

Abstract
Background
Monoclonal antibodies (mAbs) are laboratory‐produced molecules derived from the B cells of an infected host. They are being investigated as potential prophylaxis to prevent coronavirus disease 2019 (COVID‐19).
Objectives
To assess the effects of SARS‐CoV‐2‐neutralising mAbs, including mAb fragments, to prevent infection with SARS‐CoV‐2 causing COVID‐19; and to maintain the currency of the evidence, using a living systematic review approach.
Search methods
We searched the Cochrane COVID‐19 Study Register, MEDLINE, Embase, and three other databases on 27 April 2022. We checked references, searched citations, and contacted study authors to identify additional studies.
Selection criteria
We included randomised controlled trials (RCTs) that evaluated SARS‐CoV‐2‐neutralising mAbs, including mAb fragments, alone or combined, versus an active comparator, placebo, or no intervention, for pre‐exposure prophylaxis (PrEP) and postexposure prophylaxis (PEP) of COVID‐19. We excluded studies of SARS‐CoV‐2‐neutralising mAbs to treat COVID‐19, as these are part of another review.
Data collection and analysis
Two review authors independently assessed search results, extracted data, and assessed risk of bias using Cochrane RoB 2. Prioritised outcomes were infection with SARS‐CoV‐2, development of clinical COVID‐19 symptoms, all‐cause mortality, admission to hospital, quality of life, adverse events (AEs), and serious adverse events (SAEs). We rated the certainty of evidence using GRADE.
Main results
We included four RCTs of 9749 participants who were previously uninfected and unvaccinated at baseline. Median age was 42 to 76 years. Around 20% to 77.5% of participants in the PrEP studies and 35% to 100% in the PEP studies had at least one risk factor for severe COVID‐19. At baseline, 72.8% to 82.2% were SARS‐CoV‐2 antibody seronegative.
We identified four ongoing studies, and two studies awaiting classification.
Pre‐exposure prophylaxis
Tixagevimab/cilgavimab versus placebo
One study evaluated tixagevimab/cilgavimab versus placebo in participants exposed to SARS‐CoV‐2 wild‐type, Alpha, Beta, and Delta variant. About 39.3% of participants were censored for efficacy due to unblinding and 13.8% due to vaccination. Within six months, tixagevimab/cilgavimab probably decreases infection with SARS‐CoV‐2 (risk ratio (RR) 0.45, 95% confidence interval (CI) 0.29 to 0.70; 4685 participants; moderate‐certainty evidence), decreases development of clinical COVID‐19 symptoms (RR 0.18, 95% CI 0.09 to 0.35; 5172 participants; high‐certainty evidence), and may decrease admission to hospital (RR 0.03, 95% CI 0 to 0.59; 5197 participants; low‐certainty evidence). Tixagevimab/cilgavimab may result in little to no difference on mortality within six months, all‐grade AEs, and SAEs (low‐certainty evidence). Quality of life was not reported.
Casirivimab/imdevimab versus placebo
One study evaluated casirivimab/imdevimab versus placebo in participants who may have been exposed to SARS‐CoV‐2 wild‐type, Alpha, and Delta variant. About 36.5% of participants opted for SARS‐CoV‐2 vaccination and had a mean of 66.1 days between last dose of intervention and vaccination. Within six months, casirivimab/imdevimab may decrease infection with SARS‐CoV‐2 (RR 0.01, 95% CI 0 to 0.14; 825 seronegative participants; low‐certainty evidence) and may decrease development of clinical COVID‐19 symptoms (RR 0.02, 95% CI 0 to 0.27; 969 participants; low‐certainty evidence). We are uncertain whether casirivimab/imdevimab affects mortality regardless of the SARS‐CoV‐2 antibody serostatus. Casirivimab/imdevimab may increase all‐grade AEs slightly (RR 1.14, 95% CI 0.98 to 1.31; 969 participants; low‐certainty evidence). The evidence is very uncertain about the effects on grade 3 to 4 AEs and SAEs within six months. Admission to hospital and quality of life were not reported.
Postexposure prophylaxis
Bamlanivimab versus placebo
One study evaluated bamlanivimab versus placebo in participants who may have been exposed to SARS‐CoV‐2 wild‐type. Bamlanivimab probably decreases infection with SARS‐CoV‐2 versus placebo by day 29 (RR 0.76, 95% CI 0.59 to 0.98; 966 participants; moderate‐certainty evidence), may result in little to no difference on all‐cause mortality by day 60 (R 0.83, 95% CI 0.25 to 2.70; 966 participants; low‐certainty evidence), may increase all‐grade AEs by week eight (RR 1.12, 95% CI 0.86 to 1.46; 966 participants; low‐certainty evidence), and may increase slightly SAEs (RR 1.46, 95% CI 0.73 to 2.91; 966 participants; low‐certainty evidence). Development of clinical COVID‐19 symptoms, admission to hospital within 30 days, and quality of life were not reported.
Casirivimab/imdevimab versus placebo
One study evaluated casirivimab/imdevimab versus placebo in participants who may have been exposed to SARS‐CoV‐2 wild‐type, Alpha, and potentially, but less likely to Delta variant. Within 30 days, casirivimab/imdevimab decreases infection with SARS‐CoV‐2 (RR 0.34, 95% CI 0.23 to 0.48; 1505 participants; high‐certainty evidence), development of clinical COVID‐19 symptoms (broad‐term definition) (RR 0.19, 95% CI 0.10 to 0.35; 1505 participants; high‐certainty evidence), may result in little to no difference on mortality (RR 3.00, 95% CI 0.12 to 73.43; 1505 participants; low‐certainty evidence), and may result in little to no difference in admission to hospital. Casirivimab/imdevimab may slightly decrease grade 3 to 4 AEs (RR 0.50, 95% CI 0.24 to 1.02; 2617 participants; low‐certainty evidence), decreases all‐grade AEs (RR 0.70, 95% CI 0.61 to 0.80; 2617 participants; high‐certainty evidence), and may result in little to no difference on SAEs in participants regardless of SARS‐CoV‐2 antibody serostatus. Quality of life was not reported.
Authors' conclusions
For PrEP, there is a decrease in development of clinical COVID‐19 symptoms (high certainty), infection with SARS‐CoV‐2 (moderate certainty), and admission to hospital (low certainty) with tixagevimab/cilgavimab. There is low certainty of a decrease in infection with SARS‐CoV‐2, and development of clinical COVID‐19 symptoms; and a higher rate for all‐grade AEs with casirivimab/imdevimab.
For PEP, there is moderate certainty of a decrease in infection with SARS‐CoV‐2 and low certainty for a higher rate for all‐grade AEs with bamlanivimab. There is high certainty of a decrease in infection with SARS‐CoV‐2, development of clinical COVID‐19 symptoms, and a higher rate for all‐grade AEs with casirivimab/imdevimab.
Although there is high‐to‐moderate certainty evidence for some outcomes, it is insufficient to draw meaningful conclusions. These findings only apply to people unvaccinated against COVID‐19. They are only applicable to the variants prevailing during the study and not other variants (e.g. Omicron). In vitro, tixagevimab/cilgavimab is effective against Omicron, but there are no clinical data. Bamlanivimab and casirivimab/imdevimab are ineffective against Omicron in vitro.
Further studies are needed and publication of four ongoing studies may resolve the uncertainties.
Plain language summary
Are laboratory‐made COVID‐19‐specific monoclonal antibodies effective to prevent COVID‐19 in adults?
Key messages
Pre‐exposure prophylaxis of COVID‐19:
– tixagevimab/cilgavimab probably reduces number of people infected with COVID‐19 and development of COVID‐19 symptoms, and may reduce number of people admitted to hospital;
– casirivimab/imdevimab may reduce number of people infected with COVID‐19 and development of COVID‐19 symptoms, and may increase number of unwanted effects (any severity) slightly.
Postexposure prophylaxis of COVID‐19:
– bamlanivimab probably reduces number of people infected with COVID‐19;
– casirivimab/imdevimab reduces number of people infected with COVID‐19 and development of COVID‐19 symptoms, and reduces number of unwanted effects (any severity).
What are 'monoclonal' antibodies?
The body makes antibodies to defend against disease. However, they may be produced in a laboratory from cells taken from people who have recovered from a disease.
Antibodies that target only one specific protein – in this case, a protein on the SARS‐CoV‐2 virus (that causes COVID‐19) – are 'monoclonal'. They attach to the virus and stop it from entering and replicating in human cells to prevent and fight infection. They are relevant for people who do not respond or respond poorly to vaccinations.
What did we want to find out?
We wanted to know if COVID‐19‐specific monoclonal antibodies are effective at preventing COVID‐19 in people exposed to the virus or are at high risk of being exposed to the virus. We looked at:
– confirmed COVID‐19 infections;
– development of COVID‐19 symptoms;
– death from any cause;
– hospital admission;
– quality of life;
– unwanted effects, such as infections and cardiac disorders;
– serious unwanted effects, such as life‐threatening, hospitalisation, disability, or death.
What did we do?
We searched for studies comparing monoclonal antibodies (e.g. bamlanivimab, tixagevimab/cilgavimab, and casirivimab/imdevimab) with placebo (sham treatment), another treatment, or no treatment to prevent COVID‐19 in people of any age, gender, or ethnicity.
We summarised data and rated our confidence in the evidence, based on factors such as study methods and size.
What did we find?
Four studies including 9749 people; two investigated people before being exposed to SARS‐CoV‐2 (pre‐exposure prophylaxis) and two investigated people who were in contact with an infected person (postexposure prophylaxis). Studies were conducted before the emergence of the Omicron variant and before or during widespread vaccine roll‐out. Participants were unvaccinated at baseline.
Pre‐exposure prophylaxis
One study (5197 people) compared tixagevimab/cilgavimab to placebo. Participants had been exposed to wild‐type, Alpha, Beta, and Delta variants.
Tixagevimab/cilgavimab:
– reduces development of symptoms;
– probably reduces number of people infected;
– may reduce number of hospital admissions;
– may have little or no effect on deaths from any cause, unwanted effects (any severity), and serious unwanted effect.
We found no data for quality of life, or mild and severe unwanted effects.
One study (969 people) compared casirivimab/imdevimab to placebo. Participants may have been exposed to wild‐type, Alpha, and Delta variants.
Casirivimab/imdevimab:
– may reduce number of people infected and development of symptoms;
– resulted in no deaths;
– may increase unwanted effects (any severity) slightly;
– we are uncertain whether casirivimab/imdevimab may have an effect on severe and serious unwanted effects.
We found no data for number of people with COVID‐19 within 30 days, development of symptoms within 30 days, hospital admissions within 30 days, quality of life, and mild unwanted effects.
Postexposure prophylaxis (two studies)
One study (966 people) compared bamlanivimab to placebo. Participants may have been exposed to wild‐type variants.
Bamlanivimab:
– probably reduces number of people infected;
– may have little or no effect on deaths from any cause;
– may increase unwanted effects (any severity) and serious unwanted effects slightly.
We found no data for number of people who developed symptoms, hospital admissions, quality of life, mild, and severe unwanted effects.
One study (2617 people) compared casirivimab/imdevimab to placebo. Participants may have been exposed to wild‐type, Alpha, and potentially, but unlikely Delta variants.
Casirivimab/ imdevimab:
– reduces number of people infected, development of symptoms, and unwanted effects (any severity);
– may slightly reduce severe unwanted effects;
– may have little or no effect on deaths, hospital admissions, and serious unwanted effects.
We found no data for quality of life and mild unwanted effects.
What are the limitations of the evidence?
One study reported each comparison. All participants were unvaccinated at the beginning of the studies, which were conducted prior to the occurrence of Omicron. Our confidence in the evidence is high to very low. Although we have high confidence in the evidence found for relevant outcomes, the findings are not transferable to vaccinated people and variants that occurred outside the study periods, such as Omicron. Although laboratory studies have shown that tixagevimab/cilgavimab is effective against Omicron, there are no data from clinical studies. The other monoclonal antibodies included in this review were ineffective against Omicron in laboratory studies.
Four ongoing studies are likely to change our conclusions and may help us understand how new variants and COVID‐19 vaccines affect the effectiveness of monoclonal antibodies in preventing COVID‐19.
How up to date is this evidence?
The evidence is up‐to‐date to 27 April 2022.
Summary of findings
Background
Description of the condition
The clinical syndrome novel coronavirus disease 2019 (COVID‐19) is a rapidly spreading infectious disease caused by severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2; WHO 2020a). Declared a pandemic on 11 March 2020, COVID‐19 is unprecedented in comparison to previous coronavirus outbreaks, such as severe acute respiratory syndrome (SARS) with 813 deaths by July 2003 (WHO 2003), and Middle East respiratory syndrome (MERS) with 882 deaths by January 2021 (WHO 2021a).
Despite intensive international efforts to contain its spread and record‐speed vaccine roll‐out and distribution, it has resulted in more than 265 million confirmed cases and more than 5.2 million deaths worldwide up to 7 December 2021 (WHO 2021b; WHO 2021c), impacting severely on healthcare facilities, healthcare workers, and medical equipment. Weekly COVID‐19 hospitalisation rates (e.g. CDC 2021a) and case fatality rates (Johns Hopkins 2021) fluctuated largely between countries and in time due to different demographics, dominant virus variant, national public health policies, and the way data are collected (i.e. testing frequency, reporting lags, and case definition).
The median incubation time is estimated to be five to six days, and 97.5% of symptomatic cases develop symptoms within 11.5 days of exposure (Lauer 2020). The most common signs and symptoms are fever or chills, cough, fatigue, myalgia or arthralgia, headache, anosmia or dysgeusia (loss of sense of smell or taste), shortness of breath, sore throat, nausea or vomiting, diarrhoea, and nasal congestion (CDC 2021b; Struyf 2020). Around 80% of infections are mild (Wu 2020), 20% of these remain asymptomatic (Buitrago‐Garcia 2020). In contrast, around 14% of infections are classified as severe disease, and 5% develop into critical disease with intensive care unit (ICU) admittance due to respiratory failure, septic shock, or multiple organ dysfunction (Wu 2020).
Multiple risk factors can worsen the course of the disease and increase the risk of mortality, such as old age (WHO 2020a), cardiovascular disease (CVD), obesity, hypertension, diabetes, chronic respiratory disease (Chen 2020; Huang 2020; WHO 2020a), and an immunocompromised state (i.e. through malignancy or solid organ transplant) (Fung 2020; Liang 2020). As effective treatment options are still sparse (Kluge 2021; National COVID‐19 Clinical Evidence Taskforce 2020), and vaccines may not work for all populations equally well, especially for immunocompromised people (Di Fusco 2021; Galmiche 2022), additional prophylactic strategies, which includes both pre‐exposure (PrEP) and postexposure (PEP), would be welcome. PrEP applies to people who do not have SARS‐CoV‐2 infection and who have not been recently exposed to an individual with SARS‐CoV‐2 infection and who may have an inadequate immune response to COVID‐19 vaccination or are unable to be fully vaccinated. PrEP is used to prevent new infections among those at greatest risk. PEP means taking medicine to prevent SARS‐CoV‐2 infection after a possible exposure.
While some substances have already shown no benefit (e.g. hydroxychloroquine, Boulware 2020), other studies are still ongoing (e.g. hyperimmune immunoglobulins, NCT04383548; convalescent plasma, NCT04836260). Another promising option is monoclonal antibodies (mAbs).
Description of the intervention
mAbs are laboratory‐produced molecules derived from natural antibodies of hosts who have been exposed to the antigen of interest. The selection of specific mAbs is commonly based on their virus neutralising capacity. Treatment with mAbs can be regarded as passive immunisation, as substituted antibodies are supposed to mimic a person's own humoral immune response in order to support recovery and viral clearance (Bayer 2019). While other passive antibody therapies, such as convalescent plasma or hyperimmune immunoglobulin, use antibodies derived from more than one white cell clone (polyclonal), mAbs and mAb fragments target a single, predetermined epitope and no additional plasma components. Consequently, they generally cause fewer adverse events (AE) such as serum sickness, anaphylaxis (Marston 2018), and thromboembolic events (Driggin 2020). The standardised production allows comprehensive characterisation in vitro, enabling precise control of dosage and composition of a mAb cocktail, which increases effectiveness.
A variety of mAbs have been developed against both endogenous targets (i.e. in oncology and immunology) (Kaplon 2020; Lu 2020a), as well as exogenous targets (i.e. palivizumab against respiratory syncytial virus (RSV) infection (Andabaka 2013), a mAb against HIV‐1 (Jaworski 2021), and ansuvimab and REGN‐EB3 against Ebola (Mulangu 2019)). As of April 2021, at least 135 mAbs that target different epitopes on the SARS‐CoV‐2 virus, especially the Spike protein of the virus, have been developed or tested in clinical trials to treat COVID‐19 (The Antibody Society 2021; Yang 2020).
For example, in treatment with mAbs, bamlanivimab monotherapy and the combination of casirivimab/imdevimab has shown no benefit for the general population of hospitalised patients but may improve outcomes when provided earlier (i.e. to outpatients or people who had not yet produced their own antibodies) (Gottlieb 2021; Horby 2021; Lundgren 2021; Weinreich 2021). The frequency of AEs in the placebo and mAbs treatment groups was similar; however, there were slightly more infusion‐related reactions reported for the mAbs group (Gottlieb 2021; Lundgren 2021; Weinreich 2021).
Based on data from these trials (NCT04425629; NCT04427501), the US Food and Drug Administration (FDA) issued Emergency Use Authorizations (EUA) for treatment of certain patient groups with bamlanivimab, which was withdrawn due to inability to neutralise the Delta variant (FDA 2021a). Due to the emergence of the Omicron variant and the inability of most mAbs to retain activity against this variant, the FDA has revised the EUA for the mAb combinations bamlanivimab/etesevimab and casirivimab/imdevimab and restricted their use to people exposed to or infected with a variant where the mAb likely remains active against the variant (FDA 2022a). For sotrovimab, the FDA withdrew the EUA from use in areas where the Omicron sublineage BA.2 accounts for more than 50% of infections (FDA 2022b). Recently, the FDA has issued EUA for PrEP with tixagevimab/cilgavimab and for treatment with bebtelovimab, carrying a clear statement that those mAbs are "not intended to be a substitute for vaccination against COVID‐19" (FDA 2021b; FDA 2022c). The National Institutes of Health (NIH) COVID‐19 Treatment Guidelines Panel recommends the use of tixagevimab/cilgavimab as PrEP and recommends against the use of the combinations bamlanivimab/etesevimab and casirivimab/imdevimab as PEP (NIH 2022).
Alternatives to conventional full‐size mAbs, which are fragile and often administered intravenously or subcutaneously (Jovčevska 2020), are mAb fragments such as nanobodies and microbodies. Due to their small size and high stability, mAb fragments offer many advantages in terms of their production and neutralising activities (Custódio 2020; Hanke 2020), for instance allowing administration by inhalation (Gai 2021).
How the intervention might work
SARS‐CoV‐2 stems from the coronavirus family that is characterised by a positive‐sense, single‐stranded ribonucleic acid (RNA) (Lu 2020b). The spike proteins on its envelope, which give the virus its name, play a critical role in enabling it to enter a host cell by two mechanisms (Hoffmann 2020; Ou 2020). First, the human angiotensin‐converting enzyme 2 (ACE2) receptor on the spike protein binds to ACE2 proteins that are found throughout the body, but with higher expression in respiratory epithelial cells, type I and II alveolar cells in the lungs, oral cavity, kidney, testes, and intestines (Tolouian 2020). This activates the S proteins' fusion machinery, which inserts into the cellular plasma membrane, brings the viral membrane into proximity, and fuses them to create a portal to deposit the virus RNA genome into the host cell, where it starts replicating (Glaunsinger 2020; Tolouian 2020). The so‐called 'cytokine storm' plays a crucial role in disease severity, which can lead to acute respiratory distress syndrome (ARDS; de la Rica 2020).
The focus of research on SARS‐CoV‐2‐neutralising mAbs and mAb fragments is to block the binding of SARS‐CoV‐2 to the ACE2 receptor on human cells by targeting the receptor‐binding domain (RBD) on the spike protein of the virus (Hanke 2020; Lu 2021; Marovich 2020; Wrapp 2020). Microbodies, which are proteins with an extracellular domain of ACE2 fusioned to the Fc domain of an immunoglobulin, have been shown to inhibit the virus from attaching to the cells by trapping the spike protein (Glasgow 2020; Tada 2020). The possibility of administration by inhalation, directly at the infection site, could help control transmission of the virus (Gai 2021).
In several preclinical studies in mice, SARS‐CoV‐2‐neutralising mAbs injected before SARS‐CoV‐2 inoculation showed inhibition of viral replication and mitigation of disease severity (Alsoussi 2020; Hassan 2020; Rogers 2020; Zost 2020). In a study by Baum and colleagues, six rhesus macaques received casirivimab/imdevimab or placebo intravenously and, after three days, were exposed to the virus through intranasal and intratracheal routes. Despite similar RNA kinetics, sgRNA levels, which indicates viral replication, were lower in animals receiving casirivimab/imdevimab when compared to placebo. Their high‐ versus low‐dose comparison in four rhesus macaques with a 10‐fold higher virus inoculum showed decreased viral replication for the high‐dose compared with low‐dose antibody cocktail (Baum 2020a).
Currently, numerous variants of SARS‐CoV‐2 have emerged. Mutations in the spike protein of the virus may reduce the neutralising ability of a mAb if the mutation happens at or close to the mAb's target epitope, and may instead lead to increased severity of the infection, termed 'antibody‐dependent enhancement (ADE)' (Lee 2020). Since December 2020, five variants that developed independently of each other have been classified as of concern (WHO 2021d).
Currently circulating variants of concern:
Delta (also known as B.1.617.2) first occurred in October 2020 in India (WHO 2021d);
Omicron (also known as B.1.1.529 including its sublineages BA.1, BA.2, BA.3, BA.4, BA.5, and descendent lineages), identified in November 2021, is a new variant of concern and overtook delta as dominant variant (WHO 2022).
Previously circulating variants of concern:
Alpha (also known as B.1.1.7, 20I/501Y.V1 or VOC 202012/01), first identified in the UK with rapid local spread and later international spread, is associated with increased transmissibility and increase in disease severity (Muik 2021; Tang 2021);
Beta (also known as B.1.351, 20H/501Y.V2), initially identified in South Africa, may also be more transmissible, but may not increase disease severity (Wang 2021);
Gamma (also known as P.1), first identified in Brazil, might reduce antibody neutralisation (Wang 2021).
Since the first occurrence of Omicron in Botswana in November 2021, it has rapidly become the dominating variant circulating globally (WHO 2022). This variant includes several sublineages such as BA.1 and BA.2, which differ in several mutations in the spike protein. The BA.2 Omicron sublineage is now dominant in many regions (WHO 2022). In general, Omicron is associated with potential increased transmissibility, reduction in neutralising activity in sera, and reduced vaccine effectiveness (Rössler 2022; Vitiello 2022).
Early in vitro findings suggest that mAbs such as bamlanivimab, etesevimab, casirivimab, imdevimab, tixagevimab, cilgavimab, and sotrovimab efficiently neutralise the Alpha variant. However, against the variants Beta and Gamma, there was no neutralising effect detected for bamlanivimab and etesevimab. All of these mAbs except bamlanivimab could retain neutralising activity against Delta (Widera 2021).
To date only bebtelovimab and a combination of tixagevimab and cilgavimab have shown retained neutralising activity against both Omicron sublineages BA.1 and BA.2 (Iketani 2022; Takashita 2022). BA.1 was sensitive to adintrevimab and sotrovimab. In contrast, imdevimab showed neutralising activity against BA.2 (Bruel 2022).
Why it is important to do this review
Although numerous vaccines for the prevention of infection with SARS‐CoV‐2, such as BNT162b2, mRNA‐1273, COVID‐19 Vaccine by Johnson & Johnson (FDA 2021c), AZD1222 (AstraZeneca 2020), and CoronaVac (WHO 2021e) have been authorised and widely distributed, the number of cases and deaths worldwide is rising, with healthcare systems being at their limits.
Only a few treatment options are currently available for the treatment of COVID‐19. Those include corticosteroids for severe COVID‐19 and tocilizumab for certain patient groups (Kluge 2021; National COVID‐19 Clinical Evidence Taskforce 2020; Siemieniuk 2020). In 2021, the FDA issued EUA for the first oral antiviral agents against COVID‐19 such as molnupiravir and nirmatrelvir plus ritonavir, which were found to be effective in people with mild‐to‐moderate COVID‐19 who are at high risk for severe disease (FDA 2021d; FDA 2021e). Those are recommended for use in early infection (within five days of symptom onset) in unvaccinated individuals with one or more risk factors for progression to severe disease (National COVID‐19 Clinical Evidence Taskforce 2020).
Multiple factors play a role in the further spread of the virus: on the one hand, vaccine distribution has not yet reached all parts of the world equitably UNDP 2021; on the other hand, vaccine acceptance is not equally high everywhere (Nehal 2021). Additionally, vaccine effectiveness is lowered with the Omicron variant and booster doses are necessary to increase protection against symptomatic disease caused by the Omicron variant (Andrews 2022).
At the same time, the immune response to the vaccine decreases over time (Goldberg 2021), and immune response after vaccination can vary among individuals. For example, immunocompromised individuals may have an insufficient immune response to the vaccine, whereby protection against infection and a severe COVID‐19 course is reduced (Di Fusco 2021; Monin‐Aldama 2021). Even after a standard vaccine regimen (e.g. including two doses of mRNA vaccine and booster vaccinations), immune response of these patients is insufficient (Mair 2022). The FDA recently authorised a second booster dose of mRNA vaccines for people with immunocompromised conditions (FDA 2022d).
Although compared to mAbs, vaccines probably provide longer‐lasting immunity for those who respond (Marovich 2020), mAbs may serve as an additional immediate option to prevent infection especially for very young children, elderly people, and those with temporarily or permanently compromised immune systems, either after being exposed to a case or as PrEP.
The current systematic review will identify, describe, evaluate, and meta‐analyse all evidence for SARS‐CoV‐2‐neutralising mAbs, including mAb fragments, used as prophylaxis for SARS‐CoV‐2 infections. To provide a frequently updated status, we are planning to create this as a living systematic review.
Objectives
To assess the effects of SARS‐CoV‐2‐neutralising mAbs, including mAb fragments, to prevent infection with SARS‐CoV‐2 causing COVID‐19; and to maintain the currency of the evidence, using a living systematic review approach.
Methods
Criteria for considering studies for this review
Types of studies
We included randomised controlled trials (RCTs), because this study design, if performed appropriately, provides the best evidence for experimental therapies in highly controlled therapeutic settings. For RCT data, we used the methods recommended in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2021a). We had planned to include non‐standard RCT designs, such as cluster‐randomised studies (using methods as recommended in Chapter 23 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2021b)) and cross‐over studies, but have not identified any. In cross‐over studies, we would only consider results from the first period before cross‐over because COVID‐19 is not a chronic condition, and its exact course and long‐term effects are yet to be defined.
We included the following formats if there was sufficient information available on study design, characteristics of participants, interventions, and outcomes, or if we could obtain this via contact with study authors.
Full‐text publications.
Preprint articles.
Abstract publications.
Results published in study registries.
We decided to include preprints and conference abstracts to have a complete overview of the ongoing research activity. We applied no limitation with respect to the length of follow‐up. We screened platform trials, and we are continually following these because they may add new intervention arms.
Types of participants
We included studies examining participants without defined exposure, or with potential exposure to SARS‐CoV‐2, but who did not have a confirmed diagnosis of COVID‐19 (virus antigens or RNA detected). For PrEP, we included participants regardless of SARS‐CoV‐2 antibody serostatus and for PEP, we included SARS‐CoV‐2 antibody seronegative participants.
We did not exclude studies based on age, gender, ethnicity, or setting. We excluded studies that evaluated mAbs to prevent infection from other coronavirus diseases (e.g. SARS or MERS), or other viral diseases, such as influenza. If studies enrolled populations with mixed viral diseases, we only included these if trial authors provided subgroup data for participants with COVID‐19.
Treatment of individuals who have SARS‐CoV‐2 infection with SARS‐CoV‐2‐neutralising mAbs is covered in another review (Kreuzberger 2021).
Types of interventions
We included the following interventions.
SARS‐CoV‐2‐neutralising mAbs, including mAb fragments.
Combinations of SARS‐CoV‐2‐neutralising mAbs.
We included the following comparisons for studies with a control arm.
Any mAb prophylaxis compared with a control intervention, for example, vaccinations, drug prophylaxis (including but not limited to hydroxychloroquine, remdesivir), standard or hyperimmune immunoglobulin, convalescent plasma, other prevention strategies (e.g. protective clothing, face masks, social distancing), complementary medicine (e.g. quercetin, elderberry, zinc), or others.
Any mAb prophylaxis compared with no prophylaxis or placebo.
Co‐interventions were allowed, but these must have been comparable between intervention groups.
We included studies that compared several mAbs or mAb fragments with each other and another prophylaxis, placebo or no prophylaxis, as well as studies that compared several doses of one type of mAb or mAb fragments with another prophylaxis, placebo, or no prophylaxis.
We explicitly excluded mAbs or mAb fragments that were not specifically designed to target SARS‐CoV‐2 (e.g. nivolumab, tocilizumab, canakinumab).
Types of outcome measures
We evaluated core outcomes as predefined by the Core Outcome Set for studies evaluating public health, primary and secondary care interventions for prevention of COVID‐19 transmission (COMET 2020; Marshall 2020).
Primary outcomes
Effectiveness of SARS‐CoV‐2‐neutralising mAbs to prevent infection with SARS‐CoV‐2
Pre‐exposure prophylaxis of COVID‐19
Infection with SARS‐CoV‐2 (confirmed by positive reverse transcription polymerase chain reaction (RT‐PCR) test) within six months.
Development of clinical COVID‐19 symptoms, as defined by the study, within six months.
Postexposure prophylaxis of COVID‐19
Infection with SARS‐CoV‐2 (confirmed by positive RT‐PCR) test within 30 days.
Development of clinical COVID‐19 symptoms, as defined by the study, within 30 days.
Secondary outcomes
Effectiveness of SARS‐CoV‐2‐neutralising mAbs to prevent infection with SARS‐CoV‐2
Pre‐exposure prophylaxis of COVID‐19
All‐cause mortality at six months.
Admission to hospital within six months.
Admission to ICU within six months.
Quality of life, assessed with standardised scales (e.g. World Health Organization Quality Of Life assessment instrument (WHOQOL‐100; WHO 2020b), at up to six months and longest follow‐up available.
Postexposure prophylaxis of COVID‐19
All‐cause mortality at day 30, day 60, longest follow‐up, and time‐to‐event.
Admission to hospital within 30 days.
Admission to the intensive care unit (ICU) within 30 days.
Quality of life, assessed with standardised scales (e.g. WHOQOL‐100; WHO 2020b), at up to seven days, up to 30 days, and longest follow‐up available.
Safety of SARS‐CoV‐2‐neutralising mAbs to prevent infection with SARS‐CoV‐2
Pre‐exposure and postexposure prophylaxis of COVID‐19
Number of participants with adverse events (AE) (grade 1 to 2, grade 3 to 4, all grade) until end of follow‐up.
Number of participants with serious adverse events (SAE) until end of follow‐up.
Timing of outcome measurement
In case of time‐to‐event analysis (e.g. mortality), we included the outcome measure based on the longest follow‐up time. We also collected information on outcomes from all other time points reported in the publications.
We included AEs and SAEs occurring until the end of follow‐up. If sufficient data were available, we had grouped the measurement time points of eligible outcomes, for example, AEs and SAEs, into those measured directly after intervention (up to seven days after intervention), medium‐term outcomes (up to 15 days after intervention), and longer‐term outcomes (more than 30 days after intervention).
Search methods for identification of studies
This review is part of a COVID‐19 living systematic review project evaluating the effectiveness and safety of SARS‐CoV‐2‐neutralising mAbs. Our search addresses:
SARS‐CoV‐2‐neutralising mAbs to prevent infection with SARS‐CoV‐2; and
SARS‐CoV‐2‐neutralising mAbs to treat people with COVID‐19 (for the Cochrane Review by Kreuzberger 2021).
We performed weekly searches for completed and ongoing studies in all languages to limit language bias. We checked weekly for newly emerging mAbs and changing terminology regarding mAbs included in the search strategy, and adapted the strategy where necessary.
Electronic searches
For the identification of studies on the effectiveness and safety of SARS‐CoV‐2‐neutralising mAbs, we designed search strategies in accordance with the Cochrane Handbook for Systematic Reviews of Interventions (Lefebvre 2021). The review team's Information Specialist (IM) developed the search strategy based on input from clinicians, and Carolyn Dorée reviewed it. If the full‐text publication was published in a language outside the abilities of our team (English, German, Dutch, French, Italian, Malay, and Spanish), we involved Cochrane TaskExchange to identify people who were able to translate (taskexchange.cochrane.org). We searched the following databases from 1 January 2020 to 27 April 2022:
-
Cochrane COVID‐19 Study Register (covid-19.cochrane.org; inception 20 April 2022; Appendix 1) including:
monthly searches of the Cochrane Central Register of Controlled Trials (CENTRAL);
weekly searches of PubMed;
weekly searches of Embase.com;
daily searches of ClinicalTrials.com;
weekly searches of the World Health Organization (WHO) International Clinical Trials Registry Platform (ICTRP);
MEDLINE (via Ovid; 1 January 2020 to 20 April 2022; Appendix 2);
Embase (via Ovid; 1 January 2020 to 20 April 2022; Appendix 3);
PubMed (for epublications ahead of print only; 1 January 2020 to 20 April 2022; Appendix 4);
Epistemonikos, L*OVE List Coronavirus disease (COVID‐19) (app.iloveevidence.com/loves; inception 20 April 2022; Appendix 5);
WHO COVID‐19 Global literature on coronavirus disease (bvsalud.org/global-literature-on-novel-coronavirus-2019-ncov; inception 20 April 2022; Appendix 6).
To track ongoing research efforts and address the potential influence of publication bias on our conclusions, we searched relevant trial registries to find ongoing and completed, but not yet published, studies. If results were uploaded into the trials registry that had not been published elsewhere, we integrated these data for the current review and will add or replace data in future updates of this review in case of publication.
To identify prospectively registered platform trials for prevention of SARS‐CoV‐2 infection that may add an mAb arm during the course of the study, such as the COVER HCW trial (NCT04561063), we searched these trials (Appendix 7), listed them in the Characteristics of studies awaiting classification table, and will check regularly whether they have added additional intervention arms that include mAbs.
Searching other resources
We checked the reference lists of all identified studies, relevant review articles, and current treatment guidelines for further literature.
We contacted experts in the field, drug manufacturers, and regulatory agencies in order to retrieve information on unpublished studies.
We compared our results with results from projects that aim to track COVID‐19 intervention research (i.e. covid-nma.com/dataviz).
Data collection and analysis
Selection of studies
Two review authors (NK, KLC, LJE, NS, or CH) independently screened the search results for eligibility for this review by reading the abstracts using Covidence software (Covidence). Following the living review approach, we screened any new citations retrieved by the weekly searches immediately. We obtained the full‐text publications of any abstracts that both review authors found eligible, and also of those that they disagreed upon or rated as uncertain, for further discussion. Two review authors assessed the full‐text articles of selected studies. If the two review authors were unable to reach a consensus, we consulted all review authors who were involved in study selection to reach a final decision.
The search for platform trials was conducted monthly (from November 2020 to March 2022). To identify potential platform trials, two review authors (YSP, NK, CH) independently screened the results in Endnote X9.
We documented the study selection process in a flow chart, as recommended in the PRISMA statement (Moher 2009), and showed the total number of retrieved references, and the numbers of included and excluded studies. We listed all studies that we excluded after full‐text assessment and the reasons for their exclusion in the Characteristics of excluded studies table.
Data extraction and management
We conducted data extraction according to the guidelines proposed by Cochrane (Li 2021). Two review authors (NK, YSP) extracted data independently and in duplicate, using a customised data extraction form developed in Microsoft Excel. We solved disagreements by discussion. If no agreement was obtained, we involved a third review author to solve the disagreement.
We extracted the following information, if reported.
General information: author, title, source, publication date, country, language, duplicate publications.
Study characteristics: trial design, setting and dates, source of participants, inclusion/exclusion criteria, comparability of groups, intervention cross‐overs, compliance with assigned intervention, length of follow‐up.
Participant characteristics: age, gender, ethnicity, number of participants recruited/allocated/evaluated, additional diagnoses, severity of disease, previous interventions, concurrent interventions, complementary medicine (e.g. quercetin, elderberry, zinc).
Interventions: type of mAbs or mAb fragments, target of mAbs or mAb fragments, dose, frequency, timing, duration and route of administration, setting (e.g. inpatient, ambulant), duration of follow‐up.
Control intervention: concomitant prevention strategies, dose, frequency, timing, duration and route of administration, setting, duration of follow‐up.
Outcomes: as specified under Types of outcome measures.
Assessment of risk of bias in included studies
We assessed the risk of bias in RCTs using the RoB 2 tool (Sterne 2019), for the effect of the assignment to the intervention (the intention‐to‐treat (ITT) effect). The outcomes that we assessed were those specified for inclusion in the summary of findings tables.
Two review authors (NK, YSP, CH) independently assessed the risk of bias for each outcome. In case of discrepancies among judgements and inability to reach consensus, we consulted a third review author to reach a final decision. We assessed the following types of bias as outlined in Chapter 8 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2021c).
Bias arising from the randomisation process.
Bias due to deviations from the intended interventions.
Bias due to missing outcome data.
Bias in measurement of the outcome.
Bias in selection of the reported result.
For cross‐over studies, we had planned to use the RoB 2 tool, as outlined in Chapter 23 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2021b), because we only considered results from the first period before cross‐over.
For cluster‐randomised studies, we had planned to add an additional domain to assess bias arising from the timing of identification and recruitment of participants in relation to timing of randomisation, as recommended in the archived RoB 2 guidance for cluster‐randomised trials (Eldridge 2021), and in Chapter 23 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2021b).
