

Abstract
Purpose:
We aimed to characterize the urinary microbiome of catheterizing patients with neurogenic lower urinary tract dysfunction (NLUTD) and to evaluate differences based on type of bladder management or frequency of urinary tract infections.
Materials and Methods:
This is a prospective, cross-sectional study of urine samples from asymptomatic, catheterizing patients with (NLUTD) who used either clean intermittent catheterization (CIC) or indwelling catheters. Patients without symptoms of urinary tract infection provided a catheterized urine sample for urinalysis, culture, and bacterial community microbiome analysis.
Results:
Ninety-five patients submitted urine for analysis, of these 69 had sufficient sequence reads (>1203) for microbiome analysis. Patients with low bacterial signal amplification were associated with use of vaginal estrogen, no intradetrusor botulinum toxin A (BTX-A) use, and no growth on standard urine culture.
The most abundant operational taxonomic units (OTU) were from the phylum Proteobacteria, classified as Enterobacteriaceae and Escherichia. Alpha diversity varied among those who used indwelling catheters versus CIC, and those who underwent BTX-A injection versus not. On linear discriminate analysis, the relative abundance of the OTU identified as Pseudomonas was higher among patients using indwelling catheters relative to CIC. The OTU identified as Aerococcus was at a higher relative abundance among males compared to females.
Conclusions:
Enterobacteriaceae and Escherichia were the most abundant genera in the urinary microbiome of patients with NLUTD. Urinary microbiome diversity varied based on bladder management type. Future clinical correlations between microbiome of neurogenic patients and clinical presentation may help guide treatment strategies.
Keywords: microbiota, urine, urinary bladder, neurogenic, botulinum toxins, type a, urinary tract infection
INTRODUCTION:
The urinary microbiome consists of the microorganisms that live within the urinary track and their genes. Within the past decade, investigators have shown that the urinary microbiome is not sterile, that it is distinct from the vaginal and gut microbiome and that differences in the urinary microbiome are associated with urologic pathology such as incontinence, urinary tract symptoms and even malignancy.1–7 However the urinary microbiome of patients with neurogenic lower urinary tract dysfunction (NLUTD) remains largely unexplored.6,8
There is evidence that those with NLUTD have differing urinary microbiomes from non-NLUTD controls and that differences are correlated with gender, bladder management type and length of NLUTD diagnosis.6,7 Literature examining pediatric NLUTD found that Enterobacteriaceae was the most prevalent family in the urinary microbiome of this population, but no differences were found depending on whether children experienced urinary tract infections (UTI) or asymptomatic bacteriuria (ASB).9 These studies are limited by modest sample sizes, with the largest cohort comprising 36 patients.9 Many questions about the urinary microbiome in patients with NLUTD remain unanswered. It is unknown whether and how clinical management impacts the urinary microbiome and if those decisions affect urinary tract function and dysfunction among those with NLUTD. Efforts to describe the urinary microbiome in this population are a necessary step to generate hypotheses for future research and clinical applications.
This study aims to describe the urinary microbiome of catheterizing adults with NLUTD and evaluate for microbial differences based on demographics and clinical characteristics. We hypothesize that there will be differences in the microbiome based on gender, catheterization route, frequency of UTI, and use of prophylactic antibiotics. This study serves to bolster nascent data on the urinary microbiome of patients with NLUTD as well as to provide insights that can be leveraged for future applications in clinical urology.
METHODS:
This is a prospective, cross-sectional study approved by the University of Michigan Institutional Review Board (HUM00152141).
Recruitment:
Asymptomatic individuals at least 18 years old with diagnosis of NLUTD who performed CIC or used indwelling catheters were recruited at routine urology visits from January – September 2019. Patients who did not use catheters, those with UTI symptoms, radiation cystitis or bladder cancer were excluded. Patients provided written informed consent prior to data or sample collection.
Demographics and clinical information:
Demographic and baseline clinical information was collected, including age, gender, catheterization type and regimen, prophylactic antibiotic use (including gentamicin bladder irrigations), vaginal estrogen use, intradetrusor botulinum toxin A (BTX-A) treatment, and UTI history in the 12 months prior to the study visit. (Supplemental data)
Sample acquisition
Catheterized urine samples were provided during a routine clinic visit. If patients were presenting for intradetrusor BTX-A, then samples were obtained prior to antibiotic prophylaxis and BTX-A injection. The samples were separated into two aliquots: one for standard urinalysis and culture and another for microbiome analysis. Urine samples were refrigerated at 4°C until they were centrifuged and then they were stored at −68°C.
