

Summary
T cell exhaustion is a major impediment to anti-tumor immunity. However, it remains elusive how other immune cells in the tumor microenvironment (TME) contribute to this dysfunctional state. Here we show that the biology of tumor-associated macrophages (TAM) and exhausted T cells (Tex) in the TME is extensively linked. We demonstrate that in vivo depletion of TAM reduces exhaustion programs in tumor-infiltrating CD8+ T cells and reinvigorates their effector potential. Reciprocally, transcriptional and epigenetic profiling reveals that Tex express factors that actively recruit monocytes to the TME and shape their differentiation. Using lattice light sheet microscopy, we show that TAM and CD8+ T cells engage in unique long-lasting antigen-specific synaptic interactions that fail to activate T cells but prime them for exhaustion, which is then accelerated in hypoxic conditions. Spatially resolved sequencing supports a spatiotemporal self-enforcing positive feedback circuit that is aligned to protect rather than destroy a tumor.
Graphical Abstract
eTOC Blurb
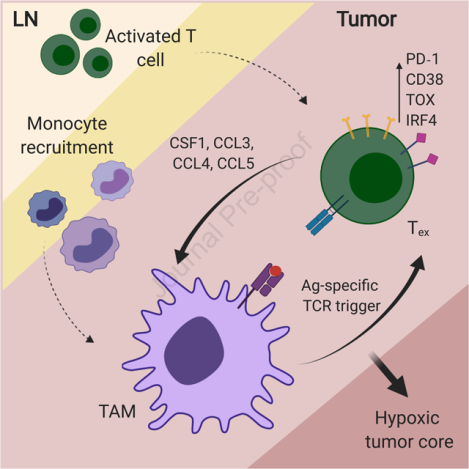
Kersten et al. demonstrate a spatiotemporal co-dependency between tumor-associated macrophages (TAM) and exhausted CD8+ T cells (Tex) in cancer. Tex shape myeloid cell recruitment and phenotype. Reciprocally, through antigen-specific stable synapses, TAM contribute to exhaustion programs in CD8+ T cells, together with hypoxia, prominent in inner regions of the tumor.
Introduction
Cancer immunotherapy – harnessing the patient’s immune system to fight cancer – has revolutionized cancer treatment strategies. However, a large proportion of patients does not show clinical response, and the mechanisms underlying resistance are still poorly understood. CD8+ T cells are critical mediators of anti-tumor immune responses, and the main target for current immunotherapy approaches. Tumor infiltration of CD8+ T cells correlates with improved prognosis and beneficial responses to immune checkpoint blockade as compared to non-infiltrated tumors (Galon et al., 2006; Tumeh et al., 2014). However, those CD8+ T cells are frequently non-functional due to their exhausted state, characterized by the expression of inhibitory molecules including PD-1, CD38 and TOX, and the loss of cytotoxic effector function (Wherry et al., 2007; Doering et al., 2012; Schietinger et al., 2016; Pauken et al., 2016; Scott et al., 2019; Khan et al., 2019). Several studies have shown that chronic antigen exposure and stimulation of the T cell receptor (TCR) are required for exhaustion programs in T cells (Utzschneider et al., 2016; Scott et al., 2019; Oliveira et al., 2021). However, how this is orchestrated in the tumor microenvironment (TME) is unclear.
The immune composition of the TME plays an important role in regulating effective anti-tumor T cell responses (Binnewies et al., 2018). Across solid tumors, the majority of immune cells in the TME is frequently comprised of antigen-presenting myeloid cells (APC), of which tumor-associated macrophages (TAM) are typically the most abundant (DeNardo et al., 2011; Ruffell et al., 2012; Broz et al., 2014). TAM abundance is correlated with poor prognosis in a variety of solid tumor types (Zhang et al., 2012; Gentles et al., 2015), and many studies report on their immunosuppressive role in cancer progression and dissemination (DeNardo and Ruffell, 2019). Conversely, some studies report immunostimulatory and anti-tumor functions through expression of TNF and iNOS, or upon treatment with CD40 agonists (Beatty et al., 2011; Klug et al., 2013). Similar to a rare population of conventional dendritic cells (cDC1), which have been described to be potent activators of anti-tumor T cells (Broz et al., 2014; Roberts et al., 2016; Salmon et al., 2016; Spranger et al., 2017), TAMs have the potential to phagocytose large amounts of tumor-associated antigens, but fail to successfully support T cell activation (Engelhardt et al., 2012; Broz et al., 2014). Interestingly, intravital imaging studies have shown that antigen-specific CD8+ T cells preferentially localize in TAM-rich areas in the TME, and form tight interactions that persist over time (Boissonnas et al., 2013; Broz et al., 2014; Peranzoni et al., 2018).
Here we dissect the molecular mechanisms of immune co-differentiation, by which TAM and exhausted CD8+ T cells (Tex) sustain each other’s maturation and presence in the TME through long-lived antigen-specific synaptic contacts. Our study reveals a mechanistic link through an antigen-driven positive feedback loop and offers a possible path whereby T cells and TAM might equally contribute to initial and sustained tumor immune evasion.
Results
CD8+ T cell exhaustion correlates with macrophage abundance in the TME
To study how myeloid immune cells contribute to CD8+ T cell exhaustion in the TME, we first focused on the concurrent events during the onset of exhaustion programs in tumor-infiltrating CD8+ T cells in mouse models of melanoma (B78ChOVA and B16ChOVA) and spontaneous breast cancer (MMTV-PyMTChOVA) (Fig. S1A). At different time points during tumor growth, we adoptively transferred Ovalbumin (OVA)-specific OT-I CD8+ T cells into tumor-bearing mice, focusing on: (1) early arrival T cells that were recently recruited to the TME (Tex d4) and (2) T cells that have resided in the TME for 14 days and have demonstrably upregulated PD-1, CD38, TOX and CD5 as assessed by flow cytometry (Tex d14) (Fig. S1B). Both of these populations demonstrated reduced production of the cytokines IFNγ and TNFα when compared to activated CD44+ endogenous CD8+ T cells in the tumor-draining lymph node (TdLN) (Fig. S1C, D). The onset of exhaustion and dysfunction in CD8+ T cells upon tumor infiltration was antigen-specific, because irrelevant LCMV-specific P14 CD8+ T cells did not acquire phenotypic markers of exhaustion when compared to OT-I CD8+ T cells (Fig. S1E–H). However, the ability to produce effector cytokines IFNγ and TNFα was blunted equivalently in endogenous, P14 and OT-I CD8+ T cells upon tumor residence (Fig. S1I, J). Loss of those markers may represent a natural decay process post-activation or universal non-antigen-specific suppression mechanisms (such as immunosuppressive cytokines like IL-10, TGFβ and iNOS) in the TME.
Tumor-associated macrophages (TAM) comprised the majority of myeloid cells in B78ChOVA melanomas in line with previous studies (Broz et al., 2014; Gentles et al., 2015; Cheng et al., 2021), and their abundance increased during tumor progression, while the fraction of CD103+ cDC1 and CD11b+ cDC2 diminished (Fig. S1K). Recognizing the stoichiometric abundance of TAM as possible APC, we sought to study the role of TAM in the onset of CD8+ T cell exhaustion by subjecting tumor-bearing mice to antibody-mediated blockade of CSF1-CSF1R signaling (Fig. 1A and Fig. S1O, R). Blockade of CSF1-CSF1R induced a significant reduction of the proportion of CD11b+F4/80+ TAM in tumors (Fig. 1B,C and Fig. S1L, P, S) and a modest increase of neutrophils (Fig. S1L). Of note, CSF1R blockade did not affect the phenotype of TAM as determined by flow cytometric analysis of the positive fraction (Fig. S1M) and expression levels of a variety of phenotypic markers on TAM (Fig. S1N).
Figure 1. CD8+ T cell exhaustion correlates with macrophage abundance in the TME.
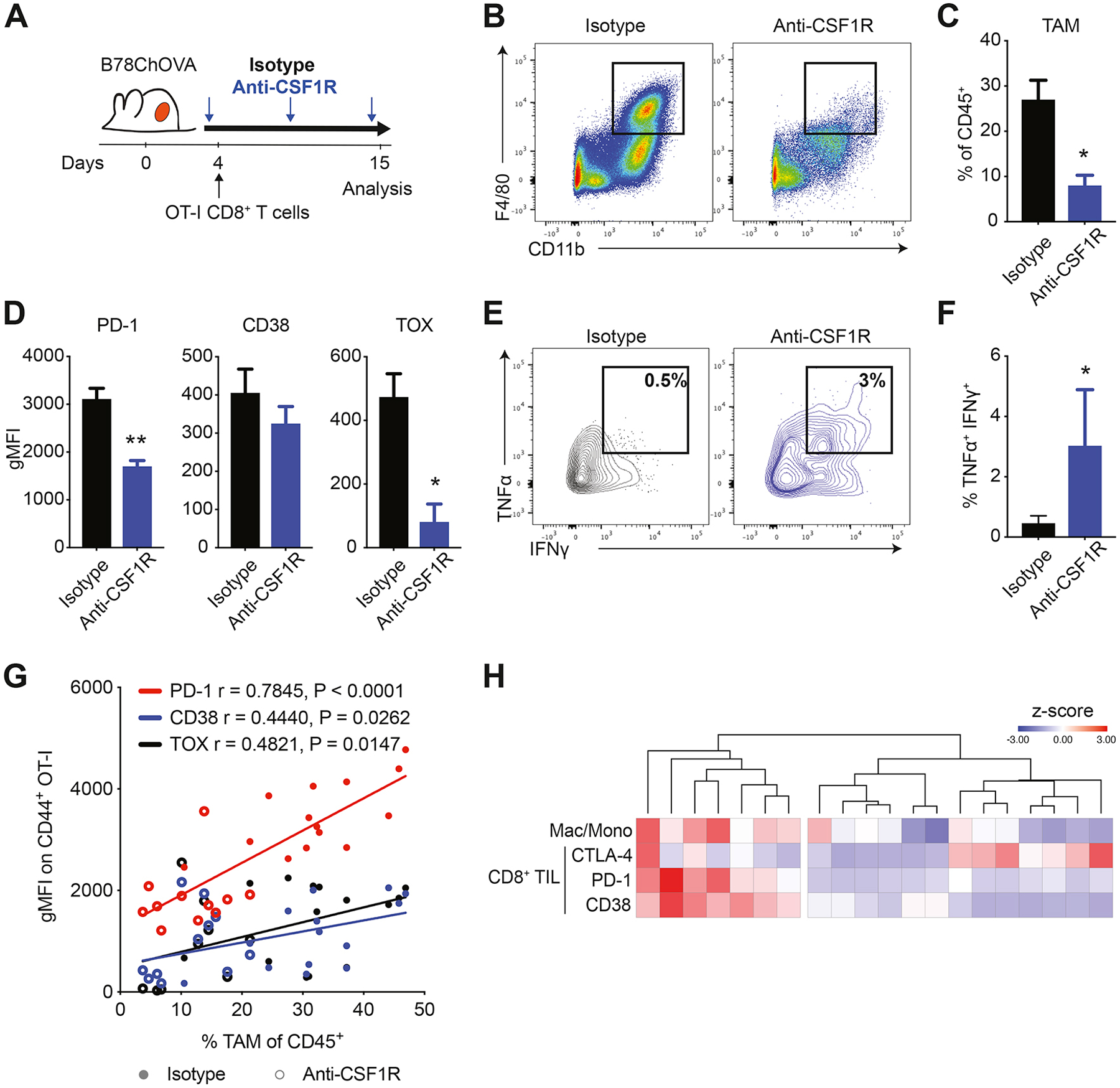
A) Experimental setup. Weekly anti-CSF1R or isotype antibody treatment was initiated 1–2 days after B78ChOVA tumor inoculation. OVA-specific OT-I CD8+ T cells were adoptively transferred 2–3 days after tumor inoculation. Mice were sacrificed at day 15 and tumors were harvested for analysis. B-C) Representative flow plots (B) and quantification (C) of CD11b+ F4/80+ macrophages in isotype and anti-CSF1R-treated tumors. N=5 mice/group. D) Surface (PD-1 and CD38) and intracellular (TOX) expression on intratumoral CD44+ OT-I CD8+ T cells from isotype and anti-CSF1R-treated mice. N=5 mice/group. E-F) Representative contour plots (E) and quantification (F) of IFNγ+TNFα+ polyfunctional CD44+ OT-I CD8+ T cells in tumors of isotype and anti-CSF1R-treated mice. N=8–9 mice/group. Pooled data from two independent experiments. G) Spearman correlation between gMFI of PD-1, CD38 and TOX expression on CD44+ OT-I CD8+ T cells and % of TAM of CD45+ cells in tumors treated with isotype (solid) or anti-CSF1R antibody (open). N=11–14 mice/group. H) Heatmap showing clustering of normalized z-scores of CTLA-4, PD-1 and CD38 expression on CD8+ TIL and macrophage/monocyte ratio in 20 fresh human kidney renal cell carcinoma samples (rows) determined by flow cytometry. All data are mean ± SEM. ** p < 0.01, * p < 0.05 as determined by Mann-Whitney U-test. See also Figure S1.
Interestingly, without affecting the proportion of infiltrating T cells (Fig. S1L) acute TAM-depletion resulted in a concurrent reduction in the expression of exhaustion markers PD-1, CD38 and TOX on tumor-infiltrating CD44+ OT-I CD8+ T cells (Fig. 1D and Fig. S1Q, T). Moreover, the resultant CD44+ OT-I CD8+ T cells produced higher levels of IFNγ and TNFα in anti-CSF1R compared to isotype-treated mice (Fig. 1E, F), but only modestly affected tumor size (Fig. S1U). We found that the expression of PD-1, CD38 and TOX on tumor-infiltrating CD44+ OT-I CD8+ T cells positively correlates with the abundance of TAM in the TME, measured across 25 mice in three independent experiments subjected to anti-CSF1R or isotype treatment (Fig. 1G). In line with this, flow cytometric profiling of CD8+ tumor-infiltrating lymphocytes (TIL) in a cohort of 20 patients with renal cell carcinomas – which are rich in myeloid and T cells (Combes et al., 2022; Mujal et al., 2022) –, demonstrated a strong association between PD-1 and CD38 (but not CTLA-4) expression and the degree to which patient’s myeloid cells had differentiated toward macrophages as compared to monocytes in the TME (Fig. 1H). We used this ratio as the pure number of macrophages did not show this association (data not shown).
CD8+ Tex express monocyte/macrophage-related factors upon prolonged residence in the TME
To test whether there is a mechanistic link between TAM abundance and CD8+ T cell exhaustion, we isolated early (Tex d4) and late (Tex d14) exhausted OT-I CD8+ T cells from B78ChOVA tumors and compared their transcriptional profile to that of splenic naïve CD44− OT-I CD8+ T cells by RNA-seq. As expected, Tex d14 showed enhanced expression of known markers associated with exhaustion (Sade-Feldman et al., 2018), including but not limited to Cd44, Pdcd1, Cd38, Tox, Irf4, Havcr2, Lag3, while expression of naïve precursor genes Sell, Tcf7 and Il7r were enriched in Tnaïve cells (Fig. 2A). Interestingly, pathway enrichment analysis of genes with a fold enrichment >5 in Tex revealed dramatic enrichment of pathways involved in “abnormal cytokine secretion” and “impaired macrophage chemotaxis” (Fig. 2B). A closer analysis of individual genes demonstrated that expression of genes associated with naïve precursor T cell states decreased and genes previously associated with exhaustion increased in Tex d14 vs Tex d4 (Fig. 2C), consistent with previous reports (Pauken et al., 2016; Schietinger et al., 2016; Sade-Feldman et al., 2018). Interestingly, a large set of myeloid-related genes was highly upregulated in exhausted CD8+ T cells and most of these increased with prolonged residence in the TME (Fig. 2C and Fig. S2A). Interestingly, the majority of the myeloid-related genes upregulated in exhausted CD8+ T cells are known regulators of monocyte/macrophage biology, while the expression of Flt3L – a formative cytokine for CD103+ cDC1 (Barry et al., 2018) – was downregulated in Tex (Fig. 2C). Increased expression of several of these myeloid-related genes was confirmed on independent sample sets transcriptionally by qRT-PCR (Fig. S2B) and by quantification of secreted protein (Fig. S2C). Of note, the majority of these changes was not observed in effector CD8+ T cells (Teff), suggesting a unique exhaustion-related expression profile in CD8+ T cells that is acquired upon prolonged residence in the TME. Increased expression of these myeloid-related genes was detected in both endogenous and OT-I exhausted CD8+ T cells, and thus seems independent of antigen-reactivity.
Figure 2. Exhausted CD8+ T cells express myeloid-related factors.
