

Abstract
WD repeat domain 5 (WDR5), an integral component of the MLL/KMT2A lysine methyltransferase complex, is critically involved in oncogenesis and represents an attractive onco-target. Inhibitors targeting protein-protein interactions (PPIs) between WDR5 and its binding partners, however, do not inhibit all of WDR5-mediated oncogenic functions and exert rather limited antitumor effects. Here, we report a cereblon (CRBN)-recruiting proteolysis targeting chimera (PROTAC) of WDR5, MS40, which selectively degrades WDR5 and the well-established neo-substrates of immunomodulatory drugs (IMiDs):CRBN, the Ikaros zinc finger (IKZF) transcription factors IKZF1 and IKZF3. MS40-induced WDR5 degradation caused disassociation of the MLL/KMT2A complex off chromatin, resulting in decreased H3K4me2. Transcriptomic profiling revealed that targets of both WDR5 and IMiDs:CRBN were significantly repressed by treatment of MS40. In MLL-rearranged leukemias, which exhibit IKZF1 high expression and dependency, co-suppression of WDR5 and Ikaros by MS40 is superior in suppressing oncogenesis to the WDR5 PPI inhibitor, to MS40’s non-PROTAC analog controls (MS40N1 and MS40N2, which do not bind CRBN and WDR5, respectively), and to a matched VHL-based WDR5 PROTAC (MS169, which degrades WDR5 but not Ikaros). MS40 suppressed the growth of primary leukemia patient cells in vitro and patient-derived xenografts in vivo. Thus, dual degradation of WDR5 and Ikaros is a promising anti-cancer strategy.
INTRODUCTION
Overexpression of WD40 repeat domain 5 (WDR5) occurs frequently in cancers such as leukemia [1], pancreatic cancer [2] and neuroblastoma [3], correlating with aggressive phenotypes and poor outcomes. WDR5 serves as an integral component of the Mixed Lineage Leukemia (MLL; also known as KMT2A) family of histone methyltransferase complexes, which modulate histone modification and gene transcription partly via methylation of histone H3 lysine 4 (H3K4) [4–6]. In human leukemias carrying MLL rearrangement (MLL-r), the WDR5:MLL complex cooperates with the MLL-r chimeric oncoproteins to maintain an oncogenic gene-expression program, and depletion of WDR5 suppressed growth of MLL-r leukemias [7–9]. Furthermore, WDR5 has a scaffolding role in promoting the chromatin association of non-MLL partners such as cMyc, which is also critically involved in tumorigenesis [2, 3, 10, 11]. Thus, inhibiting multifaceted cancer-promoting actions of WDR5 represents an attractive means for developing therapeutics.
Multiple protein-protein interaction (PPI) sites within WDR5 have been identified. The WDR5 interaction motif (WIN) and WDR5 binding motif (WBM) mediate WDR5 binding to MLL [7–9] and RBBP5, another core subunit of MLL complex [12, 13], respectively. A number of small-molecule inhibitors that block the WDR5 PPIs have been reported [14–27]. In particular, OICR-9429 [18, 20] was developed as a chemical probe to disrupt the WDR5:MLL PPI by occupying WIN. However, these WDR5 PPI inhibitors are often ineffective in killing cancer cells and are not efficacious in vivo. Such relatively weak antitumor activities are likely because: (1) WDR5 PPI inhibitors, which rely on receptor occupancy pharmacology, do not fully and durably block the interactions between WDR5 and partners, and (2) more importantly, these inhibitors target only some but not all of WDR5’s oncogenic functions. A new therapeutic strategy that can eliminate all of WDR5’s oncogenic functions is needed.
Proteolysis targeting chimeras (PROTACs) are promising new therapeutic modalities [28–30]. Two WDR5-targeting PROTACs, both of which were based on the E3 ligase VHL [31, 32], have been reported recently, including MS67 developed by us [31]. Here, we report the development of MS40, a novel cereblon (CRBN)-based WDR5 PROTAC, which can induce simultaneous degradation of WDR5 as well as Ikaros (IKZF1) and Aiolos (IKZF3), two well-established neo-substrates of IMiDs:CRBN. On one hand, the MS40-induced WDR5 degradation significantly repressed the function of MLL:WDR5 complex, leading to MLL disassociation off chromatin and a global decrease of H3K4me2. On the other hand, MS40 also suppressed activity of IKZFs, the validated drug targets of certain cancers. Gene targets of both IKZF1/3 and WDR5 were found significantly repressed by MS40. Transcriptome-modulatory effect of MS40 is superior to that of WDR5 PPI inhibitors. Consequently, MS40 treatment exhibited superior antitumor effects to WDR5 PPI inhibitors and MS40’s non-PROTAC analogues (MS40N1 and MS40N2, which do not bind CRBN and WDR5, respectively) in a set of tumor models exhibiting WDR5 and/or IKZF dependency. In particular, co-targeting WDR5 and IKZF1/3 by MS40 in MLL-r leukemias, which show high IKZF1 expression and exhibit IKZF1 dependency, is superior to a VHL-based counterpart of MS40 we designed, termed MS169, which does not degrade IKZF1/3 and selectively degrades WDR5. MS40 effectively suppressed the growth of primary leukemia cells in vitro and patient-derived xenograft (PDX) in mice. In summary, we report the discovery of a novel PROTAC that degrades both WDR5 and IKZF1/3 as a potential anti-cancer therapeutic. We also present a set of active and inactive WDR5 PROTAC analogs, which represent highly useful chemical tools for studying WDR5-mediated gene regulation in biology and medicine.
RESULTS
Discovery and biochemical characterization of MS40, a CRBN-based WDR5 PROTAC
OICR-9429 (Supplementary Fig. 1A) is a highly potent and selective WDR5 inhibitor (Ki = 64 nM), which occupies the WIN site of WDR5 to disrupt the PPIs between WDR5 and its binding partners [18]. To design WDR5 PROTACs, we analyzed the cocrystal structure of OICR-9429 in complex of WDR5 and found that the morpholine moiety is likely solvent accessible (PDB ID: 4QL1) [20]. To facilitate the linker attachment, we replaced this morpholine group with a piperazine moiety attached with a short ethylamine group (Fig. 1A). We synthesized a series of heterobifunctional compounds by conjugating this modified WDR5-binding moiety to pomalidomide (POM), which engages the CUL4A E3 ligase, CRBN, using linkers with various lengths and composition. By screening these compounds for their ability to reduce the WDR5 protein levels via immunoblotting, we identified MS40 (Fig. 1A), which bears a flexible aliphatic linker with seven methylene units, as an effective WDR5 degrader. We also designed and synthesized a negative control, MS40N1, in which the nitrogen of the pomalidomide glutarimide moiety was methylated to abolish CRBN-binding (Fig. 1B). Next, using isothermal titration calorimetry (ITC), we evaluated the binding affinities of OICR-9429, MS40 and MS40N1 to the WDR5 and CRBN’s C-terminal thalidomide binding domain (CRBN-TBD) [33, 34] (Fig. 1C; Supplementary Fig. 1B). As expected, OICR-9429 displayed high binding affinity to WDR5 (KD = 29 ± 1.5 nM), confirming that OICR-9429 is a potent WDR5 binder (Supplementary Fig. 1B). Compared to OICR-9429, MS40 (KD = 125 ± 8 nM) and MS40N1 (KD = 130 ± 1 nM) displayed 4-fold lower binding affinities to WDR5 (Fig. 1C top; Supplementary Fig. 1C). However, it is important to note that the binding affinities of MS40 and MS40N1 to WDR5 are still excellent, supporting our design hypothesis that the introduction of an E3 ligase ligand and linker to the solvent-exposed morpholino group does not profoundly affect WDR5 binding. As expected, MS40 exhibited moderate affinity (KD = 14.8 ± 1.3 μM) to CRBN-TBD, similar to pomalidomide (KD = 16 ± 4 μM) [33], while no binding was observed for the designed CRBN-inactive MS40N1 (Fig. 1C middle versus Fig. 1D). Furthermore, we assessed the cooperativity of MS40-mediated ternary complex formation by titration of WDR5 into the saturated CRBN-TBD:MS40 complex, with titration of WDR5 into MS40 as a reference. Notably, a significant enhancement was observed in WDR5 binding to CRBN-TBD:MS40 binary complex (α = 1.96), suggesting that MS40 favorably induces WDR5:MS40:CRBN ternary complex formation (Fig. 1C bottom).
Fig. 1. Design of the CRBN-based WDR5 PROTAC MS40 and measurement of MS40 binding to WDR5 and Cereblon (CRBN).
