

Summary
A virulence bacterium, Helicobacter pylori, evolved parallel to its host human, therefore, can work as a marker for tracing the human migration. We found H. pylori strains indigenous in the southernmost islands of Japanese Archipelago, Okinawa, and defined them as hspOkinawa and hpRyukyu. Genome data of the strains revealed that hspOkinawa diverged from other East Asian strains about 20,000 years ago, and that hpRyukyu diverged about 45,000 years ago. The closest strains of hpRyukyu were found from Afghanistan, Punjab, and Nepal, which suggest this strain originated in the central Asia and traveled across the Eurasian continent during Paleolithic era. The divergence date of hpRyukyu corresponds with human fossil records in Okinawa. Although it is controversial from human DNA analyses whether descendants of the Paleolithic migrants remain in the modern Japanese population, this study reveals that the bacterium of Paleolithic origin remains in the stomachs of current Japanese.
Subject areas: Gastroenterology, Bacteriology, Medical Microbiology, Microbial genetics, Phylogeny, Evolutionary history
Graphical abstract
Highlights
-
•
H. pylori strains in Okinawa, Japan, have a different origin from the main island
-
•
One of the Okinawa-specific strains originate in Western Asia in the Paleolithic era
-
•
Our results suggest ancient human migration from west to the east end of Eurasia
Gastroenterology; Bacteriology; Medical Microbiology; Microbial genetics; Phylogeny; Evolutionary history
Introduction
H. pylori adapted to the human stomach before the Out-of-Africa movement of anatomically modern humans around 70,000 years ago, and now approximately half of the global population carries H. pylori (Covacci et al., 1999). This bacterium transmits mainly from family members in childhood, and the infection continues throughout life unless eradicated (Kuipers et al., 2000). Because the evolution of H. pylori parallels that of humans, we can infer human migrations through genetic analyses of H. pylori (Linz et al., 2007; Moodley et al., 2009).
Conventionally, population and phylogenetic analyses of H. pylori have been conducted based on a multi-locus sequence typing (MLST) method utilizing concatenated sequences of seven housekeeping genes (atpA, efp, mutY, ppa, trpC, ureI, and yphC) (Maiden et al., 1998). Seven major bacterial populations of H. pylori (hpAfrica1, hpAfrica2, hpNEAfrica, hpEurope, hpAsia2, hpEastAsia, and hpSahul) (Falush et al., 2003b; Linz et al., 2007; Moodley et al., 2009) were identified, and the phylogenetic relationship of these seven populations illuminated the modern human dispersal from Africa to the Eurasian continent (Linz et al., 2007), to the Americas (Falush et al., 2003b; Moodley and Linz, 2009), and to Oceania (Moodley et al., 2009). HpEastAsia is further divided into three subpopulations: hspEAsia that distributes in the eastern area of the Eurasian continent, hspMaori that expanded from Taiwan to Oceania, and hspIndigenousAmericas (Moodley et al., 2021) that migrated to North and South America via the Bering Land Bridge.
In our previous study based on the MLST method, we found two H. pylori populations specific to Okinawa: one of the populations diverged earlier than any of the East Asian strains, and the other one diverged more recently but makes a distinct cluster within hpEastAsian strains (Matsunari et al., 2012). Okinawa area consists of the southmost islands of the Japanese Archipelago and ruled by Ryukyu dynasty before they were merged by the main island government in 1879. In this study, we name the group with the older divergence as hpRyukyu and the group with more recent divergence as hspOkinawa, regarding this group as a sub-population of hpEastAsia. HpRyukyu and hspOkinawa strains account for 16% and 14% of current Okinawa strains, respectively, and the rest belong to hspEAsia, the same group of strains on the main island of Japan.
Our previous study also revealed that hpRyukyu and hspOkinawa strains are less virulent than hspEAsia strains because of the difference of CagA, one of the major virulence genes of H. pylori (Matsunari et al., 2012). HpRyukyu strains possess CagA whose amino acid motif is similar to that of hpEurope strains. However, the CagA motif of hpRyukyu was originally found only from Okinawa but no other area. Therefore, it was named as J-Western CagA as “J” stands for “Japan.” HspOkinawa strains lack CagA, so the least virulent.
Mostly, all of strains in the main island of Japan belong to hspEAsia, which are also common in China and Korea. It was probably brought by people from the continent with rice cultivation. In contrast, the origin of hpRyukyu and hspOkinawa was enigmatic. We investigated virulence genes and seven housekeeping genes only in our previous study but advancement of DNA sequencers enabled us to obtain the whole genomes data of H. pylori. We investigated not only the whole genomes of Okinawa strains but also the genomes of global strains to speculate when and from where the host human population arrived at Japan.
