

Abstract
Skin cancer is the most diagnosed type of cancer in the United States, and while most of these malignancies are highly treatable, treatment costs still exceed $8 billion annually. Over the last 50 years, the annual incidence of skin cancer has steadily grown; therefore, understanding the environmental factors driving these types of cancer is a prominent research-focus. A causality between ultraviolet radiation (UVR) exposure and skin cancer is well-established, but exposure to UVR alone is not necessarily sufficient to induce carcinogenesis. The emerging field of circadian biology intersects strongly with the physiological systems of the mammalian body and introduces a unique opportunity for analyzing mechanisms of homeostatic disruption. The circadian clock refers to the approximate 24-hour cycle, in which protein levels of specific clock-controlled genes (CCGs) fluctuate based on the time of day. Though these CCGs are tissue specific, the skin has been observed to have a robust circadian clock that plays a role in its response to UVR exposure. This in-depth review will detail the mechanisms of the circadian clock and its role in cellular homeostasis. Next, the skin’s response to UVR exposure and its induction of DNA damage and mutations will be covered—with an additional focus placed on how the circadian clock influences this response through nucleotide excision repair. Lastly, this review will discuss current models for studying UVR-induced skin lesions and perturbations of the circadian clock, as well as the impact of these factors on human health.
Keywords: Circadian clock, circadian rhythms, skin clock, DNA damage, ultraviolet radiation, nucleotide excision repair, mutagenesis, skin cancer, carcinogenesis
1. Introduction
In a similar manner to how humans have a basic schedule of responsibilities throughout the course of each day, the cells in most organisms have a pre-wired schedule of biological events that occur based upon the reception of environmental cues. This pre-wired schedule is commonly referred to as the circadian rhythm or circadian clock. The term “circadian” is Latin in origin and translates to “around a day.” The circadian clock is driven by environmental entrainment factors called zeitgebers that fluctuate based upon the rotation of the Earth. This creates an approximately 24-hour cycle within an organism that drives cellular processes ranging from chromatin state and protein expression to metabolic and behavioral patterns [1]. It has been postulated that this 24-hour rhythm began with the early metazoans and may have evolved independently in each of the four major kingdoms [2,3]. These findings suggest that circadian fluctuation, on both a cellular and organismal level, is an advantageous trait related to organismal survival [2].
As many biological processes are tied to the rhythms of molecular clocks throughout the body, healthy circadian oscillation is crucial for maintaining homeostasis. Conflicting external cues can offset the endogenous molecular oscillation of core clock proteins in relation to day/night cycles of the Earth, which upsets homeostatic balance and leads to circadian disruption [2]. For instance, people who engage in chronic night shift work and invert their sleep schedules to daytime often experience fatigue, impaired cognitive function, and increased susceptibility to various pathologies (such as cancer) [4]. The exact mechanism behind this phenomenon, however, is unknown [5,6]. Additionally, jet lag induced by rapidly switching time zones can desynchronize the circadian clock and cause similar symptoms [7]. Maintaining healthy circadian oscillations through exposure to environmental cues at the correct time of day helps to preserve homeostatic biological processes that are critical to responding to damaging agents and maintaining genomic integrity [8,9].
Light, both visible and ultraviolet (UV), is highly interconnected with the circadian rhythm. It is the key zeitgeber and likely one of the main environmental factors that drove evolutionary development of circadian fluctuations. UV light has three subtypes: UVA (320–400 nm), UVB (280–320 nm) and UVC (100–280 nm). These subtypes are differentiated based upon the wavelength at which they travel, with UVA having the longest wavelength and UVC the shortest. The ozone layer conveniently guards life on Earth from the most damaging wavelengths of UV radiation (UVR), as all of UVC and most of UVB light is prevented from reaching the terrestrial surface. The UV light that reaches the Earth is comprised of approximately 95% UVA and 5% UVB [10]. UV light typically causes DNA damage in the form of either cyclobutane pyrimidine dimers (CPDs) and (6–4) pyrimidine-pyrimidone photoproducts (6–4 PPs) [11,12]. Unrepaired or improper repair of the DNA damage caused by UVR can lead to mutations in the DNA sequence and subsequent consequences for overall human health— especially in concert with circadian disruption [8].
This review will focus on the circadian clock in relation to UVR exposure and UVR-induced DNA damage and mutations. First, an in-depth overview of the circadian rhythm will be covered—topics like exposure to zeitgebers and inner workings of the core molecular clock. From there, the role of the circadian clock in relation to the cell cycle will be discussed. Next, the review will detail UV light and skin cellular responses to UVR exposure. The role of the circadian clock in both UVR-induced damage and nucleotide excision repair (NER) will be presented extensively here. Finally, this review will touch on the most frequently used model systems for studying UVR and the circadian rhythm, as well as known impacts of the two factors on human health.
2. The Molecular Circadian Clock
The molecular circadian time-keeping system centers around a series of self-perpetuating transcription-translation feedback loops (TTFLs) that anticipate the arrival of day and night. The core mechanism relies on two basic helix-loop-helix (bHLH)/PAS-containing transcription factors, Brain-and-Muscle-ARNT-like 1 (BMAL1) and Circadian Locomotor Output Cycles Kaput (CLOCK), which heterodimerize to drive rhythmic transcription of a number of clock-controlled genes (CCGs). Three Period (PER1–3) and two Cryptochrome (CRY1–2) genes are examples of CCGs that are imperative to the proper timing of the circadian rhythm [13–17]. Per and Cry mRNAs are translated to proteins in the cytoplasm, after which they heterodimerize and interact with casein kinase 1δ (CK1δ) and CK1ε, before translocating back into the nucleus for direct repressive contact with BMAL1 or CLOCK to close the primary negative feedback loop [18–21]. This core rhythmic interaction is further reinforced by CLOCK-BMAL1 activation of nuclear receptors REV-ERBɑ (NR1D1) and REV-ERBβ (NR1D2), which repress BMAL1 transcription through competitive binding against transcriptional activators RORɑ and RORγ to the ROR-response element in its promoter (Figure 1) [22,23].
Figure 1: From Environmental to Molecular Cues: An Overview of the Circadian Clock Pathway.
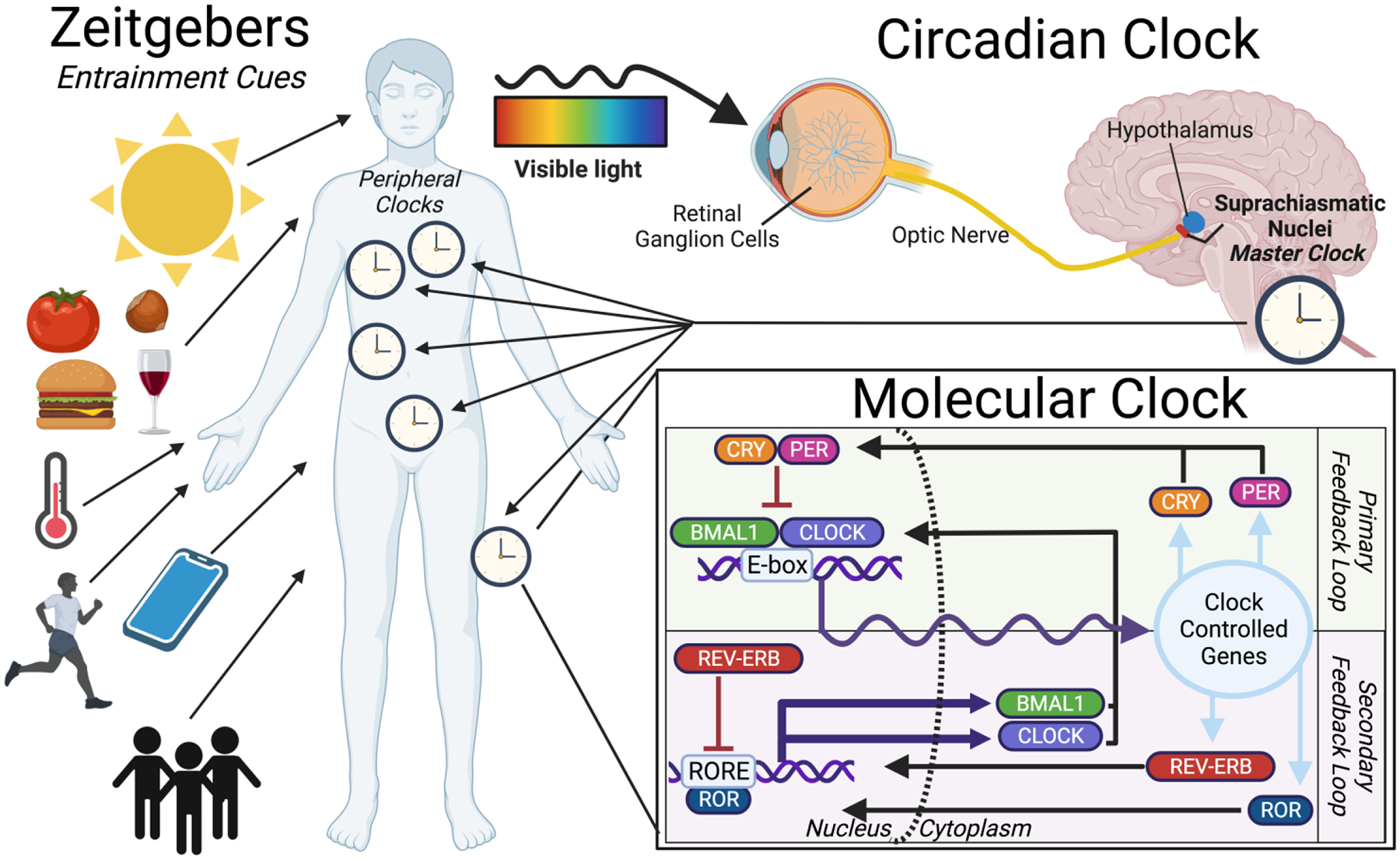
The clock begins with environmental cues (Zeitgebers) such as light, food, temperature, exercise, electronics, or social interactions. These signals program/align the “master clock” found in the SCN, which then signals the peripheral body clocks. The molecular clock pathway is shown in the inset, with the primary and secondary feedback loops. The gene-protein interactions create an oscillating rhythm of clock-controlled gene expression based on the time of day. A subset of these controlled genes then reintegrates into the feedback system.
In nocturnal animals such as mice, CLOCK-BMAL1 heterodimer activation and Per/Cry mRNA accumulation occurs during the subjective day, followed by translation and translocation of PER-CRY protein into the nucleus at night to facilitate an endogenous oscillation program of approximately 24 hours [20]. This program is reversed in humans, with peak CLOCK-BMAL1 expression and function during the subjective night and PER-CRY translocation into the nucleus during early morning hours and daytime [24]. However, despite opposite behavioral niches, the phase reversal anticipated between diurnal and nocturnal species is not a universal property and should be evaluated on a tissue-specific basis [25]. For instance, in one study that examined diurnal and nocturnal rodents, while a similar peak phase of PER2 expression in the suprachiasmatic nuclei (SCN) was observed between the two species, other brain regions saw a 180° anti-phase difference. This ultimately suggests that behavioral patterns do not necessarily define the molecular clock and should be assessed with caution [26].
The coordination and integrity of circadian rhythmic oscillations hinge upon synchronization with an individual’s environment and daily routine. From an evolutionary perspective, coherence between external factors and internal physiology/behavior optimizes an organism’s adaptation to periodic phenomena and increases overall fitness [27]. Circadian rhythms are entrained to environmental time cues or “zeitgebers”, such as seasonal light-dark cycles [28], temperature [29], exercise [30], and even food availability [31]. Each organism that relies on circadian entrainment has developed key pathways that sense these environmental zeitgebers, translates the electrochemical signals into the TTFLs previously described, and then transmits these signals throughout the organism (Fig. 1).
2.1. The Master Clock
In terrestrial organisms, light is the primary zeitgeber to which the circadian clock actively adjusts, and entrainment is initiated in the suprachiasmatic nuclei (SCN)—origin of the master circadian pacemaker. Mammalian vertebrates rely on a connection between retinal photoreceptors and the SCN of the anterior hypothalamus for acclimation to light-dark transitions [32]. Photoreceptors, rods, and cones are canonically responsible for the transduction of photons into an electrophysiological signal. In a simplified summary, rods (dim light receptors) and cones (bright light receptors) encode photic information, which is channeled through bipolar and amacrine circuity, before synaptically converging onto conventional retinal ganglion cells (RGCs) [33,34]. These RGCs then project, forming an optic nerve fiber, onto the lateral geniculate nucleus (LGN) and midbrain for visual processing (Fig. 1) [35].
In parallel with this, an additional circuit detects ambient illumination and drives subconscious physiological responses such as circadian photoentrainment, pupillary reflex, and melatonin release [36,37]. The circadian element derives from a minority of RGCs (4–5% of total) known as intrinsically photosensitive retinal ganglion cells (ipRGCs). ipRGCs express the photopigment melanopsin and capably respond to light in the presence or absence of rod/cone signaling [38]. ipRGC axon projections have been identified into other brain regions including the SCN, subparaventricular zone, ventral lateral geniculate, olivary pretectal nucleus, and intergeniculate leaflet, which have previously been acknowledged for roles in regulation of circadian rhythmicity and pupillary reflex [39,40]. This atypical RGC network was initially thought to be homogenous and exclusive to non-image-forming processes, but recent evidence has revealed five unique subtypes, four of which are implicated in image-forming pathways of the dorsal LGN. One subtype in particular, overlaps completely with a conventional RGC subtype, suggesting a role for melanopsin in pattern vision [41–43].
Outside of the brain, additional timekeeping elements have been identified in nearly every tissue and organ, down to the single-cell level [44] (see Richards et al. (2012) [45] for more detailed Review). The SCN, presiding over this circadian hierarchy, is presumed to drive synchronization and entrainment of peripheral clocks with solar day-night cycles through a combination of autonomic and neuroendocrine signaling pathways [46]. The precise mechanism and essentiality of this interaction, however, has remained a controversial topic due to the intricacies of non-invasive time-series experiments [47]. Unlike SCN neurons, which maintain rhythmicity independently, several in vitro studies note a partial or complete dampening of rhythmicity for peripheral tissue explants, casting doubt on peripheral clock capacity to self-sustain oscillations [48,49]. In vitro systems can show synchronicity for a few days after culturing and can be synchronized with media changes or serum shock treatments; however, a loss of functional coupling between nearby cells in culture quickly leads to the observed dampening effects in oscillator synchronization [50]. After several days without an entrainer, the variability in intrinsic cellular periods could lead to a dephasing and desynchronization among independent oscillators, which then facilitates a gradual dampening of the overall culture’s rhythm—something to consider in experimental design. This effect is not unique to in vitro work, and evidence of a similar amplitude reduction in vivo following SCN ablation is also observed.
Anticipated reliance on the SCN for body peripheral clock synchronization has led to the coining of the term “slave oscillators”. This generalization, however, may fail to capture the system’s full complexity. For instance, in SCN-lesioned mice with no external zeitgebers, though small amplitude reductions and an overall loss of phase coherence occurred between organs, circadian gene expression within peripheral tissues persisted [47,51,52]. In this case, maintenance of tissue integrity appears to allow for retention of at least some peripheral synchrony. Sinturel et al. (2021) attempted a similar assessment of circadian gene expression in peripheral tissues using Per2∷luciferase in freely-moving mice over several weeks [53]. They observed a reduction in whole-body bioluminescence rhythms in SCN-lesioned mice but also a retention of partial synchronization between liver hepatocytes. These observations lend additional support to the notion that the SCN is responsible for maintaining phase coherence between peripheral organs but that internal drivers of circadian rhythmicity are likely SCN independent. A certain degree of peripheral autonomy is consistent with previous studies that attribute rhythmic gene expression to a dynamic of local oscillations and systemic circadian signaling [54]. Furthermore, several papers have detailed direct entrainment of peripheral clocks to the environment, independently of the SCN or light-dark cycle (e.g. feeding [55], cortisol [56], stress [57], and exercise [58]). As these data point toward a potential paradigm shift, further research is necessary to confirm the dynamics of interaction between master and peripheral body clocks.
2.2. The Skin Clock
Local circadian control occurs in a variety of peripheral contexts, such as epidermal, cardiovascular, metabolic, endocrine, immune, and reproductive tissues. In particular, the circadian clock plays a significant role in the skin and its response to UVR exposure [59–61]. Different skin cell types (e.g., melanocytes and keratinocytes) have various processes that are controlled by the oscillation of clock proteins, and up to 7% of protein-coding genes in mouse skin are rhythmically expressed [60–62]. The skin represents an interface between the external environment and the body, so healthy oscillation of its various functions is particularly important for anticipating and responding to daily stressors. This characteristic also means the skin both receives and generates signals for circadian timing and further implies that, while many skin actions are controlled by central oscillations, they can be locally altered by external signals from the environment [63].
Cutaneous rhythms are, at least in part, operated by the master clock in the SCN. For example, a 2009 study by Tanioka et. al. revealed that lesions in the SCN abolish PER2 expression patterns found in the keratinocytes of healthy mice [60]. There is also strong evidence that robust autonomous clocks exist across different skin cell types—for instance, intrinsic PER1 oscillations have been discovered in primary dermal fibroblasts [64]. BMAL1 may play a significant role in controlling metabolism and cellular proliferation within hair follicles and the epidermis. Time-of-day patterns of cellular proliferation in mice hair follicles were dependent on BMAL1, and a BMAL1 knockdown specific to keratinocytes eliminated this pattern [62]. In addition, the S-phase activity in these cells peaks at night, which appears to be anti-phasic to metabolism peaking in mid-day. This interaction serves to decrease proliferation during times-of-day when ROS production and cellular stress is high [62]. Furthermore, melanin synthesis in circadian synchronized human melanocytes and melanoma cells is clock-controlled through BMAL1, which is responsible for transcriptional regulation of microphthalmia-associated transcription factor (MITF)—a protein involved in melanin synthesis [65].