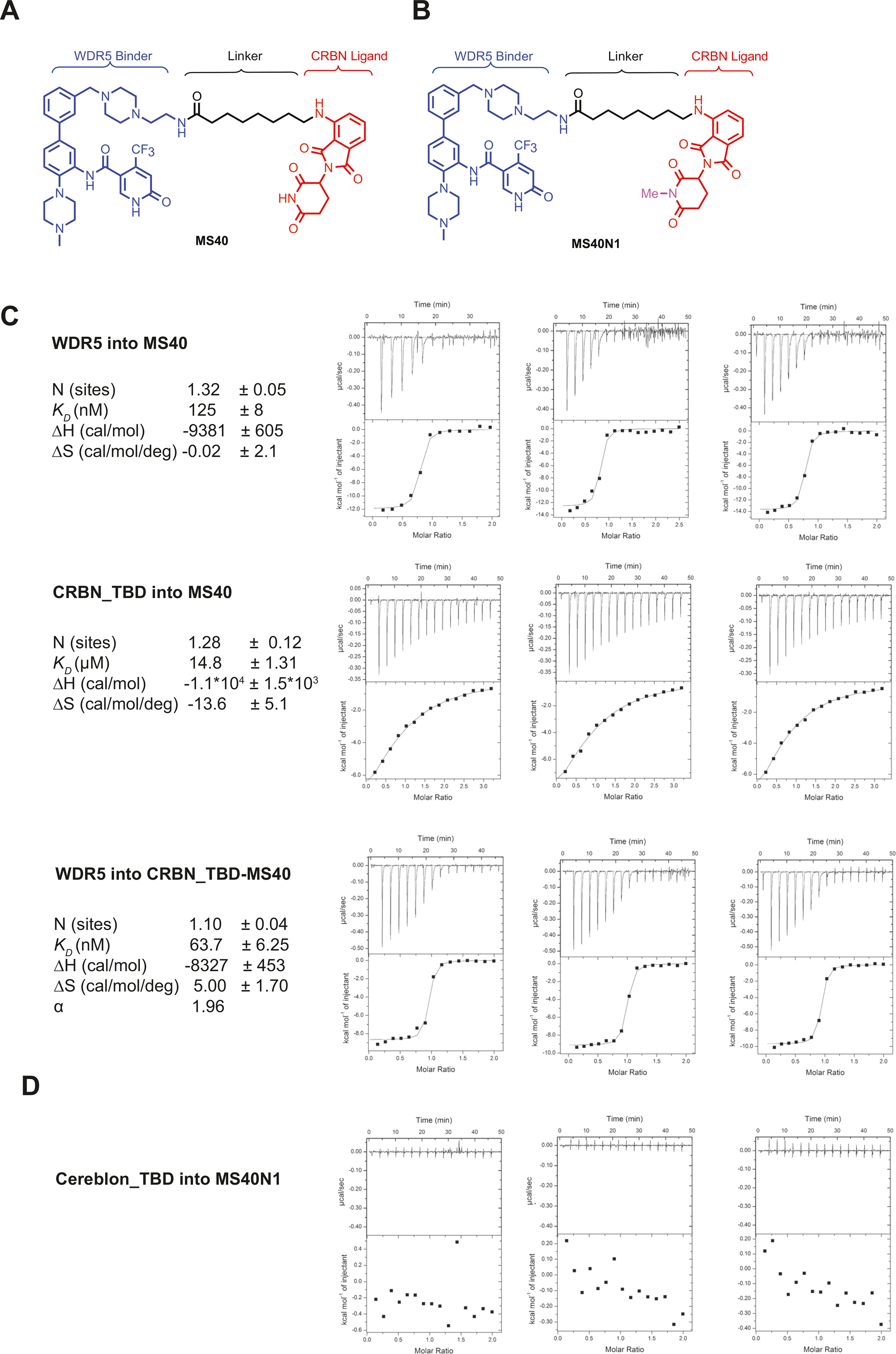
A, B Chemical structures of MS40 A and its CRBN-inactive analogue MS40N1 B. C Replicated inverse ITC titrations of WDR5 into MS40 (top), CRBN’s thalidomide-binding domain (TBD) into MS40 (middle) and WDR5 into the complex of CRBN TBD and MS40 (bottom) for measuring the binding kinetics and cooperativity (α) for MS40. The calculated values represent the mean ± SD from three independent experiments. First injection has been removed from the fitting. D Replicated inverse ITC titrations of CRBN TBD into MS40N1 for measuring binding kinetics.
MS40 selectively degrades WDR5 and CRBN’s neo-substrates, IKZF1 and IKZF3
Next, we evaluated the MS40-induced degradation in MV4;11 cells, a MLL-rearranged acute myeloid leukemia (MLL-r AML) cell line, and found that MS40, but not MS40N1, reduced the WDR5 protein level in a concentration-dependent manner, albeit with a slight hook effect observed at higher concentrations (Fig. 2A, B). The half-maximal degradation concentration (DC50) of MS40 mediated-WDR5 degradation was determined to be 42 ± 41 nM, with the maximal level of degradation (Dmax) measured as 77 ± 12% (Fig. 2C). MS40-induced WDR5 degradation was found to be time-dependent, with obvious degradation detected as early as 2 h and significant effect sustained to 24 h (Fig. 2D).
Fig. 2. Mechanism-of-action (MOA) studies of MS40-mediated WDR5 degradation.
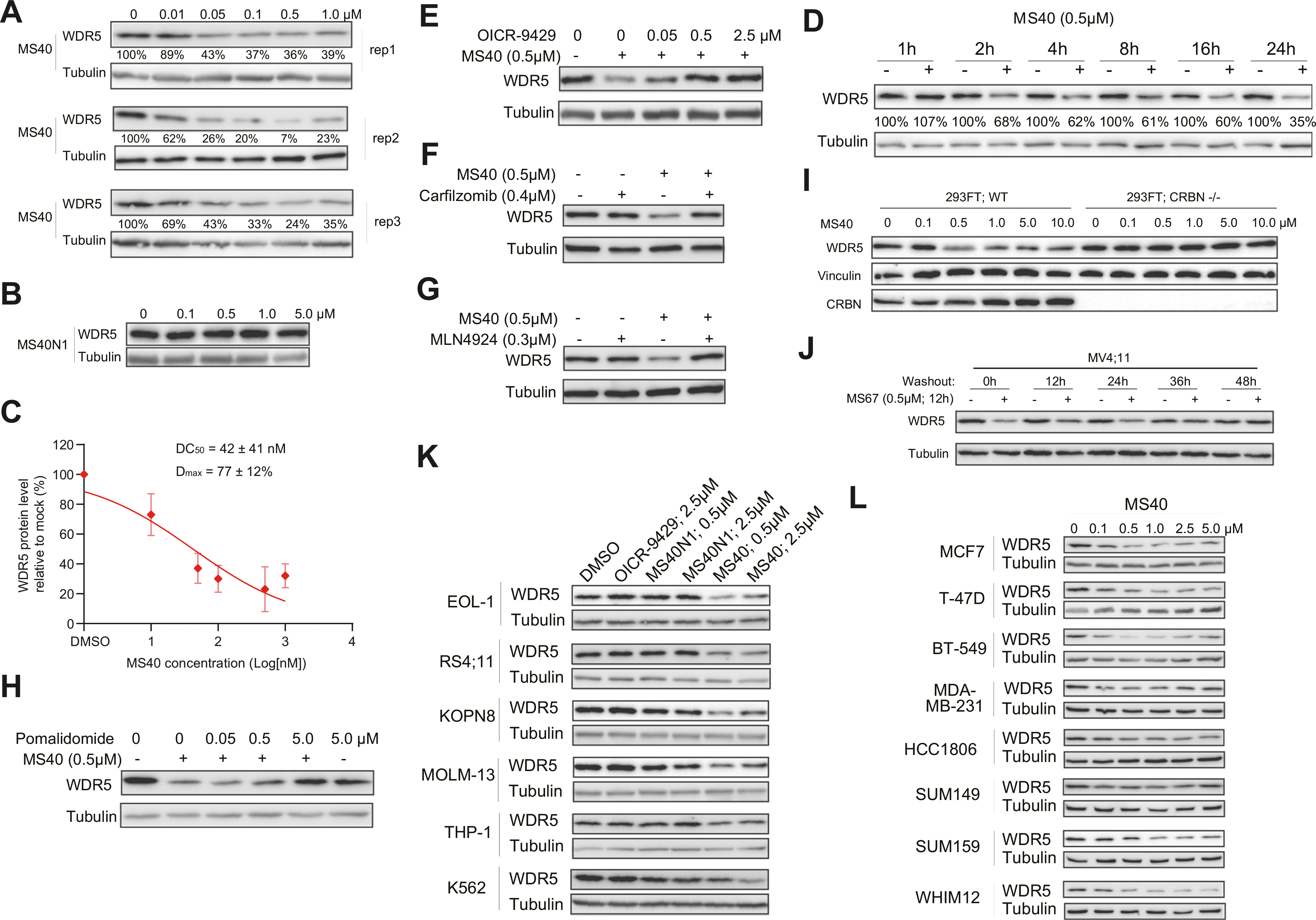
A, B Immunoblotting for WDR5 (A; three replicated experiments) in MV4;11 cells treated with the indicated concentrations of MS40 A or MS40N1 B for 18 h. Tubulin was used as the loading control. C Half maximal degradation (DC50; 42 ± 41 nM) and maximal level of degradation (Dmax; 77 ± 12%) of MS40, measured based on WDR5 degradation seen in MV4;11 cells treated with the indicated concentrations of MS40 for 18 h, with WDR5 immunoblotting signals in Fig. 2A determined by ImageJ software (n = 3 independent experiments; mean ± S.D.). D Immunoblotting for WDR5 in MV4;11 cells treated with 0.5 μM of MS40 for the indicated time. E Immunoblotting for WDR5 and tubulin in MV4;11 cells after a two-hour pre-treatment with DMSO or the indicated concentration of OICR-9429, followed by a six-hour treatment with 0.5 μM of MS40. F–H Immunoblotting for WDR5 and tubulin in MV4;11 cells after a two-hour pre-treatment with DMSO, carfilzomib (0.4 μM; F), MLN4924 (0.3 μM; G) or pomalidomide (0.05 μM, 0.5 μM, 5.0 μM; H), followed by a 6-hour treatment with 0.5 μM of MS40. I Immunoblotting for WDR5, vinculin and CRBN post-treatment of 293FT cells, either wildtype (WT) or with CRISPR/Cas9-mediated CRBN knockout (CRBN−/−), with the indicated concentrations of MS40 for 18 h. J Immunoblotting for WDR5 and tubulin in MV4;11 cells, first treated with 0.5 μM of MS40 for 12 h and then subjected to washout of MS40 for the indicated time. K, L Immunoblotting for WDR5 after an 18-hour treatment of different cell lines of leukemia K or breast tumor L with DMSO or the indicated concentrations of OICR-9429, MS40N1 or MS40.
We next determined the mechanism of action (MOA) for MS40-induced WDR5 degradation. First, pre-treatment with OICR-9429 concentration-dependently diminished the MS40-induced WDR5 degradation (Fig. 2E). Second, pre-treatment with the proteasome inhibitor carfilzomib, neddylation inhibitor MLN4924 or CRBN ligand pomalidomide also suppressed the MS40-induced degradation of WDR5 (Fig. 2F–H). Furthermore, the WDR5 degradation induced by MS40 was completely abrogated in cells lacking CRBN, compared with wild-type controls (Fig. 2I). Moreover, the WDR5 protein level was almost fully recovered 36 h after MS40 washout (Fig. 2J), suggesting that MS40-induced WDR5 degradation is reversible. Lastly, the MS40-induced WDR5 degradation effects were consistently observed in a large panel of cell lines, including five lines of MLL-r AML (Fig. 2K) and eight lines of breast cancer (Fig. 2L). In contrast, OICR-9429 and MS40N1 did not induce notable WDR5 reduction (Fig. 2K). Collectively, these results demonstrated that MS40 is a bona fide WDR5 PROTAC and consistently depletes cellular WDR5 in a WDR5-, CRBN- and proteasome-dependent manner.