Deoxyribonucleic acid (DNA) sequencing and microbiome analysis
DNA isolation, library preparation, and sequencing were performed by the University of Michigan Microbial Systems Molecular Biology Laboratory. DNA was isolated with a MagAttract PowerMicrobiome DNA/Ribonucleic acid (RNA) Kit (Qiagen) using an epMotion 5075 liquid handling system. The V4 region of the 16S rRNA gene was amplified from 1, 2 or 5μl of undiluted DNA and sequenced as described previously (500-cycles) was used.10 The 16S rRNA gene sequence data was processed and analyzed using the software package mothur (v.1.42.3).11,12 Chimeras were identified and removed. After sequence processing and alignment to the SILVA reference alignment (release 132), sequences were binned into operational taxonomic units (OTUs) based on 97% sequence similarity using the OptiClust method.13,14 The OTUs were taxonomically classified within mothur using a modified version of the Ribosomal Database Project (RDP) training set (version 16).15,16 After sequencing, the range of sequences per sample was evaluated and a cut-off mark for low sample sequences was established by rank ordering all samples by number of sequences and excluding the bottom 25% of patients, representing 23 patients; this corresponded to samples with less than 1203 sequences. We did not perform analysis on samples with less than 1,203 sequences. In samples with more than 1,203 sequences, we randomly subsampled 1,203 sequences for analysis. The 16S rRNA gene sequences are available for download from the National Library of Medicine Sequence Read Archive (Accession: PRJNA764758 and ID: 764758).
Statistical analysis:
We grouped samples based on urologic management and patient demographic to determine whether distinct microbial signatures were associated with these characteristics. The five subgroups of interest were: bladder management strategy (CIC versus indwelling catheter), gender, use of antibiotic prophylaxis (oral versus gentamicin irrigations versus none), UTI frequency (≥ 3 versus < 3 per year), and BTX-A treatment. UTI frequency was based on patient reports of symptomatic UTI and verified with culture data when available.
The Inverse Simpson Index, an alpha diversity metric indicating the richness and evenness of OTUs within each sample, was calculated. We also assessed the richness of OTUs by evaluating the number of unique OTU per sample. We compared alpha diversity between groups using the non-parametric Mann-Whitney test, or Kruskal-Wallis test for more than two groups. Beta diversity measures were used to compare bacterial community composition between groups. For Beta diversity, we used the Yue and Clayton dissimilarity index (θYC distances), a metric that takes relative abundances of both shared and non-shared OTUs between communities and uses analysis of molecular variance (AMOVA).17,18 AMOVA calculates the F statistic (Fs), the ratio of the mean variance of microbiome within a subgroup compared to the total population.19 In this way, it is possible to determine if there are statistically significant differences between the microbiota of the patient groups. Principal coordinates analysis (PCoA) was used to visualize the θYC distances between samples and to observe potential clustering of samples. Linear discriminant analysis (LDA) effect size (LEfSe) was used to determine if specific OTUs were differentially abundant in different groups.20
Mann-Whitney and Kruskal-Wallis tests were performed using Stata (version 15, Stata, College Station, Tx). Community composition bar plots were made using the web-based program Calypso using genus level data from the taxonomy summary files generated within mothur21 AMOVA and LEfSe were run using mothur (version 1.42.3, University of Michigan, Ann Arbor, Michigan, USA). The significance level for all tests was set < 0.05. P values were not adjusted for multiple comparisons.
RESULTS
Ninety-five patients with a diagnosis of NLUTD were recruited and submitted urine samples. The median number of sequence reads per sample was 6418 (IQR 1064, 11071). We rank-ordered samples by the number of sequences and excluded the lowest 25th percent of samples (n=23), which corresponded to all samples with less than 1203 sequences per sample. Three samples were collected but were not able to be included in microbiome assay.
Participant demographics, clinical history and the number of samples included in microbiome analyses are detailed in Table 1. More patients with samples included in the microbiome analysis underwent BTX-A injection (55% vs 31%, p=0.04) and neither sample from the two patients using vaginal estrogen was included in the microbiome analysis. Supplemental Table 1 describes differences in demographics and clinical characteristics based on the five subgroups of interest. Indwelling catheter users were older (66 vs 53, p=0.006) and had higher BMI (35 vs 27, p< 0.001) and fewer underwent BTX-A (55% vs 22%, p=0.01) compared to CIC users.
