

Abstract
Atopic dermatitis (AD) is a common, complex trait, arising from the interplay of multiple genetic and environmental factors. This review provides an overview of developments in the field of AD genetics. AD shows high heritability; strategies to investigate genetic risk include linkage, candidate gene studies, genome-wide association and animal modelling. Loss-of-function mutations in FLG, encoding the skin barrier protein filaggrin, remain the strongest genetic risk factor identified for AD, but variants influencing skin and systemic immune function are also important. AD is at the forefront of genetic research, from large-scale population studies to in vitro models and detailed molecular analyses. An understanding of genetic risk factors has considerably improved knowledge of mechanisms leading to atopic skin inflammation. Together this work has identified avenues for therapeutic intervention, but further research is needed to fully realise the opportunities of personalised medicine for this complex disease, to optimise patient benefit.
Key words: atopic dermatitis, eczema, filaggrin, genetic, genome-wide, risk, phenotype, transcriptome
Atopic dermatitis (AD), synonymous with atopic eczema, is a common chronic inflammatory skin disorder with a lifetime prevalence of 10–20% in developed countries (1, 2). AD is considered to be a genetically “complex disease”, with interactions of multiple genetic, biological and environmental factors leading to skin barrier dysfunction and altered immunological response. Having AD has a severely negative impact on health-related quality of life, including self-confidence and sleep; it also implies a socioeconomic burden (3).
AD has been known from ancient times. According to the Roman biographer Suetonius, the Emperor Augustus suffered from symptoms and signs of atopic diseases ”…noting a number of hard, dry patches suggesting ringworm, caused by an itching of his skin” as well as “seasonal disorders,” noticing that he experienced in the early spring “a tightness of the diaphragm; and when the sirocco blew, catarrh” (4).
SIGNIFICANCE
Atopic dermatitis (also called eczema) often runs in families, showing that this disease occurs partly because of inherited genetic risk. Research to understand the genetic variation that contributes to an individual’s risk of atopic dermatitis has improved our understanding of mechanisms in the skin that can lead to a leaky barrier and inflammation. Already this knowledge has been applied to treatment and eventually it is hoped that these insights will lead to personalised medicine, in which treatment is tailored to a patient’s genetic make-up and their individual type of atopic dermatitis.
This review aims to provide readers with a historical perspective on the progression of genetic studies in AD over recent decades, the rapid escalation of molecular techniques and a view to future opportunities in the field.
WHAT HAVE WE LEARNED ABOUT ATOPIC DERMATITIS GENETICS OVER THE PAST 100 YEARS?
Heritability of AD: family and twin studies
It can clearly be observed that atopic diseases show a familial aggregation, with clustering of affected individuals within families, demonstrating the importance of genetic heritability. The term ‘heritability’ refers to the proportion of variation within a clinical feature that is attributable to genetic factors (5). A family history of atopic diseases, in particular AD, is the strongest of all risk factors. The presence of any atopic disease in one parent is estimated to increase a child’s risk of developing AD 1.5-fold, whereas the risk is increased ˜3-fold and ˜5-fold, respectively, if one or both parents have AD (6, 7). Familial aggregation can be due to shared environment and/or shared genes and a way to address the genetic component is to study twins. These studies have shown a concordance rate of 72–86% in monozygotic twins and 21–23% in dizygotic twins (8, 9). These data demonstrate that the genetic contribution to the development of AD is substantial and this heritability has been estimated at 70–80% (10, 11) – a high heritability for a complex trait (12). For comparison, psoriasis heritability is approximately 68% (13) whilst other inflammatory barrier diseases show heritability of 7–38% for periodontitis (14) and approximately 67% in ulcerative colitis (15).
Strategies for the investigation of genetic risk
Various different strategies have been used to study genetic components in complex diseases such as AD. In broad genomic analyses (genome-wide linkage, genome-wide association studies) a pre-existing knowledge of the function of genes is not required, nor the biology of the trait in question; it is a ‘hypothesis-free’ approach. In contrast, directed genetic analysis such as a candidate gene approach is a strategy in which certain loci or genes considered to be of interest for the phenotype are selected for study. The selection can be based on earlier studies, “educated guesses” or knowledge of the pathogenesis and function of previously identified genes or loci; this is a ‘hypothesis-driven’ approach. Each of these strategies has been used to provide insight into AD.
Linkage studies
Genetic linkage is a method for mapping genes. It exploits the fact that a marker (often a microsatellite marker such as repeated DNA sequences, mostly di-, tri-, and tetra-nucleotide repeats) show variation between individuals. Informative markers have many alleles and are distributed at known locations throughout the genome. The first genome-wide study in AD identified a major susceptibility locus on chromosome 3q21 (16). During the following years, additional genome-wide studies in AD were performed and several more loci were identified including 1q21,3p,17q, 18q,11.13q. However, these loci were often too wide and they required labour intensive fine-mapping. Genome-wide association studies (GWAS) have subsequently replaced genome-wide linkage (17). The technique of GWAS is described in more detail below.
Candidate genes
Filaggrin (FLG). Using a candidate gene approach, and the link between ichthyosis vulgaris and AD, the FLG gene was identified as a susceptibility gene for AD in 2006 (18). This was a major breakthrough and also established the impaired skin barrier function as having a key role in the development of AD. Filaggrin is involved in the development of keratinocytes to maintain epidermal integrity and it is an important marker of keratinocyte differentiation. During keratinocyte differentiation, profilaggrin is dephosphorylated and degraded into monomers, which condense in the cytoskeleton of keratin to form an intensive protein-lipid matrix. Consequently, these filaggrin monomers are degraded into amino acids, which contribute to the natural moisturising factors, maintaining skin hydration, a low pH and other aspect of the barrier function of the stratum corneum (Fig. 1).
Fig. 1.

Filaggrin expression and processing in the epidermis. The pro-protein profilaggrin is cleaved in a stepwise process into filaggrin monomers which are then degraded to release amino acids, contributing to ‘natural moisturising factors’ in the stratum corneum (19, 20). Filaggrin is an important marker of keratinocyte differentiation. SC: stratum corneum; SG: stratumgranulosum; SS: stratumspinosum; SB: stratum basale.
Loss-of-function mutations in FLG are present in up to 10% in the Northern European population. They cause the common monogenetic dry skin disorder ichthyosis vulgaris. The most common loss-of-function mutations in Europe are R501X, 2282del4, R2447X and S3247X. Together these 4 null mutations account for >90% of null mutations in the population (21). Among European patients with moderate to severe AD up to 40% of the patients carry a FLG null mutation. In meta-analysis the risk of getting AD in a mutation carrier is increased 3-fold (odds ratio 3.12) (22, 23). However, among Europeans only ˜20% of patients with mild-to-moderate AD carry FLG null mutations and >50% of individuals carrying FLG mutations do not develop any atopic disease and this indicates that FLG mutations are neither necessary nor sufficient to cause AD (24).