Results
Population classification and genome tree
First, we defined hpRyukyu and hspOkinawa by the MLST method using STRUCTURE (Falush et al., 2003a) and DAPC (Jombart et al., 2010). The results are shown in Figures S1–S5 and Tables S1 and S2. We obtained genome data of H. pylori from Okinawa (hpRyukyu and hspOkinawa), Alaska, Colombia, Thailand, Bhutan, and Nepal sequenced by a PacBio, and reconstructed the complete whole genome totaling 21 strains (Table S3). Alaskan and Colombian strains were those collected from aboriginal Americans. We integrated our genome data with the data obtained from the public database. We concatenated a sequence of 619 orthologous genes totaling 88 strains (495,102 bp) and constructed a phylogenetic tree (Figure 1).
Figure 1.

Phylogenetic tree using genome data of 88 strains
NJ-tree (Kimura 2-parameter) based on concatenated sequences of 619 genes (alignment length 495,102 bp. Encircled strains are those we sequenced by PacBio for this study. The squares on the right side of the tree represent population classification by STRUCTURE analysis of MLST data. The marks on the right of the boxes represent cagA genotype: ▵ cagA (−), ∗ J-Western cagA, a: Amerind cagA, e: East Asian cagA, w: Western cagA.
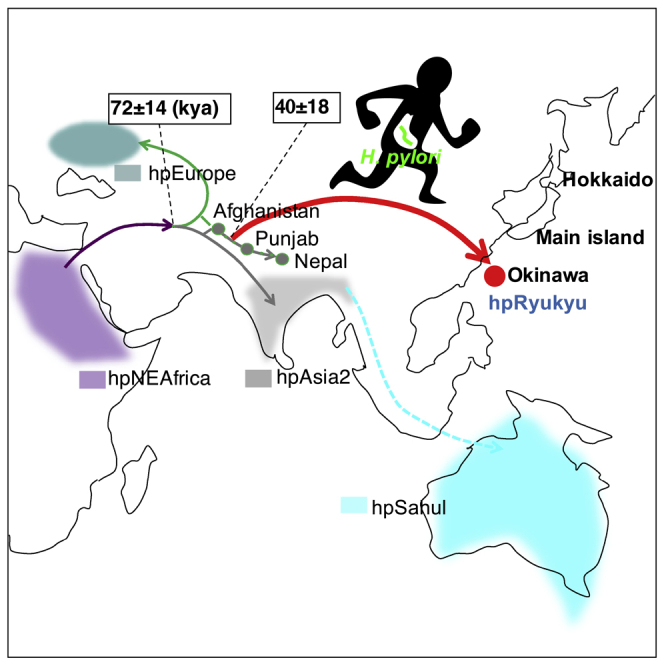
Of the 88-sequence data, HP14065, UM211, UM139, and HP15060 were not complete genomes but contig sequences (Chua et al., 2019). First, we could find only one strain that is close to hpRyukyu, a Nepalian strain NP05-124, in the complete genome data, then we explored H. pylori contig sequences deposited in GenBank and found the above four strains. We asked the author of the paper about the origin of these strains and were informed that HP14065, UM211, UM139, and HP15060 were from Afghanistan, unknown origin, Punjab, and Nepal, respectively.
The hspOkinawa sub-branch was located between hspEAsia and hspIndigenousAmericas strains, as displayed by the colored squares on the right of the branches. The hspIndigenousAmericas strains were further divided into North (Alaska and Canada) and South (Peru, Colombia, and Venezuela) American sub-branches. Hokkaido strains, which were isolated from northern aboriginal Japanese, Ainu, belonged to the North American branch.
Figure 1 also shows the genotypes of cagA, one of the well-known H. pylori virulence genes. CagA is one of the member genes of cag pathogenicity island (cagPAI) and classified as Western cagA, East Asian cagA, Amerind cagA, and J-Western cagA (Figure S6). Because cagPAI is not essential for survival, some strains lack cagPAI (cagA (−)).
HspOkinawa strains are cagA (−) and so are the North American and Hokkaido strains. HpRyukyu has J-Western cagA whose amino acid motifs are similar to the Western type but discriminated by other features (Matsunari et al., 2012). The “J” of J-Western came from Japan because this genotype was first observed only in Okinawa, Japan. However, the Afghanistan and Nepal strains also have J-Western cagA, as well as some European and African strains. Our previous study on Nepalese strains reported that there were three bacterial populations: one was typical Asia2 strains, the second was recombinants between hpAsia2 and hpEurope strains, and the third was recombinants between hpAsia2 and Nepal-specific strains (Miftahussurur et al., 2015). The Nepal-specific strains may have relation with hpRyukyu. In the third population, strains containing high ratio of the Nepal-specific component was rare, which coincident with the fast that we investigated a lot of Nepalese strains but only a few of them were close to hpRyukyu.
Scarce exogenous influence on Okinawa strains
H. pylori imports exogenous DNA frequently, and therefore its genome contains many recombined regions (Yahara et al., 2012, 2013). We estimated the intensity of DNA exchange from one strain to another using ChromoPainterv2 (Falush et al., 2003a) and fineSTRUCTURE (Falush et al., 2003a), in which the genome of one H. pylori cell is regarded as a series of chunks imported from other H. pylori cells through recombination. The result suggests that hpRyukyu and hspOkinawa had very little influence from other populations, even between the two Okinawa populations (Figure S7). This observation suggests that hpRyukyu and hspOkinawa had different origins and co-existed in Okinawa without hybridization.
