

Abstract
GLP-1 was described as an incretin over 30 years ago. GLP-1 is encoded by the preproglucagon gene (Gcg), which is expressed in the intestine, the pancreas, and the central nervous system. GLP-1 activates GLP-1 receptors (GLP-1r) on the β-cell to induce insulin secretion in a glucose-dependent manner. GLP-1 also inhibits α-cell secretion of glucagon. As few, if any, GLP-1r are expressed on α-cells, indirect regulation, via β- or δ-cell products has been thought to be the primary mechanism by which GLP-1 inhibits glucagon secretion. However, recent work suggests that there is sufficient expression of GLP-1r on α-cells for direct regulation as well. Although the predominant source of circulating GLP-1 is the intestine, the α-cell becomes a source of GLP-1 when the islet is metabolically stressed. Recent work suggests the possibility that this source of GLP-1 is also be important in regulating nutrient-induced insulin secretion in a paracrine fashion. More work is also accumulating regarding the role of glucagon, another Gcg-derived protein produced by the α-cell, in stimulating insulin secretion by acting on GLP-1r. Altogether, these data clearly demonstrate the important role of Gcg-derived peptides in regulating insulin secretion. Because of GLP-1’s important role in glucose homeostasis, it has been implicated in the success of bariatric surgery and has been successfully targeted for the treatment of type 2 diabetes mellitus.
Introduction: History of GLP-1 as an Incretin
After the discovery of insulin and the development of the radioimmunoassay to assess plasma insulin levels, a debate ensued over whether glucose was the only stimulus for insulin secretion. Spurred on by work demonstrating that glucose removal from the blood was more rapid with oral versus intravenously (IV) glucose in dogs (211), McIntyre et al. (153) found that glucose was lower but insulin was higher after isocaloric loads of glucose administered into the jejunum versus IV in man. Months later, a second study had similar findings with oral versus IV glucose (61). These data in conjunction with multiple reports in the 1920s and 1930s that intestinal mucosa had hypoglycemic properties led the authors of both papers to hypothesize that a gut-derived humoral substance contributed to the regulation of insulin secretion (Figure 1). Yet, it was not until the 1970s when the first “incretin” was discovered. When gastric inhibitory polypeptide (GIP; aka glucose-dependent insulinotropic peptide) was infused into humans IV, insulin immunoreactivity increased and glucose tolerance was improved (54). However, while gut extracts containing GIP increased insulin secretion, removal of GIP from the extracts only blunted the insulin response by 30% suggesting the presence of additional incretins (56).
Figure 1.
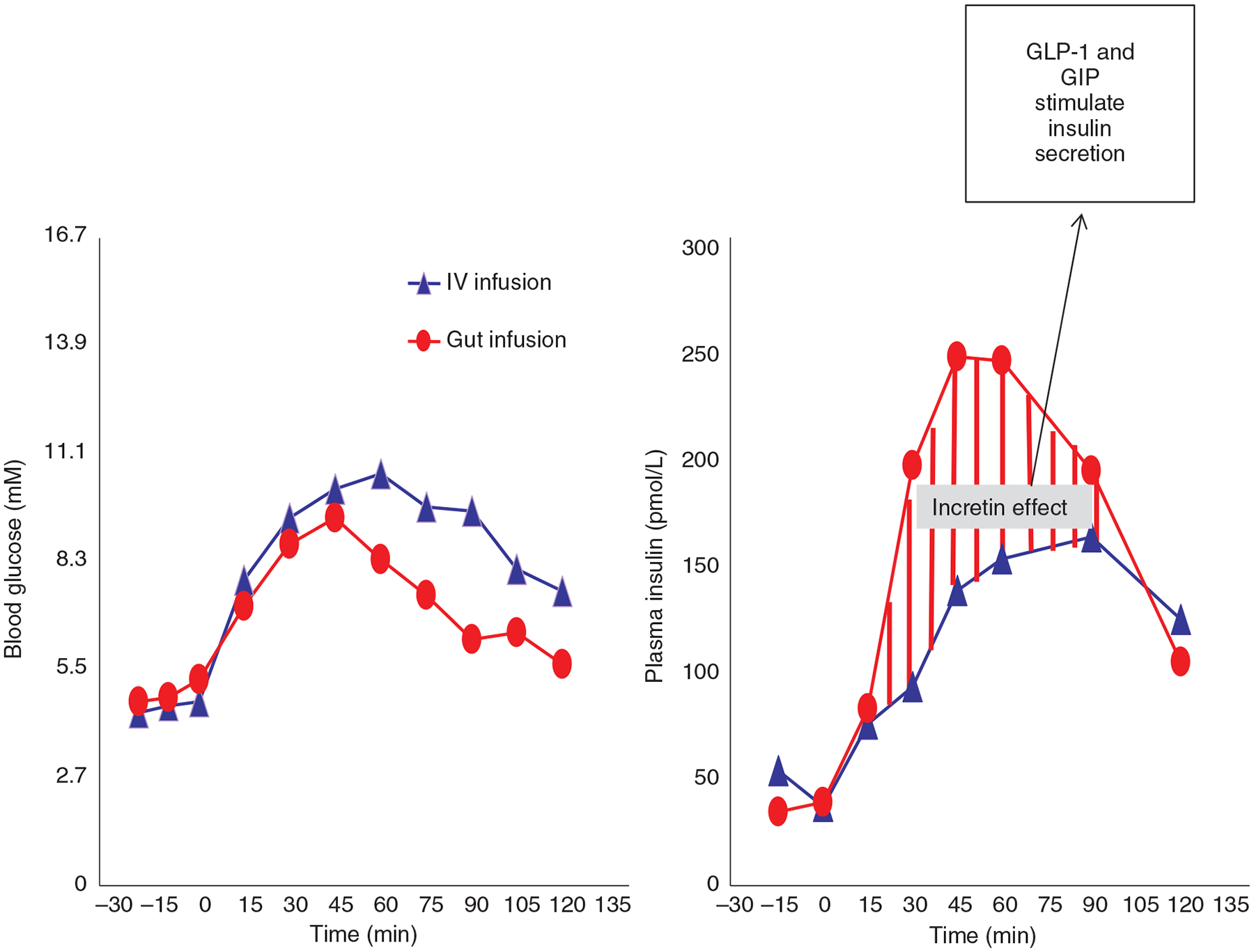
The incretin effect: Glucose levels are lower while insulin levels are higher when the same dose of glucose is administered directly into the gut versus when administered intravenously (IV). This difference in insulin between the gut and venous infusion is the “incretin effect” which occurs in response to GLP-1 and GIP secreted from the distal gut. Adapted, with permission, from McIntyre N, et al. (153).
With the discovery of glucagon and in the pursuit of understanding its form and function, two other peptides were found on the same mRNA (10, 142). These peptides were glucagon-like peptide-1 (GLP-1) and glucagon-like peptide-2 (GLP-2). At this time, the biological function of these peptides was unknown but later work demonstrated that GLP-1, but not GLP-2, stimulated insulin release in rat islets (210) and when infused in humans (127). This latter study found that GIP was less effective at increasing insulin levels and concluded that GLP-1 was a physiological incretin in man. We now know that GLP-1 has a wide array of physiological functions, yet its role in the pancreas is still the most widely studied. The purpose of this article is to discuss the role and mechanisms associated with GLP-1 and glucagon-like peptide-1 receptor (GLP-1r) signaling in the regulation of pancreatic hormone secretion and consequently glucose homeostasis. Consideration is also given to its role in bariatric surgery and the current state of GLP-1 pharmaceuticals.
Regulation of Preproglucagon (Gcg) Expression
We now know that preproglucagon (Gcg) is the gene that codes for GLP-1 and it is expressed in a specific population of intestinal enteroendocrine cells called L-cells. Gcg is also expressed within pancreatic islet α-cells, and in a distinct set of neurons within the nucleus of the solitary tract (NTS) (81, 108). Gcg undergoes tissue-specific posttranslational modification by prohormone convertases (PC). In intestinal L-cells and neurons of the NTS, a specific isoform of PC, PC1/3, is predominantly expressed and yields GLP-1, oxyntomodulin, and GLP-2 as the physiologically relevant products (134, 228, 238). In contrast, the α-cell predominantly expresses another PC isoform, PC2, which yields glucagon (92). Gcg codes for other bioactive proteins including the proglucagon fragments glicentin, glicentin-related pancreatic polypeptide (GRPP), and major proglucagon fragment (MPGF), but the functional significance of these peptides is unclear.
Nutrient status is clearly an important factor in regulating Gcg expression across all three organs for which it is expressed. Re-feeding after fasting (49, 109), dietary fibers (15, 49, 170), long-chain fatty triglycerides (109), and peptones (171, 198), all increase intestinal Gcg expression, and amino acids stimulate α-cell hyperplasia and glucagon secretion (215). Interestingly, in vitro work in cell lines suggest that physiological stimuli such as peptones act via cyclic adenosine monophosphate (cAMP) response element-binding protein (CREB) signaling to regulate transcriptional responses of Gcg (73, 250). In fact, increases in cAMP lead to increases in Gcg expression in both the pancreas and intestine (4, 21, 49, 141). Downstream of cAMP but a pathway that is distinct from protein kinase A (PKA), activation of exchange protein activated by cAMP 2 (EPAC) also increases Gcg transcription in both α- and L-cells (101, 140). In contrast, specific to the L-cell, Gcg expression has been shown to be downstream of the canonical Wnt signaling pathway (a specific signaling transduction pathway), β-catenin and transcription factor 7 like 2 (TCF7L2) (28). Although the mechanisms are unclear, it is also well established that bowel resection or injury causes a large increase in intestinal Gcg expression (125). The function is likely related to the increase in GLP-2, which functions as an intestinal growth factor (see Ref. 13 for review). Distinct regulatory mechanisms for Gcg transcriptional control between tissues is in parallel with the distinct patterns of prohormone processing in the major cell types producing Gcg peptides.
Regulation of Intestinal GLP-1 Secretion
Within the intestine, the density of Gcg-expressing cells increases from the proximal to distal gut with the expression being highest in the colon (7). L-cells within the intestinal epithelium and have apical processes that extend into the gut lumen allowing direct access to ingested nutrients (58), and all three macronutrients (carbohydrate, fat, protein) individually stimulate GLP-1 secretion (120, 249).
Greater numbers of enteroendocrine cells that express Gcg within the lower intestine suggests that postprandial GLP-1 secretion is derived from the ileum and colon. Indeed, nutrient infusions directly into the ileum cause significantly greater increases in portal vein (the major blood vessel that collects intestinal secretions) concentrations of GLP-1 (90). Interestingly, in ex vivo studies from human tissue, the duodenum and ileum but not the colon were found to be glucose responsive (218) which is different than what is reported in mouse colon (178, 191) but are consistent with data where patients that have had colon resection have normal GLP-1 responses to glucose (185). An argument has been made that duodenal secretion of GLP-1 is responsible for early phase GLP-1 secretion (222) but no direct link has been found. However, the anatomic distribution of the L-cells (highest number in distal gut) and the rapid increase in postprandial circulating GLP-1 (35) that occurs before nutrients reach the distal gut (8, 63) is evidence supportive of neural, endocrine and/or paracrine mechanisms being more critical to nutrient-induced increases in circulating GLP-1 (164).
One possibility is that there are feed-forward neural or paracrine signals from the upper gut to the ileum that stimulates the release of GLP-1 (14, 83). Transection of the gut below the duodenum resulted in a delayed GLP-1 response to nutrients compared to when the gut is simply ligated, the latter being a procedure that preserves neural innervation (196). Further support for neural regulation of GLP-1, bilateral subdiaphragmatic vagotomy in conjunction with intestinal transection completely abolishes nutrient-induced GLP-1 secretion (196). In rodents, human enteroendocrine cells, and in perfused porcine ileum, cholinergic agonists, which bind to receptors in both the enteric and parasympathetic nervous system (PNS), stimulate GLP-1 secretion (4, 14, 84, 192). Conversely, norepinephrine and α-adrenergic agonists inhibit (84) and β-adrenergic agonists stimulate (52, 182) GLP-1 secretion suggesting that sympathetic neural innervation of the gut is also important in the regulation of GLP-1 secretion. In humans, atropine, which is a PNS agonist, blunts glucose-induced increases plasma GLP-1 (8). Together these data lend support to the idea that neuronal influences are at play in the regulation of postprandial GLP-1 secretion.
Despite the anatomic limitations, there are several potentially overlapping nutrient-sensing mechanisms that have been found to regulate intestinal GLP-1 secretion. Both passive glucose transport through the sodium-glucose cotransporter 1 (SGLT1) (158, 191) and active glucose transport through glucose transporter 2 (GLUT2) have been found to regulate intestinal GLP-1 secretion as inhibitors for each transporter blocks GLP-1 release (218). Similar to pancreatic β-cells that secrete insulin, potassium adenosine tri-phosphate (KATP) channels and its associated sulfonylurea receptor are also expressed in L-cells (173, 190). Data suggest that increased glucose metabolism within L-cells leads to an increase in ATP and consequently depolarization of KATP channels triggering GLP-1 secretion (179, 190, 191). Interestingly, GLUT2 is linked to KATP channels (145) while SGLT1 is linked to voltage-gated sodium and calcium channels in GLP-1 secretion (218). However, in animal models pharmacological (128) or genetic (197) blockade of SGLT1 had a bigger impact on blunting GLP-1 secretion compared to the same manipulations towards GLUT2. In contrast, ex vivo studies in human tissue suggest GLUT2 may be more important (218). It is unknown whether these differences are simply due to the species or the experimental conditions; in particular for the ex vivo experiments.
Early work demonstrating that PKA activators could stimulate GLP-1 secretion from cell lines suggested that G-coupled protein receptors (GPCR) were involved in the regulation of GLP-1 secretion (12, 16, 192). Gαs and Gαq-coupled receptors responding to luminal bile acids and long-chain fatty acids have demonstrated potent effects on GLP-1 secretion (87). Bile acids signaling through the Gαs protein-coupled bile acid receptor 1, or TGR5, stimulate GLP-1 secretion (87, 180). Bile acids also signal through farnesoid X receptor (FXR), a nuclear transcription factor, but TGR5 is the primary bile acid receptor that can drive GLP-1 secretion (115). Offering clinical promise, in vitro data using primary enteroendocrine cells and an intestinal cell line indicated that pharmacologic TGR5 agonists are more potent GLP-1 secretagogues and are better at enhancing L-cell responses to calcium and glucose-induced GLP-1 secretion compared to naturally occurring bile acids (180).
Long-chain fatty acids and their derivatives specifically activate multiple types of GPCRs including, GPR119, GPR120, and GPR40 (96), and this activation also stimulates GLP-1 secretion. GPR119 is a Gαs-coupled protein receptor and it is highly co-localized to L-cells within the GI tract. GPR119 agonists increase GLP-1 secretion (29) and GPR119 knockout mice have a reduced GLP-1 response to an oral glucose load (132). However, a randomized double-blind placebo controlled trial revealed that GPR119 agonism was not as effective at increasing total and active GLP-1 responses to glucose or a mixed liquid meal nor was it as effective at improving glucose control over a 2-week study period (116). In contrast to GPR119, a Gαs-coupled receptor, GPR120, and GPR40 are Gαq-coupled receptors that signal through protein kinase C. Activation of both GPR40 and GPR120 result in increased GLP-1 secretion (57, 91, 99, 143); however, their role may be more pharmacological than physiological. While members of these GPCR families were under active investigation as drug targets for the treatment of type 2 diabetes mellitus (T2DM), due to lack of efficacy, the number of drugs in the pipeline has been drastically reduced.
Another type of GPCR that is expressed in the GI tract and has been found to be linked to GLP-1 secretion is “sweet taste receptors” (102). These same receptors are expressed in the tongue and are integral in sweet taste perceptions. Their function in the GI tract is less clearly understood. Nonnutritive agonists of sweet taste receptors do increase GLP-1 levels and mice null for one such sweet taste receptor, α-gustducin, have reduced GLP-1 responses to an oral glucose load (102). Some data suggest that pharmacological inhibition of sweet taste receptors blunt GLP-1 responses to nutrients (217), but other studies found no impact of sweet taste receptor agonists on GLP-1 secretion in humans (146, 178) or in in vivo or ex vivo studies in rats (208).
Altogether it seems that many molecular signaling pathways have been linked to GLP-1 secretion (Figure 2), and it is possible that all of these pathways are involved in order to provide an integrated response that also allows for fine tuning of GLP-1 secretion. One example of this is recent work that found a synergistic action of pharmacological TGR5 and GPR40 activation on the electrophysiological properties of L-cells (76). However, while data in mouse models are promising, there are currently no therapies currently in clinical trials focused on GPCR signaling to increase plasma GLP-1. Whether this is because these GPCRs are not important or whether increasing intestinal GLP-1 secretion, alone, is sufficient for obesity and/or T2DM treatment remains to be determined.
Figure 2.
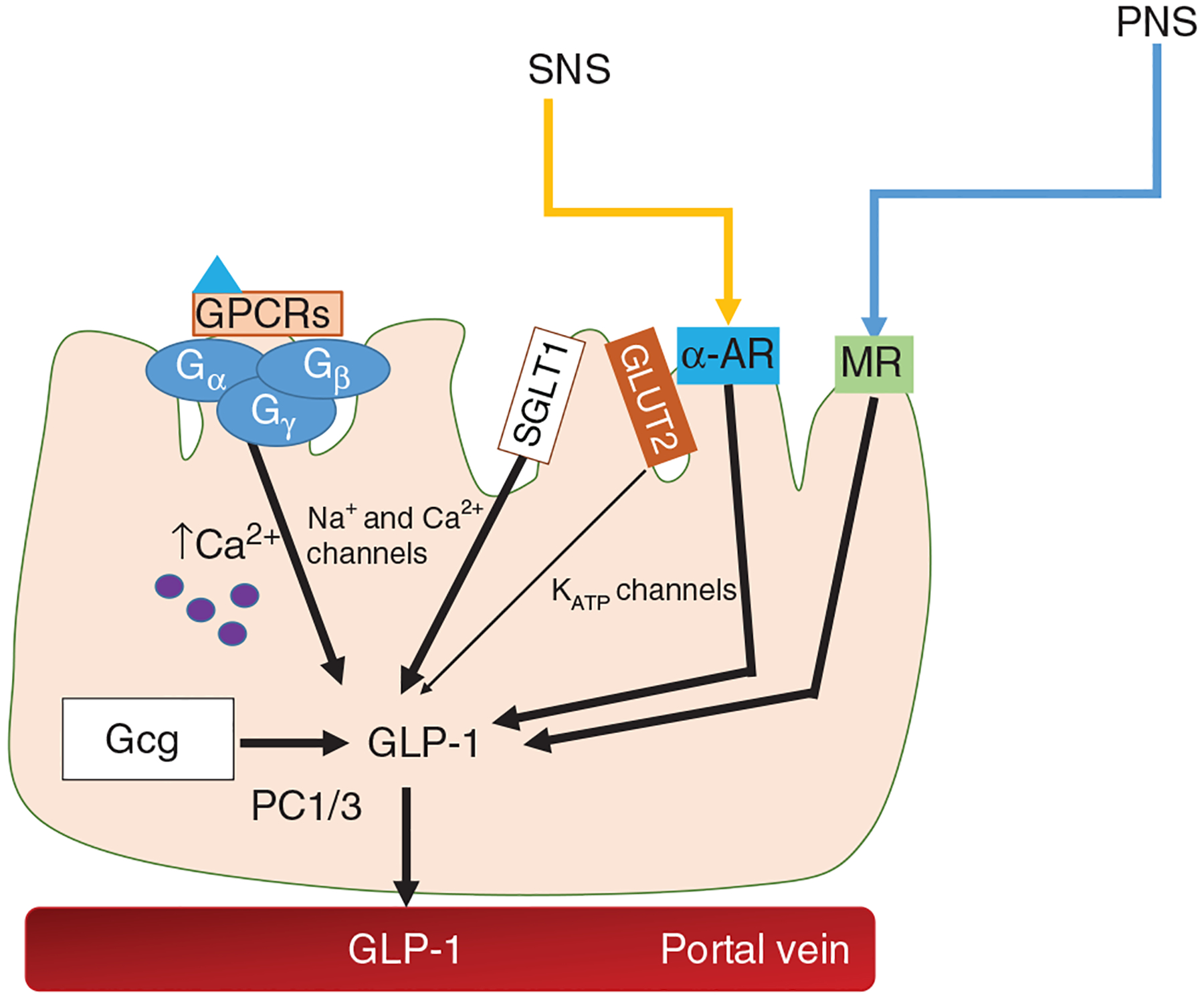
Intestinal GLP-1 secretion: Several factors have been linked to GLP-1 secretion. The parasympathetic nervous system (PNS) stimulates GLP-1 secretion via cholinergic muscarinic receptors (MR). Activation of α-adrenergic (AR) receptors stimulates while activation of β-adrenergic receptors inhibits GLP-1 release. Various GPCRS including ones activated by bile acids and various fatty acids stimulate GLP-1 through PKA signaling and increases in calcium-induced exocytosis. Lastly, direct glucose sensing, predominantly via SGLT1 in humans, activates sodium (Na+), and calcium (Ca2+) voltage-gated channels to lead to the release of GLP-1.
