

SUMMARY
To combat infectious diseases, it is important to understand how host cells interact with bacterial pathogens. Signals conveyed from pathogen to host, and vice versa, may be either chemical or mechanical. While the molecular and biochemical basis of host-pathogen interactions has been extensively explored, relatively less is known about mechanical signals and responses in the context of those interactions. Nevertheless, a wide variety of bacterial pathogens appear to have developed mechanisms to alter the cellular biomechanics of their hosts in order to promote their survival and dissemination, and in turn many host responses to infection rely on mechanical alterations in host cells and tissues to limit the spread of infection. In this review, we present recent findings on how mechanical forces generated by host cells can promote or obstruct the dissemination of intracellular bacterial pathogens. In addition, we discuss how in vivo extracellular mechanical signals influence interactions between host cells and intracellular bacterial pathogens. Examples of such signals include shear stresses caused by fluid flow over the surface of cells and variable stiffness of the extracellular matrix on which cells are anchored. We highlight bioengineering-inspired tools and techniques that can be used to measure host cell mechanics during infection. These allow for the interrogation of how mechanical signals can modulate infection alongside biochemical signals. We hope that this review will inspire the microbiology community to embrace those tools in future studies so that host cell biomechanics can be more readily explored in the context of infection studies.
KEYWORDS: cellular biomechanics, cytoskeletal mechanics, extracellular matrix mechanics, intracellular bacterial pathogens
INTRODUCTION
Cells in our body are constantly sensing and responding to a variety of biochemical and mechanical cues from their environment (1). Examples of the latter include variations in shear fluid flow (2) and in the stiffness of the extracellular matrix (ECM) on which cells are anchored (3, 4). Cells are also continuously generating and transducing forces to each other and to their extracellular surroundings. Collectively, this active mechanical behavior allows for the maintenance of barrier function and tissue integrity (5, 6). Appreciation of biomechanics as being central to cell function and human health has grown significantly over the past few decades and has lent key insights into the fields of cancer pathogenesis, stem cell biology, and developmental biology (7, 8). However, the importance of mechanics in governing how host cells interact with bacterial pathogens is only beginning to be uncovered (9–12). Recent studies reveal that bacterial pathogens have evolved sophisticated ways of hijacking host cell forces to propagate through their host. At the same time, host cells are able to respond to infection by altering their mechanics and physical characteristics to limit systemic spread. Interestingly, such paradigms are not limited to human cells and tissues but are also observed in other multicellular organisms (13) and unicellular amoebae (14), arguing that many of these biomechanical processes might be generalizable or even universal in nature.
In light of these discoveries, there has been a shift toward developing assays and organotypic models that enable the study of infection in vitro with host cells and pathogens subject to physiologically relevant mechanical cues (12, 15). The precision built into these devices allows for the modulation of one parameter at a time, which cannot be achieved in vivo (16, 17). Moreover, it is now possible to conduct biomechanical measurements in real time and in multiple parallel assays to witness the cross talk between host cells and their surroundings. Misregulation of this biomechanical cross talk during bacterial infections can reveal mechanisms by which pathogens and host cells manipulate physical forces to modulate the infection process.
Several quantitative biomechanical measurement methods that were originally developed to address other questions can be directly adapted for the study of host-pathogen interactions. For example, traction force microscopy (TFM) enables the direct measurement of forces that adherent cells exert on their ECM through the engagement of their focal (cell-ECM) adhesions and cytoskeletal actomyosin-driven contractility (18, 19) (Fig. 1A). The elastic matrices to which cells adhere in vitro are microfabricated using two-dimensional (2D) polymer gels whose elasticity can be tuned to match native cellular (patho)physiological conditions. Alternatively, cells can be seeded on micropillar arrays and cellular forces can be calculated that cause these arrays to deflect when cells engage their focal adhesions (Fig. 1B). Monolayer stress microscopy (MSM) facilitates indirect calculation of the forces that cells in confluent monolayers exert on one another, shedding light on the regulation of intercellular communication and barrier integrity (Fig. 1C) (20). Förster resonance energy transfer (FRET)-based force sensors constitute an alternative technique for measuring cell-matrix and intercellular forces with increased spatial resolution at the single-molecule level (Fig. 1D) (21). At the multicellular level, measuring the recoil that cells experience upon laser wounding can reveal the intercellular tension built within a monolayer of cells (Fig. 1E) (22). Atomic force microscopy (AFM) is often used to measure changes in cell stiffness in living cells, which can provide key insights into the organization of load-bearing structures of the cell cytoskeleton (Fig. 1F) (23, 24). Live-cell monolayer rheometry allows for the measurement of the strength of adhesion between host cells and attached bacteria (Fig. 1G) (25). These are just a few examples of existing biomechanical techniques that could be used to study the mechanics of host-pathogen interactions. Readers are directed to recent extensive reviews on additional ways of measuring cell-generated mechanical forces and evaluating the physical characteristics of cells and tissues (26–28).
FIG 1.

Biomechanical characterization of host-pathogen interactions. (A) Traction force microscopy (TFM) to measure cellular traction stresses on 2D elastic planar matrices; (B) micropillar array for measuring cellular forces; (C) monolayer stress microscopy for inferring intracellular tensions; (D) FRET-based force sensors; (E) laser ablation wounding for monolayer tension estimation; (F) atomic force microscopy (AFM) to measure cellular stiffness; (G) live-cell monolayer rheometer to measure adhesion strength between bacteria and the apical surface of cells.
In this review, we present emerging paradigms that demonstrate the importance of mechanics in governing how intracellular bacterial pathogens interact with human host cells. We first discuss strategies that bacteria employ to hijack host cellular forces to facilitate their own spread. In the second section, we address complementary paradigms, where host cells obstruct bacterial dissemination by actively changing their biomechanics in response to infection. Finally, in the third section of this review, we focus on how extracellular mechanical signals guide the interaction of host cells with intracellular bacterial pathogens. We have decided to focus our attention on intracellular bacterial pathogens (obligate and facultative) for two reasons. First, due to their intracellular nature, they have developed sophisticated strategies for manipulating their hosts, including modulating the mechanics of different host cell types to spread systemically. Second, since they survive within a host cell, they are invested in keeping it alive and hence can serve as valuable tools for probing host cell and tissue mechanobiology over long periods of time.
INTRACELLULAR BACTERIA HIJACK HOST CELL MECHANICS
Intracellular bacterial pathogens rely on their host cells to support different stages of their intracellular life cycle, including invasion, immune evasion, and spread. In many cases, the pathogens employ sophisticated strategies of host manipulation to facilitate these processes, and studying them can shed light on fundamental host cell biology as well as on bacterial pathogenesis. Previous reviews have focused on mechanisms by which intracellular bacteria modify host cell signaling pathways and cytoskeletal components to promote their survival and spread (29, 30). Here, we discuss recent studies that illustrate how pathogens subvert host cell mechanics and force transduction to achieve similar ends. With that focus, we highlight instances where bacterial pathogens employ diverse strategies to modulate host cell biomechanics to benefit their own dissemination.
Manipulation of Intercellular Forces Promotes Bacterial Cell-to-Cell Spread
Intracellular bacterial pathogens such as Listeria monocytogenes, Shigella flexneri, and Rickettsia parkeri hijack host cell actin to form “comet tails” that enable the bacterium to propel itself through the host cell cytoplasm. Using actin-based motility (ABM), the bacterium reaches the donor cell membrane, where it forms a protrusion that is engulfed by the recipient cell (29) (Fig. 2). This process of cell-to-cell spread allows for bacterial dissemination through cell monolayers and evasion of the humoral immune response (31). Although protrusion formation and cell-to-cell spread were initially thought to be solely dependent on force generated via ABM (32), recent studies suggest that modulation of mechanics at cell-cell interfaces (i.e., intercellular forces, cortical tension, membrane flexibility) also facilitates spread (9, 10, 33). Interestingly, all three pathogens weaken tension at the donor cell membrane to promote their intercellular spread, although the molecular mechanisms they employ are distinct and are discussed below.
FIG 2.

Intracellular bacterial pathogens hijack host cell mechanics to spread from cell to cell. (A) L. monocytogenes InlC interacts with host cell Tuba to reduce cortical tension and with components of the exocyst pathway to reduce plasma membrane tension at cell-cell interfaces, facilitating bacterial cell-to-cell spread. Inspired by reference 11. (B) S. flexneri IpaC interacts with β-catenin at cell-cell junctions, loosening them to facilitate bacterial intercellular spread. (C) R. parkeri Sca4 competes with α-catenin for vinculin binding to reduce mechanotransduction at the cell-cell junction, thus promoting bacterial intercellular spread. Inspired by reference 9. Note that drawings are not to scale.
L. monocytogenes relieves tension at the donor side of cell contacts.
L. monocytogenes enhances host cell membrane flexibility by relieving tension at the donor side of cell contacts to facilitate its intercellular spread (Fig. 2A). It does so by secreting the virulence factor internalin C (InlC), which competes at cell-cell junctions with the Wiskott-Aldrich syndrome protein (N-WASP) for Tuba binding (11). Tuba is an activator of the small GTPase Cdc42 (34), which, like the GTPases Rho and Rac, regulates the actomyosin cytoskeleton by interacting with actin nucleators (35). One such nucleator is N-WASP, which regulates the nucleation activity of the actin related protein 2/3 complex (Arp2/3), which in turn promotes the assembly of branched actin filament networks (36). In uninfected settings, N-WASP enhances junctional tension by stabilizing actin filaments, which would otherwise undergo stress-induced turnover, and promoting their incorporation into the apical actin rings of the adherens junctions (protein complexes at cell-cell junctions) (37, 38). Tuba assists N-WASP in fulfilling this role by destabilizing its autoinhibited closed state and may also participate in the recruitment of N-WASP to cell junctions (39). Infection with L. monocytogenes or expression of InlC displaces N-WASP from Tuba and renders epithelial cell boundaries curvy and cell-cell junctions slack, facilitating bacterial spread (11).
In contrast, during infection with ΔinlC L. monocytogenes, cell-cell interfaces are more linear and taut. Possibly due to elevated tension at host cell junctions, bacteria produce fewer protrusions and are less effective at spreading from cell to cell (11). Consistent with this hypothesis, the spread defect of this mutant strain is rescued by pharmacologically reducing actomyosin contractility in the host cells. These results suggest that secretion of InlC allows L. monocytogenes to overcome tension faced at sites of protrusion formation and therefore spread more easily to a recipient cell.
L. monocytogenes also downregulates Cdc42 activity to reduce tension at the cell-cell interface (40). Like Tuba, Cdc42 enhances N-WASP function by destabilizing its closed state and recruiting N-WASP to cell-cell junctions (41). Expression of a dominant negative Cdc42 in epithelial cells produces curved junctions and restores levels of protrusion formation in ΔinlC L. monocytogenes infections that are comparable to those in the wild-type (WT) bacterium. This further supports the proposition that intercellular spread of L. monocytogenes is facilitated by relieving tension at the host cell boundary, although biophysical measurements of tension at cell boundaries were not conducted. Although there is a correlative relationship between membrane curvature and membrane or cortical tension, deduction of causal relationships between these physical parameters is more complex (42, 43).
A complex formed by Cdc42, the polarity protein 6 (Par6), and atypical protein kinase 6 (aPKC) has been shown to participate in local adherens junction remodeling by controlling endocytosis that occurs in an Arp2/3-dependent fashion (44). Not only does L. monocytogenes reduce endocytosis of adherens junctions by diminishing Cdc42 activity (40), but it also antagonizes this role of Cdc42 by promoting exocytosis at sites of protrusion formation, which often coincide with adherens junctions (45). InlC recruits an exocyst component, Exo70, to the plasma membrane, which leads to an enrichment of exocytosis at L. monocytogenes-generated membrane protrusions. By delivering additional membrane to protrusions, exocytosis might relieve the tension faced by L. monocytogenes at the plasma membrane (Fig. 2A). Alternatively, this process might transport a tension-modulating protein to the site of protrusion formation (45). The full rescue of the ΔinlC L. monocytogenes defect in protrusion formation upon inhibition of actomyosin contractility increases the likelihood of these possibilities (11), but direct measurement of intercellular tension in control and Exo70 knockdown monolayers could further support this hypothesis. Intercellular tension could be measured indirectly via laser ablation wounding (Fig. 1E) (46) or monolayer stress microscopy (MSM), techniques which can be used to deduce intercellular tension within a monolayer based on measurements of cell-ECM traction forces (Fig. 1C) (20). Direct measurements of intercellular tension can also be performed using FRET-based force sensors, which are preferred for measuring those forces at the cellular rather than supracellular scale (Fig. 1D) (47).
S. flexneri reduces tension at the boundary of the infected donor cell.
S. flexneri also alleviates tension at the donor cell to promote efficient cell-to-cell spread, using molecular mechanisms that are distinct from those employed by L. monocytogenes to achieve similar ends. Once in the host cell cytoplasm, S. flexneri secretes the virulence factor invasion plasmid antigen C (IpaC), which competes with E-cadherin at cell-cell junctions for α-catenin binding (Fig. 2B) (33). α-Catenin is a key mechanosensor that couples the “E-cadherin–α-catenin” complex to the actin cytoskeleton in a force-dependent manner (48, 49). Thus, in uninfected cells, α-catenin transduces tension at adherens junctions (48, 49). IpaC-mediated disruption of the interaction between α-catenin and E-cadherin could reduce cortical tension (the sustained contraction of the cortical cytoskeleton) to promote protrusion formation and subsequent cell-to-cell spread. Indeed, the junctions of host cells infected with ΔipaC S. flexneri are quite taut, while bacteria are less likely to be found in protrusions and show a defect in spread (33). Similar to ΔinlC L. monocytogenes, the protrusion formation defect of ΔipaC S. flexneri is rescued by inhibition of host cell actomyosin contractility, further supporting the idea that tension alleviation at cell-cell interfaces is important for intercellular bacterial spread. It is worth noting that S. flexneri could also alleviate host cell tension to promote its initial uptake into host cells, in addition to reducing intercellular tension at cell-cell interfaces (50, 51). S. flexneri secretes the virulence factor invasion plasmid antigen A (IpaA) upon host cell contact, which interacts with focal adhesion proteins vinculin (50) and talin (51) to cause local depolymerization of F-actin filaments. The resulting depolymerization of F-actin is expected to alleviate host cell tension to facilitate bacterial uptake, although tension measurements have not yet been performed.
R. parkeri hijacks intercellular force transduction to spread.
Similar to S. flexneri, R. parkeri also disrupts the donor cell’s adherens junctions to achieve efficient cell-to-cell spread, again using a distinct molecular mechanism (Fig. 2C). Specifically, R. parkeri secretes surface cell antigen 4 (Sca4), which competes with adherens junction component α-catenin for vinculin binding (9). In uninfected cells, α-catenin recruits vinculin to stabilize and strengthen cell-cell adhesions and allow for intercellular force transduction. By preventing this recruitment, R. parkeri reduces intercellular tension at the donor side of cell contacts as measured indirectly via MSM. This leads to an increase in membrane curvature that allows the recipient cell to engulf the protrusion at a higher rate. Interestingly, while L. monocytogenes, S. flexneri, and R. parkeri all weaken tension at the donor cell membrane to promote spread, L. monocytogenes and S. flexneri do so to enhance protrusion initiation while R. parkeri does so to promote protrusion resolution. Moreover, unlike L. monocytogenes and S. flexneri, R. parkeri appears to be tail-less at protrusion sites, suggesting that the force exerted by ABM at the donor cell membrane is not required for cell-to-cell spread (9). Thus, R. parkeri enables spread by solely manipulating host cell tension through secretion of Sca4, although additional bacterial effectors (yet to be identified) might also contribute to this process. It is worth noting that R. parkeri generates shorter protrusions in A549 lung epithelial cells (up to ∼3 μm) than those created by L. monocytogenes (up to ∼17 μm) and S. flexneri (no precise measurements available for A549 cells but generally comparable to L. monocytogenes protrusions in other host cell types), which implies that R. parkeri might not need to exert as much force on the cell boundary as the other two pathogens (9, 52). It would be interesting to investigate why L. monocytogenes and S. flexneri form longer protrusions than R. parkeri and if the mechanism by which R. parkeri resolves protrusions could explain this difference in protrusion length. In fact, protrusion elongation appears to promote intercellular spread of L. monocytogenes and S. flexneri, as perturbations which decrease protrusion length all impair spread (53–55), including knockdown of diaphanous formins (proteins that nucleate the formation of long, unbranched F-actin filaments [56]), Myosin-X (a molecular motor that acts on actin filaments where actin is bundled, such as at filopodia or tails of intracellular pathogens [57]), cyclophilin A (CypA, a cytosolic protein required for stabilizing N-WASP to allow for actin filament nucleation and initiation of actin polymerization [58]), and inactivation of ERM proteins (namely, ezrin, radixin, and moesin, which play an important role in organizing the cellular membrane since they interact both with transmembrane proteins and the underlying cytoskeleton [59]).
Bacterial actin tails generate force during cell-to-cell spread.
Successful spread requires that L. monocytogenes and S. flexneri form rigid actin tails behind them, by inducing polymerization of host cell actin monomers at the bacterial surface. These actin comet tails are able to generate enough force to overcome the tension they face at the membrane to produce protrusions. In the cytoplasm, these pathogens have tails made of branched actin filaments nucleated by Arp2/3 (60). However, within protrusions, their tails are composed primarily of parallel formin-nucleated actin filaments (61, 62), which do not generate as much propulsive force as filaments that are oriented obliquely with respect to the pathogen (63). Nevertheless, these parallel formin-nucleated actin filaments form a more rigid bundle than branched actin filaments and are thus less likely to buckle in response to the tension faced at the membrane (64). Actin tails within protrusions, but not actin tails in the bulk cytoplasm, are also able to recruit the actin-stabilizing protein calponin-2 and the ERM proteins, which confer stability by linking the actin comet tail to the plasma membrane (49, 65). Inactivation of ERM proteins results in the formation of short, collapsed protrusions (54). It is not yet clear whether these two pathogens use secreted virulence factors to induce these adaptations of actin comet tails in protrusions, or whether these might simply be consequences of assembling dense actin structures adjacent to the plasma membrane that are completely mediated by normal host cell mechanisms. Either way, they do confer additional tail rigidity at the membrane, which R. parkeri, being tail-less, does not require.
Manipulation of Host Cell Mechanics Facilitates Bacterial Transcellular or Paracellular Translocation
In addition to spreading laterally between neighboring cells in an epithelium, many intracellular pathogens travel vertically, following either a transcellular route (through host cells) or a paracellular route (between host cells). Why pathogens might opt for an intercellular, transcellular, or paracellular route of spread and whether they are guided by biochemical and/or mechanical signals are very interesting but still open questions. Many of the facultative intracellular pathogens mentioned in the above sections have been shown to undergo both transcellular and paracellular translocation. For example, foodborne pathogens like L. monocytogenes, which initially occupy the gut lumen, translocate across the intestinal barrier to infect intestinal epithelial cells at their basolateral surface (66, 67) or to access the underlying tissue and spread systemically (68, 69). To do so, they often commandeer host cell mechanics by subverting trafficking (i.e., endocytosis, intracellular vesicle transport, and trans-endocytosis processes) within host cells (70, 71), compromising barrier integrity between neighboring host cells (69), decreasing tension at the basal surface of host cells (10), or altering host cell morphology (68). Along with enabling bacterial spread, these changes can increase barrier permeability, resulting in dysregulation of ion and fluid flow (72) and in increased immune cell transmigration (70).
L. monocytogenes weakens host cell traction stresses to transmigrate from the epithelial layer across the basement membrane.
