

Summary
Whole-organ mapping was used to study molecular changes in the evolution of bladder cancer from field effects. We identified more than 100 dysregulated pathways, involving immunity, differentiation, and transformation, as initiators of carcinogenesis. Dysregulation of interleukins signified the involvement of inflammation in the incipient phases of the process. An aberrant methylation/expression of multiple HOX genes signified dysregulation of the differentiation program. We identified three types of mutations based on their geographic distribution. The most common were mutations restricted to individual mucosal samples that targeted uroprogenitor cells. Two types of mutations were associated with clonal expansion and involved large areas of mucosa. The α mutations occurred at low frequencies while the β mutations increased in frequency with disease progression. Modeling revealed that bladder carcinogenesis spans 10–15 years and can be divided into dormant and progressive phases. The progressive phase lasted 1-2 years and was driven by β mutations.
Subject areas: Molecular biology, Cell biology, Cancer
Graphical abstract
Highlights
-
•
Dysregulation of multiple ILs plays a role in bladder cancer initiation
-
•
Altered HOX genes and oncogenic pathways complement dysregulated immunity
-
•
Bladder carcinogenesis comprises dormant and progressive phases
-
•
The dormant and progressive phases are driven by α and β mutations respectively
Molecular biology; Cell biology; Cancer
Introduction
Understanding the mechanisms that promote cancer initiation may facilitate the development of strategies to intercept and prevent carcinogenesis in its early phases before it evolves to intractable, clinically aggressive, and often uncurable disease. Common epithelial cancers evolve from microscopically recognizable precursor lesions such as dysplasia and carcinoma in situ. These lesions develop from poorly understood incipient events in microscopically intact tissue referred to as field effects (Saavedra, 2001). Comprehensive understanding of these initiating mechanisms is not possible unless they are analyzed in the context of the entire organ affected by the disease. Bladder cancer is a particularly useful model for such studies as the simple anatomy of the organ permits the mapping of preneoplastic lesions and field effects in the adjacent microscopically normal mucosa across the entire organ. It originates in the epithelial lining of the urinary tract, referred to as transitional epithelium or urothelium because its functional and architectural features lie between stratified multi-layered and simple non-stratified epithelia. Because of its direct and virtually constant contact with urine, the urothelium is exposed to a wide range of metabolic products and environmental factors that are potentially carcinogenic. In fact, smoking and environmental exposures to carcinogens are strong risk factors for the development of bladder cancer and it can be initiated in rodents by the nitrosamines from cigarette smoke (Czerniak et al., 2016). These factors along with infectious agents and chronic inflammation induce molecular changes in microscopically normal-appearing urothelium that can initiate carcinogenesis.
Recent studies based on limited sampling of the microscopically normal urothelium in areas of bladder mucosa adjacent to cancer disclose a complex mutational landscape with remarkable heterogeneity across clones and individuals. The majority of these mutational changes are showing a low variant allele frequency, but they involve cancer driver genes including chromatin remodeling genes (Lawson et al., 2020; Li et al., 2020; Strandgaard et al., 2020; Thomsen et al., 2017). They also revealed that the commitment to either luminal or basal subtype occurs early in the intraurothelial precursor lesions such as carcinoma in situ (Wullweber et al., 2021).
Herein we used whole-organ histologic and genomic mapping (WOHGM) to analyze the molecular profile of bladder cancer evolution from mucosal field effects on a whole-organ scale (Majewski et al., 2019). In this mapping we combined spatial microscopic assessment of the entire bladder mucosa with multi-platform genomic analyses of mRNA, methylation, copy-number variation (CNVs), and exome mutational profiles to visualize genetic and epigenetic changes in bladder cancer evolving from mucosal field effects through its intrinsic molecular luminal and basal tracks. This whole-organ–based genomic analytical algorithm provided in depth molecular characterizations of early events initiating bladder carcinogenesis.
Results
Preparation of whole-organ cystectomy maps
To molecularly characterize the evolution of bladder cancer from field effects, we collected geographically annotated mucosal samples from human cystectomies performed in patients with bladder cancer. The clinical and pathological data of nine cystectomy specimens used in this study are summarized in Table S1. For whole-organ mapping, we opened nine resected human bladders with invasive high-grade UC along the anterior wall and pinned them down to paraffin blocks. We then applied a mapping grid separating the mucosal areas into 1 × 2cm sealed wells allowing DNA and RNA to be extracted and preserving the urothelium for microscopic examination from which we reconstructed a histologic map of the entire bladder mucosa (Figures 1A–1G). We used whole transcriptome RNASeq with bulk RNA isolated from representative mucosal samples and tumors to designate them as the basal or luminal molecular subtypes using basal to luminal transition (BLT) scores as described previously (Figure S1) (Guo et al., 2020a). The results revealed that six cystectomies contained luminal cancers and that the remaining three contained basal cancers. We selected one bladder containing a luminal tumor (map 24) and one containing a basal tumor (map 19) for multi-platform whole-organ–based molecular profiling. The two histologic maps (maps 24 and 19) selected for multi-platform whole-organ molecular profiling and their geographic coordinates as well as the examples of microscopically classified urothelial areas and invasive cancer are shown in Figures S2A andS2B. The samples were of microscopically normal urothelium (NU), in situ preneoplastic conditions referred to as low and high-grade intraurothelial neoplasia (LGIN and HGIN, respectively), and urothelial carcinoma (UC). Geographically oriented samples of these two cystectomy specimens were analyzed by multiple genomic platforms using whole-transcriptome RNA sequencing, whole-genome methylation array hybridization, whole-exome DNA sequencing, and genome-wide single nucleotide polymorphism-based DNA copy number changes (Figure S2C). Microscopically NU harvested from ureters of nephrectomy specimens from patients without urothelial neoplasia was used as a reference for RNASeq and methylation. Germline DNA from peripheral blood samples was used as a reference for DNA sequencing and copy number analyses.
Figure 1.

Preparation of whole-organ maps for multi-platform genomic profiling
(A) Top view of the mapping grid for whole-organ sampling.
(B) Diagram showing open cystectomy mounted on a paraffin block.
(C) Diagram showing the details of the mapping grid preserving the surface urothelium for histologic mapping and facilitating simultaneous DNA/RNA extraction as well as its quality assessment of cytologic preparations. The four preparation steps are described in the material and methods section.
(D) Top view of the mapping grid superimposed over bladder mucosa.
(E) Impression of the mapping grid on the bladder surface.
(F) A whole-organ histologic map prepared by sampling the entire bladder mucosa in a luminal map (map 24).
(G) A whole-organ histologic map prepared by sampling the entire bladder mucosa in a basal map (map 19). The histologic map code is as follows: NU, normal urothelium; MD, mild dysplasia; MdD, moderate dysplasia; SD, severe dysplasia; CIS, carcinoma in situ; UC, urothelial carcinoma. For analytical purposes, samples of MD and MdD were combined and referred to as LGIN. Samples of SD and CIS were combined and referred to as HGIN.
(H) Expression patterns for luminal and basal markers and BLT scores for mucosal and tumor samples obtained from a cystectomy specimen with luminal bladder cancer in map 24.
(I) Expression patterns for luminal and basal markers and BLT scores for mucosal and tumor samples obtained from a cystectomy specimen with basal bladder cancer in map 19.
RNA expression changes in progression from field effects to carcinoma
Because the origins of luminal and basal tumors are unknown, we used whole-transcriptome expression based on RNA sequencing to characterize the changes in gene expression associated with progression of these tumors. We found that luminal cancer developed from luminal field effects whereas the basal cancer developed from basal field effects (Figures 1H and 1I). Quantitative analyses identified positive and negative BLT scores across the entire bladder mucosa for cancers developing along the basal and luminal tracks, respectively. This strongly suggested that the intrinsic molecular subtypes of bladder cancer are determined de novo and are consistent with animal models data implicating that the luminal and basal subtypes of urothelial cancer have distinct cells of origin (Shin et al., 2014; Van Batavia et al., 2014). This was confirmed by immunohistochemical analyses of signature luminal (GATA3) and basal (KRT5/6) markers. These analyses revealed high level of GATA3 expression in NU/LGIN through HGIN and UC samples in the luminal map (Figure S3A). On the other hand, there was an overexpression of KRT5/6 in NU/LGIN samples in the basal map which continued through the progression of neoplasia to HGIN and UC (Figure S3B).
We used unsupervised analyses to compare the gene expression changes that accompanied progression of bladder cancer along the luminal and basal tracks. Overall, 5,913 and 7,257 genes were differentially expressed in at least one luminal and one basal map sample, respectively, when compared with that in NU samples obtained from patients without urothelial neoplasia. Then we identified the abnormally expressed genes (n = 3,339 in the luminal map and n = 3,565 in the basal map) which showed continuous dysregulation at the transition involving mucosal field effects (NU/LGIN) and progressing to HGIN and finally to UC referred to as monotonically altered (Table S2). For both molecular subtypes hierarchical clustering using these genes separated all mucosal samples into two major clusters. The first cluster, the larger of the two, consisted of all (luminal map) or nearly all (basal map) NU/LGIN samples (Figures S4A andS4B). The second cluster consisted of all HGIN and UC samples in the luminal map and exclusively the UC samples in the basal map. Some of the UC and HGIN samples in the basal map co-clustered with NU/LGIN samples. There were two major waves of gene expression changes in progression of neoplasia from field effects to invasive carcinoma. The first group of these changes (n = 1,408 in luminal and n = 2,379 in basal maps) contained abnormally expressed genes in samples related to NU/LGIN that retained their abnormal expression patterns with the development of HGIN and progression to UC. The second group of genes (n = 1,931 in luminal and n = 1,186 in basal maps) were abnormally expressed at the transition from NU/LGIN to HGIN and UC. The most monotonically-dysregulated categories of these genes are shown in Figures 2A and S5A. The first of these categories had aberrant expression patterns in early phases of bladder carcinogenesis and formed upregulated or downregulated plaques involving large areas of bladder mucosa. Examples of these genes mapping to chromosome 10 in the luminal map and to chromosome 11 in the basal map with their geographic relationships with field effects and in situ precursor lesions are shown in Figures 2B–2D and S5B–S5D.
Figure 2.

Evolution of gene expression changes from field effects through HGIN to UC in bladder cancer developing along the luminal track in map 24
(A) Hierarchical clustering of mucosal and tumor samples using the top 60 most downregulated and overexpressed genes showing monotonic expression changes in samples corresponding to NU/LGIN through HGIN to UC, HGIN and UC, and UC only.
(B) Whole-organ expression map of chromosome 10 showing a chromosomal diagram and the expression pattern for genes in individual cystectomy samples classified as NU/LGIN, HGIN, and UC.
(C) Expression pattern for downregulated and overexpressed genes with monotonic changes involving NU/LGIN, HGIN, and UC in chromosome 10.
(D) Three-dimensional (3D) pattern of downregulated and overexpressed genes in mucosal and tumor samples as it relates to the whole-organ histologic map of a cystectomy sample filtered as in C.
In examining these two categories of genes we focused on the 10 most upregulated and downregulated genes which had monotonically aberrant expression patterns in samples of NU/LGIN through HGIN to UC. Among the top 10 overexpressed genes in the luminal map were DKK1,which regulates beta-catenin-dependent Wint signaling promoting proliferation and invasion (Tai et al., 2015), FGFBP1, which cooperates with the luminal growth factor receptor, FGFR3 to promote cell proliferation (Cronauer et al., 2003), and HOXB13, germline mutations of which were associated with hereditary prostate cancer (Ewing et al., 2012). Upregulated genes in the basal map included CEACAM5/7, which was implicated to have a role in metastatic outgrowth of basal/triple negative breast cancer (Williams et al., 2018), SERPINB3/4, also known as squamous cell carcinoma antigen, which is regulated by the basal transcription factor, STAT3, and is overexpressed in the sera of patients with squamous cell carcinomas (Naher et al., 2012), and CXCL1, which is overexpressed in aggressive bladder cancers (Miyake et al., 2013). Downregulated genes in the luminal map also included GSTM1, which encodes glutathione S-transferase, and genetic polymorphisms that have been linked with increased bladder cancer risk in association with smoking and exposure to environmental contaminants (Matic et al., 2013). Downregulated genes in the basal map included the UPK1/2-encoding uroplakins, which are major components of the asymmetric unit that forms the apical plaques of umbrella cells and are markers of terminal urothelial differentiation. Their downregulation in association with field effects signifies the aberrant differentiation program in bladder mucosa setting up the organ for basal cancer development. We also observed downregulation of MUC15 and CRTAC1, both of which encode extracellular glycoprotein complexes but their role in urothelial biology is uncertain (Steck et al., 2007; Yang et al., 2021; Yue et al., 2020; Zhang et al., 2020b).
To answer the question of how frequently the genes with abnormal expression levels identified in the field effects were dysregulated in bladder carcinogenesis we performed the validation studies in which we compared the expression levels for the 10 most upregulated and downregulated genes identified in the field effects of the luminal and basal maps with those in a cohort of bladder cancer patients in The Cancer Genome Atlas (TCGA). Specifically, we analyzed the expression levels for these genes in the two molecular subtypes of bladder cancer. According to the expression levels for these genes the luminal and basal subtypes can be separated into two clusters (Figures S6A–S6D). The first cluster of both molecular subtypes was characterized by high expression of these genes, whereas the second cluster was characterized by their downregulation. In general, the expression levels for the top 10 upregulated and downregulated genes in the early field effects were similar for both molecular subtypes. Overall, they were frequently dysregulated genes in bladder cancer that may be used as effective early diagnostic, prognostic, and therapeutic targets (Figures S6E–S6L).
