

Abstract
Pancreatic ductal adenocarcinoma (PDAC) is a highly metastatic disease with few effective treatments. Here we show that the mitochondrial calcium uniporter (MCU) promotes PDAC cell migration, invasion, metastasis, and metabolic stress resistance by activating the Keap1-Nrf2 antioxidant program. The cystine transporter SLC7A11 was identified as a druggable target downstream of the MCU-Nrf2 axis. Paradoxically, despite the increased ability to uptake cystine, MCU-overexpressing PDAC demonstrated characteristics typical of cystine-deprived cells and were hypersensitive to cystine deprivation-induced ferroptosis. Pharmacological inhibitors of SLC7A11 effectively induced tumor regression and abrogated MCU-driven metastasis in PDAC. In patient-derived organoid models in vitro and patient-derived xenograft models in vivo, MCU-high PDAC demonstrated increased sensitivity to SLC7A11 inhibition compared to MCU-low tumors. These data suggest that MCU is able to promote resistance to metabolic stress and drive PDAC metastasis in a cystine-dependent manner. MCU-mediated cystine addiction could be exploited as a therapeutic vulnerability to inhibit PDAC tumor growth and prevent metastasis.
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is one of the most lethal tumor malignancies. Even among resectable early stage PDAC patients, 75% will develop liver metastasis within 1–2 years after complete removal of primary tumor (1). It is urgent to understand molecular mechanisms underlying PDAC metastasis and to develop novel therapeutic strategies for metastatic PDAC.
Mitochondria are crucial in pancreatic cancer tumorigenesis and progression (2–4). In addition to their roles in cell metabolism, mitochondria are essential for Ca2+ signaling and redox balance (5). The mitochondrial calcium uniporter (MCU) is the pore-forming subunit of the MCU complex, which consists of MCU, the scaffold protein EMRE and the calcium sensitive inhibitory regulatory subunits MICU1 and MICU2 (6, 7). MCUb is a paralog of MCU that serves as a dominant negative regulator of MCU (8). The activities of MCU can be regulated by change in the ratios between MCU and MCUb and the binding of MICU1/2 to Ca2+. In addition to MCU, the mitochondrial Ca2+ levels are regulated by NCLX, a Na+-Ca2+ exchanger responsible for mCa2+ efflux (9). Ca2+ uptake by MCU is important for buffering cytosolic Ca2+ increase and regulating mitochondrial OXPHOS (6, 7, 10, 11). On the other hand, the MCU-mediated mitochondrial Ca2+ overload might lead to cell death due to opening of the mitochondrial permeability transition pores (mPTP) and excessive production of reactive oxygen species (ROS) (7, 11–13). Paradoxically, there are reports of MCU up-regulation or NCLX down-regulation in metastatic cancer, implicating a pro-metastasis role for mitochondrial Ca2+ signaling during tumor progression (14–17).
Cancer cells are characterized by simultaneous increase in oxidative stress and antioxidant response (18). The Keap1-Nrf2 pathway is the predominant anti-oxidant response and xenobiotic detoxification mechanism in mammalian cells (19–21). Keap1 is a cytosolic oxidative stress sensor that sequesters Nrf2 for ubiquitination and proteasomal degradation (19). Under oxidative stress, the oxidation of multiple Cys residues of Keap1 blocks its binding to Nrf2 (20), which allows the stabilization of Nrf2 protein and the transcription of Antioxidant-Response-Element (ARE)-harboring genes (22–24). This pathway is upregulated in PDAC (25, 26) due to oncogenic KRAS-promoted transcription of Nrf2 (26). The activation of Keap1-Nrf2 promotes cell migration, invasion and oxidative stress resistance in metastatic cancer cells (21, 27–29).
Ferroptosis is a type of iron and lipid peroxidation-dependent cell death (30–34). Cancer cells rely on cystine uptake for glutathione (GSH) synthesis, which is required for glutathione peroxidase 4 (GPX4) to reduce lipid peroxidation (33, 34). Upon cystine deprivation or GPX4 inhibition, iron rapidly catalyzes the accumulation of lipid peroxide through Fenton reaction, which eventually compromises membrane integrity and leads to cell death (30–32, 34). Mitochondrial membrane hyperpolarization driven by OXPHOS has recently been implicated in promoting ferroptosis induced by cystine deprivation (35). However, the role of mitochondrial Ca2+ signaling in ferroptosis remained unclear.
In this study we investigated the role of MCU in pancreatic cancer metastasis and progression. Our data supported that MCU promotes PDAC progression through activating the Keap1-Nrf2 circuit and the cystine addiction in MCU overexpressing PDAC is a therapeutic vulnerability that could be exploited to prevent metastatic recurrence.
Methods
Patients and Human tissue specimens
Tumor samples which include lymph node samples were obtained from 132 PDAC patients treated at the Tianjin Cancer Hospital from 2014 and 2018. All 132 patients underwent radical pancreaticoduodenectomy with R0 margins confirmed by 2 pathologists. None of the patients had received chemotherapy or radiotherapy at the time of sample collection. All patients were treated with systemic gemcitabine-based chemotherapy after operation for 6 cycles after surgery. No radiation therapy was given before or after surgery. Postoperative follow-up of patients were conducted every three months initially. Overall survival (OS) was defined as the time interval from the date of surgery to that of death due to any cause or that of the last follow-up. Relapse free survival (RFS) was calculated from the date of surgery to that of local recurrence or metastasis. Local recurrence or metastasis was diagnosed by radiological examination (contrast-enhanced CT or MRI scanning). All patients provided written informed consent. The study protocols were approved by the Ethics Committee of the Tianjin Cancer Institute and Hospital.
Immunohistochemistry analysis
Consecutive sections of formalin-fixed, paraffin-embedded (FFPE) tumors were subjected to IHC analysis for MCU and NRF2 using a DAB substrate kit (Maixin, Fuzhou, China) according to the manufacturer’s instructions. The slides were deparaffinized in xylene and rehydrated through graded ethanol to water before staining. All sections were treated with EDTA (pH 8.0) for antigen retrieval and with 3% H2O2 for the inactivation of endogenous peroxidase. Sections were incubated with anti-MCU (sigma, PA5-109304, at 1:500 dilution) antibodies or anti-NRF2 (Abcam, ab-62352, at 1:350 dilution) overnight at 4°C. After wash, the sections were stained with a secondary antibody for 30 minutes at room temperature. Phosphate-buffered saline was substituted for each primary antibody as a negative control. Five random fields were examined under a light microscope. Immunoreactivity was semi-quantitatively scored according to the estimated percentage of positive tumor cells as previously described. IHC-staining results were blindly and independently performed by two pathologists who were blinded to the clinical data. Staining intensity was scored 0 (negative), 1 (low), 2 (medium), and 3 (high). Staining extent was scored 0 (0% stained), 1 (1%–25% stained), 2 (26%–50% stained), and 3 (51%–100% stained). The final score was determined by multiplying the intensity scores with staining extent and ranged from 0 to 9. Final scores (intensity score ×percentage score) less than or equal 4 were considered as low staining, more than 4 were high staining. Antibodies used in IHC staining can be found in Table S5.
ROS measurement
Mitochondrial ROS (superoxide anion) were detected using 5 μM MitoSox Red staining (Invitrogen, Carlsbad, CA, USA) for 30 min at 37 °C according to the manufacturer’s instructions and analyzed using a FACS Calibur flow cytometer (BD Biosciences, San Jose, CA, USA). Forward and side scatter data were collected (10, 000 events per sample).
Animal experiments
All animal experiments were performed according to protocols approved by IACUC at the Tianjin Cancer Institute and Hospital or the Penn State College of Medicine.
For the orthotopic tumor models, 1×106 luciferase labeled PDAC cells (Panc-1, SW1990 or PDX677) were re-suspended in 40 μl 0.25 mg/ml Matrigel (Corning) with PBS buffer, then orthotopically injected into the carefully exposed pancreas of 5 week-old female nude mice (36, 37). The pancreas was then returned to the peritoneal cavity, the abdominal wall and the skin was closed with skin clips. For drug treatments, mice were treated with either saline vehicle (Veh.), sulfasalazine (SAS, 100 mg/kg) or IKE (20mg/kg) everyday via i.p. starting from day 7. Noninvasive bioluminescent imaging and analysis were performed as described previously using Xenogen IVIS 200 (38) starting from 1 week post-surgery. Tumor growth were analyzed by bioluminescent imaging (BLI). Six to Seven weeks post-surgery, mice were euthanized and the BLI signals in primary tumor (pancreas) or distant metastatic sites (liver, peritoneal cavity) were determined through ex vivo BLI imaging. The visible metastatic lesions in the gut, mesentery, and liver of each mouse were counted. The primary and metastatic pancreatic tumor were excised, fixed in formalin, and embedded in paraffin. Half of the tissues were subjected to hematoxylin and eosin and IHC staining as described for human PDAC tumors. Histological staining on paraffin sections (hematoxylin and eosin) were carried out using standard protocols. Following digital capture on Olympus BX51, histology images were processed using Affinity Photo software, using three filters/adjustments applied evenly across the entire image: unsharp mask, white balance, and levels adjustment.
For patient-derived xenograft (PDX) experiments, 4 PDX lines with MCU-high or MCU-low expression levels were used. The MCU expression levels were determined according to the IHC staining of the patient tumor tissues and PDX tissues. PDX tumors were cut into small pieces (about 8 mm3) and implanted into 5 week old female NSG mice subcutaneously (10 mice for each PDX line). When the tumor volume reached 50–70 mm3, mice were randomized into vehicle control group or IKE group. 100 μl IKE (20 mg/kg) or saline (vehicle control) were administrated daily via i.p.. Tumor growth was monitored every five days using a caliper and tumor volumes were calculated by the following formula: Volume= 1/2 ×L1× (L2)2, where L1 is the long axis and L2 is the short axis. Mice were sacrificed after 8 weeks and tumors were harvested. Tumor tissues were immediately fixed in buffered formalin and embedded in paraffin. Tissue slides (5μm) were prepared and Hematoxylin & Eosin (H&E) staining were performed for histopathological analysis according to instructions. IHC of 4-HNE (abcam, ab48560) staining were performed to evaluate lipid peroxidation status of tumor tissues.
Biotin labeling of oxidized KEAP1
Cells were lysed for 15 min on ice in biotin labeling lysis buffer (BLLB: 50 mM Tris-HCl pH 7.0, 5 mM EDTA, 120 mM NaCl, 0.5% Igepal-630) containing protease inhibitors and 100 mM maleimide (Sigma-129585). Insoluble material was then removed by centrifugation at 20,000g for 10 min at 4°C, the cleared supernatant was transferred to a fresh Eppendorf tube and protein concentration was determined by the Bradford assay. Protein concentration was adjusted to 1 μg/μl with BLLB, SDS was added from a 10% stock to a final concentration of 1% and the cell lysates were incubated at room temperature for 2 hours rotating. To remove unreacted maleimide, proteins were subsequently precipitated by adding 5 volumes of acetone pre-equilibrated at −20°C and incubated for 20 min at −20°C. The preparations were centrifuged at 20,000g for 10 min at 4°C, supernatants removed and discarded and precipitated protein pellet was air-dried. The pellet was then resuspended in 200 μl BLLB containing 1% SDS, 10 mM DTT and 0.1 mM biotin-maleimide (Sigma-B1267, stock dissolved in dimethylformamide) to reduce the remaining, previously oxidized, sulfhydryl groups and allow their reaction with biotin-maleimide. Proteins were again precipitated with 5 volumes of methanol (−20°C) as above, the dried pellet was resuspended in 500 μl of BLLB, incubated with 10 μl of a 50% slurry of Pierce™ Streptavidin Agarose (Thermo Scientific™ 20353) rotating at 4°C for 2h. The beads were then washed 4 times with BLLB and resuspended in SDS-PAGE loading buffer for SDS-PAGE analysis and western blotting with the KEAP1 antibodies.
