

Abstract
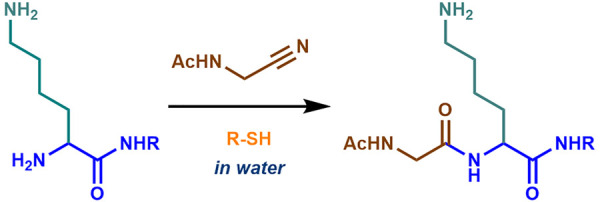
The prebiotic origin of catalyst-controlled peptide synthesis is fundamental to understanding the emergence of life. Building on our recent discovery that thiols catalyze the ligation of amino acids, amides, and peptides with amidonitriles in neutral water, we demonstrate the outcome of ligation depends on pH and that high pKa primary thiols are the ideal catalysts. While the most rapid thiol catalyzed peptide ligation occurs at pH 8.5–9, the most selective peptide ligation, that tolerates all proteinogenic side chains, occurs at pH 7. We have also identified the highly selective mechanism by which the intermediate peptidyl amidines undergo hydrolysis to α-peptides while demonstrating that the hydrolysis of amidines with nonproteinogenic structures, such as β- and γ-peptides, displays poor selectivity. Notably, this discovery enables the highly α-selective protecting-group-free ligation of lysine peptides at neutral pH while leaving the functional ε-amine side chain intact.
Peptide synthesis is one of the most important processes in chemistry and biology.1 Peptide biosynthesis is a highly evolved system,2,3 that could not have spontaneously appeared in its current form,4 but what nonenzymatic chemistry preceded it and how these reactions influenced the structure of biological peptides remains unknown. We recently reported that α-peptidyl nitriles 1 are activated for biomimetic peptide synthesis5 and that the in-built reactivity of 1(6) can drive catalytic peptide ligation (CPL; Figure 1). CPL requires no activating agent to ligate 1 with amino acid derivatives (2)5b and is a rare example of organocatalysis in water.7 The nitrile’s kinetic stability means ligation must be thiol catalyzed, and so catalyst-gated reactivity, which is an essential feature of biochemistry, is observed. As a mechanism for prebiotic peptide synthesis CPL has several appealing characteristics: it uses simple prebiotic reactants; is selective for α-amidonitriles, and therefore proteinogenic peptides; generates high peptide yields under conditions where peptides are very stable;8 and is orthogonal to (biological) phosphate activation,9 which would in principle enable independent catalytic modulation of both peptide and nucleic acid synthesis. Intriguingly, CPL produces amidines 3 when amino acids are the nucleophilic coupling partner (Figure 1; 2, X = OH), whereas peptides 5′ are formed when α-amino amides or peptides are used (Figure 1; 2′, X = NHR4).5b With this in mind, we set out to explore the conditions under which CPL delivers the highest selectivity for α-peptide formation.10
Figure 1.
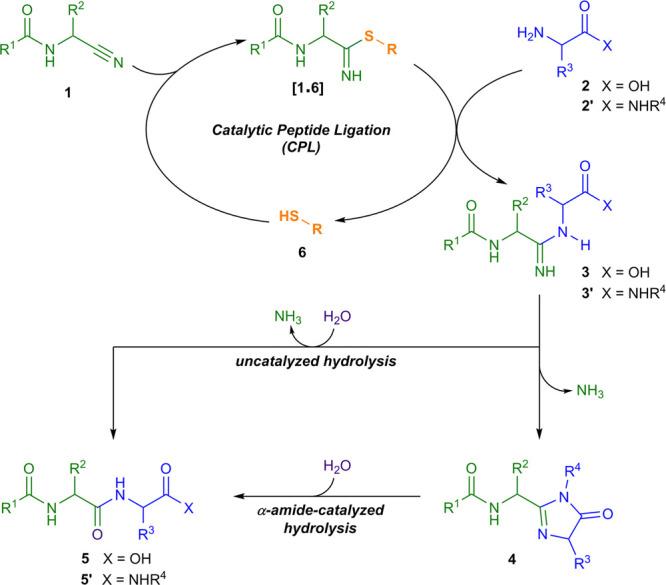
Catalytic peptide ligation (CPL) in water. Thiol-catalyzed coupling of peptide nitriles (1) with amines (2, X = OH or 2′, X = NHR4). R = alkyl or aryl; R1 = peptide or alkyl; R2 and R3 = aminoacyl side chain; R4 = H or peptide; X = OH, NH2 or peptide. Compounds 2–5 and 2′–5′ are labeled with subscripts corresponding to the single letter amino acid code.
