

Abstract
The study and application of transition metal hydrides (TMH), an active area of chemical research since the early 1960’s1, has been broadly bifurcated into fields focused on energy storage through the reduction of protons to generate hydrogen2,3 and in organic synthesis for the functionalization of unsaturated C–C, C–O, and C–N bonds.4,5 In the former instance, electrochemical means for driving such reactivity has been commonplace since the 1950’s6 but the use of stoichiometric exogenous organic- and metal-based reductants to harness the power of TMHs in synthetic chemistry remains the norm. In particular, Co-based TMHs have found widespread use for the derivatization of olefins and alkynes in complex molecule construction, often via a net hydrogen atom transfer (HAT).7 Here, we show how an electrocatalytic approach inspired by decades of energy storage precedent can be leveraged in the context of modern organic synthesis. This strategy not only offers benefits in terms of sustainability and efficiency but also enables enhanced chemoselectivity and unique and tunable reactivity. Ten different reaction manifolds across dozens of substrates are thus exemplified, along with detailed mechanistic insight on this scalable electrochemical entry to Co-H generation that uniquely takes place through a low-valent intermediate.
TMH species have been a vibrant topic for exploration in organic and organometallic synthesis.1,8 Pioneering studies in this field have led to a deep understanding of metal hydrides9 that has allowed synthetic chemists to establish these species as selective mediators for HAT chemistry.7,10 Such insights have led to the discovery of unique selectivity for known transformations11 along with the development of new chemical reactivity.12 HAT, the concerted migration of a proton and an electron from a TM-H bond to an acceptor molecule, has emerged as one of the most useful chemical processes for the hydrofunctionalization of alkenes.4 In its common manifestation, the generation of a TMH involves the exposure of an appropriate metal complex to a stoichiometric amount of reductant, such as a silane. Its subsequent reaction with an olefin leads to the formation of a C–H bond at the less electronically stabilized position along with a carbon-centred radical on the adjacent position. This intermediate can then be trapped with various reagents to form new C–C, C–N, C–O, and C–X bonds.13–18 While the overall process formally requires only the addition of a proton and an electron to form the active TMH catalytic species, exogenous chemical oxidants, are often required to elicit this reactivity. The application of HAT chemistry on a large scale could be problematic due to the need for an excess amount of external reductants with or without oxidants, resulting in poor atom economy and scalability concerns implicit in the use of organic reductants and oxidants in the same flask (Figure 1A).19 Given the growing documented utility of such reactions in organic synthesis, it is clear that more practical and universal variants are required.20,21
Figure 1:
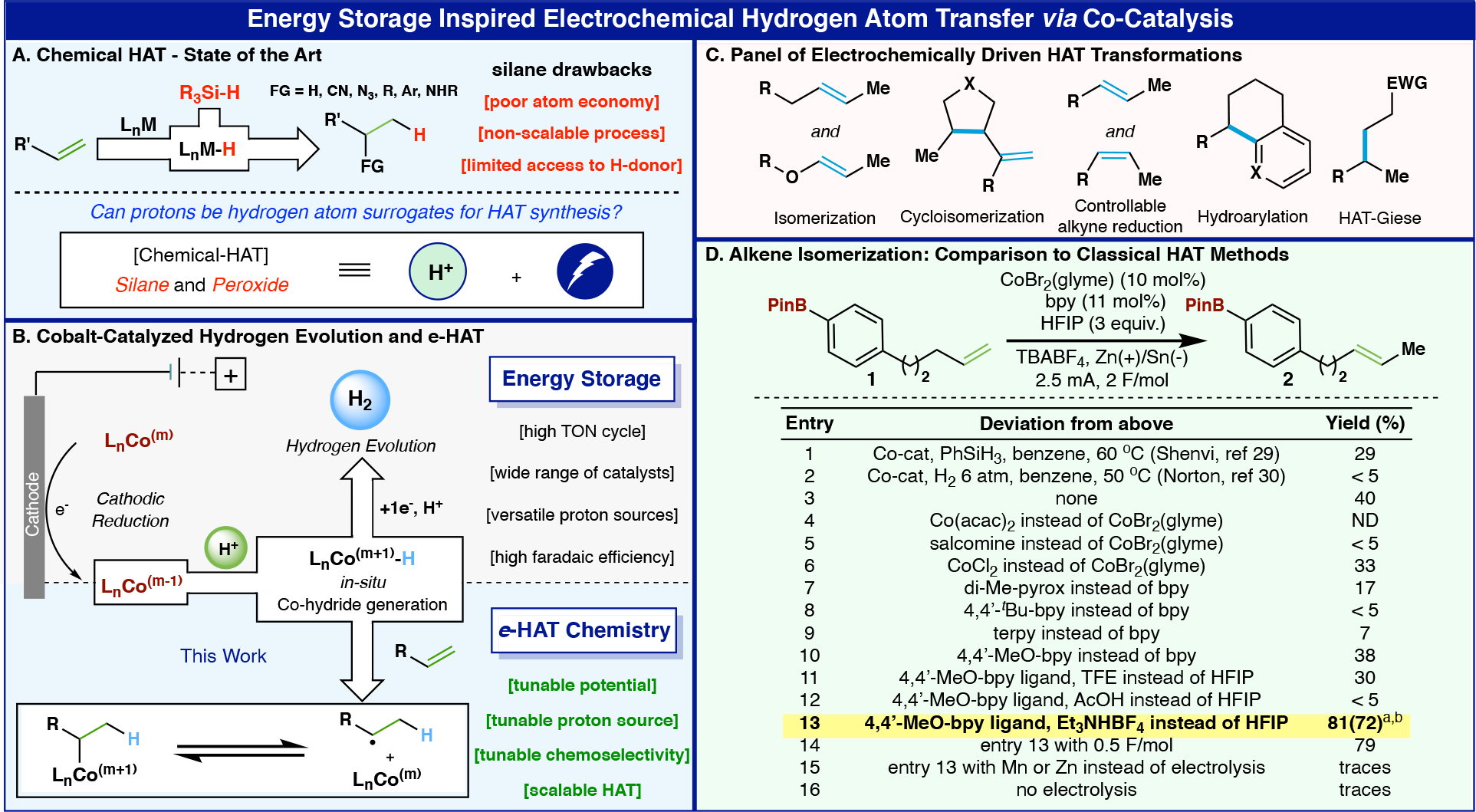
Energy Storage Inspired Electrochemical Hydrogen Atom Transver via Co-Catalysis A) Classical HAT chemistry. B) Cobalt-catalyzed H2 evolution and e-HAT. C) e-HAT transformation. D) Optimization table of alkene isomerization. Yields were determined by 1H NMR of the crude mixture. a isolated yield b E/Z ratio = 4/1.
