

Abstract
Recent advances in CRISPR-Cas9 techniques, especially the discovery of base and prime editing, have significantly improved our ability to make precise changes in the genome. We hypothesized that modulating certain endogenous pathway cells could improve the action of those editing tools in mammalian cells. We established a reporter system in which a small fragment was integrated into the genome by prime editing (PE). With this system, we screened an in-house small-molecule library and identified a group of histone deacetylase inhibitors (HDACi) increasing prime editing. We also found that HDACi increased the efficiency of both cytosine base editing (CBE) and adenine base editing (ABE). Moreover, HDACi increased the purity of cytosine base editor products, which was accompanied by an upregulation of the acetylation of uracil DNA glycosylase (UNG) and UNG inhibitor (UGI) and an enhancement of their interaction. In summary, our work demonstrated that HDACi improves Cas9-mediated prime editing and base editing.
Keywords: MT: RNA/DNA Editing, CRISPR-Cas9, HDAC inhibitors, prime editing, base editing, UNG, product purity
Graphical abstract
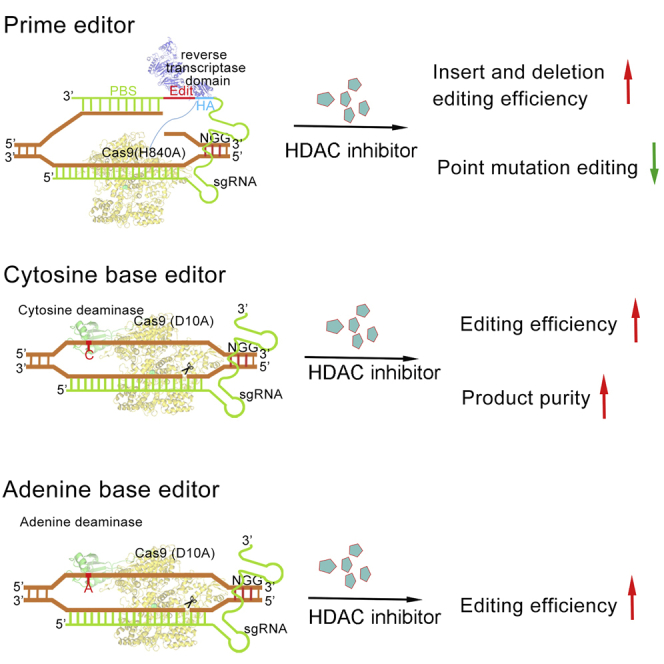
CRISPR-Cas9-derived prime editors and base editors are powerful tools in precise genome editing. We demonstrate that HDAC inhibition increases the efficiency of prime editors and base editors, which advances our understanding of regulating endogenous mechanisms to improve genome editing.
Introduction
The CRISPR systems, containing CRISPR-associated (Cas) proteins and their interacting RNAs, enable microbes to prevent the invasion of foreign phages and plasmids.1 The CRISPR systems are easily reprogrammable for genome-editing purposes, providing powerful tools for basic biomedical research and clinical translation.2,3 Recently, the development of base editing (BE) and prime editing (PE) technique enables targeted base conversions or the introduction of small genetic changes, in an efficient and irreversible way, without causing robust double-stranded DNA breaks (DSBs), making it practicable to correct small pieces of genetic lesions in inherited diseases.4, 5, 6
Although the resulting edits of each editor are quite predictable and repeatable, detailed mechanisms underlying the editing process have remained largely unknown. Especially in the case of editing mammalian genomes, which are frequently occupied with a multitude of DNA-binding factors and folded into various degrees of compactions, the entry of CRISPR editors to their target loci is not straightforward.7 Several parameters, including chromatin folding and histone occupation and modification, are known to have an effect on the efficiency of wild-type Cas9-mediated genome editing.8, 9, 10 Beyond binding to and cleaving their targets, Cas9 proteins could be also engineered to produce local base deamination (base editing) and single-stranded DNA break (SSB) and the subsequent single-stranded DNA (ssDNA) extension (prime editing).4, 5, 6 In most cases, these genetic lesions are repaired by various DNA damage repair mechanisms.11, 12, 13 Depending on the types of the lesions, a given mechanism could increase or decrease the desired editing results. For example, inhibition of the non-homologous end-joining (NHEJ) pathway decreases wild-type Cas9-induced indel formation while increasing homology-directed repair (HDR)-mediated fragment insertion.14,15 Therefore, it is reasonable to hypothesize that Cas9-mediated base or prime editing could be improved by modulating endogenous pathways.
Here, we adopt a simple reporter system to screen candidates of small molecules that might affect the editing efficiency of prime editors in HEK293T cells. In the reporter system, a small fragment was inserted into the genome through prime editing, the efficiency of which can be easily read out by regular polymerase chain reaction (PCR). We conducted a focused screen of small molecules that target chromatin modifiers, cell-cycle progression, mitosis, DNA damage response, or DNA repair. The screening identified a group of HDAC inhibitors (HDACi) that obviously increased the efficiency of the PE reporter system. HDAC inhibitors are known to be able to increase the acetylation levels of histones or other non-histone proteins, promoting an open chromatin state.16 In-depth testing with additional target loci and multiple PE strategies revealed that the improvement induced by HDAC inhibitors was dependent on different targets and editing types. We also tested the effect of HDAC inhibitors on base editing and found that HDAC inhibition generally increased the efficiency of both cytosine base editing (CBE) and adenine base editing (ABE), which agrees with recent findings.17,18 In addition, we observed that HDAC inhibition increased the purity of CBE products. Mechanistically, we found that HDAC inhibition upregulated the acetylation levels of both uracil DNA glycosylase inhibitor (UGI) and uracil DNA glycosylase (UNG) and enhanced their interaction, which possibly inhibited the action of UNG to remove urine in the edited DNA strand. Collectively, our results reveal a group of clinically safe molecules improving the efficiency of prime and base editors, which should improve the application of these tools.