Our preliminary study of CPL focused on reactions at neutral pH, but we envisaged that pH would have a profound effect on CPL, as the nucleophile and catalyst could both deprotonate at higher pH. Pleasingly, we observed that ligation of alanine (H-Ala-OH, 2A) to nitrile 1 catalyzed by Ac-Cys-OH (6a) is more rapid at pH 8.5 than at pH 7 (Table 1). In line with our prediction, we observed negligible reactivity at pH 5 but, surprisingly, slow and low yielding CPL at pH 10. This is likely due to suppressed thioimidate [1·6] protonation at pH 10. Accordingly, the optimal rate for 6a-catalyzed CPL was observed at pH 8.5–9.0.
Table 1. Effect of pH and Catalyst on Amidine 3 Formation10.


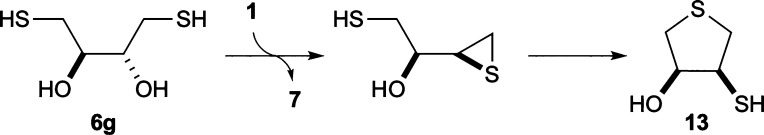
3-Mercaptopropionic acid (6b) and thioglycolic acid (6c) promoted CPL faster than 6a, giving 85–90% amidine (3A) after 36 h, at pH 8.5 and rt (Table 1). Low pKa thiols (e.g., 6d and 6e) are sluggish, with 6d only furnishing 4% amidine 3A after 36 h. Sterically hindered 6f also retarded the rate of CPL. Limited hydration of 1 to Ac-Gly-NH2 (7) (∼5%) was observed with most catalysts (Figure S12), but 6g yielded significant amide 7 (25%). We suspect 6g undergoes S-to-O acyl transfer, followed by thiirane formation, leading to 7 (Figures S13 and S14).11 These results demonstrate high-pKa primary thiols are best suited as CPL-catalysts. This stands in stark contrast to thioester ligations (e.g., native chemical ligation), which are accelerated by low pKa thiols such as 6d.12
High amidine 3 yields (82–95%) were observed in H-Gly-OH (2G), H-Ala-OH (2A), H-Leu-OH (2L), H-Ile-OH (2I), H-Phe-OH (2F), H-Met-OH (2M), H-Val-OH (2V), H-Arg-OH (2R), H-Glu-OH (2E), H-Asp-OH (2D), H-Gln-OH (2Q), H-Trp-OH (2W), H-Pro-OH (2P), and H-His-OH (2H) couplings (Figures S16–S51). H-Cys-OH (2C) coupling does not require catalysis, due to the thiol side chain.5b H-Lys-OH (2K, 86%) exhibited moderate α-selectivity (2:1, α/ε) with 60% α-amidine formation (Figure S37), while β-hydroxy amino acids (i.e., H-Ser-OH, 2S and H-Thr-OH, 2T) yielded peptides rather than amidines. We have postulated that this is due to the formation and hydrolysis of oxazoline 8 (Figure 2A). Here, at room temperature, we observed 8 as the major product (8S (55%) and 8T (67%); Figures S45 and S49). Oxazoline 8 formed rapidly from nitrile 1 and 2S or 2T, but its hydrolysis is slow at 25 °C. However, heating 8S and 8T at 60 °C for 12 h resulted in high yielding conversion to 5S and 5T.
Figure 2.