To address these types of bias, we used the signalling questions recommended in RoB 2 and make a judgement using the following options.
'Yes': if there was firm evidence that the question was fulfilled in the study (i.e. the study was at low or high risk of bias for the given direction of the question).
'Probably yes': a judgement was made that the question was fulfilled in the study (i.e. the study was at low or high risk of bias given the direction of the question).
'No': if there was firm evidence that the question was unfulfilled in the study (i.e. the study was at low or high risk of bias for the given direction of the question).
'Probably no': a judgement was made that the question was unfulfilled in the study (i.e. the study was at low or high risk of bias given the direction of the question).
'No information': if the study report did not provide sufficient information to allow any judgement.
We used the algorithms proposed by RoB 2 to assign each domain one of the following levels of bias.
Low risk of bias.
Some concerns.
High risk of bias.
Subsequently, we derived an overall risk of bias rating for each prespecified outcome in each study in accordance with the following suggestions.
'Low risk of bias': we judged the trial at low risk of bias for all domains for this result.
'Some concerns': we judged the trial to raise some concerns in at least one domain for this result, but not to be at high risk of bias for any domain.
'High risk of bias': we judged the trial at high risk of bias in at least one domain for the result, or we judged the trial to have some concerns for multiple domains in a way that substantially lowered confidence in the results.
We used the RoB 2 Excel tool to implement RoB 2 (available from riskofbias.info), and stored and presented our detailed RoB 2 assessments as supplementary online material (available at doi.org/10.5281/zenodo.6541295).
Measures of treatment effect
For continuous outcomes, we planned to record the mean, standard deviation (SD), and total number of participants in both intervention and control groups. Where continuous outcomes use the same scale, we planned to use the mean difference (MD) with 95% confidence intervals (CIs). For continuous outcomes measured with different scales, we planned to use the standardised mean difference (SMD). For interpreting SMDs, we planned to re‐express SMDs in the original units of a particular scale with the most clinical relevance and impact.
For dichotomous outcomes, we recorded the number of events and the total number of participants in both intervention and control groups. We reported the pooled risk ratio (RR) with a 95% CI (Deeks 2021). We had planned to use Peto odds ratio (OR) if the number of observed events was small (less than 5% of sample per group, Deeks 2021). However, because there was very little difference in the effect estimate whether RR or Peto OR was used, for consistency and interpretability we decided to report RRs.
If available, we had extracted and reported hazard ratios (HRs) for time‐to‐event outcomes (e.g. time to death). If HRs were not available, we planned to make every effort to estimate the HR as accurately as possible from available data using the methods proposed by Parmar 1998 and Tierney 2007. If sufficient studies provided HRs, we planned to used HRs rather than RRs or MDs in a meta‐analysis, as they provide more information.
Unit of analysis issues
The aim of this review was to summarise studies that analysed data at the level of the individual. For cluster‐randomised studies, we accepted, for example, medical practices as the unit of analysis. We collated multiple reports of one study so that the study, and not the report, was the unit of analysis.
Studies with multiple intervention groups
As recommended in Chapter 6 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2021a), for studies with multiple intervention groups of the same intervention (i.e. dose, route of administration), we evaluated whether study arms were sufficiently homogeneous to be combined. When arms could not be pooled, we compared each arm with the common comparator separately. For pairwise meta‐analyses, we had planned to split the 'shared' group into two or more groups depending on the number of intervention arms and included two or more (reasonably independent) comparisons. For this purpose, for dichotomous outcomes, both the number of events and the total number of participants would be divided, and for continuous outcomes, the total number of participants would be divided with unchanged means and SDs.
Dealing with missing data
Chapter 10 of the Cochrane Handbook for Systematic Reviews of Interventions suggests a number of potential sources for missing data, which we took into account: at study level, at outcome level, and at summary data level (Deeks 2021). At all levels, it is important to differentiate between data 'missing at random', which may often be unbiased, and 'not missing at random', which may bias study and thus review results.
Whenever possible, we contacted the original investigators to request missing data. We contacted the study authors from the included RCTs (BLAZE‐2; Isa 2021; O'Brien 2021), and requested data and additional information for our prioritised outcomes. We assumed data to be missing at random when the rate of missingness across arms was comparable and the characteristics of participants with missing data were comparable to the characteristics of participants without missing data. Otherwise, we assumed data not to be missing at random. For the primary analysis, we conducted a complete‐case analysis by excluding participants with missing outcome data from the meta‐analysis. We planned to perform sensitivity analyses to assess how robust results are to worst‐case scenario assumption. We addressed the potential impact of missing data under Potential biases in the review process.
Assessment of heterogeneity
We planned to assess the heterogeneity of intervention effects between trials using a Chi² test with a significance level at P < 0.1. We used the I² statistic (Higgins 2003) to quantify possible heterogeneity (I² statistic between 30% and 60% may signify moderate heterogeneity, I² statistic between 50% and 90% may signify substantial heterogeneity, and I² > 75% may signify considerable heterogeneity; Deeks 2021). If we considered heterogeneity to be above I² > 75%, we had planned to explore potential causes through sensitivity and subgroup analyses. If we could not find a reason for heterogeneity, we would not have performed a meta‐analysis but had planned to comment on results from all studies and present these in tables.
Assessment of reporting biases
As mentioned above, we searched trial registries to identify completed trials that have not been published elsewhere, to minimise publication bias. We intended to explore potential publication bias by generating a funnel plot and statistically testing this by conducting a linear regression test for meta‐analyses involving at least 10 trials (Sterne 2019). We considered P < 0.1 as significant for this test.
Data synthesis
If the clinical and methodological characteristics of individual studies were sufficiently homogeneous, we pooled the data in a meta‐analysis. We planned to conduct separate meta‐analyses for different mAbs, as each mAb is a different molecule with a different target epitope or polytopes. One review author entered the data into Review Manager Web software (Review Manager Web 2021), and a second review author checked the data for accuracy. We performed analyses according to the recommendations of the Cochrane Handbook for Systematic Reviews of Interventions (Deeks 2021). We conducted separate meta‐analyses for each proposed comparison, that is, separate meta‐analyses for different comparators, except if a group of drugs had been shown to be sufficiently homogeneous (e.g. corticosteroids). We planned to treat placebo and no intervention as the same intervention.
We planned to use random‐effects model for all analyses as we anticipated that true effects would be related, but not be the same for included studies. We assessed the effects of potential biases in sensitivity analyses (see Sensitivity analysis). For binary outcomes, we based the estimation of the between‐study variance on the calculation performed using the Mantel‐Haenszel method. We used the inverse variance method for continuous outcomes, outcomes that included data from cluster‐randomised trials, or outcomes where HRs were available.
Subgroup analysis and investigation of heterogeneity
To explore heterogeneity, we planned to perform subgroup analyses with tests for interaction in Review Manager Web of the following characteristics (Review Manager Web 2021).
Age of participants (divided into applicable age groups, e.g. children; 18 to 65 years; older than 65 years).
Pre‐existing condition versus without any pre‐existing condition.
Variants of SARS‐CoV‐2 detected in case of infection.
Antibodies detected at baseline.
Sensitivity analysis
We had planned sensitivity analyses to examine the influence of the following characteristics for our primary outcomes.
Risk of bias assessment components (studies with a low risk of bias or some concerns versus studies with a high risk of bias).
Comparison of preprints of COVID‐19 interventions versus peer‐reviewed articles.
Comparison of premature termination of studies with completed studies.
Missing outcome data: worst‐case scenario assumption to assess how robust results are.
Summary of findings and assessment of the certainty of the evidence
We created summary of findings tables and evaluated GRADE for interventions evaluated in RCTs using GRADEpro GDT software (Schünemann 2021). We created a separate table per mAb type. We had planned to only present summary of findings tables for the three most clinically important mAbs or mAb fragments, and decided to keep tables for PrEP and PEP separate.
For time‐to‐event outcomes, we planned to calculate absolute effects at specific time points, as recommended in the GRADE guidelines 27 (Skoetz 2020).
Here are the prioritised outcomes for the summary of findings tables.
Pre‐exposure prophylaxis of COVID‐19
Infection with SARS‐CoV‐2 (positive RT‐PCR test) within six months.
Development of clinical COVID‐19 symptoms, as defined by the study, within six months.
All‐cause mortality at six months.
Admission to hospital within six months.
Quality of life, assessed with standardised scales (e.g. WHOQOL‐100; WHO 2020b), at longest follow‐up.
Number of participants with AEs (grade 1 to 2, grade 3 to 4, all grade) until end of follow‐up.
Number of participants with SAEs until end of follow‐up.
Postexposure prophylaxis of COVID‐19
Infection with SARS‐CoV‐2 (positive RT‐PCR test) within 30 days.
Development of clinical COVID‐19 symptoms, as defined by the study, within 30 days.
All‐cause mortality at longest follow‐up and greater than 60 days (or preferably time‐to‐event estimate). If not reported, we included all‐cause mortality at day 60 followed by day 30.
Admission to hospital within 30 days.
Quality of life, assessed with standardised scales (e.g. WHOQOL‐100; WHO 2020b), at longest follow‐up.
Number of participants with AEs (grade 1 to 2, grade 3 to 4, all grade) until end of follow‐up.
Number of participants with SAEs until end of follow‐up.
Assessment of the certainty in the evidence
We used the GRADE approach to assess the certainty in the evidence for the outcomes listed in the previous section. The GRADE approach uses five domains (risk of bias, consistency of effect, imprecision, indirectness, and publication bias) to assess the certainty in the body of evidence for each prioritised outcome. We followed the current GRADE guidance for these assessments in their entirety, as recommended in Chapter 14 of the Cochrane Handbook for Systematic Reviews of Interventions (Schünemann 2021).
We used the overall risk of bias judgement, derived from the RoB 2 Excel tool, to inform our decision on downgrading for risk of bias. We phrased the findings and certainty in the evidence as suggested in the guidance on informative statements (Santesso 2020).
Living systematic review considerations
Our Information Specialist (IM) provides us with the new records each week, which two review authors are screening following the guidance on Cochrane living systematic reviews (Living Evidence Network 2019).
We will wait until the accumulating evidence changes one or more of the following components before republishing the review.
The findings of one or more prioritised outcomes.
The credibility (e.g. GRADE rating) of one or more prioritised outcomes.
New settings, populations, interventions, comparisons, or outcomes studied.
We will review the review scope and methods approximately monthly, in light of potential changes in COVID‐19 research (e.g. when additional comparisons, interventions, subgroups, outcomes, or new review methods become available).
Results
Description of studies
Results of the search
We searched all databases and screened the resulting records weekly up to 27 April 2022. Based on newly developing SARS‐CoV‐2‐specific mAbs, we have added terms to our search strategy; see Differences between protocol and review for the changes that have been implemented. Our searches retrieved 20,821 records for the mAbs‐specific searches. After removing duplicates, we screened 13,802 records based on their titles and abstracts. We excluded 13,727 records that did not meet the prespecified inclusion criteria. Of the remaining 75 records, we included 23 records on prevention in this review.
Four RCTs (18 records) are included for prevention; of these one preprint.
Four RCTs (four records) on three different mAbs or mAb combinations were ongoing.
One RCT (one record) awaiting classification.
Our platform trial search yielded 16,319 records. After removing 2892 duplicates, we screened 13,427 during title and abstract screening and looked at 69 registry records in more detail. Of these, we excluded 68 studies (272 records). We identified:
no platform trials on prevention with at least one mAb arm as experimental intervention;
one ongoing platform trial (one record) on prevention as awaiting classification that may potentially add a mAb during the course of the study.
The study flow diagram in Figure 1 illustrates the study selection process according to PRISMA guidelines (Moher 2009).
1.
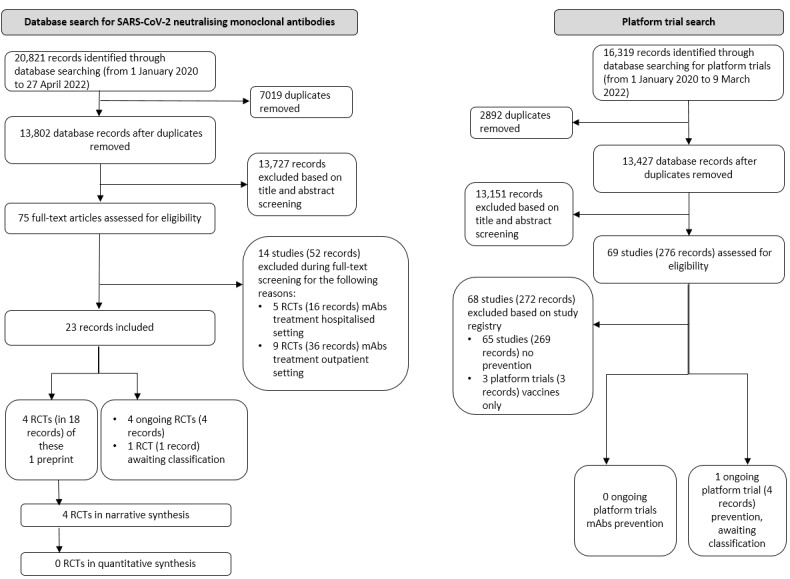
PRISMA flow diagram. mAb: monoclonal antibody; RCT: randomised controlled trial.
Included studies
See Characteristics of included studies table for full details.
Designs of the studies and sample size
We included four RCTs, involving 9749 randomised participants (BLAZE‐2; Isa 2021; O'Brien 2021; PROVENT). All four studies were double‐blind and placebo‐controlled. None of the included studies followed a cross‐over or cluster RCT design. One study randomised participants in a 2:1 ratio (PROVENT), while the other three studies followed a 1:1 ratio (BLAZE‐2; Isa 2021; O'Brien 2021). One study, published in November 2021 as a preprint, was a phase 1 study with ongoing follow‐up (Isa 2021). Three studies were published as full‐text publications (BLAZE‐2; O'Brien 2021; PROVENT), of which one study provided additional information in a conference abstract (O'Brien 2021). One was a phase 3 study with ongoing follow‐up (PROVENT). Two were multipart phase 3 studies completed on 20 May 2021 (BLAZE‐2) and 4 October 2021 (O'Brien 2021). O'Brien 2021 conducted an initial assessment of the first 554 participants, which were not included in the efficacy analysis, but analysed together with the other included participants in the safety analysis set. The studies did not report characteristics of participants with missing data. We received additional information for BLAZE‐2 regarding the measured time points of the safety outcomes and clarification regarding the incidence of SARS‐CoV‐2 infection by day 29. O'Brien 2021 provided requested data on SARS‐CoV‐2 seronegative participants for the outcomes mortality and emergency department or hospital admission. From Isa 2021, we requested the study protocol but received no answer.
Setting and participants
Two studies included participants for PrEP (Isa 2021; PROVENT), and two studies included participants after being exposed to SARS‐CoV‐2 (PEP) (BLAZE‐2; O'Brien 2021). BLAZE‐2 and O'Brien 2021 were conducted before widespread vaccine roll‐out, while the study period of Isa 2021 and PROVENT overlapped with first vaccine roll‐out.
PROVENT was a multicentre study conducted at different sites in Belgium, Spain, the UK, and the US. The study recruited unvaccinated participants with an increased risk of an inadequate immune response to COVID‐19 vaccination, an increased risk of SARS‐CoV‐2 exposure (e.g. healthcare workers, workers in industrial settings such as meatpacking plants, military personnel, students living in dormitories), or both, between 21 November 2020 to 22 May 2021. At screening, participants were required to have a negative point‐of‐care SARS‐CoV‐2 serological test and RT‐PCR testing was performed at baseline. Of the included participants, 96.3% were RT‐PCR test negative, 0.5% positive, and 3.2% unknown. Baseline SARS‐CoV‐2 antibody serostatus was measured at several time points according to the trial protocol, but not reported in the publication. Unblinding to assigned treatment was allowed if participants wanted to consider COVID‐19 vaccination. Unblinded or vaccinated participants were censored for efficacy at the date of unblinding or vaccination. About 39.3% of the participants were censored due to unblinding and 13.8% due to vaccination. A preplanned primary analysis was conducted after 30% of the participants have become unblinded. Data cut‐off for the primary analysis was 5 May 2021, with a median follow‐up of 83 days. An additional data cut‐off with an extended six‐month follow‐up was performed on 29 August 2021 with a median follow‐up of 196 days. During the study, the SARS‐CoV‐2 variants Alpha and Beta were detected in the study population as well as the Delta variant. The median age was 53.5 years and 46.1% of participants were female. Any high‐risk factor for severe COVID‐19 was present in 77.5% of the participants including obesity (41.7%), hypertension (35.9%), smoking (21.0%), diabetes (14.1%), asthma (11.1%), CVD (8.1%), cancer (7.4%), chronic obstructive pulmonary disease (COPD) (5.3%), chronic kidney disease (5.2%), chronic liver disease (4.6%), receipt of immunosuppressive therapy (3.3%), immunosuppressive disease (0.5%), sickle cell disease (less than 0.1%). In total, 73.3% of the participants had an increased risk of inadequate immune response to COVID vaccination (including factors such as older age (60 years of age or older); obesity (body mass index (BMI) 30 or greater); immunocompromised; unable to receive vaccines without adverse effects; or congestive heart failure, COPD, chronic kidney disease, or chronic liver disease) and 52.5% of participants had an increased risk of exposure to SARS‐CoV‐2.
Isa 2021 included uninfected adult volunteers who were healthy or had chronic stable medical conditions. It was a multicentre study conducted at different facilities in the USA. Recruitment started in July 2020, data cut‐off was 21 May 2021. Participants may have been exposed to multiple variants such as wild‐type, Alpha, and Delta (WHO 2021d). The study period overlapped with first vaccine roll‐out. Of 969 randomised participants, 98.9% were RT‐PCR negative. Participants were included regardless of SARS‐CoV‐2 antibody serostatus. At baseline, 85.1% of participants were seronegative for SARS‐CoV‐2 anti‐spike immunoglobulin (Ig)G and IgA and anti‐nucleocapsid IgG antibodies, 10.4% seropositive for any of the SARS‐CoV‐2 anti‐spike IgG or IgA or anti‐nucleocapsid IgG antibodies (or a combination), and 4.4% borderline or sero‐undetermined. During the study, 35.1% of participants in the intervention group and 40.8% in the placebo group opted for SARS‐CoV‐2 vaccination with a mean of 66.1 days between last dose of study drug and vaccination. They were discontinued from further intervention or placebo, censored for efficacy, and entered follow‐up. The median age was 48.0 years and 44.9% of the participants were female. At least one risk factor for severe COVID‐19 according to Centers for Disease Control and Prevention (CDC) criteria for high‐risk for severe SARS‐CoV‐2 infection, including immunosuppression, chronic disease, and obesity was observed in 20% of the participants.
BLAZE‐2 was a multicentre study conducted at different facilities in the USA. Participants were recruited from August to November 2020 and the study was probably conducted during wild‐type SARS‐CoV‐2 predominance (WHO 2021d). Unvaccinated residents and staff of skilled nursing and assisted living facilities with at least one confirmed index case were recruited. Participants were screened within seven days of reporting of the index case. RT‐PCR and SARS‐CoV‐2 serology tests were performed at baseline and participants were randomised and treated before the results were available. Anti‐nucleocapsid antibodies of IgA, IgG, and IgM combined were assessed. Of 1175 randomised participants, 966 (82.2%) were SARS‐CoV‐2 RT‐PCR negative and seronegative at baseline. Participants who were SARS‐CoV‐2 RT‐PCR positive and serology negative were evaluated as a treatment population in a separate publication. The median age ranged from 75 to 76 years (range 31 to 104 years) for residents in the intervention and placebo group and from 42 to 43 years (range 18 to 82 years) for staff in the intervention and placebo group. About 59.7% of residents and 81.5% of staff were female. All residents were at high risk of severe COVID‐19 and 41.3% of staff had one or more risk factor risk factors such as age 65 years or older, obesity, chronic kidney disease, diabetes, and immunosuppressive disease.
O'Brien 2021 was a multicentre study conducted at different sites in the USA, Romania, and Moldova. Participants were recruited from July 2020 to January 2021 and may have already been exposed to the Alpha variant and potentially, but unlikely, to the Delta variant (WHO 2021d). O'Brien 2021 randomised unvaccinated household contacts (adolescents and adults) of an index case within 96 hours after a positive test result for the index case. RT‐PCR and SARS‐CoV‐2 serology tests was performed at baseline and participants were stratified according to the result of the serology test and age at randomisation. A total of 2067 out of 2475 (83.5%) randomised participants were RT‐PCR negative at baseline and of these, 1505 (72.8%) participants were seronegative, 475 (22.1%) were seropositive, and 105 (5.1%) were sero‐undetermined at baseline for SARS‐CoV‐2 anti‐spike IgG and IgA and anti‐nucleocapsid IgG antibodies. Among the 554 participants from the initial assessment, 409 (73.8%) participants were SARS‐CoV‐2 RT‐PCR and seronegative. The mean age of the participants was 42.9 years (range 12 to 92 years) and 54.1% were female. Relevant risk factors for COVID‐19 were present in 30.5% of the participants. The proportion of participants with immunosuppressive disease was 0.5% and 1% of participants received immunosuppressive therapy.
Interventions and comparators
Isa 2021 assigned 729 participants to receive repeated subcutaneous administration of casirivimab 600 mg plus imdevimab 600 mg (total 1.2 g) every four weeks for up to six doses. PROVENT assigned 3460 participants to receive two consecutive intramuscular injections, one each of tixagevimab 150 mg and cilgavimab 150 mg (total 300 mg).
BLAZE‐2 assigned 588 participants to receive a single dose of bamlanivimab 4.2 g intravenously, and O'Brien 2021 assigned 1240 participants to receive a single dose of a combination of casirivimab 600 mg plus imdevimab 600 mg (total 1.2 g) subcutaneously.
A total of 3799 control group participants received placebo (BLAZE‐2; Isa 2021; O'Brien 2021; PROVENT). BLAZE‐2 prohibited SARS‐CoV‐2 vaccines prior and during the evaluation period. O'Brien 2021 prohibited concomitant medications such as SARS‐CoV‐2 vaccines, passive antibodies, and hydroxychloroquine for prophylaxis of SARS‐CoV‐2 infection as well as remdesivir or anti‐SARS viral agents during the efficacy analysis and follow‐up period.
Outcome measures
In PROVENT, the primary efficacy outcome was symptomatic COVID‐19 (SARS‐CoV‐2 infection confirmed by RT‐PCR) after administration of study drug on or before day 183. See Characteristics of included studies table for definition for symptomatic COVID‐19. Secondary efficacy endpoints were incidence of participants who were SARS‐CoV‐2 antibody seronegative at baseline, but seropositive at any time postbaseline, incidence of SARS‐CoV‐2 RT‐PCR positive severe or critical symptomatic illness postdose, and incidence of COVID‐19 related emergency department visits postdose. Primary safety endpoints included incidence of AEs, SAEs, medically attended AEs, and AEs of special interest. Further safety endpoints were deaths, laboratory evaluations, vital signs and physical examinations.
In Isa 2021, the primary outcomes were incidence of adverse events of special interest (AESIs), including grade 3 or greater injection‐site reactions or hypersensitivity reactions occurring within four days of administration of casirivimab/imdevimab or placebo, and serum concentration of casirivimab/imdevimab over time. AEs were rated according to the National Cancer Institute‐Common Terminology Criteria for Adverse Events (NCI‐CTCAE) v. 5.0. Participants who reported two or more events with the same preferred term were counted only once for that term. If a participant opted for COVID‐19 vaccination, only events before the vaccination were included. Secondary endpoints were proportion of participants with treatment‐emergent adverse events (TEAEs), proportion of participants who achieved or exceeded the target concentration of the study drug in serum at the end of each four‐week dosing interval, and immunogenicity measured by antidrug antibodies (ADAs). Preplanned exploratory efficacy endpoint was the incidence of SARS‐CoV‐2 infection, determined either clinically or by laboratory confirmation. Additional outcomes were AEs after COVID‐19 vaccination. Safety outcomes and the exploratory efficacy endpoint were analysed in the safety population, including all randomised participants who received any study drug and regardless of SARS‐CoV‐2 antibody serostatus.
In BLAZE‐2, the primary outcome was infection with mild or worse severity within eight weeks of randomisation, which was defined as a SARS‐CoV‐2 RT‐PCR positivity and presence of mild or worse disease within 21 days. See Characteristics of included studies table for definitions. Secondary endpoints were infection with moderate or worse severity, and infection within four and eight weeks of randomisation, COVID‐19‐related mortality, admission to hospital, or death due to COVID‐19. Safety outcomes were reported as treatment‐emergent AEs by week eight.
In O'Brien 2021, the primary outcome was infection with symptomatic disease by day 28 (strict term definition), which was subsequently revised based on an initial assessment of the first 409 seronegative participants to a broad‐term definition. See Characteristics of included studies table for definition. Secondary outcomes were infection with symptomatic disease by day 28 (CDC definition), infection with symptomatic disease by day 28 (strict term definition), viral load, duration of symptomatic infection (broad‐term definition), duration of high viral load, and duration of any symptomatic or asymptomatic infection. The primary and secondary outcomes were analysed hierarchically. Safety outcomes included AEs, reported as REAEs, AESIs, and SAEs. The safety analysis set included participants regardless of SARS‐CoV‐2 antibody serostatus and 554 participants from the initial analysis for efficacy.
Three studies reported AEs and SAEs as TEAEs, which means that they were not present at baseline or worsened in severity after baseline; therefore, we treated these as regular AEs and SAEs (BLAZE‐2; Isa 2021; O'Brien 2021).
Funding and conflicts of interest
Pharmaceutical companies funded all four studies. Eli Lilly and Co funded BLAZE‐2; Regeneron Pharmaceuticals funded Isa 2021 and O'Brien 2021; and AstraZeneca and the US Government funded PROVENT. BLAZE‐2 and O'Brien 2021 were conducted in partnership with the National Institute of Allergy and Infectious Diseases (NIAID) and the COVID‐19 Prevention Network (CoVPN). All authors reported their conflicts of interest.
Excluded studies
We excluded 14 studies (52 records) on SARS‐CoV‐2‐neutralising mAbs to treat people with COVID‐19 (see Characteristics of excluded studies table).
Five RCTs (16 records) investigated SARS‐CoV‐2‐neutralising mAbs to treat people with COVID‐19 in the hospitalised setting (ACTIV‐3; NCT04411628; NCT04426695; NCT04931238; RECOVERY).
Nine RCTs (36 records) investigated SARS‐CoV‐2‐neutralising mAbs to treat people with COVID‐19 in the outpatient setting (ACTIV‐2; BLAZE‐1; BLAZE‐4; COMET‐ICE; Eom 2021; Kim 2021; NCT04666441; OPTIMISE‐C19; Weinreich).
Studies awaiting classification
Two studies are awaiting classification (CROWN CORONATION; NCT04894474; Characteristics of studies awaiting classification table). One study, planning intravenous infusion and inhalation of BI 767551 as PEP, has been withdrawn due to project termination, but has provided no information on whether participants were already enrolled (NCT04894474). The other is a platform trial that may potentially add an mAb during the course of the study (CROWN CORONATION).
Ongoing studies
We classified four studies as ongoing (NCT04625972; NCT04859517; NCT05142527; NCT05184062; Characteristics of ongoing studies table). NCT05142527 is not yet recruiting participants and the three other studies are active, but not recruiting (NCT04625972; NCT04859517; NCT05184062).
Two studies are investigating a combination of the mAbs tixagevimab and cilgavimab and indicate overall completion date between June 2022 and June 2023 (NCT04625972 for PEP of COVID‐19; NCT05184062). Actual enrolment ranges between 272 and 1121 participants. NCT04859517 plans to investigate ADG20 for both PrEP and PEP of COVID‐19 with an estimated enrolment of 6412 participants up to March 2023. NCT05142527 plans to investigate ADM03820 with an estimated enrolment of 4450 participants up to August 2023.
Risk of bias in included studies
The four studies contributed 23 study results to seven outcomes that were assessed using RoB 2 (BLAZE‐2; Isa 2021; O'Brien 2021; PROVENT). The completed RoB 2 tool with responses to all assessed signalling questions is available online at: doi.org/10.5281/zenodo.6541295.
Overall risk of bias by outcome
The following section summarises the risk of bias per outcome for all primary and secondary outcomes relevant to this review.
Pre‐exposure prophylaxis of COVID‐19
Tixagevimab/cilgavimab compared to placebo
We judged the risk of bias for PROVENT, the only study assessing tixagevimab/cilgavimab for PrEP to be of some concerns across the outcomes: development of clinical COVID‐19 symptoms within six months (Table 13), mortality within six months (Table 14), admission to hospital within six months (Table 15), all‐grade AEs (Table 16) and SAEs (Table 17), because of potentially selection of the reported results, as the extended follow‐up at a median of 193 days was not prespecified in the study protocol. For the outcome infection with SARS‐CoV‐2 within six months (Table 12), we had in addition some concerns regarding missing outcome data, because we do not know whether data were available for all participants seronegative at baseline. For the outcomes admission to hospital, all‐grade AEs, and SAEs, we had in addition some concerns regarding measurement of the outcome, because some participants and outcome assessors were probably aware of the intervention received, as unblinding was possible, but the number of vaccinated participants was comparable in both groups. The study did not report quality of life, AEs (grade 1 to 2 and grade 3 to 4), and admission to ICU.
Risk of bias for analysis 1.2 Development of clinical COVID‐19 symptoms within 6 months.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| PROVENT | Low risk of bias | Participants were randomized via random number generation and the allocation sequence was concealed. There are no baseline differences that would suggest a problem with randomization. | Low risk of bias | Participants could be unblinded to consider vaccination, but percentages of participants with data that were unblinded were balanced between the groups. The analysis was appropriate. | Low risk of bias | Data for this outcome was available for nearly all participants (5172/5197 participants randomized). | Low risk of bias | The measurement of the outcome was appropriate, and it is unlikely that it differed between intervention groups. The outcome assessors were probably unaware of the intervention received. | Some concerns | There is a trial protocol, which provided details of the pre‐specified outcomes, but the 6 months follow‐up was not pre‐specified. | Some concerns | For this outcome, there is a low risk of bias from randomisation process, deviations from intended interventions, missing outcomes, measurement of the outcome. There are some concerns for bias due to selection of reported results. |
Risk of bias for analysis 1.3 All‐cause mortality within 6 months.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| PROVENT | Low risk of bias | Participants were randomized via random number generation and the allocation sequence was concealed. There are no baseline differences that would suggest a problem with randomization. | Low risk of bias | Participants could be unblinded to consider vaccination, but percentages of participants with data that were unblinded were balanced between the groups. The analysis was appropriate. | Low risk of bias | Data for this outcome was not available for all participants (5197/5197 participants randomized). | Low risk of bias | The measurement of the outcome was appropriate, and it is unlikely that it differed between intervention groups. The outcome assessors were probably unaware of the intervention received. | Some concerns | There is a trial protocol, which provided details of the pre‐specified outcomes, but the 6 months follow‐up was not pre‐specified. | Some concerns | For this outcome, there is a low risk of bias from randomisation process, deviations from intended interventions, missing outcomes, measurement of the outcome. There are some concerns for bias due to selection of reported results. |
Risk of bias for analysis 1.4 Admission to hospital within 6 months.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| PROVENT | Low risk of bias | Participants were randomized via random number generation and the allocation sequence was concealed. There are no baseline differences that would suggest a problem with randomization. | Low risk of bias | Participants could be unblinded to consider vaccination, but percentages of participants with data that were unblinded were balanced between the groups. The analysis was appropriate. | Low risk of bias | Data for this outcome was available for all participants (5172/5172 participants randomized). | Some concerns | The measurement of the outcome was appropriate, and it is unlikely that it differed between intervention groups. Outcome assessors were probably aware of the intervention received, but the number of participants vaccinated was comparable in both arms. | Some concerns | There is a trial protocol, which provided details of the pre‐specified outcomes, but the outcome COVID‐19 related hospital admissions and the 6 months follow‐up were probably not pre‐specified. | Some concerns | For this outcome, there is a low risk of bias from randomisation process, deviations from intended interventions and missing outcomes. There are some concerns for bias due measurement of the outcome and selection of reported results. |
Risk of bias for analysis 1.5 Adverse events: all grade within 6 months.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| PROVENT | Low risk of bias | Participants were randomized via random number generation and the allocation sequence was concealed. There are no baseline differences that would suggest a problem with randomization. | Low risk of bias | Participants could be unblinded to consider vaccination, but percentages of participants with data that were unblinded were balanced between the groups. The analysis was appropriate. | Low risk of bias | Data for this outcome was not available for all participants (5197/5197 participants randomized). | Some concerns | The measurement of the outcome was appropriate, and it is unlikely that it differed between intervention groups. Outcome assessors were probably aware of the intervention received, but the number of participants vaccinated was comparable in both arms. | Some concerns | There is a trial protocol, which provided details of the pre‐specified outcomes, but the 6 months follow‐up was probably not pre‐specified. | Some concerns | For this outcome, there is a low risk of bias from randomisation process, deviations from intended interventions and missing outcomes. However, there are some concerns for bias due measurement of the outcome and selection of reported results. |
Risk of bias for analysis 1.6 Serious adverse events within 6 months.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| PROVENT | Low risk of bias | Participants were randomized via random number generation and the allocation sequence was concealed. There are no baseline differences that would suggest a problem with randomization. | Low risk of bias | Participants could be unblinded to consider vaccination, but percentages of participants with data that were unblinded were balanced between the groups. The analysis was appropriate. | Low risk of bias | Data for this outcome was not available for all participants (5197/5197 participants randomized). | Some concerns | The measurement of the outcome was appropriate and it is unlikely that it differed between intervention groups. Outcome assessors were probably aware of the intervention received, but the number of participants vaccinated was comparable in both arms. | Some concerns | There is a trial protocol, which provided details of the pre‐specified outcomes, but the 6 months follow‐up was probably not pre‐specified. | Some concerns | For this outcome, there is a low risk of bias from randomisation process, deviations from intended interventions and missing outcomes. However, there are some concerns for bias due measurement of the outcome and selection of reported results. |
Risk of bias for analysis 1.1 Infection with SARS‐CoV‐2 within 6 months.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| PROVENT | Low risk of bias | Participants were randomized via random number generation and the allocation sequence was concealed. There are no baseline differences that would suggest a problem with randomization. | Low risk of bias | Participants could be unblinded to consider vaccination, but percentages of participants with data that were unblinded were balanced between the groups. The analysis was appropriate. | Some concerns | Data for this outcome was not available for all participants (4685/5197 participants randomized). There is no evidence that the result was not biased by missing outcome data. However, it is not likely that missingness in the outcome depended on its true value. | Low risk of bias | The measurement of the outcome was appropriate, and it is unlikely that it differed between intervention groups. The outcome assessors were probably unaware of the intervention received. | Some concerns | There is a trial protocol, which provided details of the pre‐specified outcomes, but the 6 months follow‐up was not pre‐specified. | Some concerns | For this outcome, there is a low risk of bias from randomisation process, deviations from intended interventions, and measurement of the outcome. However, there are some concerns for bias due to missing outcomes and selection of reported results. |
Casirivimab/imdevimab compared to placebo
We judged the risk of bias for Isa 2021, the only study assessing casirivimab/imdevimab for PrEP in uninfected volunteers, to be of some concerns across the outcomes: infection with SARS‐CoV‐2 within six months (Table 18), development of clinical COVID‐19 symptoms within six months (Table 19), mortality at six months (Table 20), AEs: grade 3 or greater (Table 21), all‐grade AEs (Table 22), and SAEs (Table 23), because there was no information provided on the randomisation process and allocation concealment and the study protocol was not available. The study did not report admission to hospital, quality of life, AEs (grade 1 to 2), and admission to ICU.
Risk of bias for analysis 2.1 Infection with SARS‐CoV‐2 within 6 months.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Isa 2021 | Some concerns | Participants were probably allocated randomly to either the casirivimab/imdevimab group or placebo group, but there were no further information given on the randomization process. There is no information on the allocation concealment. There are no baseline differences that would suggest a problem with randomisation. | Low risk of bias | Both participants and those delivering the intervention were probably unaware of the assigned intervention received, and the analysis was appropriate. | Low risk of bias | Data for this outcome was available for all participants who were SARS‐CoV‐2 antibody seronegative at baseline (casirivimab/imdevimab: 617 participants, placebo = 208 participants). | Low risk of bias | The measurement of the outcome was appropriate, and it is unlikely that it differed between intervention groups. The outcome assessors were probably unaware of the intervention received. | Some concerns | There was a trial register entry, which provided details of the pre‐specified outcomes, but the outcome was not pre‐specified at trial registration, but in the preprint described as a preplanned exploratory endpoint. | Some concerns | For this outcome, there is a low risk of bias from deviations from intended interventions, missing outcomes and measurement of the outcome. However, there are some concerns for bias due to randomisation process and selection of reported results. |
