

Abstract
Relevance
Spliceosome machinery plays important roles in cell biological processes, and its alterations are significantly associated with cancer pathophysiological processes and contribute to the entire healthcare process in the framework of predictive, preventive, and personalized medicine (PPPM/3P medicine).
Purpose
To understand the expression and mutant status of spliceosome genes (SGs) in common malignant tumors and their relationship with clinical characteristics, a pan-cancer analysis of these SGs was performed across 27 cancer types in 9070 patients to discover biomarkers for cancer early diagnosis and prognostic assessment, effectively stratify patients, and improve the survival and prognosis of patients in 3P medical practice.
Methods
A total of 150 SGs were collected from the KEGG database. The Python and R language were combined to process the transcriptional data of SGs and clinical data of 27 cancer types in The Cancer Genome Atlas (TCGA) database. Mutations of SGs in 27 cancer types were analyzed to identify the most common mutated SGs, as well as survival-related SGs. Different SGs were screened out, and SGs with survival significance in different types of tumors were found. Furthermore, TCGA and GTEx datasets were used to further confirm the expressions of SGs in different tumors. Western blot assay was performed to verify the expression of SNRPB protein in colon cancer and lung adenocarcinoma. Three SGs were screened out to establish the Bagging model for tumor diagnosis.
Results
Among 150 SGs, THOC2, PRPF8, SNRNP200, and SF3B1 had the highest mutation rate. The survival time of mutant THOC2 and SF3B1 was better than that of wild type, respectively. The differential expression analysis of 150 SGs between 674 normal tissue samples and 9,163 tumor tissue samples with 27 cancer types of 9070 patients showed that 13 SGs were highly expressed and 1 was low-expressed. For all cancer types, the prognosis (survival time) of the low-expression group of three SGs (SNRPB, LSM7, and HNRNPCL1) was better than the high expression group, respectively (p < 0.05). Cox hazards model showed that male, over 60 years old, clinical stages III–IV, and with highly expressed SNRPB and HNRNPCL1 had a poor prognosis. GEPIA2 website analysis showed that SNRPB and LSM7 were highly expressed in most tumors but not in LAML, showing low expression. Compared with the control group, the expression of SNRPB protein in colon cancer was increased by Western blot (p < 0.05). Enrichment analysis showed that the differential SGs were mainly enriched in RNA splicing and binding. The average error of 10-fold cross-validation of the Bagging model for diagnosed cancer was 0.093, which demonstrates that the Bagging model can effectively diagnose cancer with a small error rate.
Conclusions
This study provided the first landscape of spliceosome changes across 27 cancer types in 9070 patients and revealed that spliceosome was related to tumor progression. Spliceosome may play important an important role in cancer biological processes. These findings are the important scientific data to demonstrate the common and specific changes of spliceosome genes across 27 cancer types, which is a valuable biomarker resource to under the common or specific molecular mechanisms among different cancer types and establish biomarkers and therapeutic targets for the common or specific management of different types of cancer patients to benefit the research and practice of 3P medicine in cancers.
Supplementary Information
The online version contains supplementary material available at 10.1007/s13167-022-00279-0.
Keywords: Spliceosome, Spliceosome genes (SGs), GEPIA2, Mutation, Bagging model, Pan-cancer analysis, Survival time, Prognosis, Biomarker, Therapeutic target, Predictive preventive personalized medicine (PPPM/3P medicine)
Introduction
Structure and functions of spliceosome
Alternative splicing of RNA is a process in which the precursor mRNA (pre-mRNA) is processed into mature mRNA. During mRNA maturation, introns are removed and exons are joined to form mRNA as a template for protein translation. The alterative splicing process is completed with the participation of the spliceosomes [1], which cut introns from pre-mRNA by a two-step transesterification reaction. Spliceosomes are polyribonucleoprotein micronucleosomes composed of protein-associated small nuclear RNAs (snRNAs) that are responsible for removing introns from pre-mRNA and producing mature mRNA. There are five snRNAs (U1, U2, U4, U5, and U6), which bind to a number of proteins to form five corresponding small nuclear ribonucleoprotein particles (snRNPs; U1, U2, U4, U5, and U6). These snRNPs further assemble with many non-snRNPs to form splicesome complexes [2], which act like a master transcriptome tailor [3, 4]. To date, mass spectrometry has identified more than 200 co-protein factors that interact with human spliceosome complexes [5]. RNA splicing is a basic process of gene maturation in eukaryotes, and accurate RNA splicing is essential for cell survival [6]. The spliceosome protein complex is composed of more than 100 proteins, among which the core components are U2 snRNP proteins, U2A0, U2B00, and splicing factor 3A and 3B sub-complexes (SF3A and SF3B) [7].
Associations of spliceosome and cancers
Core components of the spliceosome affect RNA processing of specific genes to varying degrees [8]. During tumor progression, carcinogenic splicing events may occur [9]. Abnormal expression of splicing factor can promote the occurrence and development of human malignant tumors. For example, USP39, a component of the spliceosome, is often overexpressed in high-grade serous ovarian cancer, and an elevated USP39 level is associated with poor prognosis, which is a potential therapeutic target for ovarian cancer [10]. The UNC5B splicing isoform, known as UNC5B-Δ8, is abnormally expressed in the colon cancer vascular system and is associated with tumor angiogenesis and poor patient outcomes [11]. Mutations in the core components of the spliceosome are associated with cell- or tissue-specific phenotypes and diseases such as cancer [12]. Damage to many oncogenes leads to deregulate RNA splicing, often resulting in tumor hypersensitivity to targeted therapy of spliceosomes [13]. Abnormal splicing is an important source of novel cancer biomarkers, and the spliceosomal mechanism is a novel and attractive target for drug therapy [14]. SF3B1 is an important splicing factor, overexpressed in HCC, involved in the progression of cancer cells, or can be a therapeutic target for HCC [15]. With the use of spliceosome as a therapeutic target, many small molecule inhibitors have been developed, and some of them have entered clinical trials. Antisense oligonucleotides have been widely used to successfully target mRNA molecules to disrupt splicing and achieve the goal of antitumor therapy [16]. Spliceosome-targeting therapy has become an effective anticancer strategy for cancer patients with splicing defects [12]. Clinical drugs targeting KRAS4A splicing can effectively inhibit tumor stem cells [17].
Working hypothesis
RNA splicing in gene regulation and alterations in this spliceosome pathway have been implicated in many human cancers, which has been evidenced with large-scale genomic studies to uncover a spectrum of splicing machinery mutations that contribute to tumorigenesis [5]. It demonstrates that spliceosome genes (SGs) play important roles in cancers, and SG changes and its regulatory factors can affect the occurrence and development of cancer. However, there might be common alterations in SGs among different cancer types and also specific changes in SGs for a given cancer type. We hypothesize that the SG pattern changes among different cancer types to obtain the common and/or specific SG alterations, which will be the potential targets to establish biomarkers for patient stratification, predictive diagnosis, prognostic assessment, and personalized medical services, and develop therapeutic drugs for targeted prevention and personalized therapy in cancer.
Study design
The transcriptomics data of SGs combined with clinical information were collected from 9070 patients with 27 cancer types in TCGA database. The expression difference and mutation pattern of SGs were analyzed across 27 cancer types. The relationships between these SGs and survival time, SGs and clinical parameters were also analyzed, and also SG-mediated signaling pathways were studied.
Expected impacts in the framework of predictive, preventive, and personalized medicine
We expect that the changed SGs and signaling pathways are the important targets to construct clinical biomarkers for patient stratification, predictive diagnosis, prognostic assessment, and personalized medical services and develop therapeutic drugs for targeted prevention and personalized therapy to guide management of cancer patients in the context of predictive, preventive, and personalized medicine (3P medicine (PPPM)). PPPM is an effective strategy to improve treatment outcomes and patient prognosis [18]. PPPM needs to use a variety of effective molecular biomarkers, including early diagnosis and prognosis biomarkers, which can help clinicians identify patients who need early treatment [19]. Specifically speaking, we expect to identify important SGs and build a mathematical model that can diagnose cancer and make a good diagnosis. Therefore, with the reduction of sequencing cost, the establishment of artificial intelligence model can diagnose tumors and serve for tumor prediction and prevention. We also expect to find high-expression SGs associated with poor prognosis of cancer and use these altered SGs as potential targets for cancer treatment.