Sunburn apoptotic, erythemal, and genotoxic stress responses to UVR exposure are also controlled by circadian processes. These skin reactions trend toward greater severity in mice after early morning rather than evening exposures [66]. This time-of-day effect can be attributed to reduced DNA repair and increased proliferation in the early morning—factors that may exacerbate replication stress and promote p53 activity [59,62]. Interestingly, this apoptotic effect in response to UVB is reduced in mice with eliminated Cry1/2 genes, and another study showed a similar effect in BMAL1 and CLOCK-depleted human keratinocyte cultures [66,67]. DNA damage regulates the stability of CRY, and genetic depletion of Cry1 or Cry2 results in either enhancement or suppression of stress-induced Cdkn1a (p21), respectively [68]. Furthermore, BMAL1 activity is vital to activation of genotoxic stress-induced pathways like that of Hsf1 and p53 following UVR exposure [69]. The interconnectedness of these systems is also shown via observations of p53 suppression and growth acceleration in mouse embryonic fibroblasts (MEFs) harboring missense mutations in Cry2 [70]. This interactive effect between apoptosis, stress, UVR, and the circadian clock is likely due to the impact of the circadian clock on the cell cycle and will be discussed in greater detail in the next section. Additionally, the oscillation of clock genes in the skin can be locally altered by external stimuli. Multiple studies show that a low dosage of UVB radiation induces rhythmic expression of core clock genes in primary human keratinocytes [67,71]. In humans, narrow-band UVB radiation has been reported to decrease CRY2 expression in epidermal and dermal skin but increase CRY1 expression in subcutaneous adipose tissue [72]. Furthermore, erythema doses for UVB on human skin induce higher erythemal responses after evening exposures compared to morning exposures, with the latter resulting in higher p53 activity 24 hours after treatment [73]. Differing CRY2 basal expressions were reported to affect the severity of these responses. A study by Wang et. al. reveals that restricting feeding times in mice to early or mid-day results in a phase-shift and decreased amplitude for Per2 expression in the skin and completely reverses time-of-day dependent susceptibility to UV damage through nucleotide excision repair [74]. Though one study reports a possible increase in the periodicity of PER1 oscillation in adult mouse skin fibroblasts as a result of higher ambient temperatures, there is not much evidence supporting temperature as a regulatory factor in cutaneous rhythms [63,64]. An in-depth exploration on the different rhythms that are present in the skin can be found in a 2016 review by Matsui et. al. [63].
2.3. The Circadian Clock and The Cell Cycle
Mirroring principles of the circadian clock, proteins essential to cell cycle progression were initially observed to undergo daily oscillations in level to drive the cell through each cell cycle phase. Given the similarities between these two cellular mechanisms, subsequent discoveries of the interconnected nature of the circadian rhythm and the cell cycle came as little surprise. The basic cell cycle is comprised of oscillating cyclin proteins that interact with cyclin dependent kinases (CDKs) to induce progression from one cell cycle phase to the next. The levels of these cell cycle proteins, as well as their stability and function, can be altered by circadian proteins (though this interaction is tissue-specific) [75]. In epithelial tissues, such as skin and the intestinal tract, where cells are required to proliferate frequently, the clock appears essential to proper alignment of proliferation timing and stem cell differentiation [76,77]. This functionality is of particular importance for peripheral tissues in responding to cellular damage [78–81]. However, in non-proliferative tissues such as the brain, circadian clock proteins, BMAL1 and PER2, play an additional role as regulators of cellular entry and exit from the cell cycle. Despite the current evidence, however, the degree of interdependence between the circadian clock and cell cycle control may yet prove elusive. Though approximately 7% of all circadian-controlled genes regulate either cell death or cell cycle components, organisms without functional clock proteins are still viable [75,82,83]. Therefore, while circadian control does not appear to be required for cell cycle progression, optimum functionality of these processes is likely connected, provided their similar mechanisms and the overlapping networks of protein interactions. This section and Figure 2 will review the current understanding of this dynamic.
Figure 2: The Interaction of the Circadian Clock and the Cell Cycle.
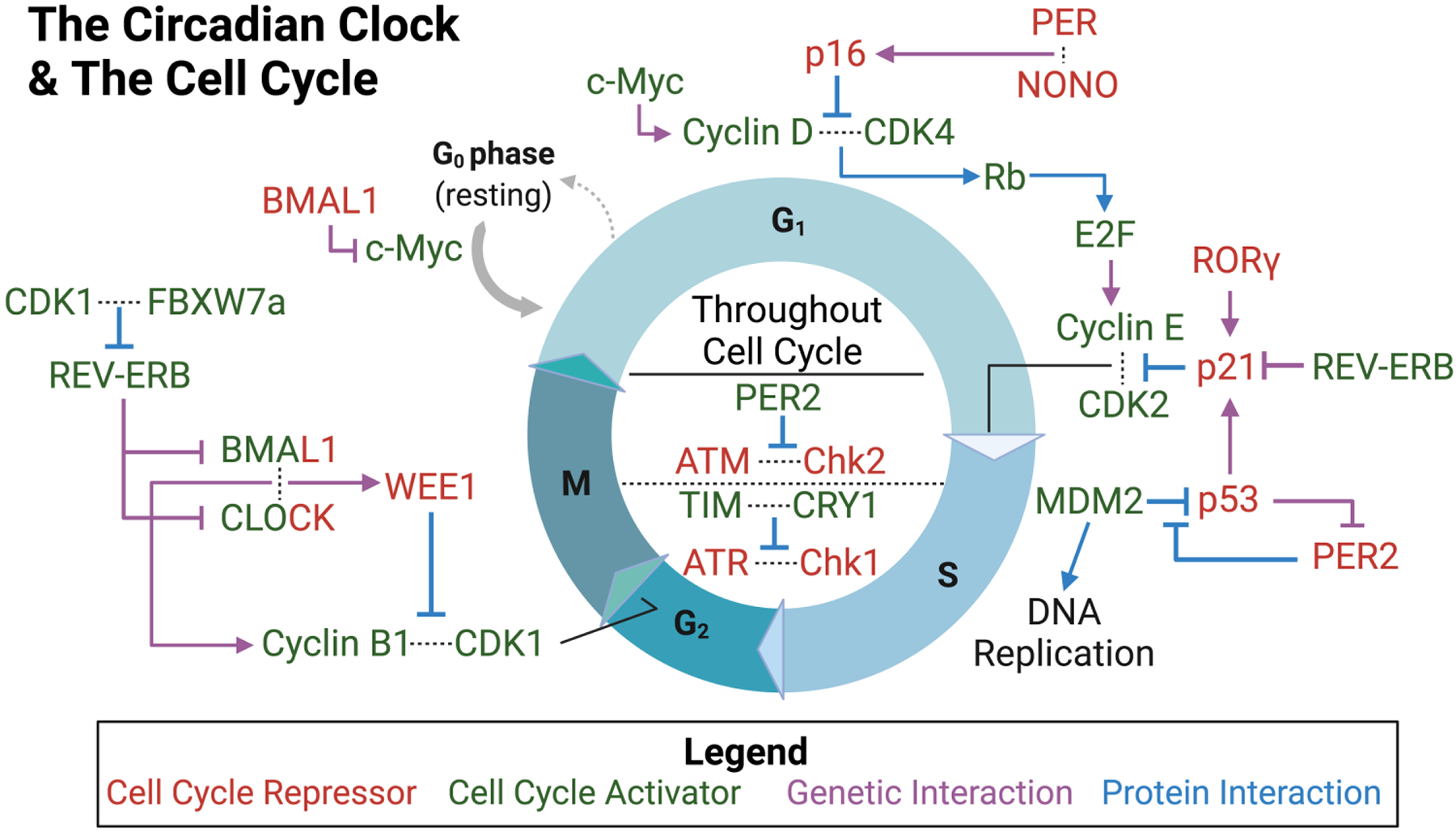
Protein and gene interactions that modify the cell cycle are adjacent to the cell cycle phase in which they function. Interactions within the circle are present throughout all phases of the cell cycle. Proteins that repress the cell cycle are shown in red, whereas those that encourage cell cycle progression are shown in green. Note: CLOCK and BMAL1 are both activators and repressors of the cell cycle. If the interaction is genetic, the arrow for the pathway is purple, and if the interaction is proteomic in nature, the arrow is blue.
When a cell is not undergoing active proliferation, it remains in the gap/growth 0 (G0) phase of the cell cycle. During this phase, the cell only undergoes basic metabolic functions. To kickstart cell proliferation, the transcription factor c-Myc, which is under BMAL1-dependent circadian control [84], initiates transcription of genes key to progression of the G1 phase. During G1, the cell is preparing for DNA synthesis by increasing the level of proteins needed for replication and by increasing cell size for the future phases. To transition from G1 phase to synthesis (S) phase, the c-Myc-induced protein cyclin D interacts with CDK4 to phosphorylate retinoblastoma (Rb) protein [84,85]. Phosphorylation of Rb allows the transcription factor E2F to activate transcription of cyclin E [86,87]. Cyclin E then interacts with CDK2 to permit cell cycle progression into S phase [85] S phase involves DNA replication, which also contains elements under circadian control—for further review on this topic see citation [88]. Key factors in the G1/S checkpoint have also been shown to be under circadian regulation.
p16 and p21 are two proteins essential to regulating the G1 phase and its transition. p16 represses CDK4 and CDK6 to prevent cyclin D binding, thereby blocking cyclin E production. [80] Alternatively, p21 binds and inhibits cyclin E/CDK2 preventing DNA replication [89]. The protein NONO is known to activate the transcription of p16 in a PER-dependent manner. Without PER or NONO, p16 shows deficient activation, which leads to decreased cellular senescence and increased cellular proliferation [80]. Conversely, BMAL1-deficient cells show p21 overexpression through decreased REV-ERB levels or increased RORγ levels, leading to decreased cellular proliferation rates [89]. Another factor that plays a critical role in cell cycle regulation, p53, has been shown to be under indirect circadian control. Upon DNA damage, there is an activation of p53 which leads to arrest of the cell cycle until repair occurs. If damage is too extensive, p53 mediates apoptosis [87]. MDM2 is a factor that facilitates p53 degradation, but when p53 is bound by PER2, this interaction is prevented and allows p53 to activate transcription of key cell cycle and DNA repair genes (such as p21) [83,90,91]. It is important to note that p53 can also act to regulate the circadian clock in a makeshift feedback loop. p53 binds to the PER2 promoter preventing CLOCK-BMAL1 binding leading to repression of PER2 transcription [92].
Alongside p53, there are two other DNA damage response proteins capable of stalling the cell cycle following DNA damage. These proteins, Ataxia–Telangiectasia Mutated (ATM) and ATM-Rad3-related (ATR), are also tied to the circadian clock. ATM interacts with Checkpoint Kinase 2 (Chk2) to stall the cell cycle after DNA double-strand breaks, typically caused by ionizing radiation (IR). PER1, however, can interfere with this interaction and prevent cell cycle arrest [93,94]. Similarly, ATR stalls the cell cycle through its interaction with Checkpoint Kinase 1 (Chk1) after UV damage in an attempt to begin DNA damage repair. Timeless (TIM), an accessory clock protein, interacts with CRY and this ATR/Chk1 complex. When TIM is downregulated under conditions of circadian misalignment, the ATR/Chk1 pathway is disrupted, preventing proper cell cycle regulation [93,95,96]. Furthermore, ATM/Chk2 and ATR/Chk1 are essential for proper maintenance of genomic integrity. These complexes can function throughout all cell cycle phases and modulate p53 or other factors to induce cell death if damage is extensive enough.
The circadian rhythm also influences cell cycle through regulation of the G2 to mitosis (G2/M) transition. The G2/M transition is highly regulated by multiple kinases and other cyclic factors, such as WEE1 and cyclin B1/CDK1. CLOCK-BMAL1 plays a role in regulating both WEE1 and cyclin B1, so it can act as both an inhibitor and activator of this cell cycle transition [97,98]. Its activator functions, however, do appear stronger based upon evidence of increased cell cycle length following CLOCK-BMAL1 knockdown [85]. During this checkpoint, WEE1 delays entry into mitosis by suppressing activity of the cyclin B1/CDK1 complex. Cyclin B1 levels increase with time spent in G2, and once cyclin B1/CDK1 reaches a certain threshold, the complex’s activity bypasses the suppressive effects of WEE1. Cyclin B1 and WEE1 both appear to be CCGs, given that removal of BMAL1 or CLOCK reduces the levels of these proteins [97,98]. Additionally, CDK1 has another function in the circadian clock-cell cycle interface. Similar to p53’s function in repressing PER2 expression mentioned above, the protein FBXW7α, in correlation with CDK1, works to regulate REV-ERB. Phosphorylation of REV-ERB by CDK1 allows FBXW7a to direct REV-ERB to the proteosome for degradation, thereby increasing the expression of CLOCK and BMAL1 [99]. The cell cycle and circadian rhythm are vital to maintaining normal cellular function and proliferation—as such, both processes comprise multiple fluctuating components under strict regulation. Given the number of reciprocal interactions and regulatory effects the proteins of these two pathways are responsible for, it is likely that disruption to any one of these components could have detrimental effects, even at an organismal level. The extent of harm, however, is ultimately dependent upon the level of disruption and other environmental factors that may come into play.
3. UV Radiation Exposure and the Circadian Clock
UVR, per the electromagnetic spectrum, is grouped by wavelength into three classes with increasing photic energy—UVA (320–400 nm), UVB (280–320 nm), and UVC (100–280 nm), respectively. Atmospheric ozone and molecular oxygen absorb the entirety of short-wavelength UVC and permit only 5%−10% of UVB to reach terrestrial surfaces (termed “global radiation”)[100]. The intensity of UVR exposure is further modulated by solar altitude and a combination of geographic latitude, cloud cover, air pollution, time-of-season, and time-of-day [101] (Fig. 3).
Figure 3: The Impact of UV Radiation on Human Skin.
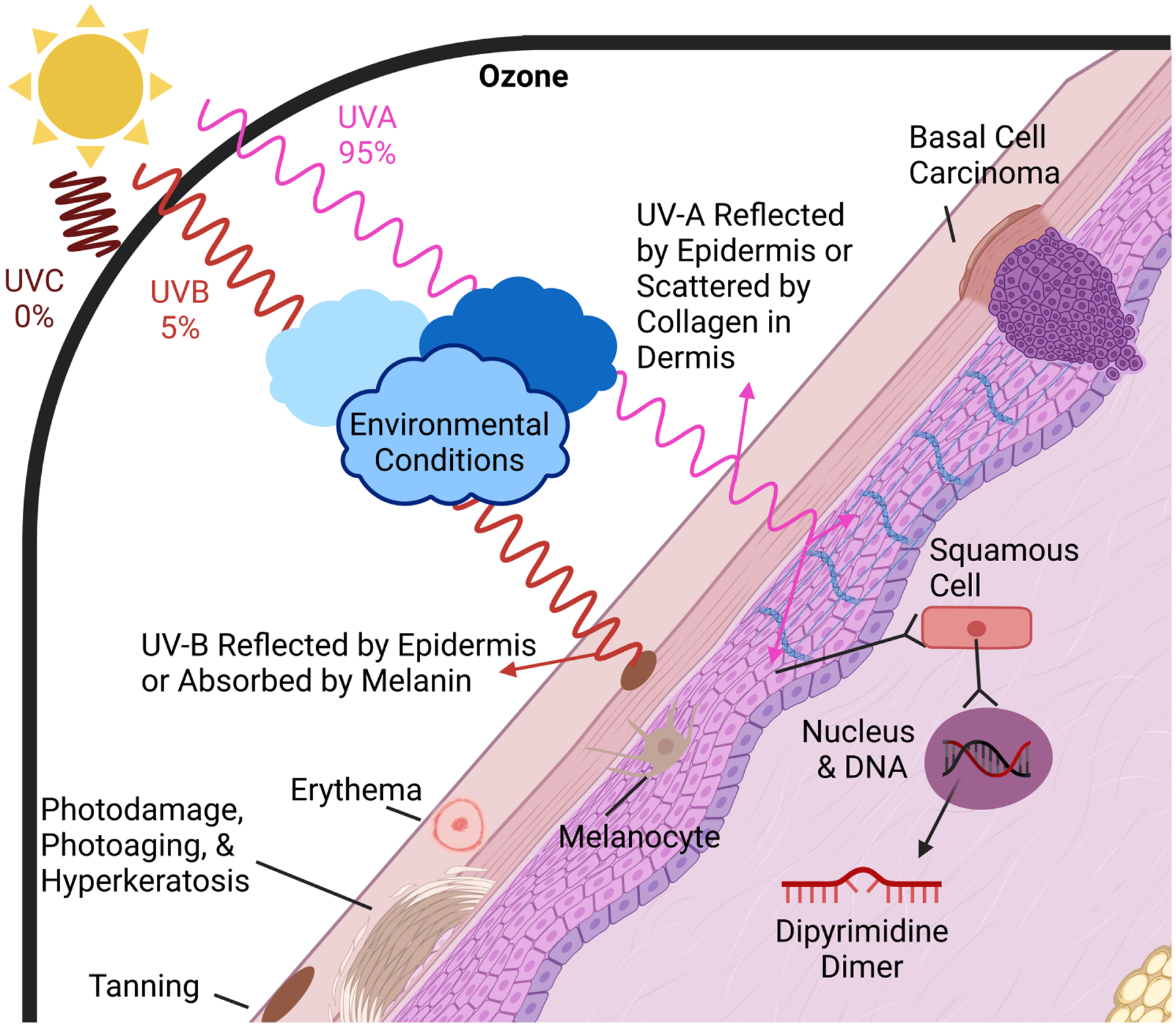
The three forms of UV light are shown: UVA and UVB passes through the atmosphere, while UVC is prevented entry by the Ozone layer (the surrounding black line). The amount of UVA and UVB that reaches the skin is dependent upon environmental conditions as depicted by the clouds. Upon reaching the skin, UVA either reflects or penetrates deeper into the skin, whereas UVB is primarily reflected or absorbed by melanin. Effects of UV light on the skin and skin cells are represented throughout the figure.
3.1. Skin Exposure to UV Radiation
Whereas UVB penetrates superficial epidermal layers of skin for delayed tanning and erythema, UVA reaches deeper into the dermis for activation of melanin synthesis, more immediate tanning effects, and lipid peroxidation [100,102]. Photodamage, hyperkeratosis, erythema, tanning, mutation, immunosuppression, and vitamin D synthesis are all acute effects of UVR exposure, while photoaging and photocarcinogenesis constitute more chronic responses [100,103–105]. In response to UV photons, skin may reflect, absorb, or scatter incoming light. The stratum corneum and subcutaneous tissue layers are spectrally reflective, whereas penetrations of UVB and UVA into the epidermis and dermis are absorbed and scattered, respectively. Absorption primarily occurs in the epidermis due to the melanin pigments present, and scattering predominates in the dermis due to its density of collagen fiber networks [65,106] (Fig. 3).