Results
Identification of HDAC inhibitors enhancing prime editing in reporter
As a first step toward the screening, we established a PE-based reporter system in which a 40 bp DNA fragment containing loxP sequence was knocked into the HEK3 loci in HEK293T cells using PE3 strategies (Figure 1A). The efficiency of the knockin was readily visualized by PCR amplification and subsequent gel electrophoresis and ImageJ analysis. On the basis of this reporter system, we screened an in-house compound library containing 54 small molecules targeting epigenetics, cell cycle, replication, and DNA damage repair (Table S1). The screening identified several molecules that increased the knockin frequency (Table S1). Interestingly, a group of HDAC inhibitors, including nexturastat A (DO1-f-2), abexinostat (DO1-d-4), and vorinostat (DO1-b-9), were among the efficient molecules that gave rise to remarkable improvement of the knockin (Figure 1B). The efficiency of loxP knockin was improved by nexturastat A, abexinostat, and vorinostat by 2.06, 1.60, and 1.57 times, respectively (Figures 1B and 1C). Therefore, these HDAC inhibitors were selected for further analysis.
Figure 1.

Screening of small molecules capable of improving prime editing
(A) Schematic diagram showing the screening strategy. The prime editor was designed to knock in a 40 bp DNA fragment into HEK3 site, and the resulting insertion can be amplified using PCR and quantified using gel electrophoresis. (B) Representative gel electrophoresis and ImageJ analysis of the effect of small molecules on the 40 bp knockin. Knockin bands were distinguishable from wild-type bands by gel electrophoresis (upper panel), and efficiency of the knockin was calculated using ImageJ software (lower panels). Nexturastat A, which improved knockin efficiency, is marked in red. (C) Summary of the effect of 54 small molecules on knockin. Each molecule was used at a concentration of 10 μM different drugs. The improvement ratios were calculated relative to DMSO. Three HDAC inhibitors, nexturastat A (DO1-f-2), abexinostat (DO1-d-4), and vorinostat (DO1-b-9), that significantly improved the efficiency of knockin are labeled by red boxes. Editing efficiencies were measured using gel electrophoresis and grayscale analysis.
HDAC inhibition improves PE-mediated insertion and deletion but not point mutation
As prime editing can produce various types of editions depending on the design of prime editing guide RNAs (pegRNAs), including targeted insertion, deletion, and point mutation, we therefore sought to verify whether these HDAC inhibitors are generally effective for different types of editions in HEK293T cells. We designed all three types of editions for HEK3 loci and examined the effects of HDAC inhibitors on targeted insertion, deletion, and point mutation editing efficiency via high-throughput sequencing (HTS) and Sanger sequencing, respectively. In consistent with the above results generated by gel electrophoresis and ImageJ analysis, we found that HDACi increased the efficiency of 40 bp knockin by 137% (Figure 2A). HDACi also showed an average of 130% increase in 40 bp deletion (Figure 2A). However, to our surprise, HDAC inhibitors did not improve efficiency of PE-mediated point mutation in HEK3 loci but rather inhibited it. Compared with the DMSO-treated group, HDAC inhibitor treatment led to an average 42.7% reduction in point mutation (15.7% versus 9%), with the most significant reduction up to 6% (Figure 2A).
Figure 2.

The effect of HDAC inhibitors on PE-mediated targeted insertions, deletions, and point mutations
(A) Four sites, HEK3, β-actin, VEGFA, and DNMT1, were used to investigate the effect of HDAC inhibitors on three different types of PE, including targeted insertion, deletion, and point mutation. (B) The relative PE efficiency of HDAC inhibitors to DMSO. Data are represented as mean ± SD; asterisks indicate statistically significant differences between DMSO-treated cells and HDAC inhibitor-treated cells (∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001). Knockin and deletion efficiencies were measured using high-throughput sequencing (HTS), and point mutation efficiencies were analyzed using Sanger sequencing and EditR calculating.
Next, we tested the effects of HDACi on an additional 3 target loci, including β-actin, VEGFA, and DNMT1. Consistent with the above observations on HEK3 loci, we found a distinct effect of HDACi on different types of editing. Overall, HDACi has a positive effect on PE-mediated insertion and deletion editions but has a negative effect on point mutation. On β-actin loci, HDACi treatment increased the efficiencies of insertion and deletion editing by 12% and 46%, respectively (Figure 2A). On VEGFA loci, the efficiencies of insertion and deletion were increased by HDACi by 10% and 20%, respectively (Figure 2A). On DNMT1 loci, HDACi treatment increased the insertion efficiency by 35% but did not obviously increase deletion efficiency (Figure 2A). In contrast, HDACi treatment inhibited the editing of point mutation across all 3 target loci (Figures 2A and 2B). The most extensive inhibition was observed on β-actin loci, the editing efficiency of which was reduced by nearly 42%. Therefore, together with the observations on HEK3 loci, these results suggested that HDACi treatment improved the action of prime editing in targeted insertion and deletion but not point mutation.
HDAC inhibition improves both cytosine and adenine base editing
Next, we tested whether those HDAC inhibitors were effective in improving the action of the base editing system in HEK293T cells. Cytosine base editing of four well-characterized endogenous loci was used to determine the effects of HDAC inhibitors. As shown in Figure 3A, C-to-T conversions of targetable positions across all four loci were improved by those inhibitors. Overall, nexturastat A, abexinostat, and vorinostat increased the editing efficiency of the cytosine base editor BE3 by an average of 0.8, 0.73, and 0.93 times, respectively. Taking site 29 as an example, HDACi improved the editing of all targetable cytosines in the putative editing window (Figure S1).
Figure 3.

HDAC inhibitor treatment increased the editing efficiency of both BE3 and ABE7.10
(A) Effect of nexturastat A, abexinostat, and vorinostat on C-to-T editing of BE3 at four loci. The control group was treated with DMSO. (B) Effect of nexturastat A and abexinostat on A-to-G editing of ABE7.10 at 7 loci. (C) Relative efficiency of the editing of BE3 or ABE7.10 by nexturastat A and abexinostat treatment to that by DMSO treatment. The efficiencies of both cytosine and adenosine base editing were quantified using Sanger sequencing and were analyzed using EditR. Each experiment was repeated at least three times. Data are represented as mean ± SD; asterisks indicate statistically significant differences between DMSO-treated cells and HDAC inhibitor-treated cells (∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001). Base editing efficiencies were analyzed using Sanger sequencing and EditR calculation.41
Next, we sought to dissect which HDAC was responsible for the improvement. We first analyzed the targets of those positive inhibitors and found that although nexturastat A was a highly selective HDAC6 inhibitor, both abexinostat and vorinostat had a wide range of targets (Figure S2A). It seemed, at this stage, that these small molecules might improve the editing efficiency of BE3 C-to-T conversion by inhibiting HDAC6. However, the concentration of nexturastat A used in the screening experiments was 10 μM, which could also inhibit other HDACs, such as HDAC1 (3.02 μM) and HDAC2 (6.92 μM), beyond HDAC6. In order to rule out the possibility that nexturastat A improved editing efficiency by inhibiting HDAC1 or HDAC2, we conducted a series of tests with different concentrations of nexturastat A. It turned out that nexturastat A can improve the base editing of BE3 only when its concentration exceeded 3 μM (Figure S2B), suggesting that inhibition of HDAC1 and HDAC2 but not HDAC6 was responsible for the improvement by Nexturastat A.