Materials and methods
Samples and datasets
Transcriptional, mutation, and clinical data of 27 cancer types in 21 anatomical sites from 9070 patients were obtained from The Cancer Genome Atlas (TCGA) website (Table 1). These cancer types were adrenocortical carcinoma (ACC) (cancer: n = 79; control: n = 0) and pheochromocytoma and paraganglioma (PCPG) (cancer: n =150; control: n = 3) in adrenal gland, bladder urothelial carcinoma (BLCA) in bladder (cancer: n = 409; control: n = 19), breast invasive carcinoma (BRCA) in breast (cancer: n = 1104; control: n = 113), cervical squamous cell carcinoma and endocervical adenocarcinoma (CESC) in cervix uteri (cancer: n = 306; control: n = 3), colon adenocarcinoma (COAD) in colon (cancer: n = 464; control: n = 41), esophageal carcinoma (ESCA) in esophagus (cancer: n = 160; control: n = 11), glioblastoma multiforme (GBM) (cancer: n = 168; control: n = 5) and brain lower-grade glioma (LGG) (cancer: n = 529; control: n = 0) in brain, pan-kidney cohort KICH (cancer: n = 65; control: n = 24) + KIRC (cancer: n = 535; control: n = 72) + KIRP (cancer: n = 289; control: n = 32) in kidney, acute myeloid leukemia (LAML) in hematopoietic system (cancer: n = 150; control: n = 0), laryngo carcinoma (HNSC) in larynx (cancer: n = 111; control: n = 12), liver hepatocellular carcinoma (LIHC) (cancer: n = 374; control: n = 50) and cholangiocarcinoma (CHOL) (cancer: n = 32; control: n = 8) in liver, lung adenocarcinoma (LUAD) (cancer: n = 526; control: n = 59) and lung squamous cell carcinoma (LUSC) (cancer: n = 501; control: n = 49) in lung, ovarian serous cystadenocarcinoma (OV) in ovary (cancer: n = 379; control: n = 0), pancreatic adenocarcinoma (PAAD) in pancreas (cancer: n = 178; control: n = 4), prostate adenocarcinoma (PRAD) in prostate (cancer: n = 499; control: n = 52), rectum adenocarcinoma (READ) in rectum(cancer: n = 95; control: n = 3), skin cutaneous melanoma (SKCM) in skin (cancer: n = 471; control: n = 1), stomach adenocarcinoma (STAD) in stomach (cancer: n = 375; control: n = 32), testicular germ cell tumors (TGCT) in testis (cancer: n = 156; control: n = 0), thyroid carcinoma (THCA) in thyroid gland (cancer: n = 510; control: n = 58), and uterine corpus endometrial carcinoma (UCEC) in corpus uteri (cancer: n = 548; control: n = 23). The json file with clinical information was downloaded from TCGA database and used the Python language (https://www.python.org/) and import pandas (https://pandas.pydata.org/) data analysis package to extract important clinical information. A total of 150 SGs were collected from the KEGG database (https://www.kegg.jp/pathway/map03040). The “clusterProfiler” software package of function bitr() was used to convert the ENTREZID or Ensembl of the genes into the SYMBOL type of the genes (Supplemental Table 1). The specific order is: bitr(geneID, fromType=“SYMBOL”, toType=c(“ENTREZID”,”ENSEMBL”), OrgDb=“org.Hs.eg.db”, drop = TRUE).
Table 1.
A total of 9070 patients including 9163 cancer tissues and 674 control tissues were analyzed
| Cancer types | Tumors | Controls |
|---|---|---|
| ACC | 79 | 0 |
| BLCA | 409 | 19 |
| BRCA | 1104 | 113 |
| CESC | 306 | 3 |
| CHOL | 32 | 8 |
| COAD | 464 | 41 |
| ESCA | 160 | 11 |
| GBM | 168 | 5 |
| HNSC | 111 | 12 |
| KICH | 65 | 24 |
| KIRC | 535 | 72 |
| KIRP | 289 | 32 |
| LAML | 150 | 0 |
| LGG | 529 | 0 |
| LIHC | 374 | 50 |
| LUAD | 526 | 59 |
| LUSC | 501 | 49 |
| OV | 379 | 0 |
| PAAD | 178 | 4 |
| PCPG | 150 | 3 |
| PRAD | 499 | 52 |
| READ | 95 | 3 |
| SKCM | 471 | 1 |
| STAD | 375 | 32 |
| TGCT | 156 | 0 |
| THCA | 510 | 58 |
| UCEC | 548 | 23 |
| Total | 9163 | 674 |
Mutation analysis of SGs
R packet “maftools” (https://www.bioconductor.org/packages/release/bioc/html/maftools.html) was used to analyze the mutations of 150 SGs. The different groups was compared, and we found SGs with different mutation rates. The short survival group was defined as an eventual survival of less than 2 years. The long survival group was defined as ultimate survival greater than 2 years. The mutations of each SG were compared between the long- and short-survival groups and between the male and female groups. The mutation rates of the two groups were compared by the mafcompare() function. The forestPlot() function was used for the forest analysis diagram and drawing between the two groups. MafSurvGroup() function completed the survival analysis and graph drawing of gene or genome mutation. If the mutation rate of the gene is greater than 5%, the survival comparison between the mutant and the wild types is analyzed.
Correlation analysis of SGs and age
The correlation was analyzed between SGs and age. The cor() function calculates the correlation coefficient. The correlation coefficient was calculated with the cor.test() function with the significance level of p < 0.05. If the correlation coefficient was positive or negative 0.1–0.3, it was considered a weak correlation; if the correlation coefficient was 0.3–0.5, it was considered a medium correlation; if the correlation coefficient was greater than 0.5, it was considered a strong correlation. Scatter plots of age and SGs were constructed with “ggplot2” packets (https://cran.r-project.org/web/packages/ggplot2/index.html).
Differential expression analysis of SGs
R language was used to load “edger” packets (https://www.bioconductor.org/packages/release/bioc/html/edgeR.html) for differential gene screening of SGs. An ANOVA-like test was done by glmQLFTest() function. The quasi-likelihood method was used for differential expression analyses of pan-cancer RNA-seq data with a stricter error rate control by accounting for the uncertainty in dispersion estimation. The criterion for screening gene differences was performed in SGs compared to the normal group. The up-regulated SG was defined with logFC>2/3, FC fold change >1.59 fold, and p value <0.05. The downregulated SG was defined with LogFC<-2/3, FC fold change <0.63 fold, and p value <0.05.
Standardization and heat map of differentially expressed SGs
The heat map of differentially expressed SGs (DESGs) was mapped with the “ComplexHeatmap” packet (https://www.bioconductor.org/packages/release/bioc/html/ComplexHeatmap.html) that mapped the genetic big data. First, all DESGs were normalized. For each gene expression in each sample, the formula X-min/Max-Min was used to standardize the corresponding data. X was for every observation. Min represented the minimum count of a gene in all samples. Max represented the maximum counts of a gene in all samples. Through such a transformation, the value of a gene was mapped to the (0, 1) interval. After the transformation, all samples of a gene data distributed between 0 and 1. There were 674 normal controls and 9163 cancers. Heatmap() function was used to draw gene heat map.
Functional enrichment analysis of DESGs
“ClusterProfiler” packet was used for enrichment analysis of DESGs, with the enrichGO function; for example, BP <-enrichGO (de, “org.Hs.eg.db”, ont=“BP”, pvalueCutoff=0.05). The Web-based Gene Set Analysis Toolkit (http://www.webgestalt.org/#) was used for enrichment analysis of DESGs as well.
Construction of machine learning models based on DESGs
The 14 DESGs screened above were included in the stepwise regression to further reduce the dimension. SPSS version 26 (IBM Corp, Armonk, NY) software was used for stepwise regression to screen meaningful DESGs. Stepwise regression was performed with a linear regression model. Linear regression equation: y = a*DESGs1 + b *DESGs2 + c *DESGs3……+ Cx (a, b, and c are parameters, and Cx is a constant term. p <0.05 is considered statistically significant after test). Finally, the above screened DESGs were used to establish machine learning model with Weka 3.8.5 software (https://www.cs.waikato.ac.nz/ml/weka/) to diagnose cancer, and 10-fold cross-validation was adopted to evaluate the diagnostic performance of the model. Three different types of machine learning models (Logical regression, J48, Bagging) were established based on DESGs to diagnose cancer. Through 10-fold cross-validation, the model with good diagnostic performance was selected. The average absolute error was calculated; the smaller the error, the better the model performance. If the mean absolute error was less than 0.2, the diagnostic performance of the model was considered to be good. If the mean absolute error was less than 0.1, the model was considered to be very superior. If the mean absolute error was less than 0.01, then the model was quite excellent and should even be considered for clinical application as it was comparable to the superior pathologist. The average accuracy of the prediction was also calculated [prediction = true positive / (true positive + false positive)].
Survival analysis of DESGs
The transcriptomic data of DESGs were divided into high-expression group (H group) and low-expression group (L group) based on the median. Each DESG was used for survival analysis between the H and L groups. The jointly screened three DESGs and the clinical information of patients were used for Cox multivariate survival analysis. The “survminer” packet (https://cran.r-project.org/web/packages/survminer/index.html) was used to perform univariate and multivariate survival analyses.