Estimation of the divergence dates
Based on the populations defined by fineSTRUCTURE, we selected representative strains to estimate divergence dates. We detected recombination signals by using the 4-gamete test and eliminated recombinant regions. Then, we constructed a Bayesian tree using BEAST2 (Bouckaert et al., 2014) and estimated the divergence dates. The divergence date of hspOkinawa was estimated around 20 thousand years ago (kya), and that of the cluster including hpRyukyu and Afghanistan-Nepal strains was estimated around 45 kya (Figure 2).
Figure 2.

Bayesian tree of representative 24 strains
The colored circles on the map indicate geographic regions of Japan and correspond to the colored circles on the H. pylori strains. Bacterial populations determined by STRUCTURE are shown by red letters. Calibration point C-1 was set at the divergence of non-African strains as lower 50 kya and upper 100 kya according to archaeological information (Bae et al., 2017; Sikora et al., 2017).
We chose oki422 as the representative strain of hpRyukyu. In Figure 2, oki422 branched out after the Afghanistan strain, although Figure 1 shows that the four hpRyukyu strains formed a sub-branch outside the Afghanistan-Nepal cluster. In either case, hpRyukyu diverged earlier than Punjab and Nepal. This suggests that hpRyukyu did not come through the extension of the Punjab-Nepal line.
Gene flow from hpRyukyu to others
The area from Afghanistan to Nepal is far away from Japan and the path from the area to Okinawa is unknown. We considered a north path that runs the north of Himalayas and a south path that runs southern coastal line. To evaluate which path is more probable, we analyzed the gene flow from hpRyukyu to others using f-statistics (fhom) (Martin et al., 2015) (Table S1). The f-statistic is a similar index to the ABBA-BABA-test (d-statistics). These statistics compare four taxa that represent three populations and one outgroup, for example, ((H1, H2). H3), H4) as H4 is the outgroup. The basic idea is that alleles shared only between H1 and H4 should be as equally frequent as that of between H2 and H4, if the allele sharing arose from incomplete lineage sorting without gene flow. Then, a skew of the sharing frequency between H1-H4 and H2-H4 is indicative of gene flow. Though the formulation of f-statistics and d-statistics differs slightly, the basic idea is the same.
We used SouthAfrica20 as the base (H4). H3 to H1 were set by branching order in Figure 2. H3 is hpRyukyu and Afghanistan-Nepal strains. SNT49 and PNG84A in H2 are strains of hpAsia2 and hpSahul, respectively. If the gene flow from H3 to H2 is stronger than that from H3 to H1, the value is positive, and vice versa. EA-Am in H1 represents 13 strains in East Asia and the Americas (strains from 51 to shi470 in Figure 2), and the f-statistics is the average of the values calculated on each EA-Am strain.
The f-statistic values were positive when SNT49 (hpAsia2 strain from India) was set to H2. This means the gene flow from the H3 strains (hpRyukyu and Afghanistan-Nepal strains) to the Indian strain is stronger than the gene flow to H1 strains (PNG84A, hpSahul strain from Papua New Guinea, and East Asian strains). This implies that the gene flow was [H3] -> [India] -> [Oceania, East Asia].
In contrast, f-statistics values were all negative when the hpSahul strain was set to H2. This result suggests that gene flow from hpRyukyu to EA-Am was stronger than that from hpRyukyu to hpSahul, i.e., the gene flow skipped hpSahul. HpSahul is considered to have expanded via coastal line to reach Oceania (Moodley et al., 2009). If the ancestral hosts of hpRyukyu moved toward the south, there should be gene flow between hpRyukyu and hpSahul before hpRyukyu reached East Asia. The weaker gene flow from hpRyukyu to hpSahul than that from hpRyukyu to East Asia implies that the ancestral hosts of hpRyukyu might not take a southern course. Rather, hpRyukyu might go to the north circumventing the Himalayas.
Discussion
Paleolithic migration to the east end
Integrating the results obtained, we assumed H. pylori migration to Japan (Figure 3). According to the results of f-statistics, hpRyukyu may have reached Okinawa via an inland path rather than the coastal route that hpSahul might have taken (Figure 3A). One candidate pathway is the Wakhan Corridor, which connects northeast Afghanistan and southwest China.
Figure 3.

Putative migration path to Japan
Putative migration path of hpRyukyu (A) and hspOkinawa (B). The line from Afghanistan to Nepal was colored green and gray since those strains belong to hpEurope or hpAsia2. Because hpSahul strains are not observed anywhere in the Eurasian continent, its putative path is indicated by a dotted light blue line. During the last ice age, Hokkaido was connected to the Eurasian continent via Sakhalin (A). The Okinawa islands were isolated all the time (B). HspEAsia and hspMaori spread with rice agriculture and advanced navigation systems, respectively (C).