GLP-1 Receptor Distribution and Signaling
Direct regulation of insulin secretion
The GLP-1r is a GPCR that is expressed within the pancreas, lung, adipose tissue, kidney, heart, vascular smooth muscle, and in a number of specific nuclei within the CNS (21, 74). Most of what we know about GLP-1r signaling is derived from studying the β-cell population of GLP-1r. When glucose is transported into the β-cell, its metabolism generates ATP, which provides energy for the closure of KATP channels and consequently an increase in intracellular calcium; the latter being necessary for exocytosis of insulin. Binding of the GLP-1r produces cAMP and consequently activation of PKA and EPAC2 (94), which potentiates glucose-stimulated insulin release (94, 144, 200). With fasting and when intracellular ADP is increased, PKA activation hyperpolarizes the β-cell membrane by increasing KATP channel conductance. When ADP levels are reduced with glucose metabolism, PKA phosphorylates a subunit of the KATP channel leading to channel closure and depolarization (136). This extends to pharmaceutical activation of the GLP-1r in that GLP-1r agonists are weak insulin secretagogues at basal glucose levels. Further, the ability of GLP-1 to suppress feeding, an action dependent upon CNS receptors, is blunted in fasted conditions (159, 243) and dependent upon nutrient-dependent intracellular pathways (88) indicating that CNS GLP-1r signaling is also dependent upon nutrient availability.
Although pharmacological data make it very clear that GLP-1 regulates insulin secretion, genetic data suggest that the system is more complex than a simple endocrine model of insulin regulation. Genetic models demonstrate that although GLP-1 regulates insulin secretion through the β-cell GLP-1r (131), the necessity of these receptors depends on the route of delivery of nutrients. Mice with an inducible knockout of the β-cell GLP-1r have normal oral, but impaired IP glucose tolerance (214). This is interesting as even whole body GLP-1r KO mice have greater impairments in IP versus oral glucose tolerance. Importantly, these responses could also be explained by the redundancy of insulinotropic signals from the gut or nervous system during oral versus IP glucose loads, but in the end still demonstrate that β-cell GLP-1r, in and of themselves, are not necessary for oral glucose tolerance.
GLP-1r regulation of insulin secretion independent of the β-cell GLP-1r
If GLP-1 has a role for regulating insulin secretion independent of its β-cell receptor, what would the population of receptors be? Given the rapid postprandial increase in insulin, one possibility is the nervous system GLP-1r. Indeed, direct administration of GLP-1 into the 3rd cerebral ventricle (ICV) increases insulin secretion (122, 209); an effect that is maintained in mice fed a high-fat diet (17). In both mice and rats, administration of ICV exendin-4(9–39) (Ex9), a potent GLP-1r antagonist, during oral glucose impairs glucose tolerance and reduces insulin levels (104, 209, 229).
There are no detectable GLP-1r on the liver or on skeletal muscle and yet GLP-1 has repeatedly been shown to not just increase insulin secretion but also to improve insulin sensitivity. Despite the lack of hepatic GLP-1r expression, intravenous GLP-1 inhibits hepatic glucose production independent of islet hormones in humans (184). CNS GLP-1r activation may also explain this finding. When administered directly into the arcuate nucleus of rats, GLP-1 decreases hepatic glucose production under clamped conditions where glucose and insulin were held constant (209). The specific population of neurons responsible for this effect is not clear as deletion of GLP-1r within the hypothalamus or even more specifically on pro-opiomelanocortin neurons with the arcuate nucleus of the hypothalamus also do not impact normal glucose regulation (18). GLP-1 activates vagal afferent neuronal activity and administration of GLP-1r antagonists into the portal vein impairs glucose tolerance (234) suggesting that the peripheral nervous system is also important in mediating GLP-1 effects on glucose homeostasis. However, neither genetic deletion of CNS nor vagal neuronal GLP-1r is not necessary for normal glucose regulation (213), but CNS (213) and specifically glutamatergic excitatory (vs. GABAergic inhibitory) neurons (1) that express GLP-1r are necessary for the weight-reducing effects of liraglutide, a long-acting GLP-1 agonist. Lastly, in obese mice and in humans, administration of GLP-1 agonists reduces hepatic steatosis (42, 78, 176, 220). Whether these effects are also due to CNS activation is unclear but they do seem to be independent of the effect of chronic GLP-1 administration to reduce body weight. It remains to be seen whether the discrepancy between the genetic versus pharmacological manipulation of CNS GLP-1r signaling and the impact on glucose regulation is due to a species difference or a development compensation in the mice. The CNS distribution of the GLP-1r is dispersed, and different populations of receptors have sometimes very different functions (see Ref. 119 for review). As neuroscience technology becomes more sophisticated, the capability to tease this system apart becomes more promising.
GLP-1r Regulation of β-cell Mass
In addition to stimulating insulin secretion, GLP-1r activation benefits β-cell survival and importantly does so in the presence of multiple apoptotic conditions including hyperglycemia, hyperlipidemia, inflammatory cytokines, and oxidative stress (47). The exact signaling mechanisms that drive β-cell growth and differentiation are still being resolved. However, phosphatidylinositol 3-kinase (PI3K) rather than PKA activation seems to be the principle signaling mechanism by which the GLP-1r controls β-cell growth and apoptosis. GLP-1 also induces a rapid cAMP-dependent activation of extracellular signaling kinase, ERK1/2, and a delayed β-arrestin [an adaptor protein necessary for GLP-1r signaling (216)]-dependent increase of ERK1/2 signaling (186). Providing a direct link, β-arrestin has been found to be necessary for the antiapoptotic effects of GLP-1 (186) and GLP-1 administration to a β-cell line reduces H2O2-induced apoptosis through both cAMP and PI3K (but not ERK1/2) signaling with independent but additive effects (97). Liraglutide also protects against apoptosis via PI3K signaling and the consequent phosphorylation of AKT (114). Recent work has demonstrated that GLP-1-induced activation of PI3K is through activation of epidermal growth factor receptor 1 (EGFR), a tyrosine kinase receptor. EGFR has been found to be necessary for the ability of exendin-4 (Ex4), another long-acting GLP-1r agonist, to regulate β-cell mass and proliferation (68). Because EGFR directly activates PI3K, these data provide a link between GLP-1r signaling, PI3K, and β-cell proliferation. Downstream of AKT phosphorylation in the activation of β-cell proliferation is Wnt signaling, a pathway established in cancer biology to be critical for cell proliferation and survival. Activation AKT leads to accumulation of cytosolic β-catenin and subsequent translocation to the nucleus where it forms a complex with TCF7L2 (139), a transcription factor that activates expression of Wnt target genes (242). siRNA silencing of β-catenin and a dominant negative insertion of TCF7L2 in INS-1 cells (a β-cell line) blunted the ability of Ex4 to stimulate β-cell proliferation (139), indicating that Wnt signaling is necessary for GLP-1-induced β-cell proliferation.
These data illustrate the wide-ranging signaling pathways induced by GLP-1r activation to regulate β-cell mass and function. There is still much debate about the relevance of this impact of GLP-1 signaling in cell lines and in mice versus in humans. While it was found that short-term incretin therapies do not expand β-cell mass in young male mice (31), In a model that enables assessment of human β-cell replication in vivo, it was found that Ex-4 induced proliferation occurred only in juvenile, but not adult islets (36). This work provides an important advance in our understanding of the decline in β-cell proliferation that occurs with aging and indicates that even pharmacological GLP-1 signaling may not be critical in driving proliferation in humans.
Impact of GLP-1 on Glucagon Secretion
GLP-1-induced inhibition of glucagon secretion has been demonstrated in a variety of species including humans (175, 195). Activation of the GLP-1r also inhibits glucagon release from isolated islets or in perfused pancreas studies (53). While the data are clear that GLP-1 inhibits glucagon, the mechanisms are debated as some report low (148, 193), if any (176, 226), expression of the GLP-1r on α-cells. One possibility is that the impact of GLP-1 on glucagon release is indirect through the release of glucagon-regulating hormones from nearby β- and/or δ-cells (53, 77). While somatostatin secreted from δ-cells inhibited glucagon secretion, a somatostatin antagonist only partially blunted the GLP-1-induced decrease in glucagon (89) suggesting additional mechanisms at play.
GLP-1 also acts on β-cells to secrete insulin, which is known to suppress glucagon (233). However, co-secreted with insulin are amylin, zinc (Zn2+) and GABA; all of which have also been shown to individually suppress the release of glucagon. For example, GABA released from β-cells enhances glucose inhibition of glucagon secretion by acting via an Akt kinase-dependent pathway (246). Co-secreted with insulin in hyperglycemic conditions, Zn2+ has been found to have inhibitory action on glucagon release from α-cells (79, 147, 252) and α-TC6 cells (an α-cell line) (79). The potential role of Zn2+ has found increasing interest due to the fact that genome-wide association studies (GWAS) have revealed that rare variants of a Zn2+ transporter gene are associated with improved glucose homeostasis and protection from T2DM (65). To determine whether the impact of Zn2+ was independent of insulin, streptozotocin-treated (to kill β-cells) rats were studied during pancreatic perfusion studies (252). In these rats, disruption of the intrapancreatic infusion of insulin bound to Zn2+, but not of insulin unbound to Zn2+, accelerated glucagon secretion, indicating that Zn2+ but not insulin inhibits glucagon secretion. The thinking is that Zn2+ inhibits pyruvate-induced glucagon secretion via the opening of KATP channels and subsequent inhibition of α-cell electrical activity (100). It is important to note that contrasting data exist suggesting that Zn2+ does not regulate glucagon (188). However, given the connection to the GWAS data, at a minimum Zn2+ is important for glucose control and a logical link for that is through inhibiting the release of glucagon.
In addition to Zn2+, insulin is co-secreted with amylin, and amylin has also been found to dose-dependently suppress arginine-mediated glucagon secretion in rats (71) while pharmacological inhibition of amylin signaling enhances glucagon secretion (70). Pramlintide, an amylin receptor agonist, improves glycemic control in T2DM patients and at least part of that effect is via inhibition of postprandial glucagon secretion (64). A caveat to all of these studies demonstrating that β-cell products could play an indirect role in GLP-1-mediated regulation of glucagon is that GLP-1 maintains the ability to inhibit glucagon secretion in type 1 diabetic patients who have little to no endogenous insulin (34, 117) demonstrating that GLP-1 inhibition of glucagon secretion does not fully depend on β-cell products.
Besides the low expression levels of the GLP-1r on the α-cell, another caveat to the potential direct role of GLP-1 on glucagon regulation is that generally, GLP-1r activation generates cAMP and increases in cAMP are associated with increased, rather than decreased, glucagon release. Recently, an α-cell GLP-1r KO mouse was generated by crossing a loxP flanked humanized GLP-1r mouse with a Gcg-Cre mouse (251). Theoretically, this will eliminate the GLP-1r only from α-cells as neither L-cells nor Gcg-expressing neurons express the GLP-1r. In these mice, the glucagon response to increasing glucose loads was increased rather than decreased in the α-cell GLP-1r KO mice. While ad lib fed glucagon levels were higher in the α-cell GLP-1r KO mice, a curious finding was that these mice had impaired intraperitoneal IP glucose tolerance and increased glucagon response to an IP glucose load, a condition that does not stimulate gut-derived GLP-1 secretion. Interestingly, the α-cell GLP-1r KO mice also showed decreased glucagon levels during hypoglycemia but had significantly higher glucagon levels postprandially. These data suggest that GLP-1 has a bi-directional regulatory role in glucagon secretion that is dependent upon nutrient status. Regardless, a couple of studies have provided a basis for an evolving story on how GLP-1 might mechanistically inhibit glucagon levels. The idea is that there are low numbers of GLP-1r on α-cells but these receptors have enough capacity to generate proportionately small amounts of cAMP (148). This small amount of cAMP mediates suppressive effects glucagon secretion through discrete inhibition of high voltage N-type calcium channels in mice (148) and via P/Q-type voltage-gated calcium channels in humans (187). In their hands, GLP-1 retained this inhibitory effect with either insulin or somatostatin antagonists onboard (148, 187). However, it would be interesting to know if somatostatin and insulin signaling are synergistic in the paracrine effect; that is if both antagonists were given, would GLP-1 still inhibit glucagon release. Regardless, this model explains how low and high intracellular cAMP concentrations with the α-cell could have opposing actions on glucagon secretion. Thus, while there is much to be learned about the signaling that drives GLP-1-induced inhibition of glucagon secretion, there are multiple indirect and direct mechanisms at play. The inhibitor action of GLP-1 on glucagon levels is often over-looked in favor of its role as an insulin secretagogue, but it is clear that pharmacologically this is one mechanism by which GLP-1r agonists improve glycemia in T2DM patients (86).
Role of α-cell Produced Gcg in Insulin Secretion
α-Cell GLP-1 during metabolic stress
PC1/3, which processes GLP-1 (and GLP-2 and oxyntomodulin) from Gcg, is more predominantly expressed in the gut and CNS, but α-cell PC1/3 activity and/or expression is found in embryonic and neonatal mice, with pregnancy, and in models of prediabetes and diabetes (118, 172, 224, 245). Although previous work has demonstrated that 1% to 5% of preproglucagon is processed to GLP-1 in the islets (126, 156), α-cell GLP-1 clearly increases when the pancreas is under metabolic stress (27, 60, 227). Although the incretin model that intestinally secreted GLP-1 is the functional source of GLP-1, these data do suggest that the α-cell pool of GLP-1 also has a functional role in the pancreas.
Streptozotocin (STZ), a β-cell toxin, is used to model diabetes in animals. In rats administered STZ, there is an acute increase in islet Gcg and PC1/3 expression that leads to an increased processing of α-cell Gcg to GLP-1 (172). While the function of α-cell GLP-1 under metabolic stress conditions is unknown, glucagon receptor (GcgR) KO animals have a developmentally driven increase in pancreatic GLP-1 production and are also, interestingly, resistant to STZ-induced diabetes (30). Further, blockade of the GLP-1r in GcgR deficient mice prevented the improved glucose tolerance seen in the mice (174). Additionally, mice with a cre-inducible α-cell KO of PC1/3 (although the extent to which intestinal and CNS PC1/3 expression was intact is unknown) had reduced levels of GLP-1 in the islet and greater impairments of glucose and insulin in response to STZ (227). Altogether these data suggest that α-cell GLP-1 production provides a protective effect on β-cell function during times of stress. Further examples of this are that in cultured α-cell lines or isolated islets, high media glucose concentrations increase PC1/3 expression and cellular GLP-1 content (152, 240). α-Cell hyperplasia also occurs with high-fat diet and this precedes β-cell mass expansion (59), and in both human and mouse islets there is positive correlation between islet levels of GLP-1 and adiposity (138). Together these data suggest a role for α-cell GLP-1 production in the adaptation to metabolic disease. Whether this increase is as a protective factor that eventually fails or a part of the pathophysiology is unknown.
Although our focus here is on pancreatic GLP-1, GLP-1 in the circulation, presumably due to intestinally secreted GLP-1, also increases with inflammation and the mechanism by which this occurs has been linked to one particular cytokine, interleukin 6 (IL-6). IL-6 is increased in inflammatory conditions including exercise and obesity (59, 60). Under severe inflammatory conditions such as sepsis, or when induced by exogenous lipopolysaccharide (LPS) administration, there is a systemic increase in GLP-1 that is dependent on IL-6 (111, 181). In response to LPS, the increase in GLP-1 is dose dependent, and GLP-1 levels remain elevated for 8-h (111). Whereas in IL-6 knockout mice, there is no increase in GLP-1 after LPS administration (111). With 90 min of exercise in mice, there is an acute increase in IL-6 and GLP-1 and again this increase in GLP-1 was not seen in IL-6 knockout mice (60). To look at α-cell production specifically, IL-6 injections were given twice daily for over a week and the protein content of pancreatic GLP-1 and insulin were both increased (60). Despite the evidence showing that IL-6 increases GLP-1 secretion from the α-cell, the function, and whether it relates to glucose regulation or not, during this kind of inflammatory state is unknown. After several hours, LPS treatment causes elevated IL-6 levels and hypoglycemia to develop and Ex4 administration blunts this hypoglycemic effect in rats suggesting that the effect of GLP-1 is related to glucose control (247) although the direction goes opposite of what we normally think of as GLP-1-mediated glucose control. Another leading hypothesis is that IL-6 induced GLP-1 is a part of a negative-feedback loop to inhibit or restrain inflammatory responses. Rat islets treated with liraglutide showed both decreased pro-inflammatory cytokine levels (IL-6 and TNF-α) and the islets had improved function (133). Both the glucose and anti-inflammatory effect of GLP-1 point to α-cell GLP-1 acting locally rather than centrally. However, whether these pharmacological agonists have the same function as endogenously secreted GLP-1 from the α-cell remains an important unresolved question.
Thus, both metabolic (hyperglycemia, STZ-induced, diabetes) and physiological (exercise, inflammation) stress conditions influence IL-6 circulating levels which may be the factor that triggers GLP-1 secretion from the α-cell (Figure 3). However, the role of increased GLP-1 secreted from the α-cell under inflammatory conditions, how it impacts overall glucose homeostasis, and how this may be targeted pharmacologically is unknown.
Figure 3.
