

Abstract
The acylation reactivity of RNA 2’-OH groups has proven broadly useful for labeling and mapping RNA. Here we perform kinetics studies to test the mechanisms governing this reaction, and we find strong steric and inductive effects modulating reactivity. The results shed light on new strategies for improved conjugation and mapping.
RNA acts as a mediator of genetic information from DNA to the ribosomal protein machinery at the molecular level1. However, the role of the biopolymer goes far beyond genetic encoding, as many studies have established the versatility of RNAs in numerous biological functions, some yet to be discovered2. Thus, it is important to develop robust chemical methods to explore the function and properties of RNA3. A major advance has been the development of covalent modification at 2’-OH for monitoring folded structure; the method has the appealing feature of yielding information at every position of the polymer4. Although acylation at 2’-OH groups was reported in early studies of RNA5, the method developed more recently by Weeks, selective hydroxyl acylation analyzed by primer extension (SHAPE), has since been employed in numerous studies for RNA structure analysis based on the elevated reactivity of unpaired nucleotides relative to those in helices6–8. Early studies were carried out with short-lived isatoic anhydride reagents, and more recently isatoic anhydride and acylimidazole reagents with improved solubility and increased half-lives in water have been developed9, 10.
In addition to mapping structure, recent studies have been aimed at developing acylation of 2’-OH groups for high-yield conjugation of RNA11. By increasing aqueous lifespan and solubility, reagents can react in stoichiometric and superstoichiometric yields12, 13. The reaction can be carried out in stochastic fashion, giving random reaction along the strand8, or can be directed to specific locations by inducing locally unpaired structure14. Goals in this high-yield application include the development of specialised reagents for applications such as RNA pulldown10, 15,16, fluorescent labeling17, and caging18–20, as well as methods for installation of acyl groups at specific nucleotides in longer RNAs of interest21.
This reaction is notable from the chemical standpoint, as it involves the unusual reactivity of an alcohol in water. Relatively little is known about why this 2’-OH group is reactive toward acyl electrophiles in comparison to other hydroxyls, such as DNA terminal hydroxyl groups, which are less hindered. Hypotheses include the effect of a lower pKa of the 2’-OH group, or influences of furanose ring conformations, or effects of neighbouring groups to catalyze reaction23, 24. Interestingly, one survey of reactivity in a biologically derived RNA noted lower average levels of reaction at cytidine 2’-OH over other nucleotides25, although this effect has not yet been measured in single nucleotides to determine of it occurs in the absence of folded structure. In addition, in selected structural contexts, certain nucleotides in folded RNAs are observed to be hyper-reactive; this has been ascribed in one study to general base catalysis by nucleobases or phosphate23. Third, while steric effects seem likely to play a role in reactivity of a secondary alcohol, surveys of reactivity in structured RNAs have to date shown little correlation with solvent accessibility23. Thus, multiple questions remain about this reactivity. Understanding the intrinsic reactivity of the 2’-OH group and the factors that modulate it is important for multiple reasons; for example, it can aid in the design of modified RNAs as well as reagents and methods for functionalising them. In addition, it could help in interpreting positional reactivity of folded RNAs as well.
In this work, we examined the kinetics of acylation at the ribose 2’-OH position in structurally varied mono- and dinucleotides. Reaction rates were measured for acylation by a well-established RNA acylating reagent, 2-methylnicotinic acid imidazolide (NAI), which can react at high yields for RNA conjugation and also at trace yields necessary for structure mapping3,10. We find strong steric and inductive effects modulating the reactivity in opposition.
Prior to performing kinetics studies, we measured the rate of hydrolysis of NAI, to test whether its decay might affect the analysis on the timescale of acylation. Using a deuterated buffer, the half-life was found by NMR measurements to be 173 min at room temperature (Fig S3–4). This is likely longer than the half-life in H2O, given the known solvent isotope effect on hydrolysis of similar species26.