Consistent with this notion, previous studies have found that HDAC1 and HDAC2 inhibitors increased the indel frequencies of wild-type Cas9. To test this possibility, we selected another HDAC inhibitor, E14, that inhibits HDAC1, HDAC2, and HDAC3 at a half maximal inhibitory concentrations (IC50) of 1.8, 3.6, and 3.0 nM, respectively, in cell-free assays.19 We found that E14 treatment also significantly increased C-to-T base editing across the above four target loci (Figure S2C). In addition, this observation was further confirmed by a more specific HDAC inhibitor, romidepsin, which is highly selective for HDAC1 and HDAC2, with IC50 values of 36 nM for HDAC1 and 47 nM for HDAC2 in cell-free assays20 (Figure S2C). Therefore, these analyses suggested that inhibition of HDAC1 and HDAC2 but not HDAC6 was responsible for the improvement of base editing. In addition, a parallel comparison of these HDAC inhibitors revealed that their improvement effects on base editing were similar (Figure S2D). We also compared the cytotoxic effects of these inhibitors and found that except for nexturastat A, they are all cytotoxic to HEK293T cells (Figure S3).
To determine how general the improvement is, we tested the performance of HDAC inhibitors on a wide range of loci edited by various cytosine base editors and adenosine base editors. First, we used 7 additional endogenous loci to further characterize the action of HDAC inhibitors on BE3. The results showed that the C-to-T base editing of all loci was extensively improved by HDAC inhibitors (Figure S4). Compared with the DMSO-treated group, nexturastat A treatment showed an average 0.86 times increase and abexinostat showed a 1.22 times increase in BE3-mediated cytosine base editing. Together with observations on the above four target loci, these results suggested that HDAC inhibition could generally improve the editing of BE3. Next, we determined whether HDAC inhibition was also effective for adenosine base editing. We found that the adenosine editing of all 8 target loci tested was improved by HDAC inhibition, among which 7 loci were significant (Figure 3B). On average, nexturastat A increased the editing of ABE by 0.46 times and abexinostat by 0.43 times (Figure 3C). Notably, the improvement in ABE by both inhibitors was lower than that in CBE (Figure 3C). Collectively, these data suggested that HDAC inhibition generally improved the action of both cytosine and adenosine base editing.
Next, we sought to determine if the improvement was also effective on other types of cytosine base editors. As shown in Figure 4A, both inhibitors increased the editing of SaCas9-derived CBE across all 5 loci, with an average increasement of 0.53 times for nexturastat A and 0.51 times for abexinostat (Figures 4A and S5). In addition, the improvement was also obvious in SpCas9-derived non-classical base editors, including AID-,21 target AID-,22 and A3A-fused23 base editors (Figure 4B), single guide RNA (sgRNA)-modified sgBEs24 (Figures S6A and S6B), internally inlaid BE-PIGS25 (Figure S6C), and the simultaneous cytosine and adenine editor CABE (Figures S6D and S6E). To further explore the general utility of HDACi in improving base editing, we tested the effects of HDACi on CBE in different cells, including HCT116 (colorectal cells), hepG2 (hepatocytes), and HeLa (cervical cells). The results showed that HDACi also improved the efficiency of CBE in these cells, although the improvement levels varied by cell type (Figure S7). Taken together, these data demonstrated that the improvement by HDAC inhibition was universal for base editing.
Figure 4.

HDAC inhibitor treatment generally increased a wide range of CBEs
(A) The effect of nexturastat A and abexinostat treatment on the editing of the SaCas9-derived base editor BE3-SaKKH. The control group was treated with DMSO. (B) The effect of nexturastat A and abexinostat treatment on different cytosine deaminase-derived base editors, including AID-BE3, target-AID, and A3A-BE3. The control group was treated with DMSO. The editing efficiency was quantified using Sanger sequencing and was analyzed using EditR. Each experiment was repeated at least three times. Data are represented as mean ± SD; asterisks indicate statistically significant differences between DMSO-treated cells and HDAC inhibitor-treated cells (∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001). Base editing efficiencies were analyzed using Sanger sequencing and EditR calculation.
As HDAC inhibition generally increased base editing, it is therefore conceivable that it also increases off-target editing. To elucidate this, we investigated the performance of HDAC inhibitors on sequence-dependent off-target editing of two well-characterized target sites, HEK4 site and site 31, whose off-target sites were extensively studied.6 As shown in Figures S8A–S8C, HDAC inhibitor treatment increased the editing of the HEK4 and site 31 off-target sites, the level of which was comparable with that of on-target sites. Similarly, we also found that HDAC inhibitor treatment increased the efficiency of sequence-independent off-target editing as determined by R-loop assay26,27 (Figures S8D and S8E).
HDAC inhibition suppressed C-to-G editing by enhancing the interaction between UNG and UGI
Product purity is one of the main features of cytosine base editors, as some deaminated cytosines were converted to G or A other than T (in most cases G), leading to impure editing products. In previous experiments, we noticed that HDAC inhibitor treatment increased the product purity of site 28 (i.e., the ratio of C-to-T conversion to all conversions in HEK293T cells) (Figure 5A). To determine whether this phenomenon is common to different targets, we tested an additional 3 targets that were prone to yield impure products. As shown in Figures 5B and 5C, inhibition of HDAC did increase product purity across all target sites tested. On average, nexturastat A and abexinostat increased the ratio of C-to-T conversion by 0.45 and 0.35 times, respectively, which were accompanied by the abatement of C-to-G conversion. The ratio of C-to-G/A conversion was decreased by 0.44 times (nexturastat A) and 0.35 times (abexinostat).
