

Summary
RNA quality control relies on co-factors and adaptors to identify and prepare substrates for degradation by ribonucleases such as the 3′ to 5′ ribonucleolytic RNA exosome. Here, we determined cryogenic electron microscopy structures of human Nuclear Exosome Targeting (NEXT) complexes bound to RNA that reveal mechanistic insights to substrate recognition and early steps that precede RNA handover to the exosome. The structures illuminate ZCCHC8 as a scaffold, mediating homodimerization while embracing the MTR4 helicase and flexibly anchoring RBM7 to the helicase core. All three subunits collaborate to bind the RNA, with RBM7 and ZCCHC8 surveying sequences upstream of the 3′ end to facilitate RNA capture by MTR4. ZCCHC8 obscures MTR4 surfaces important for RNA binding and extrusion as well as MPP6-dependent recruitment and docking onto the RNA exosome core, interactions that contribute to RNA surveillance by coordinating RNA capture, translocation and extrusion from the helicase to the exosome for decay.
Keywords: RNA surveillance, helicase, SF2, DExH-box, RNA-protein complex, translocase, exosome
In Brief
Cryo-EM analysis of human Nuclear Exosome Targeting (NEXT) complex bound to RNA substrate unveils mechanistic insights into RNA quality control
Graphical Abstract
Introduction
RNA quality control pathways ensure the integrity of the transcriptome (Doma and Parker, 2007; Wolin and Maquat, 2019). Defective, unprocessed or spurious coding and non-coding transcripts are destroyed to prevent production of unwanted proteins, their aberrant accumulation or their incorporation into R-loops or essential ribonucleoprotein complexes e.g. ribosome, spliceosome and telomerase. In eukaryotes, the transcriptome is subject to surveillance by the 3′ to 5′ ribonucleolytic RNA exosome to maintain a functional pool of RNA (Houseley et al., 2006; Puno et al., 2019). Besides quality control, the exosome is responsible for general RNA turnover and maturation of precursor RNAs. The nuclear exosome consists of a nine-subunit core (EXOSC1-9) bound to a distributive ribonuclease EXOSC10/RRP6 and a processive ribonuclease DIS3 (Gerlach et al., 2018; Liu et al., 2006; Makino et al., 2015; Wasmuth et al., 2014; Weick and Lima, 2021; Weick et al., 2018; Zinder and Lima, 2017).
To correctly target diverse substrates, the RNA exosome requires guidance and associates with RNA adaptor complexes, typically composed of a helicase bound to auxiliary RNA binding proteins, and co-factors such as MPP6 and/or C1D that recruit helicases to the exosome (Schuch et al., 2014; Wasmuth et al., 2017). In human nuclei, known adaptor complexes include the Nuclear Exosome Targeting (NEXT) complex, the Poly(A) Exosome Targeting (PAXT) connection or Polysome Protector complex (PPC) and the TRF4-2-ZCCHC7-MTR4 polyadenylation (TRAMP) complex (Lubas et al., 2011; Meola et al., 2016; Ogami et al., 2017; Das et al., 2021; LaCava et al., 2005). While NEXT and PAXT target similar substrates, PAXT prefers polyadenylated RNA whereas NEXT generally targets non-polyadenylated transcripts to the exosome for decay (Lubas et al., 2011; Meola et al., 2016; Ogami et al., 2017; Wu et al., 2020). NEXT substrates include RNA produced by pervasive transcription at intergenic loci and regulatory elements e.g. enhancers to transcripts generated by abortive and faulty transcription (Collins et al., 2021; Liu et al., 2020; Lubas et al. 2015). NEXT-mediated turnover of promoter upstream transcripts (PROMPTS) ensures unidirectional mRNA output from bidirectional promoters (Ntini et al., 2013; Preker et al., 2008). In addition, NEXT is involved in processing and quality control of precursor telomerase RNA (Gable et al., 2019) and microRNAs (Bajczyk et al., 2020) and participates in the resolution of RNA/DNA hybrid structures generated during asymmetric DNA strand mutagenesis in immunoglobulin diversification (Lim et al., 2017; Nair et al., 2020). Functional inadequacy in NEXT poses detrimental effects to health. In mice, NEXT deficiency leads to reduced fertility and shortened lifespan due to a progressive and fatal neurodevelopmental pathology (Gable et al., 2019; Wu et al., 2019) whereas in zebrafish, defects in motor neurons and cerebellar structures are reported (Giunta et al., 2016). Mutations in components of NEXT are also linked to human disorders that range from neurological diseases e.g. spinal motor neuropathy (Giunta et al., 2016), intellectual disability (Najmabadi et al., 2011), to cancer (Coccé et al., 2016; Creelan, 2018) and short telomere disease (Gable et al., 2019).
NEXT is composed of three core subunits, namely MTR4, RBM7, and ZCCHC8 (Figure 1A) (Lubas et al., 2011). MTR4 is a superfamily 2 DExH-box 3′ to 5′ RNA helicase intimately tied to functions of the nuclear exosome (Fairman-Williams et al., 2010; Gao and Yang, 2020; Weick and Lima, 2021) and is a central component of several RNA adaptor complexes including TRAMP (LaCava et al., 2005) and PAXT or PPC (Meola et al., 2016; Ogami et al., 2017). MTR4 has a helicase core characterized by a ring-like arrangement of two RecA-like ATPase modules (RecA1 and RecA2) with winged helix (WH) and helical bundle (HB) domains (Jackson et al., 2010; Weir et al., 2010). Uniquely, MTR4 contains an arch-like protrusion with a KOW domain connected to the helicase core by an elongated anti-parallel coiled-coil motif (Stalk). The helicase activity of human MTR4 is comparatively weak but is enhanced when bound to nuclear exosomes or in complexes with RBM7 and ZCCHC8 (Puno and Lima, 2018; Weick et al., 2018). RBM7 is an RNA binding protein initially described to associate with splicing factors, presumably to target intronic RNA for decay (Guo et al., 2003). It has a single RNA Recognition Motif (RRM) that binds single-stranded RNA. While RBM7 can interact with other RNA sequences, it prefers uridine-rich motifs about 20 nucleotides upstream of the non-polyadenylated or short polyadenylated 3′ ends of NEXT substrates (Hrossova et al., 2015; Lubas et al., 2015; Puno and Lima, 2018). As U-rich motifs are not strictly required, the observed specificity may reflect a conformational preference for flexible single-stranded RNA. RBM7 is recruited to MTR4 by ZCCHC8, a protein with a single zinc knuckle (ZK) motif, a proline-rich domain in spliceosome-associated protein (PSP) module that binds RBM7 (Falk et al., 2016), and a C-terminal domain that interacts with MTR4 and stimulates its helicase and ATPase activities (Puno and Lima, 2018). To date, structures are limited to individual subunits or binary complexes of peptides or domains and there are no available structures for intact nuclear exosome adaptor complexes bound to RNA thus making it difficult to understand how these complexes work to identify and capture RNA for delivery to the exosome.
Figure 1. Cryo-EM structure of human NEXT complex bound to RNA substrate 1.

(A) NEXT subunits and their domain organization.
(B) Composite map of human NEXT homodimer generated from local 3D refinement.
(C) Model of human NEXT homodimer bound to RNA substrate 1 in two orientations. Each subunit is labeled and colored uniquely.
(D) Solvent-excluded surface representation of NEXT protomer A in two orientations with each domain labeled and colored as in (A). See also Figure S1-3 and Table S1.
Here, we illuminate roles for human NEXT in RNA surveillance by presenting biochemical and structural evidence that RBM7 and ZCCHC8 surveil upstream RNA motifs to provide specificity and to facilitate capture of free 3′ ends by MTR4. Cryogenic electron microscopy (cryo-EM) structures of NEXT reveal a dimeric architecture that contributes to RNA translocation in vitro and to RNA decay in vivo. Consistent with ZCCHC8-mediated NEXT dimerization, we show that cancer-associated ZCCHC8-ROS1 fusion dimerizes and activates the kinase. Finally, our structures reveal that the ZCCHC8 scaffold covers surfaces important for RNA translocation and MTR4 interaction with the nuclear exosome, suggesting hierarchical interactions that need to be remodeled before NEXT could release RNA for decay.
Results
Molecular architecture of NEXT
To determine the architecture and basis for RNA engagement by NEXT, the human NEXT core complex (referred to as NEXT for simplicity) was reconstituted using full-length MTR4, RBM7 RRM residues 6–86, and the ZCCHC8 core with a low-complexity region (residues 416–507) truncated. The NEXT core complex exhibits similar helicase activity as one containing full-length proteins (Puno and Lima, 2018). A complex between the NEXT core and a 3′ tailed RNA stemloop (substrate 1) was prepared in the presence of ATP (Figures S1A and S1B). A 2′-amino-butyryl-pyrene-conjugated uridine (Upy) was included upstream of the 3′ end to present a bulky chemical group to stall NEXT and the helicase as it tracks along RNA. Insertion of Upy in the 3′ tail of a duplex RNA suppressed helicase activity of NEXT (Figure S1C). Cryo-EM data were collected, and data processing yielded a consensus three-dimensional (3D) map with a nominal resolution of 4.06 Å (Figures S1D-F, S2A-D).
The overall reconstruction revealed a homodimer with a flexible asymmetric bilobed structure (Figure S2A). Each lobe includes a complex of ZCCHC8-RBM7-MTR4 bound to RNA. The reconstruction revealed two regions of asymmetry, one at the dimer interface and another proximal to the helicase core that includes RBM7 and elements of ZCCHC8. To resolve structural heterogeneity and enhance local features, rounds of focused 3D refinement and classification were performed across regions of the complex that culminated in five reconstructions with resolutions ranging from 3.26 Å to 4.40 Å (Figures S2D-F; Table S1). Maps were combined to generate a composite map (Figures 1B and S2A) that was used to build and refine a model for NEXT (Figures 1C, 1D, S2A-G and S3A; Table S1). To determine if RNA was responsible for bringing two ZCCHC8-RBM7-MTR4 complexes together, data were collected on reconstituted apo NEXT that produced a reconstruction with a similar asymmetric homodimeric architecture to the RNA bound complex, albeit at lower resolution (8.36 Å), showing that NEXT dimerization is not dependent on RNA (Figures S3B-D, Table S1).
Structures of MTR4 are similar between protomers with respect to helicase core domains (RecA1, RecA2, WH, and HB) with each forming a compact fold and RNA-binding channel (Figures 1C, 1D, and S4A). The arch module protrudes from the helicase core and adopts a closed conformation where the KOW domain rests adjacent to RecA2 in proximity to the entry site of the RNA channel (Weick et al., 2018). Nucleotide was not observed in the MTR4 ATP-binding cleft and presumably dissociated during purification. The configuration of MTR4 helicase core domains in NEXT resembles those observed for MTR4 bound to ATP (Wang et al., 2019), ADP (Puno and Lima, 2018), or the ATP analog AMPPNP with a translocating RNA (Weick et al., 2018) (Figures S4A-D), perhaps consistent with the notion that, unlike DEAD-box helicases, the ATPase modules of DExH-box helicases generally do not undergo large domain movements in the course of ATPase cycle (Ozgur et al., 2015).
ZCCHC8 scaffolds the architecture of NEXT. The N-terminal region of ZCCHC8 contains a homodimerization domain (HD, residues 1-175) that features a parallel coiled-coil domain and α helices flanking a strand-exchanged β sheet core (Figures 2A and 2B). The strand-exchanged β sheet core appears symmetric, but the parallel coiled-coil domain does not adhere to the 2-fold axis as illustrated by our ability to separate two distinct classes during structure determination (Figure S1F). A Dali search of ZCCHC8 HD showed no significant similarity to known protein folds (Holm, 2020). Homotypic interactions between ZCCHC8 HD domains include hydrogen-bonding between exchanged β strands and an interface that is largely hydrophobic (Figures 2B and 2C).
Figure 2. Structure of ZCCHC8 HD and KID and their contacts to MTR4 KOW.

(A) Overview of ZCCHC8 HD and KID and their interaction with MTR4 KOW. Boxed areas indicate positions of magnified views in (B) and (D-F).
(B) Close-up of the strand-exchanged β-sheet core of ZCCHC8 HD.
(C) Homotypic interactions between ZCCHC8 HD. Protomer A ZCCHC8 HD in cartoon with side chains of hydrophobic residues shown. Protomer B ZCCHC8 HD in surface representation colored based on molecular lipophilicity potential (MLP) calculated in ChimeraX (Ghose et al., 1998; Laguerre et al., 1997; Pettersen et al., 2021).
(D-E) Magnified views (upper panel) and EM maps with model overlaid (lower panel) of protomer A MTR4 contacts to protomer A ZCCHC8 AIM (D) and protomer B ZCCHC8 HD (E).
(F) Magnified views (upper panel) and EM maps with model overlaid (lower panel) of protomer A MTR4 contacts to protomer A ZCCHC8 KID.
The ZCCHC8 homodimer orients two MTR4 helicases head-to-head by interacting with the MTR4 KOW domain via ZCCHC8 residues 176-216 (KOW interacting domain, KID) and through contacts with ZCCHC8 HD of the other protomer (Figures 2A, 2B and 2D-F). The structure shows that the ZCCHC8 KID includes a single eight amino acid arch-interaction motif (AIM) spanning residues 177-184 that is defined by a consensus sequence xωxxD(x)1/2G/P (Thoms et al., 2015; Lingaraju et al., 2019) where ω is an aromatic residue (Phe178 in ZCCHC8) (Figures 2D and S4E). ZCCHC8 Phe178 makes hydrophobic contacts to MTR4 Val766 while ZCCHC8 Asp181 interacts with MTR4 Arg743. Consistent with contacts observed in NEXT, mutations in ZCCHC8 residues Phe178 and Asp181 as well as MTR4 Arg743 impair ZCCHC8 AIM peptide interactions with MTR4 KOW (Lingaraju et al., 2019). Unlike extended unstructured AIMs observed for NVL (Lingaraju et al., 2019) and NRDE2 (Wang et al., 2019), the ZCCHC8 AIM is embedded in the β sheet of the ZCCHC8 dimer preconfigured to bind the MTR4 KOW (Figures 2B and S4E). The ZCCHC8 KID extends well beyond the AIM and includes residues 185-216 that interact within a mostly aliphatic interface of the MTR4 KOW before extending to a side of the KOW domain that faces the RNA entry site of the helicase (Figures 2A and 2F).
The middle region of ZCCHC8 (amino acids 269-403) interacts with RBM7 to tether it to the helicase core of MTR4 (Figure 3A). Contacts between ZCCHC8 and the RBM7 RRM are established by the ZCCHC8 PSP domain between residues 285 and 324 (Falk et al., 2016). Extending from the PSP in both directions are elements that reach two distinct surfaces of the MTR4 RecA2 domain through ZCCHC8 residues 270-275 (MTR4 anchor 1, MA1) and residues 328-337 (MTR4 anchor 2, MA2) (Figure 3A). ZCCHC8 MA1 binds MTR4 via ZCCHC8 Arg273 interaction with an acidic patch formed by MTR4 Glu400 and Glu990 and via ZCCHC8 Tyr274 that packs against a hydrophobic crevice in MTR4 (Figure 3B). ZCCHC8 MA2 forms a strand that extends the β sheet of MTR4 RecA2 and contains aliphatic residues Leu335 and Tyr336 that contact MTR4 L328 and H329, respectively (Figure 3C). ZCCHC8 residues 355-403 return from MTR4 to interact with the backside of the PSP domain (Figure 3A). The last portion of ZCCHC8 observed is the C-terminal domain (CTD) that includes residues 659-701. Consistent with our prior structure (Puno and Lima, 2018), the ZCCHC8 CTD extends across the base of the helicase core and RNA exit channel (Figure 1C).
Figure 3. ZCCHC8 anchors RBM7 to MTR4 helicase core.

(A) Overview of ZCCHC8 interactions with RBM7 and MTR4 in in two orientations. Locations of MA1 and MA2 regions in boxes.
(B-C) Magnified views of ZCCHC8 MA1 (B) and MA2 (C) contacts to MTR4. The corresponding EM map of each view is shown in the lower panel.
Based on these structures, it is clear that several interactions between ZCCHC8 and MTR4 are mutually exclusive with MTR4 interactions observed in complexes with the nuclear RNA exosome (Weick et al., 2018) or other exosome co-factors in complexes with TRAMP (Falk et al., 2014) and NRDE2 (Wang et al., 2019) (Figures S4F-J). NRDE2, a negative regulator of the exosome, is reported to inhibit the interaction of MTR4 with ZCCHC8 (Wang et al., 2019). Consistent with this model, NRDE2 and ZCCHC8 interact with surfaces that overlap on the MTR4 KOW and RecA2 domains (Figures S4F-J), but unlike ZCCHC8, NRDE2 binding was proposed to be incompatible with binding to RNA (Wang et al., 2019). Superposing NRDE2 and NEXT structures suggest that RNA could be accommodated between the RecA domains in the NRDE2 complex, however NRDE2 coordinates the arch and KOW in an even more closed configuration relative to NEXT, potentially blocking RNA ingress in the NRDE2 complex (Figures S4G and S4H). The conserved MTR4 RecA2 binding motifs of S. cerevisiae TRAMP subunits Air2 and Trf4 structurally resemble those of ZCCHC8 MA1 and MA2, respectively (Figures S4I and S4J) (Falk et al., 2014), but the ZCCHC8 MA2 motif is inverted relative to the Trf4 RecA2 binding motif, making it difficult to predict in the absence of structure. Because surfaces that contribute to NEXT, TRAMP and NRDE2 interactions with MTR4 are overlapping or mutually exclusive, these observations support a pivotal role for these co-factors in specifying the identity and function of MTR4 in these complexes.
RNA path in NEXT
Clear densities for RNA (substrate 1) are evident in the MTR4 helicase core and proximal to RBM7 RRM in protomers A and B (Figures 1B and S3A). Compared to translocated RNA in structures of the human MTR4-bound nuclear exosome (Weick et al., 2018), the RNA 3′ end is not extruded but is rather captured between the RecA domains of the helicases in both protomers of NEXT (Figures S4A and S4D). While portions of the RNA substrate are visible, the inherent flexibility of the substrate and RNA binding modules obscured the path for RNA, especially between MTR4 and RBM7. To improve density for RNA, RBM7 and elements of ZCCHC8, NEXT was loaded with a palindromic RNA duplex with 3′ tails on both strands (substrate 2) and cryo-EM data produced reconstructions that better defined an RNA path (Figures S5A-E). This RNA induced formation of a tetramer in ~20% of particles with two NEXT homodimers cross-braced by two RNA substrates (Figure 4A). Once the tetramer was identified, signal from each protomer was isolated by signal subtraction, recentered, and combined to generate reconstructions of RNA-bound NEXT at an overall resolution of 3.62 Å resolution after post-processing (Figures S6A-D). Five focused refinements generated 3D reconstructions with overall resolutions ranging from 3.34 Å to 3.94 Å that were combined into a composite map to aid model building and refinement (Figures 4B, 4C, S6A and S6D-G; Table S1).