Risk of bias for analysis 2.2 Development of clinical COVID‐19 symptoms within 6 months.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Isa 2021 | Some concerns | Participants were probably allocated randomly to either the casirivimab/imdevimab group or placebo group, but there were no further information given on the randomization process. There is no information on the allocation concealment. There are no baseline differences that would suggest a problem with randomisation. | Low risk of bias | Both participants and those delivering the intervention were probably unaware of the assigned intervention received, and the analysis was appropriate. | Low risk of bias | Data for this outcome was available for all participants who were SARS‐CoV‐2 antibody seronegative at baseline (casirivimab/imdevimab: 617 participants, placebo = 208 participants). | Low risk of bias | The measurement of the outcome was appropriate, and it is unlikely that it differed between intervention groups. The outcome assessors were probably unaware of the intervention received. | Some concerns | There was a trial register entry, which provided details of the pre‐specified outcomes, but the outcome was not pre‐specified at trial registration, but in the preprint described as a preplanned exploratory endpoint. | Some concerns | For this outcome, there is a low risk of bias from deviations from intended interventions, missing outcomes and measurement of the outcome. However, there are some concerns for bias due to randomisation process and selection of reported results. |
Risk of bias for analysis 2.3 All‐cause mortality within 6 months.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Isa 2021 | Some concerns | Participants were probably allocated randomly to either the casirivimab/imdevimab group or placebo group, but there were no further information given on the randomization process. There is no information on the allocation concealment. There are no baseline differences that would suggest a problem with randomisation. | Low risk of bias | Both participants and those delivering the intervention were probably unaware of the assigned intervention received, and the analysis was appropriate. | Low risk of bias | Data for this outcome was available for all treated participants (casirivimab/imdevimab: 729 participants, placebo = 240 participants). | Low risk of bias | The measurement of the outcome was appropriate, and it is unlikely that it differed between intervention groups. The outcome assessors were probably unaware of the intervention received. | Some concerns | There was a trial register entry, which provided details of the pre‐specified outcomes, but mortality was not pre‐specified at trial registration, but probably assessed as part of the safety assessment. | Some concerns | For this outcome, there is a low risk of bias from deviations from intended interventions, missing outcomes and measurement of the outcome. However, there are some concerns for bias due to randomisation process and selection of reported results. |
Risk of bias for analysis 2.4 Adverse events: grade 3 to 4 within 6 months.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Isa 2021 | Some concerns | Participants were probably allocated randomly to either the casirivimab/imdevimab group or placebo group, but there were no further information given on the randomization process. There is no information on the allocation concealment. There are no baseline differences that would suggest a problem with randomisation. | Low risk of bias | Both participants and those delivering the intervention were probably unaware of the assigned intervention received, and the analysis was appropriate. | Low risk of bias | Data for this outcome was available for all treated participants (casirivimab/imdevimab: 729 participants, placebo = 240 participants). | Low risk of bias | The measurement of the outcome was appropriate, and it is unlikely that it differed between intervention groups. The outcome assessors were probably unaware of the intervention received. | Low risk of bias | The data that produced this result was reported as planned at trial registration stage. | Some concerns | For this outcome, there is a low risk of bias from deviations from intended interventions, missing outcomes, measurement of the outcome and selection of reported results. However, there are some concerns for bias due to randomisation process. |
Risk of bias for analysis 2.5 Adverse events: all grade within 6 months.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Isa 2021 | Some concerns | Participants were probably allocated randomly to either the casirivimab/imdevimab group or placebo group, but there were no further information given on the randomization process. There is no information on the allocation concealment. There are no baseline differences that would suggest a problem with randomisation. | Low risk of bias | Both participants and those delivering the intervention were probably unaware of the assigned intervention received, and the analysis was appropriate. | Low risk of bias | Data for this outcome was available for all treated participants (casirivimab/imdevimab: 729 participants, placebo = 240 participants). | Low risk of bias | The measurement of the outcome was appropriate and it is unlikely that it differed between intervention groups. The outcome assessors were probably unaware of the intervention received. | Low risk of bias | The data that produced this result was reported as planned at trial registration stage. | Some concerns | For this outcome, there is a low risk of bias from deviations from intended interventions, missing outcomes, measurement of the outcome and selection of reported results. However, there are some concerns for bias due to randomisation process. |
Risk of bias for analysis 2.6 Serious adverse events within 6 months.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Isa 2021 | Some concerns | Participants were probably allocated randomly to either the casirivimab/imdevimab group or placebo group, but there were no further information given on the randomization process. There is no information on the allocation concealment. There are no baseline differences that would suggest a problem with randomisation. | Low risk of bias | Both participants and those delivering the intervention were probably unaware of the assigned intervention received, and the analysis was appropriate. | Low risk of bias | Data for this outcome was available for all treated participants (casirivimab/imdevimab: 729 participants, placebo = 240 participants). | Low risk of bias | The measurement of the outcome was appropriate, and it is unlikely that it differed between intervention groups. The outcome assessors were probably unaware of the intervention received. | Some concerns | There was a trial register entry, which provided details of the pre‐specified outcomes, but serious adverse events was not pre‐specified at trial registration. | Some concerns | For this outcome, there is a low risk of bias from deviations from intended interventions, missing outcomes and measurement of the outcome. However, there are some concerns for bias due to randomisation process and selection of reported results. |
Postexposure prophylaxis of COVID‐19
Bamlanivimab compared to placebo
We judged the risk of bias for BLAZE‐2, the only study assessing bamlanivimab for PEP, to be low across the outcomes: infection with SARS‐CoV‐2 by day 29 (Table 24), mortality by day 57 (Table 25), all‐grade AEs by week eight (Table 26), and SAEs by week eight (Table 27). The study did not report development of clinical COVID‐19 symptoms, admission to hospital, quality of life, AEs (grade 1 to 2 and grade 3 to 4), and admission to ICU.
Risk of bias for analysis 3.1 Infection with SARS‐CoV‐2 by day 30.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| BLAZE‐2 | Low risk of bias | Participants were randomised via an Interactive Web Response System (IWRS) and the allocation sequence was concealed. There are no baseline differences that would suggest a problem with randomisation. | Low risk of bias | Both participants and those delivering the intervention were unaware of the assigned intervention received, and the analysis was appropriate. | Low risk of bias | Data for this outcome was available for all participants with negative SARS‐CoV‐2 serology and RT‐PCR at baseline (484 treatment group and 482 placebo group randomised and analysed). | Low risk of bias | The measurement of the outcome was appropriate, and it is unlikely that it differed between intervention groups. The outcome assessors were unaware of the intervention received. | Low risk of bias | The data that produced this result was analysed in accordance with the pre‐specified analysis plan. The outcome was presented in various ways. | Low risk of bias | For this outcome, there is a low risk of bias for all the domains. |
Risk of bias for analysis 3.2 All‐cause mortality by day 60.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| BLAZE‐2 | Low risk of bias | Participants were randomised via an Interactive Web Response System (IWRS) and the allocation sequence was concealed. There are no baseline differences that would suggest a problem with randomisation. | Low risk of bias | Both participants and those delivering the intervention were unaware of the assigned intervention received, and the analysis was appropriate. | Low risk of bias | Data for this outcome was available for all participants with negative SARS‐CoV‐2 serology and RT‐PCR at baseline (484 treatment group and 482 placebo group randomised and analysed). | Low risk of bias | The measurement of the outcome was appropriate and did not differ between intervention groups. The outcome assessors were unaware of the intervention received. | Low risk of bias | The data that produced this result was analysed in accordance with the pre‐specified analysis plan and the outcome was reported as planned in the protocol. | Low risk of bias | For this outcome, there is a low risk of bias for all the domains. |
Risk of bias for analysis 3.3 Adverse events: all grade.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| BLAZE‐2 | Low risk of bias | Participants were randomised via an Interactive Web Response System (IWRS) and the allocation sequence was concealed. There are no baseline differences that would suggest a problem with randomisation. | Low risk of bias | Both participants and those delivering the intervention were unaware of the assigned intervention received, and the analysis was appropriate. | Low risk of bias | Data for this outcome was available for all participants with negative SARS‐CoV‐2 serology and RT‐PCR at baseline (484 treatment group and 482 placebo group randomised and analysed). | Low risk of bias | The measurement of the outcome was appropriate, and it is unlikely that it differed between intervention groups. The outcome assessors were unaware of the intervention received. | Low risk of bias | The data that produced this result was analysed in accordance with the pre‐specified analysis plan and the outcome was reported as planned in the protocol. | Low risk of bias | For this outcome, there is a low risk of bias for all the domains. |
Risk of bias for analysis 3.4 Serious adverse events.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| BLAZE‐2 | Low risk of bias | Participants were randomised via an Interactive Web Response System (IWRS) and the allocation sequence was concealed. There are no baseline differences that would suggest a problem with randomisation. | Low risk of bias | Both participants and those delivering the intervention were unaware of the assigned intervention received, and the analysis was appropriate. | Low risk of bias | Data for this outcome was available for all participants with negative SARS‐CoV‐2 serology and RT‐PCR at baseline (484 treatment group and 482 placebo group randomised and analysed). | Low risk of bias | The measurement of the outcome was appropriate, and it is unlikely that it differed between intervention groups. The outcome assessors were unaware of the intervention received. | Low risk of bias | The data that produced this result was analysed in accordance with the pre‐specified analysis plan and the outcome was reported as planned in the protocol. | Low risk of bias | For this outcome, there is a low risk of bias for all the domains. |
Casirivimab/imdevimab compared to placebo
We judged the risk of bias for O'Brien 2021, the only study assessing casirivimab/imdevimab for PEP, to be low across outcomes: infection with SARS‐CoV‐2 by day 28 (Table 28), mortality by day 28 (Table 30), and admission to hospital by day 28 (Table 31). In the publication, the results for these outcomes were reported in the safety analysis set, including SARS‐CoV‐2 seronegative (72.8%), seropositive (22.1%), and sero‐undetermined (5.1%) participants and 554 participants from an initial analysis for efficacy. We judged the risk of bias to be low, because the study authors provided us with the requested data for SARS‐CoV‐2 seronegative participants during the efficacy analysis period. For the outcome development of clinical COVID‐19 symptoms, we had some concerns regarding risk of bias due to selection of the reported results, because the outcome was changed based on the assessment of the first 409 participants (Table 29). For the safety outcomes AEs: grade 3 or greater (Table 32), all‐grade AEs (Table 33), and SAEs (Table 34), we had no concerns regarding risk of bias. The study did not report quality of life, AEs (grade 1 to 2), and admission to ICU.
Risk of bias for analysis 4.1 Infection with SARS‐CoV‐2 by day 30.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| O'Brien 2021 | Low risk of bias | Participants were randomised via an Interactive Web Response System (IWRS) and the allocation sequence was concealed. There are no baseline differences that would suggest a problem with randomisation. | Low risk of bias | Both participants and those delivering the intervention were unaware of the assigned intervention received, and the analysis was appropriate. | Low risk of bias | Data for this outcome was available for all participants SARS‐CoV‐2 RT‐PCR and serology negative at baseline (753 treatment group and 752 placebo group randomised and analysed). | Low risk of bias | The measurement of the outcome was appropriate, and it is unlikely that it differed between intervention groups. The outcome assessors were unaware of the intervention received. | Low risk of bias | The data that produced this result was analysed in accordance with the pre‐specified analysis plan and the outcome was reported as planned in the protocol. | Low risk of bias | For this outcome, there is a low risk of bias for all the domains. |
Risk of bias for analysis 4.3 All‐cause mortality by day 30.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| O'Brien 2021 | Low risk of bias | Participants were randomised via an Interactive Web Response System (IWRS) and the allocation sequence was concealed. There are no baseline differences that would suggest a problem with randomisation. | Low risk of bias | Both participants and those delivering the intervention were unaware of the assigned intervention received, and the analysis was appropriate. | Low risk of bias | Data for this outcome was available for all participants SARS‐CoV‐2 RT‐PCR and serology negative at baseline (753 treatment group and 752 placebo group randomised and analysed). | Low risk of bias | The outcome measurement was appropriate, and it is unlikely that it differed between intervention groups. The outcome assessors were unaware of the intervention received. | Low risk of bias | The data that produced this result was analysed in accordance with the pre‐specified analysis plan and the authors provided data for the efficacy population (SARS‐CoV‐2 seronegative participants) upon request. | Low risk of bias | For this outcome, there is a low risk of bias for all the domains. |
Risk of bias for analysis 4.4 Admission to hospital by day 30.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| O'Brien 2021 | Low risk of bias | Participants were randomised via an Interactive Web Response System (IWRS) and the allocation sequence was concealed. There are no baseline differences that would suggest a problem with randomisation. | Low risk of bias | Both participants and those delivering the intervention were unaware of the assigned intervention received, and the analysis was appropriate. | Low risk of bias | Data for this outcome was available for all participants SARS‐CoV‐2 RT‐PCR and serology negative at baseline (753 treatment group and 752 placebo group randomised and analysed). | Low risk of bias | The measurement of the outcome was probably appropriate, and it is unlikely that it differed between intervention groups. The outcome assessors were unaware of the intervention received. | Low risk of bias | The data that produced this result was in accordance with the pre‐specified analysis plan. Data for the safety analysis set was reported in the publication, but the authors provided data for the efficacy population (SARS‐CoV‐2 seronegative participants) upon request. | Low risk of bias | For this outcome, there is a low risk of bias for all the domains. |
Risk of bias for analysis 4.2 Development of clinical COVID‐19 symptoms (broad‐term).
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| O'Brien 2021 | Low risk of bias | Participants were randomised via an Interactive Web Response System (IWRS) and the allocation sequence was concealed. There are no baseline differences that would suggest a problem with randomisation. | Low risk of bias | Both participants and those delivering the intervention were unaware of the assigned intervention received, and the analysis was appropriate. | Low risk of bias | Data for this outcome was available for all participants SARS‐CoV‐2 RT‐PCR and serology negative at baseline (753 treatment group and 752 placebo group randomised and analysed). | Low risk of bias | The measurement of the outcome was appropriate, and it is unlikely that it differed between intervention groups. The outcome assessors were unaware of the intervention received. | Some concerns | The data that produced this result was measured and reported in accordance with the pre‐specified analysis plan. However, the outcome was changed based on the assessment of the first 409 participants. | Some concerns | For this outcome, there is a low risk of bias from the randomisation process, deviations from intended interventions, missing outcomes and measurement of the outcome. However, there are some concerns for bias due to selection of the reported result. |
Risk of bias for analysis 4.5 Adverse events: grade 3 to 4.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| O'Brien 2021 | Low risk of bias | Participants were randomised via an Interactive Web Response System (IWRS) and the allocation sequence was concealed. There are no baseline differences that would suggest a problem with randomisation. | Low risk of bias | Both participants and those delivering the intervention were unaware of the assigned intervention received, and the analysis was appropriate. | Low risk of bias | Data of nearly all SARS‐CoV‐2 seropositive, negative and sero‐undetermined participants at baseline were analysed. | Low risk of bias | The measurement of the outcome was appropriate, and it is unlikely that it differed between intervention groups. The outcome assessors were unaware of the intervention received. | Low risk of bias | The data that produced this result was analysed in accordance with the pre‐specified analysis plan and the outcome was reported as planned in the protocol. Participants including SARS‐CoV‐2 seropositive and sero‐undetermined participants were analysed together. | Low risk of bias | For this outcome, there is a low risk of bias for all the domains. |
Risk of bias for analysis 4.6 Adverse events: all grade.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| O'Brien 2021 | Low risk of bias | Participants were randomised via an Interactive Web Response System (IWRS) and the allocation sequence was concealed. There are no baseline differences that would suggest a problem with randomisation. | Low risk of bias | Both participants and those delivering the intervention were unaware of the assigned intervention received, and the analysis was appropriate. | Low risk of bias | Data of nearly all SARS‐CoV‐2 seropositive, negative and sero‐undetermined participants at baseline were analysed. | Low risk of bias | The measurement of the outcome was appropriate, and it is unlikely that it differed between intervention groups. The outcome assessors were unaware of the intervention received. | Low risk of bias | The data that produced this result was analysed in accordance with the pre‐specified analysis plan and the outcome was reported as planned in the protocol. Participants including SARS‐CoV‐2 seropositive and sero‐undetermined participants were analysed together. | Low risk of bias | For this outcome, there is a low risk of bias for all the domains. |
Risk of bias for analysis 4.7 Serious adverse events.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| O'Brien 2021 | Low risk of bias | Participants were randomised via an Interactive Web Response System (IWRS) and the allocation sequence was concealed. There are no baseline differences that would suggest a problem with randomisation. | Low risk of bias | Both participants and those delivering the intervention were unaware of the assigned intervention received, and the analysis was appropriate. | Low risk of bias | Data of nearly all SARS‐CoV‐2 seropositive, negative and sero‐undetermined participants at baseline were analysed. | Low risk of bias | The measurement of the outcome was appropriate, and it is unlikely that it differed between intervention groups. The outcome assessors were unaware of the intervention received. | Low risk of bias | The data that produced this result was analysed in accordance with the pre‐specified analysis plan and the outcome was reported as planned in the protocol. Participants including SARS‐CoV‐2 seropositive and sero‐undetermined participants were analysed together. | Low risk of bias | For this outcome, there is a low risk of bias for all the domains. |
Effects of interventions
See: Table 1; Table 2; Table 3; Table 4
Summary of findings 1. Tixagevimab/cilgavimab compared to placebo for pre‐exposure prophylaxis of COVID‐19.
| Tixagevimab/cilgavimab compared to placebo in previously uninfected and unvaccinated people with increased risk of exposure to SARS‐CoV‐2 or increased risk for inadequate immune response to vaccination, or both | ||||||
|
Patient or population: SARS‐CoV‐2 uninfected people without defined exposure, or with potential exposure to SARS‐CoV‐2Setting: preventive measures Intervention: tixagevimab/cilgavimab Comparison: placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | N° of participants (studies) | Certainty in the evidence (GRADE) | Comments | |
| Risk with placebo | Risk with tixagevimab/ cilgavimab | |||||
| Infection with SARS‐CoV‐2 within 6 months | 27 per 1000 |
12 per 1000 (8 to 19) |
RR 0.45 (0.29 to 0.70) |
4685 (1 RCT) |
⊕⊕⊕⊝ Moderatea |
Tixagevimab/cilgavimab probably decrease infection with SARS‐CoV‐2 within 6 months. Participants were censored at unblinding or vaccination. Participants had been exposed to SARS‐CoV‐2 wild‐type and variants such as Alpha, Beta and Delta. Extended follow‐up data cut‐off was 29 August 2021. |
| Development of clinical COVID‐19 symptoms within 6 months | 18 per 1000 |
3 per 1000 (2 to 6) |
RR 0.18 (0.09 to 0.35) |
5172 (1 RCT) |
⊕⊕⊕⊕ High |
Tixagevimab/cilgavimab decrease development of clinical symptoms within 6 months. Participants were censored at unblinding or vaccination. Participants had been exposed to SARS‐CoV‐2 wild‐type and variants such as Alpha, Beta and Delta. Extended follow‐up data cut‐off was 29 August 2021. |
| All‐cause mortality within 6 monthsb | 4 per 1000 |
3 per 1000 (1 to 7) |
RR 0.64 (0.24 to 1.73) |
5197 (1 RCT) |
⊕⊕⊝⊝ Lowc,d |
Tixagevimab/cilgavimab may result in little to no difference on mortality within 6 months in participants regardless of RT‐PCR SARS‐CoV‐2 status at baseline. Participants had been exposed to SARS‐CoV‐2 wild‐type and variants such as Alpha, Beta and Delta. Extended follow‐up data cut‐off was 29 August 2021. |
| Admission to hospital within 6 monthsb | 4 per 1000 |
0 per 1000 (0 to 2) |
RR 0.03 (0.00 to 0.59) |
5197 (1 RCT/) |
⊕⊕⊝⊝ Lowe |
Tixagevimab/cilgavimab may decrease admission to hospital within 6 months in participants regardless of RT‐PCR SARS‐CoV‐2 status at baseline. Participants had been exposed to SARS‐CoV‐2 wild‐type and variants such as Alpha, Beta and Delta. Extended follow‐up data cut‐off was 29 August 2021. |
| Quality of life at longest follow‐up | — | — | — | — | — | We identified no studies reporting quality of life. |
| Adverse events: grade 1 to 2 | — | — | — | — | — | We identified no studies reporting grade 1 to 2 adverse events. |
| Adverse events: grade 3 to 4 within 6 months | — | — | — | — | — | We identified no studies reporting grade 3 to 4 adverse events. |
| Adverse events: all grade within 6 monthsb | 455 per 1000 |
455 per 1000 (428 to 487) |
RR 1.00 (0.94 to 1.07) |
5197 (1 RCT) |
⊕⊕⊝⊝ Lowf |
Tixagevimab/cilgavimab may result in little to no difference on the occurrence of all‐grade adverse events within 6 months in participants regardless of RT‐PCR SARS‐CoV‐2 status at baseline. Participants had been exposed to SARS‐CoV‐2 wild‐type and variants such as Alpha, Beta and Delta. Extended follow‐up data cut‐off was 29 August 2021. |
| Serious adverse events within 6 monthsb | 33 per 1000 |
37 per 1000 (28 to 51) |
RR 1.12 (0.83 to 1.52) |
5197 (1 RCT) |
⊕⊕⊝⊝ Lowg |
Tixagevimab/cilgavimab may result in little to no difference on the occurrence of serious adverse events within 6 months in participants regardless of RT‐PCR SARS‐CoV‐2 status at baseline. Participants had been exposed to SARS‐CoV‐2 wild‐type and variants such as Alpha, Beta and Delta. Extended follow‐up data cut‐off was 29 August 2021. |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk on the comparison group and the relative effect of the intervention (and its 95% confidence interval). CI: confidence interval; mAb: monoclonal antibody; RCT: randomised controlled trial; RR: risk ratio; RT‐PCR: reverse transcription polymerase chain reaction; SARS‐CoV‐2: severe acute respiratory syndrome coronavirus 2. | ||||||
|
GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is the possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||||
aDowngraded one level for serious risk of bias (missing outcome data and potentially selection of the reported result). bThe safety population included participants with negative, positive and unknown RT‐PCR SARS‐CoV‐2 status at baseline. cDowngraded two levels for very serious imprecision, because of very low number of events and wide confidence intervals. dWe did not downgrade for serious risk of bias (measurement of the outcome) because for this outcome it is irrelevant whether participants were aware of the intervention received, and the number of people vaccinated was comparable in both arms. eDowngraded two levels for very serious imprecision because of very low number of events. fDowngraded one level for serious imprecision, because sample size did not meet optimal information size (6,435,640 participants) and one level for serious risk of bias (measurement of the outcome and potentially selection of the reported result). gDowngraded one level for serious imprecision, because sample size did not meet optimal information size (55,674 participants) and one level for serious risk of bias (measurement of the outcome and potentially selection of the reported result).
Summary of findings 2. Casirivimab/imdevimab compared to placebo for pre‐exposure prophylaxis of COVID‐19.
| Casirivimab/imdevimab compared to placebo in previously uninfected and unvaccinated people | ||||||
|
Patient or population: SARS‐CoV‐2 uninfected people without defined exposure, or with potential exposure to SARS‐CoV‐2 Setting: preventive measures Intervention: casirivimab/imdevimab Comparison: placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | N° of participants (studies) | Certainty in the evidence (GRADE) | Comments | |
| Risk with placebo | Risk with casirivimab/imdevimab | |||||
| Infection with SARS‐CoV‐2 within 6 monthsa | 96 per 1000 |
1 per 1000 (0 to 13) |
RR 0.01 (0.00 to 0.14) |
825 (1 RCT) |
⊕⊕⊝⊝ Lowb,c |
Casirivimab/imdevimab may decrease infection with SARS‐CoV‐2 within 6 months in participants SARS‐CoV‐2 antibody seronegative at baseline. Participants may have been exposed to SARS‐CoV‐2 wild‐type and variants such as Alpha and Delta. |
| Development of clinical COVID‐19 symptoms within 6 months | 42 per 1000 |
1 per 1000 (0 to 11) |
RR 0.02 (0.00 to 0.27) |
969 (1 RCT) |
⊕⊕⊝⊝ Lowb,c |
Casirivimab/imdevimab may decrease development of clinical COVID‐19 symptoms within 6 months. Participants may have been exposed to SARS‐CoV‐2 wild‐type and variants such as Alpha and Delta. |
| All‐cause mortality within 6 months | 1 study reported mortality by week 24. There were no deaths. | Not estimable | 969 (1 RCT) |
⊕⊝⊝⊝ Very lowb,d |
The evidence is very uncertain about the effect of casirivimab/imdevimab on mortality. Participants may have been exposed to SARS‐CoV‐2 wild‐type and variants such as Alpha and Delta. |
|
| Admission to hospital within 6 months | — | — | — | — | We identified no studies reporting admission to hospital. | |
| Quality of life at longest follow‐up | — | — | — | — | We identified no studies reporting quality of life. | |
| Adverse events: grade 1 to 2 | — | — | — | — | We identified no studies reporting grade 1 to 2 adverse events. | |
| Adverse events: grade 3 to 4 within 6 months | 13 per 1000 |
6 per 1000 (1 to 24) |
RR 0.44 (0.10 to 1.95) |
969 (1 RCT) |
⊕⊝⊝⊝ Very lowb,e |
The evidence is very uncertain about the effect of casirivimab/imdevimab on the occurrence of grade 3 to 4 adverse events within 6 months. Participants may have been exposed to SARS‐CoV‐2 wild‐type and variants such as Alpha and Delta. |
| Adverse events: all grades within 6 months | 483 per 1000 |
551 per 1000 (474 to 633) |
RR 1.14 (0.98 to 1.31) |
969 (1 RCT) |
⊕⊕⊝⊝ Lowb,f |
Casirivimab/imdevimab may increase the occurrence of all‐grade adverse events within 6 months slightly. Participants may have been exposed to SARS‐CoV‐2 wild‐type and variants such as Alpha and Delta. |
| Serious adverse events within 6 months | 8 per 1000 |
7 per 1000 (1 to 35) |
RR 0.82 (0.16 to 4.21) |
969 (1 RCT) |
⊕⊝⊝⊝ Very lowb,e |
The evidence is very uncertain about the effect of casirivimab/imdevimab on the occurrence of serious adverse events within 6 months. Participants may have been exposed to SARS‐CoV‐2 wild‐type and variants such as Alpha and Delta. |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk on the comparison group and the relative effect of the intervention (and its 95% confidence interval). CI: confidence interval; mAb: monoclonal antibody; RCT: randomised controlled trial; RR: risk ratio; SARS‐CoV‐2: severe acute respiratory syndrome coronavirus 2. | ||||||
|
GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is the possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||||
aThe outcome was assessed in participants SARS‐CoV‐2 antibody seronegative at baseline. bDowngraded one level for serious risk of bias (missing information regarding randomisation process and allocation concealment). cDowngraded one level for serious imprecision, because of low number of events. dDowngraded two levels for very serious imprecision, because there were no events, effect not estimable. eDowngraded two levels for very serious imprecision, because of very low number of events and very wide confidence intervals. fDowngraded one level for very serious imprecision, because of wide confidence intervals.
Summary of findings 3. Bamlanivimab compared to placebo for postexposure prophylaxis of COVID‐19.
| Bamlanivimab compared to placebo in previously uninfected and unvaccinated residents and staff of skilled nursing and assisted living facilities with at least one confirmed index case | ||||||
|
Patient or population: SARS‐CoV‐2 uninfected people without defined exposure, or with potential exposure to SARS‐CoV‐2 Setting: preventive measures Intervention: bamlanivimab Comparison: placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | N° of participants (studies) | Certainty in the evidence (GRADE) | Comments | |
| Risk with placebo | Risk with bamlanivimab | |||||
| Infection with SARS‐CoV‐2 by day 30 | 232 per 1000 |
176 per 1000 (137 to 228) |
RR 0.76 (0.59 to 0.98) |
966 (1 RCT) | ⊕⊕⊕⊝ Moderatea |
Bamlanivimab probably decreases infection with SARS‐CoV‐2 by day 30. Participants may have been exposed to variants such SARS‐CoV‐2 wild‐type. |
| Development of clinical COVID‐19 symptoms within 30 days | — | — | — | — | — | We identified no studies reporting development of clinical COVID‐19 symptoms. |
| All‐cause mortality by day 60 | 12 per 1000 |
10 per 1000 (3 to 34) |
RR 0.83 (0.25 to 2.70) |
966 (1 RCT) | ⊕⊕⊝⊝ Lowb |
Bamlanivimab may result in little to no difference on mortality by day 60. Participants may have been exposed to variants such SARS‐CoV‐2 wild‐type. |
| Admission to hospital within 30 days | — | — | — | — | — | We identified no studies reporting admission to hospital. |
| Quality of life at longest follow‐up | — | — | — | — | — | We identified no studies reporting quality of life. |
| Adverse events: grade 1 to 2 | — | — | — | — | — | We identified no studies reporting grade 1 to 2 adverse events. |
| Adverse events: grade 3 to 4 | — | — | — | — | — | We identified no studies reporting grade 3 to 4 adverse events. |
| Adverse events: all grade by week 8 | 178 per 1000 |
200 per 1000 (153 to 260) |
RR 1.12 (0.86 to 1.46) |
966 (1 RCT) | ⊕⊕⊝⊝ Lowc |
Bamlanivimab may increase the occurrence of all‐grade adverse events by week 8. Participants may have been exposed to variants such SARS‐CoV‐2 wild‐type. |
| Serious adverse events by week 8 | 27 per 1000 |
39 per 1000 (20 to 78) |
RR 1.46 (0.73 to 2.91) |
966 (1 RCT) | ⊕⊕⊝⊝ Lowb |
Bamlanivimab may increase the occurrence of serious adverse events by week 8 slightly. Participants may have been exposed to variants such SARS‐CoV‐2 wild‐type. |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk on the comparison group and the relative effect of the intervention (and its 95% confidence interval). CI: confidence interval; mAb: monoclonal antibody; RCT: randomised controlled trial; RR: risk ratio; SARS‐CoV‐2: severe acute respiratory syndrome coronavirus 2. | ||||||
|
GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is the possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||||
aDowngraded one level for serious imprecision, because sample size did not meet optimal information size (2046 participants). bDowngraded two levels for very serious imprecision, because of low number of events and wide confidence intervals. cDowngraded two levels for serious imprecision, because sample size did not meet optimal information size (12,078 participants) and wide confidence intervals.
Summary of findings 4. Casirivimab/imdevimab compared to placebo for postexposure prophylaxis of COVID‐19.
| Casirivimab/imdevimab compared to placebo in previously uninfected and unvaccinated household contacts of infected people | ||||||
|
Patient or population: SARS‐CoV‐2 uninfected people without defined exposure, or with potential exposure to SARS‐CoV‐2 Setting: preventive measures Intervention: casirivimab/imdevimab Comparison: placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | N° of participants (studies) | Certainty in the evidence (GRADE) | Comments | |
| Risk with placebo | Risk with casirivimab/imdevimab | |||||
| Infection with SARS‐CoV‐2 by day 30 | 142 per 1000 |
48 per 1000 (33 to 68) |
RR 0.34 (0.23 to 0.48) |
1505 (1 RCT) |
⊕⊕⊕⊕ High |
Casirivimab/imdevimab decreases infection with SARS‐CoV‐2 by day 30. Participants may have been exposed to SARS‐CoV‐2 wild‐type and variants such as Alpha and potentially, but unlikely, to Delta. |
| Development of clinical COVID‐19 symptoms within 30 days | 78 per 1000 |
15 per 1000 (8 to 27) |
RR 0.19 (0.10 to 0.35) |
1505 (1 RCT) |
⊕⊕⊕⊕ High |
Casirivimab/imdevimab decreases development of clinical COVID‐19 symptoms within 30 days (broad‐term definition). Participants may have been exposed to SARS‐CoV‐2 wild‐type and variants such as Alpha and potentially, but unlikely, to Delta. |
| All‐cause mortality by day 30 | 1 per 1000 |
2 per 1000 (0 to 74) |
RR 3.00 (0.12 to 73.43) |
1505 (1 RCT) |
⊕⊕⊝⊝ Lowa |
Casirivimab/imdevimab may result in little to no difference on mortality by day 30. Participants may have been exposed to SARS‐CoV‐2 wild‐type and variants such as Alpha and potentially, but unlikely, to Delta. |
| Admission to hospital by day 30 | 5 per 1000 |
1 per 1000 (0 to 11) |
RR 0.11 (0.01 to 2.06) |
1505 (1 RCT) |
⊕⊕⊝⊝ Lowa |
Casirivimab/imdevimab may result in little to no difference on admission to hospital by day 30. Participants may have been exposed to SARS‐CoV‐2 wild‐type and variants such as Alpha and potentially, but unlikely, to Delta. |
| Quality of life at longest follow‐up | — | — | — | — | We identified no studies reporting quality of life. | |
| Adverse events: grade 1 to 2 | — | — | — | — | We identified no studies reporting grade 1 to 2 adverse events. | |
| Adverse events: grade 3 to 4 until data cut‐offb | 17 per 1000 |
8 per 1000 (4 to 17) |
RR 0.50 (0.24 to 1.02) |
2617 (1 RCT) |
⊕⊕⊝⊝ Lowc |
Casirivimab/imdevimab may decrease slightly the occurrence of grade 3 to 4 adverse events in participants regardless of the SARS‐CoV‐2 antibody serostatus. Participants may have been exposed to SARS‐CoV‐2 wild‐type and variants such as Alpha and potentially, but unlikely, to Delta. |
| Adverse events: all grade until data cut‐offb | 290 per 1000 |
203 per 1000 (177 to 232) |
RR 0.70 (0.61 to 0.80) |
2617 (1 RCT) |
⊕⊕⊕⊕ High |
Casirivimab/imdevimab decrease the occurrence of all‐grade adverse events in participants regardless of the SARS‐CoV‐2 antibody serostatus. Participants may have been exposed to SARS‐CoV‐2 wild‐type and variants such as Alpha and potentially, but unlikely, to Delta. |
| Serious adverse events until data cut‐offb | 11 per 1000 |
8 per 1000 (3 to 17) |
RR 0.66 (0.30 to 1.47) |
2617 (1 RCT) |
⊕⊕⊝⊝ Lowc |
Casirivimab/imdevimab may result in little to no difference on the occurrence of serious adverse events in participants regardless of the SARS‐CoV‐2 antibody serostatus. Participants may have been exposed to SARS‐CoV‐2 wild‐type and variants such as Alpha and potentially, but unlikely, to Delta. |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk on the comparison group and the relative effect of the intervention (and its 95% confidence interval). CI: confidence interval; mAb: monoclonal antibody; RCT: randomised controlled trial; RR: risk ratio; SARS‐CoV‐2: severe acute respiratory syndrome coronavirus 2. | ||||||
|
GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is the possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||||
aDowngraded two levels for very serious imprecision, because of very low number of events and very wide confidence intervals. bThe safety population included at baseline SARS‐CoV‐2 antibody seropositive, seronegative and sero‐undetermined participants. Data cut‐off date was 11 March 2021. cDowngraded two levels for very serious imprecision, because of low number of events and wide confidence intervals.
SARS‐CoV‐2‐specific mAbs for pre‐exposure prophylaxis of COVID‐19
Tixagevimab/cilgavimab compared to placebo
We present our certainty in the evidence for prioritised outcomes for the comparison tixagevimab/cilgavimab and placebo for PrEP of COVID‐19 in Table 1. The outcomes all‐cause mortality, admission to hospital, all‐grade AEs, and SAEs were assessed with the safety analysis set including at baseline 96.3% SARS‐CoV‐2 RT PCR status negative, 0.5% positive, and 3.2% unknown participants. The outcomes quality of life (up to six months and longest follow‐up), AEs grades 1 to 2 and grades 3 to 4, and admission to ICU within six months were not reported (PROVENT). We included only one study for this comparison (PROVENT), and, therefore, we could not perform any subgroup analyses or any sensitivity analyses. The study did not report characteristics of participants with missing data. As we identified fewer than 10 studies and did not pool the data in a meta‐analysis, we did not generate a funnel plot.
Participants had been exposed to SARS‐CoV‐2 wild‐type, Alpha, Beta, and Delta variants.
Primary outcomes
Infection with SARS‐CoV‐2 within six months
We defined infection with SARS‐CoV‐2 as confirmed by positive RT‐PCR, but also considered a positive SARS‐CoV‐2 antibody test as a laboratory‐confirmed infection. PROVENT reported SARS‐CoV‐2 anti‐nucleocapsid seroconversion in 4685 participants who were seronegative at baseline to median follow‐up of day 193. Seroconversion was defined as seronegative at baseline and seropositive at any time after baseline up to a median follow‐up at 193 days.
Tixagevimab/cilgavimab probably decreases infection with SARS‐CoV‐2 compared to placebo within six months (2.7% in the placebo group versus 1.2% in the tixagevimab/cilgavimab group; RR 0.45, 95% CI 0.29 to 0.70; 1 RCT, 4685 participants; moderate‐certainty evidence; Analysis 1.1). At unblinding or receipt of COVID‐19 vaccine, data were censored. Our main reason for downgrading certainty in the evidence was serious risk of bias (missing outcome data and potentially selection of the reported result) because the number of participants seronegative at baseline was unclear, and the reported six‐month follow‐up was not prespecified in the study protocol.
1.1. Analysis.