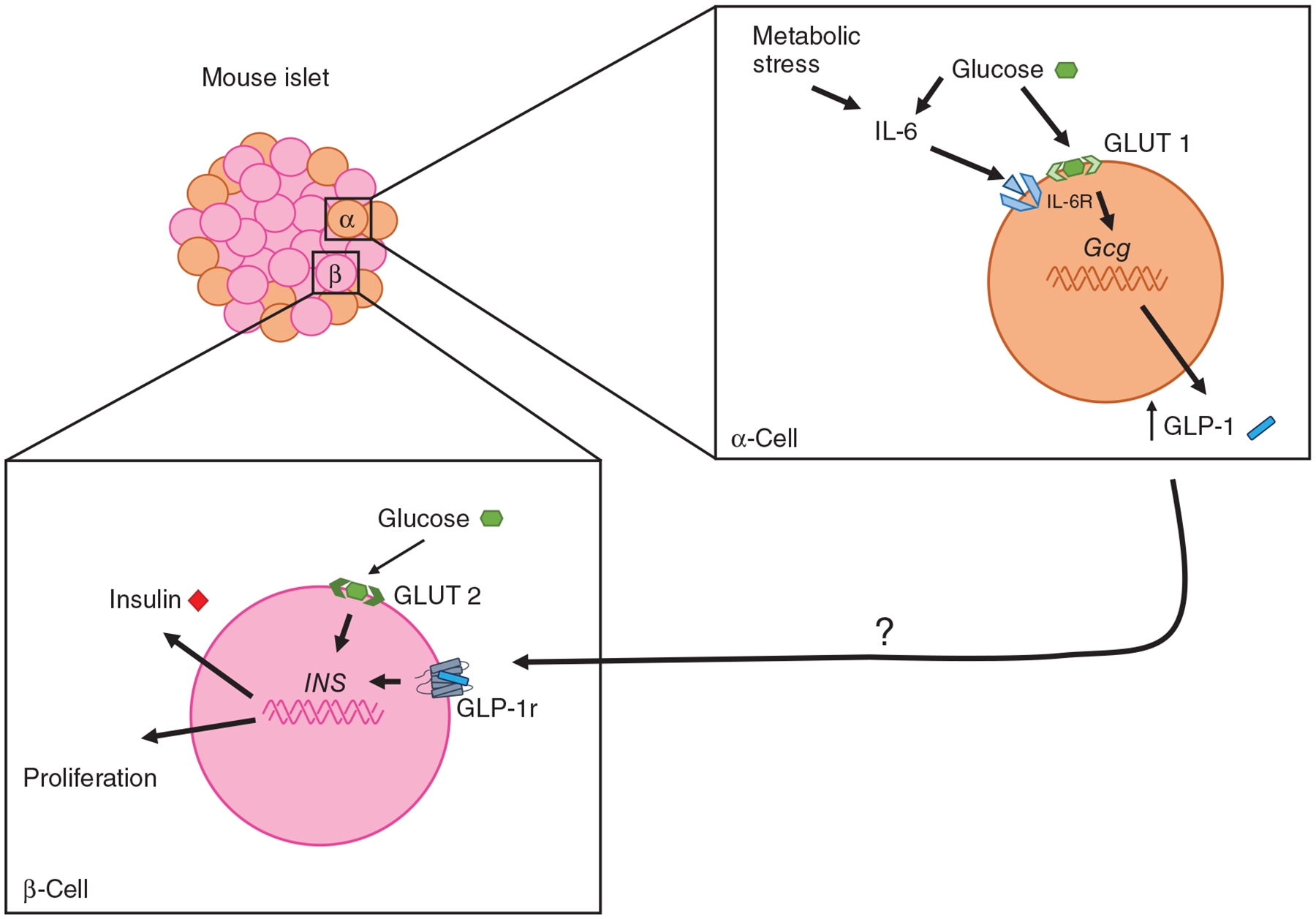
Factors that impact α-cell GLP-1 production. Metabolic stress, systemic inflammation, exercise, hyperglycemia, obesity, and diabetes stimulate α-cell GLP-1 production. IL-6 seems to be a primary factor that leads to this increase. The function of this increase is unknown but regulation of β-cell mass and function is a likely endpoint.
α-Cell GLP-1 in normal glucose regulation
The accepted dogma of GLP-1 secreted from the intestine and acting on the pancreas in an endocrine manner is difficult to reconcile given the observations that GLP-1 is rapidly degraded by dipeptidylpeptidase 4 (DPP4) and very little, in fact, only approximately 10% of intestinally secreted GLP-1 reaches the circulation (39, 82, 93). While the role of α-cell-secreted GLP-1 became established during states of metabolic stress, the question remains whether or not it has a role in normal glucose control. Although controversial, pancreatic GLP-1 has been found in normal islets and its expression increases with increasing glucose concentrations (152, 227, 240). In isolated human islets, the amount of GLP-1 was low under basal conditions and was only present in the cell lysates, not the culture medium in one study (240). However, others have found pancreatic GLP-1 to be higher in human versus mouse islets (227). In addition, PC1/3 activity can also be up-regulated by activating a bile acid receptor (TGR5) known to regulate GLP-1 secretion (145). These data suggest that the conditions by which α-cell GLP-1 is assessed may be important in the ability to detect GLP-1 levels.
Recently, the role of pancreatic Gcg, the gene that encodes GLP-1, was explored using a Cre lox-P mouse model that selectively reactivated the endogenous Gcg gene in the pancreas versus the intestine while the remaining tissues remained devoid of Gcg (27). To understand the role of GLP-1r activation specifically, glucose responses to Ex9, a GLP-1r antagonist, was examined. Ex9 had no impact on Gcg deficient animals indicating that Ex9 was a true GLP-1r antagonist, in vivo. This indicates that the presence of GLP-1 is necessary for the ability of Ex9 to impair glucose. Interestingly, animals that only expressed pancreatic, but not animals that expressed only intestinal Gcg had impaired glucose tolerance (whether oral or IP) in response to Ex9. Thus, the source of the GLP-1 ligand necessary for the ability of Ex9 to impair glucose tolerance was pancreatic and not intestinal GLP-1.
However, there is an important caveat to this work. It has been known for quite some time that glucagon increases insulin secretion (233) but whether this was physiological or pharmacological was debated. Further, whether this was through glucagon action on glucagon or GLP-1r was also studied. At the time studies had indicated that glucagon acted on the GLP-1r only at pharmacological concentrations (223) although other data suggested a more complex relationship and that glucagon could act on both glucagon and GLP-1r to regulate insulin secretion (155). Recently, three independent islet perfusion studies demonstrated that glucagon increases insulin not by acting on GcgRs but by acting on GLP-1r (22, 221, 253). At a first pass, these data suggest the possibility that it was pancreatic glucagon rather than pancreatic GLP-1 that was responsible for the ability of Ex9 to impair glucose tolerance in the previous study (27). However, the ability of glucagon to bind to the GLP-1r is extraordinarily less potent than GLP-1 (221). In addition, it would mean that the entirety of Ex9’s action is by impairing glucagon action on β-cell GLP-1r since Ex9 had no impact on glucose tolerance in animals that had fully restored intestinal Gcg expression and postprandial circulating GLP-1 levels (27). Lastly, the experimental conditions that lead to the increase in glucagon and glucagon action on the GLP-1r were specific to having both elevations in glucose and amino acids. The in vivo experiments described above (27) were only done with oral glucose.
Other mouse models have been derived in an attempt to separate the role of α-cell glucagon and GLP-1. One used a diphtheria toxin-inducible α-cell KO of Gcg and this mouse had a small impairment of age-induced IP glucose tolerance (227). Administration of DPP4 inhibitor, but not glucagon, restored glucose tolerance in these mice. The authors suggest this provides evidence that intestinally derived GLP-1 can compensate for the lack of α-cell GLP-1. However, DPP4 inhibitors increase bioactive GLP-1 AND GIP and previous work demonstrates that these drugs can fully improve glucose tolerance even if only one of the incretins have intact signaling (66). Isolated islets from another mouse model with α-cell KO of PC1/3 had reduced levels of GLP-1 in the islet and reduced glucose-stimulated insulin secretion (227). These mice also had impaired intraperitoneal, but not oral glucose tolerance (227). Similarly, β-cell GLP-1r KO mice have normal oral but not intraperitoneal, glucose tolerance (131, 209). Thus, a model where paracrine, rather than endocrine action of preproglucagon peptides in regulating insulin emerges. Because of the nature of preproglucagon processing, it will be difficult to distinguish between the impact of α-cell derived glucagon versus GLP-1 on local GLP-1r. However, this work clearly demonstrates that the conventionally accepted role of GLP-1 biology is inadequate. The combined impact factors driving an increase in α-cell GLP-1 and glucagon are summarized in Figure 4.
Figure 4.
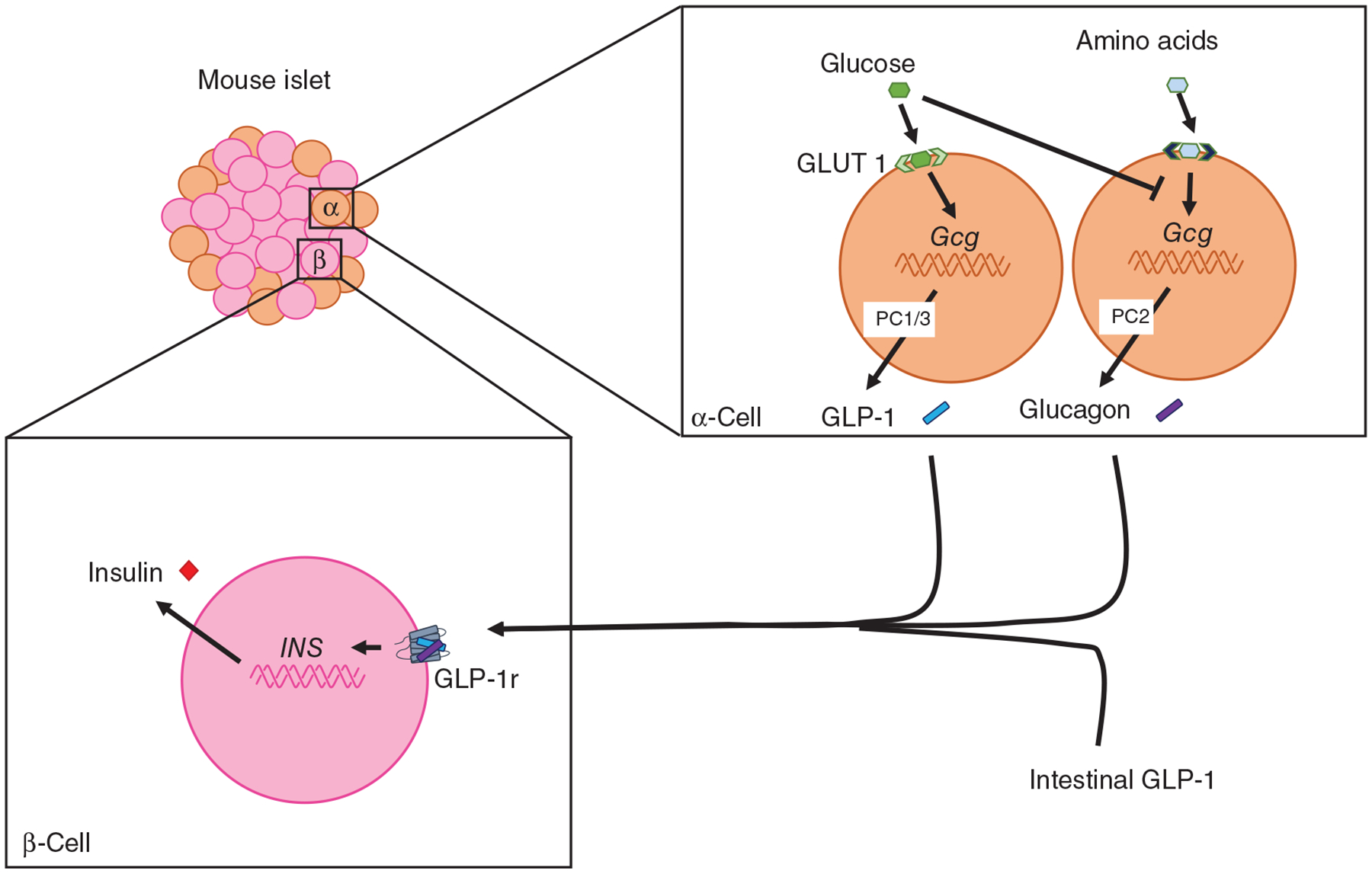
Recent work highlights a more complexity to the role of Gcg-derived peptides in the incretin effect. While historical work suggests intestinal GLP-1 is important in regulating glucose homeostasis, there may be a role for α-cell derived GLP-1 as well. In addition, in response to amino acids, glucagon is secreted and acts on local GLP-1r to regulate insulin secretion.
If we ignore the intestinal versus pancreatic source of GLP-1 topic and just focus on the fact that GLP-1 regulates insulin secretion by acting directly on β-cell GLP-1rs and either directly or indirectly suppresses glucagon then under what circumstance is glucagon important for β-cell GLP-1r signaling? Of the components of a mixed meal (lipids, carbohydrates, and proteins), free fatty acids, if anything decreases glucagon levels in man (72), carbohydrates potently suppress glucagon, and proteins (amino acids) potently stimulate glucagon secretion (232). Interestingly, free fatty acids suppress the ability of arginine to stimulate glucagon secretion in man (72) suggesting that even in a mixed meal situation, increases in glucagon levels are restrained. In addition to GLP-1 suppressing glucagon, insulin and somatostatin, which also increase during a meal, also suppress glucagon levels. Many questions arise from these observations. Is the system set-up to suppress redundant signals? Is the increase in glucagon during a high protein meal necessary to increase insulin? Is GLP-1 versus glucagon necessary for different phases of insulin secretion? Are both GLP-1 and glucagon synergistic or additive in insulin control? All of these are possibilities. However, in animals devoid of both GLP-1 and glucagon, insulin response to an intravenous and oral glucose load appear to be normal suggesting that GLP-1 and glucagon, together, are not necessary for insulin secretion and/or that redundant in vivo mechanism are able to compensate (27). As has been suggested before, it could be that glucagon offers an additional redundant signal that allows for fine-tuning of glucose control in the face of metabolic stress whether it is exercise, hypoglycemia, or postprandial glucose control.
Targeting α-cell production of GLP-1, specifically, in T2DM therapeutics is an idea that is being explored. One study used adenovirus-mediated expression of PC1/3 in α-cells to increase islet production of GLP-1 and was able to improve glucose-stimulated insulin secretion in a mouse model of type 1 diabetes (241). In addition, pharmaceutical activation of GPR142, a GCPR that is expressed in pancreatic islets and that has previously been shown to enhance glucose-dependent insulin secretion (225, 239), also increases GLP-1 secretion from the α-cell (137). Moreover, using isolated mouse islets treated with Ex9, the researchers showed that insulin secretion induced by GPR142 activation is dependent on GLP-1 (137). Thus, regardless of our understanding of the physiology or pathophysiology of islet produced GLP-1, these data suggest that this pool of GLP-1 could be targeted to treat T2DM and avoid some of the CNS side-effects (i.e., nausea) associated with long-acting GLP-1 agonists.
Targeting GLP-1 in Pharmacology
GLP-1 agonists and DPP4 inhibitors
The development of GLP-1-based drugs has been one of the major advances in diabetes medicine in recent years. The currently approved pharmaceutical strategies targeted to the GLP-1 system are aimed at either increasing endogenous GLP-1 levels with inhibitors for the protease that inactivates GLP-1 or long-acting GLP-1r agonists resistant to DPP4 cleavage (2, 51). DPP4 inhibitors are effective at stimulating insulin and reducing glucagon actions; attributes that are credited to GLP-1r signaling (46). However, DPP4 acts on GIP as well as 40 additional substrates (160). GIP is the only additional substrate of note for glucose regulation and works in both humans (5, 167) and mice (66, 85) suggest that both GIP and GLP-1 signaling are targets for the improved glucose control with these drugs.
There are now multiple GLP-1r agonists currently available for the management T2DM (20, 48, 51). Various strategies are used to extend the half-lives of these agonists compared to native GLP-1 including using a synthetic analog of Ex4 (Byetta) and the addition of a fatty acid side chain to native GLP-1 to facilitate albumin binding (Liraglutide/Victoza). In an effort to improve convenience and compliance (149), modifications of these drugs are also being made to extend the half-life to allow for once-weekly injections (Bydureon and semaglutide/Ozembic). Besides being more convenient for the patient, there seem to be added benefits of creating a more stable pharmacodynamics profile (i.e., reducing the peaks and troughs of drug action) as these drugs induce less nausea, a common side-effect of rapid-acting compounds (19, 50, 98, 194). An added benefit of these drugs compared to DPP4 inhibitors is the added weight loss (236) and for some specific GLP-1r agonists (liraglutide and semaglutide) there is a reduction in cardiovascular events (168). Because of the impact of these drugs on weight loss, there are now multiple formulations approved to specifically treat obesity independent of T2DM. Interestingly, although T2DM patients do see improvements in body mass, there is a larger benefit for the obese nondiabetic patient for weight loss.
The neural pathways leading to the weight loss effects of these drugs are being actively pursued in preclinical work. Part of the reason for this effort is to determine whether the neural circuitry that drives the weight loss effect is distinct from the circuitry that drives the nausea effect of these drugs. In animal work, we know that some neuronal regions regulating the impact of GLP-1 on food intake are distinct from those regions that regulate nausea (121, 129). As discussed above, previous preclinical work in mice and rats established that the CNS (113, 213), but not the peripheral nervous system (213) is critical in mediating the impact of long-acting GLP-1r agonists on body mass. With newer advances in neuroscience techniques, we now know that glutamatergic rather than GABAergic neurons (1) are the specific type of neurons necessary for the ability of liraglutide to induce weight loss. However, given that neither hypothalamic (Nkx2.1 neurons), PVN (Sim1 neurons), nor POMC neurons were not necessary for the ability of long-acting agonists to reduced body mass (18), the hunt is still on for the specific population of neurons responsible for the pharmaceutical reduction in body mass. Better understanding of the neural mechanisms of these processes would benefit the therapeutic utility of these agents.
Poly-agonists
An exciting recent pharmaceutical strategy to the treatment of obesity and T2DM has been the development of hybrid peptides that activate more than one receptor to generate an effect (202). Given that obesity and T2DM are diseases with integrated pathology, a multifaceted approach is likely necessary for more effective treatment. These agents are touted as mimicking the broad range of peptide increases seen after bariatric surgery. Some of the first compounds developed using this strategy were glucagon/GLP-1 co-agonists, peptides engineered to activate the cognate receptors of both peptides in different relative potencies (37, 38). The rationale behind this line of drug development is that both glucagon and GLP-1 bind specific and distinct receptor populations in the brain to cause satiety (9, 103), and activating both receptors could lead to synergistic effects. Peptides with equal agonism for each of the target drugs (e.g., GLP-1/glucagon) seem to have the most therapeutic promise (37, 38). In the case of glucagon, greater glucagon potency would lead to greater energy expenditure and suppression of food intake, but there seems to be a threshold beyond which glucose control worsens despite weight loss. The results for combined agonism in mice and rats are promising with improved glucose tolerance but also greater reductions in body weight and fat with the dual agonists compared to GLP-1r agonism alone (38, 183). Although there may be subtleties in the formulation that lead to species differences, the results in humans have not been as exciting as in mice. Specifically, a recent phase 2a clinical trial found reductions in body weight and improvements in glucose control but the degree of the improvements seemed to be within the range of what is seen with long-acting GLP-1r agonists alone (3, 189). Interestingly, a GIP receptor/GLP-1r dual agonist in phase 2 trials caused greater improvements in both HbA1c% and weight loss compared to the GLP-1 agonist alone (67). Given that GIP is thought to be important for lipogenesis, the mechanism for this effect of the dual agonist is unclear. Regardless, this drug shows great promise in improving glucose control and weight loss in T2DM patients.