The frequency of FLG null mutations diverges in different populations and >50 have been characterized worldwide (25, 26). In Asian countries, the prevalence of mutation varies from 3% to over 50%, and many mutations are family-specific (25, 27–30). Research in people of African ancestry has been relatively limited to date and the prevalence of FLG null mutations appears to be less than 1% (31–33). Studies on African Americans have shown a slightly higher frequency of FLG mutations and FLG2 has also been identified as a possible susceptibility gene (34, 35).
To address the question of why FLG mutations are so prevalent in the white European population, it has been hypothesized that this is due to an evolutionary advantage. The increased skin barrier permeability in filaggrin-deficient skin may enhance immunity to infections, conferring ‘natural vaccination’ to individuals with FLG mutations during European pandemics (36). Additionally, filaggrin deficiency may confer an evolutionary advantage in higher latitudes (i.e. Northern Europe) through its role in increasing vitamin D biosynthesis. Vitamin D3 levels are 10% higher in German and Danish individuals with FLG null mutations, which may be due to a reduction in filaggrin’s role as an endogenous UVB filter in the skin (37).
Besides mutations there are intragenic repetitive gene sequences or ‘copy number variations’ in FLG that determine the amount of filaggrin monomer expressed in the skin. Having more repeats (12 compared to 10 on each allele) is associated with reduced risk of AD (38) by a dose-dependent effect within this repetitive gene sequence. The effects of cytokines, such as IL-4, IL-13, IL-17A, IL-22, IL-25, IL-31, and TNF-α have also been shown to suppress filaggrin expression in the skin, resulting in additional barrier impairment (39, 40).
Even though FLG mutations and the filaggrin protein are extremely important in AD pathogenesis, there must be yet unknown, additional factors/genes or functions of gene involved in AD development that still are to be found.
Some other candidate genes in atopic dermatitis. Other genes that has been detected through a candidate gene approach, supported by knowledge of AD biology, and replicated by GWAS are genes involved in the Th2 immune response, for instance IL-4 located on chromosome 5q31.1, the IL-4 receptor located on chromosome 16p12.1-p11.2 and IL-13 on chromosome 5q31.1 (24, 41).
More candidate genes have been detected through the study of monogenic diseases that have features that resemble AD. Netherton syndrome (OMIM #256500) is a rare monogenic disease with AD-like lesions in the skin and increased IgE levels. The gene mutation underlying Netherton is in the Serine Protease Inhibitor Kazal-Type 5 gene (SPINK5) located on chromosome 5q32. SPINK5 encodes a 15-domain protease inhibitor Lymphoepithelial Kazal-Type-Related Inhibitor (LEKTI) which is expressed in epithelial and mucosal surfaces and in the thymus. In several studies, there has been an association between SPINK5 variants and AD, also in different populations (42–45). Other candidate genes will be studied as a result of new approaches to assessing monogenic disorders and extreme phenotypes, as discussed below.
Animal models
Animal models have the advantages that one can more easily control the environment and create genetic homogeneity. Apart from humans, dogs have spontaneous AD that has been studied and documented (46).
There are also several AD mouse models that have been described and generated over the years, each focusing on one or more aspects of human AD. The mouse models can be divided into 3 main categories: (i) Inbred strains of mice that develop AD-like phenotypes. The most well-known of these are the flaky tail mouse and the NC/Nga mouse (47, 48). The Flaky tail (ft) recessive mouse mutation arose spontaneously on the background of a recessive matted (ma) trait (49). The ft mutation has been identified as a 1-bp deletion in the Flg gene resulting in a premature stop codon (50), analogous to the human FLG mutations. More recently the ma trait has been separated from the flaky tail mouse and identified as a nonsense mutation in the novel gene Matt encoding the protein mattrin which is also postulated to have a role in skin barrier biology (51). (ii) Genetically engineered models, in which genes can be silenced or be overexpressed, for example the claudin-1 (52) and Flg knockout mice (53). (iii) Models that can be induced by exogenous agents with for example the allergens ovalbumin and house dust mite (as recently reviewed (54)).
ATOPIC DERMATITIS IS AT THE FOREFRONT OF CURRENT GENETIC TECHNOLOGY AND ITS APPLICATION
The prevalence of AD and the accessibility of disease-relevant tissues – both skin and blood – has allowed AD research to be at the forefront of applying new technologies. This has been powerfully facilitated by the active collaboration of large consortia across Europe and throughout the world. The advance of genetic and genomic analysis techniques has occurred at a rapid pace over recent decades. Large-scale and more focused molecular analysis techniques provide complimentary information; an overview of these approaches is given in Fig. 2 and each is described below.
Fig. 2.
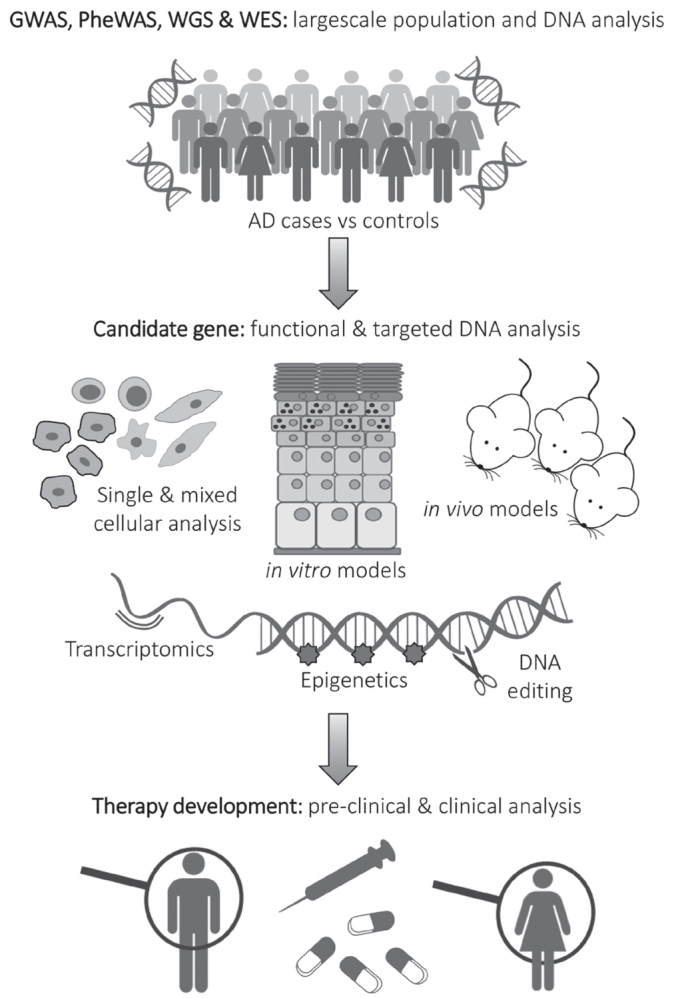
Complimentary strategies for genetic analysis leading to therapy development. GWAS, genome-wide association study; PheWAS, phenome-wide association study; WGS, whole genome sequencing; WES, whole exome sequencing; AD: atopic dermatitis.