Figure 4. Cryo-EM structure of NEXT bound to substrate 2 and RNA interactions within the complex.

(A) EM map of two NEXT homodimers cross-braced by two molecules of substrate 2 (colored uniquely).
(B-C) Composite map (B) from focused reconstructions used to build and refine the model
(C) of NEXT protomer bound to substrate 2.
(D) Schematic diagram of interactions between NEXT and RNA substrate 2. Stacking and polar interactions are indicated by solid and dashed lines, respectively. Nucleotides in gray are absent in the final model.
(E) RBM7 interaction with substrate 2 with EM density for RNA and ZCCHC8 shown and colored uniquely.
(F) RBM7 (protomer A) interaction with substrate 1 with EM density for RNA shown.
(G) MTR4 β hairpin wedges between the incoming A25 nucleotide and A26. EM density overlaid with the model is shown.
(H) Magnified view (left panel) of RNA interactions within MTR4 helicase core. Dashed lines indicate polar interactions. The corresponding EM map is shown in the right panel.
(I) Comparison of RNA-protein and MTR4 contacts in NEXT and the human MTR4-exosome complex (Weick et al., 2018).
(J) Clipped surface representations comparing the RNA path through MTR4 in NEXT and MTR4-exosome complex. See also Figures S5, S6 and Table S1.
Densities for RNA are evident, from the A-form duplex to the single-stranded 3′ overhang (Figures 4B and 4C). RBM7 RRM and ZCCHC8 ZK domains flank the oligo(U) tract (Figures 4C-E). Three nucleotides (A15-U17) dock to RBM7 RRM, with the U16 nucleobase proximal to Phe13. Similar contacts between the RBM7 RRM and RNA were observed for the NEXT-substrate 1 complex (Figure 4F). The succeeding U18-U19 nucleotides point toward the ZCCHC8 ZK. A23-A25 protrude from the MTR4 RNA entry site and the last five nucleotides A26-A30 are accommodated within the RNA channel of MTR4 mainly through polar interactions to the RNA backbone (Figures 4D and 4G-H). Consistent with conformational selection and specificity for flexible single-stranded RNA upstream of the 3′ end, the U-rich motif and RNA path in NEXT is kinked relative to the RNA path in the human MTR4-exosome complex (Figures 4i). Our structures are also consistent with studies inferring contacts with isolated RRM (Hrossova et al., 2015; Lubas et al., 2015) and crosslinking studies using intact NEXT that mapped interactions, 5′ to 3′, between RNA and RBM7, ZCCHC8 or MTR4 (Puno and Lima, 2018).
A hallmark of DExH-box helicases is a β hairpin that facilitates strand separation (Büttner et al., 2007; Gao and Yang, 2020; Ozgur et al., 2015). The MTR4 β hairpin includes Phe504, a residue near the channel entrance that wedges between A26 and the incoming A25 nucleotide (Figure 4G). The observed RNA path in the helicase core of MTR4 resembles that of other MTR4-exosome complexes, but several other interactions are absent or different. For instance, MTR4 KOW domain surfaces that contact structured RNA in MTR4-exosome (Weick et al., 2018) and MTR4-ribosome complexes (Schuller et al., 2018) are occluded by the ZCCHC8 KID (Figure 4I). Like RNA in the structure of dimeric NEXT, upstream RNA is directed away from the MTR4 KOW domain and is coordinated between RBM7 and ZCCHC8. Furthermore, the C-terminal domain of ZCCHC8 blocks the RNA exit path and occludes MTR4 surfaces required for interaction with the exosome via contacts to EXOSC2 and MPP6, a nuclear co-factor that tethers MTR4 to the exosome (Wasmuth et al., 2017; Gerlach et al., 2018; Weick et al., 2018) (Figures 4I and 4J). Collectively these contacts suggest that our structures represent early steps in RNA recognition after the 3′ end is captured but before it is extruded from MTR4 in a process that is likely dependent on ZCCHC8 CTD remodeling and/or exosome recruitment.
NEXT homodimers are more productive helicases
Our structures reveal that NEXT is a homodimer mediated by ZCCHC8. To determine if ZCCHC8 can form homodimers in human cells, we immunoprecipitated mCherry-tagged ZCCHC8 as prey from cells expressing wild-type (WT) GFP-ZCCHC8 or HD-deleted GFP-ZCCHC8ΔHD as bait. WT GFP-ZCCHC8 but not GFP-ZCCHC8ΔHD co-precipitated mCherry-ZCCHC8 (Figure S7A), consistent with HD-driven dimerization of ZCCHC8. Analysis of endogenous NEXT by size exclusion chromatography indicated an apparent molecular weight of ~1000 kiloDalton (kDa) (Ogami et al., 2017), a size that is larger but consistent with the apparent molecular weight of ~800 kDa for recombinant NEXT (Figures S7B and S7C). No smaller species were evident for endogenous or recombinant complexes, suggesting that NEXT is dimeric.
Each NEXT protomer of RBM7, ZCCHC8 and MTR4 is bound to RNA, suggesting that each protomer works independently. To determine if dimerization contributes to NEXT activities, we employed electromobility shift assay (EMSA) to measure binding at steady state and a molecular beacon helicase assay (MBHA) to measure kinetics of unwinding under pre-steady state conditions. In general, equilibrium binding constants from EMSA agree with apparent substrate affinities obtained from kinetic plots. EMSA data reveal that NEXT binds the U-rich tailed RNA with a KD of 42 nM and a Hill coefficient close to 2 indicating cooperative binding, while deletion of ZCCHC8 HD (NEXTΔHD) yields a 2-fold defect in RNA binding and loss of cooperativity (Figures 5A and 5B). Comparing RNA unwinding activities reveals 7-fold lower maximal unwinding rate (Vmax) for NEXTΔHD compared to NEXT (Figures 5C, 5D, and S7D). To determine if each MTR4 helicase must be active in the dimer to maximize activity, differentially tagged MTR4 and catalytically inactive MTR4 with a glutamine substitution of Glu253 residue (denoted as “EQ”) required for ATP hydrolysis and RNA unwinding were used to reconsitute a heterodimeric NEXTWT/EQ complex (Puno and Lima, 2018). NEXTWT/EQ activities are more similar to NEXTΔHD, with slightly worse than expected kinetics for strand displacement based on comparison to a stoichiometric mixture of WT (NEXTWT/WT) and catalytically inactive (NEXTEQ/EQ) homodimers (Figures 5C, 5D, and S7D).
Figure 5. Biochemical, qPCR, and RNAseq analysis of NEXT and variants.

(A) EMSA plots comparing NEXT and NEXTΔHD binding to 3′ A5U5A10-tailed RNA duplex substrate. Data points represent mean ± SD from three technical replicates.
(B) Bar graphs of dissociation constants KD (mean ± SEM) and Hill coefficients h (mean ± SEM) obtained from fitting the EMSA data in (B) to Hill equation.
(C) Graph of initial strand displacement rate (v0) at varying protein concentration (plotted as equivalent molar concentration of MTR4) for WT NEXT, NEXT with ZCCHC8 HD deleted (NEXTΔHD), heterodimeric NEXTWT/EQ containing wild-type MTR4 and MTR4 E253Q mutant, and a stoichiometric mixture of wild-type NEXTWT/WT and helicase-dead NEXTEQ/EQ. Assays were performed using 3′ A5U5A10-tailed RNA duplex substrate. Data points are shown as mean ± SD from three separate reactions.
(D) Bar graphs of Vmax (mean ± SEM) obtained by fitting the data in (A) to equation v0 = Vmax*[E]/(K1/2 + [E]*/(1+[E]/K′1/2)).
(E) EMSA plots comparing NEXT with RBM7 F13A/F52A mutations (NEXTFAFA) and NEXT with ZCCHC8 ZK deleted (NEXTΔZK) binding to 3′ A5U5A10-tailed RNA duplex substrate.
(F) Bar graphs of KD (mean ± SEM) and h (mean ± SEM) obtained from fitting the EMSA data in (E) to Hill equation.
(G) EMSA plots comparing NEXT, NEXTFAFA, and NEXTΔZK binding to 3′ A20-tailed RNA substrate. Data points in (E) and (G) represent mean ± SD from three technical replicates.
(H) Bar graphs of KD (mean ± SEM) and h (mean ± SEM) obtained from fitting the EMSA data in (G) to Hill equation.
(I) v0 at varying protein concentration (plotted as equivalent molar concentration of MTR4) for NEXT, NEXTFAFA, NEXTΔZK, and MTR4. Assays were performed with 3′ A5U5A10-tailed RNA substrates.
(J) Bar graphs of Vmax (mean ± SEM) obtained by fitting the data in (I) to equation v0 = Vmax*[E]/(K1/2 + [E]*/(1+[E]/K′1/2)).
(K) v0 at varying protein concentration (plotted as equivalent molar concentration of MTR4) for NEXT, NEXTFAFA, NEXTΔZK, and MTR4. Assays performed with 3′ A20-tailed RNA substrates. Data points for (I) and (K) are shown as mean ± SD from three separate reactions.
(L) Bar graphs of Vmax (mean ± SEM) obtained by fitting the data in (K) to equation v0 = Vmax*[E]/K1/2 + [E]*/(1+[E]/K′1/2)) for NEXTΔZK or v0 = Vmax [E]/K1/2 for WT NEXT and NEXTFAFA.
(M) Bar graph of relative RNA levels (mean ± SD) obtained from qPCR analysis of PROMPTS in parental HAP1 cells, ZCCHC8 CRISPR knockout HAP1 cells (HAP1 Z8 KO), and stable clones of ZCCHC8 knockout HAP1 cells complemented with GFP-tagged ZCCHC8 (GFP-Z8), ZCCHC8 with HD deleted (GFP-Z8ΔHD) or ZCCHC8 with ZK deleted (GFP-Z8ΔZK). Individual data points from three biological replicates are shown as solid circles. Data were normalized relative to RNA levels in parental line. Statistical analysis was performed using two-tailed t test. P values are indicated by *p < 0.05, **p <0.01, ***p <0.001; ****p < 0.0001.
(N) Density profiles of RNA seq (−) strand reads around protein-coding genes and long intergenic RNA transcript transcription start sites (TSSs) within 3 kb upstream and downstream of PROMPTS.
(O) Read density plot upstream and downstream of eRNAs TSSs. Both (+) and (−) strand reads are shown.
(P) Read density 500 bp upstream and 1 kb downstream of 3′ ends (EAG) of all snRNAs. Each sample in Figures 5O-5Q is labeled and colored uniquely. See also Figure S8.
RBM7 RRM and ZCCHC8 ZK contribute to substrate discrimination
Human MTR4 binds 3′ A20- and U20-tailed substrates with similar affinities whereas NEXT exhibits better binding and unwinding activity for poly(U)-containing 3′ tailed substrates that is dependent on RBM7 RRM (Hrossova et al., 2015; Lubas et al., 2015; Puno and Lima, 2018). Indeed, RBM7 RRM contacts the upstream oligo(U)-rich motif in our structures (Figures 4D-F). Consistent with sequence specificities or a preference for flexible single-stranded RNA upstream of the 3′ end, NEXT exhibits ~10-fold better affinity (EMSA KD) and 3-fold higher (Vmax) for RNA with an oligo(U)-containing 3′ tail (A5U5A10) relative to one with a poly(A) 3′ tail (A20) (Figures 5E-L and S7E-F). Unlike U-rich motifs, poly(A) forms a more rigid helical structure (Brahms et al., 1966; Hashizume and Imahori, 1967; Tang et al., 2019) so poly(A) would need to bend in NEXT to adopt the kinked RNA path observed in our structures with U-rich RNA.
To probe determinants that impart specificity for U-rich motifs, alanine substitutions of RBM7 RRM Phe13 and Phe52 (F13A/F52A, denoted as “FAFA”) were introduced to NEXT to generate NEXTFAFA, mutations previously shown to reduce binding affinity to pyrimidine-rich oligonucleotides for isolated RBM7 RRM (Hrossova et al., 2015). This complex exhibited a 3-fold defect in binding affinity and a 5-fold lower maximal unwinding rate for 3′-(A5U5A10)-tailed substrate compared to NEXT (Figures 5E, 5F, 5I, 5J, and S7E). For comparison, NEXTFAFA displayed ~2-fold defect in similar assays with 3′ A20-tailed substrate (Figures 5G, 5H, 5K, 5L, and S7F). So while RBM7 prefers oligo(U)-rich motifs, these data suggest that RBM7 also contributes to binding oligo(A) single-stranded RNA. ZCCHC8 ZK is proximal to the U-rich tract in our structures. To determine if it contributes to RNA substrate specificity, ZCCHC8 ZK was deleted (ΔZK) and NEXTΔZK was reconstituted. NEXTΔZK exhibited slightly slower rates for unwinding 3′-(A5U5A10) or 3′-(A20) relative to WT NEXT (Figures 5I-L, S7E, and S7F), but it unexpectedly resulted in a 4-fold improvement in binding to the 3′ A20-tailed RNA (Figures 5G and 5H). As affinities for 3′-(A5U5A10) tailed RNA remained similar between NEXTΔZK and NEXT, these data suggest that the ZCCHC8 ZK discriminates against poly(A) tails, perhaps by conformational selection of non-structured single-stranded RNA or a kinked RNA path as observed in our structures. Apparent substrate affinities (K1/2) derived from helicase assays follow similar trends to that observed in equilibrium binding assays (Figures 5F, 5H, and S7G).
ZCCHC8ΔHD alters NEXT target levels
ZCCHC8 contributes to NEXT activities in vitro as evidenced by a reduction in strand displacement activity for the HD mutant and improved binding to poly(A) RNA for the ZK mutant. To evaluate functional contributions of ZCCHC8 HD and ZK deletions in cells, ZCCHC8 CRISPR-Cas9 knockout HAP1 cells were transfected with doxycycline-inducible constructs of GFP-ZCCHC8, GFP-ZCCHC8ΔHDor GFP-ZCCHC8ΔZK. Stable clones were isolated, confirmed for expression and nuclear localization of ectopic ZCCHC8 constructs, and sorted for GFP expression prior to analysis of NEXT-targeted PROMPT RNAs using qPCR (Figures 5M and S8A-C). ZCCHC8 null cells accumulated PROMPT RNAs that could be rescued by expression of WT GFP-ZCCHC8 and GFP-ZCCHC8ΔZK. In contrast, cells expressing GFP-ZCCHC8ΔHD exhibited a defect in suppressing PROMPT RNA levels, consistent with reduced helicase activity observed for ZCCHC8ΔHD in vitro. RNA seq analysis confirms accumulation of PROMPT and to lesser extent enhancer RNA (eRNA) levels in cells expressing GFP-ZCCHC8ΔHD compared to GFP-ZCCHC8, while a marginal change was observed for 3′ extended small nuclear RNA (snRNA) levels (Figures 5N-P, S8D, and S8E). Consistent with global analysis, differences are also observed in coverage snapshots of select NEXT targets (Figure S8E). To confirm these results, we generated CRISPR-Cas9-edited ZCCHC8 ΔHD HAP1 cells (Figures S9A and S9B), however genomic deletion of HD resulted in reduced mRNA and protein levels (Figure S9C-E).
ZCCHC8-ROS1 leads to dimerization and increased kinase activity
Mutations in NEXT subunits are associated with various diseases and our structures provide a three-dimensional context to interpret their effects. Genetic lesions in ZCCHC8 map to either HD or KID (Figure 6A). A nonsense mutation L90X in HD results in a truncated protein and is linked to autosomal recessive intellectual disability (Najmabadi et al., 2011). Leucine substitution of Pro186 (ZCCHC8P186L) is observed in a family with 3 members afflicted with an autosomal dominant pulmonary fibrosis, a common premature aging disorder characterized by shortening of the telomeres (Gable et al., 2019). Pro186 is situated in a loop region in KID, adjacent to the AIM sequence (Figure 6B). While the side chain of Pro186 faces the dimerization domain of the other ZCCHC8 chain (Figure 6A), it lies in the interface between ZCCHC8 and the MTR4 KOW (Figure 6B). P186L may prevent NEXT assembly, perhaps contributing to reduced steady-state levels observed in patient cells (Gable et al., 2019).
Figure 6. ZCCHC8-ROS1 fusion homodimerization and increased kinase activity.

(A) Locations of L90X and P186L mutations in ZCCHC8 (orange spheres).
(B) Magnified view of Pro186 and neighboring residues. Corresponding EM density shown in right panel.
(C) Structures of ROS1-fused ZCCHC8 residues 1-80 and 1-105 and protein schematics of ZCCHC8-ROS1 fusion associated with several forms of cancer.
(D) Immunoprecipitation of GFP-tagged (bait) and mCherry-tagged (prey) ZCCHC81-80-ROS1kinase (Z81-80-ROS1kinase) and ROS1 kinase. Representative immunoblots from three replicates shown.
(E) Anti-ROS1 immunoblot analysis of Phos-tag-separated lysates of cells expressing GFP fusion constructs of ZCCHC81-80-ROS1kinase (GFP-Z81-80-ROS1kinase), ZCCHC81-80-ROS1kinase(dead) with kinase-inactivating ROS1 K1980M mutation (GFP-Z81-80- ROS1kinase(dead)) or ZCCHC81-80dm-ROS1kinase with C56E/L63E/L70E dimer-disrupting mutations (GFP-Z81-80dm-ROS1kinase). Representative immunoblot from three replicates shown.
(F) Representative histogram from flow cytometry analysis of phosphorylated ROS1 (pROS1) in HAP1 cells expressing GFP-Z81-80-ROS1kinase, GFP-Z81-80-ROS1kinase(dead), GFP- ZCCHC81-80dm-ROS1kinase, and GFP-ROS1kinase.
(G) Bar graph showing relative pROS1 levels (mean ± SD). The individual pROS1 median from five biological replicates are shown as solid circles. Data were normalized relative to cells expressing GFP-ROS1kinase. Statistical analysis was performed using two-tailed t test. P values are indicated by **p <0.01. See also Figure S9.