Bariatric Surgery and GLP-1
Why does GLP-1 increase with surgery?
Bariatric surgery is currently the most effective strategy at the treatment of obesity and its comorbidities. There are many types of bariatric surgeries. Roux-en-Y gastric bypass (RYGB; a small gastric pouch is formed and the jejunum is connected directly to the small pouch) used to be the most widely performed bariatric surgeries but its utilization has been reduced to about 20% of the procedures in the last couple of years (40). Currently, the most common surgery in the US is vertical sleeve gastrectomy (VSG; 80% of the stomach along the greater curvature is removed), which comprises about 60% of performed bariatric procedures. The switch is likely due to the fact that VSG is surgically more simplistic, leads to fewer long-term malabsorptive issues, and although dogma persists that it is less effective, randomized clinical trials demonstrate similar efficacy between VSG and RYGB (see Ref. 112 for meta-analysis).
Among the many similar physiological effects between these two surgeries is about a 10-fold increase in postprandial levels of GLP-1; something observed in both patients and rodent models of surgery (26, 105, 107, 231). A long-standing hypothesis for why GLP-1 (and other gut peptides for that matter) are increased after RYGB is that the shorter length of small bowel leads to more rapid nutrient delivery further down into the GI tract where the majority of L-cells are located (62). VSG also increases nutrient delivery to the distal gut thanks to a restricted stomach size that increases gastric pressure (26) and consequently gastric emptying rate (26, 154). Indeed, speed of nutrient delivery may be important after RYGB as the increase in nutrient-induced GLP-1 was eliminated if nutrients were delivered to the bypassed limb (169). However, in rats after VSG, glucose infused slowly and directly into the duodenum caused similar increases in GLP-1 as when the same glucose load was delivered orally (26). These data suggest that intestinal L-cells are either increased in number or in nutrient-sensitivity after VSG. While one study demonstrated that VSG in rats increased L-cell number (24) another did not (161). Differences in diet, as well as the control groups utilized (pair-fed vs. ad lib fed sham groups), could lead to differential structural and functional changes in the gut nutrient-sensing pathways (11). On the other hand, RYGB is more consistently associated with intestinal hypertrophy regardless of dietary exposure (24, 162). The intestine is considered a major site of glucose disposal after RYGB and this may provide energy for intestinal metabolic pathways to support tissue growth (203). In humans, this increase in glucose absorption after RYGB was associated with the exaggerated release of insulin and GLP-1 (43). Thus, the anatomical differences between the surgeries may lead to different adaptations in either the morphological or mechanical function of the GI tract and either of these adaptations can regulate prandial GLP-1 responses.
Another common physiological response to both RYGB and VSG is the significant increase in total and various sub-species of plasma bile acids (55, 124, 212). Bile acids have demonstrated effects on stimulating GLP-1 secretion from L-cells by acting through a specific G protein-coupled receptor (TGR5) versus their other common receptor a nuclear transcription factor, FXR (115). However, surgery-induced increases in bile acids have been demonstrated to be important for the increase in postprandial GLP-1 in one (41), but not another study (151). Further, these two studies had divergent results on the necessity of TGR5 for surgery-induced weight loss and improvements in glucose homeostasis (41, 151). Conversely, FXR seems to be necessary for the full effects of VSG, independent of GLP-1 (201). Thus, although this will require future validation, it seems that the surgery-induced increase in GLP-1 is due to intestinal responses to nutrient delivery. With RYGB, this is more acute, but with VSG, chronic adaptations are more critical.
GLP-1 as mechanism for the metabolic success of surgery
Although T2DM is thought to be chronic and progressive, bariatric surgery leads to large improvements in insulin secretion and sensitivity which results in a remission of T2DM for many patients. The consistency of the finding that GLP-1 increases with surgery in addition to this increase being associated with greater prandial insulin release (231) and greater weight loss (199) after surgery has led to the suggestion that GLP-1 is an integral mechanism for the success of surgery (Figure 5). While the postprandial GLP-1 response may be required for the insulin and glucose responses to a meal after bariatric surgery, whether GLP-1 is responsible for the resolution of T2DM is less clear. One study found that postprandial GLP-1 responses were a significant predictor of T2DM remission after RYGB (163), yet another found similar postprandial GLP-1 responses 2 years after VSG whether the patients had postoperative remission, relapse, or lack or remission of T2DM (107).
Figure 5.
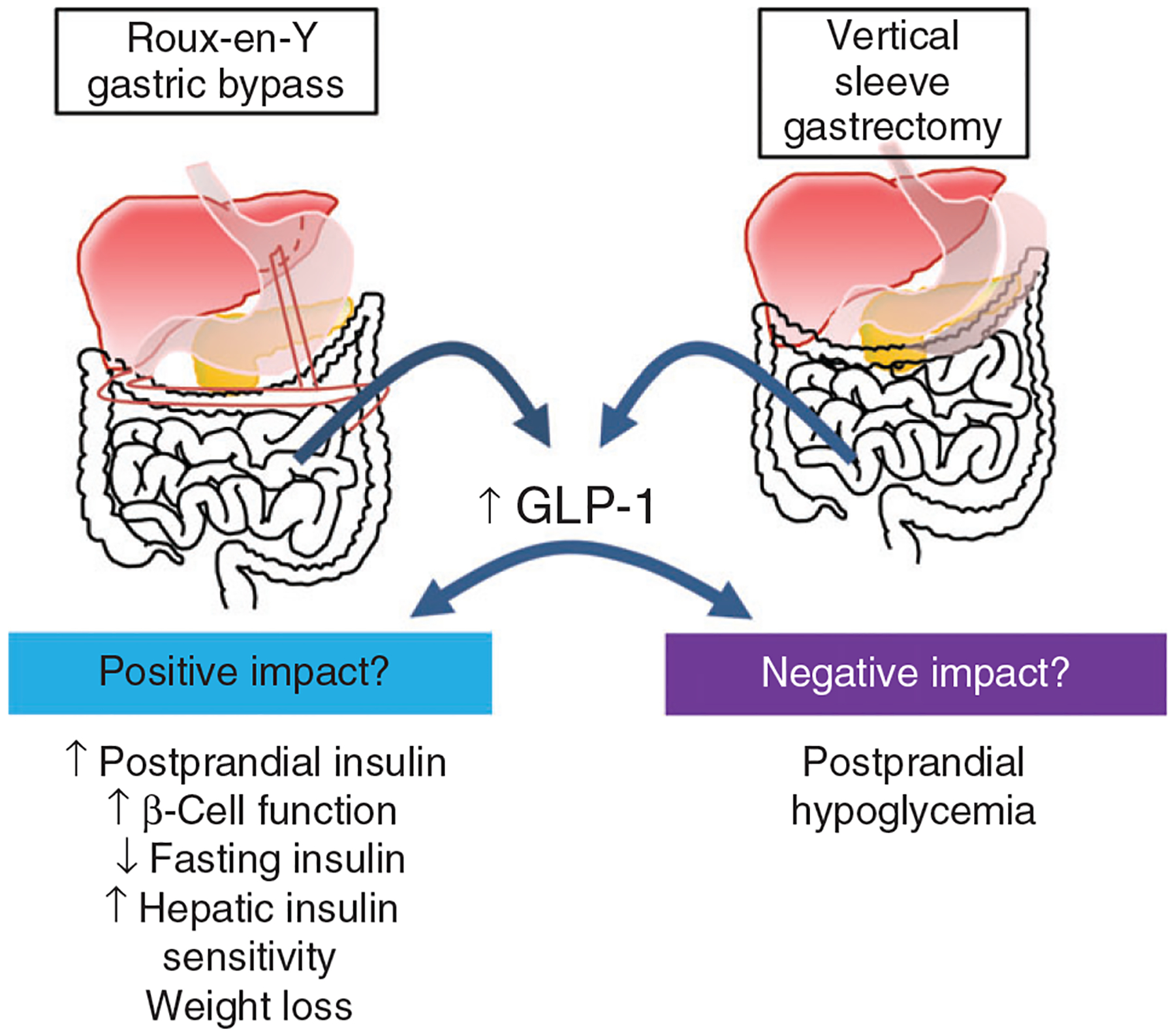
Postprandial GLP-1 increases several-fold after bariatric surgery and has been implicated as a mechanism in both positive and negative impacts of bariatric surgery. On the positive side, GLP-1 has been implicated in increasing postprandial insulin levels to restrain post-prandial glucose homeostasis, improvements in insulin sensitivity lead to reductions in fasting insulin, improved hepatic insulin sensitivity, and overall improved β-cell function. However, on the negative side, the increase in postprandial insulin is thought to contribute to post-bariatric hypoglycemia which occurs in as much as 30% of surgery patients.
In support of a role for GLP-1, multiple studies have demonstrated that administration of the GLP-1r antagonist, Ex9, reduces the insulin response to a glucose load in both humans and rodents (25, 106, 110, 206). However, these data come with an interpretative problem (235). There is no dispute that GLP-1 regulates postprandial insulin. The same dose of Ex9 impairs glucose and reduces insulin in control subjects (107) and rats (25). The degree of this impairment is similar between surgery and control conditions. If GLP-1 was more important in glucose control after surgery, the degree of impairment should be greater. Thus, the question is whether what we are seeing after surgery is reflective of the normal response or is reflective of greater importance of GLP-1 during surgery.
The incretin effect is credited to both GLP-1 and GIP (166, 237) with each thought to contribute equally to insulin secretion in nonobese, non-T2DM subjects (166). Both T2DM and to a lesser extent, obesity, reduce (123, 165), while bariatric surgery enhances, the incretin effect (130). However, there is little agreement as to whether bariatric surgery leads to an increase in GIP suggesting that the extent to which GIP contributes to the enhanced incretin effect is debatable. In an effort to determine the importance of GIP after RYGB, RYGB patients were given a DPP4 inhibitor to increase the bioavailability of both GIP and GLP-1 and then combined this with Ex9 to block GLP-1r signaling; an experimental condition that would isolate the impact of GIP signaling on glucose tolerance (219). The DPP4 inhibitor failed to improve glucose tolerance or β-cell function while GLP-1r signaling was blocked in RYGB patients. In contrast, T2DM patients that had not undergone bariatric procedures fully responded to the DPP4 inhibitor with improved glucose tolerance and insulin secretion even when combined with Ex9 (167). Together these data suggest that RYGB shifts the balance of the incretin effect toward GLP-1 and away from GIP.
Qualitatively, rodents and humans respond similarly to bariatric surgery. Both have substantial weight loss, elevated gastric emptying rate, and increased postprandial insulin and GLP-1 levels. While human work is limited to acute pharmacological intervention, preclinical work offers the additional ability to genetically manipulate the GLP-1 system and test its role in the metabolic success of surgery. One would hope that this would lead to less interpretive issues. Whole-body GLP-1r KO mice have similar weight loss and improvements in glucose tolerance compared to littermate controls after both VSG (157, 244) and RYGB (248) suggesting a limited role of GLP-1r in surgical success. Central nervous system GLP-1r has been shown to be important for regulating body weight and glucose homeostasis in rodents (18, 122, 209) and may be a target population for the impact of surgically induced GLP-1 to act in regulating body mass and glucose. However, Ex9 infused directly into the CNS of rats during RYGB or sham surgeries (248) had no significant impact on the surgery-induced reductions in body mass. Thus far, the data would seem to be in agreement that GLP-1r signaling is not necessary for the metabolic success of surgery. However, in the last few years, conflicting reports have been published. To examine the specific role of β-cell GLP-1r, two slightly different versions of an inducible Cre-loxP strategy were used to knock out these receptors and VSG was performed. One study found that β-cell GLP-1r were necessary for surgery-induced improvements in glucose tolerance and glucose-stimulated insulin secretion, but not weight loss (69); while the other found not only that these receptors were not necessary for VSG-induced improvements in glucose tolerance but those glucose responses were essentially normalized to the WT levels (44). Differences in diet, mouse models, and/or some other factor may contribute either independently or in combination to the differences in these experimental findings. Regardless, we are still left with no solid conclusion as to whether GLP-1 is necessary for the success of surgery.
The increase in GLP-1 after surgery may also have a trophic effect on β-cell mass. GLP-1 increases β-cell mass in rodents and has also been suggested to increase β-cell function in humans following bariatric surgery (80). In isolated islets, VSG mice had changes in their genetic and functional signature favoring calcium signaling and insulin secretion (45). In a pooled group of VSG and RYGB patients, pancreatic fat deposition as assessed by PET imaging was found to be reduced alongside improvements in β-cell function (95). These data suggest the possibility that the impact of surgery on the pancreas could be due to the weight loss itself and is independent of GLP-1’s trophic effect.
With both human and rodent work, the clear finding is that the increase in postprandial GLP-1 drives acute glucose responses to a meal after bariatric surgery but whether or not they are required for long-term improvements in glucose control, T2DM remission, and/or for weight loss are debatable. However, this may be difficult to determine as the degree of β-cell destruction prior to surgery may be more critical in determining whether those β-cells can recover sufficiently to resolve T2DM (6).
GLP-1 in post-bariatric hypoglycemia
One increasingly recognized surgery complication is post-bariatric hypoglycemia (PBH) (207). This is reflected in a subset of bariatric patients and is associated with symptoms of postprandial “dumping syndrome” characterized not only by hypoglycemia, but also hyperinsulinemia, sweating, nausea or vomiting, and heart palpitations. Given that adrenergic and cholinergic symptoms in the postprandial state can be nonspecific, PBH has recently been redefined as the presence of neuroglycopenic symptoms (difficulty thinking, weakness, fatigue) with concomitant hypoglycemia (<54 mg/dL) (207) that is relieved within minutes of carbohydrate ingestion. Initial reports indicated a prevalence of less than 1% for hypoglycemia requiring hospitalization, but 10% for clinically recognized hypoglycemia (135, 150). However, the use of continuous glucose monitoring (CGM) has highlighted that hypoglycemia occurs much more frequently (closer to 30%) and is observed with similar frequency in both RYGB and VSG (23, 177, 230). This condition threatens the safety of affected patients as hypoglycemia impairs cognition and increases the risk for syncope, cardiac arrhythmias, seizures, coma, and even death.
Hypoglycemia is a complication for T1DM and T2DM, but for those patients, medications can be adjusted to minimize the occurrence. The mechanism for PBH is unknown and creates a difficult therapeutic challenge. The dominance of postprandial timing indicates that hypoglycemia is partly due to the exaggerated systemic appearance of ingested glucose secondary to altered anatomy and subsequent disproportionate insulin response to a meal. One hypothesis is that this phenomenon is caused by the exaggerated postprandial GLP-1 and consequently insulin levels. Postprandial glucose, GLP1, and insulin have been found to be even higher in patients susceptible to PBH (75). Multiple studies have now demonstrated that administration of Ex9 can lower postprandial insulin and prevent hypoglycemia in RYGB patients (32, 33, 204, 205). However, patients with PBH still have higher peak glucose levels after GLP-1r antagonist treatment compared to asymptomatic patients. In addition, the rapid time course of the postsurgical increase in postprandial GLP-1 and insulin secretion (days) does not mirror the delayed development of PBH (years). Importantly, although GLP-1 is likely not a mechanism for PBH, Ex9 therapies may still be a way to treat PBH.
In conclusion, GLP-1 increases with bariatric surgery are likely due to acute and/or chronic responses to rapid nutrient entry seen with RYGB and VSG, respectively. This increase in GLP-1 likely plays an important role in the acute glucose and insulin responses to a given meal. However, whether these increases are responsible for the overall improvements in glucose homeostasis, body mass, or in the onset of PBH is something that is still debated.
Conclusions
GLP-1 was suggested to be an incretin over 32 years ago. Indeed, GLP-1 actions in the islet are implicated in the success of surgery and have been exploited for effective glucose control in T2DM patients. However, the incretin model is much too simple for the complexity of the system. GLP-1 has a wide array of physiological effects that go beyond the β-cell. Further, GLP-1 and glucagon released from the α-cell may be important for β-cell proliferation and function suggesting that paracrine regulation of the β-cell needs to be incorporated into our thinking surrounding GLP-1 function. These interesting interactions and/or overlapping functions of GLP-1 and glucagon and GPCR signaling requires further exploration. What is true, is that GLP-1r agonists are safe and effective therapies for obesity and T2DM and will remain an active area of exploration.
Didactic Synopsis.
Major teaching points
Glucagon-like peptide-1 (GLP-1) is an important post-prandial stimulus of pancreatic insulin secretion and regulator of glucose homeostasis.
GLP-1 is coded by the preproglucagon gene which is expressed in the gut, brain, and α-cells of the pancreas.
Due to tissue-specific posttranslational processing, GLP-1 is predominantly made in the gut and brain but can also be made in the pancreatic α-cells in response to nutrients and under times of stress.
Recent work suggests that α-cell GLP-1 and glucagon, another preproglucagon-derived peptide, can stimulate insulin secretion through the GLP-1r.
GLP-1 has been implicated as a cause of the metabolic success of surgeries performed to correct obesity but research in genetic mouse models challenges this assumption.
GLP-1 has been targeted very successfully for the treatment of both type 2 diabetes and obesity.
