

Abstract
Aging is the strongest risk factor for degenerative bone and joint diseases. Clinical therapies for age-related musculoskeletal disorders face significant challenges as their pathogenic mechanisms remain largely unclear. This review article focuses on the recent advances in the understanding of regulatory mechanisms of musculoskeletal aging and their clinical relevance. We begin with the prevalence and socioeconomic impacts of major age-related musculoskeletal disorders such as sarcopenia, osteoporosis, osteoarthritis, and degenerative tendinopathy. The current understanding of responsible biological mechanisms involved in general aging is then summarized. Proposed molecular, cellular, and biomechanical mechanisms relevant to the clinical manifestations of aging in the musculoskeletal system are discussed in detail, with a focus on the disorders affecting muscle, bone, articular cartilage, and tendon. Although musculoskeletal aging processes share many common pathways with the aging of other body systems, unique molecular and cellular mechanisms may be involved in the aging processes of musculoskeletal tissues. Advancements in the understanding of regulatory mechanisms of musculoskeletal aging may promote the development of novel treatments for age-related musculoskeletal disorders. Finally, future research directions for major musculoskeletal tissues including functional interaction between the tissues and their clinical relevance to age-related musculoskeletal disorders are highlighted in the future prospects section.
Keywords: musculoskeletal aging, sarcopenia, osteoporosis, osteoarthritis, tendinopathy
1. Introduction
Aging is a complex process that is characterized by a progressive loss of physiological function over time. This correlates with an increased susceptibility to many diseases that span all organs systems. In fact, age is considered the greatest risk factor for many neurodegenerative, metabolic, cardiovascular, and musculoskeletal conditions. Understanding the link between aging and disease could have major treatment and socioeconomic impacts as the population is aging at an unprecedented rate. Worldwide, the 65 years and older age group is expected to nearly triple from 617 million in 2015 to 1.57 billion while the 80 years and older age group is expected to more than triple from 126 million to 446 million by 2050.1 This puts a huge burden on the healthcare economy as utilization and spending increase with patient age.2 Identifying possible therapeutic targets that link aging and disease may be able to lengthen the “healthspan” of patients and limit healthcare costs.
The effect of aging in the musculoskeletal system impacts a large proportion of society and is primarily manifested in muscle, bone, cartilage, and tendon. Sarcopenia is a complex condition with age-related loss of muscle mass that results in decreased strength and physical endurance. The estimated direct healthcare costs of sarcopenia was $18.5 billion in 2000 in the United States alone.3 Additionally, low muscle mass and strength makes elderly patients more likely to fall and suffer fractures from low energy mechanisms due to poor bone quality and postural control.4 Osteoporosis is highly prevalent in the elderly population as it is estimated that more than 10 million Americans are affected while another 43 million have low bone density.5 Roughly one out of two Caucasian women and one in five Caucasian men will have an osteoporosis-related fracture during their life. On an annual basis, this translates into more than two million fractures and 400,000 hospitalizations.6 It is estimated that osteoporosis will be responsible for more than $25 billion in annual health care spending by 2025 in the United States.6
There is a direct correlation between increasing age and the incidence of osteoarthritis (OA), though the exact pathogenesis is largely unclear. OA is one of the top causes of disability worldwide7 and represents nearly $185 billion in total treatment costs and lost wages each year in the United States.8 No treatments are currently available to prevent or permanently reverse OA despite multiple basic science and medical innovations, including numerous drug trials.9 As such, surgical joint replacement or fusion remains the only option for patients with end-stage OA. Tendons undergo age-associated changes including disorientation of collagen fibers, thinning of fibers, myxoid degeneration and calcification. These changes are clinically evident by the increased prevalence of many tendinopathies with age. Asymptomatic full thickness rotator cuff tears are present in approximately 13% of patients over 50 years and 50% of patients over 80 years.10 Furthermore, there is an increased incidence of biceps tendinitis and ruptures, lateral epicondylitis, and Achilles tendinitis with age.11
This review will discuss the current understanding of the molecular mechanisms involved in the general aging process and specific mechanisms and clinical manifestations of aging in the musculoskeletal system along with possible therapeutic interventions.
2. Biological Mechanisms of General Aging
Aging is a multifactorial process that results from both genetic and environmental factors. Recent advances in the understanding of the molecular mechanisms of general aging are summarized below with 10 subheadings, which are primarily based on the “Hallmarks of Aging” as proposed by López-Otín et. al. in their landmark review article.12
2.1. Genomic alterations
A long-purposed theory of aging centers on the accumulation of genetic damage (DNA crosslinking, point mutations, translocations, etc.) over time from both exogenous (e.g. radiation, smoking) and endogenous (e.g. reactive oxygen species, DNA replication errors) sources which alters gene expression and intracellular signaling.13, 14 These genetic errors accumulate despite organisms’ complex DNA repair machinery. This theory of aging is supported by progeroid syndromes such as Werner’s syndrome in which defects in DNA repair machinery lead to accelerated aging.15 Ultimately, these genetic changes can limit stem cell renewal, trigger pathologic inflammatory processes or even cellular senescence, apoptosis, or unregulated expansion.13
2.2. Telomere shortening
Telomeres, highly specialized repetitive nucleotide sequences, protect the ends of chromosomes from deterioration. In humans, telomere length is reduced with age in many tissue/cell populations such as liver, kidney, spleen, peripheral blood cells, dermal fibroblast, and mucosal keratinocytes and has been linked to stem cell exhaustion.16, 17 Telomerase, a reverse transcriptase, attempts to counteract this shortening by adding new DNA onto the telomeres and is highly expressed in cells with unlimited proliferative potential.18 Telomerase deficiency in humans has been linked to the early onset of multiple clinical syndromes including pulmonary fibrosis, dyskeratosis congenital, and aplastic anemia19. Conversely, mice expressing elevated amounts of telomerase have an extended maximum lifespan compared to wild-type littermates.20
2.3. Altered epigenetic modifications
Epigenetics is the alteration of gene expression without modification to the underlying genetic code; these changes include DNA methylation, histone modifications, and non-coding RNA (ncRNA). Age-related epigenetic modifications occur due to ROS, nutrient dysregulation, chronic inflammation, and telomere shortening.21 DNA methylation occurs so predictably with age that it can accurately predict a patient’s age and is thus known as the “epigenetic clock.”22 Other epigenetic alterations, to include histone modification and ncRNA, have been shown to affect the lifespan of several model organisms.23, 24 The role of epigenetics in aging and osteoarthritis has recently been reviewed.25, 26 Epigenetics is a growing field secondary to the potential reversibility, and therefore treatability, of the aging-related changes.
2.4. Loss of proteostasis
Cell function depends on protein homeostasis, proper protein folding and degradation of misfolded/non-functional proteins. Protein chaperons play a role in folding while proteolytic systems help degrade proteins. These processes may be disrupted with aging, leading to the accumulation of damaged, aggregated, and/or dysfunctional proteins.27 This accumulation has been linked to age-associated disorders such as cataracts, Alzheimer’s disease, and Parkinson’s disease.28
Autophagy, a mechanism used to remove misfolded or aggregated proteins and eliminate intracellular pathogens, is a highly regulated cytoprotective process that plays an important role in cell growth, development and homeostasis.29 The expression of specific proteins required for autophagy induction is reduced in aged tissues and age-related diseases, including OA.30–32 Moreover, mitochondrial dysfunction, which contributes to cellular aging, occurs in part because of the inability of cells to remove their damaged mitochondria.33
2.5. Deregulated nutrient sensing
Caloric restriction is one of the most well studied mechanisms of increased lifespan. Limiting caloric intake has been shown to increase the lifespan of multiple species, including humans.34–37 The responsible molecular signaling pathways remain unclear; however, evidence suggests that the Insulin/Insulin-like growth factor-1 (IGF-1) pathway and its downstream effector, mTor, are involved via nutrient “sensing” molecules. Genetic mutations of this pathway and its downstream effects have been linked to longevity.38, 39 Sirtuins, an NAD-dependent family of proteins, play a crucial role in nutrient “sensing” and are linked to increased longevity with caloric restriction in multiple model organisms ranging from yeast to mammals.40 Additionally, pharmacologically targeting sirtuins with activating molecules has shown promise for increasing disease resistance and longevity41. Caloric restriction may also extend lifespan by preventing the age-associated decline in autophagy.42
2.6. Mitochondrial dysfunction and reactive oxygen species
Reactive oxygen species (ROS) are a group of diverse reactive chemical species involving oxygen which have both physiologic and pathologic roles. Most are generated in mitochondria during the process of oxidative phosphorylation but exogenous sources such as pollutants and radiation also produce ROS.43 Physiologically, ROS play a major role in cellular signaling and infection prevention. However, they also have many deleterious effects on cell metabolism and are linked to the development of several age-related diseases including Alzheimer’s, obesity, and diabetes.44, 45 The accumulation of ROS over time is thought to result from mitochondrial dysfunction and a reduction in ROS scavenging mechanisms such as superoxide dismutases, a class of enzymes that act as endogenous antioxidants.46
2.7. Cellular senescence
Cellular senescence results when cells no longer progress through the cell cycle. Hayflick first described cellular senescence in cultured fibroblast in the 1960s47. Cellular senescence is known to increase with aging48 and can result from multiple mechanisms including telomere shorting49 and the accumulation of genomic damage50. It is unknown if cellular senescence is protective by preventing replication of damaged cells or pathologic.
Senescence-associated secretory phenotype (SASP) refers to senescent cells that accumulate and secrete inflammatory chemical mediators including cytokines and matrix degrading proteases. SASP cells have been implicated in the aging process and documented in multiple cell types,51, 52 including chondrocytes.53 The removal of senescent cells in transgenic mice is associated with an increased lifespan and reduced morbidity, highlighting their role in aging.54
2.8. Stem cell exhaustion
Stem cells are the pluripotent cells of the body and represent the major repository of regenerative potential within organ systems. Stem cell reserves decrease with age across multiple tissues including skin, immune, hematopoietic, musculoskeletal, and nervous systems.55 Within the hematopoietic system, aged stem cells have lower rates of replication and there is an increase in anemia and myeloid cancers with age.56, 57 Similar to cellular senescence, stem cell exhaustion appears to be the result of the accumulation of multiple aging mechanisms including genomic instability, loss of proteostasis, telomere attrition, and mitochondrial dysfunction.