Given that concomitant hydrolysis of the acylating agent NAI would obfuscate kinetics of acylation over longer periods, we employed initial rate measurements to keep the effects of reagent decay to a minimum. The NAI solution in DMSO (40 mM) was added to a mixture containing 0.4 mM mononucleotide, 50 mM NaCl and 10 mM MOPS (pH 7), and product yields within 25–55 sec were analyzed by HPLC. To understand how the rate of reaction is dependent on the components, we determined the kinetic order of the acylation reaction with 3’-AMP as a model (see ESI, Figs S5–7). We confirmed an apparent second-order reaction mechanism, where the rate is proportional to the concentrations of both NAI and mononucleotide (Fig 2). Control reactions with nucleotides lacking the 2’-OH functional group (Fig 3A and Figs. S11–13) confirmed that there was no detectable product formation, establishing that 2’-OH acylation is considerably more efficient than reaction at the 5’-OH or exocyclic amines (see below).
Fig 2.
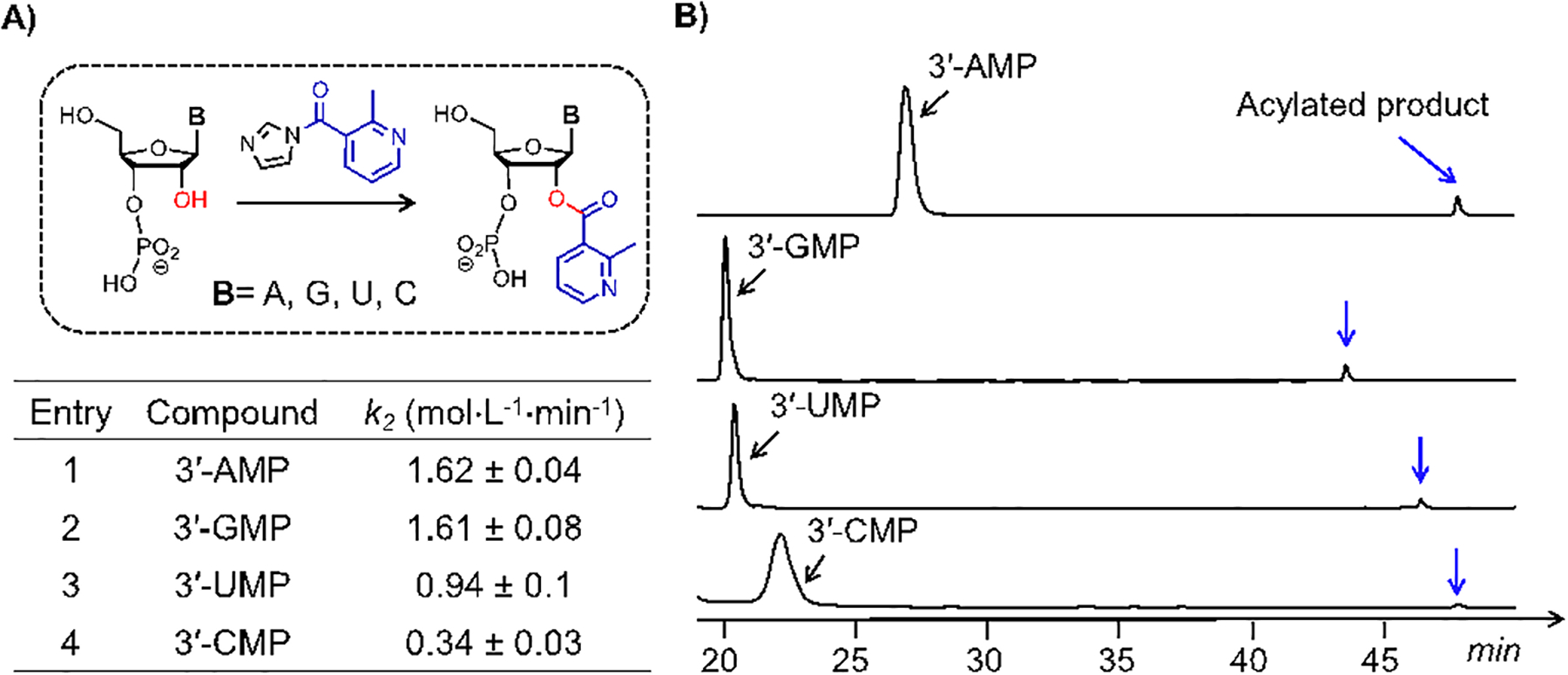
A) Kinetics of ester bond-forming reactions (rate constants shown as k2) involving mononucleotides and NAI in aqueous buffer; A) Stacked representation of HPLC chromatograms from acylation assays of the four canonical mononucleotides and NAI, recorded during initial rate measurements at low conversion. Blue arrows indicate the acylated products of the corresponding mononucleotides. See ESI for details. Means and standard deviations are shown from 3–5 independent experiments. Conditions: 0.4 mM mononucleotide, 40 mM NAI, in a buffer containing 50 mM NaCl and 10 mM MOPS (pH 7), and 5% (v/v) DMSO (23°C).