Table 1:
Patient demographics and urologic management stratified by whether samples were included or excluded from microbiome analysis.
| Excluded from Microbiome Analysis | Included in Microbiome | |||
|---|---|---|---|---|
| Total (n=95) | (n=26) | analysis (n=69)** | p | |
| Age (mean, SD) | 56 (17) | 59 (15) | 55 (18) | 0.31 |
| Gender - Female | 52 (55%) | 13 (50%) | 39 (57%) | 0.57 |
| Race | 0.091 | |||
| Caucasian | 83 (87%) | 22 (85%) | 61 (88%) | |
| Asian | 2 (2%) | 2 (8%) | 0 (0%) | |
| African American | 3 (3%) | 0 (0%) | 3 (4%) | |
| Unknown | 7 (7%) | 2 (8%) | 5 (7%) | |
| BMI (mean, SD) | 29 (8) | 28 (6) | 29 (9) | 0.63 |
| Etiology of NLUTD | 0.57 | |||
| Spinal cord injury | 43 (45%) | 11 (42%) | 32 (46%) | |
| Multiple sclerosis | 11 (12%) | 4 (15%) | 7 (10%) | |
| Myelomeningocele | 5 (5%) | 2 (8%) | 3 (4%) | |
| Non-neurogenic urinary retention | 10 (11%) | 3 (12%) | 7 (10%) | |
| Other | 30 (32%) | 7 (27%) | 23 (33%) | |
| Bladder management | 0.14 | |||
| Intermittent catheterization | 76 (80%) | 23 (88%) | 53 (77%) | |
| Indwelling catheter | ||||
| Urethral | 15 (16%) | 2 (8%) | 13 (19%) | |
| Suprapubic | 3 (3%) | 0 (0%) | 3 (4%) | |
| 3 or more UTI over 12 months | 0.49 | |||
| Yes | 22 (23%) | 0 | 22 | |
| Missing | 25 (27%) | 25 (96%) | 1 (1%) | |
| Antibiotic Prophylaxis | 0.11 | |||
| Daily oral prophylaxis | 7 (7%) | 1 (4%) | 6 (9%) | |
| Gentamicin bladder irrigation | 30 (32%) | 5 (19%) | 25 (36%) | |
| None | 57 (60%) | 19 (73%) | 38 (55%) | |
| Both | 1 (1%) | 1 (4%) | 0 (0%) | |
| Non-Antibiotic Prophylaxis * | ||||
| Cranberry | 11 (12%) | 5 (19%) | 6 (9%) | 0.15 |
| D-mannose | 3 (3%) | 2 (8%) | 1 (1%) | 0.12 |
| Methanamine | 7 (7%) | 1 (4%) | 6 (9%) | 0.42 |
| Probiotics | 8 (8%) | 2 (8%) | 6 (9%) | 0.88 |
| Vaginal Estrogen | 2 (2%) | 2 (8%) | 0 (0%) | 0.02 |
| Bladder BTX-A Use | 46 (48%) | 8 (31%) | 38 (55%) | 0.035 |
Samples with low sequence counts (less than 1203 sequence reads per sample) (n=23) or samples that were unable to be included in microbiome assay (n=3) were excluded.
More than one therapy per participant may be counted
all participant samples within the category included in microbiome analyses, unless otherwise indicated.
Urine culture Results
Urine cultures were available for 77% of samples, and most resulted in >100,000 Colony Forming Units (26%) with the following bacteria E. coli, Klebsiella and Enterococcus. Samples with low signal were associated with no growth on urine culture (42% vs 13%, p < 0.001). (Table 2)
Table 2:
Results of urine cultures stratified by whether samples were included or excluded from microbiome analysis.
| Total (n=95) | Excluded from Microbiome Analysis (n=26) | Included in Microbiome analysis (n=69) | p | |
|---|---|---|---|---|
| Urine Culture Results | <0.001 | |||
| Negative | 20 (21%) | 11 (42%) | 9 (13%) | |
| <10K Colony forming units | 9 (9%) | 3 (12%) | 6 (9%) | |
| 10–100K Colony forming units | 20 (21%) | 4 (15%) | 16 (23%) | |
| >100K Colony forming units | 25 (26%) | 0 (0%) | 25 (36%) | |
| Missing | 21 (22%) | 8 (31%) | 13 (19%) | |
| Cultured Bacteria | 0.66 | |||
| Escherichia coli | 18 (19%) | 1 (4%) | 17 (25%) | |
| Klebsiella species | 11 (12%) | 2 (8%) | 9 (13%) | |
| Enterococcus species | 9 (9%) | 2 (8%) | 7 (10%) | |
| Proteus species | 5 (5%) | 0 (0%) | 5 (7%) | |
| Group B Streptococcus | 3 (3%) | 1 (4%) | 2 (3%) | |
| Mixed organisms (greater than 3) | 2 (2%) | 0 (0%) | 2 (3%) | |
| Other | 8 (8%) | 1 (4%) | 7 (10%) |
Microbiome composition
We obtained a total of 611,767 quality-filtered 16S rRNA reads from 92 samples, clustered into 261 unique OTUs. Analysis of the bacterial community revealed 2 to 87 OTUs (97% identity) per sample. There was a substantial amount of variability in the microbial composition of the urine of catheter-dependent individuals with NLUTD, as demonstrated in the community composition stacked bar plot. (Figure 1 and Supplemental Figure 1) Despite this variability, the community consisted of only a handful of dominant genera, with urine samples often predominated by a single genus of bacteria. Among the entire study population, the most abundant bacterial OTUs were from the phylum Proteobacteria, classified as Enterobacteriaceae and Escherichia.