UVR’s capacity to excite loosely-bound valence electrons leads to alteration of the chromophores within the skin. Chromophores are the bonds within either organic unsaturated repeats or ring structures that can absorb the energy from UVR for use in photochemical reactions. Epidermal chromophores include nucleic acids (DNA), urocanic acid, aromatic amino acids (tyrosine/tryptophan), melanins (eumelanin/pheomelanin), and various precursor molecules [107]. Dermal chromophores include hemoglobin, oxyhemoglobin, and bilirubin [108]. Aromatic side chains are particularly susceptible to absorbing wavelengths under 315 nm and serve as a primary molecular target for UV [109]. Cutaneous effects of UVR overexposure are largely gated by the absorption spectrum of a given chromophore. UVB, for example, can absorb into the nucleic acid of epidermal cells and induce DNA damage directly as point mutations or chromosomal aberrations, or indirectly as strand breaks and indels during replication arrest and attempted repair [110,111].
The most abundant UVB-induced DNA lesions are cyclobutane pyrimidine dimers (CPDs) and pyrimidine-pyrimidone (6–4) photoproducts (6–4 PPs) at dipyrimidine sites. Under subsequent photon absorption, 6–4 PPs may further be converted into a third type of photoproduct known as Dewar valence isomers [112]—though this occurs infrequently since 6–4 PPs are quickly repaired [113]. All of these photolesions distort the DNA double-helix and may interfere with progression of both replication and transcriptional machinery, if not excised. UVA is weakly absorbed by DNA and, while it does cause thymine dimers, it also operates indirectly through the production of reactive oxygen species (ROS) to cause damage through oxidative stress [114,115]. Mechanisms of photosensitized oxidation reactions include induction by singlet oxygen (1O2; Type II), electron/hydrogen abstraction (Type I), and hydroxyl radicals [116]. Common oxidative lesions include 8-oxo-7,8-dihydro-2’-deoxyguanosine (8-oxoG), 5’,8-cyclo-purine adducts, 5-methylcytosine (5mC), and the products of their subsequent oxidations. Oxidative base lesions are described as weakly mutagenic but can still induce spontaneous DNA mutagenesis through base substitutions/transitions (G>T), mispairings due to conformational change, and ineffective repair mechanisms [117]. In cultured fibroblasts and keratinocytes, UVA-induced CPD formation still dominates as a more efficient mode of energy transfer than photooxidation [118].
UVR is a driver of genomic instability and possesses a characteristic mutational signature of C>T substitutions at dipyrimidine sites or CC>TT double-base changes. Under 4 kJ m−2 UVB, global CPD distribution ratios for the four possible dipyrimidine sites in cellulo are 27:27:25:21 for TT:CC:TC:CT [119]. Unfortunately, Translesion Synthesis (TLS) is error-prone, and these CPD lesions may not be corrected, leading to increased endogenous mutational burden in healthy tissues. These mutations may cause one of two types of mutations in cells that can become cancerous. The first are called driver mutations, which refer to mutations that confer a fitness or growth advantage for tumor cells and experience positive selection in the microenvironment. The second, passenger mutations, do not contribute directly to cancer development—together, these mutational types constitute a tumor’s mutated gene set.
According to Saini et al. (2021), healthy skin cells possess a baseline range of 402 to 14,029 base substitutions, 7 to 71 indels, and 1 to 14 structural variants. Despite using sun-shielded hip tissue, UV-induced DNA damage was still prevalent and attributed to an ID8-like indel mutational signature, translesion bypass over substitutions, and an accumulation of templated insertions that spawned from error-prone DSB-repair. They further identified 61 Tier 1 driver mutations from the 21 cancer-free individuals, though these were at low allele frequencies (range of 0.012–0.412; average of 0.0725) [120]. Thus, it is plausible, that progression from healthy skin to malignancy is reflective of selection for a series of driver mutations that accumulate over time, leading to genomic instability and an increase in the global prevalence of somatic mutations due to UV irradiation. The landscape of cancer genomics, however, is rapidly evolving and requires further research to disentangle the precise mechanisms of disease onset.
The cumulative mutational effects of UVR exposure drastically increase risk of malignancy in the skin. Key driver mutations associated with development of non-acral cutaneous melanoma include BRAF, NRAS, MITF, PTEN, PREX2, TERT, RAC1, RPS27, TP53, and CDKN2A [121]. Drivers for basal-cell carcinoma (BCC) include PTCH1, TP53, SMO, MYCN, PPP6C, PTPN14, STK19, LATS1, and promoters of TERT and DPH3 [122,123]. Lastly, some of the more frequent squamous-cell carcinoma (SCC) somatic drivers include NOTCH1/2, TP53, CDKN2A, ARID2, FAT1, CASP8, EP300, and CREBBP [124]. It is also worth noting that cutaneous melanomas have a higher somatic mutation rate than almost all other solid tumor types, and while this is attributable to UV-induced mutagenesis, the highly recurrent oncogenic drivers, like those in BRAF and NRAS, do not reflect a UV signature [125].
Unsurprisingly, skin cancer is the most diagnosed type of cancer in the United States and exceeds $8 billion annually in treatment costs [126]. Keratinocyte or non-melanoma skin cancers (NMSCs), such as BCC and SCC, are 18–20 times more frequent than cutaneous melanoma, but ultimately all three common forms of skin cancer can be traced back to solar irradiation (~65% of melanomas, ~90% of NMSCs)[127,128]. This is further reinforced by an inversely proportional relationship between skin cancer incidence and the degree of skin pigmentation—though darker-pigmented individuals still experience greater morbidity and mortality than Caucasians [129]. Of particular concern is the progressively increasing trends in annual skin cancer incidence over the past fifty years [130,131]. Largely attributed to the social normalization of sun-exposure, this expanding burden has additional ties to an aging populace, given the considerably higher incidence rates among elderly individuals [132]. Degree of UVR exposure, history of sunburn, and number of nevi are notable risk factors in the development of skin cancer that also appear to influence the type of malignancy that develops [133]. Furthermore, it is highly probable that the efficacy of UV DNA damage repair contributes to malignancy development. To support this hypothesis, individuals with defective DNA damage repair disorders, such as xeroderma pigmentosum (XP), are 1000 times more prone to melanoma [134–136].
3.2. Circadian Regulation of UVR-induced DNA Damage and Repair
Human skin relies innately upon melanocytes in the epidermis for synthesis of melanin pigments, as well as thickening of keratin in the stratum corneum, to absorb and scatter UVR to protect the cells from damage. Subsequent pathway activation in response to internal cellular damage that bypassed the skin barrier includes DNA repair, apoptosis, and antioxidant enzyme release [137]. Given the propensity for UVR to cause CPDs and 6–4 PPs, it is necessary for the cell to have mechanisms to repair these bulky base lesions to protect genomic stability. Though there are a number of repair mechanisms—base excision repair (BER), mismatch repair, and double-strand break repair—that can deal with DNA damage caused by both endogenous and exogenous factors including UV-induced double-strand breaks, the only known form of repair for dipyrimidine dimers caused by UV exposure in many organisms is nucleotide excision repair (NER) [93,138,139]. Since humans respond to these UVR-induced dimers via this sole mechanism, efficient activity of NER is vital for defending against sun-induced skin damage.
NER involves incision of the DNA strand on both sides of the damage site to remove bulky lesions, followed by DNA synthesis and nick sealing to repair the remaining gap in the DNA backbone [138,140]. NER has two major mechanisms that are differentiated only by their initial step where the lesion is detected. The first is Transcription-coupled NER (TC-NER), which occurs only in transcriptionally active regions of the genome when RNA polymerase (Pol) becomes stalled at the bulky lesions [134,141]. The second is Global-genome NER (GG-NER), which occurs throughout the genome including in the non-transcribed regions that TC-NER cannot repair [134,142]. In TC-NER, the lesion and stalled RNA Pol are detected by the cockayne syndrome group A (CSA) and CSB proteins [134,141], whereas with GG-NER, the damage is first sensed by xeroderma pigmentosum C (XPC) and XPE (otherwise known as DDB2) in complex with DNA damage binding protein 1 (DDBP1) and HR23B [114] (Fig. 4).
Figure 4: The Nucleotide Excision Repair Pathway.
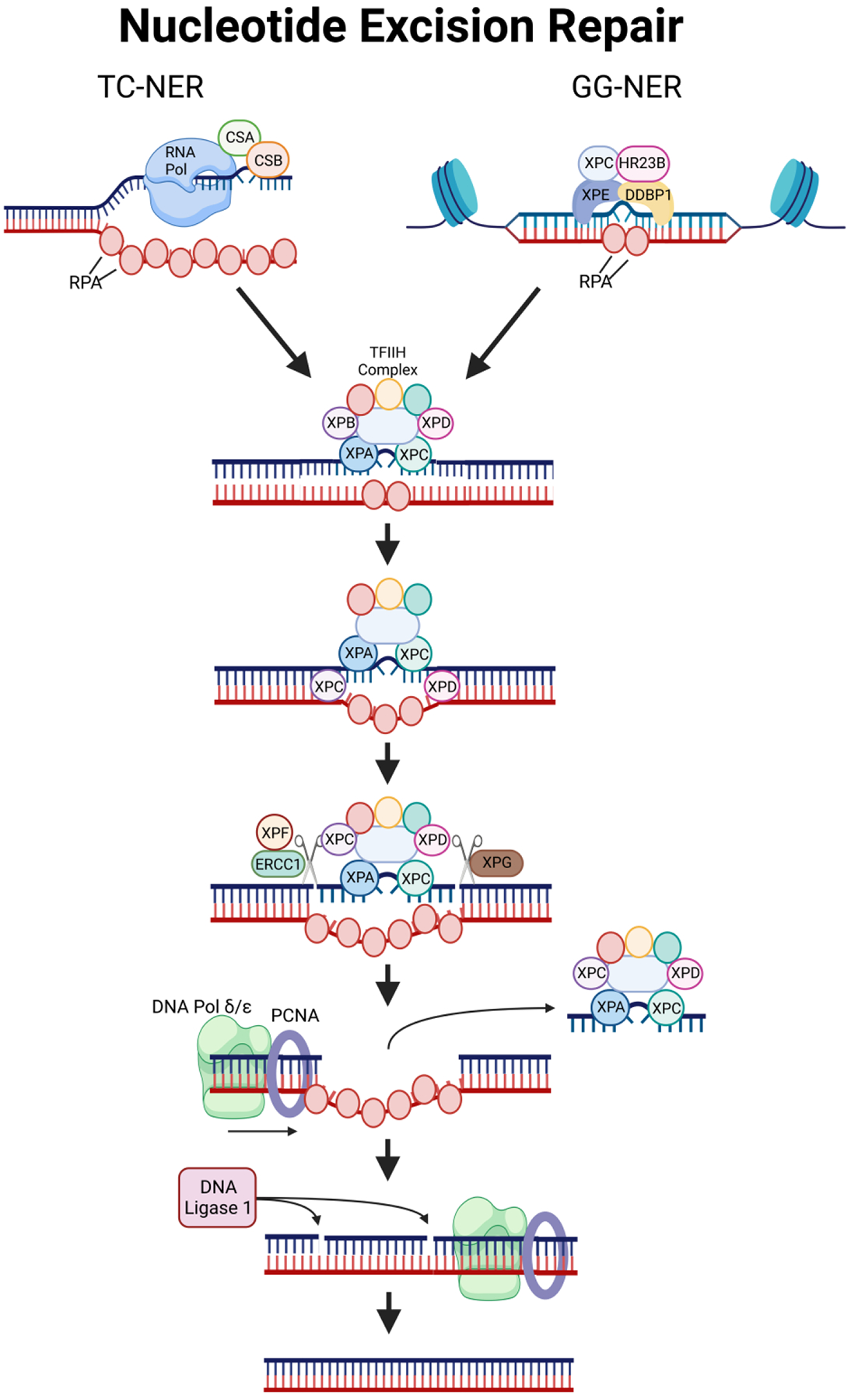
Transcription-coupled or Global genome NER is initiated depending upon how the damage is detected. CSA, CSB, RNA Pol, and RPA detect the damage during transcription in TC-NER, while XPC, XPE, HR223B, and DDBP1 detect the damage in all other instances. From there, the pathway is unified with the TFIIH complex binding to the site of damage and recruiting factors such as XPF and XPG that excise around the damage site. Finally, DNA Pol and PCNA fill the leftover DNA gap, and DNA ligase seals the new DNA into the strand.
After recognition of the damage, the process for repair in both NER pathways occurs as follows: (1) replication protein A (RPA) binds to single-stranded DNA to prevent base pairing. While XPC and XPA attract the transcription factor II H (TFIIH) complex to the site, (2) XPB and XPD helicases, which are part of the TFIIH complex, unwind the DNA. (3) XPG, also known as excision repair cross-complementation group 5 (ERCC5), and the XPF-ERCC1 complex cleave the DNA at sites surrounding the damage (as marked by XPA and RPA). (4) Next, the old lesion-bearing DNA sequence and all proteins, except RPA, are released. (5) Proliferating-cell nuclear antigen (PCNA) and DNA Pol δ/ε are attracted by RPA and synthesize new DNA to replace the removed sequence. (6) Finally, the remaining nicks between the newly synthesized DNA and old backbone are sealed by DNA ligase 1 [93,114] (Fig. 4).
There is evidence of time-of-day fluctuations in NER activity that suggest this UV-focused repair pathway is tied to the circadian clock [59,143]. More specifically, a rate-limiting component of NER, the XPA protein, has been observed to undergo daily oscillation in mouse skin. These oscillations were further associated with the efficacy of NER activity [59]. This link between the circadian clock and the NER pathway was clearly demonstrated when cells treated with cisplatin (a damaging agent that activates NER) and inhibitors of circadian clock proteins (REV-ERB and CRY) produced an increase in level of XPA and, consequently, enhanced repair activity [144]. When this mechanism is properly coordinated with an organism’s behavioral pattern, the peak of NER protein expression should coincide with the time-of-day said organism experiences the greatest amount of exposure to UVR. In line with this, exposure to UV light at unusual times (e.g., subjective night in humans) may elicit a stronger toxic response due to decreased XPA levels and therefore reduced NER activity. Interestingly, these effects may also be dependent upon the DNA strand or gene impacted by UVR. In kidney and liver tissues, there is evidence that repair of cisplatin DNA adducts is dependent upon the transcriptional status of the gene and whether the transcribed or non-transcribed strand is affected. If the gene is transcriptionally active, the transcribed strand is subject to TC-NER, whereas if the gene is not active or if the DNA adduct is present on the non-transcribed strand, the damage is repaired through the GG-NER pathway. TC-NER is the more efficient of these two pathways and does not appear to be dependent on the level of XPA within the cell; however, the GG-NER pathway is dependent on XPA levels and therefore on the circadian rhythm [145].
It is important to note that XPA level is not the sole mechanism of circadian regulation in the NER pathway. The circadian clock also acts to control NER indirectly through modulation of DNA damage response (DDR) signaling and cell cycle progression. Upon sensing UV damage, the cell undergoes a series of phosphorylation cascades to both recruit proteins to the site of damage and prevent the cell from proceeding through cell cycle stages with unrepaired damage. These processes are collectively known as the DDR. As mentioned in 2.3 The Circadian Clock and the Cell Cycle, the circadian clock regulates activation of ATR, which is recruited to the site of UV damage by RPA to stall the cell cycle until repair can occur [96,146]. Thus, control of ATR through TIM and CRY is yet another way the circadian clock mediates the response to UV-induced DNA damage signaling [96]. Similarly, a recent paper by Anabtawi et al., showed a reversal of the anti-proliferative effects of cisplatin with the addition of REV-ERB and CRY inhibitors. REV-ERB and CRY inhibition was linked to increased levels of the cell cycle regulators WEE1 and p21, which allowed the cells to proceed through the cell cycle. These findings suggest that REV-ERB and CRY may have a role in modulating cell cycle progression after NER-mediated DNA damage repair [144]. These data support the hypothesis of circadian regulation of NER through indirect mechanisms of DNA damage response (DDR) signaling and cell cycle progression in addition to the ability to regulate through XPA.
Though outside the scope of this review, recent evidence suggests that the circadian rhythm also influences DNA damage repair and response via pathways outside of NER. For instance, in humans maintaining a night shift schedule, the rhythmicity of DNA repair genes, such as PARP1 and RAD50, was lost when compared to a day shift schedule. This observation was further connected to an increased sensitivity to DNA-damaging agents in night shift workers [9]. Another study examining shift workers observed a decrease in the expression levels of BRCA1 and BRCA2 genes compared to day shift workers, which may correlate with breast cancer susceptibility [147]. Other examples include circadian modulation of BER through the key recognition and rate-limiting protein 8-Oxoguanine DNA glycosylase (OGG1) [148], depletion of CRY1 potentially impacting mismatch repair and homologous recombination genes [149], as well as the observation that REV-ERBα (NR1D1) inhibits the recruitment of repair proteins—SIRT6, pNBS1, and BRCA1—to the site of double-strand breaks [150]. This could further have ramifications in carcinogenesis since other types of damage, such as double-strand breaks, are possible when UV-induced DNA damage is not repaired in a timely manner. The circadian rhythm may also operate indirectly through modulation of other compounds like melatonin that protect the cell from damage or stimulate damage repair. For additional information on the key roles of melatonin, please see the review by Majidinia et. al. [151].
4. The Circadian Clock and UV Radiation in Research and Human Health
The wealth of epidemiological records detailing associations between skin pathologies, UVR, and circadian perturbation, while informative, are not substantial indicators of causality. To assess the impact of a potential carcinogen on human physiology, experimental data is required to establish a causal relationship and uncover underlying biological mechanisms. These experiments are not commonly performed on human subjects and require application of animal models. In the context of comparative medicine, mice (Mus musculus) and rats (Rattus norvegicus) are historically purposed for the recapitulation of normal and diseased human physiology. Albino rats were among the first species ever domesticated for scientific use, with published works as early as the 19th century [152]. Beyond key anatomical and physiological similarities, rodent models share up to 95% of their genomes with humans and offer several, more practical research advantages—such as small body sizes, short gestation times, high fertility rates, rapid sexual maturity, and shorter lifespans [153]. The latter-half of the 20th century saw rapid expansion of molecular techniques for manipulation of the mouse genome and availability of inbred strains, which ultimately tipped research consensus in favor of the smaller, cheaper-to-maintain species. The larger size and increased sociability of rats, however, allow for generation of more complex disease models and are often prioritized in the cardiovascular, pharmacology/toxicology, and behavioral science fields [154]. As the availability of rat genomic resources increases, criteria for model selection will hopefully shift emphasis from molecular tools to the appropriateness of either mouse or rat physiology with the disease phenotype being assessed.
