

Abstract
Single-cell multiomics data continues to grow at an unprecedented pace. Although several methods have demonstrated promising results in integrating several data modalities from the same tissue, the complexity and scale of data compositions present in cell atlases still pose a challenge. Here, we present scJoint, a transfer learning method to integrate atlas-scale, heterogeneous collections of scRNA-seq and scATAC-seq data. scJoint leverages information from annotated scRNA-seq data in a semisupervised framework and uses a neural network to simultaneously train labeled and unlabeled data, allowing label transfer and joint visualization in an integrative framework. Using atlas data as well as multimodal datasets generated with ASAP-seq and CITE-seq, we demonstrate that scJoint is computationally efficient and consistently achieves substantially higher cell-type label accuracy than existing methods while providing meaningful joint visualizations. Thus, scJoint overcomes the heterogeneity of different data modalities to enable a more comprehensive understanding of cellular phenotypes.
Advances in single-cell technologies have enabled comprehensive studies of cell heterogeneity, developmental dynamics and cell communications across diverse biological systems at unprecedented resolution. There are a variety of protocols profiling transcriptomics, as exemplified by single-cell RNA sequencing (scRNA-seq). In addition, several technologies have been developed for other molecular measurements in individual cells towards building a more holistic view of cell functions, including chromatin accessibility, protein abundance and methylation1.
In particular, single-cell ATAC-seq (scATAC-seq) is an epigenomic profiling technique for measuring chromatin accessibility to discover cell-type-specific regulatory mechanisms2,3. scATAC-seq offers a complementary layer of information to scRNA-seq, and together they provide a more comprehensive molecular profile of individual cells and their identities. However, it has been noted that the extreme sparsity of scATAC-seq data often limits its power in cell-type identification4. In contrast, large amounts of well-annotated scRNA-seq datasets have been curated as cell atlases5,6, motivating us to transfer cell-type information from scRNA-seq to scATAC-seq for better classification of cell types in an integrative analysis framework.
Several methods exist to denoise, batch correct and perform integration of single-omics data across several experiments for both transcriptomic data7–12 and scATAC-seq data13. However, direct applications of these methods to multiomics data integration are computationally challenging and often suboptimal, since different modalities have vastly different dimensions and sparsity levels. Recently, a growing number of methods has been proposed to address the need for integrative analysis across different modalities. When the data consist of simultaneous multimodal measurements in the same cell14,15, methods like scAI16 and MOFA+ (ref. 17) have been developed based on factor analysis and joint clustering. In general, these paired measurements are technically more challenging and costly to perform.
More commonly, different modalities are derived from different cells taken from the same or similar populations. In this setting, most existing methods can be divided broadly into four categories: manifold alignment18–20, matrix factorization (Liger21, coupled-NMF22), using correlations to identify nearby cells across modalities (Conos23, Seurat24) or neural-network approaches, each with its own limitations when facing complex data compositions as typically seen in cell atlases. Manifold alignment methods have demonstrated promising results in integrating several modalities from the same tissue. However, requiring distributions to match globally is often too restrictive when different modalities are derived from different tissues and cell types. Furthermore, matrix factorization and correlation-based methods designed for unpaired data require a separate feature selection step before integration for dimension reduction, and the method’s performance is sensitive to which genes are selected. Most existing neural-network methods for multiomics integration are based on autoencoders, which, with a few exceptions25, require paired data. In general, unsupervised training of several autoencoders simultaneously can be very challenging without pairing information across different modalities, with finding a common embedding manifold becomes more difficult as the complexity of the data increases. Hence, current methods are limited in their ability to handle the complexity and scale that characterize multiomics atlas data.
Here, we present a new scalable transfer learning method, scJoint, that effectively integrates atlas-scale scRNA-seq and scATAC-seq data using a neural-network approach (Fig. 1a). We achieve this by taking advantage of the increasing amount of scRNA-seq data with high quality annotations, and incorporating the cell-type label information into a semisupervised paradigm to train unlabeled scATAC-seq. scJoint is able to meet the challenges in integrating multiomics atlas data through the use of (1) a new loss function to explicitly incorporate dimension reduction as part of the feature engineering process in transfer learning, allowing the low-dimensional features to be revised throughout training and removing the need for selecting highly variable genes; (2) a similarity loss that adds flexibility to the alignment of modalities when their cell types do not fully overlap and (3) weight sharing across encoders for different modalities resulting in stable training.
Fig. 1 |. overview of scJoint.
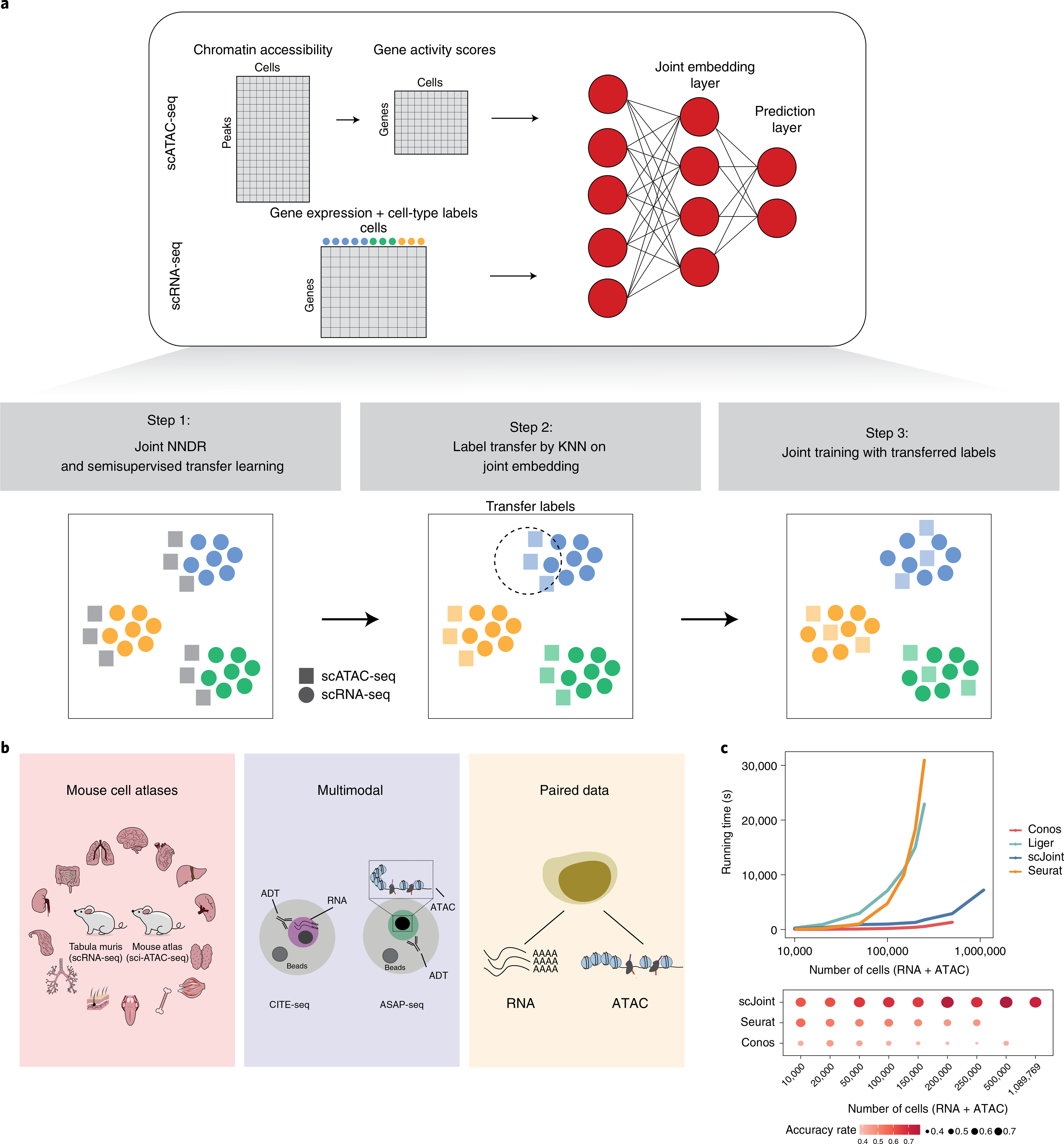
a, Overview of scJoint. The input of scJoint consists of one (or several) gene activity score matrix, calculated from the accessibility peak matrix of scATAC-seq, and one (or several) gene expression matrix including cell-type labels from scRNA-seq experiments. The three main steps of the method are illustrated. b, Three data collections analyzed in detail in this study: (1) mouse cell atlases; (2) multimodal data from PBMC; (3) paired data from adult mouse cerebral cortex data generated by SNARE-seq. c, Computation time required by different methods to integrate scRNA-seq and scATAC-seq (top) and their label transfer accuracy (bottom, computed for methods with label transfer functionality). The benchmark datasets were subsampled from 54 cell types in the human fetal atlases27,28, where the total number of RNA and ATAC cells ranges from 10,000 to 1,089,769. Seurat and Liger were terminated for out-of-memory error on datasets with 500,000 cells and more, and Conos was terminated on the 1 million cell dataset.
We illustrate scJoint’s performance in terms of label transfer accuracy, quality of joint visualizations, scalability and capacity to generalize. In particular, we highlight the scalability of scJoint through the integration of two mouse atlases5,26 and two human fetal atlases27,28. In the latter case, scJoint required only 2 h to integrate more than a million cells (Fig. 1c) while maintaining consistently high accuracy rates. The generalizability of scJoint to other types of single-cell data is demonstrated through multimodal data with paired protein measurements (CITE-seq and ASAP-seq; Fig. 1b).