Figure 1:
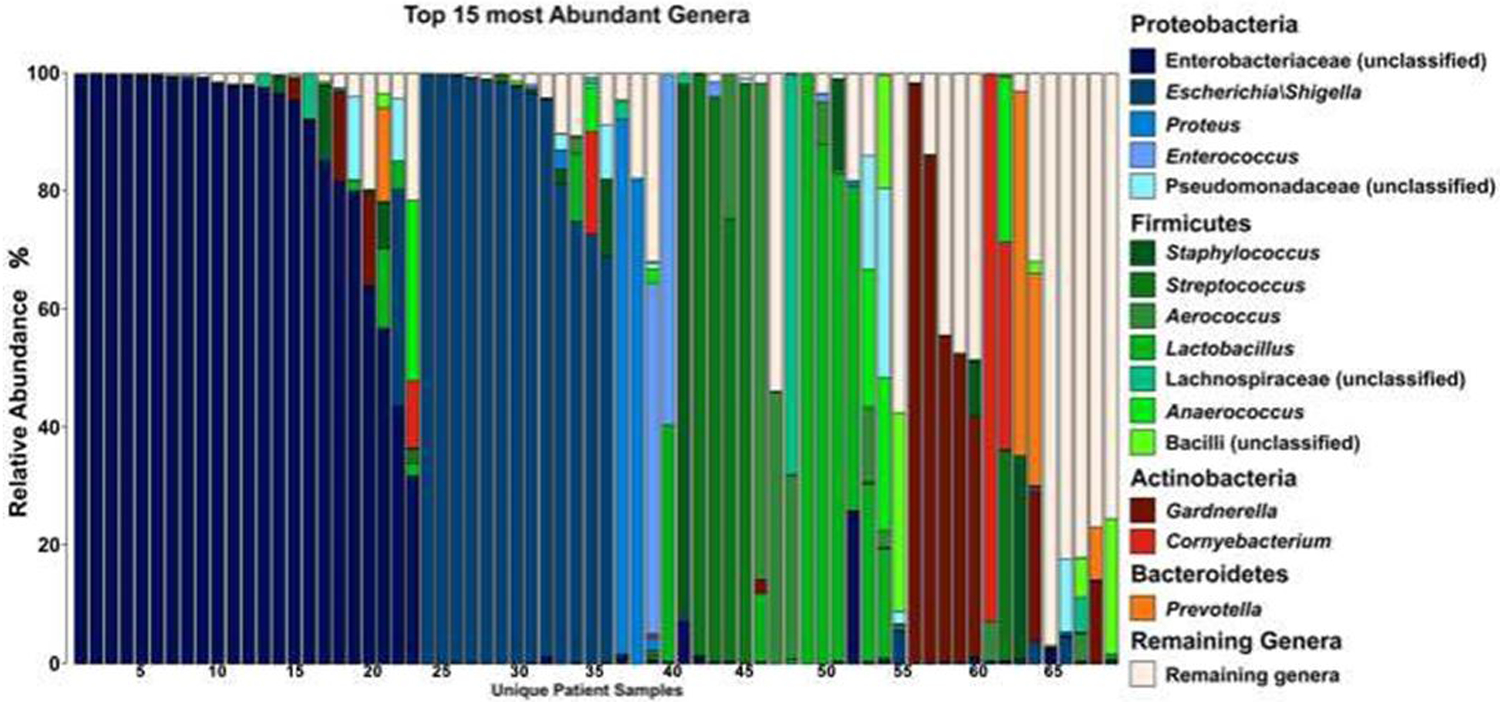
Stacked bar graph of the 15 most abundant genera among patients with neurogenic bladder. Each bar represents a different patient sample.
Alpha Diversity Measurement
Alpha diversity (Inverse Simpson Index) of the urine microbial community varied significantly based on catheterization type and BTX-A use. There was lower diversity on average among those using CIC versus indwelling catheters (1.85 versus 1.16 p=0.01) and those who underwent BTX-A injections compared to not (1.13 versus 1.71, p = 0.026). (Figure 2) We did not see differences in alpha diversity within other subgroups.
