-c) data are representative of three independent experiments.
Extended Data Fig 2. |. Alignment of human and mouse GSDMAs.
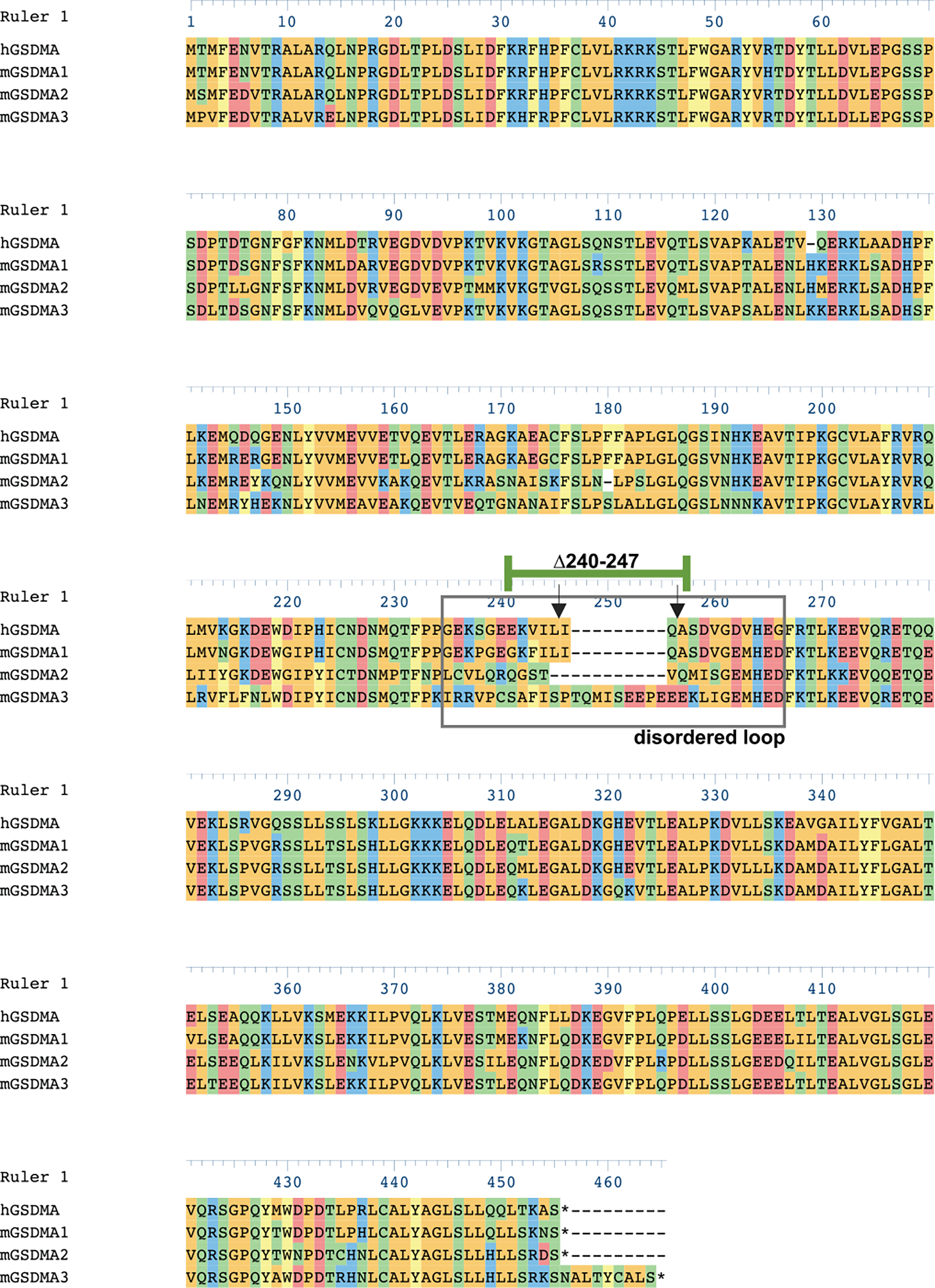
Human GSDMA (Q96QA5), mouse GSDMA_1 (Q9EST1), Mouse GSDMA_2 (Q32M21), mouse GSDMA_3 (Q5Y4Y6) aligned with Clustal Omega algorithm in DNASTAR. Disordered solvent accessible loop (gray box). Inverted arrows identify cleavage sites of SpeB on hGSDMA. Residues 240–247 of hGSDMA are identified (green bar).