Fig. 3.
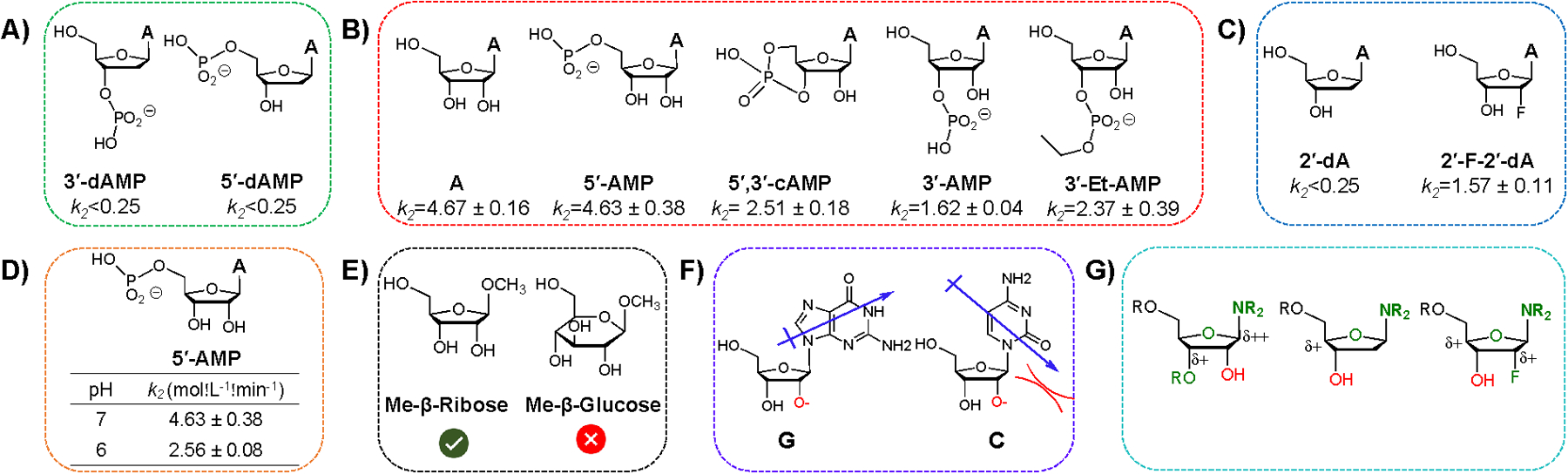
Additional compounds employed to study the mechanism of 2’-OH acylation with NAI. Second-order rate constants (k2) are shown for A-D.a For cases where k2<0.25, no acylation was observed, and this is an estimated maximum based on the smallest measurable peak area. A) Deoxynucleotide control compounds, which are unreactive; B) Nucleotides testing the role of neighbouring phosphate; C) Testing the role of inductive effects with a 2’-F group; D) Experiments at two pH values, showing rate enhancement at higher pH; E) Structures of sugar 1’-methylglycosides tested with NAI; F) Possible electronic effects on reactivity: hypothesised dipole effect on acidity of the 2’-OH. Base dipole in G electrostatically favors oxyanion, whereas the dipole in C disfavors it30; G) Schematic illustrating inductive effects of neighbouring O, N, F atoms (in green) on 2’/3’-OH (red) acidity. Conditions: 0.4 mM mononucleotide, 40 mM NAI in DMSO, containing 50 mM NaCl and 10 mM buffer (MOPS for pH 7, MES for pH 6), 23°C. DMSO content: 5% (v/v). Mean and standard deviation are shown from at least three independent experiments. For analytical data, see ESI Fig S11–24.