Figure 5.

HDAC inhibitor treatment increased product purity of BE3
(A) Representative Sanger sequencing results and EditR analysis showing the effect of HDAC inhibitors on the editing of site 28 by BE3. (B) The effects of HDAC inhibitor treatment on the C-to-T (left panel) and C-to-G/A (right panel) conversions of BE3 at 4 genomic loci. (C) Quantification of the product distribution of (B). Base editing efficiency was quantified using Sanger sequencing and analyzed using EditR. Each experiment was repeated at least three times. Data are represented as mean ± SD; asterisks indicate statistically significant differences between DMSO-treated cells and HDAC inhibitor-treated cells (∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001). Base editing efficiencies were analyzed using Sanger sequencing and EditR calculation.
Then we sought to elucidate how HDAC inhibition affects the purity of base editing. The conversion from C to G or A was likely due to the inaccurate repair of the target strand (the strand complementary to the spacer of sgRNA) that was nicked by Cas9 HNH domain during base editing. And possibly, when repairing the nick (ssDNA break), the uridine in the non-target strand that was deaminated from cytosine was supposed to be removed by UNG, generating an abasic site, which in turn induced trans-lesion synthesis of the target strand, leading to incorporation of C or T in positions opposite to abasic site. Finally, the inaccurately incorporated C or T was passed to the non-target strand as G or A by DNA repair or DNA replication mechanisms. Therefore, the formation of abasic site was the first step toward the generation of C-to-G or C-to-A conversion. Consistent with this notion, in cells lacking UNG, C-to-G or C-to-A conversion disappeared.28 Because the CBEs we used contained UGI, a bacterial inhibitor of UNG, our investigation was started with the effect of HDAC inhibition on CBE lacking UGI. We removed UGI from the CBE and tested the effect of HDAC inhibitor on the editing purity of site 28. As expect, the resulting BE-NO-UGI produced much more impure editing than editors harboring UGI. Surprisingly, HDAC inhibition could not improve the product purity of BE-NO-UGI any longer (Figure 6A). This observation was further confirmed by an additional 3 target sites (Figures 6B and 6C).
Figure 6.

The improvement of HDAC inhibitor treatment on product purity of BE3 required the presence of UGI
(A) Representative Sanger sequencing results and EditR analysis showing the effect of HDAC inhibitors on the editing of site 28 by BE-NO-UGI. (B) The effects of HDAC inhibitor treatment on the C-to-T (left panel) and C-to-G/A (right panel) conversions of BE-NO-UGI. (C) Quantification of the product distribution of (B). Each experiment was repeated at least three times. Data are represented as mean ± SD; asterisks indicate statistically significant differences between DMSO-treated cells and HDAC inhibitor-treated cells (∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001). Base editing efficiencies were analyzed using Sanger sequencing and EditR calculation.
Moreover, we also tested the effect of HDAC inhibition on BE-NO-UGI editing two target sites that are not inclined to generate impure product, site 29 and DNMT1 (Figure S9). The results showed that HDAC inhibition slightly improved the editing efficiency of BE-NO-UGI, whereas the improvement was much lower than that of BE3. Together, these observations suggested that UGI was required for the improvement of both editing efficiency and product purity by HDAC inhibition.
It is well known that acetylation modification is a key regulatory mechanism for DNA repair pathways, and many repair players,29 including UNG, were found to contain acetylation modification.30,31 The fact that the improvement of HDAC inhibition on product purity depended on the presence of UGI led us to hypothesize that HDAC inhibition would promote the acetylation of UNG or UGI or both to enhance their interaction, thereby strengthening the locally inhibitory effects of UNG-mediated BER. In fact, when we looked into the acetylation status of UNG in HEK293T cells, we observed a considerable increasement of total lysine acetylation (Figure 7A). Strikingly, we also found that UGI could be acetylated in its lysine residues, the level of which was also increased by HDACi treatment (Figure 7B). Next, we sought to investigate if the increasement in acetylation level of UNG and UGI enhanced their interaction. We performed NanoLuc binary technology (NanoBiT) assay to quantify their interaction (Figure 7C). As shown in Figure 7D, NanoBiT assay revealed that HDACi treatment indeed enhanced the interaction between UNG and UGI, in a dose-dependent manner. Taken together, these results demonstrated that HDAC inhibition promotes the product purity of CBE through increasing the acetylation of UNG and UGI.
Figure 7.

HDAC inhibition increased the acetylation level of UNG and UGI and enhanced their interaction
(A) Western blotting (WB) showing the lysine acetylation level of UNG and UGI. WB analysis for the co-immunoprecipitation (co-IP) of whole-cell proteins from HEK293T control cells or cells transfected with empty or FLAG-UNG plasmids (top panels). WB analysis for input whole-cell proteins (bottom panels). The transfected cells received DMSO or romidepsin treatment as indicated. (B) Quantification of the relative acetylation level of UNG and UGI of cells treated with romidepsin to that of DMSO-treated cells. (C) Schematic diagram showing the NanoBiT assay. NanoLuc topology model 9 consisting of a 10-stranded beta-barrel (left panel). The split point (red arrow) occurs between residues 156th and 157th amino acids, generating a large fragment of 156 amino acids, referred to as 11S, and a small fragment of 11 amino acids, referred to as 114. The interaction of fusion proteins pulled 11S and 114 together to form a functional luciferase (right panel). (D) Dose-dependent enhancement of romidepsin to the interaction between UNG and UGI. Data are represented as mean ± SD; asterisks indicate statistically significant differences between DMSO-treated cells and HDAC inhibitor-treated cells (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001).
Discussion
In this work, we used a prime editing-mediated knockin system to screen a panel of clinically safe drugs that targeted chromatin modifiers, cell-cycle progression, mitosis, DNA damage response, and DNA repair. The screening identified several HDAC inhibitors that were able to increase the editing of the reporter system. Detailed evaluations with other types of base editing and additional loci revealed that HDAC inhibition did not generally increase prime editing efficiency. Rather, its effects were loci and editing type dependent. In addition, we also found that HDAC inhibition generally increased the editing efficiency of various base editing tools, including cytosine, adenosine base editors, and their modified derivates.