Results
scJoint for cotraining labeled and unlabeled data.
The core of scJoint is a semisupervised approach to cotrain labeled (scRNA-seq) and unlabeled (scATAC-seq) data, where we address the main challenge of aligning these two distinct data modalities via a common lower dimensional space. scJoint consists of three main steps (Fig. 1a). Step 1 performs joint dimension reduction and modality alignment in a common embedding space through a new neural-network-based dimension reduction (NNDR) loss and a cosine similarity loss respectively. The NNDR loss extracts orthogonal features with maximal variability in a vein similar to PCA, while the cosine similarity loss encourages the neural network to find projections into the embedding space so that most parts of the two modalities can be aligned. The embedding of scRNA-seq is further guided by a cell-type classification loss, forming the semisupervised part. In Step 2, treating each cell in scATAC-seq data as a query, we identify the k-nearest neighbors (KNN) among scRNA-seq cells by measuring their distances in the common embedding space, and transfer the cell-type labels from scRNA-seq to scATAC-seq via majority vote. In Step 3, we further improve the mixing between the two modalities by using the transferred labels in a metric learning loss. Joint visualization of the datasets is obtained from the final embedding layer using standard tools, including tSNE29 and UMAP30. scJoint requires simple data preprocessing, with the input dimension equal to the number of genes in the given datasets after appropriate filtering. Chromatin accessibility in scATAC-seq data is first converted to gene activity scores31,32 allowing for the use of a single encoder with weight sharing for both RNA and ATAC.
We next compared scJoint with methods developed and applied recently to the integration of scRNA-seq and scATAC-seq, including Seurat v.3 (ref. 24), Conos23 for label transfer accuracy and Liger21 (as a representative matrix factorization method) for evaluating the joint embedding of the two modalities.
scJoint shows accurate and robust performance on atlas data.
We demonstrate the performance of scJoint in a complex scenario, where the heterogeneity of cell types and tissues in atlas data poses substantial challenges to data integration. We applied our method to integrate two mouse cell atlases: the Tabula Muris atlas5 for scRNA-seq data and the atlas in Cusanovich et al.26 for scATAC-seq data, containing 73 (96,404 cells from 20 organs, two protocols) and 29 (81,173 cells from 13 tissues) cell types, respectively (the last including a group annotated as ‘unknown’), of which 19 cell types are common. We focus our initial evaluation on the subset of the atlas data containing 101,692 cells from the 19 overlapping cell types only. Here, we transferred cell-type labels from scRNA-seq to scATAC-seq and compared the results with the original labels in Cusanovich et al.26 for accuracy; these original labels were also used to evaluate the quality of joint visualizations. An inspection of the tSNE plots shows that our method effectively mixes the three protocols (fluorescence-activated cell sorting (FACS), droplet, ATAC) while providing a better grouping of the cells in terms of previously defined cell types than the other methods (Fig. 2a and Supplementary Fig. 1). This observation is confirmed by the quantitative evaluation metrics, with scJoint showing substantially higher cell-type silhouette coefficients than all the other methods, and similar modality silhouette coefficients as Seurat24 and Liger21. Overall, scJoint has the highest median F1 score of silhouette coefficients, achieving a better trade-off between removing the technological variations in modalities and maintaining the cell-type signals (Fig. 2b and Supplementary Fig. 2). In terms of label transfer accuracy, scJoint assigned 84% of the cells to the correct type, 14% and 13% higher than Seurat24 and Conos23 (Fig. 2d and Supplementary Fig. 3).
Fig. 2 |. Analysis of mouse cell atlas subset data containing 19 overlapping cell types from RNA and ATAC.
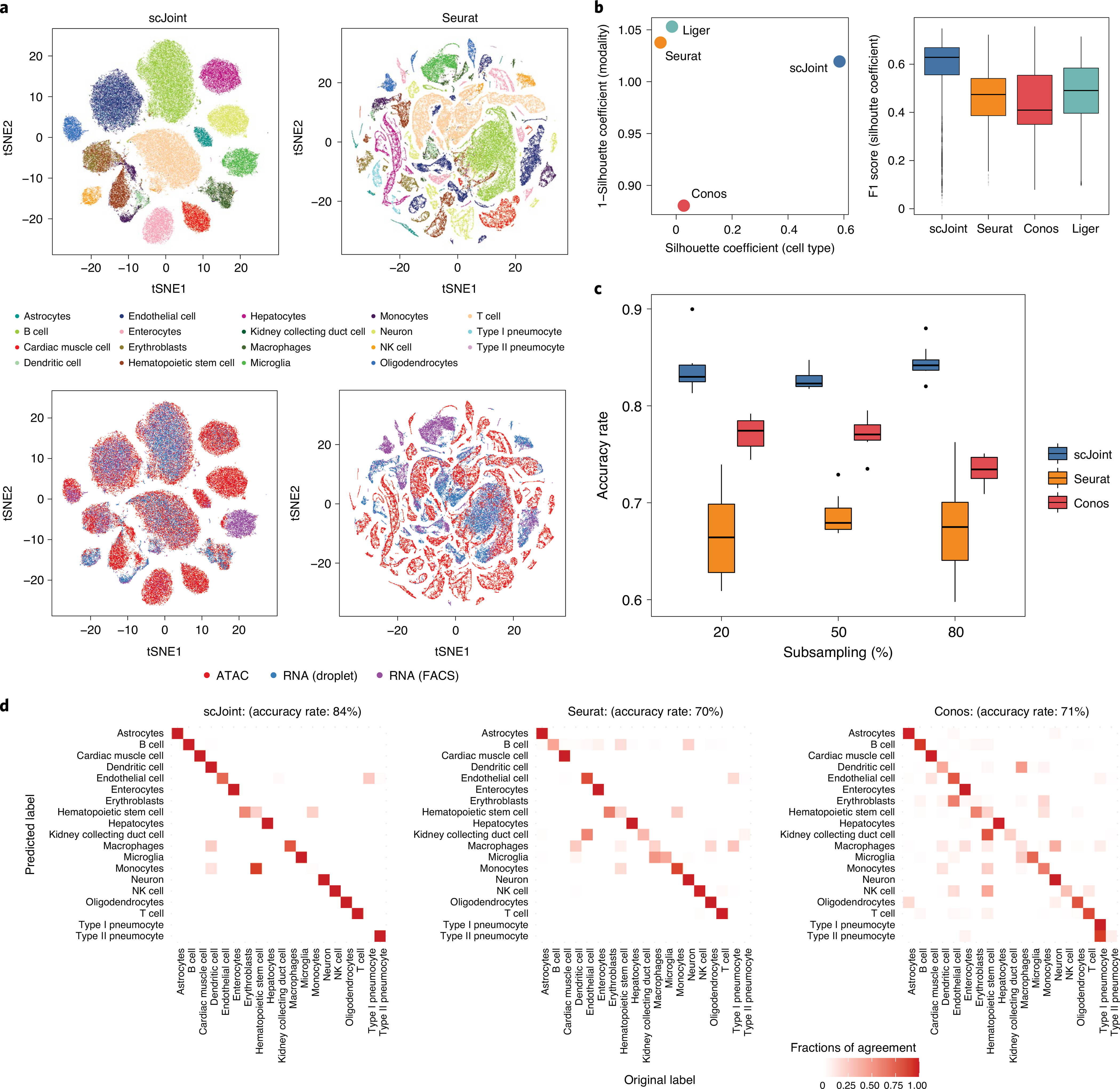
a, tSNE visualization of scJoint (left column) and Seurat (right column), colored by cell types defined in Cusanovich et al.26 (first row) and three protocols (second row). b, Scatter plot of mean silhouette coefficients for scJoint, Liger, Seurat and Conos (left panel), where the x axis shows the mean cell-type silhouette coefficients and the y axis shows 1 – mean modality silhouette coefficients; ideal outcomes would lie in the top right corner. Boxplots of F1 scores of silhouette coefficients for scJoint, Liger, Seurat and Conos (n = 101,692) (right panel). Each boxplot ranges from the upper and lower quartiles with the median as the horizontal line and whiskers extend to 1.5 times the interquartile range. c, Accuracy rates of scJoint, Seurat and Conos using 20%, 50% and 80% of cells from scRNA-seq data as training data. Ten random subsamplings were performed for each setting to generate the variance. Each boxplot ranges from the upper and lower quartiles with the median as the horizontal line and whiskers extend to 1.5 times the interquartile range. d, Predicted cell types and their fractions of agreement with the original cell types given in Cusanovich et al.26 for scJoint (left panel), Seurat (middle panel) and Conos (right panel). Clearer diagonal structure indicates better agreement.
To assess the robustness of the label transfer results, we first performed a stability analysis on this subset of atlas data by subsampling 80%, 50% and 20% of the cells from scRNA-seq as the training data. Even when only 20% of the cells were used for training, scJoint maintained a high accuracy and small variance (Fig. 2c).
To examine whether scJoint is robust to mislabelling, we randomly shuffled 5%, 10% and 20% of the cell-type labels in scRNA-seq as the training data. scJoint maintained stable and high accuracy (~82% label transfer accuracy) even when 20% of labels were shuffled (Supplementary Fig. 4). Together, these analyses suggest that scJoint is robust when applied to scRNA-seq databases with partial labels and labeling errors.