The oldest Homo sapiens fossil in Okinawa, the Yamashita Cave Man, was estimated to be around 32,000 B.P. through radiocarbon dating (Kobayashi et al., 1971). The complete skeletons of 27,000 B.P. (Shiraho-Saonetabaru) (Shinoda and Adachi, 2017) and 2,2000 B.P. (Minatogawa) (Baba and Narasaki, 1991) were also discovered from Okinawa. This suggests that some Paleolithic human populations resided in Okinawa. The divergence dates of hpRyukyu and hspOkinawa correspond with these fossil records. However, human genome analyses indicate that the modern Okinawa population was more closely associated with the Japan main island than the Ainu people (Jinam et al., 2015; Kanzawa-Kiriyama et al., 2017).
One of the possible reasons for the discrepancy between human and H. pylori analyses is that ancient traces in the autosomal DNA are attenuated by admixture with new migrants. Actually, mitochondria DNA haplogroup M7a, which was found in 3 of 5 Paleolithic Shiraho-Saonetabal individuals (Shinoda and Adachi, 2017), is still observed in about 20% of the current Okinawa population.
Another possibility is that hpRyukyu in Okinawa was transmitted from the Paleolithic migrants to the later migrants without human admixture. Then, the old migrants went extinct and only the Paleolithic H. pylori remained in the new migrants. It is controversial whether the Paleolithic migrants contributed to the modern Japanese population, although at least the Paleolithic H. pylori survived up to now.
H. pylori of hunter-gatherers and farmers
The Japanese population is thought to contain at least two layers of migrations: early hunter-gatherers (Jomon people) and later farmers (Yayoi people) (Hanihara, 1991). As the farmer population expanded in the main island of Japan, a trace of the Jomon people remained in the north (Hokkaido) and south (Okinawa) of the Japanese Archipelago.
The divergence date of hspOkinawa and Ainu strains corresponded to the Jomon era. Both hspOkinawa and Ainu strains lack cagA (designated as cagA (−)). CagA (−) strains are often observed in Africa and Europe but are rare in Asia. As the farmer population expanded on the main island of Japan, the trace of Jomon people remained in the north (Hokkaido) and south (Okinawa) of the Japanese Archipelago.
However, hspOkinawa and Ainu strains belong to different sub-branches (Figures 1 and 2). In addition, hspOkinawa has similarity with the southern strains (hspMaori) in the STRUCTURE analysis (Figures S1 and S2). We assume that the hosts of hspOkinawa were in contact with people from Southeast Asia who might carry ancestral strains of hspMaori. From these observations, we assume that ancestral hspIndigenousAmericas from the north route and ancestral hspMaori from a southern route reached the east end of the Eurasian continent and hybridized to form hspOkinawa, so hspOkinawa has both characteristics (Figure 3B).
Current H. pylori in Japan
Most of the current patients with H. pylori in the main island of Japan are infected with hspEAsia strains. HspEAsia distributes broadly in the countries such as China, Korea, Vietnam, Thailand, and Bhutan. HspEAsia evolved in the Eurasian continent and migrated to Japan with rice farmers, while the hosts of hspMaori developed an advanced navigation system and expanded via the ocean from Taiwan to New Zealand (Figure 3C). If hpRyukyu or hspOkinawa strains existed in East Eurasia or in the main island of Japan among hunter-gatherers, it might be taken over by hpEAsia strains carried by farmers, like hspIndigenousAmericas was taken over by hpEurope in the Americas and is observed only among aboriginal people in remote areas. HpRyukyu and hspOkinawa strains might have remained in Okinawa because there are many isolated islands.
The scarce admixture between hpRyukyu and hspOkinawa suggests that they co-existed independently for a long time. This independence may reflect geographic isolation because the Okinawa area consists of many islands.
Probability of influence from recent human migration
When we found Okinawa-specific strains (Matsunari et al., 2012), we considered influence from recent human migration, especially from people in the U.S. army bases located in Okinawa. However, we think the probability is low that hpRyukyu and hspOkinawa derive from recent migrants as described below.
There were hpEurope strains that might be transmitted from Western people but its ratio was less than 5% of the strains obtained from Okinawa. On the contrary, the hpRyukyu and hspOkinawa strains accounted for 16% and 14%, respectively. H. pylori does not transmit easily between adults but mostly transmit from parents to children by close contacts while the immune system is not fully developed, therefore lineage expansion takes generations. Considering the substantial inflow of foreign population to Okinawa started only after 1945, it is unlikely that hpRyukyu and hspOkinawa strains derive from recent foreign sources.
Furthermore, strains that are close to hpRyukyu were found only from Afghanistan to Nepal, although we investigated a wide variety of Asian strains and public genome data of Western and African strains. Because current human flow from Afghanistan or Nepal to Okinawa is scarce, the similarity between Okinawa strains and Afghanistan or Nepal strains is likely due to common ancestry of the host human populations.