During the preparation of this report, it came to our attention that results similar to part of our observations were reported, showing that HDAC inhibition enhances both adenosine and cytosine base editing.17,18 The underlined mechanisms were found to be attributable mainly to increasing the expression levels of proteins and target accessibility.17 Similar mechanisms of HDAC inhibition had also been observed on wild-type Cas9-mediated gene editing, including induction of indels by NHEJ and fragment knockin by HDR.8,9 These results collectively suggest that HDAC inhibition has a general positive effect on Cas9-derived base editing tools, perhaps also ZFN and TALEN.
However, the fact that HDAC inhibition specifically decreased PE-mediated point mutation while increasing PE-mediated insertion and deletion at the same sites (with identical pegRNA and nick sgRNA) indicated that mechanisms other than protein expression and target accessibility might take part. One reasonable possibility would be that the repair mechanism of PE-mediated point mutation is different from that of PE-mediated insertion or deletion and that of base editing. And the mechanism responsible for the repair of PE-mediated point mutation was adjusted by HDAC inhibition against the desired point mutation. And if this is true, HDACi inhibiting PE-mediated point mutation also predicts that the level of the adjustment surpasses the effect of HDACi on protein expression and target accessibility. To the best of our knowledge, DNA lesions created by PE had never been observed under natural conditions, and the mechanisms underlying the repair of these lesions were poorly understood. Only speculation from not so similar scenarios such as ssDNA invasion32 and flap removal33 is far from reaching an overall view.14 Recently, the laboratory of David Liu found that mismatch repair pathway had a negative effect on PE.34 Inhibition of key players of the mismatch repair pathway (MMR), including MSH2, MSH6, MLH1, and PMS2, increased the efficiency of PE. Interestingly, the degree of increase was found to correlate with the type of PE, as it declined as the length of edits increased. This result was consistent with our observation that HDACi has varied effects on different types of PEs. Considering that two different protein complexes were responsible for censoring MMR substrates, with MutSα recognizing 1 nt mismatch or loop and MutSβ recognizing more than 2 nt mismatch or loop,35,36 we speculated that HDACi might have different effects on these two sensors and thus have different impacts on different types of PEs. In fact, accumulating evidence suggests that acetylation modification is an important mechanism in the regulation of MMR activity.37 However, the effects were somewhat controversial. HDAC6 deacetylates and ubiquitinates MSH2, causing decreased MMR activity,37 while the deacetylation of MSH2 by HDAC10 might promote MMR.38 Further investigations are required in order to dissect the exact mechanisms of the effects of HDACi on PE-induced lesions.
Interestingly, in this work, we observed that HDACi treatment significantly inhibited the transition from C to G or A in cytosine base editing. And the inhibition was likely due to HDACi inducing hyper-acetylation of UNG and/or UGI, which in turn led to increased interaction between UNG and UGI. This result was consistent with the observations of acetylation modification regulating the functions of DNA repair proteins, including DNA binding ability, enzymatic activity, and protein-protein interaction.29 In addition, as UNG inhibition plays an important role in increasing the efficiency of cytosine base editing,6,28 it is also reasonable to deduce that HDACi-induced UNG inhibition also contributes to the improvement of CBE. In agreement with this notion, the improvement level of HDAC on CBE was dependent largely on the presence of UGI (Figure S6).
It is noteworthy that these observations made by Shin et al.17 and ourselves were all obtained from cultured cells that underwent robust proliferation. Whether the effect of HDAC inhibition also functions in adult animals remains unclear. Especially considering that the major target organs of gene therapy, such as liver, muscle, and retina, typically exhibit low levels of cell proliferation in adults, future investigation is needed to determine if HDACi can improve base and prime editing in vivo in adult animals. Of note, HDACi also increased off-target efficiencies of base editing. Such effect should be considered when using HDACi to improve base editing, especially for gene therapy purpose.
Materials and methods
Design and construction of plasmid
Plasmids encoding base editors (BE3 #73021, ABE7.10 #102919, and BE3-SaKKH #85170) and prime editor (PE2 #132775) were obtained from Addgene. The sgRNA plasmids were constructed by inserting spacers into Bbs1-digested empty sgRNA plasmids. Oligos used to generate spacers are listed in Table S3. pegRNA fragments were constructed by PCR-mediated elongation of each sgRNA. Sequences of sgRNA and pegRNA constructs are listed in Tables S2 and S3. AID-BE3, target-AID, and A3A-BE3 plasmids have been described previously.23,28,39 The remaining plasmids, including CABE base editor, nSaCas9, BE-NO-UGI, pVAX1-FLAG-UNG, pVAX1-FLAG-UGI, and NanoBiT, are constructed by seamless cloning (ClonExpress II One Step Cloning Kit; Vazyme Biotech). All plasmids were verified using Sanger sequencing. Sequence encoding NanoLuc was optimized for mammalian cells and synthesized by TSINGKE Biology. NanoLuc was split into two NanoBiT subunits, 114 and 11S, as previously described.40
Grayscale analysis
To determine the genome targeting knockin efficiency of prime editing at HEK3 site, PCR was performed using 2×Rapid Taq Master Mix (Vazyme) with primers 5′-TGCATTTGTAGGCTTGATGC-3′ and 5′-GTCAACCAGTATCCCGGTGC-3′ for amplification. Then the PCR product DNA fragments were detected using gel electrophoresis. The efficiencies of PE were calculated by analyzing the intensity of bands corresponding to wild-type or edited alleles using ImageJ software.
Cell culture
HEK293T, HeLa, HepG2, and HCT116 cells were cultured in DMEM (Gibco by Life Technologies), supplemented with 10% fetal bovine serum (FBS) (Life Technologies) and 1% penicillin/streptomycin (Boster Biological Technology) at 37°C and 5% CO2.
Plasmid transfection and drug treatments
In order to measure the editing efficiency at endogenous targets, cells were plated into 24-well plates 12–24 h before transfection, and each well was seeded with 2 × 105 cells. When cells reached a confluence of ∼80% they were transfected with plasmids encoding BEs (600 ng) and plasmids encoding sgRNAs (200 ng), or plasmids encoding PE2 (550 ng), plasmids encoding pegRNAs (185 ng), and plasmids encoding sgRNAs (60 ng) using Transeasy (Forgene). After 12–16 h, the transfected cells were trypsinized and plated into 96-well plates and treated with HDAC inhibitors or DMSO (negative control). After 48 h of treatment, the cells were collected, and the cell genome was extracted for subsequent editing efficiency verification.
