

Abstract
Influenza virus infection causes considerable morbidity and mortality, but current therapies have limited efficacy. We hypothesized that investigating the metabolic signaling during infection may help to design innovative antiviral approaches. Using bronchoalveolar lavages of infected mice, we here demonstrate that influenza virus induces a major reprogramming of lung metabolism. We focused on mitochondria‐derived succinate that accumulated both in the respiratory fluids of virus‐challenged mice and of patients with influenza pneumonia. Notably, succinate displays a potent antiviral activity in vitro as it inhibits the multiplication of influenza A/H1N1 and A/H3N2 strains and strongly decreases virus‐triggered metabolic perturbations and inflammatory responses. Moreover, mice receiving succinate intranasally showed reduced viral loads in lungs and increased survival compared to control animals. The antiviral mechanism involves a succinate‐dependent posttranslational modification, that is, succinylation, of the viral nucleoprotein at the highly conserved K87 residue. Succinylation of viral nucleoprotein altered its electrostatic interactions with viral RNA and further impaired the trafficking of viral ribonucleoprotein complexes. The finding that succinate efficiently disrupts the influenza replication cycle opens up new avenues for improved treatment of influenza pneumonia.
Keywords: antiviral, influenza, metabokine, signaling, virus
Subject Categories: Immunology; Metabolism; Microbiology, Virology & Host Pathogen Interaction
The energy‐production metabolite succinate protects from pulmonary viral infection via post‐translational modification‐dependent interruption of the influenza replication cycle.
Introduction
In 1918, more than 50 million people died from severe influenza viral pneumonia. One century later, and despite considerable progress in medicine, annual epidemics are still estimated to result in 3–5 million cases of severe illness worldwide and up to 650,000 deaths (Taubenberger et al, 2019; Lampejo, 2020). Influenza viruses are the etiological agents and are classified into four types (A, B, C, and D) of which influenza A virus (IAV) is clinically the most significant.
Vaccination against influenza constitutes the most effective preventive strategy. However, the short duration of vaccine‐induced immunity coupled with the intrinsic antigenic drift of IAV negatively affects vaccine efficiency (Jang & Seong, 2019). Besides, drugs targeting IAV neuraminidase (NA) are currently recommended by the WHO but their efficacy is largely disputed (WHO, 2009; Jefferson et al, 2014; Duwe, 2017). Hence, the development of innovative antiviral drugs is required to better treat IAV pneumonia (Pizzorno et al, 2019). To this end, we need a better understanding of the mechanisms of IAV––host cell interactions that may lead to IAV‐triggered lung hyper‐inflammation. Indeed, several studies suggest that such an excessive and deleterious inflammatory response is responsible for the life‐threatening “flu” syndrome (Le Goffic et al, 2006; Taubenberger et al, 2019).
Interestingly, accumulating data support the view that metabolic pathways may promote or inhibit immune and inflammatory responses (Rambold & Pearce, 2018; Williams & O’Neill, 2018; Infantino et al, 2019), in addition to their role in energy production and macromolecular biosynthesis. Succinate is of special interest in this context. It is an intermediate of the tricarboxylic acid (TCA) cycle in mitochondria. Succinate is mainly produced from succinyl coenzyme A by succinyl coenzyme A synthetase (Grimolizzi & Arranz, 2018). It can also connect intracellular metabolic status and intercellular communication, as it can be released into the extracellular space through plasma membrane transporters of the SLC13 family (Grimolizzi & Arranz, 2018; Murphy & O’Neill, 2018). As a result, succinate can accumulate extracellularly in certain pathophysiological situations such as chronic inflammatory diseases (Ryan et al, 2019). Thus, the relationship between succinate and inflammatory signaling in leukocytes is increasingly studied (Rubic et al, 2008; Tannahill et al, 2013; Jha et al, 2015; Littlewood‐Evans et al, 2016; Mills et al, 2016; Zasłona & O’Neill, 2020). In contrast, such effects of succinate on nonimmune cells such as the epithelial cells that line the airway mucosa have not been explored to a similar extent. This is important since airway epithelial cells are the main site of infection for respiratory pathogens such as IAV, and their role in controlling immune and inflammatory responses is pivotal. Furthermore, we and others have previously demonstrated that these cells are the source of major inflammatory mediators contributing to IAV‐related lung injury (Simmons & Farrar, 2008; Si‐Tahar et al, 2009; Gregory & Kobzik, 2015; Denney & Ho, 2018).
In view of the pro‐inflammatory properties of succinate (Rubic et al, 2008; Tannahill et al, 2013; Jha et al, 2015; Littlewood‐Evans et al, 2016; Mills et al, 2016; Zasłona & O’Neill, 2020), we initially speculated that succinate could contribute to the deleterious immunopathological response associated with severe IAV pneumonia. Strikingly, the opposite situation was observed. Using in vitro and in vivo approaches, we reveal that succinate restricts IAV replication and downstream inflammatory signaling. The underlying mechanism involves a specific posttranslational modification and nuclear retention of the IAV nucleoprotein (NP). Hence, we demonstrate that succinate is a key player in the antiviral defense of the lung mucosa.
Results
Influenza infection increases succinate levels in airways
Despite the growing interest in immunometabolism (Pearce & Pearce, 2018), little is known about metabolic reprogramming upon IAV infection (Smallwood et al, 2017; Tian et al, 2019; Bahadoran et al, 2020; Keshavarz et al, 2020; Wendt et al, 2021). Here, we conducted an unbiased quantification of the lung metabolome of mice four days after the infection by a sublethal dose of IAV (A/Scotland/20/74 (H3N2)). From the nuclear magnetic resonance (NMR) spectra of the bronchoalveolar lavage (BAL) fluids, we focused on succinate for two main reasons: (i) it was among the top ten most altered metabolites in the BAL fluids of IAV‐infected mice, compared to control animals (fold change = 2.2, FDR‐adjusted P‐value = 8 × 10‐7, Fig 1A) and (ii) succinate is increasingly recognized as a potent immunoregulatory mediator (Rubic et al, 2008; Tannahill et al, 2013; Jha et al, 2015; Littlewood‐Evans et al, 2016; Mills et al, 2016; Zasłona & O’Neill, 2020).
Figure 1. Influenza virus infection increases succinate levels in airways.
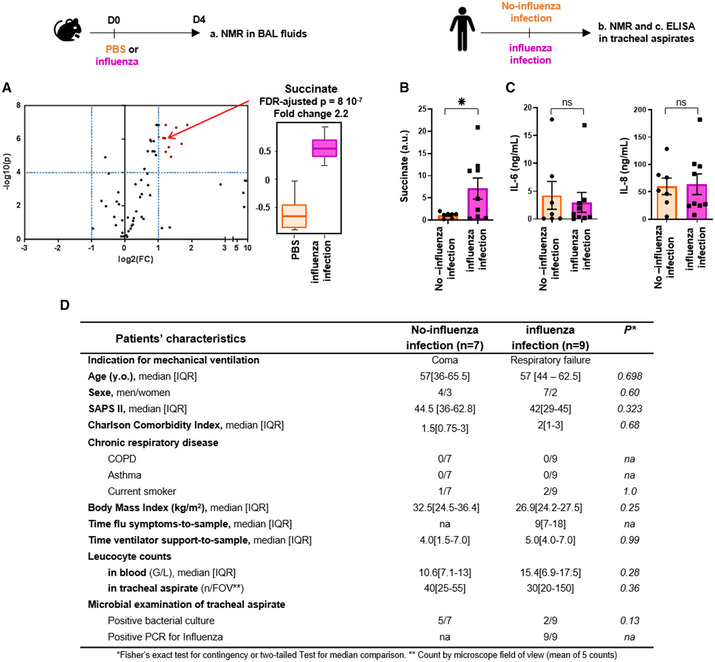
- C57Bl/6 mice were infected (n = 9) or not (n = 8) intranasally with 150 pfu of influenza A virus (IAV; A/Scotland/20/74 (H3N2)). Bronchoalveolar lavage fluids (BALs) were collected 4 days postinfection. Samples were further processed for metabolomic analysis by NMR. Left panel shows a volcano‐plot representing the comparison of “IAV‐infected” versus “control, non‐infected” animals. The graph plots the −log (P‐value) against the fold change (log2 (ratio)) for individual metabolites. Vertical dashed blue lines mark the two‐fold change and horizontal dashed blue line marks the cut‐off P‐value of 0.0001. Metabolites in the upper right square in red are significantly more abundant in “IAV‐infected” compared to “control, non‐infected” samples. Succinate (annotated by a red arrow) is one of these compounds with a FC = 2.2 and a FDR‐adjusted P‐value = 8 × 10−7. The boxplots whiskers correspond to the minimum and maximum values, while the central band is the mean. The first and third quartiles (boxes) are merely the average values between the median and the extrema.
- Shows succinate quantification by NMR from tracheal aspirates collected in mechanically ventilated patients with (pink bar, n = 9) or without (orange bar, n = 7) diagnostic of IAV pneumonia. *P < 0.05.
- IL‐6 and IL‐8 measurements by ELISA from these same tracheal aspirates.
- Main baseline characteristics of the patients included in the study.
Data information: Data are mean ± SEM and statistical analysis was performed using the Mann–Whitney U‐test.
In agreement with the data from the mouse experiments (Fig 1A), succinate level in respiratory fluids (i.e., tracheal aspirates) was significantly higher in critically ill patients with influenza infection than in patients not infected by IAV but with similar severity and lung inflammation (Fig 1B). This second group of patients included critically ill individuals ventilated for coma (hypoxic‐ischemic coma, stroke, or drug overdose) with similar comorbidities and severity score, as assessed by IL‐6 and IL‐8/CXCL8 measurements in the respiratory fluids (Fig 1C), the leucocyte count in respiratory fluids and the regular presence of positive bacterial culture (Fig 1D). The heterogeneity of succinate levels in the fluids of IAV‐infected patients can partly be ascribed to the variability at the time of hospital admission and the subsequent symptoms‐to‐sampling time (median [IQR] = 9 [7; 18] days). Overall, we demonstrated that IAV infection was associated with high levels of succinate in lungs of mice and humans.
Succinate limits the secretion of inflammatory mediators and restores metabolic dysregulation in IAV‐infected lung epithelial cells
To decipher the potential role of succinate in IAV‐infected lungs, we first compared at 20 h postinfection (pi) the gene expression profiles of IAV‐infected human bronchial epithelial cells, exposed or not to succinate. The volcano plot in Fig 2A (left panel) shows that IAV infection increased the expression of multiple gene pathways, including canonical immune and inflammatory pathways. The volcano plot in the right panel indicates that succinate treatment of IAV‐infected cells results in a drastic downregulation of those pathways. As illustrated in Fig 2A and in Appendix Fig S1, those altered pathways included, but were not limited to, “inflammasome signaling,” “TREM1 signaling,” “acute phase response signaling,” “role of PRR in recognition of bacteria and viruses,” “iNOS signaling,”, etc.). Of note is that succinate does not induce any cell cytotoxicity at the doses we used (Appendix Fig S2A and B).
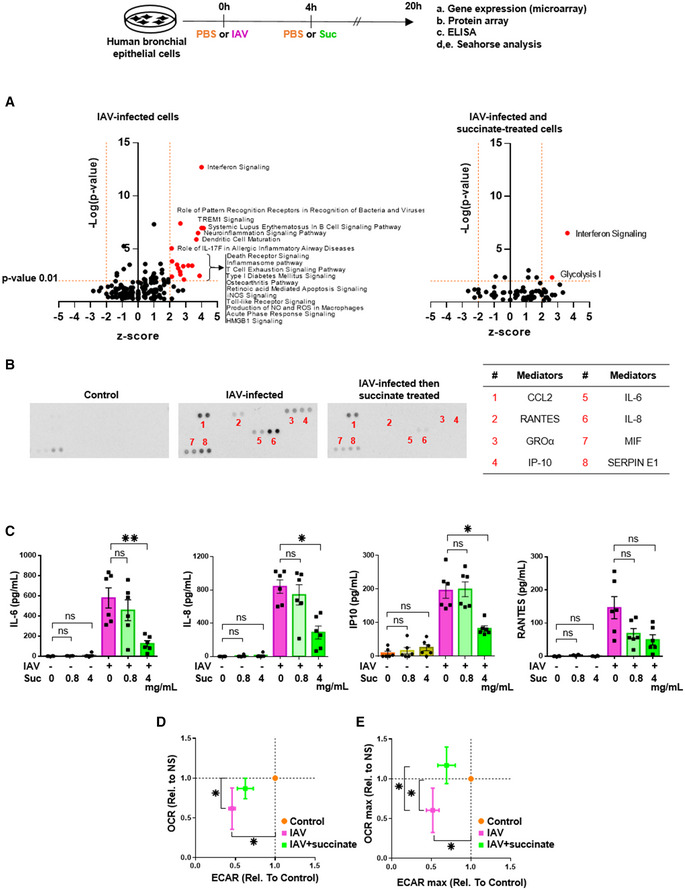
Figure 2. Succinate reverses the inflammatory response and the metabolic modifications induced by influenza virus.