2.9. Inflammation and altered intercellular communication
The major change in intercellular communication with aging is associated with inflammation. Levels of inflammatory mediators typically increase with age even in the absence of acute infection or other physiologic stress.58 While levels are still in the sub-acute range, this chronic pro-inflammatory state underlies many aging-related conditions59 and has been termed “inflammaging.”60 Proposed causes include the accumulation of tissue damage, an inability of the immune system to clear pathogens, and the proinflammatory phenotype of SASPs cells, as discussed previously. In addition, a reduction in nutrient “sensing” sirtuins may impact aging through altered intracellular communication and increased inflammation.61
2.10. Abnormal crosslinking
Crosslinking refers to the accumulation of abnormal bonds between molecules, especially biomacromolecules such as collagens as well nucleic acids and sugars. It is one of the deleterious effects of ROS in aging.62, 63 The addition of sugar molecules to proteins and nucleic acids, a process known as non-enzymatic glycosylation, leads to the production of advanced glycosylation end products (AGEs) which can promote crosslinking. AGEs have been implicated in tissue and organ system dysfunction in aging and diabetes.64 Age-related manifestations of crosslinking are the loss of skin elasticity, stiffening of blood vessels, changes in the lens of the eye, and delayed wound healing.65, 66
The possible mechanisms of general aging which impacts all organ systems are summarized in Table 1. Some of these mechanisms also play a role in the aging of the musculoskeletal system.
Table 1.
Possible mechanisms of general aging
| Risk Factors | Possible mechanisms | References |
|---|---|---|
| Genomic alterations | Genetic errors accumulate over time which ultimately leads to cellular apoptosis/senescence, limited stem cell renewal, and altered cytokine secretion. | 12–15 |
| Telomere shortening | Progressive telomere shortening occurs with cellular division which leads to decreased cellular proliferation, function, and possibly lifespan. | 16–20 |
| Altered epigenetic modification | Epigenetic modifications alter gene expression and cellular function in an age-dependent manner and alter cellular function over time. | 21–26 |
| Loss of proteostasis | Protein misfolding and decreased activity of proteolytic and autophagy systems leads to an accumulation of dysfunctional proteins altering cellular metabolism. | 27–33 |
| Deregulated nutrient sensing | Caloric restriction increases lifespan via activation of nutrient “sensing” molecules like sirtuins and modify the Insulin/IGF-1/mTor signaling pathway. | 34–42 |
| Mitochondrial dysfunction and ROS | Mitochondrial dysfunction increases reactive oxygen species that damage nucleic acids and oxidize fatty acids/proteins leading to genetic alterations and cellular dysfunction. | 43–46 |
| Cellular senescence | Aged cells no longer replicate and adopt a “senesence-associated secretory phenotype” that promotes inflammation and aging. | 47–54 |
| Stem cell exhaustion | Stem cell levels decrease with age, reducing the repair and regenerative capacity of tissues over the lifespan. | 55–57 |
| Inflammation and altered intercellular communication | Changes in cellular communication and inflammatory mediators lead to tissue dysfunction via multiple pathways including DNA damage, ROS, protein dysfunction, and altered autophagy. | 58–61 |
| Abnormal crosslinking | Abnormal intra- and inter-molecular bonding leads to altered transcription, translation and tissue properties. | 62–66 |
3. Mechanisms and Manifestations of Musculoskeletal Aging
In the musculoskeletal system, the primary tissues affected with age are muscle, bone, cartilage and tendon. Age-related changes and disorders in the musculoskeletal system, which represent some of the most disabling diseases, are summarized in Figure 1.
Figure 1:
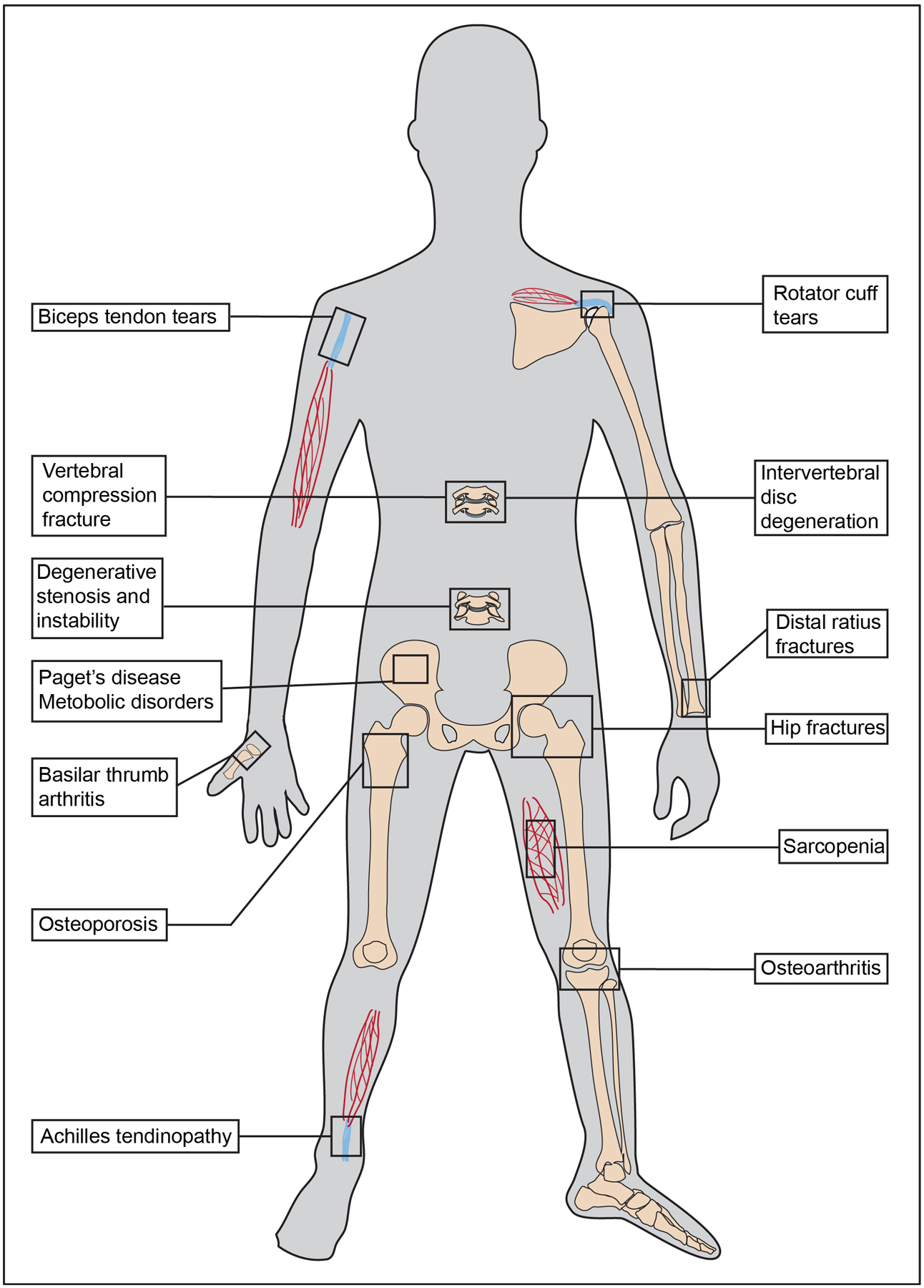
Age-associated conditions of the musculoskeletal system. Sarcopenia is a decrease in muscle mass and function with age. Tendons undergo degenerative changes with age and contribute to increased rotator cuff tears, biceps tendinitis and Achilles tendinitis. The vertebral column undergoes multiple age-related changes that contribute to increased disc herniations, spinal stenosis, foraminal stenosis, instability and deformity. Age-dependent changes in bone metabolism and structure lead to osteoporosis and increased risk of fragility fractures, specifically vertebral compression fractures, hip fractures, and distal radius fractures. Alterations in subchondral bone and overlying cartilage with age increase the risk for osteoarthritis.
3.1. Muscle
Sarcopenia is the loss of muscle mass, strength, and function with age due to changes in muscle fiber type/size, alterations of the motor nerve unit and fatty infiltration of muscle. Longitudinal studies have shown a 1–1.5% reduction in muscle strength per year in elderly patients67 with a 30–50% accumulated decrease in muscle mass by age 80.68 This results in increased frailty, fall risk, and loss of independence. The molecular and cellular mechanisms underlying these changes may include loss of repair mechanisms, accumulated damage, and altered signaling.
Failure of regeneration and repair of accumulated damage is a recognized cause for decreased muscle function. Satellite cells, the regenerative cells of skeletal muscle, are reduced in number, proliferative capacity, and differentiation ability with age.69, 70 Furthermore, satellite cell signaling in response to growth factors, such as fibroblast growth factor, is blunted with age which appears to be driven by epigenetic changes.71, 72 Additional epigenetic changes in muscle include several ncRNA that play roles in muscle aging via alterations of SIRT1, telomerase, and IGF-1/TGF-β anabolic pathways.73 Reduction in autophagy, an important cell maintenance function, has also been demonstrated in aged muscle fibers. The function of autophagy is important for muscle health and is a proposed mechanism for the beneficial effects of physical exercise in elderly patients.74
Accumulation of damage to the muscle cell machinery for contraction and energy production also results in decreased function. Post-translational changes accumulate on myosin which limit coupling with actin resulting in decreased force generation with age.75 Muscle mitochondria, the power plant of the cell, also accumulate damage with age, resulting in decreased ATP production and increased ROS.76
Muscle mass loss with age appears to be partly driven by a reduction in muscle fiber number. Aged humans lose approximately 50% of their muscle fibers77 which is primarily mediated by apoptosis.78 This apoptosis may be induced by ROS damage to mitochondrial DNA and primarily affects the large type II fast fibers. It is often accompanied by infiltration of fibrous or fatty tissue. Additional proposed mechanisms of muscle mass loss are the denervation of motor neurons and loss of trophic support.79 Sixty-year-old patients have 25–50% less motor neurons than 20-year-old patients,80 and these changes may be governed by neural redox signaling.81
Signaling pathways, particularly protein kinase B (PKB, also known as AKT) and mechanistic target of rapamycin (mTOR), are important mediators of overall lifespan and muscle function. AKT phosphorylation is decreased with age82 and the contraction induced activation of AKT/mTOR is blunted in mice with increasing age.83 These changes may decrease protein synthesis, disrupt the repair and regenerative balance of muscle, and decrease overall muscle size and strength.
Myostatin is a negative regulator of muscle mass that was first described in 1997.84 Myostatin knockout mice have a 2–3 times increase in body weight from muscle hypertrophy and hyperplasia. Agents and antibodies targeting myostatin and its downstream receptor, activin, are showing promise for sarcopenia treatment.85 Interestingly, one aspect of myostatin intra-cellular signaling is via the Akt/mTor pathway, an important mediator of aging.86
The role of caloric restriction in aging muscle is not well defined. Reduction in the Insulin/IGF-1 signaling pathway seen in caloric restriction is associated with preserved muscle mass as aged non-human primates increased muscle mass by 30% with caloric restriction.87 It has been shown that caloric restriction results in less mitochondrial dysfunction, less ROS generation, and decreased activation of apoptotic pathways in muscle.88 There is also a reduction in the inflammatory cascade in muscle from aged rats with caloric restriction.89 Conversely, muscle specific overexpression of IGF-1 in transgenic mice prevents age-associated muscle atrophy.90 As such, more mechanistic, dose-dependent, and longitudinal studies are needed to further characterize the exact role of caloric restriction and Insulin/IGF-1 signaling in muscle aging.