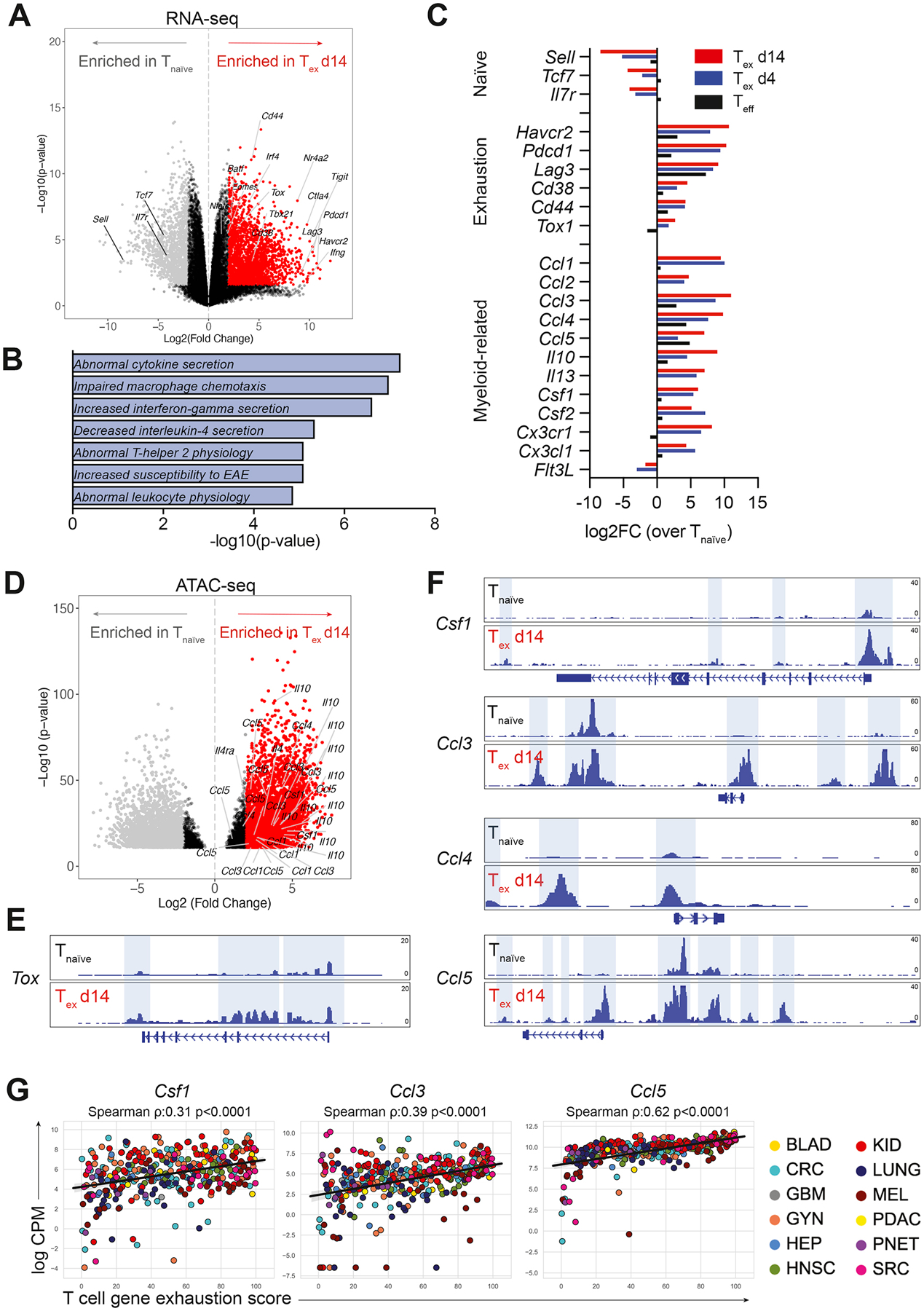
A) Volcano plot showing differential gene expression in tumor-infiltrating CD44+ OT-I CD8+ Tex d14 cells (red) compared to splenic CD44− OT-I CD8+ Tnaïve cells (grey) by RNA-seq. Colored dots (grey and red) represent genes with a log2FC>2 and FDR<0.05. B) Gene set enrichment analysis of DEGs (log2FC>5 and p-value<0.05) enriched in Tex d14 versus Tnaïve using the MGI Mammalian Phenotype Level 4 library. C) Average gene expression in Teff (black), Tex d4 (blue) and Tex d14 (red) normalized to Tnaïve as determined by RNA-seq. D) Volcano plot showing differential chromatin accessibility at transcriptional start sites in loci of myeloid genes in tumor-infiltrating CD44+ OT-I CD8+ Tex d14 cells compared to splenic CD44− OT-I CD8+ Tnaïve cells by ATAC-seq. Colored dots (grey and red) represent genes with a log2FC>2 and FDR<0.05. E-F) ATAC-seq signal tracks at the Tox (E), Csf1, Ccl3, Ccl4 and Ccl5 loci (F) highlighting differential chromatin accessibility peaks in CD44+ OT-I CD8+ Tex cells (d14) compared to splenic CD44− OT-I CD8+ Tnaïve cells. G) Correlation of normalized expression of Csf1, Ccl3 and Ccl5 transcripts and exhaustion score in FACS sorted human intratumoral T cells across multiple human cancer indications. See also Figure S2.
Epigenetic profiling using assay for transposase-accessible chromatin with sequencing (ATAC-seq) confirmed a significant enhancement of overall chromatin accessibility near the transcription start site of the genes encoding these myeloid-related genes in Tex d14 versus Tnaïve cells (Fig. 2D). A more detailed analysis of signal tracks of chromatin accessibility peaks at different gene loci revealed that Tox – a major transcriptional and epigenetic regulator of T cell exhaustion (Alfei et al., 2019; Khan et al., 2019; Scott et al., 2019; Yao et al., 2019) – as well as other well-known genes associated with exhaustion programs (Pdcd1, Cd38, Havcr2, Ctla4, Lag3 and Entpd1 (Pauken et al., 2016; Philip et al., 2017)) showed increased chromatin accessibility at promoter regions in Tex when compared to Tnaïve (Fig. 2E and Fig. S2D). This enhanced accessibility was also observed for myeloid-related genes Csf1, Ccl3, Ccl4 and Ccl5 in Tex when compared to Tnaïve (Fig. 2F). In addition, utilizing transcriptional profiles of T cells isolated from human cancers, we found that increased expression of a T cell exhaustion score correlated significantly with the expression of Csf1, Ccl3 and Ccl5 in T cells in a dataset comprising hundreds of patients across a dozen cancer indications (Combes et al., 2022) (Fig. 2G).
CD8+ Tex shape the myeloid compartment in mouse melanoma
To directly study the functional significance of chemokine gene expression by Tex, we adopted a transwell experimental system using OT-I T cells with varying activation states in the bottom well, and bone marrow-derived monocytes in the upper transwell insert (Fig. 3A). After 24 hrs of culture, significantly more monocytes had migrated through the transwell membrane towards Tex when compared to Teff, Tnaïve or no T cells (Fig. 3B), demonstrating that Tex actively secrete factors that recruit monocytes while T cells in other activation states do not. Phenotypic analysis by flow cytometry revealed that monocytes co-cultured for 2 days with Tex also show increased uniformity and/or magnitude of expression of CD80, CD86, H2kB and MHC-II when compared to Teff, Tnaïve or no T cells (Fig. 3C), suggesting that Tex-derived factors augment antigen-presentation potential in differentiating myeloid cells.
Figure 3. Exhausted CD8+ T cells recruit monocytes to the TME and shape macrophage phenotype.
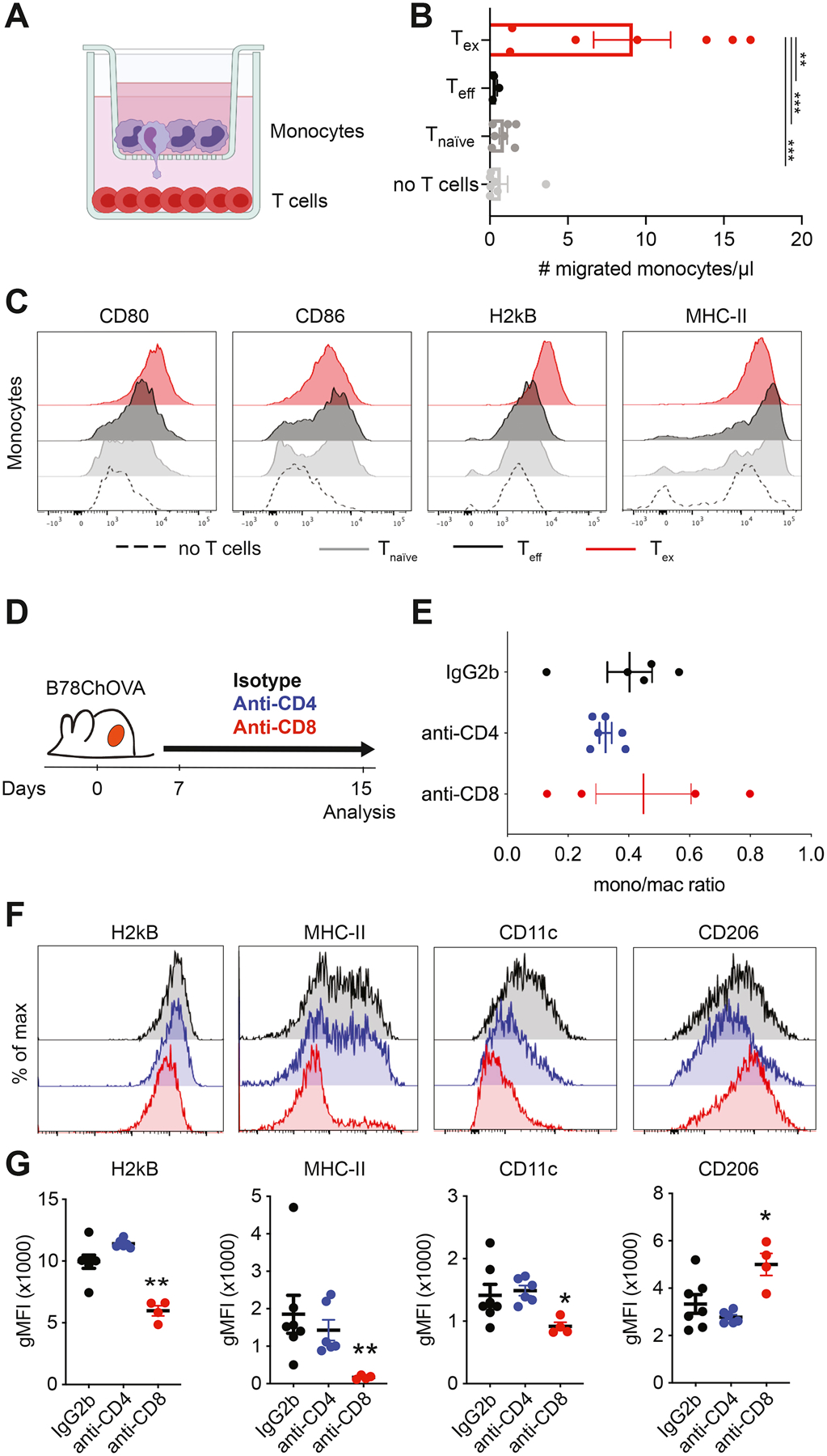
A) Experimental setup of in vitro recruitment assay. Bone marrow-derived monocytes are cultured on transwell inserts (5μm pore size) and T cells (OT-I Tnaïve, Teff and Tex) are plated in the bottom well. B) Quantification of recruited monocytes after 24 hours. Data combined from two independent experiments. Statistical significance was determined by one-way ANOVA with Holm-Sidak’s multiple testing correction. C) Representative histograms of expression of surface markers on monocytes after 48 hours of co-culture with Tnaïve, Teff and Tex cells. D) Experimental set-up of in vivo CD4+ and CD8+ T cell depletion in B78ChOVA-bearing mice. Treatment with anti-CD4/CD8 antibodies or isotype was initiated 7 days after tumor inoculation and continued until mice were sacrificed. E-F) Monocyte/macrophage ratio of the proportion of Ly6Chi monocytes and F4/80+ macrophages (gated of CD45+ cells) in the B78ChOVA TME after isotype, anti-CD4 and anti-CD8 treatment. F-G) Representative histograms (F) and quantification (G) of H2kb, MHC-II, CD11c and CD206 expression on CD11b+F4/80+ macrophages in B78ChOVA tumors after isotype, anti-CD4 and anti-CD8 treatment. Statistical significance was determined using the Mann-Whitney U test. All data are mean ± SEM. * p < 0.05, ** p < 0.01, *** p < 0.001. See also Figure S3.
To assess whether Tex actively shape the myeloid compartment in tumors in vivo, we were faced with a dearth of available models that allow for specific (conditional) deletion of exhausted T cells from the TME. Therefore, we decided to use a more widely used approach to systemically deplete CD4+ or CD8+ T cells from B78ChOVA-bearing mice using depleting antibodies (Fig. 3D). Interestingly, while the ratio of monocytes to macrophages did not change upon CD4+ or CD8+ T cell depletion (Fig. 3E), their phenotype was drastically affected by CD8+ T cell depletion in ways consistent with the results of our in vitro co-cultures. CD11b+F4/80+ TAM showed significantly reduced expression of H2kB, MHC-II and CD11c in the absence of CD8+ T cells, but not CD4+ T cells (Fig. 3F, G). In addition, CD8+ T cell depletion resulted in increased expression of ‘pro-tumorigenic M2-marker’ CD206 on TAM (Fig. 3F, G), suggesting that CD8+ T cells specifically shape myeloid cell phenotype in the TME favoring an ‘M1-like’ antigen-presenting state.
To test whether the production of CSF1 by lymphocytes had any functional relevance for myeloid composition in the TME, we generated mixed bone marrow chimeras in which Rag1−/− bone marrow was mixed 50:50 with either Csf1op/op or Csf1op/+ bone marrow and transferred into lethally irradiated Rag1−/− recipient mice (Fig. S3A). After a recovery period of 6–10 weeks, these mice were inoculated with B78ChOVA melanomas for 21 days after which the myeloid compartment in the TME was analyzed by flow cytometry. CSF1-deficiency in lymphocytes (Rag1−/−:Csf1op/op chimeras) did not affect primary tumor growth (Fig. S3B) and modestly reduced the influx of total CD45+ leukocytes compared to control animals (Rag1−/−:Csf1op/+) (Fig. S3C). In the myeloid compartment, the proportion of Ly6C+ monocytes was significantly enriched in Rag1−/−:Csf1op/op chimeras, while macrophage proportions were lower, resulting in an increased monocyte/macrophage ratio in Rag1−/−:Csf1op/op chimeras (Fig. S3D). The proportion of CD103+ cDC1 and CD11b+ cDC2 were not appreciably modulated by CSF1-deficiency in lymphocytes (Fig. S3E). In line with the results presented in Fig. 3F and G, the levels of H2kB, MHC-II and CD11c on CD11b+F4/80+ TAM were lower in Rag1−/−:Csf1op/op chimeras when compared to Rag1−/−:Csf1op/+ chimeras (Fig. S3F), while expression of CD86 and CD206 was modestly increased. Despite the large biological variation among these samples, the results support the notion that exhausted CD8+ T cells, at least partially through expression of CSF1, contribute to TAM maturation and induce an antigen-presentation phenotype in the TME.
Macrophages and CD8+ T cells engage in unique long-lived interactions and synapse formation
Since our data suggests that CD8+ Tex shape myeloid cell phenotype towards an antigen-presenting state, we took a more detailed look at the interactions between TAM and CD8+ T cells in the TME. Using 2-photon microscopy, we have previously shown that newly infiltrated antigen-specific CD8+ T cells in the TME preferentially localize in TAM-rich areas, and are captured in prolonged interactions with TAM that result in the onset of exhaustion programs (Engelhardt et al., 2012; Broz et al., 2014; Boldajipour et al., 2016). In line with these results, ex vivo coupling assays using single cell suspensions (enriched for CD45+ cells) from B16F10 and B16ChOVA tumors confirmed that OT-I CD8+ T cells preferentially form doublets with myeloid cells, and specifically TAM, in an antigen-dependent manner (Fig. S4A–C). Moreover, after adoptive transfer of both OT-I and P14 CD8+ T cells in B78ChOVA-bearing mice, we found that (after enzymatic digestion) both the proportion of total T cells doublets and the proportion of those that are coupled to a TAM are significantly higher among OT-I versus P14 or endogenous CD8+ T cells (Fig. S4D), suggesting the preferential formation of antigen-specific TAM-T cell doublets. Conventional wide field imaging demonstrated that, outside of the context of the TME, previously activated OT-I CD8+ T cells interact significantly longer with TAM sorted from OVA-expressing tumors as compared to in vitro generated bone marrow-derived dendritic cells (BMDC) unloaded or loaded with the cognate peptide SIINFEKL (SL8) (Fig. 4A, B), demonstrating a persistent interaction between TAM and CD8+ T cells despite their consistent inability to stimulate T cell proliferation.
Figure 4. TAM uniquely engage CD8+ T cells in antigen-specific long-lived synaptic interactions.