To evaluate scJoint’s computational efficiency on atlas-sized data, we further considered two human fetal atlases27,28 and created benchmark datasets by subsampling from 15 organs with 54 cell types common between scRNA-seq and scATAC-seq. The size of the datasets ranged from 10,000 to 1,089,769 cells. scJoint was substantially faster than Seurat and Liger, being the only method capable of handling more than 1 million cells (Fig. 1c and Supplementary Fig. 5). scJoint consistently achieved much higher accuracy than the other methods, with an average 20% improvement for 100,000 or more cells (Fig. 1d). Together, these results illustrate that scJoint scales well to large atlas data in terms of both computational efficiency and quality of results.
Refining scATAC-seq annotations in heterogeneous atlas data.
We next performed the more challenging task of integrating full atlas data, using the mouse atlases as an example. Since the scRNA-seq atlas contains more cell types than the scATAC-seq atlas, we use this application to illustrate how transferred labels can refine and provide new annotations to ATAC cells. To compare with the original labels, tSNE plots were constructed in the same way as in Cusanovich et al.26, using singular value decomposition of the term frequency-inverse document frequency (TF-IDF) transformation of scATAC-seq peak matrix (Fig. 3a). We observe that scJoint labels cells close together in this ATAC visualization space in a more consistent way than the other methods. Quantitatively, this is supported by scJoint’s higher silhouette coefficients (Supplementary Fig. 6) and higher overall accuracy rate (77% compared with 60% for Seurat and 55% for Conos).
Fig. 3 |. Analysis of mouse cell atlas full data.
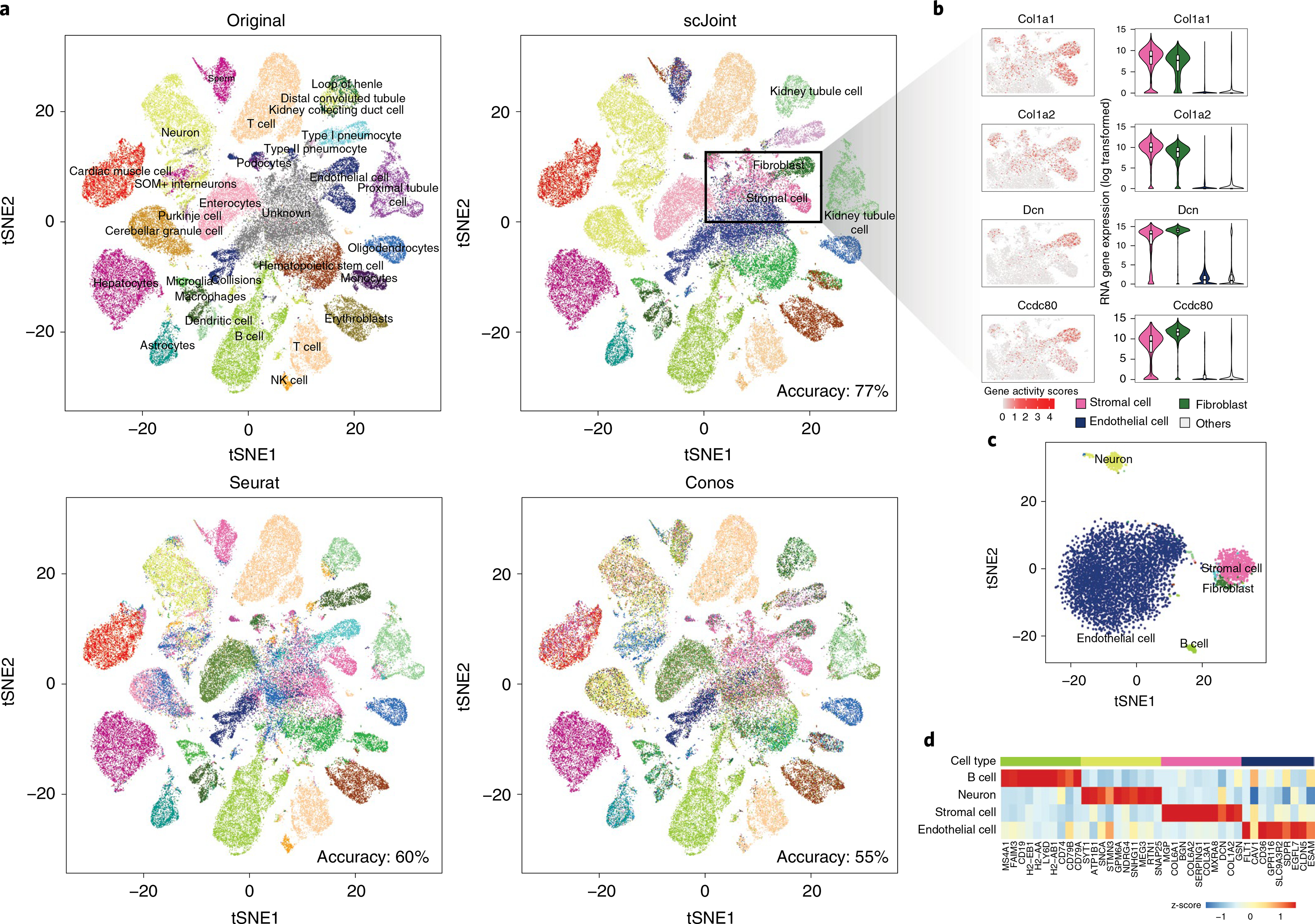
a, A 2 × 2 panel of tSNE plots generated from the top 100 dimensions of singular value decomposition of the TF-IDF transformed ATAC-seq data, colored by the original labels (top left), scJoint transferred labels (top right), Seurat transferred labels (bottom left) and Conos transferred labels (bottom right). b, Marker expressions in stromal cells and fibroblasts: Col1a1, Col1a2, Dcn and Ccdc80. The left column shows the gene activity scores of the markers in ATAC-seq data (4,352 stromal cells and 1,602 fibroblasts). The right column shows the log-transformed gene expression of the markers in stromal cells, fibroblasts and endothelial cells versus others; all cells here are taken from the FACS scRNA-seq data (n = 1,363, 2,152, 3,794 and 34,656 for stromal cells, fibroblasts, endothelial cells and others, respectively). Each boxplot ranges from the upper and lower quartiles with the median as the horizontal line and whiskers extend to 1.5 times the interquartile range. c, tSNE plot of cells originally labeled as ‘unknown’ and annotated by scJoint with probability scores greater than 0.80, colored by predicted cell types (5,931 cells). d, Heatmap of z-scores of average gene activity scores, calculated from cells aggregated by predicted cell types in ATAC. The rows indicate the top four predicted cell types by size. The columns indicate the top differential expressed genes of the corresponding cell type in RNA.
Examining the transferred labels further, we find scJoint labels a group of cells (originally labeled as ‘unknown’ or ‘endothelials’) as ‘stromal cells’ (4,352 cells) and ‘fibroblasts’ (1,602 cells), which are two cell types not present in the original ATAC labels. These cells show high gene activity scores for Col1a1, Col1a2, Dcn and Ccdc80, all of which are markers with high expression levels in stromal cells and fibroblasts, but low expression levels in endothelial cells from the scRNA-seq data (Fig. 3b). Hence, the new annotations are more consistent with the marker expression levels.
We note that scJoint allows us to annotate 5,931 cells labeled as ‘unknown’ in Cusanovich et al.26 with probability score greater than 0.80. These cells are clearly clustered into groups in the tSNE visualization of scJoint’s embedding space (Fig. 3c), with the main groups being endothelial cells, stromal cells, neurons and B cells. Using cell-type markers identified from the scRNA-seq data, the aggregated gene activity scores of these ATAC cells show clear differential expression patterns (Fig. 3d).
Integration of multimodal data across biological conditions.
We demonstrate that scJoint is capable of incorporating further modality information to RNA-seq and ATAC-seq, and is applicable to experiments with different underlying biological conditions. We consider multimodal measurements profiling gene expression levels or chromatin accessibility simultaneously with surface protein levels, which can be obtained via CITE-seq33 and ASAP-seq34. We analyzed CITE-seq and ASAP-seq data from a T cell stimulation experiment in Mimitou et al.34, which sequenced cells with these two technologies in parallel. A total of 18,088 cells were studied under two conditions: one with stimulation of anti-CD3/CD28 in the presence of IL-2 for 16 h and the other without stimulation as a control. We first clustered and annotated these cells using CiteFuse35. Compared with the cell-type labels in the original study, we were able to identify cellular subtypes with CiteFuse, further annotating five subgroups in T cells. Next, we performed integration analysis of CITE-seq and ASAP-seq by concatenating gene expression or gene activity vectors with protein measurements. We performed the analysis in two scenarios: in the stimulated and control condition separately and across the two conditions.
In both scenarios, scJoint generated better joint visualization of the two technologies (Fig. 4a and Supplementary Figs. 7 and 8). In particular, in the case where stimulated and control cells are combined, subtypes of T cell (for example, naive CD8+, effector CD8+, naive CD4+ and effector CD4+) are clearly separated, whereas cells from the two technologies are well mixed (Fig. 4a,b). The median cell-type silhouette coefficient of scJoint is 0.51, outperforming the other three methods by a large margin (Seurat 0.11, Conos 0.13 and Liger −0.06). With the highest silhouette coefficient F1 scores (median F1 score: 0.59) representing a 16–28% improvement over the other methods, scJoint demonstrates the best balance between removing technical variations and preserving biological signals (Fig. 4c and Supplementary Fig. 9).