Prior work has shown that the acylating reagent NAI reacts with all four canonical nucleotides in RNA strands25. Here, we initially tested the intrinsic reactivity of the four isolated mononucleotides, eliminating effects of folded structure. We found that the intrinsic reactivities of the 2’-OH groups of purine mononucleotides (3’-AMP and 3’-GMP) are almost identical and are greater than those of 3’-UMP and 3’-CMP by factors of 1.7- and 5.0-fold, respectively (Fig 2). The relatively low reactivity of the cytidine nucleotide is surprising, but is in accordance with a previously report of reactivity of the nucleotides in a folded RNA with an isatoic anhydride reagent in a folded RNA25, as well as in a large transcriptome-wide analysis of 16,000 RNAs using a close analogue of NAI15. The difference in reactivity in the current experiments might be explained based on the acidities of the 2’-OH groups of the mononucleotides, although these are reported to vary by only very small amounts27 (see below).
Apart from the 2’-OH position of a ribonucleotide, acylating reagents might reasonably be expected to react with the exocyclic amines of the nucleobases. Indeed, it has been reported that the exocyclic amine of cytidine reacts almost quantitatively with acetic anhydride in an organic solvent such as DMF28, 29. However, our experiments with 5’-CMP in aqueous buffer resulted in only 2’- and 3’- regioisomeric ester adducts, as determined by NMR (Figs. S8–10). No base-acylated products were observed, and a similar test with 9-methyladenine also showed no reaction at the N6 exocyclic amine (Fig. S13).
An earlier study proposed a catalytic effect of a neighbouring phosphodiester group on the rate of 2’-OH acylation by an anhydride in a large folded RNA23. To explore whether a neighboring phosphate affects acylation by NAI in smaller unconstrained substrates, we studied monomeric nucleosides and nucleotides with varied phosphate substitution (Fig. 3B). The results revealed several effects that are of interest (see also Fig S15–17, Table S2). The initial rates of reaction of A and 5’-AMP are nearly identical, but almost 3-fold higher than 3’-AMP with phosphate next to the reaction center. When the phosphate is constrained away from the 2’-OH (5’-3’-cAMP), the rate increases in comparison to 3’-AMP, suggestive of a possible unfavourable steric effect of the 3’-phosphate on the nearby 2’-OH group, and arguing against a catalytic effect of 3’-phosphate. Moreover, when a 3’-phosphomonoester (pKa’s of 1.5 and 7) is converted to a phosphodiester (pKa ~ 1.5), the initial rate does not decrease, but rather increases by a small amount (albeit slower than in the absence of phosphate). While in principle a phosphomonoester group with its near-ideal pKa of ~7.0 could catalyze neighbouring acylation by deprotonation of the hydroxyl via a general base mechanism, a diester is expected to be orders of magnitude less effective due to its low basicity. Thus, these results establish that a neighbouring phosphate is not effective at promoting acylation, and in fact appears to slow it moderately, possibly due to steric occlusion or to the unfavorable electrostatic effect (see mechanisms in Fig S18).