In parallel, the same types of species have been efficiently and sustainably generated in the hydrogen production field with a proton as the hydride source (Figure 1B).2 For example, hydrogen evolution via electrochemically generated Co–H species was known as early as 1985.22 The field was dormant for over two decades until recently, with interest in cobalt-catalyzed electrochemical hydrogen evolution for green energy storage being the subject of a large body of studies spanning hundreds of publications.23 These robust Co–H based processes feature high turnover numbers and have been optimized to high levels with >90% efficiency of H2 production from simple protic systems suggesting their commercial implementation is immanent. From a mechanistic standpoint, Co–H is formed in-situ by the protonation of low valent Co(I)/Co(0) intermediates after direct cathodic reduction.24,25 Subsequently, it can react via two different pathways to form hydrogen and regenerate the catalyst. In the first suggested mechanism, the generated Co(II)–H species decomposes by proton attack and evolves hydrogen via an intermediate dihydrogen metal complex. Alternatively, Co(III)–H can be reduced to Co(II)–H, which is followed by a similar protonation step. Interestingly, the described process operates with high faradaic efficiency in aqueous or non-aqueous mediums and various types of proton sources, such as water, acids, and alcohols. Amongst the many cobalt complexes enlisted, many do not require complex ligand architectures.26,27
Inspired by the well-established Co-electrocatalytic hydrogen evolution chemistry precedent outlined above, disclosed herein (Figure 1B) is a set of chemoselective, tunable electrochemical HAT (e-HAT) protocols free of either chemical reductants and oxidants (e.g. silanes, peroxides) or rigorous experimental protocols (moisture tolerant, no glove-box). Thus, a versatile range of tunable reactivities with alkenes and alkynes—such as isomerization, selective reduction, and hydrofunctionalization (Figure 1C)—can be realized with unmatched efficiency and chemoselectivity beyond that observed under purely chemical conditions. In support of these claims, this electrochemically-enabled reactivity is benchmarked with several of the most popular and recently disclosed chemical methods. Additionally, in-depth mechanistic analysis using cyclic voltammetry, UV-VIS spectroelectrochemistry, computation, and kinetics provides insight into e-HAT and rationalizes the observed selectivity. Finally, the scalability of this process is demonstrated in both batch and flow [gram-centigram (0.05 – 0.8 mole) scale].
As a proof of concept, alkene isomerization was selected as a model transformation since only a substoichiometric amount of Co–H is required for the reaction to proceed with complete conversion.28 Alkene 1 was selected for the initial optimization of the e-HAT reaction. Trial runs using the literature precedents for classical HAT isomerization with alkene 1 provided poor conversion to the desired product. For example, the use of 50 mol% of silane and 10 mol% of cobalt catalyst in benzene gives 29% yield of the desired product and 7% of other chain-walking isomers (entry 1, Shenvi’s protocol).29 The method of Norton, which relies on a high pressure of hydrogen gas, delivered only traces of product (entry 2).30 First forays into e-HAT isomerization followed the guiding principles31 from prior findings in electrochemistry32,33 and HAT chemistry34 to aid in the selection of proper ligands, cathodic materials and proton sources. An abbreviated summary of >200 experiments is depicted in Figure 1D (see SI for an extensive list). First, the cobalt catalyst screening (entries 3–6) revealed that CoBr2(glyme) was optimal, resulting in efficient Co–H generation with the highest yield for the alkene isomerization. However, the mass balance of these reactions consisted of a mixture of chain-walking and reduction byproducts. Thus, ligand screening was performed to cleanly obtain the desired single isomerization product (entries 7–10). Although 4,4’-dimethoxy-bipyridine cleanly afforded the desired selective 1-position isomerization, the conversion was low (38% yield + 33% recovered 1, entry 10). In order to improve the conversion while retaining the ligand-controlled selectivity, various proton sources within a wide range of pKa were explored (entries 10–13); the use of inexpensive triethylamine hydrotetrafluoroborate (3 equiv., entry 13) as a proton source emerged as optimum, providing the desired product 2 in 72% isolated yield. This unique proton source was employed due to its ability to function as a supporting electrolyte as well; its inclusion was crucial to the reproducibility and robustness of the reaction. The final set of e-HAT conditions tolerates moisture, leads to completion with catalytic amounts of electricity (0.5 F/mol, entry 14), and can be set up in minutes using a simple undivided cell and a commercial potentiostat. Interestingly, similar reactivity was not observed when conventional reductants, such as zinc and manganese, were used (entries 15–16). Of all cathodes evaluated, tin, Ni-foam, glassy-carbon, and stainless steel could be employed, but a tin cathode gave the highest yield across a broad range of substrates (see SI).
With these results in hand, the scope of the e-HAT isomerization of monosubstituted olefins was investigated (Figure 2A). A wide range of functionalities was tolerated, including free and protected amines (3, 6), anilines (5), amides (4), lactams (7), alcohols (13), and aliphatic nitriles (14) with over 80% yield on average. In addition, the e-HAT isomerization exhibited a broad scope across a range of different arenes. Under optimized conditions, alkenes were isomerized in the presence of pyridines (8), thiophenes (9), electron-deficient indoles (10), redox-active aryl bromides (11), and aryl-BPin (2). Remarkably, this method can deliver the isomerization of an allylic ether to the corresponding enol ether adduct (16). Terminal disubstituted olefins were, however, untouched by the bipyridine complex (Conditions A). As a result, another round of optimization was conducted revealing that commercially available CoII(t-Bu,t-Bucyclohexylsalen) (Co(salen)-1 – see SI for exact structure) could be employed to exclusively isomerize such olefins to the thermodynamic trisubstituted alkenes by using HFIP (hexafluoroisopropanol) as the proton source and a Ni-foam cathode in acetone (Conditions B, see SI for optimization). As e-HAT relies on the in-situ formation of a cobalt-hydride, chemoselective reactions are thus possible simply by tuning conditions. A similarly broad scope was observed for this isomerization as well (17-25). The ability to achieve olefin isomerizations in the presence of free phenols, pyridines, anilines, nitriles, and epoxides is without precedent to the best of our knowledge. Selected examples of the scope have been directly compared to existing conventional Co-H isomerization methods to show the utility of the e-HAT chemoselecticity (see SI for the comparisons – Comparison Section). In addition, the generation of β,γ-unsaturated amides has not been previously reported by isomerization methods, presumably due to a tendency to isomerize into conjugation.
Figure 2:
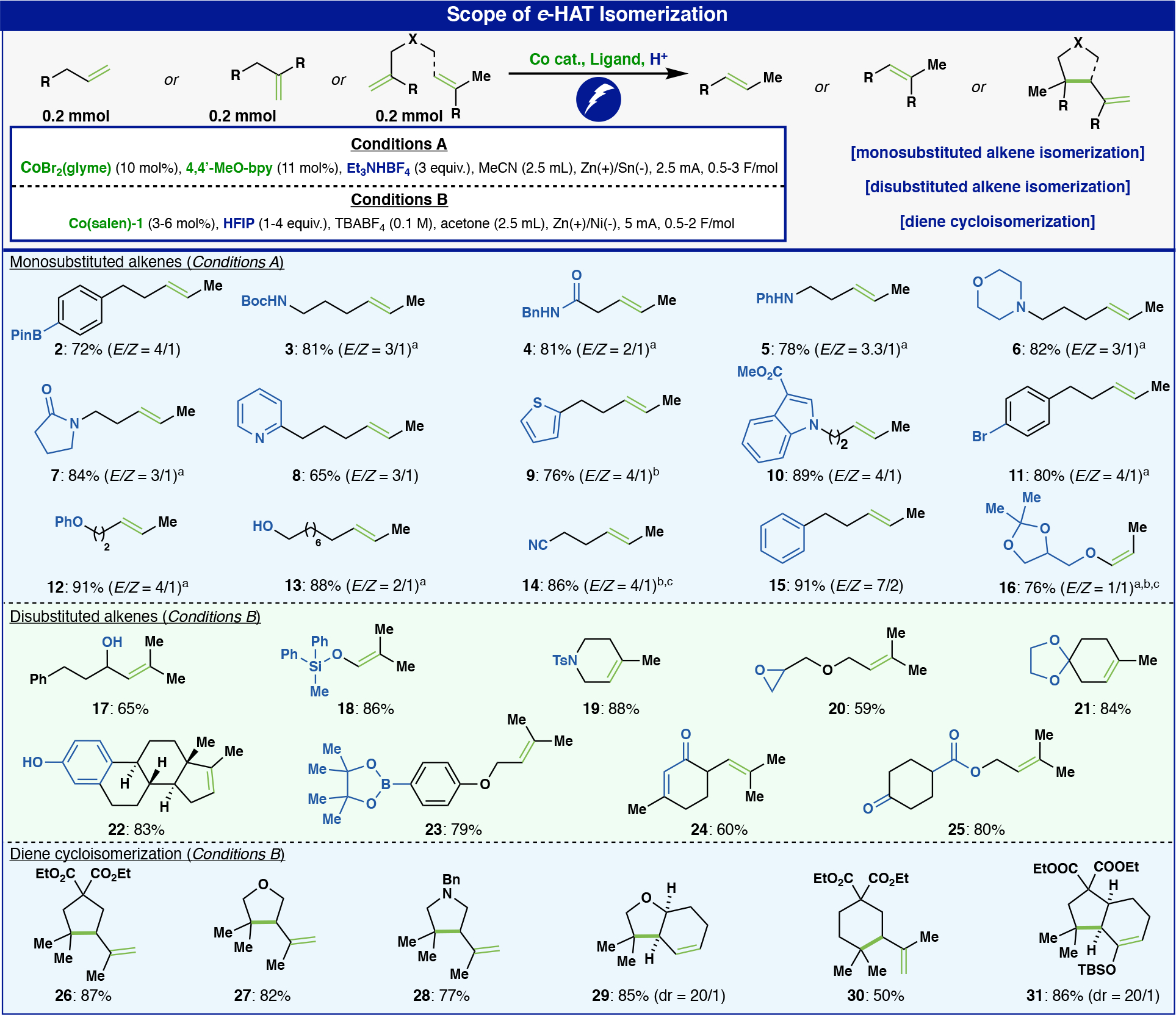
Scope of e-HAT isomerization a Mg was used as the anode. b 20 mol% of catalyst was used. c NMR yield.