L. monocytogenes has the ability to cross the placental barrier when it infects pregnant women, which is the origin of one of the most serious sequelae of L. monocytogenes infection, late-stage fetal death. At the feto-maternal barrier, L. monocytogenes modulates host cell mechanics to traverse the basement membrane of placental cells (73). After entry into syncytial trophoblasts in the placenta, the bacterium escapes from the endocytic vacuole and utilizes ABM to eventually cross through to the underlying tissue. Successful translocation in this context depends on the virulence factor internalin P (InlP) (74). InlP binds to the cytoskeletal protein afadin, leading to a decrease in the traction (cell-ECM) stresses that host cells exert onto soft elastic hydrogels as measured by traction force microscopy (TFM), suggesting that they may similarly decrease traction stress on their basement membrane in vivo (10). To conduct TFM, cells are placed on protein-coated hydrogels embedded with fluorescent tracer beads (Fig. 1B). Cells pull on their matrix, displacing the tracer beads in the process, to an extent that depends on how well organized cell-ECM adhesions are and how strongly they are connected to the underlying cytoskeleton. Based on those displacements, one can calculate traction stresses exerted by the cells on their ECM. The findings of this study suggest that InlP acts analogously to InlC but in a different subcellular location, allowing L. monocytogenes to more easily form protrusions at the base of the host cell by reducing tension at the cell-ECM interface. This is expected to facilitate bacterial transfer from placental cytotrophoblasts to the fetal stroma underneath. Though InlP expression was shown to increase bacterial transmigration from the epithelial layer across the underlying basement matrix by reducing the traction stresses exerted by host cells on the matrix, basal protrusions were not directly observed in this study. The fact that mutants lacking the ActA protein (which are unable to form actin tails) were very inefficient in transmigrating hints at a possible role for actin tails produced by bacteria in this process. Visualizing bacteria within protrusions and determining whether they maintain their actin comet tails at this site would help us understand which forces the pathogen usurps to achieve this type of transmigration.
Elevated host cell contractility disrupts cell-cell junctions to aid paracellular translocation of bacteria.
L. monocytogenes can also increase epithelial permeability to translocate across the intestine using a paracellular route (69). Here, the interaction between a bacterial virulence factor, Listeria adhesion protein (LAP), and the host cell surface receptor, Hsp60, activates NF-κB innate immune signaling, which leads to an increase in the myosin II-dependent contractile force generated by the circumferential actomyosin belt (F-actin–myosin II bundles located along the apical cell-cell junctions of an epithelial monolayer). An increase in cellular contractility leads to a decrease in transepithelial electrical resistance, an indication that barrier integrity of the epithelium is compromised (69, 75). Salmonella enterica serovar Typhimurium and S. flexneri employ similar strategies to travel paracellularly between epithelial cells to infect intestinal cells at their base, thereby avoiding apical microvilli (66, 76, 77). In both cases, infection results in enhanced actomyosin contractility of the apical epithelial belt, which alters both intercellular force propagation and protein localization at cell-cell junctions.
Pathogenic tick-borne Rickettsia also disrupts the barrier integrity of endothelial cell monolayers by attenuating the forces between cell-cell junctions (78). In vivo, this can lead to fluid accumulation in unwanted locations such as alveolar spaces and can culminate in serious diseases like vasogenic cerebral edema. Specifically, R. rickettsii disrupts cell-cell junctions of endothelial cells by inducing the mislocalization of α-catenin and promoting the redistribution of p120 catenin, which normally stabilizes VE-cadherin at the plasma membrane in uninfected cells (72). A 2012 study used AFM to measure forces at VE-cadherin-based cell-cell junctions in uninfected and R. rickettsia-infected endothelial cells (78). This was achieved by coating the cantilever tip of the AFM with VE-cadherin. The tip was then brought toward the host cells until contact was established and adhesions formed between the VE-cadherin-functionalized cantilever and the VE-cadherin receptors on the host cell surface. Then, the AFM tip was moved away from the cells, and the force to break the established bonds was measured. This study showed that infected endothelial cells exhibited reduced binding affinity to the VE-cadherin-functionalized cantilever, since the work that was required to break bonds between the two was 80% less in the infected condition than in uninfected cells (78). Therefore, reduction of force transduction due to weakening of the VE-cadherin homotypic interaction is an effective strategy to increase endothelial permeability. Interestingly, this strategy is also imitated by other pathogens like L. pneumophila (79).
Intracellular Bacteria Hijack Host Cell Extrusion To Enhance Dissemination
Bacterial infection often stimulates intestinal epithelial cell extrusion (shedding) (80–82). To extrude a single cell from a cell monolayer, its neighbor cells first form a ring composed of F-actin and myosin II around it. These surrounding neighbors contract the actomyosin ring, which squeezes the infected cell until it is expelled into the lumenal space (83). This “purse-string” mechanism is often used to extrude unfit or damaged cells, or to eliminate excess cells from a crowded monolayer (84, 85). Some pathogenic bacteria hijack cell extrusion to invade intestinal cells or disseminate widely through the host (86, 87). Other intracellular bacteria suppress extrusion to prevent the removal of infected cells from an intact monolayer and enhance intercellular spread (88). Below, we discuss studies that highlight how bacteria modulate cellular forces to promote or obstruct host cell extrusion, ultimately favoring bacterial spread.
S. enterica promotes host cell extrusion to get released into the lumen.
Salmonella enterica is an intracellular bacterial pathogen that induces cell extrusion to disseminate through its host. Enterocytes containing cytosolic S. enterica extrude more frequently than uninfected cells or cells with vacuolar S. enterica. This is caused by cytosolic S. enterica inducing pyroptosis, a class of programmed cell death (86). Epithelial cells in the gut are programmed to push out dying cells, such as the S. enterica-infected cell, and to do so via the “purse-string” mechanism (86, 89). S. enterica remains invasion competent in the cytoplasm of extruded cells and can escape to infect another cell. As it cannot spread laterally within the monolayer via ABM, S. enterica might benefit more from host cell extrusion than other intracellular pathogens (86).
L. monocytogenes benefits from homeostatic host cell extrusion to invade intestinal cells.
Intracellular pathogens can hijack host cell extrusion to promote invasion as well as dissemination. L. monocytogenes mediates bacterial entry into enterocytes by expressing the surface protein internalin A (InlA), which binds to E-cadherin and triggers bacterial uptake (90). As E-cadherin is located below tight junctions on the lateral surface of host cells, it is inaccessible to L. monocytogenes under normal conditions. However, during homeostatic “purse-string”-driven cell extrusion, tight junctions are remodeled in a manner that exposes E-cadherin, which can now act as a receptor for InlA (87). Because L. monocytogenes coopts cell extrusion to invade intestinal cells, it preferentially infects the tips of intestinal villi, where extrusion is upregulated. The explanation for why intestinal epithelial cell extrusion occurs primarily at the tips of villi is still obscure and might involve extracellular mechanics (e.g., differential fluid shear stresses or ECM stiffness experienced by the cells on the villi versus the crypts) (91). While L. monocytogenes benefits from extrusion to enter intestinal cells, its infectious cycle can also be hindered by extrusion if the bacterium does not spread to a neighboring cell before its initial host cell is expelled (92). The bacterium overcomes this challenge by spreading in a heterogenous manner, where it undergoes smaller displacements to reach immediate neighbors and larger ones to reach cells that are farther away. In this way, rather than choosing between extrusion and lateral cell-to-cell spread, the bacterium strikes an optimal balance (92).
S. flexneri limits host cell extrusion by reinforcing host cell adhesions.
Host cell extrusion can also be an innate immune mechanism utilized by the host to prevent pathogens from effectively colonizing epithelial tissues. S. flexneri, which uses ABM to spread between adjacent cells, downregulates extrusion by blocking host cell detachment from the basement membrane of the intestinal epithelium (93). Here, the bacterial virulence factor, outer surface protein E (OspE), interacts with integrin-linked kinase, a protein that couples the ECM to the host cell actin cytoskeleton, to increase the number of cell-ECM adhesions, inhibit their disassembly, and delay turnover. Although not measured in this study, the magnitude and the dynamics of traction stresses exerted by infected host cells on their ECM are expected to increase and slow down, respectively, in comparison to uninfected cells. This would result in attenuation of cell movement and detachment. Indeed, a scratch-wound assay to interrogate how OspE alters the behavior of epithelial cells revealed that OspE blocks host cell motility, a consequence of the cell’s inability to detach from the basement membrane, which is required for forward motion (93, 94).
Infection with WT S. flexneri also reduced cell rounding compared to ΔospE S. flexneri, once again linking OspE’s role at cell-ECM adhesions to the functional consequence of extrusion suppression. By suppressing host cell extrusion through expression of OspE, S. flexneri can more efficiently spread between neighboring cells than the ΔospE mutant (93). Though S. flexneri differs from S. enterica in favoring lateral cell-to-cell spread over extrusion, the strategies employed by both pathogens result in enhanced dissemination. Other intracellular bacterial pathogens, like Neisseria gonorrhoeae, also achieve efficient spread via suppression of host cell extrusion, although the mechanical changes that host cells experience require further investigation (95).
Intracellular Bacteria Alter Host Cell Motility To Enable Greater Spread
In addition to inducing host cell extrusion, intracellular pathogens can increase the motility of their host cells (96) or alter the directionality of their movement (97) to promote greater dissemination. Furthermore, certain pathogens induce angiogenesis (formation of new blood vessels), which requires increased host endothelial cell migration and proliferation (98). Angiogenesis expands the reservoir of cells the bacterium can inhabit (98) and enables bacteria to travel farther within the host organism and establish a larger number of colonies (99). To enhance host cell migration, pathogens induce formation of host cell lamellipodia (100, 101), modulate host cell-ECM adhesion (96, 101), and stimulate secretion of ECM-degrading enzymes to clear the path forward for the moving host cell (102).
Yersinia enhances T-cell motility via alterations in cell-ECM adhesion.
Yersinia pseudotuberculosis initially infects the intestine but can travel to secondary sites to cause diseases such as reactive arthritis (103). Its strategy for systemic spread involves hitching a ride in motile cells of the immune system, including T cells. The Yersinia virulence factor invasin (Inv), which binds β1 integrins, weakens adhesion of T lymphocytes to their ECM and induces formation of pseudopodia, leading to a more migratory host cell phenotype (96). In addition, Inv mimics the activity of other β1 integrin ligands like fibronectin and collagen IV, inducing host cell haptotaxis (migration toward a gradient of immobilized ECM ligands) (98). In fact, Inv is a potent haptotactic trigger, since more cells migrate directionally in response to Inv than to fibronectin or collagen IV gradients. Addition of soluble Inv also enhances migration of T cells already seeded on collagen IV and fibronectin matrices (96). This in vitro finding could be furthered by interrogating Inv’s effect on T cell migration in a more physiological context, particularly in the presence of shear flow. In the absence of shear flow, chemokines that have been immobilized onto adhesive matrices behave similarly to Inv and induce T cell migration. Under conditions of high shear, the same chemokines activate integrins to increase cell-ECM adhesion and arrest motility (104). As Inv can activate β1 integrin, it would be interesting to study whether it can also trigger arrest (105). When T cells migrate across the endothelium, they arrest in areas of high shear prior to transmigrating through the endothelium (106). Leukocytes typically follow both chemotactic and haptotactic gradients to transmigrate across epithelial monolayers. In this scenario, not only could Inv increase the speed of T cells crawling on ECM or other cell types, but it could also promote traversal across the intestine’s basement membrane, leading to the establishment of secondary infection sites (107).
S. Typhimurium infection alters electric fields in tissues to guide macrophages.
One way in which S. Typhimurium directs infected macrophages away from the infected intestine to spread infection systemically is by altering their electrical properties (Fig. 3A). When S. Typhimurium infects microfold or M cells of the follicle-associated epithelium, it compromises their barrier integrity, as measured through reduced transepithelial resistance (97, 108). The resulting change in transepithelial potential generates directional electric fields which induce the persistent migration of macrophages toward the intestine to help clear infection. Directional cell migration in response to chemical signals (i.e., electrotaxis or galvanotaxis) has been studied extensively in other contexts, e.g., during wound healing (109). In the context of infection with S. Typhimurium, the authors show that once macrophages take up bacteria at the site of infection, they often switch their direction of migration away from the intestine. Charged macromolecules at the surface of cells have long been known to be involved in electrotaxis (110). Accordingly, in this study, authors attribute this unexpected switch in migration direction to be caused by an S. Typhimurium-induced decrease of sialic acid, a negatively charged sugar present on the macrophage surface, which they find to be involved in driving galvanotaxis. The reversal in infected macrophage polarity and directionality to move away from the epithelium is proposed to assist the pathogen in disseminating more widely through the host (97).
FIG 3.

Changes in host cell motility promote bacterial dissemination. (A) Macrophages’ migration toward and away from the infected epithelium is driven by electrical signals. S. enterica hijacks the galvanotaxis-mediated response of macrophages to better disseminate. The color gradient represents the gradient in the electric field. Adapted from reference 97. (B) B. bacilliformis promotes angiogenesis and enhanced endothelial cell sprouting and migration in part through ECM remodeling.
Angiogenesis promotes bacterial dissemination.
Another strategy intracellular pathogens can employ to propagate through their host is angiogenesis, particularly in the context of chronic infection. For example, persistent infection with Chlamydia pneumoniae causes age-related macular degeneration (AMD) (111), with choroidal blood vessels invading the retina (111, 112), and atherosclerosis, which is characterized by blood vessel stiffening and neovascularization in plaques (113, 114). In vitro too, infection with C. pneumoniae promotes angiogenesis of vascular endothelial cells (VECs) (Fig. 3B). Here, the bacterium increases actin polymerization at the leading edge of infected VECs, giving rise to lamellipodial protrusions (100). As a result, VECs exhibit enhanced migration and sprout into new blood vessels (115). In vascular smooth muscle cells (VSMCs) located in the middle layer (tunica media) of the blood vessel, C. pneumoniae infection can cause atherosclerosis and neovascularization. Not only are C. pneumoniae-infected VSMCs more adept at migration, but they also exhibit increased cell-ECM adhesion which allows for persistent migration (116). Moreover, C. pneumoniae induces host cell expression of the metalloproteinases MMP3 and MMP9, matrix-degrading enzymes which remodel the ECM to change its stiffness and porosity (117). This is a well-known strategy used by metastatic cells to facilitate their dissemination through the ECM (118). MMP-driven changes in ECM mechanics could thus open a path for VSMCs to form new blood vessels, thus enabling greater dissemination of the intracellular pathogen (102).
Bartonella is another genus of intracellular bacteria that induce angiogenesis in vivo (119). Clinical samples infected with Bartonella bacilliformis revealed high levels of VEGFR1, VEGFR2, and angiopoietin-2, proteins known to stimulate angiogenesis by promoting the assembly of cell-ECM adhesions and the formation of filipodia and lamellipodia, structures which enhance host cell motility (120). Angiogenesis, as approximated by capillary-like tubes in vitro, was observed upon infection of human umbilical vein endothelial cells (HUVEC) with B. bacilliformis, as was secretion of EGF (121), which could stimulate endothelial cell motility by inducing an endothelial-to-mesenchymal transition (122). Moreover, Bartonella henselae secretes BadA to activate Hif-1, a transcription factor that coordinates angiogenesis and mediates the binding of VECs to ECM components, possibly via β1 integrins (123). As binding to β1 integrin allows Yersinia Inv to enhance the migratory behavior of its host cell (96), it would be interesting to compare the kinematics (e.g., speed, directionality) and dynamics (e.g., traction stresses) of VECs infected with B. henselae that do or do not express BadA. Further studies on the biomechanics of B. henselae-infected host cells could reveal the mechanisms driving their highly migratory phenotype.
Intracellular Bacteria Dampen Adaptive Immune Responses by Changing Host Cell Cortical Stiffness, Motility, and Adhesion Forces
Adaptive immunity provides long-term protection against specific pathogens and involves the participation of T cells and antigen-presenting cells (APCs). APCs engulf a pathogen, degrade it, and present an antigen at their cell surface. These antigens are then recognized by T cells, which are activated to secrete cytokines, stimulate immune cells, or kill cells infected with the specific pathogen (124). Intracellular pathogens can alter the motility of both T cells and APCs to lower the frequency with which they interact to form T cell-APC conjugate pairs, thereby disarming the adaptive immune response. They do so by decreasing host cell migratory speed (125–127), preventing cell polarization to impede directional movement (128–131), impairing adhesion to prohibit persistent migration (14, 131), and reducing the cell’s ability to sense a chemokine gradient and migrate directionally toward infected regions (130, 131). Intracellular pathogens can further hamper the adaptive immune response by changing host cell stiffness to reduce the efficiency with which T cells and APCs engage upon establishment of contact (127).
Intracellular bacteria impair the kinematics of infected immune cells.
S. flexneri dampens the host’s adaptive immune response such that immunity is established only after multiple reinfections and is short-lived (132). In mice, S. flexneri infects T cells in the lymph nodes, where adaptive immunity is initiated (125). T cells exhibit impaired motility in comparison to uninfected cells, which reduces their proficiency in scanning for APCs and forming T cell-APC conjugate pairs (127). This is achieved through the type 3 secretion system effector, IpgD which inactivates host cell ERM proteins, cross-linkers of the plasma membrane cortical actin network. As only activated ERM proteins redistribute in response to a chemoattractant, S. flexneri obstructs plasticity and polarization of infected T cells, hindering their chemokine-directed migration (126) (Fig. 4A).
FIG 4.
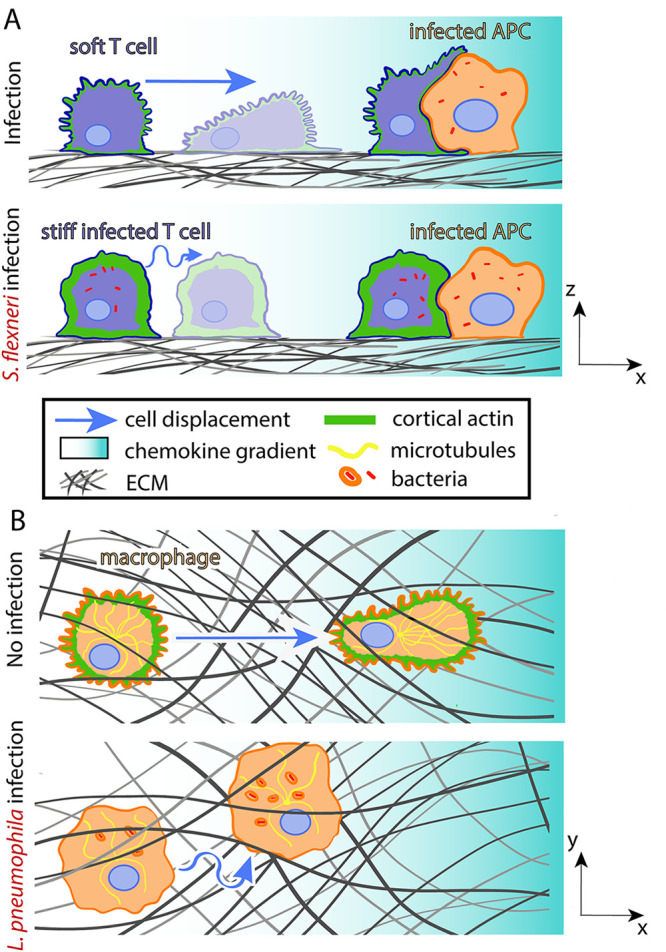
Bacteria hijack host cell mechanics during adaptive immune responses. (A) S. flexneri-infected T cells stiffen and migrate slower and with less persistence toward APCs. (B) L. pneumophila-infected macrophages soften, weaken matrix adhesion, and lose migration directionality when performing chemotaxis.
Another pathogen that reduces the formation of conjugate pairs is Legionella pneumophila, which impairs the motility of macrophages (class of APCs) (129). In response to the quorum sensing signal LAI-1, L. pneumophila inactivates host cell Cdc42, leading to a reduction of microtubules in the host cell and a dissipation of actin from the cell cortex (129) (Fig. 4B). These changes in cytoskeletal organization and possibly in cell mechanics could explain the altered kinematics of host cells, including their decreased migration directionality.
Dendritic cells (class of APCs) infected with S. Typhimurium also exhibit impaired directionality without a loss of speed (Fig. 4B) (130). Cells infected with the wild-type bacterium make more frequent turns than uninfected cells or cells lacking the virulence factor Salmonella secreted effector I (SseI). The host scaffold protein IQGAP-1, which normally localizes to lamellipodial protrusions, binds to SseI, and this interaction plays a role in hindering cell migration (131). In addition, SseI causes the persistent activation of heterotrimeric Gi proteins, which can suppress the activity of the signaling GTPase Rac and prevent the accumulation of PIP3 at the leading edge of the host cell. In this way, S. Typhimurium behaves similarly to S. flexneri and L. pneumophila, blocking cell polarization to impede chemokine-directed migration; however, S. Typhimurium employs an additional strategy. As most chemokine receptors are coupled to heterotrimeric Gi proteins, constitutive activation of Gi proteins prohibits sensing of the chemokine gradient (133). The number of CD4+ T and dendritic cells found in the spleen are reduced when mice are infected with WT rather than ΔsseI S. Typhimurium, suggesting that these strategies of impairing host cell kinematics prevent T cells from reaching extraintestinal organs where adaptive immune response processes take place and lessen the aggressiveness of the humoral response (131).
Stiffening of S. flexneri-infected T cells obstructs interactions with APCs.