Genome-wide association studies
GWAS is a technique in which very large numbers of single nucleotide polymorphisms across the genome are compared between large numbers of cases and controls, to identify differences that are associated with disease status. GWAS have been conducted in several different populations worldwide, and a recent meta-analysis has synthesized these studies (55). Over 30 loci (regions of DNA) have been identified as showing association with AD risk. Some loci include well established genetic effects, such as the epidermal differentiation complex on chromosome 1q21.3 (which includes FLG) and the cytokine cluster on chromosome 5. Many of the other regions are between genes, meaning that their functions require detailed follow-up work to ascertain a functional mechanism. One example is the region on chromosome 11q13.5 which interacts with a gene, EMSY, >30 kilo-bases away; EMSY has recently been shown to have an effect on skin barrier formation and function of relevance to AD (56). Another gene, LRRC32, >60 kilobases away from the same locus on chromosome 11q13.5, may also play a role in AD pathogenesis (57), demonstrating the pleiotropic effects that arise from genetic variation.
Further, larger, meta-GWAS studies are on-going, because larger sample sizes allow the detection of additional risk loci, although their effect sizes are likely to be smaller.
Phenome-wide association studies
Phenome-wide association (PheWAS) is a technique in which large numbers of phenotypic traits are tested for association with single genetic variants. For example, a loss of function variant in FLG shows strong association with atopic phenotypes including AD, asthma, allergic rhinitis and food allergy in a PheWAS study, as expected (58). Unexpected or previously unknown associations with genotypes may be revealed using PheWAS and the technique may also be applied to drug repositioning (59).
Whole exome sequencing and whole genome sequencing
Whole exome sequencing (WES) is a technique that studies the genetic sequence of the DNA in exons that code for proteins, and also exonic regions in non-coding RNAs. WES focuses on exons because they are most likely to have a direct functional effect; however, each variant requires careful assessment to define which may lead to loss-of-function or other functional effect.
WES in 22 Ethiopian people with AD and ichthyosis vulgaris has revealed rare variants in FLG and several other genes within the epidermal differentiation complex, as well as nonsense and missense mutations in previously unreported candidate genes including GTF2H5, ADAM33, EVPL and NLRP1 (60). Some of these findings indicate population-specific variation rather than disease-associated variants. There was no evidence of recurrently-mutated causal genes in this population and AD appears to show considerable heterogeneity in genetic susceptibility (60).
Whole genome sequencing (WGS) sequences inter-genic regions as well as exons, because many of the regulatory mechanisms are situated in intergenic DNA. WGS generates more data and is potentially more powerful than WES, but the interpretation of non-coding variants on a large scale remains very challenging as their functional effects are not well defined. The cost of WGS is also a limiting factor to sample size and to date no large WGS have been reported in AD.
Epigenetic studies
‘Epigenetic’ refers to heritable changes in gene expression that occur without alteration to the DNA sequence. In the context of AD, there are multiple environmental and pathophysiological effects which could impact on skin cells via epigenetic mechanisms, ranging from maternal factors in utero, to early life exposures, irritant and allergic effects. Two important epigenetic mechanisms are histone modifications and DNA methylation. These regulate chromatin structure and DNA accessibility to transcription factors and polymerases (61). Specific histone modifications can be used to predict and delineate regulatory features such as promotors and enhancers in the genome. Epigenetic mechanisms are central to the precise control of skin development and homeostasis (reviewed (62)). A number of studies have linked abnormal epigenetic control of the immune system and skin barrier to AD pathogenesis (63). Key differences in DNA methylation are observed between lesional and non-lesional AD epidermis and these correlate with changes in the expression of skin barrier and innate immune genes (64). Non-coding RNA including micro RNAs (miRNAs) confer an additional level of epigenetic control by regulating mRNA translation or degradation. Differential expression of number of miRNAs has been reported in lesional AD skin (63). Considerable further work is needed to fully understand epigenetic control in AD.
Three-dimensional DNA analyses
DNA may be represented diagrammatically as if it were a straight linear molecule, but in vivo it is extensively folded and wrapped around protein structures in three-dimensional space. Due to this folding, genomic regions that are far from each other in the linear DNA are brought in close proximity in the 3D genome (65). This complex and dynamic process facilitates long range control of gene expression by bringing distant promotor and enhancer elements together (66). Recent technological advances including chromosomal conformation capture (5C) and Hi-C or Hi-Cap, have allowed these interacting regions to be delineated. The techniques crosslink DNA with formaldehyde prior to digestion and sequencing so that interacting regions are sequenced together (65, 67). HiCap uses probes to capture promoters across the genome and regions important in gene regulation such as enhancers. Then, selected promoter–enhancer interactions can be sequenced. This analysis is performed in different cell lines and at different timepoints to reveal the dynamic process and identify candidate genes (68). Importantly, since the 3D interactions are cell-type as well as cell-state-specific, Hi-C analysis has been applied to differentiating keratinocytes, to characterise spacial control of promotor-enhancer interactions likely to be of relevance to AD (56, 67).
Transcriptome analysis
Transcriptomic analysis describes the study of RNA molecules that are present in a cell or tissue, having recently been transcribed from DNA. These molecules include protein-coding messenger RNA (mRNA), ribosomal RNA, transfer RNA, long non-coding RNA (lncRNA), micro RNA (miRNA) and others; their half-lives range from seconds to minutes. The transcriptome is a highly dynamic system and it is cell-type and cell state-dependent; differentiated cells show different gene expression compared to undifferentiated cells. Transcriptome analysis performed on skin itself is most relevant for dermatological conditions, but transcriptomics of serum or blood may also provide valuable insight into skin-related inflammatory conditions, including AD. Transcriptomic analyses are very sensitive; skin biopsy samples from so-called ‘non-lesional’ (clinically uninflamed) skin from an AD patient show profound abnormalities in the transcriptome, including barrier impairment, dysregulation of lipid metabolism and an activated stress response (69). The AD lesional skin transcriptome shows a disease signature (70) that improves after treatment (71).
