

Abstract
One of the keys to achieving skin regeneration lies within understanding the heterogeneity of neonatal fibroblasts, which support skin regeneration. However, the molecular underpinnings regulating the cellular states and fates of these cells are not fully understood. To investigate this, we performed a parallel multi-omics analysis by processing neonatal murine skin for single-cell ATAC-sequencing (scATAC-seq) and single-cell RNA-sequencing (scRNA-seq) separately. Our approach revealed that fibroblast clusters could be sorted into papillary and reticular lineages based on transcriptome profiling, as previously published. However, scATAC-seq analysis of neonatal fibroblast lineage markers, such as, Dpp4/CD26, Corin, and Dlk1 along with markers of myofibroblasts, revealed accessible chromatin in all fibroblast populations despite their lineage-specific transcriptome profiles. These results suggests that accessible chromatin does not always translate to gene expression and that many fibroblast lineage markers reflect a fibroblast state, which includes neonatal papillary, reticular, and myofibroblasts. This analysis also provides a possible explanation as to why these marker genes can be promiscuously expressed in different fibroblast populations under different conditions. Our scATAC-seq analysis also revealed that the functional lineage restriction between dermal papilla and adipocyte fates are regulated by distinct chromatin landscapes. Finally, we have developed a webtool for our multi-omics analysis: https://skinregeneration.org/scatacseq-and-scrnaseq-data-from-thompson-et-al-2021-2/.
Introduction
The dermis of the skin is composed of three distinct anatomical layers with their own fibroblast subtypes. These layers are called the papillary dermis, the reticular dermis, and the hypodermis/Dermal White Adipose Tissue (DWAT) (Driskell et al. 2014; Driskell et al. 2014; Driskell and Watt 2015; Griffin et al. 2020; Sorrell and Caplan 2004). Utilizing neonatal skin as a model, recent studies have proposed a functional fibroblast lineage hierarchy that groups dermal papillae, dermal sheath, arrector pili, and papillary fibroblasts into the so-called papillary fibroblast lineage (Driskell et al. 2013). The reticular fibroblast lineage was proposed to consist of the reticular fibroblasts, pre-adipocyte precursors, pre-adipocytes, and adipocytes (Driskell et al. 2013). The lineage relationship between all fibroblast subtypes in the dermis is important to consider during development, homeostasis, aging, and wound repair, due to their unique and restricted functions (Driskell et al. 2013; Mascharak et al. 2021; Plikus et al. 2017). For example, it has been hypothesized that hair follicles do not normally regenerate in wounds because of the lack of the fibroblast lineage that can differentiate into the dermal papilla (Driskell et al. 2013; Plikus et al. 2017). Additionally, the age and developmental status of skin when using markers of fibroblast heterogeneity, such as, Cd26/Dpp4, Hic1, En1, Lrig1, and Crabp1, will influence the specificity of marker expression (Abbasi et al. 2020; Driskell et al. 2013; Guerrero-Juarez et al. 2019; Jiang et al. 2018; Rinkevich et al. 2015; Salzer et al. 2018).
Neonatal murine skin holds the key to achieve hair follicle regeneration in wounds (Rognoni et al. 2016). Developing murine skin at post-natal day 0–2 is the most fully characterized for marker expression with regards to fibroblast lineage functionality. In addition, these neonatal fibroblasts have the ability to support hair follicle regeneration in wounds and chamber grafting assays (Driskell et al. 2013; Ge et al. 2020; Jensen et al. 2010; Rognoni et al. 2016). Consequently, understanding the molecular underpinnings of neonatal fibroblast lineages is an important step to develop methods to transform scarring wounds into regenerative wounds (Gomes et al. 2021; Jiang and Rinkevich 2021; Phan et al. 2020; Plikus et al. 2021).
The states and fates of fibroblasts throughout development, homeostasis, and repair are only now beginning to be defined (Abbasi et al. 2020; Ascensión et al. 2020; Gay et al. 2020; Guerrero-Juarez et al. 2019; He et al. 2020; Joost et al. 2020; Phan et al. 2020; Philippeos et al. 2018; Solé-Boldo et al. 2020; Tabib et al. 2018; Vorstandlechner et al. 2020). Importantly, in neonatal skin two distinct cell fates can be identified – the dermal papilla and adipocyte (Driskell etal. 2013; Plikus et al. 2017). However, whether neonatal papillary and reticular fibroblasts are a cellular state or fate is not clearly understood. Finally, it is possible that as fibroblasts age and undergo differentiation during wound repair, changes will occur to their developmentally established states and fates as recently shown (Abbasi et al. 2020; Foster et al. 2021).
It has been proposed that single cell methodologies are critical for distinguishing between cell fates and states in tissues (Trapnell 2015). Recently, scRNA-seq has provided an unprecedented insight into the specific transcriptional profiles of cell subpopulations in skin, particularly giving rise to new knowledge of fibroblasts and their relationships with other cells in the dermis (Abbasi et al. 2020; Ascensión et al. 2020; Gay et al. 2020; Griffin et al. 2021; Guerrero-Juarez et al. 2019; Gupta et al. 2019; He et al. 2020; Joost et al. 2020; Phan et al. 2020; Philippeos et al. 2018; Salzer et al. 2018; Shook et al. 2020; Solé-Boldo et al. 2020; Tabib et al. 2018; Vorstandlechner et al. 2020). Recently, ATAC-seq has been adopted into a single-cell approach allowing for the investigation of cellular heterogeneity based on chromatin accessibility profiles (Buenrostro et al. 2015; Buenrostro et al. 2013). These techniques have recently been used to probe fibroblasts from wounds to ascertain the cellular states during wound repair (Abbasi et al. 2020; Foster et al. 2021). However, to the best of our knowledge, an investigation into neonatal fibroblasts chromatin architecture at the single cell level has not been reported. In this study, we have investigated the relationship between gene expression and chromatin accessibility using a parallel single-cell multi-omics approach in post-natal day 0 (P0) murine skin. Through our scATAC-seq and scRNA-seq analysis, we identified genes with specific accessibility profiles that defined cellular states and fates of fibroblasts in neonatal skin. Furthermore, we have also shared our the data of the transcriptional and chromatin states of neonatal murine skin through our easily accessible webtools: https://skinregeneration.org/scatacseq-and-scrnaseq-data-from-thompson-et-al-2021-2/.
Results:
A parallel single cell multi-omics approach to analyze chromatin accessibility and gene expression in unsorted cells from neonatal skin.
To investigate cellular heterogeneity within the transcriptome and chromatin architecture at the single cell level, we dissected the skin from the entire trunk of neonatal mice (P0) to perform a scRNA-seq experiment on 13,991 unsorted P0 cellsand, in parallel from the same cell preparation, we isolated 7020 unsorted nuclei to perform a scATAC-seq experiment (Figure 1a).
Figure 1: Establishing a parallel single-cell-multi-omics approach to study unsorted cell populations from neonatal skin.
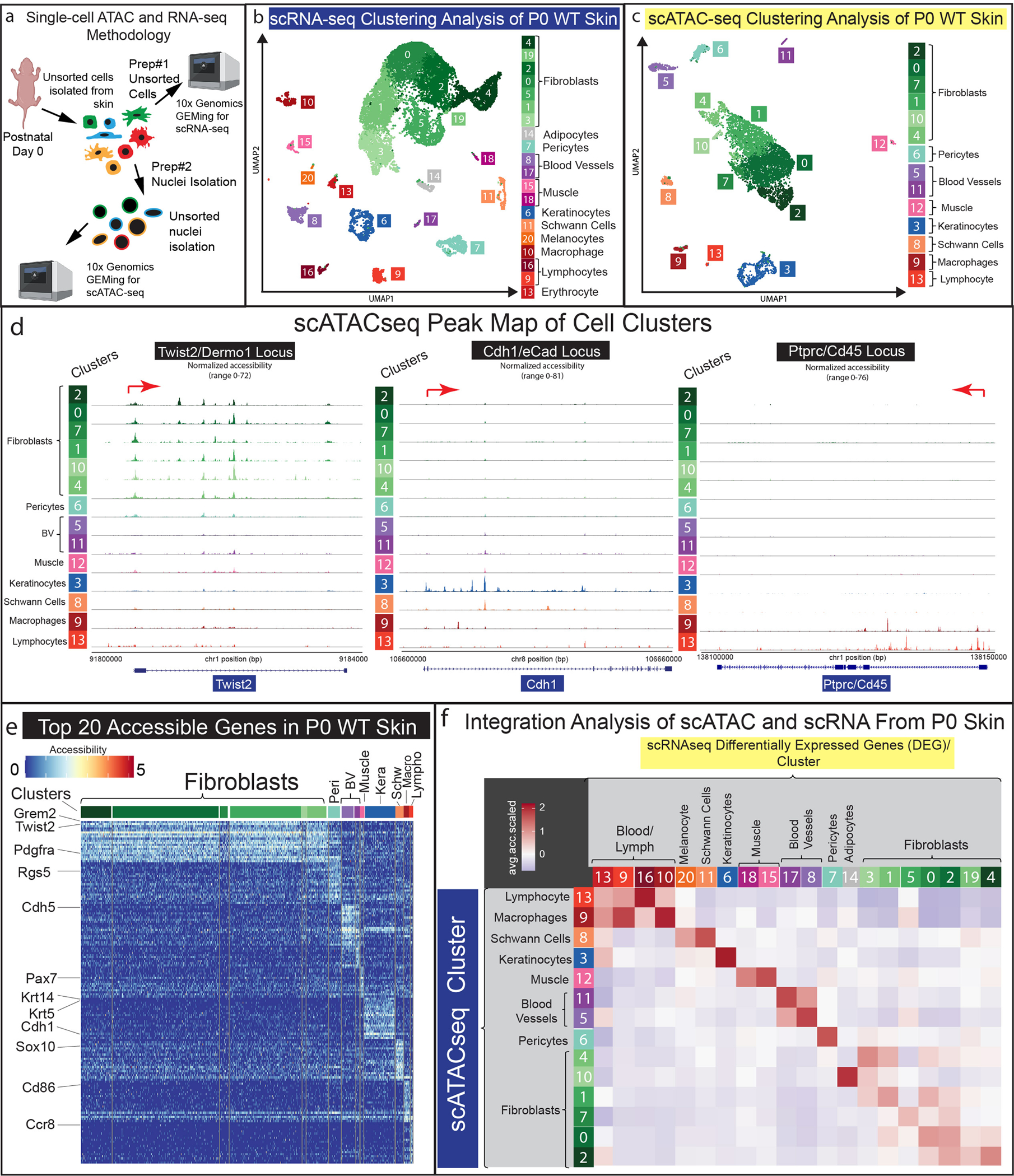
(a) Visual schematic of parallel multi-omics approach utilizing Post-natal-day 0 (P0) mouse skin. (b-c) UMAP representation of all cells processed from scRNA-seq (b) and scATAC-seq (c) data. (d) Peak maps of clusters from all P0 cells of scATAC-seq data representing Twist2/Dermo1, Cdh1/eCad, and Ptprc/Cd45. (e) Heatmap of Top-20 differentially accessible genes from scATAC-seq clusters. (f) Computational correlation analysis of scRNA-seq and scATAC-seq data.