-
AVolcano‐plot showing the most significantly regulated canonical pathways determined by microarray analysis compared with mock‐treated cells. Each dot represents a specific canonical pathway as determined by GSEA. Pathway representations result from the magnitude of regulation (z‐score, x‐axis) and significance (−log10 adjusted P‐value, y‐axis). The dashed horizontal line indicates the statistical significance threshold (P ≤ 0.01 after adjustment with the Bonferroni correction). The two vertical dashed lines show the z‐score threshold (−2: repressed; 2: induced). Colored spots characterize the most highly regulated canonical pathways endowed with statistical significance.
-
BRepresentative inflammatory protein‐array blots obtained from the supernatants of control‐ or IAV‐infected‐ or IAV‐infected and succinate‐treated cells. The table on the right side indicates the 8 most regulated mediators by succinate.
-
CQuantification by ELISA of IL‐6, IL‐8, IP‐10, and RANTES in the supernatants of cells infected or not by IAV and treated or not with succinate.
-
D, EPanels show the basal replotted ECAR and OCR (D), and glycolytic capacity (ECAR max) and maximal respiration (OCR max) (E) in cells infected with IAV and treated (green symbols) or not (pink symbols) with succinate. Data were normalized with respect to mock‐infected, untreated cells (orange symbols), and were also normalized for DNA content.
We considered that the decrease of inflammatory pathways mediated by succinate in the context of IAV infection was an important observation for two reasons: first, because inflammation is a key promoter of IAV pneumonia and lung function impairment (Simmons & Farrar, 2008; Si‐Tahar et al, 2009; Gregory & Kobzik, 2015; Denney & Ho, 2018); second, because so far succinate was mostly considered to be pro‐inflammatory (Rubic et al, 2008; Tannahill et al, 2013; Jha et al, 2015; Littlewood‐Evans et al, 2016; Mills et al, 2016; Zasłona & O’Neill, 2020). Using a semiquantitative approach (i.e. protein‐array blots), we confirmed that succinate limits the secretion of various inflammatory mediators (Fig 2B). Then, we quantified the diminution of critical inflammatory cytokines by succinate and found that this metabolite decreases down to 75% IAV‐mediated secretion of IL‐6, IL‐8, IP‐10/CXCL10. and RANTES/CCL5 (Fig 2C). Consistently, malonate (i.e., a selective inhibitor of succinate dehydrogenase (SDH)), which favors endogenous succinate accumulation (Potter & Dubois, 1943; Mills et al, 2016) also inhibited IAV‐induced inflammatory response (as assessed by IL‐6 quantification; not illustrated).
Because viruses are known to take control of the host metabolism to replicate more efficiently (Keshavarz et al, 2020), we also wanted to examine whether succinate could prevent IAV‐induced metabolic reprogramming. We first measured the IAV‐induced changes in glycolysis and mitochondrial oxidative phosphorylation. We observed that IAV significantly impaired the oxygen consumption rate (OCR) (i.e., a key metric of mitochondrial oxidative phosphorylation) and the extracellular acidification rate (ECAR) (i.e., an index of the glycolytic activity) (Fig 2D and E, pink dots). After succinate treatment, IAV‐induced metabolic dysregulation tended to be corrected, with a trend toward the normalization of both glycolysis and mitochondrial oxidative phosphorylation (Fig 2D and E, green dots).
Together, these results showed that succinate inhibited both the inflammatory and metabolic perturbations secondary to IAV infection.
Succinate blocks the multiplication of IAV in lung epithelial cells
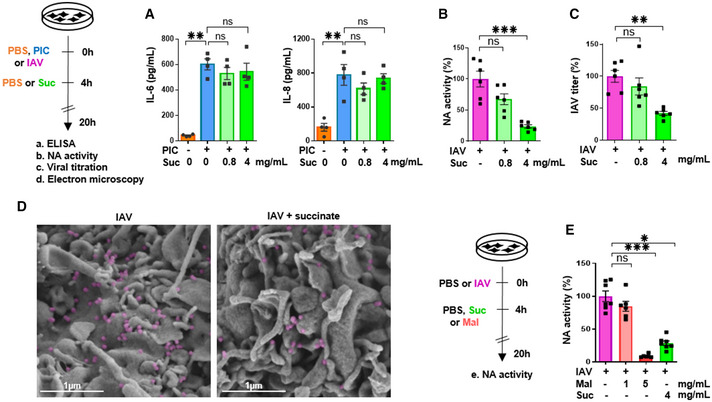
Next, we wondered whether the effects of succinate in IAV‐infected cells were due to its immunomodulatory rather than its anti‐infective properties. To answer this question, we challenged lung epithelial cells with synthetic double‐stranded RNA poly(I:C) (PIC), which is a potent agonist of TLR3 signaling that mimics IAV‐triggered immune responses (Guillot et al, 2005). Poly(I:C) induced IL‐6 and IL‐8 secretion at a similar or even higher level than IAV does. However, succinate did not inhibit poly(I:C)‐induced secretion of these cytokines and chemokines (Fig 3A). We further compared the production of viral particles in the supernatants of IAV‐infected epithelial cells exposed or not to succinate, in terms of viral neuraminidase (NA) enzymatic activity or viral titers. We observed a marked reduction of IAV load upon succinate treatment in distinct human airway epithelial cells (such as BEAS‐2B (Fig 3B and C), 16HBE14o‐ and A549 cells (Appendix Fig S4)). These results were further confirmed by a decrease of neo‐virion budding and release, as assessed by electronic microscopy (Fig 3D and Appendix Fig S3). Interestingly, the treatment of IAV‐infected lung epithelial cells with succinate or malonate resulted in a similar reduction of NA activity (Fig 3E). We confirmed and extended these findings by showing that succinate impairs IAV multiplication even more strongly in multicycle replication condition (using a MOI = 10‐3; ~5‐fold decrease) than in single cycle replication condition (using a MOI = 1; ~2‐fold decrease; Fig EV1A and B). Overall, we demonstrated that succinate inhibits IAV multiplication.
Figure 3. Succinate does not inhibit the secretion of inflammatory mediators induced by Poly(I:C), but blocks the multiplication of influenza virus in lung epithelial cells.
-
ALevels of IL‐6 and IL‐8/CXCL8, as measured by ELISA in the supernatants of cells stimulated with (PIC) and subsequently treated or not with succinate.
-
B–EA neuraminidase (NA) activity test (B), a Plaque‐Forming Units assay (C) and scanning electron microscopy (D) were used to assess the production of physical (B, D) and infectious (C) viral particles in the supernatants of IAV‐infected cells, treated or not with succinate. IAV particles budding are colored in purple. Scale bar: 1 µm. (E) A neuraminidase activity test was also applied in cells infected or not by IAV and treated or not with malonate (a succinate dehydrogenase inhibitor), in comparison with succinate.
Figure EV1. Anti‐influenza effect of succinate is more potent in multicycle replication condition than in single cycle replication condition.
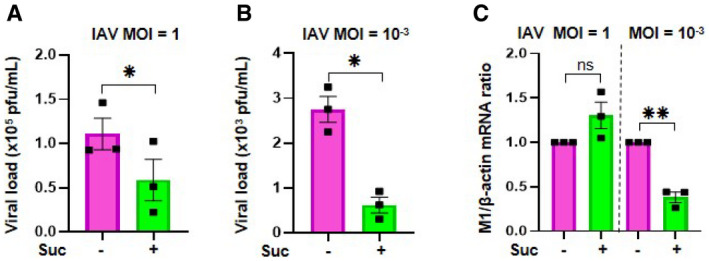
-
A, BPlaque‐Forming Unit assays determined the production of infectious viral particles in cell supernatants.
-
CThe effect of succinate on IAV transcription was assessed by RT‐qPCR to quantify M1 viral mRNA. Data information: Data are represented as the mean ± SEM of 3 independent experiments. Statistical analysis was performed using paired t‐test (A, B) or one sample t‐test (C), (*P < 0.05 and **P < 0.01).
Succinate protects mice from IAV pneumonia
We further explored whether we could confirm in vivo the anti‐influenza effect of succinate that we revealed in vitro. Mice were infected intranasally with a LD50 of A/Scotland/20/74 (H3N2) IAV and received or not succinate (4 mg/animal). Fig 4A–C shows that viral load and viral protein expression in lung tissues were lower in succinate‐treated mice at 4 days pi. To assess concomitantly the possible modulation of the immune/inflammatory response by succinate, we first evaluated the relative expression of 111 mediators in the BAL compartment (Fig 4D and Appendix Fig S5). Eighty‐four of these mediators were increased upon IAV infection, and almost all, including chemokines, cytokines, and metalloproteases, were markedly reduced in succinate‐treated animals (Fig 4D). A more accurate quantification of IL‐6 and KC by ELISA confirmed the semiquantitative protein‐array findings (Fig 4E). Consistently, the amount of inflammatory cells such as neutrophils and myeloperoxidase (a protein primarily released by neutrophils) were significantly reduced in succinate‐treated animals; Fig 4F and G; see also Appendix Fig S6 for the leukocyte gating strategy).
Figure 4. Succinate protects mice from influenza infection.
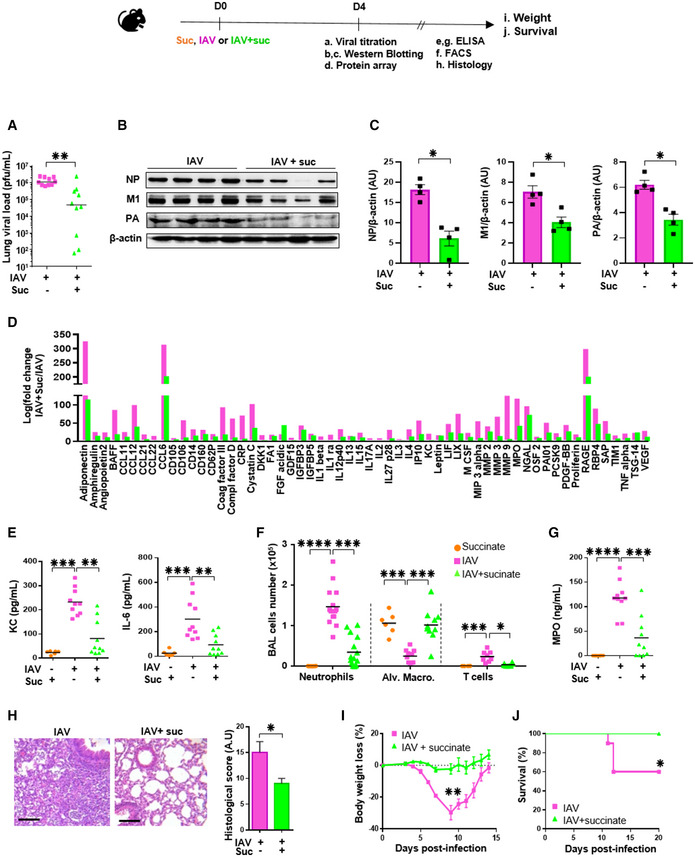
-
A–HSome mice were euthanized at 4 days postinfection and lungs as well as BAL fluids were collected to determine: (A) the viral load by a plaque forming unit assay; (B) the viral protein expression by Western blotting followed by a relative quantification (C), (D) the levels of 111 additional mediators using a specific protein array; (E, G) the levels of KC, IL‐6 and MPO by ELISA; (F) the number of immune and inflammatory cells by flow cytometry; (H) tissue lesions in lung sections stained with hematoxylin‐eosin and further assessed by microscopy. Scale bar: x6/20 µm. All data are represented as the mean or the mean ± SEM and are cumulative of 2 independent experiments with 5 animals each.
-
I, JIn separate experiments, body weight loss (n = 8) (I) and animal survival (n = 10) (J) were monitored daily. Data are represented as the mean ± SEM.
IAV‐mediated damage of the lung mucosa results from a combination of intrinsic viral pathogenicity and an inappropriate regulation of host immune mediators (Le Goffic et al, 2006; Simmons & Farrar, 2008; Blanc et al, 2016). Therefore, by histopathological scoring, we explored the effect of succinate on lung tissue aspect. We observed a significant decrease of IAV‐induced alveolar wall thickening, and hyaline membrane and epithelial necrosis in mice that received succinate compared to control, infected animals (Fig 4H). Finally, mice infected by a LD50 of IAV started to lose weight at 5 days pi (Fig 4I) and ultimately 40% succumbed (Fig 4J). Mice that received succinate did not lose weight and all survived.
The anti‐IAV effect of succinate involves a retention of the NP protein in the nuclear compartment
To better understand how succinate inhibits the production of viral particles, we studied its impact on the different stages of the replication cycle of IAV by first using experimental conditions yielding a single round of IAV infection (MOI = 1). After entry, the virus cycle starts by the transcription of viral mRNAs (Fig 5A). These viral mRNAs serve as a template for the translation of viral proteins. We observed a comparable amount of viral M1 mRNAs in nontreated and succinate‐treated cells, as assessed by RT‐qPCR (Fig 5B). By contrast, when lung epithelial cells were infected under experimental conditions that support multicycle IAV replication (MOI = 10−3), succinate induces less viral M1 transcripts due to a lower number of re‐infecting virus particles (Fig EV1, panel C).