The utility of the H. pylori genome data
Currently, evolution of modern humans is investigated using genome-wide human SNP data (Japanese Archipelago Human Population Genetics Consortium et al., 2012; Jinam et al., 2015) or whole human genome sequence data (Pagani, 2017; Pagani et al., 2016). Genome sequences of H. pylori are much smaller than those human genome data, and one may think that these small data are no longer useful for elucidating the history of human evolution. However, genome analysis of H. pylori answered three questions about human evolution in the Japanese Archipelago: (1) Are there traces of Paleolithic migrants who reached Okinawa around 30,000 years ago observable in the modern Okinawa population? (2) Are there common features between Okinawa and Ainu people originating in the Jomon era? (3) Did the ancestors of modern Ainu people receive some influence from people in North Asia? We demonstrated the effectiveness of H. pylori genome data for the study of modern human evolution in East Eurasia.
Limitations of the study
The Okinawan H. pylori in this study were all isolated from patients of the University of the Ryukyus Hospital located in the south of Okinawa main island. Okinawa area covers many islands but there was no information about the birth places of the patients. We therefore cannot clarify the geographic distribution of hpRyukyu and hspOkinawa. The host human DNA was not available because of the lack of the informed consent, therefore we cannot analyze the genetics of the hosts. These are limitations of our study and we hope to clarify these points in a future study.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited data | ||
| Genome sequences of H. pylori (Genbank accession number) | This paper | CP006820 - CP006827, CP058279 - CP058288, and CP058250 - CP058252. See Table S3 for details. |
| Software and algorithms | ||
| MEGA (v.10.1.8) | Tamura et al. (2013) | https://www.megasoftware.net/ |
| ClonalFrameML (v.1.12) | Didelot and Wilson (2015) | https://github.com/xavierdidelot/ClonalFrameML |
| jModeltest (v.2.1.10) | Posada (2008) | http://evomics.org/learning/phylogenetics/jmodeltest/ |
| BEAST2 (v2.5.0) | Bouckaert et al. (2014) | https://www.beast2.org/ |
| Chromopainter (v.2) | Lawson et al. (2012) | https://people.maths.bris.ac.uk/%7Emadjl/finestructure-old/chromopainter_info.html |
| fineSTRUCTURE (v0.1.0) | Lawson et al. (2012) | http://www.paintmychromosomes.com/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Yoshio Yamaoka (yyamaoka@oita-u.ac.jp).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Biopsy specimens of gastric mucosa were collected from H. pylori-infected patients and the bacterial cells were isolated from the biopsy specimens using standard culture methods. From each sample, a single colony was expanded on confluent plates and cultured under microaerobic conditions (12% CO2) at 37°C. Bacterial DNA was extracted from the plates using a commercially kit (QIAGEN Inc., Valencia, CA, USA).
Method details
Population structure analysis of the MLST data
Conventional MLST analysis uses 3,406 bp sequences concatenated from seven housekeeping genes. We integrated our original MLST sequences (51, 64, 21, 20, 51, 72, and 119 strains from Bangladesh, Bhutan, Laos, Myanmar, Nepal, Okinawa Japan, and Thailand, respectively) with 691 sequences from pubMLST (https://pubmlst.org/). We analyzed population structure using the model-based clustering software STRUCTURE (Falush et al., 2003a) (v2.3.4) on the dataset of total strains by changing the population number (K) from 2 to 10 and executing 10 runs for each K. We tried both no-admixture and linkage models. The results of the 10 runs for each K were integrated by CLUMPAK server (http://clumpak.tau.ac.il/distruct.html) (Kopelman et al., 2015). The DAPC (Jombart et al., 2010) package of statistics software R (v3.4.3) was applied on the same dataset.
Genome sequencing
Biopsy specimens of gastric mucosa were collected from H. pylori-infected patients in Okinawa, Japan (oki102, oki112, oki128, oki154, oki422, oki673, oki828, oki898), Hokkaido, Japan (HK711, Hk721, Hk840), Alaska, U.S.A. (AL02, AL03, AL04, AL05), Colombia (CO1681, CO1766, CO1768), Thailand (UBN18), and Bhutan (BT302), as previously described (Abe et al., 2011; Matsunari et al., 2012; Moodley et al., 2009). H. pylori were isolated from the biopsy specimens using standard culture methods, as previously described (Yamaoka et al., 1998). Genomic DNA was extracted from confluent plate cultures expanded from a single colony using a commercially available kit (QIAGEN Inc., Valencia, CA, USA). DNA samples of the 20 strains were sequenced by PacBio. Genome assembly was performed by the HGAP Pipeline to obtain the complete genome data.