4.1. Animal Models in UV Radiation Research
The field of photobiology has an extensive history applying rodent models to the question of UVR exposure and skin cancer development. Clinical observations of sun-induced skin lesions originate from the “seaman’s skin” phenomenon as early as 1896 and inspired a wave of investigators across Northern Europe, North America, and Australia to note the effects of prolonged sun exposure in human populations [155]. The late 1920s and early 1930s then saw application of these epidemiological observations toward experimental trials in animal models [156–161]. Abrikossoff (1926) and Findlay (1928) were among the first to publish the effect of UVR on animal skin epithelium and did so in the rabbit and albino mouse, respectively [156,157]. Apart from “precancerous” changes and a single putative BCC, however, only Findlay (1928) was able to demonstrate clear evidence of UVR-induced tumor formation. In a series of three experiments, Findlay challenged chemically-depilated albino mice with a combination of carcinogenic tar and UVR from a quartz mercury-vapor lamp. Treatment with both tar and radiation drastically accelerated carcinogenesis, compared to tar-only mice. The final experiment then assessed the impact of chronic daily UVR exposure alone over 58 weeks and observed development of papillomata and malignant growths in surviving mice post-217 days into the trial. However, it is worth noting that these significant results were not replicated a year later [162], and Putschar and Hotlz (1931) likely provided a more definitive confirmation in rats of chronic UVR-induced skin cancer, without chemically depilating the animals [158].
Further evolutions of the rodent model also factored differences in skin coloration and importance of exposed skin for susceptibility to UV. Rusch and Baumann (1939), for example, observe fewer tumors and later overall tumor development for irradiated C57 black mice, with respect to their albino counterparts [163]. This mirrors trends in skin cancer incidence among human populations of different races and/or skin pigmentation [129,164]. Roffo (1936) also notes that irradiated rats tend to develop epithelial tumors in naturally hairless or artificially depilated regions (e.g. ears or conjunctiva) [161]. These findings are further supported by more recent studies detailing the photoprotective role of hair against UVR [165].
In the early 1920s, fascination with hairless mice was in part due to its initial discovery in wild populations. Researchers were motivated by the general rarity of a mutation to radically alter a stock’s phenotype and a desire to improve physiological deficiencies of the initial strain [166]. The null phenotype was believed, and more recently confirmed, to be caused by a single autosomal recessive point mutation in the coding region of the hairless (Hr) gene [167]. Modeling this natural phenomenon, modern dermatological research primarily utilizes the SKH1 hairless, immunocompetent, and unpigmented mouse stock derived in the 1960s. This outbred strain is characterized by a stable retroviral insertion into intron 6 of hairless (Hrhr/Hrhr) that leads to development of alopecia and skin wrinkling after growing the first coat of fur or by three weeks of age [168]. HR protein functions as a nuclear receptor co-repressor and mediates timing of epithelial cell fate decisions. It is believed to transcriptionally repress keratinocyte terminal differentiation markers, thereby permitting hair follicle development [169]. Keratinocyte progenitors, in the absence of HR, instead differentiate into epidermis and sebaceous glands to facilitate the hairless phenotype [170]. SKH1, though non-pedigreed, is invaluable for studies of photobiology and photocarcinogenesis, as it abrogates the need for depilation, allows for continuous visibility of tumor stage and morphology, and avoids other modulating effects of the hair cycle on skin physiology. These mice further develop UVR-induced skin tumors (almost exclusively epidermal skin carcinomas) that are morphologically and molecularly similar to human lesions and serve as an appropriate disease-risk model for UVR-induced skin carcinogenesis [168,171].
4.2. Models in Circadian Biology
The current field of circadian biology adopts a similar framework to dermatology in the use of rodent models toward understanding perturbations of sleep/wake cycles. However, the majority of circadian components were first identified and cloned in Drosophila [172]. The 2017 Nobel Prize in Physiology or Medicine was awarded to Jeffrey C. Hall, Michael Rosbash, and Michael W. Young for their work in germline transformation of the Period gene [173,174] and elucidation of the core TTFL mechanism between CYCLE/CLOCK and PER/TIM in fruit flies [175–177]. The transition to mice stems from a greater overall complexity and genetic redundancy in the rodent TTFL, which constitutes a more adequate proxy for human physiology [178]. In general, mutation of a single core clock gene in mice or humans will not completely abolish circadian rhythms—with the exception of Bmal1 [14]. The first approach in circadian research involves basic mechanistic studies, where genetic or pharmacological disruption of core clock genes (e.g., Bmal1 [14], Clock [179], Cry1-2 [180], and Per1-3 [181–183]) is induced in stable inbred mouse strains or cells in culture. The second approach involves behavioral animal modeling of the circumstances that cause environmental circadian dysfunction in humans (e.g., jet lag and rotating shift schedules) (Fig. 5). Within this second approach, there are also occasional studies that evaluate these dysregulating conditions in human samples either through controlled laboratory studies or with donated samples from shift workers [9,184].
Figure 5: Model Systems to Research Circadian Disruption.
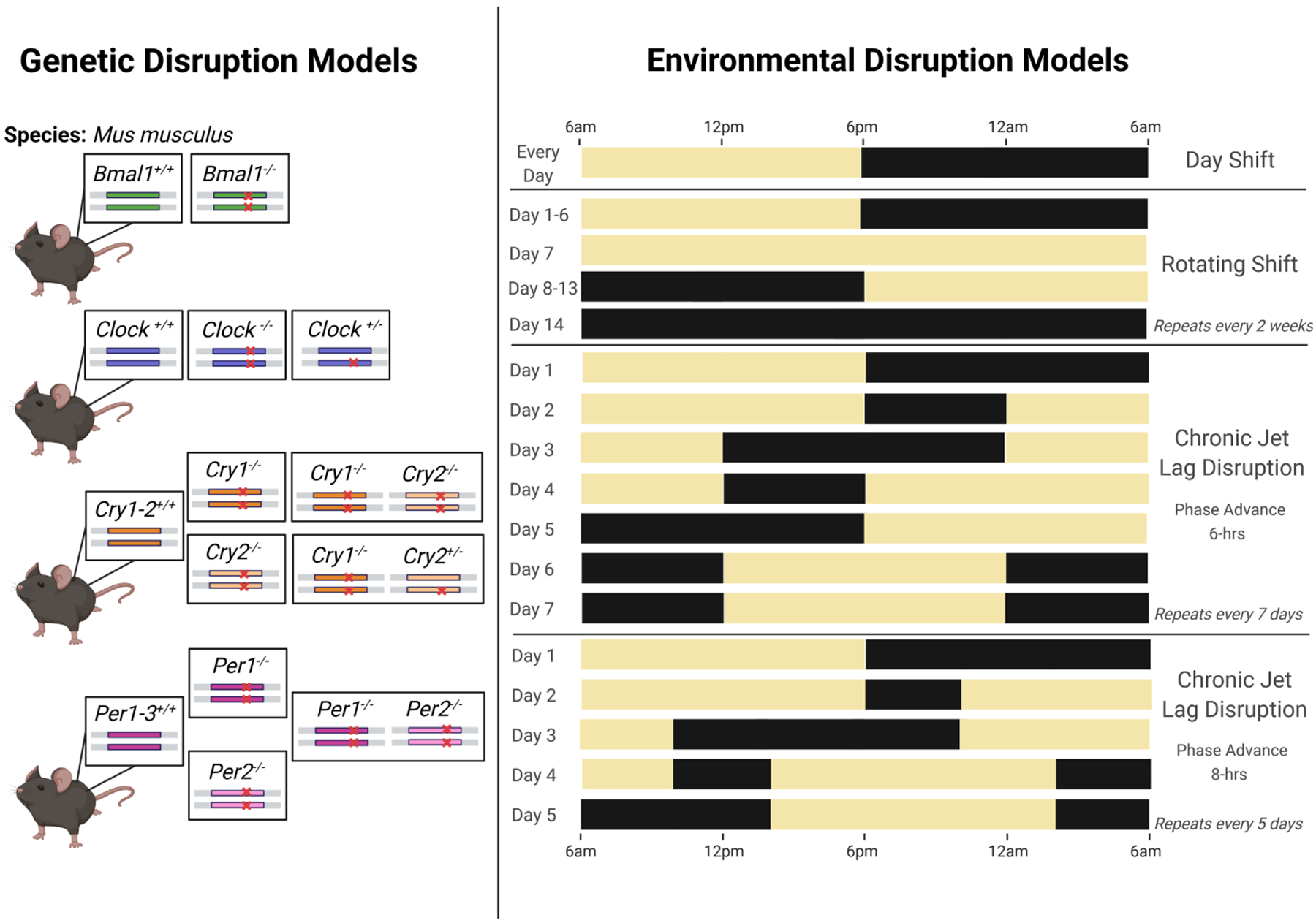
Genetic models of clock gene disruption are displayed on the left, with representations of the most common core clock gene mutations. Environmental clock disruptions are shown on the right, with schematics of the most frequently utilized light schedules to induce circadian dysregulation. Lights-on is represented by the yellow boxes and lights-off with black boxes.
Prevalent forms of circadian rhythm alterations in modern society include shift-work, trans-meridian flight, or altered day lengths [185]. Professions like hospital care, law enforcement, and factory-work are well-known for schedules catering to all hours of the day and often require individuals to rotate working day, afternoon, or night shifts. Emergency medicine is particularly notorious for the random scheduling of mandatory night shifts, and the American College of Emergency Physicians has asserted since 1994 that a continuous rotating-shift schedule adversely affects well-being and is the primary motivation for early attrition from the specialty [186]. This was later followed by the International Agency for Research on Cancer categorizing shift-work involving circadian disruption as a Group 2A carcinogen or probably carcinogenic to humans in 2007 and 2019 [6,187]. The human circadian clock cannot instantaneously adapt to perturbations of the 24-hour cycle, and persistent disruption effectuates a lag-time in which the individual is out-of-phase with entraining cues of the environment.
Shift-work that disrupts the circadian clock consists of exposure to zeitgebers—such as light, meal timing, or physical activity—during an anticipated sleep period. This desynchronization (also known as dysrhythmia or shift-lag) leads to a series of unfavorable neurologic, digestive, metabolic, social, and sleep disturbances [188]. “Shift-work” is a somewhat non-descriptive term and broadly accounts for schedules that fall outside of the 7:00 AM to 6:00 PM work-day—this includes progressive rotations between day, evening, and night shifts. A wide variety of designs for shift-work exist in the circadian literature, and the lack of a unified model may complicate generalizations about observed data and explain contradictory results [189]. In rodent models, one philosophy to recapitulate the shift-work phenomena involves repetitive manipulation of the environmental light-dark (LD) cycle from once-to-several times weekly across extended experimental intervals [190–195]. Van Dyke et al. (2015), for example, implements a rotating shift-work protocol over 17 months by extending the light or dark phase to 24 hours at the end of each week to invert the LD cycle [191] (Fig. 5). Other proposed models prioritize feeding restriction [196,197] and/or forced locomotion [189,198–200]. These alternative methodologies specifically target meal timing and activity behavior of humans, who are forced to work continuously for ~8-hour shifts during a normal rest period.
The average work schedule, however, is rarely static from week to week. McGowan and Coogan (2013) attempt three different experimental cohorts in CD-1 mice to mimic possible real-world rotating shift conditions [201]. Beginning with baseline of 12 light hours and 12 dark hours (12L:12D), the first group received a forward-rotating or clockwise protocol in which animals were exposed over a six-day period to 8-hour phase delays every two days, followed by two days in constant darkness (DD) to represent days-off. The second group was given a backwards-rotating or counterclockwise schedule that mirrored the first group’s but instead had 8-hour phase advances every two days. The last group was then provided with an alternating day and night-shift schedule within the same week by exposure to a 12-hour phase-shift every third day (24-hours of constant light or dark) without any days-off. Some researchers, however, argue that procedural alterations to LD alone are insufficient to recapitulate the complexity of human shift-work and more closely represent conditions of chronic jet-lag. Shift-workers, for example, must acclimate both to changes in work and sleep schedules, as well as the loss of daytime familial and social interactions. Returning to a diurnal routine on off-days may further delay physiological adjustment to the shift-work schedule.
Similar negative health effects are also observed with a temporaneous sleep disorder coined “jet-lag”, which arise following trans-meridian travel across multiple time zones and altitude ascent [202]. These symptoms, however, are more subtle and temporary. For instance, airline travelers are likely assisted by the entrainment cues of social interactions and local diurnal sleep/wake activity cycles to reset their clocks immediately after arrival [203,204]. Flying east-to-west requires a phase delay of the circadian system and an adjustment period proportional in hours to the number of time-zones crossed in a single day. Eastward travel, on the other hand, constitutes a phase advance and reduction in day length. The latter scenario is more disruptive to human rhythms and elicits more profound jet-lag symptoms as the clock attempts to re-align an advanced sleep schedule with the earlier time-zone [205,206]. These disturbances are a particular risk for international business-people, airline crews, and military personnel. The Defense Advanced Research Projects Agency, for example, recently committed to resolving issues of circadian disruption and gastrointestinal disorders in service members through the ADAPTER initiative—a five-year, multi-phase program for the design of an implantable/ingestible bioelectronic pharmaceutical device [207].
To study this phenomenon, Filipski and colleagues published one of the first simulated jet-lag models in rodents in 2004 [208]. They first synchronized B6D2F1 mice under standard 12-hour light, 12-hour dark conditions for three weeks, then subjected a random subset to a 10-day stint of serial 8-hour advances of the LD cycle every two days. This jet-lag schedule has been adopted by several experimental studies, with only slight variations in the normal LD acclimation period, experimental endpoint, and frequency of phase advance [209–211]. Other interpretations of the jet-lag schedule include a weekly 8-hr phase advance on Monday, followed by an 8-hour phase delay on Thursday [212,213], or a 6-hour phase advance every two [214,215] or seven [216] days (Fig. 5).
4.3. The Circadian Clock and UVR in Human Health
Perturbations of the circadian clock and exposure to UVR can have a multitude of negative consequences on human health, in both an independent and synergistic manner. Disruptions to the circadian rhythm, either by environmental factors or clock protein mutations, can cause fatigue and impaired cognitive function, as well as detrimental impacts to cutaneous, cardiovascular, metabolic, endocrine, immune, and reproductive functions [8,217]. For instance, metabolic disorders with disrupted glucose metabolism, such as diabetes and obesity, have been linked to circadian disruption [218,219]. Components of cardiovascular function (e.g., heart rate, blood volume, and blood pressure) are under circadian control, which justifies why the majority of cardiac disturbances occur in the morning [220,221]. Data linking time-of-day to proinflammatory cytokine levels and symptoms of immunological disorders have also been observed [222,223]. These examples represent only a few of the systems subject to disequilibrium by circadian dysregulation. For a more holistic review of the impacts of clock disruption, see the 2017 review by Ballesta et. al. [224].
In summary of the prior sections, the short-term impacts of UVR exposure are photodamage, hyperkeratosis, sunburn/erythema, tanning, immunosuppression, and mutation; however, more chronic conditions, such as photoaging and photocarcinogenesis, can occur due to damage built up over years of exposure [103–105]. UV exposure has been linked to a specific mutational signature of C:T or CC:TT substitutions at dipyrimidine sites, and the mutagenic potential of UV light implicates it in both non-melanoma and malignant melanoma skin cancers—ultimately justifying its classification as a Class I carcinogen [225]. When UVR induces direct (substitutions) or indirect (ROS production) DNA damage, mutations persist primarily because of illegitimate DNA damage repair. Upon accumulation of these mutations (particularly in key oncogenes), these cells may escape cell cycle regulatory mechanisms and experience uncontrolled proliferation. The mutated cells then undergo transformation and begin excreting factors that alter their microenvironment in favor of growth. The circadian clock likely plays a mechanistic role in all of these stages, so tracking integration of clock factors into each leg of the pathways facilitating malignant transformation could be key for future prevention and treatment of skin cancer (Fig. 6).
Figure 6: The Influence of the Circadian Clock and UVR on Mutation and Carcinogenesis.
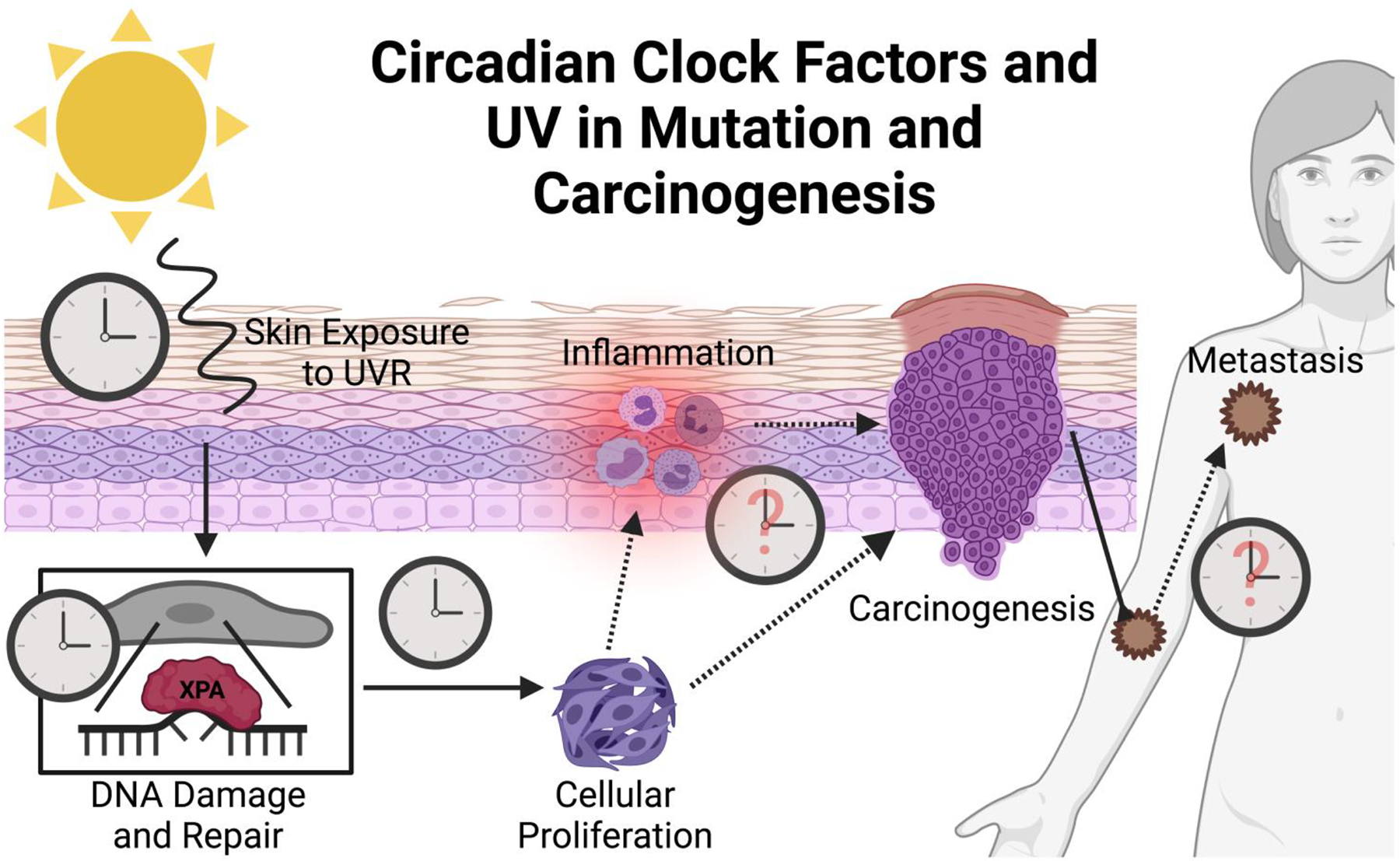
Clocks visualized in this figure represent known or putative influences of the circadian clock. The circadian clock is known to play a role in how skin and skin cells respond to UVR damage, as well as in how efficiently the NER pathway repairs DNA damage. Additionally, the clock impacts cell growth and proliferation through the cycle cycle. However, though it is hypothesized, there is very little known about clock responsibilities in inflammation, cellular carcinogenic transformation, and metastasis.