Comparison 1: Tixagevimab/cilgavimab compared to placebo to prevent COVID‐19 (pre‐exposure prophylaxis), Outcome 1: Infection with SARS‐CoV‐2 within 6 months
Development of clinical COVID‐19 symptoms within six months
PROVENT reported the first case of symptomatic illness to median follow‐up of day 193 for 5172 participants. SARS‐CoV‐2 infection was confirmed by positive SARS‐CoV‐2 RT‐PCR testing. Definitions of symptomatic disease and severe or critical disease are provided in the Characteristics of included studies table.
Tixagevimab/cilgavimab decreases development of clinical symptoms within six months (2% in the placebo group versus 0% in the tixagevimab/cilgavimab group; RR 0.18, 95% CI 0.09 to 0.35; 1 RCT; 5172 participants; high‐certainty evidence; Analysis 1.2). At unblinding or receipt of COVID‐19 vaccine, data were censored.
1.2. Analysis.

Comparison 1: Tixagevimab/cilgavimab compared to placebo to prevent COVID‐19 (pre‐exposure prophylaxis), Outcome 2: Development of clinical COVID‐19 symptoms within 6 months
Of those who developed clinical COVID‐19 symptoms, severe or critical COVID‐19 occurred in 0/3441 participants in the tixagevimab/cilgavimab group compared to 5/1731 participants in the placebo group. In the PROVENT study, two of our predefined subgroups were reported and the relative risk reduction (RRR) calculated. In the age group under 60 years, 8/1945 participants in the tixagevimab/cilgavimab group and 19/976 participants in the placebo group experienced development of symptomatic COVID‐19 infection within six months (RRR 79.6%, 95% CI 53.5 to 91.1). In the age group 60 years and older, 3/1496 participants in the tixagevimab/cilgavimab group and 12/755 participants in the placebo group developed symptomatic COVID‐19 infection within six months (RRR 87.8%, 95% CI 56.9 to 96.6).
Development of symptomatic COVID‐19 infection within six months occurred in 0/1126 participants without any coexisting condition in the tixagevimab/cilgavimab group and in 12/541 participants in the placebo group (RRR 100%). In participants with at least one coexisting condition, 11/2315 participants in the tixagevimab/cilgavimab group and 19/1190 participants in the placebo group experienced development of symptomatic COVID‐19 infection within six months (RRR 71.3%, 95% CI 39.8 to 86.4).
Secondary outcomes
All‐cause mortality at six months
PROVENT reported all AEs with outcome of death to median follow‐up of day 193 for 5197 participants. The outcome was analysed in the safety analysis set and included participants with negative, positive, and unknown RT‐PCR SARS‐CoV‐2 status at baseline. One participant randomised to placebo received incorrectly the intervention tixagevimab/cilgavimab and therefore was analysed in the tixagevimab/cilgavimab group.
Tixagevimab/cilgavimab may result in little to no difference on mortality within six months in participants regardless of RT‐PCR SARS‐CoV‐2 status at baseline (RR 0.64, 95% CI 0.24 to 1.73; 1 RCT, 5197 participants; low‐certainty evidence; Analysis 1.3). Our main reason for downgrading certainty in the evidence was very serious imprecision, because of very low number of events and wide CIs. For this outcome, we did not downgrade for serious risk of bias (measurement of the outcome), because for all‐cause mortality it is irrelevant whether participants were aware of the intervention received, and the number of people vaccinated was comparable in both arms.
1.3. Analysis.

Comparison 1: Tixagevimab/cilgavimab compared to placebo to prevent COVID‐19 (pre‐exposure prophylaxis), Outcome 3: All‐cause mortality within 6 months
Admission to hospital within six months
PROVENT reported COVID‐19‐related hospitalisations to median follow‐up of day 193 for 5197 participants. The outcome was analysed in the safety analysis set and included participants with negative, positive, and unknown RT‐PCR SARS‐CoV‐2 status at baseline. One participant randomised to placebo received incorrectly the intervention tixagevimab/cilgavimab and therefore was analysed in the tixagevimab/cilgavimab group.
Tixagevimab/cilgavimab may decrease admission to hospital within six months regardless of RT‐PCR SARS‐CoV‐2 status at baseline (RR 0.03, 95% CI 0.00 to 0.59; 1 RCT, 5197 participants; low‐certainty evidence; Analysis 1.4). Our main reason for downgrading certainty in the evidence were very serious imprecision, because of very low number of events.
1.4. Analysis.

Comparison 1: Tixagevimab/cilgavimab compared to placebo to prevent COVID‐19 (pre‐exposure prophylaxis), Outcome 4: Admission to hospital within 6 months
Admission to intensive care unit within six months
We identified no studies reporting admission to ICU within six months.
Quality of life (at six months, longest follow‐up)
We identified no studies reporting quality of life.
Adverse events (grade 1 to 2, grade 3 to 4, all grade)
PROVENT reported all‐grade AEs to median follow‐up of day 193 for 5197 participants. The safety analysis set included participants with negative, positive, and unknown RT‐PCR SARS‐CoV‐2 status at baseline. One participant randomised to placebo received incorrectly the intervention tixagevimab/cilgavimab and therefore was analysed in the tixagevimab/cilgavimab group.
Tixagevimab/cilgavimab may result in little to no difference on the occurrence of all‐grade AEs within six months regardless of RT‐PCR SARS‐CoV‐2 status at baseline (RR 1.00, 95% CI 0.94 to 1.07; 1 RCT, 5197 participants; low‐certainty evidence; Analysis 1.5). Our main reasons for downgrading certainty in the evidence were serious imprecision, because sample size did not meet optimal information size (6,435,640 participants) and serious risk of bias (measurement of the outcome), because participants were probably aware of the intervention received, as unblinding was possible.
1.5. Analysis.

Comparison 1: Tixagevimab/cilgavimab compared to placebo to prevent COVID‐19 (pre‐exposure prophylaxis), Outcome 5: Adverse events: all grade within 6 months
PROVENT did not report AEs grade 1 to 2 and grade 3 to 4. However, mild, moderate, and severe AEs were reported separately. Mild AEs occurred in 761/3461 (22.0%) participants in the tixagevimab/cilgavimab group compared to 539/1736 (21.3%) in the placebo group, moderate AEs occurred in 387/3461 (11.2%) in the tixagevimab/cilgavimab group compared to 191/1736 (11%) in the placebo group, and severe AEs in 64/3461 (1.8%) in the tixagevimab/cilgavimab group compared to 27 of 1736 (1.6%) in the placebo group.
Serious adverse events
PROVENT reported SAEs to median follow‐up of day 193 for 5197 participants. The safety analysis set included participants with negative, positive, and unknown RT‐PCR SARS‐CoV‐2 status at baseline. One participant randomised to placebo received incorrectly the intervention tixagevimab/cilgavimab and therefore was analysed in the tixagevimab/cilgavimab group.
Tixagevimab/cilgavimab may result in little to no difference on the occurrence of SAEs within six months regardless of RT‐PCR SARS‐CoV‐2 status at baseline (RR 1.12, 95% CI 0.83 to 1.52; 1 RCT, 5197 participants; low‐certainty evidence; Analysis 1.6). Our main reasons for downgrading certainty in the evidence were serious imprecision, because sample size did not meet optimal information size (55,674 participants) and serious risk of bias (measurement of the outcome), because participants were probably aware of the intervention received, as unblinding was possible.
1.6. Analysis.

Comparison 1: Tixagevimab/cilgavimab compared to placebo to prevent COVID‐19 (pre‐exposure prophylaxis), Outcome 6: Serious adverse events within 6 months
Casirivimab/imdevimab compared to placebo
We present our certainty in the evidence for prioritised outcomes for the comparison casirivimab/imdevimab and placebo for PrEP of COVID‐19 in Table 2. The population assessed for all but one reported outcome included at baseline 10.4% SARS‐CoV‐2 antibody seropositive, 85.1% seronegative, and 4.4% sero‐undetermined participants. The outcome infection with SARS‐CoV‐2 was assessed in seronegative participants at baseline. The outcomes admission to hospital within six months, quality of life (up to six months and longest follow‐up), AEs grades 1 to 2, and admission to ICU within six months were not reported (Isa 2021). We included only one study for this comparison (Isa 2021), and therefore we could not perform subgroup analysis or sensitivity analysis. Characteristics of participants with missing data were not reported in the studies. As we identified fewer than 10 studies and did not pool the data in meta‐analysis, we did not generate a funnel plot.
Participants may have been exposed to SARS‐CoV‐2 wild‐type, alpha, and Delta variants.
Primary outcomes
Infection with SARS‐CoV‐2 within six months
We defined infection with SARS‐CoV‐2 as confirmed by positive RT‐PCR, but also considered a positive SARS‐CoV‐2 antibody test as a laboratory‐confirmed infection. Isa 2021 reported SARS‐CoV‐2 anti‐nucleocapsid seroconversion in 825 participants seronegative at baseline at six months. At baseline, SARS‐CoV‐2 anti‐N and anti‐S protein antibodies (anti‐S1 domain of spike protein IgG and IgA antibodies) and anti‐nucleocapsid IgG antibodies were measured. Baseline seronegativity was defined as negative for any of the measured antibodies. At the end of treatment period, anti‐nucleocapsid IgG was measured. Seroconversion was defined as seronegative at baseline and anti‐nucleocapsid IgG seropositive at the end of the treatment period.
Casirivimab/imdevimab may decrease infection with SARS‐CoV‐2 within six months compared to placebo (10% in the placebo group versus 0% in the casirivimab/imdevimab group; RR 0.01, 95% CI 0.00 to 0.14; 1 RCT, 825 participants; low‐certainty evidence; Analysis 2.1). Our main reasons for downgrading certainty in the evidence were serious risk of bias, because of missing information regarding randomisation process, and allocation concealment and serious imprecision, because of low number of events.
2.1. Analysis.

Comparison 2: Casirivimab/imdevimab compared to placebo to prevent COVID‐19 (pre‐exposure prophylaxis), Outcome 1: Infection with SARS‐CoV‐2 within 6 months
Development of clinical COVID‐19 symptoms within six months
Isa 2021 reported symptomatic SARS‐CoV‐2 infection for 969 participants by week 24. Infections were confirmed by serology testing or central RT‐PCR testing.
Casirivimab/imdevimab may decrease development of clinical COVID‐19 symptoms within six months compared to placebo regardless of the SARS‐CoV‐2 antibody serostatus (4% in the placebo group versus 0% in the casirivimab/imdevimab group; RR 0.02, 95% CI 0 to 0.27; 1 RCT, 969 participants; low‐certainty evidence; Analysis 2.2). Our main reasons for downgrading certainty in the evidence were serious risk of bias, because of missing information regarding randomisation process, and allocation concealment and serious imprecision, because of low number of events.
2.2. Analysis.

Comparison 2: Casirivimab/imdevimab compared to placebo to prevent COVID‐19 (pre‐exposure prophylaxis), Outcome 2: Development of clinical COVID‐19 symptoms within 6 months
Secondary outcomes
All‐cause mortality at six months
Isa 2021 reported mortality by week 24 for 969 participants. The outcome was assessed as part of the safety assessment. None of the participants had died at six months.
We are uncertain whether casirivimab/imdevimab has any impact on mortality in participants regardless of the SARS‐CoV‐2 antibody serostatus (effect estimate not estimable; 1 RCT, 969 participants; very low certainty‐evidence; Analysis 2.3). Our main reasons for downgrading certainty in the evidence were serious risk of bias, because of missing information regarding randomisation process, and allocation concealment and very serious imprecision due to no events observed in any of the groups.
2.3. Analysis.

Comparison 2: Casirivimab/imdevimab compared to placebo to prevent COVID‐19 (pre‐exposure prophylaxis), Outcome 3: All‐cause mortality within 6 months
Admission to hospital within six months
We identified no studies reporting admission to hospital within six months.
Admission to intensive care unit within six months
We did not identify any study reporting this outcome.
Quality of life (at six months, longest follow‐up)
We identified no studies reporting quality of life.
Adverse events (grade 1 to 2, grade 3 to 4, all grade)
Isa 2021 reported AEs grade 3 or greater and all‐grade AEs for 969 participants. The evidence is uncertain about the effect of casirivimab/imdevimab on the occurrence of grade 3 or greater AEs compared to placebo at six months in participants, regardless of the SARS‐CoV‐2 antibody serostatus (RR 0.44, 95% CI 0.10 to 1.95; 1 RCT, 969 participants; very low certainty‐evidence; Analysis 2.4). Our main reasons for downgrading certainty in the evidence were serious risk of bias, because of missing information regarding randomisation process, and allocation concealment and very serious imprecision because of very low number of events and very wide CIs.
2.4. Analysis.

Comparison 2: Casirivimab/imdevimab compared to placebo to prevent COVID‐19 (pre‐exposure prophylaxis), Outcome 4: Adverse events: grade 3 to 4 within 6 months
Casirivimab/imdevimab may increase the occurrence of all‐grade AEs slightly compared to placebo by six months in participants regardless of the SARS‐CoV‐2 antibody serostatus (RR 1.14, 95% CI 0.98 to 1.31; 1 RCT, 969 participants; low‐certainty evidence; Analysis 2.5). Our main reasons for downgrading certainty in the evidence were serious risk of bias, because of missing information regarding randomisation process, and allocation concealment and serious imprecision because of wide CIs.
2.5. Analysis.

Comparison 2: Casirivimab/imdevimab compared to placebo to prevent COVID‐19 (pre‐exposure prophylaxis), Outcome 5: Adverse events: all grade within 6 months
AEs were reported separately for vaccinated participants. After receiving COVID‐19 vaccination, one (0.4%) participant in the casirivimab/imdevimab versus none in the placebo arm experienced at least one grade 3 or greater AE, 39 (15.2%) participants in the casirivimab/imdevimab versus 18 (18.4%) participants in the placebo arm experienced at least one all‐grade AE (Isa 2021).
Isa 2021 did not report AEs grades 1 to 2.
Serious adverse events
Isa 2021 reported SAEs for 969 participants. The evidence is very uncertain about the effect of casirivimab/imdevimab on the occurrence of SAEs compared to placebo by week 24 in participants regardless of the SARS‐CoV‐2 antibody serostatus (RR 0.82, 95% CI 0.16 to 4.21; 1 RCT, 969 participants; very low‐certainty evidence; Analysis 2.6). Our main reasons for downgrading certainty in the evidence were serious risk of bias, because of missing information regarding randomisation process, and allocation concealment and very serious imprecision because of very low number of events and very wide CIs.
2.6. Analysis.

Comparison 2: Casirivimab/imdevimab compared to placebo to prevent COVID‐19 (pre‐exposure prophylaxis), Outcome 6: Serious adverse events within 6 months
SAEs were reported separately for vaccinated participants. After receiving COVID‐19 vaccination, one (0.4%) participant in the casirivimab/imdevimab arm versus none in the placebo arm experienced at least one SAE (Isa 2021).
SARS‐CoV‐2‐specific mAbs for postexposure prophylaxis of COVID‐19
Bamlanivimab compared to placebo
We present our certainty in the evidence for prioritised outcomes for the comparison bamlanivimab and placebo for PEP of COVID‐19 in Table 3. The population assessed for all outcomes included 82.2% seronegative participants at baseline (BLAZE‐2). We included only one study for this comparison (BLAZE‐2), and therefore we could not perform subgroup or sensitivity analyses. The study did not report characteristics of participants with missing data. As we identified fewer than 10 studies and did not pool the data in meta‐analyses, we did not generate a funnel plot.
Participants may have been exposed to SARS‐CoV‐2 wild‐type.
Primary outcomes
Infection with SARS‐CoV‐2 within 30 days
BLAZE‐2 reported data by day 29 for 966 participants. Bamlanivimab probably decreases infection with SARS‐CoV‐2 compared to placebo by day 30 (RR 0.76, 95% CI 0.59 to 0.98; 1 RCT, 966 participants; moderate‐certainty evidence; Analysis 3.1). Our main reason for downgrading certainty in the evidence was serious imprecision because the sample size did not meet the optimal information size (2046 participants).
3.1. Analysis.

Comparison 3: Bamlanivimab compared to placebo to prevent COVID‐19 (postexposure prophylaxis), Outcome 1: Infection with SARS‐CoV‐2 by day 30
Additionally, the study reported a Kaplan‐Meier graph including the number of events per day up to day 57, which we used to calculate cumulative frequency of the incidence of SARS‐CoV‐2 infection by week eight (day 57). A total of 114/484 (23.6%) participants in the bamlanivimab group experienced an infection with SARS‐CoV‐2 (wild‐type) compared to 168/482 (34.9%) participants in the placebo group (RR 0.68, 95% CI 0.55 to 0.83).
Development of clinical COVID‐19 symptoms within 30 days
We identified no studies reporting development of clinical COVID‐19 symptoms within 30 days.
BLAZE‐2 reported the incidence of infection with mild or worse symptoms and moderate or worse symptoms within eight weeks. Definitions for disease severity are provided in the Characteristics of included studies section. About 8.5% of the participants who received bamlanivimab experienced an infection with mild or worse symptoms compared to 15.2% of the participants who received placebo (study‐reported OR 0.43, 95% CI 0.28 to 0.68; absolute risk difference −6.6%, 95% CI −10.7 to −2.6). In the bamlanivimab group, 8.3% compared with 14.1% in the placebo group experienced an infection with moderate or worse symptoms (OR 0.46, 95% CI 0.29 to 0.73; absolute risk difference −5.8 percentage points, 95% CI −9.8 to −1.8).
Secondary outcomes
All‐cause mortality (at day 30, 60, longest follow‐up)
BLAZE‐2 reported all‐cause mortality by day 57 including 966 participants. Bamlanivimab may result in little to no difference for all‐cause mortality by day 57 when compared to placebo (RR 0.83, 95% CI 0.25 to 2.70; 1 RCT, 966 participants; low‐certainty evidence; Analysis 3.2). Our main reason for downgrading certainty in the evidence was very serious imprecision due low number of events and wide CIs.
3.2. Analysis.

Comparison 3: Bamlanivimab compared to placebo to prevent COVID‐19 (postexposure prophylaxis), Outcome 2: All‐cause mortality by day 60
BLAZE‐2 did not report all‐cause mortality at day 30 and longest follow‐up.
Admission to hospital within 30 days
We identified no studies reporting admission to hospital within 30 days.
Admission to intensive care unit within 30 days
We identified no studies reporting admission to ICU within 30 days.
Quality of life (at day seven, 30, longest follow‐up)
We identified no studies reporting quality of life.
Adverse events (grade 1 to 2, grade 3 to 4, all grade)
BLAZE‐2 reported all‐grade AEs by week eight for 966 participants. Bamlanivimab may increase the occurrence of all‐grade AEs compared to placebo (RR 1.12, 95% CI 0.86 to 1.46; 1 RCT, 966 participants; low‐certainty evidence; Analysis 3.3). Our main reasons for downgrading certainty in the evidence were very serious imprecision, because sample size did not meet optimal information size (12,078 participants) and wide CIs.
3.3. Analysis.

Comparison 3: Bamlanivimab compared to placebo to prevent COVID‐19 (postexposure prophylaxis), Outcome 3: Adverse events: all grade
BLAZE‐2 did not report AEs grades 1 to 2 and grades 3 to 4.
Serious adverse events
BLAZE‐2 reported SAEs by week eight for 966 participants. Bamlanivimab may increase the occurrence of SAEs slightly compared to placebo (RR 1.46, 95% CI 0.73 to 2.91; 1 RCT, 966 participants; low‐certainty evidence; Analysis 3.4). Our main reasons for downgrading certainty in the evidence were very serious imprecision, because of low number of events, and wide CIs.
3.4. Analysis.

Comparison 3: Bamlanivimab compared to placebo to prevent COVID‐19 (postexposure prophylaxis), Outcome 4: Serious adverse events
Casirivimab/imdevimab compared to placebo
We present our certainty in the evidence for prioritised outcomes for the comparison casirivimab/imdevimab and placebo for PEP of COVID‐19 in Table 4. The population assessed for safety outcomes included at baseline 72.8% SARS‐CoV‐2 antibody seropositive, 22.1% seronegative, and 5.1% sero‐undetermined participants (O'Brien 2021). We included only one study for this comparison (O'Brien 2021), and therefore we could not perform subgroup or sensitivity analyses. The study did not report characteristics of participants with missing data. As we identified fewer than 10 studies and did not pool the data in meta‐analysis, we did not generate a funnel plot.
Participants may have been exposed to SARS‐CoV‐2 wild‐type, Alpha and potentially, but unlikely Delta variant.
Primary outcomes
Infection with SARS‐CoV‐2 within 30 days
O'Brien 2021 reported data by day 28 for 1505 participants. Casirivimab/imdevimab decreases infection with SARS‐CoV‐2 compared to placebo by day 28 (RR 0.34, 95% CI 0.23 to 0.48; 1 RCT, 1505 participants; high‐certainty evidence; Analysis 4.1).
4.1. Analysis.

Comparison 4: Casirivimab/imdevimab compared to placebo to prevent COVID‐19 (postexposure prophylaxis), Outcome 1: Infection with SARS‐CoV‐2 by day 30
Development of clinical COVID‐19 symptoms within 30 days
O'Brien 2021 reported infection with symptomatic disease by day 28 for 1505 participants. Symptomatic disease was defined by broad‐term, strict term, and CDC definition. All definitions are provided in the Characteristics of included studies table. We decided to use data of the broad‐term definition, because this includes any mild‐to‐moderate symptoms. Casirivimab/imdevimab decreases symptomatic infection with SARS‐CoV‐2 (broad‐term definition) compared to placebo by day 28 (RR 0.19, 95% CI 0.10 to 0.35; 1 RCT, 1505 participants; high‐certainty evidence; Analysis 4.2).
4.2. Analysis.

Comparison 4: Casirivimab/imdevimab compared to placebo to prevent COVID‐19 (postexposure prophylaxis), Outcome 2: Development of clinical COVID‐19 symptoms (broad‐term)
The reported proportion of symptomatic SARS‐CoV‐2 infection was 2/753 participants in the casirivimab/imdevimab arm compared to 22/752 participants in the placebo arm according to the strict‐term definition, and 6/753 participants in the casirivimab/imdevimab arm compared to 46/752 participants in the placebo arm according to the CDC definition (O'Brien 2021).
In a conference abstract, the results of a posthoc analysis, assessing the efficacy of casirivimab/imdevimab on the development of symptomatic SARS‐CoV‐2 infection in seronegative participants with CVD or diabetes, were reported separately as RRRs. In the overall seronegative study population (1505 participants), the RRR was 81.4% with casirivimab/imdevimab of developing symptomatic infection. In 332 participants with CVD, the RRR was 54.9% and in 103 participants with diabetes the RRR was 69.0% (O'Brien 2021).
Secondary outcomes
All‐cause mortality (at day 30, 60, longest follow‐up)
O'Brien 2021 provided data for all‐cause mortality by day 28 for SARS‐CoV‐2 seronegative participants after request. Casirivimab/imdevimab may result in little to no difference on mortality by day 30, but the evidence is uncertain (RR 3.00, 95% CI 0.12 to 73.43; 1 RCT, 1505 participants; low‐certainty evidence; Analysis 4.3). Our main reasons for downgrading certainty in the evidence were very serious imprecision, because of very low number of events, and very wide CIs.
4.3. Analysis.

Comparison 4: Casirivimab/imdevimab compared to placebo to prevent COVID‐19 (postexposure prophylaxis), Outcome 3: All‐cause mortality by day 30
In the journal publication, O'Brien 2021 reported all‐cause mortality measured outside the efficacy analysis period in the safety analysis set, including in addition to SARS‐CoV‐2 seronegative participants, seropositive, and sero‐undetermined participants, and 544 participants from the initial assessment. Two of 1311 participants had died in the casirivimab/imdevimab group and 2/1,306 participants in the placebo group.
O'Brien 2021 did not report all‐cause mortality at day 60 and longest follow‐up.
Admission to hospital within 30 days
O'Brien 2021 provided data for emergency department or hospital admission by day 28 for SARS‐CoV‐2 seronegative participants after request. Casirivimab and imdevimab may result in little to no difference on admission to hospital by day 28 (RR 0.11, 95% CI 0.01 to 2.06; 1 RCT, 1505 participants, low‐certainty evidence; Analysis 4.4). Our main reasons for downgrading certainty in the evidence were very serious imprecision, because of very low number of events and very wide CIs.
4.4. Analysis.

Comparison 4: Casirivimab/imdevimab compared to placebo to prevent COVID‐19 (postexposure prophylaxis), Outcome 4: Admission to hospital by day 30
In the journal publication, O'Brien 2021 reported emergency department or hospital admission in the safety analysis set, including seropositive and sero‐undetermined participants, and 544 participants from the initial assessment in addition to SARS‐CoV‐2 seronegative participants. In the casirivimab/imdevimab group, 2/1311 participants had died and in the placebo group, 2/1306 participants had died.
Admission to intensive care unit within 30 days
We identified no studies reporting admission to ICU within 30 days.
Quality of life (at day seven, 30, longest follow‐up)
We identified no studies reporting quality of life.
Adverse events (grade 1 to 2, grade 3 to 4, all grade)
O'Brien 2021 reported AEs grade 3 and greater and all‐grades AEs in 2617 participants. Casirivimab/imdevimab may decrease slightly the occurrence of grade 3 or greater AEs compared to placebo in participants regardless of the SARS‐CoV‐2 antibody serostatus (RR 0.50, 95% CI 0.24 to 1.02; 1 RCT, 2617 participants; low‐certainty evidence; Analysis 4.5). Our main reasons for downgrading certainty in the evidence were very serious imprecision, because of low number of events, and wide CIs.
4.5. Analysis.