Unlike L. pneumophila-infected macrophages, which lose cortical actin relative to uninfected macrophages, S. flexneri-infected CD4+ T cells show a 2-fold increase in their F-actin content compared to uninfected cells. As a result, their cortical stiffness increases significantly compared to uninfected cells (probed through microindentation) (127). This increased stiffness causes infected CD4+ T cells to resemble naive T cells and to form small immunological synapses that are not very responsive to antigenic stimulation (127). Activated T cells are generally softer than naive T cells and therefore more capable of spreading onto APCs. Upon contact, they soften further due to the role of cofilin in severing actin filaments. In naive T cells, enhanced Rho-associated protein kinase (ROCK) activity inactivates cofilin to prevent softening upon formation of the conjugate pair (134). It would be valuable to study whether S. flexneri-infected T cells are also prevented from altering their stiffness upon contacting their target APCs. Since dephosphorylation of ERM proteins is another mechanism through which activated T cells deform upon antigen recognition (135), one might hypothesize that ERM proteins, which are already dephosphorylated during infection, prevent efficient engagement of T cells with APCs.
Decreased cell-ECM adhesion impairs kinematics of infected immune cells.
Macrophages infected with S. Typhimurium and dendritic cells infected with Yersinia pestis are more likely to detach from the surface on which they are migrating than uninfected cells (131, 136). Similarly, when L. pneumophila infects the protozoan Acanthamoeba castellani, it causes its host to round up, become less adherent, and lower its migration speed. In particular, infection decreases the formation of acanthapodia, spine-like structures that allow for greater surface adhesion (14). By removing Ras homology family member A (RhoA, a GTPase that regulates critically the actin cytoskeleton [137]) from the membrane of infected macrophages, Y. enterocolitica also prevents the formation of adhesive structures, in this case, podosomes (138). It would be interesting to know how much the increase in cortical stiffness of S. flexneri-infected CD4+ T cells prevents the formation of similar protrusive structures that might aid in both the crawling movement of T cells during the scanning phase and their ability to spread onto the surface of an APC.
HOST CELLS CHANGE THEIR MECHANICS TO LIMIT BACTERIAL SPREAD
Bacterial pathogens often manipulate host cell mechanics (e.g., their migration speed, persistence, traction forces, intercellular forces, and cellular stiffness, etc.) to efficiently spread through tissues, but host cells themselves can also alter their mechanics in response to infection to impede the spread of pathogens. These biomechanical changes are often driven by innate immune signaling and are beneficial for the host in limiting bacterial spread, as discussed below (139, 140).
Host Cell Mechanics Drives Bacterial Phagocytosis
Phagocytes are best known for their ability to recognize and eliminate bacterial pathogens. As with many other aspects of bacterial pathogenesis, a great deal of work has gone into identifying the molecular pathways associated with pathogen recognition and downstream signaling for successful uptake, but relatively less is known about the mechanical forces that bacteria use to elicit their uptake and, conversely, the forces that host cells use to promote bacterial engulfment (141, 142). Typically, bacteria first engage and cluster host cell receptors, which leads to a remodeling of the cellular actin cytoskeleton. This actin reorganization presumably provides the necessary mechanical force for the creation of membrane protrusions or ruffles, which allow the membrane to wrap around the phagocytic target (143). Indeed, macrophages have been reported to employ a mechanobiological process that resembles a “hook and shovel” mechanism, where they use actin-driven membrane extensions to lift bacteria from underneath and build a phagocytic cup to engulf them (127). The protrusive force of these membrane extensions, such as filopodia, membrane ruffles, and podosomes, has been measured though protrusion force microscopy, which consists of measuring by AFM the deformation induced by these cell structures onto compliant Formvar sheets (polymer elastic porous membranes inert to chemicals [144, 145). These forces range from 3 to 100 pN and are similar to those that facilitate the engagement of phagocytic targets, including pathogens like Escherichia coli, that adhere strongly to the ECM (125). Recent technical advancements such as microparticle TFM have enabled measurement of subcellular force exertion patterns when immune cells interact with and ingest polyacrylamide beads (146). While this technique has increased our understanding of the forces required to engulf soft elastic deformable polyacrylamide-based targets, it is unclear how complementary forces involved in the engulfment of much stiffer bacterial pathogens would be measured. Technical advancements in this area would enable us to infer what forces host cells must exert to efficiently engulf bacterial targets and how these forces change depending on the shape and morphology of the bacterial target and on the receptors engaged. It could also inform us on how these host cell forces that promote bacterial engulfment are modulated when host cells encounter a dynamic extracellular mechanical environment where shear flow and ECM stiffness vary.
Inflammasome Activation Drives Stiffening of Host Cells to Limit Infection
Inflammasomes are multimeric protein complexes that assemble in the cytoplasm of infected cells and provide innate immune protection by activating caspase-1 (147). This leads to the secretion of proinflammatory cytokines and sometimes causes pyroptotic cell death. When these processes are regulated, they function to protect the host by restricting the bacterial burden in host cells (147). For example, inflammasome activation is critical in limiting S. enterica infection in macrophages (140). Here, actin polymerization in response to S. enterica infection activates the NLRC4 inflammasome, which induces a 2-fold increase in cellular stiffness as measured by a cell optical stretcher. As stiffer cells are less adept at engulfing bacteria, the inflammasome prevents further bacterial uptake by infected macrophages. Moreover, the increase in cellular stiffness might impede bacterial dissemination by limiting macrophage motility, although this was not examined explicitly. Overall, this work suggests that innate immunity can alter the cytoskeletal mechanics and motility of macrophages to restrict spread of S. enterica infection (140).
Further studies support a link between inflammasome activation and cytoskeletal dynamics in infection, although explicit measurements of cell or cytoskeletal mechanics are lacking (148, 149). For example, the pyrin inflammasome is often initiated in response to fluctuations in cytoskeletal dynamics and acts as an immune sensor to detect pathogen-induced changes in Rho GTPase activity (148). Various intracellular bacterial pathogens (e.g., Burkholderia cenocepacia, Clostridium difficile) modulate Rho GTPase activity to cause actin cytoskeletal defects that suppress host immune responses such as phagocytosis (150–152). Pyrin specifically senses modifications in RhoA GTPase activity and protects against infection by activating caspase-1. Accordingly, loss of the pyrin inflammasome elevates intracellular bacterial growth (153). Moreover, components of the inflammasome including caspase-1 modulate the actin machinery that promotes fusion of the phagosome with the lysosome, a key host defense mechanism against many bacterial pathogens, including L. pneumophila (154). In response to caspase-1 activation, the ratio of filamentous to monomeric actin increases in infected macrophages, and this probably also augments cell stiffness, which helps to constrain bacterial infection. While the role of inflammasome activation in altering host cell mechanics is beginning to be understood, the mechanisms by which cytoplasmic and cytoskeletal mechanics and dynamics lead to inflammasome activation are still to be uncovered.
Antimicrobial Peptides, Commensal Bacteria, and Probiotics Alter Cell Mechanics and Barrier Function
Host cell-produced antimicrobial peptides (AMPs), also known as host defense peptides, are short peptides often produced in response to infection. Most AMPs kill microbial pathogens directly, whereas others act indirectly by modulating various host cell processes, including cell mechanics and barrier function. Antimicrobial agents produced by commensal bacteria and probiotics can also participate in the maintenance and strengthening of the epithelial barrier by acting as key defenders against infections by intracellular bacteria (155–157).
LL-37 increases stiffness and barrier function of cells restricting infection.
The LL-37 cathelicidin is a human AMP that exhibits antibacterial activity (158) and induces actin polymerization to modify the host cell cortex in lung epithelial and endothelial cells (159). This increase in cortical actin polymerization leads to cellular stiffening (measured by AFM) and decreases epithelial permeability by strengthening cell-cell junctions. This combination of cellular stiffening and increased barrier function has been shown to obstruct Pseudomonas aeruginosa internalization and spread (159). Thus, AMPs not only kill bacteria but also limit bacterial spread by changing host cell mechanics. Further studies suggest that AMPs regulate the barrier function of epithelial cells in additional tissues, such as the skin (160). For instance, disruption of the cutaneous physical barrier leads to induction of the murine ortholog of LL-37 (CRAMP), which restores tissue integrity (161). However, the dynamic interplay between AMP production and modulation of intercellular forces necessary for proper barrier function of epithelia is still to be uncovered.
AMPs enhance cell motility and reepithelization in response to infection.
Not only do AMPs promote restoration of the physical barrier of epithelial tissues, but they can also contribute to wound healing in response to injury or infection (162, 163). For example, when infection with P. aeruginosa damages the airway epithelium, creating wounds, certain AMPs can enhance epithelial cell migration to accelerate the healing of these wounds (163). This process is mediated in part by epidermal growth factor receptor (EGFR)-driven enhancement of epithelial cell migration and increased production of metalloproteases (MMPs). Specifically, MMP9, a matrix-degrading enzyme previously linked to ECM remodeling and changes in ECM stiffness, is upregulated (117). If MMP9 drives changes in ECM stiffness during infection, it might also play a role in promoting the epithelial-to-mesenchymal transition, which gives rise to an increase in the collective motility of epithelial cells (164, 165). A more detailed mechanistic understanding is still pending. Given the increasing development of microbial resistance to conventional antibiotics worldwide, multiple efforts are focused on the potential clinical usage of AMPs to fight infection. As such, it is pertinent to study the effect of AMPs on the mechanics and motility of host cells and on restoring barrier function.
Host cell mechanics are modulated in the presence of commensal bacteria and probiotics.
Probiotics, commensal microbial populations, and their metabolites can regulate the physical properties and reinforce the barrier function of epithelial tissues, ultimately defending the host against infection (157, 158, 166). For example, urolithin A is a gut microbiota-generated metabolite that both reduces inflammation and enhances barrier function (166). Lactobacillus and Bifidobacterium genera and other probiotics enhance intestinal barrier function by increasing expression of epithelial tight junction proteins, possibly strengthening intercellular force transduction (167–169). This effect has been attributed to production of specific bacterial metabolites (e.g., secreted extracellular proteins, organic acids) and to bacterial surface layer proteins (e.g., flagellin, pili) that can directly modulate host cell signaling, impacting barrier function (170). Bacterial sphingolipids can also affect host cell mechanics and regulate barrier integrity (171). Studies have shown that the host cell’s plasma membrane is highly packed with sphingolipids that allow host cells to resist mechanical stress through signaling orchestrated by sphingosine 1-phosphate (S1P), S1P receptor (S1PR), and Rho-associated protein kinase (ROCK). Together, their activity helps increase actomyosin contractility (172, 173). It is still not well understood how sphingolipids produced by the microbiota might alter host cell mechanics and provide additional resistance to mechanical stress, thus potentially protecting against bacterial pathogens.
Changes in Cell Contractility, Adhesion, and Motility Lead to the Elimination of Infected Cells and Epithelial Remodeling
As we discussed in an earlier section, pathogenic bacteria can hijack intestinal extrusion to promote their own dissemination. However, in some cases, cell extrusion can also act as a host defense mechanism. For example, in the intestine, bacterial infection often stimulates the shedding of infected cells en masse, which is followed by stem cell proliferation and migration of surrounding uninfected cells to restore epithelial homeostasis (13). The mechanics driving this process are distinct from those of single-cell extrusion and do not involve the classical actomyosin purse-string contraction (139). Below, we discuss recent studies that showcase infected cell extrusion as a defense mechanism to eject infected cells from host tissue and restore homeostasis.
Infected cell extrusion and cell motility restore epithelial homeostasis.
A 2010 study revealed that infection of Drosophila melanogaster with bacterial pathogen Erwinia carotovora leads to a large-scale epithelial remodeling in the gut that is followed by restoration of gut homeostasis (13). At early time points postinfection, roughly half of the enterocytes present in the gut are extruded en masse, shortening the gut to 60% of its initial length. As the infection proceeds, the gut elongates due to increased proliferation of enteroblasts, differentiation of enteroblasts into enterocytes, and enhanced collective motility of these enterocytes. These processes allow the gut to return to its original dimensions 2 days postinfection, preventing excessive extrusion from leading to loss of barrier function. Here, the EGFR pathway promotes both infected cell extrusion and wound healing-driven restoration of epithelial homeostasis (174). Enterocytes in the fruit fly’s midgut employ similar mechanisms when infected with Pseudomonas entomophila (175). These studies demonstrate that extrusion of infected cells accompanied by collective migration and proliferation of uninfected cells following infection can restore gut homeostasis in the fruit fly. What the precise mechanical events might be that facilitate infected cell extrusion in this context and the restoration of gut homeostasis are still open questions.
Innate immunity-driven mechanical competition drives extrusion of infected cells.
A more recent 2021 in vitro study on L. monocytogenes-infected epithelial cells led to similar observations of infected cell extrusion en masse, driven by collective migration of uninfected bystander cells surrounding the infected domains (139). This study added to previous works by showing that this process results from a mechanical battle between uninfected bystanders and infected cells, where the former win and the latter lose and, therefore, get extruded (Fig. 5). To come to this conclusion, epithelial cell monolayers sparsely infected with L. monocytogenes were monitored over the course of several days. Using TFM and AFM, it was discovered that infected host cells become softer over time and exert less traction on the deformable matrices underneath them. In contrast, the uninfected cells surrounding the infection focus become stiffer and actively move toward the site of infection, collectively squeezing the infected cells up into an extruding mound over the course of a day. Bacteria in mounds are less able to spread laterally in the monolayer, potentially just due to geometry (i.e., because most bacteria are contained in the extruded cells and thus are no longer in close physical proximity to cells still attached to the substratum), limiting the growth of the infection focus. Meanwhile, mounded cells die over time and no longer serve as viable hosts for the bacteria. Both in vitro and in silico (finite element analysis) experiments indicate that the driving forces of this mechanical battle are directional migration of highly contractile surrounding uninfected cells, the presence of intact cell-cell junctions, and innate immune signaling through NF-κB activation. It is worth noting that infection mounds were observed in infected animal cells (MDCK, Vero) and human epithelial cells (A431D, human ileum-derived intestinal epithelial cells). Moreover, infection mounds occurred in response to infection by L. monocytogenes and R. parkeri mutants that lack the outer membrane protein B (OmpB), both of which activate NF-κΒ signaling, but extrusion was not observed with WT R. parkeri, which does not activate NF-κΒ. These findings suggest that the mechanical competition that leads to infected cell extrusion en masse is triggered by NF-κΒ activation and is not specific to L. monocytogenes infection. In addition, they underline the dynamic capability of epithelial tissue to remodel in response to infection and also propose a mechanical mechanism driven by innate immunity signals that leads to infected cell extrusion, quickly limiting the local spread of infection (139). This strategy appears to be employed broadly to defend against pathogens that activate NF-κΒ, while some pathogens (e.g., WT R. parkeri) might trick the host by suppressing NF-κB activation to avoid the mechanical battle that leads to extrusion of infected cells.
FIG 5.

Innate immunity-driven changes in host cell mechanics limit bacterial spread. Innate immune signaling drives a mechanical competition between uninfected and L. monocytogenes-infected cells. Softer and less contractile infected cells get extruded en masse out of the cellular monolayer due to the active migration and squeezing triggered by surrounding uninfected cells. Adapted from reference 139.
NF-κB activation facilitates rapid remodeling of E-cadherin cell-cell junctions and uniform collective migration of epithelial cells in response to injury or damage induced by infection (176). In mice infected intragastrically with S. flexneri, L. monocytogenes, and S. Typhimurium, waves of NF-κΒ and MAPK/ERK signals that propagated from infected cells to uninfected bystanders rendered mice resistant to infection (177). At early time points, autophagy-induced shedding of infected cells repressed subsequent bacterial invasion and pathological inflammation (178). Within the course of a day, the whole intestinal epithelium was regenerated as a result of these propagating signaling waves. The precise cellular dynamics leading to reepithelization have not yet been characterized. It would be interesting to study how these waves of NF-κΒ and MAPK/ERK signaling are correlated with changes in cell mechanics and cell-cell communication and how they give rise to mechanical competition between infected cells and bystanders. Recent work during wound healing of epithelial cells revealed ERK-mediated mechanochemical waves to emerge during collective cell migration of epithelial sheets. These waves are generated by the stretching of leading cells, which elevates ERK activity and leads to contraction of those cells and extension of the cells just behind them (179, 180). Hence, a mechanical stimulus (cell strain) leads to a biochemical response (increase in ERK activity), which leads to a mechanical response (contraction of cell). Strain rate and ERK activity oscillate with the same frequency but are slightly out of phase, with the strain rate preceding. These waves allow for long-range cell communication. Whether such waves arise during infection and what is the precise stimulus are still to be uncovered.
EXTRACELLULAR MECHANICS INFLUENCES INFECTION
A central element in mechanobiology is cellular “mechanosensing” (181). Cells actively probe the stiffness of the ECM on which they reside by exerting stresses on it via transmembrane proteins named integrins (1). The mechanism by which cells sense matrix stiffness is an active area of investigation. Likewise, cells often experience fluid shear flows at their apical surface which they can sense through specialized receptors, leading to changes in gene expression and morphological organization (182). Additional mechanical cues that cells experience include pressure-driven cellular compressions (e.g., epithelial cells in the lungs) and peristalsis-driven cellular stretching (e.g., epithelial cells in the intestine). Transduction of this mechanical information typically produces a cellular response that involves alterations in cell functions and fate, including motility (18), differentiation (183), gene expression, and protein activity (184). Recently, many studies have focused on the effect of these forces in the interaction of host cells with bacterial pathogens.
Stiffness of Extracellular Matrix Affects Infection
ECM stiffness varies in different parts of the body and over time due to aging and (patho)physiological conditions (e.g., arteriosclerosis, hypertension, fibrotic diseases, cancer) (185–187). Clinical in vivo studies show a positive correlation between atherogenesis and susceptibility to bacterial infection (188, 189), yet it is unclear if infections lead to atherosclerosis (190, 191) or if endothelial cells in stiff atherosclerotic regions are more prone to infection (192). The importance of ECM stiffness on altering host cell behavior and interaction with bacterial pathogens is just beginning to be considered, and novel mechanisms of action are being discovered (Fig. 6) (193). In the sections below, we discuss findings that illustrate how ECM stiffness can modulate host-pathogen interactions. However, additional properties of the ECM, such as ECM porosity, protein tethering, surface tension, and creep compliance, have recently emerged as important mechano-regulators of cell function as well (194, 195). Due to the absence of studies assessing the role of these mechanical cues in infection, they are not discussed below but are likely to contribute to the outcome of infection.
FIG 6.

Host cells experience extracellular mechanical cues during infection. Extracellular mechanical cues (input, left) imposed on host cells (system, middle) by their environment and the different host cell-pathogen interaction processes they could modulate (output, right). Extracellular mechanical cues include (1) ECM stiffness, (2) luminal shear flows, (3) cellular deformation due to ECM stretch, and (4) pressure-driven cellular compression. Host-pathogen interactions that could be modulated by extracellular mechanics include (i) adhesion of bacteria onto the host cell surface, (ii) uptake of bacteria within host cells (internalization), (iii) intracellular bacteria cell-to-cell spread, (iv) bacterial transmigration from the epithelial layer across the basement membrane, and (v) heterotypic bacterial spread from infected immune cells and infected immune cell transmigration through epithelia or endothelia.
ECM stiffening enhances L. monocytogenes uptake by host cells.
A 2018 study showed that L. monocytogenes is twice as likely to infect endothelial cells residing on stiff matrices than on softer ECM (Fig. 7A) (193). This finding suggests that ECM stiffness impacts infection of host cells and calls into question the physiological relevance of in vitro infection assays, which are performed on glass coverslips that are ∼6 orders of magnitude stiffer than most tissues (3 GPa versus ∼10 kPa). This study showed that elevated subendothelial matrix stiffness leads to an increase in pFAK397, that is, phosphorylation at Tyr-397 of focal adhesion kinase (FAK), a known mechano-transducer present at host cell focal adhesions (196). Higher pFAK397 levels lead to increased levels of surface vimentin, a receptor used by L. monocytogenes to adhere to endothelial cells. Additional biochemical or biophysical factors, such as changes in glycocalyx components or a rougher host cell surface, might also facilitate the increased adhesion and uptake of bacteria into host cells residing on stiffer matrices (Fig. 7A). Moreover, it is unknown if the effect of ECM stiffness on infection susceptibility represents a universal phenomenon suggestive of a generalized phagocytosis-like process or if this relationship differs depending on host cell type or bacterial species (193).
FIG 7.

