

ABSTRACT
Osteopenia is common in phenylalanine hydroxylase deficient phenylketonuria (PKU). PKU is managed by limiting dietary phenylalanine. Osteopenia in PKU might reflect a therapeutic diet, with reduced bone forming materials. However, osteopenia occurs in patients who never received dietary therapy or following short-term therapy. Humans and animal studies find no correlation between bone loss, plasma hyperphenylalaninemia, bone formation, and resorption markers. Work in the Pahenu2 mouse recently showed a mesenchymal stem cell (MSC) developmental defect in the osteoblast pathway. Specifically, Pahenu2 MSCs are affected by energy dysregulation and oxidative stress. In PKU, MSCs oximetry and respirometry show mitochondrial respiratory-chain complex 1 deficit and over-representation of superoxide, producing reactive oxygen species affecting mitochondrial function. Similar mechanisms are involved in aging bone and other rare defects including alkaptonuria and homocysteinemia. Novel interventions to support energy and reduce oxidative stress may restore bone formation PKU patients, and in metabolic diseases with related mechanisms.
KEYWORDS: Phenylketonuria, Pahenu2 mouse, osteopenia, oxidative stress, mitochondrial respiratory complex 1
Discovery of phenylketonuria and subsequent success of dietary management revealed secondary problems
The Norwegian physician Følling discovered correlation of phenylketones in the urine with heritable mental impairment in 1934.1 Phenylalanine hydroxylase (PAH) deficient PKU was the initial treatable inborn error of metabolism, with response to dietary phenylalanine (Phe) restriction shown in 1953, an important success that made PKU survivable with major problems of intellectual impairment largely controlled.2 Dietary substrate restriction (i.e. Phe restriction) would be the interventional standard for more than 60 years.
Newer interventions, in brief, include pharmacological doses (i.e. 20 mg/kg/day) of the PAH cofactor tetrahydrobiopterin (Commercially known as Kuvan), are effective in mild disease retaining partial enzyme activity.3 Recently, the US Food and Drug Administration approved alternative pathway enzyme therapy with polyethylene glycol-conjugated phenylalanine ammonia lyase (the commercial enzyme therapy Palynziq), which has broad efficacy.4 Unlike tetrahydrobiopterin therapy, Palynziq is effective in classical PKU; however, the requirement for daily injections has tempered patient enthusiasm for this intervention.
Prospective newborn screening enables early intervention to eliminate the most severe elements of neurologic disease. However, even early-identified, continuously treated patients have reduced IQ, executive function deficit, seizures, Parkinsonism, neuropsychiatric phenotypes, and osteopenia.5,6 The pathophysiology of PKU neurologic disease is incompletely characterized but involves asymmetric blood:brain barrier amino acid transport secondary to Phe over-representation in blood. Elements including neurotransmitter paucities, reduced cerebral protein synthesis, and direct Phe tissue toxicity are among hypothesized neuropathology drivers.7 Recent investigation indicates neuropathology involves unanticipated complexities involving energy dysregulation, oxidative stress, and secondary manifestations thereof.8–10
While neurologic disease is the most prominent and widely reported PKU disease presentation, other phenotypes include cardiovascular, optic, and abnormalities of the skeleton, which appear to reflect different mechanisms.11,12 Here we focus on skeletal abnormalities, specifically on reduced bone mass in PKU.13
Osteopenia in PKU
Feinberg and Fisch described osteopenia in phenylketonuric children in 1962.13 This was confirmed by Murdoch and Holman shortly thereafter,14 with many subsequent consistent reports as recently reviewed in detail.15 In brief, bone mineral density Z scores of −2.0 in the lumbar spine are common in early identified and continuously treated patients.16 Similarly, reduced total body bone mineral density occurs.17 Osteopenia occurs in well managed and in therapy noncompliant patients. Nonetheless, the pathophysiology of PKU osteopenia remains poorly characterized.