RNA sequencing
Three biological replicates each of control or MCE OE Panc-1 cells were cultured in glucose limited medium (1mM Glc) for overnight. The mRNA was extracted with RNeasy kit (Qiagen) and used for the preparation of RNA sequencing library. Directional (stranded) libraries for the paired-end sequencing of PANC-1 cells were generated with an Illumina platform. After filtering low expression genes, raw read count data were used to calculate false discovery rate (FDR) and p value by applying edgeR method.
Ferroptosis treatment
Erastin (Selleck, S7242), Z-VAD-FMK (Selleck, S7023), necrostatin-1 (Selleck, S8037), the mitoTEMPO (Millipore, C906P50), ferrostatin-1 (Cayman, 17729), CGP37157 (Sigma, 75450-34-9) and Cyclosporine A (Selleck, S2286) were each dissolved in dimethyl sulfoxide (DMSO, Sigma). Deferoxamine (Sigma, 138-14-7) was dissolved in deionized water. For ferroptosis assay cells were seeded in 6-well plates and treated with cystine-deprivation (cystine-free medium), glutamate (50 mM) or Erastin (4 μM) in the presence or absence of Z-VAD-FMK (ZVAD, 50 μM), Necrostatin-1 (Nec-1, 10 μM), cyclosporine A (CsA, 10 μM), Deferoxamine (DFO, 50 μM) or ferrostatin-1 (Fer-1, 1 μM). The effect of mitoTEMPO (mT, 2 μM), NCLX inhibitor CGP37157 (2 μM), Deferoxamine (DFO, 50 μM), ferrostatin-1 (Fer-1, 1 μM) treatment on cystine-deprivation or Erastin-induced ferroptosis were determine in some experiments. In most experiments PDAC cells were treated between 16–24 hours except for AsPC-1 cells, which were treated for 48 hours.
Measurement of lipid peroxidation
Lipid peroxide was analyzed by flow cytometry. Cell lines were plated in quadruplicate at the cell numbers indicated previously for the 12-well plate format. Cells were seed overnight and were subjected to various compound treatments for indicated times. Cells were then incubated for 30 minutes in live-cell imaging solution containing the pertinent ROS dye at the following concentrations: C-11 BODIPY (Invitrogen) (2 μM). Cells were then washed with PBS, trypsinized with 0.25% trypsin, and neutralized with 10% FBS in PBS. A minimum of 10,000 cells were analyzed per condition. For C-11 BODIPY, signal was analyzed in the FITC channel. Software analysis and histogram generation was carried out using FlowJo v10. For Lipid peroxidation confocal imaging, Cells were seeded at a density of 2.5×105 per well on coverslips placed in a 6-well dish and grown overnight in DMEM. Cells were treated with cystine-free medium for 16 hours, the coverslips were then mounted to a Teflon chamber. C11-BODIPY(Invitrogen) (5 μM) were added to each well 15 min before measurements. Reduced and oxidized C11-BODIPY were detected at emission 590 nm and 510 nm, respectively.
Organoid culture and experiment
Human pancreatic tissues were obtained from patients undergoing surgery for pancreatic cancer who consented to donate their samples for research. The collection of samples was approved by the Ethics Committee of the Tianjin Cancer Institute and Hospital. The study is compliant with all relevant ethical regulations regarding research involving human participants. Human PDAC organoid cultures were established and cultured as previously described (39). To ensure purity of the sorted cells, the sorters were set to “single cell” mode. CK19 positive Single cells were sorted into cooled, Matrigel-covered wells of 96-well plates (Costar) in at least technical triplicate. After sorting, 10μl/well undiluted Matrigel was allowed to solidify in an incubator before organoid media was added. For human organoids, Human Complete Feeding Medium (hCPLT) contained Human Wash Medium with BSA (Advanced DMEM/F-12, 10mM HEPES, 1 × GlutaMAX Supplement, 100μg/ml Primocin and 0.1% BSA), 10mM Nicotinamide, 1 × Wnt3a-Conditioned Medium, 1 X R-spondin1-Conditioned Medium, 100ng/ml mNoggin, 1 × B27 Supplement, 100μg/ml Primocin, 100ng/ml hFGF, 1.25mM N-acetylcysteine, 1μM PGE2, 50ng/ml hEGF, 10μM Y27632, 10nM hGastrin and 500nM A 83-01. PDAC organoid (3 × 103 cells/well) were seeded in 96-well plates. After 7 days in culture, the medium was replaced with hCPLT, Erastin (10μM), CGP37157 (5μM) or Erastin+CGP37157, followed by incubation for 3 days. Cell growth was analyzed by microscopy and by measuring ATP concentrations with Cell Titer-Glo® 3D Viability assay kit (Promega Corporation, WI, USA).
Glutamine uptake and Glutamate export.
1.2 × 105 cells were plated onto 12 well plate and allowed to attach for 12 hours. The cells were fed with fresh medium. After 6 hour incubation, the conditioned medium were collected and the concentration of glutamine and glutamate in the conditioned medium and fresh medium were measured using YSI 2700 Biochemistry Analyzer (Marshal Scientific). The glutamine uptake and glutamate export by PDAC cells were calculated based on the decreases in glutamine and increases in glutamate concentrations in conditioned medium relative to fresh medium. For H2O2 treatment, 100 μM H2O2 was added.
FITC-cystine uptake assay
3×105 Cells were plated onto 6 well plate for overnight. The cells were then incubated with fresh medium containing 5 μM FITC-cystine (SCT047, Sigma). Incubate the cells in a 37°C CO2 incubator for 30 minutes. The cells were resuspended in ice cold PBS containing 1% FBS after trypsinization and used for flow cytometry analysis.
Statistical analysis
Statistical analyses were performed with SPSS 18.0 software and GraphPad Prism version 5.0.3. Student’s t test or ANOVA for unpaired data was used to compare mean values. The Student’s t test for paired data was used to compare mean values for in vivo data. A Spearman rank correlation coefficient test was carried out for testing the association between ordinal variables. Kaplan-Meier survival curves were made according to these cut points; the log-rank test was used to obtain a P value for the significance of Kaplan–Meier curves’ divergence. All P-values less than 0.05 were considered statistically significant. * indicate P value < 0.05.
Data availability
The RNA sequencing data generated in this study are publicly available in Gene Expression Omnibus (GEO) at GSE199692.
Results
MCU overexpression in PDAC correlates with poor prognosis and metastatic progression
Calcium signaling is frequently dysregulated during tumorigenesis and cancer progression (40, 41). To understand the role of mitochondrial Ca2+ signaling in PDAC progression, we determined the expression levels of MCU complex subunits in paired PDAC tissue (T) and para-tumor normal pancreatic tissue (N) from 6 PDAC patients (Fig. 1A–1B). MCU protein (Fig. 1A and 1B) levels were elevated in tumor tissues by approximately 4-fold when compared to paired normal tissues, while the levels of MCUb and the inhibitory subunit MICU2 were decreased. The levels of MICU1 were decreased by approximately 2-fold while the levels of EMRE didn’t change significantly (Fig. 1A and 1B). The upregulation of MCU in PDAC was confirmed in another cohort of 10 pairs of PDAC patients (Fig. S1A and S1B).
Figure 1. MCU overexpression in PDAC correlates with poor patient prognosis and metastatic progression.
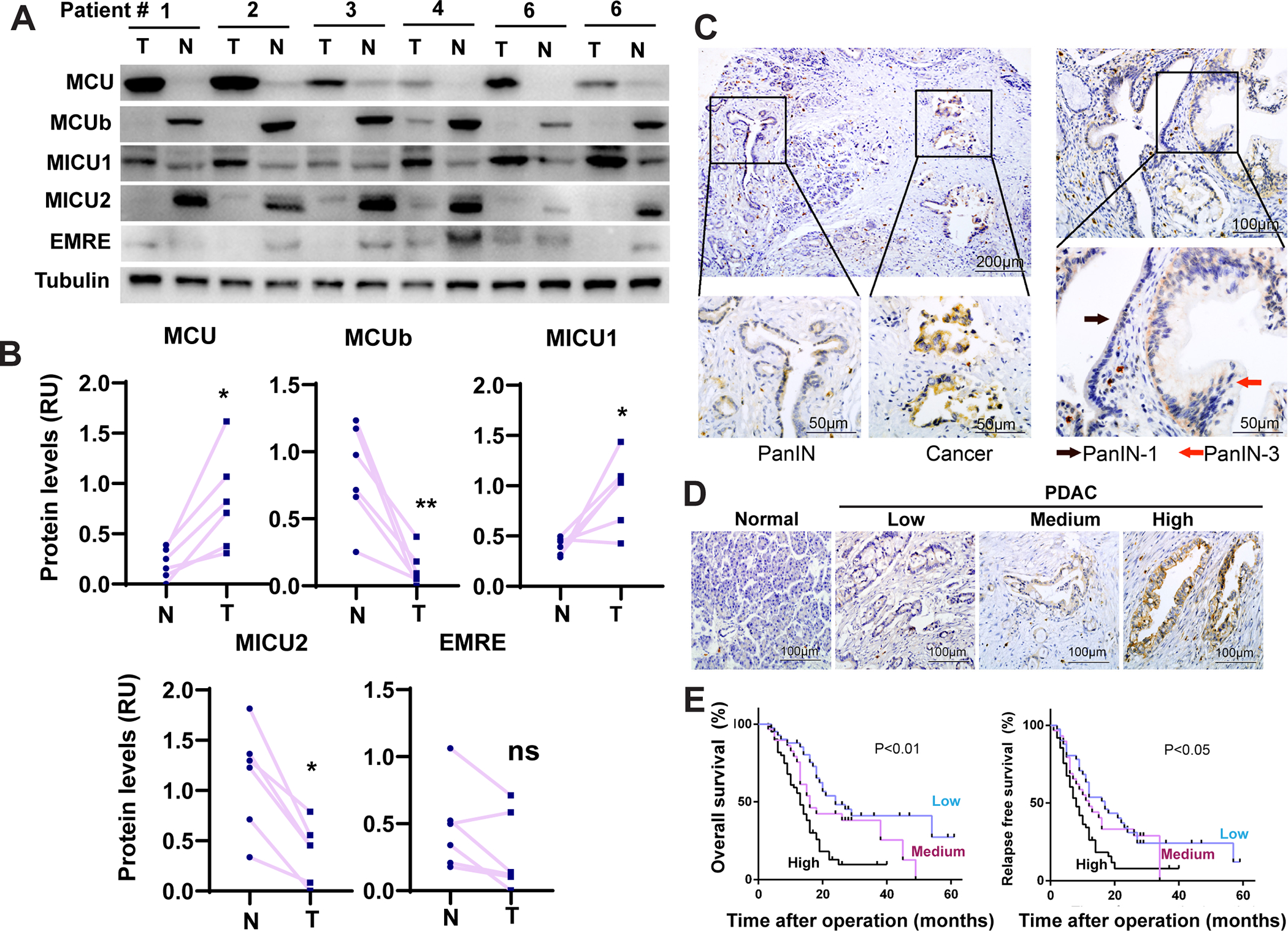
A-B, The expression levels of MCU, MCUb, MICU1, MICU2 and EMRE in resected tumor tissues (T) and paired para-tumor normal tissues (N) from 6 PDAC patients as determined by Western blotting. (B) is densitometry quantitation of data in (A).
C, IHC analysis of PDAC patient tissues showing increased expression of MCU from PanIN to cancer and from PanIN-1 to PanIN-3.
D, IHC staining of MCU expression in normal pancreatic tissue and tumor tissues from PDAC patients (E).
E, The correlation between MCU protein expression levels and overall survival or relapse free survival among a cohort of 132 PDAC patients.