Figure 5. Succinate impairs the intracellular trafficking of influenza virus.
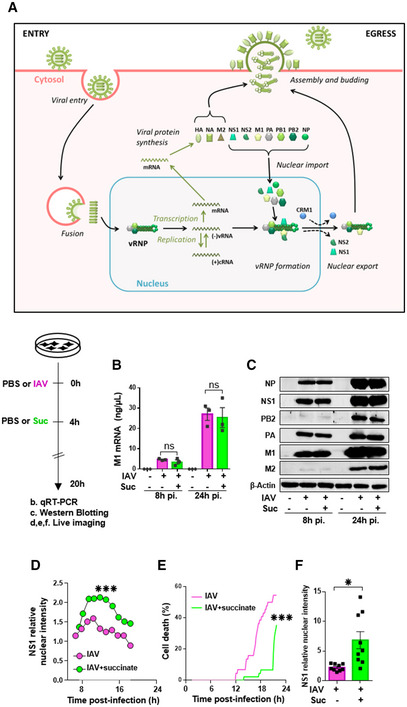
-
ASchematic representation of the IAV replication cycle. After endocytosis, the viral ribonucleoprotein (vRNP) complexes are transported into the nucleus. Viral RNAs (vRNAs) serve as templates for the synthesis of messenger RNAs (mRNAs) and complementary RNAs (cRNAs) are used for the replication of vRNAs. mRNAs are exported in the cytoplasm for translation. Some proteins are transported back to the nucleus to form new vRNPs with neosynthesized vRNAs. Newly synthesized vRNPs are exported in the cytoplasm via the CRM1 protein. HA, NA, M2 proteins and the vRNPs are transported to the plasma membrane for assembly and budding of the progeny virions.
-
B–FHuman bronchial epithelial BEAS‐2B cells were infected with A/Scotland/20/74 (H3N2) virus (IAV) at MOI = 1. After 4 h, cells were treated or not with succinate (Suc; 4 mg/ml) up to 20 h. The effect of succinate on IAV transcription (B) and protein expression (C) were assessed by RT‐qPCR to quantify the M1 viral mRNA and Western blotting to detect viral proteins (β‐actin was used as a loading control), respectively. (D–F) Human alveolar epithelial A549 cells were infected with the recombinant influenza A/WSN/33 virus expressing a fusion NS1‐eGFP protein at MOI = 0.5 for 4 h, then treated with 4 mg/ml of succinate. A549 cells were monitored for 24 h using a BioStation IM‐Q device. (D) Single‐cell dynamics of the nuclear/cytoplasmic fluorescence ratio. (E) Single‐cell dynamics of cell death as assessed by morphological analysis. (F) Quantification of the nuclear/cytoplasmic fluorescence ratio measured at 13 h postsuccinate treatment. Data are represented as the mean ± SEM of 3 biological replicates (B, C) or 3 independent experiments with 3 technical replicates each (D–F). Statistical analysis was performed using the Mann–Whitney test (B–E) or t‐test (F), (*P < 0.05 and ***P < 0.001).
The effect of succinate on the subsequent stages of the viral cycle was examined using single replication conditions. A similar viral protein expression pattern was found in nontreated and succinate‐treated cells, as assessed by western‐blot analysis of the viral NP, NS1, PB2, PA, M1, and M2 proteins (Fig 5C). The lack of a succinate effect on IAV mRNA and protein expression led us to analyze the impact of such metabolite on viral proteins trafficking. We especially examined the effect of succinate on (i) the nuclear import (or nucleo‐cytoplasmic shuttling) of newly synthetized viral proteins and (ii) the nuclear export of newly assembled viral ribonucleoprotein (vRNP) complexes (Fig 5A). We performed real‐time imaging using a NS1‐GFP‐tagged recombinant IAV (A/WSN/33 (H1N1)), which shows similar replication and virulence properties to its wild‐type counterpart (not illustrated). In the absence of succinate treatment, we typically observed a fluorescent signal in the cytosol of IAV‐infected lung epithelial cells at ~6 h pi. This was followed by progressive nuclear accumulation of the signal (at ~10 h pi). Ultimately, we observed a diffuse fluorescence signal, as well as vacuolization accompanying cell death at > 22 h pi (Fig 5D and E; see also Movie EV1 for visualization of the complete process). In contrast, in succinate‐treated cells, the fluorescent signal seemed to be more restricted to the nuclear compartment and cytoplasmic vacuolization accompanying cell death was reduced (Fig 5D and E and Movie EV2). Thorough quantification carried out on 60 cells confirmed that the nuclear/cytoplasmic fluorescence ratio was significantly higher in succinate‐treated cells than in control cells (Fig 5F).
We next studied whether the nuclear retention of NS1 by succinate could be extended to other viral proteins. Using confocal microscopy, we monitored at 20 h pi the intracellular localization of NS1 as well as of PA, PB2, M2, M1, and NP proteins in lung epithelial cells infected by IAV (A/Scotland/20/74 (H3N2)) and exposed or not to succinate. We first confirmed that NS1 shows a selective nuclear accumulation in succinate‐treated cells. Remarkably, we extended this observation to NP by confocal microscopy (Fig 6A) as well as by flow cytometry (Fig EV2). Those findings were further supported by the accurate quantification of NS1 as well as of NP relative nuclear intensities (Fig 6B). In contrast, PB2, PA, M2, M1, and proteins were essentially localized in the cytosolic compartment in both control‐ and succinate‐treated lung epithelial cells (Fig 6C).
Figure 6. The anti‐influenza virus effect of succinate involves the nuclear retention of NP and NS1 proteins.
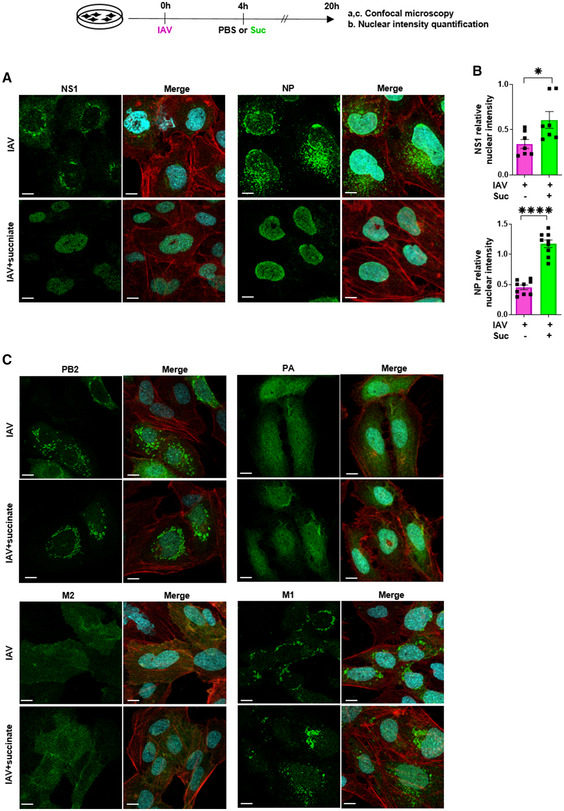
-
A–CHuman bronchial epithelial BEAS‐2B cells were infected with influenza A/Scotland/20/74 (H3N2) virus (IAV) at MOI = 1 for 4 h, then washed and treated or not with succinate (Suc; 4 mg/ml) for 20 h. (A, C) Expression of IAV proteins (i.e., PA, PB2, M2, M1, NP, NS1) was analyzed by confocal microscopy using specific antibodies. Viral proteins are stained in green, DNA in blue and actin in red. For a given antibody, pictures were taken on the exact same day with the same laser power settings, Pictures are representative of five independent experiments. Scale bar 10 µM. (B) Relative nuclear intensity of NS1 or NP was determined by using the Intensity Ratio Nuclei Cytoplasm Tool. Seven (NS1) or nine (NP) random images were collected per treatment with at least 5 cells per field. Data are represented as the mean ± SEM. Statistical analysis was performed using the Mann–Whitney test, (*P < 0.05 and ****P < 0.0001).
Figure EV2. Nucleoprotein localization determined by flow cytometry.
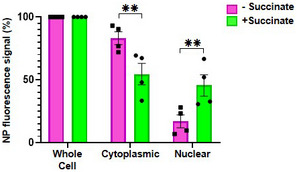
BEAS‐2B cells were infected with influenza A/Scotland/20/74 (H3N2) virus (IAV)) at MOI 1 for 4 h and treated or not with succinate (Suc) for 20 h. Nucleoprotein (NP) expression was assessed by flow cytometry using two different permeabilization buffers, one allowing whole cell fluorescent staining of NP, the other only the NP‐associated to the cytoplasmic compartment (by subtraction, nuclear‐associated NP signal was extrapolated). Data are represented as the mean ± SEM of 4 independent experiments. Statistical analysis was performed using ANOVA with Sidak’s posttest (**P < 0.01).
Interestingly, when lung epithelial cells were infected by a distinct IAV strain, that is, A/PR/8/34 (H1N1), succinate caused nuclear retention of NP but not of NS1 (Fig 7A) while it did inhibit viral multiplication, as assessed by the measurement of NA activity (P < 0.05; Fig 7B). Succinate also caused nuclear retention of NP in cells infected with a NS1‐deficient PR8 strain of virus (i.e., “PR8 ∆NS1”; Fig 7C). Taken together, these data suggested that NP retention, and not NS1, is key in the anti IAV‐effect of succinate.
Figure 7. Nuclear retention of NP—and not NS1—is a key mechanism underlying the anti‐IAV effect of succinate.
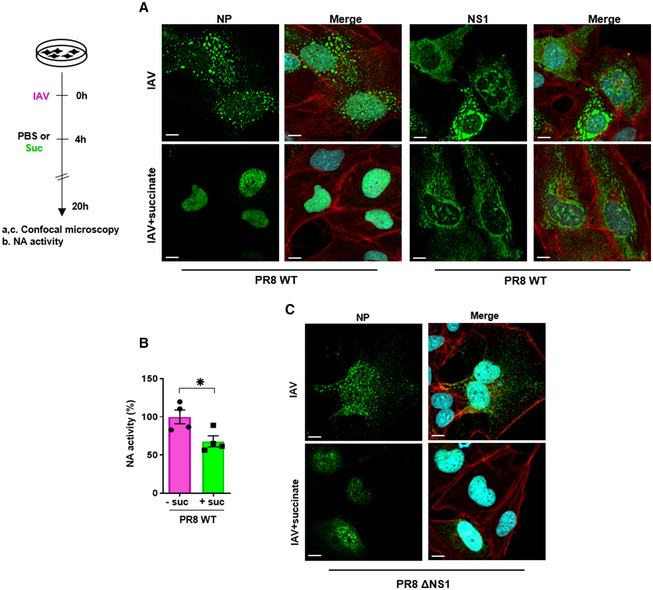
-
A–CHuman bronchial epithelial BEAS‐2B cells were infected for 4 h with influenza A/PR/8/34 (H1N1) virus, either wild‐type (PR8 WT; at MOI = 10 (A, B)) or carrying a deletion of the NS1 coding sequence (PR8ΔNS1; at MOI = 4 (C)), and subsequently treated or not for 20 h with 4 mg/ml of succinate (Suc). Expression of NP (A, C) and NS1 (A) proteins was analyzed by confocal immunofluorescence microscopy. Viral proteins are stained in green, DNA in blue and actin in red. For a given antibody, pictures were taken on the exact same day with the same laser power settings. They are representative of three independent experiments; scale bar 10 µM. (B) Neuraminidase (NA) activity in the supernatants of epithelial cells infected by PR8 WT treated or not with succinate. Data are represented as the mean ± SEM of 4 independent experiments. Statistical analysis was performed using the Student’s t‐test, (*P < 0.05).
Succinate reduces the assembly of vRNPs but does not alter the CRM1‐dependent transport pathway
Since NP has been shown to interact with the CRM1‐mediated nuclear export pathway (Elton et al, 2001), we examined whether succinate could affect CRM1 protein expression or function. As shown in Fig EV3A, CRM1 levels in IAV‐infected cells remained the same, whether these cells were exposed or not to succinate. Next, we compared the effect of succinate and leptomycin B (LMB), a specific CRM1 inhibitor, on the trafficking of the viral NEP/NS2 protein. This last protein is known to be translocated into the cytosol in a CRM1‐dependent manner (Kudo et al, 1998, 1999). As expected, nuclear export of NEP/NS2 was inhibited by LMB. In contrast, succinate did not alter NEP/NS2 localization, indicating that succinate does not mimic the CRM1‐dependent effects of LMB (Fig EV3B). Hence, these data suggest that succinate causes nuclear retention of NP, in a CRM1‐independent way.
Figure EV3. Succinate does not alter the CRM1‐dependent transport pathway or the NP oligomerization.