Initial studies on the mechanism of osteopenia in PKU
Osteopenia in PKU was, in early work, generally considered an event secondary to therapeutic diet with bone components, including calcium and phosphorous, reduced or biologically unavailable. Several studies have shown that this is not, at least in major part, the case. Osteopenia may be present from an early age without clear relationship to therapy.18 In some PKU affected children, bone mineral density is lower despite higher calcium, phosphorus, and magnesium intake.19 In patients with PKU with reduced calcium and magnesium concentrations and decreased bone formation, there was no relation of bone mass to serum Phe, protein, or mineral intake, and no differences in bone resorption.20
Additional studies, considered overall, did not show a systematic mechanism for reduced PKU bone mass based on conventional bone studies. In particular, the obvious potential correlates, including serum Phe, did not strongly relate to bone density. Indeed, some studies show negative correlations of Phe and bone mass.16,21,22 Markers of bone formation, bone resorption, and related chemistry showed no clear reasons for low bone mass in PKU.21–23
Early pathophysiological investigation of PKU osteopenia considered this due to imbalanced bone formation and resorption, with which it is difficult to disagree, although this points to no specific mechanism. Two studies investigated osteoclastogenesis in PKU patient peripheral blood. These showed that spontaneous and stimulated (macrophage colony stimulating factor, M-CSF, and receptor activator of nuclear factor kB ligand, RANKL) osteoclastogenesis was greater in PKU patient mixed leukocyte cultures, assayed by tartrate resistant acid phosphatase (TRAP) activity.24,25 In the recent decade, there has been no further evidence to substantiate this as a major disease mechanism. Although the results are not disputed, these studies do not point to a comprehensive mechanism of osteopenia in PKU.
Recent studies using mouse models
While osteopenia is not fully penetrant in patients, the Paheun2 classical PKU mouse is universally osteopenic making it ideal to investigate this phenotype. Specifically, Dobrowolski et al. used Pahenu2 (C57bl/6 background) to study Phe management on bone differentiation in vitro and in vivo by cytology, histomorphometry (static, dynamic), and biochemistry.26 This showed that Pahenu2 bone density was decreased 33% relative to wild type. Bone volume and total volume were decreased, with increased trabecular spacing. Calcein labeling showed a 25% decrease in mineral apposition.
Biochemical measurements showed no clear reason for decreased bone formation in the Paheun2 PKU mouse model, with plasma cortisol, adrenocorticotropic hormone, and 25-hydroxyvitamin D unaffected. There was a modest increased plasma PTH in Pahenu2 mice relative to controls, and plasma calcium and phosphate were reduced, consistent with a mineralization defect but not pointing to an underlying causative event.
In vitro bone-derived mesenchymal stem cells (MSCs) were used to assay developmental competence in osteoblast development. Compared to C57bl/6 MSCs, Pahenu2 MSCs displayed significantly lower bone mineralization when cultured at physiologic Phe concentration and this effect was exacerbated by hyperphenylalaninemic conditions (1200 μM Phe, the concentration that defines classical PKU), where mineralization was further reduced. These data provide the first evidence that PKU osteopenia involves an MSC developmental defect in the osteoblast pathway. In Pahenu2 MSCs, expression of Col1A1 and Rankl were suppressed, in keeping with reduced bone formation and bone turnover. Interestingly, like the brain which also host PKU disease phenotypes, MSCs are functionally PAH deficient as they neither express the PAH gene nor hydroxylate Phe. These data suggest PKU biochemical insult affects MSC differentiation in the osteoblast pathway.
Further mechanistic elements of Pahenu2 MSC osteoblast developmental deficit were recently described.27 Applying Phe restriction to Pahenu2 mice effectively reduces plasma Phe to ~200 μM (untreated Pahenu2 2,000–2200 μM). Two months of Phe management did not improve bone density relative to untreated animals by histomorphometry, suggesting mechanisms of PKU osteopenia involve more that Phe insult.
In contrast, oxidative stress has previously been recognized in PKU patients and in animal models.28–31 Evaluating Pahenu2 MSCs with MitoSox Red labeling, a specific superoxide stain, identified oxidative stress compared to controls. Since superoxide might be a product of a mitochondrial proton leak, differentiating Pahenu2 MSCs were assessed by oximetry (using the Seahorse XF, Agilent Instruments, Santa Clara, CA) where a pattern consistent with mitochondrial stress was identified including reduced maximal respiration and respiratory reserve.
Respirometry (Oroboros Oxigraph 2 K platform, Innsbruck, Austria) identified a partial respiratory-chain complex 1 deficit as being consistent with the superoxide oxidative stress. Energy dysregulation is, thus, the probable cause, at least in part, of Pahenu2 MSC dysfunction (Figure 1). Further investigation of the role of oxidative stress applied the stilbene derivative polyphenol antioxidant resveratrol in the course of Pahenu2 MSC differentiation.32 Resveraltrol increased the Pahenu2 MSC mitochondrial mass, and partially restored alkaline phosphatase activity in situ during osteoblast differentiation. This reflects that oxidative stress reduction improves energy-intensive bone formation where energy supports type I collagen synthesis and matrix mineralization, in large part due to phosphate transport.33 Related findings include mitochondrial dysfunction with bone loss in aging,34 altered redox status and mitochondrial function in alkaptonuria,35 and elevated homocysteine,36 among many others.