Data in B were analyzed using two-tailed, two sample paired Student’s t-test. * indicated p<0.05.
The immunofluorescence staining of MCU colocalized with NDUFB8 in PDAC tissues, which is consistent with the function of MCU as a mitochondrial inner membrane channel (Fig. S1C). In immunohistochemistry (IHC) staining (Fig. 1C–1D), MCU expression was very low in the normal pancreas but elevated in pancreatic intraepithelial neoplasia (PanIN) and carcinoma tissues. MCU levels were increased in carcinoma when compared PanIN in the same patient; PanIN-3 had stronger MCU staining than PanIN-1 (Fig. 1C). MCU overexpression significantly correlated with differentiation, lymph node metastasis and TNM stage (Fig. 1D, Table S1). MCU overexpressing group had significantly poorer overall survival (OS) and relapse-free survival (RFS) in our cohort of 132 PDAC patients (Fig. 1E). The correlation between MCU expression and patient survival was also confirmed in in the TCGA RNAseq database (Fig. S1D). Cox’s proportional hazard analysis indicates that MCU expression levels strongly correlates with OS and RFS in univariate analysis and is an independent predictor of OS in multivariate analysis (Table S2). Taken together, our data indicate that MCU expression levels increased during PDAC initiation and progression.
MCU promotes metabolic stress resistance and PDAC metastasis
To understand the role of MCU in PDAC progression and metastasis, we took either overexpress (OE) or knockout (KO) MCU in PDAC cell lines (Fig. S2A). MCU KO abrogated mitochondrial Ca2+ uptake without affecting Thapsigargin-induced ER Ca2+ release or store-operated calcium entry (Fig. 2A and Fig. S2B), while MCU OE was sufficient to promote mitochondrial Ca2+ uptake (Fig. 2B). Mitochondrial calcium uptake activities strongly correlated with MCU protein levels in cell lines and PDX lines (Fig. 2C, 2D and S2C). MCU KO in PDAC cells remarkably inhibited cell migration, invasion and soft agar colony formation (Fig. S2D–S2E). The decrease in migration and invasion in MCU KO cells could be rescued by the ectopic MCU (Fig. S2F and S2G). Conversely, MCU OE promoted cell migration and invasion (Fig. S2H).
Figure 2. MCU is required for metabolic stress resistance and PDAC metastasis.
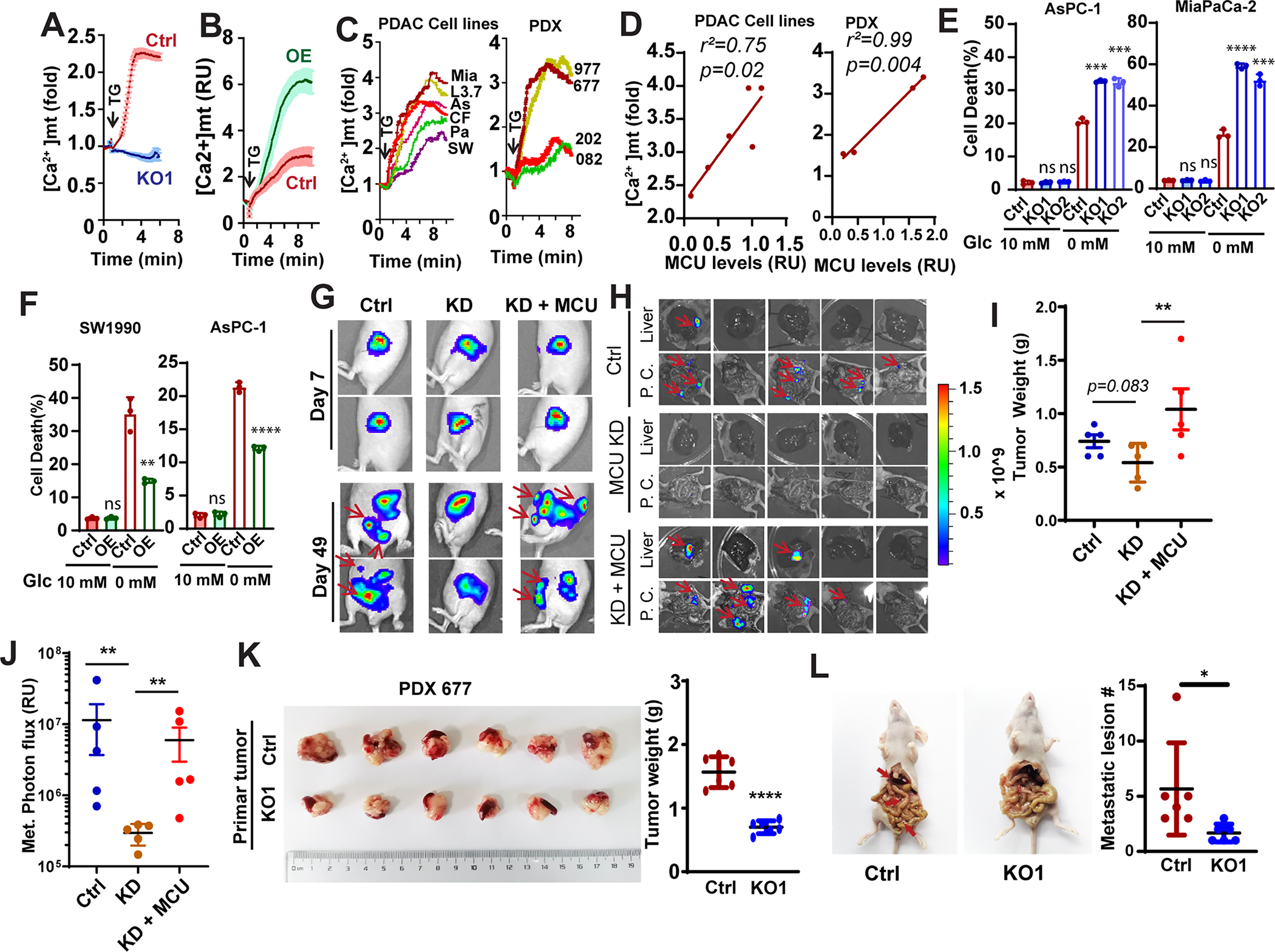
A and B, the effects of MCU KO (A) and OE (B) on mitochondrial calcium uptake in Panc-1 cells. Data are shown as mean ± SD from 20–30 cells.
C, the mitochondrial calcium uptake activities in a panel of PDAC cell lines (left panel, MiaPaCa-2, L3.7, AsPC-1, CFPAC-1, Panc-1 and SW1990) or PDX derived cell lines (PDX977, PDX677, PDX202 and PDX082) with different MCU protein expression levels.
D, linear regression to showed the correlation between mitochondrial Ca2+ uptake and protein levels of MCU in PDAC cell lines (left panel) or PDX lines (right panel).
E and F, the effects of MCU KO (E) or OE (F) on PDAC cell death when cultured under glucose replete (10 mM Glc) or glucose deprived (0 mM Glc) conditions.
G-J, the effect of MCU knockdown (KD) and KD + MCU rescue on orthotopic tumor growth and metastasis. G, representative images showing BLI imaging at day 7 and day 49. H, ex vivo BLI imaging showing metastases lesions in the liver and peritoneal cavity (P. C.). I, the weight of tumors harvested from mice implanted with Panc-1 cells. J, quantitation of BLI signals from metastases in the liver and the peritoneal cavity.
K and L, the effects of MCU KO in the MCU-high PDX PDX677 on primary tumor growth (K) and metastasis (L) in a orthotopic metastasis model.
Data in E, F, I, J, K and L were analyzed using two-tailed, two sample unpaired Student’s t-test. ** indicated p<0.01.
The glucose concentration in the tumor microenvironment could be as much as 10 times lower than in the blood circulation. Although MCU KO had no effect on cell survival when cultured under physiological concentration of glucose, it increased cell death when PDAC cells were exposed to glucose limitation (1mM Glc) (Fig. S2I). The metabolic stress sensitivities in MCU KO cells were more pronounced in glucose-free conditions (Fig. 2E), which could be rescued by ectopic MCU (Fig. S2G). Conversely, MCU OE increased resistance to glucose deprivation (Fig. 2F).
Cell migration and invasion are critical for metastatic dissemination, while metabolic stress resistance promotes metastatic colonization(42). Therefore, we evaluated the role of MCU in PDAC metastasis in an orthotopic xenograft model. We used shRNA to knockdown (KD) MCU and used MCU-EGFP to rescue the KD in Panc-1 cells (Fig. S2J). After orthotopic implantation of luciferase labeled Panc-1 cells, visible development of multiple luminescence foci were detected on day 49 in the control and rescue groups (Fig. 2G, red arrow and Fig. S2K), which was indicative of metastasis. Ex vivo examination confirmed the development of multiple metastatic lesions in the liver and the peritoneal cavity in control and MCU rescue groups (Fig. 2H and Fig. S2K–S2N). Although MCU KO only modestly decreased the tumor weight of orthotopic Panc-1 tumor (Fig. 2I, p=0.083), the development of metastases was completely abrogated (Fig. 2J). Ectopic expression of MCU in KD cells was able to restore metastasis to liver, peritoneal cavity and promote orthotopic tumor growth (Fig. 2G–2J, Fig. S2J–S2N). To further evaluate the role of MCU in PDX metastasis, we knocked out MCU in the MCU-high PDX 677 line. As shown in Fig. 2K, 2L, S2O and S2P, the orthotopically injected PDX677 was able to established primary tumor in the pancreas and metastasis to the liver and peritoneal cavity. MCU KO reduced the growth of primary tumor (Fig. 2K) and the average number of metastatic lesions (Fig. 2L) and the number of mice that developed liver metastasis (Fig. S2O–S2Q). The lack of liver metastasis in the KO group was confirmed by H&E staining (Fig. S2Q). These data support that MCU is required for PDAC metastasis.
MCU activates Keap1-Nrf2 antioxidant response in PDAC by increasing mROS
It has been previously reported that MCU promotes breast cancer cell migration and invasion by activation of HIF1α (16). However, we found that MCU KO or OE had no effects of HIF1α protein levels in PDAC (Fig. S3A). To understand the mechanisms underlying MCU-mediated PDAC metastasis, we interrogated the TCGA RNAseq database to examine genes that correlated with MCU in PDAC patients. Interestingly, MCU expression highly correlated with multiple Nrf2 target genes containing ARE (Antioxidant-Response Element) (Fig. 3A). The overexpression of MCU in PDAC cells was sufficient to increase Nrf2 levels (Fig. 3B), which was further enhanced by Glc-limitation. Conversely, MCU KO remarkably decreased the levels of Nrf2 protein under Glc-limitation (Fig. 3C). MCU overexpression also increased the mRNA levels of classical Nrf2 target genes (Fig. 3D) and promoted the binding of Nrf2 to ARE elements (Fig. S3B). ARE luciferase reporter assay indicated that MCU KO inhibited, while MCU OE enhanced, Nrf2 transcriptional activities (Fig. S3C). Taken together, our data indicate robust activation of Nrf2 by MCU in PDAC cells.
Figure 3. MCU activates Nrf2-mediated antioxidant response in PDAC.