Fig. 4 |. integration of multimodal PBMC data across biological conditions: with (stimulation) or without (control) T cell activation.
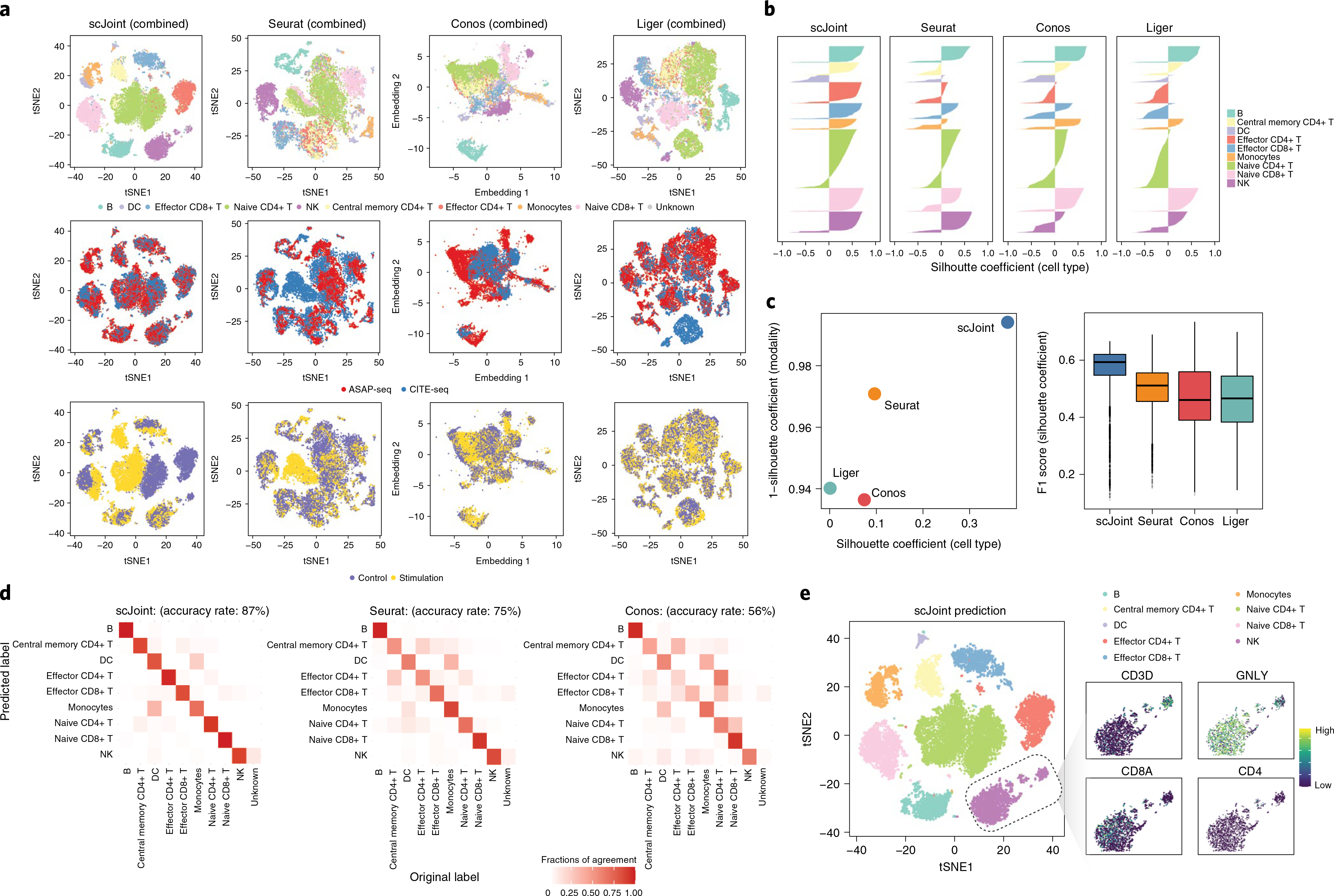
a, tSNE visualization of scJoint (first column), Seurat (second column), Conos (third column) and Liger (fourth column) of PBMC data generated from CITE-seq and ASAP-seq, colored by cell type obtained from CiteFuse and manual annotations (first row), technology (second row) and biological condition (third row). b, Barplots of cell-type silhouette coefficients for scJoint, Seurat, Conos and Liger for all cells, colored by cell type. Larger values on the x axis indicate better grouping. c, Scatter plot of mean silhouette coefficients for scJoint, Seurat, Conos and Liger (left), where the x axis denotes the mean cell-type silhouette coefficients, and the y axis denotes 1 – mean modality silhouette coefficients; ideal outcomes would lie in the top right corner. Boxplots of F1 scores of silhouette coefficients for scJoint, Liger, Seurat and Conos (n = 18,088) (right). Each boxplot ranges from the upper and lower quartiles with the median as the horizontal line and whiskers extend 1.5 times the interquartile range. d, Heatmaps comparing the original labels and the transferred labels of scJoint, Seurat and Conos. Clearer diagonal structure indicates better agreement. e, tSNE visualization of scJoint colored by the predicted cell types with gene expression levels of CD3D, NKG7, CD8A and CD4 in NK cells.
Moreover, scJoint achieves higher accuracy in label transfer under all scenarios (93% in control, 84% in stimulation and 87% in the combined case), compared with Seurat (84% in control, 79% in stimulation and 75% combined) and Conos (55% in control, 67% in stimulation and 56% in combined) (Fig. 4d and Supplementary Fig. 10). In addition, the transferred labels of scJoint from the two scenarios (control/stimulation alone or combined) are highly consistent, with 95% of cells having the same annotation, substantially greater than Seurat (84%) and Conos (59%) (Supplementary Fig. 11).
Further biological signals captured by scJoint.
In the combined analysis of stimulation and control, we find that the joint embedding generated by scJoint contains further information that allows for the identification of a cellular subtype. In the CiteFuse annotation of ASAP-seq data, we labeled one cluster of 142 cells with ambiguous marker expression as ‘unknown’. In the joint visualization of scJoint, while these ‘unknown’ cells are labeled as ‘natural killer (NK)’ cells by label transfer, they are still clearly separated from most NK cells and form a small cluster together with cells from CITE-seq. We then examined the gene and protein expression amounts of NK cell and T cell markers in this subgroup. We find that these cells have high expression of CD3 and GNLY at gene level as well as CD3, CD56, CD57 and CD244 at protein level, but low expression of CD8 (CD8A) and CD4 (CD4). This suggests these cells may be natural killer T cells, a minority of the immune cells in peripheral blood mononuclear cells (PBMC) samples (Fig. 4e and Supplementary Fig. 12)36. By contrast, although these cells lack CD8 expression, the other methods are unable to distinguish them from effector CD8+ T cells in their visualizations (Fig. 4e and Supplementary Fig. 13).
Finally, by appropriately aligning the two technologies in the embedding space, scJoint is able to show the biological difference between stimulation and control in the same cell type. In the joint visualization of scJoint, three subtypes of T cell (naive CD4+, naive CD8+ and effector CD4+) are less well mixed between the two conditions than the other cell types, consistent with the stimulation experiment aiming to activate T cells. In particular, the naive CD4+ T cells show the most notable separation between the two conditions (Fig. 4a). We then performed differential expression analysis of the scRNA-seq part of CITE-seq in each cell type across the two conditions using MAST37. We find that the naive CD4+ T cells have the largest number of unique differentially expressed genes (false discovery rate < 0.01) (Supplementary Fig. 14a). Similarly, differential proteins analysis of both CITE-seq and ATAC-seq using Wilcoxon rank-sum test on the log-transformed protein abundances also suggests that naive CD4+ T cells have the most unique differential proteins compared with other cell types (false discovery rate < 0.01) (Supplementary Fig. 14b,c).
scJoint shows versatile performance on paired data.
Although scJoint is designed for integrating unpaired data, it is still directly applicable to paired data. Such an application also enables us to use the pairing information to validate the label transfer results. For this reason, the pairing information was not used in any of the unpaired methods under comparison. We consider the integration of adult mouse cerebral cortex data generated by SNARE-seq14—a technology that can profile gene expression and chromatin accessibility in the same cell. In addition to Seurat and Liger, we compared scJoint with three other methods designed specifically for paired data, scAI16, MOFA+ (ref. 17) and Seurat (WNN)38. In our assessment, all the unpaired methods (scJoint, Seurat, Liger) treat the RNA and ATAC parts of SNARE-seq as two separate datasets, while the paired methods take the pairing information into account. Despite not using this information, the semisupervised framework of scJoint using RNA label information and its loss function designs (Supplementary Note) still produce tight groupings of cells according to cellular subtypes (Fig. 5a and Supplementary Fig. 15). scJoint achieves comparable cell-type silhouette coefficients (Fig. 5b) compared with the class of paired methods. This suggests that scJoint is versatile enough to be applied to paired data, which are becoming increasingly popular.
Fig. 5 |. Analysis of paired gene expression and chromatin accessibility data from SNARe-seq.