3.2. Bone
Age-dependent changes in bone may result in Paget’s disease of bone91, osteoporosis, and increased fracture risk. Osteoporosis, the most common bone disease in humans, is characterized by low bone mass and disruption of bone architecture with subsequent compromised bone strength and an increased risk of fracture. The prevalence of osteoporosis increases with age. Bone mass peaks in early adulthood and then declines with age due to an imbalance in bone formation and resorption.92
The most common osteoporotic fragility fractures are hip, vertebral compression, and distal radius fractures. Elderly patients with hip fractures consistently have decreased activities of daily living93 and a 30 percent mortality rate at 1 year following fracture.94 Osteoporotic vertebral compression fractures are the most common fragility fracture with a prevalence of 5–10% in patients aged 50–60 years and greater than 40% in patients over 80 years.95 This results in over 70,000 hospitalizations96 and $13.8 billion in annual healthcare costs.97 Even in the absence of fracture, other age related degenerative changes of the spine lead to significant morbidity due to radiculopathy, central stenosis, spondylolisthesis, and degenerative scoliosis.98
Multiple mechanisms drive age related changes to bone. Osteoblasts are the bone-forming cells derived from undifferentiated mesenchymal stem cells (MSCs). Osteoblasts proliferation, maturation, and subsequent bone matrix mineralization are decreased with increased ROS, accumulation of DNA damage, and reduced autophagy activity.99–101 MSCs also undergo several age-related changes which limit their differentiation and regenerative capacity. First, MSCs proliferate at slower rates,102 have reduced autophagy,103 and preferentially differentiate to adipocytes versus osteoblasts with aging104 leading to increased adiposity of bone marrow. This change in cell differentiation along with delayed periosteal reaction and prolonged endochondral calcification results in delayed bone healing of fractures from aged mice.105 Second, MSC telomeres play an important role in bone aging. Transgenic mice with reduced telomerase function have reduced bone mass secondary to uncoupling of osteoblast and osteoclast function.106, 107 Conversely, MSC telomerase activation maintains differentiation in vitro and increases bone mass in vivo.108, 109 Third, cellular responses of MSCs to anabolic signaling pathways decreases with age. For example, Wnt10b deficiency results in age-dependent loss of bone mass and progressive reduction of mesenchymal progenitor cells.110 Wnt-β-catenin signaling via mechanoreceptors is also involved in MSC differentiation to osteoblasts.111 Furthermore, MSC response to anabolic transforming growth factor beta (TGF-beta)/bone morphogenetic protein (BMP) signaling decreases with aging.112, 113 Together, these changes highlight the importance of MSC role in maintaining aging bone health.
Pharmacological interventions based on aging mechanisms that have shown promise include inhibitors of wnt antagonist,114 suppression of peroxisome proliferator-activated receptor gamma,115 anti-oxidant treatment,116 and recent evidence suggesting that SIRT1 activation via both pharmacological and caloric restriction can increase bone mass.117
Additional factors leading to changes in bone density are dysregulation of hormones (e.g. estrogen and parathyroid hormone) on osteoblasts, osteoclasts, and osteocyte activities, alterations in cellular response to hormones, and vitamin D metabolism.118, 119 These changes may increase intra-cortical porosity through remodeling and coalescence of existing cortical canals.92 Additionally, osteocyte signaling in response to mechanical load is reduced and the osteoprotective effect of the muscle factor β-aminoisobutyric acid (BAIBA) is lost with age. Theses alterations may further contribute to the progression of osteoporosis and to the attenuated effect of exercise on bone with aging.120, 121
Epigenetic modification has multiple roles in bone aging. First, DNA methylation controls Receptor activator of nuclear factor kappa-B ligand (RANKL) and osteoprotegerin (OPG) expression.122 Second, transgenic mice with SIRT1 knockout have a reduction in bone mass similar to aged animals.123 Third, non-coding RNA (ncRNA) have been shown to regulate osteoblast cell function and bone density in vivo.124–126 Finally, rapamycin inhibition of mTOR, which is known to have lifespan prolonging effects, reduces osteoclastic activity and bone loss in mice.127
3.3. Articular Cartilage
The incidence of osteoarthritis (OA) increases with age. Articular cartilage is avascular and has limited capacity for repair. Chondrocytes reside within the cartilage to secrete and maintain the extracellular matrix (ECM). With aging, the number of chondrocytes decrease with an overall decrease in ECM synthesis.128 Like aging in other tissues, SASP cells have been found in cartilage. SASP chondrocytes increase expression of inflammatory mediators and matrix degrading proteases, similar to the periarticular environment in OA.129 This increase in basal inflammation may explain why surgical models of OA show more rapid progression in aged mice as compared to young mice.130 Chondrocyte senescence has also been associated with telomere shortening.131
The accumulation of ROS has been shown to affect chondrocyte apoptosis, senescence, ECM synthesis, and signaling through nitric oxide (NO).132 NO alters singling through tyrosine activated receptors in chondrocytes from both aged mice and mice with OA.133 Additional chondrocyte changes with age include decreased mechanical stimulation and blunted responsiveness to growth factors.134 These changes tip the anabolic/catabolic ratio in favor of catabolic breakdown and lower the threshold for developing OA.135, 136
The ECM of articular cartilage undergoes age-associated changes. Articular cartilage ECM is primarily composed of water, type II collagen, and proteoglycans. Overtime, there is a reduction in the water content, increased stiffness, and loss of tensile strength leading to increased brittleness and risk of failure. Many of these changes are attributed to passive glycation and crosslinking of the ECM, primarily from AGEs.137 Changes to the ECM alter the hydrostatic biomechanics of cartilage leading to a reduction in the capacity for deformation, which increases the risk of injury. With aging, there is also increased collagenase-mediated breakdown of type II collagen, an increased ratio of keratan sulfate to chondroitin sulfate, and smaller disorganized aggrecan molecules.138 Many of these changes have been found to be under epigenetic control via DNA methylation of matrix metalloproteinases promoters139 and ncRNA regulation of anabolic signaling.140
NFAT1, a member of the nuclear factor of activated T-cells (NFAT) family of transcription factors, has recently been discovered as a regulator of joint homeostasis by controlling expression of multiple anabolic and catabolic genes.141 NFAT1 knockout mice display down-regulation of anabolic genes and up-regulation of catabolic genes at the initiation stage, leading to cartilage breakdown.142 Furthermore, NFAT1 is highly expressed in young adult articular cartilage but declines with age in correlation with cartilage degeneration, as compared to SOX9 which is primarily expressed in developing cartilage.143–145 This spontaneous decrease in NFAT1 expression with age is regulated primarily by epigenetic histone modifications.145 Further understanding of the connection between epigenetics, cartilage aging, and OA is important for the development of therapeutic strategies and has recently been reviewed in depth.26, 143
The advancement of mechanistic studies on OA pathogenesis has substantially improved our understanding of the link between biological changes and clinical manifestations of articular cartilage aging and OA. For example, increased expression of catabolic genes encoding proinflammatory cytokines and matrix metalloproteinases (MMPs) in articular cartilage is linked to degeneration and loss of cartilage, causing joint stiffness. Synovial inflammation and subchondral bone marrow edema with increased pressure in the bone marrow cavity may irritate the nerves in the synovium and periarticular bone, respectively, causing joint pain.
3.4. Tendon
Tendons are highly organized structures that transmit muscle forces to bone to produce movement. They are comprised primarily of type I collagen organized into fibrils. Tenocytes are the specialized fibroblast-like cells of tendons that maintain tendon structure, synthesize the ECM, and assist in tissue repair. Many tendinopathies increase with age. The structural changes associated with these degenerative changes have been extensively studied; however, the molecular mechanisms have not been well defined.
Changes in tendon structure with age include decreased fiber size with reduced collagen density and disorganization of the collagen fibrils. Other documented changes in tendon with age include decreased water content, decreased proteoglycans, increased calcification and lipid accumulation and an increase in crosslinking.146 Together these changes contribute to increased stiffness and decreased tensile strength, ultimately lowering the fatigue failure threshold.147–149 Inflammation and the expression of matrix metalloproteinases (MMPs) in tendons increases with age, which may contribute to these observed structural changes and clinical manifestations.150 151 Treatment with MMP inhibitors has been shown to prevent reduction in tendon strength in vitro.152
Age associated changes have been documented in 2 cell populations within tendons, tenocytes and the recently described tendon stem/progenitor cells (TSC). Tenocytes decrease in number and their morphology changes to a thinner and spindle shape with increased nuclear to cytoplasm ratio with age. Tenocytes in aged animals have decreased proliferation rates and increased cellular senescence.61 Recent evidence also suggests that aging tenocytes also have a pro-inflammatory senescent phenotype, similar to previously described SASP cells.153 TSCs are a distinct population of progenitor cells within tendons that contribute to healing and regeneration.154 Application of TSC sheets in a rat model of Achilles injury improved biomechanical and histological appearance of the tendons as compared to controls.155 There is a documented 70% reduction in TSCs with age from poor self-renewal and limited differentiation capacity causing stem cell exhaustion.156 This contributes to weaker tendon healing at the tendon-bone junction. Aged TSC also have decreased protein synthesis, less growth, and less regenerative potential with higher cellular senescence.156, 157 New evidence also demonstrates a role for epigenetic modification in tendon aging. Modulation of TSC senescence is regulated by the ncRNA, miR-135a, while TSC differentiation is regulated via microRNA-217.158, 159 Along the “altered inter-cellular communication” aging pathway, recent evidence in TSC have shown that increased signaling via ephrin receptors may have anti-aging properties by promoting cell-cell communication.160
Changes in vascularity are a documented cause of aging- or injury-mediated tendon degeneration. The supraspinatus161 and Achilles tendons162 have a hypovascular region that correlates with the location of common tears. Immobilization appears to reduce blood flow to tendons secondary to under stimulation.163 Histopathology of tendon tears shows blood vessel narrowing that is more significant in older patients,164 possibly from crosslinking within the vessel walls.
Similar to other parts of the musculoskeletal systems, exercise and mechanical stimulation has beneficial effects on tendon. In tendon, moderate exercise and mechanical loading negates some of the “stem cell exhaustion” associated with aging. TSC show increased expression of stem cell markers and proliferation with mechanical loading and AGEs are reduced in aged Achilles tendons after training.165
These discussed structural and molecular changes cumulatively result in clinical diseases such as tendinopathy in the biceps, rotator cuff, and Achilles tendons which can lead to overt tears that may require surgical intervention. Better understanding of the biomechanical aspects of tendon aging will hopefully lead to improved patient quality of life and function.