Given the radical nature of intermediates in HAT-based reactions, Conditions B not surprisingly initiated intramolecular radical cyclizations of dienes to form new C–C bonds via cycloisomerization. Accordingly, methallyl prenyl malonate can undergo intramolecular cycloisomerization to yield the corresponding trisubstituted cyclopentane (26) in a high yield with no isomerization side products. The malonate can be exchanged by an ether (27) or an amine (28) without compromising the high efficiency of the transformation. Endocyclic alkenes can be similarly used as effective cyclization partners to form the cis five-six bicyclic systems with high diastereoselectivity (29). Even a cyclic enol ether can be used as the radical acceptor (31). As shown in Figure 2, the reaction shows high efficiency for 5-membered ring formation but is less suitable for formation of 6-membered rings (30), which gave only 50% yield along with a linear isomerization side-product.
The previously discussed isomerization reactions are net-redox-neutral transformations. Therefore, a substoichiometric amount of cobalt hydride is needed to proceed efficiently since the active catalyst is regenerated during the reaction pathway (vide infra). Alternatively, cobalt-hydride chemistry can be used to reduce unsaturated systems. Such reactions, by definition, will require “stoichiometric” electrons to be added with the right tuning of the proton source and cobalt complex to achieve the desired transformation. As a proof of concept, using e-HAT logic, a new e-HAT set of conditions for Z-selective alkyne semi-reduction using HFIP as the hydride surrogate was developed. To place this into context, the most frequently employed reagents (i.e., outside a glove-box) to achieve such a reaction on unactivated (non-conjugated) alkynes involve the use of Pd (Lindlar) catalysis and diimide. For such selectivity, the 6,6’-dimethyl-bpy ligand combined with CoBr2 revealed the best reactivity delivering high Z-selectivity and minimal over-reduction (Conditions C, see SI for optimization). With this new set of conditions in hand, a range of substituted alkynes could be reduced with e-HAT to provide Z-alkenes in good yield (Figure 3) rapidly. Boc-protected amines (32), pyridines (33), ethers (34), free and silyl-protected alcohols (36, 41), aryl chlorides (37), aryl-BPin (38), carbonyls (39, 42), alkenes (39), and alkyl phosphates (40) were all tolerated. The highest Z/E selectivity was observed with primary-, secondary-, and tertiary carbons adjacent to the alkyne moiety. However, the process was less selective with quaternary carbons adjacent to the alkyne (see 43 and 44), presumably due to the increased steric hindrance surrounding the putative cobalt-alkene intermediate.
Figure 3:
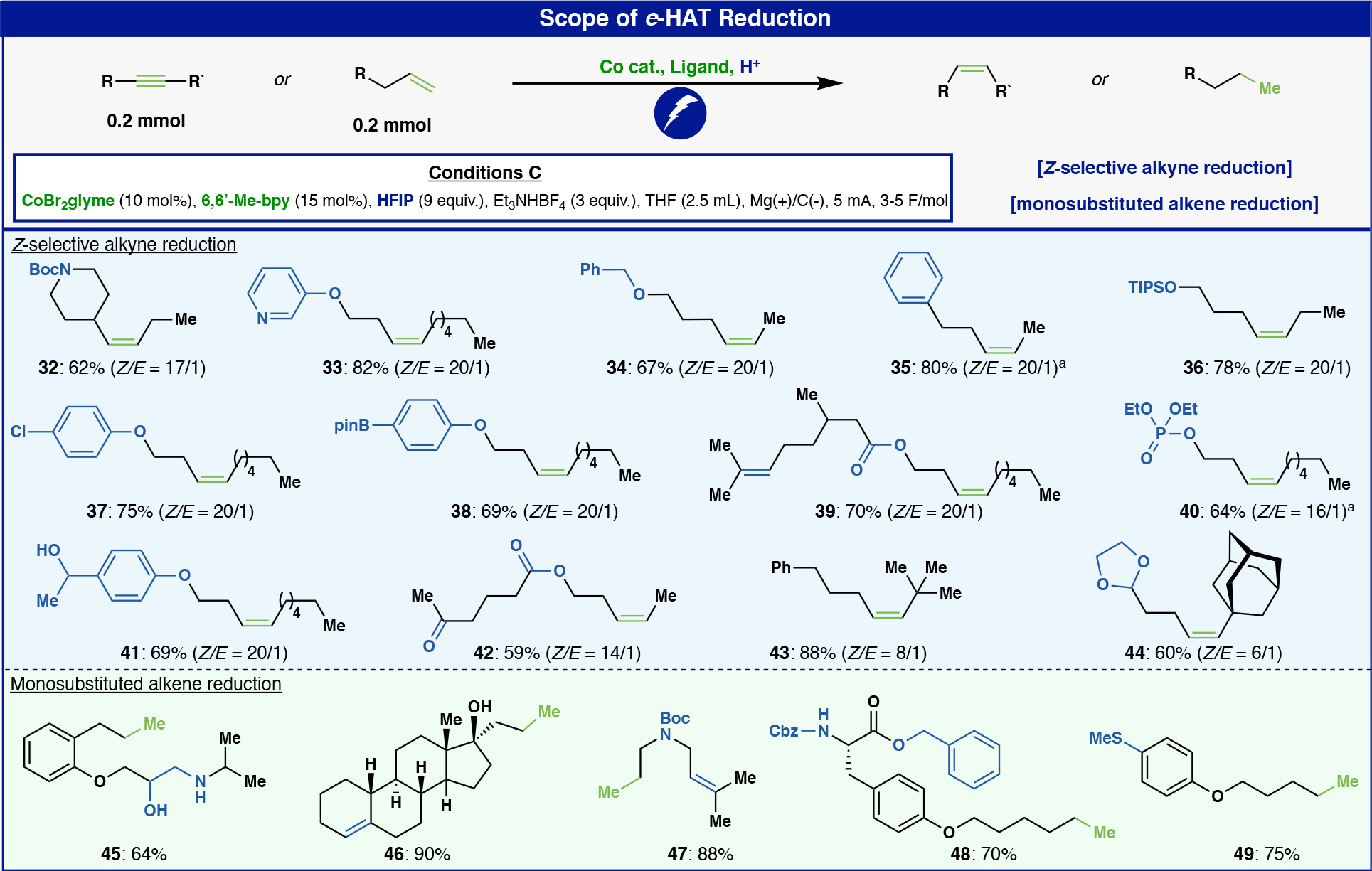
Scope of e-HAT reduction a NMR yield.