In addition to ZCCHC8 mutations, aberrant chromosomal translocations lead to fusion of ZCCHC8 HD residues 1-80 or 1-105 to the receptor tyrosine kinase (RTK) domain of ROS1 (residues 1927-2347) (Figure 6C). ROS1 is a proto-oncogenic RTK with unknown physiological function in humans (Drilon et al., 2021) yet several ROS1 gene fusions occur in cancer, most involving a diverse fusion partner to the C-terminal kinase domain (Davies and Doebele, 2013; Drilon et al., 2021). ZCCHC8-ROS1 fusion is observed in several cancers including congenital glioblastoma multiforme (Coccé et al., 2016), non-small cell lung adenocarcinoma (Creelan, 2018; Zhu et al., 2018), and spitzoid melanoma (Wiesner et al., 2014). Based on the ZCCHC8 structure, the fusion is predicted to disrupt plasma membrane localization and induce dimerization of the ROS1 kinase, perhaps further activating it through trans-autophosphorylation (Drilon et al., 2021).
To determine if ZCCHC8 residues 1-80 are sufficient to dimerize the ZCCHC81-80-ROS1kinase fusion, immunoprecipitation was performed. While GFP-ZCCHC81-80-ROS1kinase immunoprecipitated mCherry-ZCCHC81-80-ROS1kinase, no interaction was detected between GFP-ROS1kinase and mCherry-ROS1kinase (Figure 6D). Three mutations (C56E/L63E/L70E, denoted as “dm”) were introduced in the ZCCHC81-80 coiled-coil to disrupt the dimer interface (Figure S9F). GFP-ZCCHC81-80dm-ROS1kinase failed to precipitate mCherry-ZCCHC81-80-ROS1kinase (Figure S9G). We next sought to determine if ZCCHC81-80-ROS1kinase interacts with ZCCHC8 to form NEXT-ROS1 heterodimers, however GFP-ZCCHC81-80-ROS1kinase did not immunoprecipitate mCherry-ZCCHC8 (Figure S9H). These data suggest that ZCCHC81-80-ROS1kinase can self-oligomerize in human cells but does not form heterodimers with full-length ZCCHC8.
Related RTKs can be activated by oligomerization (Du and Lovly, 2018; Schlessinger, 2000), so autophosphorylation of the ROS1 kinase domain was analyzed as a surrogate for kinase activation using Phos-tag gels. A band shift was observed for GFP-ZCCHC81-80-ROS1kinase but not for GFP-ZCCHC81-80dm-ROS1kinase and GFP-ZCCHC81-80-ROS1kinase(dead) with kinase inactivating K1980M mutation (Figure 6E), supporting a model for homodimerization-dependent autophosphorylation within the ZCCHC81-80-ROS1kinase fusion. Phospho-specific flow cytometry analysis further showed ~3-fold greater levels of phosphorylated ROS1 (pROS1) in cells expressing GFP-ZCCHC81-80-ROS1kinase relative to cells expressing GFP- ROS1kinase and GFP-ZCCHC81-80dm-ROS1kinase (Figures 6F, 6G, S9I, and S9J). Although phosphorylated GFP-ZCCHC81-80dm-ROS1kinase was not detected in Phos-tag gels, phospho-flow analysis showed similar levels of pROS for cells expressing GFP-ROS1kinase and GFP-ZCCHC81-80dm-ROS1kinase compared to cells expressing GFP-ZCCHC81-80-ROS1kinase(dead). Together, these data suggest that dimerization of ZCCHC81-80-ROS1kinase could further activate the ROS1 kinase by trans-autophosphorylation.
Discussion
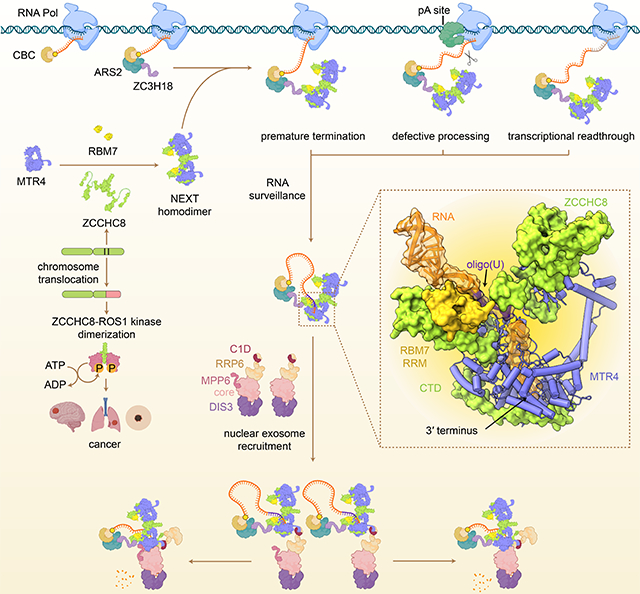
RNA quality control begins as nascent transcripts emerge from cellular transcription machinery (Figure 7). The nuclear cap-binding CBP20-CBP80 complex (CBC) first sequesters the 5′ cap and participates in dynamic interactions with different factors that determine RNA fate (Rambout and Maquat, 2020). ARS2 and ZC3H18 then associate with CBC-bound transcripts and co-transcriptionally recruit RNA adaptor complexes that surveil unprotected 3′ termini of coding and non-coding transcripts to ultimately choreograph their degradation by the exosome (Andersen et al., 2013; Giacometti et al., 2017). While structures of RNA-loaded MTR4-exosome complexes reflect the final stages of MTR4-assisted RNA decay pathway (Gerlach et al., 2018; Weick et al., 2018), prior steps pertaining to substrate recognition or modification by intact RNA bound nuclear exosome adaptor complexes remain structurally uncharacterized.
Figure 7. Model for NEXT substrate recognition and delivery to the exosome.

CBC, ARS2 and ZC3H18 deposit on 5′ capped nascent transcript and recruit NEXT that captures unprotected 3′ ends of RNA produced by purposeful, spurious or faulty transcription (Andersen et al., 2013; Giacometti et al., 2017; Rambout and Maquat, 2020). While polyadenylated exosome substrates are directed to the exosome via PAXT or PPC (Meola et al., 2016; Ogami et al., 2017), targets of the NEXT are mostly non-polyadenylated (Wu et al., 2020). NEXT forms a homodimer with each protomer capable of binding substrate. MTR4 sequesters the 3′ end whereas RBM7 RRM and ZCCHC8 ZK bind upstream sequences, with RBM7 RRM exhibiting a preference for U-rich motifs. ZCCHC8 CTD wraps around the base of the MTR4 helicase core to occlude the exit path for RNA and surfaces important for docking on the exosome core via EXOSC2 and for recruitment by MPP6. Because MPP6 interaction surfaces are occluded, recruitment to the exosome may first occur through MTR4 contacts to RRP6 and C1D. While the order of events remains unclear, surfaces required for RNA extrusion from the helicase and binding to the exosome core are occluded by ZCCHC8, so remodeling of ZCCHC8 would be required before MTR4 could dock on the RNA exosome.
Here we illuminate cryo-EM structures of NEXT that suggest how NEXT subunits interact and cooperate to target RNA substrates for decay. NEXT assembles into a homodimer with two active MTR4 helicases, each capable of accommodating a 3′ end of RNA. ZCCHC8 mediates homodimerization, wrapping around MTR4 to occlude surfaces important for RNA binding while positioning the RNA binding module of RBM7 next to the helicase core for recognition of single-stranded RNA upstream of the 3′ end. Beyond its role as a scaffold, ZCCHC8 interacts with RNA through its zinc knuckle motif, perhaps to ensure a kinked RNA path that discriminates against poly(A) tailed RNA substrates. Furthermore, ZCCHC8 covers surfaces of the MTR4 KOW that can interact with structured RNA (Schuller et al., 2018; Weick et al., 2018), potentially favoring interactions with single-stranded 3′ tails over other structured RNAs. The discrimination against poly(A) and preference for U-rich sequences may guide NEXT to RNA containing U-rich elements such as those adjacent to transcription termination or cleavage sites (Di Giammartino et al., 2011; Hrossova et al., 2015; Lubas et al., 2015; Nojima et al., 2013; Ntini et al., 2013).
In addition to cooperative binding, higher than expected activities are observed for the NEXT dimer suggesting that the two helicases work together rather than independently (Figures 5 and S7). How the two helicases communicate within NEXT remains unclear, but it is notable that 3D reconstructions of NEXT dimers reveal asymmetry with respect to densities corresponding to RBM7-ZCCHC8 and the coiled-coil domain at the dimer interface (Figures S2A and S3D). The contribution of ZCCHC8 to NEXT activities is underscored by a human disease ZCCHC8 mutation P186L that lies at the interface between the two ZCCHC8 chains and MTR4, although it remains unclear if phenotypes are due to disruption of the dimer or to reduced expression in these patients or a combination of both (Gable et al., 2019).
The discovery and structural basis for ZCCHC8 homodimerization sheds light on the functional consequences of the ZCCHC8-ROS1 fusion found in a variety of cancers. Many receptor tyrosine kinases undergo dimerization upon ligand binding, subsequently leading to autophosphorylation and kinase activation (Della Corte et al., 2018; Du and Lovly, 2018; Schlessinger, 2000). The ZCCHC8-ROS1 fusion truncates the extracellular domains, essentially making it insensitive to any ligands. Here we have shown that ZCCHC8-ROS1 fusion induces dimerization and autophosphorylation, potentially activating mechanisms that drive oncogenesis. Our structures provide a framework to evaluate a growing number of genetic mutations in subunits of the NEXT complex.
Structures of NEXT reveal an intriguing architecture that opens several questions related to its interactions with the exosome. ZCCHC8 CTD binding to MTR4 occludes surfaces important for exosome association with the helicase via MPP6 and EXOSC2. ZCCHC8 CTD also presents a steric occlusion to the RNA exit path through MTR4. Hence, ZCCHC8 CTD displacement or remodeling appear necessary before NEXT can deliver RNA to the exosome. It is intriguing that an analogous mechanism might contribute to substrate recognition and decay in the cytoplasm by the SKI-exosome complex (Kögel et al., 2022). Subunit interactions within NEXT provide a means to couple interactions with the exosome to RNA extrusion from the helicase, but only after NEXT is fully engaged by a nuclear RNA exosome. It is noteworthy that the ZCCHC8 CTD and MPP6 both stimulate helicase-dependent activities of NEXT and the exosome, respectively (Puno and Lima, 2018; Weick et al., 2018), perhaps to establish quality control by facilitating repeated attempts at RNA translocation and delivery while maintaining contacts between NEXT, the exosome and RNA. The multivalency of contacts to MTR4 in NEXT and the nuclear exosome suggest that NEXT recruitment and RNA translocation could proceed through parallel or hierarchical paths, perhaps expanding potential mechanisms to regulate the process.
The diversity of RNA substrates in the nucleus is staggering, so it is perhaps remarkable that a single motor protein, MTR4, can be utilized by the RNA exosome, NEXT, TRAMP, PAXT and other upstream co-factors to identify, discriminate, and eventually feed substrates to the nuclear exosome for processing or decay. MTR4 is granted this exceptional versatility by its ability to interact with protein co-factors and adaptors via mutually exclusive surfaces, thus ensuring specificity and perhaps a sequential hierarchy to these interactions. The single-engine, multiple-adaptor strategy is a recurrent theme in biology and is perhaps reminiscent of the Cdc48/p97 motor and its various adaptors that combine to promote specificity in processes ranging from disaggregation to processing and/or degradation by the proteasome (Boom and Meyer, 2018; Hänzelmann and Schindelin, 2017). As there are comparatively few complexes identified thus far that target RNAs for decay in the nucleus, it seems likely that additional factors, perhaps deposited on specific nascent RNAs, will contribute to RNA quality control and its regulation in the nucleus.
Limitations of the study
Our studies advance an understanding of how NEXT subunits work together to capture RNA, but many questions remain. Complexes containing full-length proteins may be required to define roles for the RBM7 C-terminal domain and ZCCHC8 low complexity region. Additional structures are needed to reveal catalytic cycles including ATP-dependent RNA translocation and extrusion, perhaps by isolating NEXT-RNA complexes in various nucleotide-bound states. Deletion of HD significantly impairs NEXT, but its cellular effect is less severe than ZCCHC8 knockout so it remains unclear how dimerization contributes to NEXT activities in cells. Perhaps defects are ameliorated by association with the nuclear exosome. With that said, it is worth noting that acute depletion of NEXT subunits results in milder accumulation of RNA targets compared to knockouts, presumably due to secondary effects (Gockert et al., 2022). Our cellular studies used fusions to a GFP variant with a monomerizing A206K mutation, but the impact of GFP fusions to ZCCHC8 warrant further analysis. As CRISPR-Cas9 deletion of HD resulted in diminished mRNA and protein levels, further work will be required to uncover mutations that impair dimerization without disrupting expression. Endogenous targets of NEXT are typically capped and bound to proteins (Figure 7), however our biochemical and structural studies use synthetic RNA, so it remains unclear how other proteins impact NEXT activities. Finally, structural and functional studies of NEXT-exosome complexes are needed to understand molecular determinants and the order of events associated with NEXT-dependent RNA decay such as ZCCHC8 CTD and/or MPP6 remodeling, the role of C1D and RRP6 in NEXT tethering to the exosome, and contributions of NEXT dimerization in exosome-mediated decay.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Christopher D. Lima (limac@mskcc.org).
Materials availability
All stable cell lines and plasmids generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and code availability
Cryo-EM density maps are deposited in the Electron Microscopy Data Bank (EMDB) for NEXT-substrate 1 complex, NEXT-substrate 2 complex, and apo-NEXT with accession numbers: EMDB: EMD-24882, EMDB: EMD-24883, and EMDB: EMD-24884, respectively. Atomic coordinates are deposited in Protein Data Bank (PDB) for NEXT-substrate 1 complex and NEXT-substrate 2 complex with accession numbers: PDB: 727B and PDB: 7S7C, respectively. RNAseq datasets generated in this study are deposited are accessible at GEO: GSE185374.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODELS and SUBJECT DETAILS
Bacterial strains
Chemically competent Escherichia coli E. cloni 10G cells (Lucigen) grown in Luria Bertani (LB) broth or agar were used for molecular cloning of NEXT subunits and variants. Recombinant proteins were produced using E. coli BL21-CodonPlus(DE3) RIL (Agilent Technologies) cells cultured in Superbroth at 37 °C with shaking (220 rpm for Innova 44R).
Expi293F cell culture and transfections for immunoprecipitation
Human embryonic kidney 293 cells (Expi293F, Thermo Fisher Scientific) used for immunoprecipitation were maintained in serum-free Expi293 medium (Thermo Fisher Scientific) at 37°C, 8% CO2 and 80% humidity with a shake speed of 125 rpm according to manufacturer’s recommendations. All stably transfected Expi293F cells were generated using the piggyBac system. Piggybac-compatible plasmid constructs were subcloned from PB-TAG-ERPE (Kim et al., 2015). 9.5 ml cell suspension with a density of 2.9 × 106 cells per ml were co-transfected with 7 μg of piggybac-compatible plasmids with the gene of interest under a tetracycline/doxycycline-inducible promoter, 1.5 μg plasmid containing a hyperactive piggybac transposase (pRP[Exp]-mCherry-CAG>hyPBase) (VectorBuilder), and 1.5 μg plasmid containing reverse tetracycline-controlled transactivator (rtTA) with downstream internal ribosome entry site (IRES) and puromycin resistance gene (PB-rtTA-IRES-Puro). 24 hours post-transfection, cells were subcultured into fresh media to a final density of 6 × 105 cells per ml, grown for another 24 hours before stable integrants were batch-selected by treatment with 2 μg/ml puromycin for 1 week. After selection, cells were grown in antibiotic free medium for 1 week and protein expression was induced by treatment with 1 μg/ml doxycycline for at least 24 hours prior to immunoprecipitation experiments.
Generation of HAP1 cells expressing GFP fusion constructs of ZCCHC8, ZCCHC8ΔHD, and ZCCHC8ΔZK for qPCR and RNA sequencing studies.
Human HAP1 cells (Horizon Discovery) were grown in Iscove’s Modified Dulbecco’s medium (IMDM, Thermo Fisher Scientific) supplemented with 10% v/v fetal calf serum (Sigma) at 37°C and 5% CO2. HAP1 cells were authenticated using short tandem repeat (STR) profiling at MSK Integrated Genomics Operations. Plasmid transfections were carried out using Lipofectamine 3000 (Thermo Fisher Scientific) following manufacturer’s protocol. 1.5 × 105 HAP1 ZCCHC8 CRISPR knockout cells (Horizon Discovery) were seeded into 2 ml medium and allowed to adhere in a 6-well plate overnight. Cells were co-transfected with 2 μg of piggybac-compatible plasmid with the gene construct downstream of a tetracycline/doxycycline-inducible promoter, 0.5 μg hyperactive piggybac transposase plasmid (pRP[Exp]-mCherry-CAG>hyPBase) (VectorBuilder), and 0.5 μg PB-rtTA-IRES-Puro. Stable integrants were batch-selected by treatment with 3-5 μg/ml puromycin for 1 week. Puromycin-selected cells were used for RNA sequencing experiments. For qPCR analysis of PROMPTs, stable clones were isolated as follows. Batch-selected cells were sorted for cells expressing similar levels of GFP using BD FACSAria and single cells were seeded into each well of a 24-well plate. Individual cells were grown and analyzed for ectopically-expressed wild-type and mutant ZCCHC8 levels. Stable clones expressing similar levels of GFP fusion constructs were treated with 5-10 ng/ml doxycycline for at least 24 hours and sorted for GFP-positive cells using BD FACSAria prior to RNA extraction and qPCR analysis.
Generation of HAP1 cells expressing GFP fusion constructs of ZCCHC81-80-ROS1kinase, ZCCHC81-80-ROS1kinase(dead), ZCCHC81-80dm-ROS1kinase and ROS1kinase for Phos-tag gel and phospho-flow analysis
HAP1 cells (Horizon Discovery) were cultured in IMDM (Thermo Fisher Scientific) supplemented with 10% v/v fetal calf serum (Sigma) at 37°C and 5% CO2. 1.5 × 105 cells were grown overnight in a 6-well plate and were co-transfected with 2 μg piggyBac-compatible plasmids with GFP fusion constructs under the control of tetracycline/doxycycline-inducible promoter, 1.5 μg pRP[Exp]-mCherry-CAG>hyPBase (VectorBuilder), and 1.5 μg PB-rtTA-IRES-Puro using Lipofectamine 3000 kit (Thermo Fisher Scientific). Stably transfected cells were selected by treatment with 2 μg/ml puromycin for 1 week. Puromycin-selected cells were used for analysis of ROS1 autophosphorylation using Phos-tag gels and by flow cytometry.