4. Future Prospects
Mechanisms of aging and degenerative musculoskeletal conditions are closely linked. Understanding the molecular and cellular mechanisms of musculoskeletal aging will promote the development of targeted treatments for age-related disorders of musculoskeletal tissues (Figure 2). Indeed, certain areas have already shown promise. However, there remains a disconnect between the described mechanisms of aging and current treatment modalities of age associated musculoskeletal disorders. The gaps between basic science results and clinical treatments of diseases in muscle, bone, articular cartilage, and tendon are highlighted below.
Figure 2:
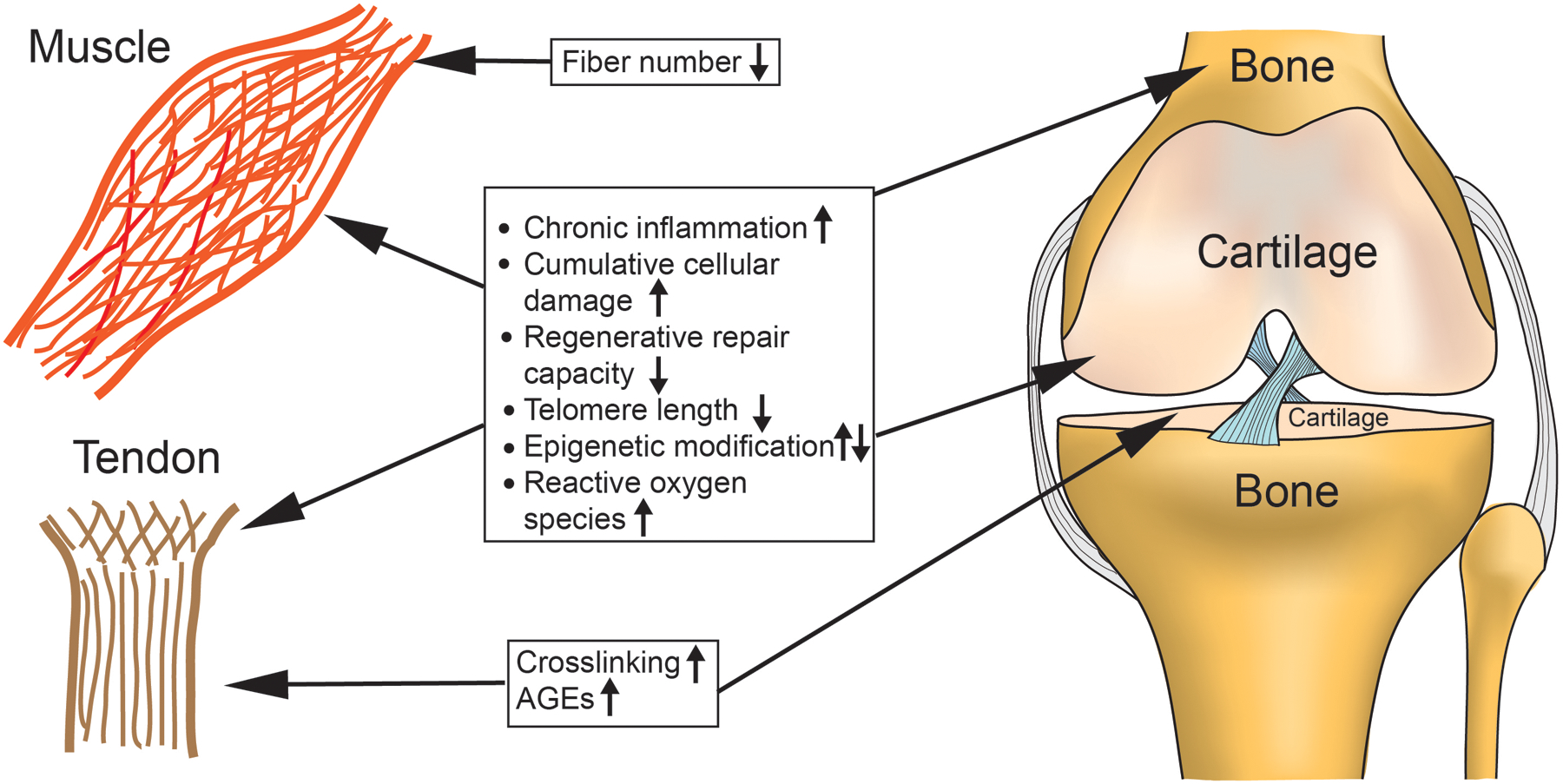
Proposed mechanisms of musculoskeletal aging. Chronic inflammation and catabolic pathway activation, cumulative cellular damage, decreased repair/regeneration, telomere shortening, epigenetic modifications and increased reactive oxygen species (ROS) are common pathways amongst many tissues of the musculoskeletal system. Age-associated fiber loss is unique to muscle aging, while crosslinking affects primarily tendons and cartilage.
4.1. Muscle
Despite the substantial clinical impact of sarcopenia, treatment options are lacking secondary to poor understanding of the involved molecular mechanisms. Resistance exercise along with supplementation of vitamin D and protein is the current treatment of choice.166, 167 Currently there are no FDA approved pharmacologic treatments for sarcopenia. Growth hormone/IGF1 and anabolic steroids have been tried with moderate success and several medications are currently being tested including ghrelin agonist, β-adrenergic agonist/antagonist, fast skeletal muscle troponin activators, biologic anti-inflammatories (infliximab, tocilizumab) and angiotension converting enzyme inhibitors. The major gap in sarcopenia treatment is an unclear understanding of the interplay between the aging mechanisms and a disconnect between the pathogenesis and treatment options. Furthermore, treatments need to be able to impact multiple mechanisms, as aging and sarcopenia are multifactorial conditions. Finally, animal models that more accurately simulate human sarcopenia will provide more robust preclinical studies.168
4.2. Bone
Osteoporosis is an increasing worldwide concern secondary to the increased aging population. Current treatment involves weight-bearing exercise, vitamin D and calcium supplementation with the addition of a pharmacological agent. Current pharmacological treatment of osteoporosis primarily targets reducing osteoclast bone resorption.169 The most popular and successful drug class is the bisphosphonates, but increasing concerns are being raised about “adynamic” bone and atypical femur fractures after long-term use. Additional anti-resorptive medications include hormone-based therapies and denusomab, a RANK-L decoy receptor. Currently the only anabolic agent available is teriparatide, a parathyroid analogue that targets osteoblasts and bone formation. Several new drugs are being developed including cathepsin K inhibitors, src kinase inhibitors, calcilytics, and inhibitors of Wnt antagonist.169
Patients with osteoporosis are often not diagnosed until they sustain a fragility fracture, or on screening at age 65, at which time bone density may be severely decreased. Gaps within osteoporosis research and treatment include delayed diagnosis, limited medications focusing on prevention, and a lack of treatments targeting the multifactorial nature of osteoporosis. Continued studies to highlight the changes in bone biology and mechanical response with age will help provide new treatment options and provide opportunities to develop earlier diagnosis and preventative measures.
4.3. Articular cartilage
OA is the most common form of joint disease and the major cause of chronic disability in middle-aged and older populations.170, 171 Cartilage degradation is a major hallmark of OA, though multiple joint tissues may be involved in the progression of OA. Many risk factors, such as aging, obesity, joint trauma, abnormal mechanical loading, genetic predisposition and sex hormone alterations, have been proposed to increase the susceptibility to OA.172–174 However, the precise mechanisms underlying OA pathogenesis remain unclear. Current pharmacologic management of OA pain with analgesics and non-steroidal anti-inflammatory drugs (NSAIDs) may relieve pain temporarily but does not cure the disease. Development of disease-modifying OA drugs (DMOADs) has been aimed to halt or reverse progression of articular cartilage damage. Many candidate DMOADs have been tested, but none have been approved by food and drug regulatory agencies in the United States or other countries.175, 176 The lack of effective non-surgical OA therapy currently drives about a million Americans to joint replacement surgery annually,177 which is not an ideal treatment modality for younger individuals who desire to preserve their natural joints. Therefore, a better understanding of OA pathogenesis is paramount to development of effective non-surgical OA therapy.
Our recent studies indicate that NFAT1 overexpression in cultivated articular chondrocytes from aged mice can significantly reverse the imbalanced chondrocyte anabolic/catabolic activities and is under epigenetic control.145 This novel finding needs to be further confirmed in preclinical and clinical studies. Epigenetic modifications with histone deacetylase inhibitors, ncRNAs, and NAD-dependent deacetylase sirtuin-1(SIRT1) activators have been shown to improve chondrocyte function and protect against age-associated OA.178, 179 Targeting these reversible aging mechanisms in cartilage could be an additional pathway to developing treatments for cartilage aging and OA.
4.4. Tendon
Anti-inflammatories, corticosteroid injections and physical therapy remain the mainstay of treatment for tendinopathy, but refractory cases may require surgical debridement.180 Physical therapy modalities include stretching/strengthening, therapeutic ultrasound, iontophoresis, low level laser treatment, and phonophoresis. Additional treatment options that have been utilized include extracorporeal shock wave therapy, nitric oxide treatments via glyceryl trinitrate patches, hyperthermia, and sclerotherapy. Growth factor therapy, especially in the form of platelet rich plasma (PRP) injections, has recently gained significant attention and has shown some positive results, but more stringent studies are needed to determine efficacy, timing and dosing.181
The current repertoire of treatment options does not include any medications targeting mechanisms of tendinopathy or tendon degeneration and represents a major gap between the current treatment options and mechanistic basic science studies. Translational studies of aging mechanisms may highlight new target options, particularly with the discovery TSCs. Continued efforts to develop preclinical models for aging and tendinopathy will also greatly improve our understanding of these conditions and promote development of new treatment modalities.
4.5. Crosstalk and interaction of musculoskeletal tissues
Musculoskeletal tissues are closely associated in both anatomy and function, but the mechanisms that coordinate their functional interactions are not well defined. Recent studies have shed some light on the crosstalk between tissues/cells. For example, crosstalk between MLO-Y4 osteocytes and C2C12 muscle cells is found to be mediated by the Wnt/β-catenin pathway,182 while muscle-derived myostatin inhibits osteoblastic differentiation by suppressing osteocyte-derived exosomal microRNA-218.183 Additionally, epigenetic mechanisms regulate the aging and healing process of the tendon-bone junction.156 Articular cartilage trophic support is provided by the surround synovial fluid and underlying subchondral bone. Subchondral bone support of cartilage decreases with age, which consequently compromises the metabolism and mechanical properties of articular cartilage.130 Furthermore, age-related comorbidities, such as chronic obstructive pulmonary disease or congestive heart failure, can limit mobility resulting in decreased bone density and muscle mass thus propagating changes already occurring in the musculoskeletal system.