Comparison 4: Casirivimab/imdevimab compared to placebo to prevent COVID‐19 (postexposure prophylaxis), Outcome 5: Adverse events: grade 3 to 4
Casirivimab/imdevimab decreases the occurrence of all‐grade AEs compared to placebo (RR 0.70, 95% CI 0.61 to 0.80; 1 RCT, 2617 participants; high‐certainty evidence; Analysis 4.6).
4.6. Analysis.

Comparison 4: Casirivimab/imdevimab compared to placebo to prevent COVID‐19 (postexposure prophylaxis), Outcome 6: Adverse events: all grade
O'Brien 2021 did not report AEs grade 1 to 2.
Serious adverse events
O'Brien 2021 reported SAEs for 2617 participants. Casirivimab/imdevimab may result in little to no difference on the occurrence of SAEs compared to placebo in participants regardless of the SARS‐CoV‐2 antibody serostatus (RR 0.66, 95% CI 0.30 to 1.47; 1 RCT, 2617 participants; low‐certainty evidence; Analysis 4.7). Our main reasons for downgrading certainty in the evidence were very serious imprecision, because of low number of events, and wide CIs.
4.7. Analysis.

Comparison 4: Casirivimab/imdevimab compared to placebo to prevent COVID‐19 (postexposure prophylaxis), Outcome 7: Serious adverse events
Discussion
Summary of main results
This review aimed to assess the effectiveness and safety of SARS‐CoV‐2‐neutralising mAbs, including mAb fragments, to prevent infection with SARS‐CoV‐2 causing COVID‐19. We included four RCTs (BLAZE‐2; Isa 2021; O'Brien 2021; PROVENT). The studies were published as preprint only (Isa 2021) or as journal publications (BLAZE‐2; O'Brien 2021; PROVENT). The studies involved 9749 randomised participants, comparing casirivimab/imdevimab versus placebo (Isa 2021), and tixagevimab/cilgavimab (PROVENT) for PrEP of COVID‐19; bamlanivimab versus placebo (BLAZE‐2), and casirivimab/imdevimab versus placebo (O'Brien 2021) for PEP of COVID‐19.
SARS‐CoV‐2‐specific mAbs for pre‐exposure prophylaxis of COVID‐19
Tixagevimab/cilgavimab compared to placebo
The study included participants with an increased risk of an inadequate immune response to COVID‐19 vaccination, an increased risk of SARS‐CoV‐2 exposure, or both (PROVENT). The SARS‐CoV‐2 variants Alpha, Beta, and Delta were detected in the study population. Participants were unvaccinated at the start of the study. Unblinding to assigned treatment was allowed if participants wanted to consider COVID‐19 vaccination. Unblinded or vaccinated participants were censored for efficacy at the date of unblinding or vaccination. At baseline, 96.3% of participants were RT‐PCR test negative, 0.5% positive, and 3.2% unknown. Tixagevimab/cilgavimab as PrEP probably decreases infection with SARS‐CoV‐2 within six months (RR 0.45, 95% CI 0.29 to 0.70; moderate‐certainty evidence) and decreases development of clinical symptoms within six months (0.18, 95% CI 0.09 to 0.35; high‐certainty evidence). Tixagevimab/cilgavimab may result in little to no difference on mortality and may decrease admission to hospital within six months in participants regardless of RT‐PCR SARS‐CoV‐2 status at baseline (mortality: RR 0.64, 95% CI 0.24 to 1.73; admission to hospital: RR 0.03, 95% CI 0.00 to 0.59; low‐certainty evidence). However, tixagevimab/cilgavimab may result in little to no difference on the occurrence of all‐grade AEs and SAEs (all‐grade AEs: RR 1.00, 95% CI 0.94 to 1.07; SAEs: RR 1.12, 95% CI 0.83 to 1.52; low‐certainty evidence). We identified no studies reporting quality of life, AEs grade 1 to 2, and AEs grade 3 to 4.
Casirivimab/imdevimab compared to placebo
The study was conducted during the emergence of multiple variants such as wild‐type, Alpha, and Delta (Isa 2021). At baseline, 10.4% of participants were SARS‐CoV‐2 antibody seropositive, 85.1% seronegative, and 4.4% sero‐undetermined. Participants were unvaccinated at the start of the study, but some received vaccination during the course of the study. Casirivimab/imdevimab may decrease infection with SARS‐CoV‐2 within six months (RR 0.01, 95% CI 0 to 0.14; low‐certainty evidence) and may decrease development of clinical COVID‐19 symptoms within six months regardless of the SARS‐CoV‐2 antibody serostatus (RR 0.02, 95% CI 0 to 0.27; low‐certainty evidence). The evidence is very uncertain about the effect of casirivimab/imdevimab on mortality. Casirivimab/imdevimab may increase the occurrence of all‐grade AEs slightly within six months regardless of the SARS‐CoV‐2 antibody serostatus. The evidence is very uncertain about the effect of casirivimab/imdevimab on the occurrence of grade 3 to 4 AEs and SAEs compared to placebo by week 24 regardless of the SARS‐CoV‐2 antibody serostatus. We identified no studies reporting admission to hospital, quality of life, and AEs grades 1 to 2.
SARS‐CoV‐2‐specific mAbs for postexposure prophylaxis of COVID‐19
Bamlanivimab compared to placebo
The study was probably conducted during wild‐type SARS‐CoV‐2 predominance in unvaccinated participants (BLAZE‐2). At baseline, 82.2% of participants were seronegative. Bamlanivimab as PEP probably decreases infection with SARS‐CoV‐2 compared to placebo by day 30 (RR 0.76, 95% CI 0.59 to 0.98; moderate‐certainty evidence) and may result in little to no difference on all‐cause mortality by day 60 (RR 0.83, 95% CI 0.25 to 2.70; low‐certainty evidence). However, it may increase the occurrence of all‐grade AEs and may increase the occurrence of serious SAEs slightly. We identified no studies reporting development of clinical COVID‐19 symptoms, all‐cause mortality at day 30 and longest follow‐up, admission to hospital, quality of life, AEs grade 1 to 2, and AEs grade 3 to 4.
Casirivimab/imdevimab compared to placebo
The study included unvaccinated participants, who may have already been exposed to the Alpha variant and potentially, but unlikely, to the Delta variant (O'Brien 2021). At baseline, 72.8% of participants were SARS‐CoV‐2 antibody seropositive, 22.1% seronegative and 5.1% sero‐undetermined. Casirivimab/imdevimab decreases infection with SARS‐CoV‐2 (RR 0.34, 95% CI 0.23 to 0.48; high‐certainty evidence) and development of clinical COVID‐19 symptoms (broad‐term definition; RR 0.19, 95% CI 0.10 to 0.35; high‐certainty evidence), and may result in little to no difference on mortality, and admission to hospital by day 30. Casirivimab/imdevimab may slightly decrease the occurrence of grade 3 to 4 AEs (RR 0.50, 95% CI 0.24 to 1.02; low‐certainty evidence), decreases the occurrence of all‐grade AEs (RR 0.70, 95% CI 0.61 to 0.80; high‐certainty evidence), and may result in little to no difference on the occurrence of SAEs in participants regardless of the SARS‐CoV‐2 antibody serostatus. We identified no studies reporting all‐cause mortality at day 60 and longest‐follow‐up, quality of life, and AEs grades 1 to 2.
Overall completeness and applicability of evidence
The evidence summarised in this review applies to the combination of tixagevimab/cilgavimab for PrEP, bamlanivimab for PEP, and the combination of casirivimab/imdevimab for both PrEP and PEP of COVID‐19, but as yet no other SARS‐CoV‐2‐neutralising mAbs or mAb fragments.
Only one study assessed SARS‐CoV‐2 variants (PROVENT). The variants Alpha, Beta, and Delta were identified in the study population and, therefore, the findings of tixagevimab/cilgavimab are applicable to people infected with these variants. The currently circulating Omicron variant was not detected in the study population. For the remaining three included studies, we can only assume which variants the participants were exposed to based on the timing of the studies (BLAZE‐2; Isa 2021; O'Brien 2021). Findings from casirivimab/imdevimab for PrEP of COVID‐19 are probably transferable to multiple variants such as wild‐type, Alpha, and Delta. In contrast, the findings from bamlanivimab for PEP of COVID‐19 can only be transferred to the wild‐type and casirivimab/imdevimab for PEP probably to Alpha and Delta. None of the evidence in this review is applicable to the most recently occurring variant Omicron. Protection against SARS‐CoV‐2 infection by SARS‐CoV‐2‐neutralising mAbs depends on the SARS‐CoV‐2 variant as the variants can escape due to mutations in the RBD. Therefore, the FDA withdrew the EUA for bamlanivimab, as bamlanivimab monotherapy is ineffective in neutralising most of the circulating SARS‐CoV‐2 variants of concern (FDA 2021f). Casirivimab combined with imdevimab remains active against previously predominant variants, including or Alpha, Beta, Delta, and Gamma, in vitro and in vivo (Baum 2020b; Copin 2021). Early findings suggest that SARS‐CoV‐2‐neutralising mAbs such as bamlanivimab and etesevimab in combination or monotherapy, casirivimab, and regdavimab are ineffective against Omicron sublineages BA.1 and BA.2, while the combination of tixagevimab and cilgavimab retains neutralising activity in vitro. Bebtelovimab is another mAb that has shown neutralising activity against Omicron. Adintrevimab remains effective against BA.1, while efficacy is reduced for sotrovimab. Imdevimab also showed neutralising activity against BA.2 (Bruel 2022). However, identified no relevant studies investigating mAbs for the prevention of infections caused by the Omicron variant. Even though tixagevimab/cilgavimab remains effective against Omicron in vitro, clinical studies are needed to assess the effectiveness specific to this variant.
All four studies included unvaccinated participants (BLAZE‐2; Isa 2021; O'Brien 2021; PROVENT). Both studies on PEP were conducted before widespread vaccine roll‐out and prohibited COVID‐19 vaccination prior and during the study period. The study period of Isa 2021 and PROVENT overlapped with first vaccine roll‐out. In Isa 2021, 35.1% of the participants in the intervention group and 40.8% in the placebo group opted for SARS‐CoV‐2 vaccination during the study period. To avoid potential interaction of the mAbs and vaccine‐induced immune responses, the CDC recommends waiting 90 days after receiving mAbs for treatment and 30 days after receiving mAbs for PEP, before SARS‐CoV‐2 vaccination (CDC 2021c). Participants received vaccination after a mean of 66.1 days from the last administration of the study drug and were then discontinued from further doses of intervention or placebo and followed for safety outcomes. They were censored for efficacy. In PROVENT, unblinding to assigned treatment was allowed if participants wanted to consider COVID‐19 vaccination. Unblinded or vaccinated participants were censored for efficacy at the date of unblinding or vaccination. About 39.3% of the participants were censored due to unblinding and 13.8% due to results not being reported separately for vaccinated individuals. After the worldwide admission of SARS‐CoV‐2 vaccines, there is no evidence available and the impact of vaccination on the efficacy and safety of SARS‐CoV‐2‐neutralising mAbs to prevent COVID‐19 remains unclear.
SARS‐CoV‐2‐neutralising mAbs for PrEP or PEP of COVID‐19 are particularly relevant for people who are at high risk for severe COVID‐19, such as immunocompromised people who do not mount an effective immune response to SARS‐CoV‐2 vaccines (i.e. through malignancy, chemotherapy, immunotherapy, or solid organ transplant) (Fung 2020; Liang 2020). Administration of mAbs could passively immunise them and protect them from SARS‐CoV‐2 infection. In Isa 2021 and BLAZE‐2, the proportion of immunocompromised participants was unclear, since the studies reported the proportion of participants with at least one risk factor including factors such as old age, immunosuppression, chronic disease, obesity, or a combination of these. Around 20% of the participants in the PrEP study (Isa 2021), and between 40% and 100% of participants in the PEP study (BLAZE‐2), had at least one risk factor for severe COVID‐19. In O'Brien 2021, the proportion of participants with immunosuppressive disease was 0.5%, and 1% of participants received immunosuppressive therapy. PROVENT focused on participants with an increased risk of an inadequate immune response to COVID‐19 vaccination, an increased risk of SARS‐CoV‐2 exposure, or both. In total, 77.5% of the participants had at least one risk factor for severe COVID‐19, of these, 7.4% had cancer, 3.3% received immunosuppressive therapy, and 0.5% had an immunosuppressive disease. However, the effect in participants with impaired immunity and ineffective immune response to SARS‐CoV‐2 vaccines is yet unclear, as O'Brien 2021 did not report results for this vulnerable subgroup and is likely underpowered to show an effect in this specific subgroup. Nevertheless, the FDA issued an EUA and in the UK, the Medicines and Healthcare products Regulatory Agency (MHRA) approved the mAb combination tixagevimab and cilgavimab for PrEP in immunocompromised people, based on the findings of the PROVENT study (Wise 2022). Several non‐RCTs investigating sotrovimab (NCT05135650; NCT05210101) and tixagevimab combined with cilgavimab (NCT05216588; NCT05234398) in immunocompromised people (some after SARS‐CoV‐2 vaccination) are underway and may provide more useful insights.
The proportion of SARS‐CoV‐2 anti‐spike IgG and IgA and anti‐nucleocapsid IgG antibody seronegative participants at baseline ranged between 72.8% and 85.1% (Isa 2021; O'Brien 2021). BLAZE‐2 analysed only seronegative participants. The vast majority of the participants had no pre‐existing immunity to SARS‐CoV‐2 at baseline. Seronegative individuals probably have a higher benefit of PrEP if the effects of treatment with SARS‐CoV‐2‐neutralising mAbs are analogised, because a higher effect to avoid the serious disease can be expected in uninfected or unvaccinated people. PROVENT did not report SARS‐CoV‐2 antibody serostatus.
In BLAZE‐2, participants were randomised and treated before the results of RT‐PCR and SARS‐CoV‐2 serology tests were available. This also reflects clinical practice, in which serostatus is not necessarily checked before administration of the intervention. For PEP, the mAb should be administered immediately after exposure to avoid a specific episode of infection. To date, there are no reliable surrogate correlates for disease or infection (or both) protection available. In seropositive people, for example, it is unclear which titres are associated with which assay and which level of protection. Therefore, the humoral vaccination response is used in relevant immunodeficiency due to its easy availability (Krammer 2021; Perry 2022). With lower mortality due to the Omicron variant and overall increasing immunocompetence in the general population, whether through vaccination or recovery, a greater focus on risk groups increases efficacy and thus reduces the number needed to treat (NNT).
None of the included studies measured quality of life. There are four ongoing studies and two studies awaiting classification, of which one had been withdrawn due to project termination and the other study is an ongoing platform trial that may potentially add a mAb intervention during the course of the study.
Quality of the evidence
SARS‐CoV‐2‐specific mAbs for pre‐exposure prophylaxis of COVID‐19
The certainty of the evidence for prioritised outcomes presented in the Table 1 ranged from high to low. We have high certainty in the identified evidence for the outcome development of clinical COVID‐19 symptoms and moderate certainty for the outcome infection with SARS‐CoV‐2 within six months. Our main concern was serious risk of bias (missing outcome data and potentially selection of the reported result, because the number of participants seronegative at baseline was unclear, and the reported six‐month follow‐up was not pre‐specified in the study protocol). For the outcomes mortality within six months and admission to hospital within six months, we rated the certainty of the evidence as low. Our main concerns were very serious imprecision, because of very low number of events and wide CIs. Also, for the safety outcomes, all‐grade AEs and SAEs within six months, we rated the certainty of the evidence as low. Here, our main concerns were serious imprecision, because the sample sizes did not meet the optimal information size (all‐grade AEs: 6,435,640 participants; SAEs: 55,674 participants) and serious risk of bias (measurement of the outcome and potentially selection of the reported result), because participants were probably aware of the intervention received, as unblinding was possible and the reported six‐month follow‐up was not prespecified in the study protocol.
The certainty of evidence for prioritised outcomes presented in the Table 2 ranged from low to very low. Each outcome was downgraded one level for serious risk of bias, because of missing information regarding randomisation process and allocation concealment. We have low‐certainty evidence for the outcomes infection with SARS‐CoV‐2 and development of clinical COVID‐19 symptoms within six months, as we downgraded a further level for serious imprecision because of low number of events. We have very‐low certainty for the outcome mortality within six months. Our main reason for further downgrading was very serious imprecision because there were no events, and therefore the effect was not estimable. For the safety outcome all‐grade AEs within six months, we have low‐certainty evidence. Our main reason for further downgrading was serious imprecision, because of wide CIs. For the outcomes grade 3 to 4 AEs and SAEs within six months, we have very low‐certainty evidence. Our main reasons for further downgrading were very serious imprecision, because of very low number of events and very wide CIs.
SARS‐CoV‐2‐specific mAbs for postexposure prophylaxis of COVID‐19
The certainty of evidence for prioritised outcomes presented in Table 3 ranged from moderate to low. We have moderate certainty for the outcome infection with SARS‐CoV‐2 by day 30. Our main concern was serious imprecision, because sample size did not meet optimal information size. We have low certainty in the identified evidence for the outcome mortality by day 60. Our main concern was very serious imprecision, because of low number of events and wide CIs. For the safety outcomes, we have low certainty for the outcome all‐grade AEs by week eight and SAEs by week eight. Our main concerns for all‐grade AEs were very serious imprecision, because sample size did not meet optimal information size (12,078 participants) and wide CIs, and for SAEs were very serious imprecision, because of low number of events and wide CIs.
The certainty of evidence for prioritised outcomes presented in the Table 4 ranged from high to low. We have high certainty in the identified evidence for the outcomes infection with SARS‐CoV‐2 by day 30 and development of clinical COVID‐19 symptoms (broad‐term definition). For the outcomes mortality up to the longest follow‐up and admission to hospital, we have low certainty in the identified evidence. Our main concerns were very serious imprecision, because of very low number of events and very wide CIs. For the safety outcome all‐grade AEs, we have high certainty in the identified evidence and low certainty for the outcomes grade 3 or greater AEs and SAEs. Our main reasons for concerns were very serious imprecision, because of low number of events and wide CIs.
Potential biases in the review process
An experienced medical information specialist (IM) developed an all‐encompassing search strategy, which was peer‐reviewed by another information specialist. We included all identified published studies, but also kept track of studies that were either still ongoing or labelled as completed in the study registry, to avoid overseeing any upcoming evidence. The sensitive search included relevant electronic databases as well as clinical trial registries. In addition to peer‐reviewed full‐text articles, we also included preprints. We are confident that we identified all relevant studies to date and will monitor ongoing studies as well as full publication of preprints closely after the publication of this first version of the review. In cases of missing data, we contacted study authors for additional data or relevant details, if we needed more information. We have received answers from the contacted study authors for BLAZE‐2 and O'Brien 2021. The author of Isa 2021 did not reply. We assumed results were not biased or impacted by missing outcome data given the low rate of missing data.
In contrast to the protocol, we differentiated between PrEP and PEP after we have included a study on PrEP and defined a separate outcome set with longer time points for PrEP, as a 30‐day interval was considered too short to measure PrEP. Furthermore, we changed the definition of one of our outcomes covered by the WHO Progression Scale. In light of the changing scale, we faced difficulties in obtaining a clear definition for the outcome development of clinical COVID‐19 symptoms and therefore used the definitions as provided by the studies (Differences between protocol and review). We do not think this has introduced bias because we made the changes based on clinical interest rather than the effect estimate.
For the outcome infection with SARS‐CoV‐2, next to the emergence of SARS‐CoV‐2 variants, there are other relevant factors that might influence the outcome. Depending on the local incidence and course of the pandemic, the risk for SARS‐CoV‐2 infection varies and could differ within and between studies. If few infections occur in a study, this cannot automatically be attributed to the intervention. It may have been because the local incidence was low and the risk of infection was much lower compared with a high local incidence. Individual factors, such as underlying diseases or therapies, also play an important role in SARS‐CoV‐2 infections. Most people infected have mild disease, but multiple risk factors can worsen the course of the disease and increase the risk of mortality (WHO 2020a).
All studies included unvaccinated participants. Therefore, we could not assess whether PrEP or PEP offer any additional benefit for SARS‐CoV‐2 vaccinated people. This should be addressed in future updates of the review.
Agreements and disagreements with other studies or reviews
We found one review of the evidence published as the Australian clinical guideline, which included data from the published full‐texts by O'Brien 2021 and PROVENT (National COVID‐19 Clinical Evidence Taskforce 2020). The results of O'Brien 2021 for the outcomes symptomatic and confirmed COVID‐19 infection and AEs and SAEs are in line with this review, but the assessment of the certainty in the evidence differs. For the outcomes symptomatic and confirmed COVID‐19 infection and all‐grade AEs, the certainty in the evidence was rated moderate and downgraded one level due to serious imprecision as the results were based on a single study. For the outcome all‐cause mortality, the data assessed in the safety population, including SARS‐CoV‐2 seropositive, seronegative, and sero‐undetermined participants was analysed. In contrast, we analysed requested data for seronegative participants. With regard to SAEs, we also rated the certainty of the evidence as low. The guideline included one additional safety outcome: discontinuation due to AEs, but did not consider the outcome admission to hospital. For tixagevimab/cilgavimab as PrEP we cannot compare our findings, as data of the primary data cut‐off date with a median follow‐up of 83 days was included in the guideline. In contrast, we included data for this review from the additional extended data cut‐off with a median follow‐up of 193 days, as this period was closer to our outcome definition of six months. For PEP, the guideline recommends the use tixagevimab/cilgavimab in research settings only, as no clinical data have been published to date. The guideline did not consider data for bamlanivimab from BLAZE‐2 for PEP or Isa 2021 for PrEP.
Authors' conclusions
Implications for practice.
The evidence for each comparison results from one study only. All studies were conducted in unvaccinated participants.
We rated the certainty in the evidence for tixagevimab combined with cilgavimab for pre‐exposure prophylaxis to prevent COVID‐19 (SARS‐CoV‐2 wild‐type and variants such as Alpha, Beta, and Delta) high to low. We have high certainty of a decrease in development of clinical COVID‐19 symptoms and moderate certainty of a decrease in infection with SARS‐CoV‐2 in the tixagevimab/cilgavimab group. We have low certainty of a decrease in admission to hospital.
We rated the certainty in the evidence for casirivimab combined with imdevimab for pre‐exposure prophylaxis to prevent COVID‐19 (probably SARS‐CoV‐2 wild‐type and variants such as Alpha and Delta) low to very low. We have low certainty of a decrease in infection with SARS‐CoV‐2, development of clinical COVID‐19 symptoms, and a higher rate for all‐grade adverse events in the casirivimab/imdevimab group.
For bamlanivimab as postexposure prophylaxis to prevent COVID‐19 (probably SARS‐CoV‐2 wild‐type), we rated the certainty in the evidence as moderate to low. We have moderate certainty of a decrease in infection with SARS‐CoV‐2 and low certainty of a higher rate of all‐grade adverse events and serious adverse events in the bamlanivimab group.
We rated the certainty in the evidence for casirivimab combined with imdevimab for postexposure prophylaxis to prevent COVID‐19 (probably SARS‐CoV‐2 wild‐type and variants such as Alpha and potentially, but unlikely, Delta) high to low. We have high certainty of a decrease in infection with SARS‐CoV‐2, development of clinical COVID‐19 symptoms, and a higher rate for all‐grade adverse events in the casirivimab/imdevimab group. We have low certainty of a lower rate of adverse events grade 3 to 4 in the casirivimab/imdevimab group.
We have high to moderate certainty in the evidence for a few highly relevant outcomes, the identified evidence is mostly not sufficient to draw meaningful conclusions regarding pre‐exposure prophylaxis or postexposure prophylaxis with any specific monoclonal antibody (mAb), because the identified evidence is only transferable to the variants prevailing in the respective study period and is not applicable to other variants (e.g. Omicron). In vitro, the combination of tixagevimab/cilgavimab has shown to remain effective against Omicron, but no clinical data are available to date. The other mAb bamlanivimab and mAb combination casirivimab/imdevimab included in this review are ineffective against Omicron in vitro. Findings can also not be transferred to SARS‐CoV‐2 vaccinated people, as only unvaccinated participants were included in the studies.
Implications for research.
We did not conduct meta‐analyses, because studies were heterogeneous with regard to different mAbs used for prophylaxis. There are currently no relevant studies examining mAbs that remain active against Omicron for the prevention of infections caused by the Omicron variant. Future research needs to examine whether the occurrence of SARS‐CoV‐2 variants such as Omicron affects the effectiveness of SARS‐CoV‐2‐neutralising mAbs and mAb fragments to prevent COVID‐19. In addition, after world‐wide vaccine roll‐out, the impact of SARS‐CoV‐2 vaccines on the effectiveness of mAbs should also be considered in future studies. None of the included studies reported quality of life.
History
Protocol first published: Issue 5, 2021
Notes
Parts of the review's background and methods sections are reproduced from the published review about SARS‐CoV‐2‐neutralising mAbs for treatment of COVID‐19 (Kreuzberger 2021).
Risk of bias
Acknowledgements
We thank Carolyn Dorée for reviewing the search strategy.
The following people conducted the editorial process for this article.
Sign‐off Editor (final editorial decision): Toby Lasserson, Deputy Editor‐in‐Chief, Cochrane Evidence Production & Methods Directorate.
Managing Editor (selected peer reviewers, provided comments, collated peer‐reviewer comments, provided editorial guidance to authors, edited the article): Lara Kahale, Cochrane Central Editorial Service.
Editorial Assistant (conducted editorial policy checks and supported editorial team): Lisa Wydrzynski, Cochrane Central Editorial Service.
Copy Editor (copy‐editing and production): Anne Lawson, Copy Edit Support, Cochrane.
Peer‐reviewers (provided comments and recommended an editorial decision): Godelieve J de Bree, Department of Internal Medicine and Infectious Diseases, Amsterdam University Medical Center (clinical review); Sanjay Bhagani, Royal Free London NHS Trust/UCL, UK (clinical review); Etienne Ngeh, Research Organisation for Health Education and Rehabilitation‐Cameroon (ROHER‐CAM), Centre for Health and Social Care Research, Sheffield Hallam University, Sheffield, UK (consumer review); Nuala Livingstone, Cochrane Evidence Production & Methods Directorate (methods review), and Robin Featherstone, Cochrane Central Editorial Service (search review).
The research was part of a project supported by the German Federal Ministry of Education and Research (NaFoUniMedCovid19, funding number: 01KX2021; part of the projects 'COVIM' and 'CEOsys'). The contents of this document reflect only the authors' views and the German Ministry is not responsible for any use that may be made of the information it contains.
The research was supported by NHS Blood and Transplant and the National Institute for Health Research (NIHR) Oxford Biomedical Research Centre (BRC). The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR, or the Department of Health.
Appendices
Appendix 1. Search strategy: Cochrane Covid‐19 Study Register
"Ly‐3832479" OR Ly3832479 OR "LY‐3832479" OR LY3832479 OR "LY‐CoV016" OR "REGN‐COV2" OR "REGEN‐COV2" OR REGN10933 OR REGN10987 OR REGEN10933 OR REGEN10987 OR casirivimab OR imdevimab OR "LY‐3819253" OR LY3819253 OR "LY‐CoV555" OR Bamlanivimab OR Banlanivimab OR "VIR‐7831" OR VIR7831 OR GSK4182136 OR "GSK‐4182136" OR sotrovimab OR AZD7442 OR "AZD‐7442" OR AZD8895 OR "AZD‐8895" OR tixagevimab OR "COV2‐2196" OR COV22196 OR AZD1061 OR "AZD‐1061" OR cilgavimab OR "COV2‐2130" OR COV22130 OR DXP593 OR "DXP‐593" OR "BGB‐DXP‐593" OR BGBDXP593 OR JS016 OR "JS‐016" OR etesevimab OR TY027 OR "TY‐027" OR CTP59 OR "CTP‐59" OR "CT‐P59" OR regdanvimab OR STI1499 OR "STI‐1499" OR "COVI‐shield" OR COVIshield OR "COVI‐guard" OR COVIguard OR BRII196 OR "BRII‐196" OR SCTA01 OR "SCTA‐01" OR MW33 OR "MW‐33" OR BRII198 OR "BRII‐198" OR HFB30132A OR "HFB‐30132A" OR ADM03820 OR "ADM‐03820" OR HLX70 OR "HLX‐70" OR STI2020 OR "STI‐2020" OR COVIAMG OR "COVI‐AMG" OR DZIF10c OR "DZIF‐10c" OR BI767551 OR "BI‐767551" OR "COV2‐2381" OR COV22381 OR "ABBV‐47D11" OR 47D11 OR ABBV47D11 OR "COR‐101" OR COR101 OR "STE90‐C11" OR "DXP‐604" OR DXP604 OR "BGB‐DXP604" OR BGBDXP604 OR "BGB‐DXP‐604" OR "chicken egg antibody" OR "egg yolk antibody" OR IgY* OR "VIR‐7832" OR VIR7832 OR GSK4182137 OR "GSK‐4182137" OR IDB003 OR MD65 OR "MTX‐COVAB"
Appendix 2. Search strategy: MEDLINE via Ovid
# Searches
1 "spike protein, SARS‐CoV‐2".af.
2 Coronavirus Infections/ or Coronavirus/
3 SARS‐CoV‐2/ or COVID‐19/