We processed the raw sequencing data using default parameters in the Seurat/Signac computational pipeline (https://github.com/DriskellLab/Thompson-et-al.-2021) (Stuart et al. 2021). The processed data revealed 14 distinct cell clusters in the scATAC-seq data and 21 distinct clusters in the scRNA-seq data that were all identifiable utilizing canonical markers such as, Pdgfra and Twist2 to mark fibroblast populations (Collins et al. 2011; Joost et al. 2020), Rgs5 to mark pericytes (Cho et al. 2003), Ptprc/Cd45 to identify immune cell populations (Collins et al. 2011), Msc and Ttn to mark muscle cells (Chauveau et al. 2014; Robb et al. 1998), Sox10 to mark Schwann cells (Rinwa et al. 2021), Dct to mark melanocytes (Belote et al. 2021), and Pecam1/Cd31 to mark vasculature (Collins et al. 2011) (Figure 1b–d). We then performed a differential accessibility analysis by grouping the fibroblast clusters together and plotting the top 20 genes from each cluster (Figure 1e). Through this analysis, we found common canonical fibroblast markers, such as Pdgfra and Twist2, were most accessible in the fibroblast clusters (Figure 1e, green subset). Canonical markers such as Rgs5 (pericytes), Cdh5 (vasculature),Pax7 (muscle/smooth muscle), Cdh1 (keratinocytes), Sox10 (Schwann/melanocytes) (Nonaka et al. 2008), Cd86 (macrophages) (Ryncarz and Anasetti 1998), and Ccr8 (lymphocytes) (Soler et al. 2006), were also highly accessible in distinct clusters. This result indicates that most cell typescan be identified by their chromatin accessibility profiles.
To test the correlation between the transcriptomic and chromatin accessibility data in our parallel multi-omics study, we probed the scATAC-seq data using the top 5% differentially expressed genes (DEGs) (Supplementary Table S1) identified for each of the 20 clusters in the scRNA-seq data. Specifically, we calculated a single mean-relative-chromatin-accessibility value (See Methods) for all the genes identified in the scRNA-seq clusters within the scATAC-seq clusters. We plotted these calculations as a correlation plot (Figure 1f). By plotting each scATAC-seq cluster’s average relative accessibility for the top transcriptional markers, the cluster-by-cluster correlation suggests cell types that highly express a gene may also have the highest accessibility. scATAC clusters of a distinct cell lineage had positive accessibility scores for their corresponding scRNA clusters. For example, the greatest accessibility for the genes in the scATAC-seq blood/lymph clusters had the highest correlation with the scRNA-seq blood/lymph clusters (Figure 1f). Curiously, the fibroblast scATAC clusters exhibited lower positive correlation scores for the fibroblast scRNA clusters, which may reflect fibroblast plasticity (Figure 1f). We conclude that a parallel single-cell-multi-omics approach reveals a correlation between gene accessibility that directly relates to transcriptional activity in P0 skin.
The specific expression of neonatal fibroblast lineage markers represents a transient cell state.
The distinct expression patterns of a molecular marker in fibroblast populations have been used to identify different functional fibroblast lineages across multiple time points of development and wound repair. However, sometimes the same gene has marked fibroblast populations with different functions. For example, Dpp4/Cd26 has been identified as a papillary lineage marker in neonatal fibroblasts, which support hair follicle regeneration in wounds (Driskell et al. 2013). However, the expression of Dpp4/Cd26 in adult tissue has been associated with increased scarring and fibrogenic potential in adult skin (Rinkevich et al. 2015; Shook et al. 2018).
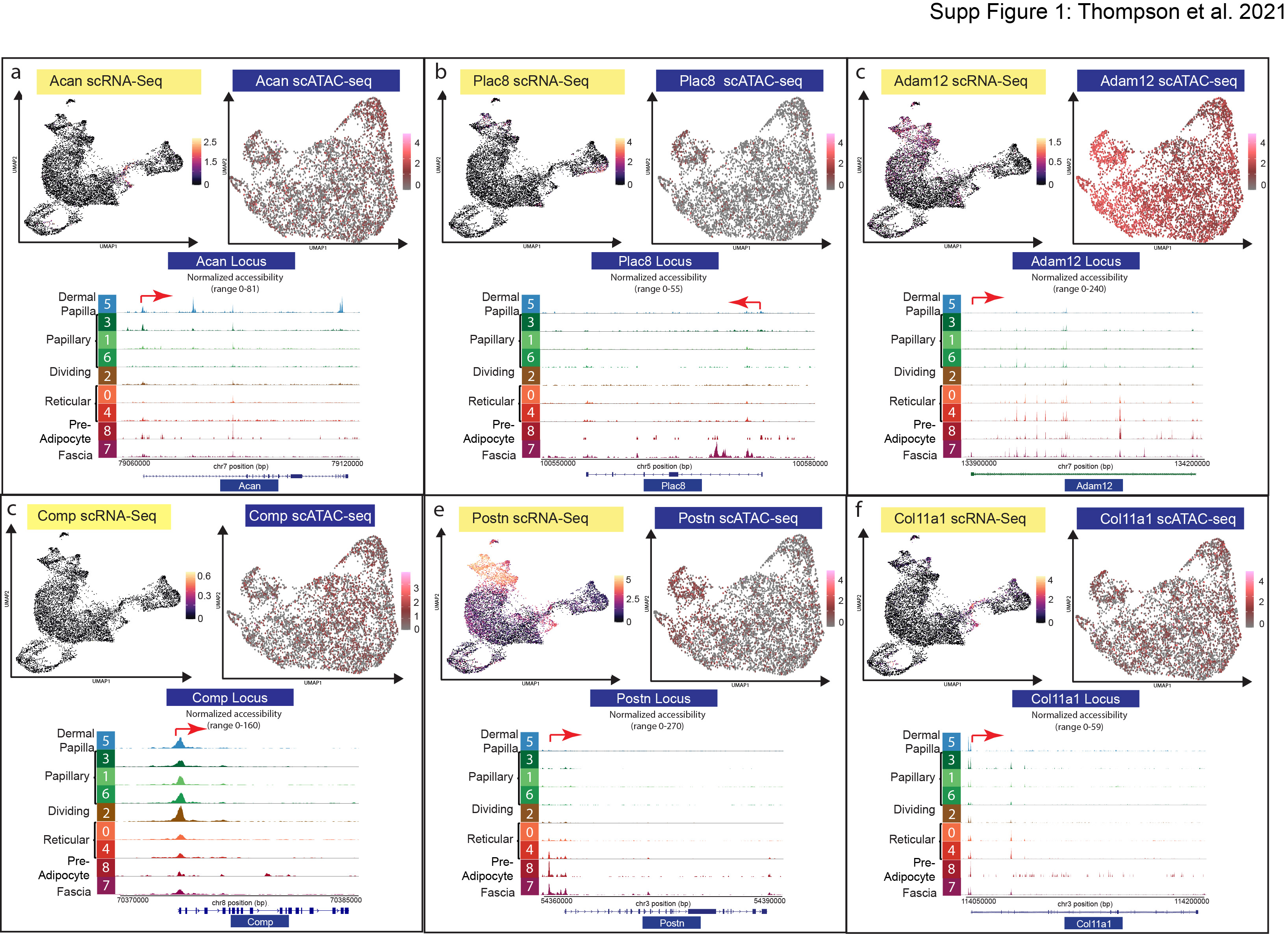
We hypothesized that all neonatal fibroblast lineages contained broadly accessible chromatin for neonatal transcriptional markers despite specific gene expression profiles. The broadly accessible chromatin would allow for each fibroblast lineage to potentially express the gene within specific environments, such as during aging or in a wound. To test this hypothesis, we examined our multi-omics data by computationally subsetting the 9723 fibroblasts from the scRNA-seq dataset and the 5326 fibroblasts from the scATAC-seq dataset for a comparative analysis. The subset data were processed in separate parallel Seurat and Signac pipelines (Figure 2a–b) (https://github.com/DriskellLab/Thompson-et-al.-2021). Our computations revealed 10 fibroblast clusters from the scRNA-seq dataset and 8 clusters from the scATAC-seq dataset (Figure 2a, d). We utilized previously defined markers of fibroblast heterogeneity to label the scRNA-seq UMAP (Figure 2a–b) (Driskell et al. 2013; Phan et al. 2020). For example, Corin expression defined the dermal papilla (Enshell-Seijffers et al. 2008), Dpp4 defined the papillary dermis, Dlk1 and Ly6a expression defined the reticular dermis and hypodermis (Figure 2a–b). In addition, we used Plac8 to identify fascia (Supplementary Figure S1) (Joost et al. 2020), while the expression of the dermal sheath marker, Acan, was confined to cluster 3 (Supplementary Figure S1).
Figure 2: Fibroblast lineage markers have broad chromatin accessibility in all scATAC-seq clusters.