5. Conclusions
Aging has a profound effect on all systems in the body, including the musculoskeletal system. This review summarizes the current understanding of molecular, cellular, and biomechanical mechanisms relevant to the clinical manifestations of musculoskeletal aging, with a focus on the disorders affecting muscle, bone, articular cartilage, and tendon. Interplay between the organs/tissues may magnify the clinical manifestations of age. Signaling crosstalk and functional interaction between the musculoskeletal tissues may further increase the risk of falls and fractures, coupled with a reduced healing capacity, contributing to significant morbidity and mortality in elderly patients. Improved understanding of the responsible mechanisms underlying age-related musculoskeletal disorders will promote the development of better treatment modalities and achieve more active, pain free, and healthy aging.
Acknowledgements
This work was supported by the U.S. National Institute of Health (NIH) Grant R01 AR059088 (to J. Wang), the U.S. Department of Defense Research Grant W81XWH-12-1-0304 (to J. Wang), and the Mary A. and Paul R. Harrington Distinguished Professorship Endowment. The authors thank Dr. Yi Feng for his expert drawing of illustrations.
Footnotes
Conflicts of interest: The authors declare no conflicts of interest.
References
- 1.Wan He, G. D, and Kowal Paul, An Aging World: 2015, U.S.C. Bureau, Editor 2016: U.S. Government Publishing Office, Washington, DC. p. P95/16–1. [Google Scholar]
- 2.Alemayehu B and Warner KE, The lifetime distribution of health care costs. Health Serv Res, 2004. 39(3): p. 627–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Janssen I, Shepard DS, Katzmarzyk PT, et al. , The healthcare costs of sarcopenia in the United States. J Am Geriatr Soc, 2004. 52(1): p. 80–5. [DOI] [PubMed] [Google Scholar]
- 4.King GW, Abreu EL, Cheng AL, et al. , A multimodal assessment of balance in elderly and young adults. Oncotarget, 2016. 7(12): p. 13297–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wright NC, Looker AC, Saag KG, et al. , The recent prevalence of osteoporosis and low bone mass in the United States based on bone mineral density at the femoral neck or lumbar spine. J Bone Miner Res, 2014. 29(11): p. 2520–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.in Bone Health and Osteoporosis: A Report of the Surgeon General 2004: Rockville (MD). [PubMed] [Google Scholar]
- 7.Hootman JM, C. J, Theis KA, Armour BS., Prevalance and most common causes of disability among adults—United States, 2005, in Morbidity and Mortality Weekly Report 2009: Center for Disease Control and Prevention. [Google Scholar]
- 8.Kotlarz H, Gunnarsson CL, Fang H, et al. , Insurer and out-of-pocket costs of osteoarthritis in the US: evidence from national survey data. Arthritis Rheum, 2009. 60(12): p. 3546–53. [DOI] [PubMed] [Google Scholar]
- 9.Qvist P, Bay-Jensen AC, Christiansen C, et al. , The disease modifying osteoarthritis drug (DMOAD): Is it in the horizon? Pharmacol Res, 2008. 58(1): p. 1–7. [DOI] [PubMed] [Google Scholar]
- 10.Tempelhof S, Rupp S, and Seil R, Age-related prevalence of rotator cuff tears in asymptomatic shoulders. J Shoulder Elbow Surg, 1999. 8(4): p. 296–9. [DOI] [PubMed] [Google Scholar]
- 11.McCarthy MM and Hannafin JA, The mature athlete: aging tendon and ligament. Sports Health, 2014. 6(1): p. 41–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lopez-Otin C, Blasco MA, Partridge L, et al. , The hallmarks of aging. Cell, 2013. 153(6): p. 1194–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hoeijmakers JH, DNA damage, aging, and cancer. N Engl J Med, 2009. 361(15): p. 1475–85. [DOI] [PubMed] [Google Scholar]
- 14.Rodier F, Coppe JP, Patil CK, et al. , Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol, 2009. 11(8): p. 973–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gray MD, Shen JC, Kamath-Loeb AS, et al. , The Werner syndrome protein is a DNA helicase. Nat Genet, 1997. 17(1): p. 100–3. [DOI] [PubMed] [Google Scholar]
- 16.Geserick C and Blasco MA, Novel roles for telomerase in aging. Mech Ageing Dev, 2006. 127(6): p. 579–83. [DOI] [PubMed] [Google Scholar]
- 17.Djojosubroto MW, Choi YS, Lee HW, et al. , Telomeres and telomerase in aging, regeneration and cancer. Mol Cells, 2003. 15(2): p. 164–75. [PubMed] [Google Scholar]
- 18.Jiang J, Chan H, Cash DD, et al. , Structure of Tetrahymena telomerase reveals previously unknown subunits, functions, and interactions. Science, 2015. 350(6260): p. aab4070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Armanios M and Blackburn EH, The telomere syndromes. Nat Rev Genet, 2012. 13(10): p. 693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gonzalez-Suarez E, Geserick C, Flores JM, et al. , Antagonistic effects of telomerase on cancer and aging in K5-mTert transgenic mice. Oncogene, 2005. 24(13): p. 2256–70. [DOI] [PubMed] [Google Scholar]
- 21.Pal S and Tyler JK, Epigenetics and aging. Sci Adv, 2016. 2(7): p. e1600584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Horvath S, DNA methylation age of human tissues and cell types. Genome Biol, 2013. 14(10): p. R115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rogina B and Helfand SL, Sir2 mediates longevity in the fly through a pathway related to calorie restriction. Proc Natl Acad Sci U S A, 2004. 101(45): p. 15998–6003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kanfi Y, Naiman S, Amir G, et al. , The sirtuin SIRT6 regulates lifespan in male mice. Nature, 2012. 483(7388): p. 218–21. [DOI] [PubMed] [Google Scholar]
- 25.Zhang M, Egan B, and Wang J, Epigenetic mechanisms underlying the aberrant catabolic and anabolic activities of osteoarthritic chondrocytes. Int J Biochem Cell Biol, 2015. 67: p. 101–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang M, Lygrisse K, and Wang J, Role of MicroRNA in Osteoarthritis. J Arthritis, 2017. 6(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tomaru U, Takahashi S, Ishizu A, et al. , Decreased proteasomal activity causes age-related phenotypes and promotes the development of metabolic abnormalities. Am J Pathol, 2012. 180(3): p. 963–72. [DOI] [PubMed] [Google Scholar]
- 28.Powers ET, Morimoto RI, Dillin A, et al. , Biological and chemical approaches to diseases of proteostasis deficiency. Annu Rev Biochem, 2009. 78: p. 959–91. [DOI] [PubMed] [Google Scholar]
- 29.Levine B and Klionsky DJ, Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell, 2004. 6(4): p. 463–77. [DOI] [PubMed] [Google Scholar]
- 30.Carames B, Taniguchi N, Otsuki S, et al. , Autophagy is a protective mechanism in normal cartilage, and its aging-related loss is linked with cell death and osteoarthritis. Arthritis Rheum, 2010. 62(3): p. 791–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Alvarez-Garcia O, Matsuzaki T, Olmer M, et al. , Regulated in Development and DNA Damage Response 1 Deficiency Impairs Autophagy and Mitochondrial Biogenesis in Articular Cartilage and Increases the Severity of Experimental Osteoarthritis. Arthritis Rheumatol, 2017. 69(7): p. 1418–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lipinski MM, Zheng B, Lu T, et al. , Genome-wide analysis reveals mechanisms modulating autophagy in normal brain aging and in Alzheimer’s disease. Proc Natl Acad Sci U S A, 2010. 107(32): p. 14164–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu JJ, Quijano C, Chen E, et al. , Mitochondrial dysfunction and oxidative stress mediate the physiological impairment induced by the disruption of autophagy. Aging (Albany NY), 2009. 1(4): p. 425–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Greer EL and Brunet A, Different dietary restriction regimens extend lifespan by both independent and overlapping genetic pathways in C. elegans. Aging Cell, 2009. 8(2): p. 113–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weindruch R, Walford RL, Fligiel S, et al. , The retardation of aging in mice by dietary restriction: longevity, cancer, immunity and lifetime energy intake. J Nutr, 1986. 116(4): p. 641–54. [DOI] [PubMed] [Google Scholar]
- 36.Colman RJ, Anderson RM, Johnson SC, et al. , Caloric restriction delays disease onset and mortality in rhesus monkeys. Science, 2009. 325(5937): p. 201–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Redman LM, Smith SR, Burton JH, et al. , Metabolic Slowing and Reduced Oxidative Damage with Sustained Caloric Restriction Support the Rate of Living and Oxidative Damage Theories of Aging. Cell Metab, 2018. 27(4): p. 805–815 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Harrison DE, Strong R, Sharp ZD, et al. , Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature, 2009. 460(7253): p. 392–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Slack C, Giannakou ME, Foley A, et al. , dFOXO-independent effects of reduced insulin-like signaling in Drosophila. Aging Cell, 2011. 10(5): p. 735–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nakagawa T and Guarente L, SnapShot: sirtuins, NAD, and aging. Cell Metab, 2014. 20(1): p. 192–192 e1. [DOI] [PubMed] [Google Scholar]
- 41.Bonkowski MS and Sinclair DA, Slowing ageing by design: the rise of NAD(+) and sirtuin-activating compounds. Nat Rev Mol Cell Biol, 2016. 17(11): p. 679–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Morselli E, Maiuri MC, Markaki M, et al. , Caloric restriction and resveratrol promote longevity through the Sirtuin-1-dependent induction of autophagy. Cell Death Dis, 2010. 1: p. e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Quinlan CL, Perevoshchikova IV, Hey-Mogensen M, et al. , Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox Biol, 2013. 1: p. 304–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dai DF, Chiao YA, Marcinek DJ, et al. , Mitochondrial oxidative stress in aging and healthspan. Longev Healthspan, 2014. 3: p. 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.West AP, Brodsky IE, Rahner C, et al. , TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature, 2011. 472(7344): p. 476–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Afonso V, Champy R, Mitrovic D, et al. , Reactive oxygen species and superoxide dismutases: role in joint diseases. Joint Bone Spine, 2007. 74(4): p. 324–9. [DOI] [PubMed] [Google Scholar]