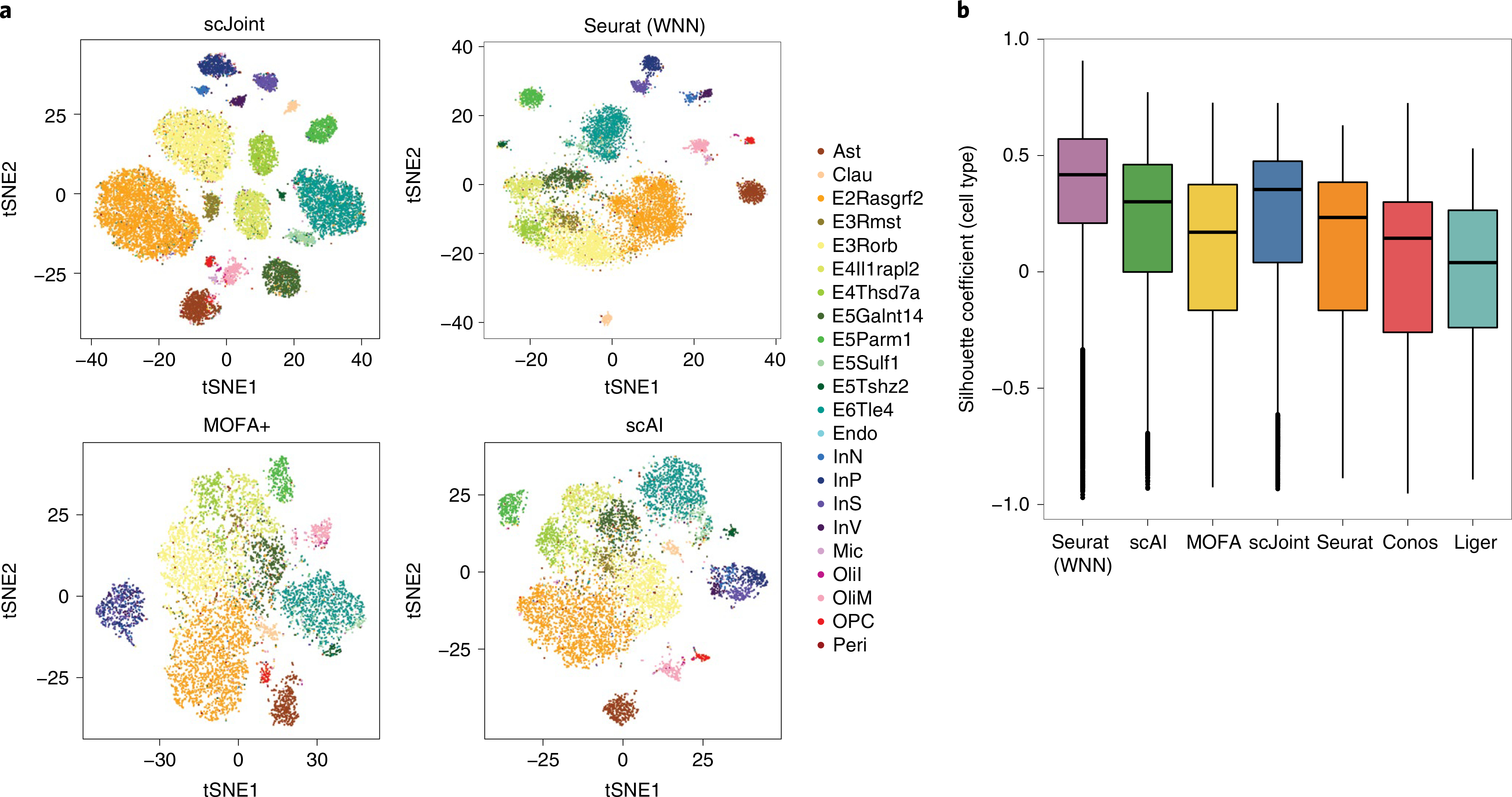
a, tSNE visualization of SNARE-seq data for scJoint, Seurat (WNN), MOFA+ and scAI, colored by cell types given in Chen et al.14. All unpaired methods treat the RNA and ATAC parts of SNARE-seq as two separate datasets. b, Boxplots of cell-type silhouette coefficients for Seurat (WNN), scAI, MOFA+, scJoint, Seurat, Conos and Liger, colored by method (n = 9,190). Each boxplot ranges from the upper and lower quartiles with the median as the horizontal line and whiskers extend to 1.5 times the interquartile range.
Comparing the performance among the unpaired methods, scJoint has the highest medians in cell-type silhouette coefficients and F1 scores (Fig. 5b and Supplementary Fig. 16). For label transfer, scJoint achieves an accuracy rate of 70.9%, retaining better performance than the other two methods (70.1% for Seurat and 49.5% for Conos). Looking closer at the performance in each cell type, scJoint performs the best in 10 out of 22 cell types in terms of F1 scores for classification (Supplementary Fig. 17). Together, these results suggest that scJoint performs the best among the unpaired methods and on par with the paired methods, despite treating paired data as separate.
Discussion
scJoint approaches the integration of scRNA-seq and scATAC-seq as a domain adaptation problem in transfer learning, using the same neural network to cotrain labeled data from the source domain (RNA) and unlabeled data from the target domain (ATAC) following a different distribution. scRNA-seq data serve as a natural source domain for transferring information to other modalities due to rapidly growing collections of annotated public data and RNA-focused computational tools that can output accurate classifications39. Using several cell atlases and multimodal data with protein measurements, we demonstrate that scJoint achieves substantially higher label transfer accuracy and provides better joint visualizations than other methods, even when (1) the data is highly complex and heterogeneous and (2) meaningful biological conditions are mixed with technical variations. We have shown that integrative analysis of single-cell multiomics data by scJoint facilitates reannotation of cell types in scATAC-seq and discovery of new subtypes not present in training data. scJoint is also flexible enough to be applied to developmental data with a simple change in training Step 3 (Methods). We applied scJoint to the human hematopoiesis data with several lineages generated by scRNA-seq and scATAC-seq40, and demonstrate that scJoint is able to show trajectory structures from continuous biological processes (Supplementary Note).
scJoint provides a concise training framework with one main tuning parameter in the construction of cosine similarity loss. As shown in Supplementary Fig. 18a, our results are stable with respect to the choice of this parameter. Similar to other methods based on neural networks, the number of hidden nodes in the architecture and other optimization details can be considered tunable as well, although they do not seem to affect our results (Supplementary Fig. 18b).
The superior performance and robustness of scJoint illustrate its utility as a tool to automatically label cells from other modalities given an annotated scRNA-seq database. By embedding all cells in a common lower dimensional space, scJoint assigns a probability score to a cell-type prediction by combining the softmax probabilities of its nearest neighbors. As we vary the amount of cutoff, the accuracy of scJoint still consistently outperforms the other methods (Supplementary Fig. 19). The robustness of scJoint was demonstrated through subsampling experiments, where the stability of our results implies the method can be applied to partially labeled databases. Despite being a semisupervised method guided by labeled data, the dimension reduction component in our design lends it sufficient flexibility to preserve implicit data signals, including biological variations induced by experimental conditions and other cellular subtypes. One can conceivably extend scJoint to an unsupervised setting, replacing the softmax prediction layer with a decoder minimizing reconstruction loss.
Although designed for unpaired data, scJoint is still directly applicable to paired data and generates joint visualizations with cells coherently grouped by cell types. In the current training scheme, the pairing information between RNA and ATAC is used only to validate the label transfer results. We expect that adapting scJoint to take paired vectors during training would enhance its performance on this type of data, and this would be especially useful in the unsupervised setting mentioned above.
We have focused on scATAC-seq as an example of epigenomic data but, in principle, scJoint extends to other modalities such as methylation data, provided the input can be summarized as gene-level scores. While the gene-level scores are amenable to generalization and widely adopted by unpaired integration methods, this step itself is also a limitation as improper aggregation can incur loss of information important for identifying subtle cell states present in the ATAC data. Extending scJoint to handle epigenomic data directly at locus level will require designing a separate encoder that is suitable for the high dimensionality and remains easy to train. With the increasing availability of paired scRNA-seq and scATAC-seq data and other single-cell multimodal technologies, methods such as BABEL41, which trains on paired data to impute gene-level signals from scATAC-seq data, can potentially be used to provide more comprehensive gene-level features than gene activity scores alone. We will pursue this for future work.
In summary, we have developed scJoint as a generalizable transfer learning method for performing integrative analysis of atlas-scale single-cell multiomics data. scJoint was shown to effectively integrate several types of measurement from both unpaired or paired profiling, outperforming other methods in label transfer accuracy and providing joint visualizations that remove technical variations while preserving meaningful biological signals. scJoint’s ability to integrate multiomics data by capturing various aspects of cell characteristics unique to different data modalities will facilitate a more comprehensive view of cell functions and cell communications.
online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41587-021-01161-6.
Methods
Architecture and training of scJoint.
The neural network in scJoint consists of one input layer and two fully connected layers. The input layer has dimension equal to the number of genes common to the expression matrix of scRNA-seq and the gene activity matrix of scATAC-seq, after simple filtering (Data preprocessing). Now that the two modalities have matching input features, we cotrain them using the same encoder, which is equivalent to weight sharing. The first fully connected layer has 64 neurons with linear activation and serves as the joint low-dimensional embedding space that captures aligned features from all cells. Visualizations of clustering structure can be obtained by applying tSNE or UMAP to the output of the embedding layer. The second fully connected layer has dimension equal to the number of cell types in scRNA-seq data. Through a softmax transformation, this layer outputs a probability vector for cell-type prediction. For cells in scRNA-seq, this layer can be trained in a supervised fashion using the cross entropy loss.