Extended Data Fig 3. |. Lytic activity, SpeB production, and binding of GAS strains.
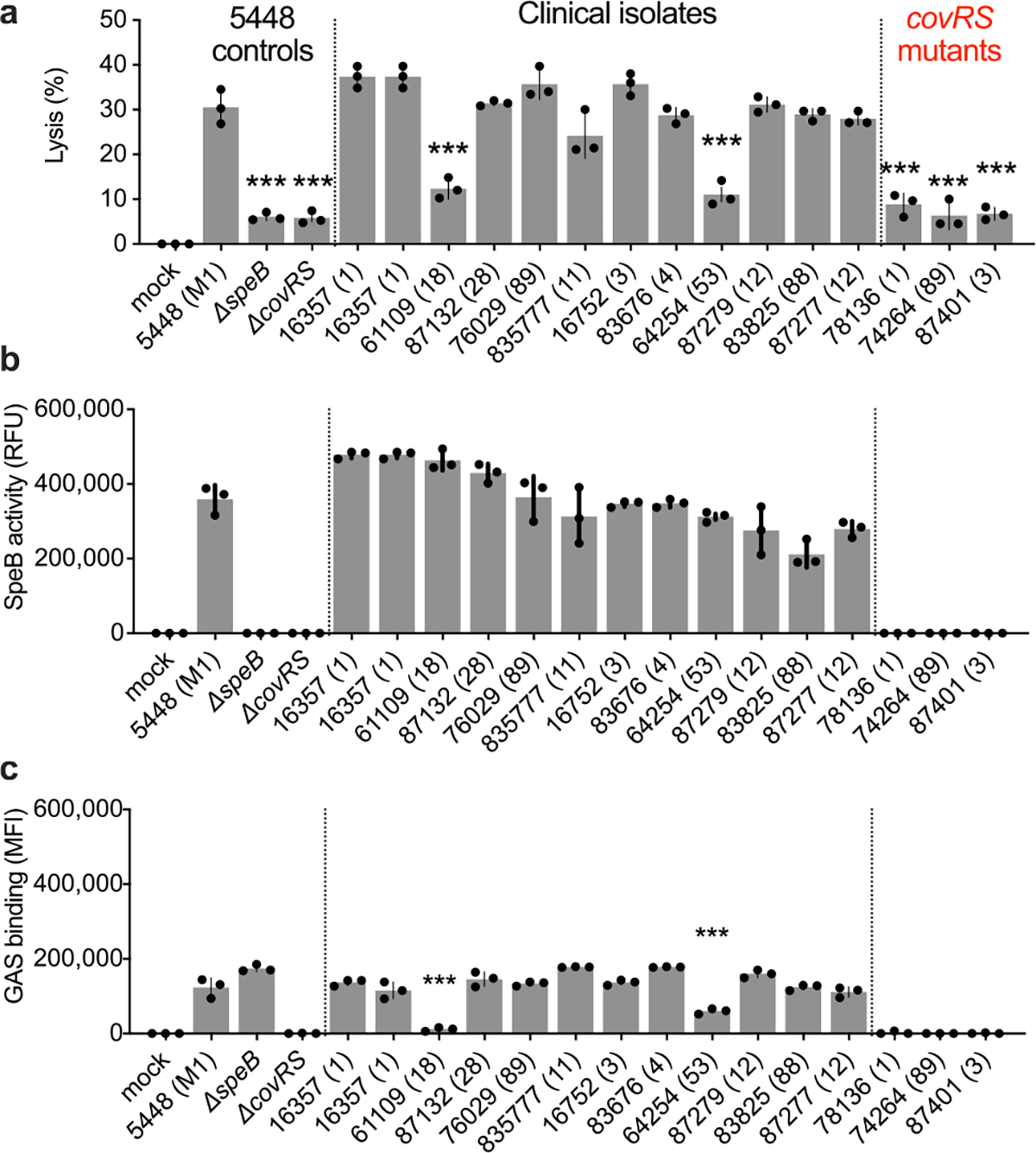
a,b, Keratinocytes were infected (MOI=100) with GAS 5448, isogenic mutant controls, or clinical isolates for 4 h and (a) lysis measured by LDH release and (b) SpeB activity measured using specific substrate Mca-IFFDTWDNE-Lys-Dnp. c, GAS labeled with Bocillin was incubated with keratinocytes (MOI=100) 1 h, washed, and adherence measured by fluorescence. Data are representative of three independent experiments with 3 technical replicates and are presented as the mean±s.d. P values were calculated by two-way ANOVA compared to 5448 (M1) control; P<0.0001.