Given that acylation is preferred at the 2’-OH group in adenosine over the more unencumbered primary 5’-OH group, we hypothesised that inductive effects might play a role at the secondary hydroxyl groups of ribose, by increasing acidity of the group and thus increasing the population of the reactive oxyanion form. The pKa values of 2’-OH groups in ribonucleotides have been measured at ca. 12.527. There is one electronegative oxygen located within three bonds’ distance from the 5’-OH group (Fig. 3G), while there are three electronegative groups within the same distance of 2’-OH. To test the inductive effect, we employed 2’-F-2’-deoxyadenosine in comparison to 2’-deoxyadenosine, where the fluorine has the possibility of increasing the acidity of the 3’-OH (Fig 3C). We found substantial reaction of the fluoronucleoside, but no reaction with dA, for a rate difference of at least 6-fold (Fig. 3C, for analytical data, see Fig. S19–21). Thus we confirm that inductive effects are likely to contribute substantially to efficient acylation of 2’-OH groups.
If, as expected, oxyanion formation at the 2’-OH is a key preequilibrium step affecting acylation rate (Fig 1), the pH of the medium is expected to influence the rate significantly. Thus, we tested acylation of 5’-AMP at two different pH values (pH 6.0 and 7.0; Fig S22). The data show that lowering pH by one unit reduces rate by nearly 2-fold, adding support to the central role of the oxyanion species (Fig. 3D).
Fig 1.
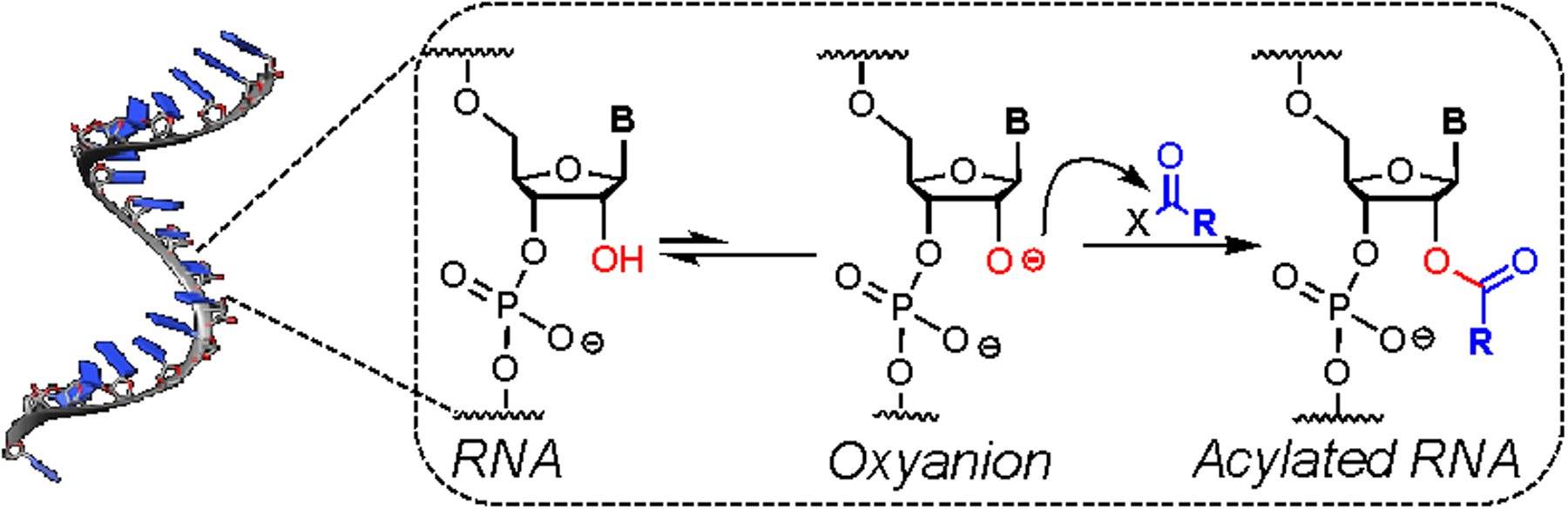
Schematic of the acylation at 2’-OH groups in RNA with activated carbonyl species, resulting in functionalized RNA. A rapid preequilibrium to form the oxyanion precedes acylation. Crystal structure of RNA was taken from22 PDB entry 1KFO.