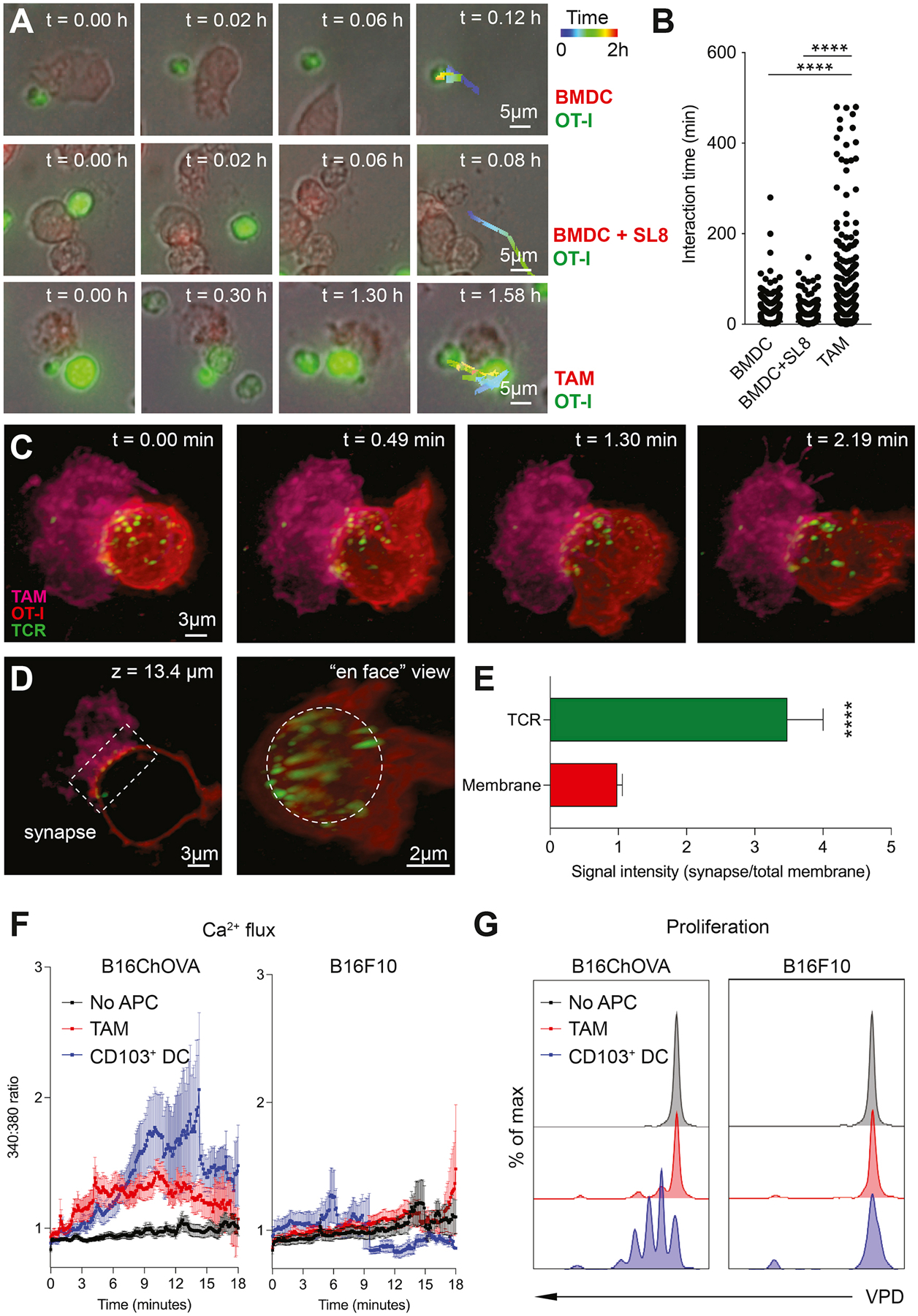
A) Representative images of ex vivo interactions between mTomato+ APC (BMDC, BMDC+SL8 or B78ChOVA-derived TAM) and CFSE-labeled previously activated CD8+ OT-I T cells over time using conventional wide field microscopy. B) Quantification of interaction time. n = 3452 TAM, n = 6134 BMDC, n = 3320 BMDC+SL8. Statistical significance was determined using the one-way ANOVA test with Holm-Sidak’s multiple comparison correction. C) Representative images of the interaction between mTomato+ TAM sorted from B78ChOVA melanomas (magenta) and previously activated CD8+ OT-I T cell labeled with CD45-AF647 (red), with the H57 TCRβ labeled with AF488 by lattice light sheet imaging. Scale bar, 3μm. D) Z-slice (left) and ‘en face’ view (right) of the TAM-CD8+ T cell interaction site showing TCR clustering in the immunological synapse (box (left) and dotted circle (right)). E) Quantification of polarized TCR clustering by determining the ratio of signal intensity of the red (membrane) or green channel (TCR) at the synapse site normalized to the entire membrane. N = 12 T cells. Statistical significance was determined using the Mann-Whitney U test. F-G) Quantification of immediate Ca2+ flux by FURA-2AM imaging (F) and proliferation after 72 hours by dilution of Violet Proliferation Dye (VPD) (G) in previously activated CD8+ OT-I T cells after interaction with TAM or CD103+ DC isolated from B16ChOVA or B16F10 tumors. Negative control represents CD8+ OT-I T cells that did not touch an APC (no APC). All data are mean ± SEM. * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001. See also Movie S1 and Figure S4.
Using lattice light sheet microscopy, we found that this stable interaction between TAMs and CD8+ T cells results in small-scale clustering of TCR at the TAM-interaction site on CD8+ T cells (Movie S1, Fig. 4C–E and Fig. S4E), consistent with this being a signaling interaction. Calcium imaging revealed that, unlike CD103+ cDC1 – potent inducers of CD8+ T cell activation (Broz et al., 2014; Roberts et al., 2016; Salmon et al., 2016; Spranger et al., 2017) – that trigger a transient flux, B16ChOVA-derived TAM induce a weak, but long-lasting Ca2+ flux in CD8+ T cells upon recognition of cognate antigen (Fig. 4F (left)). This flux was likely antigen-dependent since TAM and CD103+ cDC1 isolated from B16F10 melanomas did not induce a TCR trigger in OT-I CD8+ T cells (Fig. 4F (right)). While the transient Ca2+ flux triggered by CD103+ cDC1 is sufficient to induce proliferation of CD8+ T cells, the antigen-specific trigger provided by TAM fails to support proliferation (Fig. 4G). Thus, despite actively and profoundly engaging T cells in a unique long-lasting antigen-specific synaptic interaction, TAM fail to fully support CD8+ T cell activation and proliferation.
Unique TCR engagement by TAM induces exhaustion programs in CD8+ T cells
To take a more detailed look at the TAM-induced ‘dysfunctional’ TCR trigger, we examined ex vivo co-cultures of previously activated OT-I CD8+ T cells with TAM or CD103+ cDC1 isolated from B16ChOVA and B16F10 melanomas, or in vitro generated BMDC devoid of antigen as a negative control. As expected, after 3 days of co-culture only CD8+ T cells that had encountered CD103+ cDC1 expressing their cognate antigen displayed a CD44hi IRF4hi fully activated phenotype (Fig. 5A). Interestingly, when compared to the successful signal provided by CD103+ cDC1, TAM only modestly induced expression of activation marker CD44, while providing a similar strength in TCR trigger as suggested by the level of IRF4 expression (Fig. 5A). In chronic viral infections, IRF4 has been implicated as a main regulator of transcriptional circuits inducing and sustaining T cell exhaustion (Man et al., 2017). In line with this notion, TAM induced a significant increase in expression of PD-1 and TOX in CD8+ T cells in an antigen-specific manner and similar to CD103+ cDC1 (Fig. 5A), but nevertheless fail to support proliferation (Fig. 4G). Addition of exogenous SIINFEKL peptide (SL8) to TAM was unable to rescue CD8+ T cell activation and proliferation (Fig. S5A). Moreover, the TAM-induced exhaustion phenotype resulted in a failure to produce effector cytokines IFNγ and TNFα (Fig. S5B), suggesting that these cells are indeed dysfunctional.
Figure 5. TAM engagement contributes to induction of exhaustion programs in CD8+ T cells in an antigen-specific manner.
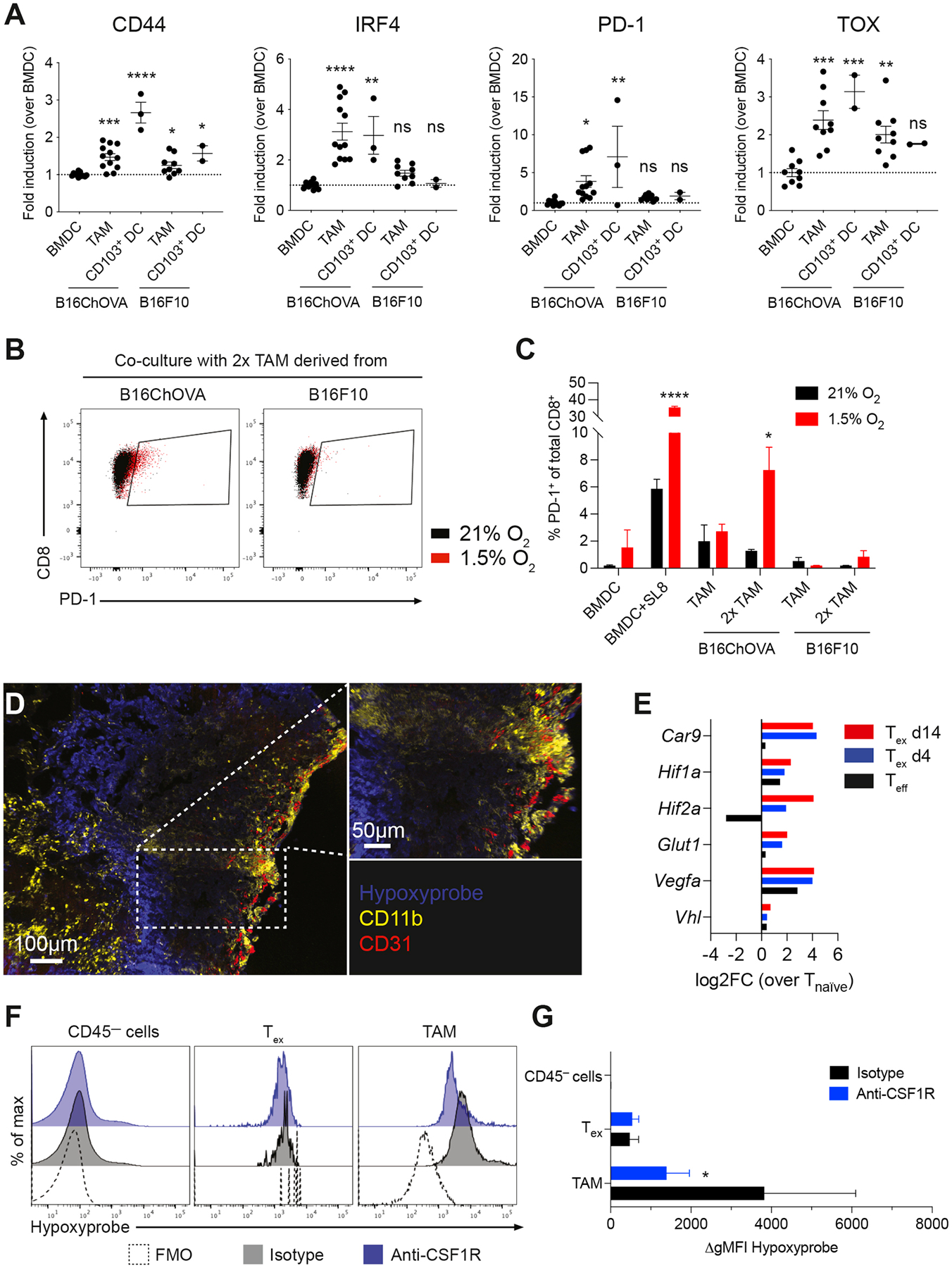
A) Flow cytometric analysis of CD44, IRF4, PD-1 and TOX expression in previously activated CD8+ OT-I T cells co-cultured for 72 hours with in vitro generated BMDC, and TAM or CD103+ DC isolated from B16ChOVA or B16F10 tumors. Data presented as fold induction over BMDC. Cumulative data from 4 independent experiments. All data are plotted as mean ± S.E.M. One-way ANOVA with Holm-Sidak correction for multiple comparisons. B-C) Representative dot plots (B) and quantification (C) of PD-1+ expression on previously activated CD8+ OT-I T cells after co-culture with in vitro generated BMDC±SL8, and TAM isolated from B16ChOVA or B16F10 tumors. Ratio of APC:T cell was 1:4 or 1:2 (2x TAM). Plates were incubated in normoxic (21% O2) and hypoxic (1.5% O2) conditions for 3 days. Statistical significance was determined using the Unpaired t-test. D) Immunofluorescence of B78ChOVA melanomas stained with pimonidazole (Hypoxyprobe) Pacific Blue, CD11b-AF594 (yellow) and CD31-AF647 (red). E) Average expression of hypoxia-related genes in Teff (black), Tex d4 (blue) and Tex d14 (red) normalized to Tnaïve as determined by RNA-seq. F-G) Representative histograms (F) and quantification (G) of hypoxyprobe staining in CD45— cells, CD44+ OT-I CD8+ T cells and CD11b+F4/80+ TAM in B78ChOVA melanomas treated with isotype (black) or anti-CSF1R antibodies (blue). Statistical significance was determined using the Mann-Whitney U test. N = 4–5 mice/group. Representative of two independent experiments. All data are mean ± SEM. * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001. See also Figure S5.
Previous work has shown that hypoxia – in combination with chronic antigenic stimulation but not in the absence of stimulation – is required to obtain a ‘full blown’ exhausted phenotype in CD8+ T cells in vitro (Scharping et al., 2021). To study whether the signals from TAM-Tex interactions are sufficient to ‘prime’ T cells for exhaustion despite being deficient at inducing their proliferation, we performed co-culture experiments under hypoxic (1.5% O2) and normoxic (21% O2) conditions. Interestingly, the proportion of PD-1+ CD8+ T cells induced by B16ChOVA-derived TAM was much more pronounced when cultured in hypoxic conditions when compared to normoxia, especially when TAM were numerically in excess (Fig. 5B, C). Together these data demonstrate that TAM can prime the onset of exhaustion programs in CD8+ T cells, a process that is exacerbated in hypoxic conditions.
Using the hypoxia tracer pimonidazole, we found that B78ChOVA melanomas show highly hypoxic areas towards the inner regions of the tumor, and away from CD31+ blood vessels (Fig. 5D). Moreover, we find that these hypoxic regions are surrounded by patches of CD11b+ macrophages (Fig. 5D). In line with this observation, expression of hypoxia-related genes (Car9, Hif1a, Hif2a, Glut1, Vegfa, Vhl) was upregulated in exhausted CD8+ T cells upon prolonged residence in the TME (Fig. 5E), suggesting that tumor-infiltrated Tex experience severe hypoxia in the TME. Interestingly, flow cytometric analysis revealed that TAM experience more severe levels of hypoxia compared to exhausted CD8+ T cells and CD45— tumor cells (Fig. 5F, G). Moreover, the degree of hypoxia was dramatically reduced in residual TAM, but not in Tex, after CSF1R blockade (Fig. 5F, G).
ZipSeq mapping reveals spatial coordination of TAM-Tex interaction dynamics in the TME
To better understand the spatial coordination of the dynamic interplay between TAM and Tex in the TME, we utilized ZipSeq, a spatial transcriptomics approach that allows us to map gene expression patterns in single cells based on their localization in the TME by printing barcodes directly onto cells in tissue (Hu et al., 2020). We used a Cd206-LSL-Venus-DTR mouse model in which expression of a Venus fluorescent reporter is driven by the endogenous Cd206 promoter (Fig. S6A). When crossed to the Csf1rCre strain, we found Venus-labeling of the majority of tumor-associated myeloid cells in B78ChOVA melanomas (data not shown). We utilized this model to define distinct regions in the tumor such as the outer rim, middle and inner compartment based on the mCherry signal in cancer cells and the Venus-expression in CD206+ myeloid cells (Fig. 6A) and apply unique Zipcodes to the surface of immune cells in each of those regions. After dissociation of tumors, we sorted CD45+ immune cells and encapsulated them for our modified 10x Genomics scRNA-seq workflow (Hu et al., 2020).
Figure 6. Spatial delineation of TAM-Tex interaction dynamics in the TME.
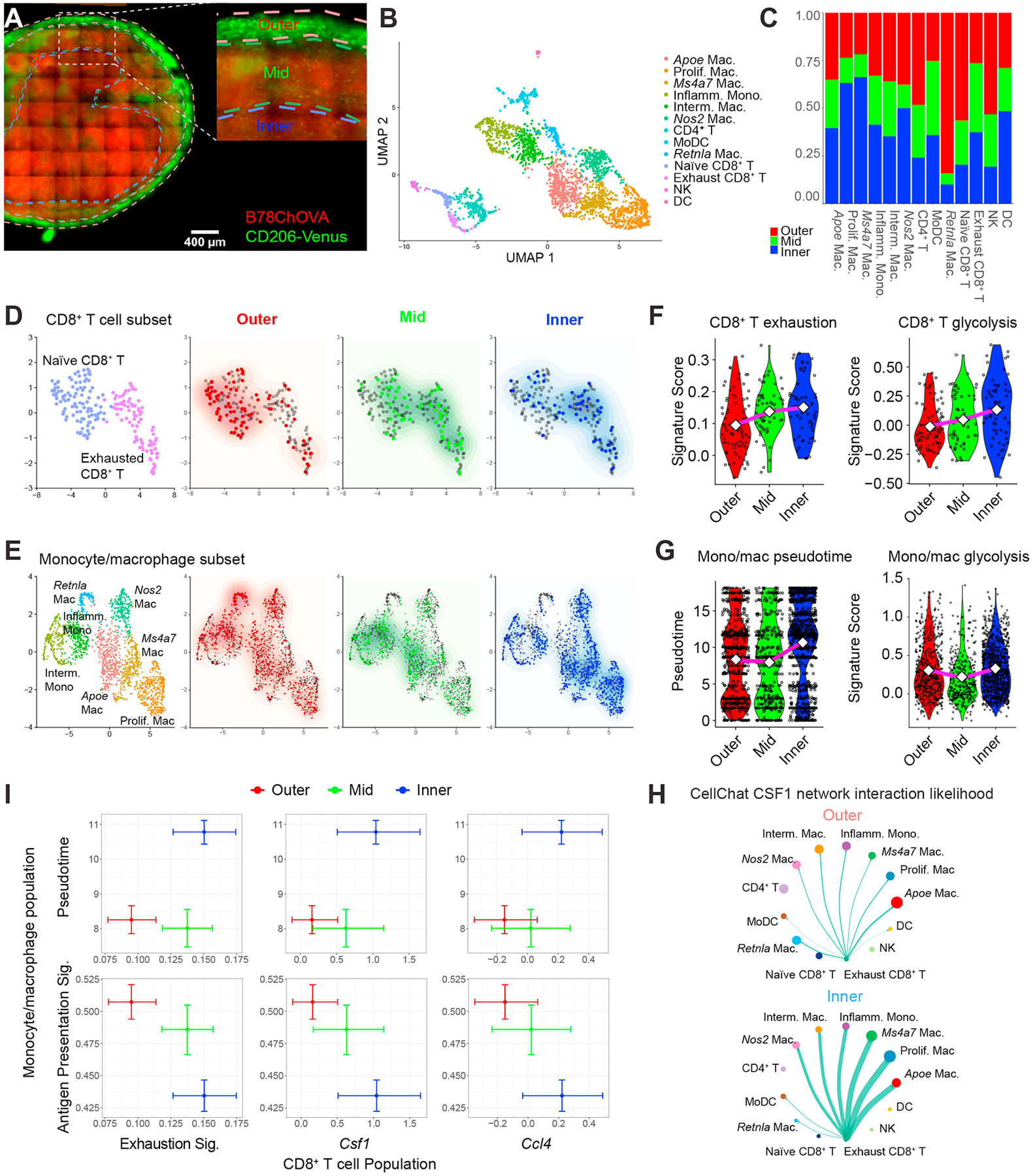
A) Imaging of a 150μm-thick live B78ChOVA melanoma section with ROI demarcation of outer, mid and inner compartments used for subsequent ZipSeq. Red channel denotes mCherry signal from B78ChOVA cancer cells and green channel indicates expression of mVenus in CD206+ macrophages in the Csf1RCre;LSL-Cd206-Venus-DTR mouse model. Scale bar = 400μm. B) UMAP representation of sorted CD45+ cells following 10X Genomics scRNA-seq workflow (n = 2765 cells with n = 427/394/335/288/275/244/220/170/120/108/91/62/31 for clusters as listed). C) Stacked bar charts representing regional distribution of distinct populations identified in B. D-E) UMAP of subsampled CD8+ T cell subset (n = 199 cells) (D) and monocyte/macrophage subset (n = 2083 cells) (E) overlaid with their regional localization. F) Violin plots representing CD8+ T cell exhaustion score (left; Outer: 0.31 / 0.10 / −0.07; Mid: 0.35 /0.14 / −0.05; Inner: 0.32 / 0.15 / −0.01) and CD8+ T cell glycolytic score (right; Outer; 0.45 / −0.01 / −0.37; Mid: 0.60 / 0.05 / −0.31; Inner: 0.69 / 0.14 / −0.45) in distinct regions in the TME. Values max / average (diamond) /min. G) Violin plots representing monocyte/macrophage pseudotime signature score (left; Outer: 18.1 / 8.3 /0.00; Mid: 18.1 / 8.0 / 0.00; Inner: 18.1 / 10.0 / 0.00) and monocyte/macrophage glycolytic score (right; Outer: 1.35 / 0.29 / −0.33; Mid: 1.39 / 0.21 / −0.33; Inner: 1.39 / 0.33 / −0.33). Values max / average (diamond) / min. H) CellChat interaction likelihood analysis for CSF1 network in outer (top) and inner (bottom) regions of the TME. Thickness of green arrows represents interaction likelihood between populations. I) Cross-whisker plots comparing expression of exhaustion signature and normalized single gene (Csf1 and Ccl4) expression in CD8+ T cells (x-axis), and pseudotime score and antigen-presentation signature in the monocyte/macrophage population (y-axis) in distinct regions in the TME. Error bars represent 95% CI as computed by bootstrap resampling. See also Figure S6 and Table S1.