Supplementary Material
Acknowledgements
We thank Victor Nizet for his insights and bacterial strains. This study received materials from the Emory Investigational Clinical Microbiology Core (supported by the Department of Medicine, Division of Infectious Diseases) and the Emory Mouse Transgenic and Gene Targeting Core (subsidized by the Emory University School of Medicine and receives support from NIH UL1TR000454). C.N.L. received support from NIH grants AI130223 and AI155885. The content is solely the responsibility of the authors and does not necessarily reflect the official views of the National Institutes of Health.
Footnotes
Competing Interests D.L.L. and C.N.L. are named inventors on a patent application that describes GSDMA activities.
Supplementary Information is available for this paper.
Data availability
All data generated or analysed during this study are included in this published article and its supplementary information files.
References
- 1.Broz P, Pelegrín P & Shao F The gasdermins, a protein family executing cell death and inflammation. Nat. Rev. Immunol. 20, 143–157 (2019) [DOI] [PubMed] [Google Scholar]
- 2.Zhou Z et al. Granzyme A from cytotoxic lymphocytes cleaves GSDMB to trigger pyroptosis in target cells. Science 368, eaaz7548 (2020). [DOI] [PubMed] [Google Scholar]
- 3.Aglietti RA et al. GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proc. Natl. Acad. Sci. 113, 7858–7863 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ding J et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 538, 111–116 (2016) [DOI] [PubMed] [Google Scholar]
- 5.Liu X et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 535, 153–158 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kayagaki N et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signaling. Nature 526, 666–671 (2015) [DOI] [PubMed] [Google Scholar]
- 7.Shi J et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665 (2015) [DOI] [PubMed] [Google Scholar]
- 8.Wang Y et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a Gasdermin. Nature 547, 99–103 (2017) [DOI] [PubMed] [Google Scholar]
- 9.Sborgi L et al. GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. EMBO J. 35, 1766–1778 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tamura M et al. Members of a novel gene family, Gsdm, are expressed exclusively in the epithelium of the skin and gastrointestinal tract in a highly tissue-specific manner. Genomics 89, 618–629 (2007). [DOI] [PubMed] [Google Scholar]
- 11.Ralph AP & Carapetis JR Group A Streptococcal Diseases and Their Global Burden. Curr. Top. Microbiol. Immunol. 1–27 (2012). [DOI] [PubMed] [Google Scholar]
- 12.Okada N, Liszewski MK, Atkinson JP & Caparon M Membrane cofactor protein (CD46) is a keratinocyte receptor for the M protein of the group A streptococcus. Proc. Natl. Acad. Sci. 92, 2489–2493 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nakagawa I et al. Autophagy defends cells against invading group A Streptococcus. Science 306, 1037–1040 (2004). [DOI] [PubMed] [Google Scholar]
- 14.Sil P, Wong S-W & Martinez J More Than Skin Deep: Autophagy Is Vital for Skin Barrier Function. Front. Immunol. 9, 1376 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barnett TC et al. The Globally Disseminated M1T1 Clone of Group A Streptococcus Evades Autophagy for Intracellular Replication. Cell Host Microbe 14, 675–682 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nelson DC, Garbe J & Collin M The cysteine proteinase SpeB from Streptococcus pyogenes–a potent modifier of immunologically important host and bacterial proteins. Biol. Chem. 392, 1077–1088 (2011). [DOI] [PubMed] [Google Scholar]
- 17.LaRock DL, Russell R, Johnson AF, Wilde S & LaRock CN Group A Streptococcus Infection of the Nasopharynx Requires Proinflammatory Signaling Through the Interleukin-1 Receptor. Infect. Immun. 88, e00356–20 (2020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.LaRock CN et al. IL-1β is an innate immune sensor of microbial proteolysis. Sci. Immunol 1, eaah3539 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cookson BT & Brennan M Pro-inflammatory programmed cell death. Trends Microbiol. 3, 113–114 (2001). [DOI] [PubMed] [Google Scholar]