To test whether ribose sugar has privileged reactivity relative to other sugars, we tested the acylation of ribofuranose and glucose with the 1’-OH groups blocked as methyl glycosides (Fig 3E). The results showed 18% conversion with the ribose compound, but no detectable product for glucose case after 10 min (Fig. S23, Table S4). Since glucose 2-OH has a similar arrangement of electronegative atoms relative to that in ribose, this suggests that ring and/or hydroxyl configurations can have a substantial influence on reactivity. Further work is needed to better understand these structural effects.
The above mononucleotide studies provide information about the inherent reactivity of monomers, but a true RNA context is likely to be affected significantly by neighbouring bases31, as the backbone and nucleotide to the 3’-side of a 2’-OH group is nearby in space (Fig 4A). Thus we extended our investigation with a series of dinucleotides to provide a more RNA-like context; no dinucleotides have been studied previously in this regard. The chimeric dinucleotides have a single acylation site (see Figs 4A and S24), and clean products at the ribose 2’-OH were observed for the series.
Fig 4.
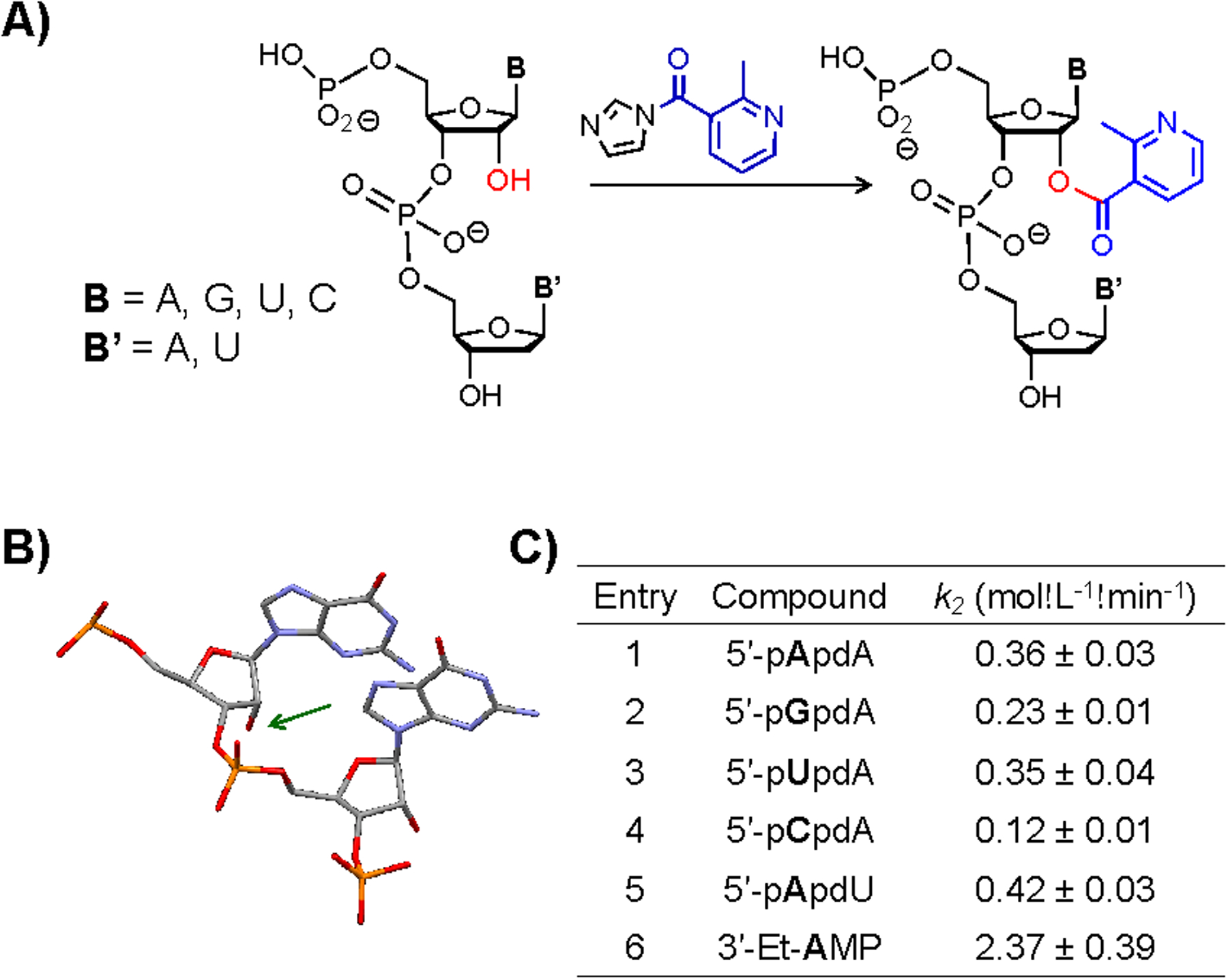
Considerations of steric effects of the 3’-adjacent nucleotide on RNA acylation. A) Structural representation of the acylation at 2’-OH (red) of dinucleotides with NAI; B) Conformation of a dinucleotide in A-form, from a crystal structure of a 14 bp RNA duplex33 (PDB entry 433D), showing steric congestion around the 2’-OH (marked by green arrow); C) Second-order rate constants for acylation reactions involving dinucleotides and NAI. Reacting nucleotide is shown in bold. Conditions: 30 μM dinucleotide, 100 mM NAI, 50 mM NaCl and 10 mM MOPS (pH 7), rt. DMSO content: 5% (v/v). Mean and standard deviation from 3–5 independent experiments.