Figure 1.
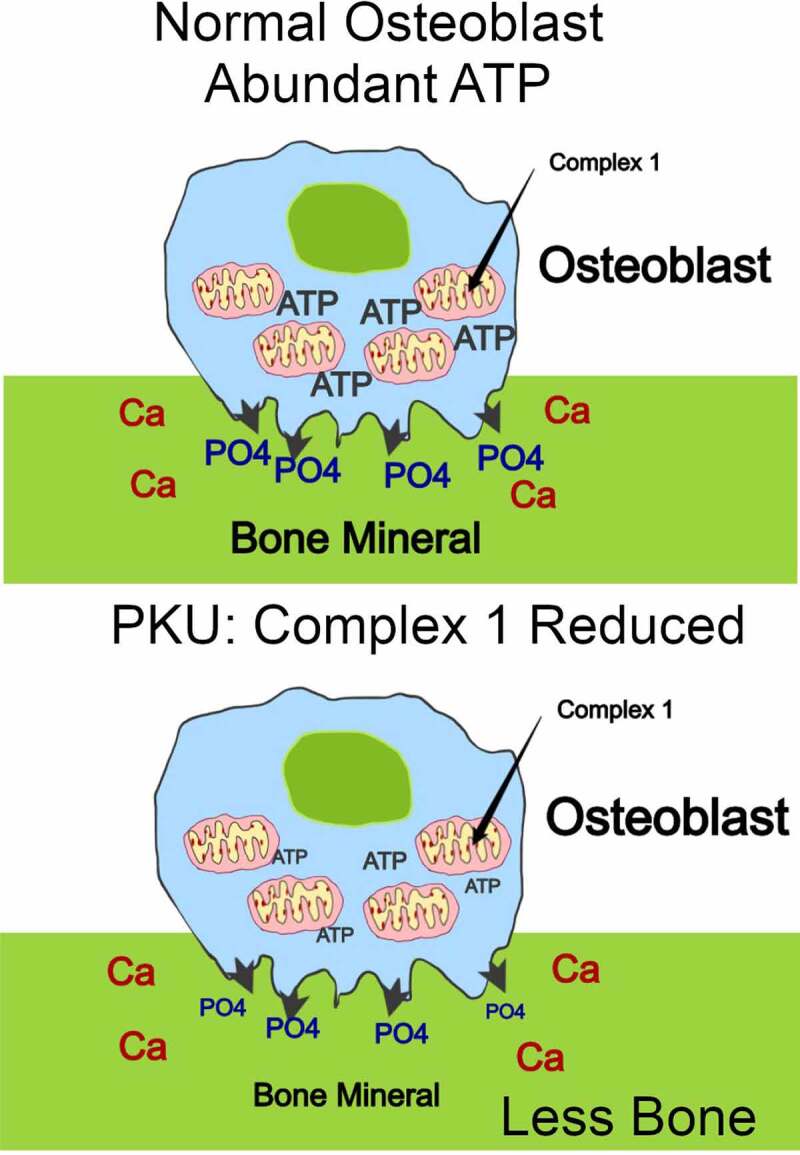
Bone formation is reduced in PKU at least in part due to energy deficit. Bone formation requires production of massive ATP from oxidative phosphorylation, energy required for production of the Type I collagen matrix and for mineralizing the matrix, dependent in large part on phosphate transport collagen synthesis,33 diagrammed as green collagen matrix and as phosphate transport (top panel). In PKU, complex 1 activity is significantly compromised with reduced ATP and bone matrix formation (bottom panel).27 Similar energy dependent effects occur in aging and in other metabolic diseases, the detailed mechanisms of which are not yet defined.34–36
Specific mitochondrial components will be investigated for more specific causes of the mitochondrial function defect. These might include expression of mitochondrial subunits,37 which might reflect mitochondrial genome or nuclear elements of mitochondrial subunits. The detailed mechanism may be valuable in treatment of PKU and related diseases with bone loss.
Conclusions and perspectives
Overall, it appears that impaired MSC differentiation in Pahenu2 at least in part is due to an energy deficit.