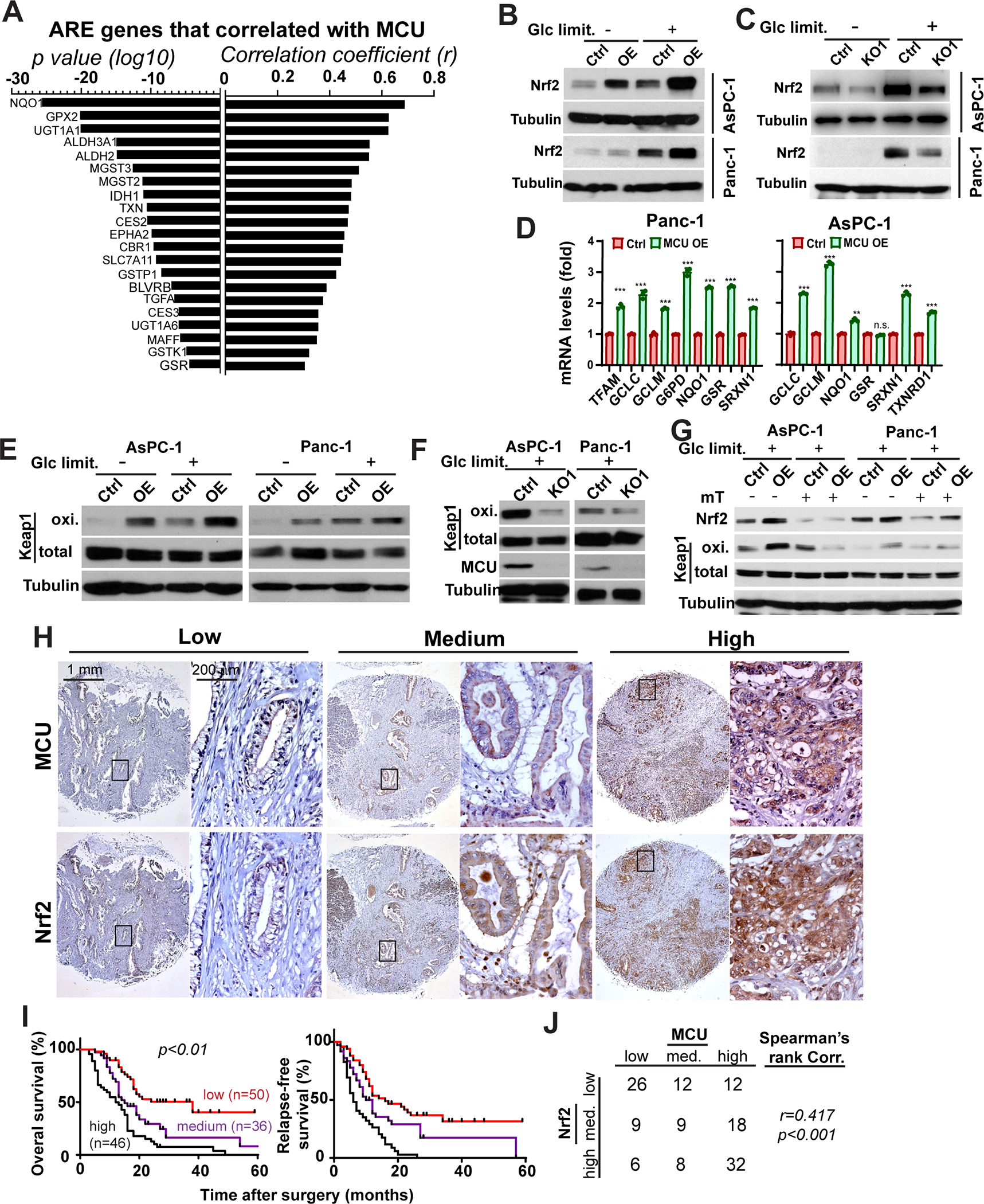
A, waterfall plot showing the correlation between MCU and ARE (antioxidant response element) gene in the TCGA PDAC RNAseq dataset. p values and correlation coefficients (r) were determined using Spearman’s rank correlation test.
B and C, the effects of MCU OE (B) or KO (C) on Nrf2 protein expression levels under regular cell culture conditions (Glc limit. −) or Glc limited (1mM Glc, Glc limit. +) conditions in Panc-1 and AsPC-1 cells.
D, the effects of MCU on the mRNA transcript levels of Nrf2 target genes in Panc-1 and AsPC-1 cells.
E and F, the effects of MCU OE (E) or KO (F) on the levels of Keap1 oxidation in PDAC cells.
G, the effects of mitoTEMPO treatment on Keap1 oxidation and Nrf2 activation in control or MCU OE PDAC cells.
H, MCU and Nrf2 IHC staining showing correlation between the expression levels of the two proteins in consecutive sections from a PDAC tissue microarray.
I, the association between Nrf2 expression levels and overall patient survival in a cohort of 132 patients. p<0.01 as determined by two-tailed log-rank test.
J, quantitation of the correlation between MCU and Nrf2 expression levels in the cohort of 132 PDAC patients in O and P. p<0.001 as determined by Spearman’s rank correlation test.
Data in D were presented as mean ± SD from three biological replicates and analyzed using two-tailed, two sample unpaired Student’s t-test. *, **, *** and **** indicated p<0.05, 0.01, 0.001 and 0.0001, respectively.
We found no difference in Nrf2 mRNA levels after MCU OE or KO (Fig. S3D). However, there was significant promotion of Keap1 oxidation in MCU OE PDAC cells (Fig. 3E). Conversely, MCU KO remarkably inhibited Keap1 oxidation (Fig. 3F). It has been previously reported that MCU-mediated mitochondrial Ca2+ uptake increased mROS (16). We confirmed elevated mROS levels in MCU OE PDAC cells (Fig. S3E–S3F) and depressed mROS levels in MCU KO cells (Fig. S3G–S3H). MCU levels in PDX lines correlated with basal mROS levels, (Fig. S3I). Treatment of Panc-1 and AsPC-1 cells with mitoTEMPO abrogated MCU-mediated Keap1 oxidation and Nrf2 accumulation (Fig. 3G). Taken together, our data suggest that mROS is required for MCU-mediated Nrf2 activation and Keap1 oxidation.
MCU expression correlates with Nrf2 levels in PDAC patients.
To determine whether MCU might indeed regulate Nrf2 among PDAC patients, we used IHC to evaluate the correlation between MCU and Nrf2 staining in PDAC patients (Fig. 3H). Strong Nrf2 staining was remarkably associated with poor OS, RFS, differentiation status and pTNM stages among PDAC patients (Fig. 3I and Table S3). Importantly, MCU-high patients also had stronger Nrf2 staining (Fig. 3H and 3J). Our data support that MCU promotes PDAC progression through Nrf2 upregulation and activation in PDAC patients.
The Keap1-Nrf2 circuit is required for MCU-mediated metastasis.
To determine whether Nrf2 is responsible for the pro-metastasis effects of MCU, we used two independent shRNA to knockdown Nrf2 expression in control and MCU OE cells (Fig. S4A). Nrf2 knockdown reduced the migration speed of individual cells without affecting cell viability under glucose replete conditions (Fig.S4B–S4D, supplementary video 1–4). Nrf2 knockdown abrogated MCU-mediated increase in cell migration, invasion (Fig. 4A–4C, S4E–S4G) and resistance to glucose deprivation (Fig.4D). Conversely we activated Nrf2 in MCU KO AsPC1 and MiaPaCa2 cells by knocking down Keap1 (Fig. 4E). Keap1 knockdown was able to increase Nrf2 and rescue Nrf2 levels in MCU KO cells (Fig. 4E). Nrf2 activation with Keap1 depletion rescued cell migration, invasion and restore resistance to glucose deprivation in MCU KO cells (Fig. 4F–4I). Our previous data showed that mROS was required for MCU to activate the Keap1-Nrf2 signaling. Therefore, we further investigated the effect of mitoTEMPO treatment in PDAC cells. mitoTEMPO treatment inhibited cell migration, invasion and wound closure in control and MCU OE cells (Fig. S4H–S4K).
Figure 4. MCU promotes cell migration / invasion, metabolic stress resistance and PDAC metastasis through activation of Nrf2.
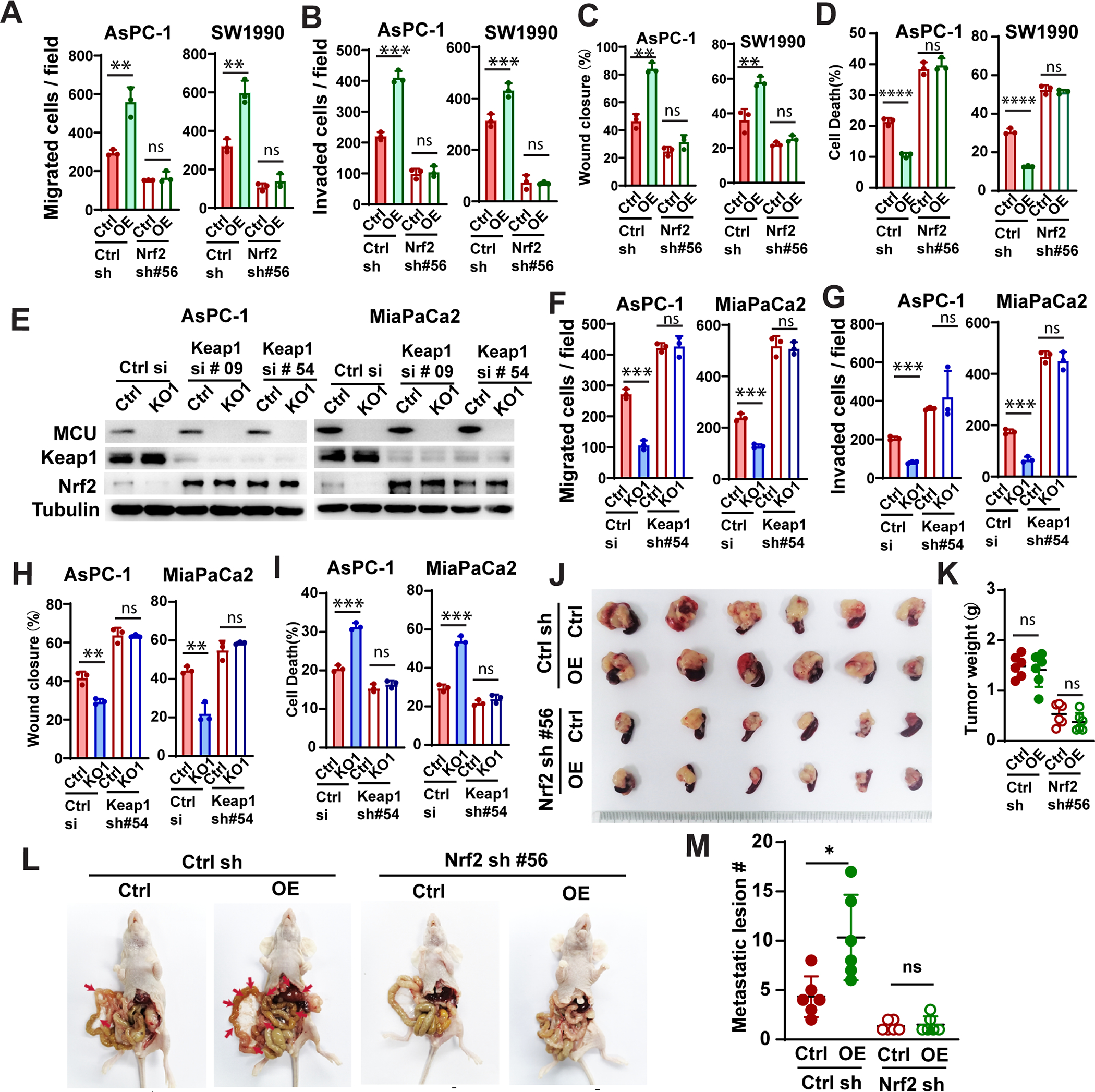
A-D, the effects of Nrf2 knockdown on cell migration (A), invasion (B), wound closure (C) and glucose deprivation-induced cell death (D) by in AsPC-1 and SW1990 cells, as determined using transwell assays (A and B), wound healing assays (C) and PI staining (D).
E, the effects of Keap1 knockdown on Nrf2 protein levels in control or MCU KO PDAC cells (AsPC1 and MiaPaCa-2).