The selective reduction of monosubstituted alkenes was similarly achieved by relying on e-HAT (Conditions C). Canonical hydrogenation with H2 over Pd/C can accomplish this type of reduction; however, the chemoselectivity of that method can be poor when competing reductively labile functionalities are present.35 Accordingly, the e-HAT was tested with substrates that can be challenging with such well-established reduction protocols. Thus, olefins containing amine-diol functionalities (45), tri-substituted alkenes (46, 47), Cbz-protected amines (48), benzyl-protected carboxylic acids (48), and thioanisole moieties (49), which can all be problematic under typical hydrogenation conditions, were smoothly reduced.
Although classical HAT chemistry has been studied for over 30 years, precise control of chemoselectivity when multiple olefins are present in a substrate has remained underdeveloped, due to the complexity of tuning the hydride donor, oxidant, and catalyst.36,37 The modularity of e-HAT can potentially address such a challenge to achieve unique and useful selectivity with polyunsaturated systems and was thus pursued using the three distinct e-HAT conditions (A-C) disclosed above. Compound 50, which contains two different mono- and disubstituted terminal alkenes, was chosen as a case study (Figure 4A–1). By applying Conditions B (Co(salen)-1/HFIP), the exocyclic alkene was selectively isomerized to form the thermodynamically favoured trisubstituted alkene product 51 in 59% yield. In contrast, exposing compound 50 to Conditions A (CoBr2/4,4’-MeO-bpy/Et3NHBF4) led to the formation of disubstituted alkene 52 in 92% yield with an E:Z isomeric ratio of 3:1. This result of e-HAT could be placed into context through a direct comparison with prior art. Various canonical Co, Fe, Pd, Ru isomerization methods resulted in an inseparable mixture of double isomerized and reduced products (see SI for detailed product distribution – Comparison Section). Similarly, evaluation of the e-HAT alkene reduction displayed exquisite selectivity towards terminal monosubstituted alkenes. Triene 53 was subjected to Conditions C that selectively reduced the desired alkene – over the 1,1- disubstituted and endocyclic olefins – in 95% isolated yield (Figure 4A–2). Likewise, diene 55 was also examined under the same conditions and exhibited similar chemoselectivity towards the monosubstituted olefin in the presence of an internal disubstituted alkene to furnish 56 in 81% yield. Using H2 with Pd/C, dimide, PET (Co-Salen with Ru catalyst and light)38, Co/H+-based39, and Co-hydride-based40 systems as direct comparison resulted in an inseparable mixture of reduced products (see SI for detailed product distribution – Comparison Section), highlighting the singular efficacy of the e-HAT method.
Figure 4:
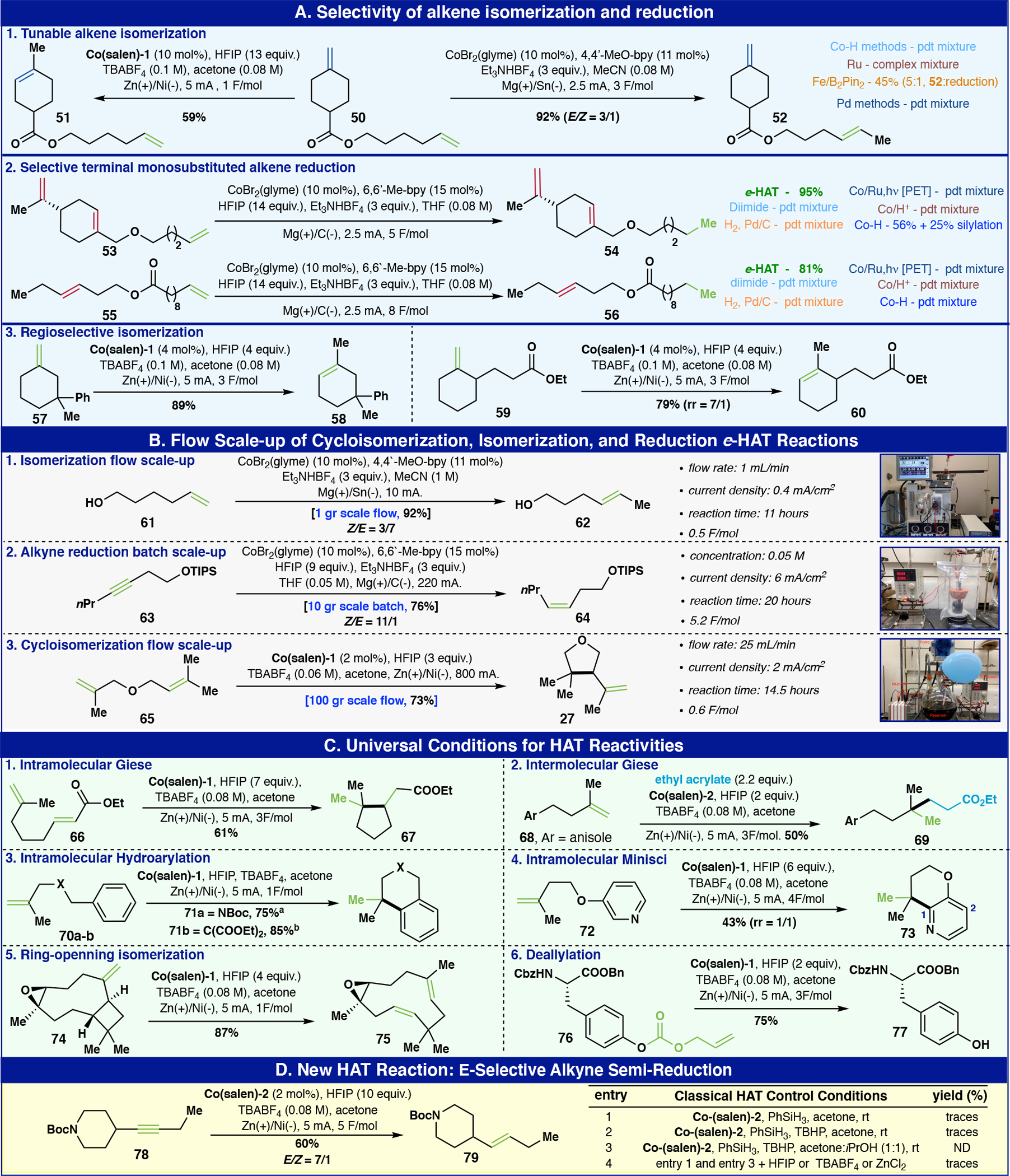
Selectivity, scalability, and reactivity of e-HAT. a HFIP (7 equiv.), TBABF4 (0.08 M) b HFIP (2 equiv.), TBABF4 (0.08 M).
A different and useful case of e-HAT chemoselectivity is exemplified with the isomerization of 1,1-disubstituted olefins to afford the less hindered isomerization products (Figure 4A–3). In the case of compound 57, the double bond migration process has essentially two unpredictable directions to form different trisubstituted alkenes. By applying the Conditions B (Co(salen)-1/HFIP) conditions with compound 57, the regioselectivity control exclusively furnished product 58 (89%). Such regioselectivity could be explained through steric discrimination as guided by the catalyst. To strengthen this hypothesis, compound 59 was also tested under the same conditions; although there is a driving force to form the tetrasubstituted thermodynamic alkene, the kinetically favored trisubstituted alkene was identified as the major product with a 7:1 isomeric ratio.