References
- 1.Adams JM, Pei H, Sandoval DA, Seeley RJ, Chang RB, Liberles SD, Olson DP. Liraglutide modulates appetite and body weight through glucagon-like peptide 1 receptor–expressing glutamatergic neurons. Diabetes 67: 1538–1548, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ahren B, Schmitz O. GLP-1 receptor agonists and DPP-4 inhibitors in the treatment of type 2 diabetes. Horm Metab Res 36: 867–876, 2004. [DOI] [PubMed] [Google Scholar]
- 3.Ambery P, Parker VE, Stumvoll M, Posch MG, Heise T, Plum-Moerschel L, Tsai L-F, Robertson D, Jain M, Petrone M, Rondinone C, Hirshberg B, Jermutus L. MEDI0382, a GLP-1 and glucagon receptor dual agonist, in obese or overweight patients with type 2 diabetes: A randomised, controlled, double-blind, ascending dose and phase 2a study. Lancet 391: 2607–2618, 2018. [DOI] [PubMed] [Google Scholar]
- 4.Anini Y, Hansotia T, Brubaker PL. Muscarinic receptors control post-prandial release of glucagon-like peptide-1: In vivo and in vitro studies in rats. Endocrinology 143: 2420–2426, 2002. [DOI] [PubMed] [Google Scholar]
- 5.Aulinger BA, Bedorf A, Kutscherauer G, De Heer J, Holst JJ, Göke B, Schirra J. Defining the role of GLP-1 in the enteroinsulinar axis in type 2 diabetes using DPP-4 inhibition and GLP-1 receptor blockade. Diabetes 63: 1079–1092, 2014. [DOI] [PubMed] [Google Scholar]
- 6.Aung L, Lee W-J, Chen SC, Ser K-H, Wu C-C, Chong K, Lee Y-C, Chen J-C. Bariatric surgery for patients with early-onset vs late-onset type 2 diabetes. JAMA Surg 151: 798, 2016. [DOI] [PubMed] [Google Scholar]
- 7.Baggio LL, Drucker DJ. Biology of incretins: GLP-1 and GIP. Gastroenterology 132: 2131–2157, 2007. [DOI] [PubMed] [Google Scholar]
- 8.Balks HJ, Holst JJ, Brabant G, Medizin ZI, Endokrinologie AK, Hannover MH. Rapid oscillations in plasma glucagon-like peptide-1 (GLP-1) in humans: Cholinergic control of GLP-1. J Clin Endocrinol Metab 82: 786–790, 2014. [DOI] [PubMed] [Google Scholar]
- 9.Barrera JG, Sandoval DA, D’Alessio DA, Seeley RJ. GLP-1 and energy balance: An integrated model of short-term and long-term control. Nat Rev Endocrinol 7: 507–516, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bell GI, Santerre RF, Mullenbach GT. Hamster preproglucagon contains the sequence of glucagon and two related peptides. Nature 302: 716–718, 1983. [DOI] [PubMed] [Google Scholar]
- 11.Brandsma E, Houben T, Fu J, Shiri-Sverdlov R, Hofker MH. The immunity-diet-microbiota axis in the development of metabolic syndrome. Curr Opin Lipidol 26: 73–81, 2015. [DOI] [PubMed] [Google Scholar]
- 12.Brubaker PL. Control of glucagon-like immunoreactive peptide secretion from fetal rat intestinal cultures. Endocrinology 123: 220–226, 1988. [DOI] [PubMed] [Google Scholar]
- 13.Brubaker PL. Glucagon-like peptide-2 and the regulation of intestinal growth and function. In: Comprehensive Physiology. Hoboken, NJ, USA: John Wiley & Sons, Inc., 2018, p. 1185–1210. [DOI] [PubMed] [Google Scholar]
- 14.Brubaker PL, Anini Y. Direct and indirect mechanisms regulating secretion of glucagon-like peptide-1 and glucagon-like peptide-2. Can J Physiol Pharmacol 81: 1005–1012, 2003. [DOI] [PubMed] [Google Scholar]
- 15.Brubaker PL, Vranic M. Glucagon-like immunoreactive peptides in a rat ileal epithelial cell line (IEC-18). Endocr Res 13: 229–241, 1987. [DOI] [PubMed] [Google Scholar]
- 16.Buchan AM, Barber DL, Gregor M, Soll AH. Morphologic and physiologic studies of canine ileal enteroglucagon-containing cells in short-term culture. Gastroenterology 93: 791–800, 1987. [DOI] [PubMed] [Google Scholar]
- 17.Burmeister MA, Ferre T, Ayala JE, King EM, Holt RM, Ayala JE. Acute activation of central GLP-1 receptors enhances hepatic insulin action and insulin secretion in high-fat-fed, insulin resistant mice. Am J Physiol Endocrinol Metab 302: E334–E343, 2012. [DOI] [PubMed] [Google Scholar]
- 18.Burmeister MA, Ayala JE, Smouse H, Landivar-Rocha A, Brown JD, Drucker DJ, Stoffers DA, Sandoval DA, Seeley RJ, Ayala JE. The hypothalamic glucagon-like peptide 1 receptor is sufficient but not necessary for the regulation of energy balance and glucose homeostasis in mice. Diabetes 66: 372–384, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Buse JB, Nauck M, Forst T, Sheu WH-H, Shenouda SK, Heilmann CR, Hoogwerf BJ, Gao A, Boardman MK, Fineman M, Porter L, Schernthaner G. Exenatide once weekly versus liraglutide once daily in patients with type 2 diabetes (DURATION-6): A randomised, open-label study. Lancet 381: 117–124, 2013. [DOI] [PubMed] [Google Scholar]
- 20.Campbell JE, Drucker DJ. Pharmacology, physiology, and mechanisms of incretin hormone action. Cell Metab 17: 819–837, 2013. [DOI] [PubMed] [Google Scholar]
- 21.Campos RV, Lee YC, Drucker DJ. Divergent tissue-specific and developmental expression of receptors for glucagon and glucagon-like peptide-1 in the mouse. Endocrinology 134: 2156–2164, 1994. [DOI] [PubMed] [Google Scholar]
- 22.Capozzi ME, Svendsen B, Encisco SE, Lewandowski SL, Martin MD, Lin H, Jaffe JL, Coch RW, Haldeman JM, MacDonald PE, Merrins MJ, D’Alessio DA, Campbell JE. β Cell tone is defined by proglucagon peptides through cAMP signaling. JCI Insight 4, 2019. DOI: 10.1172/jci.insight.126742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Capristo E, Panunzi S, De Gaetano A, Spuntarelli V, Bellantone R, Giustacchini P, Birkenfeld AL, Amiel S, Bornstein SR, Raffaelli M, Mingrone G. Incidence of hypoglycemia after gastric bypass versus sleeve gastrectomy: A randomized trial. J Clin Endocrinol Metab 103: 2136–2146, 2018. [DOI] [PubMed] [Google Scholar]
- 24.Cavin J-B, Couvelard A, Lebtahi R, Ducroc R, Arapis K, Voitellier E, Cluzeaud F, Gillard L, Hourseau M, Mikail N, Ribeiro-Parenti L, Kapel N, Marmuse J-P, Bado A, Le Gall M. Differences in alimentary glucose absorption and intestinal disposal of blood glucose following Roux-en-Y gastric bypass vs sleeve gastrectomy. Gastroenterology, 2016. DOI: 10.1053/j.gastro.2015.10.009. [DOI] [PubMed] [Google Scholar]
- 25.Chambers AP, Jessen L, Ryan KK, Sisley S, Wilson-Perez HE, Stefater MA, Gaitonde SG, Sorrell JE, Toure M, Berger J, D’Alessio DA, Woods SC, Seeley RJ, Sandoval DA. Weight-independent changes in blood glucose homeostasis after gastric bypass or vertical sleeve gastrectomy in rats. Gastroenterology 141: 950–958, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chambers AP, Smith EP, Begg DP, Grayson BE, Sisley S, Greer T, Sorrell J, Lemmen L, Lasance K, Woods SC, Seeley RJ, D’Alessio DA, Sandoval DA. Regulation of gastric emptying rate and its role in nutrient-induced GLP-1 secretion in rats after vertical sleeve gastrectomy. Am J Physiol Endocrinol Metab 306: E424–E432, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chambers AP, Sorrell JE, Haller A, Roelofs K, Hutch CR, Kim K-S, Gutierrez-Aguilar R, Li B, Drucker DJ, D’Alessio DA, Seeley RJ, Sandoval DA. The role of pancreatic preproglucagon in glucose homeostasis in mice. Cell Metab 25: 927–934, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chiang Y-T, Ip W, Jin T. The role of the Wnt signaling pathway in incretin hormone production and function. Front Physiol 3: 273, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chu Z-L, Carroll C, Alfonso J, Gutierrez V, He H, Lucman A, Pedraza M, Mondala H, Gao H, Bagnol D, Chen R, Jones RM, Behan DP, Leonard J. A role for intestinal endocrine cell-expressed g protein-coupled receptor 119 in glycemic control by enhancing glucagon-like Peptide-1 and glucose-dependent insulinotropic Peptide release. Endocrinology 149: 2038–2047, 2008. [DOI] [PubMed] [Google Scholar]
- 30.Conarello SL, Jiang G, Mu J, Li Z, Woods J, Zycband E, Ronan J, Liu F, Roy RS, Zhu L, Charron MJ, Zhang BB. Glucagon receptor knockout mice are resistant to diet-induced obesity and streptozotocin-mediated beta cell loss and hyperglycaemia. Diabetologia 50: 142–150, 2007. [DOI] [PubMed] [Google Scholar]
- 31.Cox AR, Lam CJ, Rankin MM, Rios JS, Chavez J, Bonnyman CW, King KB, Wells RA, Anthony D, Tu JX, Kim JJ, Li C, Kushner JA. Incretin therapies do not expand β-cell mass or alter pancreatic histology in young male mice. Endocrinology 158: 1701–1714, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Craig CM, Liu L-F, Deacon CF, Holst JJ, McLaughlin TL. Critical role for GLP-1 in symptomatic post-bariatric hypoglycaemia. Diabetologia 60: 531–540, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Craig CM, Liu L, Nguyen T, Price C, Bingham J, McLaughlin TL. Efficacy and pharmacokinetics of subcutaneous exendin (9–39) in patients with post-bariatric hypoglycaemia. Diabetes Obes Metab 20: 352–361, 2018. [DOI] [PubMed] [Google Scholar]
- 34.Creutzfeldt WOC, Kleine N, Willms B, Orskov C, Holst JJ, Nauck MA. Glucagonostatic actions and reduction of fasting hyperglycemia by exogenous glucagon-like peptide I(7–36) amide in type I diabetic patients. Diabetes Care 19: 580–586, 1996. [DOI] [PubMed] [Google Scholar]
- 35.D’Alessio D, Lu W, Sun W, Zheng S, Yang Q, Seeley R, Woods SC, Tso P. Fasting and postprandial concentrations of GLP-1 in intestinal lymph and portal plasma: Evidence for selective release of GLP-1 in the lymph system. Am J Physiol Regul Integr Comp Physiol 293: R2163–R2169, 2007. [DOI] [PubMed] [Google Scholar]
- 36.Dai C, Hang Y, Shostak A, Poffenberger G, Hart N, Prasad N, Phillips N, Levy SE, Greiner DL, Shultz LD, Bottino R, Kim SK, Powers AC. Age-dependent human β cell proliferation induced by glucagon-like peptide 1 and calcineurin signaling. J Clin Invest 127: 3835–3844, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Day JW, Gelfanov V, Smiley D, Carrington PE, Eiermann G, Chicchi G, Erion MD, Gidda J, Thornberry NA, Tschöp MH, Marsh DJ, SinhaRoy R, DiMarchi R, Pocai A. Optimization of co-agonism at GLP-1 and glucagon receptors to safely maximize weight reduction in DIO-rodents. Biopolymers 98: 443–450, 2012. [DOI] [PubMed] [Google Scholar]
- 38.Day JW, Ottaway N, Patterson JT, Gelfanov V, Smiley D, Gidda J, Findeisen H, Bruemmer D, Drucker DJ, Chaudhary N, Holland J, Hembree J, Abplanalp W, Grant E, Ruehl J, Wilson H, Kirchner H, Lockie SH, Hofmann S, Woods SC, Nogueiras R, Pfluger PT, Perez-Tilve D, DiMarchi R, Tschöp MH. A new glucagon and GLP-1 co-agonist eliminates obesity in rodents. Nat Chem Biol 5: 749–757, 2009. [DOI] [PubMed] [Google Scholar]
- 39.Deacon CF, Nauck MA, Toft-Nielsen M, Pridal L, Willms B, Holst JJ. Both subcutaneously and intravenously administered glucagon-like peptide I are rapidly degraded from the NH2-terminus in type II diabetic patients and in healthy subjects. Diabetes 44: 1126–1131, 1995. [DOI] [PubMed] [Google Scholar]
- 40.Dimick JB, Nicholas LH, Ryan AM, Thumma JR, Birkmeyer JD. Bariatric surgery complications before vs after implementation of a national policy restricting coverage to centers of excellence. JAMA 309: 792–799, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ding L, Sousa KM, Jin L, Dong B, Kim B-W, Ramirez R, Xiao Z, Gu Y, Yang Q, Wang J, Yu D, Pigazzi A, Schones D, Yang L, Moore D, Wang Z, Huang W. Vertical sleeve gastrectomy activates GPBAR-1/TGR5 to sustain weight loss, improve fatty liver, and remit insulin resistance in mice. Hepatology 64: 760–773, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ding X, Saxena NK, Lin S, Gupta NA, Gupta N, Anania FA. Exendin-4, a glucagon-like protein-1 (GLP-1) receptor agonist, reverses hepatic steatosis in ob/ob mice. Hepatology 43: 173–181, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dirksen C, Bojsen-Møller KN, Jørgensen NB, Jacobsen SH, Kristiansen VB, Naver LS, Hansen DL, Worm D, Holst JJ, Madsbad S. Exaggerated release and preserved insulinotropic action of glucagon-like peptide-1 underlie insulin hypersecretion in glucose-tolerant individuals after Roux-en-Y gastric bypass. Diabetologia 56: 2679–2687, 2013. [DOI] [PubMed] [Google Scholar]
- 44.Douros JD, Lewis AG, Smith EP, Niu J, Capozzi M, Wittmann A, Campbell J, Tong J, Wagner C, Mahbod P, Seeley R, D’Alessio DA. Enhanced glucose control following vertical sleeve gastrectomy does not require a β-cell glucagon-like peptide 1 receptor. Diabetes 67: 1504–1511, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Douros JD, Niu J, Sdao SM, Gregg T, Fisher-Wellman KH, Bharadwaj MS, Molina A, Arumugam R, Martin MD, Petretto E, Merrins MJ, Herman MA, Tong J, Campbell JE, D’Alessio D. Sleeve gastrectomy rapidly enhances islet function independently of body weight. JCI Insight 4: 126688, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Drucker DJ. Dipeptidyl peptidase-4 inhibition and the treatment of type 2 diabetes: Preclinical biology and mechanisms of action. Diabetes Care 30: 1335–1343, 2007. [DOI] [PubMed] [Google Scholar]
- 47.Drucker DJ. Incretin action in the pancreas: Potential promise, possible perils, and pathological pitfalls. Diabetes 62: 3316–3323, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Drucker DJ. The ascending GLP-1 road from clinical safety to reduction of cardiovascular complications. Diabetes 67: 1710–1719, 2018. [DOI] [PubMed] [Google Scholar]
- 49.Drucker DJ, Brubaker PL. Proglucagon gene expression is regulated by a cyclic AMP-dependent pathway in rat intestine. Proc Natl Acad Sci U S A 86: 3953–3957, 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Drucker DJ, Buse JB, Taylor K, Kendall DM, Trautmann M, Zhuang D, Porter L. Exenatide once weekly versus twice daily for the treatment of type 2 diabetes: A randomised, open-label, non-inferiority study. Lancet 372: 1240–1250, 2008. [DOI] [PubMed] [Google Scholar]
- 51.Drucker DJ, Nauck MA. The incretin system: Glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet 368: 1696–1705, 2006. [DOI] [PubMed] [Google Scholar]
- 52.Dumoulin V, Dakka T, Plaisancie P, Chayvialle JA, Cuber JC. Regulation of glucagon-like peptide-1-(7–36) amide, peptide YY, and neurotensin secretion by neurotransmitters and gut hormones in the isolated vascularly perfused rat ileum. Endocrinology 136: 5182–5188, 1995. [DOI] [PubMed] [Google Scholar]
- 53.Dunning BE, Foley JE, Ahren B. Alpha cell function in health and disease: Influence of glucagon-like peptide-1. Diabetologia 48: 1700–1713, 2005. [DOI] [PubMed] [Google Scholar]
- 54.Dupre J, Ross SA, Watson D, Brown JC. Stimulation of insulin secretion by gastric inhibitory peptide in man. J Clin Endocrinol Metab 37: 826–828, 1973. [DOI] [PubMed] [Google Scholar]
- 55.Dutia R, Embrey M, O’Brien S, Haeusler RA, Agénor KK, Homel P, McGinty J, Vincent RP, Alaghband-Zadeh J, Staels B, le Roux CW, Yu J, Laferrère B. Temporal changes in bile acid levels and 12α-hydroxylation after Roux-en-Y gastric bypass surgery in type 2 diabetes. Int J Obes (Lond) 39: 806–813, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ebert R, Unger H, Creutzfeldt W. Preservation of incretin activity after removal of gastric inhibitory polypeptide (GIP) from rat gut extracts by immunoadsorption. Diabetologia 24: 449–454, 1983. [DOI] [PubMed] [Google Scholar]
- 57.Edfalk S, Steneberg P, Edlund H. Gpr40 is expressed in enteroendocrine cells and mediates free fatty acid stimulation of incretin secretion. Diabetes 57: 2280–2287, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Eissele R, Göke R, Willemer S, Harthus HP, Vermeer H, Arnold R, Göke B. Glucagon-like peptide-1 cells in the gastrointestinal tract and pancreas of rat, pig and man. Eur J Clin Invest 22: 283–291, 1992. [DOI] [PubMed] [Google Scholar]
- 59.Ellingsgaard H, Ehses JA, Hammar EB, Van Lommel L, Quintens R, Martens G, Kerr-Conte J, Pattou F, Berney T, Pipeleers D, Halban PA, Schuit FC, Donath MY. Interleukin-6 regulates pancreatic alpha-cell mass expansion. Proc Natl Acad Sci U S A 105: 13163–13168, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ellingsgaard H, Hauselmann I, Schuler B, Habib AM, Baggio LL, Meier DT, Eppler E, Bouzakri K, Wueest S, Muller YD, Hansen AMK, Reinecke M, Konrad D, Gassmann M, Reimann F, Halban PA, Gromada J, Drucker DJ, Gribble FM, Ehses JA, Donath MY. Interleukin-6 enhances insulin secretion by increasing glucagon-like peptide-1 secretion from L cells and alpha cells. Nat Med 17: 1481–1489, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Elrick H, Stimmer L, Hlad CJ, Arai Y. Plasma insulin response to oral and intravenous glucose administration. J Clin Endocrinol Metab 24: 1076–1082, 1964. [DOI] [PubMed] [Google Scholar]
- 62.Falken Y, Hellstrom PM, Holst JJ, Naslund E. Changes in glucose homeostasis after Roux-en-Y gastric bypass surgery for obesity at day three, two months, and one year after surgery: Role of gut peptides. J Clin Endocrinol Metab 96: 2227–2235, 2011. [DOI] [PubMed] [Google Scholar]
- 63.Ferraris RP, Yasharpour S, Lloyd KC, Mirzayan R, Diamond JM. Luminal glucose concentrations in the gut under normal conditions. Am J Physiol 259: G822–G837, 1990. [DOI] [PubMed] [Google Scholar]
- 64.Fineman M, Weyer C, Maggs DG, Strobel S, Kolterman OG. The human amylin analog, pramlintide, reduces postprandial hyper-glucagonemia in patients with type 2 diabetes mellitus. Horm Metab Res 34: 504–508, 2002. [DOI] [PubMed] [Google Scholar]