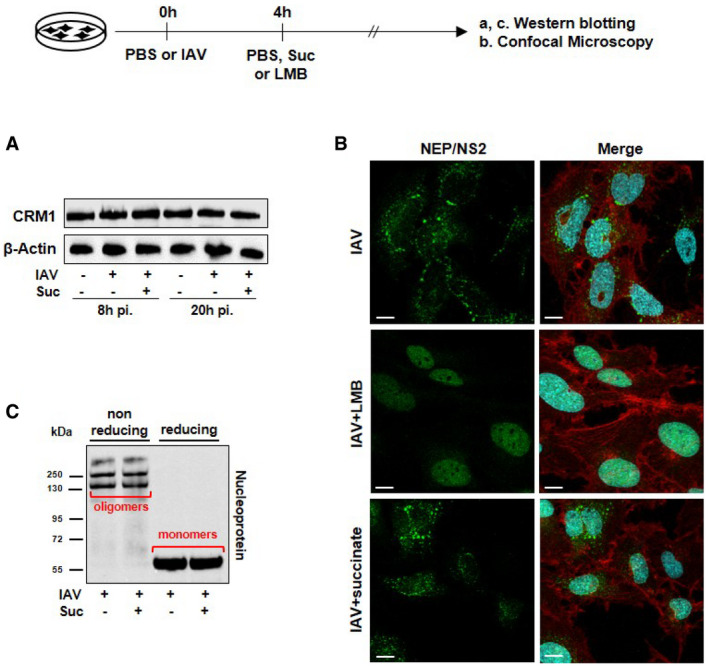
-
A, BHuman bronchial epithelial BEAS‐2B cells were infected or not with the A/Scotland/20/74 (H3N2) virus (IAV) at MOI = 1 for 4 h, and subsequently treated or not with 4 mg/ml of succinate for 20 h (A) or with 10 mM of the CRM1 inhibitor leptomycin B (LMB; middle panels in B). The expression of CRM1 was analyzed by Western blotting (A). Expression of the viral protein NEP/NS2 was analyzed by confocal fluorescence microscopy (B). NEP/NS2 is stained in green, nuclear DNA in blue and actin cytoskeleton in red. Scale bar: 10 µM.
-
CThe formation of NP oligomers and monomers was assessed by Western blotting under nonreducing and reducing conditions.
Data information: Pictures are representative of three independent experiments.
Nucleoprotein is a major structural component of vRNPs. Viral (v)RNAs are encapsidated by NP with approximately 12 nucleotides per NP molecule (Dou et al, 2018). NP oligomerization is likely to be involved in the higher order structure of vRNPs (Elton et al, 1999). We used a nondenaturing western‐blotting approach to test whether succinate could alter NP oligomerization. Fig EV3C clearly shows that this is not the case.
Succinate induces succinylation of NP at K87, a highly conserved amino acid involved in vRNA binding
Next, we evaluated the impact of succinate on the interaction of NP with the vRNA. To this end, we used confocal microscopy and a monoclonal antibody that specifically recognizes the “NP‐vRNA” complexes, but not unbound NP (Eisfeld et al, 2011), as illustrated in Fig 8A (upper right panel). Remarkably, succinate clearly reduced the fluorescence signal that marks NP‐vRNA complexes but not the fluorescence signal associated to the total form of NP (Fig 8A (lower panels) and B). These results suggested that succinate reduces the assembly of vRNPs. We further questioned what could be the underlying molecular mechanism.
Figure 8. Succinate reduces the assembly of influenza virus vRNPs by inducing succinylation of NP at K87, a highly conserved amino acid probably involved in vRNA binding.
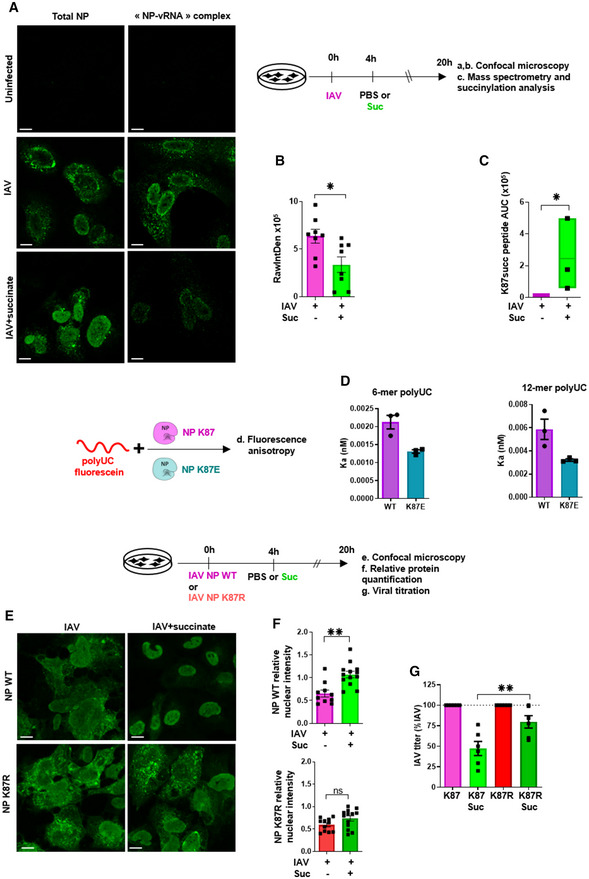
-
A, BCells were stained using a monoclonal antibody that specifically recognizes “NP‐vRNA” complexes, but not RNA‐free NP, and confocal microscopy (A) was performed (green staining). The value raw integrated density (RawIntDen, which is the sum of all pixel values in the ROI (region of interest)) was further measured (B).
-
CCells were lysed and proteins were digested to peptides with Lys‐C and trypsin. Then, succinylated peptides were isolated directly from protease‐digested protein extracts by immunoaffinity purification (IAP) using an antibody specific for the succinyl‐Lysine motif. Next, the modified peptides were further analyzed by LC‐MS/MS. Box plots show the distribution of intensities of the succinylated NP “YLEEHPSAGK87succDPK” peptide, computed as the integrated mass spectrometry peak area of the modified K87 succinyl peptide (n = 3). The boxplots whiskers correspond to the minimum and maximum values, while the central band is the median. The first and third quartiles (boxes) are merely the average values between the median and the extrema.
-
DInteraction of recombinant of wild‐type NP (NP K87) or a K87E mutant protein (NP K87E) with the indicated synthetic polyUC RNAs as measured by fluorescence anisotropy.
-
EHuman bronchial epithelial BEAS‐2B cells were infected with a A/Scotland/20/74 (H3N2) virus (IAV) carrying a wild‐type NP (NP WT) or with the corresponding mutated virus bearing a NP with a K87R substitution (NP K87R). Cells were infected by either virus at MOI = 1 for 4 h, then washed and treated or not with 4 mg/ml of succinate for 20 h. Localization of NP proteins was analyzed by confocal immunofluorescence microscopy. Scale bar: 10 µM. Pictures are representative of three independent experiments.
-
FRelative nuclear intensity of NP was determined by using the Intensity Ratio Nuclei Cytoplasm Tool.
-
GPlaque‐Forming Unit assays determined the production of infectious viral particles in cell supernatants.
As a precursor of succinyl‐coenzyme A, succinate can promote protein post‐translational modification (PTM) via succinylation on lysine residues (Xie et al, 2012; Park et al, 2013; Yang & Gibson, 2019). Lysine succinylation can cause a mass change (~100 Da) and converts a positively charged side chain into a negatively charged one, potentially causing changes in protein structure and function. Therefore, we used liquid chromatography coupled to untargeted tandem mass spectrometry (LC‐MS/MS) to examine whether succinate induced the succinylation of IAV proteins, especially NP. Notably, of all IAV proteins, only NP showed lysine succinylation in cells treated with succinate. Moreover, NP succinylation occurred at a unique residue, that is, K87 (Fig 8C and Appendix Fig S7).
In regard to these latter data as well as those highlighting the impact of succinate on vRNPs assembly (Fig 8A), it is of interest to point out that K87 residue is located in the RNA‐binding groove of NP (Ye et al, 2006). Hence, two types of experiments were further performed to assess the functional impact of succinate‐induced NP succinylation on (i) the formation of NP‐vRNA complexes and (ii) the intracellular trafficking of NP.
In line with lysine succinylation of K87, fluorescence anisotropy analysis showed that a recombinant K87E mutant NP (which bears a negative charge similar to a succinyl residue) interacts less with 6‐mer and 12‐mer polyUC RNA strands than wild‐type NP does (Fig 8D). Moreover, reverse genetic experiments showed that an IAV mutant strain bearing a K87E NP is not replicative (not illustrated), suggesting that the positively charged K87 is critical in IAV life cycle.
Conversely, we used reverse genetics and confocal microscopy to monitor the localization of NP protein in lung epithelial cells infected either by a wild‐type IAV strain (coined “NP K87”) or an IAV mutant (coined “NP K87R”; i.e., a virus which can normally replicate as assessed by PFU assay and qPCR (Appendix Fig S8) but carries a NP that cannot be succinylated at the amino acid position 87, due to the lysine‐to‐arginine substitution; Xie et al, 2012; Park et al, 2013; Yang & Gibson, 2019). We first confirmed that succinate induces a significant nuclear retention of NP in cells infected with the “NP K87” IAV strain (Fig 8E, upper panels). By contrast, succinate could not impair the intracellular trafficking of NP in cells infected by the mutant “NP K87R” IAV strain (Fig 8E, lower panels). Those findings were supported by an accurate quantification of NP relative nuclear intensities (Fig 8F). Besides, viral titer measurements in epithelial cells infected by “NP K87R” strain indicated that this mutant virus is significantly more resistant to succinate treatment than wild‐type K87 IAV (P < 0.01; Fig 8G). Altogether, our results suggest that succinate‐induced alterations of IAV life cycle involve a succinylation of NP at the lysine residue 87.
Discussion
In this study, we highlight the interplay between immunometabolism and antiviral immunity. Using a combination of in vitro and in vivo approaches, (i) we demonstrated that succinate accumulates in the airways of IAV‐infected mice as well as in patients with influenza illness; (ii) we found that this metabolite displays a potent and specific antiviral effect; (iii) we revealed that succinate induces a unique succinylation of the viral NP residue K87, which probably impairs vRNPs assembly and accounts for the observed retention of NP in the cell nucleus and disruption of the viral replication cycle; (iv) we showed that instillation of succinate in the airways protects hosts from IAV‐triggered acute pneumonia.
Hence, we have confirmed and extended previous studies that revealed profound metabolic changes in lung fluids of patients (Cui et al, 2016b) as well as in mice (Chandler et al, 2016; Cui et al, 2016a; Smallwood et al, 2017) infected by IAV. Our findings are consistent with studies suggesting that viruses can reprogram host cell metabolic pathways (Eisenreich et al, 2019). Remarkably, our PCA and multivariate statistics showed that succinate is one of the predominant metabolites that accumulate in the lungs of IAV‐infected hosts. Also, we established that succinate accumulation was restricted to the lung airspaces as succinate levels were not increased in the lung parenchyma compartment (not illustrated). This latter finding suggests that succinate modulates influenza pathogenesis by acting on cells of the top layer of the lung mucosa. However, further studies are needed to clarify which cell(s) is/are the major source of succinate accumulation in the airways and whether this is a repercussion of a dysregulated TCA catabolism and/or a higher extracellular transport of succinate.
Of note is that extracellular succinate increases in several pathological contexts with local tissue levels reaching up to 20 mM (Graham et al, 2013; Littlewood‐Evans et al, 2016). This concentration is consistent with the noncytotoxic and active amount of succinate that demonstrates an antiviral effect in our study.
Tricarboxylic acid cycle‐derived metabolites are not mere intermediates for ATP synthesis but have recently been demonstrated to play a critical immunomodulatory role (Zasłona & O’Neill, 2020). In particular, succinate has been identified as a crucial pro‐inflammatory signal that regulates the transcription factor HIF‐1α in several leukocyte subtypes (Rubic et al, 2008; Tannahill et al, 2013; Jha et al, 2015; Littlewood‐Evans et al, 2016; Mills et al, 2016; Zasłona & O’Neill, 2020). Regarding non‐leukocyte compartments, succinate is considered an ulcerative, pro‐inflammatory molecule in intestinal mucosa (Connors et al, 2018). In contrast, extracellular succinate might also mediate anti‐inflammatory responses in neural stem cells as well as in adipose tissue resident macrophages (Keiran et al, 2019). Now, we extend these findings by revealing a downregulatory effect of succinate in IAV‐triggered inflammation in lung epithelium.
Influenza A virus is known to replicate within the nucleus of lung epithelial cells (Dou et al, 2018). To this end, the viral proteins need to fold correctly, oligomerize and/or associate with each other. The neo‐synthesized vRNPs have to be properly transported across the nuclear membrane and through the cytoplasm and be packaged into new virions (Dou et al, 2018). Here, we have clearly shown that succinate induces a nuclear retention of NP, a major component of vRNPs.