Verification of three selected DESGs with different datasets
The software GraphPad Prism 8 was applied to analyze and draw the bar graph of the three selected DESGs. To more accurately compare the expression differences of the three DESGs between cancer and control groups, 655 paired data were used to compare. The differences between the two groups were compared by a t-test of paired data. The three DESGs were further verified on the GEPIA2 website (http://gepia2.cancer-pku.cn/#analysis). The Cancer Group in the GEPIA2 site was from TCGA database; the control group consisted of normal samples from TCGA database and the Genotype Tissue Expression (GTEx) database (https://commonfund.nih.gov/GTex).
Western blot
A Western blot assay was used to verify the screened gene. There were eight pairs of specimens of colon cancer and adjacent control tissues, and 5 pairs of specimens of lung adenocarcinoma and adjacent control tissues, which were collected from Tai’an Central Hospital. The proteins extracted from eight colon cancer tissues were equally mixed as a colon cancer protein sample. The proteins extracted from eight control colon tissues were equally mixed as a control protein sample. The proteins extracted from five lung adenocarcinomas were equally mixed as lung adenocarcinoma protein sample. The proteins extracted from five control lung tissues were equally mixed as the control lung protein sample. Protein concentration was determined with bicinchoninic acid (BCA) method. SDS polyacrylamide gel electrophoresis was used to isolate proteins with a protein loading amount (30 μg) per lane. The separated proteins were transferred to a PVDF membrane and incubated with the primary antibodies (rabbit anti-human SNRPB antibody and rabbit anti-human actin antibody), followed by incubation with the second antibody from Proteintech (goat anti-rabbit antibody).
Results
Overall situation of tissue samples
In TCGA database, the cancer sequencing data from 9,084 patients across 27 cancer types in 21 anatomical sites were included in this study. Of them, 674 normal tissues and 9163 cancer tissues were taken from these 9070 patients and analyzed with RNA sequencing (Supplemental Table 2). Of them, 8,859 patients had detailed clinical parameters (Fig. 1a). The drawing of the alluval diagram is completed by online software rawgraphs (https://app.rawgraphs.io/). The ratio of female to male patients was 4681:4178. The ratio of patients ≤ 60 years old to patients > 60 years old was 4510:4349. Twelve of the BRCA patients were male, accounting for 1.12% of the total BRCA. There were 23 female patients with ESCA, accounting for 14.6% of the total ESCA. There were 20 female patients with larynx cancer, accounting for 18.0% of the total larynx cancer. It can be found that there is a sex bias in the three cancers mentioned above. Among patients with TGCT tumor, 98.5% were ≤ 60 years of age. Among CESC patients, 81.4% were ≤ 60 years old. Among the lung cancer patients, 71.9% patients were over 60 years old. It can be seen that in the 3 tumors mentioned above, the onset has an age tendency. Moreover, a percentage (88.5%) of patients with larynx cancer is in the III+VI stages, and a percentage (96.0%) of pancreatic cancer patients is in the I+II stages. Therefore, laryngeal and pancreatic cancers tend to have specific clinical stages.
Fig. 1.

a Alluvial diagram of clinical parameters of 8859 cancer patients. b Waterfall map of SG mutations
Genetic mutations of SGs associated with poor survival
The first four SGs with the highest mutation rate were THOC2, PRPF8, SF3B1, and SNRNP200 (Fig. 1b; Supplemental Table 3). Among them, the mutation rate of the THOC2 gene was the highest, reaching 6.69%. The most common type of mutation was a missense mutation. The most common type of base mutation was C > T. Three mutant SGs were involved in the RTK-RAS pathway (Supplementary Figure 1).
Compared to the short survival group (Supplemental Table 4), the SGs with different mutation rates were basically distributed in the long survival group (Supplemental Table 5). For example, the mutation rate of SGs THOC2, PRPF8, SF3B1, etc. in the long survival group was significantly higher than in the short survival group (Fig. 2, p < 0.05). However, no difference in the mutated SGs was found between male and female groups.
Fig. 2.

The difference in mutation rates between the long- and short-lived groups.
Survival analysis showed that the survival time of mutant THOC2, PRPF8, and SF3B1 were better than that of wild type, respectively (Supplementary Figure 1).
Correlation between SGs and age
Correlation analysis found that 32 SGs had a weak negative correlation with age (Supplemental Table 6). No SGs were found to be positively correlated with age. Two SGs that were best associated with age were HNRNPA1L2 and HSPA1L, with correlation coefficients −0.204 and −0.245, respectively (Fig. 3).
Fig. 3.

Heatmap of correlation coefficient between age and SGs
DESG profile
Among the 150 SGs, 14 statistically significant DESGs were identified between cancer and normal control groups, including 1 significantly downregulated SG (HSPA2), and 13 significantly upregulated SGs (RNU5A-1, RNVU1-18, RNU4-2, RNU4-1, RBMXL3, RNU1-3, RNVU1-7, RNU1-4, RBMXL2, RNU1-2, HNRNPCL1, SNRPB, and LSM7) in cancer group (Fig. 4; Supplemental Table 7).
Fig. 4.

Heatmap of expression levels of 14 DESGs after normalization. The red color indicates high expression, and the blue color indicates low expression
Functional characteristics of DESGs
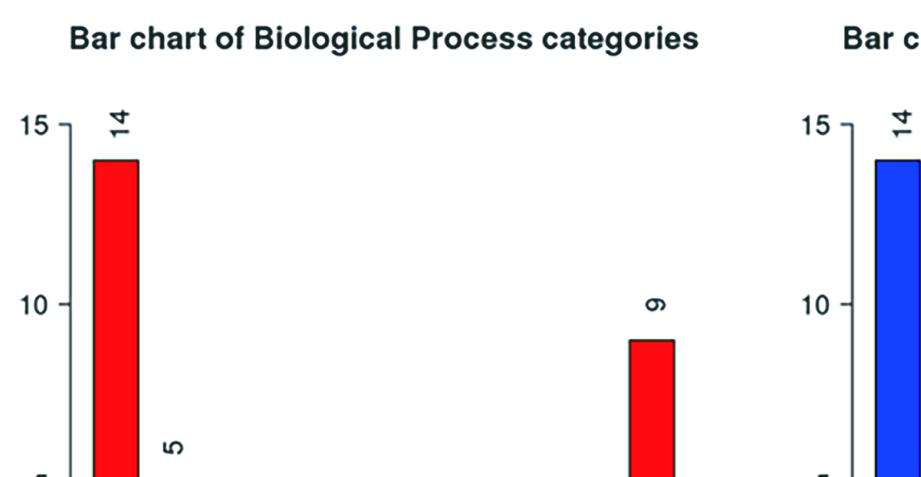
A total of 14 DESGs were used for GO enrichment analysis. For biological processes (BP), the main pathways involved in 14 DESGs were the RNA and mRNA splicing (Supplementary Figure 2). For cellular components (CC), the DESGs are mainly enriched in spliceosomal snRNP complex, small nuclear ribonucleoprotein complex, and Sm-like protein family complex (Supplementary Figure 2). For molecular function (MF), the DESGs were mainly enriched in pre-mRNA 5′-splice binding, snRNA binding, and pre-mRNA binding (Supplementary Figure 2).
GO Slim summary showed that BPs were mainly enriched in metabolic processes, CCs were mainly concentrated in the nucleus and protein-containing complex, and MFs were mainly concentrated in nucleic acid binding and protein binding functions (Supplementary Figure 3).
TRIM25 and EFTUD2 genes interacted with the most DESGs and were at the center of the network. This study found a direct interaction between SNRPB and TRIM25, SNRPB, and EFTUD2, respectively (Supplementary Figure 4).
Machine learning models of cancer diagnosis
A total of 14 DESGs were used for the stepwise regression. As a result, 3 DESGs (SNRPB, LSM7, and HSPA2) had significant characteristics. (i) For the Bagging model established with the three characteristic DESGs, 10-fold cross-validation showed that the mean absolute error was 0.094, the area under the ROC curve was 0.885, and the prediction accuracy was 0.923. (ii) For J48 (decision tree C4.5 algorithm) model (Supplementary Figure 4), 10-fold cross-validation showed that the average absolute error was 0.106, the area under the ROC curve was 0.790, and the prediction accuracy was 0.917. (iii) For the logical regression model, 10-fold cross-validation showed that the mean absolute error was 0.110, the area under the ROC curve was 0.821, and the prediction accuracy was 0.924.
Factors of poor prognosis
Excluding the samples with unknown follow-up time or zero follow-up time, a total of 8859 patients had complete sequencing data, follow-up time, and survival status (Supplemental Table 8).
Univariate survival analyses of these 14 DESGs in 8859 samples showed that the survival time of the H group of three DESGs (SNRPB, LSM7, and HNRNPCL1) was significantly different from that of the L group, and the survival time of the L group of these three DESGs was better than that of the H group (p < 0.05, Supplementary Figure 5). When specific to each cancer type, the results of survival analysis were inconsistent. The survival analysis of different cancer types showed that (i) for SNRPB, the survival of the L group of SNRPB was better than the H group in ACC, LIHC, KIRC, PIRP, and UCEC (Fig. 5); (ii) for LSM7, more cancer types had survival significance, and the survival of the L group of LSM7 was better than the H group in ACC, BLCA, CESC, KIRC, LIHC, PAAD, and STAD (Supplementary Figure 6a); and (iii) for HNRNPCL1, survival analysis of only KIRP had statistical significance between the L and H groups of HNRNPCL1 (Supplementary Figure 6b).
