

Abstract
Hepatitis B virus (HBV) infection is an important public health problem in Canada. In keeping with evolving evidence and understanding of HBV pathogenesis, the Canadian Association for the Study of Liver Disease periodically publishes HBV management guidelines. The goals of the 2018 guidelines are to (1) highlight the public health impact of HBV infection in Canada and the need to improve diagnosis and linkage to care, (2) recommend current best-practice guidelines for treatment of HBV, (3) summarize the key HBV laboratory diagnostic tests, and (4) review evidence on HBV management in special patient populations and include more detail on management of HBV in pediatric populations. An overview of novel HBV tests and therapies for HBV in development is provided to highlight the recent advances in HBV clinical research. The aim and scope of these guidelines are to serve as an up-to-date, comprehensive resource for Canadian health care providers in the management of HBV infection.
Keywords: Association of Medical Microbiology and Infectious Disease (AMMI) Canada, Canadian Association for the Study of Liver Disease (CASL), guidelines, hepatitis B
Introduction and Guideline Development Process (CS Coffin and SK Fung)
Hepatitis B virus (HBV) infection is an important public health problem in Canada. In keeping with evolving evidence and understanding of HBV pathogenesis, the Canadian Association for the Study of Liver Disease (CASL) has periodically published HBV management guidelines (1,2). In 2017, a proposal was brought forward by the HBV Guidelines panel co-chairs (CS Coffin and SK Fung) and a committee member (MM Ma) to the CASL Education Committee to update the 2012 Canadian HBV guidelines. Members of the writing committee panel were recommended by the co-chairs and approved by the chair of the CASL education and the CASL guidelines committees.
The goals of the updated 2018 HBV guidelines are as follows: (a) to highlight the continued public health impact of HBV infection in Canada and the need to improve diagnosis and linkage to care, (b) to recommend current best-practice guidelines for treatment of HBV, (c) to summarize the key HBV laboratory diagnostic tests, and (d) to review evolving evidence on HBV management, especially in special patient populations (ie, HBV-related hepatocellular carcinoma [HCC], renal failure, transplant, pregnancy, immunosuppression, and co-infection). The new guidelines outline more comprehensive details regarding management of hepatitis B in children. In addition, an overview of novel HBV diagnostic tests and new therapies for hepatitis B in development is provided to highlight the recent advances in HBV clinical research. The aim and scope of these guidelines are to serve as an up-to-date comprehensive resource for Canadian health care providers in the management of HBV infection (Table 1).
Table 1:
Agree II instrument used in assessing the 2018 Canadian Management of Hepatitis B Guidelines
| Domain | |
|---|---|
| Domain 1: scope and purpose |
Response: To develop up-to-date evidence-based guidelines on HBV screening, diagnosis, monitoring, and treatment, including special patient populations for clinicians involved in the care of patients with HBV infection. |
| Domain 2: stakeholder involvement |
Response: Representatives from 2 major Canadian medical societies formed the guideline committee, including target stakeholders from tertiary referral hepatology, liver transplant, and infectious disease clinics in 5 provincial jurisdictions. The target audience is hepatologists and infectious disease and other health care professionals involved in treating HBV infection. Guidelines were disseminated for review and feedback in annual society meetings and through society newsletter emails. |
| Domain 3: rigour of development |
|
| Domain 3: rigour of development |
Response: The writing committee utilized the Grades of Recommendation, Assessment Development and Evaluation scale (GRADE method) for grading the strength and quality of supporting evidence for each recommendation. Each specific section or recommendation was reviewed by committee members and subjected to vote and approval. The guideline draft was presented to CASL leadership and AMMI for review and feedback. A similar process for updating the guidelines has been established as per the CASL Education Committee and CASL Guidelines Writing Committee (ie, formal application and approval by the guidelines committee). |
| Domain 4: clarity of presentation |
Response: All sections have a clear recommendations and alternatives discussed as appropriate. |
| Domain 5: applicability |
Response: All sections incorporate best practice, accounting for barriers and resource limitations, as appropriate. |
| Domain 6: editorial independence |
No funding was provided to develop these guidelines. All conflict of interests by committee members were submitted to the chair of the CASL guidelines committee and pre-approved. |
Note: AMMI = Association of Medical Microbiology and Infectious Disease Canada; CASL = Canadian Association for the Study of the Liver; HBV = hepatitis B virus.
This document was presented to the membership of CASL and to the Association of Medical Microbiology and Infectious Disease Canada (AMMI) for official endorsement by both societies. The Appraisal of Guidelines for Research & Evaluation instrument was used as a framework to assess the quality of guidelines, provide a methodological strategy for the development of guidelines, and inform what and how information ought to be reported (https://www.agreetrust.org) (3) (Table 1). In addition, the strength of the recommendation and evidence for each key recommendation was rated according to the Grades of Recommendation, Assessment Development and Evaluation working scale (https://www.gradeworkinggroup.org) (4) (Table 2). A list of abbreviations commonly used in this article can be found in the Appendix.
Table 2:
Guideline development using the Grades of Recommendation, Assessment Development and Evaluation Scale (level of evidence) according to study design
| Grade | Definition |
|---|---|
| I | Randomized controlled trials |
| II-1 | Controlled trials without randomization |
| II-2 | Cohort or case-controlled studies |
| II-3 | Multiple time series, dramatic uncontrolled experiments |
| III | Opinion of respected authorities. Descriptive epidemiology |
Notes: Each recommendation was based on quality of the supporting evidence and study design and graded as class-grade: 1 (high), 2-A, 2-B, 2-C (moderate), or 3 (low) quality evidence.
1.0. PUBLIC HEALTH IMPLICATIONS OF HEPATITIS B (HH KO, MM MA, E TAM)
1.1. Epidemiology and public health burden of hepatitis B infection in Canada
HBV infection is one of the most common infections in the world. There are an estimated 257 million chronic carriers and approximately 900,000 deaths annually from cirrhosis and HCC (5–7). HBV infection prevalence is highest in the Western Pacific (6.2%) and African regions (6.1%). In Canada, hepatitis B is a notifiable disease that is reported to the Canadian Notifiable Disease Surveillance System by provincial and territorial health authorities (8). The overall acute infection rate is low in Canada and has declined from 1.0 to 0.5 per 100,000 between 2005 and 2013 (Figure 1A). This decline is largely attributable to the introduction of universal immunization programs in all provinces and territories since the early 1990s (9,10). However, it is also important to realize that the reported acute infection rate likely remains an underestimate because most people with acute HBV infection are asymptomatic and do not present for medical care or testing and remain unidentified.
Figure 1:
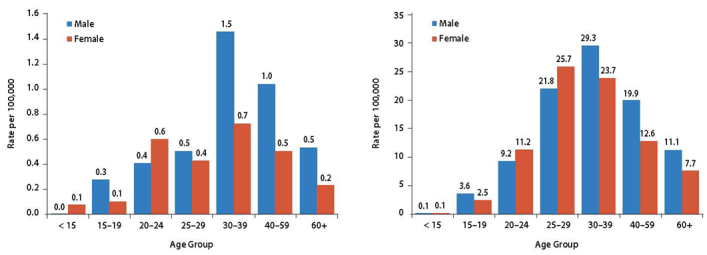
Rates of reported cases of acute (A) and chronic (B) hepatitis B virus infection in Canada by age group in 2013, according to the Canadian Notifiable Disease Surveillance System.
Government of Canada. 2017. Report on hepatitis B and C in Canada: 2014. https://www.canada.ca/en/services/health/publications/diseases-conditions/report-hepatitis-b-c-canada-2014.html
According to the Canadian Notifiable Disease Surveillance System, the rate of chronic hepatitis B (CHB) declined from 13.6 to 12.0 per 100,000 between 2009 and 2013. The highest rates were observed among men ranging in age from 30 to 39 years, followed by women ranging in age from 25 to 29 years (rates of 29.3 and 25.7 per 100,000, respectively) (Figure 1B). CHB rates higher than the national average were observed in British Columbia (23.4 per 100,000), Ontario (15.1 per 100,000), and Alberta (14.6 per 100,000) (6). CHB mostly affects individuals or populations that have not received routine immunization or are immigrants from endemic countries (5,7,8,11). In a childhood HBV surveillance study, non–Canadian-born children had a relative risk 12 times higher than that of Canadian-born children (12,13), and more than one family member is usually affected. Among Canadian-born adults, surveillance data have shown that risk factors associated with acute and chronic infection include having a chronic carrier as a family member, injection drug use, high-risk sexual activity, body piercing and tattooing, and history of blood transfusion (14–17). One review estimated the overall prevalence of HBV carriers in Canada at approximately 2%, with high-risk groups including immigrants, Indigenous populations, and street-connected individuals (11). The seroprevalence rate is highest in urban centres, and it is not uniformly distributed across the country. A recent prevalence assessment by the Canadian Liver Foundation estimated that there are 250,000–460,000 individuals with CHB infection in Canada, with approximately 50% of the carriers living in Ontario (18). Overall, the health care burden of hepatitis B remains high in Canada despite the availability of an effective vaccine for many years and implementation of a universal immunization program since the early 1990s. This high burden is related to immigration of families from countries in which hepatitis B is endemic and the incomplete success of universal and catch-up vaccination programs. Although the estimated national childhood hepatitis B vaccine coverage for more than one dose of the vaccine by age 17 years was 87.9% in 2013 (10), the prevalence of hepatitis B vaccine–induced immunity was only 52% for people aged 25–29 years and less than 30% for the age group older than 30 years (17).
CHB can progress to cirrhosis and complications of end-stage liver disease in as many as 20%–25% of individuals (19–21). The annual incidence of HCC is estimated at less than 1% in non-cirrhotic individuals and 2%–3% in those with cirrhosis (22–24). As reported in the Ontario Burden of Infectious Diseases Study, hepatitis B was the fifth-ranked pathogen causing significant health-adjusted life-years lost (25). The health burden of CHB among immigrants is even more substantial, with modelling data suggesting that immigrants with CHB lost an average of 4.6 life-years and had a higher lifetime risk of decompensated cirrhosis (12%), HCC (16%), and need for liver transplant (5%) (26).
1.2. Hepatitis B vaccination
Reducing the disease burden of CHB is contingent on preventing chronic infection through vaccination. Because the likelihood of developing CHB is greatest when exposure occurs in infancy or early childhood, the ideal strategy is to offer universal vaccination to susceptible neonates or infants. Through this strategy, the worldwide incidence of CHB in children aged younger than 5 years has fallen from 4.7% in the pre-vaccination era to 1.3% in 2015 (7). In Canada, only New Brunswick, Nunavut, and the Northwest Territories provide HBV vaccination to newborns at birth; British Columbia, Quebec, Yukon, and Prince Edward Island provide vaccination at 2 months and in pre-adolescence to capture any children who may have missed infant vaccination (i.e., new to the province or territory). Other jurisdictions do not routinely provide a birth dose of the HBV vaccine unless the neonate is deemed to be at risk through maternal screening for hepatitis B surface antigen (HBsAg), missing the opportunity to prevent early horizontal transmission (8). This interprovincial difference highlights the need for a harmonized national HBV vaccination schedule to prevent both vertical and horizontal hepatitis B transmission. For example, if a child relocates from one province or territory school system to another at a critical time, he or she may miss the opportunity to participate in school-administered vaccination programs.
Candidates for hepatitis B vaccine include all children and adolescents as well as high-risk individuals (Table 3) (8). Pre-immunization serologic screening is not routinely recommended, but it is recommended for those at high risk of infection, and HBsAg, antibody to HBsAg (anti-HBs), and antibody to HBV core (anti-HBc) testing should be done beforehand. For HIV-infected and other potentially immunocompromised adults (ie, transplant patients, dialysis patients, patients with decompensated cirrhosis), there is evidence supporting the use of a double-dose strategy for both the initial and the repeat vaccination series to increase response rate and longer durability of seroprotective responses (27,28). In adults with risk factors for reduced immune responses (including people with diabetes), newer HBV vaccine formulations may be an option, with approval of recombinant hepatitis B vaccine (HEPLISAV-B) by the US Food and Drug Administration. HEPLISAV-B is conjugated to a toll-like receptor 9 agonist adjuvant to enhance immunogenicity. In a phase 3 blinded 2:1 randomized trial of 8,374 participants, HEPLISAV-B induced significantly higher seroprotective rates after administration of two doses over 4 weeks compared with three doses of Engerix-B over a 24-week period (29).
Table 3:
Screening recommendations for hepatitis B in persons not known to be HBV immune or vaccinated
|
Note: High-risk individuals who test negative for hepatitis B surface antigen and HBV surface antibody negative should be offered the vaccine. HBV = hepatitis B virus. Adopted from the Canadian Liver Foundation (www.liver.ca) and Public Health Agency of Canada National Immunization Guide
Routine serologic testing to assess vaccine response is not recommended. After vaccination, the titre of anti-HBs decreases over time. After 10 years, more than one-third of children vaccinated during infancy will have anti-HBs titres below the accepted protective antibody level of 10 IU/L. However, hepatitis B–vaccinated adults can mount a protective immune response even 18 years after receiving a primary series of vaccinations. In those at high risk of exposure to HBV or those less likely to respond because they are immunocompromised (Table 3), post-vaccination anti-HBs serology is recommended within 1–6 months after the last dose. Routine booster vaccination (ie, single dose of the HBV vaccine) is not indicated in average-risk populations, but it may be considered in close household contacts of HBsAg-positive individuals or other high-risk individuals. For healthy adults who do not respond to the first series of vaccines, the Canadian Immunization guide recommends a second vaccine series. With completion of the second series (up to three doses), a protective anti-HBs level will be elicited in 50%–70% of individuals who failed the initial vaccine series (30).
1.3. Hepatitis B screening
CHB infection is often asymptomatic, and screening of high-risk groups is important to identify individuals for counselling to reduce the risk of transmission, monitor disease progression, identify those who require HCC surveillance, and provide antiviral therapy to those at risk of liver-related complications such as cirrhosis or HCC. The high-risk groups for whom screening should be considered are summarized in Table 3. All candidate immigrants to Canada should undergo screening for hepatitis B during their medical evaluation, providing an opportunity for those who are unaware of their hepatitis B status to be evaluated and treated, if indicated. These high-risk groups should be screened for CHB infection with HBsAg, anti-HBs, and anti-HBc. If the HBsAg test is positive, HBV e antigen (HBeAg), HBV e antibody (anti-HBe), and viral load (HBV DNA) should be checked (Tables 4–5). The interpretation of serologic tests is discussed in Sections 5.0 and 6.0.
Table 4:
Summary of HBV serological tests
| Anti-HBs | Anti-HBc | Anti-HBe | HBsAg | HBeAg | ||
|---|---|---|---|---|---|---|
| IgM | IgG/IgM total | |||||
| Immunization | A marker of immunization. Anti-HBs will be the sole seromarker present (with history of immunization) | |||||
| Acute infection | A marker of acute infection. May also indicate severe acute exacerbation of chronic infection, thus requiring clinical or epidemiological history to distinguish between acute HBV infection and severe exacerbation of chronic HBV infection. | A marker of infection or infectivity | ||||
| Previous or current (chronic) infection | A marker of previous infection (after seroconversion and in association with the presence of other HBV antibodies) | A marker of previous or current infection, depending on the presence of other serological and molecular markers of HBV infection. The presence of anti-HBc in isolation is associated with occult hepatitis B. | A marker of chronic infection indicating the phase of infection* | When positive at ≥6 months, a marker of chronic infection | A marker of viral replication and infectivity indicating the phase of infection* | |
Note: ALT = alanine aminotransferase; HB = hepatitis B; anti-HBc = antibody to HBV core; anti-HBe = antibody to HBeAg; HBeAg = HBV e antigen; anti-HBs = hepatitis B surface antibody; HBsAg = hepatitis B surface antigen; HBV = hepatitis B virus; IgG = immunoglobulin G; IgM = Immunoglobulin M
* HBeAg positive, anti-HBe negative: characterized by high HBV DNA and normal ALT (phase 1) or elevated–fluctuating ALT levels (Phase 2); HBeAg negative, anti-HBe positive: indicates the inactive carrier phase of chronic hepatitis associated with low HBV DNA and normal ALT levels (phase 3) or HBeAg-negative chronic hepatitis in the context of fluctuating HBV DNA and ALT levels, often because of the presence of mutations reducing or eliminating HBeAg expression (phase 4), according to the revised classification system.
Table 5:
Phases of chronic HBV infection according to revised and classical definitions
|
Phase 1: HBeAg + chronic infection (old terminology immune tolerance) |
Phase 2: HBeAg + chronic hepatitis (old terminology immune active) |
Phase 3: HBeAg – chronic infection (old terminology inactive carrier) |
Phase 4: HBeAg – chronic hepatitis (old terminology, HBeAg-negative chronic hepatitis) |
Phase 5: HBsAg negative or OHB |
|
|---|---|---|---|---|---|
| HBsAg | Positive | Positive | Positive | Positive | Negative |
| HBsAb | Negative | Negative | Negative | Negative | Positive or negative |
| HBeAg | Positive | Positive | Negative | Negative | Negative |
| HBV DNA IU/mL* | Often > 107 | 104–107 | Often < 2,000; sometimes > 2,000 | 103–107 | Negative or trace amount |
| ALT | Normal | Elevated or fluctuating | Normal | Often fluctuating | Normal |
| Phase | Mostly in young patients but could extend into the 4th or 5th decades | Young patients to 5th decade with active hepatitis | Variable duration with HBV immune control | Mostly in older patients with intermittent flare of hepatitis | Immune clearance of HBV or immune control of the virus with OHB |
| Non-invasive fibrosis assessment or biopsy | Normal (recent data suggesting that individuals may be at higher risk HCC) | Abnormal | Normal or mildly abnormal | Abnormal | Normal |
| Treatment | No | Yes (if no signs of spontaneous seroconversion because prolonged duration of hepatitis increases fibrosis risk) | No | Yes | No (except during immunosuppression) |
Note: All patients should be fully evaluated, including history, physical exam, liver tests, etc. (see section 4.0) to determine need for treatment. ALT = alanine aminotransferase; HBeAg = HBV e antigen; HBsAb = hepatitis B surface antibody; HBsAg = hepatitis B surface antigen; HBV = hepatitis B virus; HBV DNA = viral load; HCC = hepatocellular carcinoma; OHB = occult hepatitis B
*IU/mL = ~5 virus copies/mL
Recommendations for HBV Diagnosis and Prevention
Health Canada, The Public Health Agency, and the provinces and territories should increase support and development of uniform structural programs for hepatitis B to (a) prevent HBV infection; (b) identify infections (screening, especially in high-risk groups, including all candidate immigrants); and (c) facilitate the assessment, treatment, and education of chronically infected patients. (strong recommendation; class 2, level B)
All Canadian provinces and territories should harmonize the hepatitis B vaccination policy with universal, preferably neonatal or infant, vaccination with catch-up vaccinations for those who have not yet received adolescent vaccination. (strong recommendation; class 3)
Routine booster doses of HBV vaccine are not indicated in average-risk, immune-competent individuals who responded to the primary series of vaccine. (strong recommendation; class 2, level A)
A repeat series (three doses of vaccine) should be offered to those at high risk of exposure or those who are immmunosuppressed and who do not respond to the first series of vaccines. (strong recommendation; class 1)
All high-risk individuals should be screened for HBV infection with HBsAg, anti-HBs, and anti-HBc, and their response to the vaccine should be assessed. (strong recommendation; class 2, level B)
All high-risk individuals who are HBsAg, anti-HBc, and anti-HBs negative should receive the HBV vaccine, and their response to the vaccine should be assessed. (strong recommendation; class 1)
2.0. HBV LIFE CYCLE AND IMMUNOPATHOGENESIS (CS COFFIN)
HBV is a small, non-cytopathic DNA virus and the prototype member of the Hepadnaviridae family. The complete HBV virion or “Dane” particle contains a nucleocapsid with a circular, partially double-stranded, approximately 3,200 base-pair genome surrounded by a lipid envelope (31). The compact genome encodes four overlapping open reading frames and four RNA species that encode seven proteins: three envelope or surface proteins—large, middle, and small—forming the envelope or HBsAg, the nucleocapsid core or C protein carrying HBV core antigen specificity, the secretory HBeAg, the viral reverse transcriptase or polymerase, and the X protein. The HBeAg may be immunoregulatory, whereas the HBx protein interacts with cellular factors and modifies diverse cellular processes, promoting tumourigenesis (32,33).
HBV replication is unique; viral entry is mediated by binding of the HBV pre–surface 1 region to its specific receptor on the hepatocyte cell membrane (ie, sodium taurocholate cotransporting polypeptide, or NTCP). The nucleocapsid is released into the cytosol and transported to the nucleus, and the genomic relaxed circular (rc) DNA is converted into a covalently closed circular (ccc) DNA episome. The HBV cccDNA is the transcriptional template of all viral genes (34). The transcribed pregenomic RNA with bound polymerase is encapsidated into cores and serves as a template for reverse transcription into minus-strand DNA followed by plus-strand DNA synthesis to yield partially double-stranded rcDNA. The rcDNA is packaged into nucleocapsids surrounded by a lipoprotein envelope in the endoplasmic reticulum and exported from the cell as new infectious virions while another pool of cores with rcDNA are recycled to the nucleus (ie, intracellular cccDNA amplification). During chronic infection, up to 1012 virons can be produced daily, which together with the error-prone HBV P translates to 1010-11 point mutations per day, generating a diverse swarm of viral quasispecies. HBV diversity is constrained by overlapping open reading frames that can limit the fitness of mutants formed (35). HBV variants are selected under host and antiviral pressure, leading to immune escape and drug resistance. In some cases of HBeAg-negative hepatitis B, mutations within the HBV pre-core or core-promoter region lead to a premature stop codon that interferes with HBeAg synthesis and secretion without affecting viral replication. These mutations are associated with fulminant hepatitis and development of HCC.
In addition, the HBV genome can randomly integrate very early after virus invasion into host genes encoding proteins used in cell signalling, proliferation, and viability that can result in oncogenesis (ie, HCC). Infected hepatocytes also produce a large excess of non-infectious, subviral surface particles in spheres or filamentous forms (HBsAg) that may serve as immune decoys. It was thought that clearance of acute HBV infection triggered a vigorous innate immune response, type 1 interferons (IFNs), and development of an efficient adaptive immune (cytokine) response and inducing IFN-stimulated genes (36). However, studies in acutely infected chimpanzees (37) and using liver tissue from CHB patients show that the HBV may in fact behave as a stealth virus and evade the innate immune response (38). Despite successful immune clearance and loss of HBsAg, low-level HBV infection can persist indefinitely as occult hepatitis B (OHB), either seronegative or seropositive, with HBV DNA and cccDNA in the liver. Individuals with OHB may remain at risk of reactivation with intense immunosuppression and possibly liver disease (39).
In contrast, during chronic infection, individuals have a compromised innate and adaptive immune response characterized by suboptimal antigen presentation, exhaustion of antigen-specific T cells, and insufficient antibody production (40). HBV is a noncytopathic virus, which means that liver damage is caused not directly by the virus but rather by host immune cells infiltrating the liver. Thus, protracted immune-active flares mediated by HBV-specific T cells are important for clearing HBV from infected hepatocytes, yet they may also cause liver cell damage leading to cirrhosis. Ultimately, new treatment strategies are focusing on achieving either a functional cure (ie, HBsAg loss, immune control) or even a virological cure (targeting cccDNA) for HBV eradication (see section 18, “New HBV Therapies”) (41). A schematic of the HBV life cycle is provided in Figure 2.
Figure 2:
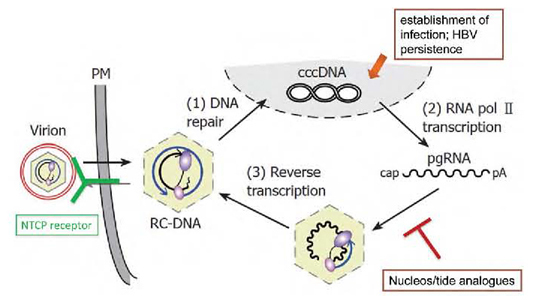
Summary of the HBV lifecycle. Key steps include the formation of intranuclear HBV minichromosome (HBV cccDNA) that is not targeted by current NA therapy. The HBV replicates through an error-prone reverse transcription of an RNA intermediate that lacks proofreading, leading to frequent mutations in the viral genome. HBV persistence is due to ineffective antiviral immune response and resilient HBV cccDNA template.
cccDNA = covalently closed circular DNA; HBV = hepatitis B virus; NA = nucleos(t)ide analogue; NTCP = sodium taurocholate cotransporting polypeptide; pgRNA = pregenomic RNA; RC-DNA = relaxed circular DNA.
3.0. NATURAL HISTORY OF HEPATITIS B INFECTION AND HBV MONITORING (E KELLY, MM MA)
Hepatitis B infection has a complex natural history (Figure 3, Tables 4–5). The outcome of acute hepatitis B infection is affected by the patient’s age and immune status at the time of exposure to the virus. In adults, the infection is often transmitted by high-risk activity such as injection drug use or through sexual contact, and after an acute infection, the infection is usually self-limited and development of lifelong immunity occurs. However, it is also recognized that despite HBsAg clearance and development of natural immunity, OHB can develop because of the persistence of HBV cccDNA, characterized by the presence of low-level HBV DNA and long-lasting HBV-specific T cell responses. These patients may be at risk of HBV reactivation with intense immunosuppression (see section 9). In rare cases (< 1%), acutely infected adults can develop severe hepatitis and fulminant liver failure, and few (< 5%) may become chronically infected with persistence (> 6 months) of serum HBsAg. Most infants or young children acquire HBV infection vertically from mother to infant in the peripartum period or horizontally (from infected family members or close household contacts) through unrecognized close contact with infectious body fluids. Population studies have documented that a significant portion of chronic HBV carriers were infected during infancy or early childhood (25,42). As noted in section 1.2, the immunization strategy of providing only adolescent vaccinations misses the opportunity to prevent early infection in infants or young children, which results in chronic infection in 90% of infants and 25%–50% of young children.
Figure 3:
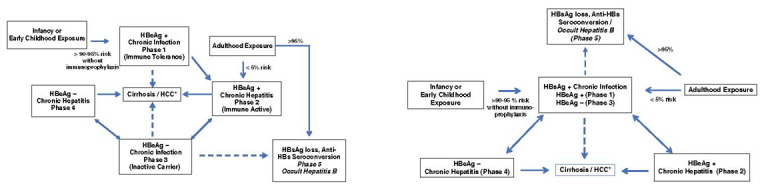
Natural history of CHB infection based on conventional definitions (A). Recent data suggest that the immune-tolerant phase is a misnomer because many patients have active HBV-specific immune responses and unrecognized hepatic necroinflammatory activity. A new classification system has been proposed, differentiating only between chronic infection versus chronic hepatitis (B).
*The risk of cirrhosis and HCC is higher in patients with high HBV DNA levels and ongoing hepatic inflammation (ie, elevated ALT)
ALT = alanine aminotransferase; anti-HBs = antibody to HBsAg; CHB = chronic hepatitis B; HBeAg = HBV e antigen; HBsAg = hepatitis B surface antigen; HBV = hepatitis B virus; HCC = hepatocellular carcinoma
CHB infection is a dynamic disease that can evolve gradually or rapidly. The natural history of HBV infection is described as progressing through distinct phases (Table 5, Figure 3A) of variable duration (42). In vertical perinatal or childhood infection, an HBeAg-positive chronic infection (phase 1; previously termed the immune-tolerant phase) is the norm, lasting 10–40 years. During phase 1, alanine aminotransferase (ALT) is normal (defined as < 19 in female patients and < 30 in male patients; see section 6.0), and the HBV DNA level is very high, with minimal fibrosis and inflammation. However, recent studies show that so-called immune-tolerant individuals show preserved HBV-specific T cell immune responses (43,44), despite normal or high-normal ALT; hence, some experts propose describing the immune-tolerant phase as “high-replicative non-inflammatory.”
The majority of chronically infected individuals can develop HBeAg-positive chronic hepatitis (phase 2; previously termed immune active) in early adulthood (~85% before age 18 years), characterized by elevated ALT and declining HBV DNA levels, that can last for less than 5 to more than 25 years. During this phase, patients seroconvert from HBeAg positive to anti-HBe positive, yet they may be at risk of cirrhosis if persistent immune-mediated necroinflammatory activity occurs. Predictors of HBeAg seroconversion are high ALT, low HBV DNA, patient age less than 40 years, and absence of cirrhosis (42,45–47).
In HBeAg-negative chronic infection (phase 3; previously termed inactive carrier), ALT decreases with low-level HBV DNA and positive anti-HBe, reflecting immune control, with improved prognosis in non-cirrhotic individuals (48). However, some HBeAg-negative patients may present with fluctuating liver enzymes and HBV DNA levels (ie, phase 4, or HBeAg-negative chronic hepatitis) and remain at high risk for fibrosis progression. In patients with HBeAg-negative chronic hepatitis, the HBV often develops immune escape mutations in the HBV pre-core and basal core promoter regions (~30% in some studies; see sections 2.0 and 5.0) (49).
Finally, only 0.5%–0.8% of CHB carriers per year show HBsAg loss (with or without anti-HBs or anti-HBc; phase 5). HBV DNA is very low or undetectable by standard clinical assays in serum with persistent HBV cccDNA and DNA in liver. HBsAg clearance is a very favourable event and deemed a functional cure of chronic infection (20,21,48,50).
The original description of HBV natural history has been challenged by new evidence showing significant HBV-specific T cell response activity in young immune-tolerant individuals (44,51) and that inactive carriers with HBV DNA of less than 2,000 IU/mL may be at risk of HCC, especially if qHBsAg levels are more than 1,000 IU/mL (see section 5.0) (52). This new terminology aims to highlight the more dynamic nature of CHB and the risk of HCC and cirrhosis even in young individuals with normal liver enzymes and high HBV DNA levels (51,53) (Table 5, Figure 3B)
The guidelines committee recommended formally adopting the revised classification systems but acknowledged that other international liver societies (54,55) as well as expert pediatric HBV specialists continue to use the “classic” HBV natural history terminology (Table 5, Figure 3A–3B).
It has been estimated that approximately 25% of those with chronic HBV infection will develop significant chronic liver disease or HCC during their lifetime. The risk for cirrhosis and liver cancer correlates with the severity of chronic injury or fibrosis, HBV DNA level, duration of infection, male sex, and concomitant liver diseases. Progression to end-stage liver disease or HCC occurs at a rate of 5%–10% per year, with an annual death rate of 20%–50% after complications develop (56,57). Thus, all HBsAg-positive patients should have regular follow-up to assess HBV DNA, liver biochemistry, and liver fibrosis. Although repeated liver biopsy may not be feasible, non-invasive testing (ie, transient elastography [TE] or serum markers) is useful in monitoring for possible fibrosis progression and need for antiviral therapy (see section 6.0).
Chronic HBV infection can cause extrahepatic syndromes, including vasculitis and a spectrum of immune complex–mediated conditions, including renal failure (section 10.0). These extrahepatic syndromes are rare but can lead to multi-organ injury and significant morbidity (58,59). It is important to make a timely diagnosis and initiate treatment to prevent multi-organ injury.
Recommendations for Monitoring of Untreated CHB Infection
-
7.
All CHB (HBsAg-positive) patients must be assessed, including a physical exam for signs of chronic liver disease, testing for baseline liver biochemistry (ALT), complete blood count, creatinine, HBV DNA, HBeAg serology, and fibrosis (either non-invasive assessment or liver biopsy). All should be evaluated for comorbid liver disease (ie, hepatitis C); screening for HIV and hepatitis delta virus (HDV) in high-risk groups (ie, persons who inject drugs, immigrants from endemic areas) should be considered. (strong recommendation; class 2, level A)
-
8.