Next, the scalability of e-HAT was evaluated in both batch and recycle flow on three different transformations; isomerization, alkyne reduction, and cycloisomerization (Figure 4B). The isomerization reaction with Conditions A was tested with compound 61 on a 1 g scale using the commercial Vapourtec ion recycle flow system (Figure 4B–1). By keeping the same parameters with 0.8 mA/cm2 current density and 1 ml/min flow rate, the product was obtained in 92% yield. In a similar fashion, the alkyne reduction was demonstrated on a 10 g scale using a batch setup to achieve a complete conversion of 63 and obtain the desired product (64) with a 76% isolated yield (11:1 of E:Z isomeric ratio, Figure 4B–2). Finally, the cycloisomerization reaction with Conditions B was conducted on a 100 g scale with compound 65 using a recycle flow apparatus containing four reaction cells (Figure 4B–3). After optimization, 100 grams of 65 were successfully converted to product 27 by keeping the same current value of 2 mA/cm2 (compared to 0.2 mmol scale) with only of 0.6 F/mol required to achieve complete conversion (73% isolated yield).
The direct hydrofunctionalization of saturated systems represents an area where HAT-based transformations have found extensive application in synthesis.7 Accordingly, the e-HAT could be implemented under a universal set of conditions to achieve a wide range of classical HAT reactions (Figure 4C). By simply using a slight modification of Conditions B, intramolecular HAT-Giese (67), hydroarylation (71), hydropyridination (73), retroisomerization of alkene-conjugated strained-rings (75), and deallylation as a general deprotection method (77) were achieved. For the intermolecular HAT-Giese with ethyl acrylate to form a quaternary carbon (50%), a modification of Conditions B was employed wherein 2 mol% of Co(tetramethyl-ethanediyl salen) (Co(salen)-2) enabled the reaction to proceed, whereas no product was observed with Co(salen)-1. This phenomenon might be explained by the efficient formation of the carbon-centred radical instead of the cobalt-carbon bond with Co(salen)-2 due to geometrical differences in the cobalt catalysts (see SI for mechanistic rationale – Computation Section).41
As a testament to the unique tunability of e-HAT, a challenging trans-selective alkyne semi-reduction was achieved using Conditions B by slightly modifying the amount of HFIP to 8 equivalents and passing through 5 F/mol of electricity. Alkyne 78 was therefore reduced to the corresponding trans-alkene in 60% isolated yield and high geometric selectivity (7:1 – E/Z). Interestingly, by performing the analogous controls with HAT chemistry using silane and the same Co(salen)-2 complex, we found that such reactivity failed to translate, once again highlighting a unique feature of the e-HAT platform (Figure 4D). The direct, E-selective reduction of alkynes can currently only be accomplished using either forcing Birch-like conditions (Li/NH3)42 or by enlisting an expensive Ru-based catalysis system.43
To further understand the e-HAT processes and support the critical steps of the proposed mechanism, cyclic voltammetry (CV), UV−vis spectroelectrochemistry, DFT computations, and kinetic analysis were employed. First, we investigated the cyclic voltammetry profiles of the Co(salen)-1 isomerization system. As shown in Figure 5B–1, the CV profile of Co(Salen)-1 at 50 mV/s showed a reversible redox peak at −1.98V (vs. Fc/Fc+) corresponding to the Co(II/I) redox couple (step A). The addition of HFIP to the Co(Salen)-1 solution resulted in the observation of heightened current response. This is expected to arise from an ECECcat process corresponding to the generation of H2 (see SI, for detailed discussion – Figure S44).24,25 Such an observation is suggestive of in-situ protonation of the low-valent Co(I) to generate the Co(III)-H (step B). The addition of both HFIP and alkene to the Co(salen)-1 solution (Conditions B) results in a slight dampening of the current response. This could indicate that the alkene is reacting with the Co-H species to catalyze the isomerization reaction, diverting the catalyst from the H2 generation cycle (step C). To further support the proposed mechanism of the Co(salen)-1 system, we sought to observe the respective catalyst resting states with in-situ UV–vis spectroelectrochemistry (Figure 5B–2). Accordingly, we examined the spectroelectrochemical properties of the Co(salen)-1 under the olefin isomerization reaction mixture. Application of a stepwise reducing potential to a Co(II)-salen solution found modest decreases in absorbance for the electronic transitions at 416 nm and 492 nm, which could result from either the direct reduction of the metal center to a Co(I)-salen (Step A) or redox non-innocence of the salen backbone (Figure 5B–2, and see SI). Reduction of Co(salen)-1 at −2.03 V in the presence of HFIP led to the complete disappearance of the Co(II)-salen electronic spectral features and appearance of a single UV-vis absorbance at 375 nm. Based on our previous observations, upon application of reducing potentials, we speculate that this new spectral feature at 375 nm could arise from the presence of a Co-hydride catalyst resting state (Step B), a commonly invoked intermediate within the field of Co-promoted HER catalysis.44,45 Intriguingly, spectroelectrochemical studies of the Co(salen)-1 in the presence of HFIP and alkene lead to the same absorbance feature at 375 nm during active electrocatalysis. Presumably this putative Co-hydride could be competent for both Co-promoted HER as well as Co-catalyzed olefin isomerization, thus appearing in both spectroelectrochemical experiments as a plausible catalyst resting state.
Figure 5:
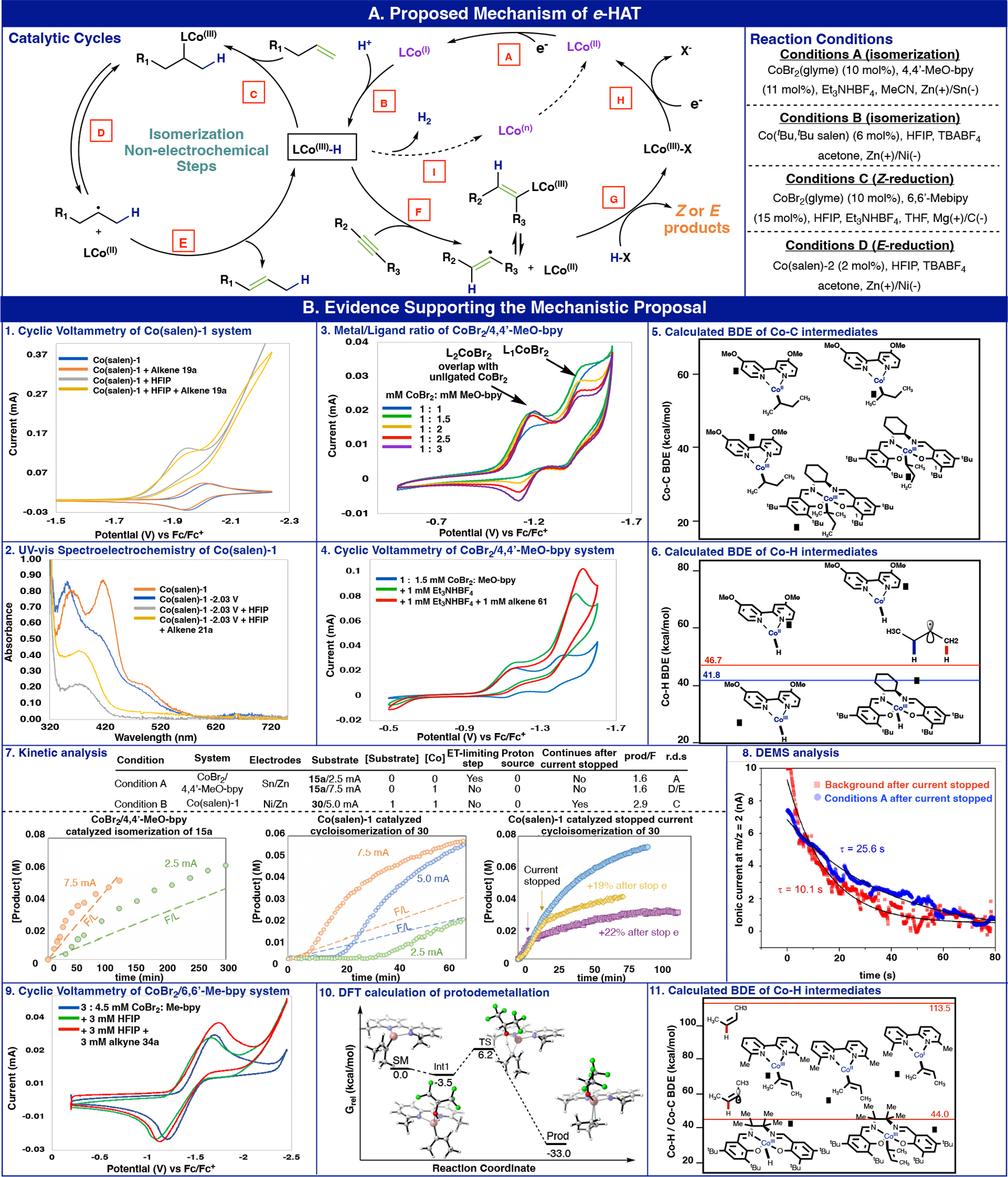
Mechanistic study (A) Proposed Mechanism pathway of e-HAT. (B) Evidence supporting the mechamstic proposal using cyclic voltammetry, UV-vis spectroelectrochemistry, kinetic analysis, DEMS, BDE and DFT calculations.