Fig. 5.

Survival analysis of SNRPB in different types of tumors in groups L and H. The genetic data of all the tumors were put together and divided into two groups according to the median value; the low value was group L, and the high value was group H
In 8859 samples, survival analysis was performed between the sexes regardless of the location of the cancer. It was found that, on the whole, the survival prognosis of women was better than that of men (Supplementary Figure 7a). If male and female sex-specific cancers were ruled out, such as prostate cancer, seminoma, breast cancer, and uterine cancer, survival analysis was carried out again. It was found that the prognosis time of female patients was still better than male patients (Supplementary Figure 7b).
A survival analysis was performed among different stages of cancer in 8859 samples. It was found that the survival time of cancer patients in stage I+II was significantly better than in stage III+IV (Supplementary Figure 7c) for most cancer types except for CHOL, PAAD, and TGCT. It means that the prognosis of advanced cancer was worse than that of early cancer for most cancer types except for CHOL, PAAD, and TGCT (Supplementary Figure 6c).
In 8859 samples, survival analysis was performed between patient ages (under 60 years old and over 60 years old). It was found that the survival of those under 60 years old was better than those over 60 years old (Supplementary Figure 7d).
Moreover, Cox proportional hazards model analysis found that being male, over 60 years old, clinical stages III–IV, and H group of SNRPB and HNRNPCL1 were the factors to indicate a poor prognosis of survival (Fig. 6, p < 0.05).
Fig. 6.

Multivariate survival analysis showed that male, over 60 years old, clinical stages III–IV, and high expression of SNRPB and HNRNPCL1, were risk factors for poor prognosis
Verification of three selected DESGs with different datasets
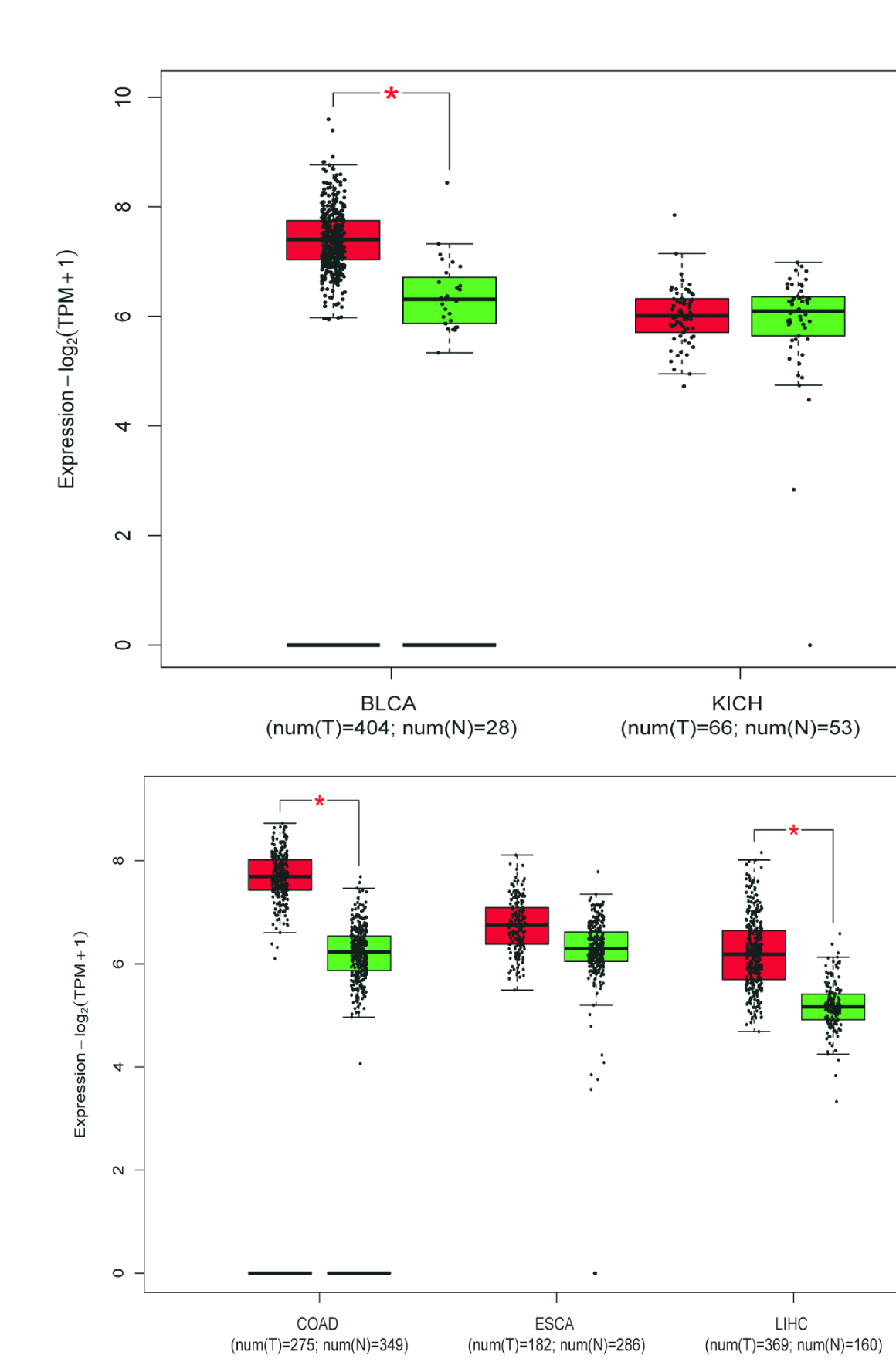
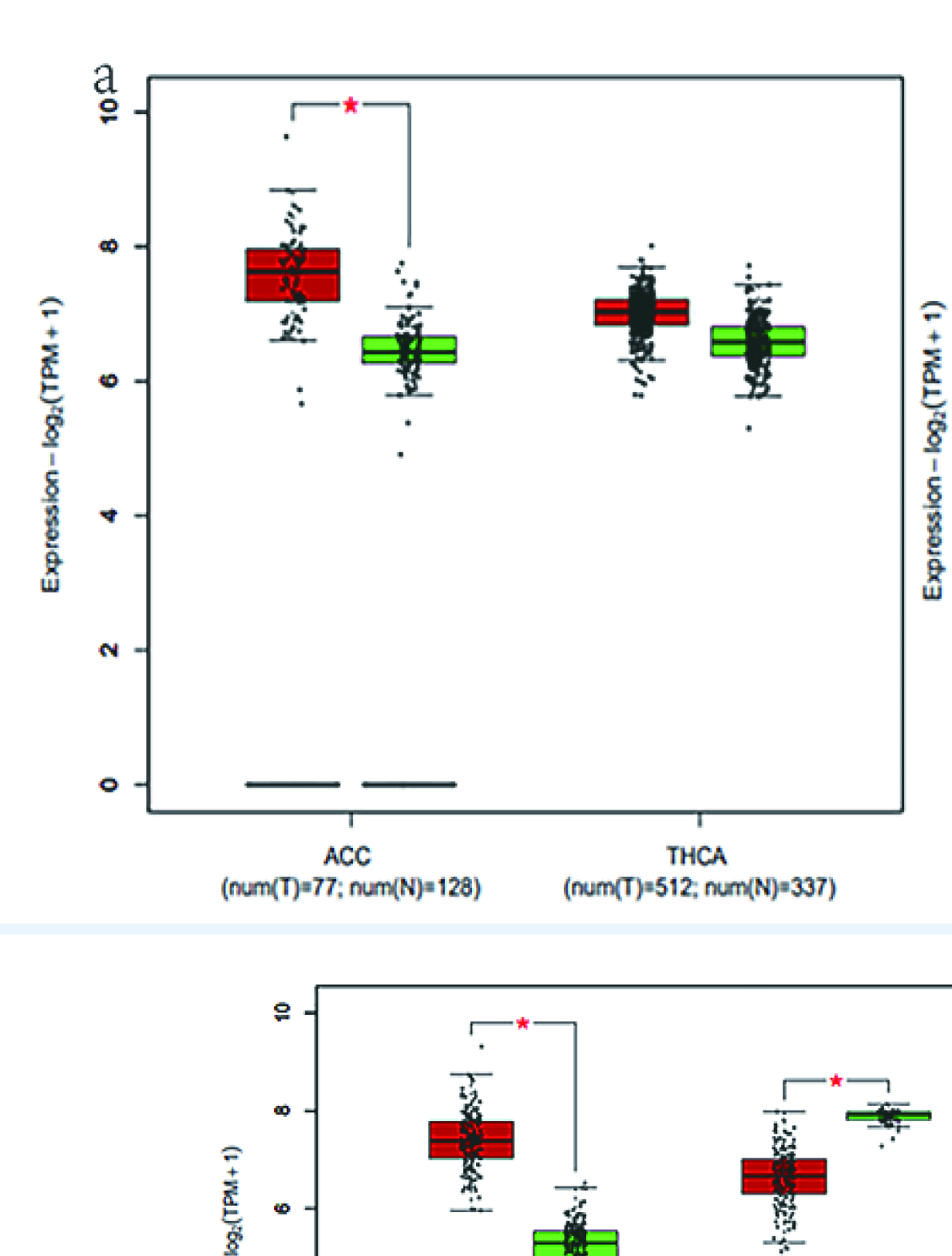
A total of 655 patients were compared who had sequencing data for both the cancer and normal groups. The expression levels of 3 DESGs (HNRNPCL1, LSM7, and SNRPB) in the cancer group were higher than those in the normal control group (p < 0.05) (Fig. 7), but HNRNPCL1 had a very low expression level. The GEPIA2 software was used to further verify these three DESGs, which divided cancer samples into six categories: digestive system, respiratory system, urinary system, gender-specific system, endocrine system, and other systems. Because the expression level of HNRNPCL1 was very low, no differences were found in each of the six systemic cancers. LSM7 gene expression was significantly higher in cancer types COAD, LIHC, PAAD, READ, STAD, LUSC, BLCA, TGCT, UCEC, ACC, LGG, and GBM compared to normal controls (Supplementary Figures 8–10). The SNRPB gene was highly expressed in most cancer types, including COAD, ESCA, LIHC, PAAD, READ, STAD, LUSC, BLCA, OV, TGCT, UCEC, ACC, LGG, and GBM (Supplementary Figures 8–10). However, surprisingly LSM7 and SNRPB were significantly lower expressed in LAML compared to controls.