Extracellular mechanics rule interaction of host cells with bacterial pathogens. (A) Sketch depicting how increased ECM matrix (black mesh) stiffness augments FAK phosphorylation (purple circles) and levels of surface vimentin (blue receptors), thus increasing the adhesion of L. monocytogenes (red rods) to host cells. Panels on the right depict additional ECM stiffness-sensitive processes that could regulate bacterial adhesion onto host cells, including variations of the host cell glycocalyx (gray) and surface roughness. (B) Cartoon depicting how catch bonds between host cell receptors and bacterial surface proteins function. Green arrows indicate the direction of applied force. (C) S. flexneri-infected epithelial cells on a gut-on-chip device compared to cells residing on static Transwells reveal that mechanical deformations and shear flows as well as ECM topography critically impact infection. Adapted from reference 12 with permission from Elsevier.
ECM softening facilitates UPEC endosomal escape.
ECM stiffness also impacts infection of bladder cells by uropathogenic Escherichia coli (UPEC) (197). After UPEC invades bladder cells in vivo, it escapes the endosome to replicate in the cytoplasm of its host (198). However, in tissue culture, UPEC is often trapped within Lamp1-positive endosomes and cannot proliferate efficiently unless the host’s actin cytoskeleton is disrupted, allowing for endosomal escape. Given that ECM stiffness is a critical regulator of actin cytoskeletal dynamics, a 2019 study examined whether ECM stiffness could modulate UPEC endosomal escape (197). Indeed, it was found that at a physiological ECM stiffness (∼300 Pa), UPEC escapes the endosome and proliferates rapidly in the cytoplasm of bladder epithelial cells. Furthermore, decreased levels of RhoB (a Rho GTPase) or RhoB effector PRK1 in cells seeded on soft ECM (as opposed to stiffer ECM or plastic) was responsible for the increase in endosomal escape (197). It would be interesting to examine the contribution of additional relevant mechanical cues, such as apically exposed fluid shear flow, on endosomal escape of UPEC from bladder cells. Shear flow could both enhance adhesion of UPEC to the surface of host bladder cells (199) and promote endosomal escape following uptake, as flow is known to alter cytoskeletal organization and dynamics in various cell types (200, 201).
ECM stiffness can affect uptake and intercellular spread of bacteria.
ECM stiffness can mediate the production of proinflammatory cytokines by immune cells (202–204). In response to lipopolysaccharide (a bacterial proinflammatory agent), macrophages residing on soft ECM produce and secrete lower amounts of proinflammatory mediators than macrophages residing on stiff ECM (205). This might be a consequence of increased Toll-like receptor 4 activity in macrophages seeded on stiff matrices, which would lead to increased activation of NF-κΒ, a transcription factor that induces secretion of proinflammatory cytokines. Interestingly, exposure of macrophages to both lipopolysaccharide and/or to specific mechanical cues (i.e., increased ECM stiffness) increases macrophage elasticity through regulation of actin polymerization and Rho GTPase activity. This then augments the macrophage’s ability to phagocytose targets (206, 207). However, most studies on phagocytosis sensitive to ECM stiffness have been performed using IgG opsonized beads as targets, so it remains unclear whether these findings would also apply during infection of macrophages with bacterial pathogens.
Other than bacterial uptake, ECM stiffness might also regulate the ability of intracellular bacteria to spread from cell to cell, as when L. monocytogenes spreads among endothelial cells (208). Intracellular bacteria undergoing ABM must overcome the tension faced at the donor cell membrane to create a protrusion that will be engulfed by a neighboring cell (209). Preliminary findings suggest that decreasing ECM stiffness reduces intracellular tension (measured via MSM), which favors L. monocytogenes intercellular spread (208). Moreover, cells residing on softer matrices tend to exhibit reduced membrane tension, which could also enhance trans-endocytosis (the process where material created in one cell undergoes endocytosis, entering another cell), thus enabling more efficient bacterial cell-to-cell spread (210–213). Indeed, evidence suggests that L. monocytogenes and S. flexneri hijack cell-cell junction communication and caveolin- or clathrin-related endocytic processes, respectively, to spread intercellularly (213, 214). However, a systematic analysis of the mechanisms by which ECM stiffness modulates intercellular force transduction and trans-endocytosis, and how they differentially impact the donor versus the recipient cell during intracellular bacterial spread, is still pending.
Apically Exposed Fluid Shear Flow Modulates Infection
Endothelial cells are constantly exposed to shear stresses due to blood flow over their surfaces and respond to these stresses with changes in gene expression and morphological organization (Fig. 6) (182). The magnitude of shear stress experienced by these cells varies widely (0 to 100 dynes/cm2) depending on the blood vessel type, size, location, age and (patho)physiological condition. While circulation in capillaries can reach very low shear stress values (<1 dynes/cm2) and can even stop for short periods of time, shear stresses in arterioles can reach 100 dynes/cm2 (215). Intestinal epithelial cells are also exposed to luminal shear flows that are much lower in magnitude (∼0.025 dynes/cm2) than those endured by endothelial cells but still have an impact on cell polarization, autophagy, villus formation, three-dimensional (3D) morphogenesis (216, 217), and even interactions between intestinal epithelial cells and the microbiome (218). Interestingly, physiological luminal flow and peristalsis-driven deformation of epithelial cells are severely disrupted in patients exhibiting various types of inflammatory bowel diseases (IBD) (218). The impact of this disruption on host-pathogen interactions has recently become an active area of research.
Bacteria turn on virulence programs upon exposure to shear flow.
Dynamic rotating wall vessel (RWV) bioreactor technology can be used to infer the role of shear stresses onto the interaction of host cells with bacterial pathogens (219). In an RWV bioreactor, host cells and/or bacterial pathogens are placed in a cylindrical vessel and are maintained in suspension by the slow rotation of the vessel. The orientation of the rotating vessel determines whether cells and/or bacteria are exposed to low or high shear stresses. This technology was used to show that fluid shear stresses alter gene expression and virulence of S. enterica grown in these vessels (219, 220). When these bacteria were then used to infect mice, mice infected with S. enterica grown in a high-shear-force environment exhibited faster disease progression and death. In another study examining infection of HeLa cells with enterohemorrhagic Escherichia coli (EHEC) residing within flow cells exposed to varying shear flow, it was also shown that EHEC responds to shear by increasing virulence through changes in gene expression (221). Here, increased infection susceptibility of host cells exposed to high shear flow by EHEC was attributed to increased expression of the locus of enterocyte (LEE) virulence factor. The magnitude of shear stresses and shear stress gradients dramatically alter host cell behavior in nonintuitive ways (222, 223). For example, exposure to high shear flow stalls endothelial cell proliferation but low shear flow augments proliferation compared to static conditions. Cells can also exhibit distinct responses to pulsatile compared to steady flow (224) or gradients of shear stress, which are often present at blood vessel branches and bifurcations (2). Can bacterial pathogens sense such subtle variations in shear and reprogram gene expression accordingly to increase efficiency of infection?
Shear flow drives adhesion of pathogens on the surface of host cells.
Shear flow also modulates the strength with which bacterial pathogens attach to receptors at the surface of host cells (15). Shear force-dependent alterations of bond strength and lifetime were first characterized in the L-selectin (a transmembrane glycoprotein)-dependent adhesion of lymphocytes to surface receptors of vascular endothelial cells (225). The strength of interaction between these two receptors increases with increasing shear stress, forming a catch bond (Fig. 7B). Catch bonds have an optimum shear stress level that leads to the highest bond lifetime (i.e., neither too low nor too high) (225). Interestingly, the surface receptors of many bacterial pathogens bond with receptors on the host cell surface, and the strength and lifetime of these bonds depend on force exertion due to fluid flow, suggesting that they may act as catch bonds (226–229). For example, the interaction between FimH adhesin found on UPEC and N-glycans on the host cell surface can switch from loose to firm with the application of a 10-fold increase in shear stress (199). EHEC, Pseudomonas aeruginosa, Borrelia burgdorferi, Staphylococcus epidermis, and Staphylococcus aureus are other bacterial pathogens that exhibit enhanced adhesion to their host cell due to catch bonds strengthened by fluid shear stresses (226–231). The precise role of shear flow in activating bacterial ligands and host cell receptors is still unclear, as are the possible effects of variations in shear stress magnitude and gradients of shear stress on these interactions. Also, whether shear stress-sensitive catch bonds ultimately increase successful invasion events of pathogens within the host has not yet been explicitly studied.
Studying infection in the presence of shear flow can reveal additional features that are not present under static conditions, as in the case of phosphoethanolamine cellulose (pEtN)-driven adhesion of UPEC to bladder epithelial cells (25). When UPEC infects bladder epithelial cells, it promotes adhesion onto host cells by producing and secreting a variety of polysaccharides, including extracellular fibers located on the surface of bacteria, such as curli (amyloid extracellular fibers) and a chemically modified form of cellulose, pEtN. Conventional tissue culture adhesion assays had not revealed a role for pEtN cellulose in UPEC adhesion to host cells. However, a live-cell monolayer microscopy rheometer that was used to measure adhesion strength between UPEC and a monolayer of bladder epithelial cells revealed that under a high-shear environment, pEtN cellulose acts as a glue to maintain curli association of bacteria with epithelial cells (25). Additional pathogens might also turn on distinct programs only when exposed to shear flows.
The outer surface of human cells is covered by a sugar coating, the glycocalyx, and with a layer of secreted mucus in the case of cells originating from the gastrointestinal, respiratory, and urogenital tracts, which are all common sites of pathogen attack. In most cases, these linings act as protective barriers for the host cells (232), although pathogens sometimes use components of the glycocalyx to promote their adhesion to host cells (233, 234). Both the glycocalyx and the mucus linings of host cells are extremely sensitive to shear flows, with their composition and rheological properties varying as a function of shear stress magnitude and/or rate of shearing (235–237). At the macroscale, mucus behaves like a non-Newtonian viscoelastic material containing both viscous (resistant to flow) and elastic (resistant to deformation) components (238). When the rate of shear is low, the viscosity of mucus is ∼3 orders of magnitude higher than that of water, while at higher shear rates, its viscosity is comparable to that of water (that is, mucus is a shear-thinning material). At the microscale relevant to the transport of pathogens, mucus can be seen as a dense heterogeneous cross-linked polymer network (237). Changes in the rheological properties of mucus may greatly affect its ability to function as a lubricant, selective barrier, and first line of defense against infection (239). The impact of shear stresses on mucus anisotropic organization and viscoelasticity is broadly unknown, as well as the effect of altering mucus properties on bacterial adhesion and subsequent uptake by host cells (237). Similarly, while the role of the host cell glycocalyx in bacterial infections is well documented (240), as is the sensitivity of glycocalyx to shear flow (235), the impact of shear flow-induced changes to the glycocalyx on host-pathogen interactions is still to be uncovered.
Shear flow modulation of membrane tension might impact infection.
Laminar shear flow typically increases host cell membrane tension, with it becoming highest near cell-cell junctions (241, 242). Increased membrane tension limits endocytic cellular trafficking by inducing rapid disassembly of caveolae (211), which are involved in intercellular bacterial spread (213). Variations in the magnitude of shear stresses and disturbances in shear flow significantly alter the cytoskeletal organization of cells, including the way they transduce forces to each other and to their environment (243, 244). After long-term exposure to shear stress, cells exhibit increased cortical stiffness, and their surface is rougher and decorated with more “ridges” (245). Experiments on endothelial cells that have been exposed to shear flow for a long time show reduced internalization of nanoparticles compared to cells under static conditions (246, 247). It is possible that shear flow could modulate the internalization and intercellular spread of pathogenic bacteria through its effect on host cell tension and intercellular force transduction, the latter of which could be evaluated using FRET-based force sensors and MSM (248).
Tensions and Compressions Experienced by Host Cells Impact Infection
Mechanical strains imposed on cells (i.e., compressions and tensions) have also emerged as key extracellular mechanical cues modulating bacterial infection. For example, intestinal epithelial cells deform in response to peristaltic waves of pressure (16). Airway epithelial cells are also exposed to mechanical compressions during normal breathing and can experience compressive stresses that are an order of magnitude higher in pathologies such as asthma-induced bronchospasm (249, 250). RNA sequencing showed that extracellular environment-triggered cellular deformations can alter gene expression (251). In this study, gene expression of duodenal organoids in tissue culture, duodenal cells placed on an organ-on-chip device (where cells are exposed to peristaltic waves of pressure), and human adult duodenal cells was compared. Gene expression of duodenal organoids in tissue culture showed considerable differences from that of the other two conditions, underlining the importance of peristalsis-driven deformations in determining the epithelial cell genotype (251). Below, we discuss recent studies on organ-on-chip devices highlighting the importance of considering these mechanical cues when performing infection assays in vitro.
Gut-on-chip devices enable observation of infection by bacterial pathogens.
Organ-on-chip microfluidic technologies have enabled scientists to conduct infection assays under conditions that more closely mimic in vivo settings than traditional tissue culture methods can provide (12, 252). For example, the organ-on-chip intestinal epithelium (gut-on-chip) enables exposure of host epithelial cells to shear flow and peristalsis-driven deformations and also takes into account the 3D topography of the intestine (16). Interestingly, this technology supports culturing commensal bacteria with intestinal cells (16). It is impossible to expose cells to a living microbiome in traditional Transwells, since bacterial overgrowth compromises the epithelium (253). However, exposing intestinal epithelial cells to physiological levels of fluid shear stresses (∼0.02 dynes/cm2) and exerting cyclic strains (10%, 0.15 Hz) that mimic peristaltic motions lead to the formation of a polarized epithelial monolayer that is rich in intestinal villi and exhibits barrier integrity. These cells can be cocultured with intestinal microbes for long periods of time (more than 1 week) (253). Moreover, by applying the two forces separately, the authors of this study discovered that peristaltic deformations rather than luminal fluid flow prevent the uncontrolled growth of commensal bacteria. This is consistent with clinical data from patients with IBD, where peristalsis is often obstructed and bacterial overgrowth has been observed (254). When intracellular enteroinvasive E. coli (EIEC) was then introduced to the gut-on-chip, the presence of probiotic bacteria protected the epithelium against infection and subsequent injury. Further study is required to understand the precise contributions and dynamics of individual species and the mechanisms they employ to regulate infection (253). It would also be interesting to decouple the impact of extracellular mechanical cues and of the microbiome from each other to directly examine the contribution of each one.
Using a similar organ-on-chip technology, a different study examined S. flexneri infection of intestinal epithelial cells under shear flow (∼0.001 dynes/cm2), in the presence of peristaltic deformations (10% at 0.15 Hz) and with 3D intestinal topography (Fig. 7C) (12). In static in vitro assays, a very high load of bacteria is required to achieve host cell infection. However, both adhesion and invasion of S. flexneri into host cells are several orders of magnitude higher in the chip than in Transwells. This is partially attributed to the fact that physiological levels of shear flow encourage bacterial adhesion to or colonization of the host cell surface and that both 3D topography and peristaltic deformations enrich bacterial invasion. The efficiency of lateral cell-to-cell and transcellular spread is also enhanced by the 3D topography and the addition of flow and/or stretch. Although this work underlines the impact of mechanical forces on epithelial cell morphology and the infection of these cells, it does not describe the molecular players that regulate these changes, which could be the focus of future studies. It is worth noting that similar to S. flexneri, when L. monocytogenes infects 2D cultures of epithelial cells that have been stretched, more bacteria adhere to host cells than to cells that have not been stretched (255). Here, cell stretching exposes E-cadherin, which the L. monocytogenes surface receptor, InlA, can more easily bind (90).
Mechanical compressions alter susceptibility of airway epithelium to infection.
The bronchial epithelium is exposed to compressive stresses during the normal respiratory cycle and experiences mechanical deformations as a result (250). Under normal conditions, bronchial epithelial cells form a physical barrier comprised of robust cell-cell adhesions that protect pulmonary airways from inhaled irritants and invading pathogens (256). Interestingly, allergens, viruses, and bacterial pathogens (e.g., Chlamydia pneumoniae, Mycoplasma pneumoniae) can induce constriction of the airways (257–259) and expose the epithelium to mechanical stresses that are up to an order of magnitude higher than they would have been otherwise (250). These stresses then induce structural, biophysical, and molecular changes in the epithelium and can lead to remodeling of the ECM and loss of barrier integrity (249). A 2016 study showed that mechanical compressive stresses increase the activity of bacterial collagenase to cause an irreversible decline in the stiffness of lung epithelial tissues, which impacts cellular behavior (260). Here, stiffness of lung epithelial tissue was measured under both uniaxial strain and cyclic mechanical loading. Interestingly, static strain of 20% is protective against ECM digestion (260), but acute mechanical forces cause deterioration in lung structure (261).
Similar to the intestinal epithelium, the lung epithelium is rich in commensal pathogens. Interestingly, pathologies like asthma or chronic obstructive pulmonary disease not only expose lung epithelia to altered mechanical cues but also lead to changes in the lung microbiome (262, 263). Clinical studies have shown that changes in both tissue mechanics and microbiome composition are correlated with the presence of bacterial infection in the lungs (264). It would be interesting to use lung-on-chip devices to study the synergistic roles of mechanical stresses and lung microbiota in protecting from or promoting bacterial infections of the lung. In particular, in vitro lung-on-chip models that take into account extracellular and intracellular mechanics might increase our understanding of how bacterial pathogens interact with lung epithelial cells (265–267). Indeed, a recent study probing how the intracellular bacterial pathogen Mycobacterium tuberculosis interacts with epithelia was conducted on a lung-on-chip device, although the contribution of extracellular mechanics to the progress of the infection was not assessed (268).
CONCLUSIONS
There are various sophisticated ways in which host cells can interact with intracellular bacterial pathogens, and these interactions can accrue either to the benefit of the pathogen (i.e., facilitate infection dissemination) or to the benefit of the host (i.e., obstruct infection spread). Bacterial pathogens can hijack host cell processes to promote their spread, and, conversely, host cells can detect infection and take action to limit it. Numerous studies on host-pathogen interactions have focused on the molecular processes governing these interactions. More recently, the importance of cellular forces and mechanics in dictating how pathogens interact with host cells has become apparent. In many cases, different intracellular bacterial species employ different bacterial effectors that target distinct host cell proteins to change a given mechanical property (e.g., to reduce host intercellular tension) to facilitate propagation through the host. For instance, reducing host intercellular tension manifests as a generalizable mechanism employed by bacteria to achieve better spread, while the molecular players involved might differ depending on the specific bacterial species or host cell type. Similarly, catch bonds strengthened by shear flow can assist many bacterial species in adhering strongly to host cells in the presence of shear flows. What all the paradigms mentioned above suggest is that it is important and pertinent to consider the role of mechanics in governing host-pathogen interactions and that it is possible that the mechanical strategies mentioned herein employed by specific bacteria or host cell types might be applicable to more systems.
In this review, we have discussed novel findings which support a critical role for cellular biomechanics in infection. Many of these studies took advantage of established techniques to measure cellular forces and quantify changes in physical characteristics during infection. However, there are various other techniques that have not been broadly used in the microbiology field but which could lend key insights into host-pathogen interactions. For instance, FRET-based force sensors could be used to characterize adhesion strength between host cell surface receptors and bacterial surface proteins. They could also be used to determine how cell-cell force transduction is locally perturbed during bacterial intercellular spread, specifically at sites of bacterial protrusions, as other tools to measure tension like MSM cannot be used to make local tension estimations. Lastly, processes involved in host-pathogen interactions can inspire the development of novel techniques in cell biomechanics. For example, the desire to learn more about the mechanics of bacterial uptake into host cells might drive the development of a novel technique to measure cellular forces and patterns during bacterial phagocytosis. Finally, the complexity of the system is further increased when one takes into account the environment in which host-pathogen interactions take place. This environment is rich in both chemical and mechanical cues. Organ-on-chip technologies have enabled recapitulation of organ-level (patho)physiology, and the performance of infection assays on such platforms has added to our understanding of how pathogenesis might occur in vivo. Further development of these platforms would enable us to make quantitative biomechanical measurements that could help identify mechanisms of action (e.g., TFM on an organ-on-chip). More recent advancements that enable fluidic coupling of multiple organs-on-chip are also expected to bring to light routes by which pathogens colonize distinct organs (269). In this way, a dialogue between different disciplines (microbiologists, bioengineers, biophysicists) and the enthusiastic adoption of interdisciplinary approaches can be very productive.
ACKNOWLEDGMENTS
We thank S. Zaver, F. Ortega, M. Footer, and members of the Theriot lab for discussions.