Orthogonal R-loop assays of independent off-target
For orthogonal R-loop assay, HEK293T cells were seeded on 24-well plates (BIOFIL). Cells at a confluence of ∼70%–80% were transfected with mixed plasmids encoding base editor (300 ng), SpCas9 on-target sgRNA (200 ng), nsaCas9 (300 ng), and SaCas9 sgRNA (200 ng) targeting a genomic locus unrelated to the on-target site. Specifically, BE3 fused with on-target sgRNA and edits at the target site. Off-target sgRNA fused with nsaCas9 to form R-loop at off-target sites to be edited by BE3. Seventy-two hours post-transfection, the genomic DNA was extracted, and on-target and off-target efficiency was detected using Sanger sequencing.
Sanger sequencing and EditR analysis
Cells were harvested 72 h post-transfection, and the genomic DNA was extracted with freshly prepared DNA extraction buffer (the lysis buffer is composed of 80% sterile water, 20% 10× PCR buffer, and 0.5% Proteinase K). The mixture was incubated at 55°C for 10 min and then was inactivated at 95°C for 10 min. Genomic regions of interest were amplified by PCR and then analyzed using Sanger sequencing. The sequences of primers are listed in Table S4. Then the base editing efficiency was quantified using EditR software (http://baseditr.com), according to the author’s description.41
Deep sequencing of genomic DNA samples and data analysis
Genomic regions of interest were amplified using High-Fidelity DNA Polymerase (Phanta Max Super-Fidelity) with primers flanked with different barcodes (Tables S5–S7). The PCR products were gel-purified and quantified using NanoDrop (Thermo Fisher Scientific). Samples were sequenced commercially using Ilumina Hiseq-2000 platform (Personal Biotechnology, Shanghai, China). The sequencing reads were extracted using custom Python scripts, and editing efficiency was manually analyzed using Excel software. The sequences of wild-type and edited alleles were used as references.
Cell viability assay
Cell viability analysis of HDAC inhibitors on HEK293T cell line was performed using the MTS, Promega #G3582 assay. HEK293T cells were digested and seeded on a 96-well plate. After 24 h, different concentrations of HDAC inhibitors were added to the DMEM, and DMSO was used as a negative control and treated with equal volumes. Cell viability determination was performed after 48 h of drug treatment. MTS reagent was diluted 1:5 with fresh medium and mixed well, and the medium was then removed and 100 μL added to each well. Absorbance was measured at 490 nm after incubating at 37°C for 1.5 h. Cell viability was compared with the DMSO treatment group. The data are obtained using GraphPad Prism 8.0.
Detection of acetylated UNG and UGI
HEK293T cells in a 60 mm2 dish were transfected with 5 μg pVAX-FLAG-UNG or pJL-SaKKH-BE3 (catalog #85170; Addgene) using Lipofectamine 2000. After transfection, cells were collected and seeded in 20 mm2 tissue culture dishes. Twenty-four hours post-transfection, cells were treated with DMSO or romidepsin (50 nM) for 6 h at 37°C. Following treatment, cells were washed once with ice-cold PBS (pH 7.4) and lysed with RIPA lysis buffer containing 2 mM PMSF and 2× protease inhibitors cocktail (catalog #HY-K0010; MedChemExpress). Then, cell lysates were further homogenized using ultrasound at 4°C and centrifuged (13,300 rpm at 4°C for 15 min). Ten microliters of anti-FLAG magnetic beads (catalog #HY-K0207; MedChemExpress) per sample were washed 4 times with washing buffer (0.1% Tween 20 in PBS) and re-suspended in 10 μL pre-cold washing buffer. Bead solution (10 μL) was added per sample and rotated 30 min at room temperature (RT). Following incubation, beads were washed with washing buffer 5 times. Proteins were eluted with 1× SDS-PAGE protein loading buffer and heated at 100°C for 10 min. The protein samples were separated using SDS-PAGE and transferred to 0.45 μm polyvinylidene fluoride (PVDF) film. Blots were blocked with 5% skim milk in TBS/T buffer (0.1% Tween 20 in TBS) with gentle shaking at RT for 2 h. Following blocking, blots were probed with anti-FLAG antibody (catalog #14793; CST) or anti-pan acetyl lysine antibody (catalog #PTM-102; PTM BioLabs) overnight at 4°C. After 3 washes with TBS/T buffer, blots were further incubated with secondary antibody with gentle shaking at RT for 1 h. Membranes were finally imaged using the BIO-LAB ChemiDoc MP imaging system with enhanced chemiluminescence substrates (Abbkine). Band intensity was quantified using ImageJ software, and the ratio of acetylated protein band to total protein band was normalized to control. All experiments were performed in triplicate, and statistical significance was determined using a two-tailed Student’s t test in GraphPad Prism 8.0 software.
NanoBiT assay
HEK293T cells in a 60 mm2 dish were transfected with 5 μg pcDNA3.1-114-HA-UNG and pcDNA3.1-11S-3×FLAG-UGI by Lipofectamine 2000. After transfection, cells were collected and re-suspended in phenol red free DMEM supplemented with 5% FBS at a density of 5 × 105 cells/mL. Ninety microliters of cell suspension was seeded in white 96-well tissue culture plates and grown in a 37°C, 5% CO2 incubator overnight. Subsequently, 10 μL of phenol red free DMEM supplemented with 5% FBS containing 9× DMSO or drugs was added to cells for 6 h. Before detection, 20 μL NanoLuc substrate (catalog #N1110; Promega) was diluted in 1 mL phenol red free DMEM containing 5% FBS. Then, cells were added with 10 μL NanoLuc substrate and incubated at RT for 15 min. Finally, luminescence was measured in a CLARIOstar microplate reader (BMG Labtech). The half maximal effective concentration (EC50) values were calculated using a dose-response model in GraphPad Prism 8.0 software. All experiments were performed in triplicate, and the values are presented as mean ± SD.
Statistical analysis
GraphPad Prism software (version 8.4.0) was used for all data analysis. All statistical comparison adjustment was performed using two-tailed Student’s t test in SPSS software (version 21.0.0.0).