The results showed strong effects of the presence of a second nucleotide 3’- to the nucleotide undergoing acylation, with rate constants suppressed 3–7-fold in the dinucleotide context. For example, the reaction of a simple diester (3’-Et- AMP) is 6-fold faster than that of the pApdA dinucleotide (Fig. 4C, Figs S25–30). Effects of varying the nucleobase on the 5’-side are smaller than in the mononucleotides; cytosine remains the slowest-reacting case (compare Fig. 2B and Fig. 4C). To test the role of the 3’-terminal nucleobase on acylation, we compared 5’-pApdA to 5’-pApdU (Fig. 4C), showing a small kinetic preference for uracil at the 3’-side, possibly reflecting the smaller size and less efficient stacking of uracil as compared to adenine32. Overall, the results show that the presence of any nucleotide 3’- to the reacting hydroxyl has a strong suppressive effect, likely from steric effects of the 3’-sugar and base.
Taken together, the data point to multiple factors that affect rates of RNA acylation. The largest effect is steric; in helical regions of RNA, the 2’-OH group is shielded by the base and sugar on the 3’-side (Fig 4A), and is positioned there relatively rigidly. This appears to be the chief reason why acylation mapping of folded structure is successful, as unpaired nucleotides are more accessible statically or dynamically6. This steric effect also makes it possible to perform high-yield conjugation of RNAs by use of a DNA complement to induce a small reactive loop21. The data suggest that the conformational constraints of such small loops aid in reactivity by preventing the stacked helical conformation, revealing the 2’-OH with less steric encumbrance. While previous studies have found no correlation of acylation reactivity and solvent accessibility in folded RNAs34, we note that H2O is far smaller than common acylation probes, and so the latter are much more likely to be subject to steric influences.
We were unable to observe catalytic effects of nearby groups on acylation rates, even with a closely situated group (phosphomonoester) with a near-ideal pKa. In general, RNA does not possess atoms that can act as efficient general bases at neutral pH, as pKa’s of basic atoms are far from ideal for proton transfer at neutral pH35. This does not rule out the possibility that certain folded structural contexts might alter pKa’s to better participate in such catalysis36.