uMAP analysis of the entire immune compartment revealed prototypical and predominant clusters of T cells and monocytes/macrophages, and smaller clusters of natural killer (NK) cells and dendritic cells (DC) (Fig. 6B and Fig. S6B, C). Some of these populations were enriched in specific regions in the TME (Fig. 6C). We further subsampled the CD8+ T cell subset and thereby revealed that CD8+ T cells with a more naïve phenotype are enriched at the outer regions of the tumor, while exhausted CD8+ T cells mainly localize deeper inside the TME (Fig. 6D). Subsampling the monocyte/macrophage subset revealed a distinct localization pattern for different subsets of macrophages; namely, RetnlaHI macrophages were exclusively found at the outer regions of the TME, while ApoeHI, Ms4a7HI and proliferating macrophages are skewed towards the interior of the TME (Fig. 6E).
In line with our previous data obtained using orthotopic implantation of MMTV-PyMTChOVA-derived breast cancer cells (Hu et al., 2020), we found that CD8+ T cells show an increased exhaustion score (Wherry et al., 2007) when located in the inner regions of the TME (Fig. 6F, left). Moreover, we applied pseudotime analysis (Cao et al., 2019) on the monocyte/macrophage subset, and specified the Ly6c2HI monocyte-like cells as the root state of the trajectory which resulted in the ApoeHI, Ms4a7HI and proliferating macrophages as the terminally states (Fig. S6D, E), consistent with our previous findings (Hu et al., 2020; Mujal et al., 2022). When we overlaid Zipseq spatial localization on our pseudotime trajectory, we found a correlation with pseudotime progressing from monocyte-like early states at the outer regions to terminally differentiated TAM states in the tumor core (Fig. S6F). This was also reflected in an advanced monocyte/macrophage pseudotime score when moving from outer towards the interior of the tumor (Fig. 6G, left). Interestingly, the positive correlation between the expression of exhaustion-related genes in CD8+ T cells and macrophage maturation towards the inner regions of the TME coincided with an increased glycolytic score (Argüello et al., 2020) in CD8+ T cells, but not in the monocyte/macrophage fraction (Fig. 6F, right and Fig. 6G, right), consistent with a more hypoxic microenvironment which correlates with this dysfunctional crosstalk between Tex and TAM.
To predict interaction likelihood between different cell types in distinct regions in the TME, we used CellChat analysis, which uses a curated database of receptor-ligand interactions to highlight likely cell-cell interactions (Jin et al., 2021). These analyses revealed that expression of the CSF1-CSF1R ligand-receptor pair is significantly enriched in likelihood, and especially in the inner region versus outer region of tumors (Fig. 6H). Interestingly, Csf1 is found to be exclusively expressed by Tex in those regions (Fig. 6H). CellChat analysis also predicted that the main receivers of Tex-derived Csf1 in the inner regions of the tumor are Ms4a7HI macrophages, proliferating macrophages and ApoeHI macrophages (Fig. 6H), pointing towards a co-dependency between TAM and Tex. In line with this, we found a positive correlation between an ‘exhaustion’ signature, as well as normalized Csf1 and Ccl4 expression in CD8+ T cells and macrophage maturation (monocyte/macrophage pseudotime score), when moving from the outer towards the inner regions of the tumor (Fig. 6I). Conversely, expression of genes associated with antigen presentation in monocyte/macrophages gradually decreased when moving closer towards the inner regions of the tumor (Fig. 6I). These data support a model in which monocytes, as they move inward and differentiate toward terminal TAM, downregulate antigen presentation in concert with the development of the exhausted state in T cells.
Discussion
We mechanistically dissected a cellular co-alignment by which tumor-associated macrophages (TAM) and exhausted CD8+ T cells (Tex) in the TME co-exist in a self-enforcing positive feedback loop in mouse and human cancers. This includes finding that the secretion of growth factors and chemokines by one induces the other, the key interaction biology — a weakly stimulatory, yet long-duration synapse that ‘primes’ T cells for exhaustion — and spatial transcriptomics that demonstrate the co-evolution of these differentiated cell states, across space, in tumor tissue. Together, this demonstrates a principle of co-evolution of immunosuppressive cell types in the TME that supports immune evasion rather than destruction of the tumor.
The presence of TAM in solid tumors often correlates with poor prognosis and failure of response to anticancer therapies (Zhang et al., 2012; De Palma and Lewis, 2013). These findings are consistent with the established role of TAM in suppressing anti-tumor T cell immunity (DeNardo and Ruffell, 2019), and recent single cell RNA sequencing studies and other immune profiling approaches have hinted towards a potential link between the presence of TAM and exhausted CD8+ T cells in several different cancer types (Bi et al., 2021; Braun et al., 2021; Combes et al., 2022; Hong et al., 2021; Hu et al., 2020; Mujal et al., 2022; O’Connell et al., 2021; Wagner et al., 2019). However, these computational predictions require experimental investigation to establish causality. Building on previous findings from our own lab and others that CD8+ T cells preferentially localize in TAM-rich areas in the TME (Boissonnas et al., 2013; Boldajipour et al., 2016; Broz et al., 2014; Engelhardt et al., 2012; Peranzoni et al., 2018), we show here that the evolution of these long considered immunosuppressive cell types in the TME is extensively linked in a causal circuit.
Macrophages are known to display a remarkable heterogeneity and plasticity that is dependent on a variety of environmental cues, some of which are derived from T cells (DeNardo and Ruffell, 2019; Guerriero, 2019). However, prior to our study, a role for Tex in shaping macrophage phenotype and function had not been reported. We find in our models that intratumoral Tex are the main immune population producing macrophage-related factors to actively recruit monocytes and modulate their differentiation trajectory favoring antigen-presentation. In our data, Csf1 is a predominant and consistent ‘exhaustion’ gene, and previous reports have identified Ccl3, 4 and 5 as among the most differentially expressed genes in chronic viral infection-associated exhaustion (Wherry et al., 2007). Interestingly, we find that Flt3L, an important cytokine for CD103+ cDC1 biology (Barry et al., 2018), is concurrently downregulated in Tex, suggesting that upon progression to exhaustion, CD8+ T cells specifically favor the recruitment of TAM (and not cDC1) to support their presence in the TME and prevent destruction of the tumor. We consider it likely that Tex express additional myeloid-modulating factors that have yet to be identified and may now be sought, based on these studies. In addition, other cell types in the TME, including tumor cells and fibroblasts, can also modulate myeloid biology through secretion of a variety of cytokines (Buechler et al., 2021), and so there may be settings in which additional cell types also contribute to the establishment of the TAM-Tex axis.
Computational analysis of our spatial ZipSeq data suggests that Tex-derived CSF1 is most likely to affect specific subpopulations of terminally differentiated macrophages that are enriched in the inner regions of the TME, including Ms4a7HI, ApoeHI, and proliferating TAM. While other scRNA-seq studies have also reported on the existence of multiple different subpopulations of monocytes and TAM in the TME (Hu et al., 2020; Katzenelenbogen et al., 2020; Molgora et al., 2020; Mujal et al., 2022), it remains to be determined whether the localization in the TME affects their polarization state. Moreover, future studies will have to determine whether these subpopulations are functionally distinct from one another in their interactions with CD8+ T cells and their ability to modulate the onset of T cell exhaustion, and how other spatially regulated factors might play a role in the occurrence of TAM-T cell interactions.
Regardless of their ability to phagocytose large amounts of antigen, TAM are often considered inferior in antigen processing and presentation as compared to conventional dendritic cells. Here we show that antigen-presenting TAM capture CD8+ T cells in uniquely long-lasting synaptic interactions characterized by the formation of variegated TCR microclusters. Despite expressing similar levels of MHC class I and II, co-stimulatory molecules and genes involved in cross-presentation as do cDC1 (Broz et al., 2014), TAM trigger only a weak TCR stimulation that fails to support proliferation, but clearly primes the onset of T cell exhaustion which is not observed in the absence of TCR-ligands presented by these TAM. Notably, blockade of immune checkpoint molecules PD-1/PD-L1 and CTLA-4 is unable to license proliferation in T cells responding to TAM (Engelhardt et al., 2012). It is clear that more work is required to better understand the fundamental nature of disparate TCR triggers and co-stimulation over time that contribute to the hyporesponsive state in T cells during tumorigenesis, as elegantly reviewed recently (Philip and Schietinger, 2021).
Recent studies have reported that development of T cell exhaustion during chronic infection and cancer occurs in a multistep fashion, revealing distinct subtypes with unique transcriptional and epigenetic dynamics, as well as their ability to respond to immune checkpoint blockade (Im et al., 2016; Philip et al., 2017; Satpathy et al., 2019; Siddiqui et al., 2019; Jansen et al., 2019; Miller et al., 2019; Beltra et al., 2020; Pritykin et al., 2021). The decision-making during this bifurcative process seems to be tightly regulated by transcription factors like IRF4 (Utzschneider et al., 2020; Chen et al., 2021; Seo et al., 2021) which we find is strongly upregulated by TAM despite only subtle other signs of TCR engagement. Furthermore, our in vitro co-culture studies suggest that TAM at least are capable of mediating the early stages of exhaustion, which is exacerbated in hypoxic conditions. Hypoxia was recently shown to be an important co-factor in the induction of exhaustion in vitro (Scharping et al., 2021), and ZipSeq transcriptomics places terminal exhaustion in vivo preferentially in hypoxic regions of the TME. In line with this, other recent studies have elegantly demonstrated how metabolic insufficiencies drive mitochondrial stress in T cells contributing to exhaustion phenotypes in chronic infections and cancer (Bengsch et al., 2016; Thommen et al., 2018; Vardhana et al., 2020; Scharping et al., 2021). Future studies are required to determine whether the induction of exhaustion programs in CD8+ T cells is spatially coordinated for example through specialized TAM-rich niches in the TME.
Taken together, our work dissects a spatiotemporal co-evolution between exhausted CD8+ T cells and TAM in the TME that supports immune evasion rather than tumor destruction. We believe that co-dependency of different lineages may explain some of the resistance of the TME to targeting — removing just one cell population will still leave the other, to influence re-establishment of the targeted one. Thus, therapeutic strategies may need to break the biology underlying the TAM-Tex axis at multiple points. Doing so may work in conjunction with existing immunotherapies to enhance anti-tumor immunity, and thereby expand the proportion of cancer patients who benefit from immunotherapies.
STAR Methods
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Matthew F. Krummel (matthew.krummel@ucsf.edu).
Materials Availability
All unique reagents generated in this study are available from the Lead Contact without restriction.
Data and Code Availability
Bulk RNA-seq, bulk ATAC-seq and single-cell RNA-seq data have been deposited at GEO (GSE201074) and are publicly available as of the date of publication. RNA-seq data from human tumors has been published previously (Combes et al., 2022) and is publicly available at GEO (GSE184398). Accession numbers are also listed in the Key Resources Table. All original code has been deposited to GitHub and is publicly available as of the date of publication. DOIs are listed in the Key Resources Table. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-mouse CD103 - BUV737 (clone 2E7) | BD BioSciences | 749393 |
| anti-mouse CD103 - PE (clone 2E7) | Biolegend | 121406 |
| anti-mouse CD103 - PerCp-Cy5.5 (clone 2E7) | Biolegend | 121416 |
| anti-mouse CD11b - AF594 (clone M1/70) | Biolegend | 101254 |
| anti-mouse CD11b - BV421 (clone M1/70) | Biolegend | 101235 |
| anti-mouse CD11b - BV605 (clone M1/70) | Biolegend | 101237 |
| anti-mouse CD11b - BV650 (clone M1/70) | Biolegend | 101239 |
| anti-mouse CD11b - BV785 (clone M1/70) | Biolegend | 101243 |
| anti-mouse CD11c - BV650 (clone N418) | Biolegend | 117339 |
| anti-mouse CD11c - PerCp-Cy5.5 (clone N418) | Biolegend | 117328 |
| anti-mouse CD206 - AF488 (clone C068C2) | Biolegend | 141710 |
| anti-mouse CD206 - PerCp-Cy5.5 (clone C068C2) | Biolegend | 141716 |
| anti-mouse CD24 - BV421 (clone M1/69) | Biolegend | 101825 |
| anti-mouse CD24 - PECy7 (clone M1/69) | Biolegend | 101822 |
| anti-mouse CD31 - AF647 (clone 390) | Biolegend | 102415 |
| anti-mouse CD38 - BV711 (clone 90/CD38) | BD BioSciences | 740697 |
| anti-mouse CD38 - FITC (clone 90/CD38) | BD BioSciences | 558813 |
| anti-mouse CD4 - BUV395 (clone RM4–5) | BD BioSciences | 563790 |
| anti-mouse CD44 - BUV737 (clone IM7) | BD BioSciences | 564392 |
| anti-mouse CD44 - AF700 (clone IM7) | eBioScience | 56-0441-80 |
| anti-mouse CD45 AF647 (clone 30-F11) | Biolegend | 103124 |
| anti-mouse CD45 - BUV395 (clone 30-F11) | BD BioSciences | 564279 |
| anti-mouse CD45 - BV421 (clone 30-F11) | Biolegend | 304032 |
| anti-mouse CD45.1 - BUV395 (clone A20) | BD BioSciences | 565212 |
| anti-mouse CD45.1 - PECy7 (clone A20) | Biolegend | 110730 |
| anti-mouse CD45R (B220) - BV785 (clone RA3–6B2) | Biolegend | 103245 |
| anti-mouse CD5 - BV510 (clone 53–7.3) | Biolegend | 100627 |
| anti-mouse CD69 - BV650 (clone H1.2F3) | Biolegend | 104541 |
| anti-mouse CD8 - AF488 (clone 53–6.7) | Biolegend | 100723 |
| anti-mouse CD8 - PerCp-Cy5.5 (clone 53–6.7) | Biolegend | 100734 |
| anti-mouse CD80 - FITC (clone 16–10A1) | eBioScience | 11-0801-82 |
| anti-mouse CD86 - APC (clone GL-1) | Biolegend | 105012 |
| anti-mouse CD86 - BV421 (clone GL-1) | Biolegend | 105031 |
| anti-mouse CD90.2 - AF700 (clone 30-H12) | Biolegend | 105320 |
| anti-mouse CD90.2 - BV785 (clone 30-H12) | Biolegend | 105331 |
| anti-mouse CSF1R (CD115) - PECy7 (clone AFS98) | Biolegend | 135524 |
| anti-mouse F4/80 - AF700 (clone BM8) | Biolegend | 123130 |
| anti-mouse F4/80 - BV510 (clone BM8) | Biolegend | 123135 |
| anti-mouse H2kB - PE (clone AF6–88.5) | Biolegend | 116507 |
| anti-mouse IFNy - PE (clone XMG1.2) | eBioscience | 12-7311-81 |
| anti-mouse IFNy - PECy7 (clone XMG1.2) | Biolegend | 505826 |
| anti-mouse IRF4 - FITC (clone 3E4) | eBioscience | 11-9858-82 |
| anti-mouse IRF4 - PECy7 (clone 3E4) | eBioscience | 25-9858-82 |
| anti-mouse Ki67 - PE eFluor610 (clone SolA15) | eBioscience | 61-5698-82 |
| anti-mouse Ly6C - BV711 (clone HK1.4) | Biolegend | 128037 |
| anti-mouse Ly6G - BV785 (clone 1A8) | Biolegend | 127645 |
| anti-mouse I-A/I-E - AF700 (clone M5/114.15.2) | Biolegend | 107622 |
| anti-mouse NK1.1 - BV785 (clone PK136) | Biolegend | 108749 |
| anti-mouse PD-1 (CD279) - BV605 (clone 29F.1A12) | Biolegend | 135219 |
| anti-mouse SiglecF - BV785 (clone E50–2440) | BD BioSciences | 740956 |