This work was supported by NIH grant no. R01AI036929 (J.A.T.), the HHMI (J.A.T.), the American Heart Association (award no. 18CDA34070047) (E.E.B.), and the Deutsche Forschungsgemeinschaft (DFG; German Research Foundation) under Germanýs Excellence Strategy–EXC 2124–390838134 (E.E.B.).
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
We declare no competing conflict of interests.
Conceptualization, writing–review & editing, E.E.B., P.R., J.A.T.; figure designing, C.K.P., E.E.B.; supervision, E.E.B., J.A.T.
Biographies

Effie E. Bastounis is a bioengineer and, since March 2021, a Junior Group Leader in the Interfaculty Institute of Microbiology and Infection Medicine at the University of Tübingen (Cluster of Excellence “CMFI”). Previously, she was an American Heart Association (AHA) postdoctoral fellow at Stanford University School of Medicine and, later, a Research Scientist Engineer III in Julie Theriot’s laboratory at the University of Washington and an AHA Career Development Awardee. She earned her B.S./M.Sc. in Electrical and Computer Engineering from the National Technical University of Athens, Greece, and pursued her Ph.D. in Bioengineering at the University of California, San Diego, where she made seminal contributions in the cell motility field by shedding light on the mechanics that power cell migration. Her current studies are focused on how mechanical forces influence the interactions between host cells and intracellular bacterial pathogens. She works on developing rigorous and reproducible culture-based methods to investigate host-pathogen interactions, drawing on biophysical, cell biological, and computational approaches.

Prathima Radhakrishnan is a Ph.D. student in the Stanford Biophysics Program who is studying Listeria monocytogenes cell-to-cell spread in the laboratory of Julie Theriot. Previously, she was an undergraduate at the University of Chicago, where she conducted research on tail-anchored membrane proteins in the molecular biophysics laboratory of Robert Keenan. In addition to her primary research on the pathogenesis of Listeria monocytogenes, she has been significantly involved in mentorship and science outreach to high school students from lower-income backgrounds. Furthermore, she has written articles explaining scientific discoveries to a lay audience and has also edited books written by scientists in a professional capacity.

Christopher K. Prinz is a postbaccalaureate biologist currently working as a Research Technician I for the Howard Hughes Medical Institute in Julie Theriot’s laboratory at the University of Washington. He earned his B.S. in Molecular, Cell, and Developmental Biology from the University of Washington in Seattle, WA. Previously, he worked on transcription factor subunit characterization at the Institute for Systems Biology in Jeff Ranish's proteomics lab and on wastewater treatment with novel ferrate ions at Seattle Central College in Esmaeel Naeemi's organic chemistry lab. His current research investigates the interplay between wound healing mechanisms in the epidermis of embryonic zebrafish.

Julie A. Theriot is currently a Professor in the Department of Biology at the University of Washington and an Investigator of the Howard Hughes Medical Institute. Prior to moving to the University of Washington, she was on the faculty of the Stanford University School of Medicine for 21 years, with appointments in the Department of Biochemistry and the Department of Microbiology & Immunology. She graduated from the Massachusetts Institute of Technology with two B.S. degrees, in Biology and Physics, and from the University of California, San Francisco, with a Ph.D. in Cell Biology. The work of her group focuses on molecular and cellular biophysics of cell motility, cell shape determination, and host-pathogen interactions. She is a coauthor of the textbook Physical Biology of the Cell.
Contributor Information
Effie E. Bastounis, Email: effie.bastounis@uni-tuebingen.de.
Julie A. Theriot, Email: jtheriot@uw.edu.
REFERENCES
- 1.Jansen KA, Donato DM, Balcioglu HE, Schmidt T, Danen EHJ, Koenderink GH. 2015. Biochim Biophys Acta 1853:3043–3052. 10.1016/j.bbamcr.2015.05.007. [DOI] [PubMed] [Google Scholar]
- 2.Ostrowski MA, Huang EY, Surya VN, Poplawski C, Barakat JM, Lin GL, Fuller GG, Dunn AR. 2016. Multiplexed fluid flow device to study cellular response to tunable shear stress gradients. Ann Biomed Eng 44:2261–2272. 10.1007/s10439-015-1500-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Birukova AA, Tian X, Cokic I, Beckham Y, Gardel M, Birukov KG. 2013. Endothelial barrier disruption and recovery is controlled by substrate stiffness. Microvasc Res 87:50–57. 10.1016/j.mvr.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Collins C, Osborne LD, Guilluy C, Chen Z, O'Brien ET, Reader JS, Burridge K, Superfine R, Tzima E. 2014. Haemodynamic and extracellular matrix cues regulate the mechanical phenotype and stiffness of aortic endothelial cells. Nat Commun 5:3984. 10.1038/ncomms4984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pannekoek W-J, de Rooij J, Gloerich M. 2019. Force transduction by cadherin adhesions in morphogenesis. F1000Res 8:F1000 Faculty Rev-1044. 10.12688/f1000research.18779.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martino F, Perestrelo AR, Vinarský V, Pagliari S, Forte G. 2018. Cellular mechanotransduction: from tension to function. Front Physiol 9:824. 10.3389/fphys.2018.00824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Discher D, Janmey P, Wang Y. 2005. Tissue cells feel and respond to the stiffness of their substrate. Science 310:1139–1143. 10.1126/science.1116995. [DOI] [PubMed] [Google Scholar]
- 8.Ghosh K, Ingber DE. 2007. Micromechanical control of cell and tissue development: implications for tissue engineering. Adv Drug Deliv Rev 59:1306–1318. 10.1016/j.addr.2007.08.014. [DOI] [PubMed] [Google Scholar]
- 9.Lamason RL, Bastounis E, Kafai NM, Serrano R, Del Alamo JC, Theriot JA, Welch MD. 2016. Rickettsia Sca4 reduces vinculin-mediated intercellular tension to promote spread. Cell 167:670–683.e10. 10.1016/j.cell.2016.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Faralla C, Bastounis EE, Ortega FE, Light SH, Rizzuto G, Nocadello S, Anderson WF, Robbins JR, Theriot JA, Bakardjiev AI. 2018. Listeria monocytogenes InlP interacts with afadin and facilitates basement membrane crossing. bioRxiv. 10.1101/242222. [DOI] [PMC free article] [PubMed]
- 11.Rajabian T, Gavicherla B, Heisig M, Muller-Altrock S, Goebel W, Gray-Owen SD, Ireton K. 2009. The bacterial virulence factor InlC perturbs apical cell junctions and promotes cell-to-cell spread of Listeria. Nat Cell Biol 11:1212–1218. 10.1038/ncb1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grassart A, Malardé V, Gobaa S, Sartori-Rupp A, Kerns J, Karalis K, Marteyn B, Sansonetti P, Sauvonnet N. 2019. Bioengineered human organ-on-chip reveals intestinal microenvironment and mechanical forces impacting Shigella infection. Cell Host Microbe 26:435–444.e4. 10.1016/j.chom.2019.08.007. [DOI] [PubMed] [Google Scholar]
- 13.Buchon N, Broderick NA, Kuraishi T, Lemaitre B. 2010. Drosophila EGFR pathway coordinates stem cell proliferation and gut remodeling following infection. BMC Biol 8:152. 10.1186/1741-7007-8-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mengue L, Richard F-J, Caubet Y, Rolland S, Héchard Y, Samba-Louaka A. 2017. Legionella pneumophila decreases velocity of Acanthamoeba castellanii. Exp Parasitol 183:124–127. 10.1016/j.exppara.2017.07.013. [DOI] [PubMed] [Google Scholar]
- 15.Barrila J, Crabbe A, Yang J, Franco K, Nydam SD, Forsyth RJ, Davis RR, Gangaraju S, Ott CM, Coyne CB, Bissell MJ, Nickerson CA. 2018. Modeling host-pathogen interactions in the context of the microenvironment: three-dimensional cell culture comes of age. Infect Immun 86:e00282-18. 10.1128/IAI.00282-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bein A, Shin W, Jalili-Firoozinezhad S, Park MH, Sontheimer-Phelps A, Tovaglieri A, Chalkiadaki A, Kim HJ, Ingber DE. 2018. Microfluidic organ-on-a-chip models of human intestine. Cell Mol Gastroenterol Hepatol 5:659–668. 10.1016/j.jcmgh.2017.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaarj K, Yoon JY. 2019. Methods of delivering mechanical stimuli to organ-on-a-chip. Micromachines (Basel) 10:700. 10.3390/mi10100700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bastounis E, Meili R, Álvarez-González B, Francois J, del Álamo JC, Firtel RA, Lasheras JC. 2014. Both contractile axial and lateral traction force dynamics drive amoeboid cell motility. J Cell Biol 204:1045–1061. 10.1083/jcb.201307106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Del Alamo JC, Meili R, Alvarez-Gonzalez B, Alonso-Latorre B, Bastounis E, Firtel R, Lasheras JC. 2013. Three-dimensional quantification of cellular traction forces and mechanosensing of thin substrata by Fourier traction force microscopy. PLoS One 8:e69850. 10.1371/journal.pone.0069850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bazellières E, Conte V, Elosegui-Artola A, Serra-Picamal X, Bintanel-Morcillo M, Roca-Cusachs P, Muñoz JJ, Sales-Pardo M, Guimerà R, Trepat X. 2015. Control of cell–cell forces and collective cell dynamics by the intercellular adhesome. Nat Cell Biol 17:409–420. 10.1038/ncb3135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chang AC, Mekhdjian AH, Morimatsu M, Denisin AK, Pruitt BL, Dunn AR. 2016. Single molecule force measurements in living cells reveal a minimally tensioned integrin state. ACS Nano 10:10745–10752. 10.1021/acsnano.6b03314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Srinivasan B, Kolli AR, Esch MB, Abaci HE, Shuler ML, Hickman JJ. 2015. TEER measurement techniques for in vitro barrier model systems. J Lab Autom 20:107–126. 10.1177/2211068214561025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Luo Q, Kuang D, Zhang B, Song G. 2016. Cell stiffness determined by atomic force microscopy and its correlation with cell motility. Biochim Biophys Acta 1860:1953–1960. 10.1016/j.bbagen.2016.06.010. [DOI] [PubMed] [Google Scholar]
- 24.Haase K, Pelling AE. 2015. Investigating cell mechanics with atomic force microscopy. J R Soc Interface 12:20140970. 10.1098/rsif.2014.0970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hollenbeck EC, Antonoplis A, Chai C, Thongsomboon W, Fuller GG, Cegelski L. 2018. Phosphoethanolamine cellulose enhances curli-mediated adhesion of uropathogenic Escherichia coli to bladder epithelial cells. Proc Natl Acad Sci USA 115:10106–10111. 10.1073/pnas.1801564115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Polacheck WJ, Chen CS. 2016. Measuring cell-generated forces: a guide to the available tools. Nat Methods 13:415–423. 10.1038/nmeth.3834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Addae-Mensah KA, Wikswo JP. 2008. Measurement techniques for cellular biomechanics in vitro. Exp Biol Med (Maywood) 233:792–809. 10.3181/0710-MR-278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gómez-González M, Latorre E, Arroyo M, Trepat X. 2020. Measuring mechanical stress in living tissues. Nat Rev Phys 2:300–317. 10.1038/s42254-020-0184-6. [DOI] [Google Scholar]
- 29.Lamason RL, Welch MD. 2017. Actin-based motility and cell-to-cell spread of bacterial pathogens. Curr Opin Microbiol 35:48–57. 10.1016/j.mib.2016.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Colonne PM, Winchell CG, Voth DE. 2016. Hijacking host cell highways: manipulation of the host actin cytoskeleton by obligate intracellular bacterial pathogens. Front Cell Infect Microbiol 6:107–107. 10.3389/fcimb.2016.00107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zenewicz LA, Shen H. 2007. Innate and adaptive immune responses to Listeria monocytogenes: a short overview. Microbes Infect 9:1208–1215. 10.1016/j.micinf.2007.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Talman AM, Chong R, Chia J, Svitkina T, Agaisse H. 2014. Actin network disassembly powers dissemination of Listeria monocytogenes. J Cell Sci 127:240–249. 10.1242/jcs.140038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Duncan JK, Wiscovitch AL, Goldberg MB, Russo BC. 2019. Shigella flexneri disruption of host cell-cell tension promotes intercellular spread. bioRxiv. 10.1101/790147. [DOI] [PMC free article] [PubMed]
- 34.Otani T, Ichii T, Aono S, Takeichi M. 2006. Cdc42 GEF Tuba regulates the junctional configuration of simple epithelial cells. J Cell Biol 175:135–146. 10.1083/jcb.200605012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Johnson DI. 1999. Cdc42: an essential Rho-type GTPase controlling eukaryotic cell polarity. Microbiol Mol Biol Rev 63:54–105. 10.1128/MMBR.63.1.54-105.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Luan Q, Zelter A, MacCoss MJ, Davis TN, Nolen BJ. 2018. Identification of Wiskott-Aldrich syndrome protein (WASP) binding sites on the branched actin filament nucleator Arp2/3 complex. Proc Natl Acad Sci USA 115:E1409–E1418. 10.1073/pnas.1716622115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu SK, Gomez GA, Michael M, Verma S, Cox HL, Lefevre JG, Parton RG, Hamilton NA, Neufeld Z, Yap AS. 2014. Cortical F-actin stabilization generates apical-lateral patterns of junctional contractility that integrate cells into epithelia. Nat Cell Biol 16:167–178. 10.1038/ncb2900. [DOI] [PubMed] [Google Scholar]
- 38.Kovacs EM, Verma S, Ali RG, Ratheesh A, Hamilton NA, Akhmanova A, Yap AS. 2011. N-WASP regulates the epithelial junctional actin cytoskeleton through a non-canonical post-nucleation pathway. Nat Cell Biol 13:934–943. 10.1038/ncb2290. [DOI] [PubMed] [Google Scholar]
- 39.Kovacs EM, Makar RS, Gertler FB. 2006. Tuba stimulates intracellular N-WASP-dependent actin assembly. J Cell Sci 119:2715–2726. 10.1242/jcs.03005. [DOI] [PubMed] [Google Scholar]
- 40.Rigano LA, Dowd GC, Wang Y, Ireton K. 2014. Listeria monocytogenes antagonizes the human GTPase Cdc42 to promote bacterial spread. Cell Microbiol 16:1068–1079. 10.1111/cmi.12260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Padrick SB, Rosen MK. 2010. Physical mechanisms of signal integration by WASP family proteins. Annu Rev Biochem 79:707–735. 10.1146/annurev.biochem.77.060407.135452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Simunovic M, Voth GA. 2015. Membrane tension controls the assembly of curvature-generating proteins. Nat Commun 6:7219. 10.1038/ncomms8219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McMahon HT, Boucrot E. 2015. Membrane curvature at a glance. J Cell Sci 128:1065–1070. 10.1242/jcs.114454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Georgiou M, Marinari E, Burden J, Baum B. 2008. Cdc42, Par6, and aPKC regulate Arp2/3-mediated endocytosis to control local adherens junction stability. Curr Biol 18:1631–1638. 10.1016/j.cub.2008.09.029. [DOI] [PubMed] [Google Scholar]
- 45.Dowd GC, Mortuza R, Bhalla M, Van Ngo H, Li Y, Rigano LA, Ireton K. 2020. Listeria monocytogenes exploits host exocytosis to promote cell-to-cell spread. Proc Natl Acad Sci USA 117:3789–3796. 10.1073/pnas.1916676117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bastounis EE, Radhakrishnan P, Prinz CK, Theriot JA. 2021. Volume measurement and biophysical characterization of mounds in epithelial monolayers after intracellular bacterial infection. STAR Protoc 2:100551. 10.1016/j.xpro.2021.100551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sim JY, Moeller J, Hart KC, Ramallo D, Vogel V, Dunn AR, Nelson WJ, Pruitt BL. 2015. Spatial distribution of cell-cell and cell-ECM adhesions regulates force balance while maintaining E-cadherin molecular tension in cell pairs. Mol Biol Cell 26:2456–2465. 10.1091/mbc.E14-12-1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kannan N, Tang VW. 2018. Myosin-1c promotes E-cadherin tension and force-dependent recruitment of α-actinin to the epithelial cell junction. J Cell Sci 131:jcs211334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huveneers S, de Rooij J. 2013. Mechanosensitive systems at the cadherin–F-actin interface. J Cell Sci 126:403–413. 10.1242/jcs.109447. [DOI] [PubMed] [Google Scholar]
- 50.Bourdet-Sicard R, Rüdiger M, Jockusch BM, Gounon P, Sansonetti PJ, Nhieu GT. 1999. Binding of the Shigella protein IpaA to vinculin induces F-actin depolymerization. EMBO J 18:5853–5862. 10.1093/emboj/18.21.5853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Valencia-Gallardo C, Bou-Nader C, Aguilar-Salvador DI, Carayol N, Quenech'Du N, Pecqueur L, Park H, Fontecave M, Izard T, Tran Van Nhieu G. 2019. Shigella IpaA binding to talin stimulates filopodial capture and cell adhesion. Cell Rep 26:921–932.e6. 10.1016/j.celrep.2018.12.091. [DOI] [PubMed] [Google Scholar]
- 52.Bishai EA, Sidhu GS, Li W, Dhillon J, Bohil AB, Cheney RE, Hartwig JH, Southwick FS. 2013. Myosin-X facilitates Shigella-induced membrane protrusions and cell-to-cell spread. Cell Microbiol 15:353–367. 10.1111/cmi.12051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fattouh R, Kwon H, Czuczman MA, Copeland JW, Pelletier L, Quinlan ME, Muise AM, Higgins DE, Brumell JH. 2015. The diaphanous-related formins promote protrusion formation and cell-to-cell spread of Listeria monocytogenes. J Infect Dis 211:1185–1195. 10.1093/infdis/jiu546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pust S, Morrison H, Wehland J, Sechi AS, Herrlich P. 2005. Listeria monocytogenes exploits ERM protein functions to efficiently spread from cell to cell. EMBO J 24:1287–1300. 10.1038/sj.emboj.7600595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dhanda AS, Lulic KT, Vogl AW, Mc Gee MM, Chiu RH, Guttman JA. 2018. Listeria membrane protrusion collapse: requirement of cyclophilin A for Listeria cell-to-cell spreading. J Infect Dis 219:145–153. 10.1093/infdis/jiy255. [DOI] [PubMed] [Google Scholar]
- 56.Daou P, Hasan S, Breitsprecher D, Baudelet E, Camoin L, Audebert S, Goode BL, Badache A. 2014. Essential and nonredundant roles for Diaphanous formins in cortical microtubule capture and directed cell migration. Mol Biol Cell 25:658–668. 10.1091/mbc.E13-08-0482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Berg JS, Cheney RE. 2002. Myosin-X is an unconventional myosin that undergoes intrafilopodial motility. Nat Cell Biol 4:246–250. 10.1038/ncb762. [DOI] [PubMed] [Google Scholar]
- 58.Calhoun CC, Lu YC, Song J, Chiu R. 2009. Knockdown endogenous CypA with siRNA in U2OS cells results in disruption of F-actin structure and alters tumor phenotype. Mol Cell Biochem 320:35–43. 10.1007/s11010-008-9896-0. [DOI] [PubMed] [Google Scholar]