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 81974238 and No. U19A2002) and 1·3·5 project for disciplines of excellence, West China Hospital, Sichuan University (ZYJC21018).
Author contributions
N.L., L.Z., G.L., and J.L. performed the experiments. Y.H., Y.W., and Y.J. analyzed the data. N.L., L.Z., S. Yang, and S. Yao designed the experiments and wrote the manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2022.05.036.
Contributor Information
Shengyong Yang, Email: yangsy@scu.edu.cn.
Shaohua Yao, Email: shaohuayao@scu.edu.cn.
Supplemental information
References
- 1.Bolotin A., Quinquis B., Sorokin A., Ehrlich S.D. Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin. Microbiology. 2005;151:2551–2561. doi: 10.1099/mic.0.28048-0. [DOI] [PubMed] [Google Scholar]
- 2.Gasiunas G., Barrangou R., Horvath P., Siksnys V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl. Acad. Sci. U S A. 2012;109:E2579–E2586. doi: 10.1073/pnas.1208507109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anzalone A.V., Randolph P.B., Davis J.R., Sousa A.A., Koblan L.W., Levy J.M., Chen P.J., Wilson C., Newby G.A., Raguram A., Liu D.R. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature. 2019;576:149–157. doi: 10.1038/s41586-019-1711-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gaudelli N.M., Komor A.C., Rees H.A., Packer M.S., Badran A.H., Bryson D.I., Liu D.R. Programmable base editing of A∗T to G∗C in genomic DNA without DNA cleavage. Nature. 2017;551:464–471. doi: 10.1038/nature24644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Komor A.C., Kim Y.B., Packer M.S., Zuris J.A., Liu D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016;533:420–424. doi: 10.1038/nature17946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Verkuijl S.A., Rots M.G. The influence of eukaryotic chromatin state on CRISPR-Cas9 editing efficiencies. Curr. Opin. Biotechnol. 2019;55:68–73. doi: 10.1016/j.copbio.2018.07.005. [DOI] [PubMed] [Google Scholar]
- 8.Liu B., Chen S., Rose A.L., Chen D., Cao F., Zwinderman M., Kiemel D., Aïssi M., Dekker F.J., Haisma H.J. Inhibition of histone deacetylase 1 (HDAC1) and HDAC2 enhances CRISPR/Cas9 genome editing. Nucleic Acids Res. 2020;48:517–532. doi: 10.1093/nar/gkz1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Park H., Shin J., Choi H., Cho B., Kim J. Valproic acid significantly improves CRISPR/Cas9-Mediated gene editing. Cells. 2020;9:1447. doi: 10.3390/cells9061447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang J.P., Yang Z.X., Zhang F., Fu Y.W., Dai X.Y., Wen W., Zhang B., Choi H., Chen W., Brown M., et al. HDAC inhibitors improve CRISPR-mediated HDR editing efficiency in iPSCs. Sci. China Life Sci. 2021;64:1449–1462. doi: 10.1007/s11427-020-1855-4. [DOI] [PubMed] [Google Scholar]
- 11.Jeggo P.A. DNA breakage and repair. Adv. Genet. 1998;38:185–218. doi: 10.1016/s0065-2660(08)60144-3. [DOI] [PubMed] [Google Scholar]
- 12.Kunz C., Saito Y., Schär P. DNA Repair in mammalian cells: mismatched repair: variations on a theme. Cell. Mol. Life Sci. 2009;66:1021–1038. doi: 10.1007/s00018-009-8739-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Symington L.S., Gautier J. Double-strand break end resection and repair pathway choice. Annu. Rev. Genet. 2011;45:247–271. doi: 10.1146/annurev-genet-110410-132435. [DOI] [PubMed] [Google Scholar]
- 14.Maruyama T., Dougan S.K., Truttmann M.C., Bilate A.M., Ingram J.R., Ploegh H.L. Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining. Nat. Biotechnol. 2015;33:538–542. doi: 10.1038/nbt.3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Robert F., Barbeau M., Ethier S., Dostie J., Pelletier J. Pharmacological inhibition of DNA-PK stimulates Cas9-mediated genome editing. Genome Med. 2015;7:93. doi: 10.1186/s13073-015-0215-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cappellacci L., Perinelli D.R., Maggi F., Grifantini M., Petrelli R. Recent progress in histone deacetylase inhibitors as anticancer agents. Curr. Med. Chem. 2020;27:2449–2493. doi: 10.2174/0929867325666181016163110. [DOI] [PubMed] [Google Scholar]
- 17.Shin H.R., See J.E., Kweon J., Kim H.S., Sung G.J., Park S., Jang A.H., Jang G., Choi K.C., Kim I., et al. Small-molecule inhibitors of histone deacetylase improve CRISPR-based adenine base editing. Nucleic Acids Res. 2021;49:2390–2399. doi: 10.1093/nar/gkab052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhao T., Li Q., Zhou C., Lv X., Liu H., Tu T., Tang N., Cheng Y., Liu X., Liu C., et al. Small-molecule compounds boost genome-editing efficiency of cytosine base editor. Nucleic Acids Res. 2021;49:8974–8986. doi: 10.1093/nar/gkab645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang Z., Shen M., Tang M., Zhang W., Cui X., Zhang Z., Pei H., Li Y., Hu M., Bai P., Chen L. Discovery of 1, 2, 4-oxadiazole-Containing hydroxamic acid derivatives as histone deacetylase inhibitors potential application in cancer therapy. Eur. J. Med. Chem. 2019;178:116–130. doi: 10.1016/j.ejmech.2019.05.089. [DOI] [PubMed] [Google Scholar]
- 20.Furumai R., Matsuyama A., Kobashi N., Lee K.H., Nishiyama M., Nakajima H., Tanaka A., Komatsu Y., Nishino N., Yoshida M., Horinouchi S. FK228 (depsipeptide) as a natural prodrug that inhibits class I histone deacetylases. Cancer Res. 2002;62:4916–4921. [PubMed] [Google Scholar]
- 21.Liu Z., Shan H., Chen S., Chen M., Zhang Q., Lai L., Li Z. Improved base editor for efficient editing in GC contexts in rabbits with an optimized AID-Cas9 fusion. FASEB J. 2019;33:9210–9219. doi: 10.1096/fj.201900476rr. [DOI] [PubMed] [Google Scholar]