CRISPR-Cas9 deletion of ZCCHC8 HD in HAP1 cell line
A day prior to transfection, 1.5 × 105 HAP1 cells (Horizon Discovery) in 2 ml IMDM with 10% (v/v) fetal calf serum were added into each well of a 6-well plate. Cells were transfected with 2.5 μg plasmid (pRP[2CRISPR]-mCherry-hCas9-U6>{hZCCHC8[gRNA#1]}-U6>{hZCCHC8[gRNA#2]}) containing two U6 promoter-driven guide RNA expression constructs, a human codon-optimized Cas9 construct and an mCherry reporter using Lipofectamine 3000 (Thermo Fisher Scientific). Transfected cells were placed in an incubator (37°C and 5% CO2) for 4 hours before treatment with 2 μM NU7441 (StemCell Technologies) for 18 hours. Individual mCherry-expressing cells were sorted using BD FACSAria, plated into a 48-well plate (Thermo Fisher Scientific), and grown for 1-2 weeks or until colonies appeared. Cells were screened for expression of ΔHD ZCCHC8 mRNA with forward primer: ATGGCCGCAGAGGTGTATTTTACT and reverse primer: TGCCTTTACTTGTATTTCTTGCCCTTC using Cells-to-CT 1-Step Power SYBR Green kit. Genomic DNA was prepared from the initial hits and was used for PCR amplification of ZCCHC8 gene fragment using forward primer: ATGCCGTCCTCCGGGCAATC and reverse primer: AGCCTAGGTTTAATTTAGAAGCGGTTGCCCCAATTTATC that flank the HD region. Amplicons were gel-purified using Qiagen gel extraction kit, cloned into pJET2.1 using CloneJet PCR cloning kit (Thermo Fisher Scientific), and submitted for Sanger DNA sequencing (Eton Biosciences). Two CRISPR-edited HAP1 ZCCHC8 ΔHD clones were identified. ZCCHC8 ΔHD mRNA expression was quantified relative to GAPDH mRNA using PowerUp SYBR Green master mix with forward primer: CAACCGCTTCTAAATGAAAACCCTC and reverse primer TTCACTTATTCGAGCAGCATTCCGAGG. ZCCHC8 ΔHD protein expression was analyzed using a Western blot analysis.
METHOD DETAILS
Protein production
Recombinant proteins were produced and purified from bacterial cells. Escherichia coli BL21-CodonPlus(DE3) RIL (Agilent Technologies) cells transformed with the expression plasmid construct were grown in Superbroth at 37 °C with shaking (220 rpm for Innova 44R) until the OD600 reached 1.8 (or >2 for MTR4 expression), then cooled in an ice-water bath for 20 min before addition of isopropyl-β-D-thiogalactoside to a final concentration of 0.25 mM. Cultures were then incubated at 18 °C with shaking for another 16-20 hours. Cells were harvested by centrifugation at 4000 x g (Beckman JLA-8.1000) for 15 min at 4°C. Cell pellets (15-20 g) were suspended in 200 ml of lysis buffer containing 50 mM Tris·HCl pH 8.0, 350 mM NaCl, 0.2 mM TCEP, 2.5 mM MgCl2, 1 μg/ml DNAse I (Sigma), 1 μg/ml lysozyme (Sigma) and disrupted using a Branson Digital Sonifier 450 Cell Disruptor (1 sec on, 1 sec off, 55-60% output, 1 min per 20 g cell mass) before ultracentrifugation at 44,000 x g (Beckman JA-20) for 30 min at 4 °C. The supernatant lysate was incubated with 5-10 ml Ni2+-NTA (Qiagen) for an hour at 4°C with rotation, poured into a gravity flow glass column (Bio-Rad), and beads were washed with 20 mM Tris·HCl pH 8.0, 350 mM NaCl, 0.2 mM TCEP, 10 mM imidazole. Bound proteins were eluted in buffer containing 20 mM Tris·HCl pH 8, 100 mM NaCl, 0.2 mM TCEP, 250 mM imidazole, passed through a 0.2 μm Pall Acrodish syringe filter and immediately applied to a 5-ml HiTrap Heparin HP column (GE Healthcare) using a 20-ml syringe. The heparin column was then connected to Akta pure chromatography system (GE Healthcare), washed with 5 column volumes of 20 mM Tris·HCl pH 8, 50 mM NaCl, 0.2 mM TCEP and the bound proteins were separated using a salt gradient (Buffer A: 20 mM Tris·HCl pH 8, 50 mM NaCl, 0.2 mM TCEP; Buffer B: 20 mM Tris·HCl pH 8, 1000 mM NaCl, 0.2 mM TCEP; Target %B = 100% over 5-10 column volumes). For MTR4, tags for affinity purification were cleaved by treatment with Ulp1 (Mossessova and Lima, 2000) and separated by size exclusion chromatography using HiLoad 26/600 Superdex 200 pg (GE Healthcare) in a buffered solution containing 20 mM Tris·HCl pH 8.0, 100 mM NaCl, and 0.1 mM TCEP. Reconstitution of the NEXT core complex was achieved by mixing a copurified heterodimer of His10-Smt3-RBM7/Smt3-ZCCHC8 constructs with 2-fold molar excess of MTR4 in the presence of Ulp1, followed by size exclusion chromatography using HiLoad 26/600 Superdex 200 pg in a buffered solution containing 20 mM Tris·HCl pH 8.0, 100 mM NaCl, and 0.1 mM TCEP. Fractions containing three subunits were pooled, concentrated using Amicon Ultra-15 50K MWCO filtration unit (used for all subsequent protein concentration step unless otherwise stated; Millipore), flash-frozen in liquid nitrogen, and stored at −80 °C until needed. PCR mutagenesis was used to generate mutations in DNA plasmids encoding NEXT subunits.
Recombinant heterodimeric NEXT containing both wild-type and helicase-dead MTR4 with E253Q mutation was reconstituted by incubating the purified His10-Smt3-RBM7/Smt3-ZCCHC8 heterodimer with an equimolar mixture of wild-type MTR4 with an N-terminal hexahistidine and non-cleavable Smt3 tag (with deletion of diglycine motif) and Strep-MTR4E253Q in 20 mM Tris·HCl pH 8.0, 100 mM NaCl, and 0.1 mM TCEP supplemented with 1 μg Ulp1 per mg total protein at 4°C for 4 hours. Ni2+-NTA beads were added to the mixture and incubated at 4°C for another hour with gentle agitation. The beads were washed with 20 mM Tris·HCl pH 8.0, 100 mM NaCl, 0.2 mM TCEP, 10 mM imidazole and bound proteins were eluted with 20 mM Tris pH 8, 100 mM NaCl, 0.2 mM TCEP, 250 mM imidazole. The eluate was then applied to a 1 ml StrepTrap HP column (GE Healthcare) connected to an Akta pure chromatography system (GE Healthcare). The column was washed with 20 column volumes of 20 mM Tris·HCl pH 8.0, 100 mM NaCl, 0.2 mM TCEP and the bound proteins were eluted with 20 mM Tris·HCl pH 8.0, 100 mM NaCl, 0.2 mM TCEP, 2.5 mM desthiobiotin. Finally, the eluate was concentrated to 500 μl and proteins were separated using Superdex 200 increase 10/300 GL (GE Healthcare) equilibrated with 20 mM Tris·HCl pH 8.0, 100 mM NaCl, and 0.1 mM TCEP.
Preparation of RNA substrates
RNA oligos were synthesized and HPLC-purified by Integrated DNA technologies (IDT) or Dharmacon Inc. Lyophilized RNA were dissolved in annealing buffer containing 20 mM Tris·HCl pH 7.0 and 100 mM potassium acetate and RNA concentrations were determined using Nanodrop 2000 spectrophotometer (Thermo Fisher Scientific). To prepare the RNA duplexes, stoichiometric amounts of oligos were mixed, heated to 95°C for 5 min, then cooled to 16°C for 10 min, and further incubated at 4°C overnight.
Electrophoretic mobility shift assay (EMSA)
Proteins at indicated concentrations were mixed with 10 nM 6-carboxyfluorescein (6-FAM)-labeled RNA substrate in solutions containing 20 mM Tris pH 7.0, 50 nM NaCl, 2.5 mM MgCl2, 1 mM AMPPNP (Sigma), 5% (v/v) glycerol, 5 mM ß-mercaptoethanol, and 0.4 U/μl human placenta RNAse inhibitor (New England Biolabs). The protein-RNA mixtures were incubated at 22 °C for 1 h before running samples (10 μl per well) in 4-20% Novex TBE gel (Thermo Fisher Scientific) with cold 0.5X Tris-Borate-EDTA (TBE) running buffer at 4 °C for 1 h under a constant voltage of 220V. Gels were imaged using a Typhoon FLA 9500 laser scanner. Band intensities were quantified using ImageJ 1.53a (Schneider et al., 2012) and RNA fraction bound was estimated by dividing the band intensities of remaining free RNA at various protein concentrations by the band intensity of total free RNA. Data were analyzed and plotted in GraphPad Prism. Dissociation constants (KDs) were obtained by fitting the data to the Hill equation: fraction bound = (Bmax[P]h)/(KDh + [P]h) where [P] is the protein concentration, h is the Hill’s coefficient, and Bmax is the maximal fraction bound.
Gel-based strand displacement assay
For reaction, a mixture of 15 μl 10X assay buffer (100 mM Tris pH 7.0, 50% (v/v) glycerol, 15 mM MgCl2), 13.5 μl water, 15 μl 50 mM ß-mercaptoethanol, 1.5 μl 40 units/μl human placenta RNAse inhibitor (New England Biolabs), 15 μl 100 nM RNA substrate and 75 μl 2X protein solution was prepared and incubated for 5 min at 22 °C. Reactions were initiated by addition of 10 μl of a pre-mixed solution of 20 mM ATP·MgCl2 (pH 7) and 4 μM DNA capture strand (5′ GCGTCTTTACGGTGCT 3′, IDT) per 90 μl reaction and mixtures were incubated at 30 °C. Aliquots were taken at various time points and mixed at 1:1 ratio with a quench solution containing 1% (w/v) SDS, 10 mM EDTA, 10% (v/v) glycerol, 0.01% (w/v) xylene cyanol supplemented with 80 units per ml Proteinase K (New England Biolabs). Quenched aliquots were incubated at 30 °C for 10 min to digest proteins. Samples (10 μl) were electrophoresed in a 20% Novex TBE gel (Thermo Fisher Scientific) using 1X TBE running buffer (prechilled to 4°C) for 1 h under a constant voltage of 220V at 4 °C. Gels were scanned using a Typhoon FLA 9500 laser scanner and rendered using ImageJ 1.53a (Schneider et al., 2012).
Molecular beacon strand displacement assay
Proteins were mixed at indicated concentrations with 10 nM RNA substrate in a solution containing 20 mM Tris·HCl pH 7.0, 50 mM NaCl, 1.5 mM MgCl2, 5% (v/v) glycerol, 5 mM ß-mercaptoethanol, and 0.4 U/μl human placenta RNAse inhibitor (New England Biolabs). Reaction mixtures (45 μl) were set up in a Corning 3693 half-area 96-well white plate, pre-incubated at 30 °C for 5 min, and started by addition of 2 mM ATP·MgCl2 (pH 7). Real time fluorescence was recorded every 30 s for 45 min at 30 °C using a SpectraMax M5 microplate reader (Molecular Devices) with excitation/emission wavelengths of 643/667 nm. The fraction of unwound substrate was calculated by dividing the baseline-corrected fluorescence value at each time point by the maximum fluorescence change achieved for a fully unwound substrate (Belon and Frick, 2008; Özeş et al., 2014). Initial rate of strand displacement was obtained by taking the slope of data points within the linear range of the reaction and kinetic data were further analyzed using GraphPad Prism. Initial rates (v0) were fitted to either
| (equation 1) |
or
| (equation 2) |
where [E] is the enzyme concentration, Vmax is the maximal strand displacement rate at enzyme saturation, and K1/2 is the half-maximal rate enzyme concentration.
Apparent molecular weight estimation
Protein standards (thyroglobulin, ferritin, aldolase, and conalbumin) and blue dextran from GE Healthcare gel filtration calibration (high molecular weight) kit were dissolved in 20 mM Tris·HCl pH 8.0, 100 mM NaCl, and 0.1 mM TCEP and mixed to a final concentration of 2 mg/ml each. 500 μl of the protein standard mixture or NEXT core complex (1 mg/ml) was injected to Superdex S200 increase 10/300 GL (GE Healthcare) connected to an Akta pure chromatography system (GE Healthcare) and eluted in 20 mM Tris·HCl pH 8.0, 100 mM NaCl, and 0.1 mM TCEP. Partition coefficient (Kav) was calculated using the equation: Kav = (Ve-V0)/(Vc-V0) where Ve is elution volume, V0 is column void volume and Vc is the geometric column volume. Apparent molecular weight was estimated using a calibration curve obtained by plotting Kav versus log(MW, kDa) of the protein standards.
Cryo-EM sample and grid preparation
RNA- and nucleotide-free NEXT (apo-NEXT) was obtained using purified recombinant core complexes with 260/280 nm absorbance ratio below 0.57 and used for subsequent preparation of NEXT in complex with RNA substrates 1 and 2. For NEXT-substrate 1 complex, 1 ml of 5 μM NEXT core complex was pre-incubated with a stoichiometric amount of RNA in 20 mM Tris·HCl pH 8.0, 50 mM NaCl, 1.5 mM MgCl2, and 0.1 mM TCEP at 22°C for 10 minutes. ATP·MgCl2 (pH 7.0) was added to a final concentration of 1 mM and the reaction mixture was incubated at 30°C for 1 hour. The RNA-loaded NEXT complex was purified by size exclusion chromatography using Superdex 200 increase 10/300 GL (GE Healthcare) in a buffered solution containing 20 mM Tris·HCl pH 8.0, 50 mM NaCl, and 0.1 mM TCEP. For the sample containing RNA substrate 2, 5 μM NEXT core complex was incubated with equimolar amount of substrate on ice for 30 minutes. RNA-protein complexes were purified by size exclusion chromatography using Superdex 200 increase 10/300 GL (GE Healthcare) in a buffered solution containing 20 mM Tris·HCl, pH 8.0, 50 mM NaCl, and 0.1 mM TCEP. Fractions from size exclusion chromatography were analyzed for protein content using NuPAGE 4-12% Bis-Tris gels (Thermo Fisher Scientific) electrophoresed with 1X MES running buffer and stained with Coomassie Brilliant Blue R-250 solution. Co-purified RNA in each fraction was detected using 20% TBE gel (Thermo Fisher Scientific) electrophoresed in 1X TBE at 220V for 1 hour at 4 °C followed by SYBR Gold staining. Protein and RNA gels were imaged using Bio-Rad ChemiDoc XRS+.
Prior to grid preparation, all RNA-protein samples were concentrated to 8 mg/ml as determined using the Bio-Rad protein assay kit. Apo-NEXT sample was concentrated to 6 mg/ml prior to use. IGEPAL CA-630 (Sigma) was added to samples to a final concentration of 0.02% (v/v) before vitrification. No crosslinking was performed for samples prior to vitrification. Approximately 3.5 μl of the concentrated samples were applied onto glow-discharged UltraAUFoil 300 mesh R1.2/1.3 grids (Quantifoil). After 30 s, grids were blotted for 2.5 s at 100% humidity and plunged into liquid ethane using an FEI Vitrobot Mark IV.
Cryo-EM data collection
For apo-NEXT and NEXT-substrate 1 complex, the grids were loaded on a Titan Krios 300kV electron microscope (FEI) equipped with a K2 Summit camera (Gatan) with a calibrated pixel size of 1.088 Å. Movies (40 frames per movie, 10 s exposure time) were collected automatically using SerialEM in super-resolution counting mode at a dose rate of 10 e-/pixel/second and a total dose of 67.6 e-/Å2/movie. For NEXT-substrate 2 complex, data acquisition was performed on a Titan Krios 300kV electron microscope (FEI) equipped with either K2 or K3 Summit direct detector (Gatan). For the data collected using the K2 camera (dataset 1), image stacks of 50 frames were recorded over 10 s in super-resolution counting mode at a dose rate of 10 e-/pixel/second for a total electron dose of 77.5 of e-/Å2/movie. For the data collected using the K3 camera (datasets 2 and 3), movies (40 frames per movie, 4 s exposure time) were recorded in super resolution mode at a dose rate of 20 e-/pixel/second and a total dose of 66 e-/Å2/movie.
Image processing
Image processing was performed in RELION 3.0 (Zivanov et al., 2018) unless otherwise stated. Movie frames were corrected for drift and dose-weighted using MOTIONCOR2 (Zheng et al., 2017). Estimation of contrast transfer function (CTF) was performed using Gctf (Zhang, 2016). Movies with an estimated resolution at or better than 4.5 Å were selected for analysis. Around 300-1000 particles were manually picked from a random subset of micrographs to obtain 2D classes that were then used as templates for automated picking of the remaining particles. Autopicked particles were extracted into 384 pixel box size and binned by 3 prior to running several rounds of 2D classification to remove junk classes. Particles were extracted and unbinned before heterogenous 3D classification.
NEXT-substrate 1 complex
Two datasets were obtained for NEXT-substrate 1 complex at MSK Richard Rifkind Center for Cryo-EM. A total of 4905 movies were collected for the first dataset which subsequently gave rise to 440,122 autopicked particles. Particles were imported to cryoSPARC (Punjani et al., 2017) to generate an ab initio model that was used as the initial reference model. After multiple rounds of 2D and 3D classifications in RELION 3.0 (Zivanov et al., 2018), 270,441 particles were selected for 3D refinement and subsequent Bayesian polishing. The same procedure was used for the second dataset (3231 movies, 520,310 autopicked particles) and a total of 347,971 were selected and combined with the selected particles from the first dataset. The combined particle stack (618,412 particles) yielded a post-processed overall map with a nominal resolution of 4.06 Å after several cycles of particle polishing and 3D refinement. All subsequent data processing steps including signal subtraction, focused refinement, and focused 3D classification were performed in RELION 3.0 (Zivanov et al., 2018). Signal subtraction using a mask encompassing the MTR4 stalk and helicase core domains of both protomers and focused 3D classifications using different regularization parameter (T) values were performed to segregate the two orientations of ZCCHC8 N-terminal coiled-coil domains. At T = 50, the two coiled-coil classes were separated. The larger class was reverted back to original particles and subjected to 3D classification with image alignment prior to a focused 3D refinement of the region encompassing ZCCHC8 HD/KID and MTR4 KOW domains of the two protomers (Z8HD/KID-MTR4KOW map in Figure S1). To improve the local resolution of the two protomers, 3D classification with image alignment was performed on the combined particle stack. A class of 242,861 particles that displayed improved density for both protomers was selected and subjected to signal subtraction and local masked 3D classification (T = 40 and T = 20 for protomers A and B, respectively). Classes with well-defined signal for MTR4 and RBM7 were refined using a local masks enclosing MTR4 from either protomers (protomer A MTR4 and protomer B MTR4 maps in Figure S1F) or pooled for another round of focused 3D classification (T = 80) on a region encompassing RBM7, ZCCHC8 PSP, and RNA prior to local refinement (protomer A MTR4KOW-Z8HD/KID-RNA and protomer B MTR4KOW-Z8HD/KID-RNA maps in Figure S1F). A composite map was generated in Phenix by combining the overall map and the focused refinement maps of Z8HD/KID-MTR4KOW CC-1, MTR4 (A and B), and MTR4KOW-Z8HD/KID-RNA (A and B) regions.