- 59.Fehon RG, McClatchey AI, Bretscher A. 2010. Organizing the cell cortex: the role of ERM proteins. Nat Rev Mol Cell Biol 11:276–287. 10.1038/nrm2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Welch MD, Way M. 2013. Arp2/3-mediated actin-based motility: a tail of pathogen abuse. Cell Host Microbe 14:242–255. 10.1016/j.chom.2013.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sechi AS, Wehland J, Small JV. 1997. The isolated comet tail pseudopodium of Listeria monocytogenes: a tail of two actin filament populations, long and axial and short and random. J Cell Biol 137:155–167. 10.1083/jcb.137.1.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Heindl JE, Saran I, Yi C-r, Lesser CF, Goldberg MB. 2010. Requirement for formin-induced actin polymerization during spread of Shigella flexneri. Infect Immun 78:193–203. 10.1128/IAI.00252-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mogilner A, Oster GF. 1996. Cell motility driven by actin polymerization. Biophys J 71:3030–3045. 10.1016/S0006-3495(96)79496-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mogilner A, Rubinstein B. 2005. The physics of filopodial protrusion. Biophys J 89:782–795. 10.1529/biophysj.104.056515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chua MD, Walker BD, Jin J-P, Guttman JA. 2018. Calponins are recruited to actin-rich structures generated by pathogenic Escherichia coli, Listeria, and Salmonella. Anat Rec (Hoboken) 301:2103–2111. 10.1002/ar.23956. [DOI] [PubMed] [Google Scholar]
- 66.Köhler H, Sakaguchi T, Hurley BP, Kase BA, Kase BJ, Reinecker H-C, McCormick BA. 2007. Salmonella enterica serovar Typhimurium regulates intercellular junction proteins and facilitates transepithelial neutrophil and bacterial passage. Am J Physiol Gastrointest Liver Physiol 293:G178–G187. 10.1152/ajpgi.00535.2006. [DOI] [PubMed] [Google Scholar]
- 67.Maldonado-Contreras A, Birtley JR, Boll E, Zhao Y, Mumy KL, Toscano J, Ayehunie S, Reinecker H-C, Stern LJ, McCormick BA. 2017. Shigella depends on SepA to destabilize the intestinal epithelial integrity via cofilin activation. Gut Microbes 8:544–560. 10.1080/19490976.2017.1339006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tahoun A, Mahajan S, Paxton E, Malterer G, Donaldson DS, Wang D, Tan A, Gillespie TL, O'Shea M, Roe AJ, Shaw DJ, Gally DL, Lengeling A, Mabbott NA, Haas J, Mahajan A. 2012. Salmonella transforms follicle-associated epithelial cells into M cells to promote intestinal invasion. Cell Host Microbe 12:645–656. 10.1016/j.chom.2012.10.009. [DOI] [PubMed] [Google Scholar]
- 69.Drolia R, Tenguria S, Durkes AC, Turner JR, Bhunia AK. 2018. Listeria adhesion protein induces intestinal epithelial barrier dysfunction for bacterial translocation. Cell Host Microbe 23:470–484.e7. 10.1016/j.chom.2018.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Agbor TA, Demma ZC, Mumy KL, Bien JD, McCormick BA. 2011. The ERM protein, Ezrin, regulates neutrophil transmigration by modulating the apical localization of MRP2 in response to the SipA effector protein during Salmonella Typhimurium infection. Cell Microbiol 13:2007–2021. 10.1111/j.1462-5822.2011.01693.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nikitas G, Deschamps C, Disson O, Niault T, Cossart P, Lecuit M. 2011. Transcytosis of Listeria monocytogenes across the intestinal barrier upon specific targeting of goblet cell accessible E-cadherin. J Exp Med 208:2263–2277. 10.1084/jem.20110560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Woods ME, Olano JP. 2008. Host defenses to Rickettsia rickettsii infection contribute to increased microvascular permeability in human cerebral endothelial cells. J Clin Immunol 28:174–185. 10.1007/s10875-007-9140-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zeldovich VB, Clausen CH, Bradford E, Fletcher DA, Maltepe E, Robbins JR, Bakardjiev AI. 2013. Placental syncytium forms a biophysical barrier against pathogen invasion. PLoS Pathog 9:e1003821. 10.1371/journal.ppat.1003821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Faralla C, Rizzuto GA, Lowe DE, Kim B, Cooke C, Shiow LR, Bakardjiev AI. 2016. InlP, a new virulence factor with strong placental tropism. Infect Immun 84:3584–3596. 10.1128/IAI.00625-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Burkholder KM, Bhunia AK. 2010. Listeria monocytogenes uses Listeria adhesion protein (LAP) to promote bacterial transepithelial translocation and induces expression of LAP receptor Hsp60. Infect Immun 78:5062–5073. 10.1128/IAI.00516-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bertelsen LS, Paesold G, Marcus SL, Finlay BB, Eckmann L, Barrett KE. 2004. Modulation of chloride secretory responses and barrier function of intestinal epithelial cells by the Salmonella effector protein SigD. Am J Physiol Cell Physiol 287:C939–C948. 10.1152/ajpcell.00413.2003. [DOI] [PubMed] [Google Scholar]
- 77.Boyle EC, Brown NF, Finlay BB. 2006. Salmonella enterica serovar Typhimurium effectors SopB, SopE, SopE2 and SipA disrupt tight junction structure and function. Cell Microbiol 8:1946–1957. 10.1111/j.1462-5822.2006.00762.x. [DOI] [PubMed] [Google Scholar]
- 78.Gong B, Ma L, Liu Y, Gong Q, Shelite T, Bouyer D, Boor PJ, Lee YS, Oberhauser A. 2012. Rickettsiae induce microvascular hyperpermeability via phosphorylation of VE-cadherins: evidence from atomic force microscopy and biochemical studies. PLoS Negl Trop Dis 6:e1699. 10.1371/journal.pntd.0001699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cai X, Yu N, Ma J, Li W-Y, Xu M, Li E, Zhang M, Wang W, Chen Y, Kang J. 2019. Altered pulmonary capillary permeability in immunosuppressed guinea pigs infected with Legionella pneumophila serogroup 1. Exp Ther Med 18:4368–4378. 10.3892/etm.2019.8102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ohsawa S, Vaughen J, Igaki T. 2018. Cell extrusion: a stress-responsive force for good or evil in epithelial homeostasis. Dev Cell 44:284–296. 10.1016/j.devcel.2018.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gudipaty SA, Rosenblatt J. 2017. Epithelial cell extrusion: pathways and pathologies. Semin Cell Dev Biol 67:132–140. 10.1016/j.semcdb.2016.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kim M, Ashida H, Ogawa M, Yoshikawa Y, Mimuro H, Sasakawa C. 2010. Bacterial interactions with the host epithelium. Cell Host Microbe 8:20–35. 10.1016/j.chom.2010.06.006. [DOI] [PubMed] [Google Scholar]
- 83.Rosenblatt J, Raff MC, Cramer LP. 2001. An epithelial cell destined for apoptosis signals its neighbors to extrude it by an actin- and myosin-dependent mechanism. Curr Biol 11:1847–1857. 10.1016/s0960-9822(01)00587-5. [DOI] [PubMed] [Google Scholar]
- 84.Saw TB, Doostmohammadi A, Nier V, Kocgozlu L, Thampi S, Toyama Y, Marcq P, Lim CT, Yeomans JM, Ladoux B. 2017. Topological defects in epithelia govern cell death and extrusion. Nature 544:212–216. 10.1038/nature21718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kocgozlu L, Saw TB, Le AP, Yow I, Shagirov M, Wong E, Mege RM, Lim CT, Toyama Y, Ladoux B. 2016. Epithelial cell packing induces distinct modes of cell extrusions. Curr Biol 26:2942–2950. 10.1016/j.cub.2016.08.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Knodler LA, Vallance BA, Celli J, Winfree S, Hansen B, Montero M, Steele-Mortimer O. 2010. Dissemination of invasive Salmonella via bacterial-induced extrusion of mucosal epithelia. Proc Natl Acad Sci USA 107:17733–17738. 10.1073/pnas.1006098107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Pentecost M, Otto G, Theriot JA, Amieva MR. 2006. Listeria monocytogenes invades the epithelial junctions at sites of cell extrusion. PLoS Pathog 2:e3. 10.1371/journal.ppat.0020003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Marteyn B, Gazi A, Sansonetti P. 2012. Shigella: a model of virulence regulation in vivo. Gut Microbes 3:104–120. 10.4161/gmic.19325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kuipers D, Mehonic A, Kajita M, Peter L, Fujita Y, Duke T, Charras G, Gale JE. 2014. Epithelial repair is a two-stage process driven first by dying cells and then by their neighbours. J Cell Sci 127:1229–1241. 10.1242/jcs.138289. [DOI] [PubMed] [Google Scholar]
- 90.Bonazzi M, Lecuit M, Cossart P. 2009. Listeria monocytogenes internalin and E-cadherin: from bench to bedside. Cold Spring Harb Perspect Biol 1:a003087. 10.1101/cshperspect.a003087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hewes SA, Wilson RL, Estes MK, Shroyer NF, Blutt SE, Grande-Allen KJ. 2020. In vitro models of the small intestine: engineering challenges and engineering solutions. Tissue Eng Part B Rev 26:313–326. 10.1089/ten.TEB.2019.0334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ortega FE, Koslover EF, Theriot JA. 2019. Listeria monocytogenes cell-to-cell spread in epithelia is heterogeneous and dominated by rare pioneer bacteria. Elife 8:e40032. 10.7554/eLife.40032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kim M, Ogawa M, Fujita Y, Yoshikawa Y, Nagai T, Koyama T, Nagai S, Lange A, Fassler R, Sasakawa C. 2009. Bacteria hijack integrin-linked kinase to stabilize focal adhesions and block cell detachment. Nature 459:578–582. 10.1038/nature07952. [DOI] [PubMed] [Google Scholar]
- 94.Nagano M, Hoshino D, Koshikawa N, Akizawa T, Seiki M. 2012. Turnover of focal adhesions and cancer cell migration. Int J Cell Biol 2012:310616. 10.1155/2012/310616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Muenzner P, Hauck CR. 2020. Neisseria gonorrhoeae blocks epithelial exfoliation by nitric-oxide-mediated metabolic cross talk to promote colonization in mice. Cell Host Microbe 27:793–808.e5. 10.1016/j.chom.2020.03.010. [DOI] [PubMed] [Google Scholar]
- 96.Arencibia I, Suárez NC, Wolf-Watz H, Sundqvist KG. 1997. Yersinia invasin, a bacterial beta1-integrin ligand, is a potent inducer of lymphocyte motility and migration to collagen type IV and fibronectin. J Immunol 159:1853–1859. [PubMed] [Google Scholar]
- 97.Sun Y, Reid B, Ferreira F, Luxardi G, Ma L, Lokken KL, Zhu K, Xu G, Sun Y, Ryzhuk V, Guo BP, Lebrilla CB, Maverakis E, Mogilner A, Zhao M. 2019. Infection-generated electric field in gut epithelium drives bidirectional migration of macrophages. PLoS Biol 17:e3000044. 10.1371/journal.pbio.3000044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lamalice L, Le Boeuf F, Huot J. 2007. Endothelial cell migration during angiogenesis. Circ Res 100:782–794. 10.1161/01.RES.0000259593.07661.1e. [DOI] [PubMed] [Google Scholar]
- 99.Kempf VAJ, Schaller M, Behrendt S, Volkmann B, Aepfelbacher M, Cakman I, Autenrieth IB. 2000. Interaction of Bartonella henselae with endothelial cells results in rapid bacterial rRNA synthesis and replication. Cell Microbiol 2:431–441. 10.1046/j.1462-5822.2000.00072.x. [DOI] [PubMed] [Google Scholar]
- 100.Wang B, Zhang L, Liu J, Ma L, Wang H, Zheng N, Chen X, Shen B, Xu Z, Zhang L. 2017. Chlamydia pneumoniae infection promotes vascular endothelial cell angiogenesis through an IQGAP1-related signaling pathway. Int J Med Microbiol 307:276–286. 10.1016/j.ijmm.2017.03.001. [DOI] [PubMed] [Google Scholar]
- 101.Zhang J, Wang H, Zhang L, Zhang T, Wang B, Li X, Wei J, Zhang L. 2014. Chlamydia pneumoniae infection induces vascular smooth muscle cell migration via Rac1 activation. J Med Microbiol 63:155–161. 10.1099/jmm.0.065359-0. [DOI] [PubMed] [Google Scholar]
- 102.Ma L, Zhang L, Wang B, Wei J, Liu J, Zhang L. 2015. Berberine inhibits Chlamydia pneumoniae infection-induced vascular smooth muscle cell migration through downregulating MMP3 and MMP9 via PI3K. Eur J Pharmacol 755:102–109. 10.1016/j.ejphar.2015.02.039. [DOI] [PubMed] [Google Scholar]
- 103.Probst P, Hermann E, Fleischer B. 1994. Role of bacteria-specific T cells in the immunopathogenesis of reactive arthritis. Trends Microbiol 2:329–332. 10.1016/0966-842x(94)90450-2. [DOI] [PubMed] [Google Scholar]
- 104.Woolf E, Grigorova I, Sagiv A, Grabovsky V, Feigelson SW, Shulman Z, Hartmann T, Sixt M, Cyster JG, Alon R. 2007. Lymph node chemokines promote sustained T lymphocyte motility without triggering stable integrin adhesiveness in the absence of shear forces. Nat Immunol 8:1076–1085. 10.1038/ni1499. [DOI] [PubMed] [Google Scholar]
- 105.Hudson KJ, Bliska JB, Bouton AH. 2005. Distinct mechanisms of integrin binding by Yersinia pseudotuberculosis adhesins determine the phagocytic response of host macrophages. Cell Microbiol 7:1474–1489. 10.1111/j.1462-5822.2005.00571.x. [DOI] [PubMed] [Google Scholar]
- 106.Luscinskas FW, Lim Y-C, Lichtman AH. 2001. Wall shear stress: the missing step for T cell transmigration? Nat Immunol 2:478–480. 10.1038/88663. [DOI] [PubMed] [Google Scholar]
- 107.Joannidis M, Truebsbach S, Bijuklic K, Schratzberger P, Dunzedorfer S, Wintersteiger S, Lhotta K, Mayer G, Wiedermann CJ. 2004. Neutrophil transmigration in renal proximal tubular LLC-PK1 cells. Cell Physiol Biochem 14:101–112. 10.1159/000076931. [DOI] [PubMed] [Google Scholar]
- 108.Fattinger SA, Böck D, Di Martino ML, Deuring S, Samperio Ventayol P, Ek V, Furter M, Kreibich S, Bosia F, Müller-Hauser AA, Nguyen BD, Rohde M, Pilhofer M, Hardt W-D, Sellin ME. 2020. Salmonella Typhimurium discreet-invasion of the murine gut absorptive epithelium. PLoS Pathog 16:e1008503. 10.1371/journal.ppat.1008503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Tai G, Reid B, Cao L, Zhao M. 2009. Electrotaxis and wound healing: experimental methods to study electric fields as a directional signal for cell migration. Methods Mol Biol 571:77–97. 10.1007/978-1-60761-198-1_5. [DOI] [PubMed] [Google Scholar]
- 110.McLaughlin S, Poo MM. 1981. The role of electro-osmosis in the electric-field-induced movement of charged macromolecules on the surfaces of cells. Biophys J 34:85–93. 10.1016/S0006-3495(81)84838-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Nakata I, Yamashiro K, Kawaguchi T, Nakanishi H, Akagi-Kurashige Y, Miyake M, Tsujikawa A, Yamada R, Matsuda F, Yoshimura N, Nagahama Study G, Nagahama Study Group. 2015. Calcium, ARMS2 genotype and Chlamydia pneumoniae infection in early age-related macular degeneration: a multivariate analysis from the Nagahama Study. Sci Rep 5:9345. 10.1038/srep09345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ambati J, Atkinson JP, Gelfand BD. 2013. Immunology of age-related macular degeneration. Nat Rev Immunol 13:438–451. 10.1038/nri3459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Fong IW, Chiu B, Viira E, Jang D, Mahony JB. 1999. De novo induction of atherosclerosis by Chlamydia pneumoniae in a rabbit model. Infect Immun 67:6048–6055. 10.1128/IAI.67.11.6048-6055.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Camaré C, Pucelle M, Nègre-Salvayre A, Salvayre R. 2017. Angiogenesis in the atherosclerotic plaque. Redox Biol 12:18–34. 10.1016/j.redox.2017.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Tomasevic N, Jia Z, Russell A, Fujii T, Hartman JJ, Clancy S, Wang M, Beraud C, Wood KW, Sakowicz R. 2007. Differential regulation of WASP and N-WASP by Cdc42, Rac1, Nck, and PI(4,5)P2. Biochemistry 46:3494–3502. 10.1021/bi062152y. [DOI] [PubMed] [Google Scholar]
- 116.Zhang LJ, Zhang LJ, Quan W, Wang BB, Shen BL, Zhang TT, Kang Y. 2011. Berberine inhibits HEp-2 cell invasion induced by Chlamydophila pneumoniae infection. J Microbiol 49:834–840. 10.1007/s12275-011-1051-z. [DOI] [PubMed] [Google Scholar]
- 117.Lachowski D, Cortes E, Rice A, Pinato D, Rombouts K, del Rio Hernandez A. 2019. Matrix stiffness modulates the activity of MMP-9 and TIMP-1 in hepatic stellate cells to perpetuate fibrosis. Sci Rep 9:7299. 10.1038/s41598-019-43759-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Gialeli C, Theocharis AD, Karamanos NK. 2011. Roles of matrix metalloproteinases in cancer progression and their pharmacological targeting. FEBS J 278:16–27. 10.1111/j.1742-4658.2010.07919.x. [DOI] [PubMed] [Google Scholar]
- 119.Garcia FU, Wojta J, Broadley KN, Davidson JM, Hoover RL. 1990. Bartonella bacilliformis stimulates endothelial cells in vitro and is angiogenic in vivo. Am J Pathol 136:1125–1135. [PMC free article] [PubMed] [Google Scholar]
- 120.Cerimele F, Brown LF, Bravo F, Ihler GM, Kouadio P, Arbiser JL. 2003. Infectious angiogenesis: Bartonella bacilliformis infection results in endothelial production of angiopoetin-2 and epidermal production of vascular endothelial growth factor. Am J Pathol 163:1321–1327. 10.1016/S0002-9440(10)63491-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hicks LD, Minnick MF. 2020. Human vascular endothelial cells express epithelial growth factor in response to infection by Bartonella bacilliformis. PLoS Negl Trop Dis 14:e0008236. 10.1371/journal.pntd.0008236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Candelle D, Carrillo LM, Arciniegas E. 2015. The endothelial cell adhesion mediated by integrins, the recruitment and activation of c-Src, EGFR, ErbB2 and involvement of endocan, influence crucial stages of endothelial-mesenchymal transition. Rev Latinoam Hipertension 10:42–45. [Google Scholar]
- 123.Riess T, Andersson SGE, Lupas A, Schaller M, Schäfer A, Kyme P, Martin J, Wälzlein J-H, Ehehalt U, Lindroos H, Schirle M, Nordheim A, Autenrieth IB, Kempf VAJ. 2004. Bartonella adhesin a mediates a proangiogenic host cell response. J Exp Med 200:1267–1278. 10.1084/jem.20040500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Pennock ND, White JT, Cross EW, Cheney EE, Tamburini BA, Kedl RM. 2013. T cell responses: naive to memory and everything in between. Adv Physiol Educ 37:273–283. 10.1152/advan.00066.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Salgado-Pabón W, Celli S, Arena ET, Nothelfer K, Roux P, Sellge G, Frigimelica E, Bousso P, Sansonetti PJ, Phalipon A. 2013. Shigella impairs T lymphocyte dynamics in vivo. Proc Natl Acad Sci USA 110:4458–4463. 10.1073/pnas.1300981110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Konradt C, Frigimelica E, Nothelfer K, Puhar A, Salgado-Pabon W, di Bartolo V, Scott-Algara D, Rodrigues CD, Sansonetti PJ, Phalipon A. 2011. The Shigella flexneri type three secretion system effector IpgD inhibits T cell migration by manipulating host phosphoinositide metabolism. Cell Host Microbe 9:263–272. 10.1016/j.chom.2011.03.010. [DOI] [PubMed] [Google Scholar]
- 127.Samassa F, Ferrari ML, Husson J, Mikhailova A, Porat Z, Sidaner F, Brunner K, Teo T-H, Frigimelica E, Tinevez J-Y, Sansonetti PJ, Thoulouze M-I, Phalipon A. 2020. Shigella impairs human T lymphocyte responsiveness by hijacking actin cytoskeleton dynamics and T cell receptor vesicular trafficking. Cell Microbiol 22:e13166. 10.1111/cmi.13166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Simon S, Wagner MA, Rothmeier E, Müller-Taubenberger A, Hilbi H. 2014. Icm/Dot-dependent inhibition of phagocyte migration by Legionella is antagonized by a translocated Ran GTPase activator. Cell Microbiol 16:977–992. 10.1111/cmi.12258. [DOI] [PubMed] [Google Scholar]