- 47.Hayflick L and Moorhead PS, The serial cultivation of human diploid cell strains. Exp Cell Res, 1961. 25: p. 585–621. [DOI] [PubMed] [Google Scholar]
- 48.Wang C, Jurk D, Maddick M, et al. , DNA damage response and cellular senescence in tissues of aging mice. Aging Cell, 2009. 8(3): p. 311–23. [DOI] [PubMed] [Google Scholar]
- 49.Bodnar AG, Ouellette M, Frolkis M, et al. , Extension of life-span by introduction of telomerase into normal human cells. Science, 1998. 279(5349): p. 349–52. [DOI] [PubMed] [Google Scholar]
- 50.Collado M, Blasco MA, and Serrano M, Cellular senescence in cancer and aging. Cell, 2007. 130(2): p. 223–33. [DOI] [PubMed] [Google Scholar]
- 51.Suzuki E, Takahashi M, Oba S, et al. , Oncogene- and oxidative stress-induced cellular senescence shows distinct expression patterns of proinflammatory cytokines in vascular endothelial cells. ScientificWorldJournal, 2013. 2013: p. 754735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bhat R, Crowe EP, Bitto A, et al. , Astrocyte senescence as a component of Alzheimer’s disease. PLoS One, 2012. 7(9): p. e45069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rose J, Soder S, Skhirtladze C, et al. , DNA damage, discoordinated gene expression and cellular senescence in osteoarthritic chondrocytes. Osteoarthritis Cartilage, 2012. 20(9): p. 1020–8. [DOI] [PubMed] [Google Scholar]
- 54.Baker DJ, Wijshake T, Tchkonia T, et al. , Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature, 2011. 479(7372): p. 232–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Boyette LB and Tuan RS, Adult Stem Cells and Diseases of Aging. J Clin Med, 2014. 3(1): p. 88–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shaw AC, Joshi S, Greenwood H, et al. , Aging of the innate immune system. Curr Opin Immunol, 2010. 22(4): p. 507–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rossi DJ, Bryder D, Seita J, et al. , Deficiencies in DNA damage repair limit the function of haematopoietic stem cells with age. Nature, 2007. 447(7145): p. 725–9. [DOI] [PubMed] [Google Scholar]
- 58.Singh T and Newman AB, Inflammatory markers in population studies of aging. Ageing Res Rev, 2011. 10(3): p. 319–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chung HY, Cesari M, Anton S, et al. , Molecular inflammation: underpinnings of aging and age-related diseases. Ageing Res Rev, 2009. 8(1): p. 18–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Franceschi C, Capri M, Monti D, et al. , Inflammaging and anti-inflammaging: a systemic perspective on aging and longevity emerged from studies in humans. Mech Ageing Dev, 2007. 128(1): p. 92–105. [DOI] [PubMed] [Google Scholar]
- 61.Kawahara TL, Michishita E, Adler AS, et al. , SIRT6 links histone H3 lysine 9 deacetylation to NF-kappaB-dependent gene expression and organismal life span. Cell, 2009. 136(1): p. 62–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chen W, Balakrishnan K, Kuang Y, et al. , Reactive oxygen species (ROS) inducible DNA cross-linking agents and their effect on cancer cells and normal lymphocytes. J Med Chem, 2014. 57(11): p. 4498–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Larios JM, Budhiraja R, Fanburg BL, et al. , Oxidative protein cross-linking reactions involving L-tyrosine in transforming growth factor-beta1-stimulated fibroblasts. J Biol Chem, 2001. 276(20): p. 17437–41. [DOI] [PubMed] [Google Scholar]
- 64.Haus JM, Carrithers JA, Trappe SW, et al. , Collagen, cross-linking, and advanced glycation end products in aging human skeletal muscle. J Appl Physiol (1985), 2007. 103(6): p. 2068–76. [DOI] [PubMed] [Google Scholar]
- 65.Aronson D, Cross-linking of glycated collagen in the pathogenesis of arterial and myocardial stiffening of aging and diabetes. J Hypertens, 2003. 21(1): p. 3–12. [DOI] [PubMed] [Google Scholar]
- 66.Rautiainen J, Nieminen MT, Salo EN, et al. , Effect of collagen cross-linking on quantitative MRI parameters of articular cartilage. Osteoarthritis Cartilage, 2016. 24(9): p. 1656–64. [DOI] [PubMed] [Google Scholar]
- 67.Frontera WR, Hughes VA, Fielding RA, et al. , Aging of skeletal muscle: a 12-yr longitudinal study. J Appl Physiol (1985), 2000. 88(4): p. 1321–6. [DOI] [PubMed] [Google Scholar]
- 68.Lexell J, Taylor CC, and Sjostrom M, What is the cause of the ageing atrophy? Total number, size and proportion of different fiber types studied in whole vastus lateralis muscle from 15- to 83-year-old men. J Neurol Sci, 1988. 84(2–3): p. 275–94. [DOI] [PubMed] [Google Scholar]
- 69.Barberi L, Scicchitano BM, De Rossi M, et al. , Age-dependent alteration in muscle regeneration: the critical role of tissue niche. Biogerontology, 2013. 14(3): p. 273–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Segales J, Perdiguero E, and Munoz-Canoves P, Regulation of Muscle Stem Cell Functions: A Focus on the p38 MAPK Signaling Pathway. Front Cell Dev Biol, 2016. 4: p. 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pawlikowski B, Vogler TO, Gadek K, et al. , Regulation of skeletal muscle stem cells by fibroblast growth factors. Dev Dyn, 2017. 246(5): p. 359–367. [DOI] [PubMed] [Google Scholar]
- 72.Liu L, Cheung TH, Charville GW, et al. , Chromatin modifications as determinants of muscle stem cell quiescence and chronological aging. Cell Rep, 2013. 4(1): p. 189–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jung HJ, Lee KP, Kwon KS, et al. , MicroRNAs in skeletal muscle aging: Current issues and perspectives. J Gerontol A Biol Sci Med Sci, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Carnio S, LoVerso F, Baraibar MA, et al. , Autophagy impairment in muscle induces neuromuscular junction degeneration and precocious aging. Cell Rep, 2014. 8(5): p. 1509–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Li M, Ogilvie H, Ochala J, et al. , Aberrant post-translational modifications compromise human myosin motor function in old age. Aging Cell, 2015. 14(2): p. 228–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Short KR, Bigelow ML, Kahl J, et al. , Decline in skeletal muscle mitochondrial function with aging in humans. Proc Natl Acad Sci U S A, 2005. 102(15): p. 5618–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lexell J, Downham D, and Sjostrom M, Distribution of different fibre types in human skeletal muscles. Fibre type arrangement in m. vastus lateralis from three groups of healthy men between 15 and 83 years. J Neurol Sci, 1986. 72(2–3): p. 211–22. [DOI] [PubMed] [Google Scholar]
- 78.Dirks A and Leeuwenburgh C, Apoptosis in skeletal muscle with aging. Am J Physiol Regul Integr Comp Physiol, 2002. 282(2): p. R519–27. [DOI] [PubMed] [Google Scholar]
- 79.Tomlinson BE and Irving D, The numbers of limb motor neurons in the human lumbosacral cord throughout life. J Neurol Sci, 1977. 34(2): p. 213–9. [DOI] [PubMed] [Google Scholar]
- 80.Stalberg E and Fawcett PR, Macro EMG in healthy subjects of different ages. J Neurol Neurosurg Psychiatry, 1982. 45(10): p. 870–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sakellariou GK, McDonagh B, Porter H, et al. , Comparison of Whole Body SOD1 Knockout with Muscle-Specific SOD1 Knockout Mice Reveals a Role for Nerve Redox Signaling in Regulation of Degenerative Pathways in Skeletal Muscle. Antioxid Redox Signal, 2018. 28(4): p. 275–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Leger B, Derave W, De Bock K, et al. , Human sarcopenia reveals an increase in SOCS-3 and myostatin and a reduced efficiency of Akt phosphorylation. Rejuvenation Res, 2008. 11(1): p. 163–175B. [DOI] [PubMed] [Google Scholar]
- 83.Funai K, Parkington JD, Carambula S, et al. , Age-associated decrease in contraction-induced activation of downstream targets of Akt/mTor signaling in skeletal muscle. Am J Physiol Regul Integr Comp Physiol, 2006. 290(4): p. R1080–6. [DOI] [PubMed] [Google Scholar]
- 84.McPherron AC, Lawler AM, and Lee SJ, Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature, 1997. 387(6628): p. 83–90. [DOI] [PubMed] [Google Scholar]
- 85.Camporez JP, Petersen MC, Abudukadier A, et al. , Anti-myostatin antibody increases muscle mass and strength and improves insulin sensitivity in old mice. Proc Natl Acad Sci U S A, 2016. 113(8): p. 2212–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Elkina Y, von Haehling S, Anker SD, et al. , The role of myostatin in muscle wasting: an overview. J Cachexia Sarcopenia Muscle, 2011. 2(3): p. 143–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Colman RJ, Beasley TM, Allison DB, et al. , Attenuation of sarcopenia by dietary restriction in rhesus monkeys. J Gerontol A Biol Sci Med Sci, 2008. 63(6): p. 556–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lopez-Lluch G, Hunt N, Jones B, et al. , Calorie restriction induces mitochondrial biogenesis and bioenergetic efficiency. Proc Natl Acad Sci U S A, 2006. 103(6): p. 1768–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Marzetti E, Carter CS, Wohlgemuth SE, et al. , Changes in IL-15 expression and death-receptor apoptotic signaling in rat gastrocnemius muscle with aging and life-long calorie restriction. Mech Ageing Dev, 2009. 130(4): p. 272–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Coleman ME, DeMayo F, Yin KC, et al. , Myogenic vector expression of insulin-like growth factor I stimulates muscle cell differentiation and myofiber hypertrophy in transgenic mice. J Biol Chem, 1995. 270(20): p. 12109–16. [DOI] [PubMed] [Google Scholar]
- 91.Corral-Gudino L, Borao-Cengotita-Bengoa M, Del Pino-Montes J, et al. , Epidemiology of Paget’s disease of bone: a systematic review and meta-analysis of secular changes. Bone, 2013. 55(2): p. 347–52. [DOI] [PubMed] [Google Scholar]
- 92.Andreasen CM, Delaisse JM, van der Eerden BC, et al. , Understanding Age-Induced Cortical Porosity in Women: The Accumulation and Coalescence of Eroded Cavities Upon Existing Intracortical Canals Is the Main Contributor. J Bone Miner Res, 2018. 33(4): p. 606–620. [DOI] [PubMed] [Google Scholar]
- 93.Magaziner J, Fredman L, Hawkes W, et al. , Changes in functional status attributable to hip fracture: a comparison of hip fracture patients to community-dwelling aged. Am J Epidemiol, 2003. 157(11): p. 1023–31. [DOI] [PubMed] [Google Scholar]