F-I, the effects of Keap1 knockdown on cell migration (F), invasion (G), wound closure (H) and glucose deprivation-induced cell death (I) by in AsPC-1 and MiaPaCa-2 cells, as determined using transwell assays (F and G), wound healing assays (H) and PI staining (I).
J-M, the effects of MCU OE and Nrf2 knockdown on SW1990 tumor growth and metastasis in a orthotopic xenograft model. J and M, images of harvested tumor (J) and quantitation of tumor weight (M) at the end of the experiment. L, representative images showing metastatic lesions (red arrow)in the peritoneal cavity and liver in mice implanted with control of MCU OE SW1990 cells with or without Nrf2 knockdown. M, quantitation of the numbers of metastatic lesions observed in L.
Data in A-D, F-I were presented as mean ± SD from three biological replicates and analyzed using two-tailed, two sample unpaired Student’s t-test. *, **, *** and **** indicated p<0.05, 0.01, 0.001 and 0.0001, respectively.
To further investigate the role of Nrf2 in MCU-mediated metastasis, we implant control or MCU OE SW1990 cells with or without Nrf2 knockdown into the pancreas of nude mice. Eight weeks after implantation, mice were euthanize, the tumor weight, number of metastatic lesions in the peritoneal cavity and the liver were evaluated (Fig. 4J–4M, S4L–S4N). MCU OE increased the average number of metastatic lesion by approximately two fold without affecting primary tumor size (Fig. 4J–4M). The shRNA depletion of Nrf2 abrogated MCU-mediated metastasis (Fig. 4L–4M, S4L–S4M) and reduced tumor growth (Fig. 4J and 4K). The lack of liver metastasis in Nrf2 knockdown groups was confirmed with H & E staining (Fig. S4N). These data support that MCU promotes pancreatic cancer cell migration, invasion and metabolic stress resistance through activation of Nrf2.
MCU-Nrf2 signaling promotes SLC7A11 expression and cystine uptake in PDAC
To identify targetable effectors responsible for MCU-mediated PDAC metastasis and progression, we evaluated MCU-mediated transcriptomic changes in pancreatic cancer cells. A total of 182 genes were upregulated (and 180 downregulated) in MCU OE Panc-1 cells under Glc-limited conditions. Upregulated genes include multiple ARE genes (Fig. S5A and Table S4), which is consistent with the notion that MCU activates Nrf2-mediated antioxidant response. 8 of the 182 upregulated genes are also positively correlated with MCU expression in the TCGA PDAC cohort (Spearman’s rank r>0.4) (Fig. 5A). The top 2 of those 8 genes are Nrf2 target genes (SLC7A11 and IDH1) (Fig. 5B). Of particular interest is SLC7A11, a functional subunit of the cystine / glutamate antiporter (system xc−) responsible for the uptake of cystine and export of glutamate (30, 43). (Fig. 5B). Although cyst(e)ine is a non-essential amino acid, cancer cells depend on SLC7A11-mediated cystine uptake for glutathione (GSH) synthesis and antioxidant response. Interestingly, SLC3A2, the regulatory unit of system xc−, and SLC38A2 (SNAT2, responsible for neutral amino acid such as glutamine transport) and GCLC / GCLM (responsible for the rate limiting step of glutathione synthesis) were among the upregulated genes in MCU OE cells (Fig. S4A–S4B and Table S4). MCU-high PDX lines (PDX 677 and 977) had higher levels of SLC7A11 than MCU-low PDX samples (PDX 202 and 081) (Fig. 5C). MCU OE increased, while MCU KO decreased the levels of SLC7A11 (Fig. 5D). MCU-mediated upregulation of SLC7A11 was abrogated by Nrf2 knockdown (Fig. 5E and 5F). MCU OE also increased the binding of Nrf2 to SLC7A11 promoter (Fig. 5G).
Figure 5. MCU-Nrf2 signaling promotes SLC7A11 expression and cystine uptake in PDAC.
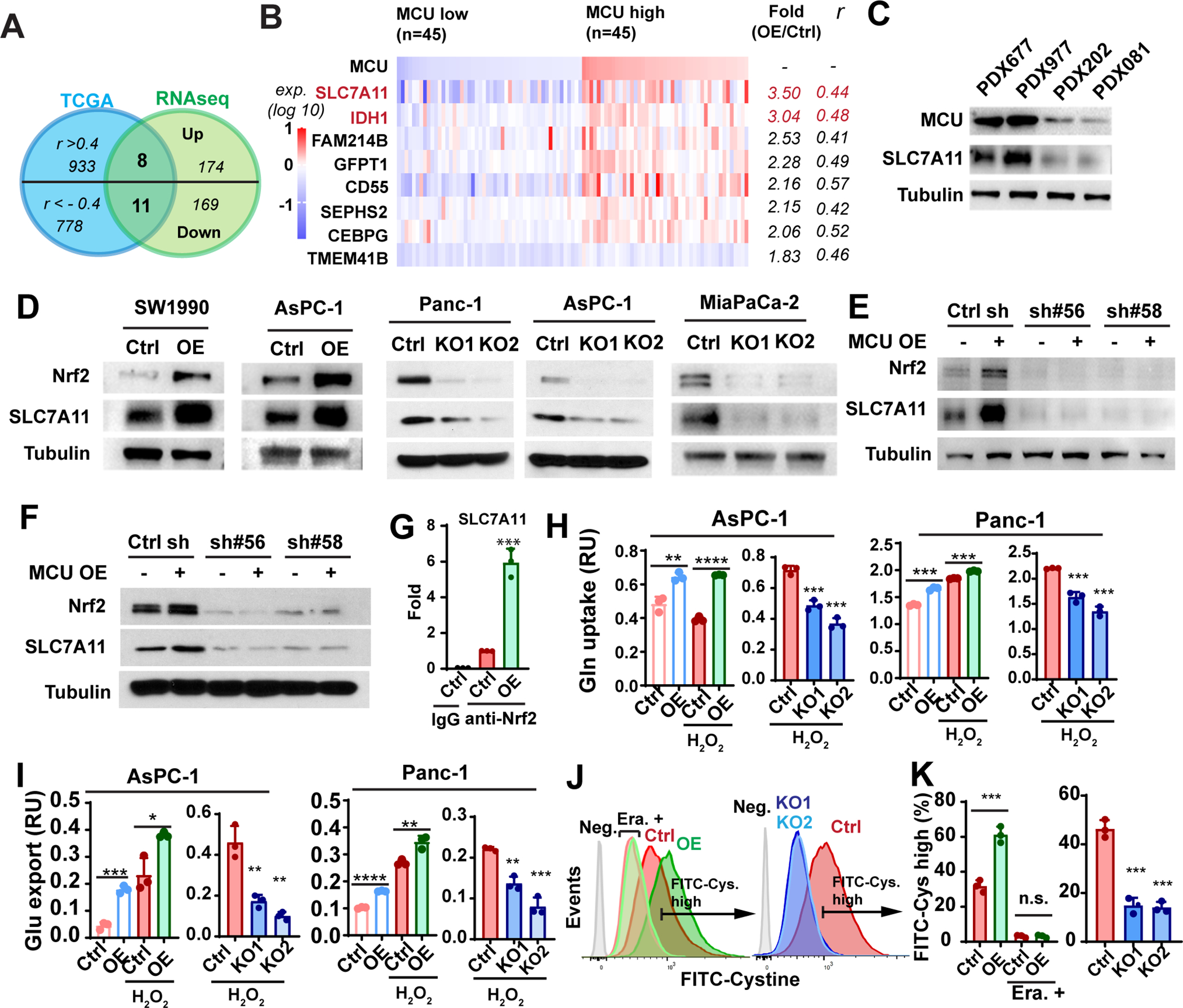
A, Venn diagram showing that 8 upregulated genes (and 11 downregulated genes) positively (or negatively correlated), with MCU in the TCGA PDAC RNAseq cohort.
B, the relative expression levels of the 8 upregulated genes in the MCU low (n=45, bottom quartile) and MCU high (n=45, top quartile) TCGA PDAC patients. Fold changes in expression levels were based on RNAseq data and Spearman’s rank correlation coefficients (r) between MCU and respective genes were calculated using the TCGA PDAC patient cohort.
C, the expression levels of MCU and SLC7A11 in MCU-high (PDX677, PDX977) and PDX-low (PDX202, PDX082) PDX-derived cell lines.
D, Western blotting experiment showing the effects of MCU KO or OE on Nrf2 and SLC7A11 protein levels in four PDAC cell lines.
E and F, Western blotting experiment showing the effects of Nrf2 knockdown on SLC7A11 expression levels in control and MCU OE AsPC-1 (E) Panc-1 (F) cells.
G, ChIP assay showing the effect of MCU OE on the binding of Nrf2 to the promoter of SLC7A11.
H and I, the effects of MCU OE and KO on glutamine (Gln) uptake (H) and glutamate (Glu) export (I) in two PDAC cell lines with or without H2O2 treatment (100 μM).
J and K, the effects of MCU OE and KO on the uptake of FITC-cystine as determined by flow cytometry. MCU-mediated cystine uptake could be abrogated by treatment with 1.5μM Erastin (Era.). (K) is the quantitation of flow cytometry experiments in (J). Cells without FITC-cystine treatment were used as negative control (Neg.).
Data in G-I and K were presented as mean ± SD from three biological replicates and analyzed using two-tailed, two sample unpaired Student’s t-test. *, **, *** and **** indicated p<0.05, 0.01, 0.001 and 0.0001, respectively. n.s. indicated not statistically significant.
System xc− exports glutamate while importing cystine. Glutamine is the main source of glutamate used by system xc− for cystine uptake (44). MCU OE increased glutamine uptake and glutamate secretion in pancreatic cancer cells, especially when cells were subjected to oxidative stress induced by H2O2 (Fig. 5H–5I). Conversely MCU KO inhibited glutamine uptake and glutamate secretion (Fig. 5H–5I). Overexpression of MCU significantly increased the uptake of FITC-cystine in Panc-1 cells (Fig. 5J–5K), which was blocked by Erastin. Conversely, MCU KO inhibited FITC-cystine uptake (Fig. 5J–5K). Taken together, our data indicate that the MCU upregulates SLC7A11 and cystine uptake through Nrf2.
Activation of mitochondrial Ca2+ signaling promotes cystine addiction and sensitize PDAC cells to ferroptosis.
Cystine deprivation induced the transcription of “Ferroptosis Signature” genes characterized by activation of the Gcn2-eIF2α-ATF4 branch of integrated stress response (43, 45). Our RNA sequencing data revealed robust upregulations of Ferroptosis Signature genes in MCU overexpressing cells (Fig. 6A–6B). We hypothesized that mROS production in MCU-upregulated PDAC might lead to increased consumption of cysteine required for antioxidant response. The high demand for cystine in MCU OE PDAC cells could lead to intracellular cysteine deficiency despite SLC7A11 upregulation. MCU OE PDAC cells indeed had lower intracellular levels of cysteine/cystine, which could be rescued by supplementation of additional cystine (1mM) (Fig. S6A). LC-MS (liquid chromatography-mass spectrometry) assay coupled with NEM (n-ethyl maleimide) derivatization showed that, MCU OE also reduced the levels of intracellular cysteine, which could be rescued by cystine supplementation (Fig. 6C).
Figure 6. Activation of mitochondrial Ca2+ signaling promotes cystine addiction and sensitize PDAC cells to ferroptosis.