Nucleoprotein is a highly basic protein which surface displays many positively charged amino acids such as lysines, which bind the negatively charged vRNAs. Therefore, NP is the main structural component of the vRNPs. X‐ray crystallographic analysis of NP structure has previously characterized an “intrinsically disordered region (IDR)‐2” (72‐DERRNKYLEEHPSAGKDPKKT‐92), located in an RNA‐binding groove that exhibits the highest conservation ratio among human, swine, and avian IAVs (Kakisaka et al, 2016). In that regard, K87 is extremely conserved (> 99.9%) among 2400 (H5N1), 23400 (H3N2), and 18800 (H1N1) IAV strains (source: Influenza research database; https://www.fludb.org; selection criteria: (i) complete coding region sequences; (ii) no geographical or temporal restrictions; (iii) no host discrimination (iv) no elimination of duplicates; not illustrated).
Influenza A virus is known to interact with the host cellular machinery, including the posttranslational modifications (PTMs) components. The most common PTMs include phosphorylation, ubiquitination, SUMOylation, acetylation, methylation, NEDDylation, and glycosylation. Many PTMs foster influenza virus infection, whereas others can contribute to antiviral defense (Hu et al, 2020).
Remarkably, succinate triggers a unique succinylation modification of K87 in NP, whereas none of the other ten major IAV proteins were found to be succinylated. Our findings further support the hypothesis that K87 is a critical amino acid in the “IDR‐2” of NP, in agreement with previous data showing that a K87A change in NP results in a decreased binding affinity for RNA (Kakisaka et al, 2016; Tang et al, 2021). Moreover, we found that (i) a NP with a K87E mutation (which bears a negative charge, mimicking a succinyl residue) interacts less efficiently than the wild‐type NP with a synthetic RNA and (ii) succinate treatment results in a lower RNA‐NP assembly. All these data are schematized in Appendix Fig S9. This is in line with the fact that IAV life cycle is not impaired by succinate‐induced lysine succinylation in epithelial cells infected by a virus strain bearing an NP K87R substitution. Taken together, these data strongly point to the fact that succinylation of the NP‐K87 could alter the electrostatic environment of the vRNA interaction site and could further impact the formation of vRNPs particles.
Concomitantly, we found that succinate treatment does not modify the accumulation of viral mRNAs or viral proteins, indicating that transcription and replication of the viral genome by the viral polymerase in the nucleus of infected cells are essentially unaffected, despite the altered NP‐RNA interaction. In the presence of succinate, the CRM1‐dependent nuclear export machinery is not inhibited either. However, NP appears to be retained in the nucleus, the levels of vRNP‐associated NP in cytoplasm are decreased, and budding of viral particles and the production of infectious virions are strongly impaired. In view of these data, we propose a hypothetical model (Fig 9) in which succinylation of NP at the highly conserved K87 residue promotes a charge shift in the RNA‐binding groove. This alters the encapsidation of vRNAs with NP in a way that preserves transcription/replication of the viral genome and the nuclear export of a vRNP’s moiety (as suggested by the cytoplasmic staining of NEP/NS2 and the polymerase subunits at late time‐points of the infection). It remains to be determined whether NP succinylation affects the packaging of the viral genome into viral particles. However, this proposal is consistent with the evidence that packaging of the segmented viral genome is governed not only by a complex network of RNA‐RNA interactions but also by specific RNA‐NP interactions (Moreira et al, 2016; Bolte et al, 2019).
Figure 9. Hypothetical model of the anti‐influenza activity of succinate.
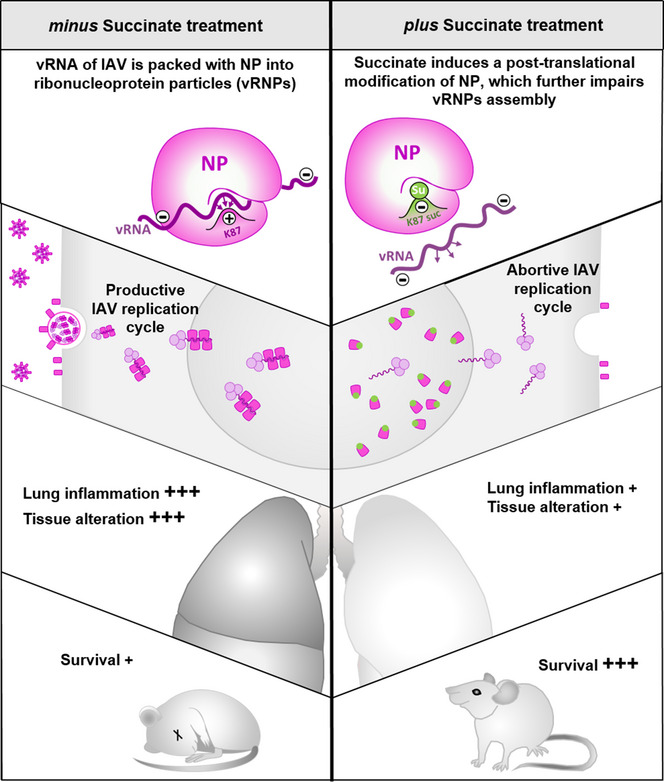
Succinate triggers a unique succinylation modification of K87 in the NP of IAV which further alters the electrostatic environment of the vRNA binding site. As a result, the formation of vRNPs particles is impaired. This further contributes to an inhibition of viral multiplication as well as viral‐induced inflammatory response. In vivo, IAV‐infected mice treated intranasally with succinate are more resistant to the development of acute pneumonia.
Regarding a limitation of our study, we have to mention a similar survival of mice infected by wild‐type IAV or by the mutant K87R IAV. This suggests that in vivo, the antiviral effect of succinate is likely the result of not only the impairment of IAV trafficking in lung epithelial cells but also the involvement of additional mechanisms, for example, inhibitory effects in other lung cell subsets such as resident and/or infiltrated leukocytes. Determination of the underlying signaling mechanisms and the cells involved in the global antiviral effect of succinate will therefore require further exploration.
In conclusion, our study demonstrates that IAV infection increases succinate levels in the airways and that this metabolite reduces IAV production through mechanisms that involve a specific posttranslational modification. This further contributes to an inhibition of viral multiplication as well as viral‐induced inflammatory response. More importantly, IAV‐infected mice treated intranasally with succinate show increased resistance to the development of acute pneumonia. Hence, our study paves the way for the development of safe and promising succinate‐based treatment of IAV infection.
Materials and Methods
Viruses
Mouse adapted‐influenza A/Scotland/20/74 (H3N2) was generously given by Pr. Sylvie van der Werf’s team (Pasteur Institute, Paris, France). The influenza A/PR/8/34 (H1N1) wild‐type and ΔNS1 strains were kindly provided by Dr. Georg Kochs (Freiburg University, Germany). Influenza A/WSN/33 (H1N1) wild‐type and NS1 flag‐tagged viruses were produced in Dr. Bernard Delmas’ team (INRAe, Jouy‐en‐Josas, France).
Animal infection and fluid collection
C57Bl/6 female mice were purchased from the Centre d'Elevage R. Janvier (Le Genest Saint‐Isle, France) and were used at about 8 weeks of age. Mice were treated in accordance with the European animal welfare regulation. Mice were bred in an animal facility in pathogen‐free conditions. Mice were fed normal mouse chow and water ad libitum and were reared and housed under standard conditions with air filtration. When needed, C57Bl/6 mice were challenged intranasally with 150 pfu of A/Scotland/20/74 (H3N2) IAV. At 4 days postinfection, airways were washed four times with 0.5 ml of PBS for BAL collection. After centrifugation, BAL fluids were stored at −80°C for subsequent measurement of inflammatory mediators and pellets were recovered in PBS 2% FBS. Erythrocytes were discarded using a red blood cell lysis buffer and leukocytes were counted and analyzed by flow cytometry. Lungs were crushed with 2 ml of PBS using Gentle MACSTM M tube and the Gentle MACS™ Octo Dissociator (Miltenyi Biotech). Lungs extracts were centrifugated 5 min at 500 g to collect supernatants which were further stored at −80°C for subsequent Western‐blotting.
Metabolomic analysis of respiratory fluids using 1H‐nuclear magnetic resonance
Freeze‐dried BAL samples were mixed with 210 µl of 0.2 M potassium phosphate buffer in deuterium oxide (D2O) 99%, pH = 7.4. Samples were spiked with 8 µl of 3‐trimethylsilylpropionic acid (0.05% wt in D2O) as an internal reference, and then samples were transferred to conventional 3 mm NMR tubes. 1H‐NMR spectra were obtained with an AVANCE III HD 600 Bruker spectrometer equipped with a TCI cryoprobe (Bruker). Standard water suppressed 1H‐NMR spectra were acquired at 298K using a “noesypr1d” pulse sequence with a relaxation delay of 20 s and 256 scans. Spectra were processed using Topspin software (Bruker). 1H‐NMR spectra were automatically reduced to ASCII files using the AMIX software package (Analysis of Mixture, version 3.8, Bruker). Spectral intensities were scaled to the total spectral intensity, and the resulting data were analyzed by multivariate and univariate statistics. The NMR assignments were done using spectra online databases such as HMDB (http://www.hmdb.ca) and ChenomX NMR Suite (version8.1 evaluation, ChenomX). The identification of succinate was carried out by supplementing a sample with exogenous succinate. Spectra analysis carried out with and without supplementation, allowed us to ensure signal specificity.
Nonsupervised multivariate principal component analyses (PCA) were performed with data generated by 1H‐NMR (BAL) using SIMCA v14 software (Umetrics). The scores were plotted in 2D versus the two first principal components to visualize possible clusters. Succinate quantification in BAL and tracheal aspirates was performed using Metaboanalyst software (www.metaboanalyst.ca).
Tracheal Aspirate collection
Aspirates were collected during routine tracheal suctioning of mechanically ventilated patients. Tracheal aspirate samples were collected from patients hospitalized at the University Hospital of Tours; 9 with influenza pneumonia and 7 not infected with influenza virus. Samples were dissociated in 3 volumes of PBS per gram, and 1 mM dithiothreitol, with stirring for 30 min. After centrifugation at 500 g for 10 min, the supernatants were stored at −80°C until use.
Cytokine array and ELISA
Cytokine array and DuoSet ELISA (Human IL‐6, IL‐8/CXCL8, IP10/CXCL10, RANTES/CCL5 and mouse IL‐6, KC, MPO) were performed according to the manufacturer’s (R&D Systems) instructions.
Cell culture
Experiments were performed using human bronchial epithelial BEAS‐2B cells, except for live imaging which used human alveolar epithelial A549 cells, plaque assays, which used Madin–Darby Canine Kidney (MDCK) cells and reverse genetics which used co‐culture of MDCK and HEK‐293T cells. All cells were mycoplasma‐free, cultured in either F‐12K Medium (BEAS‐2B), DMEM (16HBE14o‐) or MEM (A549, HEK‐293T and MDCK) supplemented with 10% FBS and 100 U/ml penicillin, 100 µg/ml streptomycin. BEAS‐2B cells were infected in medium without FBS for 4 h with A/Scotland/20/74 (H3N2) IAV at MOI = 1 (except for SEM analysis, for which an MOI = 5 was applied, and in experiments using A/PR/8/34 (H1N1) wild‐type and deltaNS1 viruses which used an MOI = 4). Cells were also stimulated in medium with FBS with 2 µg/ml of Poly (I:C). Four hours after the challenge, cells were washed with PBS and incubated for 4 h or 20 h with different concentrations of succinate or malonate, or with 10 nM of Leptomycin B. For multicycle replication analysis, BEAS‐2B cells were infected with IAV at low MOI (i.e., MOI = 10−3) for 4 h and treated or not with succinate for 20 h. 2 µg/ml of TPCK‐treated Trypsin were added simultaneously to succinate. Supernatants were centrifuged 5 min at 500 g and stored at −80°C for further analysis.
Cell proliferation and cytotoxicity assays
Cells in 96‐well plates were washed twice with PBS and incubated for 1 h at 37°C with 100 µl of MTS reagent diluted 1/5 for the cell proliferation test. Optical density was measured at 490 nm. For the cytotoxicity assay, cells were stained for 15 min at 4°C with Live/Dead or with anti‐Annexin V‐FITC (1/20) and propidium iodide (1/100) before flow cytometry analysis.
Microarrays
Profiling of human bronchial epithelial BEAS‐2B cells was performed using Agilent’s SurePrint G3 human gene expression microarray kit. A single‐color design provided two types of comparison: (i) IAV‐infected versus mock‐infected cells, and (ii) succinate‐treated infected cells versus mock‐infected cells. Cy3‐labeled cRNAs were prepared from 100 ng of total RNA using a one‐color Low Input Quick Amp labeling kit. Specific activities and cRNA yields were determined using a NanoDrop ND‐1000 instrument. For each sample, 600 ng of Cy3‐labeled cRNA (specific activity of > 9 pmol Cy3/µg cRNA) was fragmented at 60°C for 30 min. cRNA was hybridized to the microarrays for 17 h at 65°C in a rotating hybridization oven as described previously (Barthelemy et al, 2018). After hybridization, microarrays were washed and then dried immediately. After washing, the slides were scanned using a G2565CA scanner system (Agilent Technologies), with a resolution of 3 µm and a 20‐bit dynamic range. The resulting TIFF images were analyzed with Feature Extraction Software, using a GE1_107_Sep09 protocol. Identification of differentially expressed genes and functional investigations were done using GeneSpring software. Differentially expressed genes were identified using a moderated t test, and a P value cutoff of 5% was applied. A fold change cutoff of > 2 was added to select genes expressed differentially between control and treated/infected conditions. For further analysis, data files were uploaded into the Ingenuity Pathways Analysis (IPA) software (Ingenuity Systems). The right‐tailed Fisher’s exact test was used to calculate a P value for the probability that each canonical pathway assigned to that dataset is due to chance alone.