Fig. 7.

Three DESGs (HNRNPCL1, LSM7, and SNRPB) were compared with paired data in 655 patients. The expression levels of 3 DESGs in cancer group were higher than that in the normal control group with t-test, respectively (p < 0.05).
Validation of DESGs at the protein level
The Western blot assay showed that SNRPB protein was highly expressed in colon cancers compared to controls, which was consistent with the transcriptome analysis (p < 0.05, Fig. 8a). However, there was no statistical difference in the expression of SNRPB proteins between lung adenocarcinomas and controls, which was also consistent with transcriptome analysis (Fig. 8b).
Fig. 8.

Western blot analysis of SNRPB in colon cancers and lung adenocarcinomas. a The expression of SNRPB protein was significantly higher in colon cancers compared to controls (p < 0.05). b No significant difference in SNRPB protein was found in lung adenocarcinomas compared to controls (p > 0.05).
Discussion
Roles of spliceosome in cancers
Abnormal splicing events have been observed in many types of cancer [12]. Elevated splicing factor expression is a strong predictor of poor clinical outcome in neuroblastoma [20]. Spliceosomal protein Eftud2 regulates inflammatory responses in macrophages and promotes tumorigenesis [21]. The core splicing factor SF3A3 translation leads to metabolic reprogramming and stem-like characteristics that promote tumorigenesis of MYC in vivo [22]. Alternative splicing mechanisms are prevalent in various cancers and drive the production and maintenance of various cancer characteristics such as proliferation enhancement, apoptosis inhibition, invasion, and metastasis [23]. Abnormal splicing plays an important role in the evolution of myeloproliferative tumors and might be a target for specific therapeutic strategies [24]. Most eukaryotes have two different pre-mRNA splicing mechanisms: one is the main spliceosome, which removes 99% of introns; the other is the small spliceosome, which removes the rare, evolutionarily conserved introns. Mutations in noncoding genes in small introns can disrupt splices and are potential cancer drivers [25].
The associations of spliceosome alterations with cancers
Spliceosomal mutations or misalignment of RNA splicing in cancer genes are increasingly recognized as markers of cancer [26]. During development, their relative levels vary by an order of magnitude in different tissues and in different cancer samples [27]. The expressions of relevant spliceosomal components and spliceosomal factors are severely misregulated (at mRNA and protein levels) in the characteristic cohort of human high-grade astrocytomas compared to healthy brain control samples, and SRSF3, RBM22, PTBP1, and RBM3 perfectly differentiate the tumors from the control samples [9]. SRSF3 is associated with patient survival and related tumor markers, and its silencing in vivo significantly reduces tumor development and progression, possibly through PDGFRB and related oncogenic signaling pathway PI3K-Akt/ERK [9]. SM08502 inhibits the phosphorylation of serine- and arginine-rich splicing factor (SRSF) and disrupts spliceosomal activity, which is involved in the inhibition of the expression of genes and proteins associated with the Wnt pathway, thereby inhibiting cancers [28]. Targeting spliceosome therapy leads to the formation of double-stranded RNA (dsRNAs) from intron transcripts, which activates mechanisms of tumor antiviral signaling and downstream adaptive immunity [29].
Targets associated with poor prognosis
In total, 150 SGs from the KEGG database were included in this study, with a few genes missing due to the limitations of the KEGG database. After screening of DESGs, a total of 14 statistically significant DESGs were identified. After survival analysis, 3 DESGs (SNRPB, LSM7, and HNRNPCL1) were found to be significantly associated with survival. However, since the HNRNPCL1 gene sequencing value was very low, the results might be unstable or even false positive. The expression of LSM7 and SNRPB genes was abundant, and the prognosis of its low-expression group was better than its high-expression group. It suggested that these two DESGs were involved in cancer progression. A novel post-transcriptional pathway such as PAT1-LSM (LSM1 to LSM7) mRNA-binding complex regulated autophagy [30]. The study also found that increased splicing factor expression was a strong predictor of poor clinical prognosis [20]. The LSM gene could regulate circadian rhythms in plants and mammals [8]. The study has shown that LSM family members play a key role in the progression of several malignant tumors [31]. SNRPB is the core part of the spliceosome and plays a key role in the pre-splicing of mRNA [32]. SNRPB was significantly upregulated in HCC. Elevated SNRPB expression was positively correlated with invasion of adjacent organs, tumor size, serum AFP level, and poor survival in HCC patients [33]. SNRPB could promote NSCLC tumorigenesis by regulating RAB26 expression [34]. Furthermore, TCGA matching data were used, including 655 normal subjects and 655 cancer subjects, which also found that 3 DESGs (SNRPB, LSM7, and HNRNPCL1) were highly expressed in the cancer group. The different datasets from the GEPIA2 database were used for further verification, which found that LSM7 and SNRPB were highly overexpressed in most cancer types. However, LSM7 and SNRPB were underexpressed in the blood tumor LAML cancer group. The specific reasons need to be further clarified why LSM7 and SNRPB have different expressions between solid tumors and blood tumors.
A study found that spliceosome mutations promoted tumorigenesis in coordination with gene mutations [35]. Mutations in the SG PPIL1 and PRP17 lead to neurodegenerative Pons cerebellar hypoplasia with microcephaly [36]. Mutation/loss of SG ZRSR2 in human myeloid cells resulted in impaired splicing of U12 introns. ZRSR2 mutations cause loss of its function, usually in the myelodysplastic syndrome [37]. Mutations of core splicosomal proteins (SRSF2, SF3B1, and U2AF1) occur frequently in many human cancers, especially in subtypes of leukemia [38]. Mutations of spliceosomal proteins or dysregulated expression of RNA-binding protein (RBP) splice factors lead to the emergence of abnormal splice mRNA transcriptomes that suppor cancer growth [5]. Mutation analysis showed that the most common type of SG mutation was missense mutations that lead to changes in translated amino acids. The most common base change was C>T, followed by C>A, which indicated that base C was very unstable, and especially the amino group of cytosine was easily oxidized. This study found that the SGs with different mutation rates almost all appeared in the long-term survival group. For example, the prognosis of SF3B1 and THOC2 mutation group was better than wild type. The possible reason was that the mutations of SGs lead to a decrease in carcinogenic ability. SF3B1, one of the spliceosome components, could bind and stabilize snRNA U2. The binding of U2 to branch points was very important for the recognition of splicing sites. Mutations of THOC2, PRPF8, SNRNP200, and SF3B1 can also lead to other diseases. Missense THOC2 variants, which affect evolutionarily conserved amino acid residues and reduce protein stability, can lead to human neurodevelopmental disorders [39]. PRPF8 and SNRNP200 mutation can also occur in autosomal dominant retinitis pigmentosa [40, 41]. The mutation of SF3B1 has been reported in more cases, such as myelodysplastic syndromes and uveal melanoma [42–45]. The incidence of SG genetic mutations was different between men and women [46]. However, no genetic differences in genetic mutation of SGs were found between the sexes.