All CHB carriers should have regular monitoring of ALT (every 6 months) and HBV DNA (every 6–12 months), or at less frequent intervals, depending on individual baseline assessment and risk of liver disease progression. (strong recommendation; class 2, level A)
-
9.
Repeat fibrosis assessment may be indicated if persistent ALT elevation and HBV DNA are present to assess the need for treatment. (strong recommendation; class 3)
4.0. HBV-RELATED HCC (E KELLY, HH KO, MM MA)
The annual incidence of HCC in HBV-infected individuals without cirrhosis is 0.4%–0.6% in Asians, 0.2% in Alaska Natives, and approximately 0.3% in Caucasians (19,23,46). Because of limited data, the incidence of HCC among African or North American Blacks remains unclear. In cirrhotic patients, the incidence of HCC development is about 2%–3% per year, with a cumulative 5-year incidence ranging from 15% to 20% (60).
In addition to ethnicity, there are viral, host, and environmental factors that have been shown to increase the risk of cirrhosis and HCC development. Known factors associated with increased HCC risk are age older than 40 years, male, immunocompromised, positive family history, presence of cirrhosis, high serum HBV DNA (> 2,000 IU/mL or 10,000 copies/mL), elevated ALT, prolonged time to HBeAg seroconversion, genotype C, concurrent viral infections (such as hepatitis C virus [HCV], HDV, and HIV), heavy alcohol use, non-alcoholic fatty liver disease, and smoking (46). Higher HBsAg titres may also correlate with a greater risk of HCC in patients with low-level viremia and in both untreated and antiviral-treated patients (61,62), and they may predict HCC recurrence after resection for HCC (63). Serum alpha-fetoprotein (AFP) was previously used as a tumour marker for HCC, and some studies have shown AFP levels to predict tumour differentiation, size, and prognosis. However, it is important to note that HCC can occur in patients without a rise in AFP, thus measurement of AFP levels should not replace biannual ultrasound in at-risk patients (64). Both ultrasound alone and ultrasound plus AFP led to similar rates of curative treatment, with no appreciable statistical differences between the two surveillance strategies.
Several predictive scoring systems have been developed and validated to predict HBV-related HCC (65), including an easy-to-use nomogram based on the Risk Evaluation of Viral Load Elevation and Associated Liver Disease/Cancer-Hepatitis B Virus cohort and a scoring system using five predictors of HCC based on the Risk Estimation for Hepatocellular Carcinoma in Chronic Hepatitis B study (66–68). Most of these scoring systems were developed with Asian cohorts and have not shown sufficient accuracy in other populations, including Caucasians and Africans. The PAGE-B score, based on platelets, age, and gender at baseline, was recently developed and validated in Caucasian CHB patients undergoing antiviral therapy; it provides a simple scoring system using clinically available variables (69). Other studies have suggested that the PAGE-B score may also be applicable to Asian CHB patients receiving antivirals and predictive of HCC in untreated HBV (70,71). Recently, the Toronto HCC risk index was developed to predict the 10-year risk of HCC in cirrhotic patients. It is the first Canadian validated scoring system that uses readily available clinical and laboratory parameters (ie, age, sex, etiology, and platelets) to stratify patients into low, intermediate, and high risk of HCC (72).
HCC surveillance leads to an increase in detection of early-stage HCC amenable to curative therapy and improves survival. Therefore, abdominal ultrasound every 6 months is recommended for HBV-infected individuals who are at high risk of HCC. The 6-month interval was selected on the basis of tumour doubling time and cost-effectiveness analyses (73,74).
Recommendations for HCC Surveillance in HBV-Infected Individuals
-
10.Abdominal ultrasound screening every 6 months is recommended in the following patients with chronic HBV infection. Serum AFP monitoring is not recommended for HCC screening if ultrasound is available. (strong recommendation; class 2, level B)
- Asian men aged 40 years or older
- Asian women aged 50 years or older
- Persons of African origin aged 20 years or older (due to risk of HCC even in non-cirrhotic patients)
- All cirrhotic patients irrespective of age
- Family history of HCC (starting at age 40)
- All HIV-co-infected patients (starting at age 40).
5.0. LABORATORY ASSESSMENT (C Osiowy)
HBV serological markers (antigens and antibodies) diagnose infection and help to identify the clinical phase of infection (section 3.0), and molecular markers of infection (HBV DNA, cccDNA, serum RNA) indicate viral replication, transcriptional activity, and persistence of infection (section 2.0). The detection and quantification of these markers provide an assessment of the natural history of infection and guide treatment management (Table 4).
5.1. HBV serological markers
The profile of HBV serological markers provides information on a patient’s infection and immune status. The selection of screening or diagnostic serological testing is aided by patient history and clinical presentation. The presence of antibodies directed against HBV proteins (anti-HBc, anti-HBs, anti-HBe) indicates exposure and resolution or infection with the virus, or successful response to immunization. The presence of HBV antigens (HBsAg, HBeAg) in the serum indicates viral infection (Table 4). The quantification of HBV serological markers is increasingly being used as a surrogate to indicate ongoing viral replication or the presence of HBV cccDNA (discussed in the following sections).
5.2. HBV DNA
HBV DNA quantification is mandatory to guide treatment decisions and may be used to indicate the phase of infection in association with HBV serological markers. Persistently high levels of HBV DNA (> 2,000 IU/mL; 1 IU/mL = ~5 virus genome copies/mL) are a significant risk factor for liver disease progression and the development of HCC (75). Monitoring HBV DNA levels at regular intervals (3–6 months) while on treatment (especially with lower potency, first-generation nucleos(t)ide analogues [NAs]) is necessary to assess response and potential development of antiviral resistance or treatment noncompliance (76). Very sensitive molecular tests licenced by Health Canada are available for measuring HBV DNA on the basis of real-time polymerase chain reaction (PCR) methodology (Roche TaqMan HBV and Abbott RealTime HBV), with large dynamic ranges (Roche limit of detection < 6–20 IU/mL, linear range 20–1.7E+08 or ~1.0 × 108 IU/mL; Abbott limit of detection < 10–15 IU/mL, linear range 10–15 to 1E+09 or ~1.0 × 109 IU/mL). HBV DNA assays having greater sensitivity may become more relevant for guiding treatment cessation of NA, because residual low amounts of HBV DNA may be associated with a risk of relapse after treatment withdrawal (77).
OHB, defined as the presence of HBV DNA in serum or liver in the absence of detectable HBsAg (78), has been associated with the development of liver disease and is an increased risk factor for HCC. OHB is associated with reactivation of apparently resolved infection (anti-HBc positive, with or without anti-HBs, and HBsAg negative), particularly during immunosuppressive (IS) therapy or treatment of co-infecting HCV with direct-acting antivirals (DAAs; section 15.0).
The persistence of cccDNA after infection resolution or treatment-induced serum HBV DNA reduction to undetectable levels prevents complete eradication of HBV infection. This leaves the patient susceptible to reactivation and an ongoing risk of developing HCC (79). Quantification of intrahepatic cccDNA provides guidance on patient management, but as yet no practical and standardized method exists for routine clinical practice. New diagnostic tests that provide a surrogate marker of HBV replication or transcriptional activity have been identified to complement HBV DNA quantification, particularly for NA-treated patients with DNA levels reduced below detectable levels, to better understand treatment efficacy and to predict ongoing risk of HCC and relapse after withdrawal of antiviral treatment.
5.3. Quantitative serum HBV RNA
HBV pre-genomic RNA, present as non-reverse-transcribed transcripts within virus-like particles, is detectable in patient sera (80). Because pre-genomic RNA is constitutively expressed from cccDNA (Section 2.0, Figure 2), quantified levels of serum RNA can indicate cccDNA presence and activity (81) to help predict rebound after treatment withdrawal (80). Recent studies have shown that serum RNA levels are predictive of treatment response, either at baseline for the combined response to pegylated interferon (PEG-IFN) and adefovir dipivoxil (ADV) therapy in HBeAg-negative patients (82), or during NA treatment in HBeAg-positive patients with significant reductions in serum RNA levels (83). However, further studies are required to verify this predictive function, because serum RNA levels respond differently under PEG-IFN and NA therapies (82). At this time, quantitative serum HBV RNA testing is not commercially available.
5.4. qHBsAg
Quantitative HBsAg (qHBsAg) is an established marker predicting infection phase and immune control, which together with HBV DNA levels guide treatment decisions. qHBsAg testing is recommended for monitoring response to treatment and establishing chronic HBeAg-negative hepatitis (phase 4) versus HBeAg-negative infection (phase 3), as discussed next and in section 6.0. Quantitative measurement of HBsAg has been standardized to an international standard, and assays are commercially available in Canada (Abbott Architect HBsAg, Abbott Diagnostics, Mississauga, Ontario; Roche Cobas Elecsys HBsAg II quant, Roche Diagnostics, Laval, Quebec) with a broad dynamic range and limits of detection to 0.05 IU/mL. Because qHBsAg can reflect the natural history of HBV infection, it allows for putative cut-off values to predict the likelihood of moderate to severe fibrosis in treatment-naïve HBeAg-positive patients, depending on HBV genotype and ALT measurements (≥7,000–25,000 IU/mL) (84). Similarly, qHBsAg levels of less than 100 IU/mL could signify immune control and are thus associated with HBeAg-negative (Tables 4–5, Figure 3) chronic infection (phase 3) with a high likelihood of future HBsAg clearance (85), sustained virological response, and a functional cure after treatment withdrawal in HBeAg-negative patients (86).
High negative predictive values for qHBsAg levels during treatment have been determined that offer possible treatment cessation or stopping rules. HBeAg-positive patients treated with PEG-IFN who do not achieve a qHBsAg decline at week 12 or 24 and have levels remaining at more than 20,000 IU/mL have a low probability of treatment response or HBeAg seroconversion (87,88). Stopping rules for HBeAg-negative patients treated with PEG-IFN have also been established by a European study and subsequent validation studies (89,90). At week 12, a lack of decline in HBV DNA (< 2 log) and serum qHBsAg levels provides a negative predictive value of 100%, but further studies including greater HBV genotype representation are needed to confirm these rules because HBsAg kinetics during treatment vary by genotype (91).
The effect of NA treatment on HBsAg kinetics is not as pronounced as with PEG-IFN treatment because NA treatment does not directly influence immune control or inhibit cccDNA transcriptional activity. However, HBeAg-positive patients treated with third-generation NA (ie, tenofovir disoproxil fumarate [TDF] or entecavir [ETV]) for approximately 2 years demonstrated a greater qHBsAg decline than HBeAg-negative patients (92), particularly those with genotype A infection, which may simply reflect the increased HBsAg seroconversion naturally observed during genotype A infection (93). Longer treatment regimens (≥3 years) have shown that end-of-treatment qHBsAg levels of less than 100 IU/mL are associated with a low risk of relapse in HBeAg-negative patients (94).
5.5. Quantitative HBcrAg
Detection of HBV core-related antigen (HBcrAg) targets the HBV core antigen, HBeAg, and the HBeAg precursor core-related antigen, all of which share a common sequence of 149 amino acids (95). The Lumipulse G HBcrAg assay (Fujirebio US Inc., Malvern, Pennsylvania) allows quantification of HBcrAg in serum or plasma, although it is not commercially available in North America. As opposed to qHBsAg, HBcrAg quantification positively correlates with cccDNA in both HBeAg-positive and HBeAg-negative patients (96,97), providing a robust surrogate of transcriptional activity over a range of clinical phases and predictive power for HBeAg seroconversion (98). Similar to qHBsAg, HBcrAg levels accurately differentiate between those with HBeAg-negative chronic infection with immune control (ie, phase 3; Table 5, Figure 3) at a low risk of HCC and those for whom treatment is indicated (99). As with the HCC risk inherent with persistently elevated levels of HBV DNA (75), HBcrAg levels are also strongly associated with a risk for progression to HCC (100), including ongoing risk during NA therapy (101). Co-monitoring of both qHBcrAg and qHBsAg has been recommended for predicting reactivation risk in HBeAg-negative patients after treatment cessation. Virological relapse after cessation of PEG-IFN treatment with sequential or combination NA therapy was significantly greater in those having baseline HBcrAg of more than 3.5–3.8 log10 U/mL and qHBsAg of more than 3.1 log10 IU/mL (102,103).
5.6. qAnti-HBc antibody
Quantification of anti-HBc antibody (qAnti-HBc) holds promise for predictive therapeutic management. qAnti-HBc levels have been found to be closely correlated with the host immune response to HBV and thus to parallel hepatic inflammatory activity. Anti-HBc production is significantly higher and associated with liver enzyme levels during the HBeAg-positive chronic phase of infection and during HBeAg-negative hepatitis (ie, phase 4) as compared with HBeAg-negative chronic infection or low replicative phases of infection (104) (ie, phase 3; Table 5, Figure 3). Several studies have investigated the utility of baseline qAnti-HBc to predict response to treatment. Patients treated with PEG-IFN who seroconverted from HBeAg positivity to anti-HBe positivity after the end of treatment or follow-up had significantly higher levels of qAnti-HBc at baseline (105). Moreover, qAnti-HBc levels at baseline (≥4.4–4.5 log10 IU/mL) were the best independent predictor of HBeAg seroconversion after either PEG-IFN or telbivudine (TBV)–ADV treatments compared with the baseline predictive value of ALT or HBV DNA levels (106). A sandwich-based enzyme-linked immunosorbent assay validated against the World Health Organization anti-HBc standard is commercially available from Beijing Wantai Biological Pharmacy (Beijing, China) for qAnti-HBc testing (105,107). Using qAnti-HBc as a surrogate marker of the overall immune response may allow for more effective prediction of achieving response to therapy and has been shown to be independent of HBV genotype, unlike qHBsAg (108), and to have a longer half-life in serum, compared with ALT (104).
5.7. HBV sequence analysis
Molecular analysis of the HBV genome provides information on the viral genotype and the presence of mutations associated with drug resistance, immune escape, and HBeAg expression, all of which have implications for transmission, epidemiology, clinical outcome, and patient management (109). Molecular analysis is most commonly performed by sequencing, including commercial platforms (Siemens Trugene HBV genotyping assay or Abbott HBV sequencing assay) and, more recently, next-generation sequencing (110). Reverse hybridization line probe assays (INNO-LiPA, Fujirebio US Inc., Malvern, Pennsylvania) are also routinely used for HBV genotype determination and mutation detection.
5.7.1. HBV genotyping
The eight major HBV genotypes (A–H) are distinguished by 7.5% or more nucleotide divergence throughout the full genome (111) and have distinct geographic–ethnic distribution worldwide (109,112). Countries with a high rate of immigration, such as Canada, exhibit a diverse blend of all genotypes (113).
Genotypic differences during the natural progression of infection have been observed, such that genotype A (HBV/A) is highly associated with HBsAg persistence after acute infection (114) but with an apparent higher rate of spontaneous HBsAg clearance (93) than other HBV genotypes. HBV/C infection is significantly associated with delayed HBeAg seroconversion (115), resulting in persistently high HBV DNA levels that, together with a higher frequency of the double basal core promoter mutation (A1762T/G1764A) (116), leads to HBV/C being an independent risk factor in the development of HCC (117). Other HBV genotypes have been significantly associated with HCC risk, such as HBV/F in Alaska Native patients (118) or subgenotype HBV/A1 in patients in southwestern India or Sub-Saharan Africa (119,120). However, this observation may be complicated by the frequent specific ethnic association with certain genotypes. For example, circumpolar Inuit or Alaska Native people infected with subgenotype HBV/B5 (previously B6) experience no observable advanced liver disease or HCC (121,122).
Although most evidence suggests that HBV genotypes do not have an effect on the response to NA treatment or the development of resistance, HBsAg decline or clearance during PEG-IFN with (122,123) or without (124) NA therapy is highly associated with genotype A infection, regardless of HBeAg status (125). Genotype-specific PEG-IFN end-of-treatment qHBsAg cut-off values have been recommended to predict sustained response in HBeAg-negative patients (HBV/A, < 400 IU/mL; HBV/B, < 50 IU/mL; HBV/C, < 75 IU/mL; HBV/D, < 1,000 IU/mL) (91).
5.7.2. Mutation testing
Mutations within the precore (HBeAg) coding region (G1896A) and basal core promoter region (A1762T/G1764A) result in elimination or reduction, respectively, of HBeAg expression. Core promoter mutations increase intracellular core protein and allow ongoing HBV replication, resulting in continuing inflammation (126). These mutations have been established as a significant risk factor for HCC development (117,127). Similarly, deletions in the pre-surface genomic region are also an independent predictor of progression to HCC (128), likely because of modification of surface antigen–associated immune control and involvement of endoplasmic reticulum-stress pathways (129,130). Testing for G1896A or A1762T/G1764A mutations may be warranted for a better understanding of the clinical phase of patients suspected to have HBeAg-negative hepatitis (ie, Phase 4).
Detection of mutations resulting in resistance to NA treatments is necessary to understand lack of response to or increasing HBV DNA during treatment and to differentiate patient noncompliance from genotypic resistance. Subsequent NA treatment decisions may also be guided by the presence of NA-specific or cross-reactive resistance mutations (131). Such testing may not be possible in patients who have a persistently low HBV DNA level or failure to achieve undetectable HBV DNA while on treatment, because of the limits of detection of commercial tests or sequence analysis. Next-generation sequencing methodologies have been investigated for detection of HBV drug resistance mutations (132); however, the clinical relevance of very low-level mutant populations is still not fully understood (133,134).
Mutations in the HBsAg and pre-surface coding regions can have significant clinical and diagnostic consequences. These mutations may result in reduced or modulated immunogenicity or epitope-specific antibody binding affinity (135) and defective HBsAg production and secretion (130,136). Screening for HBsAg mutations is indicated for suspected diagnostic escape (eg, discordance in serological results: negative or low HBsAg levels in the presence of high HBV DNA levels or HBsAg–anti-HBs co-positivity), vaccine escape, or OHB.
Recommendations for HBV Laboratory Testing
-
11.
Clinicians should have access to regular HBV serological (ie, HBsAg, HBeAg) and quantitative HBV DNA testing to assist in patient management. (strong recommendation; class 1)
-
12.
Specialized mutation and genotype testing may be used selectively by specialists to help direct treatment and management decisions. (moderate recommendation; class 2, level B)
-
13.
Serological testing should be available from regional or provincial laboratories, and molecular testing should be available through provincial laboratories or reference laboratories (the Guide to Services provided by the National Microbiology Laboratory can be found at https://cnphi.canada.ca/gts/faces/public/index.xhtml). (moderate recommendation; class 2, level C)
-
14.
As evidence becomes available for the clinical utility of new tests to provide surrogate measures of HBV replication and the presence of cccDNA, these tests should be considered for incorporation into provincial and reference laboratories. (weak recommendation; class 3)
6.0. TREATMENT OF HBV INFECTION (SK FUNG, E TAM)
6.1. Selection of patients for treatment
The objective of treatment in CHB is to prevent the development of cirrhosis and its consequences, liver failure and HCC (75,137,138). However, not all patients infected with hepatitis B will develop these complications. The challenge is to identify those who are at risk for the development of complications and to offer them treatment. Conversely, identifying those who will not progress may spare some patients lifelong treatment. However, the tools to achieve this goal are imperfect. As noted, the factors that predict high risk of adverse outcomes include serum HBV DNA, age, hepatic fibrosis, and ALT (section 3.0) (50,139). Other factors may include quantitative HBsAg levels, HBV genotypes, and some HBV mutations (section 5.0). Of these, HBV DNA has been best studied. Several large-scale long-term prospective studies have now correlated HBV DNA at recruitment with outcome (75,137,138). They have all come to the same conclusion, that is, that the risk of developing cirrhosis and HCC and liver-related mortality increases with higher HBV DNA concentration at recruitment and with persistence of high HBV DNA during follow-up. However, in considering the role of HBV DNA as a marker of prognosis, it is important to be aware of the specific populations in these studies, which did not include patients younger than age 25 years, and the number younger than age 30 years was small. The proportion of HBeAg-negative patients ranged from about 50% to 80%. Thus, in patients older than age 30, and in particular in those who are HBeAg-negative, HBV DNA is a good predictor of risk of adverse outcomes. This is likely also true in patients older than age 40 years who are HBeAg-positive, but this is not the case in younger patients.
Other studies also show a correlation between ALT and outcome, but the association was not as strong as that for HBV DNA (140,141). In particular, patients with ALT within the laboratory normal range were also at risk for the development of cirrhosis and HCC as long as the HBV DNA concentration was higher than 10,000 copies per millilitre (~2,000 IU/mL). ALT is an imperfect marker of liver disease in CHB patients. Several studies from East Asia clearly demonstrate that higher ALT is correlated with worse liver disease outcomes. In a Hong Kong study of 3,233 untreated HBV patients, those with normal and even subnormal ALT levels were found to have the lowest risk of HBV complications (140). These and other studies suggest that the upper limit of normal for ALT used in many laboratories may be too high for Asians with CHB. Therefore, several expert societies now endorse an upper limit of normal for men and women with CHB of 35 U/L and 25 U/L, respectively (53,54,142). HBV patients with normal or near-normal ALT may still harbour significant liver disease and warrant treatment.
In an attempt to reduce the likelihood of treating patients who may never develop significant liver disease, if the HBV DNA is high and the ALT is normal, other indicators of significant liver disease should be present before starting therapy. These indicators may be noninvasive markers of fibrosis such as TE (ie, FibroScan) or serum-based fibrosis markers (ie, FibroTest), ultrasound evidence of cirrhosis, or biopsy evidence of at least moderate fibrosis or inflammation. TE higher than 10 kPa has been correlated with cirrhosis in HBV patients, and FibroTest higher than 0.8 is thought be a marker of advanced fibrosis (higher than stage 3 fibrosis) (143,144). Transient ALT elevations, particularly if mild, may not be associated with significant disease. However, prolonged ALT elevation is more likely to be associated with significant injury. Thus, contrary to previous practice, assessment of patients with normal ALT may be required to determine the presence of liver disease and to inform treatment decisions.
For both HBeAg-positive and HBeAg-negative patients, treatment should be considered once the HBV DNA is higher than 2,000 IU/mL. Previous studies have suggested that progressive liver damage occurs once HBV DNA increases above a level of 10,000 copies/mL (~2,000 IU/mL) (50,57). Although liver injury is uncommon if the HBV DNA is below 2,000 IU/mL, some patients may have HBV-induced liver disease at lower viral loads. Liver biopsy may be needed to exclude alternative diagnoses and to confirm viral-induced liver injury. Moreover, HBV DNA may fluctuate, so that repeat measurements are required. HBeAg-negative CHB is associated with more advanced liver disease and never completely remits spontaneously; thus, treatment may be necessary in patients with HBV DNA of more than 2,000 IU/mL.
Young adults who are HBeAg positive usually have very high viral loads (> 107 IU/mL), with variable ALT levels (145,146). Most often, these individuals have no or minimal liver disease on biopsy. Immediate treatment may not be necessary, even with elevated ALT. It is very difficult to predict whether these individuals will undergo spontaneous HBeAg seroconversion with remission of disease before the development of significant liver injury. Figure 4 provides an algorithm for identifying individual patients who require treatment. In summary, the decision to treat requires the consideration of several factors: patient age, level of viral replication, HBeAg status, and evidence of significant liver disease in the form of persistent elevation of ALT, fibrosis or inflammation on biopsy, or non-invasive assessment of hepatic fibrosis.
Figure 4:
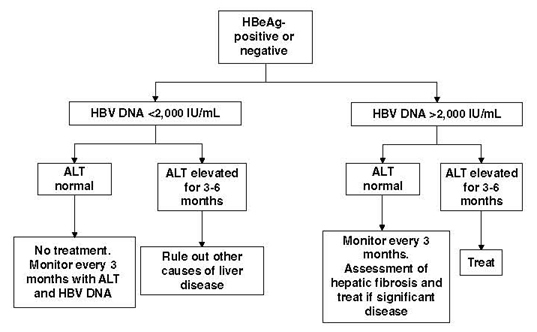
Proposed clinical algorithm for selecting HBV patients for antiviral therapy. In general, patients at risk for liver disease and needing therapy have persistently elevated ALT (normal, < 25 U/L in women, < 35 U/L in men) and elevated HBV DNA. However, normal ALT may not rule out liver disease risk and can fluctuate over time. Because of increased HCC risk with age, some experts are suggesting treating older patients (aged > 35 or 40 years) with high viral load regardless of ALT levels or fibrosis status. Any person with significant fibrosis regardless of ALT level should be considered for treatment, as per recommendation #16.
ALT = alanine aminotransferase; HBV = hepatitis B virus
6.2. Drugs to treat hepatitis B and their use
This section provides information on the specific antiviral agents licenced to treat hepatitis B in Canada. These include lamivudine (LAM), ADV, TBV, ETV, TDF, tenofovir alafenamide (TAF), and PEG-IFN alpha. A comparison of the efficacy of the different agents is illustrated in Figure 5 and Table 6.
Figure 5:
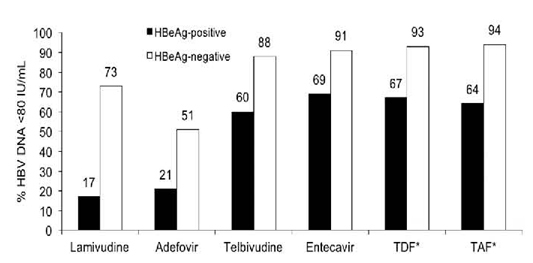
Relative potencies of different hepatitis B oral antivirals at 48–52 weeks of therapy. LAM has been compared with ETV and with TBV in two separate randomized controlled trials. ADV has not been compared directly with the other agents. TDF was compared with TAF.
* For TDF and TAF, results reported as HBV DNA < 29 IU/ml
ADV = adefovir dipivoxil; ETV = entecavir; LAM = lamivudine; TAF = tenofovir alafenamide; TBV = telbivudine; TDF = tenofovir disoproxil fumarate.
Table 6:
Seroconversion rates with hepatitis B antiviral therapy
| Therapy and duration of treatment | HBeAg seroconversion rate, % |
|---|---|
| Pegylated interferon | |
| 24–48 weeks | 29–32 |
| Lamivudine | |
| 1 year | 17–20 |
| 3 years | 40 |
| Adefovir | |
| 1 year | 12 |
| 3 years | 43 |
| Entecavir | |
| 1 year | 21 |
| 3 years | 39 |
| Telbivudine | |
| 1 year | 22 |
| 2 year | 33 |
| Tenofovir disoproxil fumarate | |
| 1 year | 21 |
| 7 years | 40 |
| Tenofovir alafenamide | |
| 1 year | 10 |
| 2 years | 18 |
6.2.1. IFNs
IFNs have both antiviral and immunomodulatory properties that make them effective in inducing long-term immunological control. Potential advantages of IFN over NA include finite-duration therapy and no concern with respect to viral resistance. The major disadvantages, however, are the associated systemic side effects and route of administration (subcutaneous injection).
Initial datasets evaluated the use of standard IFN alpha at a subcutaneous dose of 10 mIU three times per week or 5 mIU daily for 16–24 weeks (147–151). However, PEG-IFN has supplanted standard IFN use in clinical practice given its once-weekly dosing schedule and some evidence of superior efficacy. IFNs are contraindicated in decompensated cirrhosis but may be considered for patients with compensated cirrhosis (preserved hepatic synthetic function without evidence of portal hypertension). PEG-IFN may be considered for both HBeAg-positive patients (phase 2) and carefully selected HBeAg-negative patients (phase 4).
(a) HBeAg-positive chronic hepatitis (phase 2)
HBeAg seroconversion occurs in 25%–40% of treated patients (147–151). IFN-induced HBeAg seroconversion is durable in 70%–80% of patients with up to 8 years of follow-up (152–156). Delayed HBsAg clearance occurs more frequently in IFN-treated patients than in untreated controls, but this is seen in only a minority (approximately 6%–8%) (155). PEG-IFN alpha-2a (180 μg) for 24 weeks was compared with standard IFN, with PEG-IFN alpha–induced HBeAg seroconversion occurring in 28% (180 μg) versus 12% for standard IFN (157). Another study investigated the use of PEG-IFN alpha-2a with or without LAM for 48 weeks, with an HBeAg seroconversion rate of 32% in the PEG-IFN alpha-2a monotherapy group at the end of the 24-week follow-up period (158). Eight patients (3%) in this group experienced HBsAg loss at the same time point. A similar study evaluated PEG-IFN alpha-2b (100 μg for weeks 1–31; 50 μg for weeks 32–52) with or without LAM for 52 weeks and demonstrated an HBeAg seroconversion rate of 29% at the end of 26 weeks of follow-up (159). Nine patients (7%) experienced HBsAg loss. Although defining optimal treatment duration is difficult given available evidence, 48 weeks is currently considered standard of care.
IFN-induced HBeAg seroconversion is associated with improved overall survival and complication-free survival (160–162). Although data regarding the impact of PEG-IFN alpha therapy on the incidence of HCC are heterogeneous, most reports conclude that the incidence of HCC is reduced, with some evidence suggesting lower incidence of HCC than in those receiving oral antiviral therapy (163).
(b) HBeAg-negative chronic hepatitis (phase 4)
PEG-IFN alpha-2a (180 μg) with or without LAM for 48 weeks was demonstrated to be effective in HBeAg-negative patients (164). Suppression of HBV DNA to less than 400 copies/mL (~80 IU/mL) at the end of treatment was achieved in 63% of patients on PEG-IFN alpha-2a monotherapy, but only 19% continued to have durable viral suppression 24 weeks after stopping therapy. The combined endpoint of ALT normalization and HBV DNA of less than 4,000 IU/mL (~20,000 copies/mL) was achieved in 36% of those in the PEG-IFN alpha-2a group at end of treatment, with the same proportion achieving this endpoint at 24-week follow-up. HBsAg loss occurred in 4% at week 72.
(c) Predictors of Peg-Ifn response and stopping rules
Given the modest response rates and potential systemic side effects associated with PEG-IFN alpha therapy, there is significant value in identifying predictors of response in order to select optimal treatment candidates. In addition, robust stopping rules may prevent futile extension of therapy in patients unlikely to respond. PEG-IFN alpha is less effective in inducing HBeAg seroconversion in patients with high viral loads (HBV DNA > 2 × 107 IU/mL). Seroconversion rates are also reduced in those with low ALT (less than two times the upper limit of normal); therefore, PEG-IFN alpha is not recommended for treatment of patients with high viral load or low ALT. Other predictors of poor response include age older than 40 years, male sex, and cirrhosis. Genotypes A and B are associated with higher rates of HBeAg seroconversion.
Quantitative HBsAg on treatment has thus far been demonstrated to be the best on-treatment predictor of response to PEG-IFN alpha. In HBeAg-positive patients at week 24 of PEG-IFN alpha therapy, HBsAg of more than 20,000 IU/mL is associated with a 99% negative predictive value for response (defined as HBeAg loss with HBV DNA < 2,000 IU/mL), validated in a cohort of 803 HBeAg-positive patients (165). Thus, HBsAg levels of more than 20,000 IU/mL at week 24 of therapy in HBeAg-positive individuals should trigger immediate discontinuation of therapy. At this time, no widely applicable and robustly validated stopping rules are available for the HBeAg-negative patient undergoing treatment with PEG-IFN alpha. Careful selection of these individuals on the basis of clinical factors at baseline remains the best approach. A useful online calculator (PEG-IFN HBV Treatment Index; https://www.liver-GI.nl/peg-ifn) is available to predict treatment response in HBeAg-positive patients (166).