To assess both degradation effect and selectivity of MS40 at a proteome-wide level, we next performed an unbiased mass spectrometry (MS)-based quantitative proteomic analysis and found that, out of a total of 5016 identified proteins, WDR5, Ikaros (IKZF1) and LIM Domain Containing 1 (LIMD1) were the only three proteins showing the significantly decreased level in MV4;11 cells treated with 0.5 μM of MS40 for 6 h, compared to mock (Fig. 3A; Supplementary Table S1). It was not surprising that MS40 degraded IKZF1 in addition to WDR5 in this study because it is known that CRBN-recruiting PROTACs can degrade IMiD:CRBN neo-substrates including IKZF1 and (IKZF3) [35, 36]. We next confirmed the degradation effect of MS40, but not OICR-9429 or MS40N1, on WDR5, IKZF1, IKZF1-related IKZF3 (when expressed) and LIMD1 in MV4;11 and RS4;11 cells by immunoblotting (Fig. 3B). Interestingly, treatment with pomalidomide, alone or together with OICR-9429, led to degradation of IKZF1/3 as expected, but not LIMD1, indicating that degradation of LIMD1 is unique to MS40 (Fig. 3B). These results suggest that LIMD1 could be an off-target neo-substrate of the modified pomalidomide moiety of MS40. Other previously reported neo-substrates of IMiDs:CRBN-based PROTACs, GSPT1 and ZFP91 [37–39], were not affected by MS40 (Fig. 3B). Consistent with its general high expression among hematological cancers (Supplementary Fig. 2A), IKZF1 promotes oncogenic growth in multiple myeloma [35, 36] and myeloid malignancies [40, 41] such as those with MLL-r (Supplementary Fig. 2B). We thus further examined the oncotarget IKZF1 and found that MS40-induced IKZF1 degradation in a concentration- and time-dependent fashion in multiple cell lines (Fig. 3C, D). IKZF3 was not further examined due to the lack of its expression in cell lines such as MV4;11 (Fig. 3B). Moreover, depletion of LIMD1 had little effect on AML cell proliferation (Supplementary Fig. 2C); thus, it was not further investigated. Together, these results demonstrated that MS40 selectively degrades WDR5 and CRBN’s neo-substrates such as the IKZF1 oncoprotein in tumor cells.
Fig. 3. MS40 selectively degrades WDR5 and the CRBN neo-substrates (IKZF1 and IKZF3) in cells.
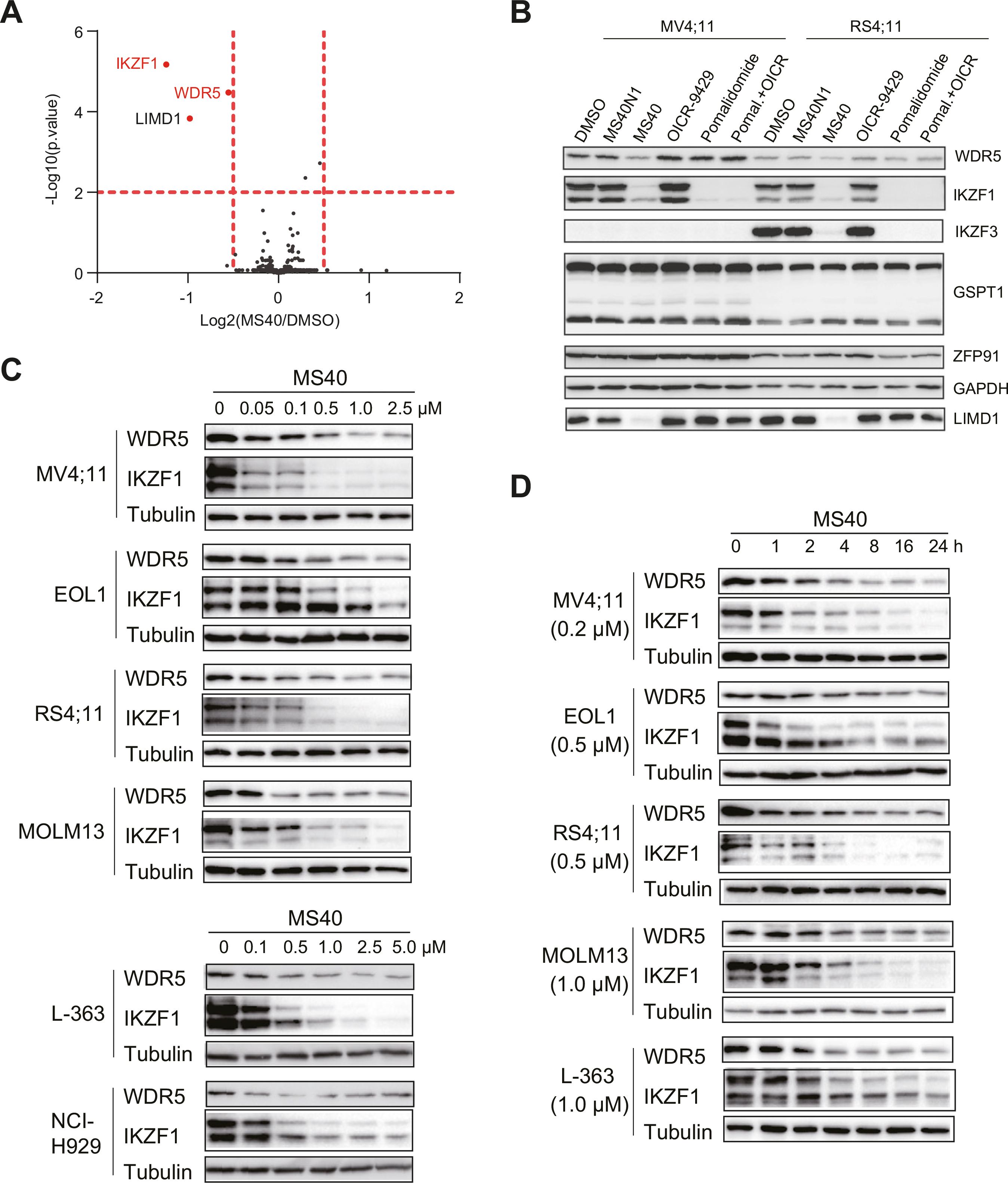
A Multiplexed quantitative proteomics analysis of MV4;11 cells (n = 5 replicates) post-treatment with 0.5 μM of MS40 versus DMSO for 6 h, which revealed WDR5, IKZF1 and LIMD1 to be most significantly downregulated, with a cut-off of P value less than 0.01 (defined by Student’s t test) and Log2 (fold-change or FC) over 0.5. B Immunoblotting for the indicated protein in MV4;11 and RS4;11 cells treated with DMSO or 0.5 μM of MS40N1, MS40, OICR-9429, pomalidomide, or OICR-9429 plus pomalidomide (1:1 molar ratio at 0.5 μM) for 18 h. C, D MS40 induced WDR5 and IKZF1 degradation is dose- and time-dependent in a panel of indicated cell lines.
MS40 depletes MLL complexes off chromatin and decreases global H3K4me2
WDR5 acts as a subunit of the MLL:RBBP5:Menin complex for depositing H3K4 methylation, a histone mark associated with transcriptional competence and activation [17, 42–44]. Treatment of MS40, but not OICR-9429 or MS40N1, caused a mild and significant decrease of H3K4me2, but not other examined histone methylation marks such as H3K4me1, H3K4me3, H3K9me3, H3K27me3 and H3K36me3 (Fig. 4A; Supplementary Fig. 3A). Comparable treatment with pomalidomide (the E3 ligand portion of MS40), either alone or together with OICR-9429 (the WDR5-binding module of MS40), did not affect global H3K4me2 (Fig. 4A), suggesting that WDR5 degraders are superior to WDR5 PPI inhibitors. Total protein levels of MLL complex components RBBP5 and Menin did not show significant changes, but the MLL protein level was reduced slightly in MV4;11 and RS4;11 cells treated with MS40, compared to MS40N1, OICR-9429 or pomalidomide (Fig. 4B; Supplementary Fig. 3B). Notably, the chromatin-bound levels of MLL complex components such as MLL, RBBP5, and/or Menin were significantly decreased post-treatment with MS40, relative to MS40N1, OICR-9429 or pomalidomide (Fig. 4B; Supplementary Fig. 3B). These results support that the observed decrease of chromatin-bound MLL:RBBP5:Menin complexes is due to MS40-induced degradation of WDR5, but not IKZFs (neo-substrates of MS40 or pomalidomide). ChIP-seq for H3K4me2 (Fig. 4C, D) and MLL (Fig. 4E, F) further substantiated their decreases in MV4;11 cells treated with MS40, relative to DMSO. As expected, a vast majority of MLL peaks overlapped H3K4me2 (Supplementary Fig. 3C). We conclude that, compared to WDR5 PPI inhibitors, WDR5 PROTACs have unique, much stronger effects on suppressing functionality of the MLL complex, as manifested by the complex displacement off chromatin and decreases of H3K4me2.
Fig. 4. MS40 treatment causes a significant loss of the chromatin-bound MLL complex components, concurrent with a decrease of H3K4me2.