Enrichment analysis of SGs showed that the main pathway involved in BP was RNA and mRNA splicing. A secondary enriched pathway was involved in the assembly of ribonucleoprotein complexes. These pathways were related to the formation and function of ribonucleoprotein complex. Protein interaction network analysis found that TRIM25 was the center of the network involved in DESGs, which suggests that TRIM25 is the hub molecule with important biological roles, and TRIM25 might be directly or indirectly involved in RNA splicing.
In addition, this study analyzed the changes of SGs between sexes, between cancer stages, and between ages. It found that when a large enough tumor sample size was included, women had better survival than men. Except for CHOL, PAAD, and TGCT tumors, the survival of the remaining tumors at stages III–IV was worse than that at stages I–II. It might be because the overall prognosis of CHOL, PAAD, and TGCT was poor due to the high degree of malignancy. The survival of cancer patients with <60 years of age was better than that of cancer patients with >60 years of age. RNA-seq analysis confirmed the selective splicing changes of SGs with age [47]. In this present study, 32 SGs were found to have a weak negative correlation with age, and no SGs were found to have a positive correlation with age. The expression level of some relevant SGs decreased gradually with the increase of age.
Predictive and preventive medicine: machine learning models for cancer diagnosis
Three DESGs (SNRPB, LSM7, and HSPA2) were screened out from 14 DESGs with the stepwise regression method. These three DESGs were used to build a machine learning model to diagnose cancer, no matter what type of cancer it was. The 10-fold cross-validation resulted in a very small mean error. The mean absolute error of the Bagging model was 0.094, which showed that the Bagging model was able to effectively diagnose cancer with a small error. With the continuous progress of artificial intelligence (AI) technology, machine learning methods have also been added to the diagnosis of diseases. Our genetic screening process of SGs provided useful insights into the future of AI in diagnosing disease. It can be seen that this machine learning model constructed with the selected DESGs can well predict the tumor, which can act as the prerequisite work for the prevention of tumor.
Personalized medicine: promising as a therapeutic target
Splicing isomers influenced the response of chemotherapeutic agents to anticancer therapy [48]. Understanding the molecular mechanisms that drive tumor formation and the underlying insights into cancer phenotypes required ones to go beyond DNA to investigate the effects of pre-mRNA treatment on cancer development and drug resistance. Small molecules of targeted spliceosome SF3B complex were effective inhibitors of cancer cell growth, which affected the assembly of spliceosome at an early stage [49]. It was believed that SGs were a promising therapeutic target for personalized treatment in the framework of PPPM.
Conclusion and expert recommendation in the context of 3P medicine
The pan-cancer analysis revealed the alteration landscape of spliceosome genes across 27 cancer types in 9070 patients, which demonstrated the common and specific changes of spliceosome genes among different cancer types. THOC2, PRPF8, SNRNP200, and SF3B1 were the high-mutation-rate SGs. Among them, the prognosis of mutant THOC2, PRPF8, and SF3B1 was better than wild type. Furthermore, this study found that SNRPB and LSM7 were highly expressed in multiple cancer types. In multisite cancers, high expression of SNRPB and LSM7 were associated with a poor prognosis. The Western blot assay confirmed that SNRPB protein was highly expressed in colon cancer compared with the control group (p < 0.05). Enrichment analysis showed that DESGs were mainly enriched in RNA and mRNA splicing pathways and were involved in RNA binding. HNRNPA1L2 and HSPA1L were negatively correlated with age. The Bagging model established with three DESGs (SNRPB, LSM7, and HSPA2) was able to effectively diagnose cancer.
We recommend focusing the study on spliceosome alterations in different cancer types. Spliceosome composition could affect RNA processing of specific genes, which could affect protein translation. This study found that SNRPB and LSM7 were highly expressed in many common tumors. It is worthy of further revealing how they participate in the occurrence and development of tumors. The present data demonstrate that spliceosomes play important roles in cancer pathogenesis. Integrative omics analysis of SGs will find the common and specific changes of SGs across different cancer types, which are resources to discover effective therapeutic targets to benefit personalized medical services and effective biomarkers for patient stratification, predictive diagnosis, and prognostic assessment in the research and practice of 3P medicine in cancers.
Supplementary Information
(PDF 1075 kb)
{kind=link}
(PNG 209 kb)
{kind=link}
(PNG 52 kb)
(PDF 2335 kb)
(PDF 852 kb)
(PDF 3358 kb)
(PDF 1098 kb)
{kind=link}
(PNG 166 kb)
(PDF 1632 kb)
{kind=link}
(PNG 172 kb)
(CSV 4 kb)
(CSV 6804 kb)
(CSV 1331 kb)
(CSV 186 kb)
(CSV 754 kb)
(CSV 268 kb)
(CSV 9 kb)
(CSV 951 kb)
Acknowledgements
The author acknowledge the financial support from the Shandong First Medical University Talent Introduction Funds (to X.Z.), Shandong First Medical University High-level Scientific Research Achievement Cultivation Funding Program (to X.Z.), the Shandong Provincial Natural Science Foundation (ZR202103020356/ZR2021MH156 to X.Z.), and the Academic Promotion Program of Shandong First Medical University (2019ZL002).
Abbreviations
- ACC
adrenocortical carcinoma
- AI
artificial intelligence
- BCA
bicinchoninic acid
- BLCA
bladder urothelial carcinoma
- BRCA
breast invasive carcinoma
- CESC
cervical squamous cell carcinoma and endocervical adenocarcinoma
- CHOL
cholangiocarcinoma
- COAD
colon adenocarcinoma
- ESCA
esophageal carcinoma
- GBM
glioblastoma multiforme
- GTEx
genotype tissue expression
- H group
high expression group
- KICH
kidney chromophobe
- KIRC
kidney renal clear cell carcinoma
- KIRP
kidney renal papillary cell carcinoma
- LAML
acute myeloid leukemia
- LGG
lower-grade glioma
- L group
low expression group
- LIHC
liver hepatocellular carcinoma
- LUAD
lung adenocarcinoma
- LUSC
lung squamous cell carcinoma
- OV
ovarian serous cystadenocarcinoma
- PAAD
pancreatic adenocarcinoma
- PCPG
pheochromocytoma and paraganglioma
- PRAD
prostate adenocarcinoma
- premRNA
precursor mRNA
- READ
rectum adenocarcinoma
- SGs
spliceosome genes
- SKCM
skin cutaneous melanoma
- SPL
spliceosomes
- STAD
stomach adenocarcinoma
- TCGA
The Cancer Genome Atlas
- TGCT
testicular germ cell tumors
- THCA
thyroid carcinoma
- UCEC
uterine corpus endometrial carcinoma
Author contributions
Z.Y. collected and analyzed data, prepared the figures and tables, and designed and wrote manuscript. A.B., S.Z., and S.Y participated in data analysis. X. Z. conceived the concept, designed manuscript, wrote and critically revised the manuscript, and was responsible for its corresponding works.
Funding
This work was supported by the Shandong First Medical University Talent Introduction Funds (to X.Z.), Shandong First Medical University High-level Scientific Research Achievement Cultivation Funding Program (to X.Z.), the Shandong Provincial Natural Science Foundation (ZR202103020356/ZR2021MH156 to X.Z.), and the Academic Promotion Program of Shandong First Medical University (2019ZL002).
Data availability
All data and materials are available in current manuscript and supplementary materials.
Code availability
The software application or custom code is provided in the current manuscript.
Declarations
Ethics approval
For Western blot analysis, the use of human tumor tissues has been approved by the Ethics Committee of Shandong First Medical University (Ethics Number: R202111050188 & 202201170002).