| anti-mouse TCF1 - PE (clone S33–966) | BD BioSciences | 564217 |
| anti-mouse TCRb - AF488 (clone H57–597) | Biolegend | 109215 |
| anti-mouse TCRb V8.1 - PE (clone MR5–2) | BD BioSciences | 553186 |
| anti-mouse TNFa - BV421 (clone MP6-XT22) | Biolegend | 506327 |
| anti-mouse TNFa - PE (clone MP6-XT22) | Biolegend | 506306 |
| anti-mouse TOX - APC (clone REA473) | Miltenyi Biotec | 130-118-335 |
| anti-mouse CD16/32 (clone 2.4G2) | BioXCell | BE0307 |
| anti-mouse CD4 InVivoMab (clone GK1.5) | BioXCell | BE0003–1 |
| anti-mouse CD8 InVivoMab (clone 2.43) | BioXCell | BE0061 |
| anti-mouse CSF1 InVivoMab (clone 5A1) | BioXCell | BE0204 |
| anti-mouse CSF1R InVivoMab (clone AFS98) | BioXCell | BE0213 |
| Rat IgG1, k (clone HRPN) | BioXCell | BE0088 |
| Rat IgG2b, k (clone LTF-2) | BioXCell | BE0090 |
| Rat IgG2a, k (clone 2A3) | BioXCell | BE0089 |
| Hypoxyprobe Pacific Blue kit (4.3.11.3 MAb1) | Hypoxyprobe, Inc. | hp15-100kit |
| Normal Rat Serum | Thermo Fisher | 10710C |
| Bacterial and virus strains | ||
| N/A | ||
| Biological samples | ||
| Human tumor samples | UC San Francisco | IRB# 20-31740 |
| Mouse tissue samples (LN, tumor) | UC San Francisco | IACUC: AN184232 |
| Chemicals, peptides, and recombinant proteins | ||
| Matrigel GFR | Corning | 356231 |
| Collagenase, Type I | Worthington Biochemical | LS004197 |
| Collagenase, Type IV | Worthington Biochemical | LS004189 |
| Dnase I | Millipore Sigma | 10104159001 |
| Ficoll-Paque Plus | GE Healthcare | 17-1440-02 |
| Ficoll-Paque Premum 1.084 | GE Healthcare | 17-5446-02 |
| Zombie NIR Fixable Viability Dye | Biolegend | 423106 |
| CFSE | Invitrogen | C34554 |
| Violet Proliferation Dye | BD Biosciences | 562158 |
| CMTMR | Thermo Fisher | C2927 |
| Brefeldin A (BFA) | Sigma-Aldrich | B7651 |
| Phorbol 12-myristate 12-acetate (PMA) | Sigma-Aldrich | P8139 |
| Ionomycin | Invitrogen | I24222 |
| Complete Freund’s Adjuvant | Sigma-Aldrich | F5881 |
| OVA peptide (257–264) SIINFEKL | Anaspec | AS-60193 |
| LCMV gp33 peptide (33–41) KAVYNFATC | Anaspec | AS-61669 |
| Recombinant murine IL-4 | Peprotech | 214-14 |
| Recombinant murine GM-CSF | Peprotech | 315-03 |
| Recombinant human IL-2 | Peprotech | 200-02 |
| Fibronectin, bovine plasma | EMD Millipore | 341631 |
| FURA-2AM | Thermo Fisher | F1221 |
| Critical commercial assays | ||
| Chromium Single Cell 3’ GEM Library & Gel Bead Kit V3 | 10x Genomics | PN-1000092 |
| Foxp3/ Transcription Factor Staining Buffer Kit | BD Biosciences | 554655 |
| EasySep Mouse CD8+ T cell Isolation Kit | STEMCELL Technologies | 19853 |
| EasySep Mouse Biotin Positive Selection Kit | STEMCELL Technologies | 17665 |
| EasySep Mouse Monocyte Isolation Kit | STEMCELL Technologies | 19861 |
| UltraComp eBeads Compensation Beads | Fisher Scientific | 01-2222-42 |
| Corning HTS Transwell 96 well permeable supports | Corning | CLS3388 |
| RNeasy Micro kit | Qiagen | 74004 |
| High-Capacity cDNA Reverse Transcription kit | Thermo Fisher | 4368814 |
| Mouse M-CSF Quantikine ELISA kit | R&D Systems | MMC00 |
| CBA Mouse MIP-1a Flex Set | BD BioSciences | 558449 |
| CBA Mouse MIP-1b Flex Set | BD BioSciences | 558343 |
| CBA Mouse MIP-1a Flex Set | BD BioSciences | 558345 |
| Deposited data | ||
| All bulk RNAseq data for IPI cohort | Combes et al. 2022 | GEO:GSE184398 |
| Bulk RNAseq and ATACseq on T cells | This paper | GEO:GSE201074 |
| Single cell RNAseq data from CD206R ZipSeq | This paper | GEO:GSE201074 |
| GitHub | Kenneth Hu Github | https://github.com/ken7hu/ZipSeq-Analysis-CD206R |
| Experimental models: Cell lines | ||
| B16F10 | AATCC | CRL-6475 |
| B16ChOVA | UC San Francisco | N/A |
| B78ChOVA | UC San Francisco | N/A |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6J | The Jackson Laboratory | Stock # 000664 |
| Mouse: B6 CD45.1 (B6.SJL-Ptprc Pepc/BoyJ) | The Jackson Laboratory | Stock # 002014 |
| Mouse: OT-I (C57BL/6-Tg(TcraTcrb)1100Mjb/J | The Jackson Laboratory | Stock # 003831 |
| Mouse: mTmG | The Jackson Laboratory | Stock # 007676 |
| Mouse: Rag1 KO (B6.129S7-Rag1tm1Mom/J) | The Jackson Laboratory | Stock # 002216 |
| Mouse: CSF1op/op (B6;C3Fe a/a-Csf1op/J) | The Jackson Laboratory | Stock # 000231 |
| Mouse: Csf1rCre (C57BL/6-Tg(Csf1r-cre)1Mnz/J) | The Jackson Laboratory | Stock # 029206 |
| Mouse: LCMV P14 (crossed to B6 background) | Michael Waterfield, UC San Francisco | MGI: 2665105 |
| Mouse: MMTV-PyMTChOVA | Matthew Krummel, UC San Francisco | MGI: 5436574 |
| Mouse: CD206-LSL-Venus-DTR | Matthew Krummel, UC San Francisco | N/A |
| Oligonucleotides | ||
| Taqman probe: Ccl3 | Life Technologies | Mm00441259_g1 |
| Taqman probe: Ccl5 | Life Technologies | Mm01302427_m1 |
| Taqman probe: Csf1 | Life Technologies | Mm00432686_m1 |
| Taqman probe: Gapdh | Life Technologies | Mm99999915_g1 |
| Recombinant DNA | ||
| N/A | ||
| Software and algorithms | ||
| Imaris | Bitplane | https://imaris.exinst.com/ |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| FlowJo | Becton Dickinson | https://flowjo.com/ |
| CellRanger 4.0.0 | 10X Genomics | 10xgenomics.com |
| Seurat | Satija et al. 2015 | https://satijalab.org/seurat |
| STAR | Dobin et al. 2013 | https://code.google.com/archive/p/rna-star/ |
| R: The Project for Statistical Computing | N/A | http://r-project.org |
| CellChat | Jin et al. 2021 | http://www.cellchat.org/ |
| Other | ||
| N/A | ||
Experimental Model and Subject Details
Human tumor samples
Flow cytometry on kidney renal clear cell carcinoma (KID) samples: samples were transported from various cancer operating rooms or outpatient clinics. All patients consented by the UCSF IPI clinical coordinator group for tissue collection under a UCSF IRB approved protocol (UCSF IRB# 20–31740). Samples were obtained after surgical excision with biopsies taken by Pathology Assistants to confirm the presence of tumor cells. Patients were selected without regard to prior treatment. Freshly resected samples were placed in ice-cold DPBS or Leibovitz’s L-15 medium in a 50 mL conical tube and immediately transported to the laboratory for sample labeling and processing. The whole tissue underwent digestion and processing to generate a single-cell suspension. In the event that part of the tissue was sliced and preserved for imaging analysis, the remaining portion of the tissue sample was used for flow cytometry analysis as described in Combes et al. (Combes et al., 2022).
Samples from the following tumor types were used for RNA-seq on FACS-isolated cell fractions performed as described previously (Combes et al., 2022): Bladder cancer (BLAD), colorectal cancer (CRC), glioblastoma multiforme (GBM), endometrial and ovarian cancer (GYN), hepatocellular carcinoma (HEP), head and neck squamous cell carcinoma (HNSC), kidney renal clear cell carcinoma (KID), lung adenocarcinoma (LUNG), skin cutaneous melanoma (MEL), pancreatic ductal adenocarcinoma (PDAC), pancreatic neuroendocrine tumors (PNET), sarcoma (SRC).
Mice
All mice were treated in accordance with the regulatory standards of the National Institutes of Health and American Association of Laboratory Animal Care and were approved by the UCSF Institution of Animal Care and Use Committee. The following mice were purchased for acute use or maintained under specific pathogen-free conditions at the University of California, San Francisco Animal Barrier Facility: C57BL6/J, C57BL6/J CD45.1, OT-I, P14 LCMV, Rag1−/−, Csf1op/op, mTmG. With the exception of Csf1op/op, all mice used in experimentation were bred to a C57BL6/J background. Mice of either sex ranging in age from 6–12 weeks were used for experimentation. For experiments using the transgenic MMTV-PyMTChOVA strain (Engelhardt et al., 2012), only mammary tumor-bearing females were used ranging in age from 12–20 weeks. Treatments in MMTV-PyMTChOVA mice were started when mammary tumors reached ~25mm2 in size. Csf1rCreCd206-LSL-Venus-DTR mice (Ray, A. et al. in preparation) were generated and used for ZipSeq. Food and water were provided ad libitum.
Tumor cell lines
Tumor cell lines B16F10 (CRL-6475, ATCC), B16ChOVA (Roberts et al., 2016; Binnewies et al., 2019) and B78ChOVA (Engelhardt et al., 2012; Broz et al., 2014) were cultured under standard conditions 37°C in 5% CO2 in DMEM (GIBCO), 10% FCS (Benchmark), 1% Pen/Strep/Glut (Invitrogen).
Method Details
Tumor growth experiments
For tumor studies, adherent tumor cells were grown to confluency and harvested using 0.05% Trypsin-EDTA (GIBCO) and washed 3x with PBS (GIBCO). 1.0×105 – 2.5×105 cells in PBS were resuspended in a 1:1 ratio with Growth Factor Reduced Matrigel (Corning) and a final volume of 50μl was injected subcutaneously into the flanks of anaesthetized and shaved mice. Tumors were allowed to grow for 14–21 days unless otherwise noted, before tumors and tumor-draining lymph nodes were harvested for analysis.
Adoptive T cell transfers
Inguinal, axillary, brachial and mesenteric lymph nodes (LN) or spleens were isolated from CD45.1 OT-I or P14 LCMV mice. LN and spleens were meshed through 70μm filters and treated with ACK red blood cell lysis buffer. CD8+ T cells were purified using EasySep CD8 negative selection kits (Stemcell Technologies). 1×105 (for >14 day read-out) – 2×106 T cells (for day 4 read-out) were adoptively transferred through retro-orbital injection in 100μl PBS.
For the comparison of OT-I T cells and p14 LCMV T cells, mice received a 1:1 mix of both T cells in 100μl of PBS through retro-orbital injection. The following day mice were inoculated with a bolus of CFA containing gp33-peptide (50μg/mouse; Anaspec) and SL8/SIINFEKL peptide (50μg/mouse; Anaspec) subcutaneously, to sustain both T cell populations.
In vivo antibody treatment
For macrophage depletions, mice received anti-CSF1 (clone 5A1; BioXCell), anti-CSF1R (clone AFS98; BioXCell) or corresponding isotype controls, Rat IgG1k (clone HRPN; BioXCell) and Rat IgG2a (clone 2A3; BioXCell), respectively. Antibodies were injected intraperitoneally at an initial dose of 1mg/mouse followed by 0.5mg/mouse every 7 days.
For T cell depletion studies, mice received anti-CD4 (clone GK1.5; BioXCell), anti-CD8a (clone 2.43; BioXCell) or corresponding isotype control, Rat IgG2b (clone LTF-2; BioXCell) dosed at 250 μg/mouse every 3–4 days.
Generation of mixed bone marrow chimeras
Mixed bone marrow chimeras were generated as described previously (Barry et al., 2018). Briefly, Rag1−/− mice were lethally irradiated with 1,100 rads of irradiation in two doses 3–5 hours apart. 2–5×106 bone marrow cells, consisting of 50% Rag1−/− and 50% Csf1op/op or Csf1op/+ bone marrow, were injected retro-orbitally to reconstitute irradiated mice. Chimeric mice were allowed to recover for 6–10 weeks, upon which mice were inoculated with B78ChOVA tumors subcutaneously.
Mouse tissue digestion and flow cytometry
Tumors were harvested and processed to single cell suspensions as described previously (Barry et al., 2018; Binnewies et al., 2019). Briefly, tumors were isolated and mechanically minced, followed by enzymatic digestion with 200μg/ml DNAse (Sigma-Aldrich), 100U/ml Collagenase I (Worthington Biochemical) and 500U/ml Collagenase Type IV (Worthington Biochemical) for 30 minutes at 37°C while shaking. Enzymatic activity was quenched by adding equal amounts of FACS buffer (2% FCS in PBS), and cell suspensions were filtered to obtain single cell suspensions. TdLN were isolated and meshed over 70μm filters in PBS to generate single cell suspensions. For each sample, 5–10×106 cells were used for staining for flow cytometry. Cells were washed with PBS prior to staining with Zombie NIR Fixable live/dead dye (Biolegend) for 20 min at 4°C. Cells were washed in PBS followed by surface staining for 30 min at 4°C with directly conjugated antibodies diluted in FACS buffer containing anti-CD16/32 (BioXCell) to block non-specific binding. Cells were washed again with FACS buffer. For intracellular staining, cells were fixed for 20 min at 4°C using the FOXP3 Fix/Perm kit (BD Biosciences), and washed in permeabilization buffer. Antibodies against intracellular targets were diluted in permeabilization buffer and cells were incubated for 30 min at 4°C followed by another wash prior to read-out on a BD LSR Fortessa SORP cytometer.
Fluorescence Activated Cell Sorting
Single cell suspensions from tumors were prepared as described above. For T cell isolations, single cell suspensions were enriched for mononuclear cells using Ficoll-Paque Premium 1.084 (GE Healthcare). For isolation of myeloid cells, single cell tumor suspensions were enriched for CD45+ cells using EasySep biotin positive selection kit (Stemcell Technologies). Enriched cells were stained for 30 min at 4°C with directly conjugated antibodies diluted in FACS buffer containing anti-CD16/32 (BioXCell) to block non-specific binding. Cells were washed again with FACS buffer and filtered over a 70μm mesh. Immediately prior to sorting, DAPI was added to exclude dead cells. Cells were sorted on a BD FACSAria Fusion and BD FACSAria2. Sorted T cells were collected directly in lysis buffer (Invitrogen) for RNA sequencing or in RPMI (GIBCO), 10% FCS (Benchmark), Pen/Strep/Glut (Invitrogen) and 50μM β-mercaptoethanol (GIBCO) at 4°C for further use ex vivo. Sorted myeloid cells were collected in DMEM (GIBCO), 10% FCS (Benchmark), Pen/Strep/Glut (Invitrogen) at 4°C for further use ex vivo.
Intracellular T cell cytokine analysis
For analysis of cytokine production by endogenous and adoptively transferred T cells, 5–10×106 LN and tumor cells were re-stimulated for 3–5 hours in RPMI (GIBCO), 10% FCS (Benchmark), Pen/Strep/Glut (Invitrogen), 50μM β-mercaptoethanol (GIBCO) containing PMA (50ng/ml; Sigma-Aldrich), ionomycin (500ng/ml; Invitrogen) and brefeldin A (3μg/ml; Sigma-Aldrich) at 37°C in 5% CO2. Cells were washed and stained for intracellular flow cytometric analysis.
ELISA and Cytometric Bead Array
Endogenous CD8+CD44− (naïve) and CD8+CD44+ (effector) T cells were isolated from lymph nodes, and CD8+CD44+ (exhausted) T cells were isolated from tumors of B78ChOVA-bearning mice using FACS. Antigen-specific OT-I CD8+CD44− (naïve) T cells were isolated directly from lymph nodes of tumor-free OT-I transgenic mice, or activated in vitro with antigen-pulsed splenocytes to generate effector OT-I T cells. Congenically labeled (CD45.1) OT-I CD8+CD44+ T cells were isolated from B78ChOVA tumors >14 days after adoptive transfer (exhausted T cells). Isolated T cells (3×105) were cultured ex vivo in RPMI (GIBCO), 10% FCS (Benchmark), Pen/Strep/Glut (Invitrogen), 50μM β-mercaptoethanol (GIBCO) for 24 hours. Levels of secreted CSF1 in supernatant was measured by ELISA (R&D Systems). Levels of secreted CCL3, CCL4 and CCL5 in supernatant were measured by Cytometric Bead Array (CBA) CCL3/MIP-1α, CCL4/MIP-1β and CCL5/RANTES Flex Sets (BD BioSciences) according to manufacturer’s recommendations.
RNA sequencing
mRNA from cells were isolated using DynaBead Direct and then converted into amplified cDNA using the Tecan Ovation RNA-Seq System V2 kit, following the manufacturer guidelines. The dsDNA is tagmented, amplified and undergoes clean up with AMPure XP bead, using the Illumina Nextera XT DNA Library Prep Kit. The resulting sequencing library is QC’d using an Agilent Bioanalyzer HS DNA chip to assess fragment size distribution and concentration. Libraries were pooled prior to single-end sequencing on and Illumina MiSeq/MiniSeq to ensure quantify library complexity. Libraries with less than 10 percent of the reads aligned to coding regions, or fewer than 1,000 unique reads in total were rejected. The validated libraries were re-pooled based on the percentage of reads in coding regions and submitted to the UCSF Center for Advanced Technology for 150bp paired end sequencing on an Illumina NovaSeq 6000.