To investigate the CoBr2/4,4’-dimethoxybipyridine isomerization system (Conditions A), the ligation state of the active cobalt catalyst was first identified (Figure 5B–3). A 1:1 mixture of CoBr2 and 4,4’-dimethoxybipyridine resulted in a CV profile with two distinct reduction peaks for Co(II) to Co(I) (Figure 5A, step A). These peaks were preliminarily assigned as the bisligated L2CoBr2 (which overlaps with unligated CoBr2) for the less negative reduction peak and the monoligated L1CoBr2 for the more negative reduction peak. This assignment is consistent with previous studies showing that the reduction potential of bisligated cobalt complexes is in general, less negative than its corresponding monoligated species.46 Next, CV studies were performed with adding concentration of 4,4’-dimethoxybipyridine. At 1:1.5 Co/ligand ratio, both reduction peaks increased. The addition of more ligand caused the peak assigned as monoligated complex to decrease. Due to the overlapping peak on the reduction wave associated with the bisligated system, the oxidation wave was examined (since the reduction of unligated CoBr2 is irreversible, see SI – Figure S50), wherein an increased oxidation peak was observed upon increasing ligand concentration beyond 1.5 equiv. These studies provide evidence for the preliminary assignment outlined above. Next, the effect of the proton source and the alkene substrate on the CV behavior of the Cobalt catalyst was studied. Using a 1:2 mM ratio of CoBr2/ligand where both the mono and bisligated species were present, the effect of adding 1 mM Et3NHBF4 was investigated (Figure 5B–4). A significant increase in peak current was observed for the monoligated cobalt complex, and a negligible change was observed for the bisligated species. These denote that the monoligated cobalt complex is active towards reaction with Et3NHBF4 to generate a Co-hydride intermediate (Figure 5A, step B). The addition of monosubstituted alkene to this solution resulted in a heightened current response indicative of a chemical reaction with alkene that regenerates the active cobalt catalyst. We therefore propose this as a series of chemical steps for the isomerization of alkenes (Figure 5A, steps C to E).
DFT computational analysis provided additional evidence for the proposed mechanism of olefin isomerization. Based on the divergent reactivity of the two complexes throughout the synthetic studies, it stands to reason that the salen derived catalysts operate with a different general mechanism than that of the bipyridine system. To explore this, the bond dissociation energies of various possible intermediates in such isomerization processes were calculated for both systems. According to the previous analysis, the reaction is initiated via Co–H generation, followed by a HAT onto the olefin to form an alkyl radical which may undergo a radical pair collapse to form an alkyl-cobalt species. The alkyl-cobalt intermediates of salen ligands contain weak Co–C bonds (27 kcal/mol for secondary carbon, 20 kcal/mol for tertiary carbon, Figure 5B–5) that resemble diradicals.47 Accordingly, we propose that steps C and E (Figure 5A) proceed via H-atom transfer with Co(salen)-1 system. Similarly, Figure 5B–6 shows that the strength of the Co–H bond in the Co(salen)–hydride complex indeed matches the C–H bonds of the substrate, which further support the thermodynamic feasibility of the HAT pathway.38 In the case of the Co-bipyridine system (Conditions A), the strong Co–C bonds of the putative intermediates (Figure 5B–6) are perhaps more consistent with an organometallic process of inner-sphere migratory insertion and β-hydride elimination. Consistent with this conclusion, the bipyridine Co-hydride species potentially involved in an HAT version of step C contain Co–H bonds much stronger than the substrates C–H bonds (CoI and CoII, Figure 5B–5) while those possibly involved in step E contain Co–H bonds much weaker than the substrate’s C–H bonds (CoIII, Figure 5B–5). Both assertions point to a thermodynamically unfavorable HAT pathway in the presence of bipyridine ligands.
Kinetic studies carried out on the isomerization of 5-phenyl-1-pentene (15a) using the Co-bipyridine system (Conditions A) and the cycloisomerization of 30 using the Co(salen)-1 system (Conditions B) revealed intriguing differences between the two cases (Figure 5B–7). For Conditions B, the reaction exhibits first order kinetics in [substrate], while for Conditions A, the reaction is zero order in [substrate]. Neither Conditions A nor Conditions B are influenced by the concentration of the proton source. These observations suggest that the rate-determining step for Conditions A is step D/E and for Conditions B is step C. The reaction is first order in [Co] under Conditions B, while under Conditions A, the reaction exhibits an unusual zero-order dependence on [Co] at low-current (2.5 mA). No evidence of deposition of Co on the electrode was found, precluding the zero-order dependence being due to an active surface bound Co species. Another possible explanation could be that at low current the electrons released are insufficient to fully engage all the solution Co species, in which case increasing the catalyst concentration would not influence rate. This hypothesis was confirmed by carrying out reactions at higher current, where the reaction under Conditions A becomes first order in [Co].
The reaction rate under Conditions A is not influenced by current from 5.0–7.5 mA, while under Conditions B, the rate increases proportionally with increasing current from 2.5 to 5.0 mA and is not influenced by current from 5.0–10 mA. Both catalyst systems exhibit an induction period, the length of which decreases with increasing current (middle plot in Figure 5B–7). For Conditions B, the induction period could be removed by pre-activating the catalyst with electrolysis approximately proportional to the catalyst concentration. Neither system requires 1F/mol substrate reacted, and thus both are substoichiometric in electrons (left and middle plots, Figure 5B–7).