4 ("2019 nCoV" or 2019nCoV or coronavir* or coronovir* or COVID or COVID19 or HCoV* or "nCov 2019" or "SARS CoV2" or "SARS CoV 2" or SARSCoV2 or "SARSCoV 2").tw,kf.
5 "severe acute respiratory syndrome coronavirus 2".tw,kf,nm.
6 ((corona* or corono*) adj1 (virus* or viral* or virinae*)).tw,kf.
7 or/2‐6
8 *Antibodies, Monoclonal/
9 Antibodies, Neutralizing/
10 Antibodies, Viral/
11 ((antibod* or mAb* or nAb*) adj2 (therap* or treatment* or neutrali?ing or prevent* or protect* or prophylax*)).tw,kf.
12 Spike Glycoprotein, Coronavirus/
13 Binding, Competitive/
14 (compet* adj1 bind*).tw,kf.
15 ("spike protein*" or "s protein*" or "Spike (S) protein").mp.
16 ((cocktail* or mixture* or comb*) adj3 (mAb* or antibod* or nAb*)).tw,kf.
17 ("two mab" or "mabs" or "two nab" or "two nabs").tw,kw.
18 (LY3832479* or LY‐CoV016* or LY‐3832479* or LYCoV016* or JS016* or JS‐016* or etesevimab*).mp.
19 (REGN‐COV* or REGN10933* or REGN10987* or REGN‐10933* or REGN‐10987* or REGEN‐COV* or REGEN10933* or REGEN10987* or REGEN‐10933* or REGEN‐10987* or ronapreve* or casirivimab* or imdevimab*).mp.
20 (LY3819253* or LY‐3819253* or LY‐CoV555* or LYCoV555* or bamlanivimab* or banlanivimab*).mp.
21 (VIR7831* or VIR‐7831* or GSK4182136* or GSK‐4182136* or sotrovimab*).mp.
22 (AZD7442* or AZD‐7442* or AZD8895* or AZD‐8895* or tixagevimab* or COV2‐2196 or COV22196* or AZD1061* or AZD‐1061* or cilgavimab* or COV2‐2130* or COV22130*).mp.
23 (DXP‐593* or DXP593* or BGB‐DXP593* or BGBDXP593* or BGB‐DXP‐593*).mp.
24 (TY027* or TY‐027*).mp.
25 (CTP59* or CTP‐59* or CT‐P59* or regdanvimab* or regkirona*).mp.
26 (STI1499* or STI‐1499* or COVI‐GUARD* or COVIguard*).mp.
27 (BRII196* or BRII‐196*).mp.
28 (SCTA01* or SCTA‐01*).mp.
29 (MW33* or MW‐33*).mp.
30 (BRII198* or BRII‐198*).mp.
31 (HFB30132A* or HFB‐30132A*).mp.
32 (ADM03820* or ADM‐03820*).mp.
33 (HLX70* or HLX‐70*).mp.
34 (STI2020* or STI‐2020* or COVIAMG* or COVI‐AMG*).mp.
35 (DZIF10c* or DZIF‐10c* or BI767551* or BI‐767551*).mp.
36 (COV2‐2381* or COV22381*).mp.
37 (ABBV47D11* or AbbVie47D11* or 47D11* or AbbVie2B04* or ABBV2B04* or 2B04*).af.
38 (COR‐101* or COR101* or STE90‐C11* or CORAT*).mp.
39 (DXP‐604* or DXP604* or BGB‐DXP604* or BGBDXP604* or BGB‐DXP‐604*).mp.
40 (Chicken egg antibod* or egg yolk antibod* or IgY* or avian antibod*).mp.
41 (VIR‐7832 or VIR7832 or GSK4182137* or GSK‐4182137*).mp.
42 (IDB003 or MD65 or MTX‐COVAB).mp.
43 (C144‐LS or C144LS or C‐135‐LS or C135‐LS or C135LS or SAB‐185 or SAB185 or JMB2002 or JMB‐2002 or TATX‐03 or TATX03 or NOVOAB‐20 or NOVOAB20 or NOVO‐AB‐20 or ABP‐300 or ABP300 or MW05 or MW‐05 or MW07 or MW‐07 or ACmab1 or covimax or adimab or CPI‐006 or CPI006 or ADG20 or "ADG20" or "CT‐P‐63" or "CT‐P63" or MAD0004J08* or CORAT* or avian antibod* or CT‐P‐63* or CT‐P63* or AF47D11* or AbbVie47D11* or 47D11* or AbbVie2B04* or ABBV2B04* or 2B04* or STI‐2099* or STI2099* or covi‐drop* or covidrop* or REGN14256*).af.
44 Single‐Chain Antibodies/
45 (nmabs or diabod* or dia‐bod* or nanobod* or nano‐bod* or microbod* or micro‐bod* or monobod* or mono‐bod*).tw,kf,nm.
46 (scFv* or "single‐chain*" or "heavy chain*" or HCAbs or SCAbs or FAB).tw,kf,nm.
47 or/8‐46
48 randomized controlled trial.pt.
49 controlled clinical trial.pt.
50 randomi?ed.ab.
51 placebo.ab.
52 drug therapy.fs.
53 randomly.ab.
54 trial.ab.
55 groups.ab.
56 or/49‐55
57 exp animals/ not humans/
58 56 not 57
59 clinical trial, phase III/
60 ("Phase 3" or "phase3" or "phase III" or P3 or "PIII").ti,ab,kw.
61 (59 or 60) not 57
62 58 or 61
63 7 and (1 or 47) and 62
64 limit 63 to yr="2020‐Current"
Appendix 3. Search strategy: Embase via Ovid
# Searches
1 coronavirinae/ or coronaviridae/ or coronaviridae infection/
2 coronavirus disease 2019/
3 Coronavirus infection/
4 sars‐related coronavirus/
5 "Severe acute respiratory syndrome coronavirus 2"/
6 ((corona* or corono*) adj1 (virus* or viral* or virinae*)).tw,kw.
7 ("2019 nCoV" or 2019nCoV or coronavir* or coronovir* or COVID or COVID19 or HCoV* or "nCov 2019" or "SARS CoV2" or "SARS CoV 2" or SARSCoV2 or "SARSCoV 2").tw,kw.
8 "Severe acute respiratory syndrome coronavirus 2".mp.
9 or/1‐8
10 Antibodies, Monoclonal/
11 Antibodies, Neutralizing/
12 Antibodies, Viral/
13 ((antibod* or mAb* or nAb*) adj2 (therap* or treatment* or neutrali?ing or prevent* or protect* or prophylax*)).tw,kw.
14 Spike Glycoprotein, Coronavirus/
15 Binding, Competitive/
16 (compet* adj1 bind*).tw,kw.
17 ("spike protein*" or "s protein*" or "Spike (S) protein").mp.
18 ((cocktail* or mixture* or comb*) adj3 (mAb* or antibod* or nAb*)).tw,kw.
19 ("two mab" or "mabs" or "two nab" or "two nabs").tw,kw.
20 (LY3832479* or LY‐CoV016* or LY‐3832479* or LYCoV016* or JS016* or JS‐016* or etesevimab*).mp.
21 (REGN‐COV* or REGN10933* or REGN10987* or REGN‐10933* or REGN‐10987* or REGEN‐COV* or REGEN10933* or REGEN10987* or REGEN‐10933* or REGEN‐10987* or ronapreve* or casirivimab* or imdevimab*).mp.
22 (LY3819253* or LY‐3819253* or LY‐CoV555* or LYCoV555* or bamlanivimab* or banlanivimab*).mp.
23 (VIR7831* or VIR‐7831* or GSK4182136* or GSK‐4182136* or sotrovimab*).mp.
24 (AZD7442* or AZD‐7442* or AZD8895* or AZD‐8895* or tixagevimab* or COV2‐2196 or COV22196* or AZD1061* or AZD‐1061* or cilgavimab* or COV2‐2130* or COV22130*).mp.
25 (DXP‐593* or DXP593* or BGB‐DXP593* or BGBDXP593* or BGB‐DXP‐593*).mp.
26 (TY027* or TY‐027*).mp.
27 (CTP59* or CTP‐59* or CT‐P59* or regdanvimab* or regkirona*).mp.
28 (STI1499* or STI‐1499* or COVI‐GUARD* or COVIguard*).mp.
29 (BRII196* or BRII‐196*).mp.
30 (SCTA01* or SCTA‐01*).mp.
31 (MW33* or MW‐33*).mp.
32 (BRII198* or BRII‐198*).mp.
33 (HFB30132A* or HFB‐30132A*).mp.
34 (ADM03820* or ADM‐03820*).mp.
35 (HLX70* or HLX‐70*).mp.
36 (STI2020* or STI‐2020* or COVIAMG* or COVI‐AMG*).mp.
37 (DZIF10c* or DZIF‐10c* or BI767551* or BI‐767551*).mp.
38 (COV2‐2381* or COV22381*).mp.
39 (ABBV47D11* or AbbVie47D11* or 47D11* or AbbVie2B04* or ABBV2B04* or 2B04*).mp.
40 (COR‐101* or COR101* or STE90‐C11* or CORAT*).mp.
41 (DXP‐604* or DXP604* or BGB‐DXP604* or BGBDXP604* or BGB‐DXP‐604*).mp.
42 (Chicken egg antibod* or egg yolk antibod* or IgY* or avian antibod*).mp.
43 (VIR‐7832* or VIR7832* or GSK4182137* or GSK‐4182137*).mp.
44 (IDB003 or MD65 or MTX‐COVAB).mp.
45 (C144‐LS or C144LS or C‐135‐LS or C135‐LS or C135LS or SAB‐185 or SAB185 or JMB2002 or JMB‐2002 or TATX‐03 or TATX03 or NOVOAB‐20 or NOVOAB20 or NOVO‐AB‐20 or ABP‐300 or ABP300 or MW05 or MW‐05 or MW07 or MW‐07 or ACmab1 or covimax or adimab or CPI‐006 or CPI006 or ADG20 or ADG 20 or MAD0004J08 or CORAT* or avian antibod* or CT‐P‐63* or CT‐P63* or AF47D11* or AbbVie47D11* or 47D11* or AbbVie2B04* or ABBV2B04* or 2B04* or STI‐2099* or STI2099* or covi‐drop* or covidrop* or REGN14256*).mp.
46 single chain fragment variable antibody/
47 (nmabs or diabod* or dia‐bod* or nanobod* or nano‐bod* or microbod* or micro‐bod* or monobod* or mono‐bod*).mp.
48 (scFv* or "single‐chain*" or "heavy chain*" or HCAbs or SCAbs).mp.
49 or/10‐48
50 Randomized controlled trial/
51 Controlled clinical study/
52 random*.ti,ab.
53 randomization/
54 intermethod comparison/
55 placebo.ti,ab.
56 (compare or compared or comparison).ti.
57 (open adj label).ti,ab.
58 ((double or single or doubly or singly) adj (blind or blinded or blindly)).ti,ab.
59 double blind procedure/
60 parallel group$1.ti,ab.
61 (crossover or cross over).ti,ab.
62 ((assign$ or match or matched or allocation) adj5 (alternate or group$1 or intervention$1 or patient$1 or subject$1 or participant$1)).ti,ab.
63 (controlled adj7 (study or design or trial)).ti,ab.
64 (volunteer or volunteers).ti,ab.
65 trial.ti.
66 or/50‐65
67 phase 3 clinical trial/
68 ("Phase 3" or "phase3" or "phase III" or P3 or "PIII").tw,kw.
69 or/67‐68
70 (animal experiment/ or Animal experiment/) not (human experiment/ or human/)
71 (66 or 69) not 70
72 9 and 49 and 71
73 limit 72 to yr="2020 ‐Current"
74 limit 73 to medline
75 73 not 74
Appendix 4. Search strategy: PubMed (ahead of print only)
#1 2019 nCoV[tiab] OR 2019nCoV[tiab] OR corona virus[tiab] OR corona viruses[tiab] OR coronavirus[tiab] OR coronaviruses[tiab] OR COVID[tiab] OR COVID19[tiab] OR nCov 2019[tiab] OR SARS‐CoV2[tiab] OR SARS CoV‐2[tiab] OR SARSCoV2[tiab] OR SARSCoV‐2[tiab] OR "COVID‐19"[Mesh] OR "Coronavirus"[Mesh:NoExp] OR "SARS‐CoV‐2"[Mesh] OR "COVID‐19"[nm] OR "severe acute respiratory syndrome coronavirus 2"[nm]
#2 (antibod*[Title/Abstract] OR mAb[Title/Abstract] OR mAbs[Title/Abstract] OR nAb[Title/Abstract] OR nAbs[Title/Abstract]) AND (therap*[Title/Abstract] OR treat*[Title/Abstract] OR neutrali*[Title/Abstract] OR prevent*[Title/Abstract] OR protect*[Title/Abstract] OR prophylax*[Title/Abstract]) OR (compet*[Title/Abstract] AND bind*[Title/Abstract]) OR (cocktail*[Title/Abstract] OR mixture*[Title/Abstract] OR comb*[Title/Abstract]) AND (mAb[Title/Abstract] OR mAbs[Title/Abstract] OR antibod*[Title/Abstract] OR nAb[Title/Abstract] OR nAbs[Title/Abstract]) OR "spike protein*"[Title/Abstract] OR "s protein*"[Title/Abstract] OR "Spike (S) protein"[Title/Abstract]
#3 (LY‐3832479 OR LY3832479 OR LY‐CoV016 OR REGN‐COV2 OR REGN10933 OR REGN10987 OR REGN‐10933 OR REGN‐10987 OR REGEN10933 OR REGEN10987 OR REGEN‐10933 OR REGEN‐10987 OR REGN‐CoV* OR REGEN‐CoV* OR ronapreve* OR casirivimab OR imdevimab OR LY‐3819253 OR LY3819253 OR LY‐CoV555 OR Bamlanivimab OR Banlanivimab OR VIR‐7831 OR VIR7831 OR GSK4182136 OR GSK‐4182136 OR sotrovimab OR AZD7442 OR AZD‐7442 OR AZD1061 OR AZD‐1061 OR AZD8895 OR AZD‐8895 OR tixagevimab OR cilgavimab OR DXP593 OR DXP‐593 OR BGB‐DXP‐593 OR BGBDXP593 OR JS016 OR JS‐016 OR LY‐CoV016 OR etesevimab OR TY027 OR TY‐027 OR CTP59 OR CT‐P59 OR regdanvimab OR STI1499 OR STI‐1499 OR COVI‐guard OR COVIguard OR BRII196 OR BRII‐196 OR SCTA01 OR SCTA‐01 OR MW33 OR MW‐33 OR BRII198 OR BRII‐198 OR HFB30132A OR HFB‐30132A OR ADM03820 OR ADM‐03820 OR HLX70 OR HLX‐70 OR STI2020 OR STI‐2020 OR COVIAMG OR COVI‐AMG OR DZIF10c OR DZIF‐10c OR BI767551 OR BI‐767551 COV2‐2381 OR COV22381 OR ABBV‐47D11 OR 47D11 OR ABBV47D11 OR COR‐101 OR COR101 OR STE90‐C11 OR DXP‐604 OR DXP604 OR BGB‐DXP604 OR BGBDXP604 OR BGB‐DXP‐604 OR „chicken egg antibod*" OR "egg yolk antibod*" OR IgY OR IgYs OR VIR‐7832 OR VIR7832 OR GSK4182137 OR GSK‐4182137 OR IDB003 OR MTX‐COVAB OR MD65 OR TATX‐03 OR NOVOAB‐20 OR "C144‐LS" OR C144LS OR "C‐135‐LS" OR "C135‐LS" OR C135LS OR "SAB‐185" OR SAB185 OR JMB2002 OR "JMB‐2002" OR "TATX‐03" OR TATX03 OR "NOVOAB‐20" OR NOVOAB20 OR "NOVO‐AB‐20" OR "ABP‐300" OR ABP300 OR MW05 OR "MW‐05" OR MW07 OR "MW‐07" OR ACmab1 OR covimax OR adimab OR "CPI‐006" OR CPI006 OR diabod* OR nanobod* OR microbod* OR monobod* OR scFv* OR "single‐chain" OR "heavy chain" OR HCAbs OR SCAbs OR nmAbs OR ADG20 OR "ADG 20" OR MAD0004J08 OR regkirona OR CORAT* OR "avian antibody" OR "CT‐P‐63" OR "CT‐P63" OR AF47D11* OR AbbVie47D11* OR 47D11* OR AbbVie2B04* OR ABBV2B04* OR 2B04* OR "STI‐2099" OR STI2099* OR "covi‐drops" OR covidrop* OR REGN14256*)
#4 "spike protein*"[Title/Abstract] OR "s protein*"[Title/Abstract] OR "Spike (S) protein"[Title/Abstract]
#5 #1 AND (#2 OR #3)
#6 #4 OR #5
#7 (publisher[sb] OR inprocess[sb] OR pubmednotmedline[sb])
#8 (randomized controlled trial[pt] OR controlled clinical trial[pt] OR randomized[tiab] OR placebo[tiab] OR drug therapy[sh] OR randomly[tiab] OR trial[tiab] OR groups[tiab] NOT (animals [mh] NOT humans [mh])
#9 #6 AND #7 AND #8 Filters: from 2020/1/1 ‐ 3000/12/12
Appendix 5. Search strategy: Epistemonikos, L*OVE List Coronavirus disease (COVID‐19)
app.iloveevidence.com/loves/5e6fdb9669c00e4ac072701d?utm=epdb_en
the Coronavirus disease (COVID‐19) L*OVE for Prevention or treatment > Pharmacological interventions > Targeted therapies > Anti‐SARS‐CoV‐2 Mab > primary studies.
Appendix 6. Search strategy: World Health Organization COVID‐19 Global literature on coronavirus disease
Advanced search: search fields: title, abstract, subject
("monoclonal antibody" or "monoclonal antibodies" or "Ly‐3832479" or Ly3832479 or "LY‐3832479" or LY3832479 or "LY‐CoV016" or "REGN‐COV2" or "REGN‐COV‐2" or "REGN‐COV" or "REGEN‐COV2" or "REGEN‐COV‐2" or "REGEN‐COV" or REGN10933 or REGN10987 or REGEN10933 or REGEN10987 or ronapreve or casirivimab or imdevimab or "LY‐3819253" or LY3819253 or "LY‐CoV555" or Bamlanivimab or Banlanivimab or "VIR‐7831" or VIR7831 or GSK4182136 or "GSK‐4182136" or sotrovimab or AZD7442 or "AZD‐7442" or AZD8895 or "AZD‐8895" or tixagevimab or "COV2‐2196" or COV22196 or AZD1061 or "AZD‐1061" or cilgavimab or "COV2‐2130" or COV22130 or DXP593 or "DXP‐593" or "BGB‐DXP‐593" or BGBDXP593 or JS016 or "JS‐016" or etesevimab or TY027 or "TY‐027" or CTP59 or "CTP‐59" or "CT‐P59" or regdanvimab or regkirona* or STI1499 or "STI‐1499" or "COVI‐guard" or COVIguard or BRII196 or "BRII‐196" or SCTA01 or "SCTA‐01" or MW33 or "MW‐33" or BRII198 or "BRII‐198" or HFB30132A or "HFB‐30132A" or ADM03820 or "ADM‐03820" or HLX70 or "HLX‐70" or STI2020 or "STI‐2020" or COVIAMG or "COVI‐AMG" or DZIF10c or "DZIF‐10c" or BI767551 or "BI‐767551" or "COV2‐2381" or COV22381 or ABBV47D11* or AbbVie47D11* or 47D11* or AbbVie2B04* or ABBV2B04* or 2B04* or "COR‐101" or COR101 or "STE90‐C11" or CORAT* or "DXP‐604" or DXP604 or "BGB‐DXP604" or BGBDXP604 or "BGB‐DXP‐604" or "chicken egg antibody" or "egg yolk antibody" or IgY* or "avian antibody" or "VIR‐7832" or VIR7832 or GSK4182137 or "GSK‐4182137" or IDB003 or MD65 or "MTX‐COVAB" or "C144‐LS" or C144LS or "C‐135‐LS" or "C135‐LS" or C135LS or "SAB‐185" or SAB185 or JMB2002 or "JMB‐2002" or "TATX‐03" or TATX03 or "NOVOAB‐20" or NOVOAB20 or "NOVO‐AB‐20" or "ABP‐300" or ABP300 or MW05 or "MW‐05" or MW07 or "MW‐07" or ACmab1 or covimax or adimab or "CPI‐006" or CPI006 or ADG20 or "ADG 20" or diabod* or nanobod* or microbod* or monobod* or scFv* or "single‐chain" or "heavy chain" or HCAbs or SCAbs or nmAb* or "two mab" or "mabs" or "two nab" or "two nabs" or "ADG20" or "ADG 20" or "MAD0004J08" or "CT‐P‐63" or "CT‐P63" or AF47D11* or AbbVie47D11* or 47D11* or AbbVie2B04* or ABBV2B04* or 2B04* or "STI‐2099" or STI2099* or "covi‐drops" or covidrop* or REGN14256*)
AND
(random* OR placebo OR trial OR groups OR "phase 3" or "phase3" or p3 or "pIII")
Appendix 7. Search strategy: Cochrane Covid‐19 Study Register for platform trials
Cochrane COVID‐19 Study Register via the Cochrane Register of Studies crsweb.cochrane.org/
1 (adaptive clinical trial):PT AND INREGISTER 2 "master protocol" OR "master study" AND INREGISTER 3 "cohort multiple randomized controlled trial*" OR "cohort multiple randomised controlled trial*" OR "cmRCT" AND INREGISTER 4 (("multi‐factorial" OR multifactorial) NEAR7 (trial OR stud*)) AND INREGISTER 22 5 (network NEAR2 trial) AND INREGISTER 6 (network NEAR2 stud*):TI AND INREGISTER 7 (additional NEAR2 (arm* OR drug* OR agent* OR treatment* OR intervention*)): TI,AB AND INREGISTER 8 "candidate agents" AND INREGISTER 9 (new NEAR2 (arm* OR drug*)): TI,AB AND INREGISTER 10 (different NEXT (agent* OR drug* OR treatment*)): TI,AB AND INREGISTER 11 (platform NEAR2 protocol) AND INREGISTER 12 ("sub‐protocol" OR "subprotocol" OR "substudy" OR "sub‐study" OR "subtrial" OR "sub‐trial"):TI,AB AND INREGISTER 13 (multiple NEAR1 (agent* OR treatment* OR intervention*)): TI,AB AND INREGISTER 14 ("candidate specific trial" OR "specific trial protocol"):TI,AB AND INREGISTER 15 "add on":TI OR ("add on" NEAR1 (stud* OR trial OR therap* OR treatment OR intervention*)):TI,AB AND INREGISTER 16 (candicate* NEAR2 therap*):TI,AB AND INREGISTER 17 (arms NEAR2 add*):TI,AB AND INREGISTER 18 (multiplatform*):TI,AB AND INREGISTER 19 (flexible trial*):TI,AB AND INREGISTER 20 (adaptive NEAR7 (trial OR stud*)):TI,AB AND INREGISTER 21 (adaptive NEAR5 (design)) AND INREGISTER 22 #1 OR #2 OR #3 OR #4 OR #5 OR #6 OR #7 OR #8 OR #9 OR #10 OR #11 OR #12 OR #13 OR #14 OR #15 OR #16 OR #17 OR #18 OR #19 OR #20 OR #21
Data and analyses
Comparison 1. Tixagevimab/cilgavimab compared to placebo to prevent COVID‐19 (pre‐exposure prophylaxis).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1.1 Infection with SARS‐CoV‐2 within 6 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.2 Development of clinical COVID‐19 symptoms within 6 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.3 All‐cause mortality within 6 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.4 Admission to hospital within 6 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.5 Adverse events: all grade within 6 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.6 Serious adverse events within 6 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 2. Casirivimab/imdevimab compared to placebo to prevent COVID‐19 (pre‐exposure prophylaxis).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 2.1 Infection with SARS‐CoV‐2 within 6 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.2 Development of clinical COVID‐19 symptoms within 6 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.3 All‐cause mortality within 6 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.4 Adverse events: grade 3 to 4 within 6 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.5 Adverse events: all grade within 6 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.6 Serious adverse events within 6 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 3. Bamlanivimab compared to placebo to prevent COVID‐19 (postexposure prophylaxis).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 3.1 Infection with SARS‐CoV‐2 by day 30 | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3.2 All‐cause mortality by day 60 | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3.3 Adverse events: all grade | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3.4 Serious adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 4. Casirivimab/imdevimab compared to placebo to prevent COVID‐19 (postexposure prophylaxis).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 4.1 Infection with SARS‐CoV‐2 by day 30 | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4.2 Development of clinical COVID‐19 symptoms (broad‐term) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4.3 All‐cause mortality by day 30 | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4.4 Admission to hospital by day 30 | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4.5 Adverse events: grade 3 to 4 | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4.6 Adverse events: all grade | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4.7 Serious adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
BLAZE‐2.
| Study characteristics | ||
| Methods | Drug name: bamlanivimab Trial design: randomised, double‐blind, placebo‐controlled, multipart, single‐dose phase 3 trial Type of publication: journal publication (English) NCT number: NCT04497987 (date of trial registration: 4 August 2020) Number of participants:
Actual enrolment: 1374 participants Actual completion date: 20 May 2021 |
|
| Participants | Setting
Eligibility criteria
Participant characteristics
|
|
| Interventions | Intervention
Comparator
|
|
| Outcomes | Efficacy outcomes
Safety outcomes
Additional study outcomes
Definitions for COVID severitya: Mild: symptoms that could include fever, cough, sore throat, malaise, headache, muscle pain, gastrointestinal symptoms. No clinical signs indicative of moderate, severe, or critical severity Moderate: any symptom of mild illness or shortness of shortness of breath with exertion. Clinical signs suggestive of moderate illness with COVID‐19, such as
Severe: any symptom of moderate illness, shortness of breath at rest, or respiratory distress Clinical signs indicative of severe systemic illness with COVID‐19, such as
Critical: evidence of critical illness, defined by ≥ 1 of the following
aAdapted from FDA 2020. |
|
| Notes | Developer: Eli Lilly and Company, NIAID, COVID‐19 Prevention Network (CoVPN) Funding: Eli Lilly and Company, NIAID, COVID‐19 Prevention Network (CoVPN) Conflicts of interest
|
|
Isa 2021.
| Study characteristics | ||
| Methods | Drug name: casirivimab and imdevimab (REGEN‐COV) Trial design: randomised, double‐blind, placebo‐controlled, phase 1 trial Type of publication: preprint (English) NCT number: NCT04519437 (date of trial registration: 19 August 2020) Number of participants:
Actual enrolment: 974 participants Estimated study completion date: 25 October 2021 |
|
| Participants | Setting
Eligibility criteria
Participant characteristics
|
|
| Interventions | Intervention
Comparator
|
|
| Outcomes | Efficacy outcomes
Safety outcomes
Additional study outcomes
|
|
| Notes | Developer: Regeneron Pharmaceuticals, Inc, NIAID, NIH Funding: Regeneron Pharmaceuticals, Inc and F Hoffmann‐La Roche Ltd Conflicts of interest
|
|
O'Brien 2021.
| Study characteristics | ||
| Methods | Drug name: casirivimab and imdevimab (REGEN‐COV) Trial design: randomised, double‐blind, placebo‐controlled, 2‐part phase 3 trial (part A assessed) Type of publication: journal publication (English) for Part A NCT number: NCT04452318 (date of trial registration: 30 June 2020) Number of participants:
Actual enrolment: 3303 participants Actual completion date: 4 October 2021 |
|
| Participants | Setting
Eligibility criteria
Participant characteristics
|
|
| Interventions | Intervention
Comparator
|
|
| Outcomes | Efficacy outcomes
Safety outcomes
Additional study outcomes
Strict‐term definition: Fever (≥ 38 °C) plus ≥ 1 respiratory symptom (sore throat, cough, shortness of breath) OR 2 respiratory symptoms (sore throat, cough, shortness of breath) OR 1 respiratory symptom (sore throat, cough, shortness of breath) plus ≥ 2 non‐respiratory symptoms (chills, nausea, vomiting, diarrhoea, headache, conjunctivitis, myalgia, arthralgia, loss of taste or smell, fatigue or general malaise) Broad‐term definition: Fever ≥ 38 °C The signs and symptoms were feverish, sore throat, cough, shortness of breath/difficulty breathing (shortness of breath), chills, nausea, vomiting, diarrhoea, headache, red or watery eyes (conjunctivitis), body aches such as muscle pain or joint pain (myalgia, arthralgia), loss of taste/smell, fatigue (fatigue or general malaise or lethargy*), loss of appetite, confusion, dizziness, pressure/tightness in chest, chest pain, stomach ache (abdominal pain*), rash, sneezing, runny nose, sputum/phlegm, other *Signs and symptoms observed in children CDC definition: ≥ 2 of the following symptoms: fever (measured or subjective), chills, rigors, myalgia, headache, sore throat, nausea or vomiting, diarrhoea, fatigue, congestion, or runny nose OR Any 1 of the following symptoms: cough, shortness of breath, difficulty breathing, new olfactory disorder, or new taste disorder OR Severe respiratory illness with ≥ 1 of the following, clinical or radiographic evidence of pneumonia, or acute respiratory distress syndrome. |
|
| Notes | Developer: Regeneron Pharmaceuticals, Inc, NIAID, NIH Funding: Regeneron Pharmaceuticals, Inc and F Hoffmann‐La Roche Ltd Conflicts of interest
|
|
PROVENT.
| Study characteristics | ||
| Methods | Drug name: AZD7442 Trial design: randomised, placebo‐controlled, double‐blind, multicentre, single‐dose (× 2 IM injections), phase 3 study Type of publication: conference abstract NCT number: NCT04625725 (date of trial registration: 12 November 2020) Number of participants:
Actual enrolment: 5197 participants Estimated completion date: 30 November 2023 |
|
| Participants | Setting
Eligibility criteria
Participant characteristics
Definition of symptomatic COVID‐19 Participant must present with ≥ 1 of the following symptoms No minimum duration
Must be present for ≥ 2 days
Definition of severe COVID‐19 Severe COVID‐19 is characterised by a minimum of either pneumonia (fever, cough, tachypnoea, OR dyspnoea, AND lung infiltrates) or hypoxaemia (SpO2 < 90% in room air or severe respiratory distress, or both) and a WHO Clinical Progression Scale score of ≥ 5. |
|
| Interventions | Intervention Tixagevimab and cilgavimab (AZD7442)
Comparator
|
|
| Outcomes | Efficacy outcomes
Safety outcomes
Additional study outcomes
|
|
| Notes | Developer: AstraZeneca Funding: tixagevimab/cilgavimab (AZD7442) is being developed with support from the US Government; AstraZeneca, Iqvia Pty Ltd Conflicts of interest:
|
|
Ab: antibody; ACE: angiotensin‐converting enzyme; ADA: antidrug antibody; AE: adverse event; AESI: adverse events of special interest; ARB: angiotensin‐receptor blocker; AUC: area under the curve; BMI: body mass index; BP: blood pressure; CDC: Centers for Disease Control and Prevention; COPD: chronic obstructive pulmonary disease; COVID‐19: coronavirus disease 2019; EAP: efficacy assessment period; ECMO: extracorporeal membrane oxygenation; EUA: Emergency Use Authorization; hIVIG: human intravenous immunoglobulin; ICU: intensive care unit; Ig: immunoglobulin; IM: intramuscular; IMP: investigational medicinal product; IQR: interquartile range; ITT: intention‐to‐treat; IV: intravenous; LAR: legally authorised representative; mAb: monoclonal antibody; MERS: Middle East respiratory syndrome; NAT: nucleic acid test; NCT: National Clinical Trial; NIAID: National Institute of Allergy and Infectious Diseases; NIH: National Institutes of Health; NIV: non‐invasive ventilation; NR: not reported; NSAID: non‐steroidal anti‐inflammatory drug; NYHA: New York Heart Association; PaO2/FiO2: ratio of arterial partial pressure of oxygen to fraction of inspired oxygen; PCR: polymerase chain reaction; PEP: postexposure prophylaxis; PGIC: Patient Global Impression of Change of symptoms associated with COVID‐19; PGIS: Patient Global Impression of Severity of symptoms associated with COVID‐19; PrEP: pre‐exposure prophylaxis; RBD: receptor‐binding domain; RT‐PCR: reverse transcription polymerase chain reaction; RT‐qPCR: quantitative reverse transcription polymerase chain reaction; SAE: serious adverse event; SARS: severe acute respiratory syndrome; SARS‐CoV‐2: severe acute respiratory syndrome coronavirus 2; SC: subcutaneous; SD: standard deviation; SpO2: oxygen saturation; TEAE: treatment‐emergent adverse event; ULN: upper limit of normal; WHO: World Health Organization; WHOQOL‐100: World Health Organization Quality Of Life scale.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| ACTIV‐2 | SARS‐CoV‐2‐specific mAb examined for treatment. |
| ACTIV‐3 | SARS‐CoV‐2‐specific mAb examined for treatment. |
| BLAZE‐1 | SARS‐CoV‐2‐specific mAb examined for treatment. |
| BLAZE‐4 | SARS‐CoV‐2‐specific mAb examined for treatment. |
| COMET‐ICE | SARS‐CoV‐2‐specific mAb examined for treatment. |
| Eom 2021 | SARS‐CoV‐2‐specific mAb examined for treatment. |
| Kim 2021 | SARS‐CoV‐2‐specific mAb examined for treatment. |
| NCT04411628 | SARS‐CoV‐2‐specific mAb examined for treatment. |
| NCT04426695 | SARS‐CoV‐2‐specific mAb examined for treatment. |
| NCT04666441 | SARS‐CoV‐2‐specific mAb examined for treatment. |
| NCT04931238 | SARS‐CoV‐2‐specific mAb examined for treatment. |
| OPTIMISE‐C19 | SARS‐CoV‐2‐specific mAb examined for treatment. |
| RECOVERY | SARS‐CoV‐2‐specific mAb examined for treatment. |
| Weinreich | SARS‐CoV‐2‐specific mAb examined for treatment. |
mAb: monoclonal antibody; SARS‐CoV‐2: severe acute respiratory syndrome coronavirus 2.
Characteristics of studies awaiting classification [ordered by study ID]
CROWN CORONATION.
| Methods | Drug name: MR or M‐M‐R II vaccine Trial design: randomised, placebo‐controlled, Bayesian platform clinical trial NCT number: NCT04333732 (date of trial registration: 3 April 2020) Target sample size: 3545 participants Estimated completion date: 31 December 2021 |
| Participants | Setting
Eligibility criteria
|
| Interventions | Education and surveillance plus MR or M‐M‐R II vaccine
Comparator
|
| Outcomes | Efficacy outcomes
Safety outcomes
|
| Notes | Developer: Merck Funding: Washington University School of Medicine, COVID‐19 Therapeutics Accelerator Recruitment status: active, not recruiting |
NCT04894474.
| Methods | Drug name: BI 767551 Trial design: randomised, placebo‐controlled, double‐blind, parallel‐group, phase 2/3 study NCT number: NCT04894474 (date of trial registration: 20 May 2021) Target sample size: 0 participants (actual enrolment) Estimated completion date: 24 July 2022 |
| Participants | Setting
Eligibility criteria
|
| Interventions | BI 767551
Comparator:
|
| Outcomes | Efficacy outcomes
Safety outcomes
Additional study outcomes
|
| Notes | Developer: Boehringer Ingelheim Funding: Boehringer Ingelheim Recruitment status: withdrawn (project terminated) |
AE: adverse event; COVID‐19: coronavirus 2019; ICU: intensive care unit; IMP: investigational medicinal product; NCT: National Clinical Trial; RT‐PCR: reverse transcription polymerase chain reaction; SAE: serious adverse event; SARS‐CoV‐2: severe acute respiratory syndrome coronavirus 2; WHOQOL‐100: World Health Organization Quality Of Life scale.
Characteristics of ongoing studies [ordered by study ID]
NCT04625972.
| Study name | Phase III double‐blind, placebo‐controlled study of AZD7442 for post‐exposure prophylaxis of COVID‐19 in adults (STORM CHASER) |
| Methods | Drug name: tixagevimab/cilgavimab (AZD7442) Trial design: randomised, placebo‐controlled, double‐blind, multicentre, single‐dose, phase 3 study NCT number: NCT04625972 (date of trial registration: 12 November 2020) Target sample size: 1121 participants (actual enrolment) Estimated completion date: 19 June 2022 |
| Participants | Setting
Eligibility criteria
|
| Interventions | Intervention Tixagevimab/cilgavimab (AZD7442) (approximately 750 participants)
Comparator (approximately 375 participants)
|
| Outcomes | Efficacy outcomes
Safety outcomes
Additional study outcomes
|
| Starting date | 2 December 2020 |
| Contact information | Myron Levin, MD; AstraZeneca |
| Notes | Developer: AstraZeneca Funding: AstraZeneca, Iqvia Pty Ltd Recruitment status: active, not recruiting |
NCT04859517.
| Study name | Evaluation of ADG20 for the prevention of COVID‐19 (EVADE) |
| Methods | Drug name: ADG20 Trial design: randomised, placebo‐controlled, double‐blind, multicentre, phase 2/3 study NCT number: NCT04859517 (date of trial registration: 26 April 2021) Target sample size: 6412 participants (estimated enrolment) Estimated completion date: March 2023 |
| Participants | Setting
Eligibility criteria
|
| Interventions | ADG20
Comparator
|
| Outcomes | Efficacy outcomes
Safety outcomes
Additional study outcomes
|
| Starting date | 23 April 2021 |
| Contact information | Study inquiry +1 781‐819‐0080 ClinicalTrials@adagiotx.com |
| Notes | Developer: Adagio Therapeutics, Inc Funding: Adagio Therapeutics, Inc Recruitment status: active, not recruiting |
NCT05142527.
| Study name | Study to evaluate the safety and efficacy of a monoclonal antibody cocktail for the prevention of COVID‐19 |
| Methods | Drug name: ADM03820 Trial design: randomised, placebo‐controlled, double‐blind, multicentre, phase 2/3 study NCT number: NCT05142527 (date of trial registration: 2 December 2021) Target sample size: 4450 participants (estimated enrolment) Estimated completion date: August 2023 |
| Participants | Setting
Eligibility criteria
|
| Interventions |
ADM03820
Comparator
|
| Outcomes | Efficacy outcomes