Fundamentally, mutation of the DNA is responsible for initiating skin cell carcinogenesis. As described earlier in section 3.2 Circadian Regulation of DNA Damage and Repair section, XPA, one of the key rate-limiting factors for NER, is a CCG. It is inversely correlated with protein levels of CRY1, so the efficiency of NER is increased at times-of-day when CRY1 is low (early evening for mice; morning for humans). Additionally, when repair efficiency is high, the rate of cell proliferation is low [66]. In other words, the processes of DNA repair and DNA replication are anti-phase. These alternating dynamics align with the previously detailed circadian regulation of both cell cycle and DDR proteins. ATR/Chk1 complex, p53, and p21 are under clock regulation and—when activated—either stall the cell cycle for repair or induce apoptosis [83,89,93–96]. The general intent is to have the DNA repaired before replication occurs. If the clock is dysregulated, it stands to reason that escape from cell cycle checkpoints and apoptosis may be possible, especially if the cell has suffered additional mutations to its regulatory elements. Based upon these interactions, a unified hypothesis could connect UV-induced DNA damage and simultaneous dysregulation of the circadian clock to the development of carcinogenesis in the skin. In representative in vivo studies, mice with genetically disrupted circadian clocks lost rhythmicity of DNA damage repair. Furthermore, mice treated in the early morning with UVB (time of low repair efficiency in nocturnal rodents) showed earlier onset of cancer and a five-fold higher incidence of invasive carcinomas compared to evening-treated mice [59,66,226].
Finally, in order for the mutated hyper-proliferative cells to become tumorigenic and metastatic, the cells must transform their microenvironment into one favorable to growth, epithelial-mesenchymal transition, and immune evasion. Favorable microenvironments for tumors possess high levels of proinflammatory cytokines and contain extracellular matrix (ECM) remodeling proteins, such as those in the matrix metallopeptidase protein family (MMP). When DNA damage goes unrepaired, levels of proinflammatory cytokines are known to increase. The circadian clock may further play a role in the regulation of these immune elements. An emerging topic in the immunological field implicates the circadian rhythm in both cytokine levels and immune cell infiltration [66,215,222,223]. Additionally, clock-controlled factors may influence ECM composition via a mechanism in which melatonin downregulates MMP-9 to facilitate ECM degradation. This, in turn, allows for potential expansion and movement of tumor cells [227]. In summary, given the high number of CCGs involved in DNA repair, cell cycle regulation, the immune system, and tumor microenvironment management, it is highly probable that circadian clock disruption is involved in mutagenesis, carcinogenesis, and expansion of skin cancer following UV light exposure (Fig. 6). These CCGs may also have roles in the onset of other cancer types, particularly those involving the endocrine or immune systems. These additional physiological programs, however, are beyond the scope of this review, and more research is needed at mechanistic levels to support causative links.
5. Concluding Remarks
The wealth of information available detailing the associations between skin pathologies, UVR, and circadian misalignment—both from an epidemiological and basic science research standpoint—clearly support a case implicating the disruption of the circadian rhythm in the process of skin carcinogenesis. However, even with the expansive literature explored above, there is much still unknown regarding the specifics of how UV exposure leads to carcinogenesis, proliferation, and metastasis. As detailed in the final section on Human Health, the circadian clock could intersect at any stage of the tumorigenic process, and its degree of involvement may even vary on a case-by-case basis. Future development of preventative and/or treatment measures would benefit from understanding: (1) if circadian disruption has more of a direct role in generating the mutations that cultivate genomic instability and eventual carcinogenesis, or (2) if it instead has an indirect role in promoting the growth of cancerous cells, and lastly (3) how circadian disruption mechanistically leads to these deleterious effects. To make progress towards this knowledge gap, development of a unified experimental design paradigm to investigate the circadian rhythm in cellular and animal model systems would likely lead to fewer discrepancies between studies. With the variety of entrainment methods—e.g., timed feeding, exercise, and the numerous experimental LD schedules—the prevalence of contradictory and incompatible results is of no surprise. Development of a more universal methodology should increase overall reproducibility—and not just for the topic of skin carcinogenesis discussed in this review. The circadian clock impacts the entire organism, and its disruption could have pathological ramifications across nearly all major peripheral organs. As such, consistent model-use has the potential to improve the integration of research findings across circadian studies within the skin and in other major organ systems.
ACKNOWLEDGEMENTS
We acknowledge BioRender for their figure building site which allowed us to produce all the figures in this manuscript. We also thank Elizabeth Schuetz for her final editing contributions.
FUNDING SOURCE
This work was supported by National Institutes of Health grant R01ES030113 and in part by the CDMRP Peer Reviewed Cancer Research Program Award W81XWH-18-1-0061 (S.G.), and by the National Institutes of Health T32 Training Grant 5T32ES007046-41 (S.J.C.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICTS OF INTEREST
The authors declare no potential conflict of interest.
References:
- [1].Masri S, Sassone-Corsi P, Plasticity and specificity of the circadian epigenome, Nat. Neurosci 13 (2010) 1324–1329. 10.1038/nn.2668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Allada R, Bass J, Circadian Mechanisms in Medicine, N. Engl. J. Med 384 (2021) 550–561. 10.1056/nejmra1802337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Gehring W, Rosbash M, The Coevolution of Blue-Light Photoreception and Circadian Rhythms, J. Mol. Evol 57 (2003) 286–289. 10.1007/s00239-003-0038-8. [DOI] [PubMed] [Google Scholar]
- [4].James SM, Honn KA, Gaddameedhi S, Van Dongen HPA, Shift Work: Disrupted Circadian Rhythms and Sleep-Implications for Health and Well-Being., Curr. Sleep Med. Reports 3 (2017) 104–112. 10.1007/s40675-017-0071-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Lunn RM, Schwingl PJ, Atwood ST, Mehta SS, Jahnke GD, Garner SC, NTP Cancer Hazard Assessment Report on Night Shift Work and Light at Night, 2021. 10.22427/NTP-CHR-NSWLAN. [DOI] [PubMed] [Google Scholar]
- [6].IARC Monographs Vol 124 group, Carcinogenicity of night shift work., Lancet. Oncol 20 (2019) 1058–1059. 10.1016/S1470-2045(19)30455-3. [DOI] [PubMed] [Google Scholar]
- [7].Foster RG, Peirson SN, Wulff K, Winnebeck E, Vetter C, Roenneberg T, Sleep and circadian rhythm disruption in social jetlag and mental illness., Prog. Mol. Biol. Transl. Sci 119 (2013) 325–46. 10.1016/B978-0-12-396971-2.00011-7. [DOI] [PubMed] [Google Scholar]
- [8].Sancar A, Van Gelder RN, Clocks, cancer, and chronochemotherapy, Science (80-. ) 371 (2021). 10.1126/science.abb0738. [DOI] [PubMed] [Google Scholar]
- [9].Koritala BSC, Porter KI, Arshad OA, Gajula RP, Mitchell HD, Arman T, Manjanatha MG, Teeguarden J, Van Dongen HPA, McDermott JE, Gaddameedhi S, Night shift schedule causes circadian dysregulation of DNA repair genes and elevated DNA damage in humans, J. Pineal Res (2021). 10.1111/jpi.12726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Schuch AP, Garcia CCM, Makita K, Menck CFM, DNA damage as a biological sensor for environmental sunlight, Photochem. Photobiol. Sci 12 (2013) 1259–1272. 10.1039/c3pp00004d. [DOI] [PubMed] [Google Scholar]
- [11].Franklin WA, Doetsch PW, Haseltine WA, Structural determinatlon of the ultraviolet light-induced thymine-cytosine pyrimidine-pyrimidone (6–4) photoproduct, Nucleic Acids Res. 13 (1985) 5317–5325. 10.1093/nar/13.14.5317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Vink AA, Berg RJ, de Gruijl FR, Roza L, Baan RA, Induction, repair and accumulation of thymine dimers in the skin of UV-B-irradiated hairless mice., Carcinogenesis. 12 (1991) 861–4. 10.1093/carcin/12.5.861. [DOI] [PubMed] [Google Scholar]
- [13].Hogenesch JB, Gu YZ, Jain S, Bradfield CA, The basic-helix-loop-helix-PAS orphan MOP3 forms transcriptionally active complexes with circadian and hypoxia factors, Proc. Natl. Acad. Sci. U. S. A 95 (1998) 5474–5479. 10.1073/pnas.95.10.5474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Bunger MK, Wilsbacher LD, Moran SM, Clendenin C, Radcliffe LA, Hogenesch JB, Simon MC, Takahashi JS, Bradfield CA, Mop3 Is an Essential Component of the Master Circadian Pacemaker in Mammals, Cell. 103 (2000) 1009–1017. 10.1016/S0092-8674(00)00205-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].King DP, Zhao Y, Sangoram AM, Wilsbacher LD, Tanaka M, Antoch MP, Steeves TDL, Vitaterna MH, Kornhauser JM, Lowrey PL, Turek FW, Takahashi JS, Positional Cloning of the Mouse Circadian Clock Gene, Cell. 89 (1997) 641–653. 10.1016/S0092-8674(00)80245-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Gekakis N, Staknis D, Nguyen HB, Davis FC, Wilsbacner LD, King DP, Takahashi JS, Weitz CJ, Role of the CLOCK protein in the mammalian circadian mechanism, Science (80-. ) 280 (1998) 1564–1569. 10.1126/science.280.5369.1564. [DOI] [PubMed] [Google Scholar]
- [17].Kume K, Zylka MJ, Sriram S, Shearman LP, Weaver DR, Jin X, Maywood ES, Hastings MH, Reppert SM, mCRY1 and mCRY2 are essential components of the negative limb of the circadian clock feedback loop, Cell. 98 (1999) 193–205. 10.1016/S0092-8674(00)81014-4. [DOI] [PubMed] [Google Scholar]
- [18].Vitaterna MH, Selby CP, Todo T, Niwa H, Thompson C, Fruechte EM, Hitomi K, Thresher RJ, Ishikawa T, Miyazaki J, Takahashi JS, Sancar A, Differential regulation of mammalian period genes and circadian rhythmicity by cryptochromes 1 and 2, Proc. Natl. Acad. Sci. U. S. A 96 (1999) 12114–12119. 10.1073/pnas.96.21.12114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Shearman LP, Sriram S, Weaver DR, Maywood ES, Chaves I, Zheng B, Kume K, Lee CC, Van Der Horst GTJ, Hastings MH, Reppert SM, Interacting molecular loops in the mammalian circadian clock, Science (80-. ) 288 (2000) 1013–1019. 10.1126/science.288.5468.1013. [DOI] [PubMed] [Google Scholar]
- [20].Lee C, Etchegaray J-P, Cagampang FRA, Loudon ASI, Reppert SM, Posttranslational Mechanisms Regulate the Mammalian Circadian Clock, Cell. 107 (2001) 855–867. 10.1016/S0092-8674(01)00610-9. [DOI] [PubMed] [Google Scholar]
- [21].Eide EJ, Vielhaber EL, Hinz WA, Virshup DM, The circadian regulatory proteins BMAL1 and cryptochromes are substrates of casein kinase Iε, J. Biol. Chem 277 (2002) 17248–17254. 10.1074/jbc.M111466200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Preitner N, Damiola F, Luis-Lopez-Molina Zakany J, Duboule D, Albrecht U, Schibler U, The orphan nuclear receptor REV-ERBα controls circadian transcription within the positive limb of the mammalian circadian oscillator, Cell. 110 (2002) 251–260. 10.1016/S0092-8674(02)00825-5. [DOI] [PubMed] [Google Scholar]
- [23].Ikeda R, Tsuchiya Y, Koike N, Umemura Y, Inokawa H, Ono R, Inoue M, Sasawaki Y, Grieten T, Okubo N, Ikoma K, Fujiwara H, Kubo T, Yagita K, REV-ERBα and REV-ERBβ function as key factors regulating Mammalian Circadian Output, Sci. Rep 9 (2019) 1–9. 10.1038/s41598-019-46656-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Akashi M, Soma H, Yamamoto T, Tsugitomi A, Yamashita S, Yamamoto T, Nishida E, Yasuda A, Liao JK, Node K, Noninvasive method for assessing the human circadian clock using hair follicle cells, Proc. Natl. Acad. Sci. U. S. A 107 (2010) 15643–15648. 10.1073/pnas.1003878107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Yan L, Smale L, Nunez AA, Circadian and photic modulation of daily rhythms in diurnal mammals, Eur. J. Neurosci 51 (2020) 551–566. 10.1111/ejn.14172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Vosko AM, Hagenauer MH, Hummer DL, Lee TM, Period gene expression in the diurnal degu (Octodon degus) differs from the nocturnal laboratory rat (Rattus norvegicus), Am. J. Physiol. - Regul. Integr. Comp. Physiol 296 (2009) 353–361. 10.1152/ajpregu.90392.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Paranjpe DA, Sharma VK, Evolution of temporal order in living organisms, J. Circadian Rhythms 3 (2005) 1–13. 10.1186/1740-3391-3-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Stothard ER, McHill AW, Depner CM, Birks BR, Moehlman TM, Ritchie HK, Guzzetti JR, Chinoy ED, LeBourgeois MK, Axelsson J, Wright KP, Circadian Entrainment to the Natural Light-Dark Cycle across Seasons and the Weekend, Curr. Biol 27 (2017) 508–513. 10.1016/j.cub.2016.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Buhr ED, Yoo SH, Takahashi JS, Temperature as a universal resetting cue for mammalian circadian oscillators, Science (80-. ) 330 (2010) 379–385. 10.1126/science.1195262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Edgar DM, Dement WC, Regularly scheduled voluntary exercise synchronizes the mouse circadian clock, Am. J. Physiol. - Regul. Integr. Comp. Physiol 261 (1991). 10.1152/ajpregu.1991.261.4.r928. [DOI] [PubMed] [Google Scholar]
- [31].Damiola F, Le Minli N, Preitner N, Kornmann B, Fleury-Olela F, Schibler U, Restricted feeding uncouples circadian oscillators in peripheral tissues from the central pacemaker in the suprachiasmatic nucleus, Genes Dev. 14 (2000) 2950–2961. 10.1101/gad.183500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Moore RY, Reppert SM, Klein DC, Suprachiasmatic nucleus: the mind’s clock, Oxford University Press, 1991. https://books.google.com/books?hl=en&lr=&id=8fgwFsmTBwgC&oi=fnd&pg=PR1&dq=.+Suprachiasmatic+nucleus:+the+mind%27s+clock.+&ots=qVEk8cT8aU&sig=6e0DyC6-Tq9SfRH5JVan8tiDMfw#v=onepage&q=.Suprachiasmaticnucleus%3Athemind’sclock.&f=false. [Google Scholar]
- [33].Masland RH, The fundamental plan of the retina, Nat. Neurosci 4 (2001) 877–886. 10.1038/nn0901-877. [DOI] [PubMed] [Google Scholar]
- [34].Fain G, Sampath AP, Rod and cone interactions in the retina [version 1; referees: 4 approved], F1000Research. 7 (2018) 1–9. 10.12688/f1000research.14412.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Cleland BG, Dubin MW, Levick WR, Sustained and transient neurones in the cat’s retina and lateral geniculate nucleus, J. Physiol 217 (1971) 473–496. 10.1113/jphysiol.1971.sp009581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Freedman MS, Lucas RJ, Soni B, Von Schantz M, Muñoz M, David-Gray Z, Foster R, Regulation of mammalian circadian behavior by non-rod, non-cone, ocular photoreceptors, Science (80-. ) 284 (1999) 502–504. 10.1126/science.284.5413.502. [DOI] [PubMed] [Google Scholar]
- [37].Lucas RJ, Freedman MS, Muñoz M, Garcia-Fernández JM, Foster RG, Regulation of the mammalian pineal by non-rod, non-cone, ocular photoreceptors, Science (80-. ) 284 (1999) 505–507. 10.1126/science.284.5413.505. [DOI] [PubMed] [Google Scholar]