Phylogenetic tree and population structure analysis of the genome data
For genomic analyses, we used the complete genome sequences of our original 20 strains and 64 strains downloaded from GenBank. Additionally, we explored Genome Neighbour report of NCBI and downloaded contig sequences of four strains (HP14065, HP15060, UM139, and UM211) that were close to hpRyukyu. We used the prokaryotic genome annotation tool Prokka (Seemann, 2014) and the pan genome pipeline Roary (Page et al., 2015) to identify orthologous gene groups of the above 88 strains. If a multiple copy of an ortholog exists in one strain (paralogs), we did not use the gene for analysis because paralogs evolves differently from a single copy of a gene and are not suitable for phylogenetic analysis. As for gene loss, we allowed lack of genes at most 5% of the strains and used pairwise deletion option when we constructed a NJ-tree (Kimura 2-parameter) by MEGA (v.10.1.8) (Tamura et al., 2013). We obtained 619 sets of orthologous genes and aligned the DNA sequences of each gene. Then we concatenated the aligned sequences into the a sequence of totally 495,102 bp. This concatenated sequence was also used for population structure analysis by Chromopainter (v.2) (Lawson et al., 2012) and fineSTRUCTURE (v.0.1.0) (Lawson et al., 2012).
Estimation of the divergence dates
The result of fineSTRUCTURE classified the 88 strains into 25 populations. We chose one strain from each population that has the least influence from other populations. Because hpEurope strains were known to be a hybrid between hpNEAfrica and hpAsia2, we excluded seven European strains. To estimate the divergence time of hpRyukyu, we included Afghanistan-Nepal strains (HP14065, HP15060, UM139) but excluded UM211, whose origin was unclear. We used the 619 orthologous gene sets of the selected 24 strains for further analysis. We used ClonalFrameML (v.1.12) (Didelot and Wilson, 2015) to eliminated recombinant regions and applied jModeltest (v.2.1.10) (Posada, 2008) to select the substitution model (GTR). The clock model (Relaxed clock lognormal) was selected by a preliminary run of BEAST2 (v2.5.0) (Bouckaert et al., 2014) executing 10,000,000 MCMC. After this process, the divergence date was estimated executing 100,000,000 MCMC. Calibration points were set between African and non-African using Log normal model, M = 2, S = 0.1, offset = 0 in BEAUti (Lower 50 kya, upper 100 kya) referring to archaeological information (Bae et al., 2017; Sikora, 2017).
Gene flow analysis
We used an improved ABBA-BABA test, fhom, to evaluate gene flow. We set SouthAfrica20 as H4 (outlier), hpRyukyu and Afghanistan-Nepal strains as H3, Indian strain (SNT49) or Papua New Guinea strain (PNG84A) as H2, and PNG84A (SNT49 as H2) or other East Asian strains as H1. We calculated fhom and took an average when the East Asian strains were set as H1. A positive value of fhom suggests that gene flow H3 to H2 is stronger than H3 to H1, and a negative value suggests the opposite.
Data access
Genome data sequenced by us are available from GenBank under the accession numbers CP006820 - CP006827, CP058279 - CP058288, and CP058250 - CP058252.
Acknowledgments
This report is based on work supported in part by grants from the National Institutes of Health (DK62813) (Y.Y.), Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology (MEXT) of Japan (221S0002, 16H06279, 18KK0266 and 21H00346) (Y.Y.), Special Coordination Funds for Promoting Science and Technology from the Japan Science and Technology Agency (J.S.T.), and The Promotion Project of Knowledge-based Industrial Clustering of Okinawa prefecture (Commissioned Projects by Okinawa Prefectural Government). We thank Research Center for GLOBAL and LOCAL Infectious Diseases, Oita University faculty of Medicine. We thank Dr. Eng-Guan Chua for providing information about the strains HP14065, HP15060, UM139, and UM211 in response to our inquiry. We also thank Prof. Tetsuya Hayashi, Prof. Yoshitoshi Ogura, and Ms. Yoko Kudo for her excellent technical assistance.
Author contributions
R.S. analyzed data and wrote the paper; N.S. wrote the paper; O.M., S.S., T.M., J.A., N.K., and F.K. performed experiments. K.T., M.S., A.S., M.K., K.S., M.A., K.K., and Y.M. performed experiments and wrote the paper; T.H. designed experiments; Y.Y. designed experiments, analyzed data, and wrote the paper.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
We worked to ensure sex balance in the selection of non-human subjects. One or more of the authors of this paper received support from a program designed to increase minority representation in science. The author list of this paper includes contributors from the location where the research was conducted who participated in the data collection, design, analysis, and/or interpretation of the work.
Published: July 15, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.104477.
Supplemental information
Data and code availability
The H. pylori genome data we sequenced have been deposited at Genbank and are publicly available as the date of publication. Accession numbers are listed in the key resources table.
This paper does not report original code.
Any additional information about the analysis in this paper is available from the lead contact upon request.