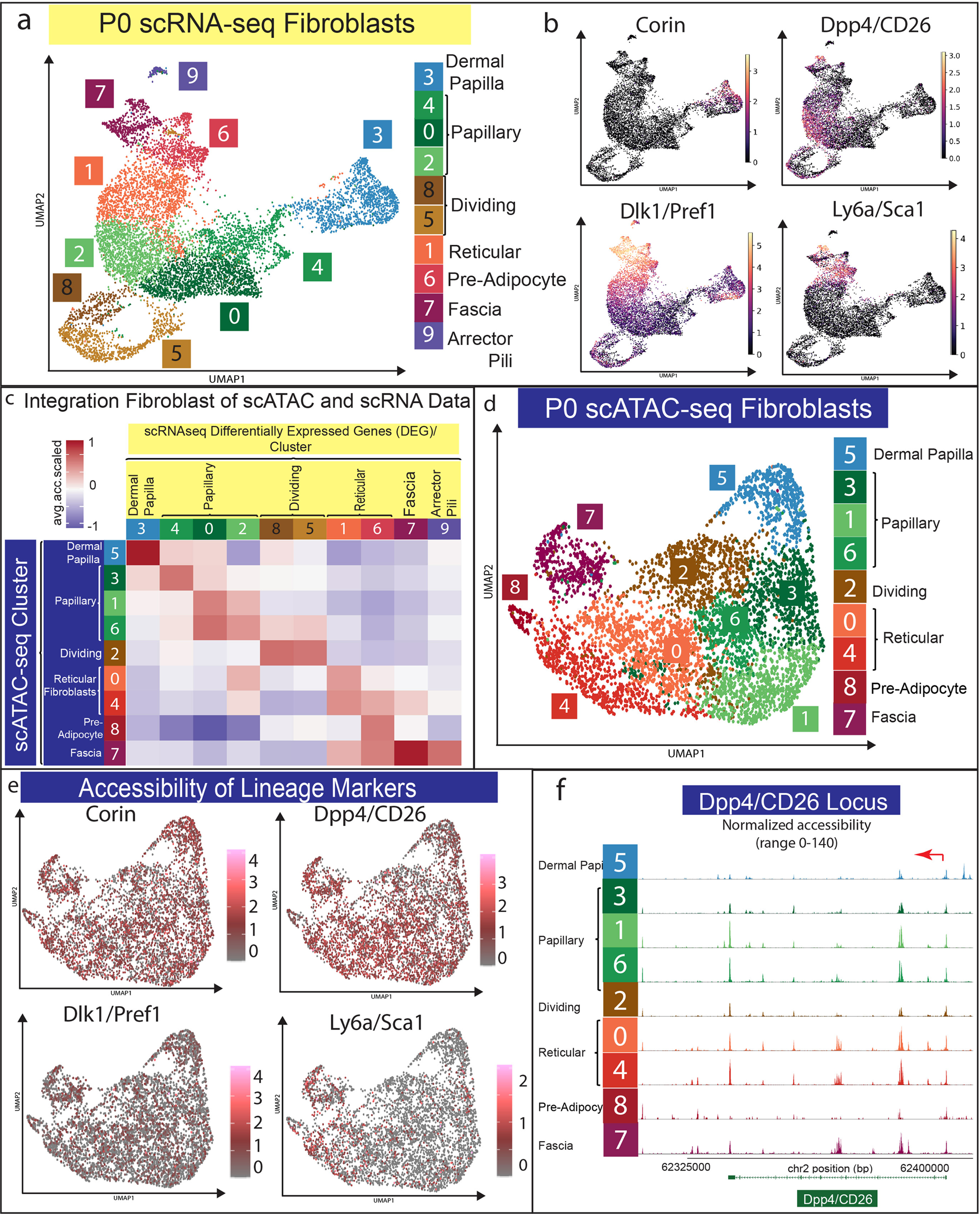
(a) UMAP of fibroblast subset from scRNA-seq data. (b) Expression of fibroblast lineage markers in scRNA-seq data. (c) Computational correlation analysis of scRNA-seq and scATAC-seq data from fibroblast subset. (d) UMAP of scATAC-seq data. (e) Accessibility of fibroblast lineage markers. (f) Peak map of fibroblast clusters from P0 skin for CD26/Dpp4 accessibility.
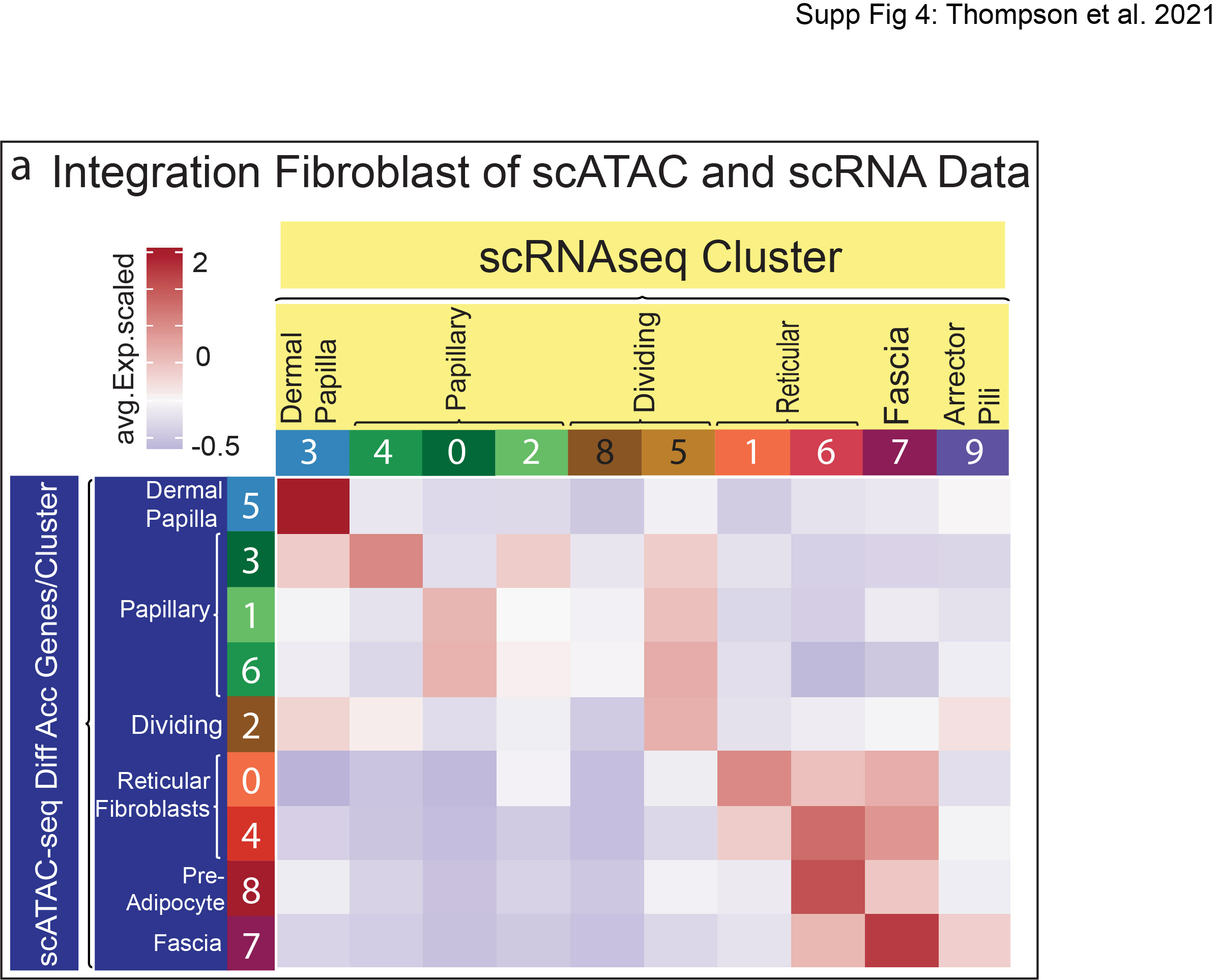
To label the 9 clusters in the scATAC-seq UMAP accurately, we performed the same correlation analysis we performed with all cells (Figure 1f) by examining the overall relative accessibility of each scATAC cluster compared to each scRNA cluster’s top 5% differentially expressed genes (DEGs) (Figure 2c; Supplementary Table S2). Using this approach, we found that the scATAC UMAP was oriented by fibroblast lineage, with the reticular lineage having negative relative accessibility for dermal papilla and papillary clusters’ DEGs while the papillary lineage clusters had negative relative accessibility for reticular fibroblast, pre-adipocyte, and fascia markers (Figure 2c). In addition, our analysis of the top 5% of the differentially accessible genes compared to the gene expression profile in each fibroblast cluster revealed potential fates for dermal papilla and pre-adipocytes/adipocytes. This suggests a positive correlation for two distinct fibroblast fates in the dermis (Supplementary Figure S4).
Intriguingly, the scATAC fibroblast clusters followed a similar UMAP orientation as the scRNA clusters, with dermal papillae and fascia/pre-adipocytes clustering on opposite ends of the fibroblast supercluster (Figure 2d). However, the fibroblast cluster in the scATAC-seq analysis was less distinct than the scRNA-seq clusters (Figure 2a, d). The accessibility of transcriptional markers of fibroblast heterogeneity did not mimic their transcriptional specificity (Figure 2e–f). For example, the neonatal papillary transcriptional marker Dpp4 was accessible in both papillary and reticular clusters while the neonatal reticular marker Dlk1 was accessible in all fibroblast clusters (Figure 2e–f). Ly6a was one of the only common transcriptional markers for the reticular fibroblast lineage to also be specifically accessible in the reticular fibroblast clusters (Figure 2e–f). These results may explain the differences in expression specificity between papillary and reticular regions when analyzed at different time points of skin development, repair, and disease (Driskell et al. 2013; Rinkevich et al. 2015). We conclude that markers of neonatal functional fibroblast lineages define a cell state.
All neonatal fibroblast populations have accessible chromatin profiles that can support transformation into a myofibroblast state.