Single cell analysis
Most of the molecular analyses on skin to date have been carried out using whole skin biopsies, or epidermal samples. However single cell analysis is now feasible, for DNA and RNA sequencing, as well as protein analysis (72). These techniques offer the prospect to study individual cells, define new cell types and gain insight into the functional and structural heterogeneity of skin as a complex organ. The Human Cell Atlas is an international collaboration to make single cell analytical data available to researchers (73) and the skin component of this atlas is eagerly awaited. Several research laboratories have already released published data and tools to allow the interrogation of skin transcriptome analysis, for example murine data from the Kasper lab (74).
CRISPR-cas9 gene editing
CRISPR (clustered regularly interspaced palindromic repeat) sequences are found in bacterial DNA and form part of their immune response to phage infection. Cas9 (CRISPR-associated protein 9) is an enzyme that cleaves DNA selectively at sequences containing the CRISPR motif. In 2012 it was reported that this mechanism can be exploited for genetic engineering; guide-RNAs are used to direct the cas9 enzyme to cleave DNA in precisely-targeted editing. Application of CRISPR-cas9 allows the effects of genetic variation to be tested directly and the technique has revolutionised molecular biology. This cost-effective and relatively easy-to-use technology has allowed researchers to precisely and efficiently target, edit, modify and mark genomic loci in a wide range of cells and organisms (75). Within dermatology, CRISPR-cas9 editing has been used to correct the genetic defects in several forms of epidermolysis bullosa and of relevance to AD, the technique can be used to investigate candidate genes in vitro (see below).
Functional analyses in vivo
Clinical observation followed-up with genetic analysis has increased our understanding of severe phenotypes which include features of AD. Following on from Netherton syndrome, these ‘human knock-out’ models include CARD11 mutations (causing systemic atopic inflammation), DSG1 and DSP mutations (causing severe dermatitis, multiple allergies and metabolic wasting) and various immunodeficiency syndromes with AD-like skin inflammation (such as Wiskott-Aldrich, caused by mutations in WAS) (76).
Functional analysis of the skin of AD patients in vivo also offers opportunities to gain understanding of the pathophysiology. Transepidermal water loss (TEWL) (77) measures the ‘inside-to-outside’ barrier function and in vivo it is proportional to skin inflammation; capacitance or conductance of the stratum corneum give a quantitative measure of water content; and tape-stripping can be used as a relatively non-invasive methods for sampling the skin transcriptome, proteome and lipids of relevance to AD (78).
Organotypic models of atopic dermatitis
Three-dimensional organotypic models of human skin bridge the gap between cultured cells in monolayer and animal models. Multi-layered organotypic models recapitulate many features of human epidermis including: morphology, spatiotemporal expression of terminal differentiation/proliferative markers and an appropriate complement of epidermal lipids (79, 80). Several organotypic models of AD have been described which generally use one of two basic approaches: the first involves the treatment of organotypic models derived from normal healthy cells with AD-relevant cytokines and the second models FLG deficient AD through gene silencing or the use of FLG-mutant keratinocytes (81). Th2 cytokines (IL-4 and IL-13) stimulate a spongiotic epidermal morphology, similar to that observed in AD (82). Organotypic models deficient in filaggrin expression broadly recapitulate many of the structural, molecular and functional defects observed in AD skin. These include a lack of keratohyalin granules, increased paracellular permeability (83, 84) and protein expression signatures consistent with AD skin (85, 86). Filaggrin deficient organotypic skin, therefore, mirrors many changes observed in the AD skin and thus represents a useful model for the study of AD disease mechanisms and therapeutic options.
Organotypic models allow the investigation of tissue-specific genetic effects and the opportunity for testing other AD candidate genes, by knockdown, over-expression, or CRISPR-cas9 editing of genes of interest.
Functional analyses in vitro
Organotypic skin models grown at the air liquid interface develop a competent bidirectional epidermal barrier with similar biophysical properties to human skin. They offer the advantage over monolayer cell cultures, that they are tractable for physiologically relevant functional analysis (87). The outside-in barrier can be quantified in organotypic models using topically applied hydrophilic dye such as Lucifer yellow. This is naturally excluded from the epidermis by the lipid-rich stratum corneum but can permeate into the deeper epidermal and dermal layers if the skin barrier is immature or impaired (83). Analogous to the in vivo situation described above, the inside-outside barrier of organotypic cultures can also be determined by measuring the rate of TEWL (56). These techniques have been used successfully to investigate both the effect of previously uncharacterized genes and the FLG deficiency on skin barrier function (56, 83, 85).
OUTSTANDING QUESTIONS AND FUTURE WORK
The rapid progress made in recent years still leaves a large amount of work to fully capitalize on novel understanding for the benefit of patients.
More detailed genetic studies
The majority of heritability in AD remains unexplained. Improvements in technology have allowed more and more detailed interrogation of the coding and non-coding regions of the genome which are likely to hold important mechanistic information. Outstanding questions involve tissue-specific effects in skin; the relative accessibility of this tissue allows dermatological studies to take advantage of direct sampling for epigenetic studies and more detailed transcriptome analyses. Copy number variation within FLG has a dose-dependent effect on AD (38) and more detailed analyses are required to assess CNV in other risk loci. On a genome-wide level, even larger numbers of cases and controls will be required to achieve the statistical power to detect gene-gene interactions and gene-environment interactions of relevance to AD. These studies remain challenging in their financial cost and computational requirements.
More inclusive genetic research
As described above, the majority of genetic research to date in AD has been conducted in people of white European ancestry. However, the clinical phenotype of AD is different in different ethnicities and studies of genetic risk in African (35) and Asian (88) populations have provided valuable complimentary insight (89). There has been a call in the field to prioritise diversity in human genomics research because this will increase the accuracy, utility and acceptability of using genomic information for clinical care (90). The International Symposium on Atopic Dermatitis (ISAD) has recently published a position statement calling for more research on AD in Africa (91).
Integration of -omics for personalised medicine
Genetic studies have given important information for understanding AD mechanisms, particularly the initial or ‘root cause’ of atopic skin inflammation. However, the combination and integration of information provided by the full complement of techniques described above will be required to increase our understanding of AD pathophysiology sufficiently to allow translation for clinical impact. Furthermore, given the complexity and diversity of this trait, further developments in machine learning and more powerful in silico analyses (76) are likely to be required to gain full benefit from the wealth of molecular data.