References
- Abe T., Kodama M., Murakami K., Matsunari O., Mizukami K., Inoue K., Uchida M., Okimoto T., Fujioka T., Uchida T., et al. Impact of Helicobacter pylori CagA diversity on gastric mucosal damage: an immunohistochemical study of East-Asian-type CagA. J. Gastroenterol. Hepatol. 2011;26:688–693. doi: 10.1111/j.1440-1746.2010.06565.x. [DOI] [PubMed] [Google Scholar]
- Baba H., Narasaki S. Minatogawa man, the oldest type of modern homo sapiens in East Asia. Quat. Res. 1991;30:221–230. doi: 10.4116/jaqua.30.221. [DOI] [Google Scholar]
- Bae C.J., Douka K., Petraglia M.D. On the origin of modern humans: Asian perspectives. Science. 2017;358:eaai9067. doi: 10.1126/science.aai9067. [DOI] [PubMed] [Google Scholar]
- Bouckaert R., Heled J., Kühnert D., Vaughan T., Wu C.H., Xie D., Suchard M.A., Rambaut A., Drummond A.J. Beast 2: a software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 2014;10:e1003537. doi: 10.1371/journal.pcbi.1003537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua E.G., Debowski A.W., Webberley K.M., Peters F., Lamichhane B., Loke M.F., Vadivelu J., Tay C.Y., Marshall B.J., Wise M.J. Analysis of core protein clusters identifies candidate variable sites conferring metronidazole resistance in Helicobacter pylori. Gastroenterol. Rep. (Oxf) 2019;7:42–49. doi: 10.1093/gastro/goy048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covacci A., Telford J.L., Giudice G.D., Parsonnet J., Rappuoli R. Helicobacter pylori virulence and genetic geography. Science. 1999;284:1328–1333. doi: 10.1126/science.284.5418.1328. [DOI] [PubMed] [Google Scholar]
- Didelot X., Wilson D.J. ClonalFrameML: efficient inference of recombination in whole bacterial genomes. PLoS Comput. Biol. 2015;11:e1004041. doi: 10.1371/journal.pcbi.1004041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falush D., Stephens M., Pritchard J.K. Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics. 2003;164:1567–1587. doi: 10.1093/genetics/164.4.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falush D., Wirth T., Linz B., Pritchard J.K., Stephens M., Kidd M., Blaser M.J., Graham D.Y., Vacher S., Perez-Perez G.I., et al. Traces of human migrations in Helicobacter pylori populations. Science. 2003;299:1582–1585. doi: 10.1126/science.1080857. [DOI] [PubMed] [Google Scholar]
- Hanihara K. Dual structure model for the population history of the Japanese. Jpn. Rev. 1991;2:1–33. [Google Scholar]
- Japanese Archipelago Human Population Genetics Consortium. Jinam T., Nishida N., Hirai M., Kawamura S., Oota H., Umetsu K., Kimura R., Ohashi J., Tajima A., et al. The history of human populations in the Japanese Archipelago inferred from genome-wide SNP data with a special reference to the Ainu and the Ryukyuan populations. J. Hum. Genet. 2012;57:787–795. doi: 10.1038/jhg.2012.114. [DOI] [PubMed] [Google Scholar]
- Jinam T.A., Kanzawa-Kiriyama H., Inoue I., Tokunaga K., Omoto K., Saitou N. Unique characteristics of the Ainu population in Northern Japan. J. Hum. Genet. 2015;60:565–571. doi: 10.1038/jhg.2015.79. [DOI] [PubMed] [Google Scholar]
- Jombart T., Devillard S., Balloux F. Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genet. 2010;11:94. doi: 10.1186/1471-2156-11-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanzawa-Kiriyama H., Kryukov K., Jinam T.A., Hosomichi K., Saso A., Suwa G., Ueda S., Yoneda M., Tajima A., Shinoda K.I., et al. A partial nuclear genome of the Jomons who lived 3000 years ago in Fukushima, Japan. J. Hum. Genet. 2017;62:213–221. doi: 10.1038/jhg.2016.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi H., Matsui Y., Suzuki H. University of tokyo radiocarbon measurements IV. Radiocarbon. 1971;13:97–102. doi: 10.1017/s0033822200000904. [DOI] [Google Scholar]
- Kopelman N.M., Mayzel J., Jakobsson M., Rosenberg N.A., Mayrose I. Clumpak: a program for identifying clustering modes and packaging population structure inferences across K. Mol. Ecol. Resour. 2015;15:1179–1191. doi: 10.1111/1755-0998.12387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuipers E.J., Israel D.A., Kusters J.G., Gerrits M.M., Weel J., van Der Ende A., van Der Hulst R.W., Wirth H.P., Höök-Nikanne J., Hook-Nikanne J., et al. Quasispecies development of Helicobacter pylori observed in paired isolates obtained years apart from the same host. J. Infect. Dis. 2000;181:273–282. doi: 10.1086/315173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson D.J., Hellenthal G., Myers S., Falush D. Inference of population structure using dense haplotype data. PLoS Genet. 2012;8:e1002453. doi: 10.1371/journal.pgen.1002453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linz B., Balloux F., Moodley Y., Manica A., Liu H., Roumagnac P., Falush D., Stamer C., Prugnolle F., van der Merwe S.W., et al. An African origin for the intimate association between humans and Helicobacter pylori. Nature. 2007;445:915–918. doi: 10.1038/nature05562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiden M.C.J., Bygraves J.A., Feil E., Morelli G., Russell J.E., Urwin R., Zhang Q., Zhou J., Zurth K., Caugant D.A., et al. Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proc. Natl. Acad. Sci. U S A. 1998;95:3140–3145. doi: 10.1073/pnas.95.6.3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin S.H., Davey J.W., Jiggins C.D. Evaluating the use of ABBA-BABA statistics to locate introgressed loci. Mol. Biol. Evol. 2015;32:244–257. doi: 10.1093/molbev/msu269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsunari O., Shiota S., Suzuki R., Watada M., Kinjo N., Murakami K., Fujioka T., Kinjo F., Yamaoka Y. Association between Helicobacter pylori virulence factors and gastroduodenal diseases in Okinawa, Japan. J. Clin. Microbiol. 2012;50:876–883. doi: 10.1128/jcm.05562-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miftahussurur M., Sharma R.P., Shrestha P.K., Suzuki R., Uchida T., Yamaoka Y. Molecular epidemiology of Helicobacter pylori infection in Nepal: specific ancestor root. PLoS One. 2015;10:e0134216. doi: 10.1371/journal.pone.0134216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moodley Y., Linz B. Helicobacter pylori sequences reflect past human migrations. Genome Dyn. 2009;6:62–74. doi: 10.1159/000235763. [DOI] [PubMed] [Google Scholar]
- Moodley Y., Brunelli A., Ghirotto S., Klyubin A., Maady A.S., Tyne W., Muñoz-Ramirez Z.Y., Zhou Z., Manica A., Linz B., Achtman M. Helicobacter pylori's historical journey through Siberia and the Americas. Proc. Natl. Acad. Sci. U S A. 2021;118 doi: 10.1073/pnas.2015523118. e2015523118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moodley Y., Linz B., Yamaoka Y., Windsor H.M., Breurec S., Wu J.Y., Maady A., Bernhoft S., Thiberge J.M., Phuanukoonnon S., et al. The peopling of the Pacific from a bacterial perspective. Science. 2009;323:527–530. doi: 10.1126/science.1166083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagani L. A Cover Story for a Nature cover: genetic signature of human expansions into Eurasia revealed by a panel of worldwide high coverage genomes. J. Anthropol. Sci. 2017;95:1–5. doi: 10.4436/JASS.95019. [DOI] [PubMed] [Google Scholar]
- Pagani L., Lawson D.J., Jagoda E., Mörseburg A., Eriksson A., Mitt M., Clemente F., Hudjashov G., DeGiorgio M., Saag L., et al. Genomic analyses inform on migration events during the peopling of Eurasia. Nature. 2016;538:238–242. doi: 10.1038/nature19792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page A.J., Cummins C.A., Hunt M., Wong V.K., Reuter S., Holden M.T., Fookes M., Falush D., Keane J.A., Parkhill J. Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics. 2015;31:3691–3693. doi: 10.1093/bioinformatics/btv421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posada D. jModelTest: phylogenetic model averaging. Mol. Biol. Evol. 2008;25:1253–1256. doi: 10.1093/molbev/msn083. [DOI] [PubMed] [Google Scholar]
- Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30:2068–2069. doi: 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- Shinoda K., Adachi N. In: New Perspectives in Southeast Asian and Pacific Prehistory. Matsumura H., Piper Philip J., Bulbeck D., editors. Australian National University Press; 2017. Ancient DNA analysis of palaeolithic Ryukyu islanders; pp. 51–60. [DOI] [Google Scholar]
- Sikora M. A genomic view of the pleistocene population history of Asia. Curr. Anthropol. 2017;58:S397–S405. doi: 10.1086/694422. [DOI] [Google Scholar]
- Sikora P., Andersson S., Winiecka-Krusnell J., Hallström B., Alsmark C., Troell K., Beser J., Arrighi R.B.G. Genomic variation in IbA10G2 and other patient-derived cryptosporidium hominis subtypes. J. Clin. Microbiol. 2017;55:844–858. doi: 10.1128/JCM.01798-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K., Stecher G., Peterson D., Filipski A., Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013;30:2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yahara K., Furuta Y., Oshima K., Yoshida M., Azuma T., Hattori M., Uchiyama I., Kobayashi I. Chromosome painting in silico in a bacterial species reveals fine population structure. Mol. Biol. Evol. 2013;30:1454–1464. doi: 10.1093/molbev/mst055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yahara K., Kawai M., Furuta Y., Takahashi N., Handa N., Tsuru T., Oshima K., Yoshida M., Azuma T., Hattori M., et al. Genome-wide survey of mutual homologous recombination in a highly sexual bacterial species. Genome Biol. Evol. 2012;4:628–640. doi: 10.1093/gbe/evs043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaoka Y., Kodama T., Kita M., Imanishi J., Kashima K., Graham D.Y. Relationship of vacA genotypes of Helicobacter pylori to cagA status, cytotoxin production, and clinical outcome. Helicobacter. 1998;3:241–253. doi: 10.1046/j.1523-5378.1998.08056.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The H. pylori genome data we sequenced have been deposited at Genbank and are publicly available as the date of publication. Accession numbers are listed in the key resources table.
This paper does not report original code.
Any additional information about the analysis in this paper is available from the lead contact upon request.