The myofibroblast fate is a key differentiated fibroblast type in the context of wound healing, scarring, and disease (Guerrero-Juarez et al. 2019; Lim et al. 2018; Plikus et al. 2021; Plikus et al. 2017; Rahmani et al. 2014). It has been previously shown that reticular/lower lineage fibroblasts express myofibroblast markers during neonatal and adult wound repair (Abbasi et al. 2020; Foster et al. 2021; Phan et al. 2020). Consequently, we investigated the expression and chromatin accessibility of key myofibroblast markers Acta2/asma and Tagln/Sm22, and Tgfbr2 in our multi-omics data set. To our surprise, our analysis revealed accessible chromatin in all neonatal fibroblast populations for Acta2/asma, Tagln/Sm22, and Tgfbr2 (Figure 3a–c). Interestingly, Acta2 showed a gradient of accessibility with the highest in the reticular/lower lineages (Figure 3a). As expected, Acta2 and Tagln were specifically expressed in the proposed dermal sheath population (Figure 3a–b), while Tgfbr2 was uniformly expressed in all fibroblast populations (Figure 3c).
Figure 3: Neonatal fibroblast populations share fibrogenic and myofibroblast chromatin accessibility profiles.
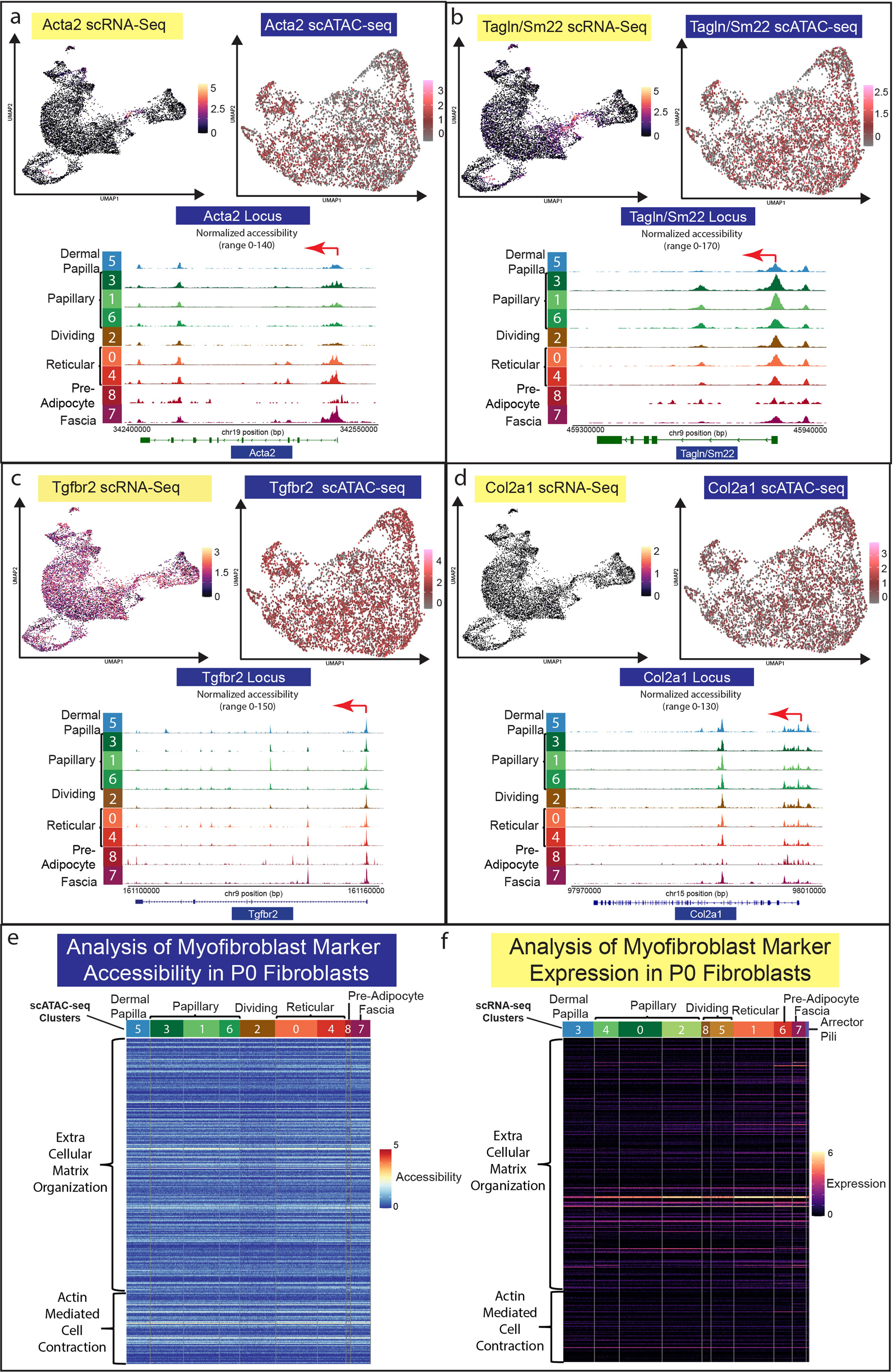
(a-d) Parallel analysis of gene expression and chromatin accessibility for Acta2/asma, Tagln/Sm22, Tgfb2r, and Col2a1. (e-f) Heatmap for accessibility (a) and gene expression (b) of myofibroblast associated GO Terms (Extracellular Matrix Organization (GO:003198) and Actin-Mediated Cell Contraction (GO:0070252)) for all fibroblast clusters in P0 skin.
Recent publications have identified genes associated with myofibroblasts in fibrosis and keloid scarring utilizing scRNA-seq (Deng et al. 2021), specifically, Col2a1, Postn, Adam12, Comp, and Col11a1. We found that the locus for Col2a1, the cartilage specific collagen, had highly accessible chromatin across all neonatal fibroblast populations, but was not detectable in the scRNA-seq analysis (Figure 3d). However, Cartilage Oligomeric Matrix Protein (Comp) revealed accessible chromatin throughout all neonatal fibroblast populations, with the highest accessibility in papillary lineages. Conversely, Comp gene expression was not detected (Supplementary Figure S1). Other genes associated with myofibroblast and keloid scarring, such as Postn, Adam12, and Col11a1 were also accessible in neonatal fibroblast subpopulations with differential expression (Supplementary Figure S1).
Since our analysis revealed accessible chromatin profiles of key myofibroblast genes, we hypothesized that all neonatal fibroblast lineages have accessible chromatin profiles to support a myofibroblast state. To investigate this hypothesis, we analyzed the chromatin accessibility and gene expression profiles in all neonatal fibroblast populations using the GO terms for Extracellular Matrix Organization (GO:003198) and Actin-Mediated Cell Contraction (GO:0070252) (Figure 3e–f). We calculated the number of accessible genes from each cluster with an accessibility score of 1 or greater for each GO term and found that almost 50% of the genes were accessible in all populations (Figure 2e) (Supplementary Table S3). Analysis of gene expression in fibroblast populations of GO terms revealed that less than 20% were expressed in fibroblasts with differential expression profiles favoring the reticular fibroblast lineages (Figure 2f) (Supplementary Table S3).
Our Go Term analysis of myofibroblast markers suggests that all neonatal fibroblasts have accessible chromatin for up to 50% of genes associated with myofibroblast activity, indicating a potential to enter a myofibroblast state particularly amongst the reticular lineage. The analysis also indicates a potential for all neonatal fibroblast populations to have chromatin profiles that support the expression of key myofibroblast markers. Our data also support the idea that myofibroblast states/phenotypes are not always associated with scarring because they are transiently observed in regenerative Acomys ear wounds (Brewer et al. 2021). However, additional studies and re-analysis of recently published scATAC-seq data (Abbasi et al. 2020; Foster et al. 2021) investigating adult and wound repair fibroblasts in the context of chromatin accessibility are now required.
Distinct chromatin landscapes define neonatal fibroblast cell fates.
It has been previously shown that neonatal papillary and reticular fibroblast lineages have restricted functional fates (Driskell et al. 2013; Mascharak et al. 2021; Plikus et al. 2017). For example, neonatal papillary fibroblast lineages do not differentiate into adipocytes, while neonatal reticular fibroblasts are restricted from forming dermal papilla. We hypothesized that the chromatin accessibility profiles would define the functional lineage restriction in neonatal fibroblasts. To investigate this hypothesis, we performed a differential accessibility analysis on the P0 scATAC-seq data for all fibroblasts clusters and examined the corresponding gene expression profiles using our scRNA-seq data (Figure 4a–c). Surprisingly, even though each cluster possessed a distinct chromatin accessibility profile, only the pre-adipocyte cluster contained uniquely accessible chromatin (Figure 4a). The pre-adipocyte cell cluster contained chromatin specifically accessible for adipogenic genes such as Adipoq, Fabp4, Adig, and Fabp12 (Figure 4c). Remarkably, the expression of these genes was not detectable in the scRNA-seq fibroblast clusters, suggesting that chromatin accessibility may be utilized to define cell fate before gene expression (Figure 4b). Gene Ontology (GO) Analysis of the accessible genes from each fibroblast cluster revealed common GO terms between dermal papilla and papillary lineages indicating associated functions, while also revealing the associated functions of adipocytes with the reticular fibroblast lineages (Supplementary Table S4). All other fibroblast clusters revealed differential accessibility profiles that were not specificto the cluster. For example, the dermal papilla cluster revealed highly accessible chromatin for Stmn2, Smoc1, Bmp3, Sobp, Lamc3, and Prex2 with lower detectable peaks in all other fibroblastclusters. In addition, RNA Velocity analysis of P0 fibroblasts revealed distinct fates for dermal papilla and adipocytes (Supplementary Figure S3). Furthermore, analysis of highly accessible genes utilizing clustered peak maps (Figure 4d) revealed that chromatin accessibility is a gradient across the clusters, with the most apparent gradients detected between clusters that were the furthest apart onthe UMAP (Figure 4d).
Figure 4: Differential chromatin accessibility analysis of fibroblast heterogeneity.