- [38].Provencio I, Rodriguez IR, Jiang G, Hayes WP, Moreira EF, Rollag MD, A novel human opsin in the inner retina, J. Neurosci 20 (2000) 600–605. 10.1523/jneurosci.20-02-00600.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Hattar S, Liao HW, Takao M, Berson DM, Yau KW, Melanopsin-containing retinal ganglion cells: Architecture, projections, and intrinsic photosensitivity, Science (80-. ) 295 (2002) 1065–1070. 10.1126/science.1069609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].HARRINGTON ME, The Ventral Lateral Geniculate Nucleus and the Intergeniculate Leaflet: Interrelated Structures in the Visual and Circadian Systems, Neurosci. Biobehav. Rev 21 (1997) 705–727. 10.1016/S0149-7634(96)00019-X. [DOI] [PubMed] [Google Scholar]
- [41].Zhao X, Stafford BK, Godin AL, King WM, Wong KY, Photoresponse diversity among the five types of intrinsically photosensitive retinal ganglion cells, J. Physiol 592 (2014) 1619–1636. 10.1113/jphysiol.2013.262782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Schmidt TM, Alam NM, Chen S, Kofuji P, Li W, Prusky GT, Hattar S, A Role for Melanopsin in Alpha Retinal Ganglion Cells and Contrast Detection, Neuron. 82 (2014) 781–788. 10.1016/j.neuron.2014.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Sonoda T, Schmidt TM, Re-evaluating the Role of Intrinsically Photosensitive Retinal Ganglion Cells: New Roles in Image-Forming Functions, Integr. Comp. Biol 56 (2016) 834–841. 10.1093/icb/icw066. [DOI] [PubMed] [Google Scholar]
- [44].Nagoshi E, Saini C, Bauer C, Laroche T, Naef F, Schibler U, Circadian gene expression in individual fibroblasts: Cell-autonomous and self-sustained oscillators pass time to daughter cells, Cell. 119 (2004) 693–705. 10.1016/j.cell.2004.11.015. [DOI] [PubMed] [Google Scholar]
- [45].Richards J, Gumz ML, Advances in understanding the peripheral circadian clocks, FASEB J. 26 (2012) 3602–3613. 10.1096/fj.12-203554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Buijs RM, Kalsbeek A, Hypothalamic integration of central and peripheral clocks, Nat. Rev. Neurosci 2 (2001) 521–526. 10.1038/35081582. [DOI] [PubMed] [Google Scholar]
- [47].Saini C, Liani A, Curie T, Gos P, Kreppel F, Emmenegger Y, Bonacina L, Wolf JP, Poget YA, Franken P, Schibler U, Real-time recording of circadian liver gene expression in freely moving mice reveals the phase-setting behavior of hepatocyte clocks, Genes Dev. 27 (2013) 1526–1536. 10.1101/gad.221374.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Yamazaki S, Numano R, Abe M, Hida A, Takahashi RI, Ueda M, Block GD, Sakaki Y, Menaker M, Tei H, Resetting central and peripheral circadian oscillators in transgenic rats, Science (80-. ) 288 (2000) 682–685. 10.1126/science.288.5466.682. [DOI] [PubMed] [Google Scholar]
- [49].Akhtar RA, Reddy AB, Maywood ES, Clayton JD, King VM, Smith AG, Gant TW, Hastings MH, Kyriacou CP, Circadian cycling of the mouse liver transcriptome, as revealed by cDNA microarray, is driven by the suprachiasmatic nucleus, Curr. Biol 12 (2002) 540–550. 10.1016/S0960-9822(02)00759-5. [DOI] [PubMed] [Google Scholar]
- [50].Welsh DK, Yoo S-H, Liu AC, Takahashi JS, Kay SA, Bioluminescence Imaging of Individual Fibroblasts Reveals Persistent, Independently Phased Circadian Rhythms of Clock Gene Expression, Curr. Biol 14 (2004) 2289–2295. 10.1016/j.cub.2004.11.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Tahara Y, Kuroda H, Saito K, Nakajima Y, Kubo Y, Ohnishi N, Seo Y, Otsuka M, Fuse Y, Ohura Y, Komatsu T, Moriya Y, Okada S, Furutani N, Hirao A, Horikawa K, Kudo T, Shibata S, In vivo monitoring of peripheral circadian clocks in the mouse, Curr. Biol 22 (2012) 1029–1034. 10.1016/j.cub.2012.04.009. [DOI] [PubMed] [Google Scholar]
- [52].Yoo SH, Yamazaki S, Lowrey PL, Shimomura K, Ko CH, Buhr ED, Siepka SM, Hong HK, Oh WJ, Yoo OJ, Menaker M, Takahashi JS, PERIOD2∷LUCIFERASE real-time reporting of circadian dynamics reveals persistent circadian oscillations in mouse peripheral tissues, Proc. Natl. Acad. Sci. U. S. A 101 (2004) 5339–5346. 10.1073/pnas.0308709101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Sinturel F, Gos P, Petrenko V, Hagedorn C, Kreppel F, Storch KF, Knutti D, Liani A, Weitz C, Emmenegger Y, Franken P, Bonacina L, Dibner C, Schibler U, Circadian hepatocyte clocks keep synchrony in the absence of a master pacemaker in the suprachiasmatic nucleus or other extrahepatic clocks, Genes Dev. 35 (2021) 329–334. 10.1101/GAD.346460.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Kornmann B, Schaad O, Bujard H, Takahashi JS, Schibler U, System-driven and oscillator-dependent circadian transcription in mice with a conditionally active liver clock, PLoS Biol. 5 (2007) 0179–0189. 10.1371/journal.pbio.0050034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Stokkan KA, Yamazaki S, Tei H, Sakaki Y, Menaker M, Entrainment of the circadian clock in the liver by feeding, Science (80-. ) 291 (2001) 490–493. 10.1126/science.291.5503.490. [DOI] [PubMed] [Google Scholar]
- [56].Mavroudis PD, Scheff JD, Calvano SE, Lowry SF, Androulakis IP, Entrainment of peripheral clock genes by cortisol, Physiol. Genomics 44 (2012) 607–621. 10.1152/physiolgenomics.00001.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Tahara Y, Shiraishi T, Kikuchi Y, Haraguchi A, Kuriki D, Sasaki H, Motohashi H, Sakai T, Shibata S, Entrainment of the mouse circadian clock by sub-acute physical and psychological stress, Sci. Rep 5 (2015) 1–11. 10.1038/srep11417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Bruns DR, Yusifova M, Marcello NA, Green CJ, Walker WJ, Schmitt EE, The peripheral circadian clock and exercise: Lessons from young and old mice, J. Circadian Rhythms 18 (2021) 1–11. 10.5334/JCR.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Gaddameedhi S, Selby CP, Kaufmann WK, Smart RC, Sancar A, Control of skin cancer by the circadian rhythm, Proc. Natl. Acad. Sci. U. S. A 108 (2011) 18790–18795. 10.1073/pnas.1115249108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Tanioka M, Yamada H, Doi M, Bando H, Yamaguchi Y, Nishigori C, Okamura H, Molecular clocks in mouse skin, J. Invest. Dermatol 129 (2009) 1225–1231. 10.1038/jid.2008.345. [DOI] [PubMed] [Google Scholar]
- [61].Duan J, Greenberg EN, Karri SS, Andersen B, The circadian clock and diseases of the skin, FEBS Lett. 595 (2021) 2413–2436. 10.1002/1873-3468.14192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Geyfman M, Kumar V, Liu Q, Ruiz R, Gordon W, Espitia F, Cam E, Millar SE, Smyth P, Ihler A, Takahashi JS, Andersen B, Brain and muscle Arnt-like protein-1 (BMAL1) controls circadian cell proliferation and susceptibility to UVB-induced DNA damage in the epidermis, Proc. Natl. Acad. Sci. U. S. A 109 (2012) 11758–11763. 10.1073/pnas.1209592109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Matsui MS, Pelle E, Dong K, Pernodet N, Biological rhythms in the skin, Int. J. Mol. Sci 17 (2016). 10.3390/ijms17060801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Sandu C, Liu T, Malan A, Challet E, Pévet P, Felder-Schmittbuhl MP, Circadian clocks in rat skin and dermal fibroblasts: Differential effects of aging, temperature and melatonin, Cell. Mol. Life Sci 72 (2015) 2237–2248. 10.1007/s00018-014-1809-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Sarkar S, Porter KI, Dakup PP, Gajula RP, Koritala BSC, Hylton R, Kemp MG, Wakamatsu K, Gaddameedhi S, Circadian clock protein BMAL1 regulates melanogenesis through MITF in melanoma cells, Pigment Cell Melanoma Res. (2021). 10.1111/pcmr.12998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Gaddameedhi S, Selby CP, Kemp MG, Ye R, Sancar A, The Circadian Clock Controls Sunburn Apoptosis and Erythema in Mouse Skin, J. Invest. Dermatol 135 (2015) 1119–1127. 10.1038/jid.2014.508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Sun Y, Wang P, Li H, Dai J, BMAL1 and CLOCK proteins in regulating UVB-induced apoptosis and DNA damage responses in human keratinocytes, J. Cell. Physiol 233 (2018) 9563–9574. 10.1002/jcp.26859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Papp SJ, Huber AL, Jordan SD, Kriebs A, Nguyen M, Moresco JJ, Yates JR, Lamia KA, DNA damage shifts circadian clock time via hausp-dependent cry1 stabilization, Elife. 2015 (2015) 1–19. 10.7554/eLife.04883.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Kawamura G, Hattori M, Takamatsu K, Tsukada T, Ninomiya Y, Benjamin I, Sassone-Corsi P, Ozawa T, Tamaru T, Cooperative interaction among BMAL1, HSF1, and p53 protects mammalian cells from UV stress, Commun. Biol 1 (2018). 10.1038/s42003-018-0209-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Chan AB, Parico GCG, Fribourgh JL, Ibrahim LH, Bollong MJ, Partch CL, Lamia KA, CRY2 missense mutations suppress P53 and enhance cell growth, Proc. Natl. Acad. Sci. U. S. A 118 (2021) 1–10. 10.1073/pnas.2101416118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Kawara S, Mydlarski R, Mamelak AJ, Freed I, Wang B, Watanabe H, Shivji G, Tavadia SK, Suzuki H, Bjarnason GA, Jordan RCK, Sauder DN, Low-dose ultraviolet B rays alter the mRNA expression of the circadian clock genes in cultured human keratinocytes, J. Invest. Dermatol 119 (2002) 1220–1223. 10.1046/j.1523-1747.2002.19619.x. [DOI] [PubMed] [Google Scholar]
- [72].Nikkola V, Miettinen ME, Karisola P, Grönroos M, Ylianttila L, Alenius H, Snellman E, Partonen T, Ultraviolet B radiation modifies circadian time in epidermal skin and in subcutaneous adipose tissue, Photodermatol. Photoimmunol. Photomed 35 (2019) 157–163. 10.1111/phpp.12440. [DOI] [PubMed] [Google Scholar]
- [73].Nikkola V, Grönroos M, Huotari-Orava R, Kautiainen H, Ylianttila L, Karppinen T, Partonen T, Snellman E, Circadian Time Effects on NB-UVB–Induced Erythema in Human Skin In Vivo, J. Invest. Dermatol 138 (2018) 464–467. 10.1016/j.jid.2017.08.016. [DOI] [PubMed] [Google Scholar]
- [74].Wang H, van Spyk E, Liu Q, Geyfman M, Salmans ML, Kumar V, Ihler A, Li N, Takahashi JS, Andersen B, Time-Restricted Feeding Shifts the Skin Circadian Clock and Alters UVB-Induced DNA Damage, Cell Rep 20 (2017) 1061–1072. 10.1016/j.celrep.2017.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Gaucher J, Montellier E, Sassone-Corsi P, Molecular Cogs: Interplay between Circadian Clock and Cell Cycle, Trends Cell Biol. 28 (2018) 368–379. 10.1016/j.tcb.2018.01.006. [DOI] [PubMed] [Google Scholar]
- [76].Weger M, Diotel N, Dorsemans AC, Dickmeis T, Weger BD, Stem cells and the circadian clock, Dev. Biol 431 (2017) 111–123. 10.1016/j.ydbio.2017.09.012. [DOI] [PubMed] [Google Scholar]
- [77].Janich P, Pascual G, Merlos-Suárez A, Batlle E, Ripperger J, Albrecht U, Cheng HYM, Obrietan K, Di Croce L, Benitah SA, The circadian molecular clock creates epidermal stem cell heterogeneity, Nature. 480 (2011) 209–214. 10.1038/nature10649. [DOI] [PubMed] [Google Scholar]
- [78].Stanger BZ, Cellular homeostasis and repair in the mammalian liver, Annu. Rev. Physiol 77 (2015) 179–200. 10.1146/annurev-physiol-021113-170255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Karpowicz P, Zhang Y, Hogenesch JB, Emery P, Perrimon N, The Circadian Clock Gates the Intestinal Stem Cell Regenerative State, Cell Rep. 3 (2013) 996–1004. 10.1016/j.celrep.2013.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Kowalska E, Ripperger JA, Hoegger DC, Bruegger P, Buch T, Birchler T, Mueller A, Albrecht U, Contaldo C, Brown SA, NONO couples the circadian clock to the cell cycle, Proc. Natl. Acad. Sci. U. S. A 110 (2013) 1592–1599. 10.1073/pnas.1213317110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Geyfman M, Kumar V, Liu Q, Ruiz R, Gordon W, Espitia F, Cam E, Millar SE, Smyth P, Ihler A, Takahashi JS, Andersen B, Brain and muscle Arnt-like protein-1 (BMAL1) controls circadian cell proliferation and susceptibility to UVB-induced DNA damage in the epidermis, Proc. Natl. Acad. Sci. U. S. A 109 (2012) 11758–11763. 10.1073/pnas.1209592109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Bouchard-Cannon P, Mendoza-Viveros L, Yuen A, Kærn M, Cheng HYM, The Circadian Molecular Clock Regulates Adult Hippocampal Neurogenesis by Controlling the Timing of Cell-Cycle Entry and Exit, Cell Rep. 5 (2013) 961–973. 10.1016/j.celrep.2013.10.037. [DOI] [PubMed] [Google Scholar]
- [83].Gotoh T, Vila-Caballer M, Santos CS, Liu J, Yang J, Finkielstein CV, The circadian factor Period 2 modulates p53 stability and transcriptional activity in unstressed cells, Mol. Biol. Cell 25 (2014) 3081–3093. 10.1091/mbc.E14-05-0993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Fu L, Pelicano H, Liu J, Huang P, Lee CC, The Circadian Gene Period2 Plays an Important Role in Tumor Suppression and DNA Damage Response In Vivo, Cell. 111 (2002) 41–50. [DOI] [PubMed] [Google Scholar]
- [85].Farshadi E, Van Der Horst GTJ, Chaves I, Molecular Links between the Circadian Clock and the Cell Cycle, J. Mol. Biol 432 (2020) 3515–3524. 10.1016/j.jmb.2020.04.003. [DOI] [PubMed] [Google Scholar]
- [86].Henley SA, Dick FA, The retinoblastoma family of proteins and their regulatory functions in the mammalian cell division cycle, Cell Div. (2012) 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Sherr CJ, Mccormick F, The RB and p53 pathways in cancer, Cancer Cell. 2 (2002) 103–112. [DOI] [PubMed] [Google Scholar]
- [88].Kolinjivadi AM, Chong ST, Ngeow J, Molecular connections between circadian rhythm and genome maintenance pathways, Endocr. Relat. Cancer 28 (2021) R55–R66. 10.1530/ERC-20-0390. [DOI] [PubMed] [Google Scholar]
- [89].Gréchez-Cassiau A, Rayet B, Guillaumond F, Teboul M, Delaunay F, The circadian clock component BMAL1 is a critical regulator of p21 WAF1/CIP1 expression and hepatocyte proliferation, J. Biol. Chem 283 (2008) 4535–4542. 10.1074/jbc.M705576200. [DOI] [PubMed] [Google Scholar]
- [90].Gotoh T, Vila-Caballer M, Liu J, Schiffhauer S, Finkielstein CV, Association of the circadian factor Period 2 to p53 influences p53’s function in DNA-damage signaling, Mol. Biol. Cell 26 (2015) 359–372. 10.1091/mbc.E14-05-0994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Gotoh T, Kim JK, Liu J, Vila-Caballer M, Stauffer PE, Tyson JJ, Finkielstein CV, Model-driven experimental approach reveals the complex regulatory distribution of p53 by the circadian factor period 2, Proc. Natl. Acad. Sci. U. S. A 113 (2016) 13516–13521. 10.1073/pnas.1607984113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Miki T, Matsumoto T, Zhao Z, Lee CC, p53 regulates Period2 expression and the circadian clock., Nat. Commun 4 (2013) 2444. 10.1038/ncomms3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Sancar A, Lindsey-boltz LA, Kang T, Reardon JT, Lee JH, Ozturk N, Circadian clock control of the cellular response to DNA damage, FEBS Lett. 584 (2010) 2618–2625. 10.1016/j.febslet.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Gery S, Komatsu N, Baldjyan L, Yu A, Koo D, Koeffler HP, The Circadian Gene Per1 Plays an Important Role in Cell Growth and DNA Damage Control in Human Cancer Cells, Mol. Cell (2006) 375–382. 10.1016/j.molcel.2006.03.038. [DOI] [PubMed] [Google Scholar]
- [95].Mullen TE, Kaufmann WK, Sancar A, Coupling of Human Circadian and Cell Cycles by the Timeless Protein, Mol. Cell. Biol 25 (2005) 3109–3116. 10.1128/MCB.25.8.3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Kang TH, Leem SH, Modulation of ATR-mediated DNA damage checkpoint response by cryptochrome 1, Nucleic Acids Res. 42 (2014) 4427–4434. 10.1093/nar/gku094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Farshadi E, Yan J, Leclere P, Goldbeter A, Chaves I, van der Horst GTJ, The positive circadian regulators CLOCK and BMAL1 control G2/M cell cycle transition through Cyclin B1, Cell Cycle. 18 (2019) 16–33. 10.1080/15384101.2018.1558638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Matsuo T, Yamaguchi S, Mitsui S, Emi A, Shimoda F, Okamura H, Control Mechanism of the Circadian Clock for Timing of Cell Division In vivo Author ( s ): Matsuo Takuya, Yamaguchi Shun, Mitsui Shigeru, Emi Aki, Shimoda Fukuko and Okamura Hitoshi, Science (80-. ) 302 (2003) 255–259. [DOI] [PubMed] [Google Scholar]