- 65.Flannick J, Thorleifsson G, Beer NL, Jacobs SBR, Grarup N, Burtt NP, Mahajan A, Fuchsberger C, Atzmon G, Benediktsson R, Blangero J, Bowden DW, Brandslund I, Brosnan J, Burslem F, Chambers J, Cho YS, Christensen C, Douglas DA, Duggirala R, Dymek Z, Farjoun Y, Fennell T, Fontanillas P, Forsén T, Gabriel S, Glaser B, Gudbjartsson DF, Hanis C, Hansen T, Hreidarsson AB, Hveem K, Ingelsson E, Isomaa B, Johansson S, Jørgensen T, Jørgensen ME, Kathiresan S, Kong A, Kooner J, Kravic J, Laakso M, Lee J-Y, Lind L, Lindgren CM, Linneberg A, Masson G, Meitinger T, Mohlke KL, Molven A, Morris AP, Potluri S, Rauramaa R, Ribel-Madsen R, Richard A-M, Rolph T, Salomaa V, Segrè AV, Skärstrand H, Steinthorsdottir V, Stringham HM, Sulem P, Tai ES, Teo YY, Teslovich T, Thorsteinsdottir U, Trimmer JK, Tuomi T, Tuomilehto J, Vaziri-Sani F, Voight BF, Wilson JG, Boehnke M, McCarthy MI, Njølstad PR, Pedersen O, Groop L, Cox DR, Stefansson K, Altshuler D, Stefansson K, Altshuler D. Loss-of-function mutations in SLC30A8 protect against type 2 diabetes. Nat Genet 46: 357–363, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Flock G, Baggio LL, Longuet C, Drucker DJ. Incretin receptors for glucagon-like peptide 1 and glucose-dependent insulinotropic polypep-tide are essential for the sustained metabolic actions of vildagliptin in mice. Diabetes 56: 3006–3013, 2007. [DOI] [PubMed] [Google Scholar]
- 67.Frias JP, Nauck MA, Van J, Kutner ME, Cui X, Benson C, Urva S, Gimeno RE, Milicevic Z, Robins D, Haupt A. Efficacy and safety of LY3298176, a novel dual GIP and GLP-1 receptor agonist, in patients with type 2 diabetes: A randomised, placebo-controlled and active comparator-controlled phase 2 trial. Lancet 392: 2180–2193, 2018. [DOI] [PubMed] [Google Scholar]
- 68.Fusco J, Xiao X, Prasadan K, Sheng Q, Chen C, Ming Y-C, Gittes G. GLP-1/Exendin-4 induces β-cell proliferation via the epidermal growth factor receptor. Sci Rep 7: 9100, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Garibay D, McGavigan AK, Lee SA, Ficorilli JV, Cox AL, Michael MD, Sloop KW, Cummings BP. β-Cell glucagon-like peptide-1 receptor contributes to improved glucose tolerance after vertical sleeve gastrectomy. Endocrinology 157: 3405–3409, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gedulin BR, Jodka CM, Herrmann K, Young AA. Role of endogenous amylin in glucagon secretion and gastric emptying in rats demonstrated with the selective antagonist, AC187. Regul Pept 137: 121–127, 2006. [DOI] [PubMed] [Google Scholar]
- 71.Gedulin BR, Rink TJ, Young AA. Dose-response for glucagonostatic effect of amylin in rats. Metabolism 46: 67–70, 1997. [DOI] [PubMed] [Google Scholar]
- 72.Gerich JE, Langlois M, Schneider V, Karam JH, Noacco C. Effects of alterations of plasma free fatty acid levels on pancreatic glucagon secretion in man. J Clin Invest 53: 1284–1289, 1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gevrey J-C, Malapel M, Philippe J, Mithieux G, Chayvialle J, Abello J, Cordier-Bussat M. Protein hydrolysates stimulate proglucagon gene transcription in intestinal endocrine cells via two elements related to cyclic AMP response element. Diabetologia 47: 926–936, 2004. [DOI] [PubMed] [Google Scholar]
- 74.Goke R, Larsen PJ, Mikkelsen JD, Sheikh SP. Distribution of GLP-1 binding sites in the rat brain: Evidence that exendin-4 is a ligand of brain GLP-1 binding sites. Eur J Neurosci 7: 2294–2300, 1995. [DOI] [PubMed] [Google Scholar]
- 75.Goldfine AB, Mun EC, Devine E, Bernier R, Baz-Hecht M, Jones DB, Schneider BE, Holst JJ, Patti ME. Patients with neuroglycopenia after gastric bypass surgery have exaggerated incretin and insulin secretory responses to a mixed meal. J Clin Endocrinol Metab 92: 4678–4685, 2007. [DOI] [PubMed] [Google Scholar]
- 76.Goldspink DA, Lu VB, Billing LJ, Larraufie P, Tolhurst G, Gribble FM, Reimann F. Mechanistic insights into the detection of free fatty and bile acids by ileal glucagon-like peptide-1 secreting cells. Mol Metab 7: 90–101, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gromada J, Franklin I, Wollheim CB. Alpha-cells of the endocrine pancreas: 35 years of research but the enigma remains. Endocr Rev 28: 84–116, 2007. [DOI] [PubMed] [Google Scholar]
- 78.Gupta NA, Mells J, Dunham RM, Grakoui A, Handy J, Saxena NK, Anania FA. Glucagon-like peptide-1 receptor is present on human hepatocytes and has a direct role in decreasing hepatic steatosis in vitro by modulating elements of the insulin signaling pathway. Hepatology 51: 1584–1592, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gyulkhandanyan AV, Lu H, Lee SC, Bhattacharjee A, Wijesekara N, Fox JEM, MacDonald PE, Chimienti F, Dai FF, Wheeler MB. Investigation of transport mechanisms and regulation of intracellular Zn2+ in pancreatic alpha-cells. J Biol Chem 283: 10184–10197, 2008. [DOI] [PubMed] [Google Scholar]
- 80.Habegger KM, Heppner KM, Amburgy SE, Ottaway N, Holland J, Raver C, Bartley E, Müller TD, Pfluger PT, Berger J, Toure M, Benoit SC, Dimarchi RD, Perez-Tilve D, D’Alessio DA, Seeley RJ, Tschöp MH. GLP-1R responsiveness predicts individual gastric bypass efficacy on glucose tolerance in rats. Diabetes, 2014. DOI: 10.2337/db13-0511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Han VKM, Hynes MA, Jin C, Towle AC, Lauder JM, Lund PK. Cellular localization of proglucagon/glucagon-like peptide 1 messenger RNAs in rat brain. J Neurosci 16: 97–107, 1986. [DOI] [PubMed] [Google Scholar]
- 82.Hansen L, Deacon CF, Ørskov C, Holst JJ. Glucagon-like peptide-1-(7–36) amide is transformed to glucagon-like peptide-1-(9–36) amide by dipeptidyl peptidase IV in the capillaries supplying the L cells of the porcine intestine. Endocrinology 140: 5356–5363, 1999. [DOI] [PubMed] [Google Scholar]
- 83.Hansen L, Holst JJ. The effects of duodenal peptides on glucagon-like peptide-1 secretion from the ileum. A duodeno–ileal loop? Regul Pept 110: 39–45, 2002. [DOI] [PubMed] [Google Scholar]
- 84.Hansen L, Lampert S, Mineo H, Holst JJ. Neural regulation of glucagon-like peptide-1 secretion in pigs. Am J Physiol Endocrinol Metab 287: E939–E947, 2004. [DOI] [PubMed] [Google Scholar]
- 85.Hansotia T, Baggio LL, Delmeire D, Hinke SA, Yamada Y, Tsukiyama K, Seino Y, Holst JJ, Schuit F, Drucker DJ. Double incretin receptor knockout (DIRKO) mice reveal an essential role for the enteroinsular axis in transducing the glucoregulatory actions of DPP-IV inhibitors. Diabetes 53: 1326–1335, 2004. [DOI] [PubMed] [Google Scholar]
- 86.Hare KJ, Vilsbøll T, Asmar M, Deacon CF, Knop FK, Holst JJ. The glucagonostatic and insulinotropic effects of glucagon-like peptide 1 contribute equally to its glucose-lowering action. Diabetes 59: 1765–1770, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hauge M, Ekberg JP, Engelstoft MS, Timshel P, Madsen AN, Schwartz TW. Gq and Gs signaling acting in synergy to control GLP-1 secretion. Mol Cell Endocrinol 449: 64–73, 2017. [DOI] [PubMed] [Google Scholar]
- 88.Hayes MR, Leichner TM, Zhao S, Lee GS, Chowansky A, Zimmer D, De Jonghe BC, Kanoski SE, Grill HJ, Bence KK. Intracellular signals mediating the food intake-suppressive effects of hindbrain glucagon-like peptide-1 receptor activation. Cell Metab 13: 320–330, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.de Heer J, Rasmussen C, Coy DH, Holst JJ. Glucagon-like peptide-1, but not glucose-dependent insulinotropic peptide, inhibits glucagon secretion via somatostatin (receptor subtype 2) in the perfused rat pancreas. Diabetologia 51: 2263–2270, 2008. [DOI] [PubMed] [Google Scholar]
- 90.Hira T, Mochida T, Miyashita K, Hara H. GLP-1 secretion is enhanced directly in the ileum but indirectly in the duodenum by a newly identified potent stimulator, zein hydrolysate, in rats. Am J Physiol Gastroin-test Liver Physiol 297: G663–G671, 2009. [DOI] [PubMed] [Google Scholar]
- 91.Hirasawa A, Tsumaya K, Awaji T, Katsuma S, Adachi T, Yamada M, Sugimoto Y, Miyazaki S, Tsujimoto G. Free fatty acids regulate gut incretin glucagon-like peptide-1 secretion through GPR120. Nat Med 11: 90–94, 2005. [DOI] [PubMed] [Google Scholar]
- 92.Holst JJ, Bersani M, Johnsen AH, Kofod H, Hartmann B, Orskov C. Proglucagon processing in porcine and human pancreas. J Biol Chem 269: 18827–18833, 1994. [PubMed] [Google Scholar]
- 93.Holst JJ, Deacon CF. Glucagon-like peptide-1 mediates the therapeutic actions of DPP-IV inhibitors. Diabetologia 48: 612–615, 2005. [DOI] [PubMed] [Google Scholar]
- 94.Holz GG. Epac: A new cAMP-binding protein in support of glucagon-like peptide-1 receptor-mediated signal transduction in the pancreatic beta-cell. Diabetes 53: 5–13, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Honka H, Koffert J, Hannukainen JC, Tuulari JJ, Karlsson HK, Immonen H, Oikonen V, Tolvanen T, Soinio M, Salminen P, Kudomi N, Mari A, Iozzo P, Nuutila P. The effects of bariatric surgery on pancreatic lipid metabolism and blood flow. J Clin Endocrinol Metab 100: 2015–2023, 2015. [DOI] [PubMed] [Google Scholar]
- 96.Hudson BD, Smith NJ, Milligan G. Experimental challenges to targeting poorly characterized GPCRs: Uncovering the therapeutic potential for free fatty acid receptors. Adv Pharmacol 62: 175–218, 2011. [DOI] [PubMed] [Google Scholar]
- 97.Hui H, Nourparvar A, Zhao X, Perfetti R. Glucagon-like peptide-1 inhibits apoptosis of insulin-secreting cells via a cyclic 5′-adenosine monophosphate-dependent protein kinase A- and a phosphatidylinositol 3-kinase-dependent pathway. Endocrinology 144: 1444–1455, 2003. [DOI] [PubMed] [Google Scholar]
- 98.Hurren KM, Pinelli NR. Drug-drug interactions with glucagon-like peptide-1 receptor agonists. Ann Pharmacother 46: 710–717, 2012. [DOI] [PubMed] [Google Scholar]
- 99.Ichimura A, Hirasawa A, Poulain-Godefroy O, Bonnefond A, Hara T, Yengo L, Kimura I, Leloire A, Liu N, Iida K, Choquet H, Besnard P, Lecoeur C, Vivequin S, Ayukawa K, Takeuchi M, Ozawa K, Tauber M, Maffeis C, Morandi A, Buzzetti R, Elliott P, Pouta A, Jarvelin M-R, Körner A, Kiess W, Pigeyre M, Caiazzo R, Van Hul W, Van Gaal L, Horber F, Balkau B, Lévy-Marchal C, Rouskas K, Kouvatsi A, Hebebrand J, Hinney A, Scherag A, Pattou F, Meyre D, Koshimizu T, Wolowczuk I, Tsujimoto G, Froguel P. Dysfunction of lipid sensor GPR120 leads to obesity in both mouse and human. Nature 483: 350–354, 2012. [DOI] [PubMed] [Google Scholar]
- 100.Ishihara H, Maechler P, Gjinovci A, Herrera P-L, Wollheim CB. Islet beta-cell secretion determines glucagon release from neighbouring alpha-cells. Nat Cell Biol 5: 330–335, 2003. [DOI] [PubMed] [Google Scholar]
- 101.Islam D, Zhang N, Wang P, Li H, Brubaker PL, Gaisano HY, Wang Q, Jin T. Epac is involved in cAMP-stimulated proglucagon expression and hormone production but not hormone secretion in pancreatic alpha- and intestinal L-cell lines. Am J Physiol Endocrinol Metab 296: E174–E181, 2009. [DOI] [PubMed] [Google Scholar]
- 102.Jang H-J, Kokrashvili Z, Theodorakis MJ, Carlson OD, Kim B-J, Zhou J, Kim HH, Xu X, Chan SL, Juhaszova M, Bernier M, Mosinger B, Margolskee RF, Egan JM. Gut-expressed gustducin and taste receptors regulate secretion of glucagon-like peptide-1. Proc Natl Acad Sci U S A 104: 15069–15074, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Jensen PB, Blume N, Mikkelsen JD, Larsen PJ, Jensen HI, Holst JJ, Madsen OD. Transplantable rat glucagonomas cause acute onset of severe anorexia and adipsia despite highly elevated NPY mRNA levels in the hypothalamic arcuate nucleus. J Clin Invest 101: 503–510, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Jessen L, Smith EP, Ulrich-Lai Y, Herman JP, Seeley RJ, Sandoval D, D’Alessio D. Central nervous system GLP-1 receptors regulate islet hormone secretion and glucose homeostasis in male rats. Endocrinology 158: 2124–2133, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Jiménez A, Casamitjana R, Viaplana-Masclans J, Lacy A, Vidal J. GLP-1 action and glucose tolerance in subjects with remission of type 2 diabetes after gastric bypass surgery. Diabetes Care 36: 2062–2069, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Jiménez A, Ceriello A, Casamitjana R, Flores L, Viaplana-Masclans J, Vidal J. Remission of type 2 diabetes after Roux-en-Y gastric bypass or sleeve gastrectomy is associated with a distinct glycemic profile. Ann Surg 261: 316–322, 2015. [DOI] [PubMed] [Google Scholar]
- 107.Jiménez A, Mari A, Casamitjana R, Lacy A, Ferrannini E, Vidal J. GLP-1 and glucose tolerance after sleeve gastrectomy in morbidly obese subjects with type 2 diabetes. Diabetes 63: 3372–3377, 2014. [DOI] [PubMed] [Google Scholar]
- 108.Jin SL, Han VKM, Simmons JG, Towle AC, Lauder JM, Lund PK. Distribution of glucagonlike peptide 1, glucagon, and glicentin in the rat brain: An immunocytochemical study. J Comp Neurol 271: 519–532, 1988. [DOI] [PubMed] [Google Scholar]
- 109.Jin T Mechanisms underlying proglucagon gene expression. J Endocrinol 198: 17–28, 2008. [DOI] [PubMed] [Google Scholar]
- 110.Jørgensen NB, Dirksen C, Bojsen-Møller KN, Jacobsen SH, Worm D, Hansen DL, Kristiansen VB, Naver L, Madsbad S, Holst JJ. Exaggerated glucagon-like peptide 1 response is important for improved β-cell function and glucose tolerance after Roux-en-Y gastric bypass in patients with type 2 diabetes. Diabetes 62: 3044–3052, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kahles F, Meyer C, Möllmann J, Diebold S, Findeisen HM, Lebherz C, Trautwein C, Koch A, Tacke F, Marx N, Lehrke M. GLP-1 secretion is increased by inflammatory stimuli in an IL-6-dependent manner, leading to hyperinsulinemia and blood glucose lowering. Diabetes 63: 3221–3229, 2014. [DOI] [PubMed] [Google Scholar]
- 112.Kang JH, Le QA. Effectiveness of bariatric surgical procedures: A systematic review and network meta-analysis of randomized controlled trials. Medicine (Baltimore) 96: e8632, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kanoski SE, Rupprecht LE, Fortin SM, De Jonghe BC, Hayes MR. The role of nausea in food intake and body weight suppression by peripheral GLP-1 receptor agonists, exendin-4 and liraglutide. Neuropharmacology 62: 1916–1927, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kapodistria K, Tsilibary E-P, Kotsopoulou E, Moustardas P, Kitsiou P. Liraglutide, a human glucagon-like peptide-1 analogue, stimulates AKT-dependent survival signalling and inhibits pancreatic β-cell apoptosis. J Cell Mol Med 22: 2970–2980, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Katsuma S, Hirasawa A, Tsujimoto G. Bile acids promote glucagon-like peptide-1 secretion through TGR5 in a murine enteroendocrine cell line STC-1. Biochem Biophys Res Commun 329: 386–390, 2005. [DOI] [PubMed] [Google Scholar]
- 116.Katz LB, Gambale JJ, Rothenberg PL, Vanapalli SR, Vaccaro N, Xi L, Sarich TC, Stein PP. Effects of JNJ-38431055, a novel GPR119 receptor agonist, in randomized, double-blind, placebo-controlled studies in subjects with type 2 diabetes. Diabetes Obes Metab 14: 709–716, 2012. [DOI] [PubMed] [Google Scholar]
- 117.Kielgast U, Asmar M, Madsbad S, Holst JJ. Effect of glucagon-like peptide-1 on alpha- and beta-cell function in C-peptide-negative type 1 diabetic patients. J Clin Endocrinol Metab 95: 2492–2496, 2010. [DOI] [PubMed] [Google Scholar]
- 118.Kilimnik G, Kim A, Steiner DF, Friedman TC, Hara M. Intraislet production of GLP-1 by activation of prohormone convertase 1/3 in pancreatic α-cells in mouse models of ß-cell regeneration. Islets 2: 149–155, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kim K-S, Seeley RJ, Sandoval DA. Signalling from the periphery to the brain that regulates energy homeostasis. Nat Rev Neurosci 19: 185–196, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kindel TL, Yang Q, Yoder SM, Tso P. Nutrient-driven incretin secretion into intestinal lymph is different between diabetic Goto-Kakizaki rats and Wistar rats. Am J Physiol Gastrointest Liver Physiol 296: G168–G174, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kinzig KP, D’Alessio DA, Herman JP, Sakai RR, Vahl TP, Figueiredo HF, Murphy EK, Seeley RJ. CNS glucagon-like peptide-1 receptors mediate endocrine and anxiety responses to interoceptive and psychogenic stressors. J Neurosci 23: 6163–6170, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Knauf C, Cani PD, Perrin C, Iglesias MA, Maury JF, Bernard E, Benhamed F, Gremeaux T, Drucker DJ, Kahn CR, Girard J, Tanti JF, Delzenne NM, Postic C, Burcelin R. Brain glucagon-like peptide-1 increases insulin secretion and muscle insulin resistance to favor hepatic glycogen storage. J Clin Invest 115: 3554–3563, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Knop FK, Aaboe K, Vilsbøll T, Vølund A, Holst JJ, Krarup T, Madsbad S. Impaired incretin effect and fasting hyperglucagonaemia characterizing type 2 diabetic subjects are early signs of dysmetabolism in obesity. Diabetes Obes Metab 14: 500–510, 2012. [DOI] [PubMed] [Google Scholar]