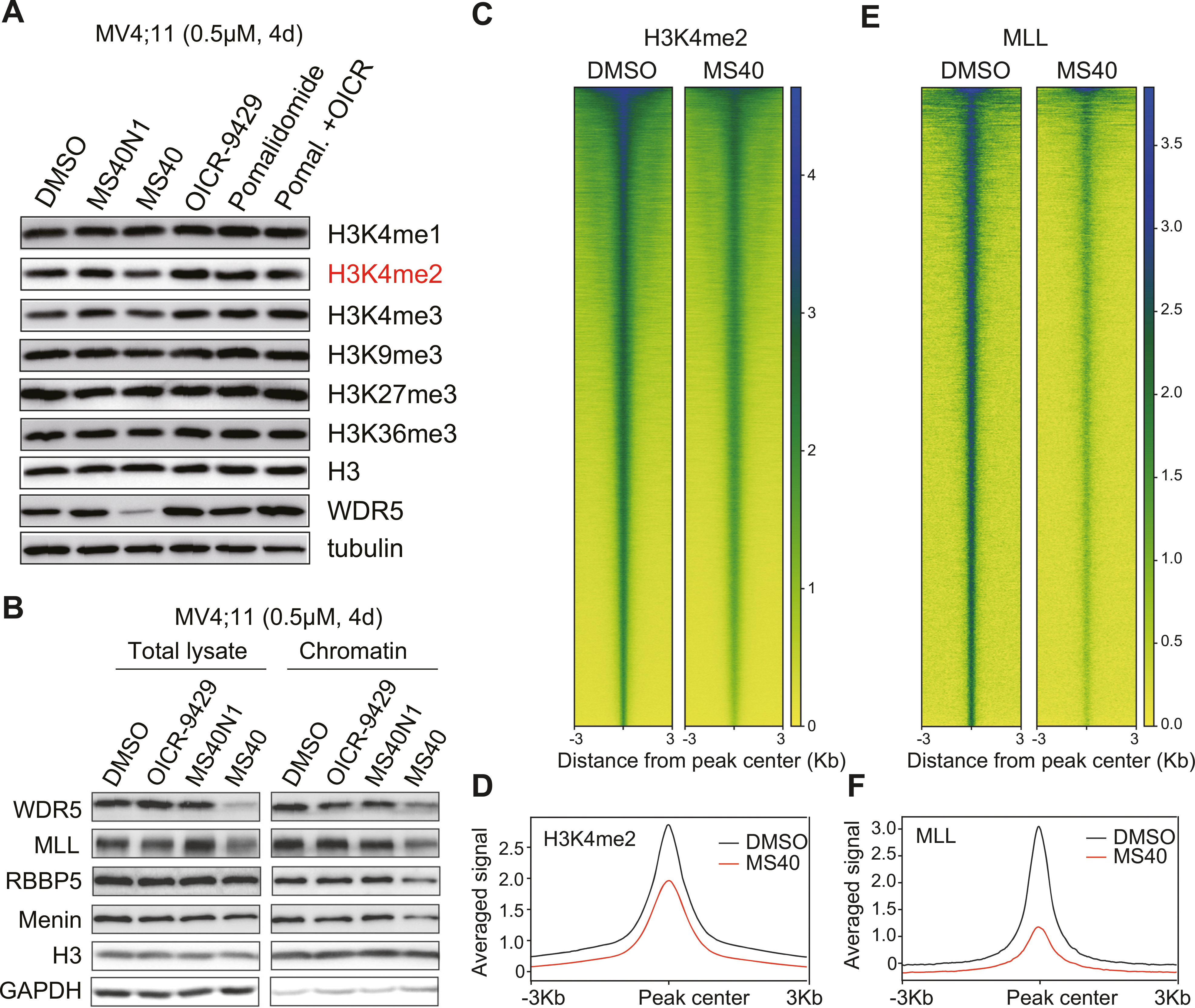
A Immunoblotting for the indicated histone modifications and WDR5 in MV4;11 cells, treated with DMSO or 0.5 μM of OICR-9429, MS40N1, MS40, pomalidomide, or OICR-9429 plus pomalidomide (1:1 molar ratio at 0.5 μM) for 4 days. B Immunoblotting for the indicated MLL complex components in total cell lysate and the chromatin-bound fraction of MV4;11 cells treated with DMSO or 0.5 μM of OICR-9429, MS40N1, or MS40 for 4 days. C–F Heatmap C, E and averaged plot D, F of H3K4me2 C, D and MLL E, F ChIP-seq signals (spike-in control normalized) in MV4;11 cells treated with 0.5 μM of MS40, in comparison to DMSO, for 4 days.
MS40 treatment effectively suppresses the transcription from target genes of WDR5:MLL complex and IKZF factors
Next, we used RNA sequencing (RNA-seq)-based transcriptomic profiling to evaluate gene-regulatory effects by MS40 versus its non-PROTAC analog (MS40N1) in MV4;11 cells. We also included knockdown (KD) of WDR5 and treatment with pomalidomide (which degrades IKZF1/3 but not WDR5) as additional controls. RNA-seq analysis revealed differentially expressed genes (DEGs) due to either compound treatment or WDR5 depletion (Supplementary Fig. 4A; Supplementary Tables S2 and S3). Consistent with the MS40-induced degradation of WDR5 and IKZFs, DEGs downregulated post-treatment with MS40 relative to DMSO overlapped with those caused by KD of WDR5 and treatment of pomalidomide (Fig. 5A, B). Gene Set Enrichment Analysis (GSEA) also revealed that DEGs down-regulated after MS40 treatment were enriched in genes down-regulated by pomalidomide treatment (Fig. 5C), the WDR5 target genes such as ribosome genes, either defined in this study or previously (Fig. 5D–G), and MLL-r-related oncogenes (Fig. 5H). In contrast, MS40N1 treatment did not result in significant gene-expression changes when compared to DMSO-treated mock (Supplementary Fig. 4A). Ribosome genes are direct targets of WDR5 [10, 24, 26]. Approximately 140 out of the total of 312 DEGs down-regulated post-treatment with MS40 relative to DMSO were directly bound by MLL based on ChIP-seq (Fig. 5I) as exemplified by oncogenes (BCL2, SNHG15, RUNX3, SMAD3 and CSNK1E) and ribosome genes (RPL27, RPL7 and RPS14), all of which showed the decreased binding of MLL and H3K4me2 and reduced gene expression following MS40 treatment (Fig. 5J; Supplementary Fig. 4B). Furthermore, we employed Global Run-on sequencing (GRO-seq), a method that allows identification of genes whose nascent transcription is affected. Based on GRO-seq, 189 genes were found to be transcriptionally suppressed by treatment with MS40, compared to DMSO (Supplementary Table S4); among these 189 genes showing decreased nascent transcription, 102 were MLL-bound and 97 were also downregulated based on RNA-seq after MS40 treatment (Fig. 5I), as seen at BCL2, RPL27 and SMAD3 (Fig. 5K; Supplementary Fig. 4C). RT-qPCR further verified the decreased gene expression of the tested oncogenes and ribosome genes after MS40 treatment relative to DMSO, which was not seen with OICR-9429 (Fig. 5L). Overall, MS40 functions as a dual degrader of WDR5 and IKZFs, inducing significant downregulation of their target genes at both nascent transcription and steady state levels.
Fig. 5. MS40 treatment effectively suppresses the transcription from target genes of WDR5:MLL complexes and IKZF factors.

A Venn diagram showing the overlap between differentially expressed genes (DEGs) downregulated in MV4;11 cells after treatment with 0.5 μM of MS40 or doxycycline-based induction of the WDR5-targeting shRNA (sh#47) for 4 days, compared to mock-treated. DEGs are determined by RNA-seq with a cut-off of the absolute value of Log2 (FC) greater than 0.5 and FDR value less than 0.05. B Venn diagram showing the overlap between DEGs downregulated in MV4;11 cells after a four-day treatment with 0.5 μM of MS40 or pomalidomide, compared to DMSO. DEGs are determined with a cut-off of the absolute value of Log2 (FC) greater than 0.5 and FDR value less than 0.05. C–H Gene set enrichment analysis (GSEA) using the above RNA-seq profiles of MV4;11 cells showing the significantly enriched gene sets after MS40 treatment versus control. I Ven diagram showing overlap between the MLL-bound genes and DEGs downregulated (defined by either RNA-seq or GRO-seq profiling) after treatment of MV4;11 cells with 0.5 μM of MS40, relative to DMSO, for 4 days. J Integrative Genomics Viewer (IGV) tracks showing the H3K4me2 and MLL ChIP-seq and RNA-seq profiles at the indicated gene in MV4;11 cells, treated with DMSO or 0.5 μM of MS40 for 4 days. K IGV tracks showing GRO-seq profiles at the indicated gene in MV4;11 cells, treated with DMSO or 0.5 μM of MS40 for 4 days. L RT-qPCR of the indicated gene after treatment of MV4;11 cells with 0.5 μM of OICR-9429, MS40, or pomalidomide, compared to DMSO. RT-qPCR signals, presented as the mean ± S.D., were normalized first to those of GAPDH and then to DMSO-treated cells (n = 3 replicated experiments).
MS40 suppresses cancer cell growth in vitro
We next assessed MS40’s anti-cancer effects. Besides MS40N1 (an E3-“dead” analog of MS40), we developed two more control compounds. First, MS40N2 (Fig. 6A) was designed to bind CRBN with a similar affinity as MS40 but not bind WDR5, which was verified by ITC-based measurements (Supplementary Fig. 5A, B). As expected, MS40N2 induced degradation of IKZF1 and IKZF3, but not WDR5 (Fig. 6B, C). As a dual degrader of WDR5 and IKZF1, MS40 is likely to display therapeutic advantages over PROTACs targeting WDR5 alone. To test this, we designed MS169 (Fig. 6D), the VHL E3 ligase-based MS40 counterpart (which does not target IKZF1/3 and has no reported neo-substrates), and found that, when compared to MS40, MS169 exhibited comparable binding affinities to WDR5 and E3 ligase (Supplementary Fig. 6A–C) and displayed a similar effect on WDR5 degradation in cells without affecting IKZF1/3 (Fig. 6E). It is worth to note that MS67 [31], which is a VHL-based WDR5 PROTAC but much more potent than MS40 (due to additional chemical optimization [31]), is not suitable for assessing whether dual degradation of WDR5 and IKZF1 is more effective than WDR5 degradation alone in inhibiting the cancer cell growth, because a VHL-based WDR5 PROTAC with similar potency as MS40 is needed for this study.
Fig. 6. Compared to its non-PROTAC analogues (MS40N1 and MS40N2) and a matched WDR5-selective PROTAC (MS169), the WDR5 and IKZF dual degrader MS40 more effectively suppresses the growth of MLL-r acute leukemia cells in vitro.