Application of genetic discoveries to drug development
The quest for understanding genetic mechanisms in AD is not merely an academic exercise. Genetic studies can provide a causative link between a sequence variant and a phenotype and drugs developed to target a pathway informed by human genetic studies have above-average chances of clinical success (92). Filaggrin deficiency remains a challenging therapeutic target, even though the genetic discovery was made more than a decade ago, but genetic studies continue to identify causal pathways for AD in increasingly precise and personalised detail. The era of ‘personalised medicine’ is expected to bring a new relationship between genomics and drug development, testing the physiological and molecular bases for disease, but success in this endeavour would ultimately transform drug development and clinical use (93).
CONCLUSION: THE FUTURE LOOKS BRIGHT
In 1952, Rosalind Franklin was the first to crystallise DNA fibres to study their structure using X-ray diffraction; in 1953 James Watson and Francis Crick reported the double helix structure of DNA; in 1990 the Human Genome Project began and in 2003 the Human Genome Project was completed, providing a sequence of the entire human genome – approximately 3 billion base pairs in length.
Since this time, we have progressed a long way in understanding more of the detail of how DNA sequence variation contributes to human health and disease. There has been a particularly rapid explosion of knowledge in the last 20 years, brought about by increased technical capacity for sequencing DNA and RNA. Whilst it is unlikely that another single gene exists with the impact of FLG upon AD risk, the future appears bright for AD patients: New techniques will refine understanding of genetic risk, with a multi-ethnic perspective, providing powerful insight to drive the development of new pharmacological interventions. These will increasingly be targeted to specific disease mechanisms for each individual patient with AD. The next 100 years is likely to see a step-change in the management of this challenging disease.
ACKNOWLEDGEMENT
MSE and SJB are funded by a Wellcome Trust Senior Research Fellowship (ref 106865/Z/15/Z) awarded to SJB. MB is funded by the Swedish Skin Foundation (Hudfonden).
Footnotes
The authors have no conflicts of interest to declare.
REFERENCES
- 1.Weidinger S, Novak N. Atopic dermatitis. Lancet 2016; 387: 1109–1122. [DOI] [PubMed] [Google Scholar]
- 2.Deckers IA, McLean S, Linssen S, Mommers M, van Schayck CP, Sheikh A. Investigating international time trends in the incidence and prevalence of atopic eczema 1990–2010: a systematic review of epidemiological studies. PLoS One 2012; 7: e39803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Drucker AM, Wang AR, Li WQ, Sevetson E, Block JK, Qureshi AA. The Burden of Atopic Dermatitis: Summary of a Report for the National Eczema Association. J Invest Dermatol 2017; 137: 26–30. [DOI] [PubMed] [Google Scholar]
- 4.Mier P. Earliest description of the atopic syndrome? Br J Dermatol 1975; 92: 359–359. [Google Scholar]
- 5.NIH . Genetics Home Reference: What is heritability? 2019. [Google Scholar]
- 6.Apfelbacher CJ, Diepgen TL, Schmitt J. Determinants of eczema: population-based cross-sectional study in Germany. Allergy 2011; 66: 206–213. [DOI] [PubMed] [Google Scholar]
- 7.Wadonda-Kabondo N, Sterne JA, Golding J, Kennedy CT, Archer CB, Dunnill MG. Association of parental eczema, hayfever, and asthma with atopic dermatitis in infancy: birth cohort study. Arch Dis Childhood 2004; 89: 917–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schultz Larsen F. Atopic dermatitis: a genetic-epidemiologic study in a population-based twin sample. J Am Acad Dermatol 1993; 28: 719–723. [DOI] [PubMed] [Google Scholar]
- 9.Larsen FS, Holm NV, Henningsen K. Atopic dermatitis. A genetic-epidemiologic study in a population-based twin sample. J Am Acad Dermatol 1986; 15: 487–494. [PubMed] [Google Scholar]
- 10.Thomsen SF, Ulrik CS, Kyvik KO, Hjelmborg JB, Skadhauge LR, Steffensen I, et al. Importance of genetic factors in the etiology of atopic dermatitis: a twin study. Allergy Asthma Proc 2007; 28: 535–539. [DOI] [PubMed] [Google Scholar]
- 11.Thomsen SF, Kyvik KO, Backer V. Etiological relationships in atopy: a review of twin studies. Twin Res Hum Genet 2008; 11: 112–120. [DOI] [PubMed] [Google Scholar]
- 12.Timpson NJ, Greenwood CMT, Soranzo N, Lawson DJ, Richards JB. Genetic architecture: the shape of the genetic contribution to human traits and disease. Nat Rev Genet 2018; 19: 110–124. [DOI] [PubMed] [Google Scholar]
- 13.Lonnberg AS, Skov L, Skytthe A, Kyvik KO, Pedersen OB, Thomsen SF. Heritability of psoriasis in a large twin sample. Br J Dermatol 2013; 169: 412–416. [DOI] [PubMed] [Google Scholar]
- 14.Nibali L, Bayliss-Chapman J, Almofareh SA, Zhou Y, Divaris K, Vieira AR. What Is the Heritability of Periodontitis? A Systematic Review. J Dent Res 2019; 98: 632–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gordon H, Trier Moller F, Andersen V, Harbord M. Heritability in inflammatory bowel disease: from the first twin study to genome-wide association studies. Inflamm Bowel Dis 2015; 21: 1428–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee YA, Wahn U, Kehrt R, Tarani L, Businco L, Gustafsson D, et al. A major susceptibility locus for atopic dermatitis maps to chromosome 3q21. Nat Genet 2000; 26: 470–473. [DOI] [PubMed] [Google Scholar]
- 17.Stemmler S, Hoffjan S. Trying to understand the genetics of atopic dermatitis. Mol Cell Probes 2016; 30: 374–385. [DOI] [PubMed] [Google Scholar]
- 18.Palmer CN, Irvine AD, Terron-Kwiatkowski A, Zhao Y, Liao H, Lee SP, et al. Common loss-of-function variants of the epidermal barrier protein filaggrin are a major predisposing factor for atopic dermatitis. Nat Genet 2006; 38: 441–446. [DOI] [PubMed] [Google Scholar]
- 19.Irvine AD, McLean WH, Leung DY. Filaggrin mutations associated with skin and allergic diseases. N Engl J Med 2011; 365: 1315–1327. [DOI] [PubMed] [Google Scholar]
- 20.Kabashima K. New concept of the pathogenesis of atopic dermatitis: interplay among the barrier, allergy, and pruritus as a trinity. J Dermatol Sci 2013; 70: 3–11. [DOI] [PubMed] [Google Scholar]