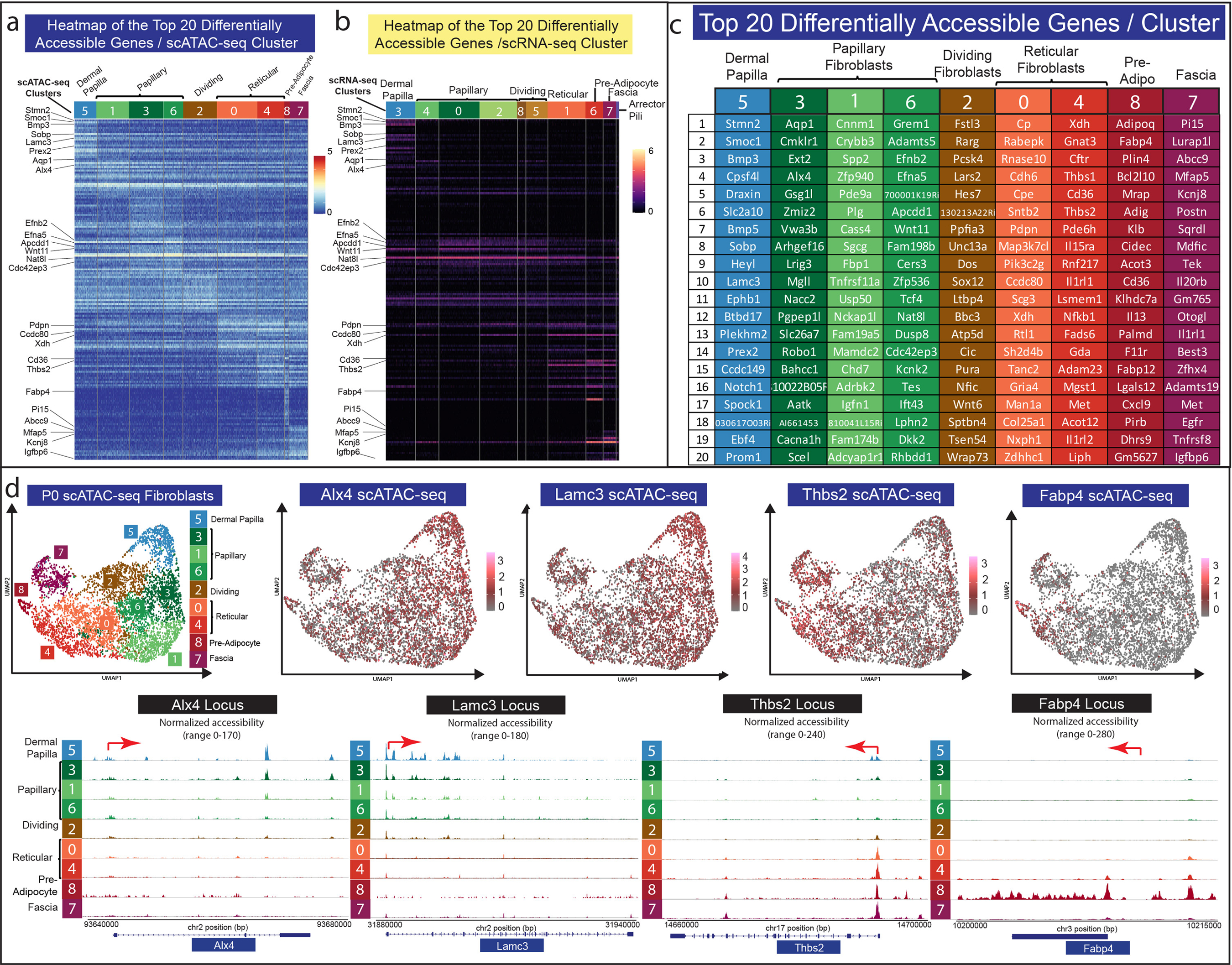
(a) Heatmap of top 20 differentially accessible genes from each fibroblast cluster. (b) Heatmap of the expression of the top 20 differentially accessible genes from each fibroblast cluster. (c) Gene list of top 20 differentially accessible clusters for each cluster. (d) Chromatin accessibility analysis of Alx4, Lamc3, Thbs2, and Fabp4.
Our investigation of the accessibility profiles of myofibroblast markers in neonatal fibroblasts has revealed that the different fibroblast subtypes could support a myofibroblast state (Figure 5a). Interestingly, recent publications investigating chromatin accessibility in fibroblast populations of wounds have revealed different subpopulations of fibroblasts associated with myofibroblast markers (Abbasi et al. 2020; Foster et al. 2021). Consequently, whether thetransition to a myofibroblast state leads to a permanent myofibroblast fate with chromatinremodeling will require further investigation using multi-omics during wound healing and in tissue with fibrosis.
Figure 5: Model of fibroblast lineages, states, and fates for P0 neonatal skin.
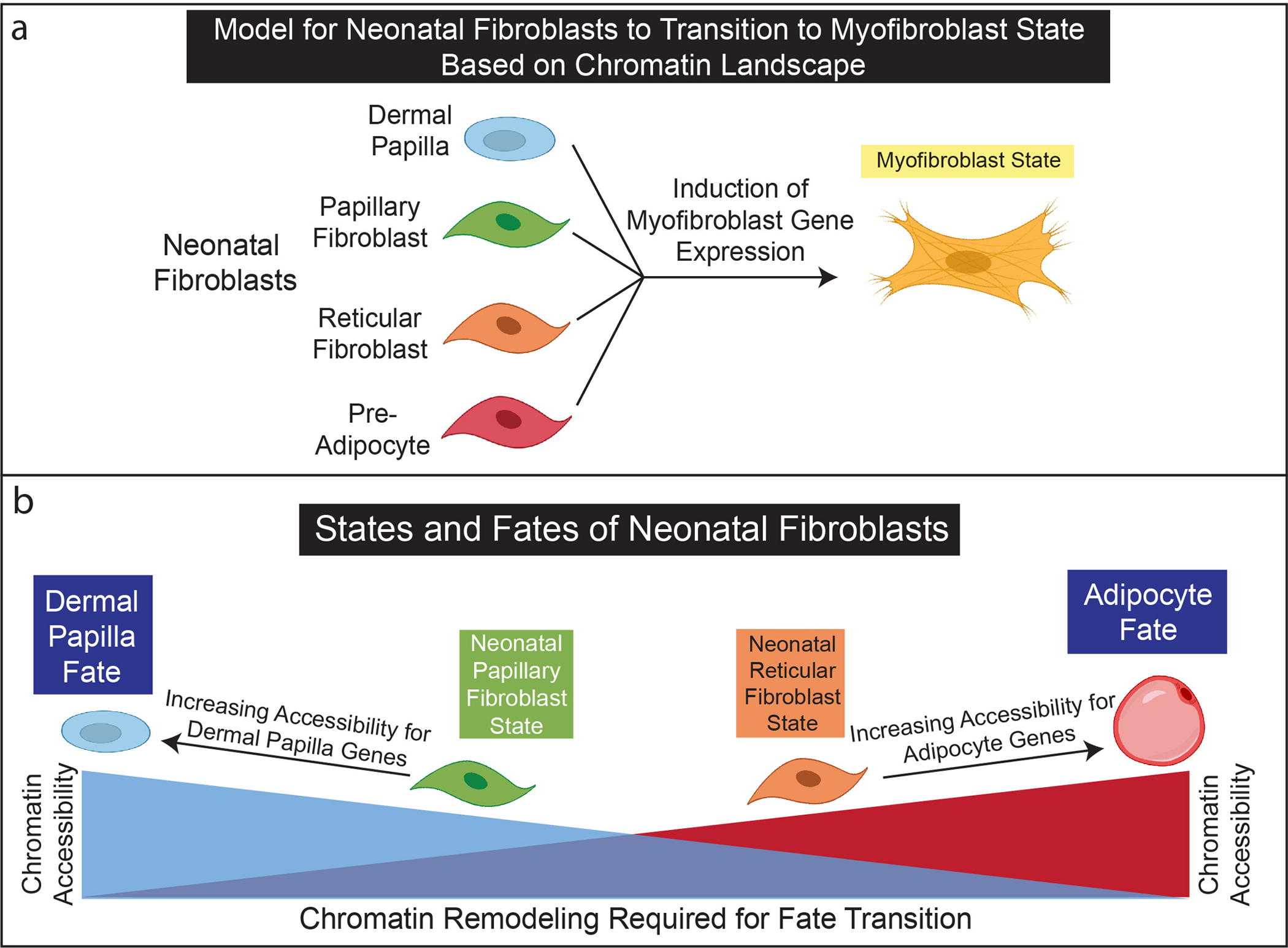
(a) Neonatal fibroblasts may transition to myofibroblasts states without rearranging chromatin landscapes. (b) The chromatin landscapes of neonatal papillary and reticular fibroblasts are poised to differentiate into dermal papilla and adipocytes. The rearrangement of chromatin landscapes between neonatal and papillary fibroblast lineages may support functional lineage restrictions.
We propose a model of neonatal fibroblast states and fates in the context of fibroblast lineages (Figure 5). It is widely considered that the dermal papilla is a cellular fate, which can be uniquely identified in neonatal skin through their support of hair follicle development (Mok et al. 2019). In addition, pre-adipocytes and their precursors are thought to develop during late embryogenesis (Driskell et al. 2014; Rivera-Gonzalez et al. 2014; Wojciechowicz et al. 2013). Our scATAC-seq analysis revealed that these fibroblast fates have distinct and exclusive chromatin landscapes. However, the neonatal papillary and reticular fibroblasts are cellular states with similar but varying degrees of chromatin accessibility reflecting a gradient toward the different specialized fates that support fibroblast lineage restriction.
A interactive webtool to share multi-omics data on https://skinregeneration.org/.