6.2.2. Tenofovir disoproxil fumarate (TDF, Viread)
TDF is an oral nucleotide reverse transcriptase inhibitor approved for chronic HBV infection. TDF has demonstrated efficacy in treatment-naïve HBeAg-positive (phase 2) and HBeAg-negative (phase 4) patients with chronic hepatitis. Long-term large registration studies of more than 430 patients report HBV DNA suppression of less than 69 IU/mL was achieved in more than 99% of HBeAg-positive and HBeAg-negative patients after 7 years (167). Normalization of ALT occurred in 80% of patients. HBeAg loss was reported in 59% of HBeAg-positive patients, and HBsAg loss was reported in 12% of patients after 7 years. At the end of 5 years of treatment, 80% of patients overall had improvement in liver histology. Seventy-five percent of patients with cirrhosis at baseline had at least a 2-point reduction in Ishak score after long-term TDF therapy (168). Nephrotoxicity and hypophosphatemia with long-term therapy were uncommonly reported (1.2% and 0.9% of patients, respectively). To date, no confirmed cases (including in vitro testing) of antiviral resistance mutation to TDF have been published after 8 years of continuous treatment (169). Interestingly, HBsAg loss occurred in 10%–15% of patients during the same period of treatment. Analysis of the Asian subset showed similar efficacy as in Caucasians. Predictors of HBsAg loss included decline in HBsAg levels on treatment, HBV genotype A, and shorter duration of chronic infection (< 4 years).
6.2.3. Tenofovir alafenamide (TAF, Vemlidy)
TAF is a novel product of tenofovir. TAF is given at a lower dose (25 mg daily) than TDF but is delivered more efficiently into hepatocytes. In phase 3 studies of 873 HBeAg-positive and 435 HBeAg-negative mainly treatment-naïve patients, TAF was compared with TDF in a blinded fashion for 2–3 years, followed by 5 years of open-label TAF (170,171). Although virologic (HBV DNA < 29 IU/mL) and serologic responses were similar between the two groups, higher rates of ALT normalization were seen in those randomized to receive TAF after 1 year of treatment. Moreover, renal function measured by serum creatinine and glomular filtration rate (GFR) as well as bone mineral density changes measured by dual-energy X-ray absorptiometry scans were significantly less common in those who received TAF. ALT flares and other adverse events were similar between the two treatment groups. Although TAF has not been studied in antiviral-resistant patients in a dedicated study, a small number of patients in the registration studies were found to have LAM-, ADV-, and ETV-resistant mutation at baseline.
Response to TAF compared with TDF was similar. On the basis of in vitro studies and case reports, TAF is expected to have significant activity against common antiviral-resistant mutations. In a recent case report from Italy, an elderly patient with LAM and ADV resistance who received rescue therapy with TDF developed Fanconi syndrome. The patient was then treated with TAF, which not only improved renal function but also maintained antiviral suppression, suggesting that TAF is a suitable alternative to TDF for multidrug-resistant HBV (172)
6.2.4. Entecavir (ETV, Baraclude)
ETV is a selective guanosine analogue and a potent inhibitor of HBV DNA replication that is well tolerated with a side effect profile similar to LAM in large clinical trials. In treatment-naïve patients, HBeAg seroconversion at 1 year is similar to other NAs at 21% after year 1 and 39% after year 3 (Table 6) (173,174). Compared with LAM-treated patients, ETV-treated patients had higher rates of HBV DNA undetectability (67% versus 36%) and histologic improvement (72% versus 62%). Only about 1%–2% of subjects developed resistance to ETV after 5 years (175). However, this is in contradistinction to resistance rates of more than 50% in those with prior LAM resistance (176).
In another study of 648 HBeAg-negative patients treated with ETV, virologic suppression and histologic improvement were significantly higher than in those treated with LAM (90% versus 72% and 70% versus 61%, respectively). In a long-term real-world cohort study of 222 Chinese patients in Hong Kong treated with 7 years of continuous ETV, 99% maintained HBV DNA suppression, 98% normalized ALT, and 82% achieved HBeAg seroconversion. Resistance to ETV was reported in only 1.2% of patients during long-term follow-up. Predictors of HBsAg loss include baseline levels less than 100 IU/mL and, on treatment, annual decline by more than 0.2 log IU/mL (177). In a study of 286 HBeAg-positive LAM-refractory patients who were treated with high-dose ETV (1 mg daily), only 20% of patients achieved undetectable HBV DNA after 1 year of treatment, and 8% of patients subsequently developed resistance to ETV; this rate increased substantially with prolonged duration of therapy (178). Thus, ETV is not recommended as salvage therapy for LAM-resistant HBV.
6.2.5. Lamivudine (LAM, Heptovir)
LAM is a pyrimidine NA inhibitor of the HBV polymerase. It was the first oral agent approved in the treatment of HBV in Canada. Many HBV patients may have received LAM in the past, especially patients who have recently immigrated from Southeast Asia where the drug is still widely used. Generally, LAM is effective at lowering HBV DNA and has an established long-term safety record (179). Compared with placebo, LAM was shown in a randomized study to reduce progression of liver disease and possibly hepatoma in HBV cirrhotic patients (180). The major disadvantage of LAM is the very high risk of developing antiviral resistance and virologic breakthrough, approaching 70% at 4 years (181). Moreover, the development of LAM resistance may lead to cross-resistance to other agents such as ETV and TBV and limit future treatment options. Therefore, LAM is not a suitable first-line treatment and is a suboptimal regimen for treatment of hepatitis B. However, LAM may still have a role in certain situations in which time-limited therapy is indicated, such as prophylaxis for those undergoing short-term immunosuppression, especially in HBsAg-negative, anti-HBc-positive cases with low or undetectable HBV DNA (Section 9.0).
6.2.6. Adefovir (ADV, Hepsera)
ADV is an NA that is rarely used because of its low potency and incomplete viral suppression in the majority of patients within the first year, possibly because of the low approved dose (10 mg daily). Side effects of ADV include nephrotoxicity, hypophosphatemia, and, rarely, Fanconi syndrome, necessitating regular monitoring of renal function (estimated GFR, or eGFR). Risk factors for ADV resistance are high baseline viral load and inadequate suppression of virus on therapy (182).
6.2.7. Telbivudine (TBV, Sebivo)
TBV is an NA that was more effective than LAM in patients with both HBeAg-positive and HBeAg-negative chronic hepatitis. In a cohort study of 196 patients in China, HBeAg seroconversion exceeded 50% after 5 years of TBV compared with ETV. However, genotypic resistance rates of 5% and 11% were reported after 1 and 2 years of TBV treatment, respectively (183). Asymptomatic rises in creatine kinase and myositis occurred in approximately 12% of patients, which has limited its use in most countries. However, TBV may have a limited role for short-term HBV treatment in pregnancy (US Food and Drug Administration class B in pregnancy) if TDF is contraindicated. In a study of 229 pregnant HBeAg-positive women with high viral load, TBV given in the second trimester of pregnancy was more effective than no antiviral treatment in reducing perinatal transmission of HBV (0% versus 8%; see section 7.0) (184,185).
6.2.8. De novo combination antiviral therapy
Although combination therapy for hepatitis B may be appropriate in certain patient populations, there are few data to support its routine use with treatment-naïve patients.
(A) PEG IFN alpha (PEG, Pegasys) plus NA
Because PEG-IFN and NA therapies have different mechanisms of action, there has been significant interest in the potential to combine the two strategies. Foreseeable benefits of a combination approach include higher rates of HBeAg seroconversion, HBsAg loss, and durable long-term viral suppression after a finite course of therapy. Multiple combination strategies have been studied, including the simultaneous initiation of PEG-IFN and nucleos(t)ide therapy, addition of PEF-IFN in individuals already virally suppressed on nucleos(t)ide therapy, and switching from nucleos(t)ide therapy to PEG-IFN. Overall, despite the theoretical benefits, results in these combination studies have been underwhelming, with little or no benefit demonstrated, and often with small sample size or methodological concerns in the study design that reduce their applicability to clinical practice (186). One notable exception is a recently published study involving 740 individuals with either HBeAg-positive or HBeAg-negative chronic hepatitis, randomized to one of four treatment arms involving PEG-IFN alpha-2a (180 μg/week, subcutaneously) or TDF (300 mg once daily): TDF plus PegIFN for 48 weeks (group A), TDF plus PEG-IFN for 16 weeks followed by 32 weeks of TDF alone (group B), TDF alone for 120 weeks (group C), or PEG-IFN alone for 48 weeks (group D). All participants were untreated, but they may have had prior exposure to NA therapy more than 24 weeks before study inclusion. All participants had elevated ALT, and those with bridging fibrosis or cirrhosis were excluded. The primary efficacy endpoint of HBsAg loss at week 72 was achieved in 9.1% of participants in group A, which was statistically significantly higher compared with 2.8%, 0%, and 2.8% in groups B, C, and D, respectively. Although HBsAg loss in group A was noted across genotypes, the highest rate of clearance occurred in genotype A (187). A subsequent analysis indicated that the only baseline factor associated with response was genotype A. HBsAg decline of more than 3.5 log IU/mL at week 24 was associated with a positive predictive value of 85% and a negative predictive value of 99% for HBsAg loss at week 72, indicating that this may represent a viable stopping rule for this combination therapy approach (188).
(B) Dual NA therapy
A number of studies have investigated combination oral nucleos(t)ide therapy to explore the potential for improved virologic and biochemical outcomes versus monotherapy. In a single-centre study in which the combination of LAM and ADV was compared with LAM alone, no difference in HBV DNA suppression, HBeAg seroconversion, or ALT normalization was observed (189). However, resistance to LAM was significantly lower in the combination group compared with the monotherapy group. In another dataset, combination LAM plus TBV was less effective than TBV alone for all endpoints (190), possibly as a result of antiviral antagonism. In a randomized open-label, multi-centre study, 379 nucleos(t)ide-naïve patients were randomized by HBeAg status to ETV plus TDF versus ETV monotherapy (191). A similar proportion of individuals achieved the primary endpoint of HBV DNA less than 50 IU/mL at week 96 of therapy in the combination (ETV plus TDF) and the ETV-alone arms (83.2% versus 76.4%; p = 0.088). However, in the subset of HBeAg-positive patients with baseline HBV DNA more than 108 IU/mL, a greater proportion of those treated with combination therapy achieved HBV DNA less than 50 IU/mL at week 96 (79% versus 62%; p = 0.018). Rates of HBeAg loss and HBeAg seroconversion were similar in the two treatment groups. No difference in the overall low rates of HBsAg loss and HBsAg seroconversion was observed. A greater proportion of individuals in the ETV-alone group achieved normal ALT levels by week 96 (81.9% versus 69.0%; p = 0.004). Although the overall role of antiviral therapy in HBeAg-positive patients with high viral load and normal ALT remains incompletely defined, treatment with TDF plus emtricitabine versus TDF alone for 192 weeks was also explored in this patient population in a randomized, double-blind study of 126 patients. Of patients in the TDF plus emtricitabine group, 76% achieved HBV DNA less than 69 IU/mL at week 192 versus 55% of those in the TDF plus placebo group (p = 0.016). Only three patients experienced HBeAg seroconversion (all in the TDF plus placebo group). No HBsAg loss occurred during the study (192).
Although combination NA therapy may result in higher rates of HBV DNA suppression in HBeAg-positive patients with high viral loads, the available data do not support the routine de novo use of this strategy, because of the lack of any indication that other clinically relevant endpoints are improved. Even so, combination therapy may remain an appropriate consideration in certain situations. In cirrhotic patients, particularly those with hepatic decompensation, the development of resistance to antiviral agents may lead to flares of liver disease that may be fatal. Therefore, combination therapy can be considered in this setting. Suggested regimens include TDF plus ETV or TDF plus emtricitabine. Figures 6 and 7 provide an algorithm to help select suitable agents to treat hepatitis B with or without cirrhosis.
Figure 6:
Algorithm for selection of specific agents for hepatitis B. The recommended first-line oral nucleos(t)ide analogue therapy is TDF, TAF, or ETV if patients have no pre-existing resistance to lamivudine. TAF or ETV may be considered in selected populations that have or are at risk for renal disease or metabolic bone disease. In selected HBeAg-positive patients (high ALT, genotype A or B, HBV DNA) may respond to finite therapy with PEG-IFN.
ALT = alanine aminotransferase; HBeAg = HBV e antigen; HBV = hepatitis B virus; ETV = entecavir; PEG-IFN = pegylated interferon; TAF = tenofovir alafenamide; TDF = tenofovir disoproxil fumarate.
Figure 7:
Suggested algorithm for management of patients with hepatitis B viral cirrhosis. Cirrhotic patients are at high risk of hepatic decompensation with virological and biochemical (hepatic) flares and progression to HCC. Thus, potent nucleos(t)ide analogue therapy is recommended with TDF or ETV (if there is no history of lamivudine resistance). Some studies indicate that initiating combination therapy is beneficial to rapidly suppress viral load in patients with cirrhosis.
ETV = entecavir; HBV = hepatitis B virus; HCC = hepatocellular carcinoma; TAF = tenofovir alafenamide; TDF = tenofovir disoproxil fumarate.
Recommendations for HBV Treatment
-
15.
HBeAg-positive (phase 2) and HBeAg-negative (phase 4) patients in whom HBV DNA is more than 2,000 IU/mL with elevated ALT (> 1 time the upper limit of normal) for 6 months should be considered for treatment. (strong recommendation; class 2, level A)
-
16.
Patients with significant inflammation or fibrosis (above stage 1) should be considered for treatment, even if HBV DNA is lower than 2,000 IU/mL or ALT is normal. (strong recommendation; class 2, level A)
-
17.
All patients with cirrhosis and detectable HBV DNA should be treated. (strong recommendation; class 2, level A)
-
18.
TAF, TDF, or ETV is first-line therapy for treatment-naïve HBV patients, because these are the most potent agents available with no or very low rates of antiviral resistance. (strong recommendation; class 1)
-
19.
TDF or TAF is first-line therapy for LAM-resistant HBV. ETV should not be used in this case because of the risk of development of ETV resistance. (strong recommendation; class 1)
-
20.
A weekly subcutaneous dose of 180 μg PEGIFN alpha-2a for 48 weeks may be used to treat non-cirrhotic HBeAg-positive CHB. (moderate recommendation; class 1)
-
21.
A weekly subcutaneous dose of 180 μg PEG-IFN alpha-2a in combination with TDF given for 48 weeks may be used to treat CHB in the absence of advanced fibrosis. (moderate recommendation; class 1)
6.3. On-treatment monitoring and discontinuation of therapy
Table 7 summarizes the definitions of antiviral treatment response.
6.3.1. IFN
Patients treated with IFN need to be monitored closely. Liver tests and a complete blood cell count should be performed monthly and thyroid-stimulating hormone should be measured every 3 months. When using PEG-IFN for HBeAg-positive patients, qHBsAg levels (if available), HBV DNA, and HBeAg–anti-HBe can be measured every 1–3 months and at the end of treatment to determine response. A lack of qHBsAg decline at 12 weeks has a strong negative predictive value for achieving a sustained virological response to PEG-IFN therapy (see sections 5.0 and 6.2.1). A 1 log10 reduction in qHBsAg levels after 3 months of PEG-IFN is predictive of HBsAg clearance. These tests should also be performed every 3–6 months for 12–18 months after treatment, because HBeAg seroconversion may occur after PEG-IFN is withdrawn. A successful outcome is defined as anti-HBe-positive with normal ALT for more than 6 months, with HBV DNA less than 2,000 IU/mL. Ideally, HBV DNA should be undetectable, because the presence of significant viremia may precede disease relapse. HBeAg-negative CHB patients treated with IFN should be monitored as for HBeAg-positive hepatitis, except that HBeAg and anti-HBe testing do not need to be repeated. Patients must continue to be screened for HCC as per current guidelines (see section 3.0), irrespective of response to PEG-IFN treatment.
6.3.2. NA
Patients treated with NAs should initially be monitored by testing for HBV DNA and ALT every 3 months while on treatment and then every 6 months once aviremia is achieved. This is to confirm an initial fall in viral load and, in the case of LAM, TBV, and ADV, to determine whether treatment with the same drug can be maintained or whether another drug should be added or substituted. HBV DNA levels must be monitored regularly to allow for early detection of viral breakthrough leading to resistance. Patients receiving TDF require 3–6 months’ monitoring of renal function and serum phosphate levels. Patients receiving TBV require monitoring of creatine kinase levels. Patients must continue to be screened for HCC per current guidelines (section 4.0), irrespective of response to antiviral treatment.
6.3.3. Post-treatment and long-term monitoring after cessation of NA therapy
There are conflicting data on the durability of response in HBeAg-positive patients who have undergone HBeAg seroconversion while on oral antiviral therapy followed by 12 months of consolidation therapy (193,194,195). Relapse is common in more than 50% of HBeAg-negative patients who discontinue oral therapy even after many years of continuous treatment. HBsAg-positive patients who have recently discontinued oral antiviral therapy should be monitored with a liver panel and HBV DNA initially every 3 months to rule out early post-treatment relapse. Virologic relapse (redetection of HBV DNA) occurs before biochemical (ALT) flares are evident. Once a patient is stable for more than 1 year after treatment, frequency of monitoring may be reduced to every 6 months. Periodic TE or FibroScan or other noninvasive tests of hepatic fibrosis can be performed every 2–3 years, and HCC screening should be done according to guidelines (section 4.0). The data on qHBsAg monitoring to guide treatment response and cessation are evolving (see section 5.0).
Recommendations for On-Treatment HBV Monitoring and Treatment Discontinuation
-
22.
The target HBV DNA level while patients are on oral NA therapy is undetectable, measured using the most sensitive test available (ie, currently Taqman PCR). (strong recommendation; class 2, level A)
-
23.
In HBeAg-positive patients who achieve HBeAg seroconversion with NA treatment, a minimum of 12 months of additional consolidation therapy is required to maximize the durability of the response before considering treatment cessation, with close monitoring for relapse thereafter. Alternatively, treatment may be continued until HBsAg loss. (moderate recommendation; class 2, level B)
-
24.
In HBeAg-negative patients, continue NA therapy until HBsAg loss. (moderate recommendation; class 2, level B)
-
25.
HBeAg-positive individuals treated with PEG-IFN alpha with HBsAg of more than 20,000 IU/mL at week 24 should discontinue therapy because of the low probability of response. (strong recommendation; class 2, level B)
-
26.
All persons with cirrhosis or HCC should be treated with NA until HBsAg loss or indefinitely. (moderate recommendation; class 2, level B)
-
27.
Patients must continue to be screened for HCC as per current guidelines irrespective of response to antiviral treatment. (strong recommendation; class 2, level B)
6.4. Resistance to antiviral therapy
6.4.1. Antiviral resistance testing
Antiviral resistance in HBV is now a minor issue owing to first-line use of agents with a high barrier to resistance such as TDF, TAF, and ETV (see Table 7 for definitions). However, LAM resistance may still occur in patients who were started on treatment in jurisdictions without access to first-line drugs. Monitoring for antiviral resistance requires regular assessment of HBV DNA levels. When resistance develops, particularly to LAM, secondary mutations may occur that may reduce susceptibility to other antivirals (196). Genotypic resistance can be detected by various methods, such as population sequencing, reverse hybridization, clonal analysis, and ultra-deep sequencing methods (see section 5.0).
Table 7:
Definitions of response to hepatitis B nucleoside antiviral agents
| Primary non-response | Less than 2-log10 IU/mL decrease in HBV DNA measured at 6 months of treatment, most commonly related to non-adherence to medication |
| Genotypic resistance | Mutation of HBV DNA polymerase known to decrease the efficacy of the antiviral agent |
| Phenotypic resistance | Defined by an in vitro assay demonstrating decreased inhibition of viral replication in the presence of the specific mutation in the polymerase gene |
| Viral breakthrough | Increase in HBV DNA of 1 log10 IU/mL or greater above the nadir, measured in two consecutive samples 1 month apart, occurring after the first 3 months of therapy; this is commonly because of genotypic resistance, but it may also be the result of lack of adherence |
| Clinical or biochemical breakthrough | A rise in ALT from its nadir during treatment associated with a rise in HBV DNA of 1 log10 IU/mL or greater; it may also be the result of either genotypic resistance or lack of adherence |
ALT = alanine aminotransferase; HBV DNA = viral load
The evidence that the benefits of viral suppression are lost when resistance occurs is considerable (197,198). Acute flares of hepatitis related to LAM- or ADV-resistant HBV can occur, and such flares may be fatal, particularly for cirrhotic patients. Therefore, the development of resistance to antivirals is a strong indication to change therapy. Early detection of antiviral resistance is important to avoid ALT flares and decompensation of liver disease.
Table 6 compares the relative potency of the different antiviral agents in non–head-to-head clinical trials (199). Table 8 shows the substitutions in the HBV polymerase gene that are associated with resistance to various agents (199). Rates of antiviral resistance from non–head-to-head studies for various agents are compared in Figure 8.
Table 8:
Mutations conferring resistance to hepatitis B nucleos(t)ide analogue therapy within the HBV polymerase/reverse transcriptase domain.
| Agent | Domain A | Domain B | Domain C | Domain D |
|---|---|---|---|---|
| Lamivudine | L80V/I | V173L, L180M | M204V/I/S | |
| Adefovir | A181V/T | N236T | ||
| Entecavir* | I169T, T184G | S202I | M250V | |
| Telbivudine | M204I |
Note: The letters before the numbers represents the wild-type amino acid. The letters after the number represents the substituted amino acid. The number refers to the amino acid position.
*Entecavir mutations only confer resistance in the presence of the M204V, M204I, and L180M mutations. In the absence of these additional mutations, the entecavir mutations do not cause resistance.
Figure 8:
Rates of resistance to antiviral agents by duration of therapy. In treatment-naïve patients, resistance rates are low with entecavir and tenofovir disoproxil fumarate. Patients with lamivudine resistance are at risk of cross-resistance and treatment failure with entecavir.
6.4.2. Management of primary nonresponse
Primary non-response is defined as a less than 1 log reduction in HBV DNA at week 12 or a less than 2 log reduction in HBV DNA at 24 weeks of antiviral therapy (Table 7) (200). Resistance testing is recommended for those receiving weaker agents such as LAM to differentiate between resistance and medication adherence. Counselling for those found to be non-adherent is recommended. For those receiving therapy with LAM or TBV, treatment can be switched at 24 weeks to a more potent agent such as TDF or TAF or, in the absence of LAM resistance, ETV and HBV DNA repeated in 3 months. Primary non-response is unusual in those receiving high-potency agents such as TDF and TAF or ETV as first-line therapy.
6.4.3. Management of resistance to specific antiviral agents
Table 9 provides a summary of the management approach for patients who develop antiviral therapy resistance.
(a) Resistance to LAM
Much is known about LAM-resistant HBV because this agent was the first antiviral to be licenced for HBV infection. Resistance to LAM monotherapy occurs very commonly: in year 1, 15%–20%; in year 2, 30%; in year 3, 50%; and in year 4, more than 75% (181). Adverse clinical outcomes such as loss of initial response, reduced HBeAg seroconversion, and hepatic decompensation have been reported in patients who develop LAM resistance. Previous studies demonstrate that the addition of ADV after virologic breakthrough when the viral load is still low is one option (201), but switching to ADV monotherapy is associated with a high rate of ADV resistance (20% after 1 year) and is not recommended (202). ETV is not an acceptable choice for LAM resistance because the response to ETV is reduced and ETV resistance is about 32% after 3 years (176). LAM-resistant HBV leads to TBV and emtricitabine cross-resistance. A randomized study of 280 chronic HBV patients with a documented LAM-resistant mutation (rtL180M ± rtM204V/I) compared treatment with TDF and treatment with TDF plus emtricitabine (Truvada) for 5 years (203). Patients had received a mean of 4 years of LAM before enrolment, and 10% had evidence of cirrhosis. Rates of virologic suppression were identical between the two groups (83%), but only 15% had HBeAg seroconversion after 5 years. HBsAg loss or seroconversion was observed in only seven patients. Bone mineral density assessed by dual-energy X-ray absorptiometry scan was performed every 6 months. A 1% and 2% reduction in bone mineral density was found at the spine and hip, respectively, over the study period, and no increase in fractures was reported. Data from this landmark study and others suggest that there is no benefit of combination antiviral treatment over TDF monotherapy, and TDF has been established as the treatment of choice for LAM-resistant HBV.
(b) Resistance to ETV
ETV resistance requires a LAM-resistant backbone (ie, a mutation in tyrosine–methionine–aspartate–aspartate locus, or YMDD). YMDD mutation alone decreases ETV potency but is not enough to produce resistance. Nonetheless, in the presence of rtM204V and rtL180M, one or more additional mutations (rt169T, rtT184G, rtS202I, rtM250V) are able to confer resistance to ETV (204) (Table 8). In registration studies of ETV in LAM-resistant patients, ETV-resistant mutations were detected in a proportion of patients at baseline before the introduction of ETV. As a result, genotypic resistance was identified in 7% of patients and viral breakthrough in 1.6% of patients at the end of the first year of therapy (178). This percentage increased to more than 30% at the end of the third year of therapy. In contrast, in nucleos(t)ide-naïve patients, resistance to ETV occurred in approximately only 1% of patients after 3 years. Therefore, patients with pre-existing LAM resistance are at risk of developing resistance to ETV (176). For this reason, ETV should not be used to rescue patients with LAM-resistant HBV.
In a study of 90 patients in South Korea with LAM and ETV resistance, TDF demonstrated similar efficacy as TDF plus ETV (HBV DNA < 15 IU/mL in 71% and 73% of patients on TDF and TDF plus ETV, respectively). While on treatment, only one TDF-treated patient developed virologic breakthrough related to medication nonadherence, and no resistant mutations other than those found at baseline were detected after 2 years (205). Thus, HBV resistance to ETV can be treated with TDF or TAF.
(c) Resistance to TDF and TAF
To date, there have been no confirmed cases (ie, with confirmatory in vitro laboratory testing) of TDF resistance in HBV-monoinfected patients after 8 years of continuous therapy (169). In fact, there is no known signature mutation for TDF in the HBV polymerase gene. A case report documented rtA194T substitution in a patient coinfected with HIV and HBV, but this mutation has not been reported in HBV-monoinfected patients and is likely not clinically significant. In registration trials of TDF, population sequencing did not reveal any conserved site changes among 4% of patients who did not achieve undetectable HBV DNA, although resistance surveillance is ongoing (206).
(d) Multidrug Resistance
The most common form of multidrug resistance in clinical practice is LAM–TBV in association with ADV resistance (rtM204V/I + rtN236T or rtA181T/V), because this combination of agents has been available for the longest period of time. In a European study of patients who harboured multidrug-resistant HBV, 75% achieved HBV DNA suppression using TDF as salvage therapy (207). In a case report of an elderly patient with multidrug resistance who developed Fanconi syndrome while receiving TDF, treatment with TAF achieved virologic suppression within 6 months with stabilization of renal function (172). In a randomized 3-year study of 189 Korean patients with ETV- or ADV-resistant HBV, virologic suppression (71%–73%) was similar in those randomized to receive TDF or TDF plus ETV (205). Although a small number of patients qualified for resistance testing, no resistant mutation to TDF was detected over the 3-year period (208). Taken together, these data suggest that TDF and TAF are safe and effective for the treatment of multidrug-resistant HBV infection.
Recommendations for Management of HBV Antiviral Resistance
-
28.
If a patient is confirmed to be treatment adherent and has increasing HBV DNA, salvage therapy should be introduced. (strong recommendation; class 1)
-
29.
Antiviral resistance testing should be used to differentiate between non-adherence and the emergence of resistant virus in patients with virologic breakthrough or persistent viremia. (moderate recommendation; class 2, level B)
-
30.
HBV DNA should initially be monitored every 3 months to allow early detection of viral resistance and every 6–12 months once aviremia is achieved if the patient is on high-potent NA or every 3 months if the patient is on LAM). (strong recommendation; class 2, level B).
-
31.
The treatment of choice for LAM-resistant HBV infection is TDF. (strong recommendation; class 1).
Management of Hepatitis B in Special Patient Populations
7.0. MANAGEMENT OF HBV AND PREGNANCY AND PREVENTION OF MOTHER-TO-CHILD TRANSMISSION (KE DOUCETTE)
7.1. Prenatal HBV screening
Although HBV prevalence in Canada is low overall, its prevalence in foreign-born and some Indigenous populations remains high and not well predicted by risk-factor–based screening. All pregnant women should therefore be screened for HBsAg in the first trimester of pregnancy; this screening is highly recommended in all Canadian health care jurisdictions (209). Those documented to be HBsAg positive require subsequent testing for HBeAg–anti-HBe, HBV DNA quantification, and ALT to assess their stage of disease. In pregnancy, the safety and accuracy of non-invasive assessments of hepatic fibrosis, such as biochemical assays or liver stiffness measurement (ie, TE or FibroScan), have not been assessed and are therefore not recommended.
7.2. HBV treatment in women of child-bearing potential or pregnancy
When making decisions regarding the initiation of antiviral therapy in women of child-bearing age or in early pregnancy, the severity of HBV-related liver disease and the safety of HBV antivirals should be considered. Most women will not have significant HBV-related disease or require immediate therapy (210–213). PEG-IFN is contraindicated in pregnancy, but it may be considered in women with favourable predictors of response (ie, low viral load, high ALT, genotype A; see section 6.0) who meet indications for therapy and are willing to defer pregnancy until after the finite duration of treatment. Pregnant women and those of child-bearing age who require immediate treatment should be initiated on an NA that is safe in pregnancy. Women who become pregnant while already on HBV therapy should continue treatment until they reach standard endpoints for the non-pregnant population; therapy should be reviewed as early as possible to ensure such patients are on a drug that is safe to use in pregnancy.
The safety of ADV, ETV, and TAF have not been adequately studied in controlled prospective trials in pregnant women. In animal studies, no adverse developmental effects have been observed when TAF was administered during organogenesis at doses of up to 50 times the recommended therapeutic dose (214). Studies in pregnant women using LAM, TBV, or TDF demonstrate no evidence of harm to the fetus (184,215–219). In addition, substantial data from the Antiretroviral Pregnancy Registry support the safety of LAM and TDF in both the first and the second and third trimesters (220). Because it has extensive safety data and a lack of resistance in HBV, TDF is the preferred NA for pregnant women and those with child-bearing potential. Although there are reasonable safety data for LAM and TBV in pregnancy, these agents are not recommended because of the risk of treatment-emergent resistance, even when used for a short duration, in those with high viral load. LAM or TBV may be considered as alternative regimens when TDF is contraindicated.
7.3. Prevention of HBV MTCT
The administration of hepatitis B immunoglobulin (HBIg) and HBV vaccine within 12 hours of birth followed by completion of the HBV vaccine series, with the second and third doses at 1 and 6 months for infants born to HBsAg-positive women, has been shown to decrease the risk of mother-to-child transmission (MTCT) from more than 90% to about 10%. The primary risk factor for immunoprophylaxis failure is high HBV DNA of more than 200,000 IU/mL (210,213,221,222).