Further analysis MSC differentiation, energy support with MSC preferred energy substrates (e.g. Gln) and effects of antioxidant treatment may identify means to manage osteopenia in PKU patients.
Because similar mechanisms are involved in other rare metabolic syndromes, the effects of antioxidant treatment might improve outcomes in several diseases.
Funding Statement
Supported in part by Department of Veterans Affairs (USA) [BX002490] and by National Institutes of Health (USA) [AR065407 and AR076146].
Highlights
Phenylketonuria, aging, and other rare inherited diseases cause reduced bone formation via reduced mitochondrial function.
Disclosure Statement
No potential conflicts of interest were disclosed.
Author contributions
ILT, CRS, and QCL performed experimental work and analyzed data, in our work cited.
SFD and HCB planned the work and wrote the paper here, and in our work cited.
References
- 1.Følling A. Uber Ausscheidung von Phenylbrenztraubensaure in den Harn als Stoffwechselanomalie in Verbindung mit Imbezillitat. Hoppe-Seylers Z Physiol Chem. 1934;277:169–76. doi: 10.1515/bchm2.1934.227.1-4.169. [DOI] [Google Scholar]
- 2.Bickel H, Gerrard J, Hickmans EM. Influence of phenylalanine intake on phenylketonuria. Lancet. 1953;265(6790):812–13. doi: 10.1016/s0140-6736(53)90473-5. [DOI] [PubMed] [Google Scholar]
- 3.Kure S, Hou DC, Ohura T, Iwamoto H, Suzuki S, Sugiyama N, Sakamoto O, Fujii K, Matsubara Y, Narisawa K, et al. Tetrahydrobiopterin-responsive phenylalanine hydroxylase deficiency. J Pediatr. 1999;135(3):375–78. doi: 10.1016/s0022-3476(99)70138-1. [DOI] [PubMed] [Google Scholar]
- 4.Longo N, Harding CO, Burton BK, Grange DK, Vockley J, Wasserstein M, Rice GM, Dorenbaum A, Neuenburg JK, Musson DG, et al. Single-dose, subcutaneous recombinant phenylalanine ammonia lyase conjugated with polyethylene glycol in adult patients with phenylketonuria: an open-label, multicentre, phase 1 dose-escalation trial. Lancet. 2014;384:37–44. doi: 10.1016/S0140-6736(13)61841-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Camp KM, Parisi MA, Acosta PB, Berry GT, Bilder DA, Blau N, Bodamer OA, Brosco JP, Brown CS, Burlina AB, et al. Phenylketonuria scientific review conference: state of the science and future research needs. Mol Genet Metab. 2014;112(2):87–122. doi: 10.1016/j.ymgme.2014.02.013. [DOI] [PubMed] [Google Scholar]
- 6.Brown CS, Lichter-Konecki U. Phenylketonuria (PKU): a problem solved? Mol Genet Metab Rep. 2015;29:8–12. doi: 10.1016/j.ymgmr.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Groot MJ, Hoeksma M, Blau N, Reijngoud DJ, van Spronsen FJ. Pathogenesis of cognitive dysfunction in phenylketonuria: review of hypotheses. Mol Genet Metab. 2010;99(Suppl 1):S86–9. doi: 10.1016/j.ymgme.2009.10.016. [DOI] [PubMed] [Google Scholar]
- 8.Rech VC, Feksa LR, Dudra-Filho CS, Wyse ATDS, Wajner M, Wannmacher CMD. Inhibition of the mitochondrial respiratory chain by phenylalanine in rat cerebral cortex. Neurochem Res. 2002;27:353–57. doi: 10.1023/a:1015529511664. [DOI] [PubMed] [Google Scholar]
- 9.Sitta A, Barschak AG, Deon M, Barden AT, Biancini GB, Vargas PR, Souza CF, Netto C, Wajner M, Vargas CR, et al. Effect of short- and long-term exposition to high phenylalanine blood levels on oxidative damage in phenylketonuric patients. Int. J. Dev. Neurosci. 2009;27:243–47. doi: 10.1016/j.ijdevneu.2009.01.001. [DOI] [PubMed] [Google Scholar]