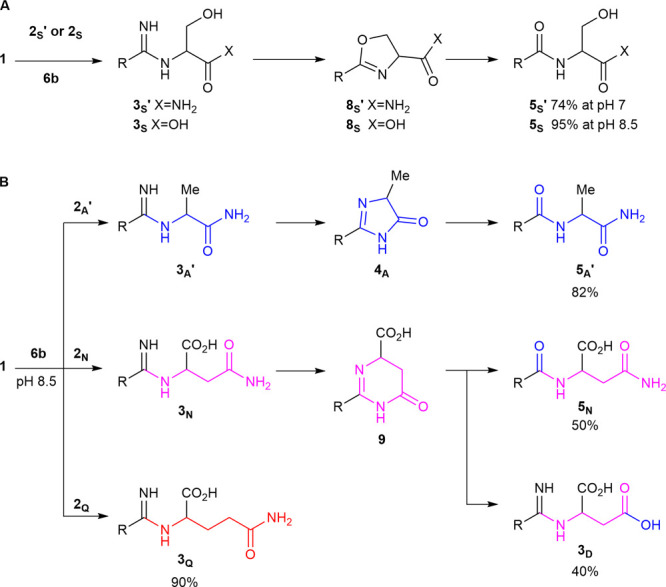
Intramolecular catalysis of amidine hydrolysis. (A) Coupling of nitrile 1 (200 mM) with serinamide (2S′, 2 equiv) yields peptide 5S′ at pH 7, via oxazoline 8, whereas serine (2S, 1 equiv) yields peptide 5S at pH 7 (ref (5b)) or pH 8.5. (B) Coupling of 1 (200 mM) with alaninamide (2A′, 2 equiv), asparagine (2N, 1 equiv), glutamine (2Q, 1 equiv) demonstrates the effect of α-, β-, and γ-amides amidine hydrolysis during CPL. R = CH2NHCOCH3.
Like 2S and 2T, α-amino amide (2′) nucleophiles directly form peptides 5′ in CPL. This selective peptide formation, promoted by the α-peptide backbone, warranted further investigation. Amidine 3′ can in principle hydrolyze through substitution of ammonia or amino amide (2′), and their similar pKaH values suggested that direct hydrolysis should yield peptide 5′ and amide 7 in comparable yields. However, peptide 5′ forms selectively, implicating intramolecular catalysis. Upon coupling H-Ala-NH2 (2A′) and nitrile 1, we observed slow hydrolysis of amidine 3A′ to 5A′. At room temperature, we also observed an imidazolone intermediate (4A) (Figure 2B, Figures S55 and S118–S120). This cyclization explains the selective formation of peptide 5′, with intramolecular substitution promoting loss of ammonia.
We speculated this selectivity would be uniquely effective for (biogenic) α-peptides. To test this, we used alaninamide (H-Ala-NH2, 2A′), asparagine (H-Asn-OH, 2N), and glutamine (H-Gln-OH, 2Q) as homologous nucleophiles (with α-, β-, and γ-amides) to investigate amide-catalyzed amidine hydrolysis (Figure 2B). α-Amides cyclize to 5-membered imidazolone 4 and hydrolyze selectively to α-peptides 5′. Although β-amides (e.g., 2N) also cyclize, they yield 6-membered dihydropyrimidone 9 that hydrolyze with poor selectivity yielding a mixture of peptide 5N (50%) and amidine 3D (40%). Thus, unlike α-amides, β-amides undergo significant β-peptide hydrolysis (Figure S22). Extending the series further inhibited cyclization completely, and γ-amide 2Q only formed amidine 3Q (Figure S28). These results demonstrate the disposition of α-amino amides (i.e., proteinogenic peptides) to catalyze selective amidine-to-peptide hydrolysis, while nonproteinogenic β- or γ-amino amides are either poor catalysts or catalytically inactive.
Cyclization of 3A′ to 4A and hydrolysis of 4A to peptide 5A′ exhibits a strong pH dependence, and both are rapid at pH 10 and sluggish at pH 7 (Figures 3, S124, and S126). However, at pH 7, both are accelerated by phosphate buffer (100 mM, Figure S123) but not by 6b (Figure 3C). This suggests that both hydrolysis and cyclization are general acid–base catalyzed. Moreover, hydrolysis can be catalyzed by a combination of intra- and intermolecular catalysis, and orthogonal catalysts (i.e., thiols and phosphates) can independently catalyze amidine ligation and hydrolysis.
Figure 3.