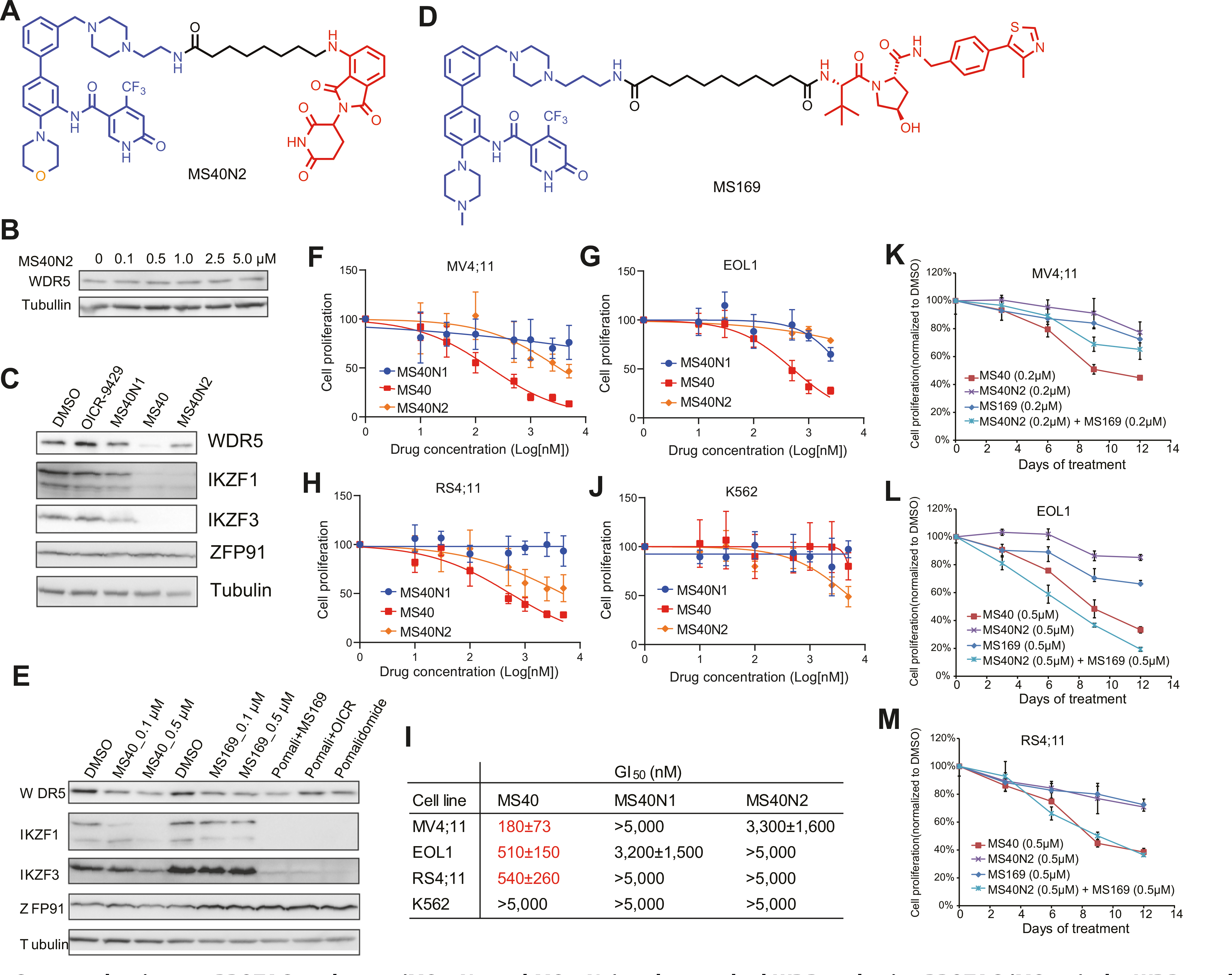
A Chemical structure of MS40N2, a MS40 analogue that can bind CRBN but not WDR5. B Immunoblotting for WDR5 and tubulin in MV4;11 cells, treated with the indicated concentrations of MS40N2 for 18 h. C Immunoblotting for WDR5, neo-substrates and tubulin post-treatment of MV4;11 cells with DMSO or 0.5 μM of MS40, MS40N1, MS40N2, or OICR-9429 for 18 h. D Chemical structure of the VHL-based WDR5 PROTAC MS169, which can degrade WDR5 but not the IMiDs:CRBN neo-substrates. E Immunoblotting for WDR5, IMiDs:CRBN’s neo-substrates and tubulin in MV4;11 cells, treated with DMSO, or the indicated concentration of MS40 (0.1 μM and 0.5 μM), MS169 (0.1 μM and 0.5 μM), pomalidomide (0.5 μM), pomalidomide plus OICR-9429 (0.5 μM, 1:1 molar ratio), or MS169 plus pomalidomide (0.5 μM, 1:1 molar ratio) for 18 h. F–J Measurement of growth inhibition effect (y-axis) after a 9-day treatment with the indicated concentration (x-axis) of MS40, MS40N1, or MS40N2, compared with DMSO, in human leukemia lines, MV4;11 F, EOL1 G, RS4;11 H and K562 J. Y-axis, presented in the mean ± SE, shows the proliferation of cells treated with compound for 9 days after normalization to that of DMSO-treated cells (n = 3 independent experiments). I Summary of GI50 values of M40, MS40N1 and MS40N2 (after a 9-day treatment) in the tested human leukemia cell lines. K–M Measurement of the growth-inhibitory effect by MS40, MS40N2, MS169, or MS40N2 plus MS169 (1:1 molar ratio) in MV4;11 K, EOL1 L, and RS4;11 M cells. Y-axis, presented in the mean ± SE, shows the proliferation of cells treated with 0.2 μM or 0.5 μM of compound for the indicated time (x-axis) after normalization to that of DMSO-treated cells (n = 3 independent experiments).
In comparison to MS40N1 or MS40N2, MS40 more effectively inhibited the in vitro growth of three acute leukemia lines harboring MLL-r (Fig. 6F–I). These compounds did not suppress growth of K562 cells, a non-MLL-r leukemia line carrying BCR-ABL, suggesting that MS40 lacks general toxicity (Fig. 6I–J). When compared to MS169 or MS40N2, MS40 generally had more profound growth-inhibitory effects on MLL-r leukemia (Fig. 6K–M). Co-treatment with MS169 and MS40N2 more closely mimicked that of MS40 (Fig. 6K–M). These results suggest that dual targeting of WDR5 and IKZF1/3 by MS40 is more effective in treating MLL-r AML than targeting of WDR5 alone (by MS169).
Furthermore, we tested anti-proliferative effects of the above compounds in BT549 and SUM149 breast cancer cells and found that they were unaffected by pomalidomide, in consistence with a general lack of IKZF1 expression and a lack of IKZF1 dependency in breast cancer (Supplementary Fig. 2A, B). Both breast cell lines were sensitive to WDR5 degradation as shown by treatment with MS40, relative to MS40N1 and OICR-9429 (Supplementary Fig. 7A, B).
Overall, MS40 significantly suppressed in vitro growth of cancer cells, which cover MLL-r AML and breast tumor. As different tumors may show WDR5 and/or IKZF dependencies, a dual WDR5 and IKZF degrader can therefore be potentially applied to multiple cancer types and be more effective than selective WDR5 degraders for treating MLL-r AML.
MS40 suppresses the in vivo growth of AML
Next, we assessed MS40’s efficacy in treating cancer models that more closely resemble the clinic. Compared to OICR-9429 or MS40N1, MS40 uniquely induced WDR5 degradation and more effectively suppressed in vitro growth of primary AML cells (Fig. 7A, B; Supplementary Fig. 8A). Furthermore, we evaluated in vivo antitumor effect of MS40 using a patient-derived xenograft (PDX) mouse model carrying MLL-AF9 + AML, after determining in vivo mouse pharmacokinetic (PK) properties of MS40 and MS169 (Fig. 7C; Supplementary Fig. 8B). Treatment of mice bearing subcutaneous PDX with MS40 (100 mg/kg via intraperitoneal injection [i.p.]; once a day, 5 days per week) significantly suppressed the tumor growth, compared to vehicle (Fig. 7D). In contrast, comparable treatment of MS169 (34 mg/kg, a comparable dose based on Cmax values of PK data shown in Fig. 7C; Supplementary Fig. 8B) or MS40N2 (100 mg/kg) showed no detected inhibitory effects on PDX tumor growth (Supplementary Fig. 8C), even though the detected concentrations of MS40N2 and MS169 in the isolated plasma and tumor samples post-treatment were higher than those of MS40 (Fig. 7E). No obvious body weight changes were found during these treatments, suggesting that these compounds including MS40 are well tolerated in vivo (Fig. 7F; Supplementary Fig. 8D). Using tumor samples isolated from the mice treated with MS40 versus vehicle, we also confirmed the expected degradation of WDR5 and the suppression of WDR5 target genes such as ribosome genes, BCL2 and RUNX3 (Fig. 7G, H).
Fig. 7. MS40 inhibits the growth of primary AML patient cells in vitro and AML PDX in vivo.
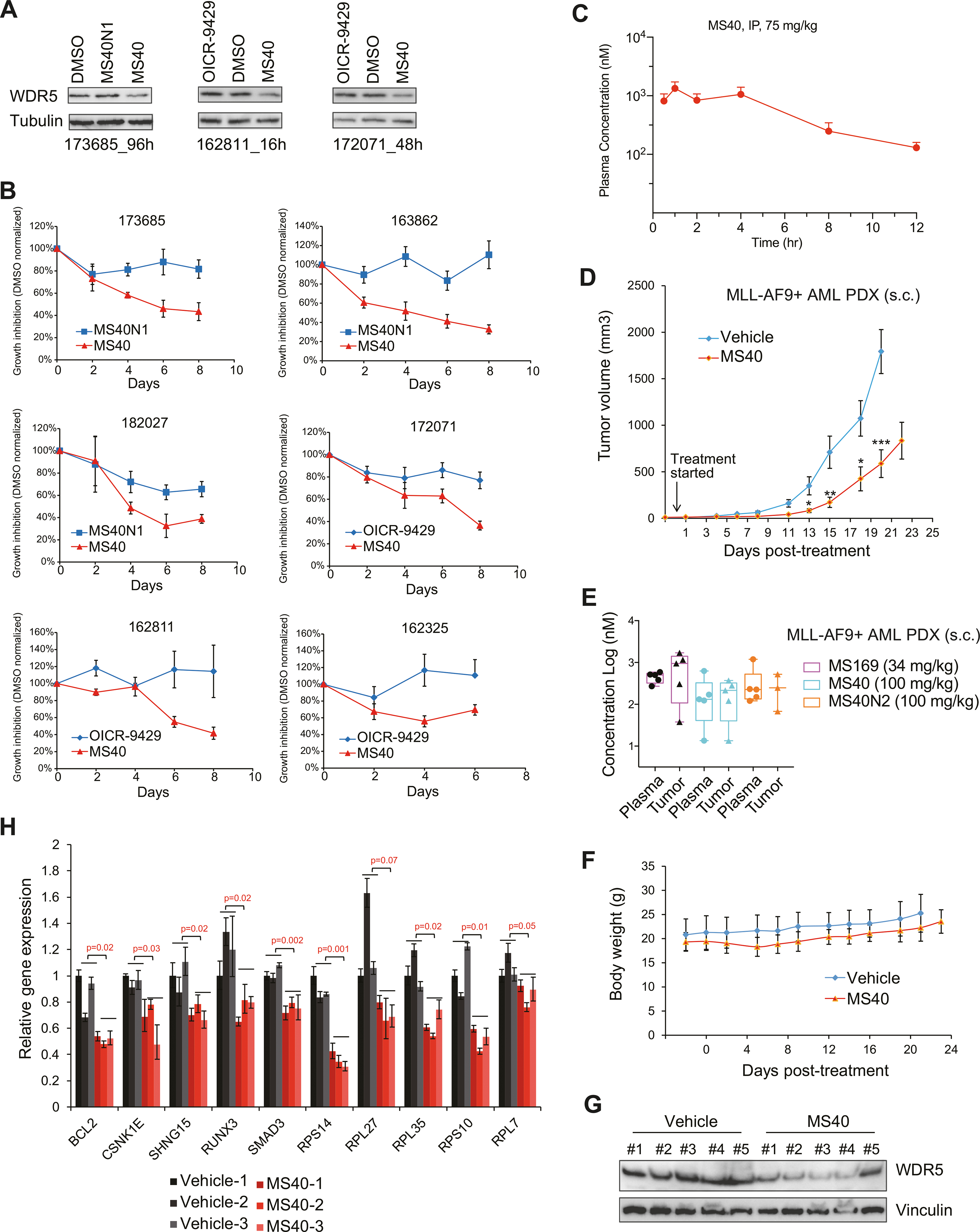
A Immunoblotting for WDR5 and tubulin in three patient-derived primary AML cells after treatment respectively for 96 h (172152, left), 16 h (162811, middle) and 48 h (172071, right) with DMSO or 5 μM of MS40, MS40N1 or OICR-9429. B Growth inhibition (y-axis) after the indicated duration (x-axis) of treatment with 5 μM of MS40, MS40N1 or OICR-9429, compared to DMSO, in six primary AML patient samples cultured in vitro. Y-axis, presented in the mean ± SE, shows the proliferation of compound-treated cells after normalization to DMSO-treated (n = 3 independent experiments). C Intra-plasma concentration of MS40 over a 12-hour period in Swiss Albino mice following a single 75 mg/kg intraperitoneal (i.p.) injection of MS40 (n = 3 mice per time point; mean ± SE). D In vivo growth of the MLL-AF9 + AML PDX tumors subcutaneously (s.c.) xenografted into the NSG-SGM3 mice, followed by treatment from the indicated starting time-point with either vehicle (n = 10 tumors) or 100 mg/kg of MS40 (n = 10; i.p. once a day for five days per week). Y-axis shows the tumor volumes and are presented in mean ± SE. *P < 0.05; **P < 0.01; ***, P < 0.001. E Compound concentrations in the plasma (left) and tumor samples (right) isolated from five NSG-SGM3 mice xenografted with the MLL-AF9 + AML PDX. Samples were collected at two hours post the last dose from the mice treated with MS40 (100 mg/kg), MS169 (34 mg/kg) or MS40N2 (100 mg/kg). F Body weight of NSG-SGM3 mice bearing the MLL-AF9 + AML PDX, treated with either vehicle (n = 5 mice) or 100 mg/kg of MS40 (n = 5, i.p. SID) for 5 days per week. G–H Immunoblotting for WDR5 and vinculin G and RT-qPCR for the indicated WDR5:MLL target genes H in the MLL-AF9 + AML PDX tumor samples isolated at two hours post the last dose from the NSG-SGM3 mice treated with vehicle or MS40. RT-qPCR signals, presented as the mean ± S.D., were normalized first to those of GAPDH and then to DMSO-treated cells (n = 3 replicated experiments).