- 10.Dobrowolski SF, Lyons-Weiler J, Spridik K, Vockley J, Skvorak K, Biery A. DNA methylation in the pathophysiology of hyperphenylalaninemia in the PAH(enu2) mouse model of phenylketonuria. Mol Genet Metab. 2016;119(1–2):1–7. doi: 10.1016/j.ymgme.2016.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Azabdaftari A, van der Giet M, Schuchardt M, Hennermann JB, Plöckinger U, Querfeld U. The cardiovascular phenotype of adult patients with phenylketonuria. Orphanet J Rare Dis. 2019;14(1):213. doi: 10.1186/s13023-019-1188-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Serfozo C, Barta AG, Horvath E, Sumanszki C, Csakany B, Resch M, Nagy ZZ, Reismann P. Altered visual functions, macular ganglion cell and papillary retinal nerve fiber layer thickness in early-treated adult PKU patients. Mol Genet Metab Rep. 2020;25:100649. doi: 10.1016/j.ymgmr.2020.100649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Feinberg SB, Fisch RO. Roentgenologic findings in growing long bones in phenylketonuria. Preliminary study. Radiology. 1962;78:394–98. doi: 10.1148/78.3.394. [DOI] [PubMed] [Google Scholar]
- 14.Murdoch MM, Holman GH. Roentgenologic bone changes in phenylketonuria. Relationship to dietary phenylalanine and serum alkaline phosphatase. Am J Dis Child. 1964;107:523–32. doi: 10.1001/archpedi.1964.02080060525013. [DOI] [PubMed] [Google Scholar]
- 15.De Castro MJ, De Lamas C, Sánchez-Pintos P, González-Lamuño D, Couce ML. Bone status in patients with phenylketonuria: a systematic review. Nutrients. 2020;12(7):2154. doi: 10.3390/nu12072154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Demirdas S, Coakley KE, Bisschop PH, Hollak CE, Bosch AM, Singh RH. Bone health in phenylketonuria: a systematic review and meta-analysis. Orphanet J Rare Dis. 2015;10:17. doi: 10.1186/s13023-015-0232-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hansen KE, Ney D. A systematic review of bone mineral density and fractures in phenylketonuria. J Inherit Metab Dis. 2014;37:875–80. doi: 10.1007/s10545-014-9735-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.de Groot MJ, Hoeksma M, van Rijn M, Slart RH, van Spronsen FJ. Relationships between lumbar bone mineral density and biochemical parameters in phenylketonuria patients. Mol Genet Metab. 2012;105:566–70. doi: 10.1016/j.ymgme.2012.01.006. [DOI] [PubMed] [Google Scholar]
- 19.Allen JR, Humphries IR, Waters DL, Roberts DCK, Lipson AH, Howman-Giles RG, Gaskin KJ. Decreased bone mineral density in children with phenylketonuria. Am J Clin Nutr. 1994;59:419–22. doi: 10.1093/ajcn/59.2.419. [DOI] [PubMed] [Google Scholar]
- 20.Hillman L, Schlotzhauer C, Lee D, Grasela J, Witter S, Allen S, Hillman R. Decreased bone mineralization in children with phenylketonuria under treatment. Eur J Pediatr. 1996;155(Suppl 1):S148–S152. doi: 10.1007/pl00014234. [DOI] [PubMed] [Google Scholar]
- 21.Adamczyk P, Morawiec-Knysak A, Płudowski P, Banaszak B, Karpe J, Pluskiewicz W. Bone metabolism and the muscle-bone relationship in children, adolescents and young adults with phenylketonuria. J Bone Miner Metab. 2011;29:236–44. doi: 10.1007/s00774-010-0216-x. [DOI] [PubMed] [Google Scholar]
- 22.Barat P, Barthe N, Redonnet-Vernhet I, Parrot F. The impact of the control of serum phenylalanine levels on osteopenia in patients with phenylketonuria. Eur J Pediatr. 2002;161:687–88. doi: 10.1007/s00431-002-1091-9. [DOI] [PubMed] [Google Scholar]
- 23.Nagasaka H, Tsukahara H, Takatani T, Sanayama Y, Takayanagi M, Ohura T, Sakamoto O, Ito T, Wada M, Yoshino M, et al. Cross-sectional study of bone metabolism with nutrition in adult classical phenylketonuric patients diagnosed by neonatal screening. J Bone Miner Metab. 2011;29:737–43. doi: 10.1007/s00774-011-0276-6. [DOI] [PubMed] [Google Scholar]
- 24.Porta F, Roato I, Mussa A, Repici M, Gorassini E, Spada M, Ferracini R. Increased spontaneous osteoclastogenesis from peripheral blood mononuclear cells in phenylketonuria. J Inherit Metab Dis. 2008;31(Suppl 2):S339–S342. doi: 10.1007/s10545-008-0907-9. [DOI] [PubMed] [Google Scholar]