Because epithelial mesenchymal transition (EMT) plays a major role in the development of many human epithelial cancers, including those that originate in the bladder, we assessed its role in the evolution of bladder cancer from mucosal field effects (Figures 3A and 3B). In solid tumors, at the core of the complex EMT circuitry are TGFB1 and p53, whose target genes positively and negatively regulate EMT. The downstream targets of this regulatory network are transcription factors that are members of the SNAIL, TWIST, ZEB, and FOX families which downregulate E-cadherin (CDH1) and other homotypic adhesion molecules including claudin-1 (CLDN1) and tight junction protein 1 (TJP1). We have also shown that p63 controls the expression of high-molecular-weight basal keratins (KRT5, KRT6, and KRT14) in urothelial cells (Choi et al., 2014; Tran et al., 2013). The central role of p63 in the regulation of EMT was confirmed in several solid tumors (Stacy et al., 2017). We previously showed that dysregulation of EMT plays a major role in the development of basal bladder cancers and their progression to highly aggressive variants such as sarcomatoid and small cell cancers (Guo et al., 2019; Yang et al., 2020). Consistent with these observations, activation of permissive components of EMT, such as the upregulation of TGFB1 and P53 target genes, was evident in field effects but widespread activation of EMT with negative EMT scores was a late event associated with progression to invasive basal bladder cancer. In contrast, we saw no major changes in EMT activation in the development of luminal bladder cancer (Figures 3A and 3B).
Figure 3.

EMT in the evolution of bladder cancer from field effects along the luminal and basal tracks
(A) Expression pattern for selected EMT-related genes and EMT scores in mucosal samples of a cystectomy specimen with luminal cancer (map 24).
(B) Expression pattern for selected EMT-related genes and EMT scores in mucosal samples of a cystectomy specimen with basal cancer (map 19).
Immune checkpoint blockade is clinically active in about 15% of patients with bladder cancer of the molecular subtypes characterized by distinct immune microenvironments. Therefore, we analyzed immune-related genes in the evolution of bladder cancer from field effects. We observed that the tumor developing along the luminal track was depleted in immune infiltration and that its cold/null phenotype was evident de novo in mucosal field effects (Figure 4A). In contrast, a tumor developing along the basal track had an increased immune signature that was already evident in mucosal field effects (Figure 4B). This was confirmed by the quantitative assessment of immune related genes calculated separately for NU/LGIN, HGIN, and UC samples and by comparing immune scores and GSEA of luminal (map 24) and basal (map 19) maps (Figures 4C–4E). The null and hot immune microenvironments of mucosal field effects in luminal and basal cancers were complemented by the downregulation and upregulation of immune regulatory genes in luminal and basal maps, respectively (Figures S7A–S7E). Moreover, the immunohistochemical analyses of CD3+ and CD8+ T lymphocytic infiltrates showed the absence of T cell infiltrates in NU/LGIN, HGIN, and UC samples of the luminal map (map 24) (Figure S8A). Brisk lymphocytic infiltrates were present in the field effects through the progression of the disease to UC in the basal map (map 19) (Figure S8B). These data support the concept that immune-null luminal cancer evolved from immune-cold field effects whereas the hot immune microenvironment of basal cancer was evident de novo in field effects, which offered early targeted, preventive, and therapeutic opportunities.
Figure 4.

Immune landscape of bladder cancer evolving from field effects along the luminal and basal tracks
(A) Expression pattern for immune-related genes in mucosal samples from a cystectomy specimen with luminal cancer (map 24).
(B) Expression pattern for immune-related genes in mucosal samples from a cystectomy specimen with basal cancer (map 19).
(C) Boxplots of immune checkpoint scores calculated using the gene expression profiles in A and B of map 24 (top) and map 19 (bottom) for subsets of samples classified as NU/LGIN, HGIN, and UC.
(D) Comparison of immune scores for luminal (map 24) and basal (map 19) cancers.
(E) GSEA for immune related genes in map 24 (luminal) and map 19 (basal). For (C) and (D) p values were calculating using Kruskal-Wallis test.
Methylation changes in progression from field effects to carcinoma
We used array hybridization to characterize whole-genome methylation changes across all samples of the cystectomies. In total, 4,333 and 5,143 genes were differentially methylated in at least one luminal and one basal map sample, respectively, when compared with that in NU samples obtained from patients without urothelial neoplasia. Then we identified the abnormally methylated genes (n = 1,380 in the luminal map and n = 1,658 in the basal map) which showed the monotonic pattern of dysmethylation in progression of neoplasia (Table S2). Hierarchical clustering according to the methylation levels for these genes separated mucosal samples from both maps into two clusters (Figures S9A andS9B). The first cluster contained the majority of NU/LGIN samples, and the second cluster contained the majority of HGIN and UC samples. Similarly to the mRNA expression levels, there were two major groups of monotonically dysmethylated genes. The first group (n = 125 in the luminal map and n = 427 in the basal map) consisted of abnormally methylated genes in samples of NU/LGIN, which retained their abnormal methylation patterns with progression to HGIN and UC. The second group (n = 1,255 genes in the luminal map and n = 1,231 genes in the basal map) was distinctively hypermethylated or hypomethylated in progression to HGIN and UC. The top monotonically hypermethylated and hypomethylated genes in both maps are shown in Figures 5A and S10A. The genes aberrantly methylated in early phases of bladder carcinogenesis formed hypermethylated or hypomethylated plaques involving large areas of bladder mucosa; examples in the luminal and basal maps are shown in Figures 5B–5D and S10B–S10D, respectively. In examining the categories of genes abnormally methylated during the development of bladder cancer we focused on the top 10 hypermethylated and hypomethylated genes that should lead to their repression or activation, which demonstrated monotonic methylation changes in samples of NU/LGIN progressing through HGIN to UCs. We expected these genes to be involved in the initiation of bladder carcinogenesis.
Figure 5.

Evolution of gene methylation changes from field effects through HGIN to UC in bladder cancer developing along the luminal track in map 24
(A) Hierarchical clustering mucosal and tumor samples using the top 42 most hypomethylated and hypermethylated genes showing monotonic expression changes in samples corresponding to NU/LGIN through HGIN to UC, HGIN and UC, and UC only.
(B) Whole-organ expression map of chromosome 18 showing a chromosomal diagram and the methylation pattern for genes in individual samples from a cystectomy classified as NU/LGIN, HGIN, and UC.
(C) Methylation pattern for hypomethylated and hypermethylated genes with monotonic changes involving NU/LGIN, HGIN, and UC in chromosome 18.
(D) 3D pattern of hypomethylated and hypermethylated genes as it relates to a whole-organ histologic map of a cystectomy sample filtered as in A.
Among the top 10 most hypermethylated genes in the luminal map were ZSCAN18 and ZNF382, which are transcription factors involved in proliferation, differentiation and apoptosis (Vedeld et al., 2015),MAP9, which encodes a microtubule associated protein involved in mitotic progression and cell migration (Zhang et al., 2020a),NID2, a member of the basement membrane protein family that binds to collagens one and four laminin (Yegin et al., 2013) and IRF4 and FBXL21, which are both involved in activation of the innate and adaptive immune systems (Alvisi et al., 2020; Shaffer et al., 2009). Among the hypomethylated (activated) genes in the early field effects were DLX6 and SP8, which are involved in embryonal development (Chung et al., 2014).HEY1, a helix-loop helix (bHLH)-type transcriptional repressor involved in the development of sarcomas (Wang et al., 2014), and TRIM31, which encodes an E3 ubiquitin-protein ligase, a negative regulator of cell growth (Liu et al., 2017). Several members of HOX gene family involved in embryonal development and body patterning were hypomethylated in field effects and included HOXA6,10,11, and 13 (Shah and Sukumar, 2010).
Among the top 10 most hypermethylated genes in the basal map were TRH, a member of the thyrotropin-releasing hormone family that controls proliferation and inhibits apoptosis of epithelial cells; the G-protein coupled receptor, GPR25; Zinc Finger Protein transcription factor, ZNF532; and MLLT6, a PHD Finger containing transcription factor involved in the regulation of several oncogenic pathways including nuclear factor κB (Liu et al., 2018; Puttipanyalears et al., 2018; Sreevalsan et al., 2020). Similar to the results in the luminal map, several members of the HOX gene family were hypermethylated in samples of NU/LGIN including HOXB2 and 4 as well as HOXD4 and 9 (Shah and Sukumar, 2010).
Similarly, to the gene expression analysis described above we compared the methylation changes in early field effects in the basal and luminal maps with those in the TCGA cohort. Using the β value which reflects the proportion of methylated DNA for these genes, we clustered the luminal and basal subtypes of bladder cancer, with enrichment of hypermethylated and hypomethylated genes into two clusters, respectively (Figures S11A–S11D). The patterns of these genes were similar in the luminal and basal subtypes. Overall, the top most dysmethylated genes identified in early field effects were frequently hypermethylated or hypomethylated in bladder cancer and thus maybe significant early diagnostic, prognostic, and therapeutic targets (Figures S11E–S11L).
Mutational landscape of field effects and their evolution to carcinoma
Whole exome sequencing of DNA from geographically mapped mucosal samples identified nonsynonymous variant alleles in 1379 and 2687 genomic loci in the luminal and basal cystectomy maps, respectively (Table S3). Heat maps of the variant allele frequencies (VAFs) for these nonsilent mutations in individual mucosal samples are shown in Figures 6A and S12A. We separated these mutations into two major clusters: Cluster A consisted of mutations with low VAFs restricted to individual mucosal samples, whereas Cluster B consisted of mutations common to multiple mucosal samples and displayed increases of VAFs with histologic progression from normal mucosa through HGIN to UC. There was an overall increase of nonsynonymous mutations that parallel the progression of disease from NU/LGIN through HGIN to UC (Figures 6B and S12B). We then restricted our analyses to variant alleles with alterations in at least three mucosal samples with VAFs≥1% in at least one sample. The results yielded 157 genes with nonsilent single nucleotide variants (SNVs) or indels in the luminal map and 198 genes with them in the basal map (Figures 6C and S12C).
Figure 6.

Enrichment of clonal genetic mutations in the evolution of bladder cancer from field effects along the luminal track in map 24
(A) Heatmap of nonsilent mutations showing VAFs in individual mucosal samples. Number of mutations in individual mucosal samples are shown in the top diagram.
(B) Boxplot analysis of number of mutations in mucosal samples classified as NU/LGIN, HGIN, and UC.
(C) Heatmap of VAFs ≥0.01 in 157 genes showing variant alleles in at least three mucosal samples. Number of α and β mutations in individual samples are shown on the top diagram.
(D) Mutant allele frequencies of α and β mutations in individual mucosal samples.
(E) Density plot showing the clonality of nonsilent VAFs in cluster α with similar low frequencies across mucosal samples that decreased with progression to HGIN and UC (Kruskal-Wallis test p value). Inset, boxplot of VAFs in three groups of samples corresponding to NU/LGIN, HGIN, and UC (Kruskal-Wallis test p value).
(F) Density plot showing the clonality of nonsilent VAFs in cluster β with a significant increase in clonality with progression to HGIN and UC. Inset, boxplot of VAFs in three groups of samples corresponding to NU/LGIN, HGIN, and UC (Kruskal-Wallis test p value). For (B) p value was calculating using ANOVA test.
Hierarchical clustering demonstrated that VAFs of these genes formed clusters α and β with distinctive behaviors in the progression to carcinoma (Figures 6C, 6D, S12C andS12D). Cluster α consisted of 80 variant alleles in the luminal map and 43 variant alleles in the basal map which demonstrated a low mutational frequency pattern across the bladder mucosa and even decreased frequencies with progression to HGIN and UC (Figures 6D, 6E,S12D andS12E). Of the 80 luminal α cluster genes 77 were altered by nonsynonymous single-nucleotide substitutions, resulting in amino acid changes (Table S4). The remaining genes were altered by an insertion (one gene) and deletions (two genes) resulting in frameshifts. In the luminal map, cluster α was enriched in mutations of genes that control invasion and migration, including RASGRF1, which stimulates the disassociation of GDP from RAS (Liao and Sharma, 2016); and DOCK7, which encodes a guanine nucleotide exchange factor that regulates the activity of the Rho family proteins (Gadea and Blangy, 2014), RAC1, RAC3 (Chan et al., 2005), DSG4, which encodes a desmosomal cadherin (Kato et al., 2015), CEACAM7, which encodes a surface glycoprotein and is a member of the carcinoembryonic antigen family(Raj et al., 2021), and ITGA7, which encodes a member of the integrin alpha chain family (Bhandari et al., 2018).
In the basal map, cluster α contained 43 variant alleles that had similar frequencies across the bladder mucosa (Figure S12C and Table S5). The most frequently mutated genes in cluster α in the basal map were involved in the transcriptional and cell cycle regulatory pathways and included genes involved in androgen receptor signaling, chromatin remodeling and DNA repair. Furthermore this cluster contained mutations of ZAP70 and POU2AF1which are major regulators of T- and B-cell development and activation of them signifies field effect involvement of immune regulatory pathways (Gonzalez-Rincon et al., 2019; Wu et al., 2019).
Clusters β, consisted of 77 genes in the luminal map and 155 genes in the basal map that exhibited a significant increase in VAFs with evolution to HGIN and UC (Figures 6D, 6F, S12D, and S12F, Tables S6 and S7). Noteworthy cell-cycle mutations included those of CDKN1A, a negative regulator of cyclin-cyclin-dependent kinase 2 that controls cell-cycle progression (Dadhania et al., 2016; Liu and Kwiatkowski, 2015), FBXW7, which binds to cyclin E and targets its ubiquitin-mediated degradation (Minella et al., 2005; Yeh et al., 2018), and APC, a tumor suppressor protein implicated to have a role in the development of colorectal cancers and that is an antagonist of the Wnt signaling pathway (Vermeulen et al., 2010; Zhang and Shay, 2017).