Collectively, MS40 has antitumor activities in primary cancer samples and a PDX mouse model and is well tolerated in vivo, suggesting that pharmacological degradation of both WDR5 and IKZF1/3 can be a more effective antitumor strategy.
DISCUSSION
Chromatin deregulation represents one of the central oncogenic pathways [45–47]. Among various chromatin-modulatory factors, WDR5 sustains oncogenesis in a range of cancers and represents an attractive onco-target for pharmacological intervention [2, 3, 7–11]. Small-molecule inhibitors that block PPIs between WDR5 and its functional partners have been developed [14–27]. However, these WDR5 PPI inhibitors are largely ineffective in inhibiting cancer growth, as we showed in this work with OICR-9429 or MS40N1. This lack of potent antitumor effects is likely due to several reasons. First, WDR5 PPI inhibitors rely on receptor occupancy pharmacology and sustained binding to PPI sites is required for achieving full and durable blockade of WDR5-mediated interaction and functionality. Second, WDR5 forms interactions with various partners [2, 3, 10, 11], although it is more widely viewed as an essential subunit of the MLL/KMT2 family lysine methyltransferase complexes [4, 5, 48]. WDR5 PPI inhibitors do not simultaneously inhibit all of WDR5’s multifaceted functions in cancer. To address these issues, we developed MS40 using the PROTAC technology. In tumor models that cover hematological cancer (AML and multiple myeloma) and breast tumor, MS40 consistently and effectively depleted WDR5 and IKZF1/3, the well-established neo-substrates of IMiDs:CRBN. The latter is due to the effects of the IMiD portion of MS40. Consequently, MS40 treatment has dual effects: it decreased the chromatin association of WDR5:MLL complexes and reduced H3K4me2, an enzymatic product of WDR5:MLL complexes, which mimics effects by WDR5 KD; and simultaneously, MS40 suppressed IKZF-mediated oncogenic activities. We corroborated such dual-targeting effects by MS-based proteomic study and by genome-wide transcriptomic profiling, which showed that MS40 simultaneously represses the expression of genes targeted by WDR5:MLL complexes and those by IMiDs. IMiDs are FDA-approved drugs for the treatment of cancers including multiple myeloma. Therefore, MS40 represents a bona fide PROTAC degrader for depleting both WDR5 and IKZF1/3. As IKZFs and WDR5 regulate different tumor-promoting programs, their dual targeting PROTAC could potentially exert more profound anti-tumor effects. Dual targeting inhibitors were reported to simultaneously target Bruton tyrosine kinase and IKZFs [49], or BRD4 and Polo-like kinase 1 [50], or EZH2 and HDACs [51].
MS40 has much superior anti-tumor effects to WDR5 PPI inhibitors. Such superior antitumor effects are consistent in different WDR5-dependent tumors such as MLL-r AML and breast cancer. Owing to its dual WDR5- and IKZF1/3-targeting nature, MS40 also offers a strategy for more effectively treating those cancer types that exhibit dependency on both WDR5 and IKZF1/3. As a proof-of-principle study, we use the MLL-r acute leukemias, which display IKZF1 high-expression and dependency, to show that MS40 is indeed more potent in treating MLL-r leukemias than MS169, a matched VHL-based PROTAC that effectively degrades WDR5 but not neo-substrates of IMiDs:CRBN. Importantly, MS40, which exhibits an excellent in vivo pharmacodynamics and pharmacokinetics (PK/PD) relationship, is more effective in treating MLL-r AML PDX in mice than MS40N1 (essentially a WDR5 PPI inhibitor) or MS169 (a degrader of WDR5 but not IKZF1/3) and lacks apparent in vivo toxicity. It is worth to note that we previously disclosed the discovery of first WDR5 PROTAC degraders including MS40 in the patent literature [52], and during the preparation of this manuscript, an independent work recently reported a WDR5 PROTAC degrader [32]. However, superiority of the reported WDR5 PROTACs [32] to WDR5 PPI inhibitors in inhibiting cancer cell growth in vitro and in vivo is generally unknown. Here, we demonstrate that MS40 is more effective than the WDR5 PPI inhibitor (MS40N1 or OICR-9429) using a suite of integrated approaches including proteomics, transcriptomics (RNAseq and GRO-seq), ChIP-seq, and in vitro and in vivo cancer treatment studies.
Furthermore, a set of paired PROTAC and non-PROTAC compounds (MS40, MS40N1, and MS40N2) are useful chemical tools for the scientific community to study WDR5 and its functional partners in biology and medicine. Future investigation is warranted to evaluate antitumor effects of MS40 (a dual degrader of WDR5 and IKZF1/3) and MS169 (a comparable WDR5-selective degrader that does not degrade IKZF1/3) in other tumors, which may rationalize potentially wider applications of the dual WDR5 and IKZF1/3-targeting strategy.
METHODS
Isothermal titration calorimetry (ITC)
VHL–Elongin C–Elongin B ternary complex (VCB), and WDR5 proteins were prepared as described previously [31] and CRBN’s thalidomide-binding domain (TBD) were dialyzed overnight in a buffer containing 50 mM potassium phosphate buffer pH 8.0, 100 mM KCl, 1 mM TCEP and 0.05% Tween20. Titrations were performed on a Microcal ITC200 instrument in the reverse mode at 25 °C, as described previously [53] with a slight modification. Briefly, Cereblon TBD or VCB (250 μM in syringe) was titrated into the PROTAC (25 μM of PROTAC in the cell). At the end of the titration, the excess of the solution was removed and the syringe was washed and dried. For the ternary titrations, the WDR5 protein (210 μM) was loaded in the syringe and titrated into either PROTAC-CRBN TBD or PROTAC-VCB complex (21 μM) in the cell. For the WDR5-PROTAC binary titrations, an initial single injection of buffer was added into the PROTAC solution in the cell (25 μM) followed by titration of WDR5 (210 μM in the syringe). The data were fitted to single binding site model using the Origin 7.0 software, supplied by MicroCal. Cooperativity values (α) were determined by calculation the ratio of the KD obtained from the binary WDR5 to PROTAC titration and KD of the ternary complex titration. The reported values represent the mean ± S.D. from three independent measurements.
Cell fractionation
Preparation of the chromatin-bound cell fraction was conducted as we described before [54]. Cells were first washed with cold PBS and then resuspended in CSK buffer (10 mM PIPES pH 7.0, 100 mM NaCl, 300 mM sucrose, 3 mM MgCl2, 0.5% Triton X-100; supplemented with sodium butyrate and inhibitors of protease and phosphatase), followed by incubation on ice for 10 minutes and centrifugation at 1500 g for 10 mins. After removing the supernatant, the remaining cell pellet was collected as the chromatin-bound fraction for immunoblotting.
Chromatin immunoprecipitation sequencing (ChIP-seq), RNA sequencing (RNA-seq) and Global Run-On sequencing (GRO-seq)
ChIP-seq was performed as previously described with slight modification [55, 56], with inclusion of Drosophila chromatin spike-in for normalization [57, 58]. As for ChIP-seq of MLL1 (Benthyl Laboratories, A300–086A), a two-step crosslinking protocol was used as described before [59]. Cells were first crosslinked with 2 mM disuccinimidyl glutarate (DSG, Sigma 80424) for 30 min with rotation at room temperature; then, cells were pelleted and washed twice with 1× PBS, followed by incubation with 1% formaldehyde for 10 min with rotation at room temperature. RNA-seq-based transcriptomic profiling was conducted as previously described [56, 60, 61]. GRO-seq was performed as before [62]. The detailed procedures of ChIP-seq, GRO-seq and RNA-seq and data analyses were provided in Supplementary Methods.