SeaHorse
Cellular oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) data were obtained using a Seahorse XF96 flux analyzer from Seahorse Bioscience (Agilent Technologies) as we described previously (Kouzi et al, 2020). Experiments were performed according to the manufacturer’s instructions. Briefly, bronchial epithelial BEAS‐2B cells were seeded in XF96 cell culture plates at 30,000 cells per well and cultured overnight, and then cells were infected with IAV for 18 h in the presence or absence of succinate. On the day of analysis, culture media were replaced with XF Base medium supplemented with glutamine (2 mM) and lacking bicarbonate (pH 7.4). Cells were then incubated at 37°C in a non‐CO2 incubator for 1 h and measurements were performed as described in the figure legends. Sequential injection of glucose (10 mM), oligomycin (1 µM), dinitrophenol (DNP) (100 µM), and rotenone/antimycine A (0.5 µM) were added according to the suppliers’ technical specifications and these permitted the determination of the main respiratory and glycolytic parameters. Data were acquired with the Seahorse Wave Controller and analyzed with Seahorse Wave Desktop Software.
Neuraminidase (NA) assay
The assay is based on the release of a 4‐methylumbelliferone fluorescent product from the 2′‐(4‐Methylumbelliferyl)‐α‐D‐N‐acetylneuraminic acid sodium salt hydrate (MU‐NANA) substrate as a measure of NA activity. 67 μl of cell supernatant was incubated with 33 µl of MU‐NANA (50 µM) in black 96‐well micro‐plates. Fluorescence was immediately measured in a kinetic assay over 1 h at Ex = 355 nm and Em = 460 nm.
IAV titration by plaque‐forming units assay
Titrations in cell supernatants, BALs, and mouse lungs were performed as previously described (Blanc et al, 2016).
Transmission and scanning electron microscopy
Cells were washed with PBS, detached using trypsin and centrifuged. Cells were fixed by incubation for 24 h in 4% paraformaldehyde, 1% glutaraldehyde in 0.1 M phosphate buffer (pH 7.2). Samples were further washed in PBS and postfixed by incubation with 2% osmium tetroxide for 1 h. Next, samples were fully dehydrated in a graded series of ethanol solutions and propylene oxide. An impregnation step was performed with a mixture of (1∶1) propylene oxide/Epon resin and then samples were left overnight in pure resin. They were further embedded in Epon resin for 48 h at 60°C. Ultra‐thin sections (90 nm) of these blocks were obtained using a Leica EM UC7 ultramicrotome (Wetzlar, Germany). Sections were stained with 2% uranyl acetate and 5% lead citrate. Observations were made with a transmission electron microscope (JEOL 1011) and analyzed with a Digital Micrograph.
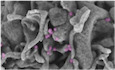
For scanning electron microscopy, samples were fully dehydrated in a graded series of ethanol solutions and dried in hexamethyldisilazane (HMDS, Sigma, St. Louis, MO). Dried samples were sprinkled onto carbon disks and coated with 40 Å platinum, with a GATAN PECS 682 apparatus (Pleasanton, CA), before observation under a Zeiss Ultra plus FEG‐SEM scanning electron microscope (Oberkochen, Germany). IAV particles were colored in pink by digital computer processing.
Western‐blotting
Cells in 6‐well plates were lysed with 150 µl of RIPA buffer (150 mM sodium chloride, 50 mM Tris–HCl, 1 mM ethylenediaminetetraacetic acid, 1% Triton X100, 1% sodium deoxycholic acid, 0.1% sodium dodecyl sulfate) and a protease inhibitor cocktail (diluted 1/200). Samples were centrifuged for 15 min at 12,000 g to eliminate debris, and then protein concentrations were measured using a PierceTM Protein BCA Assay Kit. Ten μg of total proteins were diluted with reducing or nonreducing Laemmli buffer, heated at 100°C for 5 min, then loaded onto 4/12% SDS‐PAGE. Proteins were subsequently transferred to nitrocellulose membranes, and probed with anti‐NP (1/500), anti‐NS1 (1/1,000), anti‐PA (1/1,000), anti‐PB2 (1/500), anti‐M1 (1/1,000), anti‐M2 (1/1,000), anti‐CRM1 (1/500), or anti‐β‐actin (1/5,000). Bound antibodies were revealed with an anti‐rabbit IgG for NS1 and PB2 and anti‐mouse IgG for other proteins (HRP linked) and ECL detection reagents. An automated imaging system (MF ChemiBis 3.2, DNR BioImaging Systems) was used for detection, and the FUJI FILM MultiGauge software was subsequently used for analysis and quantification.
RNA isolation and RT‐qPCR
Cells in 6‐well plates were lysed with 350 µl of “RA1” buffer (included in the Macherey–Nagel RNA extraction kit) and β‐mercaptoethanol diluted 1/100. Total RNAs from cells or from purified A/Scotland/20/74 (H3N2) IAV were extracted using the NucleoSpin® RNA kit, including a step of genomic DNA digestion with DNase. Nucleic samples were quantified using a Nanodrop 2000 UV‐visible spectrophotometer. Single‐stranded cDNA was synthesized from 500 ng total RNA for each sample with the High Capacity cDNA reverse transcription kit (Applied Biosystems), using the specific sense IAV M1 primer (5′‐TCT AAC CGA GGT CGA AAC GTA‐3′). mRNA levels were determined using quantitative real‐time PCR with a LightCycler 480 instrument (Roche Diagnostics). PCR was carried out using 10 ng of reverse‐transcribed total RNA as the template, 10 µM (each) forward and reverse primers (sense: 5′‐AAG ACC AAT CCT GTC ACC TCT GA‐3′; antisense: 5′‐CAA AGC GTC TAC GCT GCA GTC C‐3′), and 10 µl SYBR® Premix Ex Taq in a final volume of 20 µl. Each reaction was performed in duplicate in white 96‐well plates. The thermal protocol consisted of an initial denaturation step at 95°C for 30 s followed by 40 cycles of denaturation at 95°C for 5 s and primer annealing and extension at 60°C for 20 s (reading at 83°C). Melting curves were generated for each amplified cDNA to check the reaction specificity. For quantification analyses, serial dilutions of IAV cDNA (from 40 ng to 0.5 ng cDNA) were used to create a standard curve.
Flow cytometry analysis
Bronchoalveolar lavage was dispensed into round bottomed 96‐well plates and was centrifuged at 300 g at 4°C for 5 min. One well was seeded for each of the following controls: unstained cells, single‐stained cells, isotype controls. Samples were further stained using specific antibodies and appropriate isotype controls as listed in the supplementary table.
Adherent epithelial cells were removed from their substrate by treatment with Trypsin a few minutes at 37°C. Trypsin was inhibited by adding 800 µl of culture media and cells were centrifugated at 500 g for 5 min. Cell pellet was resuspended in staining buffer (50 mM EDTA, 5% FCS in PBS), dispensed into round bottomed 96‐well plates (100,000 cells in 100 µl), and were then centrifuged at 300 g at 4°C for 5 min. For nucleoprotein staining, cells were permeabilized with the Cytofix/Cytoperm™ Solution Kit or True Nuclear™ Transcription Factor Buffer Set, following manufacturer’s instructions. Samples were further incubated for 1 h at 4°C with anti‐NP‐FITC antibody diluted 1/300e or isotype control. Flow cytometry data were acquired on a MACSQuant® Analyzer and analyses were performed using the VenturiOne software.
Histopathology
Lungs were collected immediately after euthanasia and airways were washed and placed in 4% paraformaldehyde in PBS. Lung sections of approximately 4 µm thickness were cut and stained with hematoxylin–eosin. A study pathologist examined the tissue sections using light microscopy on a Leica Diaplan microscope in a blinded experimental protocol. All histopathological findings were graded in a semi‐quantitative fashion on a scale of 0–4 (0: absent, 1: mild, 2: moderate, 3: severe, 4: extremely severe). All lung preparations and analyses were performed at the LAPV (Amboise, France).
Generation of recombinant fluorescent IAV and time‐lapse imaging
The 8 plasmid reverse genetics system (Hoffmann et al, 2002) was used to generate a recombinant virus expressing an eGFP reporter protein fused to the NS1 protein of the influenza A/WSN/33 (H1N1) virus. The NS1‐eGFP‐WSN virus was rescued using a previously described procedure (Vidy et al, 2016) and was propagated in MDCK cells. For long‐term time‐lapse microscopy, A549 cells grown on cell culture in micro‐wells were infected with NS1‐eGFP‐WSN at an MOI of 0.5 pfu/cell. Pictures were taken every 10 min starting at 1 h postinfection and continuing until 24 h postinfection, on a Nikon BioStation microscope using BioStation IM software. The intensity of the GFP associated fluorescence per infected cell was quantified using Image J software.
Confocal fluorescence microscopy
Cells were grown in 12‐well plates with a cover slide in the bottom of the well. After different treatments, cells were fixed using 4% formaldehyde for 30 min at room temperature and then permeabilized in PBS 0.1% Triton X‐100 for 30 min at room temperature. Following a 1 h saturation step with PBS 1% bovine serum albumin, 0.1% Tween 20, cells were stained for 2 h at room temperature with the following antibodies: anti‐NP‐FITC (1/30), anti‐NS1 (1/200), anti‐M2 (1/150), anti‐M1 (1/150), anti‐PA (1/50), anti‐PB2 (1/200), anti‐NEP/NS2 (1/100), or anti‐NP mAb clone 3/1 (1/1,000). An anti‐rabbit‐AF488 was used for 2 h at room temperature as the secondary antibody for NS1, NEP/NS2, and PB2, and an anti‐mouse‐AF488 was used for other proteins. Then actin was stained with ActinRed 555 reagent for 30 min and nuclei were stained with the NucBlue reagent for 5 min. Samples were analyzed with a Leica SP8 confocal microscope and Leica LasX Life Sciences Software. Relative nuclear intensity was determined by using the Intensity Ratio Nuclei Cytoplasm Tool, RRID:SCR_018573).
Succinylation analysis by LC‐MS/MS
Human bronchial epithelial BEAS‐2B cells were grown in 10 culture dishes (150 mm) per condition. After different treatments, cells were washed with 5 ml of cold PBS, and then lysed with 5 ml of urea buffer (8 M urea, 200 mM ammonium bicarbonate). The lysates were clarified by centrifugation at 12,000 g for 15 min at 4°C. Protein concentrations were measured with a Nano Drop spectrophotometer to take samples corresponding to 8 mg of proteins. Reduction and alkylation of proteins were performed by incubation for 1 h at 37°C with 10 mL of DTT 10 mM (5 mM final) and then 30 min at room temperature with 4 ml of iodoacetamide 55 mM (10 mM final). After the addition of 20 ml ammonium bicarbonate 200 mM, proteins were digested for 2 h at 37°C with Trypsin/LysC diluted 1/1,000. 800 µl of TPCK‐trypsin at 1 mg/ml were added and the samples were incubated overnight at 37°C. Five mL of 50% formic acid was added to allow purification of peptides over Sep‐Pak® C18 columns. Briefly, columns were equilibrated with 3 × 2 ml of 70% CAN + 0.1% formic acid and were washed with 3 × 2 ml of 0.1% formic acid. Samples were loaded, and columns washed as previously. Elution was done with 6 ml 40% CAN + 0.1% formic acid. Eluates were frozen (−80°C) overnight and lyophilized. Immunoaffinity purification of succinylated peptides was performed with a PTMScan® Succinyl‐Lysine Motif kit according to the manufacturer’s instructions. Immuno‐isolate peptide samples were then loaded onto a homemade C18 StageTip for desalting. Peptides were eluted using 40/60 MeCN/H2O + 0.1% formic acid and vacuum concentrated to dryness before being reconstituted in injection buffer (2% MeCN/0.3% TFA) for nano‐LC‐MS/MS analysis. Liquid chromatography (LC) was performed with an RSLCnano system (Ultimate 3000, Thermo Scientific) coupled online to an Orbitrap Exploris™ 480 mass spectrometer (Thermo Scientific). Peptides were trapped on a C18 Nano Trap column with buffer A (2/98 MeCN/H2O in 0.1% formic acid) at a flow rate of 3.0 µl/min over 4 min. Separation was performed on a C18 Acclaim PepMap™ RSLC column regulated to a temperature of 50°C with a linear gradient of 2% to 25% buffer B (100% MeCN in 0.1% formic acid) at a flow rate of 300 nl/min over 91 min. MS full scans were performed in the ultrahigh‐field Orbitrap mass analyzer in m/z ranges of 375–1,500 with a resolution of 120,000 at m/z 200. The top 25 most intense ions were subjected to Orbitrap for further fragmentation via high energy collision dissociation (HCD) activation and a resolution of 15,000 with the AGC target set to 100%. We selected ions with charge state from 2+ to 6+ for screening. Normalized collision energy (NCE) was set at 30 and the dynamic exclusion was 45 s. Mass spectrometry proteomics raw data have been deposited with the ProteomeXchange Consortium via the PRIDE (Perez‐Riverol et al, 2019) partner repository with the dataset identifier PXD018419 (username: reviewer53974@ebi.ac.uk, password: hTdy0lLL). For identification, the data were searched against the Homo Sapiens (UP000005640) and the influenza A virus (UP000162741) UniProt databases using Sequest‐HT through proteome discoverer (version 2.2 or 2.4). Enzyme specificity was set to trypsin and a maximum of three‐missed cleavages sites were allowed. Oxidized methionine, N‐terminal acetylation, Succinyl‐Lysine, and carbamidomethyl cysteine were set as variable modifications. The maximum mass deviation allowed was set to 10 ppm for monoisotopic precursor ions and 0.02 Da for MS/MS peaks. The resulting files were further processed using myProMS (Poullet et al, 2007). FDR calculation employed Percolator and was set to 1% at the peptide level for the whole study. To quantify the succinylated peptide that contained lysine K87, we extracted from the MS survey of the nano‐LC‐MS/MS raw files the ion chromatogram (XIC) signal using the retention time and m/z values of the well‐characterized tryptic peptide ions using the Xcalibur software (manually). XIC areas were integrated in Xcalibur under the QualBrowser interface using the ICIS algorithm. Boxplots were constructed from two sets of three independent experiments (IAV and IAV + Succinate). Thus, three data points were used for each box. The whiskers correspond to the minimum and maximum values, while the last point is the median. The first and third quartiles are merely the average values between the median and the extrema. It should be noted that boxplots do not indicate actual distributions (there are no values inside a box, except the median), but rather they are used to show different tendencies between two conditions.