Safety outcomes
Additional study outcomes
|
| Starting date | April 2022 |
| Contact information | Angie Kimbler; angie.kimbler@resilience.com |
| Notes | Developer: Ology Bioservices Funding: Ology Bioservices Recruitment status: not yet recruiting |
NCT05184062.
| Study name | A study to evaluate the safety and tolerability of AZD7442 in Chinese adults |
| Methods | Drug name: AZD7442 Trial design: randomised, placebo‐controlled, double‐blind, phase 2 study NCT number: NCT05184062 (date of trial registration: 11 January 2022) Target sample size: 272 participants (estimated enrolment) Estimated completion date: 30 June 2023 |
| Participants | Setting
Eligibility criteria
|
| Interventions | AZD7442
Comparator
|
| Outcomes | Efficacy outcomes
Safety outcomes
Additional study outcomes
|
| Starting date | 3 December 2021 |
| Contact information | NR |
| Notes | Developer: AstraZeneca Funding: AstraZeneca Recruitment status: active, not recruiting |
Ab: antibody; ACE: angiotensin‐converting enzyme; AE: adverse event; AESI: adverse event of special interest; BMI: body mass index; COVID‐19: coronavirus disease 2019; FSH: follicle‐stimulating hormone; ICU: intensive care unit; IM: intramuscular(ly); IMP: investigational medical product; IV: intravenous; IVIG: intravenous immunoglobulin; mAb: monoclonal antibody; MERS: Middle East respiratory syndrome; NCT: National Clinical Trial; NP: nasopharyngeal; NR: not reported; PCR: polymerase chain reaction; PI: principal investigator; RBD: receptor‐binding domain; RNA: ribonucleic acid; RT‐PCR: reverse transcription polymerase chain reaction; RT‐qPCR: quantitative reverse transcription polymerase chain reaction; SAE: severe adverse event; SARS: severe acute respiratory syndrome; AE: adverse event, SARS‐CoV‐2: severe acute respiratory syndrome coronavirus 2; TIG: tetanus‐specific immunoglobulin; ULN: upper limit of normal; VZIG: varicella zoster immunoglobulin.
Differences between protocol and review
Types of participants
Together with clinicians, we specified to include for participants receiving pre‐exposure prophylaxis (PrEP) regardless of serostatus and for participants receiving postexposure prophylaxis (PEP) with negative serostatus.
Types of interventions
We decided to perform separate analyses for SARS‐CoV‐2‐specific monoclonal antibodies (mAbs) for PrEP and PEP of COVID‐19 due to differences such as frequency of administration or insulation obligation.
Types of outcome measures
In a group discussion with clinical experts, we revised the definition of the outcome 'Development of clinical COVID‐19 symptoms, assessed with the World Health Organization Clinical Progression Scale (WHO 2020c), within 30 days' in due to the changing WHO definitions on clinical progression. We decided to use the definitions as provided by the studies. For adverse events, we have added adverse events grades 1 to 2 and grades 3 to 4.
Electronic searches
We stopped using the 'Similar articles' feature on PubMed, because we did not find new relevant references with this feature. We added new identified search terms such as ronapreve, casirivimab, imdevimab, regdanvimab, regkirona, CORAT, avian antibody, CT‐P63, ABBV‐2B04, MAD0004J08, STI‐2099, covi‐drop, REGN14256, ADG 20, MAD0004J08, C144‐LS, C‐135‐LS, SAB‐185, JMB2002, TATX‐03, NOVOAB‐20, ABP‐300, MW05, MW07, ACmab1, covimax, adimab, CPI‐006, ADG20, and AbbVie47D11. These search strings were changed: #16 ((cocktail* or mixture* or comb*) adj3 (mAb* or antibod* or nAb*)) change combination* to comb*; #19: REGN‐COV2* or REGN‐COV‐2* or REGEN‐COV2* or REGEN‐COV‐2* to REGN‐COV* or REGEN‐COV*. Search lines #44 ‐ # 47 were added. We stopped comparing our results with results from the websites www.covid-trials.org and chineseantibody.org/covid-19-track, because the tracker is out of date since April 2021.
Measures of treatment effect
We had planned to use Peto OR if the number of observed events was small (less than 5% of sample per group). However, for consistency throughout the review, better interpretability, and lacking impact on the findings, we decided to report risk ratios instead.
Assessment of reporting bias
As we did not pool the data in meta‐analysis and included fewer than 10 trials, we did not generate funnel plots to assess reporting bias.
Data synthesis
We had planned to pool the data in meta‐analysis if clinical and methodological characteristics of individual studies were sufficiently homogeneous. The included studies were too heterogeneous regarding different mAb types. We decided not to perform meta‐analyses and commented on the results instead.
Subgroup analysis and investigation of heterogeneity
The subgroup analyses planned to explore:
age of participants (divided into applicable age groups, e.g. children; 18 to 65 years; older than 65 years);
pre‐existing condition versus without any pre‐existing condition;
variants of SARS‐CoV‐2 detected in case of infection;
antibodies detected at baseline.
Due to the limited number of RCTs providing relevant data and the variation of SARS‐CoV‐2‐neutralising monoclonal antibodies used across the trials, we did not perform meta‐analyses and thus no subgroup analyses.
Summary of findings table
We had planned to only present summary of findings tables for the three most clinically important mAbs or mAb fragments. Remaining mAbs or mAb fragments, we would have presented as informal summary of findings tables in the Additional tables section. Since we only created two 'Summary of Findings' tables each for PrEP and PEP, each with two different mAb types, we did not provide any additional tables.
Contributions of authors
CH: screening, data extraction, risk of bias assessment, writing the manuscript.
YSP: data extraction, risk of bias assessment, and extraction of characteristics of studies.
VP: methodological expertise and advice.
KLC: screening, clinical expertise, and advice.
LJE: screening, clinical and methodological expertise, and advice.
SS: conception of the protocol and review.
IM: development of the search strategy.
EMW: clinical expertise and advice.
CS‐O: clinical expertise and advice.
ZM: clinical expertise and advice.
CS: clinical expertise and advice.
JM: clinical expertise and advice.
MS: clinical expertise and advice.
NS: screening, methodological expertise and advice, and conception of the protocol and review.
NK: screening, data extraction, risk of bias assessment, writing the manuscript.
Sources of support
Internal sources
-
University Hospital of Cologne, Germany
Cochrane Cancer, Department I of Internal Medicine
-
NHS Blood and Transplant, UK
NHS Blood and Transplant
-
Monash University Australia, Australia
Transfusion Research Unit, Department of Epidemiology and Preventive Medicine
-
Haematology Society of Australia and New Zealand, Australia
Leukaemia Foundation and HSANZ Australia
-
Charité – Universitätsmedizin Berlin, corporate member of Freie Universität Berlin and Humboldt‐Universität zu Berlin, Berlin, Germany, Germany
Department of Infectious Diseases and Respiratory Medicine
External sources
-
Federal Ministry of Education and Research, Germany
NaFoUniMedCovid19 (funding number: 01KX2021); part of the project 'COVIM'
-
CEOsys, Germany
The CEOsys project is funded under a scheme issued by the Network of University Medicine (Nationales Forschungsnetzwerk der Universitätsmedizin (NUM)) by the Federal Ministry of Education and Research of Germany (Bundesministerium für Bildung und Forschung (BMBF))
Declarations of interest
CH: is Managing Editor at Cochrane Haematology.
YSP: is a member of staff at Cochrane Haematology.
VP: is former Managing Editor at Cochrane Haematology.
KLC: none.
LJE: is a consultant haematologist for NHS Blood and Transplant (received funds or grants) and Co‐ordinating Editor of Cochrane Haematology.
SS: I have participated in a study funded by the Federal Ministry of Education and Research, Germany (NaFoUniMedCovid19, funding number: 01KX2021; part of the project "COVIM" on kinetics and correlates of the neutralising antibody response to SARS‐CoV‐2 infection in humans. (DOI:10.1016/j.chom.2021.04.015)). In this review, I was not involved in risk of bias assessment, data extraction, or interpretation, but served as content expert.
IM: is Information Specialist at Cochrane Haematology.
EMW: none.
CS‐O: has declared to be employed by the not‐for‐profit Sanquin Blood Bank; the Blood Bank provides the plasma as raw material for the production of hyperIG by another division of Sanquin named Prothya.
ZM: is a haematologist at Monash University.
CDS: reports grants and personal fees from AstraZeneca, Janssen‐Cilag, MSD, and ViiV Healthcare; grants from Cepheid; grants, personal fees, and non‐financial support from Gilead Sciences; other from Apeiron and Eli Lilly; personal fees and non‐financial support from BBraun Melsungen; personal fees from AbbVie, BioNtech, Eli Lilly, Formycon, GSK, Molecular partners, MSD; Roche; SOBI during the conduct of the study, and Synairgen, outside the submitted work.
JJM: is an independent contractor for Atriva Therapeutics GMBH (received funds) and Gilead Sciences (received funds and travel).
MS: is an Infectious Diseases specialist at Charité University Hospital Berlin, Germany.
NS: none known; she is Co‐ordinating Editor of Cochrane Haematology, but was not involved in the editorial process for this review.
NK: is a member of staff at Cochrane Haematology.
The authors CH, YSP, VP, LJE, NS, and NK are affiliated with Cochrane Haematology but are not otherwise involved with the editorial process.
New
References
References to studies included in this review
BLAZE‐2 {published data only}
- Cohen MS, Nirula A, Mulligan M, Novak R, Marovich M, Stemer A, et al. Bamlanivimab prevents COVID-19 morbidity and mortality in nursing-home setting, 2021. www.croiconference.org/abstract/bamlanivimab-prevents-covid-19-morbidity-and-mortality-in-nursing-home-setting/ (accessed 1 June 2022).
- Cohen MS, Nirula A, Mulligan MJ, Novak RM, Marovich M, Yen C, et al. Effect of bamlanivimab vs placebo on incidence of COVID-19 among residents and staff of skilled nursing and assisted living facilities: a randomized clinical trial. JAMA 2021;326(1):46-55. [DOI: 10.1001/jama.2021.8828] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knorr J, Tuttle JL, Sabo JA, East DH, Price KL, Shen L. Innovative clinical trial design and delivery: a phase 3 COVID-19 post-exposure prophylaxis study in skilled nursing and assisted living facilities (BLAZE-2). Trials 2021;22:726. [DOI: 10.1186/s13063-021-05699-3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Isa 2021 {published data only}
- Isa F, Forleo-Neto E, Meyer J, Zheng W, Rasmussen S, Armas D, et al. Repeat subcutaneous administration of REGEN-COV in adults is well-tolerated and prevents the occurrence of COVID-19. medRxiv [Preprint] 2021. [DOI: 10.1101/2021.11.10.21265889] [DOI] [PMC free article] [PubMed]
O'Brien 2021 {published data only}
- Hassan H, Turner K, Davis J, Ganguly S, Irvin S, Partridge M, et al. P-071: pharmacokinetics and immunogenicity assessment of a single subcutaneous dose of casirivimab and imdevimab in household contacts of SARS-CoV-2 infected persons. American Society for Clinical Pharmacology and Therapeutics 111;-:S5-S80. [DOI: 10.1002/cpt.2521] [DOI] [Google Scholar]
- O'Brien M, Forleo-Neto E, Chen KC, Isa F, Heimann I, Sarkar N, et al. Casirivimab with imdevimab antibody cocktail for COVID-19 prevention: interim results, 2021. www.croiconference.org/abstract/casirivimab-with-imdevimab-antibody-cocktail-for-covid-19-prevention-interim-results/ (accessed 1 June 2022):abstract no. 123.
- O'Brien M, Forleo-Neto E, Musser B, Isa F, Chan KC, Sarkar N, et al. Subcutaneous REGEN-COV antibody combination for Covid-19 prevention. medRxiv [Preprint] 2021. [DOI: 10.1101/2021.06.14.21258567] [DOI]
- O'Brien M, Forleo-Neto E, Musser BJ, Isa F, Chan KC, Sarkar N, et al. COVID-19 prevention with subcutaneous administration of the monoclonal antibodies casirivimab and imdevimab: subgroup analysis in participants with cardiovascular disease and diabetes. American Heart Journal 2021;242:172-173, abstract no. 099. [DOI: 10.1016/j.ahj.2021.10.067] [DOI] [Google Scholar]
- O'Brien M, Forleo-Neto E, Sarkar N, Isa F, Hou P, Chan KC, et al. Subcutaneous REGEN-COV antibody combination in early SARS-CoV-2 infection. medRxiv [Preprint] 2021. [DOI: 10.1101/2021.06.14.21258569] [DOI]
- O'Brien MP, Forleo-Neto E, Sarkar N, Isa F, Hou P, Chan KC, et al. Effect of subcutaneous casirivimab and imdevimab antibody combination vs placebo on development of symptomatic COVID-19 in early asymptomatic SARS-CoV-2 infection: a randomized clinical trial. JAMA 2022;327(5):432-41. [DOI: 10.1001/jama.2021.24939] [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien M, Forleo-Neto E, Musser B, Isa F, Chan KC, Sarkar N, et al. Subcutaneous REGEN-COV antibody combination to prevent Covid-19. New England Journal of Medicine 2021;385:1184-95. [DOI: 10.1056/NEJMoa2109682] [DOI] [PMC free article] [PubMed] [Google Scholar]
PROVENT {published data only}
- American Medical Association. Tixagevimab and cilgavimab (Evusheld) for pre-exposure prophylaxis of COVID-19. JAMA 2022;327(4):384-5. [DOI: 10.1001/jama.2021.24931] [DOI] [PubMed] [Google Scholar]
- Levin MJ, Ustianowski A, De Wit S, Launay O, Avila M, Seegobin S, et al. LB5. PROVENT: Phase 3 study of efficacy and safety of AZD7442 (tixagevimab/cilgavimab) for pre-exposure prophylaxis of COVID-19 in adults. Open Forum Infectious Diseases 2021;8(Suppl 1):S810. [DOI: 10.1093/ofid/ofab466.1646] [DOI]
- Levin MJ, Ustianowski A, De Wit S, Launay O, Avila M, Templeton A, et al. Intramuscular AZD7442 (tixagevimab–cilgavimab) for prevention of Covid-19. New England Journal of Medicine 2022;386(23):2188-200. [DOI: 10.1056/NEJMoa2116620] [DOI] [PMC free article] [PubMed] [Google Scholar]
References to studies excluded from this review
ACTIV‐2 {published data only}
- Chew KW, Moser C, Daar ES, Wohl DA, Li JZ, Coombs R, et al. Bamlanivimab reduces nasopharyngeal SARS-CoV-2 RNA levels but not symptom duration in non-hospitalized adults with COVID-19. medrxiv [Preprint] 2021. [DOI: 10.1101/2021.12.17.21268009] [DOI]
- Evering TH, Giganti M, Chew KW, Hughes M, Moser C, Wohl DA, et al. LB2. Safety and efficacy of combination SARS-CoV-2 monoclonal neutralizing antibodies (mAb) BRII-196 and BRII-198 in non-hospitalized COVID-19 patients. Open Forum Infectious Diseases 2021;8:S807-8. [DOI: ] [Google Scholar]
- Evering TH, Giganti M, Chew KW, Hughes M, Moser C, Wohl DA, et al. LB2. Safety and efficacy of combination SARS-CoV-2 monoclonal neutralizing antibodies (mAb) BRII-196 and BRII-198 in non-hospitalized COVID-19 patients. Open Forum Infectious Diseases 2021;8:S807-8. [DOI: 10.1093/ofid/ofab466.1643] [DOI] [Google Scholar]
- Evering TH, Sanusi B, Jilg N, Yeh E, Moser C, Ritz J, et al. ABSTRACT O01. Prevalence and characteristics of post-acute sequelae of SARS-CoV-2 (PASC) in non-hospitalized persons with COVID-19 enrolled in a clinical trial of early treatment (ACTIV-2). Antiviral Therapy 2021;-:1-51. [DOI: 10.1177/13596535211063242] [DOI] [Google Scholar]
ACTIV‐3 {published data only}
- ACTIV-3/Therapeutics for Inpatients with COVID-19 (TICO) Study Group. Efficacy and safety of two neutralising monoclonal antibody therapies, sotrovimab and BRII-196 plus BRII-198, for adults hospitalised with COVID-19 (TICO): a randomised controlled trial. Lancet Infectious Diseases 2022;22:622-35. [DOI: 10.1016/ S1473-3099(21)00751-9] [DOI] [PMC free article] [PubMed] [Google Scholar]
- ACTIV-3/Therapeutics for Inpatients with COVID-19 (TICO) Study Group. Responses to a neutralizing monoclonal antibody for hospitalized patients with COVID-19 according to baseline antibody and antigen levels: a randomized controlled trial. Annals of Internal Medicine 2022;175:234-43. [DOI: 10.7326/M21-3507] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundgren JD, Grund B, Barkauskas CE, Holland TL, Gottlieb RL, Sandkovsky SM, et al. A neutralizing monoclonal antibody for hospitalised patients with COVID-19. New England Journal of Medicine 2021;384:905-14. [DOI: 10.1056/NEJMoa2033130] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundgren JD, Grund B, Barkauskas CE, Holland TL, Gottlieb RL, Sandkovsky U, et al. Clinical and virological response to a neutralizing monoclonal antibody for hospitalized patients with COVID-19. medrxiv [Preprint] 2021. [DOI: 10.1101/2021.07.19.21260559] [DOI] [PMC free article] [PubMed]
BLAZE‐1 {published data only}
- Chen P, Nirula A, Heller B, Gottlieb RL, Boscia J, Morris J, et al. SARS-CoV-2 neutralizing antibody LY-CoV555 in outpatients with COVID-19. New England Journal of Medicine 2020;384:229-37. [DOI: 10.1056/NEJMoa2029849] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougan M, Azizad M, Mocherla B, Gottlieb RL, Chen P, Hebert C, et al. A randomized, placebo-controlled clinical trial of bamlanivimab and etesevimab together in high-risk ambulatory patients with COVID-19 and validation of the prognostic value of persistently high viral load. Clinical Infectious Diseases 2021 Oct 28 [Epub ahead of print]. [DOI: 10.1093/cid/ciab912] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougan M, Nirula A, Azizad M, Mocherla B, Gottlieb RL, Chen P, et al. Bamlanivimab plus etesevimab in mild or moderate COVID-19. New England Journal of Medicine 2021;385(15):1382-92. [DOI: 10.1056/NEJMoa2102685] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougan M, Nirula A, Gottlieb RL, Azizad M, Mocherla B, Chen P, et al. Bamlanivimab + etesevimab for treatment of COVID-19 in high-risk ambulatory patients. Topics in Antiviral Medicine 2021;29(1):33, abstract no. 122. [Google Scholar]
- Gottlieb RL, Nirula A, Chen P, Boscia J, Heller B, Morris J, et al. Effect of bamlanivimab as monotherapy or in combination with etesevimab on viral load in patients with mild to moderate COVID-19: a randomized clinical trial. JAMA 2021;325(7):632-44. [DOI: 10.1001/jama.2021.0202] [DOI] [PMC free article] [PubMed] [Google Scholar]
BLAZE‐4 {published data only}
- Dougan M, Azizad M, Chen P, Feldman B, Frieman M, Igbinadolor A, et al. Bebtelovimab, alone or together with bamlanivimab and etesevimab, as a broadly neutralizing monoclonal antibody treatment for mild to moderate, ambulatory COVID-19. medrxiv [Preprint] 2022. [DOI: 10.1101/2022.03.10.22272100] [DOI]
COMET‐ICE {published data only}
- Gupta A, Gonzalez-Rojas Y, Juarez E, Crespo Casal M, Moya J, Falci DR, et al. Early treatment for Covid-19 with SARS-CoV-2 neutralizing antibody sotrovimab. New England Journal of Medicine 2021;385(21):1941-50. [DOI: 10.1056/NEJMoa2107934] [DOI] [PubMed] [Google Scholar]
- Gupta A, Gonzalez-Rojas Y, Juarez E, Crespo Casal M, Moya J, Falci DR, et al. Effect of the neutralizing SARS-CoV-2 antibody sotrovimab in preventing progression of COVID-19: a randomized clinical trial. medRxiv [Preprint] 2021. [DOI: 10.1101/2021.11.03.21265533] [DOI]
- Gupta A, Gonzalez-Rojas Y, Juarez E, Crespo Casal M, Moya J, Rodrigues Falci D, et al. Early COVID-19 treatment with SARS-CoV-2 neutralizing antibody sotrovimab. medRxiv [Preprint] 2021. [DOI: 10.1101/2021.05.27.21257096] [DOI] [PubMed]
- Gupta AK, Rojas YG, Juarez E, Casal MC, Moya J, Falci DR, et al. 502. Early COVID-19 treatment with SARS-CoV-2 neutralizing antibody sotrovimab. Open Forum Infectious Diseases 2021;8:S353-4. [DOI: 10.1093/ofid/ofab466.701] [DOI] [Google Scholar]
Eom 2021 {published data only}
- Eom JS, Ison M, Streinu-Cercel A, Săndulescu O, Preotescu LL, Kim YS, et al. Efficacy and safety of CT-P59 plus standard of care: a phase 2/3 randomized, double-blind, placebo-controlled trial in outpatients with mild-to-moderate SARS-CoV-2 infection [Preprint]. researchsquare.com/article/rs-296518/v1 (accessed 1 June 2022). [DOI: 10.21203/rs.3.rs-296518/v1] [DOI]
- Ison MG, Kim JY, Sandulescu O, Preotescu LL, Rivera Martinez NE, Dobryanska M, et al. Therapeutic effect of regdanvimab in patients with mild to moderate COVID-19: day 28 results from a multicentre, randomised, controlled pivotal trial. Open Forum Infectious Diseases 2021;8:S375. [DOI: 10.1093/ofid/ofab466.745] [DOI] [Google Scholar]
- Streinu-Cercel A, Săndulescu O, Preotescu LL, Kim JY, Kim YS, Cheon S, et al. Efficacy and safety of regdanvimab (CT-P59): a phase 2/3 randomized, double-blind, placebo-controlled trial in outpatients with mild-to-moderate coronavirus disease 2019. Open Forum Infectious Diseases 2022;9:ofac053. [DOI: 10.1093/ofid/ofac053] [DOI] [PMC free article] [PubMed] [Google Scholar]
Kim 2021 {published data only}
- Kim JY, Jang YR, Hong JH, Jung JG, Park J-H, Streinu-Cercel A, et al. Safety, virologic efficacy, and pharmacokinetics of CT-P59, a neutralizing monoclonal antibody against SARS-CoV-2 spike receptor-binding protein: two randomized, placebo-controlled, phase I studies in healthy individuals and patients with mild SARS-CoV-2 infection. Clinical Therapeutics 2021;43(10):1706-27. [DOI: 10.1016/j.clinthera.2021.08.009] [DOI] [PMC free article] [PubMed] [Google Scholar]
NCT04411628 {published data only}
- Chen P, Datt G, Li YG, Chien J, Price K, Chigutsa E, et al. First-in-human study of bamlanivimab in a randomized trial of hospitalized patients with COVID- 19. Clinical Pharmacology & Therapeutics 2021;110(6):1467-77. [DOI: 10.1002/cpt.2405] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims JT, Poorbaugh J, Chang CY, Holzer T, Zhang L, Engle SM, et al. Relationship between gene expression patterns from nasopharyngeal swabs and serum biomarkers in patients hospitalized with COVID-19, following treatment with the neutralizing monoclonal antibody bamlanivimab. Journal of Translational Medicine 2022;20(1):134. [DOI: 10.1186/s12967-022-03345-3] [DOI] [PMC free article] [PubMed] [Google Scholar]
NCT04426695 {published data only}
- Mylonakis E, Somersan-Karakaya S, Sivapalasingam S, Ali S, Sun Y, Bhore R, et al. LB4. Casirivimab and imdevimab for treatment of hospitalized patients with COVID-19 receiving low flow or no supplemental oxygen. Open Forum Infectious Diseases 2021;8:S809-10. [DOI: ] [Google Scholar]
- Somersan-Karakaya S, Mylonakis E, Menon VP, Wells JC, Ali S, Sivapalasingam S, et al. REGEN-COV® for treatment of hospitalized patients with Covid-19. medRxiv [Preprint] 2021. [DOI: 10.1101/2021.11.05.21265656] [DOI]
- Turner K, Davis J, Ganguly H, Hassan H, Irvin S, Partridge M. P-182. Pharmacokinetics and pharmacodynamics of casirivimab and imdevimab in hospitalized patients with COVID-19. American Society for Clinical Pharmacology and Therapeutics 2022;111:S5-80. [DOI: 10.1002/cpt.2521] [DOI] [Google Scholar]
NCT04666441 {published data only}
- Davis J, Turner K, Ganguly S, Hassan H, Irvin S, Partridge M, et al. P-049. Pharmacokinetics and pharmacodynamics of casirivimab and imdevimab in a dose-ranging study in outpatients with COVID-19. American Society for Clinical Pharmacology and Therapeutics 2022;111:S5-80. [DOI: 10.1002/cpt.2521] [DOI] [Google Scholar]
- Portal-Celhay C, Forleo-Neto E, Eagan W, Musser BJ, Davis JD, Turner KC, et al. Phase 2 dose-ranging study of the virologic efficacy and safety of the combination COVID-19 antibodies casirivimab and imdevimab in the outpatient setting. medRxiv [Preprint] 2021. [DOI: 10.1101/2021.11.09.21265912] [DOI]
NCT04931238 {published data only}
- Dong R, Jiang L, Yang T, Wang C, Zhang Y, Chen X, et al. Efficacy and safety of SARS-CoV-2 neutralizing antibody JS016 in hospitalized Chinese patients with COVID-19: a phase 2/3, multicenter, randomized, open-label, controlled trial. Antimicrobial Chemotherapy 2022;66(3):e02045-21. [DOI: 10.1128/aac.02045-21] [DOI] [PMC free article] [PubMed] [Google Scholar]
OPTIMISE‐C19 {published data only}
- Huang ST, McCreary EK, Bariola JR, Minnier TE, Wadas RJ, Shovel JA, et al. Effectiveness of casirivimab and imdevimab, and sotrovimab during Delta variant surge: a prospective cohort study and comparative effectiveness randomized trial. medRxiv [Preprint] 2021. [DOI: 10.1101/2021.12.23.21268244] [DOI] [PMC free article] [PubMed]
- Huang T, McCreary EK, Bariola JR, Wadas RJ, Kip KE, Marroquin OC, et al. The UPMC OPTIMISE-C19 (OPtimizing Treatment and Impact of Monoclonal antIbodieS through Evaluation for COVID-19) trial: a structured summary of a study protocol for an open-label, pragmatic, comparative effectiveness platform trial with response-adaptive randomization. Trials 2021;22:363. [DOI: 10.1186/s13063-021-05316-3] [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCreary EK, Bariola JR, Minnier T, Wadas RJ, Shovel JA, Albin D, et al. A learning health system randomized trial of monoclonal antibodies for Covid-19. medRxiv [Preprint] 2021. [DOI: 10.1101/2021.09.03.21262551] [DOI]
- McCreary EK, Bariola JR, Minnier T, Wadas RJ, Shovel JA, Albin D, et al. Launching a comparative effectiveness adaptive platform trial of monoclonal antibodies for COVID-19 in 21 days. Contemporary Clinical Trials 2022;113:106652. [DOI: 10.1016/j.cct.2021.106652] [DOI] [PMC free article] [PubMed] [Google Scholar]
RECOVERY {published data only}
- Horby PW, Mafham M, Peto L, Campbell M, Pessoa-Amorim G, Spata E, et al. Casirivimab and imdevimab in patients admitted to hospital with COVID-19 (RECOVERY): a randomised, controlled, open-label, platform trial. medRxiv [Preprint] 2021. [DOI: 10.1101/2021.06.15.21258542] [DOI]
Weinreich {published data only}
- Ganguly S, Turner K, Davis J, Hassan H, Irvin S, Partridge M, et al. P-061. Pharmacokinetics and pharmacodynamics of casirivimab and imdevimab in outpatients with COVID-19. American Society for Clinical Pharmacology and Therapeutics 2022;111:S5-80. [DOI: 10.1002/cpt.2521] [DOI] [Google Scholar]
- Weinreich DM, Sivapalasingam S, Norton T, Ali S, Gao H, Bhore R, et al. REGEN-COV antibody cocktail clinical outcomes study in COVID-19 outpatients. medRxiv [Preprint]. [DOI: 10.1101/2021.05.19.21257469] [DOI]
- Weinreich DM, Sivapalasingam S, Norton T, Ali S, Gao H, Bhore R, et al. REGEN-COV antibody cocktail in outpatients with COVID-19. medRxiv [Preprint] 2021. [DOI: 10.1101/2021.06.09.21257915] [DOI]
- Weinreich DM, Sivapalasingam S, Norton T, Ali S, Gao H, Bhore R, et al. REGEN-COV antibody combination and outcomes in outpatients with Covid-19. New England Journal of Medicine 2021;385(23):e81. [DOI: 10.1056/NEJMoa2108163] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinreich DM, Sivapalasingam S, Norton T, Ali S, Gao H, Bhore R, et al. REGN-COV2, a neutralizing antibody cocktail, in outpatients with COVID-19. New England Journal of Medicine 2020;384:238-51. [DOI: 10.1056/NEJMoa2035002] [DOI] [PMC free article] [PubMed] [Google Scholar]
References to studies awaiting assessment
CROWN CORONATION {published data only}EUCTR2020‐001402‐38‐GBISRCTN99916292PACTR202101794986980
- NCT04333732. CROWN CORONATION: COVID-19 research outcomes worldwide network for CORONAvirus prevenTION (CROWN CORONA). clinicaltrials.gov/ct2/show/NCT04333732 (first received 3 April 2020).
NCT04894474 {published data only}
- NCT04894474. A study to test whether BI 767551 can prevent COVID-19 in people who have been exposed to SARS-CoV-2. clinicaltrials.gov/ct2/show/NCT04894474 (first received 20 May 2021).
References to ongoing studies
NCT04625972 {published data only}
- NCT04625972. Phase III double-blind, placebo-controlled study of AZD7442 for post-exposure prophylaxis of COVID-19 in adults (STORM CHASER). clinicaltrials.gov/ct2/show/record/NCT04625972 (first received 12 November 2020).
NCT04859517 {published data only}
- NCT04859517. Evaluation of ADG20 for the prevention of COVID-19 (EVADE). clinicaltrials.gov/ct2/show/record/NCT04859517 (first received 26 April 2021).
NCT05142527 {published data only}
- NCT05142527. Study to evaluate the safety and efficacy of a monoclonal antibody cocktail for the prevention of COVID-19. clinicaltrials.gov/ct2/show/NCT05142527 (first received 2 December 2021).
NCT05184062 {published data only}
- NCT05184062. A study to evaluate the safety and tolerability of AZD7442 in Chinese adults. clinicaltrials.gov/ct2/show/NCT05184062 (first received 11 January 2022).
Additional references
Alsoussi 2020
- Alsoussi WB, Turner JS, Case JB, Zhao H, Schmitz AJ, Zhou JQ, et al. A potently neutralizing antibody protects mice against SARS-CoV-2 infection. Journal of Immunology 2020;205(4):519-22. [DOI: 10.4049/jimmunol.2000583] [DOI] [PMC free article] [PubMed] [Google Scholar]
Andabaka 2013
- Andabaka T, Nickerson JW, Rojas-Reyes MX, Rueda JD, Bacic Vrca V, Barsic B. Monoclonal antibody for reducing the risk of respiratory syncytial virus infection in children. Cochrane Database of Systematic Reviews 2013, Issue 4. Art. No: CD006602. [DOI: 10.1002/14651858.CD006602.pub4] [DOI] [PubMed] [Google Scholar]
Andrews 2022
- Andrews N, Stowe J, Kirsebom F, Toffa S, Rickeard T, Gallagher E, et al. Covid-19 vaccine effectiveness against the Omicron (B.1.1.529) variant. New England Journal of Medicine 2022;386(16):1532-46. [DOI: 10.1056/NEJMoa2119451] [DOI] [PMC free article] [PubMed] [Google Scholar]
AstraZeneca 2020
- AstraZeneca. AstraZeneca's COVID-19 vaccine authorised for emergency supply in the UK. www.astrazeneca.com/media-centre/press-releases/2020/astrazenecas-covid-19-vaccine-authorised-in-uk.html (accessed 12 January 2021).
Baum 2020a
- Baum A, Ajithdoss D, Copin R, Zhou A, Lanza K, Negron N, et al. REGN-COV2 antibodies prevent and treat SARS-CoV-2 infection in rhesus macaques and hamsters. Science 2020;370(6520):1110-5. [DOI: 10.1126/science.abe2402] [DOI] [PMC free article] [PubMed] [Google Scholar]
Baum 2020b
- Baum A, Fulton BO, Wloga E, Copin R, Pascal KE, Russo V, et al. Antibody cocktail to SARS-CoV-2 spike protein prevents rapid mutational escape seen with individual antibodies. Science 2020;369(6506):1014-8. [DOI: 10.1126/science.abd0831] [DOI] [PMC free article] [PubMed] [Google Scholar]
Bayer 2019
- Bayer V. An overview of monoclonal antibodies. Seminars in Oncology Nursing 2019;35(5):150927. [DOI: 10.1016/j.soncn.2019.08.006] [DOI] [PubMed] [Google Scholar]
Boulware 2020
- Boulware DR, Pullen MF, Bangdiwala AS, Pastick KA, Lofgren SM, Okafor EC, et al. A randomized trial of hydroxychloroquine as postexposure prophylaxis for COVID-19. New England Journal of Medicine 2020;383(6):517-25. [DOI: 10.1056/NEJMoa2016638] [DOI] [PMC free article] [PubMed] [Google Scholar]
Bruel 2022
- Bruel T, Hadjadj J, Maes P, Planas D, Seve A, Staropoli I, et al. Serum neutralization of SARS-CoV-2 Omicron sublineages BA.1 and BA.2 in patients receiving monoclonal antibodies. Nature Medicine 2022 Mar 23 [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
Buitrago‐Garcia 2020
- Buitrago-Garcia D, Egli-Gany D, Counotte MJ, Hossmann S, Imeri H, Ipekci AM, et al. Occurrence and transmission potential of asymptomatic and presymptomatic SARS-CoV-2 infections: a living systematic review and meta-analysis. PLoS Medicine 2020;17(9):e1003346. [DOI: 10.1371/journal.pmed.1003346] [DOI] [PMC free article] [PubMed] [Google Scholar]
CDC 2021a