LAM, TBV, and TDF have all been shown to further decrease the risk of MTCT in babies born to mothers with high viral load (184,215–217). In a randomized trial conducted in China of 200 pregnant women with HBV DNA of more than 200,000 IU/mL, the initiation of TDF at 30–32 weeks’ gestation until 4 weeks postpartum was compared with placebo with standard immunoprophylaxis (HBIg + three doses of HBV vaccine) administered to all infants (217). This resulted in a reduction of MTCT from 7% in the placebo group to 0% in the TDF group. In contrast, a recent double-blind clinical trial conducted in Thailand of 331 women with high viral load (median HBV DNA 1.0 × 108 IU/mL) randomized to placebo (n = 163) or TDF (n = 168) at 28 weeks of gestation showed that 3 (2%) of 147 infants included in the final analysis in the placebo group were infected compared with none in the TDF group, with no between groups statistical significance. However, in the second study, it was noted that all infants received very early immunoprophylaxis (within 1.2–1.3 hours after birth) and five doses of HBV vaccine, and TDF treatment was started earlier (at 28 versus 30–32 weeks) (223). Given the most robust published data on efficacy and high potency with a rapid reduction in DNA, along with its safety and resistance profile, TDF is the recommended agent for prevention of HBV MTCT. Therapy should be initiated at 24–32 weeks (commonly, at around 28 weeks) of gestation. Earlier initiation of therapy should be considered in women with risk factors for pre-term labour, women who are pregnant with multiples (twins or more), and women undergoing invasive procedures, such as amniocentesis. Initiation in the second trimester should also be considered in women with a viral load greater than 109 IU/mL.
The optimal duration of NA therapy postpartum, when being given strictly to prevent MTCT, is uncertain. Prolonging therapy 4–12 weeks postpartum decreases the likelihood of ALT flares from 45% to 30% (217), but these flares are generally mild and self-limited regardless. This should be considered along with the mother’s plans to breastfeed. Although TDF is excreted in breast milk, it is excreted at low concentrations, representing only 0.03% of the proposed oral infant dose of TDF (224). Hepatitis B is also detectable in breast milk; however, this does not increase the risk of MTCT, and did not even in the era before vaccination (225–227). As such, breastfeeding is not contraindicated for either women with untreated HBV or those on TDF. Because delivery by cesarean section carries inherent risk and has not been shown to further decrease the risk of MTCT, it is not recommended for HBsAg-positive mothers outside of the usual obstetrical indications (211). Prevention strategies cannot eliminate risk, so babies should be tested for anti-HBs and HBsAg starting at age 9 months and 1–4 months after the last dose of the vaccine is administered to document that they are uninfected and immune (8). Although the incidence of vaccine escape mutations is rare, case reports have been reported in fully immunized children; hence, if clinically indicated, repeat serology may be considered for the child at an older age (228,229).
Recommendations for Management of HBV and Pregnancy
-
32.
All pregnant women should be screened for HBsAg in the first trimester of pregnancy. (strong recommendation; class 1)
-
33.
HBsAg-positive pregnant women should undergo additional assessment for HBeAg, anti-HBe, HBV DNA, and ALT and be referred to a specialist for management. (strong recommendation; class 1)
-
34.
TDF is the drug of choice for pregnant women and women of child-bearing potential who require immediate treatment for HBV. (strong recommendation; class 1)
-
35.
For women planning to become pregnant who have indications for therapy and favourable predictors of response (low HBV DNA, high ALT, genotype A), PEG-IFN for 48 weeks can be considered for those who are willing to defer pregnancy to undertake therapy of finite duration (as is recommended for all chronic carriers who wish to consider finite therapy). (moderate recommendation; class 2, level B)
-
36.
Pregnant women with HBV DNA of more than 200,000 IU/mL should initiate antiviral therapy at 24–32 weeks (~28 weeks) of gestation to reduce the risk of vertical transmission; TDF is the drug of choice and may be stopped at delivery or up to 12 weeks postpartum if given strictly to prevent MTCT. (strong recommendation; class 1)
-
37.
All women should be monitored for ALT flares postpartum, with ALT testing every 4 weeks for the first 3 months and then at 6 months, followed by routine monitoring thereafter. (moderate recommendation; class 2, level B)
-
38.
All infants born to HBsAg-positive mothers should receive immunoprophylaxis with HBIg and HBV vaccine within 12 hours of birth and completion of second and third HBV vaccine doses at 1 and 6 months, respectively. (strong recommendation; Class 1)
-
39.
Babies should be tested for HBsAg and anti-HBs between 1 and 4 months after the last dose of vaccine to confirm that they are uninfected and immune. (strong recommendation; class 2, level A)
-
40.
Breastfeeding is not contraindicated in either untreated HBsAg-positive mothers or those on NA. (strong recommendation; class 1)
8.0. HBV-INFECTED HCWS (KE DOUCETTE)
Transmission of HBV from health care workers (HCWs) to patients is rare and estimated to be less than 1% by prospective studies and 0.24%–0.024% by modelling (230,231). Since the introduction of HBV vaccine in the 1980s, the global prevalence of HBV has declined, with an increasing proportion of the population immune to it. Because they are high-risk groups for exposure, all susceptible HCWs and students should be vaccinated for HBV (8).
Since the introduction of serology for HBV screening in the 1970s, there have been more than 45 reports of HBV transmission from HCWs and more than 400 patients infected (232,233). No dentist-to-patient transmission has been reported since the late 1980s, but this may in part reflect exclusion of HBsAg-positive individuals from dentistry for a period. Two cases of HCW-to-patient transmission have been reported in Canada, one by an electroencephalogram technician involving a breach in infection prevention and control (IPC) practices and the other by an infected orthopedic surgeon (234,235). Synthesis of the data suggest that HCWs and students not involved in exposure-prone procedures (EPPs) do not transmit HBV unless there is a breach in IPC practices (232,236).
In general, the risk of transmission of HBV from a HCW to a patient requires (a) sufficient infectious virus circulating in the HCW’s blood, (b) a breach in the HCW’s skin integrity (eg, needle-stick, non-intact skin because of a medical condition), and (c) exposure of a patient’s blood, wound, or mucous membranes to the HCW’s blood. The second and third criteria are what aid in defining EPPs; however, defining EPPs remains challenging because of vast, varying, and evolving surgical techniques in addition to individual patterns of practice. Essentially, EPPs are procedures that occur in difficult-to-access areas, with significant risk of a puncture wound to the HCW (237,238). Updated US Centers for Disease Control and Prevention recommendations categorize only a limited number of procedures as category I, increased risk, or EPPs for the transmission of HBV from provider to patient (236). These procedures include major abdominal, cardiothoracic, and orthopedic surgery; repair of major traumatic injuries; abdominal and vaginal hysterectomy; cesarean section; vaginal deliveries; and major oral and maxillofacial surgery, such as fracture reductions. Notably, no routine dental procedures are included as EPPs, and none of these Category I procedures is essential, or even commonly performed, by medical or dental students.
Defining an HBV DNA threshold below which EPPs can safely be performed is also challenging because of incomplete data in the literature regarding the HCW viral load at the transmission event, variability in viral load results even with commercial assays reporting less than 10 IU/mL (239), and fluctuations in viral load in untreated individuals (240). Although the lowest documented HBV DNA documented in a surgeon transmitting HBV to a patient is 4 × 104 virus copies per millilitre (6.9 × 103 IU/mL), viral load cut-offs of 200–2,000 IU/mL have been recommended in Europe (53,232) and in the United States (236,237). These thresholds aim to be well below the lowest documented HCW-to-patient transmission event considering in vitro and in vivo HBV DNA fluctuations. The Public Health Agency of Canada has set a threshold of 1,000 IU/mL, above which HCWs infected with HBV are restricted from performing EPPs. However, the province of Quebec has set this threshold at 2,000 IU/mL (241). Current antiviral therapy for HBV can reduce HBV DNA to undetectable levels in most individuals within weeks to months and is recommended for HCWs who engage in EPPs who have a viral load above the established threshold. Because a level of 1,000 IU/mL does not fully align with clinically established indications for therapy and many individuals with a viral load in this range will have fluctuations above and below this threshold, the pros and cons of initiating antiviral therapy should be discussed with any HCW performing EPPs who has detectable HBV DNA. The primary advantage is to minimize the risk of intermittent restrictions on performing EPPs should the HBV DNA increase above the threshold, with the primary disadvantage being the need for long-term antiviral therapy strictly for the continued practice of EPPs.
All HCWs with chronic HBV, regardless of whether or not they perform EPPs or are on antiviral therapy, should be under the care of a physician with expertise in HBV management and followed up according to standard recommendations in the general population. In addition, they should understand routine IPC practices to minimize the risk of transmission and be instructed in how to report any potential patient exposure in their workplace or facility.
Recommendations for the Management of HCWs with Hepatitis B
-
41.
All HCWs who perform EPPs as defined by the US Centers for Disease Control and Prevention (procedures performed on a difficult-to-access area with limited visualization and significant risk of puncture to the HCW) have an ethical obligation to know their HBV status. (strong recommendation; class 2)
-
42.
All susceptible HCWs, including students, should be immunized for HBV. (strong recommendation; class 1)
-
43.
HBV infection alone should not disqualify an infected person from study or practice in any health care field. (strong recommendation; class 2, level C)
-
44.
HCWs infected with HBV should be under the care of an independent physician with expertise in the management of HBV. (moderate recommendation; class 2, level B)
-
45.
HCWs infected with HBV who do not perform EPPs do not need restrictions placed on their practice. (moderate recommendation; class 2, level B)
-
46.
HCWs infected with HBV should be restricted from performing EPPs if their HBV DNA is above 1,000 IU/mL. (moderate recommendation; class 2, level B)
-
47.
For HCWs infected with HBV who perform EPPs, consideration should be given to treating at any viral load, after a discussion of the potential risks and benefits, given the common fluctuations in HBV DNA in untreated individuals and the risk of interruption in practice should such a fluctuation occur. (moderate recommendation; class 3)
-
48.
HCWs infected with HBV who perform EPPs should have HBV DNA monitored every 3–6 months regardless of whether or not they are on antiviral therapy. (moderate recommendation; class 2, level B)
-
49.
HCWs infected with HBV should understand routine IPC practices to minimize the risk of transmission and know how to report any potential patient exposure in their workplace or facility. (strong recommendation; class 2, level A)
9.0. MANAGEMENT OF HEPATITIS B IN IMMUNOSUPPRESSED PATIENTS (KE DOUCETTE)
Both HBsAg-positive and HBsAg-negative, anti-HBc-positive individuals undergoing IS therapy or chemotherapy are at risk for HBV reactivation. The risk is generally classified as high (> 10%), moderate (1%–10%), or low (< 1%) (53,242–244), although there are new and emerging therapies for which the risk remains somewhat uncertain. HBV reactivation may result in ALT flares, fulminant liver failure, and death. Table 10 summarizes the IS therapies and relative risks of HBV reactivation. Because HBV infection is not well predicted by risk factor–based screening, all candidates for IS or chemotherapy should be screened for HBsAg, anti-HBc, and anti-HBs before therapy. Those who are non-immune should be vaccinated in accordance with Canadian National Advisory Committee on Immunization recommendations (8).
Table 10:
Risk of HBV reactivation with immunosuppression and chemotherapy in HBsAg-Positive and HBsAg-Negative, anti-HBc positive Patients
| Risk group and HBV serology | Immunosuppressive or chemotherapy |
|---|---|
| High-risk group (> 10%) | |
| HBsAg positive OR HBsAg negative and anti-HBc positive (high risk regardless of anti-HBs titre levels) |
B-cell-depleting agents such as rituximab and ofatumumab |
| HBsAg positive | Anthracycline derivatives such as doxorubicin and epirubicin Corticosteroid therapy for ≥4 weeks (prednisone equivalent > 10–20 mg/day) |
| Moderate-risk group (1%–10%) | |
| HBsAg positive OR HBsAg negative and anti-HBc positive (may be lower risk and monitoring may be sufficient if high anti-HBs titres > 100 IU/L) |
TNF-α inhibitors: etanercept, adalimumab, certolizumab, certolizumab, infliximab Other cytokine inhibitors and integrin inhibitors: abatacept, ustekinumab, natalizumab, vedolizumab Tyrosine kinase inhibitors: imatinib, nilotinib, ibrutinib |
| HBsAg positive | Corticosteroid therapy for ≥ 4 wk (prednisone equivalent < 10 mg/day) |
| HBsAg negative and anti-HBc positive (may be lower risk and monitoring may be sufficient if high anti-HBs titres > 100 IU/L) | Corticosteroid therapy for ≥4 weeks (prednisone equivalent > 10–20 mg/day) Anthracycline derivatives: doxorubicin and epirubicin |
| Low-risk group (< 1%) | |
| HBsAg positive OR HBsAg negative and anti-HBc positive (low risk especially if high anti-HBs titres > 100 IU/L) |
Traditional immunosuppressive agents: azathioprine, 6-mercaptopurine, methotrexate Intra-articular corticosteroids Corticosteroid therapy for ≤1 week |
| HBsAg negative/anti-HBc positive (low risk especially if high anti-HBs titres > 100 IU/L) | Corticosteroid therapy for ≥ 4 wk (prednisone equivalent < 10 mg/day) |
anti-HBc = antibody to HBV core; HBsAg = hepatitis B surface antigen; HBV = hepatitis B virus; TNF = tumour necrosing factor
Figure 9 outlines the approach to management of HBsAg-positive and HBsAg-negative, anti-HBc-positive (with or without detectable anti-HBs) patients undergoing immunosuppression or chemotherapy. For those at lower risk for reactivation, monitoring is recommended, whereas for those at higher risk for reactivation, prophylactic antiviral therapy is recommended. Although the use of LAM in this setting has been extensively studied and decreases the risk of reactivation and death (245,246), more recent studies have shown that potent NAs such as ETV are superior to LAM in preventing reactivation in those at high risk for reactivation (247,248). As such, a potent NA is preferred for those who are at high risk for reactivation (ie, especially if they are HBsAg positive with detectable HBV DNA). The risk of HBV reactivation continues for at least 6–12 months after completion of immunosuppression, and even longer in those undergoing potent immunosuppression with B-cell-depleting therapy (≤2 years). Thus, NA therapy should continue for at least 6–12 months after completion of IS therapy or until immune reconstitution (249). New immune-modulating therapies are rapidly being developed, and the effect of these drugs on HBV reactivation is not always clear at the time they are introduced into clinical use. For new agents for which the risk is unknown, monitoring ALT every 1–3 months is recommended as a minimum, with HBV DNA monitoring every 3–6 months.
Figure 9:
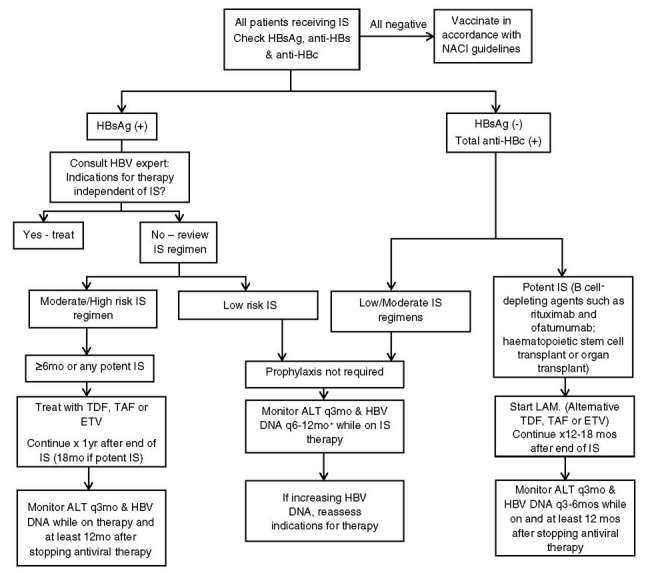
Algorithm for management of hepatitis B in immunosuppressed patients. All patients should be screened for HBV and offered vaccination if at risk. HBsAg-positive patients should receive prophylaxis with most regimens. HBsAg-negative, anti-HBc-positive patients with immunosuppression may be treated with first-generation NA (LAM) because of the lower risk of resistance in those with undetectable HBV DNA. In patients that are HBsAg negative, anti-HBc positive with high anti-HBs titres (>100 IU/L) on low-moderate risk IMS, monitoring may be acceptable.
anti-HBc = antibody to HBV core; anti-HBs = antibody to HBsAg; ETV = entecavir; HBsAg = hepatitis B surface antigen; HBV = hepatitis B virus; IS = immunosuppression; LAM = lamivudine; NA = nucleos(t)ide analogue; NACI = National Advisory Committee on Immunization; TAF = tenofovir alafenamide; TDF = tenofovir disoproxil fumarate.
Recommendations for Management of Hepatitis B in Immunosuppressed Patients
-
50.
All patients undergoing immunosuppression or chemotherapy should be screened for HBsAg, anti-HBc, and anti-HBs before therapy. (strong recommendation; class 2, level A)
-
51.
HBsAg-positive patients are at high risk of reactivation and should undergo either monitoring or prophylactic NA therapy (especially with moderate to potent immunosuppression). (strong recommendation; class 2, level A)
-
52.
HBsAg-negative, anti-HBc-positive patients may be at risk of reactivation and, based on their degree of risk, should undergo either monitoring (if they have high anti-HBs titres of > 100–1,000 IU/L) or prophylactic NA therapy (especially if they are on B-cell-depleting therapies). (moderate recommendation; class 2, level C)
-
53.
Potent NAs (ETV, TDF, or TAF) are preferred when prophylaxis is used; LAM is an alternative especially for HBsAg-negative, anti-HBc-positive cases. (strong recommendation; class 2, level A)
-
54.
After completion of IS therapy, monitoring or prophylaxis, as applicable, should continue for at least 12 months or until immune reconstitution, or longer in those who received B-cell-depleting therapies. (strong recommendation; class 2, level B)
10.0. HBV-ASSOCIATED RENAL DISEASE AND MANAGEMENT OF HEPATITIS B IN PATIENTS WITH END-STAGE RENAL DISEASE (E KELLY, MM MA)
10.1. Extrahepatic HBV and renal disease
Extrahepatic manifestations of HBV, including renal disease, may occur in patients with CHB as a result of circulating immune complex deposition and complement activation (250). Renal extrahepatic manifestations of HBV include membranoproliferative glomerulonephritis, membranous nephropathy, polyarteritis nodosa, immunoglobulin A nephropathy, mesangial proliferative glomerulonephritis, and amyloidosis (251). Polyarteritis nodosa is a rare systemic complication of chronic HBV infection that causes inflammation of the small- and medium-sized blood vessels (252). Polyarteritis nodosa can affect multiple organ systems, leading to multiorgan failure. In glomerulonephritis (GN) associated with hepatitis B infection, the deposition of circulating immune complexes in the glomeruli of kidneys cause the severe destruction of the glomeruli. GN occurs mainly in male children in HBV-endemic areas. In children, GN is usually self-limited without progression to renal failure. In adults, hepatitis B–induced GN can be severe and can progress to renal failure. An immune complex serum sickness–like syndrome can cause arthritis and dermatitis, but the syndrome tends to resolve without significant sequelae. Cryoglobulinemia has been reported in association with hepatitis B infection. In general, a kidney biopsy demonstrating HBV antigen deposition is required for diagnosis (253). Treatment with antiviral therapy should be considered, and agents with high potency but lower nephrotoxicity such as ETV or TAF are preferred.
10.2. Management of HBV in patients with renal failure
All NAs used for treatment of HBV undergo renal clearance. The NAs have been demonstrated to affect renal function in some patients on long-term HBV therapy. Mechanisms of injury include alteration in tubular transport and mitochondrial injury. Cases of severe tubular damage leading to Fanconi syndrome have been described with long-term use of TDF, mostly in individuals co-infected with HBV and HIV (254–257). In such cases, there are reports of successful improvement in renal function (eGFR) with a switch to ETV (254). Although eGFR declines are more common with TDF, other NAs have also shown a risk for progressive renal injury. Therefore, careful monitoring for all patients on antiviral therapy is indicated. Renal function should be monitored every 6 months while patients are on HBV antiviral therapy. In addition, among patients initiating TDF, monitoring serum phosphorus levels and urine protein and glucose before treatment initiation, and at least annually while on therapy, is recommended. If signs of declining renal function or development of hypophosphatemia or proteinuria occur, a switch to another agent should be considered.
Among patients with established chronic kidney disease initiating antiviral therapy, options for therapy include TDF, ETV, or TAF. TDF and ETV require dose adjustment for eGFR of less than 50 mL/min (258), whereas TAF does not require dose adjustment if eGFR is less than or equal to 15 mL/min. Although pharmacokinetic studies and case reports have not shown significant differences between patients with eGFR of less than 15 mL/min and those on renal replacement therapy (259,260), TAF has not been approved for use with those patients. In recent registered clinical trials, TAF was shown to be superior to TDF in preservation of glomerular and tubular renal function at 48 weeks and showed less progression to chronic kidney disease up to 48 weeks (170,171,260). Data have also shown reduced proteinuria, albuminuria, and tubular protein losses in individuals co-infected with HIV and HBV on TAF (261,262). Longer term clinical data on the efficacy of TAF are lacking. Either ETV or TAF is recommended as a first-line treatment for patients with impaired eGFR (< 60 mL/min), with a preference for TAF for patients with prior exposure or resistance to LAM. Among patients on hemodialysis or with eGFR of less than 15 mL/min, currently available treatment options include renally dosed ETV or TDF (see Section 6.0).
Recommendations for Management of Hepatitis B and Renal Disease
-
55.
Biopsy-proven renal disease caused by CHB is an indication for hepatitis B treatment. (strong recommendation; Class 1)
-
56.
Renal function should be monitored every 6–12 months while patients are on NA therapy; urine protein and glucose and fasting phosphate should be monitored annually. (strong recommendation; class 2, level B)
-
57.
Among patients with signs of worsening renal function (decline in eGFR) while on TDF, a switch to ETV or TAF is recommended. (strong recommendation; class 2, level A).
11.0. MANAGEMENT OF HEPATITIS B AND DECOMPENSATED CIRRHOSIS (C FOURNIER AND JP VILLENEUVE)
Worldwide, CHB infection is responsible for a third of all cases of cirrhosis and more than half a million deaths annually. In patient with chronic HBV infection, the mortality rate differs according to the study population. Patients who have cirrhosis are estimated to progress to decompensated cirrhosis at a rate of 3% annually, usually during the first 5 years after the development of cirrhosis. The 5-year survival rate is 84% in patients with compensated cirrhosis, but it decreases to 14%–35% in individuals with decompensated cirrhosis (section 2.0) (263–265)
In a known cirrhotic patient, decompensated cirrhosis is defined by at least a new-onset episode of jaundice or by the presence of ascites, hepatic encephalopathy, or variceal bleeding. Several studies have shown that patients with high levels of viral replication, demonstrated by either a positive HBeAg or a high HBV DNA serum concentration, have a four to six times increased risk of hepatic decompensation during follow-up. Besides viral replication, additional risk factors for the progression of the severity of the liver disease include ALT flares, the presence of HDV or HCV co-infection, age, male sex, and the severity of fibrosis (section 3.0). In patients with advanced cirrhosis, risk factors for short-term mortality are hepatic dysfunction (low serum albumin, low platelets, increased serum bilirubin), the presence of ascites or hepatic encephalopathy, advanced age, and signs of portal hypertension (50,53,54,264–266). Patients with decompensated cirrhosis should be referred for consideration of liver transplantation and treated with NAs as soon as possible. High Child-Pugh or Model for End-Stage Liver Disease scores are predictors of poor survival. The Model for End-Stage Liver Disease score is used to prioritize patients for liver transplantation in Canada (267,268).
In patients with decompensated cirrhosis, NA treatment must be started as soon as possible, irrespective of ALT level, HBeAg status, and HBV DNA. Studies have shown that early treatment results in better results than delayed treatment with improved 5-year survival rates (60% versus 46%, respectively). PEG-IFN alpha is contraindicated in patients with severe liver disease. Among NAs, ETV or TDF is the preferred treatment option in view of their potency and low rate of development of resistance. In patients with prior LAM resistance, TDF is the preferred choice (see section 6; Figures 6–7, Table 9). Lifelong treatment is recommended. Although NAs have a good safety profile, there are few reports on their tolerability and safety in patients with severe liver disease (171,269–273). Lactic acidosis has been reported as a very rare side effect of NAs by inhibiting mitochondrial polymerase γ in liver and muscle, which remains a concern in patients with advanced decompensated cirrhosis (274). In patients with HBV–HIV co-infection and compensated liver disease, TDF has been reported to result in a decrease in renal function and a decrease in bone mineral density, and these findings have rarely been described in HBV monoinfection (274,275). The dosage of NAs must be adjusted to renal function. Because of its favourable safety profile with regard to kidney function, TAF could be an interesting option for patients with decompensated cirrhosis, but data on the safety of TAF in this population are lacking (section 10.0) (171,272,273). Antiviral therapy with NAs significantly modifies the natural history of decompensated HBV cirrhosis. Undetectable HBV DNA can be achieved in a majority of patients. Treatment results in a clinical and biochemical improvement in 40%–50% of cases, and approximately one-third of cases can be delisted for liver transplantation (273).
Table 9:
Management of antiviral-resistant HBV
| Resistance mutation | |||
|---|---|---|---|
| LAM resistant (L180M + M204V, M204I) | ADV resistant (N236T) | ADV resistant (A181V) | |
| Mutation confers reduced sensitivity to listed drugs | ETV, TBV | TDF | LAM |
| Drugs remaining active | ADV, TDF* | LAM, ETV, TBV | TDF,* ETV |
*Studies are ongoing regarding use of tenofovir alafenamide instead of TDF for LAM or ADV resistance.
ADV = adefovir; ETV = entecavir; HBV = hepatitis B virus; LAM = lamivudine; TBV = telbivudine; TDF = tenofovir
Recommendations for Management of Hepatitis B and Decompensated Cirrhosis
-
58.
NA treatment of adults with decompensated cirrhosis should be initiated promptly, regardless of HBV DNA, HBeAg status, or ALT level. (strong recommendation; class 1)
-
59.
Potent antiviral therapy with an NA with a low rate of drug resistance must be initiated. TDF or ETV are the preferred options. (strong recommendation; class 1)
-
60.
TAF is a potential option for patients with decompensated cirrhosis, but long-term data on the safety of TAF in this population are lacking. (moderate recommendation; class 2, level B)
12.0. MANAGEMENT OF HEPATITIS B IN LIVER TRANSPLANTATION (C FOURNIER AND JP VILLENEUVE)
Because of the efficacy of NA treatment, HBV-related severe liver disease has become an uncommon indication for liver transplantation in Canada. HCC is the most common indication for liver transplant in HBV patients. Before the advent of hepatitis B immunoglobulin (HBIg) and NAs, recurrent HBV infection after liver transplantation was a major problem, but it is now very rare (276). All HBV patients considered for liver transplantation should be treated with NAs to achieve undetectable HBV DNA levels at the time of transplant. A combination of HBIg and NA is the standard of care after liver transplant and prevents reinfection of the graft in more than 95% of cases (277–279). ETV or TDF is the preferred treatment option. TDF is the best alternative for patients with prior LAM resistance. ETV (and possibly TAF) appear preferable in patients with post-transplant renal impairment (53,54,275,280,281). The route of administration, dosing, and duration of treatment with HBIg vary from one transplant centre to the other. Subcutaneous and intramuscular HBIg have an efficacy similar to that of intravenous HBIg and are usually preferred in the long term (282). The combination of HBIg and potent NAs has allowed a decrease in the dose of HBIg after transplant with the aim of achieving anti-HBs levels of about 50–100 IU/L. In selected patients with a low risk of HBV recurrence (ie, undetectable HBV DNA at transplant, HBeAg negative, no HCC, no HIV or HDV co-infection, good adherence to treatment), HBIg can be discontinued 6–12 months after transplant (279,283,284). Because HBIg is costly and inconvenient and NAs have become more efficacious, the question of whether HBIg is needed at all has been raised. Studies using prophylaxis with NAs alone suggest that this is feasible with a low rate of HBsAg recurrence (284,285). HBsAg-negative transplanted patients receiving a liver from a donor with evidence of past HBV infection (HBsAg-negative and anti-HBc-positive donor) are at risk of HBV reactivation because of immunosuppression and should receive lifelong prophylaxis with NAs. A first-generation NA (LAM) is a reasonable choice in recipients of anti-HBc-positive grafts especially if high anti-HBs titres (286,287).
Recommendations for Management of Hepatitis B and Liver Transplantation
-
61.
All HBV patients considered for liver transplantation should be treated with NAs. (strong recommendation; class 1)
-
62.
A combination of HBIg and NAs (ETV or TDF) is the standard of care after liver transplant. (strong recommendation; class 2, level A)
-
63.
In selected patients with a low risk of HBV recurrence, HBIg can be discontinued 6–12 months after transplant. (moderate recommendation; class 2, level A)
-
64.
HBsAg-negative transplanted patients receiving a liver from a donor with evidence of past HBV infection (ie, anti-HBc positive, HBsAg negative) should receive lifelong prophylaxis with NAs. (strong recommendation; class 2, level A)
13.0. MANAGEMENT OF ACUTE HEPATITIS B INFECTION (JP VILLENEUVE)
Antiviral therapy is generally not necessary for patients with acute hepatitis B, because 90%–95% of immunocompetent adults will recover spontaneously clinically and virologically, including seroconversion to anti-HBs (section 3.0). Severe or fulminant acute hepatitis B is a rare but potentially life-threatening manifestation of the disease. Severe acute hepatitis B is usually defined by the presence of coagulopathy (international normalized ratio > 1.5) or marked jaundice for more than 4 weeks; fulminant hepatitis, by the presence of hepatic encephalopathy. Two small randomized controlled trials of LAM versus placebo in patients with severe acute hepatitis B found no difference in clinical or biochemical outcomes between the two groups (288,289).
Despite the lack of benefit from these small underpowered trials, an argument has been made to treat with NA patients with severe or fulminant acute hepatitis B who are candidates for liver transplantation to reduce the risk of recurrent hepatitis B after transplant (53,54). TDF, ETV, or even LAM are reasonable choices and have been shown to be safe in acute hepatitis B.
Recommendations for Management of Acute Hepatitis B Infection
-
65.
More than 90%–95% of immunocompetent adults with acute hepatitis B do not require specific treatment because they will fully recover spontaneously. (strong recommendation; class 2, level A)
-
66.
Patients with severe or fulminant acute hepatitis B can be treated with NAs if they are considered for liver transplantation. (strong recommendation; class 2, level C).
14.0. HBV AND HIV CO-INFECTION (CL COOPER)
Approximately 6%–8% of Canadians living with HIV are co-infected with HBV (290). Co-infection is associated with more rapid progression of liver fibrosis and a heavy burden of HCC, cirrhosis, and end-stage liver disease (291). Antivirals including TDF, TAF, LAM , and emtricitabine possess activity against both HIV and HBV. As such, they should be included in HIV antiretroviral regimens. Treatment by physicians with HIV and HBV expertise should be initiated once the diagnosis of HIV and HBV is established and irrespective of CD4+ T cell count. Monitoring for HBV-specific immune reconstitution during the initial three months after anti-retroviral initiation is required (292). There is a small risk for proximal tubular nephropathy (ie, Fanconi syndrome) and osteopenia with the use of TDF. This risk is greater with TDF than with TAF. Emerging data suggest HIV and HBV efficacy as well as safety in TAF recipients co-infected with HIV and HBV (293). Therefore, TAF may represent a preferred antiviral in the context of HIV–HBV co-infection.
Some populations of HIV-infected individuals are at greater risk for exposure to HBV, for example, men who have sex with men and persons who inject drugs (294). All people living with HIV should be screened for HBV infection and immunity. If a patient is anti-HBs negative, then HBV immunization should be pursued because it is effective in preventing infection (295) (see section 1.0). HIV–HBV co-infection is not a contraindication to liver transplantation assuming all other eligibility criteria are met. Post-transplant outcomes including survival are comparable to those of people with HBV monoinfection (275,278). In addition, HBsAg-positive individuals who would potentially benefit from HIV pre-exposure prophylaxis with TDF and emtricitabine should have such prophylaxis initiated, regardless of HBV status. However, HBV status should be closely monitored for virological and biochemical flares once pre-exposure prophylaxis is no longer needed. There is no risk of anti-HBV resistance with TDF treatment interruptions.