NEXT-substrate 2 complex
Three datasets were obtained for the NEXT-substrate 2 complex. Dataset 1 was collected at NYSBC Simons Electron Microscopy Center while datasets 2 and 2 were collected at MSK Richard Rifkind Center for Cryo-EM. Movies collected in super-resolution mode were aligned and Fourier cropped to 1.1 Å prior to subsequent analysis. A total of 18,294 movies were collected that was pruned to 13,842 movies after manual inspection for ice contamination and exclusion of movies with an estimated resolution above 4.5 Å. 822,892 autopicked particles were subjected to 2D classification and 3D classification with image alignment to remove junk particles. Classes containing two NEXT homodimers cross-braced by RNA were selected and combined to a total of 368,334 particles. Several cycles of 3D refinement and Bayesian polishing were performed prior to another round of 3D classification with image alignment to identify a NEXT tetramer class (78,308 particles). At this stage, the particles were processed using RELION 3.1 (Zivanov et al., 2018). After 3D refinement, particles were subjected to signal subtraction using masks enclosing each protomer followed by recentering and global 3D refinement. This resulted in particles with isolated signal from each protomer, which were combined to a total 313,232 particles. Masked 3D refinement of the combined particle stack produced a map with a nominal resolution of 3.85 Å after post-processing. Local refinement was performed in a region encompassing MTR4 helicase core, ZCCHC8 PSP domain, RBM7, and RNA prior to 3D classification with no image alignment (T = 60). A class (252,638 particles) with improved density for RNA and RBM7 was identified that yielded a post-processed overall map with a resolution of 3.62 Å. Focused 3D refinement were subsequently performed using local masks that encompassed MTR4 core, MTR4 core-ZCCHC8 PSP-RBM7-RNA, RBM7-RNA, MTR4-ZCCHC8 HD/KID, and MTR4 KOW-ZCCHC8 HD/KID regions. These focused refinement maps and the overall map were post-processed and combined to generate a composite map in Phenix (Adams et al., 2010).
Apo-NEXT
A total of 6705 movies were collected for apo-NEXT that afforded 465,038 autopicked particles. The best 2D class averages from multiple reference-free 2D classification steps were subjected to two rounds of 3D classification with image alignment. Homogenous 3D refinement, Bayesian polishing, and per particle defocus refinement were performed using 52,777 particles from the selected 3D classes. A consensus map with an overall resolution of 8.36 Å was obtained after post-processing (Figure S3D).
Model building and refinement
The atomic model of human MTR4 and ZCCHC8 CTD was docked into densities and manually rebuilt in Coot (Emsley, 2010) using a prior crystal structure of human MTR4 helicase core bound to ZCCHC8 CTD (Puno and Lima, 2018) and a model of the arch domain based on homology to S. cerevisiae Mtr4 (Kelley et al., 2015). RBM7 RRM and ZCCHC8 PSP domains were docked into densities and manually rebuilt using the X-ray structure of a RBM7 RRM-ZCCHC8 PSP complex. ZCCHC8 HD and KID domains were built using a focused refinement map of the ZCCHC8HD/KID-MTR4KOW region. Other unknown regions of ZCCHC8 and RNA were initially built into EM densities of protomer A (NEXT-substrate 1 complex) after manual inspection using Coot (Emsley et al., 2010) and refined using PHENIX (Adams et al., 2010). Model from protomer A (NEXT-substrate 1 complex) was used to rebuild into densities encompassing protomer B (NEXT-substrate 1 complex) as well as the NEXT-substrate 2 complex. Model geometry was analyzed using Molprobity (Chen et al., 2010). Local resolution maps were generated using Phenix (Adams et al., 2010). Structures and maps were rendered using ChimeraX (Pettersen et al., 2021) or Pymol (Schrödinger, LLC).
Immunoprecipitation
Expi293F cells (5 x 107) were lysed using 2 ml of ice-cold RIPA buffer supplemented with 250 units/ml TurboNuclease (Accelagen), 2.5 mM MgCl2, and EDTA-free SigmaFAST protease inhibitor cocktail (Millipore Sigma) then clarified by centrifugation at 18,000 x g for 10 minutes at 4°C. Immunoprecipitation of GFP-tagged proteins was performed using a magnetic GFP-Trap kit (Chromotek). A 500 μl aliquot of lysate was diluted 3-fold with IP wash buffer (20 mM Tris·HCl pH 8.0, 100 mM NaCl, and 0.1 mM TCEP), transferred into a 2 ml microcentrofuge tube, and incubated with 50 μl of GFP-Trap slurry in a Thermo Scientific tube revolver/rotator spinning at 10 rpm at 4°C for 20 min. Magnetic beads were collected using a DynaMag-2 and washed with IP wash buffer 3 times. Immunoprecipitated proteins were eluted using 1X LDS sample buffer supplemented with 1% (v/v) BME and detected by a Western blot analysis.
Western blot analysis
Samples in 1X LDS sample buffer supplemented with 1-5% (v/v) BME were electrophoresed in NuPAGE 4-12% Bis-Tris polyacrylamide gel using 1X MES running buffer and blotted onto 0.2 μm PVDF membrane using a semi-dry Trans-Blot Turbo transfer system (Bio-Rad) at a constant current of 24 mA for 8-10 minutes. Western detection was performed using the iBind Flex Western apparatus (Thermo Fisher Scientific) or WesternEaze-Chemi kit (Advansta). Alternatively, immunodetection was performed as follows: After transfer, the membrane was blocked using AdvanBlock-Chemi blocking solution (Advansta) for 1 hour at room temperature and incubated with primary antibodies diluted using AdvanBlock-Chemi blocking solution overnight at 4°C. After two washes with 1X Tris-buffered saline with 0.05% (v/v) Tween (TBS-T) (Thermo Fisher Scientific), the membrane was incubated with secondary antibody diluted in AdvanBlock-Chemi blocking solution for 1 hour at 22°C and then washed again 4 times with TBS-T for 15 min each wash at 22°C. Finally, SuperSignal West Dura Extended Duration substrate (Thermo Fisher Scientific) was added to the membrane and chemiluminescence was visualized using Bio-Rad ChemiDoc XRS+. The primary antibodies used were mouse anti-ZCCHC8 (1:500, Abcam), mouse anti-GFP (1:1000, Thermo Fisher Scientific), mouse anti-mCherry (1:1000, Thermo Fisher Scientific), mouse anti-beta-actin (1:2000, Thermo Fisher Scientific), and mouse anti-ROS1 (1:1000, Origene). Sheep HRP-linked anti-mouse IgG (1:20,000 for traditional Western antibody staining or 1:4000 for iBind Western analysis, GE Healthcare) was used as secondary antibody.
Confocal imaging
GFP-ZCCHC8, GFP-ZCCHC8ΔHD, and GFP-ZCCHC8ΔZK HAP1 cells were grown in a 4-well chambered slide with a glass bottom (ibidi) and treated with 15 ng/ml doxycycline for 24 hours to induce expression of GFP-tagged proteins. An hour before imaging, the medium was supplemented with 1X NucSpot Live 650 dye (Biotium) and 100 μM verapamil (Biotium) to stain the cell nuclei. The slide was transferred to a Leica Inverted Confocal SP8 (Leica Microsystems) equipped with an environment chamber at 37°C and 5% CO2. Live cell imaging was performed with an HC PL APO CS2 63x/1.4 objective in fluorescence mode using 479 nm and 670 nm as excitation wavelengths. Confocal images were rendered using Fiji ImageJ 2.1.0. (Schneider et al., 2012)
qPCR
Total RNA was extracted from pelleted cells using RNeasy plus mini kit (Qiagen) and used to generate a cDNA library using a Superscript IV VILO master mix with ezDNAse kit (Thermo Fisher Scientific). qPCR samples were prepared using PowerUp SYBR Green master mix (Thermo Fisher Scientific) with 0.3 μM each of forward and reverse primers. Reactions were performed with an annealing temperature of 59°C and analyzed using QuantStudio 6 Pro Fast mode (Thermo Fisher Scientific). Forward and reverse primer sequences for GAPDH, proPOGZ, and proEXTI were obtained from Blasius et al., 2014 while sequences for proKLF6, proRBM39, proSERIN3, and proTTC32 were obtained from Wu et al., 2020 (see Table S2 for primer sequences).
RNA extraction and total RNA sequencing
Cell pellets were lysed in buffer supplemented with 50% (v/v) isopropanol and 0.5% (v/v) 2-mercaptoethanol and RNA was extracted from the lysate using the MagMAX mirVana Total RNA Isolation Kit (Thermo Fisher Scientific) on the KingFisher Flex Magnetic Particle Processor (Thermo Scientific catalog # 5400630) according to instructions provided by the manufacturer with 10 million cells input. The purified RNA was quantified using Ribogreen assay kit (Thermo Fisher Scientific) and assessed for quality using Agilent BioAnalyzer. One μg of total RNA with an RNA integrity number varying from 9.3-10 underwent ribosomal depletion and library preparation using the TruSeq Stranded Total RNA LT Kit (Illumina) according to the manufacturer’s protocol with 8 cycles of PCR. Samples were barcoded and run on a NovaSeq 6000 in a PE100 run, using the NovaSeq 6000 S2 Reagent Kit (200 Cycles) (Illumina). An average of 56 million paired reads were generated per sample and 63% of the data mapped to the transcriptome.
Phos-tag gel analysis of ROS1 autophosphorylation
HAP1 cells (4.5 × 106) stably transfected with doxycycline-inducible GFP fusion constructs of ZCCHC81-80-ROS1kinase, ZCCHC81-80-ROS1kinase(dead) or ZCCHC81-80dm-ROS1kinase were grown in 150 mm dish overnight and then treated with 8, 16, 8 ng/ml doxycycline for 24 hours, respectively. Cells were trypsinized using TrypLE (Thermo Fisher Scientific, washed with PBS, and resuspended in FACS buffer (PBS supplemented with 2% (v/v) fetal calf serum and 2 mM EDTA) to a final density of 20 × 106 cells per ml. Cells were passed through a Corning 70 μm cell stainer and sorted for cells expressing low GFP levels (Figure S9J) using BD FACSAria at MSK Flow Cytometry core facility. Sorted cells were collected in IMDM with 20% (v/v) fetal calf serum, pelleted by centrifugation at 500 × g and resuspended in lysis solution containing 1X Laemlli buffer (Bio-Rad) supplemented with 250 units per ml Turbonuclease (Accelagen) and 5% (v/v) ß-mercaptoethanol. 1 ml lysis solution was used per 20 million cells. 10 μl of cell lysate was loaded into each well of SuperSep Phos-tag (50 μmol/L) 100 mm × 100 mm × 8.6 mm pre-cast 12.5% gel (Wako) and electrophoresed in cold 1X Tris/Glycine/SDS running buffer (Bio-Rad) at 200 V for 2 h at 4 °C. Gels were washed three times with 30 ml 1X NuPage transfer buffer (Thermo Fisher Scientific) supplemented with 10% (v/v) methanol and 10 mM EDTA for 20 min each. Gels were subsequently washed three times with 30 ml 1X NuPage (Thermo Fisher Scientific) with 10% (v/v) methanol for 10 min each. Proteins were transferred onto 0.2 μm PVDF membrane (Bio-Rad Transfer Pack) using Trans-Blot Turbo semi-dry transfer apparatus (1.3 A, 25 V setting for 8 min) (Bio-Rad). Membranes were probed for ROS1 using Western blot analysis.
Flow cytometry analysis of ROS1 autophosphorylation
HAP1 cells stably transfected with doxycycline-inducible GFP-ZCCHC81-80-ROS1kinase, GFP-ZCCHC81-80-ROS1kinase(dead) or GFP-ROS1kinase were grown in T-75 flasks and treated with 8, 16, 8 ng/ml doxycycline for 24 hours, respectively. Cells were trypsinized, stained with LIVE/DEAD Fixable Violet Dead Cell Stain kit (Thermo Fisher Scientific), washed with PBS and fixed with 3.2% (v/v) paraformaldehyde in PBS supplemented with 5 mM EDTA for 20 min. After two washes with PBS, cells were resuspended in 0.5 ml PBS and permeabilized with 4.5 ml of ice-cold methanol for 20 min and washed two more times with PBS. Cells were transferred into 5-ml polypropylene tubes, blocked with staining solution (PBS with 5% fetal calf serum, 5 mM EDTA, 200 μg/ml IgG from human serum (Sigma)) and incubated for 1 hour in staining solution with 1 μg/ml anti-phospho(Y2274)-ROS1 (pROS1) antibody (Thermo Fisher Scientific) labeled with AlexaFluor 647 (Zip rapid antibody labeling kit – Thermo Fisher Scientific). Cells were washed three times with PBS, resuspended in staining solution and analyzed for GFP and AlexaFluor 647 fluorescence using a Fortessa (BD Biosciences) instrument operated using the BD FACSDiva software. Gates and regions used for relative cellular pROS1 quantitation are shown in Figure S9J. Compensated data were processed and rendered using FCS Express 7 Research.
Bioinformatics analysis
Bioinformatics analysis was performed using MSK Bioinformatics core facility services. The output data (FASTQ files) were mapped to human genome (hg19) using the 2-pass mapping method (Engström et al., 2013) with STAR (Dobin et al., 2013) with Gencode v18 annotations. Output SAM files were post-processed using PICARD tool AddOrReplaceReadGroups to add read groups, sort files and convert them to compressed BAM format. The expression count matrix for each feature (exon, PROMPT, eRNA) was computed from the mapped reads using HTSeq v0.5.3 (Anders et al., 2015) with a custom GTF that included Gencode annotations, PROMPT regions from Wu et al. (2020) and eRNA regions from (Andersson et al., 2014). Genome coverage normalized to reads per million was computed for each strand using STAR (Dobin et al., 2013) in run mode inputAlignmentsFromBAM and output in bedGraph format. Coverage was averaged across replicates in each sample group. Normalized coverage tracks were rendered and compared between samples using Integrative Genomics Viewer (Robinson et al., 2011).
PROMPT regions were converted from GRCh38 to hg19 using UCSC liftOver 9 (Hinrichs et al., 2006). Custom R script was used to find all Gencode annotated protein coding and lincRNA transcription start sites (TSS) within 3kb downstream of any PROMPT. If multiple PROMPTs fell within the 3kb downstream region of a single TSS, only the closest one was kept for analysis. The region 3kb upstream and 3kb downstream of these PROMPT-proximal TSSs were saved in BED format. Coverage for these 6kb regions was extracted from bedGraph files of each sample group described above. Finally, Gencode annotated exon regions were converted to BED format and removed from TSS window coverage files using bedtools subtract. Figures were generated using R showing total coverage of all PROMPTs on reverse strand, colored by sample group.
TSSs of eRNA regions were identified as the midpoint between their start and end coordinates and were filtered for those at least 2kb upstream or downstream of any Gencode annotated gene. As previously described, 6kb regions around each TSS were saved in BED format and coverage for these regions was extracted from bedGraph files of each sample group using bedtools intersect. Exon regions were removed using bedtools subtract. Figures were generated using R showing total coverage around eRNA TSSs, separated by transcription direction and colored by sample group.
snRNA transcripts were filtered from Gencode annotations and regions extending from 1kb upstream to 1kb downstream of their 3' ends were saved in BED format. Coverage for these 2kb regions were extracted from bedGraph files for each sample group using bedtools intersect. Figure was generated using R to show total coverage of all snRNA EAGs, colored by sample group.
QUANTITATION AND STATISTICAL ANALYSIS
Biochemical assays related to Figure 5A-L were performed using at least three technical replicates. EMSA (Figure 5A, 5E, and 5G) and strand displacement kinetic data (Figure 5C, 5I, and 5K) were plotted as mean ± standard deviation using GraphPad Prism. Biological experiments related to Figures 5M-P were performed in three biological replicates while Figures 6F-G in five biological replicates. PROMPT RNA levels from the qPCR assay (Figure 5M) were normalized relative to RNA levels in parental HAP1 cells. Relative pROS1 levels in Figure 6G were obtained by taking the Alexa Fluor 647 intensity median of each cell population and normalizing against the median of cells expressing GFP-ROS1. The mean and individual normalized data points were plotted along with the standard deviations represented by error bars in the figures. Statistical significance was determined using unpaired two-tailed t test in GraphPad Prism 9. P values are indicated by *p < 0.05, **p <0.01, ***p <0.001; ****p < 0.0001.
Supplementary Material
Figure S1. NEXT-substrate 1 complex preparation, cryo-EM data collection and processing, Related to Figure 1 and STAR Methods.
(A) RNA substrate 1 schematic diagram.
(B) Coomassie-stained denaturing polyacrylamide gel electrophoresis (PAGE) showing eluted fractions from gel filtration chromatography of reconstituted NEXT-substrate 1 complex. The co-purified RNA was separated using 20% TBE PAGE and visualized by SYBR Gold staining.
(C) Schematic diagram of gel-based strand displacement assay (left panel) and native PAGE gel showing time courses of strand displacement assays of NEXT using a 3′ A20 tailed RNA substrate with or without an internal 2′ pyrene-modified uridine (right panel).
(D) Sample micrograph of NEXT-substrate 1 complex. Scale bar represents 75 μm.
(E) Representative 2D class averages of NEXT-substrate 1
(F) Schematic diagram showing the single-particle analysis used to obtain the overall and composite reconstructions related to Figure 1, S2 and S3A.