Figure 2:
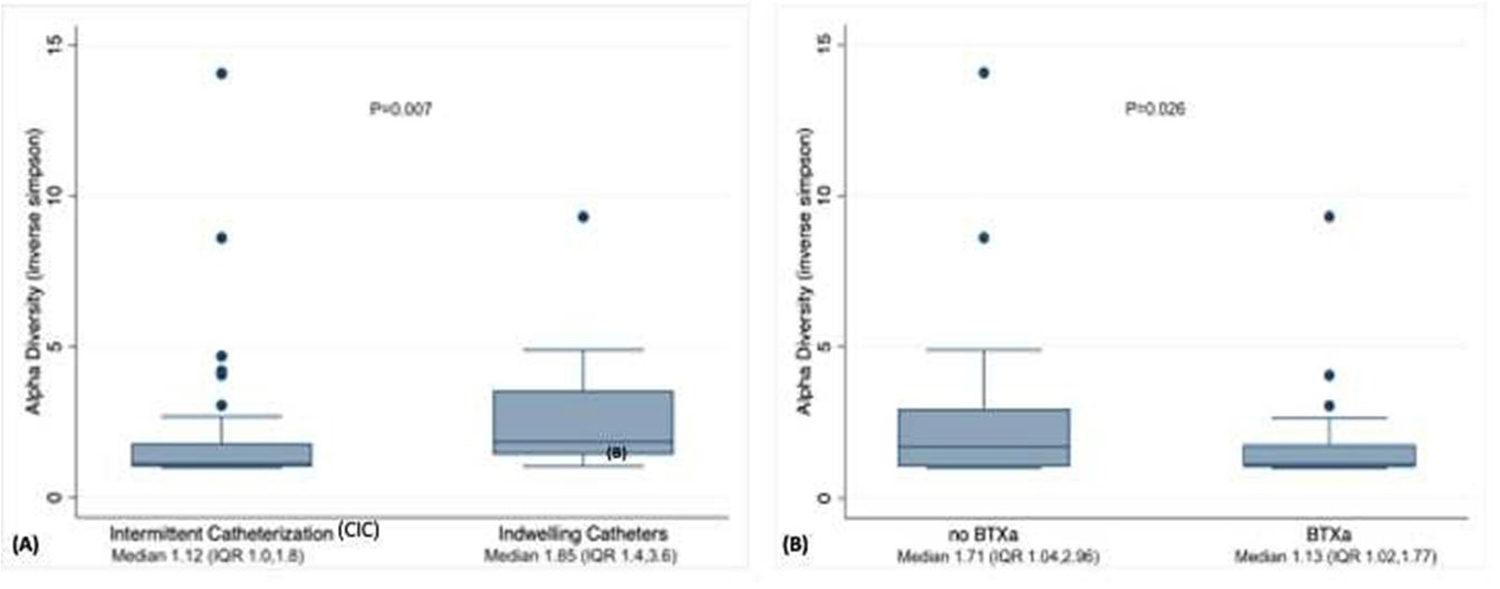
Box plot of diversity within samples (alpha diversity) of patients with neurogenic lower urinary tract dysfunction using (A) intermittent (CIC) versus indwelling catheter and use of intradetrusor (B) onabotulinumtoxin A (BTX-A) injection. Inverse Simpson Index of zero indicates no diversity and greater Inverse Simpson Index indicates greater diversity within each sample.
Beta Diversity Measurement
AMOVA detected no significant difference in urinary microbial community composition (beta diversity) based on catheterization type (Fs = 0.81, p = 0.69), gender (Fs = 0.96, p = 0.45), UTI frequency (Fs = 0.97, p = 0.42), BTX-A use (Fs = 0.91, p = 0.476) or antibiotic prophylaxis use (Fs = 1.2, p = 0.23). The θYC distances between samples based on the predefined subgroups were visualized by Principal Coordinate Analysis (PCoA) and no clustering was seen.
Linear Discriminant Analysis
LefSe was used to determine if specific OTUs were differentially abundant between groups. Pseudomonas was identified as being at a higher relative abundance in patients with indwelling catheters versus CIC (LDA score [log 10] 4.83, p=0.0003). However, the overall relative abundance of Pseudomonas was low. Aerococcus was at a higher relative abundance among males compared to females (LDA score [log 10] 4.23, p=0.009). (Figure 3) There were no other differences in relative abundance of OTUs within other pre-defined groups.