In the basal map the frequencies of 155 mutated genes in cluster β also increased with progression of neoplasia (Figures S12D and S12F and Table S7). The transcriptional regulators and genes controlling cell cycle progression made up the largest group of mutated genes in this cluster consisting of HORMAD1 (Gao et al., 2018), CDK13 (Lu et al., 2020), CDKN2A (Garcia-Perdomo et al., 2019), BTG2 (Tsui et al., 2018), BATF (Wei et al., 2019) among others. In addition to mutational change, CDKN2A is frequently inactivated in many cancers including bladder cancers via homozygous deletions and methylation (Garcia-Perdomo et al., 2019). The other major group of mutated genes comprised genes involved in signal transduction and metabolism. These included ACSL6which regulates fatty acid metabolism (Soupene et al., 2010) and BAIAP3 a member of the secretin receptor family involved in angiogenesis (Shiratsuchi et al., 1998). A distinct group of genes mutated in basal bladder cancer were involved in chromatin remodeling, DNA repair and RNA regulation including 15 unique KMT2D and 10 unique KDM6A mutations, these two genes are among the most frequently mutated chromatin-remodeling genes in bladder cancer. Notably, several of the genes mutated in basal cancer are involved in inflammatory responses. These genes included TLR4/5, toll-like receptor family members that play a fundamental role in pathogen recognition and activation of innate immune responses and PIK3AP1, which is involved in the survival of mature B cells and negatively regulates inflammatory cytokine production (Guo et al., 2020b; Zhou et al., 2012).
Overall, analyses of the geographic mutational landscapes in bladder mucosa demonstrated remarkable similarity of cancers evolving along the luminal and basal tracks. Specifically, they revealed mutational heterogeneity in mucosal field effects with significant changes in clonal expansion of a unique set of genes in the luminal and basal maps with disease progression to HGIN and UC.
We also performed validation studies of the mutations identified in the luminal and basal maps focusing on mutations in clusters α and β because they appear to be involved in the development and progression of bladder cancer. We confirmed the presence of 19 mutations from clusters α and β in both maps by using polymerase chain reaction (manual PCR-based Sanger sequencing (Table S8).
We performed additional validation analyses of mutations in clusters α and β in the TCGA cohort (Cancer Genome Atlas Research, 2014; Robertson et al., 2017). In cluster α, the most frequently mutated genes identified in the luminal map were RB1 (16%), FAT3 (15%) and DNAHR (10%) whereas those in the basal map were LRRK2 (11%) and ERCC2 (10%) (Figures S13 and S14). Among the genes in cluster β, which were associated with increased clonality in the progression of the disease, the most frequently mutated genes in the luminal map were LRP1 (10%), BSN (8%) and CDKN1A (8%) and those in the basal map were KMT2C (21%), KDM6A (24%), and BIRC6 (15%) (Figures S15 and S16).
Mechanism of mutagenesis driving the development of bladder cancer from field effects
To characterize the evolution of the mutational signatures associated with progression of bladder cancer from field effects, we analyzed six single-base nucleotide substitutions (C> A, C>G, C>T, T>A, T>C, and T>G) and their context motifs in all mucosal samples of the luminal and basal maps (Alexandrov et al., 2013). In both maps, the frequency of C>T mutations increased at the point of evolution from NU/LGIN to HGIN and UC with significant changes in 20 mutational signatures (Figures S17A–S17C and S18A–S18C). The most prominent signatures in the luminal map (signatures 1, 13, 29, and 30) are associated with aging, APOBEC, tobacco use, and an unknown etiology, respectively (Figures S17D–S17F). Similarly, signatures 1, 2, 3, 6, and 30, associated with aging, APOBEC, defective homologous recombination, defective mismatch repair, and an unknown etiology, respectively, predominated in the basal map (Figures S18D–S18F). Signatures 1, 13, and 30 were dominant in the field effects of the luminal map and contributed to the progression to carcinoma with the APOBEC and 13 signature weight scores significantly increasing with progression of the disease. A similar pattern was evident in the basal map with signatures 2, 6, 13, and 30 being dominant in the field effects and contributing to the progression to carcinoma. To evaluate the contribution of individual mutagenesis patterns to the mutational landscape of bladder mucosal samples, we performed bootstrapping analysis and calculated the pvalues to assess their significance (p<0.05 was considered statistically significant). This approach confirmed the dominance of signatures 1, 2, 6, 13, 29, and 30 in the luminal map (Figure S17G) and signatures 2, 6, 13, and 30 in the basal map (Figure S18G).
The analyses of alpha and beta mutations identified their distinctive mutational signatures. The six single-based nucleotide substitutions and their context motifs were different in clusters α and β (Figures S17H, S17I, S18H, andS18I). Similarly, it was evident that mutations alpha and beta were associated with distinct mutational signatures further confirming their development through distinct mutational mechanisms (Figures S17J, S17K, S18J, and S18K).
Evolution of copy-number changes from field effects to carcinoma
We used genome-wide SNP microarrays to analyze copy-number changes in both basal and luminal cystectomy specimens(Majewski et al., 2019). Changes in the gene copy-numbers were less evident in areas of bladder mucosa corresponding to field effects and designated as NU/LGIN than in HGIN and UC. Similar to the mutational changes described above, genome-wide copy-number alterations increased with progression from NU/LGIN to HGIN and UC (Figures S19A, S19B, S20A, and S20B). In both cystectomies they formed clearly defined plaques outlining areas of bladder mucosa that contained HGIN and UC with some adjacent areas of NU/LGIN (Figures S19C and S20C). These patterns of loco-geographic distribution indicated the involvement of wide-spread CNVs in later phases of tumor evolution associated with progression to HGIN and UC. The copy-number changes resulted in both gains and losses of multiple chromosomes that involved large segments of their p and q arms and in some instances entire chromosomes. Clustering of mucosal samples according to CNVs and the Euclidean distance revealed two major clusters of mucosal samples, with low and high numbers of CNVs (Figures S19D and S20D). Practically all of the UC and HGIN samples were contained in the second clusters. According to the Hamming distance, we detected significant differences in copy-number variability among samples classified as NU/LGIN and HGIN/UC (Figures S19E and S20E).
Modeling of bladder cancer evolution from mucosal field effects
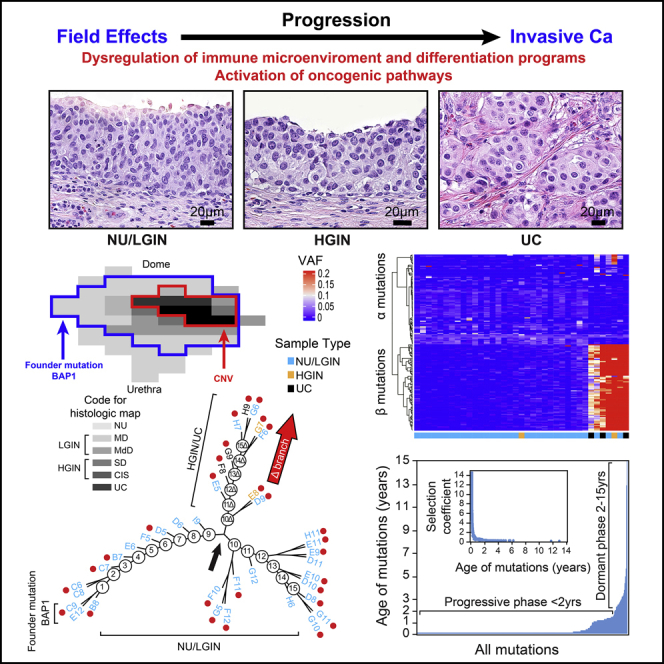
To understand the pattern of clonal evolution of cancer development from field effects, we used all nonsilent and silent mutations to construct cancer evolutionary trees. This revealed complex branching patterns reflecting multiple waves of clonal expansion represented by at least 15 and 17 nodes in luminal and basal bladder cancers, respectively (Figure 7A and S21A). The luminal map had a divergence at node 9 with the development of a branch designated as Δ comprising nodes 10Δ through 15Δ which progressed to HGIN and UC (Figure 7A). In the basal map the divergence occurred after node 12 where the main NU/LGIN clone evolved to HGIN and UC (Figure S21A). In the luminal map the main NU/LGIN clone continued to evolve after node 9 with a complex branching pattern which did not progress to HGIN or UC. In the basal map, successive clones of NU/LGIN at nodes 9, 12, 15, and 17 developed branches of clonal expansion which also did not progress to HGIN and UC. Branch Δ in the luminal map was characterized by an increase in the number of mutations and their VAFs, signifying their clonal expansion in the progression process (Figures 7B, 7C, S21B, and S21C). The increased numbers of mutations and VAFs were particularly evident in cluster β, signifying their putative driver roles in the development of HGIN and UC (Figures 7D, 7E, S21D, and S21E). Likewise, in nodes 13 through 17 in the basal map, the numbers of mutations and VAFs in cluster β increased dramatically confirming their putative driver roles in the development of HGIN and UC (Figures S21B–S21E). We identified a founder mutation of the BAP1 gene (Louie and Kurzrock, 2020) in the luminal map and one of the CAPRIN1 gene (Yang et al., 2019) in the basal map (Figures 7A and S21A). These mutations were present in the samples connected to node 1 in both maps (i.e., were present in areas of the bladder mucosa that were the farthest genetic distance from node 9 in the luminal map and node 13 in the basal map, which were the checkpoints for progression to HGIN and UC). Most importantly, they formed continuous plaques in the luminal and basal maps, which involved large areas of bladder mucosa (Figures 7F and S21F). Two founder mutations, BAP1 and CAPRIN1, were confirmed in multiple mucosal samples by blocking PCR combined with Sanger sequencing (Figures 7G, S21G, and Table S8).
Figure 7.

Evolution of mutations from field effects along the luminal track
(A) Parsimony analysis showing an evolutionary tree with nine nodes of expansion of successive clones of cells in the field effects corresponding to NU/LGIN with major branching at node nine designated the Δ branch, which evolved to HGIN and UC. All foci of HGIN and UC evolved from successive waves of clonal expansion in the Δ branch. The main NU/LGIN clone continued to evolve in successive waves of mutational changes (nodes 10-15) with a complex branching pattern that did not progress to HGIN or UC.
(B) Numbers of mutations in nodes 1-15 and the Δ branch in the evolutionary tree shown in A.
(C) VAFs of all nonsynonymous mutations in nodes 1-15 and the Δ branch in the evolutionary tree shown in A (Wilcoxon rank-sum p value).
(D) Numbers of mutations in cluster β in nodes 1-15 and the Δ branch in the evolutionary tree shown in A.
(E) VAFs of mutations in cluster β in nodes 1-15 and the Δ branch in the evolutionary tree shown in A (Wilcoxon rank-sum p value).
(F) Whole-organ histologic map showing a plaque-like mucosal area outlined by a blue line with a founder mutation of BAP1. The red line outlines an area corresponding to widespread CNV alterations.
(G) Validation of BAP1 mutational inactivation by Sanger sequencing. Variant allele of BAP1 DNA traces are shown in selected mucosal samples and compared to the wild-type sequence.
(H) Ages of all synonymous and nonsynonymous mutations predicted by mathematical modeling. Inset, the selection coefficient in relation to the predicted mutation age.
(I) Ages of the mutations in clusters α and β predicted using mathematical modeling. Inset, the selection coefficient for mutations in clusters α and β in relation to their ages.
(J) Ages of the mutations in cluster β predicted using mathematical modeling. Inset, the selection coefficient for mutations in cluster β in relation to their ages. Inset, the selection coefficient for mutations in cluster β in relation to their ages.
(K) Composite bar graph showing the distribution of all nucleotide substitutions in dormant and progressive phases of bladder cancer development.
(L) Bar plots showing the number of silent and nonsilent mutations in the dormant and progressive phases of bladder cancer development.
(M) VAF in dormant and progressive phases of bladder cancer development.
(N) Proportion of SNVs in specific nucleotide motifs for each category of substitutions in dormant and progressive phases of bladder cancer development.
(O) Weight scores for mutagenesis patterns in dormant and progressive phases of bladder cancer development. For (B) and (D) p values were calculated using a Kruskal-Wallis test. For (K) and (L) p values were calculated using a test of proportions. For (M) p values were calculated using a two sample Wilcoxon test.
To answer the question of how long bladder cancer takes to develop, we applied a mathematical modeling algorithm using the whole-organ mutational profiles and the sequences of successive clonal evolution in the nodes of the parsimony trees with a time-continuous Markov branching process (Kimmel, 2015). This approach provided an estimation of the duration of progression on a timescale of the transformation process based on maximal parsimonious principles. Initially, we performed the modeling by using all nonsynonymous and synonymous mutations (Figures 7H and S21H). These analyses showed that the tumors in the basal and luminal maps evolved over about 10–15 years, and that the age-related curves of mutations had left-skewed patterns, with the early mutations being more than 10 years old. Based on the timescale described above, the process of tumor development could be divided into two major phases. First, the older (dormant) phase, in which mutations developed gradually over about a decade, involved mutations that were characterized by low selection coefficients, which was consistent with the idea that they produced a marginal proliferative advantage. Second, the more recent (progressive) phase was less than 2 years old and was characterized by accumulation of large numbers of mutations with high selection coefficients indicating that they were associated with clonal expansion and produced a high proliferative advantage. We repeated these analyses with a focus on mutations characterized by clonal expansions (i.e., those in clusters α and β). The overall age-related pattern of mutations was similar in both maps spanning about 10 years. Again, the numberof mutations were limited and many of them were older than 2 years, corresponding to the dormant phase of tumor development characterized by a low selection coefficient with a minimal proliferative advantage. A large proportion of the mutations that were younger than 2 years with high selection coefficients corresponding to the progressive phase of the disease development (Figures 7I and S21I). When we restricted the mathematical modeling to clusters β, (i.e., the mutations characterized by increased clonality with disease progression and representing the putative drivers of the progression process), it showed that in the basal and luminal maps these mutations developed over the previous year and produced a high proliferative advantage as shown by increased selection coefficients (Figures 7J and S21J). To address the issue of potential distinctive mutational landscape involved in different phases of bladder carcinogenesis, we performed a detailed analysis of the mutational patterns in dormant and progressive phases of the disease (Figures 7K–7O and S21K–S21O). These analyses showed that in both maps the progressive phase was associated with an increase of C > T substitutions, major increase of nonsilent mutations and their VAFs with the involvement of distinct mutational signatures as compared to the dormant phase of the disease. Overall, these analyses support the concept that there are distinct mutational mechanisms that are switched on in a background of a dormant field effect driving the progression to clinically aggressive invasive bladder cancer.