Figure S2. Quality of cryo-EM reconstructions of NEXT-substrate 1 complex, Related to Figure 1 and S1.
(A) Overall and composite maps of NEXT-substrate 1 complex.
(B-C) Local resolution estimates (B) and angular distribution plots (C) for the overall reconstruction.
(D) Fourier Shell Correlation (FSC) between half maps obtained from indicated reconstructions.
(E) Local resolution estimates (upper panel) and angular distribution plots (lower panel) of focused refinement maps.
(F) Magnified view of protomer A and B RBM7-RNA regions showing local resolution maps with respective models overlaid.
(G) FSC curve between the final model and composite map.
Figure S3. NEXT-substrate 1 EM densities, Related to Figure 1 and S2, and cryo-EM data and single particle analysis of apo NEXT.
(A) Representative EM densities (gray mesh) of various regions of the NEXT-substrate 1 composite map with the respective model.
(B-D) Sample micrograph (B), representative 2D class averages (C), and single particle analysis
(D) of apo NEXT. Scale bar represents 75 μm.
Figure S5. NEXT-substrate 2 complex preparation, cryo-EM data collection and processing, Related to Figure 4 and STAR Methods.
(A) RNA substrate 2 schematic after hybridization.
(B) Coomassie-stained denaturing PAGE showing the protein profiles of peak fractions obtained from gel filtration chromatography of reconstituted NEXT-substrate 2 complex. Bound RNA was separated using 20% TBE PAGE and visualized by SYBR Gold staining.
(C-D) Representative micrograph (C) and 2D class averages (D) of NEXT-substrate 2 complex. Scale bar represents 75 μm.
(E) Single particle analysis workflow for reconstructions related to Figure 4.
Figure S6. Quality of cryo-EM reconstructions of NEXT-substrate 2 complex, Related to Figure 4 and S5.
(A) Consensus and composite maps of NEXT-substrate 2 complex.
(B-C) Local resolution estimates (B) and angular distribution plots (C) for the overall reconstruction.
(D) Fourier Shell Correlation (FSC) between half maps of overall and focused reconstructions.
(E) Local resolution estimates of focused refinement maps.
(F) Representative EM densities (gray mesh) of various regions of the composite reconstruction overlaid with the respective model.
(G) FSC curve between the final model and composite map.
Figure S7. Immunoprecipitation, gel filtration, and helicase assays, Related to Figure 5 and STAR Methods.
(A) Representative Western blots from three immunoprecipitation experiments analyzing ZCCHC8 dimerization in Expi293F cells expressing mCherry-ZCCHC8 (prey) and GFP-tagged wild-type and mutant ZCCHC8 constructs (bait).
(B) Superdex 200 increase 10/300 GL chromatogram of reconstituted human NEXT core complex.
(C) Calibration curve used to estimate the apparent molecular weight of NEXT core complex.
(D) Velocity plots used to calculate vo in Figure 5C with data (mean ± SD) from three separate reactions and the fitted linear curve shown.
(E) Velocity plots used to calculate vo in Figure 5I with data (mean ± SD) from three separate reactions and the fitted linear curve shown.
(F) Velocity plots used to calculate vo in Figure 5K with data (mean ± SD) from three separate reactions and the fitted linear curve shown.
(G) Bar graphs of K1/2 (mean ± SEM) from left to right obtained from kinetic plots in Figures 5C, 5I, and 5K, respectively.
Figure S8. Western blot, confocal imaging, FACS, and RNA seq analysis, Related to Figure 5 and STAR Methods.
(A) Anti-ZCCHC8 blot of HAP1 parental line, ZCCHC8 CRISPR knockout HAP1 cell line (HAP1Z8KO), and isolated GFP-ZCCHC8, GFP-ZCCHC8ΔHD, and GFP-ZCCHC8ΔZK stable HAP1 clones. Anti-β-actin was used as loading control.
(B) Live cell confocal analysis of GFP-ZCCHC8, GFP-ZCCHC8ΔHD and GFP-ZCCHC8ΔZK stable HAP1 clones. Scale bar represents 75 μm. Cells were treated with doxycycline for 24 hours prior to imaging and the nuclei were stained with NucSpot Live 650 dye an hour before analysis.
(C) FACS profile of GFP-ZCCHC8, GFP-ZCCHC8ΔHD, and GFP-ZCCHC8ΔZK cells used in qPCR analysis of PROMPTS.
(D) Integrative Genomics Viewer coverage snapshots of select PROMPTS, eRNAs, and snRNA 3′ readthrough regions.
(E) Magnified views of metagene plots in Figure 5N-P with average densities (solid lines) and confidence interval to the mean (shaded areas) shown.
Figure S9. CRISPR-Cas9 HD deletion, qPCR, Western blot, immunoprecipitation, and flow cytometry analysis, Related to Figure 5, 6 and STAR Methods.
(A) CRISPR-Cas9 strategy used to generate ZCCHC8 ΔHD HAP1 cells.
(B) Representative Sanger sequencing chromatogram of CRISPR-Cas9 edited ZCCHC8 ΔHD HAP1 clone.
(C) SYBR safe-stained agarose gels showing PCR analysis of ZCCHC8 transcript levels in ZCCHC8 ΔHD HAP1 clones and parental HAP1. GAPDH was used as endogenous control.
(D) Bar graph showing qPCR analysis of ZCCHC8 transcript levels (mean ± SEM from three technical replicates) in ZCCHC8 ΔHD HAP1 clones and parental HAP1.
(E) Anti-ZCCHC8 immunoblots of parental HAP1, HAP1 ZCCHC8 KO (HAP1 Z8 KO) and HAP1 ZCCHC8 ΔHD cell lysates. Anti-β-actin was used as loading control.
(F) Positions of dm mutations (C56E/L63E/L70E) in ZCCHC81-80 dimer interface.
(G) Representative immunoblots from three immunoprecipitation experiments analyzing effects of C56E/L63E/L70E triple mutations in ZCCHC81-80dm-ROS1kinase dimerization in Expi293F cells expressing mCherry-Z81-80-ROS1kinase (prey) and GFP-tagged constructs (GFP-Z81-80-ROS1kinase, and GFP-Z81-80dm-ROS1kinase) as bait.
(H) Representative immunoblots from three immunoprecipitation experiments analyzing ZCCHC81-80-ROS1kinase heterodimerization with full-length ZCCHC8 in Expi293F cells expressing mCherry-ZCCHC8 (prey) and GFP-tagged constructs (GFP-Z81-80-ROS1kinase, GFP-ROS1kinase, QFP-ZCCHC8) as bait.
(I) Anti-ROS1 blot of HAP1 cells stably transfected with doxycycline-inducible GFP-Z81-80-ROS1kinase, GFP-Z81-80- ROS1kinase(dead), GFP-ROS1kinase and GFP-Z81-80dm-ROS1kinase.
(J) Gating workflow used to analyze pROS1 levels using Phos-tag gel and flow cytometry in Figures 6E-G.
Table S1. EM data and refinement, Related to Figures 1, 4, S1-S6, and STAR Methods.
Table S2. Sequences of primers used for qPCR analysis, Related to Figure 5 and STAR Methods
Figure S4. Structural comparisons of MTR4 helicase core with different nucleotides bound and MTR4 binding motifs of ZCCHC8 and various exosome co-factors, Related to Figure 1, 2, 3 and 4.
(A-D) Superposition of the helicase core of NEXT MTR4 protomer A with the core domains of MTR4 in protomer B (A), ATP-bound MTR4 (PDB ID 6IEG) (B), ADP-bound MTR4-ZCCHC8CTD complex (PDB ID 6C90) (C), and AMPPNP-bound MTR4-exosome complex with a translocating RNA (PDB ID 6D6R) (D). Nucleotide and RNA are shown as stick models.
(E) Magnified views of AIM sequences from ZCCHC8, NVL (PDB ID 6RO1), and NRDE2 (PDB ID 6IEH) and their interaction with MTR4 (left to right).
(F) Overview of MTR4 contacts of ZCCHC8, S. cerevisiae TRAMP subunits Trf4 and Air2 (PDB ID 4U4C), and human NRDE2 (PDB ID 6IEH) (top to bottom).
(G) Structural comparison of NRDE2-MTR4 and NEXT complexes.
(H) Cut-through magnified view of the boxed region in (G) showing RNA within MTR4 helicase core.
(I) Structural comparison of ZCCHC8 MA1 with equivalent RecA2 binding motifs of Air2 and NRDE2.
(J) Structural comparison of ZCCHC8 MA2 with homologous RecA2 binding motifs of Trf4 and NRDE2.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-ROS1 (phospho-Y2274) | Thermo Fisher Scientific | Cat#PA5-64608; RRID:AB_2663812 |
| Mouse monoclonal anti-ROS1 | Origene | Cat#TA805734; RRID:AB_2627841 |
| Mouse polyclonal anti-ZCCHC8 | Abcam | Cat#ab68739; RRID:AB_1271512 |
| Mouse monoclonal anti-GFP | Thermo Fisher Scientific | Cat#MA5-15256; RRID:AB_10979281 |
| Mouse monoclonal Anti-mCherry | Thermo Fisher Scientific | Cat#MA5-32977 |
| Mouse monoclonal anti-beta-actin | Thermo Fisher Scientific | Cat#MA5-15739; RRID:AB_10979409 |
| Sheep monoclonal anti-mouse IgG | GE Healthcare | Cat#NA931V; RRID:AB_772210 |
| Bacterial and virus strains | ||
| E. coli BL21CodonPlus(DE3)-RIL competent cells | Agilent Technologies | Cat#230245 |
| E. coli E. cloni 10G competent cells | Lucigen | Cat#60106-2 |
| Chemicals, peptides, and recombinant proteins | ||
| AdvanBlock-Chemi blocking solution | Advansta | Cat#R-03726-E10 |
| Adenosine 5′(β,γ-imido) triphosphate lithium salt hydrate | Sigma | Cat#A2647 |
| GFP-Trap magnetic agarose | Chromotek | Cat#gtma-20; RRID:AB_2631358 |
| IGEPAL CA-630 | Sigma | Cat#I8896 |
| Iscove’s Modified Dulbecco’s medium | Thermo Fisher Scientific | Cat#12440053 |
| Laemmli Sample buffer (4X) | Bio-Rad | Cat#1610747 |
| Magnesium chloride hexahydrate | Sigma | Cat#M9272 |
| Tris(2-carboxyethyl)phosphine (TCEP) | Soltec Ventures | Cat#M115 |
| RIPA | Thermo Fisher Scientific | Cat#89901 |
| RNase inhibitor, human placenta | New Engaland Biolabs | Cat#M0307S |
| Proteinase K | New England Biolabs | Cat#P8107S |
| Superdex 200 Increase 10/300 GL | GE Healthcare | Cat#28990944 |
| SuperSep Phos-tag (50 μmol/L), 12.5%, 17-well | FUJIFILM Wako | Cat#195-17991 |
| SuperSignal West Dura Extended Duration substrate | Thermo Fisher Scientific | Cat#34075 |
| TBE (10X) | Thermo Fisher Scientific | Cat#AM9864 |
| NU7741 | StemCell Technologies | Cat#74082 |
| NuPAGE MES SDS Running Buffer (20X) | Thermo Fisher Scientific | Cat#NP0002 |
| NuPAGE LDS Sample Buffer (4X) | Thermo Fisher Scientific | Cat#NP0007 |
| Novex TBE gel, 20% | Thermo Fisher Scientific | Cat#EC63152BOX |
| Novex TBE gel, 4-40% | Thermo Fisher Scientific | Cat#EC62252BOX |
| NuPage 4-12% Bis-Tris protein gels | Thermo Fisher Scientific | Cat#NP0322BOX |
| NuPage Transfer Buffer (20X) | Thermo Fisher Scientific | Cat#NP006 |
| Ni-NTA agarose | Qiagen | Cat#30250 |
| HiTrap Heparin HP | GE Healthcare | Cat#17040601 |
| HiLoad 26/600 Superdex 200 pg | GE Healthcare | Cat#289289336 |
| Ulp1 protease | In-house | N/A |
| Critical commercial assays | ||
| Expi293 Expression System Kit | Thermo Fisher Scientific | Cat#A14635 |
| Gel filtration calibration kit high molecular weight | GE Healthcare | Cat#28403842 |
| iBind Flex solution kit | Thermo Fisher Scientific | Cat#SLF2020 |
| Lipofectamine 3000 transfection reagents | Thermo Fisher Scientific | Cat#L3000001 |
| LIVE/DEAD Fixable Violet Dead Cell Stain Kit | Thermo Fisher Scientific | Cat#L34955 |
| NucSpot Live 650 kit | Biotium | Cat#40082 |
| RNeasy Pus Mini Kit | Qiagen | Cat#74034 |
| Superscript IV VILO master mix | Thermo Fisher Scientific | Cat#11756050 |
| TruSeq Standard Total RNA LT Kit | Illumina | Cat#RS-122-1202 |
| WesternEaze-Chemi kit | Advansta | Cat#K-12054-010 |
| Zip Alexa Fluor 647 Rapid Antibody Labeling Kit | Thermo Fisher Scientific | Cat#Z11235 |
| Deposited data | ||
| NEXT-RNA substrate 1 complex structure | This paper | PDB: 7S7B |
| NEXT-RNA substrate 1 complex EM maps | This paper | EMDB: EMD-24882 |
| NEXT-RNA substrate 2 complex structure | This paper | PDB: 7S7C |
| NEXT-RNA substrate 2 complex EM maps | This paper | EMDB: EMD-24883 |
| Apo NEXT EM map | This paper | EMDB: EMD-24884 |
| RNA-seq data | This paper | GEO: GSE185374 Token: ytspiamilduxlsx |
| Experimental models: Cell lines | ||
| Human: Expi293F cells | Thermo Fisher Scientific | Cat#A14635 |
| Human: HAP1 | Horizon Discovery | Cat#C631 |
| Human: HAP1 ZCCHC8 knockout | Horizon Discovery | Cat#HZGHC005158c012 |
| Human: Expi293F stably transfected with PB-rtTA-Puro, PB-GFP-ZCCHC8, and PB-mCherry-ZCCHC8 | This work | N/A |
| Human: Expi293F stably transfected with PB-rtTA-Puro, PB-GFP-ZCCHC8ΔHD, and PB-mCherry-ZCCHC8 | This work | N/A |
| Human: Expi293F stably transfected with PB-rtTA-Puro, PB-GFP-ZCCHC81-80-ROS1kinase, and PB-mCherry-ZCCHC81-80-ROS1kinase | This work | N/A |
| Human: Expi293F stably transfected with PB-rtTA-Puro, PB-GFP-ZCCHC81-80dm-ROS1kinase, and PB-mCherry-ZCCHC81-80-ROS1kinase | This work | N/A |
| Human: Expi293F stably transfected with PB-rtTA-Puro, PB-GFP-ROS1kinase, and PB-mCherry-ROS1kinase | This work | N/A |
| Human: Expi293F stably transfected with PB-rtTA-Puro, PB-GFP- ZCCHC81-80-ROS1kinase, and PB-mCherry-ZCCHC8 | This work | N/A |
| Human: Expi293F stably transfected with PB-rtTA-Puro, PB-GFP- ROS1kinase, and PB-mCherry-ZCCHC8 | This work | N/A |
| Human: HAP1 ZCCHC8 CRISPR knockout stably transfected with PB-rtTA-Puro and PB-GFP-ZCCHC8 | This work | N/A |
| Human: HAP1 ZCCHC8 CRISPR knockout stably transfected with PB-rtTA-Puro and PB-GFP-ZCCHC8ΔHD | This work | N/A |
| Human: HAP1 ZCCHC8 CRISPR knockout stably transfected with PB-rtTA-Puro and PB-GFP-ZCCHC8ΔZK | This work | N/A |
| Human: HAP1 ZCCHC8 stably transfected with PB-rtTA-Puro and PB-GFP-ZCCHC81-80-ROS1kinase | This work | N/A |
| Human: HAP1 ZCCHC8 stably transfected with PB-rtTA-Puro and PB-GFP-ZCCHC81-80dm-ROS1kinase | This wok | N/A |
| Human: HAP1 stably transfected with PB-rtTA-Puro and PB-GFP-ZCCHC81-80-ROS1kinase(dead) | This work | N/A |
| Human: HAP1 stably transfected with PB-rtTA-Puro and PB-GFP-ROS1kinase | This work | N/A |
| Oligonucleotides | ||
| 6-FAM-AGCACCGUAAAGACGC (gel-based assay RNA top strand) | IDT | N/A |
| GCGUCUUUACGGUGCUAAAAAAAAAAAAAAAAAAAA (gel-based assay bottom strand for duplex with 3′ 20-nt poly(A) overhang) | IDT | N/A |
| GCGUCUUUACGGUGCUUpyAAAAAAAAAAAAAAAAA (gel-based assay bottom strand for duplex with 2′ pyrene modified uridine Upy) | Dharmacon | N/A |
| ACAUGAGGAUCACCCAUGUAAUCUCUUUCAAAAAAUpyACAAAAAAAA (cryo-EM substrate 1, Upy= 2′ pyrene modified uridine) | Dharmacon | N/A |
| GGCGCGCGCCAAAAAUUUUUAAAAAAAAAA (cryo-EM substrate 2) | Dharmacon | N/A |
| 6-FAM-AGUGCGCUGUAUCUUCAAGGCCACU (EMSA RNA top strand) | IDT | N/A |
| Iowa Black RQ-AGUGCGCUGUAUCUUCAAGGCCACU-Cy5 (molecular beacon helicase assay RNA top strand) | IDT | N/A |
| AGUGGCCUUGAAGAUACAGCGCACUAAAAAAAAAAAAAAAAAAAA (molecular beacon assay RNA bottom strand with 3′ 20-nt poly(A) overhang) | IDT | N/A |
| AGUGGCCUUGAAGAUACAGCGCACUAAAAAUUUUUAAAAAAAAAA (molecular beacon assay RNA bottom strand with 3′ A5U5A10 overhang) | IDT | N/A |
| qPCR and CRISPR screening primers | See Table S2 | N/A |
| Recombinant DNA | ||
| pET-28a-His10-Smt3-RBM7core | Puno and Lima, 2018 | N/A |
| pET-28a- His10-Smt3-MTR4 | Puno and Lima, 2018 | N/A |
| pRSF-Duet1-Smt3-ZCCHC8core | Puno and Lima, 2018 | N/A |
| pRSF-Duet1-Smt3-ZCCHC8coreΔHD | This work | N/A |
| pRSF-Duet1-Smt3-ZCCHC8coreΔZK | This work | N/A |
| pRSF-HiS6-ncSmt3-MTR4 | This work | N/A |
| pET-28a- His10-Smt3-Strep-MTR4 | This work | N/A |
| pET-28a- His10-Smt3-Strep-MTR4-E253Q | This work | N/A |
| pRP[Exp]-mCherry-CAG>hyPBase | VectorBuilder | Cat#VB160216-10057 |
| PB-TAG-ERPE | Addgene; Kim et al., 2015 | Cat#80479, RRID:Addgene_80479 |
| PB-rtTA-Puro | This work | N/A |
| PB-GFP-ZCCHC8 | This work | N/A |
| PB-GFP-ZCCHC8ΔNTD | This work | N/A |
| PB-mCherry-ZCCHC8 | This work | N/A |
| PB-GFP-ZCCHC8ΔZK | This work | N/A |
| PB-GFP-ZCCHC81-80-ROS1kinase | This work | N/A |
| PB-GFP-ZCCHC81-80-ROS1kinase(dead) | This work | N/A |
| PB-GFP-ZCCHC81-80dm-ROS1kinase | This work | N/A |
| PB-GFP-ROS1kinase | This work | N/A |
| pRP[2CRISPR]-mCherry-hCas9-U6>{hZCCHC8[gRNA#1]}-U6>{hZCCHC8[gRNA#2]}) | This work | Cat#: VB200528-1045ndr |
| Software and algorithms | ||
| BD FACSDiva software | BD Biosciences | http://www.bdbiosciences.com/instruments/software/facsdiva/index.jsp; RRID:SCR_001456 |
| Biorender | Biorender | http://biorender.com; #NI22WEQFBQ;#YI22XZN09U and #TM22XZN108 RRID:SCR_018361 |
| Coot | Emsley et al., 2010 | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/; RRID:SCR_014222 |
| FCS Express Research version 7 | De Novo software company | https://denovosoftware.com/; RRID:SCR_016431 |
| Gctf | Zhang, 2016 | https://www2.mrc-lmb.cam.ac.uk/research/locally-developed-software/zhang-software/#gctf; RRID:SCR_016500 |
| Integrative Genomics Viewer (IGV) | Robinson et al., 2011 | https://software.broadinstitute.org/software/igv/ |
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| Jalview version 2 | Waterhouse et al., 2009 | www.jalview.org/: RRID:SCR_006459 |
| Leginon data collection software | Suloway et al., 2005 | https://emg.nysbc.org/redmine/projects/leginon; RRID:SCR_016731 |
| MolProbity | Chen et al., 2010 | http://molprobity.biochem.duke.edu; RRID:SCR_014226 |
| Phenix suite | Adams et al., 2010 | https://www.phenix-online.org/; RRID:SCR_014224 |
| Prism version 9 | Graphpad | https://www.graphpad.com/scientific-software/prism/; RRID:SCR_002798 |
| Pymol version 2.4.1 | Schroedinger LLC | https://pymol.org; RRID:SCR_000305 |
| RELION | Scheres, 2012; Zivanov et al., 2018 | http://www2.mrc-lmb.cam.ac.uk/relion; RRID:SCR_016274 |
| Serial EM data collection software | Mastronarde, 2003 | http://bio3d.colorado.edu/SerialEM/; RRID:SCR_017293 |
| STAR | Dobin et al., 2013 | http://code.google.com/p/rna-star/; RRID:SCR_004463 |
| UCSF ChimeraX version 1.2.5 | Pettersen et al., 2021 | https://www.cgl.ucsf.edu/chimerax/; RRID:SCR_015872 |
| UCSF MotionCor2 | Zheng et al., 2017 | https://emcore.ucsf.edu/ucsf-software |
| Other | ||
| Quantifoil R 1.2/1.3 300 mesh gold grids | Electron Microscopy Services | Cat#Q43841 |
| Vitrobot Mark IV | FEI – Thermo Fisher | https://www.fei.com |
| Titan Krios | FEI – Thermo Fisher | https://www.fei.com |
| K2 Summit Camera | Gatan, Inc | www.gatan.com |
| K3 Camera | Gatan, Inc | www.gatan.com |
| Corning 3693 White Half-Area 96-well plate | Fisher Scientific | Cat#07-200-326 |
| Trans-blot Turbo | Bio-Rad | Cat#1704150 |
| iBind Flex Western device | Thermo Fisher Scientific | Cat#SLF2000 |
Highlights.