- 21.Sandilands A, Terron-Kwiatkowski A, Hull P, O’Regan G, Clayton T, Watson R, et al. Comprehensive analysis of the gene encoding filaggrin uncovers prevalent and rare mutations in ichthyosis vulgaris and atopic eczema. Nat Genet 2007; 39: 650–654. [DOI] [PubMed] [Google Scholar]
- 22.Brown SJ. Molecular mechanisms in atopic eczema: insights gained from genetic studies. J Pathol 2017; 241: 140–145. [DOI] [PubMed] [Google Scholar]
- 23.Rodriguez E, Baurecht H, Herberich E, Wagenpfeil S, Brown SJ, Cordell HJ, et al. Meta-analysis of filaggrin polymorphisms in eczema and asthma: robust risk factors in atopic disease. J Allergy Clin Immunol 2009; 123: 1361–1370 e1367. [DOI] [PubMed] [Google Scholar]
- 24.Weidinger S, Beck LA, Bieber T, Kabashima K, Irvine AD. Atopic dermatitis. Nat Rev Dis Primers 2018; 4: 1. [DOI] [PubMed] [Google Scholar]
- 25.Chen H, Common JE, Haines RL, Balakrishnan A, Brown SJ, Goh CS, et al. Wide spectrum of filaggrin-null mutations in atopic dermatitis highlights differences between Singaporean Chinese and European populations. Br J Dermatol 2011; 165: 106–114. [DOI] [PubMed] [Google Scholar]
- 26.O’Regan GM, Irvine AD. The role of filaggrin loss-of-function mutations in atopic dermatitis. Curr Opin Allergy Clin Immunol 2008; 8: 406–410. [DOI] [PubMed] [Google Scholar]
- 27.Pigors M, Common JEA, Wong X, Malik S, Scott CA, Tabarra N, et al. Exome sequencing and rare variant analysis reveals multiple filaggrin mutations in Bangladeshi families with atopic eczema and additional risk genes. J Invest Dermatol 2018; 138: 2674–2677. [DOI] [PubMed] [Google Scholar]
- 28.Enomoto H, Hirata K, Otsuka K, Kawai T, Takahashi T, Hirota T, et al. Filaggrin null mutations are associated with atopic dermatitis and elevated levels of IgE in the Japanese population: a family and case-control study. J Hum Genet 2008; 53: 615–621. [DOI] [PubMed] [Google Scholar]
- 29.Hamada T, Sandilands A, Fukuda S, Sakaguchi S, Ohyama B, Yasumoto S, et al. De novo occurrence of the filaggrin mutation p.R501X with prevalent mutation c.3321delA in a Japanese family with ichthyosis vulgaris complicated by atopic dermatitis. J Invest Dermatol 2008; 128: 1323–1325. [DOI] [PubMed] [Google Scholar]
- 30.Nomura T, Sandilands A, Akiyama M, Liao H, Evans AT, Sakai K, et al. Unique mutations in the filaggrin gene in Japanese patients with ichthyosis vulgaris and atopic dermatitis. J Allergy Clin Immunol 2007; 119: 434–440. [DOI] [PubMed] [Google Scholar]
- 31.Thawer-Esmail F, Jakasa I, Todd G, Wen Y, Brown SJ, Kroboth K, et al. South African amaXhosa patients with atopic dermatitis have decreased levels of filaggrin breakdown products but no loss-of-function mutations in filaggrin. J Allergy Clin Immunol 2014; 133: 280–282 e 281–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thyssen JP, Godoy-Gijon E, Elias PM. Ichthyosis vulgaris: the filaggrin mutation disease. Br J Dermatol 2013; 168: 1155–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Winge MC, Bilcha KD, Lieden A, Shibeshi D, Sandilands A, Wahlgren CF, et al. Novel filaggrin mutation but no other loss-of-function variants found in Ethiopian patients with atopic dermatitis. Br J Dermatol 2011; 165: 1074–1080. [DOI] [PubMed] [Google Scholar]
- 34.Margolis DJ, Gupta J, Apter AJ, Ganguly T, Hoffstad O, Papadopoulos M, et al. Filaggrin-2 variation is associated with more persistent atopic dermatitis in African American subjects. J Allergy Clin Immunol 2014; 133: 784–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Almoguera B, Vazquez L, Mentch F, March ME, Connolly JJ, Peissig PL, et al. Novel locus for atopic dermatitis in African Americans and replication in European Americans. J Allergy Clin Immunol 2019; 143: 1229–1231. [DOI] [PubMed] [Google Scholar]
- 36.Irvine AD, McLean WH. Breaking the (un)sound barrier: filaggrin is a major gene for atopic dermatitis. J Invest Dermatol 2006; 126: 1200–1202. [DOI] [PubMed] [Google Scholar]
- 37.Thyssen JP, Thuesen B, Huth C, Standl M, Carson CG, Heinrich J, et al. Skin barrier abnormality caused by filaggrin (FLG) mutations is associated with increased serum 25-hydroxyvitamin D concentrations. J Allergy Clin Immunol 2012; 130: 1204–1207 e1202. [DOI] [PubMed] [Google Scholar]
- 38.Brown SJ, Kroboth K, Sandilands A, Campbell LE, Pohler E, Kezic S, et al. Intragenic copy number variation within filaggrin contributes to the risk of atopic dermatitis with a dose-dependent effect. J Invest Dermatol 2012; 132: 98–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Howell MD, Kim BE, Gao P, Grant AV, Boguniewicz M, Debenedetto A, et al. Cytokine modulation of atopic dermatitis filaggrin skin expression. J Allergy Clin Immunol 2007; 120: 150–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim BE, Howell MD, Guttman-Yassky E, Gilleaudeau PM, Cardinale IR, Boguniewicz M, et al. TNF-alpha downregulates filaggrin and loricrin through c-Jun N-terminal kinase: role for TNF-alpha antagonists to improve skin barrier. J Invest Dermatol 2011; 131: 1272–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Al-Shobaili HA, Ahmed AA, Alnomair N, Alobead ZA, Rasheed Z. Molecular Genetic of Atopic dermatitis: An Update. Int J Health Sci (Qassim) 2016; 10: 96–120. [PMC free article] [PubMed] [Google Scholar]
- 42.Walley AJ, Chavanas S, Moffatt MF, Esnouf RM, Ubhi B, Lawrence R, et al. Gene polymorphism in Netherton and common atopic disease. Nat Genet 2001; 29: 175–178. [DOI] [PubMed] [Google Scholar]
- 43.Hubiche T, Ged C, Benard A, Leaute-Labreze C, McElreavey K, de Verneuil H, et al. Analysis of SPINK 5, KLK 7 and FLG genotypes in a French atopic dermatitis cohort. Acta Derm Venereol 2007; 87: 499–505. [DOI] [PubMed] [Google Scholar]
- 44.Weidinger S, Baurecht H, Wagenpfeil S, Henderson J, Novak N, Sandilands A, et al. Analysis of the individual and aggregate genetic contributions of previously identified serine peptidase inhibitor Kazal type 5 (SPINK5), kallikrein-related peptidase 7 (KLK7), and filaggrin (FLG) polymorphisms to eczema risk. J Allergy Clin Immunol 2008; 122: 560–568.e564. [DOI] [PubMed] [Google Scholar]
- 45.Asad S, Tapia-Paez I, Montano Montes A, Wahlgren CF, Bilcha KD, Nordenskjold M, et al. Evaluation of Single Nucleotide Variants in Ethiopian Patients with Atopic Dermatitis. Acta Derm Venereol 2019; 99: 101–102. [DOI] [PubMed] [Google Scholar]