Raw fastq reads were QC’d and trimmed to remove adapter contamination, and poly-G artifacts using using fastp version 0.19.6 (Chen et al., 2018). Reads with fewer than 20bp post-trimming were discarded. Trimmed reads were aligned to the GRCm38 reference sequence annotated with Gencode V25 (Frankish et al., 2019) using STAR version 2.6.1b (Dobin et al., 2013) with the following parameters (--quantMode GeneCounts–outFilterMismatchNoverLmax 0.04 --alignIntronMax 100000 --alignMatesGapMax 100000 --alignSJDBoverhangMin 10 --alignSJstitchMismatchNmax 5 −1 5 5 --chimSegmentMin 12 --chimJunctionOverhangMin 12 --chimSegmentReadGapMax 3 --chimMultimapScoreRange 10 --chimMultimapNmax 10 --chimNonchimScoreDropMin 10 --peOverlapNbasesMin 12 --peOverlapMMp 0.1) STAR-generated reads counts from each library were processed using the limma/Voom pipeline (Law et al., 2014; Smyth, 2005) using the edgeR package (Robinson et al., 2010). Briefly, the read counts are loaded into a DGEList object to generate Counts Per Million (CPM), and then filtered to retain only genes with at least 10 counts in a worthwhile number of samples and at least 15 counts across all samples. The CPM matrix is normalized using TMM Trimmed mean of M-values and processed using voom to estimate the mean-variance relationship to identify edge weights that can be used to fit to a linear model with limma lmFit. Differential gene expression between two groups of empirical Bayes moderation of the standard errors towards a global value. A list of transcriptional DEGs between Tnaïve and Tex d14 with a FC equal to or >5 was generated and gene set enrichment analysis was performed using the MGI Mammalian Phenotype Level 4 database in Enrichr (Chen et al., 2013; Kuleshov et al., 2016; Xie et al., 2021).
ATAC sequencing
ATAC-seq samples were processed according to the Omni-ATAC protocol (Corces et al., 2017). 5×104 cells per replicate were lysed in 50 μL ATAC resuspension buffer supplemented with 0.1% NP40, 0.1% Tween-20, and 0.01% Digitonin. After lysis, nuclei were transposed using 2.5 μl Tn5 transposase in a 50 μl reaction for 30 min at 37°C. Finally, the transposed DNA was purified using a commercial PCR cleanup kit and libraries were prepared for sequencing. 2×75 paired end sequencing was performed on an Illumina sequencer.
ATAC-seq computational analysis was performed as previously described (Weber et al., 2021). Briefly, read trimming and filtering was performed with fastp. Reads were mapped to the hg38 reference genome using hisat2 with the --no-spliced-alignment option. Picard was used to remove duplicates from bam files. We removed any reads not mapping to chromosomes 1–22 and chrX (ie chrY reads, mitochondrial reads, and other reads were discarded). The deduplicated and filtered fragments were then formatted into a bed file. Peaks were called using MACS2. Peaks from each sample were iteratively merged into a high confidence union peak set for all samples as previously described (Corces et al., 2018). A peak by sample matrix was created by overlapping fragments in each sample with each peak, and this matrix was used to perform differential peak analysis in DESeq2. Genome coverage files were created from the fragments file by loading the fragments into R and then exporting bigwig files normalized by reads in transcription start sites using ‘rtracklayer::export’. Normalized track files were visualized using the Integrative Genomics Viewer.
qRT-PCR
RNA was extracted from FACS-sorted immune cell populations using Qiagen RNeasy Micro kit (Qiagen) and the yield was measured using Nanodrop. cDNA first-strand synthesis was performed using High-Capacity cDNA Reverse Transcription kit (Thermo Fisher Scientific) using random primers. qRT-PCR analysis was performed using Taqman probes targeting Ccl3 (Mm00441259_g1), Ccl5 (Mm01302427_m1), Csf1 (Mm00432686_m1) and Gapdh (Mm99999915_g1). All probes were obtained from Life Technologies. For amplification reactions, iTaq Universal Probes Supermix was used according to manufacturer’s instructions. qRT-PCR was performed on a QuantStudio 12K Flex lightcycler (Applied Biosystems by Life Technologies). For quantification the delta Ct method was used: delta Ct sample — delta Ct reference gene. All transcripts were normalized to Gapdh.
Monocyte recruitment transwell assays
Bone marrow was obtained from femurs and tibia of CD45.1 mice, and monocytes were isolated using EasySep Mouse Monocyte Isolation kits (Stemcell Technologies). For transwell assays, 1×105 monocytes were added to top inserts containing 5.0 μm pore polycarbonate membrane (Corning). 0.5×105 naïve, previously activated or exhausted OT-I T cells were cultured in bottom wells in RPMI (GIBCO), 10% FCS (Benchmark), Pen/Strep/Glut (Invitrogen) and 50μM β-mercaptoethanol (GIBCO). Migration through the membrane was analyzed after 24 hours of culture. Plate was briefly centrifuged briefly at 1000rpm for 1 min to collect cells stuck to the membrane. Cells were collected for analysis by flow cytometry. Absolute counting beads (Life Technologies) were added for quantification of the number of migrated cells.
Generation of activated T cells
OT-I T cells were activated in vitro as described previously (Broz et al., 2014). Briefly, OT-I lymph node cells were stimulated with B6 splenocytes pulsed with SL8 peptide (100ng/ml; Anaspec) for 30 min at 37°C and then washed 3 times. On day 2–3, cells were expanded by adding human IL-2 (2U/ml; Peprotech) to fresh RPMI (GIBCO), 10% FCS (Benchmark), Pen/Strep/Glut (Invitrogen), 50μM β-mercaptoethanol (GIBCO). Cells were used for co-culture assays on day 4–5. Prior to use dead cells were excluded using Ficoll-Paque PLUS (GE Healthcare).
Bone marrow-derived dendritic cells
BMDC were generated as described previously (Broz et al., 2014). Briefly, bone marrow was obtained from femurs and tibia of C57BL6/J mice and cultured in DMEM (GIBCO), 10% FCS (Benchmark), Pen/Strep/Glut (Invitrogen) in the presence of 7.5 ng/ml GM-CSF (Peprotech) for 6–8 days, followed by the addition of 60ng/ml IL4 (Peprotech) for the last 2 days. Media was refreshed every 3–4 days. For co-cultures studies, BMDC were pulsed with SL8 peptide (100ng/ml; Anaspec) for 30 min at 37°C and then washed 3 times prior to use.
Quantification of APC-T cell interactions
APC (BMDC, BMDC+SL8 or sorted TAM) were obtained from mTmG mice, and previously activated OT-I T cells were stained for 15 minutes at 37°C with 2 μM CFSE (Invitrogen) in PBS and washed in RMPI prior to use. Cells were co-cultured in NUNC 8 well chamber slides (Thermo Scientific) that were coated with fibronectin (2μg/ml; EMD Millipore) in PBS at 37°C for 1 hour before use. APCs in phenol red-free RPMI were allowed to attach to the chamber slides for 20–30 minutes at 37°C and 5% CO2. Right before imaging, T cells (resuspended in 0.1% agarose) were added to the wells and slides were loaded for imaging. To visualize the interaction between different APC populations and T cells, a conventional widefield Zeiss Axiovert 200M was used with a Sutter Lambda XL illumination source, running on μMagellan software. Images were acquired every 2 minutes for 6 hours using a 20x objective. Samples were kept at 37°C using a heated robotic stage. Image analysis was performed in Imaris (Bitplane) and ImageJ.
APC-T cell coupling assay
Ex vivo coupling assays were performed as described previously (Broz et al., 2014). Briefly, single cells suspensions were enriched for CD45+ cells using EasySep biotin positive selection kit (Stemcell Technologies). Enriched cells were stained with with Zombie NIR Fixable live/dead dye (Biolegend) for 20 min at 4°C, followed by staining for 30 min at 4°C with directly conjugated antibodies diluted in FACS buffer containing anti-CD16/32 (BioXCell) to block non-specific binding. Cells were washed again and co-cultured with VPD-labeled previously activated OT-I CD8+ T cells for 1 hour at 37°C. Cells were lightly fixed in 2% PFA followed by read-out on a BD LSR Fortessa SORP cytometer.
Lattice light-sheet imaging
Lattice light-sheet (LLS) imaging was performed in a manner previously described (Cai et al., 2017). Briefly, 5 mm diameter round coverslips were cleaned by a plasma cleaner and coated with fibronectin (2μg/ml; EMD Millipore) in PBS at 37°C for 1 hour before use. TAM sorted from B78ChOVA-bearing mTmG mice were dropped onto the coverslip and incubated at 37°C, 5% CO2 for 20–30 min. Previously activated OT-I CD8+ T cells were labeled with CD45-AF647 (clone 30-F11; Biolegend) and TCRβ AF488 (clone H57–597; BioLegend) for 30 min and washed in FACS buffer. Right before imaging, T cells were dropped onto the coverslip containing TAM. The sample was then loaded into the previously conditioned sample bath and secured. Imaging was performed with a 488-nm, 560-nm, or 642-nm laser (MPBC, Canada) dependent upon sample labeling in single or two-color mode. Exposure time was 10 ms per frame leading to a temporal resolution of 4.5 s. Image renderings were created using Imaris software (Bitplane). Quantification of TCR clustering was performed using ImageJ. Briefly, channels were separated and entire T cell membrane versus TAM interaction site was outlined manually. Signal intensity for red (CD45) and green (TCR) channel was calculated. The following formula was used to determine TCR signal intensity for both channels at the synaptic TAM-T cell interaction site: signaling intensity = (intensity at synapse/intensity total membrane).
FURA-2AM Calcium imaging
TAM or CD103+ DC were sorted from B16ChOVA or B16F10 tumors as per description above, and stained with 2 μM CMTMR (Thermo Fisher Scientific) in PBS for 15 minutes at 37°C, followed by a wash in RPMI. Cells were co-cultured in NUNC 8 well chamber slides (Thermo Scientific) that were coated with fibronectin (2μg/ml; EMD Millipore) in PBS at 37°C for 1 hour before use. APCs in phenol red-free RPMI were allowed to attach to the chamber slides for 20–30 minutes at 37°C and 5% CO2. Right before imaging, previously activated OT-I T cells were labeled with FURA-2 AM (0.5μM; Invitrogen) for 15 minutes at RT. Cells were washed and resuspended in phenol red-free RPMI (GIBCO), 10% FCS (Benchmark), Pen/Strep/Glut (Invitrogen), 50μM β-mercaptoethanol (GIBCO) supplemented with 0.1% agarose and were added to the wells. Imaging was performed using an Zeiss Axiovert 200M microscope with a Sutter Lambda XL illumination source equipped with a 40x oil objective. Images were acquired every 5 seconds for 18–21 minutes. Samples were kept at 37°C using a heated robotic stage. Image analysis was performed in Imaris (Bitplane). Briefly, background subtraction was performed and surfaces were created for APCs and T cells to quantify dwell time to determine whether cells were touching (cut-off equal to or >3). Calcium2+ flux was determined by calculating the average 340/380 ratiometric fluorescence per cell after contact for each time point.
Hypoxyprobe imaging
Mice were injected with pimonidazole hydrochloride in PBS (80mg/kg; Hypoxyprobe) intraperitoneally 1.5 hours prior to sacrifice. Tissues were dissected and processed as described above. For flow cytometry studies, pimonidazole was visualized using anti-pimonidazole antibodies (Pacific Blue Mab-1 clone 4.3.11.3; Hypoxyprobe) after cells were fixed for 20 min at 4°C using the FOXP3 Fix/Perm kit (BD Biosciences), and washed in permeabilization buffer. For imaging studies, dissected tumors were embedded in OCT and sectioned into 10μm cryosections. Cryosections were stored at −80°C until further use. For immunostaining, sections were fixed in 4% PFA (Electron Microscopy Sciences) for 20 minutes at RT, followed by a rinse in PBS containing 1% BSA (Sigma). Sections were blocked in 1% BSA in PBS containing anti-CD16/32 (BioXCell) for 1 hour at RT, and washed. Sections were stained with CD11b-AF594 (clone M1/70;BioLegend), CD31-AF647 (clone 390;BioLegend) and Pacific Blue Mab-1 (clone 4.3.11.3;Hypoxyprobe) for 1 hour at RT, followed by a wash and mounted using Vectashield (Vector Laboratories) and sealed with nail polish. Images were acquired on a Leica SP8 confocal microscope. Data analysis was performed using Imaris (Bitplane).
In vitro APC-T cell co-culture assays
APC populations were sorted from tumors as described above, and co-cultured with 1×105 previously activated OT-I CD8+ T cells labeled with Violet Proliferation dye (VPD; BD Biosciences) at a 1:5 ratio (unless otherwise noted) in RPMI (GIBCO), 10% FCS (Benchmark), Pen/Strep/Glut (Invitrogen), 50μM β-mercaptoethanol (GIBCO) in 96-well round bottom plates. Cells were harvested for analysis 3 days later, unless otherwise noted. In vitro generated BMDC and BMDC+SL8 were used as negative and positive controls, respectively.
For hypoxia experiments, plates containing exact same experimental groups were incubated in an Avatar hypoxic bioreactor (XcellBio) at 1.5% O2 or under ambient 21% O2 for comparison and cells were harvested after 3 days.
ZipSeq
ZipSeq spatial transcriptomics was performed as described previously (Hu et al., 2020). Briefly, B78ChOVA cells were injected subcutaneously as described above and were harvested day 16 post-injection. Tumors were sectioned while live using a compresstome (Precisionary Instruments VFZ-310–0Z) to generate ~160 μm sections. The sectioning, imaging, spatial barcoding, tumor dissociation, sorting, 10X encapsulation and library construction were identical to the methods described in (Hu et al., 2020). The targeted number of cells for loading was 5000. With this in mind, we aimed for 30,000 reads per cell during sequencing on an Illumina S4 flowcell with a 1:10 molar ratio of Zipcode reads to gene expression reads. Resulting fastq files were processed using the CellRanger 4.0.0 pipeline, aligning to the GRCm38 Mus musculus assembly. CellRanger output thus resulted in ~359k reads for the gene expression library and ~40k reads for the Zipcode library.
Analysis of scRNA-Seq
The raw feature-barcode matrix generated by 10X CellRanger was loaded into Seurat (Satija et al., 2015). Cells with mitochondrial read % over 20% and those with less than 500 genes detected were excluded from analysis. Zipcode read counts from CellRanger were also loaded into Seurat as a separate ‘ADT’ assay and using CLR normalized counts, cells with either too few Zipcode reads or mixed Zipcode reads were also excluded from analysis. Following built-in Seurat methods for gene expression normalization and variance stabilization (Single Cell Transform (Hafemeister and Satija, 2019)), cells underwent one more round of clean-up, removing a small cluster of contaminating CD45– cells and another small cluster dominated by mitochondrial and ribosomal genes. This yielded 2765 cells. At this stage, the mean # of UMI’s and # of detected genes was: (25,566 and 4,199 respectively) while for the antibody derived Zipcode tags the mean UMI was 1,891 reads. At this stage, we also determined cluster identities using Seurat’s FindAllMarkers function performed on the log-normalized read counts which by default uses the Wilcoxon Rank Sum test.
For CellChat analysis, this cleaned object was fed into the CellChat workflow, using the built-in mouse ligand-receptor database, and a tri-mean thresholding for significance of interaction. The Seurat object was split into 3 sub-objects based on regional assignment and these 3 objects were separately analyzed using the CellChat workflow for multiple datasets and then merged. For signature score generation, we used Seurat’s built-in AddModuleScore function with gene lists for Glycolysis (Argüello et al., 2020), T cell exhaustion (Wherry et al., 2007), and Antigen Presentation (GO term 0048002) using 50 control features. Full gene lists can be found in Table S1. For pseudotime analysis, the monocyte/macrophage sub-object was passed into Monocle v3 without any changes to the UMAP dimensional reduction.
Quantification and Statistical Analysis
Statistical analysis
Unless specifically noted, all data are representative of ≥ 2 separate experiments. Experimental group assignment was determined by random designation. Statistical analyses were performed using GraphPad Prism software. Error bars represent ± standard error of the mean (SEM) calculated using Prism. Specific statistical tests used were paired t-tests, unpaired t-tests, and one-way ANOVA unless otherwise noted. P-values <0.05 were considered statistically significant. Investigators were not blinded to group assignment during experimental procedures or analysis.
Supplementary Material
Movie S1. Synapse formation between TAM and CD8+ T cells (related to Figure 4). Lattice light sheet imaging of the interaction between mTomato+ TAM sorted from B78ChOVA melanomas (magenta) and previously activated CD8+ OT-I T cell labeled with CD45-AF647 (red), with the H57 TCRβ labeled with AF488. Scale bar, 5μm.
Highlights.
Onset of T cell exhaustion in cancer is linked to TAM abundance
Tex express myeloid-related factors to shape myeloid cell recruitment and phenotype
TAM form uniquely long-lasting synapses with CD8+ T cells that license exhaustion
ZipSeq reveals spatial coordination of TAM-Tex axis in inner regions of tumors
Acknowledgements
We thank all members of the Krummel laboratory for discussion and support. We also thank the ImmunoX CoLabs at UCSF for technical assistance and support. Figures were created using BioRender.com. Funding: K.K. was supported by a Rubicon postdoctoral fellowship 019.163LW.006 from the Netherlands Organization for Scientific Research (NWO), and a Parker Scholar Award from the Parker Institute for Cancer Immunotherapy. K.H.H. was supported by a Parker Scholar Award from the Parker Institute for Cancer Immunotherapy, and a Jean Perkins postdoctoral fellowship from the American Cancer Society. A.R. and E.C. were supported by postdoctoral fellowships from the Cancer Research Institute. J.A.B was supported by a Stanford Graduate Fellowship and the National Science Foundation Graduate Research Fellowship under Grant No. DGE-1656518. A.T.S. was supported by NIH U01CA260852, a Cancer Research Institute Technology Impact Award, and a Career Award for Medical Scientists from the Burroughs Wellcome Fund. We acknowledge the UCSF Parnassus Flow Core (RRID:SCR_018206 and DRC Center Grant NIH P30 DK063720) for assistance and using their instruments and services. This work was supported by NIH/NCI U54CA163123, 5U01CA217864, R21CA191428 and R01CA197363 to M.F.K.