- 124.Kohli R, Bradley D, Setchell KD, Eagon JC, Abumrad N, Klein S. Weight loss induced by Roux-en-Y gastric bypass but not laparoscopic adjustable gastric banding increases circulating bile acids. J Clin Endocrinol Metab 98: E708–E712, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Koopmann MC, Nelson DW, Murali SG, Liu X, Brownfield MS, Holst JJ, Ney DM. Exogenous glucagon-like peptide-2 (GLP-2) augments GLP-2 receptor mRNA and maintains proglucagon mRNA levels in resected rats. JPEN J Parenter Enteral Nutr 32: 254–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kreymann B, Ghatei MA, Domin J, Kanse S, Bloom SR. Developmental patterns of glucagon-like peptide-1-(7–36) amide and peptide-YY in rat pancreas and gut. Endocrinology 129: 1001–1005, 1991. [DOI] [PubMed] [Google Scholar]
- 127.Kreymann B, Ghatei MA, Williams G, Bloom SR. Glucagon-like peptide-1 7–36: A physiological incretin in man. Lancet 2: 1300–1303, 1987. [DOI] [PubMed] [Google Scholar]
- 128.Kuhre RE, Frost CR, Svendsen B, Holst JJ. Molecular mechanisms of glucose-stimulated GLP-1 secretion from perfused rat small intestine. Diabetes 64: 370–382, 2015. [DOI] [PubMed] [Google Scholar]
- 129.Lachey JL, D’Alessio DA, Rinaman L, Elmquist JK, Drucker DJ, Seeley RJ. The role of central glucagon-like peptide-1 in mediating the effects of visceral illness: Differential effects in rats and mice. Endocrinology 146: 458–462, 2005. [DOI] [PubMed] [Google Scholar]
- 130.Laferrere B, Heshka S, Wang K, Khan Y, McGinty J, Teixeira J, Hart AB, Olivan B. Incretin levels and effect are markedly enhanced 1 month after Roux-en-Y gastric bypass surgery in obese patients with type 2 diabetes. Diabetes Care 30: 1709–1716, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lamont BJ, Li Y, Kwan E, Brown TJ, Gaisano H, Drucker DJ. Pancreatic GLP-1 receptor activation is sufficient for incretin control of glucose metabolism in mice. J Clin Invest 122: 388–402, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Lan H, Vassileva G, Corona A, Liu L, Baker H, Golovko A, Abbondanzo SJ, Hu W, Yang S, Ning Y, Del Vecchio RA, Poulet F, Laverty M, Gustafson EL, Hedrick JA, Kowalski TJ. GPR119 is required for physiological regulation of glucagon-like peptide-1 secretion but not for metabolic homeostasis. J Endocrinol 201: 219–230, 2009. [DOI] [PubMed] [Google Scholar]
- 133.Langlois A, Dal S, Vivot K, Mura C, Seyfritz E, Bietiger W, Dollinger C, Peronet C, Maillard E, Pinget M, Jeandidier N, Sigrist S. Improvement of islet graft function using liraglutide is correlated with its anti-inflammatory properties. Br J Pharmacol 173: 3443–3453, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Larsen PJ, Tang-Christensen M, Holst JJ, Orskov C. Distribution of glucagon-like peptide-1 and other preproglucagon-derived peptides in the rat hypothalamus and brainstem. Neuroscience 77: 257–270, 1997. [DOI] [PubMed] [Google Scholar]
- 135.Lee CJ, Wood GC, Lazo M, Brown TT, Clark JM, Still C, Benotti P. Risk of post-gastric bypass surgery hypoglycemia in nondiabetic individuals: A single center experience. Obesity 24: 1342–1348, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Light PE, Manning Fox JE, Riedel MJ, Wheeler MB. Glucagon-like peptide-1 inhibits pancreatic ATP-sensitive potassium channels via a protein kinase A- and ADP-dependent mechanism. Mol Endocrinol 16: 2135–2144, 2002. [DOI] [PubMed] [Google Scholar]
- 137.Lin HV, Wang J, Wang J, Li W, Wang X, Alston JT, Thomas MK, Briere DA, Syed SK, Efanov AM. GPR142 prompts glucagon-like Peptide-1 release from islets to improve β cell function. Mol Metab 11: 205–211, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Linnemann AK, Neuman JC, Battiola TJ, Wisinski JA, Kimple ME, Davis DB. Glucagon-like peptide-1 regulates cholecystokinin production in β-cells to protect from apoptosis. Mol Endocrinol 29: 978–987, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Liu Z, Habener JF. Glucagon-like peptide-1 activation of TCF7L2-dependent Wnt signaling enhances pancreatic beta cell proliferation. J Biol Chem 283: 8723–8735, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Lotfi S, Li Z, Sun J, Zuo Y, Lam PPL, Kang Y, Rahimi M, Islam D, Wang P, Gaisano HY, Jin T. Role of the exchange protein directly activated by cyclic adenosine 5′-monophosphate (Epac) pathway in regulating proglucagon gene expression in intestinal endocrine L cells. Endocrinology 147: 3727–3736, 2006. [DOI] [PubMed] [Google Scholar]
- 141.Lü F, Jin T, Drucker DJ. Proglucagon gene expression is induced by gastrin-releasing peptide in a mouse enteroendocrine cell line. Endocrinology 137: 3710–3716, 1996. [DOI] [PubMed] [Google Scholar]
- 142.Lund PK, Goodman RH, Dee PC, Habener JF. Pancreatic preproglucagon cDNA contains two glucagon-related coding sequences arranged in tandem. Proc Natl Acad Sci U S A 79: 345–349, 1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Luo J, Swaminath G, Brown SP, Zhang J, Guo Q, Chen M, Nguyen K, Tran T, Miao L, Dransfield PJ, Vimolratana M, Houze JB, Wong S, Toteva M, Shan B, Li F, Zhuang R, Lin DC-H. A potent class of GPR40 full agonists engages the enteroinsular axis to promote glucose control in rodents. PLoS One 7: e46300, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.MacDonald PE, Salapatek AMF, Wheeler MB. Glucagon-like peptide-1 receptor activation antagonizes voltage-dependent repolarizing K(+) currents in beta-cells: A possible glucose-dependent insulinotropic mechanism. Diabetes 51 (Suppl 3): S443–S447, 2002. [DOI] [PubMed] [Google Scholar]
- 145.Mace OJ, Schindler M, Patel S. The regulation of K- and L-cell activity by GLUT2 and the calcium-sensing receptor CasR in rat small intestine. J Physiol 590: 2917–2936, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Maersk M, Belza A, Holst JJ, Fenger-Grøn M, Pedersen SB, Astrup A, Richelsen B. Satiety scores and satiety hormone response after sucrose-sweetened soft drink compared with isocaloric semi-skimmed milk and with non-caloric soft drink: A controlled trial. Eur J Clin Nutr 66: 523–529, 2012. [DOI] [PubMed] [Google Scholar]
- 147.Le Marchand SJ, Piston DW. Glucose suppression of glucagon secretion: Metabolic and calcium responses from alpha-cells in intact mouse pancreatic islets. J Biol Chem 285: 14389–14398, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.De Marinis YZ, Salehi A, Ward CE, Zhang Q, Abdulkader F, Bengtsson M, Braha O, Braun M, Ramracheya R, Amisten S, Habib AM, Moritoh Y, Zhang E, Reimann F, Rosengren AH, Shibasaki T, Gribble F, Renström E, Seino S, Eliasson L, Rorsman P. GLP-1 inhibits and adrenaline stimulates glucagon release by differential modulation of N- and L-type Ca2+ channel-dependent exocytosis. Cell Metab 11: 543–553, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Marre M, Penfornis A. GLP-1 receptor agonists today. Diabetes Res Clin Pract 93: 317–327, 2011. [DOI] [PubMed] [Google Scholar]
- 150.Marsk R, Jonas E, Rasmussen F, Näslund E. Nationwide cohort study of post-gastric bypass hypoglycaemia including 5,040 patients undergoing surgery for obesity in 1986–2006 in Sweden. Diabetologia 53: 2307–2311, 2010. [DOI] [PubMed] [Google Scholar]
- 151.McGavigan AK, Garibay D, Henseler ZM, Chen J, Bettaieb A, Haj FG, Ley RE, Chouinard ML, Cummings BP. TGR5 contributes to glucoregulatory improvements after vertical sleeve gastrectomy in mice. Gut, 2017. DOI: 10.1136/gutjnl-2015-309871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.McGirr R, Ejbick CE, Carter DE, Andrews JD, Nie Y, Friedman TC, Dhanvantari S. Glucose dependence of the regulated secretory pathway in alphaTC1–6 cells. Endocrinology 146: 4514–4523, 2005. [DOI] [PubMed] [Google Scholar]
- 153.McIntyre N, Holsworth DC, Turner DS. New interpretation of oral glucose tolerance. Lancet 2: 20–21, 1964. [DOI] [PubMed] [Google Scholar]
- 154.Melissas J, Koukouraki S, Askoxylakis J, Stathaki M, Daskalakis M, Perisinakis K, Karkavitsas N. Sleeve gastrectomy—A restrictive procedure? Obes Surg 17: 57, 2007. [DOI] [PubMed] [Google Scholar]
- 155.Moens K, Flamez D, Van Schravendijk C, Ling Z, Pipeleers D, Schuit F. Dual glucagon recognition by pancreatic beta-cells via glucagon and glucagon-like peptide 1 receptors. Diabetes 47: 66–72, 1998. [DOI] [PubMed] [Google Scholar]
- 156.Mojsov S, Heinrich G, Wilson IB, Ravazzola M, Orci L, Habener JF. Pre-proglucagon gene expression in pancreas and intestine diversifies at the level of post-translational procesing. J Biol Chem 261: 11880–11889, 1996. [PubMed] [Google Scholar]
- 157.Mokadem M, Zechner JF, Margolskee RF, Drucker DJ, Aguirre V. Effects of Roux-en-Y gastric bypass on energy and glucose homeostasis are preserved in two mouse models of functional glucagon-like peptide-1 deficiency. Mol Metab 3: 191–201, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Moriya R, Shirakura T, Ito J, Mashiko S, Seo T. Activation of sodium-glucose cotransporter 1 ameliorates hyperglycemia by mediating incretin secretion in mice. Am J Physiol Endocrinol Metab 297: E1358–E1365, 2009. [DOI] [PubMed] [Google Scholar]
- 159.Mul JD, Begg DP, Barrera JG, Li B, Matter EK, D’Alessio DA, Woods SC, Seeley RJ, Sandoval DA. High-fat diet changes the temporal profile of GLP-1 receptor-mediated hypophagia in rats. Am J Physiol Regul Integr Comp Physiol 305: R68–R77, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Mulvihill EE, Drucker DJ. Pharmacology, physiology, and mechanisms of action of dipeptidyl peptidase-4 inhibitors. Endocr Rev 35: 992–1019, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Mumphrey MB, Hao Z, Townsend RL, Patterson LM, Berthoud H-R. Sleeve gastrectomy does not cause hypertrophy and reprogramming of intestinal glucose metabolism in rats. Obes Surg 25: 1468–1473, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Mumphrey MB, Patterson LM, Zheng H, Berthoud H-R. Roux-en-Y gastric bypass surgery increases number but not density of CCK-, GLP-1-, 5-HT-, and neurotensin-expressing enteroendocrine cells in rats. Neurogastroenterol Motil 25: e70–e79, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Nannipieri M, Baldi S, Mari A, Colligiani D, Guarino D, Camastra S, Barsotti E, Berta R, Moriconi D, Bellini R, Anselmino M, Ferrannini E. Roux-en-Y gastric bypass and sleeve gastrectomy: Mechanisms of diabetes remission and role of gut hormones. J Clin Endocrinol Metab 98: 4391–4399, 2013. [DOI] [PubMed] [Google Scholar]
- 164.Nauck MA, Vardarli I, Deacon CF, Holst JJ, Meier JJ. Secretion of glucagon-like peptide-1 (GLP-1) in type 2 diabetes: What is up, what is down? Diabetologia 54: 10–18, 2011. [DOI] [PubMed] [Google Scholar]
- 165.Nauck M, Stöckmann F, Ebert R, Creutzfeldt W. Reduced incretin effect in type 2 (non-insulin-dependent) diabetes. Diabetologia 29: 46–52, 1986. [DOI] [PubMed] [Google Scholar]
- 166.Nauck MA, Bartels E, Orskov C, Ebert R, Creutzfeldt W. Additive insulinotropic effects of exogenous synthetic human gastric inhibitory polypeptide and glucagon-like peptide-1-(7–36) amide infused at near-physiological insulinotropic hormone and glucose concentrations. J Clin Endocrinol Metab 76: 912–917, 1993. [DOI] [PubMed] [Google Scholar]
- 167.Nauck MA, Kind J, Köthe LD, Holst JJ, Deacon CF, Broschag M, He YL, Kjems L, Foley J. Quantification of the contribution of GLP-1 to mediating insulinotropic effects of DPP-4 inhibition with vildagliptin in healthy subjects and patients with type 2 diabetes using exendin [9–39] as a GLP-1 receptor antagonist. Diabetes 65: 2440–2447, 2016. [DOI] [PubMed] [Google Scholar]
- 168.Nauck MA, Meier JJ, Cavender MA, Abd El Aziz M, Drucker DJ. Cardiovascular actions and clinical outcomes with glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors. Circulation 136: 849–870, 2017. [DOI] [PubMed] [Google Scholar]
- 169.Nguyen NQ, Debreceni TL, Bambrick JE, Bellon M, Wishart J, Stand-field S, Rayner CK, Horowitz M. Rapid gastric and intestinal transit is a major determinant of changes in blood glucose, intestinal hormones, glucose absorption, and postprandial symptoms after gastric bypass. Obesity (Silver Spring) 22: 2003–2009, 2014. [DOI] [PubMed] [Google Scholar]
- 170.Nian M, Drucker DJ, Irwin D. Divergent regulation of human and rat proglucagon gene promoters in vivo. Am J Physiol Liver Physiol 277: G829–G837, 1999. [DOI] [PubMed] [Google Scholar]
- 171.Nian M, Gu J, Irwin DM, Drucker DJ. Human glucagon gene promoter sequences regulating tissue-specific versus nutrient-regulated gene expression. Am J Physiol Regul Integr Comp Physiol 282: R173–R183, 2002. [DOI] [PubMed] [Google Scholar]
- 172.Nie Y, Nakashima M, Brubaker PL, Li QL, Perfetti R, Jansen E, Zambre Y, Pipeleers D, Friedman TC. Regulation of pancreatic PC1 and PC2 associated with increased glucagon-like peptide 1 in diabetic rats. J Clin Invest 105: 955–965, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Nielsen LB, Ploug KB, Swift P, Ørskov C, Jansen-Olesen I, Chiarelli F, Holst JJ, Hougaard P, Pörksen S, Holl R, de Beaufort C, Gammeltoft S, Rorsman P, Mortensen HB, Hansen L. Co-localisation of the Kir6.2/SUR1 channel complex with glucagon-like peptide-1 and glucose-dependent insulinotrophic polypeptide expression in human ileal cells and implications for glycaemic control in new onset type 1 diabetes. Eur J Endocrinol 156: 663–671, 2007. [DOI] [PubMed] [Google Scholar]
- 174.Omar BA, Andersen B, Hald J, Raun K, Nishimura E, Ahrén B. Fibrob-last growth factor 21 (FGF21) and glucagon-like peptide 1 contribute to diabetes resistance in glucagon receptor-deficient mice. Diabetes 63: 101–110, 2014. [DOI] [PubMed] [Google Scholar]
- 175.Orskov C, Holst JJ, Nielsen C. Effect of truncated glucagon-like peptide-1 (proglucagon-(78–107)amide) on endocrine secretion from pig pancreas, antrum, and nonatral stomach. Endocrinology 123: 2009–2013, 1988. [DOI] [PubMed] [Google Scholar]
- 176.Panjwani N, Mulvihill EE, Longuet C, Yusta B, Campbell JE, Brown TJ, Streutker C, Holland D, Cao X, Baggio LL, Drucker DJ. GLP-1 receptor activation indirectly reduces hepatic lipid accumulation but does not attenuate development of atherosclerosis in diabetic male ApoE(−/−) mice. Endocrinology 154: 127–139, 2013. [DOI] [PubMed] [Google Scholar]
- 177.Papamargaritis D, Koukoulis G, Sioka E, Zachari E, Bargiota A, Zacharoulis D, Tzovaras G. Dumping symptoms and incidence of hypoglycaemia after provocation test at 6 and 12 months after laparoscopic sleeve gastrectomy. Obes Surg 22: 1600–1606, 2012. [DOI] [PubMed] [Google Scholar]
- 178.Parker HE, Adriaenssens A, Rogers G, Richards P, Koepsell H, Reimann F, Gribble FM. Predominant role of active versus facilitative glucose transport for glucagon-like peptide-1 secretion. Diabetologia 55: 2445–2455, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Parker HE, Reimann F, Gribble FM. Molecular mechanisms underlying nutrient-stimulated incretin secretion. Expert Rev Mol Med 12: e1, 2010. [DOI] [PubMed] [Google Scholar]
- 180.Parker HE, Wallis K, le Roux CW, Wong KY, Reimann F, Gribble FM. Molecular mechanisms underlying bile acid-stimulated glucagon-like peptide-1 secretion. Br J Pharmacol 165: 414–423, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Perl SH, Bloch O, Zelnic-Yuval D, Love I, Mendel-Cohen L, Flor H, Rapoport MJ. Sepsis-induced activation of endogenous GLP-1 system is enhanced in type 2 diabetes. Diabetes Metab Res Rev 34: e2982, 2018. [DOI] [PubMed] [Google Scholar]
- 182.Plaisancie P, Bernard C, Chayvialle JA, Cuber JC. Regulation of glucagon-like peptide-1-(7–36) amide secretion by intestinal neuro-transmitters and hormones in the isolated vascularly perfused rat colon. Endocrinology 135: 2398–2403, 1994. [DOI] [PubMed] [Google Scholar]
- 183.Pocai A, Carrington PE, Adams JR, Wright M, Eiermann G, Zhu L, Du X, Petrov A, Lassman ME, Jiang G, Liu F, Miller C, Tota LM, Zhou G, Zhang X, Sountis MM, Santoprete A, Capito’ E, Chicchi GG, Thornberry N, Bianchi E, Pessi A, Marsh DJ, SinhaRoy R. Glucagon-like peptide 1/glucagon receptor dual agonism reverses obesity in mice. Diabetes 58: 2258–2266, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Prigeon RL, Quddusi S, Paty B, D’Alessio DA. Suppression of glucose production by GLP-1 independent of islet hormones: A novel extrapancreatic effect. Am J Physiol Endocrinol Metab 285: E701–E707, 2003. [DOI] [PubMed] [Google Scholar]
- 185.Printz H, Reiter S, Samadi N, Ebrahimsade S, Kirchner R, Arnold R, Göke B. GLP-1 release in man after lower large bowel resection or intrarectal glucose administration. Digestion 59: 689–695, 1998. [DOI] [PubMed] [Google Scholar]
- 186.Quoyer J, Longuet C, Broca C, Linck N, Costes S, Varin E, Bockaert J, Bertrand G, Dalle S. GLP-1 mediates antiapoptotic effect by phosphorylating Bad through a beta-arrestin 1-mediated ERK1/2 activation in pancreatic beta-cells. J Biol Chem 285: 1989–2002, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Ramracheya R, Chapman C, Chibalina M, Dou H, Miranda C, González A, Moritoh Y, Shigeto M, Zhang Q, Braun M, Clark A, Johnson PR, Rorsman P, Briant LJB. GLP-1 suppresses glucagon secretion in human pancreatic alpha-cells by inhibition of P/Q-type Ca2+ channels. Physiol Rep 6: e13852, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Ravier MA, Rutter GA. Glucose or insulin, but not zinc ions, inhibit glucagon secretion from mouse pancreatic alpha-cells. Diabetes 54: 1789–1797, 2005. [DOI] [PubMed] [Google Scholar]