ZCCHC8 mediates NEXT homodimerization and anchors RBM7 to the helicase core of MTR4
RBM7 and ZCCHC8 provide specificity by binding RNA elements upstream of the 3′ end
ZCCHC8 blocks RNA extrusion path and MTR4 surfaces that bind MPP6 and the exosome
Oncogenic ZCCHC8-ROS1 fusion forms oligomers and increases kinase activity
Acknowledgements
We thank members of the Lima lab for advice. We thank Michael DiMattia, Laurent Cappadocia, and Jason De La Cruz of MSK Richard Rifkind Center for Cryo-EM for input at early stages of cryo-EM data collection and processing and Eva-Maria Weick for review of the manuscript. This research was supported in part by NIH National Institute of General Medical Sciences (NIGMS) Grants R35 GM118080 (to C.D.L.). We acknowledge use of the Integrated Genomics Operation Core, funded by the NCI Cancer Center Support Grant (CCSG, P30 CA008748), Cycle for Survival, and the Marie-Josée and Henry R. Kravis Center for Molecular Oncology. We acknowledge Nicholas Socci, Caitlin Jones and MSK Bioinformatics core for processing RNA sequencing data. Work was conducted in part at the Simons Electron Microscopy Center and National Resource for Automated Molecular Microscopy located at the New York Structural Biology Center, supported by grants from the Simons Foundation (349247), NYSTAR, and the NIH National Institute of General Medical Sciences (GM103310). The content is solely the responsibility of the authors and does not represent the official views of the National Institutes of Health. C.D.L. is an investigator of the Howard Hughes Medical Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare no competing financial interests.
References
- Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. (2010). PHENIX: a comprehensive python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D Biol. Crystallogr 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, and Huber W (2015). HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen PR, Domanski M, Kristiansen MS, Storvall H, Ntini E, Verheggen C, Schein A, Bunkenborg J, Poser I, Hallais M, et al. (2013). The human cap-binding complex is functionally connected to the nuclear RNA exosome. Nat. Struct. Mol. Biol 20, 1367–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson R, Gebhard C, Miguel-Escalada I, Hoof I, Bornholdt J, Boyd M, Chen Y, Zhao X, Schmidl C, Suzuki T, et al. (2014). An atlas of active enhancers across human cell types and tissues. Nature 2014 5077493 507, 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghose AK, Viswanadhan VN and Wendoloski JJ (1998). Prediction of Hydrophobic (Lipophilic) Properties of Small Organic Molecules Using Fragmental Methods: An Analysis of ALOGP and CLOGP Methods. J. Phys. Chem. A 102, 3762–3772. [Google Scholar]
- Bajczyk M, Lange H, Bielewicz D, Szewc L, Bhat SS, Dolata J, Kuhn L, Szweykowska-Kulinska Z, Gagliardi D, and Jarmolowski A (2020). SERRATE interacts with the nuclear exosome targeting (NEXT) complex to degrade primary miRNA precursors in Arabidopsis. Nucleic Acids Res. 48, 6839–6854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belon CA, and Frick DN (2008). Monitoring helicase activity with molecular beacons. Biotechniques 45, 433–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Boom J, and Meyer H (2018). VCP/p97-Mediated Unfolding as a Principle in Protein Homeostasis and Signaling. Mol. Cell 69, 182–194. [DOI] [PubMed] [Google Scholar]
- Blasius M, Wagner SA, Choudhary C, Bartek J, Jackson SP, 2014. A quantitative 14-3-3 interaction screen connects the nuclear exosome targeting complex to the DNA damage response. Genes Dev 28, 1977–1982. doi: 10.1101/gad.246272.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brahms J, Michelson AM, and Van Holde KE (1966). Adenylate oligomers in single- and double-strand conformation. J. Mol. Biol 15, 467–488. [DOI] [PubMed] [Google Scholar]
- Büttner K, Nehring S, and Hopfner K-P (2007). Structural basis for DNA duplex separation by a superfamily-2 helicase. Nat. Struct. Mol. Biol 14, 647–652. [DOI] [PubMed] [Google Scholar]
- Chen VB, Arendall WB, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC, and IUCr (2010). MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coccé MC, Mardin BR, Bens S, Stütz AM, Lubieniecki F, Vater I, Korbel JO, Siebert R, Alonso CN, and Gallego MS (2016). Identification of ZCCHC8 as fusion partner of ROS1 in a case of congenital glioblastoma multiforme with a t(6;12)(q21;q24.3). Genes, Chromosom. Cancer 55, 677–687. [DOI] [PubMed] [Google Scholar]
- Collins JW, Martin D, Core G and C.B., Wang S, and Yamada KM (2021). ZCCHC8 is required for the degradation of pervasive transcripts originating from multiple genomic regulatory features. BioRxiv 2021.01.29.428898. [Google Scholar]
- Della Corte CM, Viscardi G, Di Liello R, Fasano M, Martinelli E, Troiani T, Ciardiello F, and Morgillo F (2018). Role and targeting of anaplastic lymphoma kinase in cancer. Mol. Cancer 2018 171 17, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creelan BC (2018). Clinical Activity of Crizotinib in Lung Adenocarcinoma Harboring a Rare ZCCHC8-ROS1 Fusion. J. Thorac. Oncol 13, e148–e150. [DOI] [PubMed] [Google Scholar]
- Das M, Zattas D, Zinder JC, Wasmuth EV, Henri J, and Lima CD (2021). Substrate discrimination and quality control require each catalytic activity of TRAMP and the nuclear RNA exosome. Proc. Natl. Acad. Sci 118, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies KD, and Doebele RC (2013). Molecular Pathways: ROS1 Fusion Proteins in Cancer. Clin. Cancer Res 19, 4040–4045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Giammartino DC, Nishida K, and Manley JL (2011). Mechanisms and Consequences of Alternative Polyadenylation. Mol. Cell 43, 853–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doma MK, and Parker R (2007). RNA Quality Control in Eukaryotes. Cell 131, 660–668. [DOI] [PubMed] [Google Scholar]
- Drilon A, Jenkins C, Iyer S, Schoenfeld A, Keddy C, and Davare MA (2021). ROS1-dependent cancers — biology, diagnostics and therapeutics. Nat. Rev. Clin. Oncol 18, 35–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Z, and Lovly CM (2018). Mechanisms of receptor tyrosine kinase activation in cancer. Mol. Cancer 2018 171 17, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K, and IUCr (2010). Features and development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engström PG, Steijger T, Sipos B, Grant GR, Kahles A, Rätsch G, Goldman N, Hubbard TJ, Harrow J, Guigó R, et al. (2013). Systematic evaluation of spliced alignment programs for RNA-seq data. Nat. Methods 2013 1012 10, 1185–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairman-Williams ME, Guenther UP, and Jankowsky E (2010). SF1 and SF2 helicases: family matters. Curr. Opin. Struct. Biol 20, 313–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk S, Weir JR, Hentschel J, Reichelt P, Bonneau F, and Conti E (2014). The Molecular architecture of the TRAMP complex reveals the organization and interplay of its two catalytic activities. Mol. Cell 55, 856–867. [DOI] [PubMed] [Google Scholar]
- Falk S, Finogenova K, Melko M, Benda C, Lykke-Andersen S, Jensen TH, and Conti E (2016). Structure of the RBM7–ZCCHC8 core of the NEXT complex reveals connections to splicing factors. Nat. Commun 7, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gable DL, Gaysinskaya V, Atik CC, Conover Talbot C, Kang B, Stanley SE, Pugh EW, Amat-Codina N, Schenk KM, Arcasoy MO, et al. (2019). ZCCHC8, the nuclear exosome targeting component, is mutated in familial pulmonary fibrosis and is required for telomerase RNA maturation. Genes Dev. 33, 1381–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, and Yang W (2020). Different mechanisms for translocation by monomeric and hexameric helicases. Curr. Opin. Struct. Biol 61, 25–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlach P, Schuller JM, Bonneau F, Basquin J, Reichelt P, Falk S, and Conti E (2018). Distinct and evolutionary conserved structural features of the human nuclear exosome complex. Elife 7, e38686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giacometti S, Benbahouche NEH, Domanski M, Robert MC, Meola N, Lubas M, Bukenborg J, Andersen JS, Schulze WM, Verheggen C, et al. (2017). Mutually Exclusive CBC-Containing Complexes Contribute to RNA Fate. Cell Rep. 18, 2635–2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giunta M, Edvardson S, Xu Y, Schuelke M, Gomez-Duran A, Boczonadi V, Elpeleg O, Müller JS, and Horvath R (2016). Altered RNA metabolism due to a homozygous RBM7 mutation in a patient with spinal motor neuropathy. Hum. Mol. Genet 25, 2985–2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gockert M, Schmid M, Jakobsen L, Jens M, Andersen JS, and Heick Jensen T (2022). Rapid factor depletion highlights intricacies of nucleoplasmic RNA degradation. Nucleic Acids Res. 50, 1583–1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo TB, Boros LG, Chan KC, Hikim APS, Hudson AP, Swerdloff RS, Mitchell AP, and Salameh WA (2003). Spermatogenetic Expression of RNA-Binding Motif Protein 7, a Protein That Interacts With Splicing Factors. J. Androl 24, 204–214. [DOI] [PubMed] [Google Scholar]
- Hänzelmann P, and Schindelin H (2017). The interplay of cofactor interactions and post-translational modifications in the regulation of the AAA+ ATPase p97. Front. Mol. Biosci 4, 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinrichs AS, Karolchik D, Baertsch R, Barber GP, Bejerano G, Clawson H, Diekhans M, Furey TS, Harte RA, Hsu F, et al. (2006). The UCSC Genome Browser Database: update 2006. Nucleic Acids Res. 34, D590–D598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashizume H, and Imahori K (1967). Circular Dichroism and Conformation of Natural and Synthetic Polynucleotides. J. Biochem 61, 738–749. [DOI] [PubMed] [Google Scholar]
- Holm L (2020). Using Dali for Protein Structure Comparison. Methods Mol. Biol 2112, 29–42. [DOI] [PubMed] [Google Scholar]
- Houseley J, LaCava J, and Tollervey D (2006). RNA-quality control by the exosome. Nat. Rev. Mol. Cell Biol 7, 529–539. [DOI] [PubMed] [Google Scholar]
- Hrossova D, Sikorsky T, Potesil D, Bartosovic M, Pasulka J, Zdrahal Z, Stefl R, and Vanacova S (2015). RBM7 subunit of the NEXT complex binds U-rich sequences and targets 3′-end extended forms of snRNAs. Nucleic Acids Res. 43, 4236–4248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson RN, Klauer AA, Hintze BJ, Robinson H, Van Hoof A, and Johnson SJ (2010). The crystal structure of Mtr4 reveals a novel arch domain required for rRNA processing. EMBO J. 29, 2205–2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley LA, Mezulis S, Yates CM, Wass MN, and Sternberg MJE (2015). The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015 106 10, 845–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S-I, Oceguera-Yanez F, Sakurai C, Nakagawa M, Yamanaka S, and Woltjen K (2015). Inducible Transgene Expression in Human iPS Cells Using Versatile All-in-One piggyBac Transposons. Methods Mol. Biol 1357, 111–131. [DOI] [PubMed] [Google Scholar]
- Kögel A, Keidel A, Bonneau F, Schäfer IB, and Conti E (2022). The human SKI complex regulates channeling of ribosome-bound RNA to the exosome via an intrinsic gatekeeping mechanism. Mol. Cell 82, 756–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaCava J, Houseley J, Saveanu C, Petfalski E, Thompson E, Jacquier A, and Tollervey D (2005). RNA degradation by the exosome is promoted by a nuclear polyadenylation complex. Cell 121, 713–724. [DOI] [PubMed] [Google Scholar]
- Laguerre M, Saux M, Dubost JP, And Carpy A (1997). MLPP: A Program for the Calculation of Molecular Lipophilicity Potential in Proteins. Pharm. Pharmacol. Commun 3, 217–222. [Google Scholar]
- Lim J, Giri PK, Kazadi D, Laffleur B, Zhang W, Grinstein V, Pefanis E, Brown LM, Ladewig E, Martin O, et al. (2017). Nuclear Proximity of Mtr4 to RNA Exosome Restricts DNA Mutational Asymmetry. Cell 169, 523–537.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingaraju M, Johnsen D, Schlundt A, Langer LM, Basquin J, Sattler M, Heick Jensen T, Falk S, and Conti E (2019). The MTR4 helicase recruits nuclear adaptors of the human RNA exosome using distinct arch-interacting motifs. Nat. Commun 10, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Dou X, Chen C, Chen C, Liu C, Michelle Xu M, Zhao S, Shen B, Gao Y, Han D, et al. (2020). N6-methyladenosine of chromosome-associated regulatory RNA regulates chromatin state and transcription. Science 367, 580–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Greimann JC, and Lima CD (2006). Reconstitution, activities, and structure of the eukaryotic RNA exosome. Cell 127, 1223–1237. [DOI] [PubMed] [Google Scholar]
- Lubas M, Christensen MS, Kristiansen MS, Domanski M, Falkenby LG, Lykke-Andersen S, Andersen JS, Dziembowski A, and Jensen TH (2011). Interaction Profiling Identifies the Human Nuclear Exosome Targeting Complex. Mol. Cell 43, 624–637. [DOI] [PubMed] [Google Scholar]
- Lubas M, Andersen PR, Schein A, Dziembowski A, Kudla G, and Jensen TH (2015). The human nuclear exosome targeting complex is loaded onto newly synthesized RNA to direct early ribonucleolysis. Cell Rep. 10, 178–192. [DOI] [PubMed] [Google Scholar]
- Makino DL, Schuch B, Stegmann E, Baumgärtner M, Basquin C, and Conti E (2015). RNA degradation paths in a 12-subunit nuclear exosome complex. Nature 524, 54–58. [DOI] [PubMed] [Google Scholar]
- Mastronarde DN (2003). SerialEM: A Program for Automated Tilt Series Acquisition on Tecnai Microscopes Using Prediction of Specimen Position. Microsc. Microanal 9, 1182–1183. [Google Scholar]
- Meola N, Domanski M, Karadoulama E, Chen Y, Gentil C, Pultz D, Vitting-Seerup K, Lykke-Andersen S, Andersen JS, Sandelin A, et al. (2016). Identification of a nuclear exosome decay pathway for processed transcripts. Mol. Cell 64, 520–533. [DOI] [PubMed] [Google Scholar]
- Mossessova E, and Lima CD (2000). Ulp1-SUMO Crystal Structure and Genetic Analysis Reveal Conserved Interactions and a Regulatory Element Essential for Cell Growth in Yeast. Mol. Cell 5, 865–876. [DOI] [PubMed] [Google Scholar]
- Nair L, Chung H, and Basu U (2020). Regulation of long non-coding RNAs and genome dynamics by the RNA surveillance machinery. Nat. Rev. Mol. Cell Biol. 2020 213 21, 123–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Najmabadi H, Hu H, Garshasbi M, Zemojtel T, Abedini SS, Chen W, Hosseini M, Behjati F, Haas S, Jamali P, et al. (2011). Deep sequencing reveals 50 novel genes for recessive cognitive disorders. Nature 478, 57–63. [DOI] [PubMed] [Google Scholar]
- Nojima T, Dienstbier M, Murphy S, Proudfoot NJ, and Dye MJ (2013). Definition of RNA Polymerase II CoTC Terminator Elements in the Human Genome. Cell Rep. 3, 1080–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ntini E, Järvelin AI, Bornholdt J, Chen Y, Boyd M, Jørgensen M, Andersson R, Hoof I, Schein A, Andersen PR, et al. (2013). Polyadenylation site–induced decay of upstream transcripts enforces promoter directionality. Nat. Struct. Mol. Biol 20, 923–928. [DOI] [PubMed] [Google Scholar]