Consent to participate
Not applicable
Consent form publication
Not applicable
Conflict of interest
The authors declare no conflict of interest.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Lu B, Abdel-Wahab O. Promoting spliceosome assembly for therapeutic intent. Trends Pharmacol Sci. 2021. 10.1016/j.tips.2021.09.006. [DOI] [PubMed]
- 2.Plaschka C, Lin P, Charenton C, Nagai K. Prespliceosome structure provides insights into spliceosome assembly and regulation. Nature. 2018;559(7714):419–422. doi: 10.1038/s41586-018-0323-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhou Z, Gong Q, Wang Y, Li M, Wang L, Ding H, Li P. The biological function and clinical significance of SF3B1 mutations in cancer. Biomarker Res. 2020;8:38. doi: 10.1186/s40364-020-00220-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Borišek J, Casalino L, Saltalamacchia A, Mays S, Malcovati L, Magistrato A. Atomic-level mechanism of pre-mRNA splicing in health and disease. Acc Chem Res. 2021;54(1):144–154. doi: 10.1021/acs.accounts.0c00578. [DOI] [PubMed] [Google Scholar]
- 5.Wang E, Aifantis I. RNA splicing and cancer. Trends Cancer. 2020;6(8):631–644. doi: 10.1016/j.trecan.2020.04.011. [DOI] [PubMed] [Google Scholar]
- 6.Nguyen J, Drabarek W, Yavuzyigitoglu S, Medico Salsench E, Verdijk R, Naus N, de Klein A, Kiliç E, Brosens E. Spliceosome mutations in uveal melanoma. Int J Mol Sci. 2020;21(24). 10.3390/ijms21249546. [DOI] [PMC free article] [PubMed]
- 7.Coltri P, Dos Santos M, da Silva G. Splicing and cancer: challenges and opportunities. Wiley Interdisciplinary Reviews RNA. 2019;10(3):e1527. doi: 10.1002/wrna.1527. [DOI] [PubMed] [Google Scholar]
- 8.Perez-Santángelo S, Mancini E, Francey L, Schlaen R, Chernomoretz A, Hogenesch J, Yanovsky M. Role for LSM genes in the regulation of circadian rhythms. Proc Natl Acad Sci U S A. 2014;111(42):15166–15171. doi: 10.1073/pnas.1409791111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fuentes-Fayos A, Vázquez-Borrego M, Jiménez-Vacas J, Bejarano L, Pedraza-Arévalo S, L-López F, Blanco-Acevedo C, Sánchez-Sánchez R, Reyes O, Ventura S, Solivera J, Breunig J, Blasco M, Gahete M, Castaño J, Luque R. Splicing machinery dysregulation drives glioblastoma development/aggressiveness: oncogenic role of SRSF3. Brain. 2020;143(11):3273–3293. doi: 10.1093/brain/awaa273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang S, Wang Z, Li J, Qin J, Song J, Li Y, Zhao L, Zhang X, Guo H, Shao C, Kong B, Liu Z. Splicing factor USP39 promotes ovarian cancer malignancy through maintaining efficient splicing of oncogenic HMGA2. Cell Death Dis. 2021;12(4):294. doi: 10.1038/s41419-021-03581-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pradella D, Deflorian G, Pezzotta A, Di Matteo A, Belloni E, Campolungo D, Paradisi A, Bugatti M, Vermi W, Campioni M, Chiapparino A, Scietti L, Forneris F, Giampietro C, Volf N, Rehman M, Zacchigna S, Paronetto M, Pistocchi A, Eichmann A, Mehlen P, Ghigna C. A ligand-insensitive UNC5B splicing isoform regulates angiogenesis by promoting apoptosis. Nat Commun. 2021;12(1):4872. doi: 10.1038/s41467-021-24998-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang H, Beutler B, Zhang D. Emerging roles of spliceosome in cancer and immunity. Protein Cell. 2021. 10.1007/s13238-021-00856-5. [DOI] [PMC free article] [PubMed]
- 13.Bowling E, Wang J, Gong F, Wu W, Neill N, Kim I, Tyagi S, Orellana M, Kurley S, Dominguez-Vidaña R, Chung H, Hsu T, Dubrulle J, Saltzman A, Li H, Meena,J, Canlas G, Chamakuri S, Singh S, Simon L, Olson C, Dobrolecki L, Lewis M, Zhang B, Golding I, Rosen J, Young D, Malovannaya A, Stossi F, Miles G, Ellis M, Yu L, Buonamici S, Lin C, Karlin K, Zhang X, Westbrook T. Spliceosome-targeted therapies trigger an antiviral immune response in triple-negative breast cancer. Cell. 2021; 184 (2): 384-403.e21. 10.1016/j.cell.2020.12.031. [DOI] [PMC free article] [PubMed]
- 14.Sciarrillo R, Wojtuszkiewicz A, Assaraf Y, Jansen G, Kaspers G, Giovannetti E, Cloos J. The role of alternative splicing in cancer: from oncogenesis to drug resistance. Drug Resist Updat. 2020;53:100728. doi: 10.1016/j.drup.2020.100728. [DOI] [PubMed] [Google Scholar]
- 15.López-Cánovas J, Del Rio-Moreno M, García-Fernandez H, Jiménez-Vacas J, Moreno-Montilla M, Sánchez-Frias M, Amado V, L-López F, Fondevila M, Ciria R, Gómez-Luque I, Briceño J, Nogueiras R, de la Mata M, Castaño J, Rodriguez-Perálvarez M, Luque R, Gahete M. Splicing factor SF3B1 is overexpressed and implicated in the aggressiveness and survival of hepatocellular carcinoma. Cancer Lett. 2021;496:72–83. doi: 10.1016/j.canlet.2020.10.010. [DOI] [PubMed] [Google Scholar]
- 16.Fox R, Lytle N, Jaquish D, Park F, Ito T, Bajaj J, Koechlein C, Zimdahl B, Yano M, Kopp J, Kritzik M, Sicklick J, Sander M, Grandgenett P, Hollingsworth M, Shibata S, Pizzo D, Valasek M, Sasik R, Scadeng M, Okano H, Kim Y, MacLeod A, Lowy A, Reya T. Image-based detection and targeting of therapy resistance in pancreatic adenocarcinoma. Nature. 2016;534(7607):407–411. doi: 10.1038/nature17988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen W, To M, Westcott P, Delrosario R, Kim I, Philips M, Tran Q, Bollam S, Goodarzi H, Bayani N, Mirzoeva O, Balmain A. Targeting KRAS4A splicing through the RBM39/DCAF15 pathway inhibits cancer stem cells. Nat Commun. 2021;12(1):4288. doi: 10.1038/s41467-021-24498-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lu M, Zhan H, Liu B, Li D, Li W, et al. N6-methyladenosine-related non-coding RNAs are potential prognostic and immunotherapeutic responsiveness biomarkers for bladder cancer. EPMA J. 2021;12:589–604. doi: 10.1007/s13167-021-00259-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheng T, Zhan X. Pattern recognition for predictive, preventive, and personalized medicine in cancer. EPMA J. 2017;8:51–60. doi: 10.1007/s13167-017-0083-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shi Y, Yuan J, Rraklli V, Maxymovitz E, Cipullo M, Liu M, Li S, Westerlund I, Bedoya-Reina O, Bullova P, Rorbach J, Juhlin C, Stenman A, Larsson C, Kogner P, O'Sullivan M, Schlisio S, Holmberg J. Aberrant splicing in neuroblastoma generates RNA-fusion transcripts and provides vulnerability to spliceosome inhibitors. Nucleic Acids Res. 2021;49(5):2509–2521. doi: 10.1093/nar/gkab054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lv Z, Wang Z, Luo L, Chen Y, Han G, Wang R, Xiao H, Li X, Hou C, Feng J, Shen B, Wang Y, Peng H, Guo R, Li Y, Chen G. Spliceosome protein Eftud2 promotes colitis-associated tumorigenesis by modulating inflammatory response of macrophage. Mucosal Immunol. 2019;12(5):1164–1173. doi: 10.1038/s41385-019-0184-y. [DOI] [PubMed] [Google Scholar]
- 22.Cieśla M, Ngoc P, Cordero E, Martinez Á, Morsing M, Muthukumar S, Beneventi G, Madej M, Munita R, Jönsson T, Lövgren K, Ebbesson A, Nodin B, Hedenfalk I, Jirström K, Vallon-Christersson J, Honeth G, Staaf J, Incarnato D, Pietras K, Bosch A, Bellodi C. Oncogenic translation directs spliceosome dynamics revealing an integral role for SF3A3 in breast cancer. Mol Cell. 2021;81(7):1453–1468.e12. doi: 10.1016/j.molcel.2021.01.034. [DOI] [PubMed] [Google Scholar]
- 23.Du J, Zhu G, Cai J, Wang B, Luo Y, Chen C, Cai C, Zhang S, Zhou J, Fan J, Zhu W, Dai Z. Splicing factors: insights into their regulatory network in alternative splicing in cancer. Cancer Lett. 2021;501:83–104. doi: 10.1016/j.canlet.2020.11.043. [DOI] [PubMed] [Google Scholar]
- 24.Hautin M, Mornet C, Chauveau A, Bernard D, Corcos L, Lippert E. Splicing anomalies in myeloproliferative neoplasms: paving the way for new therapeutic venues. Cancers. 2020;12(8). 10.3390/cancers12082216. [DOI] [PMC free article] [PubMed]