Given S scRNA-seq experiments with expression matrices and T scATAC-seq experiments with gene activity score matrices, with S and T representing the number of different batches whose technical variations need to be removed. Assume suitable intersections have been taken so that all matrices have the same set of genes. Let be the expression profiles of cells after preprocessing from a scRNA-seq dataset indexed by s ∈ {1, …, S}, and be the corresponding cell-type annotations. Here each is a G-dimensional vector, where G is the number of genes; , where K is the number of cell types; Ns is the number of cells in experiment s. Similarly, let be the vectors of gene activity scores after preprocessing from the t-th scATAC-seq dataset with Nt cells (t ∈ {1, …, T}), whose cell types are unlabeled. The neural network is parametrized by a set of weights and biases, collectively denoted θ. Let , D = 64, be the output of the embedding layer when the input has gone through a transformation of f parametrized by θ. Similarly , where h denotes the output from the prediction layer that goes through the softmax transformation. Thus is a probability vector after the softmax transformation. and are defined in the same way for input from scATAC-seq.
The training of scJoint consists of three steps.
Step 1: Joint NNDR and semisupervised transfer learning.
We first perform joint dimension reduction and feature alignment by imposing suitable loss functions on the outputs of the two fully connected layers. A minibatch of data for training is constructed by sampling equal-sized subsets of cells from each dataset, that is, , where each subset (or ) has B cells.
- NNDR Loss. In a spirit similar to principal component analysis (PCA), the NNDR loss aims to capture low-dimensional, orthogonal features when projecting each data batch into the embedding space. For now we omit the dataset-specific superscript, with the understanding that this loss function is applied to each and . Given input vectors , define , and as the sample correlation matrix. The NNDR loss is:
Note that, to minimize this loss, we maximize the variability within each coordinate (inverse of the first term) and minimize the correlation between all coordinate pairs (the second term) to achieve orthogonality. The last term tries to fix the means of all coordinates near zero for model identifiability, preventing θ from drifting to unstable regions of the parameter space. - Cosine similarity loss. This loss is applied to the embedding layer outputs from and , for every t, and attempts to maximize the similarity between best aligned ATAC and RNA data pairs. Let p be the fraction of data pairs we expect to have high cosine similarity scores. Setting p < 1 accounts for situations where RNA and ATAC do not share all their cell types. We set p = 0.8 for all the results presented in the paper, and our results seem to be stable with respect to this parameter (Supplementary Fig. 18a) when the cell types fully overlap. Recall that for a pair of general vectors (u, v), the cosine similarity is defined as cos(u, v) = ⟨u, v⟩/(∥u∥∥v∥). For each with , we find the corresponding with input xi(b) that maximizes . From , we then choose the top p fraction of cells with the highest cosine score and denote the index set . ( has size ⌊Bp⌋) The loss is given by
-
Cross entropy loss. For every with cell-type annotations , we apply the cross entropy loss to the prediction layer after softmax transformation to supervise the learning of scRNA-seq datasets:
where 1(·) is an indicator function.In Step 1, the final loss function we minimize with respect to θ for a minibatch is
Step 2: Cell-type label transfer by KNN in joint embedding space.
The output of Step 1 is a joint embedding space that has aligned RNA and ATAC roughly with cells from either modality lying close if they have similar low-dimensional representations in this space. Therefore, using the embedding vectors for cells in all the datasets and calculating the Euclidean distances, we can determine the KNN among all RNA cells for each cell i in ATAC; denote this set of RNA cells . The cell-type label of i is estimated via majority vote using . All results in this paper were obtained from using 30 nearest neighbors. Let the majority cell type be k*, then the probability score of cell-type prediction for cell i in ATAC is an average of its nearest neighbors in RNA. Since for each is already a probability vector after the softmax transformation, we take pθ,j = gθ,j(k*) as the probability score of RNA cell j in the majority class . For other , we threshold the probability score as 0. The probability score of ATAC cell i is then calculated as
Step 3: Joint training with transferred cell-type labels (for well-differentiated cell types).
In the final step of the training, we refine the joint embedding space and improve mixing of cells from the same cell type using the transferred labels from Step 2. We include an further loss function used commonly in metric learning for enhancing embedded clustering structure given labeled data. The other loss functions and network architecture remain the same as Step 1, with ATAC cells and their transferred labels added to .
For each cell type k ∈ {1, …, K}, we initialize the class center randomly. We construct minibatches of cells from all the datasets in the same way as in Step 1. Now that all cells have cell-type labels (given or transferred), for convenience we will refer to cells in a minibatch without explicitly labeling which dataset they come from. For a given , we first revise the class centers by taking the average of ck and {fθ,b} with and yb = k. Let the revised centers be . As the number of minibatches grows, the influence of the initial ck becomes negligible. The metric learning loss we use is the center loss:
The total loss function we minimize in Step 3 is given by
where λ is a parameter adjusting the weight of the center loss; a larger weight encourages stronger mixing among cells from the same cell type.
We perform a final round of majority vote by KNN using distances in the embedding space. If the prediction of any ATAC cell is different from Step 2, we revise both its prediction and probability score in the same way as Step 2. Before visualization with tSNE, all embedding vectors are normalized using L2 norm.
Step 3′: Joint training with transferred cell-type labels (for developmental data).
For developmental data, the underlying cell states are more continuous and less well separated. Since the cross entropy loss is a classification loss that enforces separation between cell types, we remove it from in the overall loss when training Step 3 to enable continuous visualizations of trajectory data. Step 1 and Step 2 remain unchanged.
More detailed explanations on the role of each loss function component can be found in the Supplementary Note.
Training details.
The batch size B was set to 256 in all cases. The other training details, including learning rate and number of training epochs used in each dataset, can be found in Supplementary Tables 1 and 2. The weight λ was set to 1 for all data except the mouse atlas, which contains two scRNA-seq datasets sequenced with different technologies. In this case, λ was set to 10 to enhance mixing in the presence of batch effect; the results were very stable for any choice of λ between 10 and 40 (see Supplementary Note for details). We started all the training with the learning rate set to 0.01, since a large learning rate has the benefit of faster training. However, if the values of the loss functions were observed to have too much fluctuation, we decreased the learning rate to 0.001 for more stable training.
Data preprocessing.
Mouse atlas data. The processed gene expression matrix and the cell-type annotation of the Tabula Muris mouse data of scRNA-seq were downloaded from https://tabula-muris.ds.czbiohub.org/, which has 41,965 cells from protocol FACS and 54,439 cells from microfluidic droplets (droplet). The quantitative gene activity score matrix and the cell-type annotation of the mouse sci-ATAC-seq atlas were downloaded from https://atlas.gs.washington.edu/mouse-atac/, including 81,173 cells in total. The number of common genes between two modalities is 15,519. The labels are obtained from the original paper26, which first performed Louvain clustering on the tSNE space using the first 50 dimensions of the singular value decomposition of the TF-IDF transformed peak matrix. The same procedure was repeated within each cluster to obtain finer clusters, resulting in 85 clusters for cluster annotation. Each cluster was then assigned to a cell type by intersecting the differential gene activities with a collected set of cell-type markers. The annotations were further refined through classification and manual review, resulting in 29 cell types. We checked the cell-type annotations from both scRNA-seq and sci-ATAC-seq studies manually and reannotated the labels such that the naming convention is consistent across the datasets. For example, the cell type ‘Cardiac muscle cell’ in the sci-ATAC-seq dataset was changed to ‘Cardiomyocytes’. We also combined some of the cellular subtypes in the sci-ATAC-seq data to increase the percentage of overlapping labels between two atlases for evaluation. More specifically, we combined ‘Regulatory T cell’ and ‘T cell’ into ‘T cell’; ‘Immature B cell’, ‘Activated B cell’ and ‘B cell’ into ‘B cell’ and ‘Excitatory neurons’ and ‘Inhibitory neurons’ into ‘Neuron’.
Human fetal atlas data. The scRNA-seq data of the human fetal atlas data was downloaded from the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) accession number GSE156793, including both raw gene expression and cell-type information27. The scATAC-seq data was downloaded from GSE149683, and the gene activity matrices were extracted from the Seurat objects provided28. There are 54 cell types in common between the two human fetal atlases. In our computational benchmarking analysis, we included only cells from the common cell types, resulting in a total of 656,074 cells from the scATAC-seq data. To construct a balanced scRNA-seq training set, for cell type i with number of cells ni > 10,000, we subsampled max{0.05ni, 10, 000} cells; all cells were included for cell types with fewer than 10,000 cells. This resulted in 433,695 cells from the scRNA-seq data.
SNARE-seq data. The SNARE-seq data from adult mouse cerebral cortex was downloaded from MCBI GEO accession number GSE126074 (ref. 14), with both raw gene expression and DNA accessibility measurements available for the same cell. The fastq files were downloaded from the Sequence Read Archive for SRP183521. We first derived the fragment files from the fastq files using sinto fragments (sinto v.0.7.2), and then generated the gene activity matrix using Signac (v.1.1.0.9000)32. The cell-type information was obtained from the original study14. We filtered out the cells that were originally labeled as ‘Misc’ (cells of miscellaneous cluster), resulting in a dataset with 9,190 cells and 15,725 genes for the integrative analysis.