Integrated analyses of pathways and regulons
To identify pathways involved in the development of bladder cancer we used genome-wide expression and methylation levels of monotonically altered genes and complemented them with mutations of genes in clusters α and β. This identified multiple dysregulated canonical pathways within each platform. Subsequent integrative analysis identified 91 and 125 canonical pathways dysregulated in the luminal and basal maps, respectively in NU/LGIN samples, which were continuously dysregulated in the progression to HGIN and UC (Tables S9 and S10). These pathways can be classified into three major groups (i.e., regulating immunity and inflammation, signal transduction and differentiation and those involved in oncogenesis). Because many of these pathways were related to each other and some were altered in both maps, we compiled a list of 30 selected pathways dysregulated in field effects and performed their combined analysis in both maps (Figure 8A). The striking dominant feature of these mucosal field effects was dysregulation of multiple pathways controlling immunity and inflammation. Wide-spread dysregulation of signaling controlled by interleukins (ILs) such as IL-2, -3, -6-8, −17, and −23 was the dominant feature of field effects and was complemented by changes in B- and T-cell signaling as well as changes controlling mucosal microenvironment. Dysregulation of signal transduction pathways, many of which involved the tissue differentiation program included HIF1, VEGT, mTOR as well as EFG, NGF, and AGF signaling. Dysregulation of multiple oncogenic pathways, such as p53, glioma, melanoma, breast cancer, and general cancer signaling, signified the transformative effects synergistic with alterations in immunity and tissue differentiation programs as initiating changes in bladder carcinogenesis. We validated these findings by analyzing the enrichment scores of 30 regulons corresponding to the same pathways controlling immunity and inflammation, signal transduction and tissue differentiation as well as oncogenesis in the TCGA cohort. It confirmed that they were frequently involved in bladder cancer. The activation of many of them was enriched specifically in luminal and basal cancers (Figures 8B, 8C, and S22).
Figure 8.

Interactive analyses of molecular pathways in bladder cancer development from field effects with their validation in the TCGA cohort
(A) Combined selected monotonically dysregulated pathways in field effects of both luminal and basal maps (One-sided Fisher exact test p value).
(B) Differential enrichment scores for the regulons controlling immunity, inflammation, signal transduction/differentiation, and oncogenesis in molecular subtypes of bladder cancer in the TCGA cohort (n = 408).
(C) Expression pattern for selected genes in the regulons of ILs, epidermal growth factor, and ovarian cancer signaling in the molecular subtypes of bladder cancer in the TCGA cohort (n = 408).
Integrated analyses across the platforms showed that dysregulation of IL and HOX genes were the dominant features in the mucosal field effects of both maps. Therefore, we analyzed the expression levels of these two gene families in whole organ maps and in the TCGA cohort. In both data files we identified mRNA expression levels for 33 out of 36 IL and for all 39 HOX genes. These analyses showed that the most of IL genes were upregulated in the mucosal field effects of both luminal and basal maps regardless of their null or hot immune microenvironments (Figure S23). Similarly, the HOX genes were widely dysregulated in the mucosal field effects of both maps. (Figure S24). The analyses in the TCGA cohort showed that both groups of genes were frequently dysregulated in bladder cancers of luminal and basal subtypes (Figures S25 and S26). Taken together these analyses confirmed the important roles of the IL and HOX genes in the incipient phases of bladder carcinogenesis.
Discussion
This study provided a comprehensive characterization of mucosal field effects associated with the development of bladder cancer on a whole-organ scale. It demonstrated complex, diffuse genome-wide mucosal changes in DNA methylation and mRNA expression involving multiple pathways affecting inflammation and immune responses, urothelial differentiation, and proliferation. Surprisingly, the field effects in microscopically-intact urothelium harbored dysregulations of multiple oncogenic pathways. The study also revealed that the two major intrinsic molecular subtypes of bladder cancer are determined de novo as luminal cancer developed from luminal field effects whereas basal cancer evolved from basal field effects. The field effects associated with the development of luminal cancer demonstrated retention of the luminal differentiation program, however, it was altered and exhibited dysregulation of multiple pathways involved in the differentiation of stratified epithelia. In contrast, the basal field effects involved suppression of the luminal program with diffuse mucosal downregulation of terminal urothelial differentiation exemplified by reduced expression of uroplakins. These results are consistent with animal models data showing that these two subtypes originate from distinct uroprogenitor cells (Shin et al., 2014; Van Batavia et al., 2014). Limited mapping of human cystectomy specimens provides evidence that molecular subtype commitment occurs in carcinoma in situ (Wullweber et al., 2021). Our data support the concept that such commitment occurs much earlier and it is already evident in the field effects.
The widespread plaque-like mRNA expression and DNA methylation changes in the mucosal field effects occurred against a background of highly heterogeneous mutations with low allele frequencies, indicating that the mRNA and DNA methylation changes were the primary early events of bladder carcinogenesis. According to the loco-geographic distribution patterns of mutations and their allele frequencies, we identified three distinct groups of mutations. The first group comprised low-frequency mutations restricted to individual mucosal samples. They very likely represented the progeny of individual uroprogenitor cells. The other two groups of mutations were associated with clonal expansion and involved large areas of bladder mucosa including those with microscopically normal-appearing urothelium. The first of these two groups, referred to as cluster α mutations, were characterized by low VAFs affecting typically less than 5% of cells in the urothelium. These mutations had stable low frequencies that did not increase with disease progression. In fact, in one of the maps this clone appeared to be eliminated with the progression of NU/LGIN to HGIN and UC and maybe actually negatively selected. We hypothesize that the clones of cells with these mutations set the stage for carcinogenesis by disturbing urothelial differentiation, providing a growth advantage and dysregulating immunity. The cells with these mutations apparently co-existed harmoniously with other urothelial cells and were microscopically normal appearing. In contrast, the number and VAFs of mutations in clusters β increased substantially with disease progression to the fully malignant phenotype, supporting the concept of their positive selections and driver role in this process. Mutational and clonality analyses identified founder mutations of clones in β clusters involving the BAP1 and CAPRIN1 genes in luminal and basal maps. Consistent with their founder and contributory driver’s roles, these mutations formed continuous large plaques involving most of the bladder mucosa and their frequencies increased dramatically with progression. Time modeling revealed that bladder carcinogenesis is a prolonged and clinically occult process spanning 10-15 years. The long dormant phase of bladder carcinogenesis lasting about 10 years was succeeded by an accelerated progressive phase during the last 1-2 years before the development of clinically symptomatic invasive bladder cancer. In both maps the final progressive phase of the disease appeared to be driven by the β clone mutations with a high proliferative advantage.
Several recent communications based on limited sampling of adjacent microscopically NU disclose a complex mutational landscape of low frequency mutations with high regional and interpatient variability (Lawson et al., 2020; Li et al., 2020; Strandgaard et al., 2020; Thomsen et al., 2017). Mutations in microscopically NU adjacent to cancer widely involved chromatin remodeling genes such as KMT2D and KDM6A and rarely well-known cancer driver genes such as TP53, PIK3CA, and FGFR3 (Li et al., 2020). These studies provide important insights into the mutational process occurring in microscopically NU but due to limited sampling they cannot precisely evaluate geographic distribution of these mutations and their involvement in clonal expansion of intraurothelial precursor lesions and their progression to invasive cancer.
Of interest is the fact that clustering of bladder mucosal samples using both gene expression and methylation profiles as well as mutational landscapes was practically never perfect for their microscopic designations and some of the NU/LGIN samples co-clustered with HGIN/UC samples. In many instances, these samples were in close proximity to areas of carcinoma in situ or invasive cancer.
Our data support the concept that the widespread dysregulation of the mucosal immune environment involving multiple ILs plays an important role in bladder cancer initiation. The ILs act on a background of an altered urothelial differentiation program exemplified by dysregulation of the HOX genes and activated oncogenic pathways causing an irreversible damage to the urothelium which precipitates the development of clinically aggressive invasive bladder cancer.
Limitations of the study
Limitations of our study are related to the volume of data and the complexity of analytical approach which permitted at this phase the whole organ analysis of two representative bladder cancers evolving along the luminal and basal tracks. The molecular profile of bladder cancer evolution described in this study reflects cancer development through dysplasia carcinoma in situ sequence and does not address its development through a more common low-grade papillary pathway. Comparisons of our observations with those of TCGA cohort revealed that mucosal field changes identified in the two maps played a role in a large proportion of UCs. Therefore, even though this study is limited to two whole organ maps it represents significant advancement to our understanding of bladder carcinogenesis. The use of NU harvested from the ureters of nephrectomy specimens from patients without urothelial neoplasia is a suboptimal source for the reference baseline, as it is not identical to the NU of the bladder and can also be perceived as a limitation in our analytical algorithm (Riedel et al., 2005).
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| Human mucosal samples taken from the whole bladder of two individuals and representative mucosal samples taken from seven individuals | UT MD Anderson Cancer Center | Nine human bladder maps. See Table S1 |
| Critical commercial assays | ||
| Whole Exome Sequencing on Illumina NovaSeq 6000 Platform | Illumina | N/A |
| Illumina Infinium Omni2.5-8 BeadChips for CNV | Illumina | GPL16104 |
| Infinium MethylationEPIC BeadChip Kit | Illumina | GPL21145 |
| Deposited data | ||
| RNAseq raw and analyzed data | This paper | SRA: PRJNA736323 |
| Exom Sequencing raw and analyzed data | This paper | SRA: PRJNA736323 |
| CNV raw and analyzed data | This paper | GEO: GSE171363 |
| Methylation raw and analyzed data | This paper | GEO: GSE171363 |
| Human reference NCBI build 38, GRCh38 | Genome Reference Consortium | https://www.ncbi.nlm.nih.gov/assembly/grc/human |
| Human Genome Browser, GRCh38 | University of California Santa Cruz Genomic Institute | https://genome.ucsc.edu/cgi-bin/hgGateway |
| TCGA mutational data | Broad Institute Genome Data Analysis Centers TCGA | http://gdac.broadinstitute.org/ |
| RNASeq level 3 data, Illumina methylation array 450 and SNP6 CNV | R package TCGAGeneReport (version 2.3) | http://gdac.broadinstitute.org/ |
| Mutational signatures | Alexandrov et al., 2013 | http://cancer.sanger.ac.uk/cosmic/signatures |
| Oligonucleotides | This paper | See Table S8 |
| Software and algorithms | ||
| RSeQC software | Wang et al., 2012 | http://rseqc.sourceforge.net/ |
| FastQC software | N/A | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| FeatureCounts software in the Subread package | Liao et al., 2014 | http://subread.sourceforge.net/ |
| DESeq2 software (version 1.26.0) | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| Illumina GenomeStudio software (version 2.0) | N/A | https://support.illumina.com/downloads/genomestudio-2-0.html |
| OncoSNP software | Yau et al., 2010 | https://sourceforge.net/projects/oncosnp/ |
| minfi R package (version 1.30.0) | Aryee et al., 2014; Fortin et al., 2014 | https://bioconductor.org/packages/release/bioc/html/minfi |
| Significance analysis of microarray (SAM) | Tusher et al., 2001 | https://tibshirani.su.domains/SAM/ |
| BWA-MEM (version 0.7.12) | Li and Durbin, (2010). | http://bio-bwa.sourceforge.net/ |
| Samtools (version 1.4) | Etherington et al., 2015 | http://samtools.sourceforge.net |
| Picard (version 2.5.0) | N/A | https://broadinstitute.github.io/picard |
| GATK (version 4.1.3.0) | McKenna et al., 2010 | https://software.broadinstitute.org/gatk/download |
| MuTect2 | DePristo et al., 2011; Cai et al., 2016 | https://software.broadinstitute.org/gatk/documentation/tooldocs/3.8-0/org_broadinstitute_gatk_tools_walkers_cancer_m2_MuTect2.php |
| Oncotator (version 0.8.0) | Ramos et al., 2015 | http://portals.broadinstitute.org/oncotator |
| Parsimony method | Nixon, 1999; Paradis, 2011) | https://www.ncbi.nlm.nih.gov/Class/NAWBIS/Modules/Phylogenetics/phylo14.html |
| Ingenuity Pathway Analysis (IPA) | Jiménez-Marin et al., 2009; Krämer et al., 2014 | https://www.qiagen.com/us/products/life-science-research/research-applications/gene-expression-analysis/analysis/ingenuity-pathway-analysis/ |
| Gene set enrichment analysis (GSEA) | Subramanian et al., 2005 | https://software.broadinstitute.org/cancer/software/gsea/wiki/index.php/Main_Page |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Bogdan Czerniak (bczernia@mdanderson.org)
Materials availability
This study did not generate new unique reagents or material.
Experimental model and subject details
Human samples and clinical data were collected and archived according to a laboratory protocol approved by the Institutional Review Board of The University of Texas MD Anderson Cancer Center. Whole-organ histologic and genetic mapping was performed using radical cystectomy samples from nine male White patients with a mean age of 73.6 years (range, 55.0–86.0 years) who had high-grade muscle-invasive (T3) UC (Table S1).
Method details
Preparation of whole-organ maps and extraction of DNA/RNA
The preparation of cystectomy samples for DNA/RNA extraction and whole-organ histologic mapping followed the four steps described below and illustrated in Figure 1 (Lee et al., 2007; Majewski et al., 2008).
-
Step 1
Each cystectomy sample was opened longitudinally along the anterior wall of the bladder and pinned to a paraffin block. A mapping grid was then applied and pressed down against the bladder mucosa with mechanical screws, which provided sealed wells that separated mucosal areas and tumors into 1 x 2-cm (2-cm2) rectangles. The grooves at the bottom of the mapping grid preserved the urothelium for microscopic examination.
-
Step 2
Phosphate-buffered saline (0.4 mL) was poured into each well, and the surface urothelium was scraped with a custom-designed metallic scraper. A small proportion of this fluid containing urothelial or tumor cells (∼20 μL) was used for cytospin preparations to assess the purity of cell suspensions for DNA and RNA extraction. In areas containing visible tumor, the tissue was dissected directly from the bladder.
-
Step 3
TRIzol reagent (1.6 mL) was poured into each mapping well, and the fluid was collected into separate Eppendorf tubes labeled with numbers corresponding to individual mapping wells in the bladder mucosa. The TRIzol reagent fluid contained lysed cells and was used for DNA/RNA extraction. The samples with tumor tissue were cut into small pieces and treated with TRIzol.