Declaration of Interest
A.T.S. is a scientific founder of Immunai and founder of Cartography Biosciences and receives research funding from Merck Research Laboratories and Allogene Therapeutics. M.F.K. is a founder and shareholder in Pionyr Immunotherapeutics and in Foundery Innovations, that prosecute and develop novel immunotherapeutics respectively. The other authors declare no competing interests.
Inclusion and Diversity
One or more of the authors of this paper self-identifies as living with a disability.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alfei F, Kanev K, Hofmann M, Wu M, Ghoneim HE, Roelli P, Utzschneider DT, von Hoesslin M, Cullen JG, Fan Y, et al. (2019). TOX reinforces the phenotype and longevity of exhausted T cells in chronic viral infection. Nature 571, 265–269. 10.1038/s41586-019-1326-9. [DOI] [PubMed] [Google Scholar]
- Argüello RJ, Combes AJ, Char R, Gigan J-P, Baaziz AI, Bousiquot E, Camosseto V, Samad B, Tsui J, Yan P, et al. (2020). SCENITH: A Flow Cytometry-Based Method to Functionally Profile Energy Metabolism with Single-Cell Resolution. Cell Metab. 32, 1063–1075. 10.1016/j.cmet.2020.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry KC, Hsu J, Broz ML, Cueto FJ, Binnewies M, Combes AJ, Nelson AE, Loo K, Kumar R, Rosenblum MD, et al. (2018). A natural killer-dendritic cell axis defines checkpoint therapy-responsive tumor microenvironments. Nat. Med 24, 1178–1191. 10.1038/s41591-018-0085-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W, Huhn RD, Song W, Li D, Sharp LL, et al. (2011). CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science 331, 1612–1616. 10.1126/science.1198443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltra J-C, Manne S, Abdel-Hakeem MS, Kurachi M, Giles JR, Chen Z, Casella V, Ngiow SF, Khan O, Huang YJ, et al. (2020). Developmental Relationships of Four Exhausted CD8+ T Cell Subsets Reveals Underlying Transcriptional and Epigenetic Landscape Control Mechanisms. Immunity 52, 825–841. 10.1016/j.immuni.2020.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengsch B, Johnson AL, Kurachi M, Odorizzi PM, Pauken KE, Attanasio J, Stelekati E, McLane LM, Paley MA, Delgoffe GM, et al. (2016). Bioenergetic Insufficiencies Due to Metabolic Alterations Regulated by the Inhibitory Receptor PD-1 Are an Early Driver of CD8(+) T Cell Exhaustion. Immunity 45, 358–373. 10.1016/j.immuni.2016.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi K, He MX, Bakouny Z, Kanodia A, Napolitano S, Wu J, Grimaldi G, Braun DA, Cuoco MS, Mayorga A, et al. (2021). Tumor and immune reprogramming during immunotherapy in advanced renal cell carcinoma. Cancer Cell 39, 649–661. 10.1016/j.ccell.2021.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, Coussens LM, Gabrilovich DI, Ostrand-Rosenberg S, Hedrick CC, et al. (2018). Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med 24, 541–550. 10.1038/s41591-018-0014-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binnewies M, Mujal AM, Pollack JL, Combes AJ, Hardison EA, Barry KC, Tsui J, Ruhland MK, Kersten K, Abushawish MA, et al. (2019). Unleashing Type-2 Dendritic Cells to Drive Protective Antitumor CD4+ T Cell Immunity. Cell 177, 556–571. 10.1016/j.cell.2019.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boissonnas A, Licata F, Poupel L, Jacquelin S, Fetler L, Krumeich S, Théry C, Amigorena S, and Combadière C (2013). CD8+ tumor-infiltrating T cells are trapped in the tumor-dendritic cell network. Neoplasia N. Y. N 15, 85–94. 10.1593/neo.121572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boldajipour B, Nelson A, and Krummel MF (2016). Tumor-infiltrating lymphocytes are dynamically desensitized to antigen but are maintained by homeostatic cytokine. JCI Insight 1, e89289. 10.1172/jci.insight.89289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun DA, Street K, Burke KP, Cookmeyer DL, Denize T, Pedersen CB, Gohil SH, Schindler N, Pomerance L, Hirsch L, et al. (2021). Progressive immune dysfunction with advancing disease stage in renal cell carcinoma. Cancer Cell 39, 632–648. 10.1016/j.ccell.2021.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broz ML, Binnewies M, Boldajipour B, Nelson AE, Pollack JL, Erle DJ, Barczak A, Rosenblum MD, Daud A, Barber DL, et al. (2014). Dissecting the Tumor Myeloid Compartment Reveals Rare Activating Antigen-Presenting Cells Critical for T Cell Immunity. Cancer Cell 26, 638–652. 10.1016/j.ccell.2014.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buechler MB, Fu W, and Turley SJ (2021). Fibroblast-macrophage reciprocal interactions in health, fibrosis, and cancer. Immunity 54, 903–915. 10.1016/j.immuni.2021.04.021. [DOI] [PubMed] [Google Scholar]
- Cai E, Marchuk K, Beemiller P, Beppler C, Rubashkin MG, Weaver VM, Gérard A, Liu T-L, Chen B-C, Betzig E, et al. (2017). Visualizing dynamic microvillar search and stabilization during ligand detection by T cells. Science 356, eaal3118. 10.1126/science.aal3118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J, Spielmann M, Qiu X, Huang X, Ibrahim DM, Hill AJ, Zhang F, Mundlos S, Christiansen L, Steemers FJ, et al. (2019). The single-cell transcriptional landscape of mammalian organogenesis. Nature 566, 496–502. 10.1038/s41586-019-0969-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Zhou Y, Chen Y, and Gu J (2018). fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890. 10.1093/bioinformatics/bty560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Zander RA, Wu X, Schauder DM, Kasmani MY, Shen J, Zheng S, Burns R, Taparowsky EJ, and Cui W (2021). BATF regulates progenitor to cytolytic effector CD8+ T cell transition during chronic viral infection. Nat. Immunol 22, 996–1007. 10.1038/s41590-021-00965-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng S, Li Z, Gao R, Xing B, Gao Y, Yang Y, Qin S, Zhang L, Ouyang H, Du P, et al. (2021). A pan-cancer single-cell transcriptional atlas of tumor infiltrating myeloid cells. Cell 184, 792–809. 10.1016/j.cell.2021.01.010. [DOI] [PubMed] [Google Scholar]
- Combes AJ, Samad B, Tsui J, Chew NW, Yan P, Reeder GC, Kushnoor D, Shen A, Davidson B, Barczak AJ, et al. (2022). Discovering dominant tumor immune archetypes in a pan-cancer census. Cell 185, 184–203.e19. 10.1016/j.cell.2021.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corces MR, Trevino AE, Hamilton EG, Greenside PG, Sinnott-Armstrong NA, Vesuna S, Satpathy AT, Rubin AJ, Montine KS, Wu B, et al. (2017). An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 14, 959–962. 10.1038/nmeth.4396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corces MR, Granja JM, Shams S, Louie BH, Seoane JA, Zhou W, Silva TC, Groeneveld C, Wong CK, Cho SW, et al. (2018). The chromatin accessibility landscape of primary human cancers. Science 362, eaav1898. 10.1126/science.aav1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Palma M, and Lewis CE (2013). Macrophage regulation of tumor responses to anticancer therapies. Cancer Cell 23, 277–286. 10.1016/j.ccr.2013.02.013. [DOI] [PubMed] [Google Scholar]
- DeNardo DG, and Ruffell B (2019). Macrophages as regulators of tumour immunity and immunotherapy. Nat. Rev. Immunol 19, 369–382. 10.1038/s41577-019-0127-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeNardo DG, Brennan DJ, Rexhepaj E, Ruffell B, Shiao SL, Madden SF, Gallagher WM, Wadhwani N, Keil SD, Junaid SA, et al. (2011). Leukocyte Complexity Predicts Breast Cancer Survival and Functionally Regulates Response to Chemotherapy. Cancer Discov. 1, 54–67. 10.1158/2159-8274.CD-10-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doering TA, Crawford A, Angelosanto JM, Paley MA, Ziegler CG, and Wherry EJ (2012). Network analysis reveals centrally connected genes and pathways involved in CD8+ T cell exhaustion versus memory. Immunity 37, 1130–1144. 10.1016/j.immuni.2012.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelhardt JJ, Boldajipour B, Beemiller P, Pandurangi P, Sorensen C, Werb Z, Egeblad M, and Krummel MF (2012). Marginating dendritic cells of the tumor microenvironment cross-present tumor antigens and stably engage tumor-specific T cells. Cancer Cell 21, 402–417. 10.1016/j.ccr.2012.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankish A, Diekhans M, Ferreira A-M, Johnson R, Jungreis I, Loveland J, Mudge JM, Sisu C, Wright J, Armstrong J, et al. (2019). GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 47, D766–D773. 10.1093/nar/gky955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pagès C, Tosolini M, Camus M, Berger A, Wind P, et al. (2006). Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 313, 1960–1964. 10.1126/science.1129139. [DOI] [PubMed] [Google Scholar]
- Gentles AJ, Newman AM, Liu CL, Bratman SV, Feng W, Kim D, Nair VS, Xu Y, Khuong A, Hoang CD, et al. (2015). The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat. Med 21, 938–945. 10.1038/nm.3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerriero JL (2019). Macrophages: Their Untold Story in T Cell Activation and Function. Int. Rev. Cell Mol. Biol 342, 73–93. 10.1016/bs.ircmb.2018.07.001. [DOI] [PubMed] [Google Scholar]
- Hafemeister C, and Satija R (2019). Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 20, 296. 10.1186/s13059-019-1874-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong F, Meng Q, Zhang W, Zheng R, Li X, Cheng T, Hu D, and Gao X (2021). Single-Cell Analysis of the Pan-Cancer Immune Microenvironment and scTIME Portal. Cancer Immunol. Res 9, 939–951. 10.1158/2326-6066.CIR-20-1026. [DOI] [PubMed] [Google Scholar]
- Hu KH, Eichorst JP, McGinnis CS, Patterson DM, Chow ED, Kersten K, Jameson SC, Gartner ZJ, Rao AA, and Krummel MF (2020). ZipSeq: barcoding for real-time mapping of single cell transcriptomes. Nat. Methods 17, 833–843. 10.1038/s41592-020-0880-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Im SJ, Hashimoto M, Gerner MY, Lee J, Kissick HT, Burger MC, Shan Q, Hale JS, Lee J, Nasti TH, et al. (2016). Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 537, 417–421. 10.1038/nature19330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen CS, Prokhnevska N, Master VA, Sanda MG, Carlisle JW, Bilen MA, Cardenas M, Wilkinson S, Lake R, Sowalsky AG, et al. (2019). An intra-tumoral niche maintains and differentiates stem-like CD8 T cells. Nature 576, 465–470. 10.1038/s41586-019-1836-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin S, Guerrero-Juarez CF, Zhang L, Chang I, Ramos R, Kuan C-H, Myung P, Plikus MV, and Nie Q (2021). Inference and analysis of cell-cell communication using CellChat. Nat. Commun 12, 1088. 10.1038/s41467-021-21246-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katzenelenbogen Y, Sheban F, Yalin A, Yofe I, Svetlichnyy D, Jaitin DA, Bornstein C, Moshe A, Keren-Shaul H, Cohen M, et al. (2020). Coupled scRNA-Seq and Intracellular Protein Activity Reveal an Immunosuppressive Role of TREM2 in Cancer. Cell 182, 872–885. 10.1016/j.cell.2020.06.032. [DOI] [PubMed] [Google Scholar]
- Khan O, Giles JR, McDonald S, Manne S, Ngiow SF, Patel KP, Werner MT, Huang AC, Alexander KA, Wu JE, et al. (2019). TOX transcriptionally and epigenetically programs CD8 + T cell exhaustion. Nature 571, 211. 10.1038/s41586-019-1325-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klug F, Prakash H, Huber PE, Seibel T, Bender N, Halama N, Pfirschke C, Voss RH, Timke C, Umansky L, et al. (2013). Low-dose irradiation programs macrophage differentiation to an iNOS+/M1 phenotype that orchestrates effective T cell immunotherapy. Cancer Cell 24, 589–602. 10.1016/j.ccr.2013.09.014. [DOI] [PubMed] [Google Scholar]
- Law CW, Chen Y, Shi W, and Smyth GK (2014). voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 15, R29. 10.1186/gb-2014-15-2-r29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man K, Gabriel SS, Liao Y, Gloury R, Preston S, Henstridge DC, Pellegrini M, Zehn D, Berberich-Siebelt F, Febbraio MA, et al. (2017). Transcription Factor IRF4 Promotes CD8+ T Cell Exhaustion and Limits the Development of Memory-like T Cells during Chronic Infection. Immunity 47, 1129–1141. 10.1016/j.immuni.2017.11.021. [DOI] [PubMed] [Google Scholar]
- Miller BC, Sen DR, Al Abosy R, Bi K, Virkud YV, LaFleur MW, Yates KB, Lako A, Felt K, Naik GS, et al. (2019). Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint blockade. Nat. Immunol 20, 326–336. 10.1038/s41590-019-0312-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molgora M, Esaulova E, Vermi W, Hou J, Chen Y, Luo J, Brioschi S, Bugatti M, Omodei AS, Ricci B, et al. (2020). TREM2 Modulation Remodels the Tumor Myeloid Landscape Enhancing Anti-PD-1 Immunotherapy. Cell 182, 886–900. 10.1016/j.cell.2020.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mujal AM, Combes AJ, Rao AA, Binnewies M, Samad B, Tsui J, Boissonnas A, Pollack JL, Argüello RJ, Meng MV, et al. (2022). Holistic Characterization of Tumor Monocyte-to-Macrophage Differentiation Integrates Distinct Immune Phenotypes in Kidney Cancer. Cancer Immunol. Res canimm.0588.2021. 10.1158/2326-6066.CIR-21-0588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connell P, Hyslop S, Blake MK, Godbehere S, Amalfitano A, and Aldhamen YA (2021). SLAMF7 Signaling Reprograms T Cells toward Exhaustion in the Tumor Microenvironment. J. Immunol. Baltim. Md 1950 206, 193–205. 10.4049/jimmunol.2000300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira G, Stromhaug K, Klaeger S, Kula T, Frederick DT, Le PM, Forman J, Huang T, Li S, Zhang W, et al. (2021). Phenotype, specificity and avidity of antitumour CD8+ T cells in melanoma. Nature 596, 119–125. 10.1038/s41586-021-03704-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauken KE, Sammons MA, Odorizzi PM, Manne S, Godec J, Khan O, Drake AM, Chen Z, Sen DR, Kurachi M, et al. (2016). Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science 354, 1160–1165. 10.1126/science.aaf2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peranzoni E, Lemoine J, Vimeux L, Feuillet V, Barrin S, Kantari-Mimoun C, Bercovici N, Guérin M, Biton J, Ouakrim H, et al. (2018). Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti–PD-1 treatment. Proc. Natl. Acad. Sci 115, E4041–E4050. 10.1073/pnas.1720948115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philip M, and Schietinger A (2021). CD8+ T cell differentiation and dysfunction in cancer. Nat. Rev. Immunol 10.1038/s41577-021-00574-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philip M, Fairchild L, Sun L, Horste EL, Camara S, Shakiba M, Scott AC, Viale A, Lauer P, Merghoub T, et al. (2017). Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature 545, 452–456. 10.1038/nature22367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritykin Y, van der Veeken J, Pine AR, Zhong Y, Sahin M, Mazutis L, Pe’er D, Rudensky AY, and Leslie CS (2021). A unified atlas of CD8 T cell dysfunctional states in cancer and infection. Mol. Cell 81, 2477–2493. 10.1016/j.molcel.2021.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts EW, Broz ML, Binnewies M, Headley MB, Nelson AE, Wolf DM, Kaisho T, Bogunovic D, Bhardwaj N, and Krummel MF (2016). Critical Role for CD103+/CD141+ Dendritic Cells Bearing CCR7 for Tumor Antigen Trafficking and Priming of T Cell Immunity in Melanoma. Cancer Cell 30, 324–336. 10.1016/j.ccell.2016.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruffell B, Au A, Rugo HS, Esserman LJ, Hwang ES, and Coussens LM (2012). Leukocyte composition of human breast cancer. Proc. Natl. Acad. Sci. U. S. A 109, 2796–2801. 10.1073/pnas.1104303108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sade-Feldman M, Yizhak K, Bjorgaard SL, Ray JP, de Boer CG, Jenkins RW, Lieb DJ, Chen JH, Frederick DT, Barzily-Rokni M, et al. (2018). Defining T Cell States Associated with Response to Checkpoint Immunotherapy in Melanoma. Cell 175, 998–1013. 