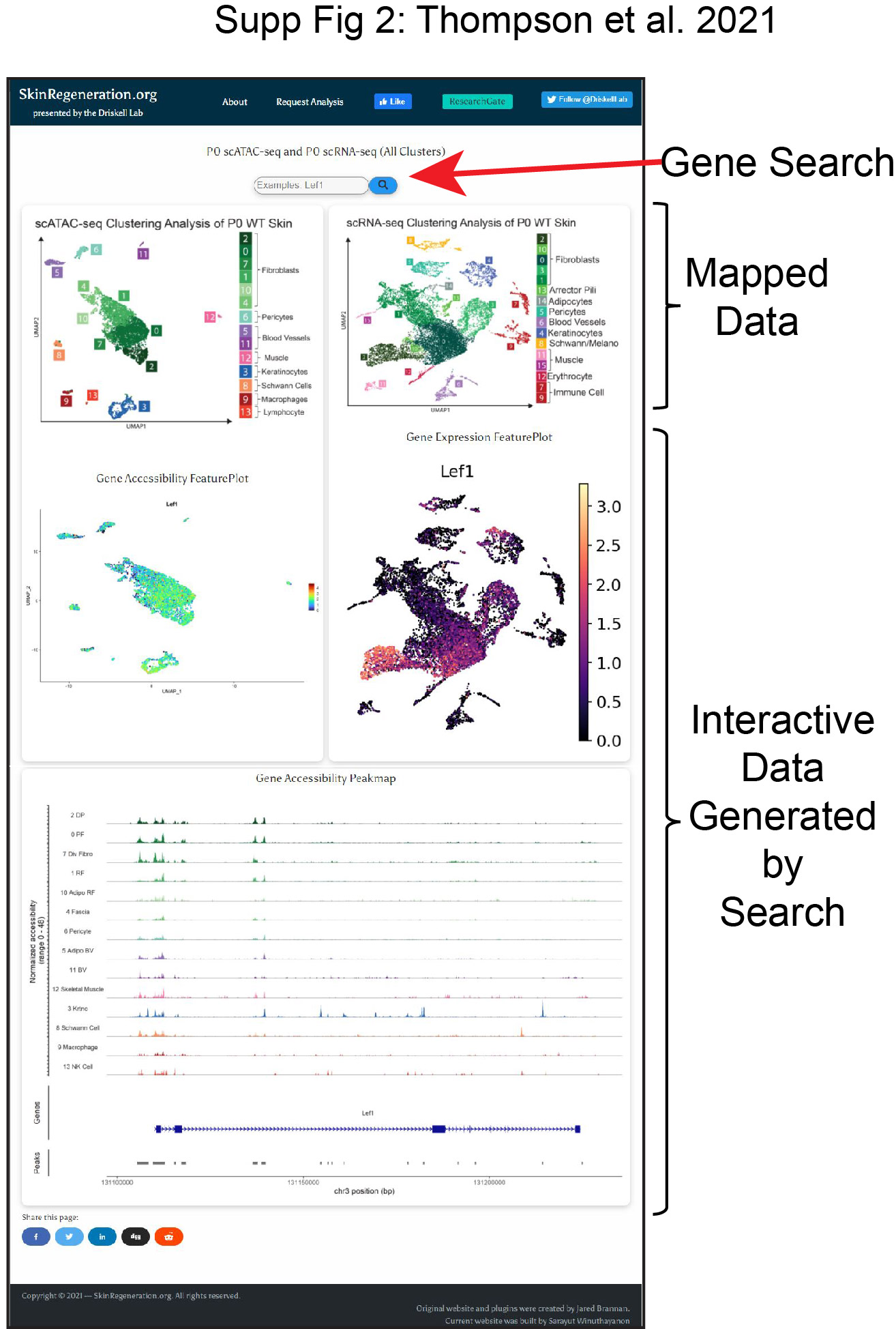
Single-cell multi-omics analysis provides an unprecedented amount of critically important data that can require computer programming skills to simply query the large datasets (Phan et al. 2021). However, the advent of cloud based webtools has the potential for providing a platform for easy gene searches across vast datasets without the user having prior coding knowledge (Joost et al. 2020; Sennett et al. 2015; Phan et al. 2021). Here we generated a webtool that allows for simple gene searches across both scRNA-seq and scATAC-seq datasets. This multi-omics webtoolexists at the following webpage: https://skinregeneration.org/scatacseq-and-scrnaseq-data-from-thompson-et-al-2021-2/. One webpage was generated for the analysis of all the cell types in our P0 skin preparation alongside one page for just the fibroblast subset. The pages consist of a search bar placedabove static images of labeled UMAPs, with interactive feature plots and clustered peak maps generated by the search function (Supplementary Figure S2). Importantly, since our datasets were generated in parallel from the same sample preparation, we present them as a non-integrated analysis. This website will be a useful tool to understand the cell states and potential fates of neonatal cells in skin.
Discussion
In this study, we utilized a parallel multi-omics approach to provide an understanding of the molecular underpinnings of fibroblast lineage restriction, cell states, and fates in neonatal skin. Neonatal skin has regenerative properties that allow for hair follicle reformation upon wounding, which is lost during the skin maturation process (Phan et al. 2020; Rognoni et al. 2016; Telerman et al. 2017). Importantly, the skin maturation process may render fibroblast populations less plastic as a consequence of development and aging (Salzer et al. 2018). In addition, neonatal fibroblasts are the critical component to support regenerating hair follicles and arrector pili muscles in skin grafting assays and during wound healing (Driskell et al. 2013; Ge et al. 2020; Jensen et al. 2010). Consequently, understanding the transcriptome and chromatin architecture profiles of the cell types in neonatal skin will provide a platform for understanding how to transform aging adult skin to be regenerative (Gomes et al. 2021; Phan et al. 2020; Plikus et al. 2021).
Our multi-omics analysis of a single time point for neonatal fibroblast lineages revealed that both papillary and reticular lineages have distinct but non-exclusive chromatin profiles. Although cell clusters in scRNA-seq and scATAC-seq could be mapped to each other through computational correlation analysis (Figure 2c), chromatin accessibility in fibroblast clusters did not always reflect gene expression. Gene expression was associated with accessible chromatin, but accessible chromatin did not always translate to gene expression. This was particularly evident when investigating neonatal fibroblast lineage markers, in addition to myofibroblast markers. For example, Cd26/Dpp4 has been reported to be expressed specifically in the regenerative neonatal papillary fibroblast lineage but also in scarring fibroblasts of the skin (Driskell et al. 2013; Rinkevich et al. 2015). Our analysis revealed that many neonatal fibroblast marker expression, including Cd26/Dpp4, were not restricted at the chromatin architecture level, but rather seem to be controlled by transcription factor networks or extrinsic signaling factors (Figure 2). Recent publications investigating chromatin accessibility of Cd26/Dpp4, Acta2/asma, and Col1a1 have suggested that chromatin remodeling may occur in fibroblasts during wound repair with differential accessibility in heterogenous fibroblasts in wounds (Abbasi et al. 2020; Foster et al. 2021). Our data also revealed that chromatin accessibility of 50% of GO-Terms associated with myofibroblast markers across multiple fibroblast subpopulations, and that the expression of these genes was not restricted at the chromatin accessibility level. Consequently, additional scATAC-seq studies of adult time points and reanalysis of published wound healing studies (Abbasi et al. 2020; Foster et al. 2021) are now required to understand chromatin remodeling that may occur during skin maturation and during wound healing and regeneration.
Our multi-omics analysis in neonatal skin also revealed two specialized fibroblast fates defined by distinct chromatin accessibility landscapes: the dermal papilla and the pre-adipocyte. These cell types in neonatal skin have distinct functions, either to support hair follicle formation or adipogenesis (Driskell et al. 2013). Interestingly, our analysis revealed that neonatal dermal papilla and pre-adipocytes also shared a strong affinity with their respected lineage precursors (Figure 2, 4). For example, the top genes that defined the dermal papilla chromatin accessibility profile such as Lamc3, Alx4, Bmp3, Sobp, and Draxin, were also found to have increased accessibility in the neonatal papillary fibroblasts. In contrast, the chromatin accessibility profile of pre-adipocytes and fascia share highly accessible chromatin with the reticular clusters,such as Thbs2, Pdpn, Xdh and Ly6a, with the expansion of accessible chromatin to Fabp4, Adipoq, Adig, Fabp12, and Cd36 in pre-adipocytes. Therefore, neonatal reticular fibroblasts are poised to follow the adipogenic programming while neonatal papillary fibroblasts may have the potential to differentiate into dermal papilla. Consequently, we propose that fate restricted fibroblast lineages are based on the amount of chromatin remodeling required to convert between dermal papilla and pre-adipocytes fates. Our results also support the previously reported de-differentiation of specialized cell states into myofibroblasts, as we have shown that the transition among different cell states is permissible by the chromatin architecture (Plikus et al. 2017; Shook et al. 2020; Shook et al. 2016). However, additional multi-omics studies of different conditions are necessary to understand the dynamic equilibrium between the transitional cell states and fates in adult skin, wound healing, and disease.
Materials and Methods
Generation of single cell suspension and single nuclei suspensions from the skin of post-natal-day 0 (P0) mice for scRNA-seq and scATAC-seq analysis.
We utilized C57B16 mice to produce pregnant females. All animal procedures performed in this study were in accordance with Washington State University IACUC approved protocols. Once the females gave birth, we immediately harvested the neonatal mice and processed the skin of neonates as previously described to generate single cell suspension ofthe dermis (Jensen et al. 2010). To generate scRNA-seq data we processed our single cell preparation directly for the 10X Genomics Chromium Single Cell 3’kit V3 to generate sequencinglibraries aiming for over 10,000 cells. Utilizing the same dermal cell preparation, we processed single nuclei according to the instructions aiming for over 10,000 nuclei to be processed utilizinga 10x Genomics Chromium kit.
scRNA-seq and scATAC-seq data pre-processing.
scRNA-seq and scATAC-seq libraries were sequenced on a NovaSeq600 (100 bp Paired-End). Demultiplexed Paired-End Fastq files were aligned to the mm10-based reference genome using 10X Genomics cellranger function (CellRanger version 5.0.0 for scRNA-seq and cellranger-atac 1.2.0 for scATAC-seq). The alignment outputs were used in downstream computational analysis.
scRNA-seq analysis.
We analyzed the P0 scRNA-seq data using the Seurat package in R (Stuart et al. 2019) and published on our github website (https://github.com/DriskellLab/Thompson-et-al.-2021) (Stuart et al. 2020). Utilizing the Seurat pipeline we filtered out cells that have expressed less than 200 genes and genes that are expressed in less than 3 cells. We also normalized and scaled the data using default arguments for Seurat’s recommended SCTransform function. To generate the scRNA-seq UMAPs we utilized the standard Seurat pipeline while clustering was performed with 1:30 PCs at a resolution of 0.5 with the SLM algorithm. Differential expression analysis on the identified clusters was performed using the FindMarkers function with a min.pct argument of 0.05 and logfold change threshold of 0.0. Genes were then ranked based on a score of the logfold change multiplied by the ratio of pct.1 to pct.2, which resulted in the selection of the top 5% of differentially expressed genes. Top 20 differentially expressed genes were found by filtering the top 5% of differentially expressed genes to genes with pct.1 column greater than or equal to 0.5 to identify differentially expressed genes that were expressed in more than 50% of the cells in the cluster.
scATAC-seq data analysis.