Figure 3:
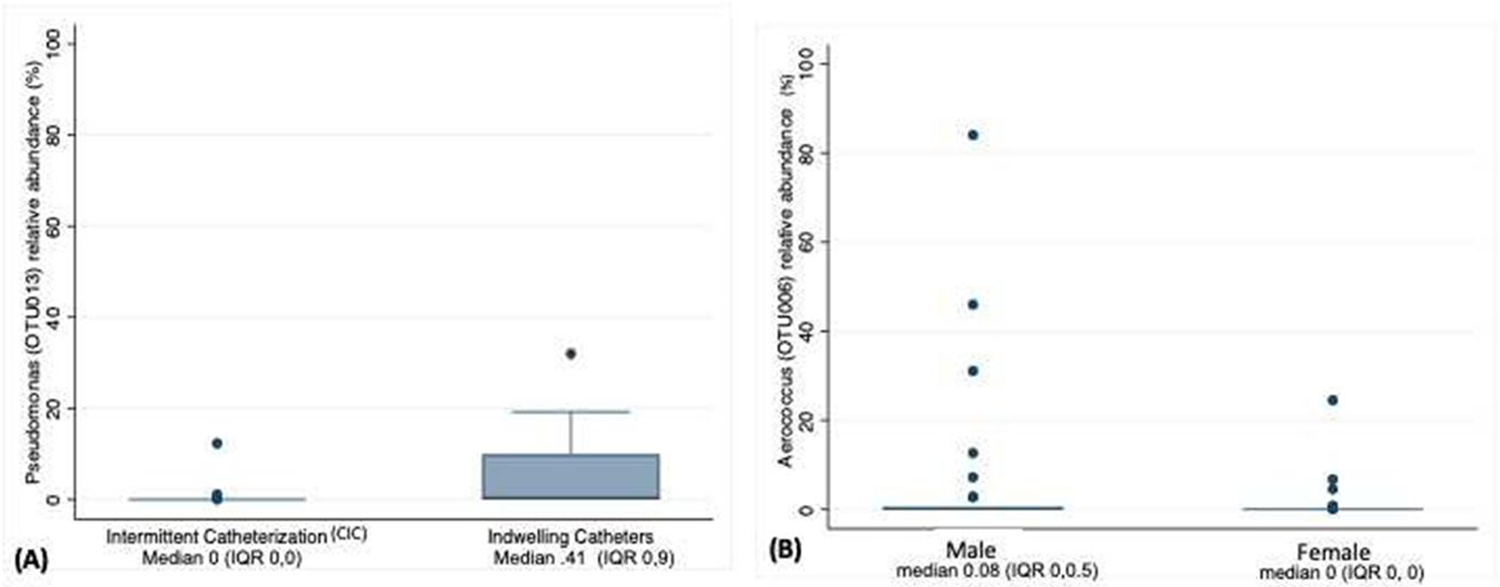
Box plot of Linear discriminant analysis (LDA) effect size (LEfSe) demonstrating that while overall relative abundance is low (A) Pseudomonas is differentially abundant among individuals utilizing indwelling catheters versus those on CIC, and (B) Aerococcus is differentially abundant among males compared to females.
DISCUSSION
In catheterized patients with NLUTD we find that the most abundant urinary bacterial OTUs were from the phylum Proteobacteria, classified as Enterobacteriaceae and Escherichia. Those with indwelling catheters had higher relative abundance of Pseudomonas compared to CIC, though the total abundance of Pseudomonas was low. Men had greater relative abundance of Aerococcus vs women. Patients with indwelling catheters also had higher microbiome diversity within each sample (alpha diversity). Conversely, patients with history of routine BTX-A had a lower alpha diversity compared to those with no history of BTX-A use. The community composition between subgroups (beta diversity) based on gender, bladder management, frequency of UTI, BTX-A use, or prophylactic antibiotic use did not vary. The findings support prior studies showing that the Proteobacteria, commonly characterized as uropathogens, predominate in the urine of catheterizing patients with NLUTD.6,7,9,22,23
Discrete patterns in the urinary microbiomes of patients with and without urinary symptoms have been found among cohorts of non-NLUTD patients.1–5 However, this has not always been the case among the few studies that studied associations between the urinary microbiome and uro-pathology in those with NLUTD.9,23 Forster and colleagues analyzed the microbiome of 36 children with NLUTD who used CIC with either UTI, ASB or negative urine cultures and found that while those with UTI or ASB had higher relative abundance of Enterobacteriaceae, there was no significant difference in the community composition of the three groups.9 Given the complexity of the urinary microbiome among patients with NLUTD, which is dominated by Proteobacteria, differences in cross sectional studies may be challenging to identify. And, as Forster and others suggest, longitudinal changes within individuals’ microbiome may be a better method to detect associations between the urinary microbiome and lower urinary tract pathology than cross sectional comparisons between symptomatic and asymptomatic patients.9,23
Another question in this population is whether urinary microbiome diversity is due to the underlying NLUTD or rather the catheter use. In 2016 Groah and colleagues showed that samples from spontaneously voiding patients with NLUTD trend towards less diversity than those using catheters.6 Within their study, the relative abundance of Enterobacteriaceae did not differ between samples from patients with NLUTD who voided spontaneously compared to non-NLUTD patients.6 Our results from a population of adults with NLUTD using indwelling or CIC support these prior findings. We found decreased alpha diversity and a lower relative abundance of the Proteobacteria Pseudomonas among those who used CIC versus indwelling catheters.