Cell proliferation assay
Flow-growing leukemia cells were seeded at the density of 2 × 105/mL in triplicate in the 24-well plates, with compounds pre-added into medium at a range of concentrations as we conducted before [31, 63]. Medium with fresh compounds were changed every three days. Cells were passaged after mixing via pipetting and then dilution to keep the cell density under 1 × 106/mL at all time. Cells were counted by a TC10 automated cell counter (Bio-Rad) every three days. The 50% of maximal growth inhibition (GI50) values were calculated using GraphPad Prism software with a nonlinear regression analysis. The mean ± SE (standard error) was calculated from three independent experiments.
In vivo efficacy studies
All experiments involving mice were performed according to the Institutional Animal Care and Use Committee (IACUC)-approved protocol. For subcutaneous (s.c.) xenograft of AML patient-derived xenograft (PDX) cells, 3 million of MLL-AF9 + AML PDX cells (DFAM-68555-V1; available from PRoXe.org) [64], suspended in a mixture (1:1, vol/vol) of 1× PBS and Matrigel (Corning, 354248), were injected subcutaneously to both flanks of each 8-week-old NSG-SGM3 mouse (NOD-SCID IL2Rgnull-3/GM/SF; JAX, stock #013062). After three days, mice were randomly distributed into different treatment groups. For the MS40 group, mice were treated with MS40 once per day (SID) by i.p. injections at a dose of 100 mg/kg for 5 days per week (from Monday to Friday). For MS169 treatment, the used dose was 34 mg/kg (SID; i.p.) for 5 days per week (from Monday to Friday). For MS40N2 treatment, the used dose was 100 mg/kg (SID; i.p.) for 5 days per week (from Monday to Friday). The matched vehicle was used as respective treatment controls. Tumor volume was recorded every 2–3 days via caliper measurement (carried out by the UNC Animal Studies Core). The study was terminated when the tumor size reached the IACUC allowed limit.
Statistics and reproducibility
Experimental data are presented as the mean ± SD of three independent experiments unless otherwise noted. Statistical analysis was performed using an unpaired two-sided Student’s t-test for comparing two sets of data with assumed normal distribution. For in vivo studies, a two-sided Student’s t-test was performed to calculate the statistical differences in tumor sizes. The quantification for immunoblots was calculated from at least three biologically independent treatments. All statistical analysis and visualizations were performed using GraphPad (Prism v8.4.2) or Excel.
Data sharing
The genomic profiling datasets such as ChIP-seq, GRO-seq, and RNA-seq related to this work have been deposited in the NCBI GEO under accession number GSE175548.
Additional methods such as chemical synthesis, protein purification, antibodies and immunoblotting, cell fractionation, detailed protocols of chromatin immunoprecipitation sequencing (ChIP-seq) and data analysis, gene KD, RNA sequencing (RNA-seq) and data analysis, Global Run-On sequencing (GRO-seq) and data analysis, quantitative reverse transcriptase polymerase chain reaction (RT-qPCR), mass spectrometry-based proteomics profiling with tandem mass tag isobaric labeling (TMT), pharmacokinetic study, bioanalysis of plasma and tumor samples, and culture of human patient-derived AML cells, can be found in supplemental materials.
Supplementary Material
ACKNOWLEDGEMENTS
We graciously thank Drs. J Bradner, W Kaelin, Y Dou, H Wen, X Shi, and H Jiang for providing reagents and cells used in the study and the Wang and Jin Laboratory members for helpful discussion and technical support. This work was supported in part by the US National Institutes of Health grants R01GM122749 (to JJ), P30CA196521 (to JJ), R01CA211336 (to GGW), R01CA215284 (to GGW), R01CA268384 (to JJ and GGW), R35GM131780 (to AKA), R24GM137786 (to AJT), P20GM121293 (to AJT), R01CA236209 (to AJT), P20GM103429 (to AJT), an NIH/Office of the Director Grant S10OD018445 (to SGM), R01GM137009 (to ZL), U54 CA217297/PRJ001 (to ZL), endowed professorships from the Icahn School of Medicine at Mount Sinai (to JJ and AKA), and grants/awards from Gabrielle’s Angel Foundation for Cancer Research (to GGW), When Everyone Survives (WES) Leukemia Research Foundation (to GGW) and UNC Lineberger Cancer Center UCRF Stimulus Initiative Grants (to GGW and LC). G.G.W. is an American Cancer Society Research Scholar, a Leukemia and Lymphoma Society Scholar, and an American Society of Hematology Scholar in Basic Science. This work utilized the NMR Spectrometer Systems at Mount Sinai acquired with funding from National Institutes of Health SIG grants 1S10OD025132 and 1S10OD028504. We thank UNC’s facilities, including High-throughput Sequencing Facility (HTSF), Bioinformatics Core, Tissue Culture Facility, Animal Studies Core, and UNC Tissue Procurement Facility, for their professional assistance in this work. The cores affiliated with the UNC Cancer Center are supported in part by the UNC Lineberger Comprehensive Cancer Center Core Support Grant P30CA016086.
Footnotes
COMPETING INTERESTS
JJ, GW, JL, XY, and DL are inventors of a patent application filed by the Icahn School of Medicine at Mount Sinai and University of North Carolina at Chapel Hill. The Jin laboratory received research funds from Celgene Corporation, Levo Therapeutics, Inc., Cullgen, Inc. and Cullinan Oncology, Inc. JJ is a cofounder, scientific advisory board member and equity shareholder in Cullgen, Inc. and a consultant for Cullgen, Inc., EpiCypher, Inc. and Accent Therapeutics, Inc.
ADDITIONAL INFORMATION
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41388-022-02340-8.
REFERENCES
- 1.Ge Z, Song EJ, Kawasawa YI, Li J, Dovat S, Song C. WDR5 high expression and its effect on tumorigenesis in leukemia. Oncotarget. 2016;7:37740–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Carugo A, Genovese G, Seth S, Nezi L, Rose JL, Bossi D, et al. In vivo functional platform targeting patient-derived xenografts identifies WDR5-Myc association as a critical determinant of pancreatic cancer. Cell Rep 2016;16:133–47. [DOI] [PubMed] [Google Scholar]
- 3.Sun Y, Bell JL, Carter D, Gherardi S, Poulos RC, Milazzo G, et al. WDR5 Supports an N-Myc Transcriptional Complex That Drives a Protumorigenic Gene Expression Signature in Neuroblastoma. Cancer Res. 2015;75:5143–54. [DOI] [PubMed] [Google Scholar]
- 4.Miller T, Krogan NJ, Dover J, Erdjument-Bromage H, Tempst P, Johnston M, et al. COMPASS: a complex of proteins associated with a trithorax-related SET domain protein. Proc Natl Acad Sci USA. 2001;98:12902–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Trievel RC, Shilatifard A. WDR5, a complexed protein. Nat Struct Mol Biol. 2009;16:678–80. [DOI] [PubMed] [Google Scholar]
- 6.Rao RC, Dou Y. Hijacked in cancer: the KMT2 (MLL) family of methyltransferases. Nat Rev Cancer. 2015;15:334–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Patel A, Dharmarajan V, Cosgrove MS. Structure of WDR5 bound to mixed lineage leukemia protein-1 peptide. J Biol Chem. 2008;283:32158–61. [DOI] [PubMed] [Google Scholar]
- 8.Patel A, Vought VE, Dharmarajan V, Cosgrove MS. A conserved arginine-containing motif crucial for the assembly and enzymatic activity of the mixed lineage leukemia protein-1 core complex. J Biol Chem. 2008;283:32162–75. [DOI] [PubMed] [Google Scholar]
- 9.Dharmarajan V, Lee JH, Patel A, Skalnik DG, Cosgrove MS. Structural basis for WDR5 interaction (Win) motif recognition in human SET1 family histone methyltransferases. J Biol Chem. 2012;287:27275–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thomas LR, Wang Q, Grieb BC, Phan J, Foshage AM, Sun Q, et al. Interaction with WDR5 promotes target gene recognition and tumorigenesis by MYC. Mol Cell. 2015;58:440–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guarnaccia AD, Tansey WP. Moonlighting with WDR5: a cellular multitasker. J Clin Med. 2018; 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alicea-Velazquez NL, Shinsky SA, Loh DM, Lee JH, Skalnik DG, Cosgrove MS. Targeted Disruption of the Interaction between WD-40 Repeat Protein 5 (WDR5) and Mixed Lineage Leukemia (MLL)/SET1 family proteins specifically inhibits MLL1 and SETd1A methyltransferase complexes. J Biol Chem. 2016;291:22357–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Odho Z, Southall SM, Wilson JR. Characterization of a novel WDR5-binding site that recruits RbBP5 through a conserved motif to enhance methylation of histone H3 lysine 4 by mixed lineage leukemia protein-1. J Biol Chem. 2010;285:32967–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Senisterra G, Wu H, Allali-Hassani A, Wasney GA, Barsyte-Lovejoy D, Dombrovski L, et al. Small-molecule inhibition of MLL activity by disruption of its interaction with WDR5. Biochem J. 2013;449:151–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bolshan Y, Getlik M, Kuznetsova E, Wasney GA, Hajian T, Poda G, et al. Synthesis, Optimization, and Evaluation of Novel Small Molecules as Antagonists of WDR5-MLL Interaction. ACS Med Chem Lett. 2013;4:353–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Karatas H, Townsend EC, Cao F, Chen Y, Bernard D, Liu L, et al. High-affinity, small-molecule peptidomimetic inhibitors of MLL1/WDR5 protein-protein interaction. J Am Chem Soc. 2013;135:669–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cao F, Townsend EC, Karatas H, Xu J, Li L, Lee S, et al. Targeting MLL1 H3K4 methyltransferase activity in mixed-lineage leukemia. Mol Cell. 2014;53:247–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grebien F, Vedadi M, Getlik M, Giambruno R, Grover A, Avellino R, et al. Pharmacological targeting of the Wdr5-MLL interaction in C/EBPalpha N-terminal leukemia. Nat Chem Biol. 2015;11:571–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhu J, Sammons MA, Donahue G, Dou Z, Vedadi M, Getlik M, et al. Gain-of-function p53 mutants co-opt chromatin pathways to drive cancer growth. Nature. 2015;525:206–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Getlik M, Smil D, Zepeda-Velazquez C, Bolshan Y, Poda G, Wu H, et al. Structure-based optimization of a small molecule antagonist of the interaction between WD repeat-containing protein 5 (WDR5) and mixed-lineage leukemia 1 (MLL1). J Med. Chem. 2016;59:2478–96. [DOI] [PubMed] [Google Scholar]