- 46.Nuttall TJ, Marsella R, Rosenbaum MR, Gonzales AJ, Fadok VA. Update on pathogenesis, diagnosis, and treatment of atopic dermatitis in dogs. J Am Vet Med Assoc 2019; 254: 1291–1300. [DOI] [PubMed] [Google Scholar]
- 47.Matsuda H, Watanabe N, Geba GP, Sperl J, Tsudzuki M, Hiroi J, et al. Development of atopic dermatitis-like skin lesion with IgE hyperproduction in NC/Nga mice. Int Immunol 1997; 9: 461–466. [DOI] [PubMed] [Google Scholar]
- 48.Vestergaard C, Yoneyama H, Matsushima K. The NC/Nga mouse: a model for atopic dermatitis. Mol Med Today 2000; 6: 209–210. [DOI] [PubMed] [Google Scholar]
- 49.Presland RB, Boggess D, Lewis SP, Hull C, Fleckman P, Sundberg JP. Loss of normal profilaggrin and filaggrin in flaky tail (ft/ft) mice: an animal model for the filaggrin-deficient skin disease ichthyosis vulgaris. J Invest Dermatol 2000; 115: 1072–1081. [DOI] [PubMed] [Google Scholar]
- 50.Fallon PG, Sasaki T, Sandilands A, Campbell LE, Saunders SP, Mangan NE, et al. A homozygous frameshift mutation in the mouse Flg gene facilitates enhanced percutaneous allergen priming. Nat Genet 2009; 41: 602–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Saunders SP, Goh CS, Brown SJ, Palmer CN, Porter RM, Cole C, et al. Tmem79/Matt is the matted mouse gene and is a predisposing gene for atopic dermatitis in human subjects. J Allergy Clin Immunol 2013; 132: 1121–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Atsugi T, Yokouchi M, Hirano T, Hirabayashi A, Nagai T, Ohyama M, et al. Holocrine secretion occurs outside the tight junction barrier in multicellular glands: lessons from claudin1-deficient mice. J Invest Dermatol 2020; 140: 298–308.e5. [DOI] [PubMed] [Google Scholar]
- 53.Kawasaki H, Nagao K, Kubo A, Hata T, Shimizu A, Mizuno H, et al. Altered stratum corneum barrier and enhanced percutaneous immune responses in filaggrin-null mice. J Allergy Clin Immunol 2012; 129: 1538–1546 e1536. [DOI] [PubMed] [Google Scholar]
- 54.Kim D, Kobayashi T, Nagao K. Research Techniques Made Simple: Mouse Models of Atopic Dermatitis. J Invest Dermatol 2019; 139: 984–990 e981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Paternoster L, Standl M, Waage J, Baurecht H, Hotze M, Strachan DP, et al. Multi-ancestry genome-wide association study of 21,000 cases and 95,000 controls identifies new risk loci for atopic dermatitis. Nat Genet 2015; 47: 1449–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Elias MS, Wright SC, Remenyi J, Abbott JC, Bray SE, Cole C, et al. EMSY expression affects multiple components of the skin barrier with relevance to atopic dermatitis. J Allergy Clin Immunol 2019; 144: 470–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Manz J, Rodriguez E, ElSharawy A, Oesau EM, Petersen BS, Baurecht H, et al. Targeted resequencing and functional testing identifies low-frequency missense variants in the gene encoding GARP as significant contributors to atopic dermatitis risk. J Invest Dermatol 2016; 136: 2380–2386. [DOI] [PubMed] [Google Scholar]
- 58.Loset M, Brown SJ, Saunes M, Hveem K. Genetics of atopic dermatitis: from DNA sequence to clinical relevance. Dermatology 2019; 235: 355–364. [DOI] [PubMed] [Google Scholar]
- 59.Rastegar-Mojarad M, Ye Z, Kolesar JM, Hebbring SJ, Lin SM. Opportunities for drug repositioning from phenome-wide association studies. Nat Biotechnol 2015; 33: 342–345. [DOI] [PubMed] [Google Scholar]
- 60.Taylan F, Nilsson D, Asad S, Lieden A, Wahlgren CF, Winge MC, et al. Whole-exome sequencing of Ethiopian patients with ichthyosis vulgaris and atopic dermatitis. J Allergy Clin Immunol 2015; 136: 507–509 e519. [DOI] [PubMed] [Google Scholar]
- 61.Bird A. Perceptions of epigenetics. Nature 2007; 447: 396–398. [DOI] [PubMed] [Google Scholar]
- 62.Kang S, Chovatiya G, Tumbar T. Epigenetic control in skin development, homeostasis and injury repair. Exp Dermatol 2019; 28: 453–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liang Y, Chang C, Lu Q. The genetics and epigenetics of atopic dermatitis-filaggrin and other polymorphisms. Clin Rev Allergy Immunol 2016; 51: 315–328. [DOI] [PubMed] [Google Scholar]
- 64.Rodriguez E, Baurecht H, Wahn AF, Kretschmer A, Hotze M, Zeilinger S, et al. An integrated epigenetic and transcriptomic analysis reveals distinct tissue-specific patterns of DNA methylation associated with atopic dermatitis. J Invest Dermatol 2014; 134: 1873–1883. [DOI] [PubMed] [Google Scholar]
- 65.Zheng H, Xie W. The role of 3D genome organization in development and cell differentiation. Nat Rev Mol Cell Bio 2019; 20: 535–550. [DOI] [PubMed] [Google Scholar]
- 66.Schoenfelder S, Fraser P. Long-range enhancer-promoter contacts in gene expression control. Nat Rev Genet 2019; 20: 437–455. [DOI] [PubMed] [Google Scholar]
- 67.Rubin AJ, Barajas BC, Furlan-Magaril M, Lopez-Pajares V, Mumbach MR, Howard I, et al. Lineage-specific dynamic and pre-established enhancer-promoter contacts cooperate in terminal differentiation. Nat Genet 2017; 49: 1522–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Anil A, Spalinskas R, Akerborg O, Sahlen P. HiCapTools: a software suite for probe design and proximity detection for targeted chromosome conformation capture applications. Bioinformatics 2018; 34: 675–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cole C, Kroboth K, Schurch NJ, Sandilands A, Sherstnev A, O’Regan GM, et al. Filaggrin-stratified transcriptomic analysis of pediatric skin identifies mechanistic pathways in patients with atopic dermatitis. J Allergy Clin Immunol 2014; 134: 82–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Suarez-Farinas M, Ungar B, Correa da Rosa J, Ewald DA, Rozenblit M, Gonzalez J, et al. RNA sequencing atopic dermatitis transcriptome profiling provides insights into novel disease mechanisms with potential therapeutic implications. J Allergy Clin Immunol 2015; 135: 1218–1227. [DOI] [PubMed] [Google Scholar]
- 71.Beck LA, Thaci D, Hamilton JD, Graham NM, Bieber T, Rocklin R, et al. Dupilumab treatment in adults with moderate-tosevere atopic dermatitis. N Engl J Med 2014; 371: 130–139. [DOI] [PubMed] [Google Scholar]
- 72.Heath JR, Ribas A, Mischel PS. Single-cell analysis tools for drug discovery and development. Nat Rev Drug Discov 2016; 15: 204–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.https://www.humancellatlas.org/. Human Skin Atlas 2019.