Among our study population, we found a higher proportion of samples from those undergoing BTX-A had sequence counts sufficient for microbiome analysis. However, in those with sufficient bacterial rRNA sequences for microbiome analysis, there was less bacterial diversity in BTX-A users compared to non-users. There is no literature to date available on the impact of BTX-A on the urinary microbiome. However, the data available on the urinary microbiome of healthy women with urgency urinary incontinence would suggest that a higher bacterial diversity may be associated with urgency urinary incontinence and may predict failure to anticholinergic therapy.24 Future studies looking at longitudinal changes within individuals’ microbiome before and after BTX-A use are necessary.
This study provides a robust, cross-sectional sample of urinary microbiome data from catheterizing patients with NLUTD. Yet, we were limited by the fact a quarter of samples were unable to be analyzed due to low sequence counts. Though we did not measure microbial biomass, our findings suggest a low urinary microbiome biomass in samples from catheterizing patients with neurogenic bladder. To prevent contamination, which can skew data from low biomass environments, all specimens were processed by a centralized microbiome core laboratory on a single plate. Further, a negative control was performed during DNA isolation and negative and positive controls were performed during sequencing. We also assessed for differences in patient characteristics between included and excluded samples and reported these within our results.
The finding that Proteobacteria predominate the urinary microbiome of catheterizing patients with NLUTD and does not vary based on frequency of UTI or use of antibiotic prophylaxis, has several clinical implications. First, it reinforces the concept that these bacteria, often perceived to be uropathogens, are not necessarily associated with adverse patient outcomes and in the absence of symptoms do not require antibiotic treatment. Next it brings to light the fact that antibiotic prophylaxis may do little to alter the urinary microbiome in these patients, and perhaps the mechanism of action for symptomatic improvement is not tied to antimicrobial properties. Finally, our findings show that further research is required to evaluate the microbiome of non-catheterizing NLUTD patients, to provide longitudinal data in these patients, and to correlate these to patients’ bladder symptoms.
CONCLUSION
In this urinary microbiome study of 95 samples from catheterized adults with NLUTD, we find that the most abundant bacterial OTUs were from the phylum Proteobacteria, classified as Enterobacteriaceae and Escherichia. Even though these are considered urinary pathogens, there was no variability in the urinary microbiome based on frequency of UTI nor use of antibiotic UTI prophylaxis. Future work that provides longitudinal patient data and correlates this to patient reported outcome measures can help guide clinical use of urinary microbiome research.
Supplementary Material
Acknowledgement:
This research was supported by work performed by The University of Michigan Microbiome Core
Source of Funding:
University of Michigan Medical School Host Microbiome Initiative, Microbiome Explorer Program Grant; T32 NIDDK Grant T32DK007782; REDCap is supported by the Michigan Institute for Clinical & Health Research UL1TR002240
Key of Definitions for Abbreviations
- AMOVA
analysis of molecular variance
- ASB
asymptomatic bacteriuria
- BTX-A
botulinum toxin A
- CIC
clean intermittent catheterization
- DNA
deoxyribonucleic acid
- LDA
Linear discriminant analysis
- LEfSe
Linear discriminant analysis (LDA) effect size
- NLUTD
neurogenic lower urinary tract dysfunction
- OTU
operational taxonomic units
- PCoA
Principal Coordinate Analysis
- RNA
ribonucleic acid
- rRNA
ribosomal ribonucleic acid
- UTI
urinary tract infection
- θYC
Yue and Clayton dissimilarity index distances
Data Availability:
16S rRNA gene sequences are available for download from the National Library of Medicine Sequence Read Archive (Accession: PRJNA764758 and ID: 764758).