These kinetic results suggest subtle differences between the two catalyst systems in the isomerization mechanism presented in Figure 5A. The mechanism consists of two coupled cycles; the product turnover is shown on the left, and the electrochemical catalyst activation cycle is shown in purple. The isomerization product cycle can theoretically be sustained in the absence of electrochemistry, consistent with the fact that the reaction with either catalyst requires less than 1 F/mol substrate. Turnover in the cycloisomerization reaction using Co(salen)-1 continues, albeit more slowly, even after the current is stopped (right plot in Figure 5B–7). However, reactions using Co-bipyridine do not proceed further when the current is stopped. Differential electrochemical mass spectrometry (DEMS) enables operando measurement with gaseous or volatile products and was employed to further investigate such phenomena in the Co-bipyridine system.48 The transient response behaviors of H2 mass spectrometric signals were studied after removing the applied current. With only a proton source, the hydrogen formation was terminated and hydrogen signals displayed an obvious exponential decay with similar relaxation time (8–10 seconds), independent of the applied currents (see SI, Figure S64). Therefore, the larger relaxation time with addition of the Co catalyst and alkene (Conditions A) compared with only a proton source suggested extra hydrogen release from Co–H intermediate after the current was stopped (Figure 5B–8). This DEMS observation served as compelling evidence for the existence of Co hydride intermediate and its conversion back to Co(II) after termination of the electrolysis. In the absence of current, LCo(III)–H cannot go through steps A and B, and the cycle becomes stalled after of Co(III)–H conversion to Co(II) via step I.
The Z-selective semi-reduction of alkynes using CoBr2/6,6’-dimethylbipyridine (Conditions C) was also investigated. Using a mixture of 1:1.5 ratio of CoBr2/ligand (predominantly monoligated, and similar to the ratio in the reaction conditions), the effect of HFIP and alkyne addition (Figure 5B–9) was studied. The addition of 1 equivalent of HFIP showed a shift in the cathodic peak, denoting a chemical step that generates a new electroactive cobalt intermediate proposed as Co(III)-H (Figure 5A, step B). The addition of alkyne to this mixture resulted in an increase in cathodic peak current with reduction potential similar to the active catalyst. This denotes a chemical reaction of the cobalt-hydride intermediate with the alkyne accompanied by regeneration of the monoligated cobalt(II) catalyst (Figure 5A, steps F to H). Computational analysis corroborated this proposed mechanism of proton migration from HFIP to the coordinated alkyne (Figure 5B–10, step G) as it demonstrated feasible energetics. The barrier for such a process is notably low (ΔG‡=9.7 kcal/mol) as it proceeds through a concerted mechanism (for discussion on multiple spin and oxidation states for cobalt complexes, see SI Computation Section). These values provide support for step G of the alkyne Z-reduction proposed catalytic cycle. A mechanistic comparison of the E-selective (Condition D) and Z-selective (Condition C) alkyne reductions was demonstrated by BDE analysis (Figure 5B–11). Consistent with our argument made for Figure 5B–5 and 6, the relatively weak Co–H (42.4 kcal/mol) and Co–C (37.8 kcal/mol) bond strength in the Co-Salen complexes (Condition D) suggest that a radical type HAT pathway is more likely than the organometallic pathway, which favors the E-reduction of the alkyne.
Cobalt-hydride species have found widespread uses for the derivatization and functionalization of unsaturated C-–C bonds in complex molecule construction, usually via a net hydrogen atom transfer. Studies in this area continue to the present day; the contribution reported herein affords a new perspective on how an electrocatalytic approach inspired by decades of energy storage precedent can be leveraged in the context of efficient cobalt-hydride generation with discreet applications in modern organic synthesis. Such an approach not only offers benefits in terms of sustainability and efficiency but also enables enhanced functional group tolerance, unique chemoselectivity, and tunable reactivity. This electroreductive protocol can be performed in an undivided cell, on multiple scales, without strict removal of air or water, and in the absence of expensive silanes/boranes or stoichiometric oxidants. Ten different reactions spanning isomerization, reduction and hydrofunctionalization manifolds across dozens of substrates demonstrate the broad scope of this electrochemical entry into Co–H chemistry. Finally, mechanistic studies including computational analysis, cyclic voltammetry, DEMS, Kinetic studies, and UV-vis measuments provide a comprehensive understanding of ligand-controlled Co–H generation, proceeding via low valent metal intermediates, and their corresponding selective e-HAT reactions.
Supplementary Material
Acknowledgement
This work was supported by the NSF Center for Synthetic Organic Electrochemistry, CHE-2002158.. S.G. thanks the Council for Higher Education, Fulbright Israel and Yad Hanadiv for the generous fellowships. A.B. thanks the Austrian Science Fund (FWF) for an Erwin Schrödinger Fellowship (J 4452-N). H.-J.Z. thanks the SIOC fellowship. C. A. M. thanks the National Institute of General Medical Sciences of the National Institutes of Health (K99GM140249). The authors are grateful to D.-H. Huang and L. Pasternack (Scripps Research) for assistance with the NMR spectroscopy, to J. Chen, B. Sanchez and E. Sturgell (Scripps Automated Synthesis Facility) for assistance with HPLC, high-resolution mass spectroscopy and LCMS. We thank Dr. S. Harwood, Dr. Y. Kawamata, Dr. K.X. Rodriguez and C. Bi for helpful advice and suggestions. We also thank Prof. Q. Liu and X. Liu (Tsinghua University) for the helpful discussion.
Footnotes
Competing interest
The authors declare no financial or non-financial competing interest.
Data availability
The data that support the findings of this study are available from the corresponding authors upon reasonable request.
References:
- 1.Norton JR, & Sowa J Introduction: metal hydrides. Chem. Rev. 116, 8315–8317 (2016). [DOI] [PubMed] [Google Scholar]
- 2.Luo GG et al. Recent progress in ligand-centered homogeneous electrocatalysts for hydrogen evolution reaction. Inorg. Chem. Front. 6, 343–354 (2019). [Google Scholar]
- 3.Margarit CG, Asimow NG, Thorarinsdottir AE, Costentin C, & Nocera DG Impactful Role of Cocatalysts on Molecular Electrocatalytic Hydrogen Production. ACS Catal 11, 4561–4567 (2021). [Google Scholar]
- 4.Shevick SL et al. Catalytic hydrogen atom transfer to alkenes: a roadmap for metal hydrides and radicals. Chem. Sci. 11, 12401–12422 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Armstrong KC, & Waymouth RM Electroreduction of Benzaldehyde with a Metal–Ligand Bifunctional Hydroxycyclopentadienyl Molybdenum (II) Hydride. Organometallics, 39, 4415–4419 (2020). [Google Scholar]
- 6.Vesborg PC, Seger B, & Chorkendorff IB Recent development in hydrogen evolution reaction catalysts and their practical implementation. J. Phys. Chem. 6, 951–957 (2015). [DOI] [PubMed] [Google Scholar]
- 7.Crossley SW, Obradors C, Martinez RM, & Shenvi RA Mn-, Fe-, and Co-catalyzed radical hydrofunctionalizations of olefins. Chem. Rev. 116, 8912–9000 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wiedner ES et al. Thermodynamic hydricity of transition metal hydrides. Chem. Rev. 116, 8655–8692 (2016). [DOI] [PubMed] [Google Scholar]