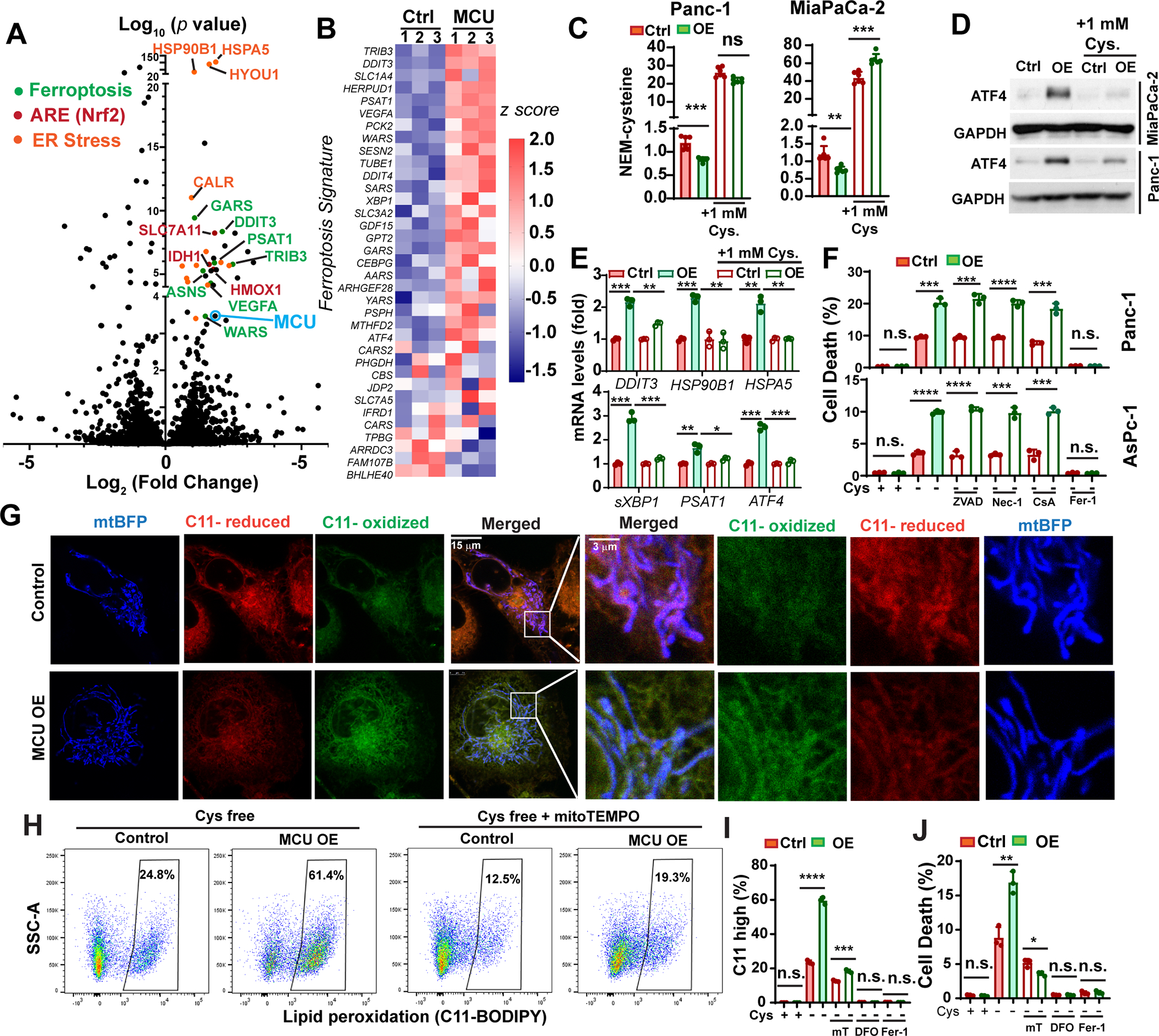
A, Volcano plot showing genes differentially regulated in MCU OE Panc1 cells when compared to control cells, as determined by RNA sequencing. Some of the ARE genes, ER-stress response genes and Ferroptosis Signature genes were labeled as indicated.
B, the differential expression of Ferroptosis Signature genes in control and MCU OE Panc-1 cells.
C, the effects of MCU OE and supplementation of extracellular cystine (1mM) on intracellular cysteine levels in two PDAC cell lines as determined by the NEM-cysteine assay and LC-MS.
D and E, the effects of MCU OE and cystine supplementation on ATF4 protein levels (D) and the mRNA transcript levels of ER-stress / Ferroptosis Signature genes (E) in two PDAC cell lines.
F, the effects of MCU OE on cell death induced by cystine-deprivation (cystine-free medium) in the presence of ZVAD (50 μM), Necrostatin-1 (Nec-1, 10 μM), cyclosporine A (CsA, 10 μM) or ferrostatin-1 (Fer-1, 1μM).
G, confocal microscopy and C11-BODIPY (C11) staining showing the levels and subcellular localization of lipid peroxide in control and MCU OE Panc-1 cells when treated with cystine-free medium for 6 hours. Reduced and oxidized C11-BODIPY were detected at emission 590 nm and 510 nm, respectively.
H, representative C11-BODIPY flow cytometry experiment showing the effect of MCU OE and mitoTEMPO treatment on lipid peroxidation when cultured in cystine-free medium for 18 hours.
I, quantitation of the effects of treatments with mitoTEMPO (mT, 2μM), DFO (Deferoxamine, 50 μM) or ferrostatin-1 (Fer-1, 1μM) on lipid peroxidation in control and MCU OE Panc-1 cells when cultured in cystine free medium (18 hours).
J, the effect of mT, DFO, Fer-1 treatment on cystine-deprivation-induced ferroptosis in control and MCU OE Panc-1 cells.
Data in C, E, F, I-K and N were presented as mean ± SD from three biological replicates and analyzed using two-tailed, two sample unpaired Student’s t-test. *, **, *** and **** indicated p<0.05, 0.01, 0.001 and 0.0001, respectively. n.s. indicated not statistically significant.
We observed marked upregulation of ATF4 protein levels and the expression Ferroptosis Signature transcripts in MCU OE cells (Fig.6D–6E). Importantly, cystine supplementation in the medium reduced the levels of ATF4 and Ferroptosis Signature genes in MCU OE cells (Fig. 6D–6E).
We hypothesized that cystine addiction in MCU overexpressing PDAC might constitute a therapeutic vulnerability. To investigate this possibility we used three different approaches (cystine-free medium, glutamate or Erastin) to suppress cystine uptake. MCU overexpressing PDAC cells were hypersensitive to cystine-deprivation induced cell death (Fig. 6F and Fig. S6B). In contrast, MCU slightly reduced sensitivities to ferroptosis induced by GPX4 inhibitor RSL3 (Fig. S6C). Conversely, MCU KO inhibited ferroptosis induced by cystine-deprivation (Fig. S6D). Cystine deprivation-induced cell death in MCU OE cells could be rescued by ferroptosis inhibitor ferrostatin-1 (Fer-1), but not by ZVAD, Nec-1 or cyclosporine A (Fig. 6F), which suggested that MCU promotes ferroptosis induced by cystine deprivation (but not by GPX4 inhibition).
It was recently reported that mROS plays a critical role in promoting ferroptosis (35, 46). We observed that MCU promoted lipid peroxidation in PDAC cells 3 to 6 hours after cystine-deprivation and some of the lipid peroxidation colocalized with mitochondria (Fig.6G and Fig. S6E). Flow cytometry analysis confirmed MCU OE elevated lipid peroxidation, while MCU KO decreased lipid peroxidation (Fig. 6H–6I and Fig. S6F–S6G) in cystine-deprived PDAC cells. The MCU-mediated lipid peroxidation could be rescued by mROS scavenging with mitoTEMPO (Fig. 6H–6I). mitoTEMPO also abrogated the ferroptosis-promoting effects of MCU (Fig. 6J). Taken together, these data support that MCU-mediated mROS is responsible for cystine addiction and promoting ferroptosis in PDAC.
It has been previously reported that inhibition of NCLX was able to increase mCa2+ and mROS levels (17, 47). As shown in Fig. S6H, inhibition of NCLX with the pharmacological inhibitor CGP37157 did not affect PDAC cell viability in the presence of cystine; however, when combined with cystine deprivation CGP37157 robustly increased ferroptosis (Fig. S6H) and lipid peroxidation (Fig. S6I–S6J). The effect of CGP37157-mediated cell death and lipid peroxidation could be abrogated by DFO or Fer-1 (Fig. S6H–S6J). Our data suggested that elevation of mCa2+ is sufficient to sensitize PDAC cells to ferroptosis.
MCU-driven cystine addiction is a therapeutic vulnerability that can be exploited to prevent PDAC metastasis
Next, we examined whether MCU-driven cystine addiction could be exploited as a therapeutic vulnerability using SLC7A11 inhibitors sulfasalazine (SAS) and Imidazole Ketone Erastin (IKE) (Fig. 7A–7B, S7A). BLI data showed that SAS and IKE has much stronger inhibitory effects on tumor growth in MCU overexpressing groups (Fig. 7A–7B). After euthanasia the primary tumor growth and distant metastases in these mice were analyzed (Fig. 7C–7F). MCU significantly promoted metastasis to both the liver and the peritoneal cavity (P. C.) (Fig. 7C–7E) and moderately increased tumor weight (Fig. 7F). The medium BLI signals from metastatic lesion in the MCU OE (vehicle) group were more than 10 times higher than that in the control group (vehicle) (Fig. 7E). These data support the notion that MCU promotes pancreatic cancer metastasis.
Figure 7. MCU-driven cystine addiction is a metabolic vulnerability that can be exploited to prevent PDAC metastasis.
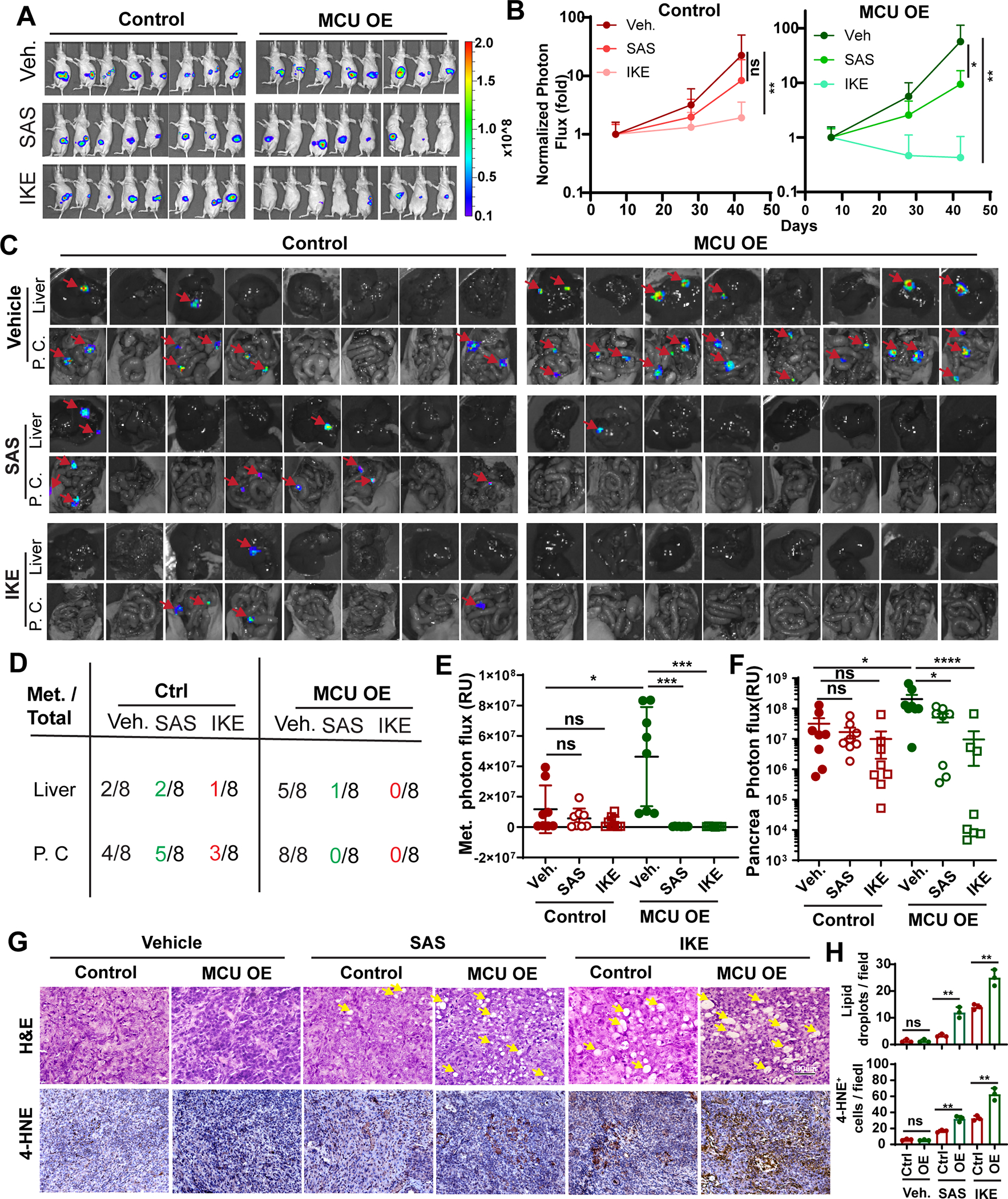
A, BLI imaging of mice orthotopically implanted with control of MCU OE Panc-1 cells 49 days after surgery. Mice were treated with either saline vehicle (Veh.), sulfasalazine (SAS, 100 mg/kg) or IKE (20mg/kg) everyday starting from day 7.