- 94.Roche JJ, Wenn RT, Sahota O, et al. , Effect of comorbidities and postoperative complications on mortality after hip fracture in elderly people: prospective observational cohort study. BMJ, 2005. 331(7529): p. 1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wong CC and McGirt MJ, Vertebral compression fractures: a review of current management and multimodal therapy. J Multidiscip Healthc, 2013. 6: p. 205–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Gehlbach SH, Burge RT, Puleo E, et al. , Hospital care of osteoporosis-related vertebral fractures. Osteoporos Int, 2003. 14(1): p. 53–60. [DOI] [PubMed] [Google Scholar]
- 97.Truumees E, Osteoporosis. Spine (Phila Pa 1976), 2001. 26(8): p. 930–2. [DOI] [PubMed] [Google Scholar]
- 98.Papadakis M, Sapkas G, Papadopoulos EC, et al. , Pathophysiology and biomechanics of the aging spine. Open Orthop J, 2011. 5: p. 335–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Nojiri H, Saita Y, Morikawa D, et al. , Cytoplasmic superoxide causes bone fragility owing to low-turnover osteoporosis and impaired collagen cross-linking. J Bone Miner Res, 2011. 26(11): p. 2682–94. [DOI] [PubMed] [Google Scholar]
- 100.Chen Q, Liu K, Robinson AR, et al. , DNA damage drives accelerated bone aging via an NF-kappaB-dependent mechanism. J Bone Miner Res, 2013. 28(5): p. 1214–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Onal M, Piemontese M, Xiong J, et al. , Suppression of autophagy in osteocytes mimics skeletal aging. J Biol Chem, 2013. 288(24): p. 17432–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Li Y, Charif N, Mainard D, et al. , Donor’s age dependent proliferation decrease of human bone marrow mesenchymal stem cells is linked to diminished clonogenicity. Biomed Mater Eng, 2014. 24(1 Suppl): p. 47–52. [DOI] [PubMed] [Google Scholar]
- 103.Ma Y, Qi M, An Y, et al. , Autophagy controls mesenchymal stem cell properties and senescence during bone aging. Aging Cell, 2018. 17(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Akune T, Ohba S, Kamekura S, et al. , PPARgamma insufficiency enhances osteogenesis through osteoblast formation from bone marrow progenitors. J Clin Invest, 2004. 113(6): p. 846–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lu C, Miclau T, Hu D, et al. , Cellular basis for age-related changes in fracture repair. J Orthop Res, 2005. 23(6): p. 1300–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Pignolo RJ, Suda RK, McMillan EA, et al. , Defects in telomere maintenance molecules impair osteoblast differentiation and promote osteoporosis. Aging Cell, 2008. 7(1): p. 23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Saeed H, Abdallah BM, Ditzel N, et al. , Telomerase-deficient mice exhibit bone loss owing to defects in osteoblasts and increased osteoclastogenesis by inflammatory microenvironment. J Bone Miner Res, 2011. 26(7): p. 1494–505. [DOI] [PubMed] [Google Scholar]
- 108.Gronthos S, Chen S, Wang CY, et al. , Telomerase accelerates osteogenesis of bone marrow stromal stem cells by upregulation of CBFA1, osterix, and osteocalcin. J Bone Miner Res, 2003. 18(4): p. 716–22. [DOI] [PubMed] [Google Scholar]
- 109.Yudoh K and Nishioka K, Telomerized presenescent osteoblasts prevent bone mass loss in vivo. Gene Ther, 2004. 11(11): p. 909–15. [DOI] [PubMed] [Google Scholar]
- 110.Stevens JR, Miranda-Carboni GA, Singer MA, et al. , Wnt10b deficiency results in age-dependent loss of bone mass and progressive reduction of mesenchymal progenitor cells. J Bone Miner Res, 2010. 25(10): p. 2138–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ozcivici E, Luu YK, Adler B, et al. , Mechanical signals as anabolic agents in bone. Nat Rev Rheumatol, 2010. 6(1): p. 50–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Moerman EJ, Teng K, Lipschitz DA, et al. , Aging activates adipogenic and suppresses osteogenic programs in mesenchymal marrow stroma/stem cells: the role of PPAR-gamma2 transcription factor and TGF-beta/BMP signaling pathways. Aging Cell, 2004. 3(6): p. 379–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Xian L, Wu X, Pang L, et al. , Matrix IGF-1 maintains bone mass by activation of mTOR in mesenchymal stem cells. Nat Med, 2012. 18(7): p. 1095–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hoeppner LH, Secreto FJ, and Westendorf JJ, Wnt signaling as a therapeutic target for bone diseases. Expert Opin Ther Targets, 2009. 13(4): p. 485–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Duque G, Li W, Vidal C, et al. , Pharmacological inhibition of PPARgamma increases osteoblastogenesis and bone mass in male C57BL/6 mice. J Bone Miner Res, 2013. 28(3): p. 639–48. [DOI] [PubMed] [Google Scholar]
- 116.Lean JM, Davies JT, Fuller K, et al. , A crucial role for thiol antioxidants in estrogen-deficiency bone loss. J Clin Invest, 2003. 112(6): p. 915–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Zainabadi K, Liu CJ, Caldwell ALM, et al. , SIRT1 is a positive regulator of in vivo bone mass and a therapeutic target for osteoporosis. PLoS One, 2017. 12(9): p. e0185236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Sahota O, Osteoporosis and the role of vitamin D and calcium-vitamin D deficiency, vitamin D insufficiency and vitamin D sufficiency. Age Ageing, 2000. 29(4): p. 301–4. [DOI] [PubMed] [Google Scholar]
- 119.Jahn K, Kelkar S, Zhao H, et al. , Osteocytes Acidify Their Microenvironment in Response to PTHrP In Vitro and in Lactating Mice In Vivo. J Bone Miner Res, 2017. 32(8): p. 1761–1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Donahue SW, Jacobs CR, and Donahue HJ, Flow-induced calcium oscillations in rat osteoblasts are age, loading frequency, and shear stress dependent. Am J Physiol Cell Physiol, 2001. 281(5): p. C1635–41. [DOI] [PubMed] [Google Scholar]
- 121.Kitase Y, Vallejo JA, Gutheil W, et al. , beta-aminoisobutyric Acid, l-BAIBA, Is a Muscle-Derived Osteocyte Survival Factor. Cell Rep, 2018. 22(6): p. 1531–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Delgado-Calle J, Sanudo C, Fernandez AF, et al. , Role of DNA methylation in the regulation of the RANKL-OPG system in human bone. Epigenetics, 2012. 7(1): p. 83–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Cohen-Kfir E, Artsi H, Levin A, et al. , Sirt1 is a regulator of bone mass and a repressor of Sost encoding for sclerostin, a bone formation inhibitor. Endocrinology, 2011. 152(12): p. 4514–24. [DOI] [PubMed] [Google Scholar]
- 124.Lian JB, Stein GS, van Wijnen AJ, et al. , MicroRNA control of bone formation and homeostasis. Nat Rev Endocrinol, 2012. 8(4): p. 212–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Li H, Xie H, Liu W, et al. , A novel microRNA targeting HDAC5 regulates osteoblast differentiation in mice and contributes to primary osteoporosis in humans. J Clin Invest, 2009. 119(12): p. 3666–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Wang FS, Chuang PC, Lin CL, et al. , MicroRNA-29a protects against glucocorticoid-induced bone loss and fragility in rats by orchestrating bone acquisition and resorption. Arthritis Rheum, 2013. 65(6): p. 1530–40. [DOI] [PubMed] [Google Scholar]
- 127.Kneissel M, Luong-Nguyen NH, Baptist M, et al. , Everolimus suppresses cancellous bone loss, bone resorption, and cathepsin K expression by osteoclasts. Bone, 2004. 35(5): p. 1144–56. [DOI] [PubMed] [Google Scholar]
- 128.Bolton MC, Dudhia J, and Bayliss MT, Age-related changes in the synthesis of link protein and aggrecan in human articular cartilage: implications for aggregate stability. Biochem J, 1999. 337 ( Pt 1): p. 77–82. [PMC free article] [PubMed] [Google Scholar]
- 129.Price JS, Waters JG, Darrah C, et al. , The role of chondrocyte senescence in osteoarthritis. Aging Cell, 2002. 1(1): p. 57–65. [DOI] [PubMed] [Google Scholar]
- 130.Huang H, Skelly JD, Ayers DC, et al. , Age-dependent Changes in the Articular Cartilage and Subchondral Bone of C57BL/6 Mice after Surgical Destabilization of Medial Meniscus. Sci Rep, 2017. 7: p. 42294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Martin JA, Klingelhutz AJ, Moussavi-Harami F, et al. , Effects of oxidative damage and telomerase activity on human articular cartilage chondrocyte senescence. J Gerontol A Biol Sci Med Sci, 2004. 59(4): p. 324–37. [DOI] [PubMed] [Google Scholar]
- 132.Carlo MD Jr. and Loeser RF, Increased oxidative stress with aging reduces chondrocyte survival: correlation with intracellular glutathione levels. Arthritis Rheum, 2003. 48(12): p. 3419–30. [DOI] [PubMed] [Google Scholar]
- 133.Loeser RF, Carlson CS, Del Carlo M, et al. , Detection of nitrotyrosine in aging and osteoarthritic cartilage: Correlation of oxidative damage with the presence of interleukin-1beta and with chondrocyte resistance to insulin-like growth factor 1. Arthritis Rheum, 2002. 46(9): p. 2349–57. [DOI] [PubMed] [Google Scholar]
- 134.Madej W, van Caam A, Davidson EN, et al. , Ageing is associated with reduction of mechanically-induced activation of Smad2/3P signaling in articular cartilage. Osteoarthritis Cartilage, 2016. 24(1): p. 146–57. [DOI] [PubMed] [Google Scholar]
- 135.Loeser RF, Shanker G, Carlson CS, et al. , Reduction in the chondrocyte response to insulin-like growth factor 1 in aging and osteoarthritis: studies in a non-human primate model of naturally occurring disease. Arthritis Rheum, 2000. 43(9): p. 2110–20. [DOI] [PubMed] [Google Scholar]
- 136.Blaney Davidson EN, Scharstuhl A, Vitters EL, et al. , Reduced transforming growth factor-beta signaling in cartilage of old mice: role in impaired repair capacity. Arthritis Res Ther, 2005. 7(6): p. R1338–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Bank RA, Bayliss MT, Lafeber FP, et al. , Ageing and zonal variation in post-translational modification of collagen in normal human articular cartilage. The age-related increase in non-enzymatic glycation affects biomechanical properties of cartilage. Biochem J, 1998. 330 ( Pt 1): p. 345–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Dejica VM, Mort JS, Laverty S, et al. , Increased type II collagen cleavage by cathepsin K and collagenase activities with aging and osteoarthritis in human articular cartilage. Arthritis Res Ther, 2012. 14(3): p. R113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Bui C, Barter MJ, Scott JL, et al. , cAMP response element-binding (CREB) recruitment following a specific CpG demethylation leads to the elevated expression of the matrix metalloproteinase 13 in human articular chondrocytes and osteoarthritis. FASEB J, 2012. 26(7): p. 3000–11. [DOI] [PubMed] [Google Scholar]