Recommendations for Management of HBV–HIV Coinfection
-
67.
All people living with HIV should be screened for HBV infection and immunity. If an individual is anti-HBs and anti-HBc negative, then HBV immunization should be pursued. (strong recommendation; class 2, level A)
-
68.
HIV treatment with dual HBV-active antivirals should be initiated once the diagnosis of HIV and HBV is established and irrespective of CD4+ T cell count. (strong recommendation; class 2, level A)
-
69.
On-treatment monitoring of HBV is the same as for people with HBV monoinfection, but HBV therapy is continued until HBsAg loss. (moderate recommendation; class 2, level C)
-
70.
Patients who have interruptions in HIV therapy that included dual-active HBV antivirals should be monitored for HBV reactivation if they are unable to continue with an anti-HBV antiviral. (strong recommendation; class 2, level B)
15.0. HBV–HCV CO-INFECTION (CL COOPER)
Because of shared risk factors, HBV and HCV can be found concurrently (296,297). Fibrosis progression is accelerated, and HCC risk is increased (298). Screening for one is recommended in the presence of the other (53,54). There is a risk of HBV flare in HBsAg-positive individuals initiating DAA therapy for hepatitis C (299). On the basis of suboptimal data, this risk appears to be low; often only HBV DNA increases but, in its most severe form, it can result in fulminant hepatitis (300,301). Co-initiation of a HBV antiviral concurrent with DAA dosing should be considered, with HBV NA therapy maintained for 12 weeks after the completion of anti-HCV DAA treatment (302). An acceptable alternative is to monitor these patients without initiating HBV antiviral therapy. Irrespective of whether an HBV antiviral is initiated, liver enzymes should be monitored at least every 4 weeks, and while a patient is on DAA therapy, HBV DNA levels should be monitored for up to 24 weeks after completion (53). Similar ALT monitoring is advised for HBsAg-negative, anti-HBc-positive individuals, especially if cirrhotic. The incidence of HBsAg and HBV DNA re-emergence with DAA treatment is uncommon (< 10% are reported to show low-level HBV viremia) in the anti-HBc-positive population (303,302,304).
Recommendations for Management of HBV–HCV Coinfection
-
71.
All HCV patients should be tested for HBsAg before HCV DAA therapy is initiated. (strong recommendation; class 2, level B)
-
72.
HBsAg-positive patients should undergo monthly ALT and every-3-month HBV DNA monitoring while on HCV DAA and until 24 weeks after treatment. HBV antiviral therapy should be initiated promptly for patients with a rise in ALT and HBV DNA (> 1 log IU/mL) during or after completion of DAA therapy. (moderate recommendation; class 2, level C)
-
73.
HBsAg-negative and anti-HBc-positive (with or without anti-HBs) patients should be monitored with ALT while on HCV DAA and until 24 weeks after treatment. HBsAg and HBV DNA should be measured if ALT does not normalize or increases during or after completion of DAA treatment. HBV antiviral therapy should be initiated promptly for patients in whom HBsAg or detectable HBV DNA with elevated ALT emerges. (moderate recommendation; class 2, level C)
16.0. HBV–HDV CO-INFECTION (CS COFFIN)
HDV is a small satellite single-stranded RNA virus. It can only occur in the presence of concomitant HBV infection and is spread by similar blood-borne or parenteral routes as well as by sexual transmission (305). HDV co-infection can occur simultaneously with acute HBV infection or as a superinfection in chronically infected HBV carriers, and it is considered the most severe form of viral hepatitis in humans. The complete HDV antigen consists of the viral genome, 2 delta isoform proteins, encapsulated by a lipid envelope embedded with HBV surface proteins. HDV entry is also mediated by binding of the HBV pre-S1 to the hepatocyte bile acid receptor (NCTP; section 2.0), thus explaining the reliance of HDV on HBV to complete its life cycle (306). It is estimated that approximately 15–20 million people worldwide have HDV co-infection, or 5% of all persons with HBV infection (307). There is significant geographic variation worldwide; HDV is endemic in parts of Western and Central Africa, South America, certain parts of Asia (India and Pakistan), Turkey, Southern Italy, and countries from the former Soviet Union. In particular, the Amazon Basin and Mongolia have the highest reported rates of HDV infection, affecting approximately 60%–65% of HBsAg-positive individuals (307). In the United States and Europe, it is estimated that about 8% of HBV carriers have HDV co-infection, especially among persons who inject drugs (305). Prevalence rates in Canada are similarly low, based on limited epidemiological studies (308,309). Commercially available diagnostic tests for HDV screening are not widely available in many regions worldwide, and HBsAg-positive individuals are not routinely screened; hence, the prevalence of HDV is likely underestimated. The recommended screening test is antibody (immunoglobulin G) to HDV antigens. To confirm active infection or viremia, HCV RNA by PCR methodology is recommended, which is quantified on the virus copies per millilitre or, more recently, an international standard of international units per milliliter (305).
Both acute HDV–HBV coinfection and HDV superinfection lead to a more severe viral hepatitis and liver disease. Compared with individuals with HBV monoinfection, co-infected individuals are more likely to progress to cirrhosis (> 80%), even within 2 years of infection, and are at higher risk of developing HCC and liver decompensation (307). As a result of complex viral interactions, HDV co-infection can lead to suppression of HBV replication and vice versa (310), with fluctuating levels of HBV DNA, quantitative HBsAg, and HDV RNA over time with active hepatitis. Non-invasive assessment of fibrosis (ie, FibroScan) and other scores are not well validated or do not perform well in patients with chronic HDV; hence, liver biopsy is considered the gold standard for assessing the stage of disease (311).
Treatment options for HDV are limited, other than liver transplantation for patients with end-stage liver disease who meet other transplant criteria. The HBV NAs are not effective against HDV. The only currently approved therapy is IFN (PEG-IFN) administered for a minimum of 48 weeks, with some studies extending therapy to 96 weeks (312). However, PEG-IFN is only effective in about 20%–30% of HDV co-infected individuals, as confirmed by serum HDV RNA negativity at the end of treatment, according to sensitive PCR (HDV RNA) tests. The most comprehensive evaluation of PEG-IFN alpha treatment with long-term outcome reporting has been conducted in two studies by the Hep-Net-International Delta Hepatitis Intervention Trial (HIDIT) study group. In the HIDIT-1 study, patients received either PEG-IFN alpha-2a and ADV or either drug alone. In the PEG-IFN treated arm, about 25% of patients achieved HDV RNA negativity that was sustained for 24 weeks after the end of treatment, compared with 0% of those treated with ADV (313). A long-term, 5-year follow-up of patients from the HIDIT–1 study showed that, despite testing HDV negative at end of treatment, 70% of patients who were treated in either of the PEG-IFN arms were positive at least once during follow-up, with 44% being HDV RNA positive at end of follow-up (314). This highlights the need for close follow-up of patients even when HDV RNA negativity is achieved. A second study (HIDIT–2; published in abstract form) aimed at determining the benefit of longer treatment (96 weeks) with PEG-IFN alpha and TDF did not demonstrate any improvement in response rates (315). However, long-term follow-up of the patients treated with high-dose IFN (mean 5.2 years, range extending to 12 years) demonstrated more frequent loss of HDV RNA, increased survival rates, and lower likelihood of clinical liver disease progression (316). Despite HDV patients’ poor response rates to IFN therapy, the potential for long-term clinical benefits in responders supports a recommendation for treatment in patients with chronic HDV infection. However, careful selection of patients for treatment, taking into consideration ALT levels, the degree of histologic fibrosis, and tolerance of side effects, is recommended. A clinical score (ie, baseline event anticipation) has been developed to predict patients who are at increased risk of liver-related complications, and should be considered in treatment decisions (317). HDV is considered an “orphan” disease; hence, new therapies are urgently needed. Clinical trials have been performed targeting HBsAg (given HDV reliance on the HBV surface to complete its life cycle); they have included entry inhibitors (MyrcludexB), antisense molecules (see Section 18.0), and nucleic acid polymers to block particle release, as well as prenylation inhibitors (318).
There is a reported risk of HBV reactivation with suppression of HDV; thus, monitoring for HBV flares is recommended during PEG-IFN therapy, and some recommend consideration of anti-HBV therapy to block residual HBV replication, especially in patients who have underlying advanced fibrosis or cirrhosis (318). All HDV–HBV co-infected patients with cirrhosis should undergo HCC surveillance—starting at recommended ages for HBV monoinfected patients of African or Asian descent—as well as consideration of early-age screening of those with more advanced liver disease (greater than stage 3 fibrosis) (319).
Recommendations for HBV and HDV Coinfection
-
74.
HBV-positive patients from HDV-endemic areas who have a history of injection drug use, abnormal ALT despite antiviral therapy, and advanced liver disease should be screened for HDV co-infection. (strong recommendation; class 2, level B).
-
75.
HDV screening should use an approved assay for HDV antibody detection followed by confirmatory HDV RNA by PCR. (strong recommendation; class 2, level B)
-
76.
PEG-IFN therapy (180 μg once weekly) for 48 weeks should be considered in individuals without contraindications to IFN treatment. (moderate recommendation; class 1)
-
77.
HBV should be monitored during PEG-IFN therapy, and anti-HBV NA should be considered to block residual viral replication, especially if patients are cirrhotic and given the risk of more aggressive disease progression. (moderate recommendation; class 3)
-
78.
Research on HDV epidemiology, including detailed long-term natural history studies and new therapies, is urgently needed. (strong recommendation; class 3)
17.0. MANAGEMENT OF HEPATITIS B IN PEDIATRIC PATIENTS (SR MARTIN AND F ALVAREZ)
17.1. Prevalence of HBV infection in children
Since 2013, no data have been available in Canada on the prevalence of either acute or chronic HBV infection in pediatric patients. However, several publications from pediatric centres throughout the country provide some information on the Canadian experience. In 2009, Statistics Canada reported a prevalence of around 0.4% (95% confidence interval: 0.2%–0.7%) in patients between ages 14 and 49 years, thus including some adolescents. Data from the government of Canada in 2013 showed that the peak of acute and chronic infection is observed around adolescence (Figure 1) (section 1.0). In 2009, higher prevalence was observed among non-White and foreign-born people. In the same age group, a higher percentage of previously resolved HBV infection was found—less than 2.7% in Canadian-born people and 9.9% in foreign-born people.
A recent report from a pediatric centre in Ontario described a study population in which 85% of children with chronic HBV infection were Asian (320). Another series from Quebec indicated that around 43% were Asian and, in most cases, were adopted children from Asia (321); this centre has lately observed no changes in the followed population with chronic HBV. Data published by the Canadian Public Health Services in 2013 are provided in Figure 1 and available online at https://www.canada.ca/en/public-health/services/publications/diseases-conditions/report-hepatitis-b-c-canada-2013.html.
17.2. Natural history of HBV infection in pediatrics
The great majority of children are asymptomatic at presentation. An exception is children who may present with acute liver failure. Asian children, mostly infected by vertical transmission, show delayed HBeAg seroconversion around puberty (321, 322), either spontaneously or induced by treatment, in comparison with those infected by horizontal transmission, in whom HBeAg seroconversion is observed before age 10 years. Other studies have reported lower rates of HBeAg seroconversion by age 18 years (~50%), especially in Asian children infected by vertical transmission (323).
Several factors predict HBeAg seroconversion, such as an increase in serum ALT levels (in general, more than two times normal values); a decrease in circulating HBV DNA or viral replication (324), and liver inflammatory activity shown by liver biopsy (indicated only in particular circumstances before the beginning of treatment). It should be noted that the presence of moderate or severe hepatitis, as well as the presence of fibrosis, on histology is infrequent in children with chronic HBV infection, although recent data suggest that approximately 25% of children in the United States and Canada who underwent liver biopsy due to a clinical indication have fibrosis higher than Stage 2 (325). Treated children show a tendency to seroconvert from HBeAg to HBe antibody faster than untreated patients, but at the end of the pediatric age range (< 18 years), the difference is not statistically significant (Figure 3A).
Approximately 20% of children seroconvert from HBsAg positive to negative and express HBs antibody before age 18 years, with no significant differences between treated and untreated children. Spontaneous clearance of HBsAg occurs mainly after horizontal transmission in children who have already shown HBeAg seroconversion (326). Extrahepatic complications such as glomerulonephritis are rare—fewer than 1% of children—but in most cases, they will require antiviral treatment (section 10.0). HDV co-infection is rare in Canadian children, but it has been associated with more severe forms of hepatitis and rapid progression to advanced fibrosis, with poor response to available treatments (327). Cirrhosis occurs in 3% of infected children, usually at a young age. In long-term follow-up, these children are at risk for development of HCC, which is rarely reported in non-cirrhotic children (328). In some cases, resolution of the infection (HBeAg and HBsAg seroconversion) can lead to involution of the fibrosis. HBeAg-negative hepatitis is observed in around 5% of children in long-term follow-up and is more common in children with HBV genotype D infection who live in countries around the Mediterranean basin (329).
17.3. Hepatitis B treatment in children
Although children with persistently normal ALT levels and low or undetectable HBV DNA usually have a favourable long-term outcome (329), the benefits of treating chronic HBV infection in children with respect to long-term complications such as cirrhosis and HCC remain to be clearly demonstrated. The spontaneous HBeAg seroconversion rate among children is high, comparable to that induced by treatment, such that treatment may not be necessary (145,321,329–332). Published trials on any of the previously mentioned therapies show 20%–24% HBeAg seroconversion versus 10% for placebo. However, by age 18 years, no difference in the rate of HBeAg seroconversion was observed between groups; hence, the long-term benefits of earlier treatment in children with ALT less than three to four times the normal level remain unclear. Accordingly, the decision to treat takes into account the presence and degree of persistent inflammatory activity associated with high levels of HBV DNA (and its surrogate, HBeAg positivity) and balances this with the cost and limited long-term data on outcomes of children exposed to treatment. Data predominantly based on studies with Asian cohorts (Taiwan) suggest that earlier treatment should be considered even for young persons with normal liver enzymes (43,51). At present, non-invasive evaluation of the liver by TE (ie, FibroScan) for children with CHB has not been validated and does not evaluate inflammatory activity; hence, in children liver biopsy may be necessary to confirm significant liver disease.
17.3.1. Summary of indications for HBV treatment in children
Persistently elevated serum ALT levels in a child with elevated HBV DNA levels (ALT > 1.5 times normal, during 1 year of observation; HBV DNA > 2,000 IU/mL; no liver biopsy required)
Children with cirrhosis, HBV-associated extrahepatic complications (glomerulonephritis, Guillain-Barré, cryoglobulinemia, polyarteritis nodosa), or HIV, HCV, or HDV co-infection and detectable HBV DNA (no liver biopsy required)
Moderate to severe liver inflammation with F2–F4-stage fibrosis on liver biopsy
Family history of HCC in children with high viral load (333)
Liver transplant in HBV-infected recipients, recipient of anti-HBc-positive donor organ and treatment with IS or cytotoxic medications in HBsAg-positive individual or anti-HBc-positive individual, as outlined in sections 9.0 and 12.0 (no liver biopsy required).
17.3.2. Summary of HBV Treatment Options in Pediatrics
IFN alpha can be used in treatment of children younger than age 1 year at a dose of 6 MIU/m2 subcutaneously three times per week (maximum of 10 mIU/dose) for 6 months. The pivotal large, multinational, randomized controlled trial of IFN alpha in children with HBeAg-positive infection found higher rates of HBeAg loss and negative HBV DNA in treated children compared with controls (26% versus 11%; p = 0.03) after 24 weeks of therapy (334). The factors associated with response were elevated ALT, low serum HBV DNA levels, female sex, and young age (< 5 years)—factors that also identify children who will spontaneously seroconvert. Long-term follow-up studies suggest that untreated children may have similar rates of HBeAg seroconversion as IFN alpha–treated children. Although treatment promotes earlier seroconversion by about 3 years, the long-term benefit in the disease’s natural history has not been demonstrated (335,336). Moreover, IFN treatment is costly and difficult to administer, and it is associated with significant side effects (growth, neurotoxicity, cytopenia, and exacerbation of autoimmune disorders) that must be balanced with finite treatment duration, long-lasting response, and no risk of inducing resistance. PEG-IFN has been evaluated for hepatitis B in pediatrics (www.clinicaltrials.gov identifier NCT01519960) and has been used to treat children with hepatitis C; hence, some providers consider using this antiviral drug.
LAM can be used for children younger than age 2 years at a dose of 3 mg/kg/day (maximum 100 mg) for 1 year. In HBeAg-positive children with elevated ALT, response rates to 1 year of LAM were similar to those to IFN alpha (23% of treated children cleared HBeAg and HBV DNA versus 13% with placebo) (337). A follow-up to this study showed the durability of virologic response to be more than 90% 2 years after stopping therapy (338). However, as with adult disease, LAM resistance developed in 19% of patients after 1 year. Although an additional 2 years of LAM increased the virologic response rates, the YMDD mutation rate increased to 64% after 3 years of therapy (339). Thus, LAM is not a suitable for use with young children because this group of patients has high viral loads and may require many years of therapy or treatment years later after a period of inactive disease. The presence of LAM resistance will severely limit future treatment choices.
ADV may be used in children older than age 12 years at a dose of 10 mg by mouth once daily for more than 1 year. In a large pediatric study, higher rates of HBeAg seroconversion versus placebo (15.9% versus 5.3%) were observed after treatment with 48 weeks of ADV (340). No benefit was found in children ages 2–11 years. Dose-related effects on renal function and bone health and concerns with the emergence of resistance in adult studies limit the use of ADV in pediatrics.
ETV has been used in children older than age 2 years at a dose of 0.015 mg/kg (maximum of 0.5 mg/day) in children weighing less than 32.5 kg and 0.5 mg/day in those weighing more than 32.6 kg for more than 1 year. HBeAg seroconversion and HBV DNA of less than 50 IU/mL were 24.2% versus 3.3% in those receiving placebo at week 48 (p < 0.0008). Emergent resistance was 0.6% and 2.6% at the end of the first and second years of treatment, respectively (341). Comparable adverse events were reported between those receiving ETV and placebo.
TDF has been used with children older than age 12 years at a dose of 300 mg daily for more than 1 year. After 42 weeks of treatment, HBeAg seroconversion was 21% versus 15% (nonsignificant) in the placebo group. No particular adverse event is recorded in treated patients; however, monitoring of renal function is suggested (342).
17.4. Monitoring of children with hepatitis B
Monitoring of children with chronic HBV infection serves to identify the status of the disease—according to the classic description of HBV natural history (ie, immune-tolerant, immune active, inactive, and reactivation phases of hepatitis B) (section 3.0, Table 3, Figure 3A)—and complications of the infection, particularly cirrhosis and HCC. At a minimum, all children with chronic HBV infection should be monitored annually with complete blood cell count, serum aminotransferases, AFP, HBV profile (HBV serology and HBV DNA), and liver ultrasound with TE or liver stiffness measurement (ie, FibroScan, if available). In general, whether HBeAg negative or positive, children with elevated ALT and HBV DNA should be monitored more frequently (ie, monthly to every 3 months depending on severity of ALT flare). Children who are on NA treatment should be monitored every 3 months, and more frequently if receiving PEG-IFN.
Recommendations for Management of HBV in Pediatric Patients
-
79.
There is no indication for treatment of children without complications in the immune-tolerant (phase 1) and inactive (phase 3) phases of chronic HBV infection. (moderate recommendation; class 2, level B)
-
80.
Children with chronic HBV infection, either HBeAg positive or negative, should be followed once a year by ultrasound examination of the liver (ideally with elastography or fibrosis assessment) and serum AFP, in addition to ALT and HBV DNA. (moderate recommendation; class 3)
-
81.
In HBeAg-positive children with serum ALT more than 1.3–2 times the normal values, monitoring every 3 months for at least 1 year, to document decreasing or low levels of serum HBV DNA, is recommended before considering treatment to account for possible spontaneous seroconversion. (strong recommendation; class 2, level C)
-
82.
Treatment should be considered for children with cirrhosis, extrahepatic disease, and co-infection. (strong recommendation; class 2, level C)
-
83.
ETV and TDF are drugs that are orally administered and exhibit a high genotypic barrier to resistance and a favourable side effect profile. Thus, ETV should be considered as a first-line therapy for children older than age 3 years and TDF for those older than age 12 years. PEG-IFN treatment may also be considered, which must be weighed against the significant side effects and potential impact on central nervous system development (strong recommendation; class 1, level A)
-
84.
NAs should be continued for 1 year after the disappearance of HBeAg and the appearance of anti-HBe (seroconversion) before attempting cessation. (strong recommendation; class 1, level A)
-
85.
After seroconversion, whether spontaneous or treatment induced, children should be followed every 3 months for at least 1 year. After confirmation of immune inactive status (phase 3, persistently normal ALT, HBV DNA < 2,000 IU/mL), monitoring should be continued every 6 months. (strong recommendation; class 2, level B)
18.0. OVERVIEW OF NEW THERAPIES (A RAMJI)
These guidelines discuss the utilization of approved regimens for the management of hepatitis B. A number of molecules in various stages of development are being investigated to treat HBV. Broadly, these can be divided into DAAs that target the virus and interfere in the replication process or DAAs that target the host immune system to promote HBV elimination. A combination of these mechanisms will likely be needed if persistent hepatitis B infection is to be cleared to achieve a functional or virological cure. A list of ongoing HBV clinical trials can be found at http://www.hepb.org and https://www.clinicaltrials.gov.
18.1. Direct-acting anti-HBV (target the viral replicative pathway; section 2.0)
Entry inhibitors: The mechanism of action is through highly specific binding and competition for the HBV NCTP receptor, thus preventing the HBV from entering into hepatocytes. Myrcludex-B (Hepatera Russia with MYR Germany) is in phase II study and has also been tested for treatment of HBV and HDV (delta coinfection).
Capsid inhibitors: These molecules target and block viral capsid assembly. There are multiple molecules in phase 1 and 2 clinical trials (ie, including agents from Janssen, Arbutus Biopharma, and Assembly Biosciences).
Antisense molecules: These agents bind to viral messenger RNA and interfere with viral protein production, including HBsAg (phase 1 clinical trials have been conducted by Ionis Pharma and Glaxo-Smith-Kline).
Silencing RNAs: These agents interfere and destroy viral RNA and have been tested in phase 1 and 2 studies (Roche Pharmaceuticals and Arbutus Biopharma).
HBsAg inhibitors: These molecules inhibit HBsAg expression and production. Two agents from Replicor Pharma have also been tested for HBV and HDV (delta co-infection) in phase 2 clinical trials.
18.2. Anti-HBV agents targeting the host immune system
Therapeutic vaccines: It is believed that adaptive and innate immune responses are important to clear chronic HBV infection. Therapeutic vaccines are being evaluated to reverse T cell exhaustion. A number of candidate molecules are in clinical study, including recombinant peptide-based vaccines, DNA-based vaccines, viral vector-based vaccines, and cell-based vaccines. These agents are in phase 1 and 2 clinical trials (GlobeImmune, Inovio, Transgene, and Ichor medical systems).
Agents that activate the innate immune system: These molecules activate various pathways of the innate system and aim to promote clearance of persistent HBV infection. Agents include toll-like receptor agonists, toll-like receptor 7 or 8 agonists, and RIG-1 and NOD-2 agonists in phase 1 and 2 clinical trials (Globeimmune, Transgene, Gilead Sciences, SpringBank Pharmaceuticals).
19.0. SUMMARY (CS COFFIN AND SK FUNG)
Despite the availability of an effective vaccine for more than three decades, hepatitis B is a serious global pathogen that leads to significant morbidity and mortality in many Canadians. Advances in HBV virological and serological diagnostic tests, non-invasive tests for assessment of liver disease, and antiviral therapy have led to improved prognosis for many patients diagnosed with hepatitis B. However, important gaps in HBV management in Canada remain, including prevention (ie, universal infant or neonatal vaccination), diagnosis (identifying those at risk for end-stage liver cancer and need for therapy), and access to effective second-generation NA therapy. The goal of the updated 2018 CASL/AMMI guidelines is to summarize best-practice management but also to highlight the need for increased resources for treatment and research on hepatitis B pathogenesis and epidemiology in Canada. With the success in DAA therapy for hepatitis C virus, there is renewed hope for a cure for CHB infection.
List of Abbreviations
- ADV–
adefovir dipivoxil
- AFP–
alpha-fetoprotein
- ALT–
alanine aminotransferase
- AGREE-II–
Appraisal of Guidelines for Research & Evaluation
- AMMI–
Association of Medical Microbiology and Infectious Disease Canada
- anti-HBc–
antibody to HBV core
- anti-HBe–
antibody to HBeAg
- anti-HBs–
antibody to HBsAg
- CASL–
Canadian Association for the Study of Liver Disease
- CHB–
chronic hepatitis B
- DAA–
direct-acting antiviral therapy
- eGFR–
estimated glomerular filtration rate
- EPPs–
exposure-prove procedures
- ETV–
entecavir
- GFR–
glomerular filtration rate
- HBcrAg–
HBV core-related antigen
- HBeAg–
HBV e antigen
- HBIg–
hepatitis B immunoglobulin
- HBsAg–
hepatitis B surface antigen
- HBV–
hepatitis B virus
- HBV cccDNA–
HBV covalently closed circular DNA
- HCC–
hepatocellular carcinoma
- HCV–
hepatitis C virus
- HCWs–
health care workers
- HDV–
hepatitis delta virus
- IFN–
interferon
- IPC–
infection prevention and control
- IS–
immunosuppressive
- LAM–
lamivudine
- MTCT–
mother-to-child transmission
- NA–
nucleos(t)ide analogue
- NCTP–
sodium taurocholate cotransporting polypeptide
- OHB–
occult hepatitis B
- PCR–
polymerase chain reaction
- PEG-IFN–
pegylated interferon
- pgRNA–
HBV pregenomic RNA
- qAnti-HBC–
quantitative anti-HBC antibody
- qHBsAg–
quantitative hepatitis B surface antigen
- rcDNA–
relaxed circular DNA
- TAF–
tenofovir alafenamide
- TBV–
telbivudine
- TDF–
tenofovir disoproxil fumarate
- YMDD–
tyrosine–methionine–aspartate–aspartate
Acknowledgements:
The authors acknowledge Dr. Morris Sherman, chairman of the Canadian Association for the Study of Liver Disease (CASL) Guidelines Committee, and CASL and Association of Medical Microbiology and Infectious Disease (AMMI) membership for their feedback and comments.
Appendix
Funding:
No funding was received for this article.
Disclosures:
Scott Fung reports speaking and teaching fees from Gilead, Merck, and Abbvie and advisory board and consulting roles with Gilead, Merck, Abbvie and Lupin. Carla Coffin reports investigator initiated research grant/research materials from GSK, Gilead Sciences, Arbutus Biopharma, and Bristol-Myers Squibb; educational grants from Merck, Gilead Sciences, and Janssen Inc.; advisory board roles with Merck, Gilead Sciences, and GSK; CTPC committee role with Springbank Pharmaceuticals; and local site PI participation in Clinical Trials with Gilead Sciences, Springbank Pharmaceuticals, and Transgene. Curtis Cooper reports Speaker fees from Gilead Sciences, Merck, and ABBVIE; advisor role with Gilead Sciences, Merck, and ABBVIE; and research/program support from Gilead Sciences, Merck, and ABBVIE. Karen Doucette reports clinical trials with Gilead, Merck, AbbVie, BMS, and Pfizer; speaker honorarium from Merck and Gilead; and educational grants from Merck, Gilead, BMS, and Astellas. Claire Fournier reports local site PI/sub-investigator participation in clinical trials for Transgene, Beckmann, Gilead, Bristol-Myers Squibb Canada, and Immune carta. Erin Kelly reports speaking fees from Lupin, Intercept, and Gilead and advisory board role with Intercept. Hin Hin Ko reports speaker fees from Merck, Intercept, Lupin, Allergan, and Procter and Gamble; advisory board role with Intercept and Lupin; and subinvestigator for studies role with Gilead, Merck, and Intercept. Mang Ma reports clinical investigator role with AbbVie, Gilead, Transgene, Sillagen, and BTG International and advisory board role with AbbVie, Gilead, and Merck. Alnoor Ramji reports clinical investigator role with Abbvie, Allergan, BMS, Gilead, Janssen, Intercept, Norvartis, and Merck; consultant role with Abbvie, Gilead, Intercept, and Lupin; speaker fees from Abbvie, Intercept, Gilead, and Merck; grants from Abbvie, Gilead, Intercept, and Merck. Edward Tam reports clinical investigator role with AbbVie, Allergan, BMS, Boehringer Ingelheim, Genfit, Gilead, Intercept, Janssen, Merck, Novartis, Roche, and Shire; advisory board role with AbbVie, BMS, Boehringer Ingelheim, Gilead, Intercept, Janssen, Lupin, Merck, PendoPharm, and Roche; and speaker honoraria from AbbVie, BMS, Gilead, Intercept, Merck, PenoPharm, and Roche. Jean Pierre Villeneuve reports clinical investigator role with Transgene, and Immune carta.
References
- 1.Sherman M,Bain V,Villeneuve JP, et al. The management of chronic viral hepatitis: a Canadian consensus conference 2004. Can J Gastroenterol 2004;18:715–728. 10.1155/2004/201031. Medline: [DOI] [PubMed] [Google Scholar]
- 2.Coffin CS,Fung SK,Ma MM. Management of chronic hepatitis B: Canadian Association for the Study of the Liver consensus guidelines. Can J Gastroenterol 2012;26:917–38. 10.1155/2012/506819. Medline: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brouwers MC,Kho ME,Browman GP, et al. AGREE II: advancing guideline development, reporting and evaluation in health care. CMAJ 2010;182:E839–42. 10.1503/cmaj.090449. Medline: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guyatt GH,Oxman AD,Vist GE, et al. GRADE: an emerging consensus on rating quality of evidence and strength of recommendations. BMJ 2008;336:924–6. 10.1136/bmj.39489.470347.AD. Medline: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schweitzer A,Horn J,Mikolajczyk RT, et al. Estimations of worldwide prevalence of chronic hepatitis B virus infection: a systematic review of data published between 1965 and 2013. Lancet 2015;386:1546–55. 10.1016/S0140-6736(15)61412-X [DOI] [PubMed] [Google Scholar]
- 6. Public Health Agency of Canada (PHAC). Brief Report: Hepatitis B Infection in Canada. Ottawa: PHAC; 2011. [Google Scholar]
- 7. World Health Organization (WHO). Hepatitis B (Fact sheet no. 204). Geneva: WHO; 2017. [Google Scholar]
- 8.Public Health Agency of Canada (PHAC). Canadian immunization guide: hepatitis B vaccine. 2017. https://www.canada.ca/en/public-health/services/publications/healthy-living/canadian-immunization-guide-part-4-active-vaccines/page-7-hepatitis-b-vaccine.html (January 2018).