Multimodal data (CITE-seq and ASAP-seq PBMC data). The ASAP-seq and CITE-seq data were downloaded from GEO accession number GSE156478 (ref. 34), which included the fragment files and antibody-derived tags (ADTs) matrices for ASAP-seq, the raw unique molecular identifier (UMI) and ADT matrices for CITE-seq, from both control and stimulated conditions. The gene activity matrices for ASAP-seq were generated by Signac. Most of the thresholds we used for quality control metrics were consistent with those in the original paper34. The control and stimulated CITE-seq were filtered based on the following criteria: mitochondrial reads greater than 10%; number of expressed genes fewer than 500; total number of UMI fewer than 1,000; total number of ADTs from the rat isotype control greater than 55 and 65 in the control and stimulated conditions, respectively; total number of UMI greater than 12,000 and 20,000 for the control and stimulated conditions, respectively and total number of ADTs fewer than 10,000 and 30,000 for control and stimulated conditions, respectively. We further filtered out cells that were classified as doublets in the original study. For the ASAP-seq data, we filtered out cells with a number of ADTs more than 10,000 and number of peaks more than 100,000. Finally, 4,502 cells (control) and 5,468 cells (stimulated) from ASAP-seq, 4,644 cells (control) and 3,474 cells (stimulated) from CITE-seq were included in the downstream analysis. The number of common genes across the four matrices is 17,441 and the number of common ADTs is 227. We used CiteFuse to integrate the peak matrix or gene expression matrix with their corresponding protein expression and obtain clustering for ASAP-seq and CITE-seq in each condition separately35. For ASAP-seq, the similarity matrices of the chromatin accessibility are calculated by applying the Pearson correlation to the TF-IDF transformation of the peak matrix. We then followed the procedure described in Maecker et al.42 to annotate the clusters.
Human hematopoiesis data. The gene expression, peak matrix and clustering results of human hematopoiesis data from healthy donors were downloaded from https://github.com/GreenleafLab/MPAL-Single-Cell-2019 (ref. 40). The gene activity matrices were generated by Signac. We excluded cells labeled as ‘Unknown’ and combined the clusters with the same cell-type annotation into one label (for example, ‘CLP.1’ and ‘CLP.2’ as ‘CLP’), resulting in 35,038 cells for scRNA-seq data and 35,582 cells for scATAC-seq data for the analysis.
For scJoint, all the gene expression matrices and gene activity score matrices were binarized as 0 or 1, with 1 representing any nonzero original values, as the final input for training. Binarization scales the two modalities so that their distributions have the same range and reduces the amount of noise in the data for easier cotraining.
Recent studies have also illustrated that dropout patterns represented by the binarization in single-cell RNA-seq data are biologically meaningful for cell-type clustering and cell-level analyses43,44. Consistent with this, we also find that binarization leads to optimal performance of scJoint in label transfer, and scJoint is robust to how the binary matrix is constructed (more details in Supplementary Note).
Settings used in other methods.
For the unpaired data (mouse cell atlases and multimodal data from CITE-seq and ASAP-seq), we benchmarked the performance of scJoint against three other methods designed for integrating unpaired single-cell multimodal data: Seurat (v.3), Conos and Liger. We compared the label transfer accuracy with Seurat and Conos and the joint visualizations with all three methods. For the paired data (SNARE-seq), we further compared joint visualizations with two methods designed specifically for paired data, scAI and MOFA+. For all the unpaired methods, we used gene activity matrices derived from the above data preprocessing step as input for scATAC-seq. For the two paired methods, we used the peak matrices of scATAC-seq data as input. Detailed settings used in each method are as follows.
-
Seurat. R package Seurat v.3.2.0 (ref. 24) was used for all the datasets. The raw count matrix of scRNA-seq and unnormalized gene activity score matrix of scATAC-seq were used as input, which were then normalized using the NormalizeData function in Seurat. Noted that for the CITE-seq and ASAP-seq data, the input was a concatenated matrix of log-transformed normalized gene expression data/gene activity score matrix and log-transformed ADTs matrix. The top 2,000 most variable genes were selected from scRNA-seq using FindVariableFeatures with vst as method. To identify the anchors between scRNA-seq and scATAC-seq data, the FindTransferAnchors function was used with ‘cca’ as reduction method. The scATAC-seq data was then imputed using TransferAnchors function, where the anchors were weighted by latent semantic indexing reduced dimension of scATAC-seq. PCA was then performed on the merged matrix of scRNA-seq data and imputed scATAC-seq data. For all the datasets, 30 principal components (PCs) were used for joint visualization with tSNE (function RunTSNE).
For the mouse cell atlas data, we first integrated the two scRNA-seq datasets (FACS and droplet) using FindIntegrationAnchors and IntegrateData, and then the integrated matrix was scaled using ScaleData and used as reference to find transfer anchors.
Conos. R package conos v.1.3.1 (ref. 23) was used for all the datasets. We used function basicP2proc in pagoda2 package (v.0.1.2) to process the raw count matrix of scRNA-seq and unnormalized gene activity score matrix of scATAC-seq. The joint graph was built using buildGraph with k=15, k.self=5 and k.self.weigh=0.01, which were set as suggested in the tutorial for integrating RNA and ATAC (http://pklab.med.harvard.edu/peterk/conos/atac_rna/example.html). The joint visualization of scRNA-seq and scATAC-seq were generated using largeVis by embedGraph, which is the default visualization in Conos.
Liger. R package liger v.0.5.0 (ref. 21) was used for the datasets. The raw count matrix of scRNA-seq and unnormalized gene activity score matrix of scATAC-seq were used as input, which were normalized using normalize function in liger. Highly variable genes were selected using the scRNA-seq. For the mouse cell atlas data, both FACS and droplet scRNA-seq data were used to select features. For all the datasets, number of factors was set to 20 in optimizeALS. tSNE was then performed on the normalized cell factors to generate the joint visualization of scRNA-seq and scATAC-seq (function runTSNE in liger).
scAI. R package scAI v.1.0.0 (ref.16) was used for the integration of SNARE-seq data. The raw count matrix of scRNA-seq and raw peak matrix of scATAC-seq were used as input. We ran scAI using run_scAI by setting the rank of the inferred factor set as 20, do.fast = TRUE, and nrun = 1, with other parameters set as default, as suggested in the pipeline in the github repository. tSNE plots were generated using reducedDims function in scAI.
MOFA+. R package MOFA2 v.1.0 (ref. 17) was used for the integration of SNARE-seq data. Following the suggested integration tutorial for SNARE-seq in the github repository, we first selected top 2,500 most variable genes using FindVariableFeatures in Seurat package with vst as method, and the top 5,000 most variable ATAC peaks with disp as method. By subsetting the counts matrix of scRNA-seq and peak matrix of scATAC-seq with the selected features, we ran MOFA+ by setting the number of factors as ten, with other parameters set as default. tSNE plots were generated using run_tsne function in MOFA2.
Seurat (WNN). R package Seurat v.4.0.2 (ref. 38) was used for the integration of SNARE-seq data. Following the tutorial in their GitHub repository, the two modalities were integrated using FindMultiModalNeighbors, where the anchors were weighted by the first 50 components of latent semantic indexing reduced dimension of scATAC-seq (with the first dimension excluded) and 50 top PCs of scRNA-seq.
Evaluation metrics.
Joint embedding evaluation—silhouette coefficients.
To evaluate whether the joint embeddings from different methods show clustering structure reflecting biological signals or technical variations, we calculated the silhouette coefficient for each cell by considering two different groupings: (1) grouping based on the modalities (scRNA-seq or scATAC-seq), called the modality silhouette coefficient (smodality); (2) grouping based on known cell types, called the cell-type silhouette coefficient (scellTypes). Note that, for the atlas data, we consider FACS and droplet in scRNA-seq as two distinct technologies and the modality silhouette coefficient has three groups (FACS, droplet, ATAC) in the calculation. For SNARE-seq, the paired methods (scAI and MOFA+) have no modality silhouette coefficients since each cell has one paired profile of RNA and ATAC. An ideal joint visualization should have low modality silhouette coefficients, suggesting removal of the technical effect, and large cell-type silhouette coefficients, indicating that the cells are grouped by cell type. The Euclidean distance for all methods except Conos is obtained from the tSNE embedding. For Conos, the distance is obtained from the largeVis embedding, which is the method’s default output.
We then summarize the two silhouette coefficients by calculating an F1 score as follows:
where s′ = (s + 1)/2. A higher F1 score indicates better performance in the alignment of the modalities as well as the preservation of biological signals.
Accuracy evaluation of transferred labels.
We evaluated the accuracy of label transfer from two aspects: (1) overall accuracy rate; (2) cell-type classification F1 score. The overall accuracy rate was computed accounting only for the common cell types between scRNA-seq and scATAC-seq data. The cell-type classification F1 score is the harmonic mean of precision and recall of each cell type.
Running time evaluation.
We evaluated running time using one core and one graphics processing unit on a research server with dual Intel (R) Xeon(R) Gold 6148 Processor (40 total cores, 768 GB total memory) and dual RTX2080TI graphics processing units. Using the preprocessed human fetal atlas data, we created benchmarking datasets with 5,000, 10,000, 25,000, 50,000, 75,000, 100,000, 125,000 and 250,000 cells from scRNA-seq and scATAC-seq data, respectively. We further ran scJoint on the whole preprocessed data with 433,695 cells from scRNA-seq and 656,074 cells from scATAC-seq. In this case, the other three methods failed to run due to an out-of-memory error. For each method, we measured total running time as the running time of feature selection, label transfer and joint embedding construction of scATAC-seq and scRNA-seq. The training details for scJoint are listed in Supplementary Table 2. Following common practice in neural-network training, we increased the batch size as the number of training datapoints increased.