- 25.Roato I, Porta F, Mussa A, D’Amico L, Fiore L, Garelli D, Spada M, Ferracini R. Bone impairment in phenylketonuria is characterized by circulating osteoclast precursors and activated T cell increase. PLoS One. 2010;5(11):e14167. doi: 10.1371/journal.pone.0014167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dobrowolski SF, Tourkova IL, Robinson LJ, Secunda C, Spridik K, Blair HC. A bone mineralization defect in the Pahenu2 model of classical phenylketonuria involves compromised mesenchymal stem cell differentiation. Mol Genet Metab. 2018;125(3):193–99. doi: 10.1016/j.ymgme.2018.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dobrowolski SF, Sudano C, Phua YL, Tourkova IL, Spridik K, Goetzman ES, Vockley J, Blair HC.. Mesenchymal stem cell energy deficit and oxidative stress contribute to osteopenia in the Pahenu2 classical PKU mouse. Mol Genet Metab. 2021. Feb 11:S1096-7192(21)00034–2. doi: 10.1016/j.ymgme.2021.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ercal N, Aykin-Burns N, Gurer-Orhan H, McDonald JD. Oxidative stress in a phenylketonuria animal model. Free Radic Biol Med. 2002;32:906–11. doi: 10.1016/s0891-5849(02)00781-5. [DOI] [PubMed] [Google Scholar]
- 29.Colomé C, Artuch R, Vilaseca MA, Sierra C, Brandi N, Lambruschini N, Cambra FJ, Campistol J. Lipophilic antioxidants in patients with phenylketonuria. Am J Clin Nutr. 2003;77:185–88. doi: 10.1093/ajcn/77.1.185. [DOI] [PubMed] [Google Scholar]
- 30.Sitta A, Barschak AG, Deon M, Terroso T, Pires R, Giugliani R, Dutra-Filho CS, Wajner M, Vargas CR. Investigation of oxidative stress parameters in treated phenylketonuric patients. Metab Brain Dis. 2006;21(4):287–96. doi: 10.1007/s11011-006-9035-0. [DOI] [PubMed] [Google Scholar]
- 31.Sirtori LR, Dutra-Filho C, Fitarelli D, Sitta A, Haeser A, Barschak AG, Wajner M, Coelho DM, Llesuy S, Belló-Klein A, et al. Oxidative stress in patients with phenylketonuria. Biochim Biophys Acta. 2005;1740(1):68–73. doi: 10.1016/j.bbadis.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 32.Schrauwen P, Timmers S. Can resveratrol help to maintain metabolic health? Proc Nutr Soc. 2014;73(2):271–77. doi: 10.1017/S0029665113003856. [DOI] [PubMed] [Google Scholar]
- 33.Schlesinger PH, Braddock DT, Larrouture QC, Ray EC, Riazanski V, Nelson DJ, Tourkova IL, Blair HC. Phylogeny and chemistry of biological mineral transport. Bone. 2020;141:115621. doi: 10.1016/j.bone.2020.115621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dobson PF, Dennis EP, Hipps D, Reeve A, Laude A, Bradshaw C, Stamp C, Smith A, Deehan DJ, Turnbull DM, et al. Mitochondrial dysfunction impairs osteogenesis, increases osteoclast activity, and accelerates age related bone loss. Sci Rep. 2020;10(1):11643. doi: 10.1038/s41598-020-68566-2. PMID: 32669663; PMCID: PMC7363892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schiavone ML, Pecorelli A, Woodby B, Ferrara F, Pambianchi E, Santucci A, Valacchi G. Mechanisms involved in the unbalanced redox homeostasis in osteoblastic cellular model of Alkaptonuria. Arch Biochem Biophys. 2020;690:108416. doi: 10.1016/j.abb.2020.108416. [DOI] [PubMed] [Google Scholar]
- 36.Behera J, Bala J, Nuru M, Tyagi SC, Tyagi N. Homocysteine as a pathological biomarker for bone disease. J Cell Physiol. 2017;232(10):2704–09. doi: 10.1002/jcp.25693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Raikhinstein M, Hanukoglu I. Mitochondrial-genome-encoded RNAs: differential regulation by corticotropin in bovine adrenocortical cells. Proc Natl Acad Sci U S A. 1993;90(22):10509–13. doi: 10.1073/pnas.90.22.10509. [DOI] [PMC free article] [PubMed] [Google Scholar]