- 129.Simon S, Schell U, Heuer N, Hager D, Albers MF, Matthias J, Fahrnbauer F, Trauner D, Eichinger L, Hedberg C, Hilbi H. 2015. Inter-kingdom signaling by the Legionella quorum sensing molecule LAI-1 modulates cell migration through an IQGAP1-Cdc42-ARHGEF9-dependent pathway. PLoS Pathog 11:e1005307. 10.1371/journal.ppat.1005307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Brink T, Leiss V, Siegert P, Jehle D, Ebner JK, Schwan C, Shymanets A, Wiese S, Nürnberg B, Hensel M, Aktories K, Orth JHC. 2018. Salmonella Typhimurium effector SseI inhibits chemotaxis and increases host cell survival by deamidation of heterotrimeric Gi proteins. PLoS Pathog 14:e1007248. 10.1371/journal.ppat.1007248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.McLaughlin LM, Govoni GR, Gerke C, Gopinath S, Peng K, Laidlaw G, Chien Y-H, Jeong H-W, Li Z, Brown MD, Sacks DB, Monack D. 2009. The Salmonella SPI2 effector SseI mediates long-term systemic infection by modulating host cell migration. PLoS Pathog 5:e1000671. 10.1371/journal.ppat.1000671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Raqib R, Qadri F, SarkEr P, Mia SMS, Sansonnetti PJ, Albert MJ, Andersson J. 2002. Delayed and reduced adaptive humoral immune responses in children with shigellosis compared with in adults. Scand J Immunol 55:414–423. 10.1046/j.1365-3083.2002.01079.x. [DOI] [PubMed] [Google Scholar]
- 133.Thelen M, Stein JV. 2008. How chemokines invite leukocytes to dance. Nat Immunol 9:953–959. 10.1038/ni.f.207. [DOI] [PubMed] [Google Scholar]
- 134.Thauland TJ, Hu KH, Bruce MA, Butte MJ. 2017. Cytoskeletal adaptivity regulates T cell receptor signaling. Sci Signal 10:eaah3737. 10.1126/scisignal.aah3737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Faure S, Salazar-Fontana LI, Semichon M, Tybulewicz VLJ, Bismuth G, Trautmann A, Germain RN, Delon J. 2004. ERM proteins regulate cytoskeleton relaxation promoting T cell–APC conjugation. Nat Immunol 5:272–279. 10.1038/ni1039. [DOI] [PubMed] [Google Scholar]
- 136.Velan B, Bar-Haim E, Zauberman A, Mamroud E, Shafferman A, Cohen S. 2006. Discordance in the effects of Yersinia pestis on the dendritic cell functions manifested by induction of maturation and paralysis of migration. Infect Immun 74:6365–6376. 10.1128/IAI.00974-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Spiering D, Hodgson L. 2011. Dynamics of the Rho-family small GTPases in actin regulation and motility. Cell Adh Migr 5:170–180. 10.4161/cam.5.2.14403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Aepfelbacher M, Trasak C, Wilharm G, Wiedemann A, Trülzsch K, Krauss K, Gierschik P, Heesemann J. 2003. Characterization of YopT effects on rho GTPases in Yersinia enterocolitica-infected cells. J Biol Chem 278:33217–33223. 10.1074/jbc.M303349200. [DOI] [PubMed] [Google Scholar]
- 139.Bastounis EE, Alcalde FS, Radhakrishnan P, Engström P, Gómez Benito MJ, Oswald MS, Yeh Y-T, Smith J, Welch M, García-Aznar JM, Theriot JA. 2021. Mechanical competition triggered by innate immune signaling drives the collective extrusion of bacterially-infected epithelial cells. Dev Cell 56:443–460. 10.1016/j.devcel.2021.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Man SM, Ekpenyong A, Tourlomousis P, Achouri S, Cammarota E, Hughes K, Rizzo A, Ng G, Wright JA, Cicuta P, Guck JR, Bryant CE. 2014. Actin polymerization as a key innate immune effector mechanism to control Salmonella infection. Proc Natl Acad Sci USA 111:17588–17593. 10.1073/pnas.1419925111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Ostrowski PP, Grinstein S, Freeman SA. 2016. Diffusion barriers, mechanical forces, and the biophysics of phagocytosis. Dev Cell 38:135–146. 10.1016/j.devcel.2016.06.023. [DOI] [PubMed] [Google Scholar]
- 142.Vorselen D, Labitigan RLD, Theriot JA. 2020. A mechanical perspective on phagocytic cup formation. Curr Opin Cell Biol 66:112–122. 10.1016/j.ceb.2020.05.011. [DOI] [PubMed] [Google Scholar]
- 143.Möller J, Lühmann T, Chabria M, Hall H, Vogel V. 2013. Macrophages lift off surface-bound bacteria using a filopodium-lamellipodium hook-and-shovel mechanism. Sci Rep 3:2884–2884. 10.1038/srep02884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Auchter E, Marquez J, Stevens G, Silva R, McCulloch Q, Guengerich Q, Blair A, Litchfield S, Li N, Sheehan C, Chamberlin R, Yarbro SL, Dervishi E. 2018. Ultra-thin and strong formvar-based membranes with controlled porosity for micro- and nano-scale systems. Nanotechnology 29:215712. 10.1088/1361-6528/aab4a4. [DOI] [PubMed] [Google Scholar]
- 145.Labernadie A, Bouissou A, Delobelle P, Balor S, Voituriez R, Proag A, Fourquaux I, Thibault C, Vieu C, Poincloux R, Charrière GM, Maridonneau-Parini I. 2014. Protrusion force microscopy reveals oscillatory force generation and mechanosensing activity of human macrophage podosomes. Nat Commun 5:5343. 10.1038/ncomms6343. [DOI] [PubMed] [Google Scholar]
- 146.Vorselen D, Wang Y, de Jesus MM, Shah PK, Footer MJ, Huse M, Cai W, Theriot JA. 2020. Microparticle traction force microscopy reveals subcellular force exertion patterns in immune cell-target interactions. Nat Commun 11:20. 10.1038/s41467-019-13804-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Storek KM, Monack DM. 2015. Bacterial recognition pathways that lead to inflammasome activation. Immunol Rev 265:112–129. 10.1111/imr.12289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Gao W, Yang J, Liu W, Wang Y, Shao F. 2016. Site-specific phosphorylation and microtubule dynamics control Pyrin inflammasome activation. Proc Natl Acad Sci USA 113:E4857–E4866. 10.1073/pnas.1601700113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Schnappauf O, Chae JJ, Kastner DL, Aksentijevich I. 2019. The pyrin inflammasome in health and disease. Front Immunol 10:1745. 10.3389/fimmu.2019.01745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Kamanova J, Kofronova O, Masin J, Genth H, Vojtova J, Linhartova I, Benada O, Just I, Sebo P. 2008. Adenylate cyclase toxin subverts phagocyte function by RhoA inhibition and unproductive ruffling. J Immunol 181:5587–5597. 10.4049/jimmunol.181.8.5587. [DOI] [PubMed] [Google Scholar]
- 151.Aubert DF, Xu H, Yang J, Shi X, Gao W, Li L, Bisaro F, Chen S, Valvano MA, Shao F. 2016. A Burkholderia type VI effector deamidates Rho GTPases to activate the pyrin inflammasome and trigger inflammation. Cell Host Microbe 19:664–674. 10.1016/j.chom.2016.04.004. [DOI] [PubMed] [Google Scholar]
- 152.Ng J, Hirota SA, Gross O, Li Y, Ulke-Lemee A, Potentier MS, Schenck LP, Vilaysane A, Seamone ME, Feng H, Armstrong GD, Tschopp J, Macdonald JA, Muruve DA, Beck PL. 2010. Clostridium difficile toxin-induced inflammation and intestinal injury are mediated by the inflammasome. Gastroenterology 139:542–552.e1-3. 10.1053/j.gastro.2010.04.005. [DOI] [PubMed] [Google Scholar]
- 153.Xu H, Yang J, Gao W, Li L, Li P, Zhang L, Gong YN, Peng X, Xi JJ, Chen S, Wang F, Shao F. 2014. Innate immune sensing of bacterial modifications of Rho GTPases by the pyrin inflammasome. Nature 513:237–241. 10.1038/nature13449. [DOI] [PubMed] [Google Scholar]
- 154.Caution K, Gavrilin MA, Tazi M, Kanneganti A, Layman D, Hoque S, Krause K, Amer AO. 2015. Caspase-11 and caspase-1 differentially modulate actin polymerization via RhoA and Slingshot proteins to promote bacterial clearance. Sci Rep 5:18479. 10.1038/srep18479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Yu LC-H, Wang J-T, Wei S-C, Ni Y-H. 2012. Host-microbial interactions and regulation of intestinal epithelial barrier function: from physiology to pathology. World J Gastrointest Pathophysiol 3:27–43. 10.4291/wjgp.v3.i1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Hiippala K, Jouhten H, Ronkainen A, Hartikainen A, Kainulainen V, Jalanka J, Satokari R. 2018. The potential of gut commensals in reinforcing intestinal barrier function and alleviating inflammation. Nutrients 10:988. 10.3390/nu10080988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Martín R, Miquel S, Ulmer J, Kechaou N, Langella P, Bermúdez-Humarán LG. 2013. Role of commensal and probiotic bacteria in human health: a focus on inflammatory bowel disease. Microb Cell Fact 12:71–71. 10.1186/1475-2859-12-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Dürr UHN, Sudheendra US, Ramamoorthy A. 2006. LL-37, the only human member of the cathelicidin family of antimicrobial peptides. Biochim Biophys Acta 1758:1408–1425. 10.1016/j.bbamem.2006.03.030. [DOI] [PubMed] [Google Scholar]
- 159.Byfield FJ, Kowalski M, Cruz K, Leszczyńska K, Namiot A, Savage PB, Bucki R, Janmey PA. 2011. Cathelicidin LL-37 increases lung epithelial cell stiffness, decreases transepithelial permeability, and prevents epithelial invasion by Pseudomonas aeruginosa. J Immunol 187:6402–6409. 10.4049/jimmunol.1102185. [DOI] [PubMed] [Google Scholar]
- 160.Borkowski AW, Gallo RL. 2011. The coordinated response of the physical and antimicrobial peptide barriers of the skin. J Invest Dermatol 131:285–287. 10.1038/jid.2010.360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Aberg KM, Man MQ, Gallo RL, Ganz T, Crumrine D, Brown BE, Choi EH, Kim DK, Schroder JM, Feingold KR, Elias PM. 2008. Co-regulation and interdependence of the mammalian epidermal permeability and antimicrobial barriers. J Invest Dermatol 128:917–925. 10.1038/sj.jid.5701099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Liu N, Guan S, Wang H, Li C, Cheng J, Yu H, Lin L, Pan Y. 2018. The antimicrobial peptide Nal-P-113 exerts a reparative effect by promoting cell proliferation, migration, and cell cycle progression. Biomed Res Int 2018:7349351. 10.1155/2018/7349351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Cappiello F, Ranieri D, Carnicelli V, Casciaro B, Chen HT, Ferrera L, Di YP, Mangoni ML. 2019. Bronchial epithelium repair by Esculentin-1a-derived antimicrobial peptides: involvement of metalloproteinase-9 and interleukin-8, and evaluation of peptides' immunogenicity. Sci Rep 9:18988. 10.1038/s41598-019-55426-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Radisky ES, Radisky DC. 2010. Matrix metalloproteinase-induced epithelial-mesenchymal transition in breast cancer. J Mammary Gland Biol Neoplasia 15:201–212. 10.1007/s10911-010-9177-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Agraval H, Yadav UCS. 2019. MMP-2 and MMP-9 mediate cigarette smoke extract-induced epithelial-mesenchymal transition in airway epithelial cells via EGFR/Akt/GSK3β/β-catenin pathway: amelioration by fisetin. Chem Biol Interact 314:108846. 10.1016/j.cbi.2019.108846. [DOI] [PubMed] [Google Scholar]
- 166.Singh R, Chandrashekharappa S, Bodduluri SR, Baby BV, Hegde B, Kotla NG, Hiwale AA, Saiyed T, Patel P, Vijay-Kumar M, Langille MGI, Douglas GM, Cheng X, Rouchka EC, Waigel SJ, Dryden GW, Alatassi H, Zhang H-G, Haribabu B, Vemula PK, Jala VR. 2019. Enhancement of the gut barrier integrity by a microbial metabolite through the Nrf2 pathway. Nat Commun 10:89. 10.1038/s41467-018-07859-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Parker A, Fonseca S, Carding SR. 2020. Gut microbes and metabolites as modulators of blood-brain barrier integrity and brain health. Gut Microbes 11:135–157. 10.1080/19490976.2019.1638722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Celikkaya ME, Akcora B, Hakverdi S, Ozer B, Ulutas KT, Duran N. 2019. Effects of probiotic use on bacterial translocation in created rat models with biliary obstructions. Eurasian J Med 51:106–111. 10.5152/eurasianjmed.2019.18426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Ohland CL, MacNaughton WK. 2010. Probiotic bacteria and intestinal epithelial barrier function. Am J Physiol Gastrointest Liver Physiol 298:G807–G819. 10.1152/ajpgi.00243.2009. [DOI] [PubMed] [Google Scholar]
- 170.Liu Q, Yu Z, Tian F, Zhao J, Zhang H, Zhai Q, Chen W. 2020. Surface components and metabolites of probiotics for regulation of intestinal epithelial barrier. Microb Cell Fact 19:23. 10.1186/s12934-020-1289-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Heaver SL, Johnson EL, Ley RE. 2018. Sphingolipids in host-microbial interactions. Curr Opin Microbiol 43:92–99. 10.1016/j.mib.2017.12.011. [DOI] [PubMed] [Google Scholar]
- 172.Zhang JN, Zhao Y, Liu C, Han ES, Yu X, Lidington D, Bolz SS, You L. 2015. The role of the sphingosine-1-phosphate signaling pathway in osteocyte mechanotransduction. Bone 79:71–78. 10.1016/j.bone.2015.05.017. [DOI] [PubMed] [Google Scholar]
- 173.Hendley AM, Wang YJ, Polireddy K, Alsina J, Ahmed I, Lafaro KJ, Zhang H, Roy N, Savidge SG, Cao Y, Hebrok M, Maitra A, Reynolds AB, Goggins M, Younes M, Iacobuzio-Donahue CA, Leach SD, Bailey JM. 2016. p120 catenin suppresses basal epithelial cell extrusion in invasive pancreatic neoplasia. Cancer Res 76:3351–3363. 10.1158/0008-5472.CAN-15-2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Apidianakis Y, Rahme LG. 2011. Drosophila melanogaster as a model for human intestinal infection and pathology. Dis Model Mech 4:21–30. . 10.1242/dmm.003970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Jiang H, Patel PH, Kohlmaier A, Grenley MO, McEwen DG, Edgar BA. 2009. Cytokine/Jak/Stat signaling mediates regeneration and homeostasis in the Drosophila midgut. Cell 137:1343–1355. 10.1016/j.cell.2009.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Carvalho L, Jacinto A, Matova N. 2014. The Toll/NF-κB signaling pathway is required for epidermal wound repair in Drosophila. Proc Natl Acad Sci USA 111:E5373–E5382. 10.1073/pnas.1408224111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Kasper CA, Sorg I, Schmutz C, Tschon T, Wischnewski H, Kim ML, Arrieumerlou C. 2010. Cell-cell propagation of NF-kappaB transcription factor and MAP kinase activation amplifies innate immunity against bacterial infection. Immunity 33:804–816. 10.1016/j.immuni.2010.10.015. [DOI] [PubMed] [Google Scholar]
- 178.Chang S-Y, Lee S-N, Yang J-Y, Kim DW, Yoon J-H, Ko H-J, Ogawa M, Sasakawa C, Kweon M-N. 2013. Autophagy controls an intrinsic host defense to bacteria by promoting epithelial cell survival: a murine model. PLoS One 8:e81095. 10.1371/journal.pone.0081095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Hino N, Rossetti L, Marín-Llauradó A, Aoki K, Trepat X, Matsuda M, Hirashima T. 2019. ERK-mediated mechanochemical waves direct collective cell polarization. bioRxiv. 10.1101/2019.12.25.888552. [DOI] [PubMed]
- 180.Aikin TJ, Peterson AF, Pokrass MJ, Clark HR, Regot S. 2019. Collective MAPK signaling dynamics coordinates epithelial homeostasis. bioRxiv. 10.1101/826917. [DOI]
- 181.Luo T, Mohan K, Iglesias P, Robinson D. 2013. Molecular mechanisms of cellular mechanosensing. Nat Mater 12:1064–1071. 10.1038/nmat3772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Dahl KN, Kalinowski A, Pekkan K. 2010. Mechanobiology and the microcirculation: cellular, nuclear and fluid mechanics. Microcirculation 17:179–191. 10.1111/j.1549-8719.2009.00016.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Engler A, Bacakova L, Newman C, Hategan A, Griffin M, Discher D. 2004. Substrate compliance versus ligand density in cell on gel responses. Biophys J 86:617–628. 10.1016/S0006-3495(04)74140-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Bastounis EE, Yeh YT, Theriot JA. 2019. Subendothelial stiffness alters endothelial cell traction force generation while exerting a minimal effect on the transcriptome. Sci Rep 9:18209. 10.1038/s41598-019-54336-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Cunningham KS, Gotlieb AI. 2005. The role of shear stress in the pathogenesis of atherosclerosis. Lab Invest 85:9–23. 10.1038/labinvest.3700215. [DOI] [PubMed] [Google Scholar]
- 186.Zieman SJ, Melenovsky V, Kass DA. 2005. Mechanisms, pathophysiology, and therapy of arterial stiffness. Arterioscler Thromb Vasc Biol 25:932–943. 10.1161/01.ATV.0000160548.78317.29. [DOI] [PubMed] [Google Scholar]
- 187.Lampi MC, Reinhart-King CA. 2018. Targeting extracellular matrix stiffness to attenuate disease: from molecular mechanisms to clinical trials. Sci Transl Med 10:eaao0475. 10.1126/scitranslmed.aao0475. [DOI] [PubMed] [Google Scholar]
- 188.Epstein SE, Zhu J, Najafi AH, Burnett MS. 2009. Insights into the role of infection in atherogenesis and in plaque rupture. Circulation 119:3133–3141. 10.1161/CIRCULATIONAHA.109.849455. [DOI] [PubMed] [Google Scholar]
- 189.Rosenfeld ME, Campbell LA. 2011. Pathogens and atherosclerosis: update on the potential contribution of multiple infectious organisms to the pathogenesis of atherosclerosis. Thromb Haemost 106:858–867. 10.1160/TH11-06-0392. [DOI] [PubMed] [Google Scholar]
- 190.Mullick AE, Tobias PS, Curtiss LK. 2005. Modulation of atherosclerosis in mice. J Clin Invest 115:3149–3156. 10.1172/JCI25482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Keller TT, Mairuhu ATA, de Kruif MD, Klein SK, Gerdes VEA, ten Cate H, Brandjes DPM, Levi M, van Gorp ECM. 2003. Infections and endothelial cells. Cardiovasc Res 60:40–48. 10.1016/s0008-6363(03)00354-7. [DOI] [PubMed] [Google Scholar]
- 192.Hayashi C, Papadopoulos G, Gudino CV, Weinberg EO, Barth KR, Madrigal Aé G, Chen Y, Ning H, LaValley M, Gibson FC, Hamilton JA, Genco CA. 2012. Protective role for TLR4 signaling in atherosclerosis progression as revealed by infection with a common oral pathogen. J Immunol 189:3681–3688. 10.4049/jimmunol.1201541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Bastounis EE, Yeh Y-T, Theriot JA. 2018. Matrix stiffness modulates infection of endothelial cells by Listeria monocytogenes via expression of cell surface vimentin. Mol Biol Cell 29:1571–1589. 10.1091/mbc.E18-04-0228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Cheng Z, Shurer CR, Schmidt S, Gupta VK, Chuang G, Su J, Watkins AR, Shetty A, Spector JA, Hui C-Y, Reesink HL, Paszek MJ. 2020. The surface stress of biomedical silicones is a stimulant of cellular response. Sci Adv 6:eaay0076. 10.1126/sciadv.aay0076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Trappmann B, Gautrot JE, Connelly JT, Strange DGT, Li Y, Oyen ML, Cohen Stuart MA, Boehm H, Li B, Vogel V, Spatz JP, Watt FM, Huck WTS. 2012. Extracellular-matrix tethering regulates stem-cell fate. Nat Mater 11:642–649. 10.1038/nmat3339. [DOI] [PubMed] [Google Scholar]
- 196.Geiger B, Bershadsky A, Pankov R, Yamada KM. 2001. Transmembrane crosstalk between the extracellular matrix-cytoskeleton crosstalk. Nat Rev Mol Cell Biol 2:793–805. 10.1038/35099066. [DOI] [PubMed] [Google Scholar]
- 197.Moorthy S, Byfield FJ, Janmey PA, Klein EA. 2020. Matrix stiffness regulates endosomal escape of uropathogenic E. coli. Cell Microbiol 22:e13196. 10.1111/cmi.13196. [DOI] [PubMed] [Google Scholar]
- 198.Eto DS, Sundsbak JL, Mulvey MA. 2006. Actin-gated intracellular growth and resurgence of uropathogenic Escherichia coli. Cell Microbiol 8:704–717. 10.1111/j.1462-5822.2006.00691.x. [DOI] [PubMed] [Google Scholar]