- 22.Nishida K., Arazoe T., Yachie N., Banno S., Kakimoto M., Tabata M., Mochizuki M., Miyabe A., Araki M., Hara K.Y., et al. Targeted nucleotide editing using hybrid prokaryotic and vertebrate adaptive immune systems. Science. 2016;353:aaf8729. doi: 10.1126/science.aaf8729. [DOI] [PubMed] [Google Scholar]
- 23.Wang X., Li J., Wang Y., Yang B., Wei J., Wu J., Wang R., Huang X., Chen J., Yang L. Efficient base editing in methylated regions with a human APOBEC3A-Cas9 fusion. Nat. Biotechnol. 2018;36:946–949. doi: 10.1038/nbt.4198. [DOI] [PubMed] [Google Scholar]
- 24.Wang Y., Zhou L., Tao R., Liu N., Long J., Qin F., Tang W., Yang Y., Chen Q., Yao S. sgBE: a structure-guided design of sgRNA architecture specifies base editing window and enables simultaneous conversion of cytosine and adenosine. Genome Biol. 2020;21:222. doi: 10.1186/s13059-020-02137-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang Y., Zhou L., Liu N., Yao S. BE-PIGS: a base-editing tool with deaminases inlaid into Cas9 PI domain significantly expanded the editing scope. Signal Transduct. Target Ther. 2019;4:36. doi: 10.1038/s41392-019-0072-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Doman J.L., Raguram A., Newby G.A., Liu D.R. Evaluation and minimization of Cas9-independent off-target DNA editing by cytosine base editors. Nat. Biotechnol. 2020;38:620–628. doi: 10.1038/s41587-020-0414-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jin S., Fei H., Zhu Z., Luo Y., Liu J., Gao S., Zhang F., Chen Y.H., Wang Y., Gao C. Rationally designed APOBEC3B cytosine base editors with improved specificity. Mol. Cell. 2020;79:728–740.e6. doi: 10.1016/j.molcel.2020.07.005. [DOI] [PubMed] [Google Scholar]
- 28.Komor A.C., Zhao K.T., Packer M.S., Gaudelli N.M., Waterbury A.L., Koblan L.W., Kim Y.B., Badran A.H., Liu D.R. Improved base excision repair inhibition and bacteriophage Mu Gam protein yields C:G-to-T:A base editors with higher efficiency and product purity. Sci. Adv. 2017;3:eaao4774. doi: 10.1126/sciadv.aao4774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li S., Shi B., Liu X., An H.X. Acetylation and deacetylation of DNA repair proteins in cancers. Front. Oncol. 2020;10:573502. doi: 10.3389/fonc.2020.573502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bhakat K.K., Hazra T.K., Mitra S. Acetylation of the human DNA glycosylase NEIL2 and inhibition of its activity. Nucleic Acids Res. 2004;32:3033–3039. doi: 10.1093/nar/gkh632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Choudhary C., Kumar C., Gnad F., Nielsen M.L., Rehman M., Walther T.C., Olsen J.V., Mann M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- 32.Caldecott K.W. Single-strand break repair and genetic disease. Nat. Rev. Genet. 2008;9:619–631. doi: 10.1038/nrg2380. [DOI] [PubMed] [Google Scholar]
- 33.Balakrishnan L., Bambara R.A. Flap endonuclease 1. Annu. Rev. Biochem. 2013;82:119–138. doi: 10.1146/annurev-biochem-072511-122603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen P.J., Hussmann J.A., Yan J., Knipping F., Ravisankar P., Chen P.F., Chen C., Nelson J.W., Newby G.A., Sahin M., et al. Enhanced prime editing systems by manipulating cellular determinants of editing outcomes. Cell. 2021;184:5635–5652.e29. doi: 10.1016/j.cell.2021.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Warren J.J., Pohlhaus T.J., Changela A., Iyer R.R., Modrich P.L., Beese L.S. Structure of the human MutSα DNA lesion recognition complex. Mol. Cell. 2007;26:579–592. doi: 10.1016/j.molcel.2007.04.018. [DOI] [PubMed] [Google Scholar]
- 36.Gupta S., Gellert M., Yang W. Mechanism of mismatch recognition revealed by human MutSβ bound to unpaired DNA loops. Nat. Struct. Mol. Biol. 2011;19:72–78. doi: 10.1038/nsmb.2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang M., Xiang S., Joo H.Y., Wang L., Williams K.A., Liu W., Hu C., Tong D., Haakenson J., Wang C., et al. HDAC6 deacetylates and ubiquitinates MSH2 to maintain proper levels of MutSα. Mol. Cell. 2014;55:31–46. doi: 10.1016/j.molcel.2014.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Radhakrishnan R., Li Y., Xiang S., Yuan F., Yuan Z., Telles E., Fang J., Coppola D., Shibata D., Lane W.S., et al. Histone deacetylase 10 regulates DNA mismatch repair and may involve the deacetylation of MutS homolog 2. J. Biol. Chem. 2015;290:22795–22804. doi: 10.1074/jbc.m114.612945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shimatani Z., Kashojiya S., Takayama M., Terada R., Arazoe T., Ishii H., Teramura H., Yamamoto T., Komatsu H., Miura K., et al. Targeted base editing in rice and tomato using a CRISPR-Cas9 cytidine deaminase fusion. Nat. Biotechnol. 2017;35:441–443. doi: 10.1038/nbt.3833. [DOI] [PubMed] [Google Scholar]
- 40.Dixon A.S., Schwinn M.K., Hall M.P., Zimmerman K., Otto P., Lubben T.H., Butler B.L., Binkowski B.F., Machleidt T., Kirkland T.A., et al. NanoLuc complementation reporter optimized for accurate measurement of protein interactions in cells. ACS Chem. Biol. 2016;11:400–408. doi: 10.1021/acschembio.5b00753. [DOI] [PubMed] [Google Scholar]
- 41.Kluesner M.G., Nedveck D.A., Lahr W.S., Garbe J.R., Abrahante J.E., Webber B.R., Moriarity B.S. EditR: a method to quantify base editing from sanger sequencing. CRISPR J. 2018;1:239–250. doi: 10.1089/crispr.2018.0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.