B, quantitation of BLI signals from experiments in (A) and day 7, 28 and 49. BLI photon flux signals on day 28 and 42 were normalized against signals from day 7 in each group. n=8. Data were analyzed using two-way ANOVA.
C, ex vivo BLI imaging showing the development of metastases lesions (red arrows) in liver and peritoneal cavity (P. C.) in mice from (A) after euthanasia at day 49. N=8
D, summary of total metastasis foci numbers from ex vivo imaging experiments in (C).
E, quantitation of BLI photon flux signals from ex. vivo imaging of metastases. n=8
F, quantitation of tumor weight harvested from orthotopic xenograft experiment in (A).
G, H&E and 4-HNE staining showing lipid-droplet like structures (yellow arrow) and lipid peroxidation (4-HNE positive cells) in harvested orthotopic tumors.
H, quantitation of the numbers of lipid droplets and 4-HNE positive cells per 40x field in three orthotopic tumors from each group. n=3
Data in E, F and H were presented as mean ± SD from 3–8 biological replicates and analyzed using two-tailed, Mann-Whitney test (E and F) or unpaired Student’s t-test. *, ** and **** indicated p<0.05, 0.01, and 0.0001, respectively. n.s. indicated not statistically significant.
The IKE treatment in the MCU OE group resulted in complete tumor regression in 50% of mice (Fig. 7A–7B, Fig. S7B) and abolished the development of liver and peritoneal metastasis (Fig. 7C–7E). SAS treatment led to strong inhibition of both liver and peritoneal metastasis in the MCU OE group (Fig. 7C–7E). The efficacies of IKE was much stronger than SAS, probably due to the fact that IKE (IC50: 34 nM) was a much more potent inducer of ferroptosis than SAS (IC50: 160 μM). Hematoxylin and eosin (H&E) staining of harvested orthotopic tumor revealed formation of lipid droplet-like structures (Fig. 7G, arrow) in SAS and IKE treated mice, which is consistent with a previous report (45). SAS and IKE-induced larger and more numerous lipid-droplets, more intense 4-HNE staining and large areas of necrosis in MCU OE groups (Fig. 7G–7H, Fig. S7C), which is suggestive of increased ferroptosis in these tumors. Taken together, these data support that MCU overexpressing PDAC are hypersensitive to cystine-deprivation and SLC7A11 inhibitors could be used to prevent metastatic recurrence among these PDAC patients.
MCU-high PDAC are more sensitive to cystine-deprivation
We prepare PDOs from 3 MCU-high and 3 MCU-low PDAC patients (Fig. S8A) and tested their sensitivities to Erastin. As shown in Fig. 8A and 8B, Erastin treatment inhibited the growth of the MCU-low and MCU-high PDO lines by 52.6±3.1% and 76.0±9.1%, respectively, which suggested MCU-high PDAC patients might be more sensitive to cystine-deprivation treatment. To further examine this notion, we implanted PDX tumors derived from 2 of the MCU-high patients (patient 677 and 977) and 2 of the MCU-low patients (patient 022 and 081) into NSG mice (Fig. S7B). After the tumors reached 50–70 mm3, mice were treated with vehicle control or IKE (20 mg/kg) for 40 days. IKE treatment inhibited the growth of the two MCU-high PDX tumor lines by 73.4±4.5% (PDX677) to 88.4±2.7% (PDX977) (Fig. 8C). In contrast, the inhibitory effects of IKE on the growth of MCU-low PDX tumors were only 14.7±12.8% (PDX202) to 31.4±8.0% (PDX081) (Fig. 8C). IKE treatment induced significantly more lipid droplets and stronger 4-HNE staining in MCU-high PDX tumors (Fig. 8D–8E and Fig. S7C–S7D). To determine whether MCU plays a causative role in cystine addiction, we used CRISPR/Cas9 to knockout MCU in cell lines derived from the MCU-high PDX677 (Fig. S8E). MCU KO abrogated the anti-tumor effects of IKE treatment in this MCU-high PDX line (Fig. 8F, 8G, S8F), which suggested that MCU is required for sensitivities to IKE. Taken together, MCU-high PDAC are more sensitive to SLC7A11 inhibition than MCU-low tumors. The cystine-addiction in MCU-high PDAC patients could be exploited as a therapeutic vulnerability to inhibit tumor growth and prevent metastasis.
Figure 8. MCU-high PDAC are more sensitive to SLC7A11 inhibitors in PDO and PDX models.
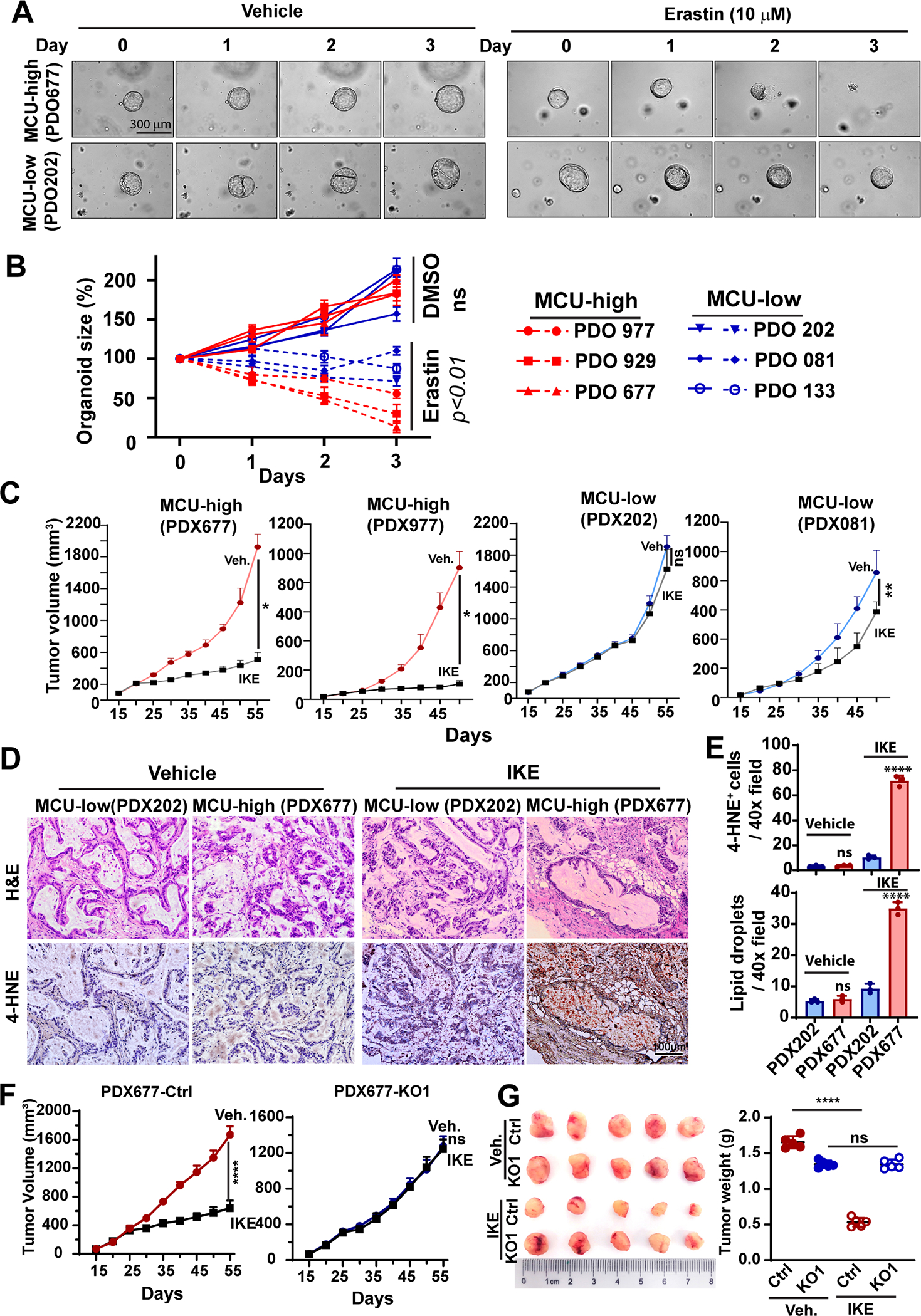
A and B, representative bright field microscopy (A) or quantitation of organoid growth over time (B) showing the effects of Erastin (10 μM) on the growth of PDOs (patient-derived orgonids) prepared from MCU-high or MCU-low PDAC patients.
C, the effects of IKE administration on the tumor growth in PDX lines derived from MCU-high (PDX677 and PDX977) or PDX-low (PDX202 and PDX081) PDAC patients. n=5 mice per group.
D and E, representative H&E staining and 4-HNE staining images (D) and quantitation (E) of the presensence of 4-HNE+ cells or large lipid droplets in PDX tumors harvested from experiments in C.
F and G, the effects of MCU KO on the response to IKE treatment in the the MCU-high PDX677, as shown by tumor volume growth curves (F) and the weight of tumors harvested at the end of experiment (G).
Data in B, C, E-G were presented as mean ± SD from 3–5 biological replicates and analyzed using two-tailed, Mann-Whitney test (E and F) or unpaired Student’s t-test. *, ** and **** indicated p<0.05, 0.01, and 0.0001, respectively. n.s. indicated not statistically significant.
Discussion
Pancreatic cancer is a highly metastatic tumor malignancy with poor prognosis and limited treatment options. Here we showed that MCU is upregulated during pancreatic cancer initiation and progression. MCU promotes cell migration, invasion and metabolic / oxidative stress resistance in cell culture and xenograft models. The mitochondria are the major source of ROS in the cell. The elevated mCa2+ increases mROS by promoting the tricarboxylic acid cycle and oxidative phosphorylation (11). ROS play complicate and context-dependent role in cancer initiation and progression. Although it is known that excessive ROS increase could lead to cell death, it is also well established that modest ROS production could promote cell migration, invasion and metastasis by activating HIF1α signaling and inhibiting protein phosphatases such as PTEN (18). Our data show that by activating the Keap1-Nrf2 antioxidant response, MCU-mediated mROS signal contribute to promoting metabolic stress resistance and metastatic progression.