The computational pipeline for our scATAC-seq analysis can be found on the Driskell lab Github webpage (https://github.com/DriskellLab/Thompson-et-al.-2021). Briefly, we processed the data by performing filtering steps involving peak fragments, percent reads, blacklist ratios, nucleosome signals, and passed filters test (https://github.com/DriskellLab/Thompson-et-al.-2021). After performing the filtering steps, a gene activity matrix was constructed by computing the average reads across the gene body and 2000bp upstream of the promoters, following Signac default parameters. All variable features (variable peaks) were used for dimensional reduction via Latent Semantic Indexing. The UMAP was generated using PCs 2:30 (because PC1 had a strong correlation to sequencing depth) with a resolution of 0.3. Differential accessibility was performed using the gene activity matrix. Differentially accessible genes were determined and ranked using the same method as with scRNA-seq data’s differentially expressed genes, and the Top 20 were determined by filtering based on pct.1 column greater than or equal to 0.66 for all the clusters and 0.5 for the fibroblast subset.
scRNA-seq and scATAC-seq correlation analysis.
To generate the integration heatmaps, we used the list of the top 5% differentially expressed genes from the scRNA-seq data to extract the scaled accessibility score from the scATAC-seq dataset. To do this we utilized the “DotPlot()” function in Seurat allowing us to generate a dataframe that contained the average relative accessibility scores for each gene across each scATAC cluster for the top 5% differentially expressed genes from the scRNA-seq data. These values were plotted as a heatmap. See Driskell lab Github for computational pipeline.
Supplementary Material
(a-c) Analysis of dermal sheath (Acan), fascia (Plac8), and myofibroblast markers (Adam12, Comp, Postn, and Col11a1) markers in scRNA-seq and scATAC-seq data. (d-f) scATAC-seq analysis of myofibroblast and fibrotic markers.
{kind=link}
Representative webpage for webtool at skinregeneration.org with static images, and interactive plots based on the search function. The webtool can be accessed at: https://skinregeneration.org/scatacseq-and-scrnaseq-data-from-thompson-et-al-2021-2/
{kind=link}
(a) RNA velocity UMAP projection of neonatal fibroblast. (b) Single gene velocity and expression analysis of the top 5 accessible genes from each scATAC-seq cluster.
{kind=link}
(a) Correlation plot of top 5% of scATAC-seq for each cluster genes plotted with gene expression data.
{kind=link}
Differentially expressed genes from scRNA-seq clusters utilized for correlation analysis.
Top5 genes were highlighted from scRNA-seq data utilized for correlation analysis.
List of the 50% of the genes from myofibroblast markers contained open chromatin in fibroblast populations.
Investigation for GO terms for specifically accessible genes for each fibroblast cluster
Acknowledgements
RRD is supported by NIH NIAMS Grants: R01AR078743-01 and R56AR073778-01A1. SMT is supported by the Barry Goldwater Scholarship. QP is supported by a Poncin Research Fellowship. The authors wish to acknowledge Blanca Biladeau Lopez, the Oatley Lab, and the Washington State University Molecular Biology and Genomics Core for assistance with the 10x Genomics single-cell platform. The authors would also like to acknowledge Jared Brannan, Jasson Makkar, and Dr. Nate Law fordiscussions and feedback. The illustrations in the figures were created using Bio-Render. The sequencing was performed at UC San Diego IGM Genomics Center utilizing an Illumina NovaSeq6000 that was purchased with funding from National Institutes of Health SIG grant (#S10 ODO26929).
Footnotes
Publisher's Disclaimer: This is a PDF file of an article that has undergone enhancements after acceptance, such as the addition of a cover page and metadata, and formatting for readability, but it is not yet the definitive version of record. This version will undergo additional copyediting, typesetting and review before it is published in its final form, but we are providing this version to give early visibility of the article. Please note that, during the production process, errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
The authors declare no conflict of interest.
Data availability.
We have generated a web-resource to share the data generated from these studies. The data can easily be queried at: https://skinregeneration.org/scatacseq-and-scrnaseq-data-from-thompson-et-al-2021-2/. Datasets related to this article can be found at https://www.ncbi.nlm.nih.gov/gds/?term=GSE189210, hosted at NIH GEO Datasets. (Edgar et al. 2002). Our source code can be found on our Github webpage https://github.com/DriskellLab/Thompson-et-al.-2021.
References
- Abbasi S, Sinha S, Labit E, Rosin NL, Yoon G, Rahmani W, et al. Distinct Regulatory Programs Control the Latent Regenerative Potential of Dermal Fibroblasts during Wound Healing. Cell Stem Cell 2020;27(3):396–412.e6 [DOI] [PubMed] [Google Scholar]
- Ascensión AM, Fuertes-Álvarez S, Ibañez-Solé O, Izeta A, Araúzo-Bravo MJ. Human dermal fibroblast subpopulations are conserved across single-cell RNA sequencing studies. J Invest Dermatol Elsevier; 2020;0(0) Available from: https://www.jidonline.org/article/S0022-202X(20)32403-9/abstract [DOI] [PubMed] [Google Scholar]
- Belote RL, Le D, Maynard A, Lang UE, Sinclair A, Lohman BK, et al. Human melanocyte development and melanoma dedifferentiation at single-cell resolution. Nat Cell Biol 2021;23(9):1035–47 [DOI] [PubMed] [Google Scholar]
- Brewer CM, Nelson BR, Wakenight P, Collins SJ, Okamura DM, Dong XR, et al. Adaptations in Hippo-Yap signaling and myofibroblast fate underlie scar-free ear appendage wound healing in spiny mice. Dev Cell 2021;56(19):2722–2740.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 2013;10(12):1213–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Wu B, Litzenburger UM, Ruff D, Gonzales ML, Snyder MP, et al. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 2015;523(7561):486–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauveau C, Rowell J, Ferreiro A. A rising titan: TTN review and mutation update. Hum Mutat 2014;35(9):1046–59 [DOI] [PubMed] [Google Scholar]
- Cho H, Kozasa T, Bondjers C, Betsholtz C, Kehrl JH. Pericyte-specific expression of Rgs5: implications for PDGF and EDG receptor signaling during vascular maturation. FASEB J 2003;17(3):440–2 [DOI] [PubMed] [Google Scholar]
- Collins CA, Kretzschmar K, Watt FM. Reprogramming adult dermis to a neonatal state through epidermal activation of β-catenin. Development Oxford University Press for The Company of Biologists Limited; 2011;138(23):5189–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng C-C, Hu Y-F, Zhu D-H, Cheng Q, Gu J-J, Feng Q-L, et al. Single-cell RNA-seq reveals fibroblast heterogeneity and increased mesenchymal fibroblasts in human fibrotic skin diseases. Nat Commun 2021;12(1):3709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driskell RR, Jahoda CAB, Chuong C-M, Watt FM, Horsley V. Defining dermal adipose tissue. Exp Dermatol 2014;23(9):629–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driskell RR, Lichtenberger BM, Hoste E, Kretzschmar K, Simons BD, Charalambous M, et al. Distinct fibroblast lineages determine dermal architecture in skin development and repair. Nature 2013;504(7479):277–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driskell RR, Watt FM. Understanding fibroblast heterogeneity in the skin. Trends in Cell Biology 2015;25(2):92–9 [DOI] [PubMed] [Google Scholar]
- Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 2002. Jan 1;30(1):207–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enshell-Seijffers D, Lindon C, Morgan BA. The serine protease Corin is a novel modifier of the Agouti pathway. Development 2008;135(2):217–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster DS, Januszyk M, Yost KE, Chinta MS, Gulati GS, Nguyen AT, et al. Integrated spatial multiomics reveals fibroblast fate during tissue repair. Proc Natl Acad Sci U S A 2021;118(41):e2110025118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gay D, Ghinatti G, Guerrero-Juarez CF, Ferrer RA, Ferri F, Lim CH, et al. Phagocytosis of Wnt inhibitor SFRP4 by late wound macrophages drives chronic Wnt activity for fibrotic skin healing. Science Advances American Association for the Advancement of Science; 2020;6(12):eaay3704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge Y, Miao Y, Gur-Cohen S, Gomez N, Yang H, Nikolova M, et al. The aging skin microenvironment dictates stem cell behavior. Proc Natl Acad Sci U S A 2020;117(10):5339–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes RN, Manuel F, Nascimento DS. The bright side of fibroblasts: molecular signature and regenerative cues in major organs. NPJ Regen Med 2021;6(1):43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin MF, Borrelli MR, Garcia JT, Januszyk M, King M, Lerbs T, et al. JUN promotes hypertrophic skin scarring via CD36 in preclinical in vitro and in vivo models. Sci Transl Med 2021;13(609):eabb3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin MF, desJardins-Park HE, Mascharak S, Borrelli MR, Longaker MT. Understanding the impact of fibroblast heterogeneity on skin fibrosis. Dis Model Mech 2020;13(6) Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7328159/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrero-Juarez CF, Dedhia PH, Jin S, Ruiz-Vega R, Ma D, Liu Y, et al. Single-cell analysis reveals fibroblast heterogeneity and myeloid-derived adipocyte progenitors in murine skin wounds. Nat Commun 2019;10(1):650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta K, Levinsohn J, Linderman G, Chen D, Sun TY, Dong D, et al. Single-Cell Analysis Reveals a Hair Follicle Dermal Niche Molecular Differentiation Trajectory that Begins Prior to Morphogenesis. Dev Cell 2019;48(1):17–31.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He H, Suryawanshi H, Morozov P, Gay-Mimbrera J, Del Duca E, Kim HJ, et al. Single-cell transcriptome analysis of human skin identifies novel fibroblast subpopulation and enrichment of immune subsets in atopic dermatitis. J Allergy Clin Immunol 2020;145(6):1615–28 [DOI] [PubMed] [Google Scholar]