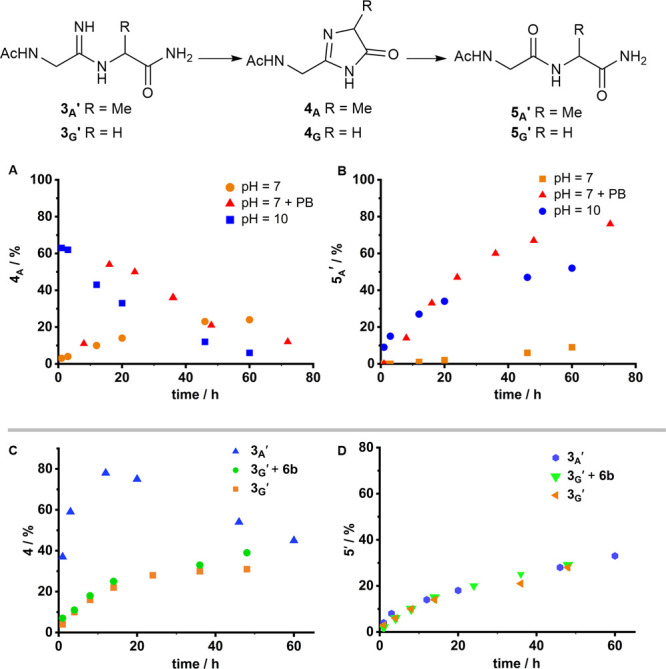
Effect of pH, buffer and catalyst on imidazolone formation and hydrolysis. Time courses to show the (A) formation of imidazolone 4A from amidine 3A′ (25 mM) at rt and pH 10 or pH 7, with and without phosphate buffer (PB, 100 mM); (B) formation of peptide 5A′ from 3A′ (25 mM) at r.t and pH 10 or pH 7, with and without PB (100 mM); (C) formation of 4A from 3A′ (25 mM) and 4G from 3G′ (25 mM) at rt and pH 9, with and without 6b (100 mM); (D) formation of peptides 5A′ from 3A′ (25 mM) and 5G′ from 3G′ (25 mM) at rt and pH 9, with and without 6b (100 mM).
To investigate the effect of side chains on amidine hydrolysis, proteinogenic amino amides (2′) were studied in CPL with 1. Peptides were formed in 70–90% yield with H-Ala-NH2 (2A′), H-Val-NH2 (2V′), H-Leu-NH2 (2L′), H-Phe-NH2 (2F′), H-Arg-NH2 (2R′), H-His-NH2 (2H′), H-Pro-NH2 (2P′), H-Tyr-NH2 (2Y′), H-Trp-NH2 (2W′), H-Glu-NH2 (2E′) and H-Asp-NH2 (2D′) (Figure S54–S101). H-Ile-NH2 (2I′) forms a mixture of diastereomers (11:9 ratio) in high yield (86%). Since H-Ile-OH (2I) forms only one amidine diastereomer, this implies racemization occurs during the cyclization-hydrolysis process. Slightly lower yields were observed with H-Gly-NH2 (2G′, 50%, Figure S70), H-Gln-NH2 (2Q′, 66%, Figure S68) and H-Met-NH2 (2M′, 63%, Figure S81). All NMR data were consistent with formation of intermediates 4. The course of the reaction was different for the amino amides (H-Ser-NH22S′, H-Thr-NH22T′ and H-Asn-NH22N′) with side chains that promote amidine hydrolysis. Asparaginamide (2N′) formed a mixture of Asn and Asp peptides in 50% yield after 10 days, alongside 7 (45%; Figure S62), but Asn peptides undergo facile hydrolysis,13 so this low yield is likely intrinsic to this side chain and no attempt was made to optimize H-Asn-NH2 coupling.