Fluorescence anisotropy measurements
The full‐length influenza virus NP (strain A/WSN/1933) gene, cloned in pET22b (Novagen), was used. The point mutation that gives the K87 > E change was introduced by PCR, by site directed mutagenesis and full‐plasmid amplification, followed by DpnI restriction digestion. Sequencing was done by Eurofins Genomics. Production in Escherichia coli BL21 RIL (DE3) cells and purification of wild‐type NP and K87E were carried out as previously described (Chenavas et al, 2013; Labaronne et al, 2016). Fluorescence anisotropy measurements were done in a 384‐well microplate, and acquisitions were performed at room temperature after a 5‐min incubation on a Clariostar plate reader (BMG Labtech) with polarization filters. NP was serially diluted in buffer (20 mM Tris‐HCl pH 7.5, 150 mM NaCl and 5 mM β‐mercaptoethanol) and titrated into 5 nM polyUC RNA (Integrated DNA Technologies) 3′‐labeled with 6‐fluorescein (FAM). After blank subtraction of the RNA alone, a triplicate of each titration was fitted to the Prism (GraphPad) binding equation “Single binding site with Hill slope (h).”
Reverse genetics
Five hundred nanograms of each of the 8 bidirectional pRF483 plasmids encoding all the genomic segments of the A/Scotland/20/74 (H3N2) virus were transfected by LTX lipofectamine (Invitrogen) in a mix of HEK293T and MDCK cells. The reconstitution of viruses with an NP bearing the wild‐type K87, the K87R, or the K87E mutations was performed in parallel. Negative controls of each virus were also prepared by transfection of cells with all genomic‐encoding plasmids, with the exception of the PB1 expression plasmid. After 24 h, the cells were washed and incubated for 48 h in MEM medium supplemented with 1 μg/ml of trypsin‐TPCK. The viruses released were further amplified in MDCK cells in MEM medium supplemented with 1 μg/ml of trypsin‐TPCK and titrated after 72 h. NP sequence of rescued viruses (K87 and K87R) were verified by sequencing using the following primers (sense: 5’‐ATG GCG TCC CAA GGC‐3’; antisense: 5’‐TTA ATT GTC GTA CTC CTC TGC‐3’; Eurofins Genomics)).
Statistical analysis
Sample size estimate for animal studies was based on the experience of our group on IAV‐infected mice model. There was no randomization or blinding procedure. Sample size for the human investigation could not be calculated because of the exploratory nature of this research but it was estimated according to previous studies with a similar design (Guillon et al, 2015, 2016, 2019). Statistical analyses were performed using GraphPad Prism. Data are reported as mean ± SEM. Statistical values, including the number of replicates (n) and the statistical test used, can be found in the figure legends. *P < 0.05, **P < 0.005, ***P < 0.0005, ****P < 0.0001. For in vitro experiments, n = the number of separate experiments. For in vivo work, n = the number of individual animals.
Study approvals
All animal experimentations were performed according to the ethical guidelines and were approved by our local and national ethics committee (CEEA.19, #201604071220401‐4885). Study of human samples was approved by the IRB of the Tours University Hospital/French national bioethics authority (CPP‐37 2012‐R21) (DC‐2014‐2285). Informed written consent was obtained from all subjects. The study was conformed to the principles set out in the VMA Declaration of Helsinki and the Department of Health and Human Service Belmont Report.
Author contributions
Antoine Guillon: Conceptualization; Formal analysis; Supervision; Validation; Writing—original draft; Writing—review and editing. Deborah Brea‐Diakite: Data curation; Formal analysis; Validation; Investigation; Visualization; Methodology. Adeline Cezard: Formal analysis; Validation; Investigation; Methodology. Alan Wacquiez: Validation; Investigation; Methodology. Thomas Baranek: Validation; Investigation; Methodology; Writing—review and editing. Jerome Bourgeais: Validation; Investigation; Methodology; Writing—review and editing. Frédéric Picou: Validation; Investigation; Methodology. Virginie Vasseur: Investigation; Methodology. Léa Meyer: Investigation; Methodology. Christophe Chevalier: Resources; Methodology; Writing—review and editing. Adrien Auvet: Formal analysis; Investigation; Methodology. José M. Carballido: Resources. Lydie Nadal Desbarats: Validation; Investigation; Methodology; Writing—review and editing. Florent Dingli: Validation; Investigation; Methodology. Andrei Turtoi: Resources; Investigation; Writing—review and editing. Audrey Le Gouellec: Investigation; Methodology; Writing—review and editing. Florence Fauvelle: Investigation; Methodology; Writing—review and editing. Amélie Donchet: Investigation; Methodology. Thibaut Crepin: Resources; Validation; Investigation; Methodology; Writing—review and editing. Pieter S. Hiemstra: Resources; Validation; Writing—review and editing. Christophe Paget: Resources; Validation; Writing—review and editing. Damarys Loew: Resources; Validation; Investigation; Methodology; Writing—review and editing. Olivier Herault: Resources; Validation; Writing—review and editing. Nadia Naffakh: Resources; Formal analysis; Validation; Writing—review and editing. Ronan Le Goffic: Resources; Formal analysis; Validation; Writing—review and editing. Mustapha Si‐Tahar: Conceptualization; Data curation; Formal analysis; Supervision; Funding acquisition; Validation; Writing—original draft; Project administration; Writing—review and editing.
Disclosure and competing interests statement
M.S.‐T. holds a French national research agency grant (ANR‐21‐CE18‐0061‐01, which started January 2022) to develop a succinate‐based therapy against influenza virus infection. The other authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Movie EV1
Movie EV2
Acknowledgements
This work was partially supported by the following grants: Inserm, Université of Tours, Région Centre‐Val de Loire FLU‐MET#2018‐00124196, VLM#RF2018052289 (to M.S.‐T.); FEDER Euro‐FERI (to M.S.‐T. and C.P.), Studium research fellowship (to P.S.H. and M.S.‐T.), French research network on influenza viruses (ResaFlu, GDR2073) financed by CNRS (to M.S.‐T, R.L.G, N.N., T.C., C.C.); SIRIC Montpellier Cancer Grant INCa_Inserm_DGOS_12553 (to A.T.); LabEx IBEID Grant No. 10‐LABX‐0062 (to N.N.); Ph.D. fellowship from INRAe – Department of animal health (to L.M.). This work used the platforms of the Grenoble Instruct‐ERIC center (ISBG; UMS 3518 CNRS‐CEA‐UGA‐EMBL) within the Grenoble Partnership for Structural Biology (PSB), supported by FRISBI (ANR‐10‐INBS‐05‐02) and GRAL, financed by the University Grenoble Alpes – Ecoles Universitaires de Recherche CBH‐EUR‐GS (ANR‐17‐EURE‐0003). The authors are very grateful to Dr Evgenia Turtoi (Université de Montpellier, France) for LC‐MS metabolomic analyses, sample preparations and measurements, to Dr. Bruno Da Costa (VIM, INRAe, Jouy‐en‐Josas, France) for his contribution to the production of wild‐type and mutant influenza viruses by reverse genetics and to Pr. Rob WH Ruigrok (IBS, Grenoble, France) for fruitful discussions. We also thank Dr Richard Webby (St. Jude Children's Research Hospital, Memphis, USA) and Dr Daniel Marc (INRAe, Nouzilly, France) for their generous gifts of the mAb 3/1 antibody and the polyclonal anti‐NS1 antibody, respectively, and Dr Georg Kochs (Freiburg University) for kindly providing the A/PR/8/34 wild‐type and ∆NS1 viruses. The authors would like to thank the Animal Facility of the University of Tours, the study nurses and all the physicians of the Critical Care Section of Tours University Hospital, for their assistance. We are also grateful to all patients who volunteered for this study.
The EMBO Journal (2022) 41: e108306
Data availability
MIAME (minimum information about microarray experiment) compliant data were deposited in Array‐Express at EMBL (E‐MTAB‐10031; https://www.ebi.ac.uk/arrayexpress/experiments/E‐MTAB‐10031/). Succinylation data provided by LC‐MS/MS were deposited in PRIDE (PXD018419; http://www.ebi.ac.uk/pride/archive/projects/PXD018419).