- [99].Zhao X, Hirota T, Han X, Cho H, Chong LW, Lamia K, Liu S, Atkins AR, Banayo E, Liddle C, Yu RT, Yates JR, Kay SA, Downes M, Evans RM, Circadian Amplitude Regulation via FBXW7-Targeted REV-ERBα Degradation, Cell. 165 (2016) 1644–1657. 10.1016/j.cell.2016.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Organization WH, Ultraviolet radiation., Environ. Heal. Criteria (1994). http://www.inchem.org/documents/ehc/ehc/ehc160.htm. [Google Scholar]
- [101].Hölzle E, Hönigsmann H, Ultraviolette Strahlung – Quellen, Spektren, Umwelteinflüsse, JDDG J. Der Dtsch. Dermatologischen Gesellschaft 3 (2005) S3–S10. 10.1111/j.1610-0387.2005.04392.x. [DOI] [PubMed] [Google Scholar]
- [102].Bruls WAG, Slaper H, Van Der Leun JC, Berrens L, Transmission of Human Epidermis and Stratum Corneum As a Function of Thickness in the Ultraviolet and Visible Wavelengths, Photochem. Photobiol 40 (1984) 485–494. 10.1111/j.1751-1097.1984.tb04622.x. [DOI] [PubMed] [Google Scholar]
- [103].Goldberg LH, Altman A, Benign skin changes associated with chronic sunlight exposure., Cutis. 34 (1984) 33–8, 40. http://www.ncbi.nlm.nih.gov/pubmed/6467973. [PubMed] [Google Scholar]
- [104].Adams JS, Clemens TL, Parrish JA, Holick MF, Vitamin-D Synthesis and Metabolism after Ultraviolet Irradiation of Normal and Vitamin-D-Deficient Subjects, N. Engl. J. Med 306 (1982) 722–725. 10.1056/NEJM198203253061206. [DOI] [PubMed] [Google Scholar]
- [105].Kripke ML, Cox PA, Alas LG, Yarosh DB, Pyrimidine dimers in DNA initiate systemic immunosuppression in UV- irradiated mice, Proc. Natl. Acad. Sci. U. S. A 89 (1992) 7516–7520. 10.1073/pnas.89.16.7516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Bauer JR, Bruins AA, Hardeberg JY, Verdaasdonk RM, A spectral filter array camera for clinical monitoring and diagnosis: Proof of concept for skin oxygenation imaging, J. Imaging 5 (2019) 1–23. 10.3390/jimaging5080066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Young AR, Physics in Medicine & Biology Chromophores in human skin Chromophores in human skin, Phys. Med. Biol 42 (1997). [DOI] [PubMed] [Google Scholar]
- [108].Anderson RR, Parrish JA, The optics of human skin, J. Invest. Dermatol 77 (1981) 13–19. 10.1111/1523-1747.ep12479191. [DOI] [PubMed] [Google Scholar]
- [109].Beaven GH, Holiday ER, Ultraviolet Absorption Spectra of Proteins and Amino Acids, in: Anson ML, Bailey K, J.T.B.T.-A. in Edsall PC (Eds.), Adv. Protein Chem, Academic Press, 1952: pp. 319–386. 10.1016/S0065-3233(08)60022-4. [DOI] [PubMed] [Google Scholar]
- [110].Chu EHY, Effects of ultraviolet radiation on mammalian cells I. Induction of chromosome aberrations, Mutat. Res. - Fundam. Mol. Mech. Mutagen 2 (1965) 75–94. 10.1016/0027-5107(65)90010-2. [DOI] [PubMed] [Google Scholar]
- [111].Limoli CL, Giedzinski E, Bonner WM, Cleaver JE, UV-induced replication arrest in the xeroderma pigmentosum variant leads to DNA doublestrand breaks, γ-H2AX formation, and Mre11 relocalization, Proc. Natl. Acad. Sci. U. S. A 99 (2002) 233–238. 10.1073/pnas.231611798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Chadwick CA, Potten CS, Nikaido O, Matsunaga T, Proby C, Young AR, The detection of cyclobutane thymine dimers, (6–4) photolesions and the Dewar photoisomers in sections of UV-irradiated human skin using specific antibodies, and the demonstration of depth penetration effects, J. Photochem. Photobiol. B Biol 28 (1995) 163–170. 10.1016/1011-1344(94)07096-7. [DOI] [PubMed] [Google Scholar]
- [113].Mitchell DL, Nairn RS, The biology of the (6–4) photoproduct., Photochem. Photobiol 49 (1989) 805–19. 10.1111/j.1751-1097.1989.tb05578.x. [DOI] [PubMed] [Google Scholar]
- [114].Shah P, He Y-Y, Molecular regulation of UV-induced DNA repair., Photochem. Photobiol 91 (2017) 254–64. 10.1111/php.12406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].D’Orazio J, Jarrett S, Amaro-Ortiz A, Scott T, UV radiation and the skin, Int. J. Mol. Sci 14 (2013) 12222–12248. 10.3390/ijms140612222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Baptista MS, Cadet J, Di Mascio P, Ghogare AA, Greer A, Hamblin MR, Lorente C, Nunez SC, Ribeiro MS, Thomas AH, Vignoni M, Yoshimura TM, Type I and Type II Photosensitized Oxidation Reactions: Guidelines and Mechanistic Pathways, Photochem. Photobiol 93 (2017) 912–919. 10.1111/php.12716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Cooke MS, Evans MD, Dizdaroglu M, Lunec J, Oxidative DNA damage: mechanisms, mutation, and disease, FASEB J. 17 (2003) 1195–1214. 10.1096/fj.02-0752rev. [DOI] [PubMed] [Google Scholar]
- [118].Courdavault S, Baudouin C, Charveron M, Favier A, Cadet J, Douki T, Larger yield of cyclobutane dimers than 8-oxo-7,8-dihydroguanine in the DNA of UVA-irradiated human skin cells, Mutat. Res. - Fundam. Mol. Mech. Mutagen 556 (2004) 135–142. 10.1016/j.mrfmmm.2004.07.011. [DOI] [PubMed] [Google Scholar]
- [119].Bastien N, Therrien JP, Drouin R, Cytosine containing dipyrimidine sites can be hotspots of cyclobutane pyrimidine dimer formation after UVB exposure, Photochem. Photobiol. Sci 12 (2013) 1544–1554. 10.1039/c3pp50099c. [DOI] [PubMed] [Google Scholar]
- [120].Saini N, Giacobone CK, Klimczak LJ, Papas BN, Burkholder AB, Li JL, Fargo DC, Bai R, Gerrish K, Innes CL, Schurman SH, Gordenin DA, UV-exposure, endogenous DNA damage, and DNA replication errors shape the spectra of genome changes in human skin, PLoS Genet. 17 (2021) 1–23. 10.1371/journal.pgen.1009302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Reddy BY, Miller DM, Tsao H, Somatic driver mutations in melanoma, Cancer. 123 (2017) 2104–2117. 10.1002/cncr.30593. [DOI] [PubMed] [Google Scholar]
- [122].Bonilla X, Parmentier L, King B, Bezrukov F, Kaya G, Zoete V, Seplyarskiy VB, Sharpe HJ, McKee T, Letourneau A, Ribaux PG, Popadin K, Basset-Seguin N, Ben Chaabene R, Santoni FA, Andrianova MA, Guipponi M, Garieri M, Verdan C, Grosdemange K, Sumara O, Eilers M, Aifantis I, Michielin O, De Sauvage FJ, Antonarakis SE, Nikolaev SI, Genomic analysis identifies new drivers and progression pathways in skin basal cell carcinoma, Nat. Genet 48 (2016) 398–406. 10.1038/ng.3525. [DOI] [PubMed] [Google Scholar]
- [123].Pellegrini C, Maturo MG, Di Nardo L, Ciciarelli V, Gutiérrez García-Rodrigo C, Fargnoli MC, Understanding the molecular genetics of basal cell carcinoma, Int. J. Mol. Sci 18 (2017). 10.3390/ijms18112485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Chang D, Shain AH, The landscape of driver mutations in cutaneous squamous cell carcinoma, BioRxiv. (2020) 2020.12.13.422581. 10.1101/2020.12.13.422581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat J-P, Nickerson E, Auclair D, Li L, Place C, DiCara D, Ramos AH, Lawrence MS, Cibulskis K, Sivachenko A, Voet D, Saksena G, Stransky N, Onofrio RC, Winckler W, Ardlie K, Wagle N, Wargo J, Chong K, Morton DL, Stemke-Hale K, Chen G, Noble M, Meyerson M, Ladbury JE, Davies MA, Gershenwald JE, Wagner SN, Hoon DSB, Schadendorf D, Lander ES, Gabriel SB, Getz G, Garraway LA, Chin L, A Landscape of Driver Mutations in Melanoma, Cell. 150 (2012) 251–263. 10.1016/j.cell.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Guy GP, Machlin SR, Ekwueme DU, Yabroff KR, Prevalence and costs of skin cancer treatment in the U.S., 2002–2006 and 2007–2011, Am. J. Prev. Med 48 (2015) 183–187. 10.1016/j.amepre.2014.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Parkin DM, Mesher D, Sasieni P, Cancers attributable to solar (ultraviolet) radiation exposure in the UK in 2010, Br. J. Cancer 105 (2011) S66–S69. 10.1038/bjc.2011.486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Armstrong BK, How sun exposure causes skin cancer: an epidemiological perspective, in: Prev. Ski. Cancer, Springer, 2004: pp. 89–116. https://books.google.com/books?hl=en&lr=&id=qeO9BwAAQBAJ&oi=fnd&pg=PA88&ots=HaGyU3E2f-&sig=EZd9423A1448_dPROyyR1e69Fy4#v=onepage&q&f=false. [Google Scholar]
- [129].Gloster HM, Neal K, Skin cancer in skin of color, J. Am. Acad. Dermatol 55 (2006) 741–760. 10.1016/j.jaad.2005.08.063. [DOI] [PubMed] [Google Scholar]
- [130].Glass AG, Hoover RN, The Emerging Epidemic of Melanoma and Squamous Cell Skin Cancer, JAMA J. Am. Med. Assoc 262 (1989) 2097–2100. 10.1001/jama.1989.03430150065027. [DOI] [PubMed] [Google Scholar]
- [131].Muzic JG, Schmitt AR, Wright AC, Alniemi DT, Zubair AS, Olazagasti Lourido JM, Sosa Seda IM, Weaver AL, Baum CL, Incidence and Trends of Basal Cell Carcinoma and Cutaneous Squamous Cell Carcinoma, Mayo Clin. Proc 92 (2017) 890–898. 10.1016/j.mayocp.2017.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Whiteman DC, Green AC, Olsen CM, The Growing Burden of Invasive Melanoma: Projections of Incidence Rates and Numbers of New Cases in Six Susceptible Populations through 2031, J. Invest. Dermatol 136 (2016) 1161–1171. 10.1016/j.jid.2016.01.035. [DOI] [PubMed] [Google Scholar]
- [133].Gandini S, Sera F, Cattaruzza MS, Pasquini P, Picconi O, Boyle P, Melchi CF, Meta-analysis of risk factors for cutaneous melanoma: II. Sun exposure, Eur. J. Cancer 41 (2005) 45–60. 10.1016/j.ejca.2004.10.016. [DOI] [PubMed] [Google Scholar]
- [134].Cleaver JE, Cancer in xeroderma pigmentosum and related disorders of DNA repair, Nat. Rev. Cancer 5 (2005) 564–573. 10.1038/nrc1652. [DOI] [PubMed] [Google Scholar]
- [135].Wang Y, Digiovanna JJ, Stern JB, Hornyak TJ, Raffeld M, Khan SG, Oh K-S, Hollander MC, Dennis PA, Kraemer KH, Evidence of ultraviolet type mutations in xeroderma pigmentosum melanomas., Proc. Natl. Acad. Sci. U. S. A 106 (2009) 6279–84. 10.1073/pnas.0812401106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Dakup P, Gaddameedhi S, Impact of the Circadian Clock on UV-Induced DNA Damage Response and Photocarcinogenesis, Photochem. Photobiol 93 (2017) 296–303. 10.1111/php.12662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].D’Errico M, Teson M, Calcagnile A, Proietti De Santis L, Nikaido O, Botta E, Zambruno G, Stefanini M, Dogliotti E, Apoptosis and efficient repair of DNA damage protect human keratinocytes against UVB [2], Cell Death Differ. 10 (2003) 754–756. 10.1038/sj.cdd.4401224. [DOI] [PubMed] [Google Scholar]
- [138].Reardon JT, Sancar A, Nucleotide Excision Repair, Prog. Nucleic Acid Res. Mol. Biol 79 (2005) 183–235. 10.1016/S0079-6603(04)79004-2. [DOI] [PubMed] [Google Scholar]
- [139].Matsumura Y, Ananthaswamy HN, Toxic effects of ultraviolet radiation on the skin, Toxicol. Appl. Pharmacol 195 (2004) 298–308. 10.1016/j.taap.2003.08.019. [DOI] [PubMed] [Google Scholar]
- [140].Schärer OD, Nucleotide excision repair in Eukaryotes, Cold Spring Harb. Perspect. Biol 5 (2013) 1–19. 10.1101/cshperspect.a012609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Svejstrup JQ, Rescue of arrested RNA polymerase II complexes, J. Cell Sci 116 (2003) 447–451. 10.1242/jcs.00271. [DOI] [PubMed] [Google Scholar]
- [142].Riedl T, Hanaoka F, Egly JM, The comings and goings of nucleotide excision repair factors on damaged DNA, EMBO J. 22 (2003) 5293–5303. 10.1093/emboj/cdg489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Kang TH, Reardon JT, Kemp M, Sancar A, Orcadian oscillation of nucleotide excision repair in mammalian brain, Proc. Natl. Acad. Sci. U. S. A 106 (2009) 2864–2867. 10.1073/pnas.0812638106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Anabtawi N, Cvammen W, Kemp MG, Pharmacological inhibition of cryptochrome and REV-ERB promotes DNA repair and cell cycle arrest in cisplatin-treated human cells., Sci. Rep 11 (2021) 17997. 10.1038/s41598-021-97603-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Yang Y, Adebali O, Wu G, Selby CP, Chiou YY, Rashid N, Hu J, Hogenesch JB, Sancar A, Cisplatin-DNA adduct repair of transcribed genes is controlled by two circadian programs in mouse tissues, Proc. Natl. Acad. Sci. U. S. A 115 (2018) E4777–E4785. 10.1073/pnas.1804493115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Auclair Y, Rouget R, Drobetsky EA, ATR kinase as master regulator of nucleotide excision repair during S phase of the cell cycle, Cell Cycle. 8 (2009) 1865–1871. 10.4161/cc.8.12.8800. [DOI] [PubMed] [Google Scholar]
- [147].Bracci M, Ciarapica V, Zabaleta ME, Tartaglione MF, Pirozzi S, Giuliani L, Piva F, Valentino M, Ledda C, Rapisarda V, Stevens RG, Santarelli L, BRCA1 and BRCA2 Gene Expression: Diurnal Variability and Influence of Shift Work, Cancers (Basel). 11 (2019). 10.3390/cancers11081146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Manzella N, Bracci M, Strafella E, Staffolani S, Ciarapica V, Copertaro A, Rapisarda V, Ledda C, Amati M, Valentino M, Tomasetti M, Stevens RG, Santarelli L, Circadian Modulation of 8-Oxoguanine DNA Damage Repair., Sci. Rep 5 (2015) 13752. 10.1038/srep13752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Shafi AA, McNair CM, McCann JJ, Alshalalfa M, Shostak A, Severson TM, Zhu Y, Bergman A, Gordon N, Mandigo AC, Chand SN, Gallagher P, Dylgjeri E, Laufer TS, Vasilevskaya IA, Schiewer MJ, Brunner M, Feng FY, Zwart W, Knudsen KE, The circadian cryptochrome, CRY1, is a pro-tumorigenic factor that rhythmically modulates DNA repair, Nat. Commun 12 (2021). 10.1038/s41467-020-20513-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Ka NL, Na TY, Na H, Lee MH, Park HS, Hwang S, Kim IY, Seong JK, Lee MO, NR1D1 recruitment to sites of DNA damage inhibits repair and is associated with chemosensitivity of breast cancer, Cancer Res. 77 (2017) 2453–2463. 10.1158/0008-5472.CAN-16-2099. [DOI] [PubMed] [Google Scholar]
- [151].Majidinia M, Sadeghpour A, Mehrzadi S, Reiter RJ, Khatami N, Yousefi B, Melatonin: A pleiotropic molecule that modulates DNA damage response and repair pathways., J. Pineal Res 63 (2017). 10.1111/jpi.12416. [DOI] [PubMed] [Google Scholar]
- [152].Kuramoto T, Nakanishi S, Ochiai M, Nakagama H, Voigt B, Serikawa T, Origins of albino and hooded rats: Implications from molecular genetic analysis across modern laboratory rat strains, PLoS One. 7 (2012). 10.1371/journal.pone.0043059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Bryda EC, The Mighty Mouse: the impact of rodents on advances in biomedical research, Mo. Med 110 (2013) 207. [PMC free article] [PubMed] [Google Scholar]
- [154].Szpirer C, Rat models of human diseases and related phenotypes: A systematic inventory of the causative genes, J. Biomed. Sci 27 (2020). 10.1186/s12929-020-00673-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Blum HF, Sunlight and cancer of the skin, J. Natl. Cancer Inst 1 (1940) 397–421. [Google Scholar]
- [156].Abrikosoff A, Über den Einfluss der ultravioletten Strahlen auf den Regenerationsprozess der Kaninchenhaut, Verhandl. Deutsch. Path. Gesel-Isch 21 (1926) 163. [Google Scholar]
- [157].Findlay GM, Ultra-violet light and skin cancer., Lancet. (1928) 1070–1073. [DOI] [PubMed] [Google Scholar]
- [158].Putschar W, Holtz F, Erzeugung von Hautkrebsen bei Ratten durch langdauernde Ultraviolettbestrahlung, Z. Krebsforsch 33 (1931) 219–260. [Google Scholar]
- [159].Herlitz CW, Jundell I, Wahlgren F, Durch Ultraviolettbestrahlung erzeugte maligne Neu-bildungen bei weissen Mäusen. 1, Acta Pædiatrica. 10 (1931) 321–347. [Google Scholar]
- [160].Roffo AH, Effect of sunlight on the development of cancer. Experimental results., Zentralbl. Allg. Pathol 62 (1935) 324–334. [Google Scholar]
- [161].Roffo AH, Role Ultra-Violet Rays in the Development of Cancer Provoked by the Sun., Role Ultra-Violet Rays Dev. Cancer Provoked by Sun (1936) 472–474. [Google Scholar]