- Ogami K, Richard P, Chen Y, Hoque M, Li W, Moresco JJ, Yates JR, Tian B, and Manley JL (2017). An Mtr4/ZFC3H1 complex facilitates turnover of unstable nuclear RNAs to prevent their cytoplasmic transport and global translational repression. Genes Dev. 31, 1257–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Özeş AR, Feoktistova K, Avanzino BC, Baldwin EP, and Fraser CS (2014). Real-time fluorescence assays to monitor duplex unwinding and ATPase activities of helicases. Nat. Protoc. 2014 97 9, 1645–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozgur S, Buchwald G, Falk S, Chakrabarti S, Prabu JR, and Conti E (2015). The conformational plasticity of eukaryotic RNA-dependent ATPases. FEBS J. 282, 850–863. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Meng EC, Couch GS, Croll TI, Morris JH, and Ferrin TE (2021). UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 30, 70–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preker P, Nielsen J, Kammler S, Lykke-Andersen S, Christensen MS, Mapendano CK, Schierup MH, and Jensen TH (2008). RNA exosome depletion reveals transcription upstream of active human promoters. Science 322, 1851–1854. [DOI] [PubMed] [Google Scholar]
- Punjani A, Rubinstein JL, Fleet DJ, and Brubaker MA (2017). cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296. [DOI] [PubMed] [Google Scholar]
- Puno MR, and Lima CD (2018). Structural basis for MTR4-ZCCHC8 interactions that stimulate the MTR4 helicase in the nuclear exosome-targeting complex. Proc. Natl. Acad. Sci. U. S. A 115, E5506–E5515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puno MR, Weick E-M, Das M, and Lima CD (2019). Snapshot: The RNA Exosome. Cell 179, 282–282. [DOI] [PubMed] [Google Scholar]
- Rambout X, and Maquat LE (2020). The nuclear cap-binding complex as choreographer of gene transcription and pre-mRNA processing. Genes Dev. 34, 1113–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, and Mesirov JP (2011). Integrative genomics viewer. Nat. Biotechnol 29, 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheres SHW (2012). RELION: Implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol 180, 519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlessinger J (2000). Cell Signaling by Receptor Tyrosine Kinases. Cell 103, 211–225. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuch B, Feigenbutz M, Makino DL, Falk S, Basquin C, Mitchell P, and Conti E (2014). The exosome-binding factors Rrp6 and Rrp47 form a composite surface for recruiting the Mtr4 helicase. EMBO J. 33, 2829–2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuller JM, Falk S, Fromm L, Hurt E, and Conti E (2018). Structure of the nuclear exosome captured on a maturing preribosome. Science 360, 219–222. [DOI] [PubMed] [Google Scholar]
- Suloway C, Pulokas J, Fellmann D, Cheng A, Guerra F, Quispe J, Stagg S, Potter CS, and Carragher B (2005). Automated molecular microscopy: The new Leginon system. J. Struct. Biol 151, 41–60. [DOI] [PubMed] [Google Scholar]
- Tang TTL, Stowell JAW, Hill CH, and Passmore LA (2019). The intrinsic structure of poly(A) RNA determines the specificity of Pan2 and Caf1 deadenylases. Nat. Struct. Mol. Biol. 2019 266 26, 433–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoms M, Thomson E, Baßler J, Gnädig M, Griesel S, and Hurt E (2015). The exosome is recruited to RNA substrates through specific adaptor proteins. Cell 162, 1029–1038. [DOI] [PubMed] [Google Scholar]
- Wang J, Chen J, Wu G, Zhang H, Du X, Chen S, Zhang L, Wang K, Fan J, Gao S, et al. (2019). NRDE2 negatively regulates exosome functions by inhibiting MTR4 recruitment and exosome interaction. Genes Dev. 33, 536–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse AM, Procter JB, Martin DMA, Clamp M, Barton GJ (Eds.), 2009. Jalview Version 2-a multiple sequence alignment editor and analysis workbench. Bioinformatics 25, 1189–1191. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasmuth EV, Januszyk K, and Lima CD (2014). Structure of an Rrp6–RNA exosome complex bound to poly(A) RNA. Nature 511, 435–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasmuth EV, Zinder JC, Zattas D, Das M, and Lima CD (2017). Structure and reconstitution of yeast Mpp6-nuclear exosome complexes reveals that Mpp6 stimulates RNA decay and recruits the Mtr4 helicase. Elife 6, e29062. doi: 10.7554/eLife.29062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weick EM, and Lima CD (2021). RNA helicases are hubs that orchestrate exosome-dependent 3′–5′ decay. Curr. Opin. Struct. Biol 67, 86–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weick E-M, Puno MR, Januszyk K, Zinder JC, Dimattia MA, and Lima CD (2018). Helicase-Dependent RNA Decay Illuminated by a Cryo-EM Structure of a Human Nuclear RNA Exosome-MTR4 Complex. Cell 173, 1663–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir JR, Bonneau F, Hentschel J, Conti E, 2010. Structural analysis reveals the characteristic features of Mtr4, a DExH helicase involved in nuclear RNA processing and surveillance. Proc. Natl. Acad. Sci 107, 12139–12144. doi: 10.1073/pnas.1004953107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiesner T, He J, Yelensky R, Esteve-Puig R, Botton T, Yeh I, Lipson D, Otto G, Brennan K, Murali R, et al. (2014). Kinase fusions are frequent in Spitz tumours and spitzoid melanomas. Nat. Commun 5, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolin SL, and Maquat LE (2019). Cellular RNA Surveillance in Health and Disease. Science 366, 822–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G, Schmid M, Rib L, Polak P, Meola N, Sandelin A, and Jensen TH (2020). A Two-Layered Targeting Mechanism Underlies Nuclear RNA Sorting by the Human Exosome. Cell Rep. 30, 2387–2401. [DOI] [PubMed] [Google Scholar]
- Wu Y, Liu W, Chen J, Wang H, Gao Y, and Gao S (2019). Nuclear Exosome Targeting Complex Core Factor Zcchc8 Regulates the Degradation of LINE1 RNA in Early Embryos and Embryonic Stem Cells. Cell Rep. 29, 2461–2472. [DOI] [PubMed] [Google Scholar]
- Zhang K (2016). Gctf: Real-time CTF determination and correction. J. Struct. Biol 193, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng SQ, Palovcak E, Armache JP, Verba KA, Cheng Y, and Agard DA (2017). MotionCor2: Anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Wang W, Xu C, Zhuang W, Song Z, Du K, Chen G, Lv T, and Song Y (2018). A novel co-existing ZCCHC8-ROS1 and de-novo MET amplification dual driver in advanced lung adenocarcinoma with a good response to crizotinib.Cancer Biol Ther. 19, 1097–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinder JC, and Lima CD (2017). Targeting RNA for processing or destruction by the eukaryotic RNA exosome and its cofactors. Genes Dev. 31, 88–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zivanov J, Nakane T, Forsberg BO, Kimanius D, Hagen WJH, Lindahl E, and Scheres SHW (2018). New tools for automated high-resolution cryo-EM structure determination in RELION-3. Elife 7, 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. NEXT-substrate 1 complex preparation, cryo-EM data collection and processing, Related to Figure 1 and STAR Methods.
(A) RNA substrate 1 schematic diagram.
(B) Coomassie-stained denaturing polyacrylamide gel electrophoresis (PAGE) showing eluted fractions from gel filtration chromatography of reconstituted NEXT-substrate 1 complex. The co-purified RNA was separated using 20% TBE PAGE and visualized by SYBR Gold staining.
(C) Schematic diagram of gel-based strand displacement assay (left panel) and native PAGE gel showing time courses of strand displacement assays of NEXT using a 3′ A20 tailed RNA substrate with or without an internal 2′ pyrene-modified uridine (right panel).
(D) Sample micrograph of NEXT-substrate 1 complex. Scale bar represents 75 μm.
(E) Representative 2D class averages of NEXT-substrate 1
(F) Schematic diagram showing the single-particle analysis used to obtain the overall and composite reconstructions related to Figure 1, S2 and S3A.
Figure S2. Quality of cryo-EM reconstructions of NEXT-substrate 1 complex, Related to Figure 1 and S1.
(A) Overall and composite maps of NEXT-substrate 1 complex.
(B-C) Local resolution estimates (B) and angular distribution plots (C) for the overall reconstruction.
(D) Fourier Shell Correlation (FSC) between half maps obtained from indicated reconstructions.
(E) Local resolution estimates (upper panel) and angular distribution plots (lower panel) of focused refinement maps.
(F) Magnified view of protomer A and B RBM7-RNA regions showing local resolution maps with respective models overlaid.
(G) FSC curve between the final model and composite map.
Figure S3. NEXT-substrate 1 EM densities, Related to Figure 1 and S2, and cryo-EM data and single particle analysis of apo NEXT.
(A) Representative EM densities (gray mesh) of various regions of the NEXT-substrate 1 composite map with the respective model.
(B-D) Sample micrograph (B), representative 2D class averages (C), and single particle analysis
(D) of apo NEXT. Scale bar represents 75 μm.
Figure S5. NEXT-substrate 2 complex preparation, cryo-EM data collection and processing, Related to Figure 4 and STAR Methods.
(A) RNA substrate 2 schematic after hybridization.
(B) Coomassie-stained denaturing PAGE showing the protein profiles of peak fractions obtained from gel filtration chromatography of reconstituted NEXT-substrate 2 complex. Bound RNA was separated using 20% TBE PAGE and visualized by SYBR Gold staining.
(C-D) Representative micrograph (C) and 2D class averages (D) of NEXT-substrate 2 complex. Scale bar represents 75 μm.
(E) Single particle analysis workflow for reconstructions related to Figure 4.
Figure S6. Quality of cryo-EM reconstructions of NEXT-substrate 2 complex, Related to Figure 4 and S5.
(A) Consensus and composite maps of NEXT-substrate 2 complex.
(B-C) Local resolution estimates (B) and angular distribution plots (C) for the overall reconstruction.
(D) Fourier Shell Correlation (FSC) between half maps of overall and focused reconstructions.
(E) Local resolution estimates of focused refinement maps.
(F) Representative EM densities (gray mesh) of various regions of the composite reconstruction overlaid with the respective model.
(G) FSC curve between the final model and composite map.
Figure S7. Immunoprecipitation, gel filtration, and helicase assays, Related to Figure 5 and STAR Methods.
(A) Representative Western blots from three immunoprecipitation experiments analyzing ZCCHC8 dimerization in Expi293F cells expressing mCherry-ZCCHC8 (prey) and GFP-tagged wild-type and mutant ZCCHC8 constructs (bait).
(B) Superdex 200 increase 10/300 GL chromatogram of reconstituted human NEXT core complex.
(C) Calibration curve used to estimate the apparent molecular weight of NEXT core complex.
(D) Velocity plots used to calculate vo in Figure 5C with data (mean ± SD) from three separate reactions and the fitted linear curve shown.
(E) Velocity plots used to calculate vo in Figure 5I with data (mean ± SD) from three separate reactions and the fitted linear curve shown.
(F) Velocity plots used to calculate vo in Figure 5K with data (mean ± SD) from three separate reactions and the fitted linear curve shown.
(G) Bar graphs of K1/2 (mean ± SEM) from left to right obtained from kinetic plots in Figures 5C, 5I, and 5K, respectively.
Figure S8. Western blot, confocal imaging, FACS, and RNA seq analysis, Related to Figure 5 and STAR Methods.
(A) Anti-ZCCHC8 blot of HAP1 parental line, ZCCHC8 CRISPR knockout HAP1 cell line (HAP1Z8KO), and isolated GFP-ZCCHC8, GFP-ZCCHC8ΔHD, and GFP-ZCCHC8ΔZK stable HAP1 clones. Anti-β-actin was used as loading control.
(B) Live cell confocal analysis of GFP-ZCCHC8, GFP-ZCCHC8ΔHD and GFP-ZCCHC8ΔZK stable HAP1 clones. Scale bar represents 75 μm. Cells were treated with doxycycline for 24 hours prior to imaging and the nuclei were stained with NucSpot Live 650 dye an hour before analysis.
(C) FACS profile of GFP-ZCCHC8, GFP-ZCCHC8ΔHD, and GFP-ZCCHC8ΔZK cells used in qPCR analysis of PROMPTS.
(D) Integrative Genomics Viewer coverage snapshots of select PROMPTS, eRNAs, and snRNA 3′ readthrough regions.
(E) Magnified views of metagene plots in Figure 5N-P with average densities (solid lines) and confidence interval to the mean (shaded areas) shown.
Figure S9. CRISPR-Cas9 HD deletion, qPCR, Western blot, immunoprecipitation, and flow cytometry analysis, Related to Figure 5, 6 and STAR Methods.
(A) CRISPR-Cas9 strategy used to generate ZCCHC8 ΔHD HAP1 cells.
(B) Representative Sanger sequencing chromatogram of CRISPR-Cas9 edited ZCCHC8 ΔHD HAP1 clone.
(C) SYBR safe-stained agarose gels showing PCR analysis of ZCCHC8 transcript levels in ZCCHC8 ΔHD HAP1 clones and parental HAP1. GAPDH was used as endogenous control.
(D) Bar graph showing qPCR analysis of ZCCHC8 transcript levels (mean ± SEM from three technical replicates) in ZCCHC8 ΔHD HAP1 clones and parental HAP1.
(E) Anti-ZCCHC8 immunoblots of parental HAP1, HAP1 ZCCHC8 KO (HAP1 Z8 KO) and HAP1 ZCCHC8 ΔHD cell lysates. Anti-β-actin was used as loading control.
(F) Positions of dm mutations (C56E/L63E/L70E) in ZCCHC81-80 dimer interface.
(G) Representative immunoblots from three immunoprecipitation experiments analyzing effects of C56E/L63E/L70E triple mutations in ZCCHC81-80dm-ROS1kinase dimerization in Expi293F cells expressing mCherry-Z81-80-ROS1kinase (prey) and GFP-tagged constructs (GFP-Z81-80-ROS1kinase, and GFP-Z81-80dm-ROS1kinase) as bait.
(H) Representative immunoblots from three immunoprecipitation experiments analyzing ZCCHC81-80-ROS1kinase heterodimerization with full-length ZCCHC8 in Expi293F cells expressing mCherry-ZCCHC8 (prey) and GFP-tagged constructs (GFP-Z81-80-ROS1kinase, GFP-ROS1kinase, QFP-ZCCHC8) as bait.
(I) Anti-ROS1 blot of HAP1 cells stably transfected with doxycycline-inducible GFP-Z81-80-ROS1kinase, GFP-Z81-80- ROS1kinase(dead), GFP-ROS1kinase and GFP-Z81-80dm-ROS1kinase.
(J) Gating workflow used to analyze pROS1 levels using Phos-tag gel and flow cytometry in Figures 6E-G.
Table S1. EM data and refinement, Related to Figures 1, 4, S1-S6, and STAR Methods.
Table S2. Sequences of primers used for qPCR analysis, Related to Figure 5 and STAR Methods
Figure S4. Structural comparisons of MTR4 helicase core with different nucleotides bound and MTR4 binding motifs of ZCCHC8 and various exosome co-factors, Related to Figure 1, 2, 3 and 4.
(A-D) Superposition of the helicase core of NEXT MTR4 protomer A with the core domains of MTR4 in protomer B (A), ATP-bound MTR4 (PDB ID 6IEG) (B), ADP-bound MTR4-ZCCHC8CTD complex (PDB ID 6C90) (C), and AMPPNP-bound MTR4-exosome complex with a translocating RNA (PDB ID 6D6R) (D). Nucleotide and RNA are shown as stick models.
(E) Magnified views of AIM sequences from ZCCHC8, NVL (PDB ID 6RO1), and NRDE2 (PDB ID 6IEH) and their interaction with MTR4 (left to right).
(F) Overview of MTR4 contacts of ZCCHC8, S. cerevisiae TRAMP subunits Trf4 and Air2 (PDB ID 4U4C), and human NRDE2 (PDB ID 6IEH) (top to bottom).
(G) Structural comparison of NRDE2-MTR4 and NEXT complexes.
(H) Cut-through magnified view of the boxed region in (G) showing RNA within MTR4 helicase core.
(I) Structural comparison of ZCCHC8 MA1 with equivalent RecA2 binding motifs of Air2 and NRDE2.
(J) Structural comparison of ZCCHC8 MA2 with homologous RecA2 binding motifs of Trf4 and NRDE2.
Data Availability Statement
Cryo-EM density maps are deposited in the Electron Microscopy Data Bank (EMDB) for NEXT-substrate 1 complex, NEXT-substrate 2 complex, and apo-NEXT with accession numbers: EMDB: EMD-24882, EMDB: EMD-24883, and EMDB: EMD-24884, respectively. Atomic coordinates are deposited in Protein Data Bank (PDB) for NEXT-substrate 1 complex and NEXT-substrate 2 complex with accession numbers: PDB: 727B and PDB: 7S7C, respectively. RNAseq datasets generated in this study are deposited are accessible at GEO: GSE185374.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.