- 21.Li DD, Chen WL, Wang ZH, Xie YY, Xu XL, Jiang ZY, et al. High-affinity small molecular blockers of mixed lineage leukemia 1 (MLL1)-WDR5 interaction inhibit MLL1 complex H3K4 methyltransferase activity. Eur J Med Chem. 2016;124:480–9. [DOI] [PubMed] [Google Scholar]
- 22.Karatas H, Li Y, Liu L, Ji J, Lee S, Chen Y, et al. Discovery of a highly potent, cell-permeable macrocyclic peptidomimetic (MM-589) targeting the WD repeat domain 5 protein (WDR5)-mixed lineage leukemia (MLL) protein-protein interaction. J medicinal Chem 2017;60:4818–39. [DOI] [PubMed] [Google Scholar]
- 23.Wang F, Jeon KO, Salovich JM, Macdonald JD, Alvarado J, Gogliotti RD. et al. Discovery of potent 2-Aryl-6,7-dihydro-5 H-pyrrolo[1,2- a]imidazoles as WDR5-WIN-site inhibitors using fragment-based methods and structure-based design. J. Med. Chem. 2018;61:5623–42.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aho ER, Wang J, Gogliotti RD, Howard GC, Phan J, Acharya P, et al. Displacement of WDR5 from Chromatin by a WIN Site Inhibitor with Picomolar Affinity. Cell Rep. 2019;26:2916–28. e2913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Macdonald JD, Chacon Simon S, Han C, Wang F, Shaw JG, Howes JE, et al. Discovery and optimization of salicylic acid-derived sulfonamide inhibitors of the WD repeat-containing protein 5-MYC protein-protein interaction. J Med Chem. 2019;62:11232–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tian J, Teuscher KB, Aho ER, Alvarado JR, Mills JJ, Meyers KM, et al. Discovery and structure-based optimization of potent and selective WD repeat domain 5 (WDR5) inhibitors containing a dihydroisoquinolinone bicyclic core. J Med Chem. 2020;63:656–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chacon Simon S, Wang F, Thomas LR, Phan J, Zhao B, Olejniczak ET, et al. Discovery of WD repeat-containing protein 5 (WDR5)-MYC inhibitors using fragment-based methods and structure-based design. J Med Chem. 2020;63:4315–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lai AC, Crews CM. Induced protein degradation: an emerging drug discovery paradigm. Nat Rev Drug Discov. 2017;16:101–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schapira M, Calabrese MF, Bullock AN, Crews CM. Targeted protein degradation: expanding the toolbox. Nat Rev Drug Discov. 2019;18:949–63. [DOI] [PubMed] [Google Scholar]
- 30.Dale B, Cheng M, Park KS, Kaniskan HU, Xiong Y, Jin J. Advancing targeted protein degradation for cancer therapy. Nat Rev Cancer. 2021;21:638–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yu X, Li D, Kottur J, Shen Y, Kim HS, Park KS, et al. A selective WDR5 degrader inhibits acute myeloid leukemia in patient-derived mouse models. Sci Transl Med. 2021;13:eabj1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dolle A, Adhikari B, Kramer A, Weckesser J, Berner N, Berger LM, et al. Design, synthesis, and evaluation of WD-repeat-containing protein 5 (WDR5) degraders. J Med Chem. 2021;64:10682–710. [DOI] [PubMed] [Google Scholar]
- 33.Akuffo AA, Alontaga AY, Metcalf R, Beatty MS, Becker A, McDaniel JM, et al. Ligand-mediated protein degradation reveals functional conservation among sequence variants of the CUL4-type E3 ligase substrate receptor cereblon. J Biol Chem. 2018;293:6187–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mori T, Ito T, Liu S, Ando H, Sakamoto S, Yamaguchi Y, et al. Structural basis of thalidomide enantiomer binding to cereblon. Sci Rep. 2018;8:1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lu G, Middleton RE, Sun H, Naniong M, Ott CJ, Mitsiades CS, et al. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science. 2014;343:305–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kronke J, Fink EC, Hollenbach PW, MacBeth KJ, Hurst SN, Udeshi ND, et al. Lenalidomide induces ubiquitination and degradation of CK1alpha in del(5q) MDS. Nature. 2015;523:183–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Matyskiela ME, Lu G, Ito T, Pagarigan B, Lu CC, Miller K, et al. A novel cereblon modulator recruits GSPT1 to the CRL4(CRBN) ubiquitin ligase. Nature. 2016;535:252–7. [DOI] [PubMed] [Google Scholar]
- 38.Ishoey M, Chorn S, Singh N, Jaeger MG, Brand M, Paulk J, et al. Translation termination factor GSPT1 is a phenotypically relevant off-target of heterobifunctional phthalimide degraders. ACS Chem Biol. 2018;13:553–60. [DOI] [PubMed] [Google Scholar]
- 39.An J, Ponthier CM, Sack R, Seebacher J, Stadler MB, Donovan KA, et al. pSILAC mass spectrometry reveals ZFP91 as IMiD-dependent substrate of the CRL4 (CRBN) ubiquitin ligase. Nat Commun. 2017;8:15398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xia R, Cheng Y, Han X, Wei Y, Wei X. Ikaros proteins in tumor: current perspectives and new developments. Front Mol Biosci. 2021;8:788440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Park I, Phan TM, Fang J. Novel molecular mechanism of lenalidomide in myeloid malignancies independent of deletion of chromosome 5q. Cancers (Basel) 2021; 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dou Y, Milne TA, Tackett AJ, Smith ER, Fukuda A, Wysocka J, et al. Physical association and coordinate function of the H3 K4 methyltransferase MLL1 and the H4 K16 acetyltransferase MOF. Cell. 2005;121:873–85. [DOI] [PubMed] [Google Scholar]
- 43.Steward MM, Lee JS, O’Donovan A, Wyatt M, Bernstein BE, Shilatifard A. Molecular regulation of H3K4 trimethylation by ASH2L, a shared subunit of MLL complexes. Nat Struct Mol Biol. 2006;13:852–4. [DOI] [PubMed] [Google Scholar]
- 44.Patel A, Vought VE, Dharmarajan V, Cosgrove MS. A novel non-SET domain multi-subunit methyltransferase required for sequential nucleosomal histone H3 methylation by the mixed lineage leukemia protein-1 (MLL1) core complex. J Biol Chem. 2011;286:3359–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhao S, Allis CD, Wang GG. The language of chromatin modification in human cancers. Nat Rev Cancer. 2021;21:413–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chi P, Allis CD, Wang GG. Covalent histone modifications-miswritten, misinterpreted and mis-erased in human cancers. Nat Rev Cancer. 2010;10:457–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dawson MA. The cancer epigenome: concepts, challenges, and therapeutic opportunities. Science. 2017;355:1147–52. [DOI] [PubMed] [Google Scholar]
- 48.Roguev A, Schaft D, Shevchenko A, Pijnappel WW, Wilm M, Aasland R, et al. The Saccharomyces cerevisiae Set1 complex includes an Ash2 homologue and methylates histone 3 lysine 4. EMBO J. 2001;20:7137–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dobrovolsky D, Wang ES, Morrow S, Leahy C, Faust T, Nowak RP, et al. Bruton tyrosine kinase degradation as a therapeutic strategy for cancer. Blood. 2019;133:952–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ciceri P, Muller S, O’Mahony A, Fedorov O, Filippakopoulos P, Hunt JP, et al. Dual kinase-bromodomain inhibitors for rationally designed polypharmacology. Nat Chem Biol. 2014;10:305–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Romanelli A, Stazi G, Fioravanti R, Zwergel C, Di Bello E, Pomella S, et al. Design of first-in-class dual EZH2/HDAC inhibitor: biochemical activity and biological evaluation in cancer cells. ACS Med Chem Lett. 2020;11:977–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jin J, Wang G, Liu J, Yu X, Li D. WD40 REPEAT DOMAIN PROTEIN 5 (WDR5) DEGRADATION/DISRUPTION COMPOUNDS AND METHODS OF USE. WO2019246570; 2019. [Google Scholar]
- 53.Gadd MS, Testa A, Lucas X, Chan KH, Chen W, Lamont DJ, et al. Structural basis of PROTAC cooperative recognition for selective protein degradation. Nat Chem Biol. 2017;13:514–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li J, Galbo PM Jr., Gong W, Storey AJ, Tsai YH, Yu X, et al. ZMYND11-MBTD1 induces leukemogenesis through hijacking NuA4/TIP60 acetyltransferase complex and a PWWP-mediated chromatin association mechanism. Nat Commun. 2021;12:1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lu R, Wang P, Parton T, Zhou Y, Chrysovergis K, Rockowitz S, et al. Epigenetic perturbations by Arg882-mutated DNMT3A potentiate aberrant stem cell gene-expression program and acute leukemia development. Cancer Cell. 2016;30:92–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cai L, Tsai YH, Wang P, Wang J, Li D, Fan H, et al. ZFX mediates non-canonical oncogenic functions of the androgen receptor splice variant 7 in castrate-resistant prostate cancer. Mol cell. 2018;72:341–54. e346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Egan B, Yuan CC, Craske ML, Labhart P, Guler GD, Arnott D, et al. An alternative approach to chip-seq normalization enables detection of genome-wide changes in histone H3 lysine 27 trimethylation upon EZH2 Inhibition. PLoS ONE. 2016;11:e0166438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ahn JH, Davis ES, Daugird TA, Zhao S, Quiroga IY, Uryu H, et al. Phase separation drives aberrant chromatin looping and cancer development. Nature. 2021;595:591–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xu J, Li L, Xiong J, DenDekker A, Ye A, Karatas H, et al. MLL1 and MLL1 fusion proteins have distinct functions in regulating leukemic transcription program. Cell Discov. 2016;2:16008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fan H, Lu J, Guo Y, Li D, Zhang ZM, Tsai YH, et al. BAHCC1 binds H3K27me3 via a conserved BAH module to mediate gene silencing and oncogenesis. Nat Genet. 2020;52:1384–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ren Z, Ahn JH, Liu H, Tsai YH, Bhanu NV, Koss B, et al. PHF19 promotes multiple myeloma tumorigenicity through PRC2 activation and broad H3K27me3 domain formation. Blood. 2019;134:1176–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li W, Notani D, Ma Q, Tanasa B, Nunez E, Chen AY, et al. Functional roles of enhancer RNAs for oestrogen-dependent transcriptional activation. Nature. 2013;498:516–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Xu B, On DM, Ma A, Parton T, Konze KD, Pattenden SG, et al. Selective inhibition of EZH2 and EZH1 enzymatic activity by a small molecule suppresses MLL-rearranged leukemia. Blood. 2015;125:346–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Townsend EC, Murakami MA, Christodoulou A, Christie AL, Koster J, DeSouza TA, et al. The public repository of xenografts enables discovery and randomized phase II-like trials in mice. Cancer Cell. 2016;29:574–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.