- 199.Thomas WE, Trintchina E, Forero M, Vogel V, Sokurenko EV. 2002. Bacterial adhesion to target cells enhanced by shear force. Cell 109:913–923. 10.1016/s0092-8674(02)00796-1. [DOI] [PubMed] [Google Scholar]
- 200.Tarbell JM. 2010. Shear stress and the endothelial transport barrier. Cardiovasc Res 87:320–330. 10.1093/cvr/cvq146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Shi Z-D, Tarbell JM. 2011. Fluid flow mechanotransduction in vascular smooth muscle cells and fibroblasts. Ann Biomed Eng 39:1608–1619. 10.1007/s10439-011-0309-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Kothapalli D, Liu S-L, Bae YH, Monslow J, Xu T, Hawthorne EA, Byfield FJ, Castagnino P, Rao S, Rader DJ, Puré E, Phillips MC, Lund-Katz S, Janmey PA, Assoian RK. 2012. Cardiovascular protection by apoE and apoE-HDL linked to suppression of ECM gene expression and arterial stiffening. Cell Rep 2:1259–1271. 10.1016/j.celrep.2012.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Bufi N, Saitakis M, Dogniaux S, Buschinger O, Bohineust A, Richert A, Maurin M, Hivroz C, Asnacios A. 2015. Human primary immune cells exhibit distinct mechanical properties that are modified by inflammation. Biophys J 108:2181–2190. 10.1016/j.bpj.2015.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Previtera ML, Sengupta A. 2015. Substrate stiffness regulates proinflammatory mediator production through TLR4 activity in macrophages. PLoS One 10:e0145813. 10.1371/journal.pone.0145813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Blakney AK, Swartzlander MD, Bryant SJ. 2012. The effects of substrate stiffness on the in vitro activation of macrophages and in vivo host response to poly(ethylene glycol)-based hydrogels. J Biomed Mater Res A 100:1375–1386. 10.1002/jbm.a.34104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Patel NR, Bole M, Chen C, Hardin CC, Kho AT, Mih J, Deng L, Butler J, Tschumperlin D, Fredberg JJ, Krishnan R, Koziel H. 2012. Cell elasticity determines macrophage function. PLoS One 7:e41024. 10.1371/journal.pone.0041024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Pageon SV, Govendir MA, Kempe D, Biro M. 2018. Mechanoimmunology: molecular-scale forces govern immune cell functions. Mol Biol Cell 29:1919–1926. 10.1091/mbc.E18-02-0120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Bastounis EE, Ortega FE, Serrano R, Theriot JA. 2018. A multi-well format polyacrylamide-based assay for studying the effect of extracellular matrix stiffness on the bacterial infection of adherent cells. J Vis Exp 2018 July 5:57361. 10.3791/57361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Gouin E, Welch MD, Cossart P. 2005. Actin-based motility of intracellular pathogens. Curr Opin Microbiol 8:35–45. 10.1016/j.mib.2004.12.013. [DOI] [PubMed] [Google Scholar]
- 210.El-Mohri H, Wu Y, Mohanty S, Ghosh G. 2017. Impact of matrix stiffness on fibroblast function. Mater Sci Eng C 74:146–151. 10.1016/j.msec.2017.02.001. [DOI] [PubMed] [Google Scholar]
- 211.Sinha B, Koster D, Ruez R, Gonnord P, Bastiani M, Abankwa D, Stan RV, Butler-Browne G, Vedie B, Johannes L, Morone N, Parton RG, Raposo G, Sens P, Lamaze C, Nassoy P. 2011. Cells respond to mechanical stress by rapid disassembly of caveolae. Cell 144:402–413. 10.1016/j.cell.2010.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Huang C, Butler PJ, Tong S, Muddana HS, Bao G, Zhang S. 2013. Substrate stiffness regulates cellular uptake of nanoparticles. Nano Lett 13:1611–1615. 10.1021/nl400033h. [DOI] [PubMed] [Google Scholar]
- 213.Sanderlin AG, Vondrak C, Scricco AJ, Fedrigo I, Ahyong V, Lamason RL. 2019. RNAi screen reveals a role for PACSIN2 and caveolins during bacterial cell-to-cell spread. Mol Biol Cell 30:2124–2133. 10.1091/mbc.E19-04-0197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Fukumatsu M, Ogawa M, Arakawa S, Suzuki M, Nakayama K, Shimizu S, Kim M, Mimuro H, Sasakawa C. 2012. Shigella targets epithelial tricellular junctions and uses a noncanonical clathrin-dependent endocytic pathway to spread between cells. Cell Host Microbe 11:325–336. 10.1016/j.chom.2012.03.001. [DOI] [PubMed] [Google Scholar]
- 215.Soyer M, Duménil G. 2011. Introducing shear stress in the study of bacterial adhesion. J Vis Exp 2011 Sep 2:e3241. 10.3791/3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Kim SW, Ehrman J, Ahn M-R, Kondo J, Lopez AAM, Oh YS, Kim XH, Crawley SW, Goldenring JR, Tyska MJ, Rericha EC, Lau KS. 2017. Shear stress induces noncanonical autophagy in intestinal epithelial monolayers. Mol Biol Cell 28:3043–3056. 10.1091/mbc.E17-01-0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Shin W, Hinojosa CD, Ingber DE, Kim HJ. 2019. Human intestinal morphogenesis controlled by transepithelial morphogen gradient and flow-dependent physical cues in a microengineered gut-on-a-chip. iScience 15:391–406. 10.1016/j.isci.2019.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Kim HJ, Li H, Collins JJ, Ingber DE. 2016. Contributions of microbiome and mechanical deformation to intestinal bacterial overgrowth and inflammation in a human gut-on-a-chip. Proc Natl Acad Sci USA 113:E7–E15. 10.1073/pnas.1522193112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Yang J, Barrila J, Roland KL, Ott CM, Nickerson CA. 2016. Physiological fluid shear alters the virulence potential of invasive multidrug-resistant non-typhoidal Salmonella Typhimurium D23580. NPJ Microgravity 2:16021–16021. 10.1038/npjmgrav.2016.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Nickerson CA, Ott CM, Mister SJ, Morrow BJ, Burns-Keliher L, Pierson DL. 2000. Microgravity as a novel environmental signal affecting Salmonella enterica serovar Typhimurium virulence. Infect Immun 68:3147–3152. 10.1128/IAI.68.6.3147-3152.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Alsharif G, Ahmad S, Islam MS, Shah R, Busby SJ, Krachler AM. 2015. Host attachment and fluid shear are integrated into a mechanical signal regulating virulence in Escherichia coli O157:H7. Proc Natl Acad Sci USA 112:5503–5508. 10.1073/pnas.1422986112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Dolan JM, Meng H, Singh S, Paluch R, Kolega J. 2011. High fluid shear stress and spatial shear stress gradients affect endothelial proliferation, survival, and alignment. Ann Biomed Eng 39:1620–1631. 10.1007/s10439-011-0267-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Park JY, White JB, Walker N, Kuo C-H, Cha W, Meyerhoff ME, Takayama S. 2011. Responses of endothelial cells to extremely slow flows. Biomicrofluidics 5:22211–22211. 10.1063/1.3576932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Kadohama T, Nishimura K, Hoshino Y, Sasajima T, Sumpio BE. 2007. Effects of different types of fluid shear stress on endothelial cell proliferation and survival. J Cell Physiol 212:244–251. 10.1002/jcp.21024. [DOI] [PubMed] [Google Scholar]
- 225.Beste MT, Hammer DA. 2008. Selectin catch-slip kinetics encode shear threshold adhesive behavior of rolling leukocytes. Proc Natl Acad Sci USA 105:20716–20721. 10.1073/pnas.0808213105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Persat A, Nadell CD, Kim MK, Ingremeau F, Siryaporn A, Drescher K, Wingreen NS, Bassler BL, Gitai Z, Stone HA. 2015. The mechanical world of bacteria. Cell 161:988–997. 10.1016/j.cell.2015.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Lecuyer S, Rusconi R, Shen Y, Forsyth A, Vlamakis H, Kolter R, Stone HA. 2011. Shear stress increases the residence time of adhesion of Pseudomonas aeruginosa. Biophys J 100:341–350. 10.1016/j.bpj.2010.11.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Claes J, Vanassche T, Peetermans M, Liesenborghs L, Vandenbriele C, Vanhoorelbeke K, Missiakas D, Schneewind O, Hoylaerts MF, Heying R, Verhamme P. 2014. Adhesion of Staphylococcus aureus to the vessel wall under flow is mediated by von Willebrand factor-binding protein. Blood 124:1669–1676. 10.1182/blood-2014-02-558890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Vileno B, Lekka M, Sienkiewicz A, Jeney S, Stoessel G, Lekki J, Forro L, Stachura Z. 2007. Stiffness alterations of single cells induced by UV in the presence of nanoTiO2. Environ Sci Technol 41:5149–5153. 10.1021/es0629561. [DOI] [PubMed] [Google Scholar]
- 230.Ebady R, Niddam AF, Boczula AE, Kim YR, Gupta N, Tang TT, Odisho T, Zhi H, Simmons CA, Skare JT, Moriarty TJ. 2016. Biomechanics of Borrelia burgdorferi vascular interactions. Cell Rep 16:2593–2604. 10.1016/j.celrep.2016.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Tchesnokova V, McVeigh AL, Kidd B, Yakovenko O, Thomas WE, Sokurenko EV, Savarino SJ. 2010. Shear-enhanced binding of intestinal colonization factor antigen I of enterotoxigenic Escherichia coli. Mol Microbiol 76:489–502. 10.1111/j.1365-2958.2010.07116.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Pavelka M, Roth J. 2010. Glycocalyx (cell coat), p 160–161. In Pavelka M, Roth J (ed), Functional ultrastructure: atlas of tissue biology and pathology. Springer, Vienna, Austria. [Google Scholar]
- 233.Henry-Stanley MJ, Hess DJ, Erickson EA, Garni RM, Wells CL. 2003. Role of heparan sulfate in interactions of Listeria monocytogenes with enterocytes. Med Microbiol Immunol 192:107–115. 10.1007/s00430-002-0165-7. [DOI] [PubMed] [Google Scholar]
- 234.García B, Merayo-Lloves J, Martin C, Alcalde I, Quirós LM, Vazquez F. 2016. Surface proteoglycans as mediators in bacterial pathogens infections. Front Microbiol 7:220. 10.3389/fmicb.2016.00220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Zeng Y, Tarbell JM. 2014. The adaptive remodeling of endothelial glycocalyx in response to fluid shear stress. PLoS One 9:e86249. 10.1371/journal.pone.0086249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Shi Z-D, Wang H, Tarbell JM. 2011. Heparan sulfate proteoglycans mediate interstitial flow mechanotransduction regulating MMP-13 expression and cell motility via FAK-ERK in 3D collagen. PLoS One 6:e15956. 10.1371/journal.pone.0015956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Figueroa-Morales N, Dominguez-Rubio L, Ott TL, Aranson IS. 2019. Mechanical shear controls bacterial penetration in mucus. Sci Rep 9:9713. 10.1038/s41598-019-46085-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Leal J, Smyth HDC, Ghosh D. 2017. Physicochemical properties of mucus and their impact on transmucosal drug delivery. Int J Pharm 532:555–572. 10.1016/j.ijpharm.2017.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Lai SK, Wang Y-Y, Wirtz D, Hanes J. 2009. Micro- and macrorheology of mucus. Adv Drug Deliv Rev 61:86–100. 10.1016/j.addr.2008.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Jinno A, Park PW. 2015. Role of glycosaminoglycans in infectious disease. Methods Mol Biol 1229:567–585. 10.1007/978-1-4939-1714-3_45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.White CR, Frangos JA. 2007. The shear stress of it all: the cell membrane and mechanochemical transduction. Philos Trans R Soc Lond B Biol Sci 362:1459–1467. 10.1098/rstb.2007.2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Melchior B, Frangos JA. 2010. Shear-induced endothelial cell-cell junction inclination. Am J Physiol Cell Physiol 299:C621–C629. 10.1152/ajpcell.00156.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Perrault CM, Brugues A, Bazellieres E, Ricco P, Lacroix D, Trepat X. 2015. Traction forces of endothelial cells under slow shear flow. Biophys J 109:1533–1536. 10.1016/j.bpj.2015.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Hur SS, del Alamo JC, Park JS, Li YS, Nguyen HA, Teng D, Wang KC, Flores L, Alonso-Latorre B, Lasheras JC, Chien S. 2012. Roles of cell confluency and fluid shear in 3-dimensional intracellular forces in endothelial cells. Proc Natl Acad Sci USA 109:11110–11115. 10.1073/pnas.1207326109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Zeng Y, Waters M, Andrews A, Honarmandi P, Ebong EE, Rizzo V, Tarbell JM. 2013. Fluid shear stress induces the clustering of heparan sulfate via mobility of glypican-1 in lipid rafts. Am J Physiol Heart Circ Physiol 305:H811–H820. 10.1152/ajpheart.00764.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Lin A, Sabnis A, Kona S, Nattama S, Patel H, Dong J-F, Nguyen KT. 2010. Shear-regulated uptake of nanoparticles by endothelial cells and development of endothelial-targeting nanoparticles. J Biomed Mater Res A 93:833–842. 10.1002/jbm.a.32592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Klingberg H, Loft S, Oddershede LB, Møller P. 2015. The influence of flow, shear stress and adhesion molecule targeting on gold nanoparticle uptake in human endothelial cells. Nanoscale 7:11409–11419. 10.1039/C5NR01467K. [DOI] [PubMed] [Google Scholar]
- 248.Tonkin ML, Boulanger MJ. 2015. The shear stress of host cell invasion: exploring the role of biomolecular complexes. PLoS Pathog 11:e1004539. 10.1371/journal.ppat.1004539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Kılıç A, Ameli A, Park J-A, Kho AT, Tantisira K, Santolini M, Cheng F, Mitchel JA, McGill M, O’Sullivan MJ, De Marzio M, Sharma A, Randell SH, Drazen JM, Fredberg JJ, Weiss ST. 2020. Mechanical forces induce an asthma gene signature in healthy airway epithelial cells. Sci Rep 10:966. 10.1038/s41598-020-57755-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Park J-A, Fredberg JJ, Drazen JM. 2015. Putting the squeeze on airway epithelia. Physiology (Bethesda) 30:293–303. 10.1152/physiol.00004.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Kasendra M, Luc R, Yin J, Manatakis DV, Apostolou A, Sunuwar L, Obrigewitch J, Hamilton GA, Donowitz M, Karalis K. 2019. Organoid-derived Duodenum Intestine-Chip enables preclinical drug assessment in a human relevant system. bioRxiv. 10.1101/723015. [DOI] [PMC free article] [PubMed]
- 252.Ronaldson-Bouchard K, Vunjak-Novakovic G. 2018. Organs-on-a-Chip: a fast track for engineered human tissues in drug development. Cell Stem Cell 22:310–324. 10.1016/j.stem.2018.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Kim HJ, Huh D, Hamilton G, Ingber DE. 2012. Human gut-on-a-chip inhabited by microbial flora that experiences intestinal peristalsis-like motions and flow. Lab Chip 12:2165–2174. 10.1039/c2lc40074j. [DOI] [PubMed] [Google Scholar]
- 254.Ghoshal UC, Ghoshal U. 2017. Small intestinal bacterial overgrowth and other intestinal disorders. Gastroenterol Clin North Am 46:103–120. 10.1016/j.gtc.2016.09.008. [DOI] [PubMed] [Google Scholar]
- 255.Ortega FE, Rengarajan M, Chavez N, Radhakrishnan P, Gloerich M, Bianchini J, Siemers K, Luckett WS, Lauer P, Nelson WJ, Theriot JA. 2017. Adhesion to the host cell surface is sufficient to mediate Listeria monocytogenes entry into epithelial cells. Mol Biol Cell 28:2945–2957. 10.1091/mbc.E16-12-0851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Santus P, Pecchiari M, Tursi F, Valenti V, Saad M, Radovanovic D. 2019. The airways' mechanical stress in lung disease: implications for COPD pathophysiology and treatment evaluation. Can Respir J 2019:3546056. 10.1155/2019/3546056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Darveaux JI, Lemanske RF, Jr.. 2014. Infection-related asthma. J Allergy Clin Immunol Pract 2:658–663. 10.1016/j.jaip.2014.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Iikura M, Hojo M, Koketsu R, Watanabe S, Sato A, Chino H, Ro S, Masaki H, Hirashima J, Ishii S, Naka G, Takasaki J, Izumi S, Kobayashi N, Yamaguchi S, Nakae S, Sugiyama H. 2015. The importance of bacterial and viral infections associated with adult asthma exacerbations in clinical practice. PLoS One 10:e0123584. 10.1371/journal.pone.0123584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Sutherland ER, Martin RJ. 2007. Asthma and atypical bacterial infection. Chest 132:1962–1966. 10.1378/chest.06-2415. [DOI] [PubMed] [Google Scholar]
- 260.Yi E, Sato S, Takahashi A, Parameswaran H, Blute TA, Bartolák-Suki E, Suki B. 2016. Mechanical forces accelerate collagen digestion by bacterial collagenase in lung tissue strips. Front Physiol 7:287. 10.3389/fphys.2016.00287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Szabari MV, Parameswaran H, Sato S, Hantos Z, Bartolák-Suki E, Suki B. 2012. Acute mechanical forces cause deterioration in lung structure and function in elastase-induced emphysema. Am J Physiol Lung Cell Mol Physiol 303:L567–L574. 10.1152/ajplung.00217.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Hiemstra PS, McCray PB, Jr, Bals R. 2015. The innate immune function of airway epithelial cells in inflammatory lung disease. Eur Respir J 45:1150–1162. 10.1183/09031936.00141514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Hansel TT, Johnston SL, Openshaw PJ. 2013. Microbes and mucosal immune responses in asthma. Lancet 381:861–873. 10.1016/S0140-6736(12)62202-8. [DOI] [PubMed] [Google Scholar]
- 264.Dickson RP, Erb-Downward JR, Huffnagle GB. 2013. The role of the bacterial microbiome in lung disease. Expert Rev Respir Med 7:245–257. 10.1586/ers.13.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Konar D, Devarasetty M, Yildiz DV, Atala A, Murphy SV. 2016. Lung-on-a-chip technologies for disease modeling and drug development. Biomed Eng Comput Biol 7:17–27. 10.4137/BECB.S34252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Huh DD. 2015. A human breathing lung-on-a-chip. Ann Am Thorac Soc 12(Suppl 1):S42–S44. 10.1513/AnnalsATS.201410-442MG. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Benam KH, Villenave R, Lucchesi C, Varone A, Hubeau C, Lee H-H, Alves SE, Salmon M, Ferrante TC, Weaver JC, Bahinski A, Hamilton GA, Ingber DE. 2016. Small airway-on-a-chip enables analysis of human lung inflammation and drug responses in vitro. Nat Methods 13:151–157. 10.1038/nmeth.3697. [DOI] [PubMed] [Google Scholar]
- 268.Thacker VV, Dhar N, Sharma K, Barrile R, Karalis K, McKinney JD. 2020. A lung-on-chip infection model reveals protective and permissive roles of alveolar epithelial cells in tuberculosis. bioRxiv. 10.1101/2020.02.03.931170. [DOI] [PMC free article] [PubMed]
- 269.Novak R, Ingram M, Marquez S, Das D, Delahanty A, Herland A, Maoz BM, Jeanty SSF, Somayaji MR, Burt M, Calamari E, Chalkiadaki A, Cho A, Choe Y, Chou DB, Cronce M, Dauth S, Divic T, Fernandez-Alcon J, Ferrante T, Ferrier J, FitzGerald EA, Fleming R, Jalili-Firoozinezhad S, Grevesse T, Goss JA, Hamkins-Indik T, Henry O, Hinojosa C, Huffstater T, Jang K-J, Kujala V, Leng L, Mannix R, Milton Y, Nawroth J, Nestor BA, Ng CF, O'Connor B, Park T-E, Sanchez H, Sliz J, Sontheimer-Phelps A, Swenor B, Thompson G, Touloumes GJ, Tranchemontagne Z, Wen N, Yadid M, Bahinski A, et al. 2020. Robotic fluidic coupling and interrogation of multiple vascularized organ chips. Nat Biomed Eng 4:407–420. 10.1038/s41551-019-0497-x. [DOI] [PMC free article] [PubMed] [Google Scholar]