References
- Bahadoran A, Bezavada L, Smallwood HS (2020) Fueling influenza and the immune response: Implications for metabolic reprogramming during influenza infection and immunometabolism. Immunol Rev 295: 140–166 [DOI] [PubMed] [Google Scholar]
- Barthelemy A, Sencio V, Soulard D, Deruyter L, Faveeuw C, Le Goffic R, Trottein F (2018) Interleukin‐22 immunotherapy during severe influenza enhances lung tissue integrity and reduces secondary bacterial systemic invasion. Infect Immun 86, e00706‐17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc F, Furio L, Moisy D, Yen H‐L, Chignard M, Letavernier E, Naffakh N, Mok CKP, Si‐Tahar M (2016) Targeting host calpain proteases decreases influenza A virus infection. Am J Physiol Lung Cell Mol Physiol 310: L689–L699 [DOI] [PubMed] [Google Scholar]
- Bolte H, Rosu ME, Hagelauer E, García‐Sastre A, Schwemmle M (2019) Packaging of the influenza virus genome is governed by a plastic network of RNA‐ and nucleoprotein‐mediated interactions. J Virol 93: e01861‐18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandler JD, Hu X, Ko E‐J, Park S, Lee Y‐T, Orr M, Fernandes J, Uppal K, Kang S‐M, Jones DP et al (2016) Metabolic pathways of lung inflammation revealed by high‐resolution metabolomics (HRM) of H1N1 influenza virus infection in mice. Am J Physiol Regul Integr Comp Physiol 311: R906–R916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chenavas S, Estrozi LF, Slama‐Schwok A, Delmas B, Di Primo C, Baudin F, Li X, Crépin T, Ruigrok RWH (2013) Monomeric nucleoprotein of influenza A virus. PLoS Pathog 9: e1003275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connors J, Dawe N, Van Limbergen J (2018) The role of succinate in the regulation of intestinal inflammation. Nutrients 11: 25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui L, Zheng D, Lee YH, Chan TK, Kumar Y, Ho WE, Chen JZ, Tannenbaum SR, Ong CN (2016a) Metabolomics investigation reveals metabolite mediators associated with acute lung injury and repair in a murine model of influenza pneumonia. Sci Rep 6: 26076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Z, Ojaghian MR, Tao Z, Kakar KU, Zeng J, Zhao W, Duan Y, Vera Cruz CM, Li B, Zhu B et al (2016b) Multiplex PCR assay for simultaneous detection of six major bacterial pathogens of rice. J Appl Microbiol 120: 1357–1367 [DOI] [PubMed] [Google Scholar]
- Denney L, Ho L‐P (2018) The role of respiratory epithelium in host defence against influenza virus infection. Biomed J 41: 218–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou D, Revol R, Östbye H, Wang H, Daniels R (2018) Influenza A virus cell entry, replication, virion assembly and movement. Front Immunol 9: 158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duwe S (2017) Influenza viruses – antiviral therapy and resistance. GMS Infect Dis 5: Doc04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenreich W, Rudel T, Heesemann J, Goebel W (2019) How viral and intracellular bacterial pathogens reprogram the metabolism of host cells to allow their intracellular replication. Front Cell Infect Microbiol 9: 42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisfeld AJ, Kawakami E, Watanabe T, Neumann G, Kawaoka Y (2011) RAB11A is essential for transport of the influenza virus genome to the plasma membrane. J Virol 85: 6117–6126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elton D, Medcalf E, Bishop K, Digard P (1999) Oligomerization of the influenza virus nucleoprotein: identification of positive and negative sequence elements. Virology 260: 190–200 [DOI] [PubMed] [Google Scholar]
- Elton D, Simpson‐Holley M, Archer K, Medcalf L, Hallam R, McCauley J, Digard P (2001) Interaction of the influenza virus nucleoprotein with the cellular CRM1‐mediated nuclear export pathway. J Virol 75: 408–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham LS, Krass L, Zariffard MR, Spear GT, Mirmonsef P (2013) Effects of succinic acid and other microbial fermentation products on HIV expression in macrophages. Biores Open Access 2: 385–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory DJ, Kobzik L (2015) Influenza lung injury: mechanisms and therapeutic opportunities. Am J Physiol Lung Cell Mol Physiol 309: L1041–L1046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimolizzi F, Arranz L (2018) Multiple faces of succinate beyond metabolism in blood. Haematologica 103: 1586–1592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillon A, Brea D, Morello E, Tang A, Jouan Y, Ramphal R, Korkmaz B, Perez‐Cruz M, Trottein F, O’Callaghan RJ et al (2016) Pseudomonas aeruginosa proteolytically alters the interleukin 22‐dependent lung mucosal defense. Virulence 8: 810–820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillon A, Brea D, Luczka E, Hervé V, Hasanat S, Thorey C, Pérez‐Cruz M, Hordeaux J, Mankikian J, Gosset P et al (2019) Inactivation of the interleukin‐22 pathway in the airways of cystic fibrosis patients. Cytokine 113: 470–474 [DOI] [PubMed] [Google Scholar]
- Guillon A, Jouan Y, Brea D, Gueugnon F, Dalloneau E, Baranek T, Henry C, Morello E, Renauld J‐C, Pichavant M et al (2015) Neutrophil proteases alter the interleukin‐22‐receptor‐dependent lung antimicrobial defence. Eur Respir J 46: 771–782 [DOI] [PubMed] [Google Scholar]
- Guillot L, Le Goffic R, Bloch S, Escriou N, Akira S, Chignard M, Si‐Tahar M (2005) Involvement of toll‐like receptor 3 in the immune response of lung epithelial cells to double‐stranded RNA and influenza A virus. J Biol Chem 280: 5571–5580 [DOI] [PubMed] [Google Scholar]
- Hoffmann E, Krauss S, Perez D, Webby R, Webster RG (2002) Eight‐plasmid system for rapid generation of influenza virus vaccines. Vaccine 20: 3165–3170 [DOI] [PubMed] [Google Scholar]
- Hu J, Zhang L, Liu X (2020) Role of post‐translational modifications in influenza A virus life cycle and host innate immune response. Front Microbiol 11: 517461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Infantino V, Pierri CL, Iacobazzi V (2019) Metabolic routes in inflammation: the citrate pathway and its potential as therapeutic target. Curr Med Chem 26: 7104–7116 [DOI] [PubMed] [Google Scholar]
- Jang YH, Seong BL (2019) The quest for a truly universal influenza vaccine. Front Cell Infect Microbiol 9: 344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jefferson T, Jones MA, Doshi P, Del Mar CB, Hama R, Thompson M, Spencer EA, Onakpoya I, Mahtani KR, Nunan DN et al (2014) Neuraminidase inhibitors for preventing and treating influenza in healthy adults and children. Cochrane Database Syst Rev CD008965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha A, Huang S‐C, Sergushichev A, Lampropoulou V, Ivanova Y, Loginicheva E, Chmielewski K, Stewart K, Ashall J, Everts B et al (2015) Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 42: 419–430 [DOI] [PubMed] [Google Scholar]
- Kakisaka M, Yamada K, Yamaji‐Hasegawa A, Kobayashi T, Aida Y (2016) Intrinsically disordered region of influenza A NP regulates viral genome packaging via interactions with viral RNA and host PI(4,5)P2. Virology 496: 116–126 [DOI] [PubMed] [Google Scholar]
- Keiran N, Ceperuelo‐Mallafré V, Calvo E, Hernández‐Alvarez MI, Ejarque M, Núñez‐Roa C, Horrillo D, Maymó‐Masip E, Rodríguez MM, Fradera R et al (2019) SUCNR1 controls an anti‐inflammatory program in macrophages to regulate the metabolic response to obesity. Nat Immunol 20: 581–592 [DOI] [PubMed] [Google Scholar]
- Keshavarz M, Solaymani‐Mohammadi F, Namdari H, Arjeini Y, Mousavi MJ, Rezaei F (2020) Metabolic host response and therapeutic approaches to influenza infection. Cell Mol Biol Lett 25: 15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzi F, Zibara K, Bourgeais J, Picou F, Gallay N, Brossaud J, Dakik H, Roux B, Hamard S, Le Nail L‐R et al (2020) Disruption of gap junctions attenuates acute myeloid leukemia chemoresistance induced by bone marrow mesenchymal stromal cells. Oncogene 39: 1198–1212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudo N, Matsumori N, Taoka H, Fujiwara D, Schreiner EP, Wolff B, Yoshida M, Horinouchi S (1999) Leptomycin B inactivates CRM1/exportin 1 by covalent modification at a cysteine residue in the central conserved region. Proc Natl Acad Sci USA 96: 9112–9117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudo N, Wolff B, Sekimoto T, Schreiner EP, Yoneda Y, Yanagida M, Horinouchi S, Yoshida M (1998) Leptomycin B inhibition of signal‐mediated nuclear export by direct binding to CRM1. Exp Cell Res 242: 540–547 [DOI] [PubMed] [Google Scholar]
- Labaronne A, Swale C, Monod A, Schoehn G, Crépin T, Ruigrok RWH (2016) Binding of RNA by the nucleoproteins of influenza viruses A and B. Viruses 8: 247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampejo T (2020) Influenza and antiviral resistance: an overview. Eur J Clin Microbiol Infect Dis 39: 1201–1208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Goffic R, Balloy V, Lagranderie M, Alexopoulou L, Escriou N, Flavell R, Chignard M, Si‐Tahar M (2006) Detrimental contribution of the Toll‐like receptor (TLR)3 to influenza A virus‐induced acute pneumonia. PLoS Pathog 2: e53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Littlewood‐Evans A, Sarret S, Apfel V, Loesle P, Dawson J, Zhang J, Muller A, Tigani B, Kneuer R, Patel S et al (2016) GPR91 senses extracellular succinate released from inflammatory macrophages and exacerbates rheumatoid arthritis. J Exp Med 213: 1655–1662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills EL, Kelly B, Logan A, Costa ASH, Varma M, Bryant CE, Tourlomousis P, Däbritz JHM, Gottlieb E, Latorre I et al (2016) Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell 167: 457–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreira ÉA, Weber A, Bolte H, Kolesnikova L, Giese S, Lakdawala S, Beer M, Zimmer G, García‐Sastre A, Schwemmle M et al (2016) A conserved influenza A virus nucleoprotein code controls specific viral genome packaging. Nat Commun 7: 12861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy MP, O’Neill LAJ (2018) Krebs cycle reimagined: the emerging roles of succinate and itaconate as signal transducers. Cell 174: 780–784 [DOI] [PubMed] [Google Scholar]
- Park J, Chen Y, Tishkoff D, Peng C, Tan M, Dai L, Xie Z, Zhang YI, Zwaans B, Skinner M et al (2013) SIRT5‐mediated lysine desuccinylation impacts diverse metabolic pathways. Mol Cell 50: 919–930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce EJ, Pearce EL (2018) Immunometabolism in 2017: driving immunity: all roads lead to metabolism. Nat Rev Immunol 18: 81–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez‐Riverol Y, Csordas A, Bai J, Bernal‐Llinares M, Hewapathirana S, Kundu DJ, Inuganti A, Griss J, Mayer G, Eisenacher M et al (2019) The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res 47: D442–D450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizzorno A, Padey B, Terrier O, Rosa‐Calatrava M (2019) Drug repurposing approaches for the treatment of influenza viral infection: reviving old drugs to fight against a long‐lived enemy. Front Immunol 10: 531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter VR, Dubois KP (1943) Studies on the mechanism of hydrogen transport in animal tissues: VI. Inhibitor studies with succinic dehydrogenase. J Gen Physiol 26: 391–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poullet P, Carpentier S, Barillot E (2007) myProMS, a web server for management and validation of mass spectrometry‐based proteomic data. Proteomics 7: 2553–2556 [DOI] [PubMed] [Google Scholar]
- Rambold AS, Pearce EL (2018) Mitochondrial dynamics at the interface of immune cell metabolism and function. Trends Immunol 39: 6–18 [DOI] [PubMed] [Google Scholar]
- Rubic T, Lametschwandtner G, Jost S, Hinteregger S, Kund J, Carballido‐Perrig N, Schwärzler C, Junt T, Voshol H, Meingassner JG et al (2008) Triggering the succinate receptor GPR91 on dendritic cells enhances immunity. Nat Immunol 9: 1261–1269 [DOI] [PubMed] [Google Scholar]
- Ryan DG, Murphy MP, Frezza C, Prag HA, Chouchani ET, O’Neill LA, Mills EL (2019) Coupling Krebs cycle metabolites to signalling in immunity and cancer. Nat Metab 1: 16–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons C, Farrar J (2008) Insights into inflammation and influenza. N Engl J Med 359: 1621–1623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Si‐Tahar M, Touqui L, Chignard M (2009) Innate immunity and inflammation–two facets of the same anti‐infectious reaction. Clin Exp Immunol 156: 194–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smallwood HS, Duan S, Morfouace M, Rezinciuc S, Shulkin BL, Shelat A, Zink EE, Milasta S, Bajracharya R, Oluwaseum AJ et al (2017) Targeting metabolic reprogramming by influenza infection for therapeutic intervention. Cell Rep 19: 1640–1653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y‐S, Xu S, Chen Y‐W, Wang J‐H, Shaw P‐C (2021) Crystal structures of influenza nucleoprotein complexed with nucleic acid provide insights into the mechanism of RNA interaction. Nucleic Acids Res 49: 4144–4154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tannahill GM, Curtis AM, Adamik J, Palsson‐McDermott EM, McGettrick AF, Goel G, Frezza C, Bernard NJ, Kelly B, Foley NH et al (2013) Succinate is an inflammatory signal that induces IL‐1β through HIF‐1α. Nature 496: 238–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taubenberger JK, Kash JC, Morens DM (2019) The 1918 influenza pandemic: 100 years of questions answered and unanswered. Sci Transl Med 11: eaau5485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian X, Zhang K, Min J, Chen C, Cao Y, Ding C, Liu W, Li J (2019) Metabolomic analysis of influenza A virus A/WSN/1933 (H1N1) infected A549 cells during first cycle of viral replication. Viruses 11: 1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidy A, Maisonnasse P, Da Costa B, Delmas B, Chevalier C, Le Goffic R (2016) The influenza virus protein PB1‐F2 increases viral pathogenesis through neutrophil recruitment and NK cells inhibition. PLoS One 11: e0165361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendt CH, Castro‐Pearson S, Proper J, Pett S, Griffin TJ, Kan V, Carbone J, Koulouris N, Reilly C, Neaton JD et al (2021) Metabolite profiles associated with disease progression in influenza infection. PLoS One 16: e0247493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO (2009) WHO guidelines for pharmacological management of pandemic (H1N1) 2009 influenza and other influenza viruses WHO . [PubMed]
- Williams NC, O’Neill LAJ (2018) A role for the Krebs cycle intermediate citrate in metabolic reprogramming in innate immunity and inflammation. Front Immunol 9: 141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Dai J, Dai L, Tan M, Cheng Z, Wu Y, Boeke JD, Zhao Y (2012) Lysine succinylation and lysine malonylation in histones. Mol Cell Proteomics MCP 11: 100–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Gibson GE (2019) Succinylation links metabolism to protein functions. Neurochem Res 44: 2346–2359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Q, Krug RM, Tao YJ (2006) The mechanism by which influenza A virus nucleoprotein forms oligomers and binds RNA. Nature 444: 1078–1082 [DOI] [PubMed] [Google Scholar]
- Zasłona Z, O’Neill LAJ (2020) Cytokine‐like roles for metabolites in immunity. Mol Cell 78: 814–823 [DOI] [PubMed] [Google Scholar]