- 9.Delage G,Carter AO. Hepatitis B infection in Canada: epidemiology and implications for control. Can Fam Physician 1992;38:2656–66. Medline: [PMC free article] [PubMed] [Google Scholar]
- 10. Public Health Agency of Canada (PHAC). Vaccine coverage in Canadian children: Results from the 2013 Childhood National Immunization Coverage Survey (CNICS). Ottawa: PHAC; 2016. [Google Scholar]
- 11.Minuk GY,Uhanova J. Chronic hepatitis B infection in Canada. Can J Infect Dis 2001;12:351–6. 10.1155/2001/650313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu HX,Andonov A,Giulivi A, et al. Enhanced surveillance for childhood hepatitis B virus infection in Canada, 1999–2003. Int J Med Sci 2005;2(4):143–6. 10.7150/ijms.2.143. Medline: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pottie K,Greenaway C,Feightner J, et al. Evidence-based clinical guidelines for immigrants and refugees. CMAJ 2011;183:E824–925. 10.1503/cmaj.090313. Medline: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang J,Zou S,Giulivi A. Epidemiology of hepatitis B in Canada. Can J Infect Dis 2001;12:345–50. 10.1155/2001/790915. Medline: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moses S,Mestery K,Kaita KD, et al. Viral hepatitis in a Canadian street-involved population. Can J Public Health 2002;93:123–8. Medline: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jafari S,Buxton JA,Afshar K, et al. Tattooing and risk of hepatitis B: a systematic review and meta-analysis. Can J Public Health 2012;103:207–12. Medline: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rotermann M,Langlois K,Andonov A, et al. Seroprevalence of hepatitis B and C virus infections: results from the 2007 to 2009 and 2009 to 2011 Canadian Health Measures Survey. Health Rep 2013;24(11):3–13. Medline: [PubMed] [Google Scholar]
- 18.Sherman M. Liver disease in Canada: a crisis in the making—an assessment of liver disease in Canada. Markham, ON: Canadian Liver Foundation; 2013. [Google Scholar]
- 19.Beasley R. Hepatitis B: the major etiology of hepatocellular carcinoma. Cancer 1988;61:1942–56. [DOI] [PubMed] [Google Scholar]
- 20.Lai CL,Ratziu V,Yuen MF, et al. Viral hepatitis B. Lancet 2003;362:2089–94. 10.1016/S0140-6736(03)15108-2 [DOI] [PubMed] [Google Scholar]
- 21.McMahon BJ. Natural history of chronic hepatitis B. Clin Liver Dis 2010;14:381–96. 10.1016/j.cld.2010.05.007. Medline: [DOI] [PubMed] [Google Scholar]
- 22.Realdi G,Fattovich G,Hadziyannis S, et al.; Investigators of the European Concerted Action on Viral Hepatitis (EUROHEP). Survival and prognostic factors in 366 patients with compensated cirrhosis type B: a multicenter study. J Hepatol 1994;21:656–66. 10.1016/S0168-8278(94)80115-0 [DOI] [PubMed] [Google Scholar]
- 23.Chiaramonte M,Stroffolini T,Vian A, et al. Rate of incidence of hepatocellular carcinoma in patients with compensated viral cirrhosis. Cancer 1999;85:2132–7. [DOI] [PubMed] [Google Scholar]
- 24.Poh Z,Goh BB,Chang PE, et al. Rates of cirrhosis and hepatocellular carcinoma in chronic hepatitis B and the role of surveillance: a 10-year follow-up of 673 patients. Eur J Gastroenterol Hepatol 2015;27:638–43. 10.1097/MEG.0000000000000341. Medline: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kwong JC,Ratnasingham S,Campitelli MA, et al. The impact of infection on population health: results of the Ontario Burden of Infectious Diseases study. PLoS One 2012;7(9):e44103. 10.1371/journal.pone.0044103. Medline: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wong WW,Woo G,Heathcote EJ, et al. Disease burden of chronic hepatitis B among immigrants in Canada. Can J Gastroenterol 2013;27:137–47. 10.1155/2013/924640. Medline: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ni JD,Xiong YZ,Wang XJ, et al. Does increased hepatitis B vaccination dose lead to a better immune response in HIV-infected patients than standard dose vaccination: a meta-analysis? Int J STD AIDS 2013;24:117–22. 10.1177/0956462412472309. Medline: [DOI] [PubMed] [Google Scholar]
- 28.Wilkins E,Nelson M,Agarwal K, et al. British HIV Association guidelines for the management of hepatitis viruses in adults infected with HIV 2013. HIV Med 2013;14(Suppl 4):1–71. 10.1111/hiv.12106 [DOI] [PubMed] [Google Scholar]
- 29.Jackson S,Lentino J,Kopp J, et al. Immunogenicity of a two-dose investigational hepatitis B vaccine, HBsAg-1018, using a toll-like receptor 9 agonist adjuvant compared with a licensed hepatitis B vaccine in adults. Vaccine 2017. 36:668–674 [DOI] [PubMed] [Google Scholar]
- 30. National Advisory Committee on Immunization (NACI). Update on the recommended use of Hepatitis B vaccine. Ottawa: Public Health Agency of Canada; 2017. https://www.canada.ca/en/public-health/services/publications/healthy-living/update-recommended-use-hepatitis-b-vaccine.html (November 1, 2018). [Google Scholar]
- 31.Gerlich WH. Medical virology of hepatitis B: how it began and where we are now. Virol J 2013;10:239. 10.1186/1743-422X-10-239. Medline: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Benhenda S,Cougot D,Buendia MA, et al. Hepatitis B virus X protein molecular functions and its role in virus life cycle and pathogenesis. Adv Cancer Res 2009;103:75–109. 10.1016/S0065-230X(09)03004-8 [DOI] [PubMed] [Google Scholar]
- 33.Slagle BL,Bouchard MJ. Hepatitis B virus X and regulation of viral gene expression. Cold Spring Harb Perspect Med 2016;6(3):a021402. 10.1101/cshperspect.a021402. Medline: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zoulim F. New insight on hepatitis B virus persistence from the study of intrahepatic viral cccDNA. J Hepatol 2005;42:302–8. 10.1016/j.jhep.2004.12.015. Medline: [DOI] [PubMed] [Google Scholar]
- 35.Valaydon ZS,Locarnini SA. The virological aspects of hepatitis B. Best Pract Res Clin Gastroenterol 2017;31:257–264. 10.1016/j.bpg.2017.04.013. Medline: [DOI] [PubMed] [Google Scholar]
- 36.Bertoletti A,Ferrari C. Innate and adaptive immune responses in chronic hepatitis B virus infections: towards restoration of immune control of viral infection. Gut 2012;61:1754–64. [DOI] [PubMed] [Google Scholar]
- 37.Wieland SF,Chisari FV. Stealth and cunning: hepatitis B and hepatitis C viruses. J Virol 2005;79:9369–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Suslov A,Boldanova T,Wang X, et al. Hepatitis B virus does not interfere with innate immune responses in the human liver. Gastroenterology 2018;154:1778–90. 10.1053/j.gastro.2018.01.034 [DOI] [PubMed] [Google Scholar]
- 39.Raimondo G,Allain JP,Brunetto MR, et al. Statements from the Taormina expert meeting on occult hepatitis B virus infection. J Hepatol 2008;49:652–7. 10.1016/j.jhep.2008.07.014. Medline: [DOI] [PubMed] [Google Scholar]
- 40.Rehermann B,Nascimbeni M. Immunology of hepatitis B virus and hepatitis C virus infection. Nat Rev Immunol 2005;5(3):215–29. 10.1038/nri1573. Medline: [DOI] [PubMed] [Google Scholar]
- 41.Block TM,Locarnini S,McMahon BJ, et al. Use of current and new endpoints in the evaluation of experimental hepatitis B therapeutics. Clin Infect Dis 2017;64(9):1283–1288. 10.1093/cid/cix129. Medline: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lok AS,Lai CL,Wu PC, et al. Spontaneous hepatitis B e antigen to antibody seroconversion and reversion in Chinese patients with chronic hepatitis B virus infection. Gastroenterology 1987;92:1839–43. 10.1016/0016-5085(87)90613-5 [DOI] [PubMed] [Google Scholar]
- 43.Bertoletti A,Kennedy PT. The immune tolerant phase of chronic HBV infection: new perspectives on an old concept. Cell Mol Immunol 2015;12(3):258–63. 10.1038/cmi.2014.79. Medline: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kennedy PTF,Sandalova E,Jo J, et al. Preserved T-cell function in children and young adults with immune-tolerant chronic hepatitis B. Gastroenterology 2012;143:637–645. 10.1053/j.gastro.2012.06.009. Medline: [DOI] [PubMed] [Google Scholar]
- 45.Liaw YF,Sheen IS,Chen TJ, et al. Incidence, determinants and significance of delayed clearance of serum HBsAg in chronic hepatitis B virus infection: a prospective study. Hepatology 1991;13:627–31. 10.1002/hep.1840130403. Medline: [DOI] [PubMed] [Google Scholar]
- 46.McMahon BJ. The natural history of chronic hepatitis B virus infection. Hepatology 2009;49(S5):S45–55. 10.1002/hep.22898. Medline: [DOI] [PubMed] [Google Scholar]
- 47.Chu CM,Liaw YF. Hepatitis B virus-related cirrhosis: natural history and treatment. Semin Liver Dis 2006;26:142–52. 10.1055/s-2006-939752. Medline: [DOI] [PubMed] [Google Scholar]
- 48.Yu MW,Hsu FC,Sheen IS, et al. Prospective study of hepatocellular carcinoma and liver cirrhosis in asymptomatic chronic hepatitis B virus carriers. Am J Epidemiol 1997;145:1039–47. 10.1093/oxfordjournals.aje.a009060 [DOI] [PubMed] [Google Scholar]
- 49.Lin CL,Liao LY,Liu CJ, et al. Hepatitis B viral factors in HBeAg-negative carriers with persistently normal serum alanine aminotransferase levels. Hepatology 2007;45:1193–8. 10.1002/hep.21585. Medline: [DOI] [PubMed] [Google Scholar]
- 50.Fattovich G,Bortolotti F,Donato F. Natural history of chronic hepatitis B: special emphasis on disease progression and prognostic factors. J Hepatol 2008;48:335–52. 10.1016/j.jhep.2007.11.011. Medline: [DOI] [PubMed] [Google Scholar]
- 51.Kennedy PTF,Litwin S,Dolman GE, et al. Immune tolerant chronic hepatitis B: the unrecognized risks. Viruses 2017;9(5):96. 10.3390/v9050096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tseng TC,Liu CJ,Yang HC, et al. Serum hepatitis B surface antigen levels help predict disease progression in patients with low hepatitis B virus loads. Hepatology 2013;57(2):441–50. 10.1002/hep.2604. Medline: [DOI] [PubMed] [Google Scholar]
- 53. European Association for the Study of the Liver. EASL 2017 Clinical Practice Guidelines on the management of hepatitis B virus infection. J Hepatol 2017;67(2):370–398. 10.1016/j.jhep.2017.03.021. Medline: [DOI] [PubMed] [Google Scholar]
- 54.Terrault NA,Bzowej NH,Chang KM, et al. AASLD guidelines for treatment of chronic hepatitis B. Hepatology 2016;63(1):261–83. 10.1002/hep.28156. Medline: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sarin SK,Kumar M,Lau GK, et al. Asian-Pacific clinical practice guidelines on the management of hepatitis B: a 2015 update. Hepatol Int 2016;10(1):1–98. 10.1007/s12072-015-9675-4. Medline: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lin X,Robinson NJ,Thursz M, et al. Chronic hepatitis B virus infection in the Asia-Pacific region and Africa: review of disease progression. J Gastroenterol Hepatol 2005;20:833–43. 10.1111/j.1440-1746.2005.03813.x. Medline: [DOI] [PubMed] [Google Scholar]
- 57.Yim HJ,Lok AS. Natural history of chronic hepatitis B virus infection: what we knew in 1981 and what we know in 2005. Hepatology 2006;43(S1):S173–81. 10.1002/hep.20956. Medline: [DOI] [PubMed] [Google Scholar]
- 58.Kappus MR,Sterling RK. Extrahepatic manifestations of acute hepatitis B virus infection. Gastroenterol Hepatol (N Y) 2013;9:123–6. [PMC free article] [PubMed] [Google Scholar]
- 59.Han SH. Extrahepatic manifestations of chronic hepatitis B. Clin Liver Dis 2004;8:403–18. 10.1016/j.cld.2004.02.003. Medline: [DOI] [PubMed] [Google Scholar]
- 60.Villeneuve JP,Desrochers M,Infante-Rivard C, et al. A long-term follow-up study of asymptomatic hepatitis B surface antigen-positive carriers in Montreal. Gastroenterology 1994;106:1000–5. 10.1016/0016-5085(94)90760-9 [DOI] [PubMed] [Google Scholar]
- 61.Yang HI,Tseng TC,Liu J, et al. Incorporating serum level of hepatitis B surface antigen or omitting level of hepatitis B virus DNA does not affect calculation of risk for hepatocellular carcinoma in patients without cirrhosis. Clin Gastroenterol Hepatol 2016;14:461–468 e2. [DOI] [PubMed] [Google Scholar]
- 62.Qu LS,Liu JX,Zhang HF, et al. Effect of serum hepatitis B surface antigen levels on predicting the clinical outcomes of chronic hepatitis B infection: a meta-analysis. Hepatol Res 2014;45(9):1004–13. 10.1111/hepr.12444 [DOI] [PubMed] [Google Scholar]
- 63.Zhou HB,Li QM,Zhong ZR, et al. Level of hepatitis B surface antigen might serve as a new marker to predict hepatocellular carcinoma recurrence following curative resection in patients with low viral load. Am J Cancer Res 2015;5(2):756–71. Medline: [PMC free article] [PubMed] [Google Scholar]
- 64.Blank S,Wang Q,Fiel MI, et al. Assessing prognostic significance of preoperative alpha-fetoprotein in hepatitis B-associated hepatocellular carcinoma: normal is not the new normal. Ann Surg Oncol 2014;21:986–94. 10.1245/s10434-013-3357-z. Medline: [DOI] [PubMed] [Google Scholar]
- 65.Liu C,Xiao GQ,Yan LN, et al. Value of α-fetoprotein in association with clinicopathological features of hepatocellular carcinoma. World J Gastroenterol 2013;19:1811–9. 10.3748/wjg.v19.i11.1811. Medline: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yang HI,Sherman M,Su J, et al. Nomograms for risk of hepatocellular carcinoma in patients with chronic hepatitis B virus infection. J Clin Oncol 2010;28(14):2437–44. 10.1200/JCO.2009.27.4456. Medline: [DOI] [PubMed] [Google Scholar]
- 67.Yang HI,Yuen MF,Chan HL, et al. Risk estimation for hepatocellular carcinoma in chronic hepatitis B (REACH-B): development and validation of a predictive score. Lancet Oncol 2011;12:568–74. 10.1016/S1470-2045(11)70077-8 [DOI] [PubMed] [Google Scholar]
- 68.Jeon MY,Lee HW,Kim SU, et al. Feasibility of dynamic risk prediction for hepatocellular carcinoma development in patients with chronic hepatitis B. Liver Int 2018. Apr;38:676–686. 10.1111/liv.13583. Medline: [DOI] [PubMed] [Google Scholar]
- 69.Papatheodoridis G,Dalekos G,Sypsa V, et al. PAGE-B predicts the risk of developing hepatocellular carcinoma in Caucasians with chronic hepatitis B on 5-year antiviral therapy. J Hepatol 2016;64:800–6. 10.1016/j.jhep.2015.11.035. Medline: [DOI] [PubMed] [Google Scholar]
- 70.Kim MN,Hwang SG,Rim KS, et al. Validation of PAGE-B model in Asian chronic hepatitis B patients receiving entecavir or tenofovir. Liver Int 2017;37:1788–1795. 10.1111/liv.13450. Medline: [DOI] [PubMed] [Google Scholar]
- 71.Brouwer WP,van der Meer AJP,Boonstra A, et al. Prediction of long-term clinical outcome in a diverse chronic hepatitis B population: role of the PAGE-B score. J Viral Hepat 2017;24:1023–31. 10.1111/jvh.12727. Medline: [DOI] [PubMed] [Google Scholar]
- 72.Sharma SA,Kowgier M,Hansen BE, et al. Toronto HCC risk index: a validated scoring system to predict 10-year risk of HCC in patients with cirrhosis. J Hepatol 2018;68:92–99. [DOI] [PubMed] [Google Scholar]
- 73.Yuen MF,Lai CL. Screening for hepatocellular carcinoma: survival benefit and cost-effectiveness. Ann Oncol 2003;14:1463–7. 10.1093/annonc/mdg400 [DOI] [PubMed] [Google Scholar]
- 74.Bruix J,Reig M,Sherman M. Evidence-based diagnosis, staging, and treatment of patients with hepatocellular carcinoma. Gastroenterology 2016;150:835–53. 10.1053/j.gastro.2015.12.041. Medline: [DOI] [PubMed] [Google Scholar]
- 75.Chen CJ,Yang HI,Su J, et al. Risk of hepatocellular carcinoma across a biological gradient of serum hepatitis B virus DNA level. JAMA 2006;295:65–73. 10.1001/jama.295.1.65. Medline: [DOI] [PubMed] [Google Scholar]
- 76.Ghany MG. Current treatment guidelines of chronic hepatitis B: the role of nucleos(t)ide analogues and peginterferon. Best Pract Res Clin Gastroenterol 2017;31:299–309. 10.1016/j.bpg.2017.04.012. Medline: [DOI] [PubMed] [Google Scholar]
- 77.Osiowy C,Coffin C,Andonov A. Review of laboratory tests used in monitoring hepatitis B response to pegylated interferon and nucleos(t)ide analog therapy. Curr Treat Options Infect Dis 2016;8(3):177–193. 10.1007/s40506-016-0080-x. Medline: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Makvandi M. Update on occult hepatitis B virus infection. World J Gastroenterol 2016;22:8720–34. 10.3748/wjg.v22.i39.8720. Medline: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kumar R,Perez-Del-Pulgar S,Testoni B, et al. Clinical relevance of the study of hepatitis B virus covalently closed circular DNA. Liver Int 2016;36 Suppl 1:72–7. 10.1111/liv.13001. Medline: [DOI] [PubMed] [Google Scholar]
- 80.Wang J,Shen T,Huang X, et al. Serum hepatitis B virus RNA is encapsidated pregenome RNA that may be associated with persistence of viral infection and rebound. J Hepatol 2016;65:700–10. 10.1016/j.jhep.2016.05.029. Medline: [DOI] [PubMed] [Google Scholar]
- 81.Giersch K,Allweiss L,Volz T, et al. Serum HBV pgRNA as a clinical marker for cccDNA activity. J Hepatol 2017;66:460–2. 10.1016/j.jhep.2016.09.028. Medline: [DOI] [PubMed] [Google Scholar]
- 82.Jansen L,Kootstra NA,van Dort KA, et al. Hepatitis B virus pregenomic RNA is present in virions in plasma and is associated with a response to pegylated interferon alfa-2a and nucleos(t)ide analogues. J Infect Dis 2016;213:224–32. 10.1093/infdis/jiv397. Medline: [DOI] [PubMed] [Google Scholar]
- 83.van Bommel F,Bartens A,Mysickova A, et al. Serum hepatitis B virus RNA levels as an early predictor of hepatitis B envelope antigen seroconversion during treatment with polymerase inhibitors. Hepatology 2015;61:66–76. 10.1002/hep.27381. Medline: [DOI] [PubMed] [Google Scholar]
- 84.Marcellin P,Martinot-Peignoux M,Asselah T, et al. Serum levels of hepatitis B surface antigen predict severity of fibrosis in patients with E antigen-positive chronic hepatitis B. Clin Gastroenterol Hepatol 2015;13:1532–9.e1. [DOI] [PubMed] [Google Scholar]
- 85.Brouwer WP,Chan HL,Brunetto MR, et al. Repeated measurements of hepatitis B surface antigen identify carriers of inactive HBV during long-term follow-up. Clin Gastroenterol Hepatol 2016;14:1481–1489.e5. [DOI] [PubMed] [Google Scholar]
- 86.Chang ML,Liaw YF,Hadziyannis SJ. Systematic review: cessation of long-term nucleos(t)ide analogue therapy in patients with hepatitis B e antigen-negative chronic hepatitis B. Aliment Pharmacol Ther 2015;42:243–57. 10.1111/apt.13272. Medline: [DOI] [PubMed] [Google Scholar]
- 87.Charatcharoenwitthaya P,Sukeepaisarnjaroen W,Piratvisuth T, et al. Treatment outcomes and validation of the stopping rule for response to peginterferon in chronic hepatitis B: a Thai nationwide cohort study. J Gastroenterol Hepatol 2016;31:1874–81. 10.1111/jgh.13378. Medline: [DOI] [PubMed] [Google Scholar]
- 88.Cornberg M,Wong VW,Locarnini S, et al. The role of quantitative hepatitis B surface antigen revisited. J Hepatol 2017;66:398–411. 10.1016/j.jhep.2016.08.009. Medline: [DOI] [PubMed] [Google Scholar]
- 89.Rijckborst V,Hansen BE,Ferenci P, et al. Validation of a stopping rule at week 12 using HBsAg and HBV DNA for HBeAg-negative patients treated with peginterferon alfa-2a. J Hepatol 2012;56:1006–11. 10.1016/j.jhep.2011.12.007. Medline: [DOI] [PubMed] [Google Scholar]
- 90.Peng H,Wei F,Liu JY, et al. Response-guided therapy of regimens based on PEG-interferon for chronic hepatitis B using on-treatment hepatitis B surface antigen quantification: a meta-analysis. Hepatol Int 2015;9:543–57. 10.1007/s12072-015-9644-y. Medline: [DOI] [PubMed] [Google Scholar]
- 91.Brunetto MR,Marcellin P,Cherubini B, et al. Response to peginterferon alfa-2a (40KD) in HBeAg-negative CHB: on-treatment kinetics of HBsAg serum levels vary by HBV genotype. J Hepatol 2013;59:1153–9. 10.1016/j.jhep.2013.07.017. Medline: [DOI] [PubMed] [Google Scholar]
- 92.Zoulim F,Carosi G,Greenbloom S, et al. Quantification of HBsAg in nucleos(t)ide-naive patients treated for chronic hepatitis B with entecavir with or without tenofovir in the BE-LOW study. J Hepatol 2015;62:56–63. 10.1016/j.jhep.2014.08.031. Medline: [DOI] [PubMed] [Google Scholar]
- 93.Sanchez-Tapias JM,Costa J,Mas A, et al. Influence of hepatitis B virus genotype on the long-term outcome of chronic hepatitis B in western patients. Gastroenterology 2002;123:1848–56. 10.1053/gast.2002.37041. Medline: [DOI] [PubMed] [Google Scholar]
- 94.Hsu YC,Mo LR,Chang CY, et al. Association between serum level of hepatitis B surface antigen at end of entecavir therapy and risk of relapse in E antigen-negative patients. Clin Gastroenterol Hepatol 2016;14:1490–1498.e3. [DOI] [PubMed] [Google Scholar]
- 95.Kimura T,Rokuhara A,Sakamoto Y, et al. Sensitive enzyme immunoassay for hepatitis B virus core-related antigens and their correlation to virus load. J Clin Microbiol 2002;40:439–45. 10.1128/JCM.40.2.439-445.2002. Medline: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chen EQ,Feng S,Wang ML, et al. Serum hepatitis B core-related antigen is a satisfactory surrogate marker of intrahepatic covalently closed circular DNA in chronic hepatitis B. Sci Rep 2017;7(1):173. 10.1038/s41598-017-00111-0. Medline: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wong DK,Seto WK,Cheung KS, et al. Hepatitis B virus core-related antigen as a surrogate marker for covalently closed circular DNA. Liver Int 2017;37:995–1001. 10.1111/liv.13346. Medline: [DOI] [PubMed] [Google Scholar]
- 98.Song G,Yang R,Rao H, et al. Serum HBV core-related antigen is a good predictor for spontaneous HBeAg seroconversion in chronic hepatitis B patients. J Med Virol 2017;89:463–468. 10.1002/jmv.24657. Medline: [DOI] [PubMed] [Google Scholar]
- 99.Gou Y,Zhao Y,Rao C, et al. Predictive value of hepatitis B core-related antigen (HBcrAg) during the natural history of hepatitis B virus infection. Clin Lab 2017;63(7):1063–70. 10.7754/Clin.Lab.2017.161034. Medline: [DOI] [PubMed] [Google Scholar]
- 100.Tada T,Kumada T,Toyoda H, et al. HBcrAg predicts hepatocellular carcinoma development: an analysis using time-dependent receiver operating characteristics. J Hepatol 2016;65:48–56. 10.1016/j.jhep.2016.03.013. Medline: [DOI] [PubMed] [Google Scholar]
- 101.Honda M,Shirasaki T,Terashima T, et al. Hepatitis B virus (HBV) core-related antigen during nucleos(t)ide analog therapy is related to intra-hepatic HBV replication and development of hepatocellular carcinoma. J Infect Dis 2016;213:1096–106. 10.1093/infdis/jiv572 . Medline: [DOI] [PubMed] [Google Scholar]
- 102.Matsumoto A,Nishiguchi S,Enomoto H, et al. Combinational use of hepatitis B viral antigens predicts responses to nucleos(t)ide analogue/peg-interferon sequential therapy. J Gastroenterol 2018;53:247–257. 10.1007/s00535-017-1360-z. Medline: [DOI] [PubMed] [Google Scholar]
- 103.Martinot-Peignoux M,Lapalus M,Maylin S, et al. Baseline HBsAg and HBcrAg titres allow peginterferon-based “precision medicine” in HBeAg-negative chronic hepatitis B patients. J Viral Hepat 2016;23:905–911. 10.1111/jvh.12565. Medline: [DOI] [PubMed] [Google Scholar]
- 104.Song LW,Liu PG,Liu CJ, et al. Quantitative hepatitis B core antibody levels in the natural history of hepatitis B virus infection. Clin Microbiol Infect 2015;21:197–203. 10.1016/j.cmi.2014.10.002. Medline: [DOI] [PubMed] [Google Scholar]
- 105.Yuan Q,Song LW,Liu CJ, et al. Quantitative hepatitis B core antibody level may help predict treatment response in chronic hepatitis B patients. Gut 2013;62:182–4. 10.1136/gutjnl-2012-302656. Medline: [DOI] [PubMed] [Google Scholar]
- 106.Fan R,Sun J,Yuan Q, et al. Baseline quantitative hepatitis B core antibody titre alone strongly predicts HBeAg seroconversion across chronic hepatitis B patients treated with peginterferon or nucleos(t)ide analogues. Gut 2016;65:313–20. 10.1136/gutjnl-2014-308546. Medline: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Li A,Yuan Q,Huang Z, et al. Novel double-antigen sandwich immunoassay for human hepatitis B core antibody. Clin Vaccine Immunol 2010;17:464–9. 10.1128/CVI.00457-09. Medline: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Yuan Q,Song LW,Cavallone D, et al. Total hepatitis B core antigen antibody, a quantitative non-invasive marker of hepatitis B virus induced liver disease. PLoS One 2015;10(6):e0130209. 10.1371/journal.pone.0130209. Medline: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lin CL,Kao JH. Hepatitis B virus genotypes and variants. Cold Spring Harb Perspect Med 2015;5(5):a021436. 10.1101/cshperspect.a021436. Medline: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gencay M,Hubner K,Gohl P, et al. Ultra-deep sequencing reveals high prevalence and broad structural diversity of hepatitis B surface antigen mutations in a global population. PLoS One 2017;12(5):e0172101. 10.1371/journal.pone.0172101. Medline: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kramvis A,Arakawa K,Yu MC, et al. Relationship of serological subtype, basic core promoter and precore mutations to genotypes/subgenotypes of hepatitis B virus. J Med Virol 2008;80:27–46. 10.1002/jmv.21049. Medline: [DOI] [PubMed] [Google Scholar]
- 112.Zhang Q,Liao Y,Chen J, et al. Epidemiology study of HBV genotypes and antiviral drug resistance in multi-ethnic regions from Western China. Sci Rep 2015;5(1):17413. 10.1038/srep17413. Medline: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Osiowy C,Giles E,Trubnikov M, et al. Characterization of acute and chronic hepatitis B virus genotypes in Canada. PLoS One 2015;10(9):e0136074. 10.1371/journal.pone.0136074. Medline: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ito K,Yoneda M,Sakamoto K, et al. Virological and clinical characteristics of hepatitis B virus genotype A. J Gastroenterol 2018;53(1):18–26. 10.1007/s00535-017-1367-5. Medline: [DOI] [PubMed] [Google Scholar]
- 115.Ni YH,Chang MH,Wang KJ, et al. Clinical relevance of hepatitis B virus genotype in children with chronic infection and hepatocellular carcinoma. Gastroenterology 2004;127:1733–8. 10.1053/j.gastro.2004.09.048 [DOI] [PubMed] [Google Scholar]
- 116.Kao JH,Chen PJ,Lai MY, et al. Basal core promoter mutations of hepatitis B virus increase the risk of hepatocellular carcinoma in hepatitis B carriers. Gastroenterology 2003;124:327–34. 10.1053/gast.2003.50053. Medline: [DOI] [PubMed] [Google Scholar]
- 117.Yang HI,Yeh SH,Chen PJ, et al. Associations between hepatitis B virus genotype and mutants and the risk of hepatocellular carcinoma. J Natl Cancer Inst 2008;100:1134–43. 10.1093/jnci/djn243. Medline: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Livingston SE,Simonetti JP,Bulkow LR, et al. Clearance of hepatitis B e antigen in patients with chronic hepatitis B and genotypes A, B, C, D, and F. Gastroenterology 2007;133:1452–7. 10.1053/j.gastro.2007.08.010. Medline: [DOI] [PubMed] [Google Scholar]
- 119.Kew MC,Kramvis A,Yu MC, et al. Increased hepatocarcinogenic potential of hepatitis B virus genotype A in Bantu-speaking Sub-Saharan Africans. J Med Virol 2005;75:513–21. 10.1002/jmv.20311. Medline: [DOI] [PubMed] [Google Scholar]
- 120.Gopalakrishnan D,Keyter M,Shenoy KT, et al. Hepatitis B virus subgenotype A1 predominates in liver disease patients from Kerala, India. World J Gastroenterol 2013;19:9294–306. 10.3748/wjg.v19.i48.9294. Medline: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Minuk GY,Macrury S,Uhanova J, et al. A paucity of liver disease in Canadian Inuit with chronic hepatitis B virus, subgenotype B6 infection. J Viral Hepat 2013;20:890–6. 10.1111/jvh.12121. Medline: [DOI] [PubMed] [Google Scholar]
- 122.Krarup HB,Andersen S,Madsen PH, et al. Benign course of long-standing hepatitis B virus infection among Greenland Inuit? Scand J Gastroenterol 2008;43:334–43. 10.1080/00365520701712198. Medline: [DOI] [PubMed] [Google Scholar]
- 123.Boglione L,Cariti G,Di Perri G, et al. Sequential therapy with entecavir and pegylated interferon in a cohort of young patients affected by chronic hepatitis B. J Med Virol 2016;88:1953–9. 10.1002/jmv.24534. Medline: [DOI] [PubMed] [Google Scholar]