- Jensen KB, Driskell RR, Watt FM. Assaying proliferation and differentiation capacity of stem cells using disaggregated adult mouse epidermis. Nat Protoc 2010;5(5):898–911 [DOI] [PubMed] [Google Scholar]
- Jiang D, Correa-Gallegos D, Christ S, Stefanska A, Liu J, Ramesh P, et al. Two succeeding fibroblastic lineages drive dermal development and the transition from regeneration to scarring. Nat Cell Biol 2018;20(4):422–31 [DOI] [PubMed] [Google Scholar]
- Jiang D, Rinkevich Y. Distinct fibroblasts in scars and regeneration. Curr Opin Genet Dev 2021;70:7–14 [DOI] [PubMed] [Google Scholar]
- Joost S, Annusver K, Jacob T, Sun X, Dalessandri T, Sivan U, et al. The Molecular Anatomy of Mouse Skin during Hair Growth and Rest. Cell Stem Cell 2020;26(3):441–457.e7 [DOI] [PubMed] [Google Scholar]
- Lim CH, Sun Q, Ratti K, Lee S-H, Zheng Y, Takeo M, et al. Hedgehog stimulates hair follicle neogenesis by creating inductive dermis during murine skin wound healing. Nat Commun 2018;9 Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6249328/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascharak S, desJardins-Park HE, Davitt MF, Griffin M, Borrelli MR, Moore AL, et al. Preventing Engrailed-1 activation in fibroblasts yields wound regeneration without scarring. Science 2021;372(6540):eaba2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mok K-W, Saxena N, Heitman N, Grisanti L, Srivastava D, Muraro M, et al. Dermal Condensate Niche Fate Specification Occurs Prior to Formation and Is Placode Progenitor Dependent. Dev Cell 2019;48(1):32–48.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonaka D, Chiriboga L, Rubin BP. Sox10: a pan-schwannian and melanocytic marker. Am J Surg Pathol 2008;32(9):1291–8 [DOI] [PubMed] [Google Scholar]
- Phan QM, Driskell IM, Driskell RR. The Three Rs of Single-Cell RNA Sequencing: Reuse, Refine, and Resource. J Invest Dermatol 2021;141(7):1627–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan QM, Fine GM, Salz L, Herrera GG, Wildman B, Driskell IM, et al. Lef1 expression in fibroblasts maintains developmental potential in adult skin to regenerate wounds. Elife 2020;9:e60066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philippeos C, Telerman SB, Oulès B, Pisco AO, Shaw TJ, Elgueta R, et al. Spatial and Single-Cell Transcriptional Profiling Identifies Functionally Distinct Human Dermal Fibroblast Subpopulations. J Invest Dermatol 2018;138(4):811–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plikus MV, Guerrero-Juarez CF, Ito M, Li YR, Dedhia PH, Zheng Y, et al. Regeneration of fat cells from myofibroblasts during wound healing. Science 2017;355(6326):748–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plikus MV, Wang X, Sinha S, Forte E, Thompson SM, Herzog EL, et al. Fibroblasts: Origins, definitions, and functions in health and disease. Cell 2021;184(15):3852–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahmani W, Abbasi S, Hagner A, Raharjo E, Kumar R, Hotta A, et al. Hair follicle dermal stem cells regenerate the dermal sheath, repopulate the dermal papilla, and modulate hair type. Dev Cell 2014;31(5):543–58 [DOI] [PubMed] [Google Scholar]
- Rinkevich Y, Walmsley GG, Hu MS, Maan ZN, Newman AM, Drukker M, et al. Skin fibrosis. Identification and isolation of a dermal lineage with intrinsic fibrogenic potential. Science 2015;348(6232):aaa2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinwa P, Calvo-Enrique L, Zhang M-D, Nyengaard JR, Karlsson P, Ernfors P. Demise of nociceptive Schwann cells causes nerve retraction and pain hyperalgesia. Pain 2021;162(6):1816–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera-Gonzalez G, Shook B, Horsley V. Adipocytes in skin health and disease. Cold Spring Harb Perspect Med 2014;4(3):a015271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robb L, Hartley L, Wang CC, Harvey RP, Begley CG. musculin: a murine basic helix-loop-helix transcription factor gene expressed in embryonic skeletal muscle. Mech Dev 1998;76(1–2):197–201 [DOI] [PubMed] [Google Scholar]
- Rognoni E, Gomez C, Pisco AO, Rawlins EL, Simons BD, Watt FM, et al. Inhibition of β-catenin signalling in dermal fibroblasts enhances hair follicle regeneration during wound healing. Development 2016;143(14):2522–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryncarz RE, Anasetti C. Expression of CD86 on human marrow CD34(+) cells identifies immunocompetent committed precursors of macrophages and dendritic cells. Blood 1998;91(10):3892–900 [PubMed] [Google Scholar]
- Salzer MC, Lafzi A, Berenguer-Llergo A, Youssif C, Castellanos A, Solanas G, et al. Identity Noise and Adipogenic Traits Characterize Dermal Fibroblast Aging. Cell 2018;175(6):1575–1590.e22 [DOI] [PubMed] [Google Scholar]
- Sennett R, Wang Z, Rezza A, Grisanti L, Roitershtein N, Sicchio C, et al. An Integrated Transcriptome Atlas of Embryonic Hair Follicle Progenitors, Their Niche, and the Developing Skin. Developmental Cell Elsevier; 2015;34(5):577–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shook B, Rivera Gonzalez G, Ebmeier S, Grisotti G, Zwick R, Horsley V. The Role of Adipocytes in Tissue Regeneration and Stem Cell Niches. Annu Rev Cell Dev Biol 2016;32:609–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shook BA, Wasko RR, Mano O, Rutenberg-Schoenberg M, Rudolph MC, Zirak B, et al. Dermal Adipocyte Lipolysis and Myofibroblast Conversion Are Required for Efficient Skin Repair. Cell Stem Cell 2020;26(6):880–895.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shook BA, Wasko RR, Rivera-Gonzalez GC, Salazar-Gatzimas E, López-Giráldez F, Dash BC, et al. Myofibroblast proliferation and heterogeneity are supported by macrophages during skin repair. Science 2018;362(6417):eaar2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solé-Boldo L, Raddatz G, Schütz S, Mallm J-P, Rippe K, Lonsdorf AS, et al. Single-cell transcriptomes of the human skin reveal age-related loss of fibroblast priming. Commun Biol 2020;3(1):188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soler D, Chapman TR, Poisson LR, Wang L, Cote-Sierra J, Ryan M, et al. CCR8 expression identifies CD4 memory T cells enriched for FOXP3+ regulatory and Th2 effector lymphocytes. J Immunol 2006;177(10):6940–51 [DOI] [PubMed] [Google Scholar]
- Sorrell JM, Caplan AI. Fibroblast heterogeneity: more than skin deep. Journal of Cell Science The Company of Biologists Ltd; 2004;117(5):667–75 [DOI] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM, et al. Comprehensive Integration of Single-Cell Data. Cell 2019;177(7):1888–1902.e21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T, Srivastava A, Lareau C, Satija R. Multimodal single-cell chromatin analysis with Signac. bioRxiv 2020;2020.11.09.373613 [Google Scholar]
- Stuart T, Srivastava A, Madad S, Lareau CA, Satija R. Single-cell chromatin state analysis with Signac. Nat Methods 2021;18(11):1333–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabib T, Morse C, Wang T, Chen W, Lafyatis R. SFRP2/DPP4 and FMO1/LSP1 Define Major Fibroblast Populations in Human Skin. J Invest Dermatol 2018;138(4):802–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telerman SB, Rognoni E, Sequeira I, Pisco AO, Lichtenberger BM, Culley OJ, et al. Dermal Blimp1 Acts Downstream of Epidermal TGFβ and Wnt/β-Catenin to Regulate Hair Follicle Formation and Growth. J Invest Dermatol 2017;137(11):2270–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C Defining cell types and states with single-cell genomics. Genome Res 2015;25(10):1491–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorstandlechner V, Laggner M, Kalinina P, Haslik W, Radtke C, Shaw L, et al. Deciphering the functional heterogeneity of skin fibroblasts using single-cell RNA sequencing. FASEB J 2020;34(3):3677–92 [DOI] [PubMed] [Google Scholar]
- Wojciechowicz K, Gledhill K, Ambler CA, Manning CB, Jahoda CAB. Development of the mouse dermal adipose layer occurs independently of subcutaneous adipose tissue and is marked by restricted early expression of FABP4. PLoS One 2013;8(3):e59811. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(a-c) Analysis of dermal sheath (Acan), fascia (Plac8), and myofibroblast markers (Adam12, Comp, Postn, and Col11a1) markers in scRNA-seq and scATAC-seq data. (d-f) scATAC-seq analysis of myofibroblast and fibrotic markers.
Representative webpage for webtool at skinregeneration.org with static images, and interactive plots based on the search function. The webtool can be accessed at: https://skinregeneration.org/scatacseq-and-scrnaseq-data-from-thompson-et-al-2021-2/
(a) RNA velocity UMAP projection of neonatal fibroblast. (b) Single gene velocity and expression analysis of the top 5 accessible genes from each scATAC-seq cluster.
(a) Correlation plot of top 5% of scATAC-seq for each cluster genes plotted with gene expression data.
Differentially expressed genes from scRNA-seq clusters utilized for correlation analysis.
Top5 genes were highlighted from scRNA-seq data utilized for correlation analysis.
List of the 50% of the genes from myofibroblast markers contained open chromatin in fibroblast populations.
Investigation for GO terms for specifically accessible genes for each fibroblast cluster
Data Availability Statement
We have generated a web-resource to share the data generated from these studies. The data can easily be queried at: https://skinregeneration.org/scatacseq-and-scrnaseq-data-from-thompson-et-al-2021-2/. Datasets related to this article can be found at https://www.ncbi.nlm.nih.gov/gds/?term=GSE189210, hosted at NIH GEO Datasets. (Edgar et al. 2002). Our source code can be found on our Github webpage https://github.com/DriskellLab/Thompson-et-al.-2021.