- 25.Inoue D, Polaski J, Taylor J, Castel P, Chen S, Kobayashi S, Hogg S, Hayashi Y, Pineda J, El Marabti E, Erickson C, Knorr K, Fukumoto M, Yamazaki H, Tanaka A, Fukui C, Lu S, Durham B, Liu B, Wang E, Mehta S, Zakheim D, Garippa R, Penson A, Chew G, McCormick F, Bradley R, Abdel-Wahab O. Minor intron retention drives clonal hematopoietic disorders and diverse cancer predisposition. Nat Genet. 2021;53(5):707–718. doi: 10.1038/s41588-021-00828-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aird D, Teng T, Huang C, Pazolli E, Banka D, Cheung-Ong K, Eifert C, Furman C, Wu Z, Seiler M, Buonamici S, Fekkes P, Karr C, Palacino J, Park E, Smith P, Yu L, Mizui Y, Warmuth M, Chicas A, Corson L, Zhu P. Sensitivity to splicing modulation of BCL2 family genes defines cancer therapeutic strategies for splicing modulators. Nat Commun. 2019;10(1):137. doi: 10.1038/s41467-018-08150-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dvinge H, Guenthoer J, Porter P, Bradley R. RNA components of the spliceosome regulate tissue- and cancer-specific alternative splicing. Genome Res. 2019;29(10):1591–1604. doi: 10.1101/gr.246678.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tam B, Chiu K, Chung H, Bossard C, Nguyen J, Creger E, Eastman B, Mak C, Ibanez M, Ghias A, Cahiwat J, Do L, Cho S, Nguyen J, Deshmukh V, Stewart J, Chen C, Barroga C, Dellamary L, Kc S, Phalen T, Hood J, Cha S, Yazici Y. The CLK inhibitor SM08502 induces anti-tumor activity and reduces Wnt pathway gene expression in gastrointestinal cancer models. Cancer Lett. 2020;473:186–197. doi: 10.1016/j.canlet.2019.09.009. [DOI] [PubMed] [Google Scholar]
- 29.Ishak C, Loo Yau H, De Carvalho D. Spliceosome-targeted therapies induce dsRNA responses. Immunity. 2021;54(1):11–13. doi: 10.1016/j.immuni.2020.12.012. [DOI] [PubMed] [Google Scholar]
- 30.Gatica D, Hu G, Liu X, Zhang N, Williamson P, Klionsky D. The Pat1-Lsm complex stabilizes ATG mRNA during nitrogen starvation-induced autophagy. Mol Cell. 2019;73(2):314–324.e4. doi: 10.1016/j.molcel.2018.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ta H, Wang W, Phan N, An Ton N, Anuraga G, et al. Potential therapeutic and prognostic values of LSM family genes in breast cancer. Cancers. 2021;13. 10.3390/cancers13194902. [DOI] [PMC free article] [PubMed]
- 32.Liu N, Chen A, Feng N, Liu X, Zhang L. SNRPB is a mediator for cellular response to cisplatin in non-small-cell lung cancer. Med Oncol. 2021;38(5):57. doi: 10.1007/s12032-021-01502-0. [DOI] [PubMed] [Google Scholar]
- 33.Zhan Y, Li L, Zeng T, Zhou N, Guan X, Li Y. SNRPB-mediated RNA splicing drives tumor cell proliferation and stemness in hepatocellular carcinoma. Aging. 2020;13(1):537–554. doi: 10.18632/aging.202164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu N, Wu Z, Chen A, Wang Y, Cai D, Zheng J, Liu Y, Zhang L. SNRPB promotes the tumorigenic potential of NSCLC in part by regulating RAB26. Cell Death Dis. 2019;10(9):667. doi: 10.1038/s41419-019-1929-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yoshimi A, Lin K, Wiseman D, Rahman M, Pastore A, Wang B, Lee S, Micol J, Zhang X, de Botton S, Penard-Lacronique V, Stein E, Cho H, Miles R, Inoue D, Albrecht T, Somervaille T, Batta K, Amaral F, Simeoni F, Wilks D, Cargo C, Intlekofer A, Levine R, Dvinge H, Bradley R, Wagner E, Krainer A, Abdel-Wahab O. Coordinated alterations in RNA splicing and epigenetic regulation drive leukaemogenesis. Nature. 2019;574(7777):273–277. doi: 10.1038/s41586-019-1618-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chai G, Webb A, Li C, Antaki D, Lee S, Breuss M, Lang N, Stanley V, Anzenberg P, Yang X, Marshall T, Gaffney P, Wierenga K, Chung B, Tsang M, Pais L, Lovgren A, VanNoy G, Rehm H, Mirzaa G, Leon E, Diaz J, Neumann A, Kalverda A, Manfield I, Parry D, Logan C, Johnson C, Bonthron D, Valleley E, Issa M, Abdel-Ghafar S, Abdel-Hamid M, Jennings P, Zaki M, Sheridan E, Gleeson J. Mutations in spliceosomal genes PPIL1 and PRP17 cause neurodegenerative pontocerebellar hypoplasia with microcephaly. Neuron. 2021;109(2):241–256.e9. doi: 10.1016/j.neuron.2020.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Madan V, Cao Z, Teoh W, Dakle P, Han L, Shyamsunder P, Jeitany M, Zhou S, Li J, Nordin H, Shi J, Yu S, Yang H, Hossain M, Chng W, Koeffler H. ZRSR1 cooperates with ZRSR2 in regulating splicing of U12-type introns in murine hematopoietic cells. Haematologica. 2021. 10.3324/haematol.2020.260562. [DOI] [PMC free article] [PubMed]
- 38.Bamopoulos S, Batcha A, Jurinovic V, Rothenberg-Thurley M, Janke H, et al. Clinical presentation and differential splicing of SRSF2, U2AF1 and SF3B1 mutations in patients with acute myeloid leukemia. Leukemia. 2020;34:2621–2634. doi: 10.1038/s41375-020-0839-4. [DOI] [PubMed] [Google Scholar]
- 39.Kumar R, Gardner A, Homan C, Douglas E, Mefford H, et al. Severe neurocognitive and growth disorders due to variation in THOC2, an essential component of nuclear mRNA export machinery. Hum Mutat. 2018;39:1126–1138. doi: 10.1002/humu.23557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu Z, Zhong M, Li M, Huang H, Liao J, et al. Mutation analysis of pre-mRNA splicing genes PRPF31, PRPF8, and SNRNP200 in Chinese families with autosomal dominant retinitis pigmentosa. Curr Mol Med. 2018;18:287–294. doi: 10.2174/1566524018666181024160452. [DOI] [PubMed] [Google Scholar]
- 41.Zhang T, Bai J, Zhang X, Zheng X, Lu N, et al. SNRNP200 mutations cause autosomal dominant retinitis pigmentosa. Front Med. 2020;7:588991. doi: 10.3389/fmed.2020.588991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Petiti J, Itri F, Signorino E, Frolli A, Fava C, et al. SF3B1 detection of p.Lys700Glu mutation by PNA-PCR clamping in myelodysplastic syndromes and myeloproliferative neoplasms. J Clin Med. 2022;11. 10.3390/jcm11051267. [DOI] [PMC free article] [PubMed]
- 43.Drabarek W, van Riet J, Nguyen J, Smit K, van Poppelen N, et al. Identification of early-onset metastasis in SF3B1 mutated uveal melanoma. Cancers. 2022;14. 10.3390/cancers14030846. [DOI] [PMC free article] [PubMed]
- 44.Neumann N, Wen K. Myelodysplastic/myeloproliferative neoplasm with ring sideroblasts, thrombocytosis, and mutated JAK2/SF3B1 without anemia. Blood. 2022;139:466. doi: 10.1182/blood.2021014276. [DOI] [PubMed] [Google Scholar]
- 45.Lieu Y, Liu Z, Ali A, Wei X, Penson A, et al. SF3B1 mutant-induced missplicing of MAP3K7 causes anemia in myelodysplastic syndromes. Proc Natl Acad Sci U S A. 2022;119. 10.1073/pnas.2111703119. [DOI] [PMC free article] [PubMed]
- 46.Li C, Prokopec S, Sun R, Yousif F, Schmitz N, Boutros P. Sex differences in oncogenic mutational processes. Nat Commun. 2020;11(1):4330. doi: 10.1038/s41467-020-17359-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ubaida-Mohien C, Lyashkov A, Gonzalez-Freire M, Tharakan R, Shardell M, Moaddel R, 46. Semba R, Chia C, Gorospe M, Sen R, Ferrucci L. Discovery proteomics in aging human skeletal muscle finds change in spliceosome, immunity, proteostasis and mitochondria. eLife. 2019; 8. 10.7554/eLife.49874. [DOI] [PMC free article] [PubMed]
- 48.Desterro J, Bak-Gordon P, Carmo-Fonseca M. Targeting mRNA processing as an anticancer strategy. Nat Rev Drug Discov. 2020;19(2):112–129. doi: 10.1038/s41573-019-0042-3. [DOI] [PubMed] [Google Scholar]
- 49.Gamboa Lopez A, Allu S, Mendez P, Chandrashekar Reddy G, Maul-Newby H, Ghosh A, Jurica M. Herboxidiene features that mediate conformation-dependent SF3B1 interactions to inhibit splicing. ACS Chem Biol. 2021;16(3):520–528. doi: 10.1021/acschembio.0c00965. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF 1075 kb)
(PNG 209 kb)
(PNG 52 kb)
(PDF 2335 kb)
(PDF 852 kb)
(PDF 3358 kb)
(PDF 1098 kb)
(PNG 166 kb)
(PDF 1632 kb)
(PNG 172 kb)
(CSV 4 kb)
(CSV 6804 kb)
(CSV 1331 kb)
(CSV 186 kb)
(CSV 754 kb)
(CSV 268 kb)
(CSV 9 kb)
(CSV 951 kb)
Data Availability Statement
All data and materials are available in current manuscript and supplementary materials.
The software application or custom code is provided in the current manuscript.