Cancer cells depend on SLC7A11-mediated cystine uptake for GSH synthesis antioxidant response (44). GSH is required for GPX4 to reduce lipid peroxidation and prevent ferroptosis (48). Although mitochondria might not be essential for ferroptosis, mitochondria-derived ROS promotes ferroptosis induced by cystine-deprivation, but not by GPX4 inhibitors (35). By increasing mROS, MCU remarkably increased lipid peroxidation and ferroptosis under cystine-deprivation. Interestingly, MCU modestly promote resistance to ferroptosis induced by GPX4 inhibitor RSL3. Among cysteine metabolites, Coenzyme A has been shown to have anti-ferroptosis activities (45).
Our data supported that the MCU-mediated antioxidant response and metastatic progression has high demand for cystine-uptake from the tumor microenvironment. When cystine is available, MCU-mediated mROS oxidize Keap1 and upregulates Nrf2 antioxidant response to promote metabolic stress resistance and pancreatic cancer metastasis. However, upon SLC7A11 inhibition or cystine deprivation, MCU-mediated mROS promotes lipid peroxidation and exacerbates ferroptosis. Therefore, the cystine addiction in MCU overexpressing PDAC could be potentially exploited as a metabolic vulnerability. There are FDA-approved drugs (e.g. sorafenib, SAS) that could be repurposed to inhibit SLC7A11 (43). More specific inhibitors with higher potencies and favorable pharmacokinetics (e.g. IKE) have also been developed (49). These inhibitors could be employed to prevent metastatic recurrence in MCU overexpressing PDAC.
Supplementary Material
Statement of Significance.
Elevated mitochondrial calcium uptake in pancreatic ductal adenocarcinoma promotes metastasis but exposes cystine addiction and ferroptosis sensitivity that could be targeted to improve pancreatic cancer treatment.
Acknowledgement
We would like to thank Dr. Katherine Aird for assistance with the use of YSI 2700 Biochemical Analyzer, Ryan Yoast for assistance with mitochondrial calcium uptake assay and Yuka Imamura for help with RNA sequencing data analysis. S. Yang is supported by grants from the National Institute of Heath (R01CA233844, R01CA256911). J. Hao is supported by National Natural Science Foundation of China (grants 81720108028, 81672431).
Footnotes
Conflict of interest:
The authors declare no conflict of interest
Reference
- 1.Groot VP, Rezaee N, Wu W, Cameron JL, Fishman EK, Hruban RH, et al. Patterns, Timing, and Predictors of Recurrence Following Pancreatectomy for Pancreatic Ductal Adenocarcinoma. Ann Surg. 2018;267(5):936–45. [DOI] [PubMed] [Google Scholar]
- 2.Sancho P, Barneda D, and Heeschen C. Hallmarks of cancer stem cell metabolism. Br J Cancer. 2016;114(12):1305–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Viale A, Pettazzoni P, Lyssiotis CA, Ying H, Sanchez N, Marchesini M, et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature. 2014;514(7524):628–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lin S, Huang C, Sun J, Bollt O, Wang X, Martine E, et al. The mitochondrial deoxyguanosine kinase is required for cancer cell stemness in lung adenocarcinoma. EMBO molecular medicine. 2019;11(12):e10849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vultur A, Gibhardt CS, Stanisz H, and Bogeski I. The role of the mitochondrial calcium uniporter (MCU) complex in cancer. Pflug Arch Eur J Phy. 2018;470(8):1149–63. [DOI] [PubMed] [Google Scholar]
- 6.Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature. 2011;476(7360):341–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.De Stefani D, Rizzuto R, and Pozzan T. Enjoy the Trip: Calcium in Mitochondria Back and Forth. Annual review of biochemistry. 2016;85:161–92. [DOI] [PubMed] [Google Scholar]
- 8.De Stefani D, Patron M, and Rizzuto R. Structure and function of the mitochondrial calcium uniporter complex. Biochim Biophys Acta. 2015;1853(9):2006–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Palty R, Silverman WF, Hershfinkel M, Caporale T, Sensi SL, Parnis J, et al. NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc Natl Acad Sci U S A. 2010;107(1):436–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pan X, Liu J, Nguyen T, Liu C, Sun J, Teng Y, et al. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat Cell Biol. 2013;15(12):1464–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Delierneux C, Kouba S, Shanmughapriya S, Potier-Cartereau M, Trebak M, and Hempel N. Mitochondrial Calcium Regulation of Redox Signaling in Cancer. Cells. 2020;9(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu JC, Liu J, Holmstrom KM, Menazza S, Parks RJ, Fergusson MM, et al. MICU1 Serves as a Molecular Gatekeeper to Prevent In Vivo Mitochondrial Calcium Overload. Cell reports. 2016;16(6):1561–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mallilankaraman K, Doonan P, Cardenas C, Chandramoorthy HC, Muller M, Miller R, et al. MICU1 is an essential gatekeeper for MCU-mediated mitochondrial Ca(2+) uptake that regulates cell survival. Cell. 2012;151(3):630–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ren T, Wang J, Zhang H, Yuan P, Zhu J, Wu Y, et al. MCUR1-Mediated Mitochondrial Calcium Signaling Facilitates Cell Survival of Hepatocellular Carcinoma via Reactive Oxygen Species-Dependent P53 Degradation. Antioxid Redox Signal. 2018;28(12):1120–36. [DOI] [PubMed] [Google Scholar]
- 15.Ren T, Zhang H, Wang J, Zhu J, Jin M, Wu Y, et al. MCU-dependent mitochondrial Ca(2+) inhibits NAD(+)/SIRT3/SOD2 pathway to promote ROS production and metastasis of HCC cells. Oncogene. 2017;36(42):5897–909. [DOI] [PubMed] [Google Scholar]
- 16.Tosatto A, Sommaggio R, Kummerow C, Bentham RB, Blacker TS, Berecz T, et al. The mitochondrial calcium uniporter regulates breast cancer progression via HIF-1alpha. EMBO molecular medicine. 2016;8(5):569–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pathak T, Gueguinou M, Walter V, Delierneux C, Johnson MT, Zhang X, et al. Dichotomous role of the human mitochondrial Na(+)/Ca2(+)/Li(+) exchanger NCLX in colorectal cancer growth and metastasis. Elife. 2020;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reczek CR, and Chandel NS. The Two Faces of Reactive Oxygen Species in Cancer. Annu Rev Canc Biol. 2017;1:79–98. [Google Scholar]
- 19.Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, et al. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999;13(1):76–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mitsuishi Y, Motohashi H, and Yamamoto M. The Keap1-Nrf2 system in cancers: stress response and anabolic metabolism. Front Oncol. 2012;2:200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jaramillo MC, and Zhang DD. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev. 2013;27(20):2179–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tonelli C, Chio IIC, and Tuveson DA. Transcriptional Regulation by Nrf2. Antioxid Redox Signal. 2018;29(17):1727–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gorrini C, Harris IS, and Mak TW. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov. 2013;12(12):931–47. [DOI] [PubMed] [Google Scholar]
- 24.Kensler TW, Wakabayashi N, and Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. 2007;47:89–116. [DOI] [PubMed] [Google Scholar]
- 25.Chio IIC, Jafarnejad SM, Ponz-Sarvise M, Park Y, Rivera K, Palm W, et al. NRF2 Promotes Tumor Maintenance by Modulating mRNA Translation in Pancreatic Cancer. Cell. 2016;166(4):963–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.DeNicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K, et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature. 2011;475(7354):106–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wiel C, Le Gal K, Ibrahim MX, Jahangir CA, Kashif M, Yao H, et al. BACH1 Stabilization by Antioxidants Stimulates Lung Cancer Metastasis. Cell. 2019;178(2):330–45 e22. [DOI] [PubMed] [Google Scholar]
- 28.Lignitto L, LeBoeuf SE, Homer H, Jiang S, Askenazi M, Karakousi TR, et al. Nrf2 Activation Promotes Lung Cancer Metastasis by Inhibiting the Degradation of Bach1. Cell. 2019;178(2):316–29 e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.DeNicola GM, Chen PH, Mullarky E, Sudderth JA, Hu Z, Wu D, et al. NRF2 regulates serine biosynthesis in non-small cell lung cancer. Nat Genet. 2015;47(12):1475–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell. 2017;171(2):273–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gaschler MM, and Stockwell BR. Lipid peroxidation in cell death. Biochem Biophys Res Commun. 2017;482(3):419–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Conrad M, Kagan VE, Bayir H, Pagnussat GC, Head B, Traber MG, et al. Regulation of lipid peroxidation and ferroptosis in diverse species. Genes Dev. 2018;32(9–10):602–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hassannia B, Vandenabeele P, and Vanden Berghe T. Targeting Ferroptosis to Iron Out Cancer. Cancer Cell. 2019;35(6):830–49. [DOI] [PubMed] [Google Scholar]
- 35.Gao M, Yi J, Zhu J, Minikes AM, Monian P, Thompson CB, et al. Role of Mitochondria in Ferroptosis. Molecular cell. 2019;73(2):354–63 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang C, Li Z, Li N, Li Y, Chang A, Zhao T, et al. Interleukin 35 Expression Correlates With Microvessel Density in Pancreatic Ductal Adenocarcinoma, Recruits Monocytes, and Promotes Growth and Angiogenesis of Xenograft Tumors in Mice. Gastroenterology. 2018;154(3):675–88. [DOI] [PubMed] [Google Scholar]
- 37.Huang C, Li N, Li Z, Chang A, Chen Y, Zhao T, et al. Tumour-derived Interleukin 35 promotes pancreatic ductal adenocarcinoma cell extravasation and metastasis by inducing ICAM1 expression. Nature communications. 2017;8:14035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang X, Li X, Wei X, Jiang H, Lan C, Yang S, et al. PD-L1 is a direct target of cancer-FOXP3 in pancreatic ductal adenocarcinoma (PDAC), and combined immunotherapy with antibodies against PD-L1 and CCL5 is effective in the treatment of PDAC. Signal Transduct Target Ther. 2020;5(1):38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tiriac H, Belleau P, Engle DD, Plenker D, Deschenes A, Somerville TDD, et al. Organoid Profiling Identifies Common Responders to Chemotherapy in Pancreatic Cancer. Cancer Discov. 2018;8(9):1112–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lu F, Li Y, Lin S, Cheng H, and Yang S. Spatiotemporal regulation of store-operated calcium entry in cancer metastasis. Biochem Soc Trans. 2021;49(6):2581–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mo P, and Yang S. The store-operated calcium channels in cancer metastasis: from cell migration, invasion to metastatic colonization. Front Biosci (Landmark Ed). 2018;23:1241–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lin S, Taylor MD, Singh PK, and Yang S. How Does Fascin Promote Cancer Metastasis? Febs J 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, Hayano M, et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife. 2014;3:e02523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Muir A, Danai LV, Gui DY, Waingarten CY, Lewis CA, and Vander Heiden MG. Environmental cystine drives glutamine anaplerosis and sensitizes cancer cells to glutaminase inhibition. Elife. 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Badgley MA, Kremer DM, Maurer HC, DelGiorno KE, Lee HJ, Purohit V, et al. Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science. 2020;368(6486):85–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gao M, Monian P, Quadri N, Ramasamy R, and Jiang X. Glutaminolysis and Transferrin Regulate Ferroptosis. Molecular cell. 2015;59(2):298–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ben-Kasus Nissim T, Zhang X, Elazar A, Roy S, Stolwijk JA, Zhou Y, et al. Mitochondria control store-operated Ca(2+) entry through Na(+) and redox signals. EMBO J. 2017;36(6):797–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156(1–2):317–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang Y, Tan H, Daniels JD, Zandkarimi F, Liu H, Brown LM, et al. Imidazole Ketone Erastin Induces Ferroptosis and Slows Tumor Growth in a Mouse Lymphoma Model. Cell Chem Biol. 2019;26(5):623–33 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA sequencing data generated in this study are publicly available in Gene Expression Omnibus (GEO) at GSE199692.