- 189.Rayner CK, Horowitz M. Agonism of receptors in the gut–pancreas axis in type 2 diabetes: Are two better than one? Lancet 391: 2577–2578, 2018. [DOI] [PubMed] [Google Scholar]
- 190.Reimann F, Gribble FM. Glucose-sensing in glucagon-like peptide-1-secreting cells. Diabetes 51: 2757–2763, 2002. [DOI] [PubMed] [Google Scholar]
- 191.Reimann F, Habib AM, Tolhurst G, Parker HE, Rogers GJ, Gribble FM. Glucose sensing in L cells: A primary cell study. Cell Metab, 2008. DOI: 10.1016/j.cmet.2008.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Reimer RA, Darimont C, Gremlich S, Nicolas-Métral V, Rüegg UT, Macé K. A human cellular model for studying the regulation of glucagon-like peptide-1 secretion. Endocrinology 142: 4522–4528, 2001. [DOI] [PubMed] [Google Scholar]
- 193.Richards P, Parker HE, Adriaenssens AE, Hodgson JM, Cork SC, Trapp S, Gribble FM, Reimann F. Identification and characterisation of glucagon-like peptide-1 receptor expressing cells using a new transgenic mouse model. Diabetes 63: 1224, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Ridge T, Moretto T, Macconell L, Pencek R, Han J, Schulteis C, Porter L. Comparison of safety and tolerability with continuous (exenatide once weekly) or intermittent (exenatide twice daily) GLP-1 receptor agonism in patients with type 2 diabetes. Diabetes Obes Metab, 2012. DOI: 10.1111/j.1463-1326.2012.01639.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Ritzel R, Orskov C, Holst JJ, Nauck MA. Pharmacokinetic, insulinotropic, and glucagonostatic properties of GLP-1 [7–36 amide] after subcutaneous injection in healthy volunteers. Dose-response-relationships. Diabetologia 38: 720–725, 1995. [DOI] [PubMed] [Google Scholar]
- 196.Rocca AS, Brubaker PL. Role of the vagus nerve in mediating proximal nutrient-induced glucagon-like peptide-1 secretion. Endocrinology 140: 1687–1694, 1999. [DOI] [PubMed] [Google Scholar]
- 197.Röder PV, Geillinger KE, Zietek TS, Thorens B, Koepsell H, Daniel H. The role of SGLT1 and GLUT2 in intestinal glucose transport and sensing. PLoS One 9: e89977, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Rouille Y, Martin S, Steiner DF. Differential processing of proglucagon by the subtilisin-like prohormone convertases PC2 and PC3 to generate either glucagon or glucagon-like peptide. J Biol Chem 270: 26488–26496, 1995. [DOI] [PubMed] [Google Scholar]
- 199.le Roux CW, Welbourn R, Werling M, Osborne A, Kokkinos A, Laurenius A, Lönroth H, Fändriks L, Ghatei MA, Bloom SR, Olbers T. Gut hormones as mediators of appetite and weight loss after Roux-en-Y gastric bypass. Ann Surg 246: 780–785, 2007. [DOI] [PubMed] [Google Scholar]
- 200.Rowlands J, Heng J, Newsholme P, Carlessi R. Pleiotropic effects of GLP-1 and analogs on cell signaling, metabolism, and function. Front Endocrinol (Lausanne) 9: 672, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Ryan KK, Tremaroli V, Clemmensen C, Kovatcheva-Datchary P, Myronovych A, Karns R, Wilson-Perez HE, Sandoval DA, Kohli R, Backhed F, Seeley RJ. FXR is a molecular target for the effects of vertical sleeve gastrectomy. Nature 509: 183–188, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Sadry SA, Drucker DJ. Emerging combinatorial hormone therapies for the treatment of obesity and T2DM. Nat Rev Endocrinol 9: 425–433, 2013. [DOI] [PubMed] [Google Scholar]
- 203.Saeidi N, Meoli L, Nestoridi E, Gupta NK, Kvas S, Kucharczyk J, Bonab AA, Fischman AJ, Yarmush ML, Stylopoulos N. Reprogramming of intestinal glucose metabolism and glycemic control in rats after gastric bypass. Science 341: 406–410, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Salehi M, Gastaldelli A, D’Alessio DA. Altered islet function and insulin clearance cause hyperinsulinemia in gastric bypass patients with symptoms of postprandial hypoglycemia. J Clin Endocrinol Metab 2014, 2014. (Epub). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Salehi M, Gastaldelli A, D’Alessio DA. Blockade of glucagon-like peptide 1 receptor corrects postprandial hypoglycemia after gastric bypass. Gastroenterology 146: 669.e2–680.e2, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Salehi M, Prigeon RL, D’Alessio DA. Gastric bypass surgery enhances glucagon-like peptide 1-stimulated postprandial insulin secretion in humans. Diabetes 60: 2308–2314, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Salehi M, Vella A, Mclaughlin T, Patti M, Society E. Hypoglycemia after gastric bypass surgery: Current concepts and controversies. J Clin Endocrinol Metab 103: 2815–2826, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Saltiel M, Kuhre R, Christiansen C, Eliasen R, Conde-Frieboes K, Rosenkilde M, Holst J. Sweet taste receptor activation in the gut is of limited importance for glucose-stimulated GLP-1 and GIP secretion. Nutrients 9: 418, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Sandoval DA, Bagnol D, Woods SC, D’Alessio DA, Seeley RJ. Arcuate glucagon-like peptide 1 receptors regulate glucose homeostasis but not food intake. Diabetes 57: 2046–2054, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Schmidt WE, Siegel EG, Creutzfeldt W. Glucagon-like peptide-1 but not glucagon-like peptide-2 stimulates insulin release from isolated rat pancreatic islets. Diabetologia 28: 704–707, 1985. [DOI] [PubMed] [Google Scholar]
- 211.Scow RO, Cornfield J. Quantitative relations between the oral and intravenous glucose tolerance curves. Am J Physiol Content 179: 435–438, 1954. [DOI] [PubMed] [Google Scholar]
- 212.Simonen M, Dali-Youcef N, Kaminska D, Venesmaa S, Käkelä P, Pääkkönen M, Hallikainen M, Kolehmainen M, Uusitupa M, Moilanen L, Laakso M, Gylling H, Patti ME, Auwerx J, Pihlajamäki J. Conjugated bile acids associate with altered rates of glucose and lipid oxidation after Roux-en-Y gastric bypass. Obes Surg 22: 1473–1480, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Sisley S, Gutierrez-aguilar R, Scott M, Alessio DAD, Sandoval DA, Seeley RJ. Neuronal GLP1R mediates liraglutide’s anorectic but not glucose-lowering effect. J Clin Invest 124: 2456–2463, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Smith EP, An Z, Wagner C, Lewis AG, Cohen EB, Li B, Mahbod P, Sandoval D, Perez-Tilve D, Tamarina N, Philipson LH, Stoffers DA, Seeley RJ, D’Alessio DA. The role of β cell glucagon-like peptide-1 signaling in glucose regulation and response to diabetes drugs. Cell Metab 19: 1050–1057, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Solloway MJ, Madjidi A, Gu C, Eastham-Anderson J, Clarke HJ, Kljavin N, Zavala-Solorio J, Kates L, Friedman B, Brauer M, Wang J, Fiehn O, Kolumam G, Stern H, Lowe JB, Peterson AS, Allan BB. Glucagon couples hepatic amino acid catabolism to mTOR-dependent regulation of α-cell mass. Cell Rep 12: 495–510, 2015. [DOI] [PubMed] [Google Scholar]
- 216.Sonoda N, Imamura T, Yoshizaki T, Babendure JL, Lu J-C, Olefsky JM. Beta-Arrestin-1 mediates glucagon-like peptide-1 signaling to insulin secretion in cultured pancreatic beta cells. Proc Natl Acad Sci U S A 105: 6614–6619, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Steinert RE, Gerspach AC, Gutmann H, Asarian L, Drewe J, Beglinger C. The functional involvement of gut-expressed sweet taste receptors in glucose-stimulated secretion of glucagon-like peptide-1 (GLP-1) and peptide YY (PYY). Clin Nutr 30: 524–532, 2011. [DOI] [PubMed] [Google Scholar]
- 218.Sun EW, de Fontgalland D, Rabbitt P, Hollington P, Sposato L, Due SL, Wattchow DA, Rayner CK, Deane AM, Young RL, Keating DJ. Mechanisms controlling glucose-induced GLP-1 secretion in human small intestine. Diabetes 66: 2144–2149, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Svane MS, Bojsen-Møller KN, Nielsen S, Jørgensen NB, Dirksen C, Bendtsen F, Kristiansen VB, Hartmann B, Holst JJ, Madsbad S. Effects of endogenous GLP-1 and GIP on glucose tolerance after Roux-en-Y gastric bypass surgery. Am J Physiol Endocrinol Metab 310: E505–E514, 2016. [DOI] [PubMed] [Google Scholar]
- 220.Svegliati-Baroni G, Saccomanno S, Rychlicki C, Agostinelli L, De Minicis S, Candelaresi C, Faraci G, Pacetti D, Vivarelli M, Nicolini D, Garelli P, Casini A, Manco M, Mingrone G, Risaliti A, Frega GN, Benedetti A, Gastaldelli A. Glucagon-like peptide-1 receptor activation stimulates hepatic lipid oxidation and restores hepatic signalling alteration induced by a high-fat diet in nonalcoholic steatohepatitis. Liver Int 31: 1285–1297, 2011. [DOI] [PubMed] [Google Scholar]
- 221.Svendsen B, Larsen O, Buur M, Gabe N, Christiansen CB, Rosenkilde MM, Drucker DJ, Juul J, Correspondence H, Holst JJ. Insulin secretion depends on intra-islet glucagon signaling. Cell Rep 25: 1127–1134, 2018. [DOI] [PubMed] [Google Scholar]
- 222.Theodorakis MJ, Carlson O, Michopoulos S, Doyle ME, Juhaszova M, Petraki K, Egan JM. Human duodenal enteroendocrine cells: Source of both incretin peptides, GLP-1 and GIP. Am J Physiol Endocrinol Metab 290: E550–E559, 2006. [DOI] [PubMed] [Google Scholar]
- 223.Thorens B Expression cloning of the pancreatic b-cell receptor for the gluco-incretin hormone glucagon-like peptide 1. Proc Natl Acad Sci U S A 89: 8641–8645, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Thyssen S, Arany E, Hill DJ. Ontogeny of regeneration of beta-cells in the neonatal rat after treatment with streptozotocin. Endocrinology 147: 2346–2356, 2006. [DOI] [PubMed] [Google Scholar]
- 225.Toda N, Hao X, Ogawa Y, Oda K, Yu M, Fu Z, Chen Y, Kim Y, Lizarzaburu M, Lively S, Lawlis S, Murakoshi M, Nara F, Watanabe N, Reagan JD, Tian H, Fu A, Motani A, Liu Q, Lin Y-J, Zhuang R, Xiong Y, Fan P, Medina J, Li L, Izumi M, Okuyama R, Shibuya S. Potent and orally bioavailable GPR142 agonists as novel insulin secretagogues for the treatment of type 2 diabetes. ACS Med Chem Lett 4: 790–794, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Tornehave D, Kristensen P, Rømer J, Knudsen LB, Heller RS. Expression of the GLP-1 receptor in mouse, rat, and human pancreas. J Histochem Cytochem 56: 841–851, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Traub S, Meier DT, Schulze F, Dror E, Nordmann TM, Goetz N, Koch N, Dalmas E, Stawiski M, Makshana V, Thorel F, Herrera PL, Böni-Schnetzler M, Donath MY. Pancreatic α cell-derived glucagon-related peptides are required for β cell adaptation and glucose homeostasis. Cell Rep 18: 3192–3203, 2017. [DOI] [PubMed] [Google Scholar]
- 228.Tucker JD, Dhanvantari S, Brubaker PL. Proglucagon processing in islet and intestinal cell lines. Regul Pept 62: 29–35, 1996. [DOI] [PubMed] [Google Scholar]
- 229.Tudurí E, Beiroa D, Porteiro B, López M, Diéguez C, Nogueiras R. Acute but not chronic activation of brain glucagon-like peptide-1 receptors enhances glucose-stimulated insulin secretion in mice. Diabetes Obes Metab 17: 789–799, 2015. [DOI] [PubMed] [Google Scholar]
- 230.Tzovaras G, Papamargaritis D, Sioka E, Zachari E, Baloyiannis I, Zacharoulis D, Koukoulis G. Symptoms suggestive of dumping syndrome after provocation in patients after laparoscopic sleeve gastrectomy. Obes Surg 22: 23–28, 2012. [DOI] [PubMed] [Google Scholar]
- 231.Umeda LM, Silva EA, Carneiro G, Arasaki CH, Geloneze B, Zanella MT. Early improvement in glycemic control after bariatric surgery and its relationships with insulin, GLP-1, and glucagon secretion in type 2 diabetic patients. Obes Surg 21: 896–901, 2011. [DOI] [PubMed] [Google Scholar]
- 232.Unger RH, Aguilar-Parada E, Müller WA, Eisentraut AM. Studies of pancreatic alpha cell function in normal and diabetic subjects. J Clin Invest 49: 837–848, 1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Unger RH, Orci L. Paracrinology of islets and the paracrinopathy of diabetes. Proc Natl Acad Sci 107: 16009–16012, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Vahl TP, Tauchi M, Durler TS, Elfers EE, Fernandes TM, Bitner RD, Ellis KS, Woods SC, Seeley RJ, Herman JP, D’Alessio DA. Glucagon-like peptide-1 (GLP-1) receptors expressed on nerve terminals in the portal vein mediate the effects of endogenous GLP-1 on glucose tolerance in rats. Endocrinology 148: 4965–4973, 2007. [DOI] [PubMed] [Google Scholar]
- 235.Vidal J, de Hollanda A, Jiménez A. GLP-1 is not the key mediator of the health benefits of metabolic surgery. Surg Obes Relat Dis 12: 1225–1229, 2016. [DOI] [PubMed] [Google Scholar]
- 236.Vilsbøll T, Christensen M, Junker AE, Knop FK, Gluud LL. Effects of glucagon-like peptide-1 receptor agonists on weight loss: Systematic review and meta-analyses of randomised controlled trials. BMJ 344: d7771, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Vilsbøll T, Krarup T, Madsbad S, Holst JJ. Both GLP-1 and GIP are insulinotropic at basal and postprandial glucose levels and contribute nearly equally to the incretin effect of a meal in healthy subjects. Regul Pept 114: 115–121, 2003. [DOI] [PubMed] [Google Scholar]
- 238.Vrang N, Hansen M, Larsen PJ, Tang-Christensen M. Characterization of brainstem preproglucagon projections to the paraventricular and dorsomedial hypothalamic nuclei. Brain Res 1149: 118–126, 2007. [DOI] [PubMed] [Google Scholar]
- 239.Wang J, Carrillo JJ, Lin HV. GPR142 agonists stimulate glucose-dependent insulin secretion via Gq-dependent signaling. PLoS One 11: e0154452, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Whalley NM, Pritchard LE, Smith DM, White A. Processing of proglucagon to GLP-1 in pancreatic α-cells: Is this a paracrine mechanism enabling GLP-1 to act on β-cells? J Endocrinol 211: 99–106, 2011. [DOI] [PubMed] [Google Scholar]
- 241.Wideman RD, Yu ILY, Webber TD, Verchere CB, Johnson JD, Cheung AT, Kieffer TJ. Improving function and survival of pancreatic islets by endogenous production of glucagon-like peptide 1 (GLP-1). Proc Natl Acad Sci U S A 103: 13468–13473, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Willert K, Jones KA. Wnt signaling: Is the party in the nucleus? Genes Dev 20: 1394–1404, 2006. [DOI] [PubMed] [Google Scholar]
- 243.Williams DL, Baskin DG, Schwartz MW. Leptin regulation of the anorexic response to glucagon-like peptide-1 receptor stimulation. Diabetes 55: 3387–3393, 2006. [DOI] [PubMed] [Google Scholar]
- 244.Wilson-Pérez HE, Chambers AP, Ryan KK, Li B, Sandoval DA, Stoffers D, Drucker DJ, Pérez-Tilve D, Seeley RJ. Vertical sleeve gastrectomy is effective in two genetic mouse models of glucagon-like peptide-1 receptor deficiency. Diabetes 62: 2380–2385, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Wilson ME, Kalamaras JA, German MS. Expression pattern of IAPP and prohormone convertase 1/3 reveals a distinctive set of endocrine cells in the embryonic pancreas. Mech Dev 115: 171–176, 2002. [DOI] [PubMed] [Google Scholar]
- 246.Xu E, Kumar M, Zhang Y, Ju W, Obata T, Zhang N, Liu S, Wendt A, Deng S, Ebina Y, Wheeler MB, Braun M, Wang Q. Intra-islet insulin suppresses glucagon release via GABA-GABAA receptor system. Cell Metab 3: 47–58, 2006. [DOI] [PubMed] [Google Scholar]
- 247.Yanay O, Bailey AL, Kernan K, Zimmerman JJ, Osborne WR. Effects of exendin-4, a glucagon like peptide-1 receptor agonist, on neutrophil count and inflammatory cytokines in a rat model of endotoxemia. J Inflamm Res 8: 129, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Ye J, Hao Z, Mumphrey MB, Townsend RL, Patterson LM, Stylopoulos N, Munzberg H, Morrison CD, Drucker DJ, Berthoud H-R. GLP-1 receptor signaling is not required for reduced body weight after RYGB in rodents. Am J Physiol Regul Integr Comp Physiol 306: R352–R362, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Yoder SM, Yang Q, Kindel TL, Tso P. Stimulation of incretin secretion by dietary lipid: Is it dose dependent? Am J Physiol Gastrointest Liver Physiol 297: G299–G305, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Yu Z, Jin T. New insights into the role of cAMP in the production and function of the incretin hormone glucagon-like peptide-1 (GLP-1). Cell Signal 22: 1–8, 2010. [DOI] [PubMed] [Google Scholar]
- 251.Zhang Y, Parajuli KR, Fava GE, Gupta R, Xu W, Nguyen LU, Zakaria AF, Fonseca VA, Wang H, Mauvais-Jarvis F, Sloop KW, Wu H. GLP-1 receptor in pancreatic α-cells regulates glucagon secretion in a glucose-dependent bidirectional manner. Diabetes 68: 34–44, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Zhou H, Zhang T, Harmon JS, Bryan J, Robertson RP. Zinc, not insulin, regulates the rat alpha-cell response to hypoglycemia in vivo. Diabetes 56: 1107–1112, 2007. [DOI] [PubMed] [Google Scholar]
- 253.Zhu L, Dattaroy D, Pham J, Wang L, Barella LF, Cui Y, Wilkins KJ, Roth BL, Hochgeschwender U, Matschinsky FM, Kaestner KH, Doliba NM, Wess J. Intra-islet glucagon signaling is critical for maintaining glucose homeostasis. JCI Insight 5, 2019. DOI: 10.1172/jci.insight.127994. [DOI] [PMC free article] [PubMed] [Google Scholar]