- [162].Kohn-Speyer A, EFFECT OF ULTRA-VIOLET RADIATION ON THE INCIDENCE OF TAR CANCER IN MICE., Lancet. 214 (1929) 1305–1306. [Google Scholar]
- [163].Rusch HP, Baumann CA, Tumor production in mice with ultraviolet irradiation, Am. J. Cancer 35 (1939) 55–62. [Google Scholar]
- [164].Montagna W, Carlisle K, The architecture of black and white facial skin, J. Am. Acad. Dermatol 24 (1991) 929–937. [DOI] [PubMed] [Google Scholar]
- [165].De Gálvez MV, Aguilera J, Bernabõ JL, Sánchez-Roldán C, Herrera-Ceballos E, Human Hair as a Natural Sun Protection Agent: A Quantitative Study, Photochem. Photobiol 91 (2015) 966–970. 10.1111/php.12433. [DOI] [PubMed] [Google Scholar]
- [166].Crew FAE, Mirskaia L, The character “hairless” in the mouse, J. Genet 25 (1931) 17–24. [Google Scholar]
- [167].Begoia Cachongonzalez M, Fennert S, Coffint JM, Morant C, Best S, Stoye JP, Structure and expression of the hairless gene of mice, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Benavides F, Oberyszyn TM, VanBuskirk AM, Reeve VE, Kusewitt DF, The hairless mouse in skin research, J. Dermatol. Sci 53 (2009) 10–18. 10.1016/j.jdermsci.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Zarach JM, Beaudoin GMJ, Coulombe PA, Thompson CC, The co-repressor hairless has a role in epithelial cell differentiation in the skin, Development. 131 (2004) 4189–4200. 10.1242/dev.01303. [DOI] [PubMed] [Google Scholar]
- [170].Zhu K, Xu C, Liu M, Zhang J, Hairless controls hair fate decision via Wnt/β–catenin signaling, Biochem. Biophys. Res. Commun 491 (2017) 567–570. 10.1016/j.bbrc.2017.07.164. [DOI] [PubMed] [Google Scholar]
- [171].de Gruijl FR, Forbes PD, UV-induced skin cancer in a hairless mouse model, Bioessays. 17 (1995) 651–660. [DOI] [PubMed] [Google Scholar]
- [172].Konopka RJ, Benzer S, Clock mutants of Drosophila melanogaster, Proc. Natl. Acad. Sci 68 (1971) 2112–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Zehring WA, Wheeler DA, Reddy P, Konopka RJ, Kyriacou CP, Rosbash M, Hall JC, P-element transformation with period locus DNA restores rhythmicity to mutant, arrhythmic Drosophila melanogaster., Cell. 39 (1984) 369–376. http://ovidsp.ovid.com/ovidweb.cgi?T=JS&CSC=Y&NEWS=N&PAGE=fulltext&D=med2&AN=6094014. [DOI] [PubMed] [Google Scholar]
- [174].Young MW, Bargiello TA, Jackson FR, Restoration of circadian bahavioural rhythms by gene transfer in Drosophila, Nature. 312 (1984) 752–754. papers2://publication/uuid/4E3D25C9-A9CF-4649-ADD0-655FF6BB01BD. [DOI] [PubMed] [Google Scholar]
- [175].Rutila JE, Suri V, Le M, So WV, Rosbash M, Hall JC, Cycle is a second bHLH-PAS clock protein essential for circadian rhythmicity and transcription of Drosophila period and timeless, Cell. 93 (1998) 805–814. 10.1016/S0092-8674(00)81441-5. [DOI] [PubMed] [Google Scholar]
- [176].Emery P, So WV, Kaneko M, Hall JC, Rosbash M, Cry, a Drosophila clock and light-regulated cryptochrome, is a major contributor to circadian rhythm resetting and photosensitivity, Cell. 95 (1998) 669–679. 10.1016/S0092-8674(00)81637-2. [DOI] [PubMed] [Google Scholar]
- [177].Allada R, White NE, So WV, Hall JC, Rosbash M, A mutant Drosophila homolog of mammalian clock disrupts circadian rhythms and transcription of period and timeless, Cell. 93 (1998) 791–804. 10.1016/S0092-8674(00)81440-3. [DOI] [PubMed] [Google Scholar]
- [178].Rosato E, Tauber E, Kyriacou CP, Molecular genetics of the fruit-fly circadian clock, Eur. J. Hum. Genet 14 (2006) 729–738. 10.1038/sj.ejhg.5201547. [DOI] [PubMed] [Google Scholar]
- [179].Vitaterna MH, King DP, Chang AM, Kornhauser JM, Lowrey PL, McDonald JD, Dove WF, Pinto LH, Turek FW, Takahashi JS, Mutagenesis and mapping of a mouse gene, Clock, essential for circadian behavior, Science. 264 (1994) 719–725. 10.1126/science.8171325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].van der Horst GTJ, Muijtjens M, Kobayashi K, Takano R, Kanno S, Takao M, de Wit J, Verkerk A, Eker APM, van Leenen D, Buijs R, Bootsma D, Hoeijmakers JHJ, Yasui A, Mammalian Cry1 and Cry2 are essential for maintenance of circadian rhythms, Nature. 398 (1999) 627–630. 10.1038/19323. [DOI] [PubMed] [Google Scholar]
- [181].Bae K, Jin X, Maywood ES, Hastings MH, Reppert SM, Weaver DR, Differential Functions of mPer1, mPer2, and mPer3 in the SCN Circadian Clock al The negative feedback loop involves the dynamic reg, 2001. [DOI] [PubMed] [Google Scholar]
- [182].Zheng B, Larkin DW, Albrecht U, Sun ZS, Sage M, Eichele G, Lee CC, Bradley A, The mPer2 gene encodes a functional component of the mammalian circadian clock, Nature. 400 (1999) 169–173. 10.1038/22118. [DOI] [PubMed] [Google Scholar]
- [183].Shearman LP, Jin X, Lee C, Reppert SM, Weaver DR, Targeted Disruption of the mPer3 Gene: Subtle Effects on Circadian Clock Function, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Skornyakov E, Gaddameedhi S, Paech GM, Sparrow AR, Satterfield BC, Shattuck NL, Layton ME, Karatsoreos I, VAN Dongen HPA, Cardiac autonomic activity during simulated shift work, Ind. Health 57 (2019) 118–132. 10.2486/indhealth.2018-0044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Winget CM, DeRoshia CW, Markley CL, Holley DC, A review of human physiological and performance changes associated with desynchronosis of biological rhythms, Aviat. Space. Environ. Med 55 (1984) 1085–1096. [PubMed] [Google Scholar]
- [186].A.C. of E. Physicians, Policy statement. Emergency physician shift work, Ann Emerg Med 43 (2017) 151–152. [Google Scholar]
- [187].Straif K, Baan R, Grosse Y, Secretan B, El Ghissassi F, Bouvard V, Altieri A, Benbrahim-Tallaa L, Cogliano V, Carcinogenicity of shift-work, painting, and fire-fighting., Lancet Oncol. 8 (2007) 1065–1066. 10.1016/s1470-2045(07)70373-x. [DOI] [PubMed] [Google Scholar]
- [188].Fritschi L, Glass DC, Heyworth JS, Aronson K, Girschik J, Boyle T, Grundy A, Erren TC, Hypotheses for mechanisms linking shiftwork and cancer, Med. Hypotheses 77 (2011) 430–436. 10.1016/j.mehy.2011.06.002. [DOI] [PubMed] [Google Scholar]
- [189].Salgado-Delgado R, Ángeles-Castellanos M, Buijs MR, Escobar C, Internal desynchronization in a model of night-work by forced activity in rats, Neuroscience. 154 (2008) 922–931. 10.1016/j.neuroscience.2008.03.066. [DOI] [PubMed] [Google Scholar]
- [190].Tsai L-L, Tsai Y-C, Hwang K, Huang Y-W, Tzeng J-E, Repeated light-dark shifts speed up body weight gain in male F344 rats Downloaded from, Am J Physiol Endocrinol Metab. 289 (2005) 212–217. 10.1152/ajpendo.00603.2004.-This. [DOI] [PubMed] [Google Scholar]
- [191].Van Dycke KCG, Rodenburg W, Van Oostrom CTM, Van Kerkhof LWM, Pennings JLA, Roenneberg T, Van Steeg H, Van Der Horst GTJ, Chronically Alternating Light Cycles Increase Breast Cancer Risk in Mice, Curr. Biol 25 (2015) 1932–1937. 10.1016/j.cub.2015.06.012. [DOI] [PubMed] [Google Scholar]
- [192].Figueiro MG, Radetsky L, Plitnick B, Rea MS, Glucose tolerance in mice exposed to light-dark stimulus patterns mirroring dayshift and rotating shift schedules, Sci. Rep 7 (2017). 10.1038/srep40661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Christie S, Vincent AD, Li H, Frisby CL, Kentish SJ, O’rielly R, Wittert GA, Page AJ, A rotating light cycle promotes weight gain and hepatic lipid storage in mice, Am J Physiol Gastrointest Liver Physiol. 315 (2018) 932–942. 10.1152/ajpgi.00020.2018.-Processes. [DOI] [PubMed] [Google Scholar]
- [194].Dakup PP, Porter KI, Gajula RP, Goel PN, Cheng Z, Gaddameedhi S, The circadian clock protects against ionizing radiation-induced cardiotoxicity, FASEB J. 34 (2020) 3347–3358. 10.1096/fj.201901850RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [195].Dakup PP, Porter KI, Gaddameedhi S, The circadian clock protects against acute radiation-induced dermatitis, Toxicol. Appl. Pharmacol. 399 (2020) 115040. 10.1016/j.taap.2020.115040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [196].Murphy HM, Wideman CH, Nadzam GR, A laboratory animal model of human shift work, Integr. Physiol. Behav. Sci 38 (2003) 316–328. [DOI] [PubMed] [Google Scholar]
- [197].Bartol-Munier I, Gourmelen S, Pevet P, Challet E, Combined effects of high-fat feeding and circadian desynchronization, Int. J. Obes 30 (2006) 60–67. 10.1038/sj.ijo.0803048. [DOI] [PubMed] [Google Scholar]
- [198].Salgado-Delgado R, Angeles-Castellanos M, Saderi N, Buijs RM, Escobar C, Food intake during the normal activity phase prevents obesity and circadian desynchrony in a rat model of night work, Endocrinology. 151 (2010) 1019–1029. 10.1210/en.2009-0864. [DOI] [PubMed] [Google Scholar]
- [199].Leenaars CHC, Kalsbeek A, Hanegraaf MAJ, Foppen E, Joosten RNJMA, Post G, Dematteis M, Feenstra MGP, Van Someren EJW, Unaltered instrumental learning and attenuated body-weight gain in rats during non-rotating simulated shiftwork, Chronobiol. Int 29 (2012) 344–355. 10.3109/07420528.2011.654018. [DOI] [PubMed] [Google Scholar]
- [200].Grønli J, Meerlo P, Pedersen TT, Pallesen S, Skrede S, Marti AR, Wisor JP, Murison R, Henriksen TEG, Rempe MJ, Mrdalj J, A rodent model of night-shift work induces short-term and enduring sleep and electroencephalographic disturbances, J. Biol. Rhythms 32 (2017) 48–63. 10.1177/0748730416675460. [DOI] [PubMed] [Google Scholar]
- [201].McGowan NM, Coogan AN, Circadian and behavioural responses to shift work-like schedules of light/dark in the mouse, J. Mol. Psychiatry 1 (2013) 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [202].Lastella M, Roach GD, Halson SL, Gore CJ, Garvican-Lewis LA, Sargent C, The effects of transmeridian travel and altitude on sleep: preparation for football competition, J. Sports Sci. Med 13 (2014) 718–720. https://pubmed.ncbi.nlm.nih.gov/25177205. [PMC free article] [PubMed] [Google Scholar]
- [203].Mrosovsky N, Phase response curves for social entrainment, J. Comp. Physiol. A 162 (1988) 35–46. [DOI] [PubMed] [Google Scholar]
- [204].Honma K, Honma S, Nakamura K, Sasaki M, Endo T, Takahashi T, Differential effects of bright light and social cues on reentrainment of human circadian rhythms, Am. J. Physiol. Integr. Comp. Physiol 268 (1995) R528–R535. [DOI] [PubMed] [Google Scholar]
- [205].Comperatore CA, Krueger GP, Circadian rhythm desynchronosis, jet lag, shift lag, and coping strategies., Occup. Med 5 (1990) 323–341. [PubMed] [Google Scholar]
- [206].Eastman CI, Burgess HJ, How to Travel the World Without Jet Lag, Sleep Med. Clin 4 (2009) 241–255. 10.1016/j.jsmc.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].Biological Technologies Office (DARPA), Broad Agency Announcement ADvanced Acclimation and Protection Tool for Environmental Readiness (ADAPTER), 2020. https://beta.sam.gov.
- [208].Filipski E, Delaunay F, King VM, Wu M-W, Claustrat B, Gréchez-Cassiau A, Guettier C, Hastings MH, Francis L, Effects of Chronic Jet Lag on Tumor Progression in Mice, 2004. [DOI] [PubMed] [Google Scholar]
- [209].Iwamoto A, Kawai M, Furuse M, Yasuo S, Effects of chronic jet lag on the central and peripheral circadian clocks in CBA/N mice, Chronobiol. Int 31 (2014) 189–198. 10.3109/07420528.2013.837478. [DOI] [PubMed] [Google Scholar]
- [210].Papagiannakopoulos T, Bauer MR, Davidson SM, Heimann M, Subbaraj L, Bhutkar A, Bartlebaugh J, Vander Heiden MG, Jacks T, Circadian Rhythm Disruption Promotes Lung Tumorigenesis, Cell Metab. 24 (2016) 324–331. 10.1016/j.cmet.2016.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [211].Hadadi E, Taylor W, Li XM, Aslan Y, Villote M, Rivière J, Duvallet G, Auriau C, Dulong S, Raymond-Letron I, Provot S, Bennaceur-Griscelli A, Acloque H, Chronic circadian disruption modulates breast cancer stemness and immune microenvironment to drive metastasis in mice, Nat. Commun 11 (2020). 10.1038/s41467-020-16890-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [212].Kettner NM, Mayo SA, Hua J, Lee C, Moore DD, Fu L, Circadian dysfunction induces leptin resistance in mice, Cell Metab. 22 (2015) 448–459. 10.1016/j.cmet.2015.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [213].Kettner NM, Voicu H, Finegold MJ, Coarfa C, Sreekumar A, Putluri N, Katchy CA, Lee C, Moore DD, Fu L, Circadian Homeostasis of Liver Metabolism Suppresses Hepatocarcinogenesis, Cancer Cell. 30 (2016) 909–924. 10.1016/j.ccell.2016.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [214].Casiraghi LP, Oda GA, Chiesa JJ, Friesen WO, Golombek DA, Forced desynchronization of activity rhythms in a model of chronic jet lag in mice, J. Biol. Rhythms 27 (2012) 59–69. 10.1177/0748730411429447. [DOI] [PubMed] [Google Scholar]
- [215].Aiello I, Mul Fedele ML, Román F, Marpegan L, Caldart C, Chiesa JJ, Golombek DA, V Finkielstein C, Paladino N, Circadian disruption promotes tumor-immune microenvironment remodeling favoring tumor cell proliferation, 2020. http://advances.sciencemag.org/. [DOI] [PMC free article] [PubMed]
- [216].Davidson AJ, Sellix MT, Daniel J, Yamazaki S, Menaker M, Block GD, Chronic jet-lag increases mortality in aged mice, Curr. Biol. CB 16 (2006) R914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [217].Ashbrook LH, Krystal AD, Fu YH, Ptáček LJ, Genetics of the human circadian clock and sleep homeostat, Neuropsychopharmacology. 45 (2020) 45–54. 10.1038/s41386-019-0476-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [218].Pan A, Schernhammer ES, Sun Q, Hu FB, Rotating night shift work and risk of type 2 diabetes: two prospective cohort studies in women., PLoS Med. 8 (2011) e1001141. 10.1371/journal.pmed.1001141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [219].Turek FW, Joshu C, Kohsaka A, Lin E, Ivanova G, McDearmon E, Laposky A, Losee-Olson S, Easton A, Jensen DR, Eckel RH, Takahashi JS, Bass J, Obesity and metabolic syndrome in circadian Clock mutant mice., Science. 308 (2005) 1043–5. 10.1126/science.1108750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [220].Smolensky MH, Hermida RC, Portaluppi F, Circadian mechanisms of 24-hour blood pressure regulation and patterning., Sleep Med. Rev 33 (2017) 4–16. 10.1016/j.smrv.2016.02.003. [DOI] [PubMed] [Google Scholar]
- [221].Portaluppi F, Hermida RC, Circadian rhythms in cardiac arrhythmias and opportunities for their chronotherapy., Adv. Drug Deliv. Rev 59 (2007) 940–51. 10.1016/j.addr.2006.10.011. [DOI] [PubMed] [Google Scholar]
- [222].Geiger SS, Fagundes CT, Siegel RM, Chrono-immunology: progress and challenges in understanding links between the circadian and immune systems., Immunology. 146 (2015) 349–58. 10.1111/imm.12525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [223].Labrecque N, Cermakian N, Circadian Clocks in the Immune System., J. Biol. Rhythms 30 (2015) 277–90. 10.1177/0748730415577723. [DOI] [PubMed] [Google Scholar]
- [224].Ballesta A, Innominato PF, Dallmann R, Rand DA, Lévi FA, Systems Chronotherapeutics., Pharmacol. Rev 69 (2017) 161–199. 10.1124/pr.116.013441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [225].Cadet J, Douki T, Formation of UV-induced DNA damage contributing to skin cancer development, Photochem. Photobiol. Sci 17 (2018) 1816–1841. 10.1039/c7pp00395a. [DOI] [PubMed] [Google Scholar]
- [226].Gaddameedhi S, Kemp MG, Reardon JT, Shields JM, Smith-Roe SL, Kaufmann WK, Sancar A, Similar nucleotide excision repair capacity in melanocytes and melanoma cells, Cancer Res. 70 (2010) 4922–4930. 10.1158/0008-5472.CAN-10-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [227].Ho H-Y, Lin C-W, Chien M-H, Reiter RJ, Su S-C, Hsieh Y-H, Yang S-F, Melatonin suppresses TPA-induced metastasis by downregulating matrix metalloproteinase-9 expression through JNK/SP-1 signaling in nasopharyngeal carcinoma., J. Pineal Res 61 (2016) 479–492. 10.1111/jpi.12365. [DOI] [PubMed] [Google Scholar]