- 124.Woo ASJ,Kwok R,Ahmed T. Alpha-interferon treatment in hepatitis B. Ann Transl Med 2017;5(7):159. 10.21037/atm.2017.03.69. Medline: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Tian Q,Jia J. Hepatitis B virus genotypes: epidemiological and clinical relevance in Asia. Hepatol Int 2016;10:854–60. 10.1007/s12072-016-9745-2. Medline: [DOI] [PubMed] [Google Scholar]
- 126.Ducancelle A,Pivert A,Bertrais S, et al. Different precore/core mutations of hepatitis B interact with, limit, or favor liver fibrosis severity. J Gastroenterol Hepatol 2016;31:1750–56. 10.1111/jgh.13338. Medline: [DOI] [PubMed] [Google Scholar]
- 127.Sung FY,Lan CY,Huang CJ, et al. Progressive accumulation of mutations in the hepatitis B virus genome and its impact on time to diagnosis of hepatocellular carcinoma. Hepatology 2016;64:720–31. 10.1002/hep.28654. Medline: [DOI] [PubMed] [Google Scholar]
- 128.Pollicino T,Cacciola I,Saffioti F, et al. Hepatitis B virus PreS/S gene variants: pathobiology and clinical implications. J Hepatol 2014;61:408–17. 10.1016/j.jhep.2014.04.041. Medline: [DOI] [PubMed] [Google Scholar]
- 129.Malve B,Eschlimann M,Galgey S, et al. Impact of deletions and mutations in Hepatitis B virus envelope proteins on serological profile and clinical evolution. Virus Res 2017;238:141–147. 10.1016/j.virusres.2017.06.028. Medline: [DOI] [PubMed] [Google Scholar]
- 130.Liu WC,Wu IC,Lee YC, et al. Hepatocellular carcinoma-associated single-nucleotide variants and deletions identified by the use of genome-wide high-throughput analysis of hepatitis B virus. J Pathol 2017;243:176–92. 10.1002/path.4938. Medline: [DOI] [PubMed] [Google Scholar]
- 131.Lim YS. Management of antiviral resistance in chronic hepatitis B. Gut Liver 2017;11:189–95. 10.5009/gnl15562. Medline: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Lowe CF,Merrick L,Harrigan PR, et al. Implementation of next-generation sequencing for hepatitis B virus resistance testing and genotyping in a clinical microbiology laboratory. J Clin Microbiol 2016;54(1):127–33. 10.1128/JCM.02229-15. Medline: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Menzo S,Vincenti D,Solmone M, et al. Low-abundance drug resistance mutations: extending the HIV paradigm to hepatitis B virus. J Infect Dis 2009;200:1798–9; author reply 1799–800. [DOI] [PubMed] [Google Scholar]
- 134.Jones LR,Sede M,Manrique JM, et al. Hepatitis B virus resistance substitutions: long-term analysis by next-generation sequencing. Arch Virol 2016;161:2885–91. 10.1007/s00705-016-2959-8. Medline: [DOI] [PubMed] [Google Scholar]
- 135.Hossain MG,Ueda K. Investigation of a novel hepatitis B virus surface antigen (HBsAg) escape mutant affecting immunogenicity. PLoS One 2017;12(1):e0167871. 10.1371/journal.pone.0167871. Medline: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kalinina T,Iwanski A,Will H, et al. Deficiency in virion secretion and decreased stability of the hepatitis B virus immune escape mutant G145R. Hepatology 2003;38:1274–81. 10.1053/jhep.2003.50484. Medline: [DOI] [PubMed] [Google Scholar]
- 137.Chen G,Lin W,Shen F, et al. Past HBV viral load as predictor of mortality and morbidity from HCC and chronic liver disease in a prospective study. Am J Gastroenterol 2006;101:1797–803. 10.1111/j.1572-0241.2006.00647.x. Medline: [DOI] [PubMed] [Google Scholar]
- 138.Iloeje UH,Yang HI,Su J, et al. Predicting cirrhosis risk based on the level of circulating hepatitis B viral load. Gastroenterology 2006;130:678–86. 10.1053/j.gastro.2005.11.016. Medline: [DOI] [PubMed] [Google Scholar]
- 139.Yu MW,Yeh SH,Chen PJ, et al. Hepatitis B virus genotype and DNA level and hepatocellular carcinoma: a prospective study in men. J Natl Cancer Inst 2005;97:265–72. 10.1093/jnci/dji043. Medline: [DOI] [PubMed] [Google Scholar]
- 140.Kim HC,Nam CM,Jee SH, et al. Normal serum aminotransferase concentration and risk of mortality from liver diseases: prospective cohort study. BMJ 2004;328:983. 10.1136/bmj.38050.593634.63. Medline: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Yuen MF,Yuan HJ,Wong DK, et al. Prognostic determinants for chronic hepatitis B in Asians: therapeutic implications. Gut 2005;54:1610–4. 10.1136/gut.2005.065136. Medline: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Terrault NA,Lok ASF,McMahon BJ, et al. Update on prevention, diagnosis, and treatment of chronic hepatitis B: AASLD 2018 hepatitis B guidance. Hepatology 2018;67:1560–99. 10.1002/hep.29800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Vigano M,Paggi S,Lampertico P, et al. Dual cut-off transient elastography to assess liver fibrosis in chronic hepatitis B: a cohort study with internal validation. Aliment Pharmacol Ther 2011;34:353–62. 10.1111/j.1365-2036.2011.04722.x. Medline: [DOI] [PubMed] [Google Scholar]
- 144.Chon YE,Choi EH,Song KJ, et al. Performance of transient elastography for the staging of liver fibrosis in patients with chronic hepatitis B: a meta-analysis. PLoS One 2012;7(9):e44930. 10.1371/journal.pone.0044930. Medline: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Lok AS,Lai CL. A longitudinal follow-up of asymptomatic hepatitis B surface antigen-positive Chinese children. Hepatology 1988;8:1130–3. 10.1002/hep.1840080527 [DOI] [PubMed] [Google Scholar]
- 146.Chang MH,Hsu HY,Hsu HC, et al. The significance of spontaneous hepatitis B e antigen seroconversion in childhood: with special emphasis on the clearance of hepatitis B e antigen before 3 years of age. Hepatology 1995;22:1387–92. 10.1016/0270-9139(95)90141-8 [DOI] [PubMed] [Google Scholar]
- 147.Brook MG,McDonald JA,Karayiannis P, et al. Randomised controlled trial of interferon alfa 2A (rbe) (Roferon-A) for the treatment of chronic hepatitis B virus (HBV) infection: factors that influence response. Gut 1989;30:1116–22. 10.1136/gut.30.8.1116. Medline: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Brunetto MR,Oliveri F,Colombatto P, et al. Treatment of HBeAg-negative chronic hepatitis B with interferon or pegylated interferon. J Hepatol 2003;39(Suppl 1):S164–7. 10.1016/S0168-8278(03)00329-5 [DOI] [PubMed] [Google Scholar]
- 149.Craxi A,Di Bona D,Camma C. Interferon-alpha for HBeAg-positive chronic hepatitis B. J Hepatol 2003;39(Suppl 1):S99–105. 10.1016/S0168-8278(03)00154-5 [DOI] [PubMed] [Google Scholar]
- 150.Perrillo RP,Schiff ER,Davis GL, et al. A randomized, controlled trial of interferon alfa-2b alone and after prednisone withdrawal for the treatment of chronic hepatitis B. N Engl J Med 1990;323:295–301. 10.1056/NEJM199008023230503. Medline: [DOI] [PubMed] [Google Scholar]
- 151.Wong DK,Cheung AM,O’Rourke K, et al. Effect of alpha-interferon treatment in patients with hepatitis B e antigen-positive chronic hepatitis B. A meta-analysis. Ann Intern Med 1993;119:312–23. 10.7326/0003-4819-119-4-199308150-00011. Medline: [DOI] [PubMed] [Google Scholar]
- 152.Lau DT,Everhart J,Kleiner DE, et al. Long-term follow-up of patients with chronic hepatitis B treated with interferon alfa. Gastroenterology 1997;113:1660–7. 10.1053/gast.1997.v113.pm9352870. Medline: [DOI] [PubMed] [Google Scholar]
- 153.Lin SM,Sheen IS,Chien RN, et al. Long-term beneficial effect of interferon therapy in patients with chronic hepatitis B virus infection. Hepatology 1999;29:971–5. 10.1002/hep.510290312. Medline: [DOI] [PubMed] [Google Scholar]
- 154.Lok AS,Chung HT,Liu VW, et al. Long-term follow-up of chronic hepatitis B patients treated with interferon alfa. Gastroenterology 1993;105:1833–8. 10.1016/0016-5085(93)91082-S [DOI] [PubMed] [Google Scholar]
- 155.Korenman J,Baker B,Waggoner J, et al. Long-term remission of chronic hepatitis B after alpha-interferon therapy. Ann Intern Med 1991;114:629–34. 10.7326/0003-4819-114-8-629. Medline: [DOI] [PubMed] [Google Scholar]
- 156.Krogsgaard K; The Long-Term Follow-up Investigator Group. The European Study Group on Viral Hepatitis (EUROHEP). Executive Team on Anti-Viral Treatment. The long-term effect of treatment with interferon-alpha-2a in chronic hepatitis B. J Viral Hepat 1998;5:389–97. 10.1046/j.1365-2893.1998.00118.x. Medline: [DOI] [PubMed] [Google Scholar]
- 157.Cooksley WG,Piratvisuth T,Lee SD, et al. Peginterferon alpha-2a (40 kDa): an advance in the treatment of hepatitis B e antigen-positive chronic hepatitis B. J Viral Hepat 2003;10:298–305. 10.1046/j.1365-2893.2003.00450.x. Medline: [DOI] [PubMed] [Google Scholar]
- 158.Lau GK,Piratvisuth T,Luo KX, et al. Peginterferon alfa-2a, lamivudine, and the combination for HBeAg-positive chronic hepatitis B. N Engl J Med 2005;352:2682–95. 10.1056/NEJMoa043470. Medline: [DOI] [PubMed] [Google Scholar]
- 159.Janssen HL,van Zonneveld M,Senturk H, et al. Pegylated interferon alfa-2b alone or in combination with lamivudine for HBeAg-positive chronic hepatitis B: a randomised trial. Lancet 2005;365:123–9. 10.1016/S0140-6736(05)17701-0. Medline: [DOI] [PubMed] [Google Scholar]
- 160.Niederau C,Heintges T,Lange S, et al. Long-term follow-up of HBeAg-positive patients treated with interferon alfa for chronic hepatitis B. N Engl J Med 1996;334:1422–7. 10.1056/NEJM199605303342202. Medline: [DOI] [PubMed] [Google Scholar]
- 161.Fattovich G,Giustina G,Realdi G, et al. Long-term outcome of hepatitis B e antigen-positive patients with compensated cirrhosis treated with interferon alfa. Hepatology 1997;26:1338–42. 10.1002/hep.510260536. Medline: [DOI] [PubMed] [Google Scholar]
- 162.Lin SM,Yu ML,Lee CM, et al. Interferon therapy in HBeAg positive chronic hepatitis reduces progression to cirrhosis and hepatocellular carcinoma. J Hepatol 2007;46:45–52. 10.1016/j.jhep.2006.08.021. Medline: [DOI] [PubMed] [Google Scholar]
- 163.Liang KH,Hsu CW,Chang ML, et al. Peginterferon is superior to nucleos(t)ide analogues for prevention of hepatocellular carcinoma in chronic hepatitis B. J Infect Dis 2016;213:966–74. 10.1093/infdis/jiv547. Medline: [DOI] [PubMed] [Google Scholar]
- 164.Marcellin P,Lau GK,Bonino F, et al. Peginterferon alfa-2a alone, lamivudine alone, and the two in combination in patients with HBeAg-negative chronic hepatitis B. N Engl J Med 2004;351:1206–17. 10.1056/NEJMoa040431. Medline: [DOI] [PubMed] [Google Scholar]
- 165.Sonneveld MJ,Hansen BE,Piratvisuth T, et al. Response-guided peginterferon therapy in hepatitis B e antigen-positive chronic hepatitis B using serum hepatitis B surface antigen levels. Hepatology 2013;58:872–80. 10.1002/hep.26436. Medline: [DOI] [PubMed] [Google Scholar]
- 166.Buster EH,Hansen BE,Lau GK, et al. Factors that predict response of patients with hepatitis B e antigen-positive chronic hepatitis B to peginterferon-alfa. Gastroenterology 2009;137:2002–9. 10.1053/j.gastro.2009.08.061. Medline: [DOI] [PubMed] [Google Scholar]
- 167.Buti M,Tsai N,Petersen J, et al. Seven-year efficacy and safety of treatment with tenofovir disoproxil fumarate for chronic hepatitis B virus infection. Dig Dis Sci 2015;60:1457–64. 10.1007/s10620-014-3486-7. Medline: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Marcellin P,Gane E,Buti M, et al. Regression of cirrhosis during treatment with tenofovir disoproxil fumarate for chronic hepatitis B: a 5-year open-label follow-up study. Lancet 2013;381:468–75. 10.1016/S0140-6736(12)61425-1. Medline: [DOI] [PubMed] [Google Scholar]
- 169.Liu Y,Corsa AC,Buti M, et al. No detectable resistance to tenofovir disoproxil fumarate in HBeAg+ and HBeAg-patients with chronic hepatitis B after 8 years of treatment. J Viral Hepat 2017;24:68–74. 10.1111/jvh.12613. Medline: [DOI] [PubMed] [Google Scholar]
- 170.Chan HL,Fung S,Seto WK, et al. Tenofovir alafenamide versus tenofovir disoproxil fumarate for the treatment of HBeAg-positive chronic hepatitis B virus infection: a randomised, double-blind, phase 3, non-inferiority trial. Lancet Gastroenterol Hepatol 2016;1:185–95. 10.1016/S2468-1253(16)30024-3. Medline: [DOI] [PubMed] [Google Scholar]
- 171.Buti M,Gane E,Seto WK, et al. Tenofovir alafenamide versus tenofovir disoproxil fumarate for the treatment of patients with HBeAg-negative chronic hepatitis B virus infection: a randomised, double-blind, phase 3, non-inferiority trial. Lancet Gastroenterol Hepatol 2016;1:196–206. 10.1016/S2468-1253(16)30107-8. Medline: [DOI] [PubMed] [Google Scholar]
- 172.Grossi G,Loglio A,Facchetti F, et al. Tenofovir alafenamide as a rescue therapy in a patient with HBV-cirrhosis with a history of Fanconi syndrome and multidrug resistance. J Hepatol 2018;68:195–8. 10.1016/j.jhep.2017.08.020. Medline: [DOI] [PubMed] [Google Scholar]
- 173.Chang TT,Gish RG,de Man R, et al. A comparison of entecavir and lamivudine for HBeAg-positive chronic hepatitis B. N Engl J Med 2006;354:1001–10. 10.1056/NEJMoa051285. Medline: [DOI] [PubMed] [Google Scholar]
- 174.Gish RG,Lok AS,Chang TT, et al. Entecavir therapy for up to 96 weeks in patients with HBeAg-positive chronic hepatitis B. Gastroenterology 2007. 133:1437–44. 10.1053/j.gastro.2007.08.025. Medline: [DOI] [PubMed] [Google Scholar]
- 175.Colonno RJ,Rose R,Baldick CJ, et al. Entecavir resistance is rare in nucleoside naive patients with hepatitis B. Hepatology 2006;44:1656–65. 10.1002/hep.21422. Medline: [DOI] [PubMed] [Google Scholar]
- 176.Tenney DJ,Rose RE,Baldick CJ, et al. Two-year assessment of entecavir resistance in lamivudine-refractory hepatitis B virus patients reveals different clinical outcomes depending on the resistance substitutions present. Antimicrob Agents Chemother 2007;51:902–11. 10.1128/AAC.00833-06. Medline: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Lam YF,Seto WK,Wong D, et al. Seven-year treatment outcome of entecavir in a real-world cohort: effects on clinical parameters, HBsAg and HBcrAg levels. Clin Transl Gastroenterol 2017;8(10):e125. 10.1038/ctg.2017.51. Medline: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Sherman M,Yurdaydin C,Sollano J, et al. Entecavir for treatment of lamivudine-refractory, HBeAg-positive chronic hepatitis B. Gastroenterology 2006;130:2039–49. 10.1053/j.gastro.2006.04.007. Medline: [DOI] [PubMed] [Google Scholar]
- 179.Lok AS,Lai CL,Leung N, et al. Long-term safety of lamivudine treatment in patients with chronic hepatitis B. Gastroenterology 2003;125:1714–22. 10.1053/j.gastro.2003.09.033. Medline: [DOI] [PubMed] [Google Scholar]
- 180.Liaw YF,Sung JJ,Chow WC, et al. Lamivudine for patients with chronic hepatitis B and advanced liver disease. N Engl J Med 2004;351:1521–31. 10.1056/NEJMoa033364. Medline: [DOI] [PubMed] [Google Scholar]
- 181.Lai CL,Dienstag J,Schiff E, et al. Prevalence and clinical correlates of YMDD variants during lamivudine therapy for patients with chronic hepatitis B. Clin Infect Dis 2003;36:687–96. 10.1086/368083. Medline: [DOI] [PubMed] [Google Scholar]
- 182.Fung SK,Chae HB,Fontana RJ, et al. Virologic response and resistance to adefovir in patients with chronic hepatitis B. J Hepatol 2006;44:283–90. 10.1016/j.jhep.2005.10.018. Medline: [DOI] [PubMed] [Google Scholar]
- 183.Liaw YF,Gane E,Leung N, et al. 2-Year GLOBE trial results: telbivudine is superior to lamivudine in patients with chronic hepatitis B. Gastroenterology 2009;136:486–95. 10.1053/j.gastro.2008.10.026. Medline: [DOI] [PubMed] [Google Scholar]
- 184.Han GR,Cao MK,Zhao W, et al. A prospective and open-label study for the efficacy and safety of telbivudine in pregnancy for the prevention of perinatal transmission of hepatitis B virus infection. J Hepatol 2011;55:1215–21. 10.1016/j.jhep.2011.02.032. Medline: [DOI] [PubMed] [Google Scholar]
- 185.Zhang Y,Hu P,Qi X, et al. A comparison of telbivudine and entecavir in the treatment of hepatitis B e antigen-positive patients: a prospective cohort study in China. Clin Microbiol Infect 2016;22:287 e1–9. 10.1016/j.cmi.2015.10.02. Medline: [DOI] [PubMed] [Google Scholar]
- 186.Vigano M,Invernizzi F,Grossi G, et al. Review article: the potential of interferon and nucleos(t)ide analogue combination therapy in chronic hepatitis B infection. Aliment Pharmacol Ther 2016;44:653–61. 10.1111/apt.13751. Medline: [DOI] [PubMed] [Google Scholar]
- 187.Marcellin P,Ahn SH,Ma X, et al. Combination of tenofovir disoproxil fumarate and peginterferon alpha-2a increases loss of hepatitis B surface antigen in patients with chronic hepatitis B. Gastroenterology 2016;150:134–44.e10. 10.1053/j.gastro.2015.09.043. Medline: [DOI] [PubMed] [Google Scholar]
- 188.Marcellin P,Ahn SH,Chuang WL, et al. Predictors of response to tenofovir disoproxil fumarate plus peginterferon alfa-2a combination therapy for chronic hepatitis B. Aliment Pharmacol Ther 2016;44:957–66. 10.1111/apt.13779. Medline: [DOI] [PubMed] [Google Scholar]
- 189.Peters MG,Hann HW,Martin P, et al. Adefovir dipivoxil alone or in combination with lamivudine in patients with lamivudine-resistant chronic hepatitis B. Gastroenterology 2004;126:91–101. 10.1053/j.gastro.2003.10.051. Medline: [DOI] [PubMed] [Google Scholar]
- 190.Lai CL,Leung N,Teo EK, et al. A 1-year trial of telbivudine, lamivudine, and the combination in patients with hepatitis B e antigen-positive chronic hepatitis B. Gastroenterology 2005;129:528–36. 10.1053/j.gastro.2005.05.053. Medline: [DOI] [PubMed] [Google Scholar]
- 191.Lok AS,Trinh H,Carosi G, et al. Efficacy of entecavir with or without tenofovir disoproxil fumarate for nucleos(t)ide-naive patients with chronic hepatitis B. Gastroenterology 2012;143:619–28.e1. 10.1053//j.gastro.2012.05.037. Medline: [DOI] [PubMed] [Google Scholar]
- 192.Chan HL,Chan CK,Hui AJ, et al. Effects of tenofovir disoproxil fumarate in hepatitis B e antigen-positive patients with normal levels of alanine aminotransferase and high levels of hepatitis B virus DNA. Gastroenterology 2014;146:1240–8. 10.1053/j.gastro.2014.01.044. Medline: [DOI] [PubMed] [Google Scholar]
- 193.Fong TL,Tien A,Jo KJ, et al. Durability of hepatitis B e antigen seroconversion in chronic hepatitis B patients treated with entecavir or tenofovir. Dig Dis Sci 2015;60:3465–72. 10.1007/s10620-015-3775-9. Medline: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Nagata N,Kagawa T,Hirose S, et al. Off-treatment durability of antiviral response to nucleoside analogues in patients with chronic hepatitis B. BMC Gastroenterol 2016;16:38. 10.1186/s12876-016-0454-z. Medline: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Reijnders JG,Perquin MJ,Zhang N, et al. Nucleos(t)ide analogues only induce temporary hepatitis B e antigen seroconversion in most patients with chronic hepatitis B. Gastroenterology 2010;139:491–8. 10.1053/j.gastro.2010.03.059. Medline: [DOI] [PubMed] [Google Scholar]
- 196.Lee YS,Suh DJ,Lim YS, et al. Increased risk of adefovir resistance in patients with lamivudine-resistant chronic hepatitis B after 48 weeks of adefovir dipivoxil monotherapy. Hepatology 2006;43:1385–91. 10.1002/hep.21189. Medline: [DOI] [PubMed] [Google Scholar]
- 197.Wang JH,Lu SN,Lee CM, et al. Fatal hepatic failure after emergence of the hepatitis B virus mutant during lamivudine therapy in a patient with liver cirrhosis. Scand J Gastroenterol 2002;37:366–9. 10.1080/003655202317284309. Medline: [DOI] [PubMed] [Google Scholar]
- 198.Fung SK,Andreone P,Han SH, et al. Adefovir-resistant hepatitis B can be associated with viral rebound and hepatic decompensation. J Hepatol 2005;43:937–43. 10.1016/j.jhep.2005.05.037. Medline: [DOI] [PubMed] [Google Scholar]
- 199.Dienstag JL,Wei LJ,Xu D, et al. Cross-study analysis of the relative efficacies of oral antiviral therapies for chronic hepatitis B infection in nucleoside-naive patients. Clin Drug Investig 2007;27:35–49. 10.2165/00044011-200727010-00003. Medline: [DOI] [PubMed] [Google Scholar]
- 200.Lok AS,Zoulim F,Locarnini S, et al. Antiviral drug-resistant HBV: standardization of nomenclature and assays and recommendations for management. Hepatology 2007;46:254–65. 10.1002/hep.21698. Medline: [DOI] [PubMed] [Google Scholar]
- 201.Lampertico P,Vigano M,Manenti E, et al. Adefovir rapidly suppresses hepatitis B in HBeAg-negative patients developing genotypic resistance to lamivudine. Hepatology 2005;42:1414–9. 10.1002/hep.20939. Medline: [DOI] [PubMed] [Google Scholar]
- 202.Rapti I,Dimou E,Mitsoula P, et al. Adding-on versus switching-to adefovir therapy in lamivudine-resistant HBeAg-negative chronic hepatitis B. Hepatology 2007;45:307–13. 10.1002/hep.21534. Medline: [DOI] [PubMed] [Google Scholar]
- 203.Fung S,Kwan P,Fabri M, et al. Randomized comparison of tenofovir disoproxil fumarate vs emtricitabine and tenofovir disoproxil fumarate in patients with lamivudine-resistant chronic hepatitis B. Gastroenterology 2014;146:980–8. 10.1053/j.gastro.2013.12.028. Medline: [DOI] [PubMed] [Google Scholar]
- 204.Tenney DJ,Levine SM,Rose RE, et al. Clinical emergence of entecavir-resistant hepatitis B virus requires additional substitutions in virus already resistant to Lamivudine. Antimicrob Agents Chemother 2004;48:3498–507. 10.1128/AAC.48.9.3498-3507.2004. Medline: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Lim YS,Byun KS,Yoo BC, et al. Tenofovir monotherapy versus tenofovir and entecavir combination therapy in patients with entecavir-resistant chronic hepatitis B with multiple drug failure: results of a randomised trial. Gut 2016;65:852–60. 10.1136/gutjnl-2014-308353. Medline: [DOI] [PubMed] [Google Scholar]
- 206.Marcellin P,Heathcote EJ,Buti M, et al. Tenofovir disoproxil fumarate versus adefovir dipivoxil for chronic hepatitis B. N Engl J Med 2008;359:2442–55. 10.1056/NEJMoa0802878. Medline: [DOI] [PubMed] [Google Scholar]
- 207.van Bommel F,Zollner B,Sarrazin C, et al. Tenofovir for patients with lamivudine-resistant hepatitis B virus (HBV) infection and high HBV DNA level during adefovir therapy. Hepatology 2006;44:318–25. 10.1002/hep.21253. Medline: [DOI] [PubMed] [Google Scholar]
- 208.Lim YS,Lee YS,Gwak GY, et al. Monotherapy with tenofovir disoproxil fumarate for multiple drug-resistant chronic hepatitis B: 3-year trial. Hepatology 2017;66:772–83. 10.1002/hep.29187. Medline: [DOI] [PubMed] [Google Scholar]
- 209. Public Health Agency of Canada (PHAC). Primary care management of hepatitis B—quick reference. Toronto: PHAC; 2014. [Google Scholar]
- 210.Singh AE,Plitt SS,Osiowy C, et al. Factors associated with vaccine failure and vertical transmission of hepatitis B among a cohort of Canadian mothers and infants. J Viral Hepat 2011;18:468–73. 10.1111/j.1365-2893.2010.01333.x. Medline: [DOI] [PubMed] [Google Scholar]
- 211.Castillo E,Murphy K,van Schalkwyk J. No. 342—Hepatitis B and pregnancy. J Obstet Gynaecol Can 2017;39:181–90. 10.1016/j.jogc.2016.11.001. Medline: [DOI] [PubMed] [Google Scholar]
- 212.Joshi SS,Wong D,Castillo E, et al. Peripartum cytokine flares in a multiethnic cohort of chronic hepatitis B carriers does not correlate with hepatitis B virus suppression or increased risk of liver disease. Am J Reprod Immunol 2017;78(4): e12707. 10.1111/aji.12707. Medline: [DOI] [PubMed] [Google Scholar]
- 213.van Schalkwyk J,Nourmoussavi M,Massey A, et al. Missed opportunities for prevention of perinatal transmission of hepatitis B: a retrospective cohort study. Can J Gastroenterol Hepatol 2014;28:525–8. 10.1155/2014/549764. Medline: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Gilead Sciences. Product monograph, including patient medication information: Vemildy™ (tenofovir alafenamide) tablets. 2017. http://www.gilead.ca/application/files/9715/3756/6953/Vemlidy_English_PM_e193066-GS-001.pdf (October 21, 2018)
- 215.Chen HL,Lee CN,Chang CH, et al. Efficacy of maternal tenofovir disoproxil fumarate in interrupting mother-to-infant transmission of hepatitis B virus. Hepatology 2015;62:375–86. 10.1002/hep.27837. Medline: [DOI] [PubMed] [Google Scholar]
- 216.Han GR,Jiang HX,Yue X, et al. Efficacy and safety of telbivudine treatment: an open-label, prospective study in pregnant women for the prevention of perinatal transmission of hepatitis B virus infection. J Viral Hepat 2015;22(9):754–62. 10.1111/jvh.12379. Medline: [DOI] [PubMed] [Google Scholar]
- 217.Pan CQ,Duan Z,Dai E, et al. Tenofovir to prevent hepatitis B transmission in mothers with high viral load. N Engl J Med 2016;374:2324–34. 10.1056/NEJMoa1508660. Medline: [DOI] [PubMed] [Google Scholar]
- 218.Xu WM,Cui YT,Wang L, et al. Lamivudine in late pregnancy to prevent perinatal transmission of hepatitis B virus infection: a multicentre, randomized, double-blind, placebo-controlled study. J Viral Hepat 2009;16:94–103. 10.1111/j.1365-2893.2008.01056.x. Medline: [DOI] [PubMed] [Google Scholar]
- 219.Samadi Kochaksaraei G,Castillo E,Osman M, et al. Clinical course of 161 untreated and tenofovir-treated chronic hepatitis B pregnant patients in a low hepatitis B virus endemic region. J Viral Hepat 2016;23:15–22. 10.1111/jvh.12436. Medline: [DOI] [PubMed] [Google Scholar]
- 220. Antiretroviral Pregnancy Registry International Interim Report: 1 January 1989 through 31 January 2017. Wilmington, NC: Registry Coordinating Centre; 2017. [Google Scholar]
- 221.Chen HL,Lin LH,Hu FC, et al. Effects of maternal screening and universal immunization to prevent mother-to-infant transmission of HBV. Gastroenterology 2012;142(4):773–81.e2. 10.1053/j.gastro.2011.12.035. Medline: [DOI] [PubMed] [Google Scholar]
- 222.Zou H,Chen Y,Duan Z, et al. Virologic factors associated with failure to passive-active immunoprophylaxis in infants born to HBsAg-positive mothers. J Viral Hepat 2012;19(2):e18–25. 10.1111/j.1365-2893.2011.01492.x. Medline: [DOI] [PubMed] [Google Scholar]
- 223.Jourdain G,Ngo-Giang-Huong N,Harrison L, et al. Tenofovir versus placebo to prevent perinatal rransmission of hepatitis B. N Engl J Med 2018;378:911–23. 10.1056/NEJMoa1708131. Medline: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Benaboud S,Pruvost A,Coffie PA, et al. Concentrations of tenofovir and emtricitabine in breast milk of HIV-1-infected women in Abidjan, Cote d’Ivoire, in the ANRS 12109 TEmAA Study, Step 2. Antimicrob Agents Chemother 2011;55:1315–7. 10.1128/AAC.00514-10. Medline: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.de Martino M,Appendino C,Resti M, et al. Should hepatitis B surface antigen positive mothers breast feed? Arch Dis Child 1985;60:972–4. 10.1136/adc.60.10.972. Medline: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Hill JB,Sheffield JS,Kim MJ, et al. Risk of hepatitis B transmission in breast-fed infants of chronic hepatitis B carriers. Obstet Gynecol 2002;99:1049–52. 10.1097/00006250-200206000-00018. Medline: [DOI] [PubMed] [Google Scholar]
- 227.Tseng RY,Lam CW,Tam J. Breastfeeding babies of HBsAg-positive mothers. Lancet 1988;2:1032. 10.1016/S0140-6736(88)90803-3. Medline: [DOI] [PubMed] [Google Scholar]
- 228.Sa-Nguanmoo P,Tangkijvanich P,Tharmaphornpilas P, et al. Molecular analysis of hepatitis B virus associated with vaccine failure in infants and mothers: a case-control study in Thailand. J Med Virol 2012;84:1177–85. 10.1002/jmv.23260. Medline: [DOI] [PubMed] [Google Scholar]
- 229.Ho MS,Lu CF,Kuo J, et al. A family cluster of an immune escape variant of hepatitis B virus infecting a mother and her two fully immunized children. Clin Diagn Lab Immunol 1995;2(6):760–2. Medline: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Bell DM,Shapiro CN,Culver DH, et al. Risk of hepatitis B and human immunodeficiency virus transmission to a patient from an infected surgeon due to percutaneous injury during an invasive procedure: estimates based on a model. Infect Agents Dis 1992;1(5):263–9. Medline: [PubMed] [Google Scholar]
- 231.LaBrecque DR,Muhs JM,Lutwick LI, et al. The risk of hepatitis B transmission from health care workers to patients in a hospital setting—a prospective study. Hepatology 1986;6:205–8. 10.1002/hep.1840060209. Medline: [DOI] [PubMed] [Google Scholar]