- 9.Hu Y, Shaw AP, Estes DP, & Norton JR (2016). Transition-metal hydride radical cations. Chem. Rev. 116, 8427–8462. [DOI] [PubMed] [Google Scholar]
- 10.Eisenberg DC, & Norton JR Hydrogen-atom transfer reactions of transition-metal hydrides. Isr. J. Chem 31, 55–66 (1991). [Google Scholar]
- 11.Puls F, Linke P, Kataeva O, & Knölker HJ Iron-Catalyzed Wacker-type Oxidation of Olefins at Room Temperature with 1, 3-Diketones or Neocuproine as Ligands. Angew. Chem, Int. Ed. (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Isayama S, & Mukaiyama T A New Method for Preparation of Alcohols from Olefins with Molecular Oxygen and Phenylsilane by the Use of Bis (acetylacetonato) cobalt (II). Chem. Lett. 18, 1071–1074 (1989). [Google Scholar]
- 13.Gui J et al. Practical olefin hydroamination with nitroarenes. Science, 348, 886–891 (2015). [DOI] [PubMed] [Google Scholar]
- 14.Lo JC, Gui J, Yabe Y, Pan CM, & Baran PS Functionalized olefin cross-coupling to construct carbon–carbon bonds. Nature, 516, 343–348 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gaspar B, & Carreira EM Mild cobalt-catalyzed hydrocyanation of olefins with tosyl cyanide. Angew. Chem. In. Ed. 46, 4519–4522 (2007). [DOI] [PubMed] [Google Scholar]
- 16.Shigehisa H, Aoki T, Yamaguchi S, Shimizu N, & Hiroya K Hydroalkoxylation of unactivated olefins with carbon radicals and carbocation species as key intermediates. J. Am. Chem. Soc. 135, 10306–10309 (2013). [DOI] [PubMed] [Google Scholar]
- 17.Leggans EK, Barker TJ, Duncan KK, & Boger DL Iron (III)/NaBH4-mediated additions to unactivated alkenes: synthesis of novel 20ʹ-vinblastine analogues. Org. Lett. 14, 1428–1431 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Song L et al. Dual electrocatalysis enables enantioselective hydrocyanation of conjugated alkenes. Nat. Chem. 12, 747–754 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wells AS On the perils of unexpected silane generation. Org. Process Res. Dev. 14, 484–484 (2010). [Google Scholar]
- 20.Yu K, Yao F, Zeng Q, Xie H, & Ding H Asymmetric Total Syntheses of (+)-Davisinol and (+)-18-Benzoyldavisinol: A HAT-Initiated Transannular Redox Radical Approach. J Am. Chem. Soc. 143, 10576–10581 (2021). [DOI] [PubMed] [Google Scholar]
- 21.Godfrey NA, Schatz DJ, & Pronin SV Twelve-step asymmetric synthesis of (−)-nodulisporic acid C. J. Am. Chem. Soc. 140, 12770–12774 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kellett RM, & Spiro TG Cobalt (I) porphyrin catalysts of hydrogen production from water. Inorg. Chem. 24, 2373–2377 (1985). [Google Scholar]
- 23.Zhang W, Cui L, & Liu J Recent advances in cobalt-based electrocatalysts for hydrogen and oxygen evolution reactions. J. Alloy. Compd. 821, 153542 (2020). [Google Scholar]
- 24.Wiedner ES, & Bullock RM Electrochemical detection of transient cobalt hydride intermediates of electrocatalytic hydrogen production. J. Am. Chem. Soc. 138, 8309–8318 (2016). [DOI] [PubMed] [Google Scholar]
- 25.Marinescu SC, Winkler JR, & Gray HB Molecular mechanisms of cobalt-catalyzed hydrogen evolution. PNAS 109, 15127–15131 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Beyene BB, Mane SB, & Hung CH Electrochemical hydrogen evolution by cobalt (II) porphyrins: effects of ligand modification on catalytic activity, efficiency and overpotential. J. Electrochem. Soc. 165, 481 (2018). [Google Scholar]
- 27.Queyriaux N, Jane RT, Massin J, Artero V, & Chavarot-Kerlidou M Recent developments in hydrogen evolving molecular cobalt (II)–polypyridyl catalysts. Coordin. Chem. Rev. 304, 3–19 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kapat A, Sperger T, Guven S, & Schoenebeck F E-Olefins through intramolecular radical relocation. Science 363, 391–396 (2019). [DOI] [PubMed] [Google Scholar]
- 29.Crossley SW, Barabé F, & Shenvi RA Simple, chemoselective, catalytic olefin isomerization. J. Am. Chem. Soc. 136, 16788–16791 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li G et al. Radical isomerization and cycloisomerization initiated by H• transfer. J. Am. Chem. Soc. 138, 7698–7704 (2016). [DOI] [PubMed] [Google Scholar]
- 31.Kingston C et al. A Survival Guide for the “Electro-curious”. Acc. Chem. Res. 53, 72–83 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gao Y et al. Electrochemical Nozaki–Hiyama–Kishi Coupling: Scope, Applications, and Mechanism. J. Am. Chem. Soc. 143, 9478–9488, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gnaim S et al. Electrochemically driven desaturation of carbonyl compounds. Nat. Chem. 13, 367–372 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lo JC et al. Fe-catalyzed C–C bond construction from olefins via radicals. J. Am. Chem. Soc. 139, 2484–2503 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Felpin FX, & Fouquet E A Useful, Reliable and Safer Protocol for Hydrogenation and the Hydrogenolysis of O-Benzyl Groups: The In Situ Preparation of an Active Pd0/C Catalyst with Well-Defined Properties. Chem. Eur. J. 16, 12440–12445 (2010). [DOI] [PubMed] [Google Scholar]
- 36.Friedfeld MR, Margulieux GW, Schaefer BA, & Chirik PJ Bis (phosphine) cobalt dialkyl complexes for directed catalytic alkene hydrogenation. J. Am. Chem. Soc. 136, 13178–13181 (2014). [DOI] [PubMed] [Google Scholar]
- 37.Liu X et al. Cobalt-catalyzed regioselective olefin isomerization under kinetic control. J. Am. Chem. Soc. 140, 6873–6882 (2018). [DOI] [PubMed] [Google Scholar]
- 38.Kamei Y et al. Silane-and peroxide-free hydrogen atom transfer hydrogenation using ascorbic acid and cobalt-photoredox dual catalysis. Nat. Commun. 12, 1–9 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Van der Puyl V, McCourt RO, & Shenvi RA Cobalt-catalyzed alkene hydrogenation by reductive turnover. Tetrahedron Lett. 72, 153047 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Raya B, Biswas S, & RajanBabu TV Selective cobalt-catalyzed reduction of terminal alkenes and alkynes using (EtO)2Si(Me)H as a stoichiometric reductant. ACS Catal 6, 6318–6323 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yin YN, Ding RQ, Ouyang DC, Zhang Q, & Zhu R Highly chemoselective synthesis of hindered amides via cobalt-catalyzed intermolecular oxidative hydroamidation. Nat. Commun. 12, 1–10 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Benkeser RA, Schroll G, & Sauve DM Reduction of organic compounds by lithium in low molecular weight amines. II. Stereochemistry. Chemical reduction of an isolated non-terminal double bond. J. Am. Chem. Soc. 77, 3378–3379 (1955). [Google Scholar]
- 43.Fürstner A trans-Hydrogenation, gem-Hydrogenation, and trans-Hydrometalation of Alkynes: An Interim Report on an Unorthodox Reactivity Paradigm. J. Am. Chem. Soc. 141, 11–24 (2018). [DOI] [PubMed] [Google Scholar]
- 44.Wiedner ES, & Bullock RM Electrochemical detection of transient cobalt hydride intermediates of electrocatalytic hydrogen production. J. Am. Chem. Soc. 138, 8309–8318 (2016). [DOI] [PubMed] [Google Scholar]
- 45.Walaijai K, Cavill SA, Whitwood AC, Douthwaite RE, & Perutz RN Electrocatalytic Proton Reduction by a Cobalt (III) Hydride Complex with Phosphinopyridine PN Ligands. Inorg. Chem. 59, 18055–18067 (2020). [DOI] [PubMed] [Google Scholar]
- 46.Hickey DP et al. Investigating the role of ligand electronics on stabilizing electrocatalytically relevant low-valent Co (I) intermediates. J. Am. Chem. Soc. 141, 1382–1392 (2019). [DOI] [PubMed] [Google Scholar]
- 47.Qi XJ, Li Z, Fu Y, Guo QX, & Liu L anti-Spin-Delocalization Effect in Co– C Bond Dissociation Enthalpies. Organometallics 27, 2688–2698 (2008). [Google Scholar]
- 48.Yang Y et al. Operando methods in electrocatalysis. ACS Catal. 11, 1136–1178 (2021). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding authors upon reasonable request.