Surprisingly, coupling with 2S′ led to decomposition at pH 8.5, forming no detectable peptide (Figure S88). Oxazoline 8S′ (Figure 2) was, however, observed at pH 7, and heating 8S′ at 60 °C led to peptide 5S′ (74%) after 36 h (Figure S89). Similarly, 2T′ was converted to 5T′ (85%) after 36 h at 60 °C and pH 7 (Figure S97) as a single diastereomer. On the other hand, nonproteinogenic O-methyl serinamide 2MeS′ decomposed rather than forming peptide even at pH 7 (Figure S142). These results demonstrate that, at pH 7, oxazoline formation overcomes the incompatibility of β-hydroxyl residues with CPL at elevated pH. Likewise, CPL with peptide nucleophiles (e.g., H-Ala-Gly-Ala-OH 2AGA; Figures S102–S106) at pH 8.5 and 60 °C only furnished tetrapeptide Ac-Gly-Ala-Gly-Ala-OH (5AGA) in 50% yield, alongside substantial 7 (30%, Figure S104), whereas, at pH 7 and 60 °C, CPL was much more selective and ligation was observed to yield 5AGA (81%) (Figure S102). Thus, though faster at pH 8.5, CPL is only universally compatible and high yielding with proteinogenic peptides at neutral pH.
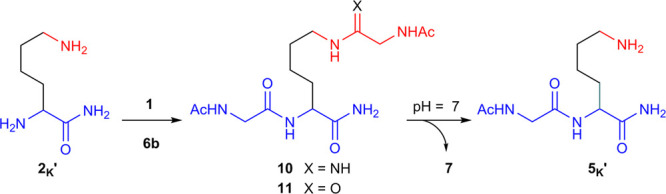
We next turned our attention to (uncatalyzed) hydrolysis of amidine 3 (X = OH). Whereas high selectivity of amidine-to-peptide hydrolysis was observed for α-amide 3A′, α-acid 3A furnished a mixture of 7 and 5A. At 80 °C, moderate selectivity for hydrolysis of 3A to 7 (2:1 7/5A) was observed at pH 7–9 (Table 2, entries 2–4). We postulated that this selectivity arose due to the difference in amine pKaH (2A = 9.7; ammonia = 9.2), with the higher pKaH amine selectively substituted. This suggested a new mechanism to effect selective α-ligation of lysine peptides in water.5a,14 The high pKaH of the ε-amine (10.8) compared to ammonia (9.2) suggested that hydrolysis of ε-lysyl amidines would selectively yield the free ε-amine. Furthermore, because α-lysyl peptide amidines (e.g., 3K′) undergo effective (intramolecular amide-catalyzed) hydrolysis to α-peptides, we envisaged α-peptide ligation and ε-hydrolysis would operate together and reinforce selectivity for proteinogenic peptide ligation.
Table 2. Hydrolysis of Amidine 3Aa.

| entry | pH | temp, °C | buffer (500 mM) | time | 3A, % | 5A, % | 2A, % |
|---|---|---|---|---|---|---|---|
| 1 | 7 | 20 | 30 days | 100 | 0 | 0 | |
| 2 | 7 | 80 | 18 h | 34 | 15 | 36 | |
| 3 | 7 | 80 | PB | 18 h | 11 | 31 | 52 |
| 4 | 9 | 80 | BB | 18 h | 0 | 38 | 61 |
PB = phosphate buffer, BB = borate buffer.
Upon coupling of H-Lys-NH2 (2K′) and 1, at pH 9, we observed that α-peptide 5K′ (20%) was a minor product, formed alongside ε-amide 11 (15%) and substantial amounts of N-acetylglycinamide 7 (59%) after 24 h at 80 °C (Table 3, entry 2 and Figure S135). This demonstrates that α-ligation is disfavored at pH 9 and the predominant product is hydration (i.e., 7). However, at neutral pH, the selectivity for α-ligation was dramatically increased. At pH 7 peptide 5K′ was the major product after 6 days, yielding 5K′ (58–65%) and only 7% ε-amide 11 (α:ε 9:1; Figure S137). Neutral pH ligation of lysyl peptides was similarly effective; H-Lys-Gly-OH (2KG) and H-Lys-Lys-OH (2KK) were ligated selectively to afford peptide Ac-Gly-Lys-Gly-OH 5KG (α:ε 7:1; Figures S138 and 139) and Ac-Gly-Lys-Lys-OH 5KK (α:ε 5:1; Figures S140–141).