- 20.Uhlen M et al. A genome-wide transcriptomic analysis of protein-coding genes in human blood cells. Science 366, eaax9198 (2019). [DOI] [PubMed] [Google Scholar]
- 21.Nizet V et al. Innate antimicrobial peptide protects the skin from invasive bacterial infection. Nature 414, 454–457 (2001). [DOI] [PubMed] [Google Scholar]
- 22.Walker MJ et al. DNase Sda1 provides selection pressure for a switch to invasive group A streptococcal infection. Nat. Med. 13, 981–985 (2007). [DOI] [PubMed] [Google Scholar]
- 23.Schrager HM, Rheinwald JG & Wessels MR Hyaluronic acid capsule and the role of streptococcal entry into keratinocytes in invasive skin infection. J. Clin. Invest. 98, 1954–1958 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.O’Seaghdha M & Wessels MR Streptolysin O and its Co-Toxin NAD-glycohydrolase Protect Group A Streptococcus from Xenophagic Killing. PLOS Pathog. 9, e1003394 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barnett TC, Bowen AC & Carapetis JR The fall and rise of Group A Streptococcus diseases. Epidemiol. Infect. 147, e4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Svensson MD et al. Role for a secreted cysteine proteinase in the establishment of host tissue tropism by group A streptococci. Mol. Microbiol. 38, 242–253 (2000). [DOI] [PubMed] [Google Scholar]
- 27.Cywes C & Wessels MR Group A Streptococcus tissue invasion by CD44-mediated cell signalling. Nature 414, 648–652 (2001). [DOI] [PubMed] [Google Scholar]
- 28.Molinari G & Chhatwal GS Invasion and Survival of Streptococcus pyogenes in Eukaryotic Cells Correlates with the Source of the Clinical Isolates. J. Infect. Dis. 177, 1600–1607 (1998). [DOI] [PubMed] [Google Scholar]
- 29.Wang JR & Stinson MW M protein mediates streptococcal adhesion to HEp-2 cells. Infect. Immun. 62, 442–448 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sakurai A et al. Specific Behavior of Intracellular Streptococcus pyogenes That Has Undergone Autophagic Degradation Is Associated with Bacterial Streptolysin O and Host Small G Proteins Rab5 and Rab7. J. Biol. Chem. 285, 22666–22675 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ruan J, Xia S, Liu X, Lieberman J & Wu H Cryo-EM structure of the gasdermin A3 membrane pore. Nature 557, 62–67 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.LaRock CN et al. Group A Streptococcal M1 Protein Sequesters Cathelicidin to Evade Innate Immune Killing. Cell Host Microbe 18, 471–477 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aziz RK et al. Invasive M1T1 group A Streptococcus undergoes a phase-shift in vivo to prevent proteolytic degradation of multiple virulence factors by SpeB. Mol. Microbiol. 51, 123–134 (2004). [DOI] [PubMed] [Google Scholar]
- 34.Klock HE & Lesley SA The Polymerase Incomplete Primer Extension (PIPE) Method Applied to High-Throughput Cloning and Site-Directed Mutagenesis. in High Throughput Protein Expression and Purification: Methods and Protocols (ed. Doyle SA) 91–103 (Humana Press, 2009). [DOI] [PubMed] [Google Scholar]
- 35.Miao EA et al. Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1β via Ipaf. Nat. Immunol. 7, 569–575 (2006). [DOI] [PubMed] [Google Scholar]
- 36.Lichti U, Anders J & Yuspa SH Isolation and short term culture of primary keratinocytes, hair follicle populations, and dermal cells from newborn mice and keratinocytes from adult mice, for in vitro analysis and for grafting to immunodeficient mice. Nat. Protoc. 3, 799–810 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jumper J et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.LaRock CN & Cookson BT The Yersinia Virulence Effector YopM Binds Caspase-1 to Arrest Inflammasome Assembly and Processing. Cell Host Microbe 12, 799–805 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analysed during this study are included in this published article and its supplementary information files.