-
Step 4
In the final step, each well was washed with phosphate-buffered saline, and the mapping grid was removed from the surface of the bladder, which was then fixed in formalin overnight. The mapping grid left a permanent impression on the bladder surface, and its grooves preserved the urothelium for histologic mapping of the entire mucosa. After formalin fixation, each piece of bladder surface along with the preserved tissue was embedded in paraffin. One section from each block was stained with hematoxylin and eosin to microscopically evaluate the distribution of NU, in situ precursor lesions, and UC. The precursor intraurothelial lesions were dichotomized into LGIN and HGIN (Lee et al., 2007). Map samples containing tumor tissue were classified according to the two-tier histologic tumor grading system of the World Health Organization and referred to as low- and high-grade (Eble JN et al., 2004). The growth pattern for papillary versus nonpapillary (or solid) and depth of invasion were also recorded. Levels of invasion were defined according to the TNM staging system (Sobin et al., 2009).
Two steps were carried out for quality control of sample and cell suspension preparations as well as DNA/RNA extraction. The first step consisted of overall assessment of the cystectomy samples in terms of representation of the full spectrum of precursor lesions and tumor samples as well as the purity of urothelial and tumor cell preparations. Only cystectomy samples with intact areas of cancer involving no more than 50% of the bladder mucosa were accepted for whole-organ mapping. Moreover, a cystectomy sample accepted for likely informative mapping had bladder mucosa with intact surface urothelium in more than 90% of the sampling wells and the full spectrum of microscopically recognizable precursor intraurothelial lesions. In addition, only samples containing more than 90% microscopically recognizable intact urothelial or tumor cells were used for DNA/RNA extraction, yielding 5–70 mg of DNA.
The second quality control step consisted of assessment of the quality of the final DNA/RNA preparations for genomic profiling. Their quality was verified using a NanoDrop spectrophotometer (Thermo Scientific), Bioanalyzer system (Agilent), and Qubit fluorometer (Invitrogen). Using these quality checks, only whole-organ cystectomies with a sufficient amount of high-quality DNA and RNA in at least 90% of mucosal samples were accepted for genomic profiling. This provided about 30-40 DNA/RNA samples per cystectomy corresponding to specific mucosal areas yielding 10–15 μg of DNA per sample and 25-30 μg of RNA per sample.
Quantification and statistical analysis
RNA sequencing and data analysis
The RNA integrity was assessed using a 2100 Bioanalyzer (Agilent Technologies). The RNA concentration was determined using RiboGreen quantification (Quant-iT RiboGreen RNA Assay Kit; Invitrogen). RNA samples meeting a quantity threshold of 1 μg and with an RNA integrity number of at least 7 were analyzed in the Advanced Technology Genomics Core at MD Anderson. Prior to RNA library construction, ribosomal RNA was removed from total RNA preparations, and cDNA synthesis using oligo d(T) and random hexamers was performed. The library was made up of random fragments representing the entire sample. It was created by shearing DNA into 150-400 base fragments that were ligated to specific adapters. Following a sample cleanup step, the resulting library was quantified using quantitative PCR and checked for quality using a TapeStation (Agilent Technologies). The analyses were performed with 79 RNA samples from 74 mucosal samples in two maps (34 samples from map 19 and 40 samples from map 24) and from 5 sex-matched control NU suspensions, which were prepared from the ureters in nephrectomy samples that were free of urothelial neoplasia (Love et al., 2014).
Quality control for RNA was conducted using RSeQC (Wang et al., 2012) and FastQC software. Sequencing reads were aligned to the GRCh38 reference genome using STAR (version 2.7.3a) (Dobin et al., 2013) with GENCODE (release 32) transcript annotations (Harrow et al., 2012). Read counts for individual genes were obtained using featureCounts software in the Subread package (Liao et al., 2014). On average, 27 million reads per sample associated with more than 58,000 unique genes, both coding and noncoding, were obtained.
From this set of genes, 44,000 with more than 10 reads in at least one sample were selected and used in the subsequent analyses. Genes differentially expressed in specific sample groups were identified using DESeq2 software (version 1.26.0) (Love et al., 2014) with the Wald test by using a design formula that included batch effect correction. Benjamini-Hochberg correction was used for multiple testing in all instances (Benjamini and Hochberg, 1995).
The normalized read counts were transformed into log2 ratios between the signal of NU/LGIN, HGIN, and UC samples, and the average signal obtained from the normal control samples. The resulting log fold-change values (LFC) were used for the detection of monotonically dysregulated genes in 3 groups of samples which followed the progression pattern of neoplasia from field effects (NU/LGIN) through HGIN to UC as follows: (1) the genes in the first group were dysregulated in early field effects (NU/LGIN) and continued their dysregulation in the same direction with progression to HGIN and UC. They were identified by their different expression levels in at least 80% of all samples and have their expression levels upregulated or downregulated by at least LFC = 1.5; (2) the genes in the second group were dysregulated in parallel to disease progression to HGIN and continued the same direction of their dysregulation in progression to UC. They were identified by their dysregulation in 80% of HGIN/UC samples with altered expression levels by at least LFC = 1.5; and (3) the genes in the third group were identified by their dysregulation of at least LFC = 1.5 in UC samples only.
For assessment of the luminal and basal phenotypes, the expression levels for 28 luminal and 20 basal marker genes identified previously were used. For quantitative assessment of these two molecular subtypes of bladder cancer, the previously developed BLT score was used (Guo et al., 2020a). In brief, for assessment of the luminal phenotype, the 14 luminal markers in the original classifier were used (Choi et al., 2014; Dadhania et al., 2016). To increase the power of analyses, these markers were complemented by 14 PPARγ target genes previously shown to be significantly enriched in luminal cancers (Choi et al., 2014; Dadhania et al., 2016). Similarly, for assessment of the basal phenotype, the 9 basal markers from the original classifier were used, and they were complemented by an additional 11 p63 target genes shown to be significantly enriched in basal cancers (Choi et al., 2014; Dadhania et al., 2016). Linear discriminant analysis was performed to determine the power of individual markers in identifying molecular subtypes of bladder cancer (Solberg, 1978). The unidimensional BLT score was defined as , where is the negative coefficient of the linear discriminant and is the expression of marker genes. Next, least absolute shrinkage and selection operator analysis was used to select the best 16 luminal and 12 basal markers to combat multicollinearity (Li et al., 2018). Specifically, least absolute shrinkage and selection operator were applied to the L1 parameter to constrain the sum of the absolute values for the model parameters. In the process, 28 genes with non-zero coefficients after the regularization process were selected for calculation of the BLT score. The TCGA cohort was used as a training set to build a linear discriminant analysis model with the 28 selected genes.
To assess the EMT status in the evolution of bladder cancer from mucosal field effects, the expression levels for signature transcription factors involved in the activation of EMT of SNAIL, TWIST, ZEB, FOX, SOX, and KLF families were analyzed, complemented by analyses of the expression of homotypic adhesion molecules such as E-cadherin, CLDN1, and tight junction protein 1. To quantitatively assess the level of EMT, the EMT score was calculated based on a 76-gene expression signature reported by Byers et al. (Byers et al., 2013; Guo et al., 2019). For each sample, the score was calculated as a weighted sum of 76 gene expression levels using , where wi is the correlation coefficient between the ith gene expression in the signature and that of E-cadherin and Gij is the ith gene’s normalized expression in the jth sample. The scores were centered by subtracting the mean for all tumor samples so that the grand mean of the score was zero.
To analyze immune gene expression signatures of bladder cancer evolution from field effects, dendrogram nodes corresponding to genes expressed in specific immune cell types were identified via DAVID Functional Annotation Clustering and Ingenuity Pathway Analysis. The immune expression signature was quantitatively assessed by calculating the immune scores for the expression profiles genes (Guo et al., 2019). The immune score for the ith sample was defined as mi-(1/n) , where mi is the median expression level across the ith sample’s immune expression profile and (1/n) is the grand mean of medians across all n samples.
Methylation and data analysis
The MethylationEPIC BeadChip method (Illumina), which facilitates quantitative interrogation of more than 850,000 methylation sites across the genome at single-nucleotide resolution, was used for methylation analysis. In brief, bisulfite conversion on 1 μg of genomic DNA was performed using an EpiTect kit (QIAGEN) according to the manufacturer’s instructions. Bisulfite-converted DNAs were enzymatically fragmented prior to hybridization to BeadChip arrays. Bead arrays were scanned using the Illumina iScan technology to produce IDAT files. The raw IDAT files for each sample were preprocessed and normalized using the minfi R package (version 1.30.0) (Aryee et al., 2014; Fortin et al., 2014). CpG sites that were on the sex chromosome, were cross-reactive, or had single-nucleotide polymorphisms were filtered out.
As there were multiple CpG sites mapped to a given gene on the EPIC array, gene-level methylation data were summarized using Spearman correlations between the CpG sites within each gene and its expression level based on the results of an external data set (TCGA bladder tumor samples). Specifically, the methylation level for each gene was represented as the β value of the single probe with the most negative correlation with mRNA expression level in the TCGA cohort by interrogating the first exon, 5′ untranslated region, or up to 1500 base pairs upstream from the transcription start site. The resulting whole-organ mapping methylation data set in total contained 14,744 genes with 36 samples in each map and 8 normal controls for downstream analysis.
A nonparametric testing method for array-based high-throughput data called significance analysis of microarray (SAM) was performed to compare the methylation β values for NU/LGIN vs. control, HGIN vs. control, and UC vs. control, respectively (Tusher et al., 2001). The number of permutations in SAM was set to 100 with pre-specified false discovery rate (FDR) error control threshold to be 0.2 across all group comparisons, i.e., any gene with FDR q-value smaller than 0.2 was considered to show a significant methylation change in a given group compared with the controls (Storey and Tibshirani, 2003). The monotonically dysregulated genes in 3 groups of samples which followed the progression pattern of neoplasia from field effects (NU/LGIN) through HGIN to UC were detected as follows: (1) The genes in the first group were dysregulated in early field effects (NU/LGIN) and continued their dysregulation in the same direction with progression to HGIN and UC. They were identified by being significant with q-value < 0.2 in the same direction in each of the three tests of NU/LGIN, HGIN, and UC compared with the controls respectively; (2) the genes in the second group were dysregulated in parallel to disease progression to HGIN and continued the same direction of their dysregulation in progression to UC. These genes were significant with q-value < 0.2 in the same direction (hyper or hypomethylated) in the tests of HGIN, and UC compared with the controls and not significant in the NU/LGIN test compared with the controls; (3) genes showing changes in UC only were those with significant q-value < 0.2 only in the test comparing UC and the controls.
For visualization in the heatmaps, the methylation levels were transformed into log2 ratios comparing the samples in the NU/LGIN, HGIN, and UC groups with the normal control samples, i.e., the log2 ratio for gene j in sample i is
Whole-exome sequencing and data analysis
A sequencing pipeline in the Functional Genomic Core at MD Anderson was used for whole-exome sequencing and was performed with an Illumina NovaSeq 6000 sequencer using a high-output flow cell with 150-nt paired-end runs and an average mean (± SD) coverage across the samples of 479 ± 71 (coverage range, 302-678). The sequencing coverage for sample groups in map 24 (luminal) and map 19 (basal) were as follows: map 24, NU/LGIN, and 496 ± 69; HGIN, 539 ± 102; UC, 472 ± 14 and map 19, NU/LGIN, and 460 ± 71; HGIN, 436 ± 29; UC, 425 ± 121. All steps, including Illumina library preparation, exome capture, Illumina sequencing, downstream exome data processing, mutation calling, and annotation, were performed essentially as outlined in recent cancer sequencing project reports (Robertson et al., 2017; Weinstein et al., 2014). In brief, for data analysis, BWA-MEM software (version 0.7.12) was used to align reads to the GRCh38 reference genome (Li and Durbin, 2010). SAMtools (version 1.4) and Picard (version 2.5.0) were used to sort and convert formats and remove duplicate reads (Etherington et al., 2015). The Genome Analysis Toolkit (version 3.4–46) was used to generate realigned and recalibrated BAM files (McKenna et al., 2010). We applied the F score β 1.0 in the function of FilterMutectCalls in GATK:4.1.3.0 to identify the mutations with low frequencies. Briefly, the F score was used to calculate the relative weights of recall to precision to disguise the signals from noise (https://www.biorxi.org/content/biorxiv/early/2019/12/02/861054.full.pdf). Mutect2 and Oncotator (version 1.8.0.0) were used to identify nonsilent and silent mutations and to produce function-based annotations of the SNVs and insertions/deletions (Cai et al., 2016; Callari et al., 2017; DePristo et al., 2011; Ramos et al., 2015). To calculate the expected sensitivity of our whole-exome sequencing results we used bam-readcount program to collect numbers of reads confirming the presence of reference and alternative alleles in each SNV per sample. Based on allele frequencies in WXS and the coverage in RNASeq we selected for validation those variant alleles for which the probability of finding at least two variant reads was higher than 90%. This provided an average sensitivity of variant allele detection for two maps of 85%.
The mutations identified in maps 19 and 24 were analyzed in the TCGA cohort, for which the mutational data were downloaded from the TCGA portal (https://portal.gdc.cancer.gov/). MutSigCV (version 1.4; https://software.broadinstitute.org/cancer/cga/mutsig) was used to identify genes that were mutated more often than expected by chance given the background mutational process. The list of significant genes determined by using a false-discovery rate (FDR) cutoff of 0.05 (Majewski et al., 2019).