- 140.Steck E, Boeuf S, Gabler J, et al. , Regulation of H19 and its encoded microRNA-675 in osteoarthritis and under anabolic and catabolic in vitro conditions. J Mol Med (Berl), 2012. 90(10): p. 1185–95. [DOI] [PubMed] [Google Scholar]
- 141.Zhang M, Lu Q, Budden T, et al. , NFAT1 protects articular cartilage against osteoarthritic degradation by directly regulating transcription of both anabolic and catabolic genes Bone and Joint Research, 2019. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Wang J, Gardner BM, Lu Q, et al. , Transcription factor Nfat1 deficiency causes osteoarthritis through dysfunction of adult articular chondrocytes. J Pathol, 2009. 219(2): p. 163–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Zhang M, Lu Q, Miller AH, et al. , Dynamic epigenetic mechanisms regulate age-dependent SOX9 expression in mouse articular cartilage. Int J Biochem Cell Biol, 2016. 72: p. 125–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Rodova M, Lu Q, Li Y, et al. , Nfat1 regulates adult articular chondrocyte function through its age-dependent expression mediated by epigenetic histone methylation. J Bone Miner Res, 2011. 26(8): p. 1974–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Zhang M, Lu Q, Egan B, et al. , Epigenetically mediated spontaneous reduction of NFAT1 expression causes imbalanced metabolic activities of articular chondrocytes in aged mice. Osteoarthritis Cartilage, 2016. 24(7): p. 1274–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Kannus P, Paavola M, & Józsa L, Aging and degeneration of tendons, in Tendon Injuries: Basic Science and Clinical Medicine 2005, Springer; London. p. 25–31. [Google Scholar]
- 147.Vafek EC, Plate JF, Friedman E, et al. , The effect of strain and age on the mechanical properties of rat Achilles tendons. Muscles Ligaments Tendons J, 2017. 7(3): p. 548–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Thornton GM, Lemmex DB, Ono Y, et al. , Aging affects mechanical properties and lubricin/PRG4 gene expression in normal ligaments. J Biomech, 2015. 48(12): p. 3306–11. [DOI] [PubMed] [Google Scholar]
- 149.Peffers MJ, Thorpe CT, Collins JA, et al. , Proteomic analysis reveals age-related changes in tendon matrix composition, with age- and injury-specific matrix fragmentation. J Biol Chem, 2014. 289(37): p. 25867–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Kietrys DM, Barr-Gillespie AE, Amin M, et al. , Aging contributes to inflammation in upper extremity tendons and declines in forelimb agility in a rat model of upper extremity overuse. PLoS One, 2012. 7(10): p. e46954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Yu TY, Pang JH, Wu KP, et al. , Aging is associated with increased activities of matrix metalloproteinase-2 and -9 in tenocytes. BMC Musculoskelet Disord, 2013. 14: p. 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Arnoczky SP, Lavagnino M, Egerbacher M, et al. , Matrix metalloproteinase inhibitors prevent a decrease in the mechanical properties of stress-deprived tendons: an in vitro experimental study. Am J Sports Med, 2007. 35(5): p. 763–9. [DOI] [PubMed] [Google Scholar]
- 153.Tsai WC, Chang HN, Yu TY, et al. , Decreased proliferation of aging tenocytes is associated with down-regulation of cellular senescence-inhibited gene and up-regulation of p27. J Orthop Res, 2011. 29(10): p. 1598–603. [DOI] [PubMed] [Google Scholar]
- 154.Leong DJ and Sun HB, Mesenchymal stem cells in tendon repair and regeneration: basic understanding and translational challenges. Ann N Y Acad Sci, 2016. 1383(1): p. 88–96. [DOI] [PubMed] [Google Scholar]
- 155.Komatsu I, Wang JH, Iwasaki K, et al. , The effect of tendon stem/progenitor cell (TSC) sheet on the early tendon healing in a rat Achilles tendon injury model. Acta Biomater, 2016. 42: p. 136–146. [DOI] [PubMed] [Google Scholar]
- 156.Zhou Z, Akinbiyi T, Xu L, et al. , Tendon-derived stem/progenitor cell aging: defective self-renewal and altered fate. Aging Cell, 2010. 9(5): p. 911–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Kohler J, Popov C, Klotz B, et al. , Uncovering the cellular and molecular changes in tendon stem/progenitor cells attributed to tendon aging and degeneration. Aging Cell, 2013. 12(6): p. 988–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Chen L, Wang GD, Liu JP, et al. , miR-135a modulates tendon stem/progenitor cell senescence via suppressing ROCK1. Bone, 2015. 71: p. 210–6. [DOI] [PubMed] [Google Scholar]
- 159.Han W, Wang B, Liu J, et al. , The p16/miR-217/EGR1 pathway modulates age-related tenogenic differentiation in tendon stem/progenitor cells. Acta Biochim Biophys Sin (Shanghai), 2017. 49(11): p. 1015–1021. [DOI] [PubMed] [Google Scholar]
- 160.Popov C, Kohler J, and Docheva D, Activation of EphA4 and EphB2 Reverse Signaling Restores the Age-Associated Reduction of Self-Renewal, Migration, and Actin Turnover in Human Tendon Stem/Progenitor Cells. Front Aging Neurosci, 2015. 7: p. 246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Ling SC, Chen CF, and Wan RX, A study on the vascular supply of the supraspinatus tendon. Surg Radiol Anat, 1990. 12(3): p. 161–5. [DOI] [PubMed] [Google Scholar]
- 162.Chen TM, Rozen WM, Pan WR, et al. , The arterial anatomy of the Achilles tendon: anatomical study and clinical implications. Clin Anat, 2009. 22(3): p. 377–85. [DOI] [PubMed] [Google Scholar]
- 163.Marqueti RC, Durigan JLQ, Oliveira AJS, et al. , Effects of aging and resistance training in rat tendon remodeling. FASEB J, 2018. 32(1): p. 353–368. [DOI] [PubMed] [Google Scholar]
- 164.Chard MD, Cawston TE, Riley GP, et al. , Rotator cuff degeneration and lateral epicondylitis: a comparative histological study. Ann Rheum Dis, 1994. 53(1): p. 30–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Thampatty BP and Wang JH, Mechanobiology of young and aging tendons: In vivo studies with treadmill running. J Orthop Res, 2018. 36(2): p. 557–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Barber L, Scicchitano BM, and Musaro A, Molecular and Cellular Mechanisms of Muscle Aging and Sarcopenia and Effects of Electrical Stimulation in Seniors. Eur J Transl Myol, 2015. 25(4): p. 231–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Morley JE, Pharmacologic Options for the Treatment of Sarcopenia. Calcif Tissue Int, 2016. 98(4): p. 319–33. [DOI] [PubMed] [Google Scholar]
- 168.Palus S, Springer JI, Doehner W, et al. , Models of sarcopenia: Short review. Int J Cardiol, 2017. 238: p. 19–21. [DOI] [PubMed] [Google Scholar]
- 169.Wilton JM, Denosumab: new horizons in the treatment of osteoporosis. Nurs Womens Health, 2011. 15(3): p. 249–52. [DOI] [PubMed] [Google Scholar]
- 170.Lawrence RC, Felson DT, Helmick CG, et al. , Estimates of the prevalence of arthritis and other rheumatic conditions in the United States. Part II. Arthritis Rheum, 2008. 58(1): p. 26–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Loeser RF, Aging and osteoarthritis: the role of chondrocyte senescence and aging changes in the cartilage matrix. Osteoarthritis Cartilage, 2009. 17(8): p. 971–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Shane Anderson A and Loeser RF, Why is osteoarthritis an age-related disease? Best Pract Res Clin Rheumatol, 2010. 24(1): p. 15–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Goldring MB and Goldring SR, Osteoarthritis. J Cell Physiol, 2007. 213(3): p. 626–34. [DOI] [PubMed] [Google Scholar]
- 174.Buckwalter JA and Martin JA, Osteoarthritis. Adv Drug Deliv Rev, 2006. 58(2): p. 150–67. [DOI] [PubMed] [Google Scholar]
- 175.Hellio le Graverand MP, Clemmer RS, Redifer P, et al. , A 2-year randomised, double-blind, placebo-controlled, multicentre study of oral selective iNOS inhibitor, cindunistat (SD-6010), in patients with symptomatic osteoarthritis of the knee. Ann Rheum Dis, 2013. 72(2): p. 187–95. [DOI] [PubMed] [Google Scholar]
- 176.Le Graverand-Gastineau MP, Disease modifying osteoarthritis drugs: facing development challenges and choosing molecular targets. Curr Drug Targets, 2010. 11(5): p. 528–35. [DOI] [PubMed] [Google Scholar]
- 177.Kurtz SM, Lau EC, Ong KL, et al. , Which Clinical and Patient Factors Influence the National Economic Burden of Hospital Readmissions After Total Joint Arthroplasty? Clin Orthop Relat Res, 2017. 475(12): p. 2926–2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Guan YJ, Li J, Yang X, et al. , Evidence that miR-146a attenuates aging- and trauma-induced osteoarthritis by inhibiting Notch1, IL-6, and IL-1 mediated catabolism. Aging Cell, 2018: p. e12752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.McGee-Lawrence ME, Bradley EW, Dudakovic A, et al. , Histone deacetylase 3 is required for maintenance of bone mass during aging. Bone, 2013. 52(1): p. 296–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Andres BM and Murrell GA, Treatment of tendinopathy: what works, what does not, and what is on the horizon. Clin Orthop Relat Res, 2008. 466(7): p. 1539–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Fitzpatrick J, Bulsara M, and Zheng MH, The Effectiveness of Platelet-Rich Plasma in the Treatment of Tendinopathy: A Meta-analysis of Randomized Controlled Clinical Trials. Am J Sports Med, 2017. 45(1): p. 226–233. [DOI] [PubMed] [Google Scholar]
- 182.Huang J, Romero-Suarez S, Lara N, et al. , Crosstalk between MLO-Y4 osteocytes and C2C12 muscle cells is mediated by the Wnt/beta-catenin pathway. JBMR Plus, 2017. 1(2): p. 86–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Qin Y, Peng Y, Zhao W, et al. , Myostatin inhibits osteoblastic differentiation by suppressing osteocyte-derived exosomal microRNA-218: A novel mechanism in muscle-bone communication. J Biol Chem, 2017. 292(26): p. 11021–11033. [DOI] [PMC free article] [PubMed] [Google Scholar]