Table 3. Selectivity for Ligation at α- or ε-Amine of Lysyl Peptidesa.
| entry | pH | ε-amidine 10, % | total amide (α + ε), % | ratio α:ε acylation |
|---|---|---|---|---|
| 1 | 9b | 0 | 35 | 1.3 |
| 2 | 7b | 19 | 67 | 3.5 |
| 3 | 7c | 0 | 72 | 9.3 |
Selective α-lysyl peptide (blue) over ε-lysyl amine (red) coupling of 200 mM 2K′ with 200 mM 1, 30 mol % 6b at 80 °C.
Yields after 1 day.
Yields after 6 days.
In conclusion, we have demonstrated that proteinogenic substrates undergo selective CPL to furnish racemic α-peptides catalyzed by the adjacent α-amide/peptide at neutral pH. β-Hydroxyl α-amides retain chirality via hydroxyl catalysis, but O-methylation inhibits peptide formation in these substrates even at neutral pH. The impact and value of peptide racemization and stereoretention, within a (likely racemic) prebiotic environment, during self-catalyzed peptidyl-amidine hydrolysis remains an open question.5b By studying the (uncatalyzed) hydrolysis of amidines we have discovered a preference for the substitution of the higher pKaH amine. This uncatalyzed reaction operates in tandem with α-amide-catalyzed hydrolysis to enhance the selectivity for lysine α-peptide synthesis at neutral pH in water while retaining the lysyl (functional) ε-amine group.
Acknowledgments
We thank K. Karu (UCL Mass Spectrometry Facility). We thank the EPSRC (EP/K004980/1, EP/M507970/1, EP/P020410/1), Leverhulme Trust (RPG-2019-214), Simons Foundation (318881FY19), and Volkswagen Foundation (94743) for financial support.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.2c03486.
Experimental procedures and spectroscopic data (PDF)
Author Contributions
† J.S. and D.W. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- a Constable D. J. C.; Dunn P. J.; Hayler J. D.; Humphrey G. R.; Leazer J. L. Jr.; Linderman R. J.; Lorenz K.; Manley J.; Pearlman B. A.; Wells A.; Zaks A.; Zhang T. Y. Key green chemistry research areas—a perspective from pharmaceutical manufacturers. Green Chem. 2007, 9, 411–420. 10.1039/B703488C. [DOI] [Google Scholar]; b Valeur E.; Bradley M. Amide bond formation: beyond the myth of coupling reagents. Chem. Soc. Rev. 2009, 38, 606–631. 10.1039/B701677H. [DOI] [PubMed] [Google Scholar]; c Pattabiraman V. R.; Bode J. W. Rethinking amide bond synthesis. Nature 2011, 480, 471. 10.1038/nature10702. [DOI] [PubMed] [Google Scholar]; d de Figueiredo R. M.; Suppo J.-S.; Campagne J.-M. Nonclassical Routes for Amide Bond Formation. Chem. Rev. 2016, 116, 12029–12122. 10.1021/acs.chemrev.6b00237. [DOI] [PubMed] [Google Scholar]; e Sabatini M. T.; Boulton L. T.; Sneddon H. F.; Sheppard T. D. A green chemistry perspective on catalytic amide bond formation. Nat. Catal. 2019, 2, 10–17. 10.1038/s41929-018-0211-5. [DOI] [Google Scholar]; f Wang X. Challenges and outlook for catalytic direct amidation reactions. Nat. Catal. 2019, 2, 98–102. 10.1038/s41929-018-0215-1. [DOI] [Google Scholar]
- a Nissen P.; Hansen J.; Ban N.; Moore P. B.; Steitz T. A. The Structural Basis of Ribosome Activity in Peptide Bond Synthesis. Science 2000, 289, 920–930. 10.1126/science.289.5481.920. [DOI] [PubMed] [Google Scholar]; b Fischbach M. A.; Walsh C. T. Assembly-line enzymology for polyketide and nonribosomal peptide antibiotics: Logic, machinery, and mechanisms. Chem. Rev. 2006, 106, 3468–3496. 10.1021/cr0503097. [DOI] [PubMed] [Google Scholar]; c Schmeing T. M.; Ramakrishnan V. What recent ribosome structures have revealed about the mechanism of translation. Nature 2009, 461, 1234–1242. 10.1038/nature08403. [DOI] [PubMed] [Google Scholar]