Mutational signatures and data analysis
Nonsilent mutations identified in maps 19 and 24 that were present in at least one sample corresponding to the base pair substitutions C > A, C > G, C > T, T > A, T > C, and T > G were used for the analysis of mutational signatures. The Fisher exact test was used to test the null hypothesis that they were equally distributed in NU/LGIN, HGIN, and UC samples. The genomic context of SNVs, referred to as fingerprints, which included the two bases flanking 5′ and -3′ sides at each position for a total of 96 possible mutational patterns, was assembled. The Wilcoxon rank sum test was used to test the hypothesis that the frequency of any fingerprint did not differ between any two groups of mucosal samples. The Benjamini-Hochberg method was applied to control the FDR (Benjamini and Hochberg, 1995). For each sample, its mutational fingerprints (V) and the quadratic programming method were used to compute a weight score (H) for each of 30 canonical mutational signatures (W) available from the Sanger Institute (https://cancer.sanger.ac.uk/cosmic/signatures). A 96 by 30 matrix of canonical signatures (W) was assembled, and given the 96 by 1 mutational profile of a sample (V), a 30 by 1 vector (H) for each of the canonical signatures’ relative contributions to the sample profile was computed by solving the optimization problem
Optimization problems were solved via quadratic programming using the R package quadprog (version 1.5–5). The Kruskal-Wallis test was used to verify the null hypothesis of no difference in weight scores among NU/LGIN, HGIN, and UC samples. Bootstrapping analysis was performed to determine the significance of the contribution of mutational signatures in individual mucosal samples. Mutational fingerprints (V) were resampled for each sample with replacement, and the weight score was computed as described above 2000 times. A one-sided empirical p value was calculated as the percentage of weight scores greater than or equal to the observed sample weight score in the resampling distribution.
Copy-number genotyping and data analysis
Quality control for and preprocessing of Illumina Infinium Omni2.5-8 microarray data were conducted using Illumina GenomeStudio software (version 2.0) with library files provided by Illumina (version 1.4-a1) and GRCh38 reference genome coordinates. In total, 64 samples (32 for map 19 and 32 for map 24) were studied as a single batch. Log R ratios and B-allele frequencies were obtained using the cnvPartition GenomeStudio plugin (version 3.2.0) with the GC wave adjustment option. The log R ratios and B-allele frequencies were exported from GenomeStudio as a single table in wide format and then split into sample-specific files using a kcolumn script, which is part of the PennCNV software tool (version 1.0.5) (Wang et al., 2007). After adjusting the column order in the data files using our custom shell script, detection of copy-number variable regions was conducted using OncoSNP software (version 1.4) (Yau et al., 2010). OncoSNP was executed for tumor-normal tissue sample pairs defined in the batch file with both stromal contamination and intratumoral heterogeneity models. The log R ratios and B-allele frequency plots were created using OncoSNP for all chromosomes. Additional visualizations of copy number-altered regions based on .cnvs and .qc result files provided by OncoSNP were created using our custom R functions.
Verification of mutations using PCR
Seventeen gene mutations in clusters α and β were verified using PCR-based Sanger sequencing PCR. The list of verified mutations and their respective primers are provided in Table S8. Briefly, Taq DNA polymerase (M0273S; New England BioLabs) was used to amplify 25 ng of genomic DNA. Fifteen mutations (PLCB3, PACS1, OTOP1, CDKN1A, APC, FBXW7, BRAF, RCC1, ZMIZ1, ERCC2, KDM6A, FBXW7-2, CDKN2A, KMT2C and SMARCA4) were verified by three-step PCR (39 cycles at 95°C for 15 seconds, 60°C for 15 seconds and 72°C for 8 seconds).
Low VAFs of BAP1 and CAPRIN1 founder mutations were verified by two-step blocking PCR (34 cycles at 95°C for 30 seconds and 58.6°C for 30 seconds to amplify BAP1 and 34 cycles at 95°C for 30 second and 62.8°C for 30 seconds to amplify CAPRIN1) (Wu et al., 2017). The amplified products were sequenced via the Sanger method using a 373Oxl sequencer (Applied Biosystems).
Phylogenetic analysis and modeling of bladder cancer evolution from field effects
To reconstruct a phylogenetic tree, the Hamming distances among mucosal samples were calculated using a binary matrix of all nonsilent and silent mutations present in at least one sample, and the maximum parsimony method was applied (Nixon, 1999; Paradis, 2011). In this representation of the phylogenetic tree, each node models a population of cells: the length of the edge connecting two nodes is proportional to the number of mutated genes, whereas a branch represents a time point in the evolution of neoplasia when two distinct populations emerge. The length of the branch is proportional to the number of mutated genes that are private in each population.
To reconstruct the time of evolution from mucosal field effects to bladder cancer, the time-continuous Markov branching process with immigration and parsimonious principles was used (Lange, 2010). In brief, a mutationj appears at time in a progenitor cell of the urinary bladder urothelial lining and gives rise to a mutant clone. Mutant cells divide at rate λ (1/year), and after division, one cell enters self-renewal and the other differentiates with probability or both cells enter self-renewal with probability sj. As a consequence, the mutant clone grows exponentially as exp (λsjt), where t is the age of the j-th mutant’s clone counted from . The secondary clones expand, involving different areas of bladder mucosa at times modeled by a stochastic Poisson process with intensity (1/yr) (Last and Penrose, 2017). If the expected cell counts in the successive -th mutant clones are denoted byi = 0, 1, 2,…, and the number of haploid genomes in normal uroprogenitor cells are denoted by 2N, the corresponding VAFs are defined as the ratios and are computed as follows (Lange, 2010):
For any mutationj of age tj, the sequence of expectations was computed to estimate the coefficients and . The coefficient c = 2N is a constant parameter representing an estimate of the number of uroprogenitor cells in the sampled area. The computations were performed for 102-105 uroprogenitor cells in the sampled mucosal area, which did not significantly change the time modeling results, but the best fit was obtained with 5 × 103 uroprogenitor cells, for which the data are presented. With a cell division rate λ and migration rate v, the parameter bj is the proxy for mutation age tj, whereas the ratio aj/bj is the proxy for selection coefficient sj. A fitting algorithm with the optimization programs fminsearch and fminbnd in the MATLAB programming language was used to estimate the sequence of mutations in tumor development (Lagarias et al., 1998), (Brent, 1972; Forsythe et al., 1977). The resulting time estimates were presented as bar diagrams representing the ages of mutations and point charts of the corresponding selection coefficients.
Integrative analysis of pathways and regulons
To identify molecular pathways involved in the development of bladder cancer, we performed the Ingenuity Pathway Analysis (IPA; Ingenuity Systems, Redwood City, CA) focusing on pathways dysregulated in the field effects which continued their dysregulation in progression to HGIN and UC and referred to as monotonically dysregulated (Jiménez-Marin et al., 2009; Krämer et al., 2014). In these analyses we used abnormally expressed and dysmethylated genes as well as mutated genes from clusters α and β. Initially, we computed the enrichment p-values for IPA canonical pathways based on a one-sided Fisher’s exact test for each of the four gene groups identified by RNAseq, methylation and mutational platforms (α and β mutational clusters). Since changes of the methylation patterns appeared to be dominant in the field effects, we identified the nominally significant pathways with the methylation enrichment p-values < 0.05. In order to perform integrated analysis of the pathways across the platforms the enrichment p-values for gene expression and mutationdata for the same pathways were cross-checked with the methylation enrichment p-values and visualized using bar plots of –log10(p-value). This provided an integrated list of 91 and 125 monotonically dysregulated pathways in the field effects of luminal and basal maps respectively (Tables S9 and S10). Out of these lists we selected 30 pathways, many of which were dysregulated in both maps, and controlled immunity/ inflammation, signal transduction/differentiation and oncogenesis. In order to address the question how frequently these pathways are involved in bladder carcinogenesis we performed the modified analysis of their regulons in the TCGA cohort (Castro et al., 2016). First, we identified the genes encoding known transcription factors (TFs) in these pathways (Lambert et al., 2018). We then considered all target genes for each TF in the given pathway as the regulon and performed a two-tailed GSEA using R package “RTN” for the TCGA bladder cancer RNA-seq data (Castro et al., 2016; Subramanian et al., 2005). This method first divided the set of target genes for each TF into positive and negative targets associated with the TF’s expression level using Spearman’s correlation coefficient. The distribution of the positive and negative targets was tested respectively, producing enrichment scores (ES) for each sample. The difference between positive ES and negative ES resulted in the differential enrichment score (dES). Since there could be more than one TF in a pathway, we calculated a pathway-level dES as follows: let dESij be the dES for sample i and TF j (i = 1, …,n samples, j = 1, …,p TFs), then the pathway-level dES for sample i was , where if the majority (≥50%) of the pairwise correlations between TF j’s dES and the other TFs’ dES was positive, otherwise .
Control and reference samples
The mutational and CNV analyses were performed in comparison with paired normal genomic DNA from the same patient. Normal genomic DNA was extracted from the buffy coat of peripheral blood after Ficoll centrifugation. Additional normal genomic DNA was extracted from bladder smooth muscle. Two or three samples of bladder smooth muscle (each measuring ∼1 cm3) were snap-frozen. The absence of tumor infiltration from control samples was confirmed via microscopic examination of hematoxylin- and eosin-stained frozen sections of samples subjected to DNA extraction. For analysis of gene expression and methylation status, NU obtained from the ureters in samples from nephrectomies performed for renal cell carcinoma confined to the kidney was used. Ureters were opened longitudinally and pinned down to a paraffin block, and the urothelial surface was scraped with a surgical blade. The quality and purity of urothelial cell suspensions were checked using cytospin preparations, and the samples were snap-frozen in phosphate-buffered saline for subsequent DNA/RNA extraction. Validation analyses were performed on the TCGA cohort (n = 408). The TCGA mutational data were downloaded in the mutation annotation format (MAF) from the GDAC website: http://gdac.broadinstitute.org/. The level 3 RNASeq, and Illumina methylation array 450 data were obtained through the R package TCGAGeneReport (version 2.3) (https://github.com/MD-Anderson-Bioinformatics/GeneSurvey). The downloaded RNASeq data were normalized and log-transformed while the methylation data comprised the β values.
General statistical analysis
Wilcoxon rank-sum test was used for two-sample comparisons, whereas the Kruskal-Wallis test was used for multiple group comparisons. Chi-squared test was used to test association between categorical variables; Fisher’s exact test was used whenever there was a cell count less than 5 in a given contingency table. All statistical tests were two-sided unless otherwise specified. False discovery rate (FDR) was used to correct for multiple hypothesis testing. An adjusted p value with FDR < 0.05 was considered statistically significant unless otherwise specified. An unadjusted p-value < 0.05 was considered nominally significant.
Acknowledgments
This study was supported by NCI Genitourinary Bladder SPORE Grant P50CA 91846 (Project 1 and Core C) to B.C., Cancer Prevention and Research Institute of Texas (CPRIT) Grant RP220021 to B.C., MDACC Nathan W. Lassiter Endowment; as well as Donor Funds to B.C; the Polish National Science Centre grants No. 2018/29/B/ST7/02550 to M.K. and 2016/23/D/ST7/03665 to R.J.; National Institute of Health grants R01HL136333 and R01HL134880 to Y.C.; and the European Union Fund grant POWR.03.02.00-00-I029 to P.K.
Author contributions
B.C., conceived and supervised the whole project as well as interpreted the data; J.B., D.C., S.L., and T.M. performed the experiments and generated the data; C.C.G. and B.C. performed pathological analysis; H.Y., R.J., Z.W., Y.C., K.N.D., L.Z., S.C., F.T., P.K. and H.C. performed data analysis; N.N., C.D., J.G., D.T., and C.L. provided cystectomy specimen and clinical data; J.N.W., W.W., P.W., and M.K. performed and supervised data analysis; D.M. interpreted the data with a focus on pathway analysis; M.K. designed the mathematical model; B.C. wrote the manuscript with input from others.
Declaration of interests
We confirm that there are no competing interests.
Published: July 15, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.104551.
Supplemental information
Data and code availability
-
•
The article contains all data sets generated or analyzed during this study. The WES and bulk RNA sequencing data generated or analyzed in this study have been deposited in NCBI Sequence Read Archive (SRA) with accession number PRJNA736323. The methylation and CNV data have been deposited in NCBI Gene Expression Omnibus (GEO) with accession number GSE171363.
-
•
The study did not generate original code.
-
•
Any additional information required to reanalyze the data included in this study will be provided by lead contact upon request.