REFERENCES
- 1.Wolfe AJ and Brubaker L: Urobiome updates: advances in urinary microbiome research. Nat. Rev. Urol 2019; 16: 73–74. Available at: 10.1038/s41585-018-0127-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Komesu YM, Richter HE, Carper B, et al. : The urinary microbiome in women with mixed urinary incontinence compared to similarly aged controls. Int. Urogynecol. J 2018; 29: 1785–1795. Available at: 10.1007/s00192-018-3683-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bajic P, Wolfe AJ and Gupta GN: The Urinary Microbiome: Implications in Bladder Cancer Pathogenesis and Therapeutics. Urology 2019; 126: 10–15. Available at: 10.1016/j.urology.2018.12.034. [DOI] [PubMed] [Google Scholar]
- 4.Bajic P, Van Kuiken ME, Burge BK, et al. : Male Bladder Microbiome Relates to Lower Urinary Tract Symptoms. Eur Urol Focus 2020; 6: 376–382. Available at: 10.1016/j.euf.2018.08.001. [DOI] [PubMed] [Google Scholar]
- 5.Thomas-White K, Forster SC, Kumar N, et al. : Culturing of female bladder bacteria reveals an interconnected urogenital microbiota. Nat. Commun 2018; 9: 1557. Available at: 10.1038/s41467-018-03968-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Groah SL, Pérez-Losada M, Caldovic L, et al. : Redefining Healthy Urine: A Cross-Sectional Exploratory Metagenomic Study of People With and Without Bladder Dysfunction. J. Urol 2016; 196: 579–587. Available at: 10.1016/j.juro.2016.01.088. [DOI] [PubMed] [Google Scholar]
- 7.Fouts DE, Pieper R, Szpakowski S, et al. : Integrated next-generation sequencing of 16S rDNA and metaproteomics differentiate the healthy urine microbiome from asymptomatic bacteriuria in neuropathic bladder associated with spinal cord injury. J. Transl. Med 2012; 10: 1–17. Available at: https://translational.medicine.biomedcentral.com/articles/10.1186/1479-5876-10-174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee KW, Song HY and Kim YH: The microbiome in urological diseases. Investig Clin Urol 2020; 61: 338–348. Available at: 10.4111/icu.2020.61.4.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Forster CS, Panchapakesan K, Stroud C, et al. : A cross-sectional analysis of the urine microbiome of children with neuropathic bladders. J. Pediatr. Urol 2020; 16: 593.e1–593.e8. Available at: 10.1016/j.jpurol.2020.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Seekatz AM, Theriot CM, Molloy CT, et al. : Fecal Microbiota Transplantation Eliminates Clostridium difficile in a Murine Model of Relapsing Disease. Infection and Immunity 2015; 83: 3838–3846. Available at: 10.1128/iai.00459-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kozich JJ, Westcott SL, Baxter NT, et al. : Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl. Environ. Microbiol 2013; 79: 5112–5120. Available at: 10.1128/AEM.01043-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schloss PD, Westcott SL, Ryabin T, et al. : Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol 2009; 75: 7537–7541. Available at: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Westcott SL and Schloss PD: OptiClust, an Improved Method for Assigning Amplicon-Based Sequence Data to Operational Taxonomic Units. mSphere 2017; 2. Available at: 10.1128/mSphereDirect.00073-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schloss PD: A high-throughput DNA sequence aligner for microbial ecology studies. PLoS One 2009; 4: e8230. Available at: 10.1371/journal.pone.0008230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang Q, Garrity GM, Tiedje JM, et al. : Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol 2007; 73: 5261–5267. Available at: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cole JR, Wang Q, Fish JA, et al. : Ribosomal Database Project: data and tools for high throughput rRNA analysis. Nucleic Acids Res. 2014; 42: D633–42. Available at: 10.1093/nar/gkt1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yue JC and Clayton MK: A Similarity Measure Based on Species Proportions. Communications in Statistics - Theory and Methods 2005; 34: 2123–2131. Available at: 10.1080/STA-200066418. [DOI] [Google Scholar]
- 18.Anderson MJ: A new method for non-parametric multivariate analysis of variance: NON-PARAMETRIC MANOVA FOR ECOLOGY. Austral Ecol. 2001; 26: 32–46. Available at: http://doi.wiley.com/10.1111/j.1442-9993.2001.01070.pp.x. [Google Scholar]
- 19.Weir BS: Estimating F-statistics: A historical view. Philos. Sci 2012; 79: 637–643. Available at: 10.1086/667904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Segata N, Izard J, Waldron L, et al. : Metagenomic biomarker discovery and explanation. Genome Biol. 2011; 12: R60. Available at: 10.1186/gb-2011-12-6-r60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zakrzewski M, Proietti C, Ellis JJ, et al. : Calypso: a user-friendly web-server for mining and visualizing microbiome–environment interactions. 2017. Available at: https://academic.oup.com/bioinformatics/article-abstract/33/5/782/2627437. [DOI] [PMC free article] [PubMed]
- 22.Bank S, Hansen TM, Søby KM, et al. : Actinobaculum schaalii in urological patients, screened with real-time polymerase chain reaction. Scand. J. Urol. Nephrol 2011; 45: 406–410. Available at: 10.3109/00365599.2011.599333. [DOI] [PubMed] [Google Scholar]
- 23.Bossa L, Kline K, McDougald D, et al. : Urinary catheter-associated microbiota change in accordance with treatment and infection status. PLoS One 2017; 12: e0177633. Available at: 10.1371/journal.pone.0177633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Govender Y, Gabriel I, Minassian V, et al. : The Current Evidence on the Association Between the Urinary Microbiome and Urinary Incontinence in Women. Front. Cell. Infect. Microbiol 2019; 9: 133. Available at: 10.3389/fcimb.2019.00133. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
16S rRNA gene sequences are available for download from the National Library of Medicine Sequence Read Archive (Accession: PRJNA764758 and ID: 764758).