Reporting Summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
All single-cell datasets used in this paper are publicly available. • Mouse atlas data. The scRNA-seq dataset was downloaded from Tabula Muris5 (https://tabula-muris.ds.czbiohub.org/). The sci-ATAC-seq dataset of Cusanovich et al.26 was downloaded from https://atlas.gs.washington.edu/mouse-atac/. • Human fetal atlas data. The scRNA-seq dataset from Cao et al.27 was downloaded from GSE156793. The scATAC-seq dataset from Domcke et al.28 was downloaded from GSE149683. • SNARE-seq data. The SNARE-seq dataset of adult mouse cerebral cortex14 was downloaded from GSE126074. • Multimodal PBMC data. The ASAP-seq and CITE-seq datasets from Mimitou et al.34 were obtained from GSE156478. • Human hematopoiesis data. The scRNA-seq and scATAC-seq datasets from Granja et al.40 were downloaded from https://github.com/GreenleafLab/MPAL-Single-Cell-2019.
Code availability
scJoint was implemented using PyTorch (v.1.0.0) with code available at https://github.com/SydneyBioX/scJoint.
Supplementary Material
Acknowledgements
We gratefully acknowledge the following funding sources: Research Training Program Tuition Fee Offset and Stipend Scholarship and Chen Family Research Scholarship to Y.L.; Australian Research Council Discovery Project grant (DP170100654) and AIR@ innoHK program of the Innovation and Technology Commission of Hong Kong to J.Y.H.Y.; Australian Research Council DECRA Fellowship (DE180101252) to Y.X.R.W; NIH grants R01 HG010359 and P50 HG007735 to W.H.W.
Footnotes
Competing interests
The authors declare no competing interests.
Additional information
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41587-021-01161-6.
Peer review information Nature Biotechnology thanks Jingshu Wang, Nancy Zhang and Qing Nie for their contribution to the peer review of this work.
Reprints and permissions information is available at www.nature.com/reprints.
References
- 1.Stuart T & Satija R Integrative single-cell analysis. Nat. Rev. Genet 20, 257–272 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Berger SL The complex language of chromatin regulation during transcription. Nature 447, 407–412 (2007). [DOI] [PubMed] [Google Scholar]
- 3.Klemm SL, Shipony Z & Greenleaf WJ Chromatin accessibility and the regulatory epigenome. Nat. Rev. Genet 20, 207–220 (2019). [DOI] [PubMed] [Google Scholar]
- 4.Pott S & Lieb JD Single-cell atac-seq: strength in numbers. Genome Biol. 16, 172 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schaum N et al. Single-cell transcriptomics of 20 mouse organs creates a tabula muris: the tabula muris consortium. Nature 562, 367 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Regev A et al. Science forum: the human cell atlas. eLife 6, e27041 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lopez R, Regier J, Cole MB, Jordan MI & Yosef N Deep generative modeling for single-cell transcriptomics. Nat. Methods 15, 1053–1058 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang J et al. Data denoising with transfer learning in single-cell transcriptomics. Nat. Methods 16, 875–878 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lin Y et al. scMerge leverages factor analysis, stable expression, and pseudoreplication to merge multiple single-cell RNA-seq datasets. Proc. Natl Acad. Sci. USA 116, 9775–9784 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Korsunsky I et al. Fast, sensitive and accurate integration of single-cell data with harmony. Nat. Methods 16, 1289–1296 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang T et al. Bermuda: a novel deep transfer learning method for single-cell RNA sequencing batch correction reveals hidden high-resolution cellular subtypes. Genome Biol. 20, 165 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Amodio M et al. Exploring single-cell data with deep multitasking neural networks. Nat. Methods 16, 1139–1145 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xiong L et al. Scale method for single-cell atac-seq analysis via latent feature extraction. Nat. Commun 10, 4576 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen S, Lake BB & Zhang K High-throughput sequencing of the transcriptome and chromatin accessibility in the same cell. Nat. Biotechnol 37, 1452–1457 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cao J et al. Joint profiling of chromatin accessibility and gene expression in thousands of single cells. Science 361, 1380–1385 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jin S, Zhang L & Nie Q scAI: an unsupervised approach for the integrative analysis of parallel single-cell transcriptomic and epigenomic profiles. Genome Biol. 21, 25 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Argelaguet R et al. MOFA+: a statistical framework for comprehensive integration of multi-modal single-cell data. Genome Biol. 21, 111 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Welch JD, Hartemink AJ & Prins JF MATCHER: manifold alignment reveals correspondence between single cell transcriptome and epigenome dynamics. Genome Biol. 18, 138 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Amodio M & and Krishnaswamy S MAGAN: aligning biological manifolds. In Proc. 35th International Conference on Machine Learning (eds. Dy J & Krause A) 215–223 (PMLR, 2018). [Google Scholar]
- 20.Liu J, Huang Y, Vert J-P & Noble WS Jointly embedding multiple single-cell omics measurements. Algorithms Bioinform. 143, 10 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Welch JD et al. Single-cell multi-omic integration compares and contrasts features of brain cell identity. Cell 177, 1873–1887 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Duren Z et al. Integrative analysis of single-cell genomics data by coupled nonnegative matrix factorizations. Proc. Natl Acad. Sci. USA 115, 7723–7728 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barkas N et al. Joint analysis of heterogeneous single-cell RNA-seq dataset collections. Nat. Methods 16, 695–698 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stuart T et al. Comprehensive integration of single-cell data. Cell 177, 1888–1902 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.DaiYang K et al. Multi-domain translation between single-cell imaging and sequencing data using autoencoders. Nat. Commun 12, 31 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cusanovich DA et al. A single-cell atlas of in vivo mammalian chromatin accessibility. Cell 174, 1309–1324 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cao J A human cell atlas of fetal gene expression. Science 370, eaba7721 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Domcke S A human cell atlas of fetal chromatin accessibility. Science 370, eaba7612 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van der Maaten L & Hinton G Visualizing data using t-SNE. J. Machine Learning Res 9, 2579–2605 (2008). [Google Scholar]
- 30.McInnes L, Healy J & Melville J UMAP: Uniform manifold approximation and projection for dimension reduction. Preprint at arXiv https://arxiv.org/abs/1802.03426 (2018). [Google Scholar]
- 31.Pliner HA et al. Cicero predicts cis-regulatory DNA interactions from single-cell chromatin accessibility data. Mol. Cell 71, 858–871 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stuart T, Srivastava A, Madad S, Lareau CA & Satija R Single-cell chromatin state analysis with Signac. Nat. Methods 18, 1333–1341 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stoeckius M et al. Simultaneous epitope and transcriptome measurement in single cells. Nat. Methods 14, 865 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mimitou EP et al. Scalable, multimodal profiling of chromatin accessibility, gene expression and protein levels in single cells. Nat. Biotechnol 39, 1246–1258 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim HJ, Lin Y, Geddes TA, Yang JYH & Yang P CiteFuse enables multi-modal analysis of CITE-seq data. Bioinformatics 36, 4137–4143 (2020). [DOI] [PubMed] [Google Scholar]
- 36.Godfrey DI, MacDonald HR, Kronenberg M, Smyth MJ & Van Kaer L NKT cells: what’s in a name? Nat. Rev. Immunol 4, 231–237 (2004). [DOI] [PubMed] [Google Scholar]
- 37.Finak G et al. MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol. 16, 278 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hao Y Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Abdelaal T et al. A comparison of automatic cell identification methods for single-cell RNA sequencing data. Genome Bol. 20, 194 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Granja JM et al. Single-cell multiomic analysis identifies regulatory programs in mixed-phenotype acute leukemia. Nat. Biotechnol 37, 1458–1465 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu KE, Yost KE, Chang HY & Zou J Babel enables cross-modality translation between multiomic profiles at single-cell resolution. Proc. Natl Acad. Sci. USA 118, e2023070118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maecker HT, McCoy JP & Nussenblatt R Standardizing immunophenotyping for the human immunology project. Nat. Rev. Immunol 12, 191–200 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Qiu P Embracing the dropouts in single-cell RNA-seq analysis. Nat. Commun 11, 1169 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jiang R, Sun T, Song D & Li JJ Zeros in scRNA-seq data: good or bad? how to embrace or tackle zeros in scRNA-seq data analysis? Preprint at bioRxiv (2020). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All single-cell datasets used in this paper are publicly available. • Mouse atlas data. The scRNA-seq dataset was downloaded from Tabula Muris5 (https://tabula-muris.ds.czbiohub.org/). The sci-ATAC-seq dataset of Cusanovich et al.26 was downloaded from https://atlas.gs.washington.edu/mouse-atac/. • Human fetal atlas data. The scRNA-seq dataset from Cao et al.27 was downloaded from GSE156793. The scATAC-seq dataset from Domcke et al.28 was downloaded from GSE149683. • SNARE-seq data. The SNARE-seq dataset of adult mouse cerebral cortex14 was downloaded from GSE126074. • Multimodal PBMC data. The ASAP-seq and CITE-seq datasets from Mimitou et al.34 were obtained from GSE156478. • Human hematopoiesis data. The scRNA-seq and scATAC-seq datasets from Granja et al.40 were downloaded from https://github.com/GreenleafLab/MPAL-Single-Cell-2019.