References
- Alexandrov L.B., Nik-Zainal S., Wedge D.C., Aparicio S.A.J.R., Behjati S., Biankin A.V., Bignell G.R., Bolli N., Borg A., Børresen-Dale A.L., et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvisi G., Brummelman J., Puccio S., Mazza E.M., Tomada E.P., Losurdo A., Zanon V., Peano C., Colombo F.S., Scarpa A., et al. IRF4 instructs effector Treg differentiation and immune suppression in human cancer. J. Clin. Invest. 2020;130:3137–3150. doi: 10.1172/JCI130426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aryee M.J., Jaffe A.E., Corrada-Bravo H., Ladd-Acosta C., Feinberg A.P., Hansen K.D., Irizarry R.A. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics. 2014;30:1363–1369. doi: 10.1093/bioinformatics/btu049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y., Hochberg Y. Controlling the false discovery rate - a practical and powerful approach to multiple testing. J. Roy. Stat. Soc. B Stat. Methodol. 1995;57:289–300. doi: 10.1111/j.2517-6161.1995.tb02031.x. [DOI] [Google Scholar]
- Bhandari A., Xia E., Zhou Y., Guan Y., Xiang J., Kong L., Wang Y., Yang F., Wang O., Zhang X. ITGA7 functions as a tumor suppressor and regulates migration and invasion in breast cancer. Cancer Manag. Res. 2018;10:969–976. doi: 10.2147/CMAR.S160379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brent R. Prentice-Hall; 1972. Algorithms for Minimization without Derivatives. [Google Scholar]
- Byers L.A., Diao L., Wang J., Saintigny P., Girard L., Peyton M., Shen L., Fan Y., Giri U., Tumula P.K., et al. An epithelial-mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin. Cancer Res. 2013;19:279–290. doi: 10.1158/1078-0432.CCR-12-1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai L., Yuan W., Zhang Z., He L., Chou K.C. In-depth comparison of somatic point mutation callers based on different tumor next-generation sequencing depth data. Sci. Rep. 2016;6:36540. doi: 10.1038/srep36540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callari M., Sammut S.J., De Mattos-Arruda L., Bruna A., Rueda O.M., Chin S.F., Caldas C. Intersect-then-combine approach: improving the performance of somatic variant calling in whole exome sequencing data using multiple aligners and callers. Genome Med. 2017;9:35. doi: 10.1186/s13073-017-0425-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research N. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature. 2014;507:315–322. doi: 10.1038/nature12965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro M.A.A., de Santiago I., Campbell T.M., Vaughn C., Hickey T.E., Ross E., Tilley W.D., Markowetz F., Ponder B.A.J., Meyer K.B. Regulators of genetic risk of breast cancer identified by integrative network analysis. Nat. Genet. 2016;48:12–21. doi: 10.1038/ng.3458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan A.Y., Coniglio S.J., Chuang Y.Y., Michaelson D., Knaus U.G., Philips M.R., Symons M. Roles of the Rac1 and Rac3 GTPases in human tumor cell invasion. Oncogene. 2005;24:7821–7829. doi: 10.1038/sj.onc.1208909. [DOI] [PubMed] [Google Scholar]
- Choi W., Porten S., Kim S., Willis D., Plimack E.R., Hoffman-Censits J., Roth B., Cheng T., Tran M., Lee I.L., et al. Identification of distinct basal and luminal subtypes of muscle-invasive bladder cancer with different sensitivities to frontline chemotherapy. Cancer Cell. 2014;25:152–165. doi: 10.1016/j.ccr.2014.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung H.A., Medina-Ruiz S., Harland R.M. Sp8 regulates inner ear development. Proc. Natl. Acad. Sci. USA. 2014;111:6329–6334. doi: 10.1073/pnas.1319301111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronauer M.V., Schulz W.A., Seifert H.H., Ackermann R., Burchardt M. Fibroblast growth factors and their receptors in urological cancers: basic research and clinical implications. Eur. Urol. 2003;43:309–319. doi: 10.1016/s0302-2838(03)00005-8. [DOI] [PubMed] [Google Scholar]
- Czerniak B., Dinney C., McConkey D. Origins of bladder cancer. Annu. Rev. Pathol. 2016;11:149–174. doi: 10.1146/annurev-pathol-012513-104703. [DOI] [PubMed] [Google Scholar]
- Dadhania V., Zhang M., Zhang L., Bondaruk J., Majewski T., Siefker-Radtke A., Guo C.C., Dinney C., Cogdell D.E., Zhang S., et al. Meta-analysis of the luminal and basal subtypes of bladder cancer and the identification of signature immunohistochemical markers for clinical use. EBioMedicine. 2016;12:105–117. doi: 10.1016/j.ebiom.2016.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo M.A., Banks E., Poplin R., Garimella K.V., Maguire J.R., Hartl C., Philippakis A.A., del Angel G., Rivas M.A., Hanna M., et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eble JN S.G., Epstein J., Sesterhenn I. International Agency for Research on Cancer; 2004. Pathology and Genetics of Tumors in the Urinary System. [Google Scholar]
- Etherington G.J., Ramirez-Gonzalez R.H., MacLean D. bio-samtools 2: a package for analysis and visualization of sequence and alignment data with SAMtools in Ruby. Bioinformatics. 2015;31:2565–2567. doi: 10.1093/bioinformatics/btv178. [DOI] [PubMed] [Google Scholar]
- Ewing C.M., Ray A.M., Lange E.M., Zuhlke K.A., Robbins C.M., Tembe W.D., Wiley K.E., Isaacs S.D., Johng D., Wang Y., et al. Germline mutations in HOXB13 and prostate-cancer risk. N. Engl. J. Med. 2012;366:141–149. doi: 10.1056/NEJMoa1110000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsythe G.E., Malcolm M.A., Moler C.B. Prentice-Hall; 1977. Computer Methods for Mathematical Computations. [Google Scholar]
- Fortin J.P., Labbe A., Lemire M., Zanke B.W., Hudson T.J., Fertig E.J., Greenwood C.M.T., Hansen K.D. Functional normalization of 450k methylation array data improves replication in large cancer studies. Genome Biol. 2014;15:503. doi: 10.1186/s13059-014-0503-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadea G., Blangy A. Dock-family exchange factors in cell migration and disease. Eur. J. Cell Biol. 2014;93:466–477. doi: 10.1016/j.ejcb.2014.06.003. [DOI] [PubMed] [Google Scholar]
- Gao Y., Kardos J., Yang Y., Tamir T.Y., Mutter-Rottmayer E., Weissman B., Major M.B., Kim W.Y., Vaziri C. The cancer/testes (CT) antigen HORMAD1 promotes homologous recombinational DNA repair and radioresistance in lung adenocarcinoma cells. Sci. Rep. 2018;8:15304. doi: 10.1038/s41598-018-33601-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Perdomo H.A., Usubillaga-Velasquez J.P., Zapata-Copete J.A., Reis L.O. Mutations in CDKN2A and the FGFR3 genes on bladder cancer diagnosis: a systematic review and meta-analysis. World J. Urol. 2019;37:2001–2007. doi: 10.1007/s00345-019-02779-7. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Rincon J., Méndez M., Gómez S., García J.F., Martín P., Bellas C., Pedrosa L., Rodríguez-Pinilla S.M., Camacho F.I., Quero C., et al. Unraveling transformation of follicular lymphoma to diffuse large B-cell lymphoma. PLoS One. 2019;14:e0212813. doi: 10.1371/journal.pone.0212813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo C.C., Bondaruk J., Yao H., Wang Z., Zhang L., Lee S., Lee J.G., Cogdell D., Zhang M., Yang G., et al. Assessment of luminal and basal phenotypes in bladder cancer. Sci. Rep. 2020;10:9743. doi: 10.1038/s41598-020-66747-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo C.C., Majewski T., Zhang L., Yao H., Bondaruk J., Wang Y., Zhang S., Wang Z., Lee J.G., Lee S., et al. Dysregulation of EMT drives the progression to clinically aggressive sarcomatoid bladder cancer. Cell Rep. 2019;27:1781–1793.e4. doi: 10.1016/j.celrep.2019.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J., Tang T., Li J., Yang Y., Quan Y., Zhang L., Huang W., Zhou M. Overexpression of miR-142-5p suppresses the progression of cervical cancer through targeting PIK3AP1 expression. Mol. Cell Biol. 2020;41:e0036320. doi: 10.1128/MCB.00363-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrow J., Frankish A., Gonzalez J.M., Tapanari E., Diekhans M., Kokocinski F., Aken B.L., Barrell D., Zadissa A., Searle S., et al. GENCODE: the reference human genome annotation for the ENCODE Project. Genome Res. 2012;22:1760–1774. doi: 10.1101/gr.135350.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiménez-Marin A., Collado-Romero M., Ramirez-Boo M., Arce C., Garrido J.J. Biological pathway analysis by ArrayUnlock and ingenuity pathway analysis. BMC Proc. 2009;3:S6. doi: 10.1186/1753-6561-3-S4-S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato M., Shimizu A., Yokoyama Y., Kaira K., Shimomura Y., Ishida-Yamamoto A., Kamei K., Tokunaga F., Ishikawa O. An autosomal recessive mutation of DSG4 causes monilethrix through the ER stress response. J. Invest. Dermatol. 2015;135:1253–1260. doi: 10.1038/jid.2015.12. [DOI] [PubMed] [Google Scholar]
- Kimmel M. 2nd Edition. Springer; 2015. Branching Processes in Biology. [Google Scholar]
- Krämer A., Green J., Pollard J., Tugendreich S. Causal analysis approaches in ingenuity pathway analysis. Bioinformatics. 2014;30:523–530. doi: 10.1093/bioinformatics/btt703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagarias J.C., Reeds J.A., Wright M.H., Wright P.E. Convergence properties of the Nelder-Mead simplex method in low dimensions. SIAM J. Optim. 1998;9:112–147. doi: 10.1137/S1052623496303470. [DOI] [Google Scholar]
- Lambert S.A., Jolma A., Campitelli L.F., Das P.K., Yin Y., Albu M., Chen X., Taipale J., Hughes T.R., Weirauch M.T. The human transcription factors. Cell. 2018;172:650–665. doi: 10.1016/j.cell.2018.01.029. [DOI] [PubMed] [Google Scholar]
- Lange K. 2nd Edition. Springer; 2010. Branching Processes. [Google Scholar]
- Last G., Penrose M. Cambridge University Press; 2017. Lectures on the Poisson Process. [Google Scholar]
- Lawson A.R.J., Abascal F., Coorens T.H.H., Hooks Y., O'Neill L., Latimer C., Raine K., Sanders M.A., Warren A.Y., Mahbubani K.T.A., et al. Extensive heterogeneity in somatic mutation and selection in the human bladder. Science. 2020;370:75–82. doi: 10.1126/science.aba8347. [DOI] [PubMed] [Google Scholar]
- Lee S., Jeong J., Majewski T., Scherer S.E., Kim M.S., Tuziak T., Tang K.S., Baggerly K., Grossman H.B., Zhou J.H., et al. Forerunner genes contiguous to RB1 contribute to the development of in situ neoplasia. Proc. Natl. Acad. Sci. USA. 2007;104:13732–13737. doi: 10.1073/pnas.0701771104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R., Du Y., Chen Z., Xu D., Lin T., Jin S., Wang G., Liu Z., Lu M., Chen X., et al. Macroscopic somatic clonal expansion in morphologically normal human urothelium. Science. 2020;370:82–89. doi: 10.1126/science.aba7300. [DOI] [PubMed] [Google Scholar]
- Li Z., Liu H., Tu W. Model selection in multivariate semiparametric regression. Stat. Methods Med. Res. 2018;27:3026–3038. doi: 10.1177/0962280217690769. [DOI] [PubMed] [Google Scholar]
- Liao W., Sharma S. Modulation of B-cell receptor and microenvironment signaling by a guanine exchange factor in B-cell malignancies. Cancer Biol. Med. 2016;13:277–285. doi: 10.20892/j.issn.2095-3941.2016.0026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y., Smyth G.K., Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- Liu B., Zhang M., Chu H., Zhang H., Wu H., Song G., Wang P., Zhao K., Hou J., Wang X., et al. The ubiquitin E3 ligase TRIM31 promotes aggregation and activation of the signaling adaptor MAVS through Lys63-linked polyubiquitination. Nat. Immunol. 2017;18:214–224. doi: 10.1038/ni.3641. [DOI] [PubMed] [Google Scholar]
- Liu R., Gao J., Yang Y., Qiu R., Zheng Y., Huang W., Zeng Y., Hou Y., Wang S., Leng S., et al. PHD finger protein 1 (PHF1) is a novel reader for histone H4R3 symmetric dimethylation and coordinates with PRMT5-WDR77/CRL4B complex to promote tumorigenesis. Nucleic Acids Res. 2018;46:6608–6626. doi: 10.1093/nar/gky461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Kwiatkowski D.J. Combined CDKN1A/TP53 mutation in bladder cancer is a therapeutic target. Mol. Cancer Ther. 2015;14:174–182. doi: 10.1158/1535-7163.MCT-14-0622-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louie B.H., Kurzrock R. BAP1: not just a BRCA1-associated protein. Cancer Treat Rev. 2020;90:102091. doi: 10.1016/j.ctrv.2020.102091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X.L., Zhan R., Zhao G.M., Qian Z.H., Gong C.C., Li Y.Q. Expression of CDK13 was associated with prognosis and expression of HIF-1α and beclin1 in breast cancer patients. J. Invest. Surg. 2020;35:442–447. doi: 10.1080/08941939.2020.1852344. [DOI] [PubMed] [Google Scholar]
- Majewski T., Lee S., Jeong J., Yoon D.S., Kram A., Kim M.S., Tuziak T., Bondaruk J., Lee S., Park W.S., et al. Understanding the development of human bladder cancer by using a whole-organ genomic mapping strategy. Lab. Invest. 2008;88:694–721. doi: 10.1038/labinvest.2008.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majewski T., Yao H., Bondaruk J., Chung W., Lee S., Lee J.G., Zhang S., Cogdell D., Yang G., Choi W., et al. Whole-organ genomic characterization of mucosal field effects initiating bladder carcinogenesis. Cell Rep. 2019;26:2241–2256.e4. doi: 10.1016/j.celrep.2019.01.095. [DOI] [PubMed] [Google Scholar]
- Matic M., Pekmezovic T., Djukic T., Mimic-Oka J., Dragicevic D., Krivic B., Suvakov S., Savic-Radojevic A., Pljesa-Ercegovac M., Tulic C., et al. GSTA1, GSTM1, GSTP1, and GSTT1 polymorphisms and susceptibility to smoking-related bladder cancer: a case-control study. Urol. Oncol. 2013;31:1184–1192. doi: 10.1016/j.urolonc.2011.08.005. [DOI] [PubMed] [Google Scholar]
- McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minella A.C., Welcker M., Clurman B.E. Ras activity regulates cyclin E degradation by the Fbw7 pathway. Proc. Natl. Acad. Sci. USA. 2005;102:9649–9654. doi: 10.1073/pnas.0503677102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyake M., Lawton A., Goodison S., Urquidi V., Gomes-Giacoia E., Zhang G., Ross S., Kim J., Rosser C.J. Chemokine (C-X-C) ligand 1 (CXCL1) protein expression is increased in aggressive bladder cancers. BMC Cancer. 2013;13:322. doi: 10.1186/1471-2407-13-322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naher L., Kiyoshima T., Kobayashi I., Wada H., Nagata K., Fujiwara H., Ookuma Y.F., Ozeki S., Nakamura S., Sakai H. STAT3 signal transduction through interleukin-22 in oral squamous cell carcinoma. Int. J. Oncol. 2012;41:1577–1586. doi: 10.3892/ijo.2012.1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon K.C. The Parsimony Ratchet, a new method for rapid parsimony analysis. Cladistics. 1999;15:407–414. doi: 10.1111/j.1096-0031.1999.tb00277.x. [DOI] [PubMed] [Google Scholar]
- Paradis E. Time-dependent speciation and extinction from phylogenies: a least squares approach. Evolution. 2011;65:661–672. doi: 10.1111/j.1558-5646.2010.01179.x. [DOI] [PubMed] [Google Scholar]
- Puttipanyalears C., Arayataweegool A., Chalertpet K., Rattanachayoto P., Mahattanasakul P., Tangjaturonsasme N., Kerekhanjanarong V., Mutirangura A., Kitkumthorn N. TRH site-specific methylation in oral and oropharyngeal squamous cell carcinoma. BMC Cancer. 2018;18:786. doi: 10.1186/s12885-018-4706-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj D., Nikolaidi M., Garces I., Lorizio D., Castro N.M., Caiafa S.G., Moore K., Brown N.F., Kocher H.M., Duan X., et al. CEACAM7 is an effective target for CAR T-cell therapy of pancreatic ductal adenocarcinoma. Clin. Cancer Res. 2021;27:1538–1552. doi: 10.1158/1078-0432.CCR-19-2163. [DOI] [PubMed] [Google Scholar]
- Ramos A.H., Lichtenstein L., Gupta M., Lawrence M.S., Pugh T.J., Saksena G., Meyerson M., Getz G. Oncotator: cancer variant annotation tool. Hum. Mutat. 2015;36:E2423–E2429. doi: 10.1002/humu.22771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedel I., Liang F.X., Deng F.M., Tu L., Kreibich G., Wu X.R., Sun T.T., Hergt M., Moll R. Urothelial umbrella cells of human ureter are heterogeneous with respect to their uroplakin composition: different degrees of urothelial maturity in ureter and bladder? Eur. J. Cell Biol. 2005;84:393–405. doi: 10.1016/j.ejcb.2004.12.011. [DOI] [PubMed] [Google Scholar]
- Robertson A.G., Kim J., Al-Ahmadie H., Bellmunt J., Guo G., Cherniack A.D., Hinoue T., Laird P.W., Hoadley K.A., Akbani R., et al. Comprehensive molecular characterization of muscle-invasive bladder cancer. Cell. 2017;171:540–556.e25. doi: 10.1016/j.cell.2017.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saavedra J.A. Oxfor University Press; 2001. Pathology of the Incipient Neoplasia. [Google Scholar]
- Shaffer A.L., Emre N.T., Romesser P.B., Staudt L.M. IRF4: immunity. Malignancy! Therapy? Clin. Cancer Res. 2009;15:2954–2961. doi: 10.1158/1078-0432.CCR-08-1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah N., Sukumar S. The Hox genes and their roles in oncogenesis. Nat. Rev. Cancer. 2010;10:361–371. doi: 10.1038/nrc2826. [DOI] [PubMed] [Google Scholar]
- Shin K., Lim A., Odegaard J.I., Honeycutt J.D., Kawano S., Hsieh M.H., Beachy P.A. Cellular origin of bladder neoplasia and tissue dynamics of its progression to invasive carcinoma. Nat. Cell Biol. 2014;16:469–478. doi: 10.1038/ncb2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiratsuchi T., Oda K., Nishimori H., Suzuki M., Takahashi E., Tokino T., Nakamura Y. Cloning and characterization of BAP3 (BAI-associated protein 3), a C2 domain-containing protein that interacts with BAI1. Biochem. Biophys. Res. Commun. 1998;251:158–165. doi: 10.1006/bbrc.1998.9408. [DOI] [PubMed] [Google Scholar]
- Sobin L.H., Gospodarowicz M.D., Wittekind C. 9 Edition. Blackwell; 2009. TNM Classification of Malignant Tumors. [Google Scholar]
- Solberg H.E. Discriminant analysis. CRC Crit. Rev. Clin. Lab. Sci. 1978;9:209–242. doi: 10.3109/10408367809150920. [DOI] [PubMed] [Google Scholar]
- Soupene E., Dinh N.P., Siliakus M., Kuypers F.A. Activity of the acyl-CoA synthetase ACSL6 isoforms: role of the fatty acid Gate-domains. BMC Biochem. 2010;11:18. doi: 10.1186/1471-2091-11-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sreevalsan S., Döring M., Paszkowski-Rogacz M., Brux M., Blanck C., Meyer M., Momburg F., Buchholz F., Theis M. MLLT6 maintains PD-L1 expression and mediates tumor immune resistance. EMBO Rep. 2020;21:e50155. doi: 10.15252/embr.202050155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stacy A.J., Craig M.P., Sakaram S., Kadakia M. ΔNp63α and microRNAs: leveraging the epithelial-mesenchymal transition. Oncotarget. 2017;8:2114–2129. doi: 10.18632/oncotarget.13797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steck E., Bräun J., Pelttari K., Kadel S., Kalbacher H., Richter W. Chondrocyte secreted CRTAC1: a glycosylated extracellular matrix molecule of human articular cartilage. Matrix Biol. 2007;26:30–41. doi: 10.1016/j.matbio.2006.09.006. [DOI] [PubMed] [Google Scholar]
- Storey J.D., Tibshirani R. Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. USA. 2003;100:9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strandgaard T., Nordentoft I., Lamy P., Christensen E., Borg H., Thomsen M.B.H., Thomsen M., Jensen J.B., Bjerggaard Jensen J., Dyrskjøt L., Dyrskjot L. Mutational analysis of field cancerization in bladder cancer. Bladder Cancer. 2020;6:253–264. doi: 10.3233/blc-200282. [DOI] [Google Scholar]
- Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai D., Wells K., Arcaroli J., Vanderbilt C., Aisner D.L., Messersmith W.A., Lieu C.H. Targeting the WNT signaling pathway in cancer therapeutics. Oncologist. 2015;20:1189–1198. doi: 10.1634/theoncologist.2015-0057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen M.B.H., Nordentoft I., Lamy P., Vang S., Reinert L., Mapendano C.K., Høyer S., Orntoft T.F., Jensen J.B., Dyrskjøt L., Dyrskjot L. Comprehensive multiregional analysis of molecular heterogeneity in bladder cancer. Sci. Rep. 2017;7:11702. doi: 10.1038/s41598-017-11291-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran M.N., Choi W., Wszolek M.F., Navai N., Lee I.L.C., Nitti G., Wen S., Flores E.R., Siefker-Radtke A., Czerniak B., et al. The p63 protein isoform DeltaNp63alpha inhibits epithelial-mesenchymal transition in human bladder cancer cells: role of MIR-205. J. Biol. Chem. 2013;288:3275–3288. doi: 10.1074/jbc.M112.408104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsui K.H., Chiang K.C., Lin Y.H., Chang K.S., Feng T.H., Juang H.H. BTG2 is a tumor suppressor gene upregulated by p53 and PTEN in human bladder carcinoma cells. Cancer Med. 2018;7:184–195. doi: 10.1002/cam4.1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tusher V.G., Tibshirani R., Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. USA. 2001;98:5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Batavia J., Yamany T., Molotkov A., Dan H., Mansukhani M., Batourina E., Schneider K., Oyon D., Dunlop M., Wu X.R., et al. Bladder cancers arise from distinct urothelial sub-populations. Nat. Cell Biol. 2014;16:982–991. doi: 10.1038/ncb3038. [DOI] [PubMed] [Google Scholar]
- Vedeld H.M., Andresen K., Andrassy Eilertsen I., Nesbakken A., Seruca R., Gladhaug I.P., Thiis-Evensen E., Rognum T.O., Muri Boberg K., Lind G.E. The novel colorectal cancer biomarkers CDO1, ZSCAN18 and ZNF331 are frequently methylated across gastrointestinal cancers. Int. J. Cancer. 2015;136:844–853. doi: 10.1002/ijc.29039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermeulen L., De Sousa E Melo F., van der Heijden M., Cameron K., de Jong J.H., Borovski T., Tuynman J.B., Todaro M., Merz C., Rodermond H., et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat. Cell Biol. 2010;12:468–476. doi: 10.1038/ncb2048. [DOI] [PubMed] [Google Scholar]
- Wang K., Li M.Y., Hadley D., Liu R., Glessner J., Grant S.F.A., Hakonarson H., Bucan M. PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 2007;17:1665–1674. doi: 10.1101/gr.6861907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.G., Wang S.Q., Li W. RSeQC: quality control of RNA-seq experiments. Bioinformatics. 2012;28:2184–2185. doi: 10.1093/bioinformatics/bts356. [DOI] [PubMed] [Google Scholar]
- Wang X., He Z., Xia T., Li X., Liang D., Lin X., Wen H., Lan K. Latency-associated nuclear antigen of Kaposi sarcoma-associated herpesvirus promotes angiogenesis through targeting notch signaling effector Hey1. Cancer Res. 2014;74:2026–2037. doi: 10.1158/0008-5472.CAN-13-1467. [DOI] [PubMed] [Google Scholar]
- Wei J., Long L., Zheng W., Dhungana Y., Lim S.A., Guy C., Wang Y., Wang Y.D., Qian C., Xu B., et al. Targeting REGNASE-1 programs long-lived effector T cells for cancer therapy. Nature. 2019;576:471–476. doi: 10.1038/s41586-019-1821-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein J.N., Akbani R., Broom B.M., Wang W.Y., Verhaak R.G.W., McConkey D., Lerner S., Morgan M., Creighton C.J., Smith C., et al. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature. 2014;507:315–322. doi: 10.1038/nature12965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams L.A., Butler E.N., Sun X., Allott E.H., Cohen S.M., Fuller A.M., Hoadley K.A., Perou C.M., Geradts J., Olshan A.F., Troester M.A. TP53 protein levels, RNA-based pathway assessment, and race among invasive breast cancer cases. NPJ Breast Cancer. 2018;4:13. doi: 10.1038/s41523-018-0067-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L.R., Chen S.X., Wu Y., Patel A.A., Zhang D.Y. Multiplexed enrichment of rare DNA variants via sequence-selective and temperature-robust amplification. Nat. Biomed. Eng. 2017;1:714–723. doi: 10.1038/s41551-017-0126-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X., Gu Z., Chen Y., Chen B., Chen W., Weng L., Liu X. Application of PD-1 blockade in cancer immunotherapy. Comput. Struct. Biotechnol. J. 2019;17:661–674. doi: 10.1016/j.csbj.2019.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wullweber A., Strick R., Lange F., Sikic D., Taubert H., Wach S., Wullich B., Bertz S., Weyerer V., Stoehr R., et al. Bladder tumor subtype commitment occurs in carcinoma in situ driven by key signaling pathways including ECM remodeling. Cancer Res. 2021;81:1552–1566. doi: 10.1158/0008-5472.CAN-20-2336. [DOI] [PubMed] [Google Scholar]
- Yang G., Bondaruk J., Cogdell D., Wang Z., Lee S., Lee J.G., Zhang S., Choi W., Wang Y., Liang Y., et al. Urothelial-to-Neural plasticity drives progression to small cell bladder cancer. iScience. 2020;23:101201. doi: 10.1016/j.isci.2020.101201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J., Fan L., Liao X., Cui G., Hu H. CRTAC1 (Cartilage acidic protein 1) inhibits cell proliferation, migration, invasion and epithelial-mesenchymal transition (EMT) process in bladder cancer by downregulating Yin Yang 1 (YY1) to inactivate the TGF-beta pathway. Bioengineered. 2021;12:9377–9389. doi: 10.1080/21655979.2021.1974645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z.S., Qing H., Gui H., Luo J., Dai L.J., Wang B. Role of caprin-1 in carcinogenesis. Oncol. Lett. 2019;18:15–21. doi: 10.3892/ol.2019.10295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yau C., Mouradov D., Jorissen R.N., Colella S., Mirza G., Steers G., Harris A., Ragoussis J., Sieber O., Holmes C.C. A statistical approach for detecting genomic aberrations in heterogeneous tumor samples from single nucleotide polymorphism genotyping data. Genome Biol. 2010;11:R92. doi: 10.1186/gb-2010-11-9-r92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yegin Z., Gunes S., Buyukalpelli R. Hypermethylation of TWIST1 and NID2 in tumor tissues and voided urine in urinary bladder cancer patients. DNA Cell Biol. 2013;32:386–392. doi: 10.1089/dna.2013.2030. [DOI] [PubMed] [Google Scholar]
- Yeh C.H., Bellon M., Nicot C. FBXW7: a critical tumor suppressor of human cancers. Mol. Cancer. 2018;17:115. doi: 10.1186/s12943-018-0857-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue Y., Hui K., Wu S., Zhang M., Que T., Gu Y., Wang X., Wu K., Fan J. MUC15 inhibits cancer metastasis via PI3K/AKT signaling in renal cell carcinoma. Cell Death Dis. 2020;11:336. doi: 10.1038/s41419-020-2518-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Huang J.Z., Zhang Y.Q., Zhang X., Zhao L.Y., Li C.G., Zhou Y.F., Wei H., Yu J. Microtubule associated protein 9 inhibits liver tumorigenesis by suppressing ERCC3. EBioMedicine. 2020;53:102701. doi: 10.1016/j.ebiom.2020.102701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L., Shay J.W. Multiple roles of APC and its therapeutic implications in colorectal cancer. J. Natl. Cancer Inst. 2017;109:djw332. doi: 10.1093/jnci/djw332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S., Zhang W., Xiao Y., Qin T., Yue Y., Qian W., Shen X., Ma Q., Wang Z. Targeting MUC15 protein in cancer: molecular mechanisms and therapeutic perspectives. Curr. Cancer Drug Targets. 2020;20:647–653. doi: 10.2174/1568009620666200601140639. [DOI] [PubMed] [Google Scholar]
- Zhou R., Zhang R., Sun Y., Platt S., Szczotka-Flynn L., Pearlman E. Innate immune regulation of Serratia marcescens-induced corneal inflammation and infection. Invest. Ophthalmol. Vis. Sci. 2012;53:7382. doi: 10.1167/iovs.12-10238. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
The article contains all data sets generated or analyzed during this study. The WES and bulk RNA sequencing data generated or analyzed in this study have been deposited in NCBI Sequence Read Archive (SRA) with accession number PRJNA736323. The methylation and CNV data have been deposited in NCBI Gene Expression Omnibus (GEO) with accession number GSE171363.
-
•
The study did not generate original code.
-
•
Any additional information required to reanalyze the data included in this study will be provided by lead contact upon request.