- Centers for Disease Control and Prevention. COVIDView – a weekly surveillance summary of U.S. COVID-19 activity (Key updates for week 4). www.cdc.gov/coronavirus/2019-ncov/covid-data/covidview/index.html (accessed 8 December 2021).
CDC 2021b
- Centers for Disease Control and Prevention. Symptoms of COVID-19. www.cdc.gov/coronavirus/2019-ncov/symptoms-testing/symptoms.html (accessed 8 December 2021).
CDC 2021c
- Centers for Disease Control and Prevention. Interim clinical considerations for use of COVID-19 vaccines currently approved or authorized in the United States. www.cdc.gov/vaccines/covid-19/clinical-considerations/covid-19-vaccines-us.html (accessed 8 December 2021).
Chen 2020
- Chen L, Xiong J, Bao L, Shi Y. Convalescent plasma as a potential therapy for COVID-19. Lancet Infectious Diseases 2020;20(4):398-400. [DOI] [PMC free article] [PubMed] [Google Scholar]
COMET 2020
- Core Outcome Measures in Effectiveness Trials (COMET). Core outcome set developers’ response to COVID-19. www.comet-initiative.org/Studies/Details/1538 (accessed 9 February 2021).
Copin 2021
- Copin R, Baum A, Wloga E, Pascal KE, Giordano S, O Fulton B, et al. The monoclonal antibody combination REGEN-COV protects against SARS-CoV-2 mutational escape in preclinical and human studies. Cell 2021;184(15):3949-61.e11. [DOI: 10.1016/j.cell.2021.06.002] [DOI] [PMC free article] [PubMed] [Google Scholar]
Covidence [Computer program]
- Veritas Health Innovation Covidence. Melbourne, Australia: Veritas Health Innovation, accessed after 21 December 2020. Available at covidence.org.
Custódio 2020
- Custódio TF, Das H, Sheward DJ, Hanke L, Pazicky S, Pieprzyk S. Selection, biophysical and structural analysis of synthetic nanobodies that effectively neutralize SARS-CoV-2. Nature Communications 2020;11:5588. [DOI: 10.1038/s41467-020-19204-y] [DOI] [PMC free article] [PubMed] [Google Scholar]
Deeks 2021
- Deeks JJ, Higgins JP, Altman DG. Chapter 10: Analysing data and undertaking meta-analyses. In: Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, et al, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 6.2 (updated February 2021). Cochrane, 2021. Available from training.cochrane.org/handbook/archive/v6.2.
de la Rica 2020
- la Rica R, Borges M, Gonzalez-Freire M. COVID-19: in the eye of the cytokine storm. Frontiers in Immunology 2020;11:2313. [DOI: 10.3389/fimmu.2020.558898] [DOI] [PMC free article] [PubMed] [Google Scholar]
Di Fusco 2021
- Di Fusco M, Moran MM, Cane A, Curcio D, Khan F, Malhotra D. Evaluation of COVID-19 vaccine breakthrough infections among immunocompromised patients fully vaccinated with BNT162b2. Journal of Medical Economics 2021;24(1):1248-60. [DOI: 10.1080/13696998.2021.2002063] [DOI] [PubMed] [Google Scholar]
Driggin 2020
- Driggin E, Madhavan Mahesh V, Bikdeli B, Chuich T, Laracy J, Biondi-Zoccai G, et al. Cardiovascular considerations for patients, health care workers, and health systems during the COVID-19 pandemic. Journal of the American College of Cardiology 2020;75(18):2352-71. [DOI: 10.1016/j.jacc.2020.03.031] [DOI] [PMC free article] [PubMed] [Google Scholar]
Eldridge 2021
- Eldridge S, Campbell M, Campbell M, Dahota A, Giraudeau B, Reeves B, et al. Revised Cochrane risk of bias tool for randomized trials (RoB 2.0). Additional considerations for cluster-randomized trials (RoB 2 CRT). www.riskofbias.info/welcome/rob-2-0-tool/rob-2-for-cluster-randomized-trials 2021.
Endnote X9 [Computer program]
- EndNote X9. EndNote. Clarivate, 2013.
FDA 2020
- US Food and Drug Administration. COVID-19: developing drugs and biological products for treatment or prevention guidance for industry. www.fda.gov/media/137926/download (accessed 7 June 2022).
FDA 2021a
- US Food and Drug Administration. Emergency Use Authorization – Coronavirus Disease 2019 (COVID-19) EUA information. www.fda.gov/emergency-preparedness-and-response/mcm-legal-regulatory-and-policy-framework/emergency-use-authorization#coviddrugs (accessed 9 December 2021). [PubMed]
FDA 2021b
- US Food and Drug Administration. Coronavirus (COVID-19) update: FDA authorizes new long-acting monoclonal antibodies for pre-exposure prevention of COVID-19 in certain individuals. www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-authorizes-new-long-acting-monoclonal-antibodies-pre-exposure (accessed 20 April 2022).
FDA 2021c
- US Food and Drug Administration. COVID-19 vaccines. www.fda.gov/emergency-preparedness-and-response/coronavirus-disease-2019-covid-19/covid-19-vaccines (accessed 12 January 2021).
FDA 2021d
- US Food and Drug Administration. Coronavirus (COVID-19) update: FDA authorizes first oral antiviral for treatment of COVID-19. www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-authorizes-additional-oral-antiviral-treatment-covid-19-certain (accessed 20 April 2022).
FDA 2021e
- US Food and Drug Administration. Coronavirus (COVID-19) update: FDA authorizes additional oral antiviral for treatment of COVID-19 in certain adults. www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-authorizes-additional-oral-antiviral-treatment-covid-19-certain (accessed 20 April 2022).
FDA 2021f
- US Food and Drug Administration. Coronavirus (COVID-19) update: FDA revokes Emergency Use Authorization for monoclonal antibody bamlanivimab. www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-revokes-emergency-use-authorization-monoclonal-antibody-bamlanivimab (accessed 20 April 2022).
FDA 2022a
- US Food and Drug Administration. Coronavirus (COVID-19) update: FDA limits use of certain monoclonal antibodies to treat COVID-19 due to the Omicron variant. www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-limits-use-certain-monoclonal-antibodies-treat-covid-19-due-omicron (accessed 20 April 2022).
FDA 2022b
- US Food and Drug Administration. FDA updates Sotrovimab Emergency Use Authorization. www.fda.gov/drugs/drug-safety-and-availability/fda-updates-sotrovimab-emergency-use-authorization (accessed 20 April 2022).
FDA 2022c
- US Food and Drug Administration. Coronavirus (COVID-19) update: FDA authorizes new monoclonal antibody for treatment of COVID-19 that retains activity against Omicron variant. www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-authorizes-new-monoclonal-antibody-treatment-covid-19-retains (accessed 20 April 2022).
FDA 2022d
- US Food and Drug Administration. Coronavirus (COVID-19) update: FDA authorizes second booster dose of two COVID-19 vaccines for older and immunocompromised individuals. www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-authorizes-second-booster-dose-two-covid-19-vaccines-older-and (accessed 20 April 2022).
Fung 2020
- Fung M, Babik JM. COVID-19 in immunocompromised hosts: what we know so far. Clinical Infectious Diseases 2020;72(2):340-50. [DOI: 10.1093/cid/ciaa863] [DOI] [PMC free article] [PubMed] [Google Scholar]
Gai 2021
- Gai J, Ma L, Li G, Zhu M, Qiao P, Li X, et al. A potent neutralizing nanobody against SARS-CoV-2 with inhaled delivery potential. MedComm 2021;2:101-13. [DOI: 10.1002/mco2.60] [DOI] [PMC free article] [PubMed] [Google Scholar]
Galmiche 2022
- Galmiche S, Nguyen LB, Tartour E, Lamballerie X, Wittkop L, Loubet P, et al. Immunological and clinical efficacy of COVID-19 vaccines in immunocompromised populations: a systematic review. Clinical Microbiology and Infection 2022;28(2):163-77. [DOI: 10.1016/j.cmi.2021.09.036] [DOI] [PMC free article] [PubMed] [Google Scholar]
Glasgow 2020
- Glasgow A, Glasgow J, Limonta D, Solomon P, Lui I, Zhang Y, et al. Engineered ACE2 receptor traps potently neutralize SARS-CoV-2. Proceedings of the National Academy of Sciences of the United States of America 2020;117(45):28046-55. [DOI: 10.1073/pnas.2016093117] [DOI] [PMC free article] [PubMed] [Google Scholar]
Glaunsinger 2020
- Glaunsinger B. Lecture 2: "Coronavirus biology", from the course "COVID-19, SARS-CoV-2 and the pandemic" [video]. biology.mit.edu/undergraduate/current-students/subject-offerings/covid-19-sars-cov-2-and-the-pandemic (accessed 4 November 2020).
Goldberg 2021
- Goldberg Y, Mandel M, Bar-On YM, Bodenheimer O, Freedman L, Haas EJ, et al. Waning immunity after the BNT162b2 vaccine in Israel. New England Journal of Medicine 2021;385:e85. [DOI: 10.1056/NEJMoa2114228] [DOI] [PMC free article] [PubMed] [Google Scholar]
Gottlieb 2021
- Gottlieb RL, Nirula A, Chen P, Boscia J, Heller B, Morris J, et al. Effect of bamlanivimab as monotherapy or in combination with etesevimab on viral load in patients with mild to moderate COVID-19: a randomized clinical trial. Journal of the American Medical Association 2021;327(7):632-44. [DOI: 10.1001/jama.2021.0202] [DOI] [PMC free article] [PubMed] [Google Scholar]
GRADEpro GDT [Computer program]
- McMaster University (developed by Evidence Prime) GRADEpro GDT. Hamilton (ON): McMaster University (developed by Evidence Prime), 2020. Available at gradepro.org.
Hanke 2020
- Hanke L, Vidakovics Perez L, Sheward DJ, Das H, Schulte T, Moliner-Morro A, et al. An alpaca nanobody neutralizes SARS-CoV-2 by blocking receptor interaction. Nature Communications 2020;11:4420. [DOI: 10.1038/s41467-020-18174-5] [DOI] [PMC free article] [PubMed] [Google Scholar]
Hassan 2020
- Hassan AO, Case JB, Winkler ES, Thackray LB, Kafai NM, Bailey AL, et al. A SARS-CoV-2 infection model in mice demonstrates protection by neutralizing antibodies. Cell 2020;182(3):744-53.e4. [DOI: 10.1016/j.cell.2020.06.011] [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2003
- Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta-analyses. BMJ 2003;327:557-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2021a
- Higgins JP, Li T, Deeks JJ. Chapter 6: Choosing effect measures and computing estimates of effect. In: Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, et al, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 6.2 (updated February 2021). Cochrane, 2021. Available from training.cochrane.org/handbook/archive/v6.2.
Higgins 2021b
- Higgins JP, Eldridge S, Li T. Chapter 23: Including variants on randomized trials. In: Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, et al, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 6.2 (updated February 2021). Cochrane, 2021. Available from training.cochrane.org/handbook/archive/v6.2.
Higgins 2021c
- Higgins JP, Savović J, Page MJ, Elbers RG, Sterne JA. Chapter 8: Assessing risk of bias in a randomized trial. In: Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, et al, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 6.2 (updated February 2021). Cochrane, 2021. Available from training.cochrane.org/handbook/archive/v6.2.
Hoffmann 2020
- Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020;181(2):271-80.e8. [DOI: 10.1016/j.cell.2020.02.052] [DOI] [PMC free article] [PubMed] [Google Scholar]
Horby 2021
- Horby PW, Mafham M, Peto L, Campbell M, Pessoa-Amorim G, Spata E, et al. Casirivimab and imdevimab in patients admitted to hospital with COVID-19 (RECOVERY): a randomised, controlled, open-label, platform trial. medRxiv [Preprint] 2021. [DOI: 10.1101/2021.06.15.21258542] [DOI]
Huang 2020
- Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020;395(10223):497-506. [DOI: 10.1016/S0140-6736(20)30183-5] [DOI] [PMC free article] [PubMed] [Google Scholar]
Iketani 2022
- Iketani S, Liu L, Guo Y, Liu L, Chan JF, Huang Y, et al. Antibody evasion properties of SARS-CoV-2 Omicron sublineages. Nature 2022;604(7906):553-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Jaworski 2021
- Jaworski JP. Neutralizing monoclonal antibodies for COVID-19 treatment and prevention. Biomedical Journal 2021;44(1):7-17. [DOI: 10.1016/j.bj.2020.11.011] [DOI] [PMC free article] [PubMed]
Johns Hopkins 2021
- Mortality analysis. www.coronavirus.jhu.edu/data/mortality (accessed 11 March 2021).
Jovčevska 2020
- Jovčevska I, Muyldermans S. The therapeutic potential of nanobodies. BioDrugs 2020;34:11-26. [DOI: 10.1007/s40259-019-00392-z] [DOI] [PMC free article] [PubMed] [Google Scholar]
Kaplon 2020
- Kaplon H, Muralidharan M, Schneider Z, Reichert JM. Antibodies to watch in 2020. MAbs 2020;12(1):e1703531. [DOI: 10.1080/19420862.2019.1703531] [DOI] [PMC free article] [PubMed] [Google Scholar]
Kluge 2021
- Kluge S, Malin JJ, Fichtner F, Müller OJ, Skoetz N, Karagiannidis C. Recommendations on the in-hospital treatment of patients with COVID-19. Deutsches Arzteblatt International 2021;118:865-71. [DOI: 10.3238/arztebl.m2021.0374] [DOI] [PMC free article] [PubMed] [Google Scholar]
Krammer 2021
- Krammer F. A correlate of protection for SARS-CoV-2 vaccines is urgently needed. Nature Medicine 2021;27(7):1147-8. [DOI: 10.1038/s41591-021-01432-4] [DOI] [PubMed] [Google Scholar]
Kreuzberger 2021
- Kreuzberger N, Hirsch C, Chai KL, Piechotta V, Valk SJ, Estcourt LJ, et al. SARS-CoV-2-neutralising monoclonal antibodies for treatment of COVID-19. Cochrane Database of Systematic Reviews 2021, Issue 1. Art. No: CD013825. [DOI: 10.1002/14651858.CD013825.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Lauer 2020
- Lauer SA, Grantz KH, Bi Q, Jones FK, Zheng Q, Meredith HR, et al. The incubation period of coronavirus disease 2019 (COVID-19) from publicly reported confirmed cases: estimation and application. Annals of Internal Medicine 2020;172(9):577-82. [DOI: 10.7326/M20-0504] [DOI] [PMC free article] [PubMed] [Google Scholar]
Lee 2020
- Lee WS, Wheatley AK, Kent SJ, DeKosky BJ. Antibody-dependent enhancement and SARS-CoV-2 vaccines and therapies. Nature Microbiology 2020;5:1185-91. [DOI: 10.1038/s41564-020-00789-5] [DOI] [PMC free article] [PubMed] [Google Scholar]
Lefebvre 2021
- Lefebvre C, Glanville J, Briscoe S, Littlewood A, Marshall C, Metzendorf M-I, et al. Chapter 4: Searching for and selecting studies. In: Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, et al, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 6.2 (updated February 2021). Cochrane, 2021. Available from training.cochrane.org/handbook/archive/v6.2.
Li 2021
- Li T, Higgins JP, Deeks JJ. Chapter 5: Collecting data. In: Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, et al, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 6.2 (updated February 2021). Cochrane, 2021. Available from training.cochrane.org/handbook/archive/v6.2.
Liang 2020
- Liang W, Guan W, Chen R, Wang Wi, Li J, Xu K, et al. Cancer patients in SARS-CoV-2 infection: a nationwide analysis in China. Lancet Oncology 2020;21(3):335-7. [DOI: 10.1016/S1470-2045(20)30096-6] [DOI] [PMC free article] [PubMed] [Google Scholar]
Living Evidence Network 2019
- Living Evidence Network. Guidance for the production and publication of Cochrane living systematic reviews: Cochrane Reviews in living mode, 2019. community.cochrane.org/review-production/production-resources/living-systematic-reviews (accessed 1 June 2022).
Lu 2020a
- Lu R, Hwang Y, Liu I, Lee C, Tsai H, Li H, et al. Development of therapeutic antibodies for the treatment of diseases. Journal of Biomedical Science 2020 2020;27(1):1. [DOI: 10.1186/s12929-019-0592-z] [DOI] [PMC free article] [PubMed] [Google Scholar]
Lu 2020b
- Lu R, Zhao X, Li J, Niu P, Yang B, Wu H, et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet 2020;395(10224):565-74. [DOI: 10.1016/S0140-6736(20)30251-8] [DOI] [PMC free article] [PubMed] [Google Scholar]
Lu 2021
- Lu Q, Zhang Z, Li H, Zhong K, Zhao Q, Wang Z, et al. Development of multivalent nanobodies blocking SARS-CoV-2 infection by targeting RBD of spike protein. Journal of Nanobiotechnology 2021;19(1):33. [DOI: 10.1186/s12951-021-00768-w] [DOI] [PMC free article] [PubMed] [Google Scholar]
Lundgren 2021
- Lundgren J, Grund B, Barkauskas CE, Holland TL, Gottlieb RL, Sandkovsky U, et al. A neutralizing monoclonal antibody for hospitalized patients with Covid-19. New England Journal of Medicine 2021;384:905-14. [DOI: 10.1056/NEJMoa2033130] [DOI] [PMC free article] [PubMed] [Google Scholar]
Mair 2022
- Mair MJ, Berger JM, Mitterer M, Gansterer M, Bathke AC, Trutschnig W, et al. Third dose of SARS-CoV-2 vaccination in hemato-oncological patients and health care workers: immune responses and adverse events – a retrospective cohort study. European Journal of Cancer 2022;165:184-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
Marovich 2020
- Marovich M, Mascola JR, Cohen MS. Monoclonal antibodies for prevention and treatment of COVID-19. Journal of the American Medical Association 2020;324(2):131-2. [DOI: 10.1001/jama.2020.10245] [DOI] [PubMed] [Google Scholar]
Marshall 2020
- Marshall JC, Murthy S, Diaz J, Adhikari NK, Angus DC, Arabi YM, et al. A minimal common outcome measure set for COVID-19 clinical research. Lancet Infectious Diseases 2020;20(8):e192-7. [DOI: 10.1016/S1473-3099(20)30483-7] [DOI] [PMC free article] [PubMed] [Google Scholar]
Marston 2018
- Marston HD, Paules CI, Fauci AS. Monoclonal antibodies for emerging infectious diseases – borrowing from history. New England Journal of Medicine 2018;378(16):1469-72. [PMID: 10.1056/NEJMp1802256] [DOI] [PubMed] [Google Scholar]
Microsoft Excel [Computer program]
- Microsoft Excel. Microsoft Corporation. Microsoft Corporation, 2018. office.microsoft.com/excel.
Moher 2009
- Moher D, Liberati A, Tetzlaff J, Altman DG. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. Journal of Clinical Epidemiology 2009;62(10):1006-12. [DOI: 10.1371/journal.pmed.1000097] [DOI] [PubMed] [Google Scholar]
Monin‐Aldama 2021
- Monin-Aldama L, Laing AG, Muñoz-Ruiz M, McKenzie DR, Molino del Barrio I, Alaguthurai T, et al. Interim results of the safety and immune-efficacy of 1 versus 2 doses of COVID-19 vaccine BNT162b2 for cancer patients in the context of the UK vaccine priority guidelines. medRxiv [Preprint] 2021. [DOI: 10.1101/2021.03.17.21253131] [DOI]
Muik 2021
- Muik A, Wallisch AK, Saenger B, Swanson KA, Muehl J, Chen W, et al. Neutralization of SARS-CoV-2 lineage B.1.1.7 pseudovirus by BNT162b2 vaccine-elicited human sera. Science 2021;371(6534):1152-3. [DOI: 10.1126/science.abg6105] [DOI] [PMC free article] [PubMed] [Google Scholar]
Mulangu 2019
- Mulangu S, Dodd LE, Davey RT, Tshiani Mbaya O, Proschan M, Mukadi D, et al. A randomized, controlled trial of ebola virus disease therapeutics. New England Journal of Medicine 2019;381(24):2293-303. [DOI: 10.1056/NEJMoa1910993] [DOI] [PMC free article] [PubMed] [Google Scholar]
National COVID‐19 Clinical Evidence Taskforce 2020
- National COVID-19 Clinical Evidence Taskforce. Caring for people with COVID-19: supporting Australia's healthcare professionals with continually updated, evidence-based clinical guidelines. covid19evidence.net.au/#living-guidelines (accessed 22 November 2021).
NCT04383548
- NCT04383548. Clinical study for efficacy of anti-corona VS2 immunoglobulins prepared from COVID19 convalescent plasma prepared by VIPS Mini-Pool IVIG medical devices in prevention of SARS-CoV-2 infection in high risk groups as well as treatment of early cases of COVID19 patients. clinicaltrials.gov/ct2/show/NCT04383548 (first received 12 May 2020).
NCT04425629
- NCT04425629. Safety, tolerability, and efficacy of anti-spike (S) SARS-CoV-2 monoclonal antibodies for the treatment of ambulatory adult and pediatric patients with COVID-19. clinicaltrials.gov/ct2/show/NCT04425629 (first received 11 June 2020).
NCT04427501
- NCT04427501. A randomized, double-blind, placebo-controlled, phase 2/3 study to evaluate the efficacy and safety of LY3819253 and LY3832479 in participants with mild to moderate COVID-19 illness. clinicaltrials.gov/ct2/show/NCT04427501 (first received 11 June 2020).
NCT04561063
- NCT04561063. COVID-19 Prophylaxis South Africa (COVER HCW) (COVER). clinicaltrials.gov/ct2/show/NCT04561063 (first received 23 September 2020). [NCT04561063]
NCT04836260
- NCT04836260. Preemptive use of convalescent plasma for high-risk patients with COVID-19. clinicaltrials.gov/ct2/show/NCT04836260 (first received 8 April 2021).
NCT05135650
- NCT05135650. Pharmacokinetics of sotrovimab as pre-exposure prophylaxis for COVID-19 in hematopoietic stem cell transplant recipients, COVIDMAB study. clinicaltrials.gov/ct2/show/NCT05135650 (first received 26 November 2021).
NCT05210101
- NCT05210101. A safety and tolerability study of sotrovimab (VIR-7831) prophylaxis against COVID-19 in immunocompromised individuals. clinicaltrials.gov/ct2/show/NCT05210101 (first received 27 January 2022).
NCT05216588
- NCT05216588. Pre-exposure prophylaxis of SARS-CoV-2 infection (COVID-19) by monoclonal antibodies with early access authorization in immunocompromised patients. A prospective cohort (PRECOVIM). clinicaltrials.gov/ct2/show/NCT05216588 (first received 31 January 2022).
NCT05234398
- NCT05234398. Tixagevimab/cilgavimab protection of Covid-19 in transplanted patients (TIXCI-TRANS). clinicaltrials.gov/ct2/show/NCT05234398 (first received 10 February 2022).
Nehal 2021
- Nehal KR, Steendam LM, Campos Ponce M, Hoevenand M, Smit GS. Worldwide vaccination willingness for COVID-19: a systematic review and meta-analysis. Vaccines 2021;9(10):1071. [DOI: 10.3390/vaccines9101071] [DOI] [PMC free article] [PubMed] [Google Scholar]
NIH 2022
- NIH COVID-19 treatment guidelines. Prevention of SARS-CoV-2 Infection. www.covid19treatmentguidelines.nih.gov/overview/prevention-of-sars-cov-2/?utm_source=site&utm_medium=home&utm_campaign=highlights (accessed 20 April 2022).
Ou 2020
- Ou X, Liu Y, Lei X, Li P, Mi D, Ren L, et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nature Communications 2020;11(1):1620. [DOI: 10.1038/s41467-020-15562-9] [DOI] [PMC free article] [PubMed] [Google Scholar]
Parmar 1998
- Parmar MK, Torri V, Stewart L. Extracting summary statistics to perform meta-analyses of the published literature for survival endpoints. Statistics in Medicine 1998;17(24):2815-34. [DOI: ] [DOI] [PubMed] [Google Scholar]
Perry 2022
- Perry J, Osman S, Wright J, Richard-Greenblatt M, Buchan SA, Sadarangani M, et al. Does a humoral correlate of protection exist for SARS-CoV-2? A systematic review. PLoS One 2022;17(4):e0266852. [DOI: 10.1371/journal.pone.0266852] [DOI] [PMC free article] [PubMed] [Google Scholar]
Review Manager Web 2021 [Computer program]
- The Cochrane Collaboration Review Manager Web (RevMan Web). Version 2.6.0. The Cochrane Collaboration, 2021. Available at revman.cochrane.org.
Rogers 2020
- Rogers TF, Zhao F, Huang D, Beutler N, Burns A, He W, et al. Isolation of potent SARS-CoV-2 neutralizing antibodies and protection from disease in a small animal model. Science 2020;369(6506):956-63. [DOI: 10.1126/science.abc7520] [DOI] [PMC free article] [PubMed] [Google Scholar]
Rössler 2022
- Rössler A, Riepler L, Bante D, Laer D, Kimpel J. SARS-CoV-2 Omicron variant neutralization in serum from vaccinated and convalescent persons. New England Journal of Medicine 2022;386(7):698-700. [DOI: 10.1056/NEJMc2119236] [DOI] [PMC free article] [PubMed] [Google Scholar]
Santesso 2020
- Santesso N, Glenton C, Dahm P, Garner P, Akl A, Alper B, et al. GRADE guidelines 26: informative statements to communicate the findings of systematic reviews of interventions. Journal of Clinical Epidemiology 2020;119:126-35. [DOI: 10.1016/j.jclinepi.2019.10.014] [DOI] [PubMed] [Google Scholar]
Schünemann 2021
- Schünemann HJ, Higgins JP, Vist GE, Glasziou P, Akl EA, Skoetz N, et al. Chapter 14: Completing 'Summary of findings' tables and grading the certainty of the evidence. In: Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, et al, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 6.2 (updated February 2021). Cochrane, 2021. Available from training.cochrane.org/handbook/archive/v6.2.
Siemieniuk 2020
- Siemieniuk R, Rochwerg B, Agoritsas T, Lamontagne F, Leo YS, Macdonald H, et al. A living WHO guideline on drugs for Covid-19. BMJ 2020;370:m3379:1-14. [DOI: 10.1136/bmj.m3379] [DOI] [PubMed] [Google Scholar]
Skoetz 2020
- Skoetz N, Goldkuhle M, Dalen EC, Akl EA, Trivella M, Mustafa RA, et al. GRADE guidelines 27: how to calculate absolute effects for time-to-event outcomes in summary of findings tables and Evidence Profiles. Journal of Clinical Epidemiology 2020;118:124-31. [DOI: 10.1016/j.jclinepi.2019.10.015] [DOI] [PubMed] [Google Scholar]
Sterne 2019
- Sterne JA, Savovic J, Page MJ, Elbers RG, Blencowe N, Boutron I, et al. RoB 2: a revised tool for assessing risk of bias in randomised trials. BMJ 2019;366:l4898. [DOI: 10.1136/bmj.l4898] [DOI] [PubMed] [Google Scholar]
Struyf 2020
- Struyf T, Deeks JJ, Dinnes J, Takwoingi Y, Davenport C, Leeflang MM, et al. Signs and symptoms to determine if a patient presenting in primary care or hospital outpatient settings has COVID-19. Cochrane Database of Systematic Reviews 2020, Issue 7. Art. No: CD013665. [DOI: 10.1002/14651858.CD013665] [DOI] [PMC free article] [PubMed] [Google Scholar]
Tada 2020
- Tada T, Fan C, Chen J, Kaur R, Stapleford KA, Gristick H, et al. An ACE2 microbody containing a single immunoglobulin Fc domain is a potent inhibitor of SARS-CoV-2. Cell Reports 2020;33(12):108528. [DOI: 10.1016/j.celrep.2020.108528] [DOI] [PMC free article] [PubMed] [Google Scholar]
Takashita 2022
- Takashita E, Kinoshita N, Yamayoshi S, Sakai-Tagawa Y, Fujisaki S, Ito M, et al. Efficacy of antibodies and antiviral drugs against Covid-19 Omicron variant. New England Journal of Medicine 2022;386(10):995-8. [DOI: 10.1056/NEJMc2119407] [DOI] [PMC free article] [PubMed] [Google Scholar]
Tang 2021
- Tang JW, Tambyah PA, Hui DS. Emergence of a new SARS-CoV-2 variant in the UK. Journal of Infection 2021;82(4):e27-e28. [DOI: 10.1016/j.jinf.2020.12.024] [DOI] [PMC free article] [PubMed] [Google Scholar]
The Antibody Society 2021
- The Antibody Society. www.antibodysociety.org/covid-19-biologics-tracker/ (accessed 8 December 2021).
Tierney 2007
- Tierney JF, Stewart LA, Ghersi D, Burdett S, Sydes MR. Practical methods for incorporating summary time-to-event data into meta-analysis. Trials 2007;8:16. [DOI: 10.1186/1745-6215-8-16] [DOI] [PMC free article] [PubMed] [Google Scholar]
Tolouian 2020
- Tolouian R, Vahed SZ, Ghiyasvand S, Tolouian A, Ardalan M. COVID-19 interactions with angiotensin-converting enzyme 2 (ACE2) and the kinin system; looking at a potential treatment. Journal of Renal Injury Prevention 2020;9(2):e19. [DOI: 10.34172/jrip.2020.19] [DOI] [Google Scholar]
UNDP 2021
- United Nations Development Programme. Global dashboard for vaccine equity. data.undp.org/vaccine-equity/ (accessed 20 April 2022).
Vitiello 2022
- Vitiello A, Ferrara F, Auti AM, Di Domenico M, Boccellino M. Advances in the Omicron variant development. Journal of Internal Medicine 2022 Mar 15 [Epub ahead of print]. [DOI: 10.1111/joim.13478] [DOI] [PMC free article] [PubMed] [Google Scholar]
Wang 2021
- Wang P, Casner RG, Nair MS, Wang M, Yu J, Cerutti G, et al. Increased resistance of SARS-CoV-2 variant P.1 to antibody neutralization. bioRxiv [Preprint] 2021. [DOI: 10.1101/2021.03.01.433466] [DOI] [PMC free article] [PubMed]
Weinreich 2021
- Weinreich DM, Sivapalasingam S, Norton T, Ali S, Gao H, Bhore R, et al. REGEN-COV antibody combination and outcomes in outpatients with Covid-19. New England Journal of Medicine 2021;385:e81. [DOI: 10.1056/NEJMoa2108163] [DOI] [PMC free article] [PubMed] [Google Scholar]
WHO 2003
- World Health Organization. Cumulative number of reported probable cases of SARS. www.who.int/csr/sars/country/2003_07_11/en (accessed 8 February 2021).
WHO 2020a
- World Health Organization. Report of the WHO–China joint mission on coronavirus disease 2019 (COVID-19). www.who.int/docs/default-source/coronaviruse/who-china-joint-mission-on-covid-19-final-report (accessed 1 June 2022).
WHO 2020b
- World Health Organization. World Health Organization Quality of Life assessment instrument (WHOQOL-100). www.who.int/tools/whoqol/whoqol-100 (accessed 5 May 2021).
WHO 2020c
- WHO Working Group on the Clinical Characterisation and Management of COVID-19 infection. A minimal common outcome measure set for COVID-19 clinical research. Lancet Infectious Diseases 2020;20(8):e192-7. [DOI: 10.1016/S1473-3099(20)30483-7] [DOI] [PMC free article] [PubMed] [Google Scholar]
WHO 2021a
- World Health Organization Eastern Mediterranean Regional Office. Middle East respiratory syndrome. www.emro.who.int/health-topics/mers-cov/mers-outbreaks.html (accessed 26 February 2021).
WHO 2021b
- World Health Organization. WHO Coronavirus Disease (COVID-19) Dashboard. covid19.who.int (accessed 8 February 2021).
WHO 2021c
- World Health Organization. Weekly epidemiological update – 27 January 2021. www.who.int/publications/m/item/weekly-epidemiological-update-27-january-2021 (accessed 7 February 2021).
WHO 2021d
- World Health Organization. Tracking SARS-CoV-2 variants. www.who.int/en/activities/tracking-SARS-CoV-2-variants/ (accessed 9 December 2021).
WHO 2021e
- World Health Organisation. WHO validates Sinovac COVID-19 vaccine for emergency use and issues interim policy recommendations. www.who.int/news/item/01-06-2021-who-validates-sinovac-covid-19-vaccine-for-emergency-use-and-issues-interim-policy-recommendations (accessed 17 November 2021).
WHO 2022
- World Health Organization. Statement on Omicron sublineage BA.2. www.who.int/news/item/22-02-2022-statement-on-omicron-sublineage-ba.2 (accessed 20 April 2022).
Widera 2021
- Widera M, Wilhelm A, Hoehl S, Pallas C, Kohmer N, Wolf T, et al. Bamlanivimab does not neutralize two SARS-CoV-2 variants carrying E484K in vitro. medRxiv [Preprint]. [DOI: 10.1101/2021.02.24.21252372] [DOI]
Wise 2022
- Wise J. Covid-19: Evusheld is approved in UK for prophylaxis in immunocompromised people. BMJ 2022;376:o722. [DOI: 10.1136/bmj.o722] [DOI] [PubMed] [Google Scholar]
Wrapp 2020
- Wrapp D, De Vlieger D, Corbett KS, Torres GM, Wang N, Breedam W, et al. Structural basis for potent neutralization of betacoronaviruses by single-domain camelid antibodies. Cell 2020;181(5):1004-15. [DOI: 10.1016/j.cell.2020.04.031] [DOI] [PMC free article] [PubMed] [Google Scholar]
Wu 2020
- Wu Z, McGoogan JM. Characteristics of and important lessons from the coronavirus disease 2019 (COVID-19) outbreak in China: summary of a report of 72 314 cases from the Chinese Center for Disease Control and Prevention. Journal of the American Medical Association 2020;323(13):1239-42. [DOI: 10.1001/jama.2020.2648] [DOI] [PubMed] [Google Scholar]
Yang 2020
- Yang L, Liu W, Yu X, Wu M, Reichert JM, Ho M. COVID-19 antibody therapeutics tracker: a global online database of antibody therapeutics for the prevention and treatment of COVID-19. Antibody Therapeutics 2020;3(3):205-12. [DOI: 10.1093/abt/tbaa020] [DOI] [PMC free article] [PubMed] [Google Scholar]
Zost 2020
- Zost SJ, Gilchuk P, Case JB, Binshtein E, Chen RE, Nkolola JP, et al. Potently neutralizing and protective human antibodies against SARS-CoV-2. Nature 2020;584(7821):443-9. [DOI: 10.1038/s41586-020-2548-6] [DOI] [PMC free article] [PubMed] [Google Scholar]
References to other published versions of this review
Hirsch 2021
- Hirsch C, Valk SJ, Piechotta V, Chai KL, Estcourt LJ, Monsef I, et al. SARS-CoV-2-neutralising monoclonal antibodies to prevent COVID-19. Cochrane Database of Systematic Reviews 2021, Issue 5. Art. No: CD014945. [DOI: 10.1002/14651858.CD014945] [DOI] [PMC free article] [PubMed] [Google Scholar]