10.1016/j.cell.2018.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmon H, Idoyaga J, Rahman A, Leboeuf M, Remark R, Jordan S, Casanova-Acebes M, Khudoynazarova M, Agudo J, Tung N, et al. (2016). Expansion and Activation of CD103+ Dendritic Cell Progenitors at the Tumor Site Enhances Tumor Responses to Therapeutic PD-L1 and BRAF Inhibition. Immunity 44, 924–938. 10.1016/j.immuni.2016.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satija R, Farrell JA, Gennert D, Schier AF, and Regev A (2015). Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol 33, 495–502. 10.1038/nbt.3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satpathy AT, Granja JM, Yost KE, Qi Y, Meschi F, McDermott GP, Olsen BN, Mumbach MR, Pierce SE, Corces MR, et al. (2019). Massively parallel single-cell chromatin landscapes of human immune cell development and intratumoral T cell exhaustion. Nat. Biotechnol 37, 925–936. 10.1038/s41587-019-0206-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharping NE, Rivadeneira DB, Menk AV, Vignali PDA, Ford BR, Rittenhouse NL, Peralta R, Wang Y, Wang Y, DePeaux K, et al. (2021). Mitochondrial stress induced by continuous stimulation under hypoxia rapidly drives T cell exhaustion. Nat. Immunol 22, 205–215. 10.1038/s41590-020-00834-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schietinger A, Philip M, Krisnawan VE, Chiu EY, Delrow JJ, Basom RS, Lauer P, Brockstedt DG, Knoblaugh SE, Hämmerling GJ, et al. (2016). Tumor-Specific T Cell Dysfunction Is a Dynamic Antigen-Driven Differentiation Program Initiated Early during Tumorigenesis. Immunity 45, 389–401. 10.1016/j.immuni.2016.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott AC, Dündar F, Zumbo P, Chandran SS, Klebanoff CA, Shakiba M, Trivedi P, Menocal L, Appleby H, Camara S, et al. (2019). TOX is a critical regulator of tumour-specific T cell differentiation. Nature 571, 270–274. 10.1038/s41586-019-1324-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo H, González-Avalos E, Zhang W, Ramchandani P, Yang C, Lio C-WJ, Rao A, and Hogan PG (2021). BATF and IRF4 cooperate to counter exhaustion in tumor-infiltrating CAR T cells. Nat. Immunol 22, 983–995. 10.1038/s41590-021-00964-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiqui I, Schaeuble K, Chennupati V, Fuertes Marraco SA, Calderon-Copete S, Pais Ferreira D, Carmona SJ, Scarpellino L, Gfeller D, Pradervand S, et al. (2019). Intratumoral Tcf1+PD-1+CD8+ T Cells with Stem-like Properties Promote Tumor Control in Response to Vaccination and Checkpoint Blockade Immunotherapy. Immunity 50, 195–211. 10.1016/j.immuni.2018.12.021. [DOI] [PubMed] [Google Scholar]
- Smyth GK (2005). limma: Linear Models for Microarray Data. In Bioinformatics and Computational Biology Solutions Using R and Bioconductor, Gentleman R, Carey VJ, Huber W, Irizarry RA, and Dudoit S, eds. (New York, NY: Springer New York; ), pp. 397–420. [Google Scholar]
- Spranger S, Dai D, Horton B, and Gajewski TF (2017). Tumor-Residing Batf3 Dendritic Cells Are Required for Effector T Cell Trafficking and Adoptive T Cell Therapy. Cancer Cell 31, 711–723. 10.1016/j.ccell.2017.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thommen DS, Koelzer VH, Herzig P, Roller A, Trefny M, Dimeloe S, Kiialainen A, Hanhart J, Schill C, Hess C, et al. (2018). A transcriptionally and functionally distinct PD-1+ CD8+ T cell pool with predictive potential in non-small-cell lung cancer treated with PD-1 blockade. Nat. Med 24, 994–1004. 10.1038/s41591-018-0057-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJM, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, et al. (2014). PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515, 568–571. 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utzschneider DT, Alfei F, Roelli P, Barras D, Chennupati V, Darbre S, Delorenzi M, Pinschewer DD, and Zehn D (2016). High antigen levels induce an exhausted phenotype in a chronic infection without impairing T cell expansion and survival. J. Exp. Med 213, 1819–1834. 10.1084/jem.20150598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utzschneider DT, Gabriel SS, Chisanga D, Gloury R, Gubser PM, Vasanthakumar A, Shi W, and Kallies A (2020). Early precursor T cells establish and propagate T cell exhaustion in chronic infection. Nat. Immunol 21, 1256–1266. 10.1038/s41590-020-0760-z. [DOI] [PubMed] [Google Scholar]
- Vardhana SA, Hwee MA, Berisa M, Wells DK, Yost KE, King B, Smith M, Herrera PS, Chang HY, Satpathy AT, et al. (2020). Impaired mitochondrial oxidative phosphorylation limits the self-renewal of T cells exposed to persistent antigen. Nat. Immunol 21, 1022–1033. 10.1038/s41590-020-0725-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner J, Rapsomaniki MA, Chevrier S, Anzeneder T, Langwieder C, Dykgers A, Rees M, Ramaswamy A, Muenst S, Soysal SD, et al. (2019). A Single-Cell Atlas of the Tumor and Immune Ecosystem of Human Breast Cancer. Cell 177, 1330–1345. 10.1016/j.cell.2019.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber EW, Parker KR, Sotillo E, Lynn RC, Anbunathan H, Lattin J, Good Z, Belk JA, Daniel B, Klysz D, et al. (2021). Transient “rest” restores functionality in exhausted CAR-T cells via epigenetic remodeling. Science 372, eaba1786. 10.1126/science.aba1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wherry EJ, Ha S-J, Kaech SM, Haining WN, Sarkar S, Kalia V, Subramaniam S, Blattman JN, Barber DL, and Ahmed R (2007). Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity 27, 670–684. 10.1016/j.immuni.2007.09.006. [DOI] [PubMed] [Google Scholar]
- Yao C, Sun H-W, Lacey NE, Ji Y, Moseman EA, Shih H-Y, Heuston EF, Kirby M, Anderson S, Cheng J, et al. (2019). Single-cell RNA-seq reveals TOX as a key regulator of CD8+ T cell persistence in chronic infection. Nat. Immunol 20, 890–901. 10.1038/s41590-019-0403-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Liu L, Gong C, Shi H, Zeng Y, Wang X, Zhao Y, and Wei Y (2012). Prognostic significance of tumor-associated macrophages in solid tumor: a meta-analysis of the literature. PloS One 7, e50946. 10.1371/journal.pone.0050946. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie S1. Synapse formation between TAM and CD8+ T cells (related to Figure 4). Lattice light sheet imaging of the interaction between mTomato+ TAM sorted from B78ChOVA melanomas (magenta) and previously activated CD8+ OT-I T cell labeled with CD45-AF647 (red), with the H57 TCRβ labeled with AF488. Scale bar, 5μm.
Data Availability Statement
Bulk RNA-seq, bulk ATAC-seq and single-cell RNA-seq data have been deposited at GEO (GSE201074) and are publicly available as of the date of publication. RNA-seq data from human tumors has been published previously (Combes et al., 2022) and is publicly available at GEO (GSE184398). Accession numbers are also listed in the Key Resources Table. All original code has been deposited to GitHub and is publicly available as of the date of publication. DOIs are listed in the Key Resources Table. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-mouse CD103 - BUV737 (clone 2E7) | BD BioSciences | 749393 |
| anti-mouse CD103 - PE (clone 2E7) | Biolegend | 121406 |
| anti-mouse CD103 - PerCp-Cy5.5 (clone 2E7) | Biolegend | 121416 |
| anti-mouse CD11b - AF594 (clone M1/70) | Biolegend | 101254 |
| anti-mouse CD11b - BV421 (clone M1/70) | Biolegend | 101235 |
| anti-mouse CD11b - BV605 (clone M1/70) | Biolegend | 101237 |
| anti-mouse CD11b - BV650 (clone M1/70) | Biolegend | 101239 |
| anti-mouse CD11b - BV785 (clone M1/70) | Biolegend | 101243 |
| anti-mouse CD11c - BV650 (clone N418) | Biolegend | 117339 |
| anti-mouse CD11c - PerCp-Cy5.5 (clone N418) | Biolegend | 117328 |
| anti-mouse CD206 - AF488 (clone C068C2) | Biolegend | 141710 |
| anti-mouse CD206 - PerCp-Cy5.5 (clone C068C2) | Biolegend | 141716 |
| anti-mouse CD24 - BV421 (clone M1/69) | Biolegend | 101825 |
| anti-mouse CD24 - PECy7 (clone M1/69) | Biolegend | 101822 |
| anti-mouse CD31 - AF647 (clone 390) | Biolegend | 102415 |
| anti-mouse CD38 - BV711 (clone 90/CD38) | BD BioSciences | 740697 |
| anti-mouse CD38 - FITC (clone 90/CD38) | BD BioSciences | 558813 |
| anti-mouse CD4 - BUV395 (clone RM4–5) | BD BioSciences | 563790 |
| anti-mouse CD44 - BUV737 (clone IM7) | BD BioSciences | 564392 |
| anti-mouse CD44 - AF700 (clone IM7) | eBioScience | 56-0441-80 |
| anti-mouse CD45 AF647 (clone 30-F11) | Biolegend | 103124 |
| anti-mouse CD45 - BUV395 (clone 30-F11) | BD BioSciences | 564279 |
| anti-mouse CD45 - BV421 (clone 30-F11) | Biolegend | 304032 |
| anti-mouse CD45.1 - BUV395 (clone A20) | BD BioSciences | 565212 |
| anti-mouse CD45.1 - PECy7 (clone A20) | Biolegend | 110730 |
| anti-mouse CD45R (B220) - BV785 (clone RA3–6B2) | Biolegend | 103245 |
| anti-mouse CD5 - BV510 (clone 53–7.3) | Biolegend | 100627 |
| anti-mouse CD69 - BV650 (clone H1.2F3) | Biolegend | 104541 |
| anti-mouse CD8 - AF488 (clone 53–6.7) | Biolegend | 100723 |
| anti-mouse CD8 - PerCp-Cy5.5 (clone 53–6.7) | Biolegend | 100734 |
| anti-mouse CD80 - FITC (clone 16–10A1) | eBioScience | 11-0801-82 |
| anti-mouse CD86 - APC (clone GL-1) | Biolegend | 105012 |
| anti-mouse CD86 - BV421 (clone GL-1) | Biolegend | 105031 |
| anti-mouse CD90.2 - AF700 (clone 30-H12) | Biolegend | 105320 |
| anti-mouse CD90.2 - BV785 (clone 30-H12) | Biolegend | 105331 |
| anti-mouse CSF1R (CD115) - PECy7 (clone AFS98) | Biolegend | 135524 |
| anti-mouse F4/80 - AF700 (clone BM8) | Biolegend | 123130 |
| anti-mouse F4/80 - BV510 (clone BM8) | Biolegend | 123135 |
| anti-mouse H2kB - PE (clone AF6–88.5) | Biolegend | 116507 |
| anti-mouse IFNy - PE (clone XMG1.2) | eBioscience | 12-7311-81 |
| anti-mouse IFNy - PECy7 (clone XMG1.2) | Biolegend | 505826 |
| anti-mouse IRF4 - FITC (clone 3E4) | eBioscience | 11-9858-82 |
| anti-mouse IRF4 - PECy7 (clone 3E4) | eBioscience | 25-9858-82 |
| anti-mouse Ki67 - PE eFluor610 (clone SolA15) | eBioscience | 61-5698-82 |
| anti-mouse Ly6C - BV711 (clone HK1.4) | Biolegend | 128037 |
| anti-mouse Ly6G - BV785 (clone 1A8) | Biolegend | 127645 |
| anti-mouse I-A/I-E - AF700 (clone M5/114.15.2) | Biolegend | 107622 |
| anti-mouse NK1.1 - BV785 (clone PK136) | Biolegend | 108749 |
| anti-mouse PD-1 (CD279) - BV605 (clone 29F.1A12) | Biolegend | 135219 |
| anti-mouse SiglecF - BV785 (clone E50–2440) | BD BioSciences | 740956 |
| anti-mouse TCF1 - PE (clone S33–966) | BD BioSciences | 564217 |
| anti-mouse TCRb - AF488 (clone H57–597) | Biolegend | 109215 |
| anti-mouse TCRb V8.1 - PE (clone MR5–2) | BD BioSciences | 553186 |
| anti-mouse TNFa - BV421 (clone MP6-XT22) | Biolegend | 506327 |
| anti-mouse TNFa - PE (clone MP6-XT22) | Biolegend | 506306 |
| anti-mouse TOX - APC (clone REA473) | Miltenyi Biotec | 130-118-335 |
| anti-mouse CD16/32 (clone 2.4G2) | BioXCell | BE0307 |
| anti-mouse CD4 InVivoMab (clone GK1.5) | BioXCell | BE0003–1 |
| anti-mouse CD8 InVivoMab (clone 2.43) | BioXCell | BE0061 |
| anti-mouse CSF1 InVivoMab (clone 5A1) | BioXCell | BE0204 |
| anti-mouse CSF1R InVivoMab (clone AFS98) | BioXCell | BE0213 |
| Rat IgG1, k (clone HRPN) | BioXCell | BE0088 |
| Rat IgG2b, k (clone LTF-2) | BioXCell | BE0090 |
| Rat IgG2a, k (clone 2A3) | BioXCell | BE0089 |
| Hypoxyprobe Pacific Blue kit (4.3.11.3 MAb1) | Hypoxyprobe, Inc. | hp15-100kit |
| Normal Rat Serum | Thermo Fisher | 10710C |
| Bacterial and virus strains | ||
| N/A | ||
| Biological samples | ||
| Human tumor samples | UC San Francisco | IRB# 20-31740 |
| Mouse tissue samples (LN, tumor) | UC San Francisco | IACUC: AN184232 |
| Chemicals, peptides, and recombinant proteins | ||
| Matrigel GFR | Corning | 356231 |
| Collagenase, Type I | Worthington Biochemical | LS004197 |
| Collagenase, Type IV | Worthington Biochemical | LS004189 |
| Dnase I | Millipore Sigma | 10104159001 |
| Ficoll-Paque Plus | GE Healthcare | 17-1440-02 |
| Ficoll-Paque Premum 1.084 | GE Healthcare | 17-5446-02 |
| Zombie NIR Fixable Viability Dye | Biolegend | 423106 |
| CFSE | Invitrogen | C34554 |
| Violet Proliferation Dye | BD Biosciences | 562158 |
| CMTMR | Thermo Fisher | C2927 |
| Brefeldin A (BFA) | Sigma-Aldrich | B7651 |
| Phorbol 12-myristate 12-acetate (PMA) | Sigma-Aldrich | P8139 |
| Ionomycin | Invitrogen | I24222 |
| Complete Freund’s Adjuvant | Sigma-Aldrich | F5881 |
| OVA peptide (257–264) SIINFEKL | Anaspec | AS-60193 |
| LCMV gp33 peptide (33–41) KAVYNFATC | Anaspec | AS-61669 |
| Recombinant murine IL-4 | Peprotech | 214-14 |
| Recombinant murine GM-CSF | Peprotech | 315-03 |
| Recombinant human IL-2 | Peprotech | 200-02 |
| Fibronectin, bovine plasma | EMD Millipore | 341631 |
| FURA-2AM | Thermo Fisher | F1221 |
| Critical commercial assays | ||
| Chromium Single Cell 3’ GEM Library & Gel Bead Kit V3 | 10x Genomics | PN-1000092 |
| Foxp3/ Transcription Factor Staining Buffer Kit | BD Biosciences | 554655 |
| EasySep Mouse CD8+ T cell Isolation Kit | STEMCELL Technologies | 19853 |
| EasySep Mouse Biotin Positive Selection Kit | STEMCELL Technologies | 17665 |
| EasySep Mouse Monocyte Isolation Kit | STEMCELL Technologies | 19861 |
| UltraComp eBeads Compensation Beads | Fisher Scientific | 01-2222-42 |
| Corning HTS Transwell 96 well permeable supports | Corning | CLS3388 |
| RNeasy Micro kit | Qiagen | 74004 |
| High-Capacity cDNA Reverse Transcription kit | Thermo Fisher | 4368814 |
| Mouse M-CSF Quantikine ELISA kit | R&D Systems | MMC00 |
| CBA Mouse MIP-1a Flex Set | BD BioSciences | 558449 |
| CBA Mouse MIP-1b Flex Set | BD BioSciences | 558343 |
| CBA Mouse MIP-1a Flex Set | BD BioSciences | 558345 |
| Deposited data | ||
| All bulk RNAseq data for IPI cohort | Combes et al. 2022 | GEO:GSE184398 |
| Bulk RNAseq and ATACseq on T cells | This paper | GEO:GSE201074 |
| Single cell RNAseq data from CD206R ZipSeq | This paper | GEO:GSE201074 |
| GitHub | Kenneth Hu Github | https://github.com/ken7hu/ZipSeq-Analysis-CD206R |
| Experimental models: Cell lines | ||
| B16F10 | AATCC | CRL-6475 |
| B16ChOVA | UC San Francisco | N/A |
| B78ChOVA | UC San Francisco | N/A |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6J | The Jackson Laboratory | Stock # 000664 |
| Mouse: B6 CD45.1 (B6.SJL-Ptprc Pepc/BoyJ) | The Jackson Laboratory | Stock # 002014 |
| Mouse: OT-I (C57BL/6-Tg(TcraTcrb)1100Mjb/J | The Jackson Laboratory | Stock # 003831 |
| Mouse: mTmG | The Jackson Laboratory | Stock # 007676 |
| Mouse: Rag1 KO (B6.129S7-Rag1tm1Mom/J) | The Jackson Laboratory | Stock # 002216 |
| Mouse: CSF1op/op (B6;C3Fe a/a-Csf1op/J) | The Jackson Laboratory | Stock # 000231 |
| Mouse: Csf1rCre (C57BL/6-Tg(Csf1r-cre)1Mnz/J) | The Jackson Laboratory | Stock # 029206 |
| Mouse: LCMV P14 (crossed to B6 background) | Michael Waterfield, UC San Francisco | MGI: 2665105 |
| Mouse: MMTV-PyMTChOVA | Matthew Krummel, UC San Francisco | MGI: 5436574 |
| Mouse: CD206-LSL-Venus-DTR | Matthew Krummel, UC San Francisco | N/A |
| Oligonucleotides | ||
| Taqman probe: Ccl3 | Life Technologies | Mm00441259_g1 |
| Taqman probe: Ccl5 | Life Technologies | Mm01302427_m1 |
| Taqman probe: Csf1 | Life Technologies | Mm00432686_m1 |
| Taqman probe: Gapdh | Life Technologies | Mm99999915_g1 |
| Recombinant DNA | ||
| N/A | ||
| Software and algorithms | ||
| Imaris | Bitplane | https://imaris.exinst.com/ |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| FlowJo | Becton Dickinson | https://flowjo.com/ |
| CellRanger 4.0.0 | 10X Genomics | 10xgenomics.com |
| Seurat | Satija et al. 2015 | https://satijalab.org/seurat |
| STAR | Dobin et al. 2013 | https://code.google.com/archive/p/rna-star/ |
| R: The Project for Statistical Computing | N/A | http://r-project.org |
| CellChat | Jin et al. 2021 | http://www.cellchat.org/ |
| Other | ||
| N/A | ||