- Knowles J. R. Enzyme catalysis: not different, just better. Nature 1991, 350, 121–124. 10.1038/350121a0. [DOI] [PubMed] [Google Scholar]
- a Ruiz-Mirazo K.; Briones C.; de la Escosura A. Prebiotic Systems Chemistry: New Perspectives for the Origins of Life. Chem. Rev. 2014, 114, 285–366. 10.1021/cr2004844. [DOI] [PubMed] [Google Scholar]; b Frenkel-Pinter M.; Samanta M.; Ashkenasy G.; Leman L. J. Prebiotic Peptides: Molecular Hubs in the Origin of Life. Chem. Rev. 2020, 120, 4707–4765. 10.1021/acs.chemrev.9b00664. [DOI] [PubMed] [Google Scholar]
- a Canavelli P.; Islam S.; Powner M. W. Peptide ligation by chemoselective aminonitrile coupling in water. Nature 2019, 571, 546–549. 10.1038/s41586-019-1371-4. [DOI] [PubMed] [Google Scholar]; b Foden C. S.; Islam S.; Fernandez-Garcia C.; Maugeri L.; Sheppard T. D.; Powner M. W. Prebiotic synthesis of cysteine peptides that catalyze peptide ligation in neutral water. Science 2020, 370, 865–869. 10.1126/science.abd5680. [DOI] [PubMed] [Google Scholar]
- a Sutherland J. D. The Origin of Life—Out of the Blue. Angew. Chem., Int. Ed. 2016, 55, 104–121. 10.1002/anie.201506585. [DOI] [PubMed] [Google Scholar]; b Islam S.; Powner M. W. Prebiotic Systems Chemistry: Complexity Overcoming Clutter. Chem. 2017, 2, 470–501. 10.1016/j.chempr.2017.03.001. [DOI] [Google Scholar]; c Islam S.; Bučar D.-K.; Powner M. W. Prebiotic selection and assembly of proteinogenic amino acids and natural nucleotides from complex mixtures. Nat. Chem. 2017, 9, 584–589. 10.1038/nchem.2703. [DOI] [Google Scholar]
- a Barbas C. F. III Organocatalysis Lost: Modern Chemistry, Ancient Chemistry, and an Unseen Biosynthetic Apparatus. Angew. Chem., Int. Ed. 2008, 47, 42–47. 10.1002/anie.200702210. [DOI] [PubMed] [Google Scholar]; b van der Helm M. P.; Klemm B.; Eelkema R. Organocatalysis in aqueous media. Nat. Rev. Chem. 2019, 3, 491–508. 10.1038/s41570-019-0116-0. [DOI] [Google Scholar]
- a Steinberg S. M.; Bada J. L. Peptide decomposition in the neutral pH region via the formation of diketopiperazines. J. Org. Chem. 1983, 48, 2295–2298. 10.1021/jo00161a036. [DOI] [Google Scholar]; b Radzicka A.; Wolfenden R. Rates of Uncatalyzed Peptide Bond Hydrolysis in Neutral Solution and the Transition State Affinities of Proteases. J. Am. Chem. Soc. 1996, 118, 6105–6109. 10.1021/ja954077c. [DOI] [Google Scholar]; c Sun Y.; Frenkel-Pinter M.; Liotta C. L.; Grover M. A. The pH dependent mechanisms of non-enzymatic peptide bond cleavage reactions. Phys. Chem. Chem. Phys. 2020, 22, 107–113. 10.1039/C9CP05240B. [DOI] [PubMed] [Google Scholar]
- a Reese C. B.; Simons C.; Pei-Zhuo Z. The synthesis of 2′-thiouridylyl-(3′→ 5′)-uridine. J. Chem. Soc., Chem. Commun. 1994, 1, 1809–1810. 10.1039/C39940001809. [DOI] [Google Scholar]; b Dantzman C. L.; Kiessling L. L. Reactivity of a 2’-thio nucleotide analog. J. Am. Chem. Soc. 1996, 118, 11715–11719. 10.1021/ja962265c. [DOI] [Google Scholar]; c Hamm M. L.; Nikolic D.; Van Breemen R. B.; Piccirilli J. A. Unconventional origin of metal ion rescue in the hammerhead ribozyme reaction: Mn2+-assisted redox conversion of 2′-mercaptocytidine to cytidine. J. Am. Chem. Soc. 2000, 122, 12069–12078. 10.1021/ja000379p. [DOI] [Google Scholar]