- 74.Joost S. The molecular anatomy of mouse skin during hair growth and rest. bioRxiv 2019. [DOI] [PubMed] [Google Scholar]
- 75.Doudna JA, Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 2014; 346: 1258096. [DOI] [PubMed] [Google Scholar]
- 76.Eyerich K, Brown SJ, Perez White BE, Tanaka RJ, Bissonette R, Dhar S, et al. Human and computational models of atopic dermatitis: A review and perspectives by an expert panel of the International Eczema Council. J Allergy Clin Immunol 2019; 143: 36–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Alexander H, Brown S, Danby S, Flohr C. Research Techniques Made Simple: Transepidermal Water Loss Measurement as a Research Tool. J Invest Dermatol 2018; 138: 2295–2300 e2291. [DOI] [PubMed] [Google Scholar]
- 78.Eui Kim B, Goleva E, Kim PS, Norquest K, Bronchick C, Taylor P, et al. Side-by-side comparison of skin biopsies and skin tape stripping highlights abnormal stratum corneum in atopic dermatitis. J Invest Dermatol 2019; 139: 2387–2389.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mertsching H, Weimer M, Kersen S, Brunner H. Human skin equivalent as an alternative to animal testing. GMS Krankenhhyg Interdiszip 2008; 3: Doc11. [PMC free article] [PubMed] [Google Scholar]
- 80.Thakoersing VS, van Smeden J, Mulder AA, Vreeken RJ, El Ghalbzouri A, Bouwstra JA. Increased presence of monounsaturated fatty acids in the stratum corneum of human skin equivalents. J Invest Dermatol 2013; 133: 59–67. [DOI] [PubMed] [Google Scholar]
- 81.Niehues H, Schalkwijk J, van Vlijmen-Willems I, Rodijk-Olthuis D, van Rossum MM, Wladykowski E, et al. Epidermal equivalents of filaggrin null keratinocytes do not show impaired skin barrier function. J Allergy Clin Immunol 2017; 139: 1979–1981 e1913. [DOI] [PubMed] [Google Scholar]
- 82.Danso MO, van Drongelen V, Mulder A, van Esch J, Scott H, van Smeden J, et al. TNF-alpha and Th2 cytokines induce atopic dermatitis-like features on epidermal differentiation proteins and stratum corneum lipids in human skin equivalents. J Invest Dermatol 2014; 134: 1941–1950. [DOI] [PubMed] [Google Scholar]
- 83.Mildner M, Jin J, Eckhart L, Kezic S, Gruber F, Barresi C, et al. Knockdown of filaggrin impairs diffusion barrier function and increases UV sensitivity in a human skin model. J Invest Dermatol 2010; 130: 2286–2294. [DOI] [PubMed] [Google Scholar]
- 84.Pendaries V, Malaisse J, Pellerin L, Le Lamer M, Nachat R, Kezic S, et al. Knockdown of filaggrin in a three-dimensional reconstructed human epidermis impairs keratinocyte differentiation. J Invest Dermatol 2014; 134: 2938–2946. [DOI] [PubMed] [Google Scholar]
- 85.Elias MS, Wright SC, Nicholson WV, Morrison KD, Prescott AR, Ten Have S, et al. Proteomic analysis of a filaggrin-deficient skin organoid model shows evidence of increased transcriptional-translational activity, keratinocyte-immune crosstalk and disordered axon guidance. [version 1; peer review: awaiting peer review]. Wellcome Open Research 2019; 4: 134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Elias MS, Long HA, Newman CF, Wilson PA, West A, McGill PJ, et al. Proteomic analysis of filaggrin deficiency identifies molecular signatures characteristic of atopic eczema. J Allergy Clin Immunol 2017; 140: 1299–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhang Z, Michniak-Kohn BB. Tissue engineered human skin equivalents. Pharmaceutics 2012; 4: 26–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Noda S, Suarez-Farinas M, Ungar B, Kim SJ, de Guzman Strong C, Xu H, et al. The Asian atopic dermatitis phenotype combines features of atopic dermatitis and psoriasis with increased TH17 polarization. J Allergy Clin Immunol 2015; 136: 1254–1264. [DOI] [PubMed] [Google Scholar]
- 89.Kaufman BP, Guttman-Yassky E, Alexis AF. Atopic dermatitis in diverse racial and ethnic groups-Variations in epidemiology, genetics, clinical presentation and treatment. Exp Dermatol 2018; 27: 340–357. [DOI] [PubMed] [Google Scholar]
- 90.Hindorff LA, Bonham VL, Brody LC, Ginoza MEC, Hutter CM, Manolio TA, et al. Prioritizing diversity in human genomics research. Nat Rev Genet 2018; 19: 175–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Schmid-Grendelmeier P, Takaoka R, Ahogo KC, Belachew WA, Brown SJ, Correia JC, et al. Position Statement on Atopic Dermatitis in Sub-Saharan Africa: current status and road-map. J Eur Acad Dermatol Venereol 2019; 33: 2019–2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kamb A, Harper S, Stefansson K. Human genetics as a foundation for innovative drug development. Nat Biotechnol 2013; 31: 975–978. [DOI] [PubMed] [Google Scholar]
- 93.Dugger SA, Platt A, Goldstein DB. Drug development in the era of precision medicine. Nat Rev Drug Discov 2018; 17: 183–196. [DOI] [PMC free article] [PubMed] [Google Scholar]