- 232.Gunson RN,Shouval D,Roggendorf M, et al. Hepatitis B virus (HBV) and hepatitis C virus (HCV) infections in health care workers (HCWs): guidelines for prevention of transmission of HBV and HCV from HCW to patients. J Clin Virol 2003;27:213–30. 10.1016/S1386-6532(03)00087-8. Medline: [DOI] [PubMed] [Google Scholar]
- 233.Mele A,Ippolito G,Craxi A, et al. Risk management of HBsAg or anti-HCV positive healthcare workers in hospital. Dig Liver Dis 2001;33:795–802. 10.1016/S1590-8658(01)80698-8. Medline: [DOI] [PubMed] [Google Scholar]
- 234. An outbreak of hepatitis B associated with reusable subdermal electroencephalogram electrodes. Hepatitis B Outbreak Investigation Team. CMAJ 2000;162:1127–31. Medline: [PMC free article] [PubMed] [Google Scholar]
- 235.Johnston BL,Conley JM. Nosocomial transmission of bloodborne viruses from infected health care workers to patients. Can J Infect Dis 2003;14:192–6. 10.1155/2003/567290. Medline: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Centers for Disease Control and Prevention. Updated CDC recommendations for the management of hepatitis B virus-infected health-care providers and students. MMWR Recomm Rep 2012;61(RR-3):1–12. Medline: [PubMed] [Google Scholar]
- 237.Henderson DK,Dembry L,Fishman NO, et al. SHEA guideline for management of healthcare workers who are infected with hepatitis B virus, hepatitis C virus, and/or human immunodeficiency virus. Infect Control Hosp Epidemiol 2010;31:203–32. 10.1086/650298. Medline: [DOI] [PubMed] [Google Scholar]
- 238.Reitsma AM,Closen ML,Cunningham M, et al. Infected physicians and invasive procedures: safe practice management. Clin Infect Dis 2005;40:1665–72. 10.1086/429821. Medline: [DOI] [PubMed] [Google Scholar]
- 239.Corden S,Ballard AL,Ijaz S, et al. HBV DNA levels and transmission of hepatitis B by health care workers. J Clin Virol 2003;27:52–8. 10.1016/S1386-6532(02)00127-0. Medline: [DOI] [PubMed] [Google Scholar]
- 240.Raven SF,de Heus B,Wong A, et al. Fluctuation of viremia in hepatitis B virus-infected healthcare workers performing exposure-prone procedures in the Netherlands. Infect Control Hosp Epidemiol 2016;37:655–60. 10.1017/ice.2016.49. Medline: [DOI] [PubMed] [Google Scholar]
- 241. Institut National de Sante Publique du Québec. Recommandations concernant l’évaluation et le suivi des soignants infectés par le virus de l’hépatite B (VHB). Quebec: Gouvernement du Québec; 2015. [Google Scholar]
- 242.Perrillo RP,Gish R,Falck-Ytter YT. American Gastroenterological Association Institute technical review on prevention and treatment of hepatitis B virus reactivation during immunosuppressive drug therapy. Gastroenterology 2015;148:221–44.e3. 10.1053/j.gastro.2014.10.038. Medline: [DOI] [PubMed] [Google Scholar]
- 243.Reddy KR,Beavers KL,Hammond SP, et al. American Gastroenterological Association Institute guideline on the prevention and treatment of hepatitis B virus reactivation during immunosuppressive drug therapy. Gastroenterology 2015;148:215–9; quiz e16–7. 10.1053/j.gastro.2014.10.039. Medline: [DOI] [PubMed] [Google Scholar]
- 244.Hwang JP,Somerfield MR,Alston-Johnson DE, et al. Hepatitis B virus screening for patients with cancer before therapy: American Society of Clinical Oncology provisional clinical opinion update. J Clin Oncol 2015;33:2212–20. 10.1200/JCO.2015.61.3745. Medline: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Martyak LA,Taqavi E,Saab S. Lamivudine prophylaxis is effective in reducing hepatitis B reactivation and reactivation-related mortality in chemotherapy patients: a meta-analysis. Liver Int 2008;28(1):28–38. 10.1111/j.1478-3231.2007.01618.x. Medline: [DOI] [PubMed] [Google Scholar]
- 246.Loomba R,Rowley A,Wesley R, et al. Systematic review: the effect of preventive lamivudine on hepatitis B reactivation during chemotherapy. Ann Intern Med 2008;148:519–28. 10.7326/0003-4819-148-7-200804010-00008. Medline: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Huang H,Li X,Zhu J, et al. Entecavir vs lamivudine for prevention of hepatitis B virus reactivation among patients with untreated diffuse large B-cell lymphoma receiving R-CHOP chemotherapy: a randomized clinical trial. JAMA 2014;312:2521–30. 10.1001/jama.2014.15704. Medline: [DOI] [PubMed] [Google Scholar]
- 248.Chen FW,Coyle L,Jones BE, et al. Entecavir versus lamivudine for hepatitis B prophylaxis in patients with haematological disease. Liver Int 2013;33:1203–10. 10.1111/liv.12154. Medline: [DOI] [PubMed] [Google Scholar]
- 249.Seto WK,Chan TS,Hwang YY, et al. Hepatitis B reactivation in patients with previous hepatitis B virus exposure undergoing rituximab-containing chemotherapy for lymphoma: a prospective study. J Clin Oncol 2014;32:3736–43. 10.1200/JCO.2014.56.7081. Medline: [DOI] [PubMed] [Google Scholar]
- 250.Shah AS,Amarapurkar DN. Spectrum of hepatitis B and renal involvement. Liver Int 2018;38:23–32. 10.1111/liv.13498. Medline: [DOI] [PubMed] [Google Scholar]
- 251.Levy M,Chen N. Worldwide perspective of hepatitis B-associated glomerulonephritis in the 80s. Kidney Int Suppl 1991;35:S24–33. Medline: [PubMed] [Google Scholar]
- 252.Michalak T. Immune complexes of hepatitis B surface antigen in the pathogenesis of periarteritis nodosa. A study of seven necropsy cases. Am J Pathol 1978;90:619–32. Medline: [PMC free article] [PubMed] [Google Scholar]
- 253.Lai KN,Li PK,Lui SF, et al. Membranous nephropathy related to hepatitis B virus in adults. N Engl J Med 1991;324:1457–63. 10.1056/NEJM199105233242103. Medline: [DOI] [PubMed] [Google Scholar]
- 254.Conti F,Vitale G,Cursaro C, et al. Tenofovir-induced Fanconi syndrome in a patient with chronic hepatitis B monoinfection. Ann Hepatol 2016;15:273–6. 10.5604/16652681.1193725. Medline: [DOI] [PubMed] [Google Scholar]
- 255.Hwang HS,Park CW,Song MJ. Tenofovir-associated Fanconi syndrome and nephrotic syndrome in a patient with chronic hepatitis B monoinfection. Hepatology 2015;62(4):1318–20. 10.1002/hep.27730. Medline: [DOI] [PubMed] [Google Scholar]
- 256.Hall AM,Hendry BM,Nitsch D, et al. Tenofovir-associated kidney toxicity in HIV-infected patients: a review of the evidence. Am J Kidney Dis 2011;57:773–80. 10.1053/j.ajkd.2011.01.022. Medline: [DOI] [PubMed] [Google Scholar]
- 257.Laprise C,Baril JG,Dufresne S, et al. Association between tenofovir exposure and reduced kidney function in a cohort of HIV-positive patients: results from 10 years of follow-up. Clin Infect Dis 2013;56:567–75. 10.1093/cid/cis937. Medline: [DOI] [PubMed] [Google Scholar]
- 258.Fontana RJ. Side effects of long-term oral antiviral therapy for hepatitis B. Hepatology 2009;49(5 Suppl):S185–95. 10.1002/hep.22885. Medline: [DOI] [PubMed] [Google Scholar]
- 259.Tartaglia A,Ferrara SM,Sica S, et al. Successful treatment with tenofovir alafenamide of a HIV/hepatitis B virus coinfected patient with HIV and hepatitis B virus drug resistance, end-stage renal disease on haemodialysis. AIDS 2017;31:2314–5. 10.1097/QAD.0000000000001623. Medline: [DOI] [PubMed] [Google Scholar]
- 260.Custodio JM,Fordyce M,Garner W, et al. Pharmacokinetics and safety of tenofovir alafenamide in HIV-uninfected subjects with severe renal impairment. Antimicrob Agents Chemother 2016;60:5135–40. 10.1128/AAC.00005-16. Medline: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Post FA,Tebas P,Clarke A, et al. Brief report: switching to tenofovir alafenamide, coformulated with elvitegravir, cobicistat, and emtricitabine, in HIV-infected adults with renal impairment: 96-week results from a single-arm, multicenter, open-label phase 3 study. J Acquir Immune Defic Syndr 2017;74:180–4. 10.1097/QAI.0000000000001186. Medline: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Pozniak A,Arribas JR,Gathe J, et al. Switching to tenofovir alafenamide, coformulated with elvitegravir, cobicistat, and emtricitabine, in HIV-infected patients with renal impairment: 48-week results from a single-arm, multicenter, open-label phase 3 study. J Acquir Immune Defic Syndr 2016;71:530–7. 10.1097/QAI.0000000000000908. Medline: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Fattovich G,Giustina G,Schalm SW, et al.; The EUROHEP Study Group on Hepatitis B Virus and Cirrhosis. Occurrence of hepatocellular carcinoma and decompensation in western European patients with cirrhosis type B. Hepatology 1995;21:77–82. 10.1016/0270-9139(95)90411-5. Medline: [DOI] [PubMed] [Google Scholar]
- 264.Chen YC,Chu CM,Yeh CT, et al. Natural course following the onset of cirrhosis in patients with chronic hepatitis B: a long-term follow-up study. Hepatol Int 2007;1:267–73. 10.1007/s12072-007-5001-0. Medline: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Fattovich G,Pantalena M,Zagni I, et al. Effect of hepatitis B and C virus infections on the natural history of compensated cirrhosis: a cohort study of 297 patients. Am J Gastroenterol 2002;97:2886–95. 10.1111/j.1572-0241.2002.07057.x. Medline: [DOI] [PubMed] [Google Scholar]
- 266.Liaw YF,Chen YC,Sheen IS, et al. Impact of acute hepatitis C virus superinfection in patients with chronic hepatitis B virus infection. Gastroenterology 2004;126:1024–9. 10.1053/j.gastro.2004.01.011. Medline: [DOI] [PubMed] [Google Scholar]
- 267.Wiesner R,Edwards E,Freeman R, et al. Model for end-stage liver disease (MELD) and allocation of donor livers. Gastroenterology 2003;124:91–6. 10.1053/gast.2003.50016. Medline: [DOI] [PubMed] [Google Scholar]
- 268.Murray KF,Carithers RL, Jr., et al. AASLD practice guidelines: evaluation of the patient for liver transplantation. Hepatology 2005;41:1407–32. 10.1002/hep.20704. Medline: [DOI] [PubMed] [Google Scholar]
- 269.Jang JW,Choi JY,Kim YS, et al. Long-term effect of antiviral therapy on disease course after decompensation in patients with hepatitis B virus-related cirrhosis. Hepatology 2015;61:1809–20. 10.1002/hep.27723. Medline: [DOI] [PubMed] [Google Scholar]
- 270.Singal AK,Fontana RJ. Meta-analysis: oral anti-viral agents in adults with decompensated hepatitis B virus cirrhosis. Aliment Pharmacol Ther 2012;35:674–89. 10.1111/j.1365-2036.2011.04990.x. Medline: [DOI] [PubMed] [Google Scholar]
- 271.Liaw YF,Sheen IS,Lee CM, et al. Tenofovir disoproxil fumarate (TDF), emtricitabine/TDF, and entecavir in patients with decompensated chronic hepatitis B liver disease. Hepatology 2011;53:62–72. 10.1002/hep.23952. Medline: [DOI] [PubMed] [Google Scholar]
- 272.Peng CY,Chien RN,Liaw YF. Hepatitis B virus-related decompensated liver cirrhosis: benefits of antiviral therapy. J Hepatol 2012;57:442–50. 10.1016/j.jhep.2012.02.033. Medline: [DOI] [PubMed] [Google Scholar]
- 273.Lok AS,McMahon BJ,Brown RS, Jr., et al. Antiviral therapy for chronic hepatitis B viral infection in adults: a systematic review and meta-analysis. Hepatology 2016;63:284–306. 10.1002/hep.28280. Medline: [DOI] [PubMed] [Google Scholar]
- 274.Kraut JA,Madias NE. Lactic acidosis. N Engl J Med 2014;371:2309–19. 10.1056/NEJMra1309483. Medline: [DOI] [PubMed] [Google Scholar]
- 275.Zhou K,Terrault N. Management of hepatitis B in special populations. Best Pract Res Clin Gastroenterol 2017;31:311–20. 10.1016/j.bpg.2017.06.002. Medline: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Todo S,Demetris AJ,Van Thiel D, et al. Orthotopic liver transplantation for patients with hepatitis B virus-related liver disease. Hepatology 1991;13:619–26. 10.1016/0270-9139(91)92554-L. Medline: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Idilman R,Akyildiz M,Keskin O, et al. The long-term efficacy of combining nucleos(t)ide analog and low-dose hepatitis B immunoglobulin on post-transplant hepatitis B virus recurrence. Clin Transplant 2016;30:1216–21. 10.1111/ctr.12804. Medline: [DOI] [PubMed] [Google Scholar]
- 278.Coffin CS,Stock PG,Dove LM, et al. Virologic and clinical outcomes of hepatitis B virus infection in HIV-HBV coinfected transplant recipients. Am J Transplant 2010;10:1268–75. 10.1111/j.1600-6143.2010.03070.x. Medline: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Radhakrishnan K,Chi A,Quan DJ, et al. Short course of postoperative hepatitis B immunoglobulin plus antivirals prevents reinfection of liver transplant recipients. Transplantation 2017;101:2079–82. 10.1097/TP.0000000000001786. Medline: [DOI] [PubMed] [Google Scholar]
- 280.Cholongitas E,Papatheodoridis GV. High genetic barrier nucleos(t)ide analogue(s) for prophylaxis from hepatitis B virus recurrence after liver transplantation: a systematic review. Am J Transplant 2013;13:353–62. 10.1111/j.1600-6143.2012.04315.x. Medline: [DOI] [PubMed] [Google Scholar]
- 281.Fung J,Wong T,Chok K, et al. Long-term outcomes of entecavir monotherapy for chronic hepatitis B after liver transplantation: results up to 8 years. Hepatology 2017;66:1036–44. 10.1002/hep.29191. Medline: [DOI] [PubMed] [Google Scholar]
- 282.Klein CG,Cicinnati V,Schmidt H, et al. Compliance and tolerability of subcutaneous hepatitis B immunoglobulin self-administration in liver transplant patients: a prospective, observational, multicenter study. Ann Transplant 2013;18:677–84. 10.12659/AOT.889269. Medline: [DOI] [PubMed] [Google Scholar]
- 283.Cholongitas E,Goulis I,Antoniadis N, et al. New nucleos(t)ide analogue monoprophylaxis after cessation of hepatitis B immunoglobulin is effective against hepatitis B recurrence. Transpl Int 2014;27:1022–8. 10.1111/tri.12370. Medline: [DOI] [PubMed] [Google Scholar]
- 284.Fox AN,Terrault NA. The option of HBIG-free prophylaxis against recurrent HBV. J Hepatol 2012;56:1189–97. 10.1016/j.jhep.2011.08.026. Medline: [DOI] [PubMed] [Google Scholar]
- 285.Fernandez I,Loinaz C,Hernandez O, et al. Tenofovir/entecavir monotherapy after hepatitis B immunoglobulin withdrawal is safe and effective in the prevention of hepatitis B in liver transplant recipients. Transpl Infect Dis 2015;17:695–701. 10.1111/tid.12434. Medline: [DOI] [PubMed] [Google Scholar]
- 286.Bortoluzzi I,Gambato M,Albertoni L, et al. Use of grafts from anti-HBc-positive donors in liver transplantation: a 5-year, single-center experience. Transplant Proc 2013;45:2707–10. 10.1016/j.transproceed.2013.07.049. Medline: [DOI] [PubMed] [Google Scholar]
- 287.Joya-Vazquez PP,Dodson FS,Dvorchik I, et al. Impact of anti-hepatitis Bc-positive grafts on the outcome of liver transplantation for HBV-related cirrhosis. Transplantation 2002;73:1598–602. 10.1097/00007890-200205270-00013. Medline: [DOI] [PubMed] [Google Scholar]
- 288.Kumar M,Satapathy S,Monga R, et al. A randomized controlled trial of lamivudine to treat acute hepatitis B. Hepatology 2007;45:97–101. 10.1002/hep.21486. Medline: [DOI] [PubMed] [Google Scholar]
- 289.Wiegand J,Wedemeyer H,Franke A, et al. Treatment of severe, nonfulminant acute hepatitis B with lamivudine vs placebo: a prospective randomized double-blinded multicentre trial. J Viral Hepat 2014;21:744–50. 10.1111/jvh.12210. Medline: [DOI] [PubMed] [Google Scholar]
- 290. Public Health Agency of Canada (PHAC). HIV/AIDS Epi Updates—Chapter 13: HIV/AIDS in Canada among people from countries where HIV is endemic. Ottawa: PHAC; 2012. [Google Scholar]
- 291.Singh KP,Crane M,Audsley J, et al. HIV-hepatitis B virus coinfection: epidemiology, pathogenesis, and treatment. AIDS 2017;31:2035–52. 10.1097/QAD.0000000000001574. Medline: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Ryom L,Boesecke C,Bracchi M, et al. Highlights of the 2017 European AIDS Clinical Society (EACS) guidelines for the treatment of adult HIV-positive persons version 9.0. HIV Med 2018. 19(5):309–15. 10.1111/hiv.12600. Medline: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Gallant J,Brunetta J,Crofoot G, et al. Brief report: Efficacy and safety of switching to a single-tablet regimen of elvitegravir/cobicistat/emtricitabine/tenofovir alafenamide in HIV-1/hepatitis B-coinfected adults. J Acquir Immune Defic Syndr 2016;73:294–8. 10.1097/QAI.0000000000001069. Medline: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294. NIH Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the use of antiretroviral agents in HIV-1-infected adults and adolescents. Bethesda, MD: U.S. Department of Health and Human Services; 2016. [Google Scholar]
- 295.Lara AN,Sartori AM,Fonseca MO, et al. Long-term protection after hepatitis B vaccination in people living with HIV. Vaccine 2017;35:4155–61. 10.1016/j.vaccine.2017.06.040. Medline: [DOI] [PubMed] [Google Scholar]
- 296.De Monte A,Courjon J,Anty R, et al. Direct-acting antiviral treatment in adults infected with hepatitis C virus: reactivation of hepatitis B virus coinfection as a further challenge. J Clin Virol 2016;78:27–30. 10.1016/j.jcv.2016.02.026. Medline: [DOI] [PubMed] [Google Scholar]
- 297.Konstantinou D,Deutsch M. The spectrum of HBV/HCV coinfection: epidemiology, clinical characteristics, viralinteractions and management. Ann Gastroenterol 2015;28(2):221–8. Medline: [PMC free article] [PubMed] [Google Scholar]
- 298.Caccamo G,Saffioti F,Raimondo G. Hepatitis B virus and hepatitis C virus dual infection. World J Gastroenterol 2014;20:14559–67. 10.3748/wjg.v20.i40.14559. Medline: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Collins JM,Raphael KL,Terry C, et al. Hepatitis B virus reactivation during successful treatment of hepatitis C virus with sofosbuvir and simeprevir. Clin Infect Dis 2015;61:1304–6. 10.1093/cid/civ474. Medline: [DOI] [PubMed] [Google Scholar]
- 300.Ende AR,Kim NH,Yeh MM, et al. Fulminant hepatitis B reactivation leading to liver transplantation in a patient with chronic hepatitis C treated with simeprevir and sofosbuvir: a case report. J Med Case Rep 2015;9:164. 10.1186/s13256-015-0630-8. Medline: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Mucke MM,Backus LI,Mucke VT, et al. Hepatitis B virus reactivation during direct-acting antiviral therapy for hepatitis C: a systematic review and meta-analysis. Lancet Gastroenterol Hepatol 2018. 3(3):172–80. 10.1016/S2468-1253(18)30002-5. Medline: [DOI] [PubMed] [Google Scholar]
- 302.Calvaruso V,Ferraro D,Licata A, et al. HBV reactivation in patients with HCV/HBV cirrhosis on treatment with direct-acting antivirals. J Viral Hepat 2018;25:72–9. 10.1111/jvh.12754. Medline: [DOI] [PubMed] [Google Scholar]
- 303.Ogawa E,Furusyo N,Murata M, et al. Potential risk of HBV reactivation in patients with resolved HBV infection undergoing direct-acting antiviral treatment for HCV. Liver Int 2018;38:76–83. 10.1111/liv.13496. Medline: [DOI] [PubMed] [Google Scholar]
- 304.Wu T,Kwok RM,Tran TT. Isolated anti-HBc: the relevance of hepatitis B core antibody—a review of new issues. Am J Gastroenterol 2017;112:1780–8. 10.1038/ajg.2017.397. Medline: [DOI] [PubMed] [Google Scholar]
- 305.Noureddin M,Gish R. Hepatitis delta: epidemiology, diagnosis and management 36 years after discovery. Curr Gastroenterol Rep 2014;16:365. 10.1007/s11894-013-0365-x. Medline: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Lempp FA,Ni Y,Urban S. Hepatitis delta virus: insights into a peculiar pathogen and novel treatment options. Nat Rev Gastroenterol Hepatol 2016;13:580–9. 10.1038/nrgastro.2016.126. Medline: [DOI] [PubMed] [Google Scholar]
- 307.Rizzetto M. The adventure of delta. Liver Int 2016;36(Suppl 1):135–40. 10.1111/liv.13018. Medline: [DOI] [PubMed] [Google Scholar]
- 308.Cheng HH,Wang DQ,Minuk GY, et al. The prevalence of antibody to delta virus in western Canada. Clin Invest Med 1986;9:156–9. Medline: [PubMed] [Google Scholar]
- 309.Ratnam S,Head CB,Butler RW. Lack of evidence of hepatitis D (delta) infection in Newfoundland and Labrador. CMAJ 1986;134:905–7. Medline: [PMC free article] [PubMed] [Google Scholar]
- 310.Schaper M,Rodriguez-Frias F,Jardi R, et al. Quantitative longitudinal evaluations of hepatitis delta virus RNA and hepatitis B virus DNA shows a dynamic, complex replicative profile in chronic hepatitis B and D. J Hepatol 2010;52:658–64. 10.1016/j.jhep.2009.10.036. Medline: [DOI] [PubMed] [Google Scholar]
- 311.Lutterkort GL,Wranke A,Yurdaydin C, et al. Non-invasive fibrosis score for hepatitis delta. Liver Int 2017;37:196–204. 10.1111/liv.13205. Medline: [DOI] [PubMed] [Google Scholar]
- 312.Triantos C,Kalafateli M,Nikolopoulou V, et al. Meta-analysis: antiviral treatment for hepatitis D. Aliment Pharmacol Ther 2012;35:663–73. 10.1111/j.1365-2036.2012.04993.x. Medline: [DOI] [PubMed] [Google Scholar]
- 313.Wedemeyer H,Yurdaydin C,Dalekos GN, et al. Peginterferon plus adefovir versus either drug alone for hepatitis delta. N Engl J Med 2011;364:322–31. 10.1056/NEJMoa0912696. Medline: [DOI] [PubMed] [Google Scholar]
- 314.Heidrich B,Yurdaydin C,Kabacam G, et al. Late HDV RNA relapse after peginterferon alpha-based therapy of chronic hepatitis delta. Hepatology 2014;60:87–97. 10.1002/hep.27102. Medline: [DOI] [PubMed] [Google Scholar]
- 315.Wedemeyer H,Yurdaydin C,Ernst S, et al. O4 prolonged therapy of hepatitis delta for 96 weeks with PEG-IFNa-2a plus tenofovir or placebo does not prevent HDV RNA relapse: the HIDIT-2 study. Journal of Hepatology. 60(1 Suppl), 2014:S2–3. 10.1016/s0168-8278(14)60006-4. [DOI] [Google Scholar]
- 316.Wranke A,Serrano BC,Heidrich B, et al. Antiviral treatment and liver-related complications in hepatitis delta. Hepatology 2017;65:414–25. 10.1002/hep.28876. Medline: [DOI] [PubMed] [Google Scholar]
- 317.Calle Serrano B,Grosshennig A,Homs M, et al. Development and evaluation of a baseline-event-anticipation score for hepatitis delta. J Viral Hepat 2014;21(11):e154–63. 10.1111/jvh.12251. Medline: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Wranke A,Wedemeyer H. Antiviral therapy of hepatitis delta virus infection—progress and challenges towards cure. Curr Opin Virol 2016;20:112–8. 10.1016/j.coviro.2016.10.002. Medline: [DOI] [PubMed] [Google Scholar]
- 319.Verme G,Brunetto MR,Oliveri F, et al. Role of hepatitis delta virus infection in hepatocellular carcinoma. Dig Dis Sci 1991;36:1134–6. 10.1007/BF01297460. Medline: [DOI] [PubMed] [Google Scholar]
- 320.Schwarz KB,Cloonan YK,Ling SC, et al. Children with chronic hepatitis B in the United States and Canada. J Pediatr 2015;167:1287–94.e2. 10.1016/j.jpeds.2015.08.021. Medline: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Marx G,Martin SR,Chicoine JF, et al. Long-term follow-up of chronic hepatitis B virus infection in children of different ethnic origins. J Infect Dis 2002;186:295–301. 10.1086/341508. Medline: [DOI] [PubMed] [Google Scholar]
- 322.Wu JF,Tsai WY,Hsu HY, et al. Effect of puberty onset on spontaneous hepatitis B virus e antigen seroconversion in men. Gastroenterology 2010;138:942–8.e1. 10.1053/j.gastro.2009.11.051. Medline: [DOI] [PubMed] [Google Scholar]
- 323.Popalis C,Yeung LT,Ling SC, et al. Chronic hepatitis B virus (HBV) infection in children: 25 years’ experience. J Viral Hepat 2013;20(4):e20–6. 10.1111/jvh.12019. Medline: [DOI] [PubMed] [Google Scholar]
- 324.Yang HI,Hung HL,Lee MH, et al. Incidence and determinants of spontaneous seroclearance of hepatitis B e antigen and DNA in patients with chronic hepatitis B. Clin Gastroenterol Hepatol 2012;10:527–34.e1-2. 10.1016/j.cgh.2011.12.019. Medline: [DOI] [PubMed] [Google Scholar]
- 325.Murray K,Rodriguez-Baez N,Kleiner D, et al. Liver histology in North American children with immune-active chronic hepatitis B infection. Boston: American Association for the Study of Liver Disease. Hepatology 2016;64(Suppl1):Abstract No 105. [Google Scholar]
- 326.Chiu YC,Liao SF,Wu JF, et al. Factors affecting the natural decay of hepatitis B surface antigen in children with chronic hepatitis B virus infection during long-term follow-up. J Pediatr 2014;165:767–72.e1. 10.1016/j.jpeds.2014.06.059. Medline: [DOI] [PubMed] [Google Scholar]
- 327.Xue MM,Glenn JS,Leung DH. Hepatitis D in children. J Pediatr Gastroenterol Nutr 2015;61:271–81. 10.1097/MPG.0000000000000859. Medline: [DOI] [PubMed] [Google Scholar]
- 328.Mogul D,Ling S,Murray K, et al. Characteristics of HBV-associated hepatocellular carcinoma in children: a multi-center study. JPGN 2018; 67(4):437–40. 10.1097/MPG.0000000000002093. Medline: [DOI] [PubMed] [Google Scholar]
- 329.Bortolotti F,Guido M,Bartolacci S, et al. Chronic hepatitis B in children after e antigen seroclearance: final report of a 29-year longitudinal study. Hepatology 2006;43:556–62. 10.1002/hep.21077. Medline: [DOI] [PubMed] [Google Scholar]
- 330.McMahon BJ,Holck P,Bulkow L, et al. Serologic and clinical outcomes of 1536 Alaska Natives chronically infected with hepatitis B virus. Ann Intern Med 2001;135:759–68. 10.7326/0003-4819-135-9-200111060-00006. Medline: [DOI] [PubMed] [Google Scholar]
- 331.Bortolotti F,Cadrobbi P,Crivellaro C, et al. Long-term outcome of chronic type B hepatitis in patients who acquire hepatitis B virus infection in childhood. Gastroenterology 1990;99:805–10. 10.1016/0016-5085(90)90972-4. Medline: [DOI] [PubMed] [Google Scholar]
- 332.Evans AA,Fine M,London WT. Spontaneous seroconversion in hepatitis B e antigen-positive chronic hepatitis B: implications for interferon therapy. J Infect Dis 1997;176:845–50. 10.1086/516538. Medline: [DOI] [PubMed] [Google Scholar]
- 333.Yu MW,Chang HC,Liaw YF, et al. Familial risk of hepatocellular carcinoma among chronic hepatitis B carriers and their relatives. J Natl Cancer Inst 2000;92:1159–64. 10.1093/jnci/92.14.1159. Medline: [DOI] [PubMed] [Google Scholar]
- 334.Sokal EM,Conjeevaram HS,Roberts EA, et al. Interferon alfa therapy for chronic hepatitis B in children: a multinational randomized controlled trial. Gastroenterology 1998;114:988–95. 10.1016/S0016-5085(98)70318-X. Medline: [DOI] [PubMed] [Google Scholar]
- 335.Vo Thi Diem H,Bourgois A,Bontems P, et al. Chronic hepatitis B infection: long term comparison of children receiving interferon alpha and untreated controls. J Pediatr Gastroenterol Nutr 2005;40:141–5. 10.1097/00005176-200502000-00011. Medline: [DOI] [PubMed] [Google Scholar]
- 336.Bortolotti F,Jara P,Barbera C, et al. Long term effect of alpha interferon in children with chronic hepatitis B. Gut 2000;46:715–8. 10.1136/gut.46.5.715. Medline: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Jonas MM,Mizerski J,Badia IB, et al. Clinical trial of lamivudine in children with chronic hepatitis B. N Engl J Med 2002;346:1706–13. 10.1056/NEJMoa012452. Medline: [DOI] [PubMed] [Google Scholar]
- 338.Jonas MM,Little NR,Gardner SD, et al. Long-term lamivudine treatment of children with chronic hepatitis B: durability of therapeutic responses and safety. J Viral Hepat 2008;15:20–7. 10.1111/j.1365-2893.2007.00891.x. Medline: [DOI] [PubMed] [Google Scholar]
- 339.Sokal EM,Kelly DA,Mizerski J, et al. Long-term lamivudine therapy for children with HBeAg-positive chronic hepatitis B. Hepatology 2006;43:225–32. 10.1002/hep.21020. Medline: [DOI] [PubMed] [Google Scholar]
- 340.Jonas MM,Kelly D,Pollack H, et al. Safety, efficacy, and pharmacokinetics of adefovir dipivoxil in children and adolescents (age 2 to <18 years) with chronic hepatitis B. Hepatology 2008;47:1863–71. 10.1002/hep.22250. Medline: [DOI] [PubMed] [Google Scholar]
- 341.Jonas MM,Chang MH,Sokal E, et al. Randomized, controlled trial of entecavir versus placebo in children with hepatitis B envelope antigen-positive chronic hepatitis B. Hepatology 2016;63:377–87. 10.1002/hep.28015. Medline: [DOI] [PubMed] [Google Scholar]
- 342.Murray KF,Szenborn L,Wysocki J, et al. Randomized, placebo-controlled trial of tenofovir disoproxil fumarate in adolescents with chronic hepatitis B. Hepatology 2012;56:2018–26. 10.1002/hep.25818. Medline: [DOI] [PubMed] [Google Scholar]