- a Portillo-Ledesma S.; Sardi F.; Manta B.; Tourn M. V.; Clippe A.; Knoops B.; Alvarez B.; Coitiño E. L.; Ferrer-Sueta G. Deconstructing the Catalytic Efficiency of Peroxiredoxin-5 Peroxidatic Cysteine. Biochemistry 2014, 53, 6113–6125. 10.1021/bi500389m. [DOI] [PubMed] [Google Scholar]; b Keire D. A.; Strauss E.; Guo W.; Noszál B.; Rabenstein D. L. Kinetics and Equilibria of Thiol/Disulfide Interchange Reactions of Selected Biological Thiols and Related Molecules with Oxidized Glutathione. J. Org. Chem. 1992, 57, 123–127. 10.1021/jo00027a023. [DOI] [Google Scholar]; c Lindley H. A. Study of the Kinetics of the Reaction between Thiol Compounds and Chloroacetamide. Biochem. J. 1960, 74, 577–584. 10.1042/bj0740577. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Johnson E. C. B.; Kent S. B. H. Insights into the Mechanism and Catalysis of the Native Chemical Ligation Reaction. J. Am. Chem. Soc. 2006, 128, 6640–6646. 10.1021/ja058344i. [DOI] [PubMed] [Google Scholar]; e Patel V. M.; Joshi J. D. Equilibrium Study on the Complex Formation of Europium-, Terbium-, Dysprosium- and Thulium(III) with Some Oxyacids, Thioacids and Phenols. J. Indian Chem. Soc. 1998, 75, 100–101. [Google Scholar]; f Singh R.; Lamoureux G. V.; Lees W. J.; Whitesides G. M.. Reagents for Rapid Reduction of Disulfide Bonds. In Methods in Enzymology; 1995; pp 167–173. [DOI] [PubMed] [Google Scholar]
-
6g has
recently been used as the catalyst for CPL on an RNA template. Based
on our data, and the observed concomitant degradation of 6g and hydration of 1, we suspect higher yields could
have been achieved using 6b. See:Müller F.; Escobar L.; Xu F.; Węgrzyn E.; Nainytė M.; Amatov T.; Chan C.; Pichler A.; Carell T.
A Prebiotically Plausible Scenario
of an RNA–Peptide World. Nature
2022, 605, 279–284. 10.1038/s41586-022-04676-3. [DOI] [PMC free article] [PubMed] [Google Scholar];
- a Dawson P.; Muir T.; Clark-Lewis I.; Kent S. Synthesis of proteins by native chemical ligation. Science 1994, 266, 776–779. 10.1126/science.7973629. [DOI] [PubMed] [Google Scholar]; b Kulkarni S. S.; Sayers J.; Premdjee B.; Payne R. J. Rapid and efficient protein synthesis through expansion of the native chemical ligation concept. Nat. Rev. Chem. 2018, 2, 0122. 10.1038/s41570-018-0122. [DOI] [Google Scholar]
- a Clarke S. Propensity for spontaneous succinimide formation from aspartyl and asparaginyl residues in cellular proteins. Int. J. Pept. Protein Res. 1987, 30, 808–821. 10.1111/j.1399-3011.1987.tb03390.x. [DOI] [PubMed] [Google Scholar]; b Friedrich M. G.; Wang Z.; Schey K. L.; Truscott R. Mechanism of protein cleavage at asparagine leading to protein-protein cross-links. Biochem. J. 2019, 476, 3817–3834. 10.1042/BCJ20190743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frenkel-Pinter M.; Haynes J. W.; Martin C.; Petrov A. S.; Burcar B. T.; Krishnamurthy R.; Hud N. V.; Leman L. J.; Williams L. D. Selective incorporation of proteinaceous over nonproteinaceous cationic amino acids in model prebiotic oligomerization reactions. Proc. Natl. Acad. Sci. U.S.A. 2019, 116, 16338–16346. 10.1073/pnas.1904849116. [DOI] [PMC free article] [PubMed] [Google Scholar]

Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.