Our data also confirm the significance of the oxyanion form of the 2’-OH in reaction (Fig. 1), which is modulated inductively. More work is needed to understand the low reactivity of cytidine, but we note that the base’s dipole30, unique among the four bases, could potentially destabilise oxyanion formation (Fig. 3F), and a factor of four in reactivity could be explained by a small pKa difference26.
Taken together, the results provide new insights into mechanisms governing selective acylation at 2’-OH groups. The data suggest that to design modified RNAs for greatest acylation efficiency, one should aim to present 2’-OH groups with the least steric encumbrance possible. This might be achieved, for example, in small constrained loop structures14,29,30. In modified RNAs, it may also be possible to enhance yield further by designing modified nucleosides having greater acidity at 2’-OH.
Supplementary Material
Acknowledgements
We thank the U.S. National Institutes of Health (GM127295 and GM130704) for support.
Footnotes
Electronic Supplementary Information (ESI) available See DOI: 10.1039/x0xx00000x
Conflicts of interest
There are no conflicts to declare.
Notes and references
- 1.Nelson D and Cox M, Lehninger Principles of Biochemistry W.H. Freeman and Company, New York, 2005. [Google Scholar]
- 2.Cech TR and Steitz JA, Cell, 2014, 157, 77–94. [DOI] [PubMed] [Google Scholar]
- 3.Velema WA and Kool ET, Nat Rev Chem, 2020, 4, 22–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Paredes E, Evans M and Das SR, Methods, 2011, 54, 251–259. [DOI] [PubMed] [Google Scholar]
- 5.Knorre DG, Pustoshilova NM, Teplova N and Shamovsk GG, Biokhimiya, 1965, 30, 1218–1224. [PubMed] [Google Scholar]
- 6.Weeks KM and Mauger DM, Acc Chem Res, 2011, 44, 1280–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mortimer SA and Weeks KM, J Am Chem Soc, 2007, 129, 4144–4145. [DOI] [PubMed] [Google Scholar]
- 8.Smola MJ, Rice GM, Busan S, Siegfried NA and Weeks KM, Nat Protoc, 2015, 10, 1643–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Spitale RC, et al. , Nat Chem Biol, 2013, 9, 18–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fessler AB, Fowler AJ and Ogle CA, Bioconjugate Chemistry, 2021, 32, 904–908. [DOI] [PubMed] [Google Scholar]
- 11.Kadina A, Kietrys AM and Kool ET, Angew Chem Int Ed Engl, 2018, 57, 3059–3063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nodin L, Noël O, Chaminade F, Maskri O, Barbier V, David O, Fossé P and Xie J, Bioorg Med Chem Lett, 2015, 25, 566–570. [DOI] [PubMed] [Google Scholar]
- 13.Velema WA, Kietrys AM and Kool ET, J Am Chem Soc, 2018, 140, 3491–3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xiao L, Habibian M and Kool ET, J Am Chem Soc, 2020, 142, 16357–16363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Spitale RC, Flynn RA, et al. , Nature, 2015, 519, 486–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ursuegui S, Chivot N, Moutin S, Burr A, Fossey C, Cailly T, Laayoun A, Fabis F and Laurent A, Chem Commun (Camb), 2014, 50, 5748–5751. [DOI] [PubMed] [Google Scholar]
- 17.Park HS, Kietrys AM and Kool ET, Chem Commun (Camb), 2019, 55, 5135–5138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Park HS, Jash B, Xiao L, Jun YW and Kool ET, Org Biomol Chem, 2021, 19, 8367–8376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ankenbruck N, Courtney T, Naro Y and Deiters A, Angew Chem Int Ed Engl, 2018, 57, 2768–2798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Resendiz MJE, Schön A, Freire E and Greenberg MM, J Am Chem Soc, 2012, 134, 12478–12481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xiao L, Jun YW and Kool ET, Angew Chem Int Ed Engl, 2021, 60, 26798–26805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lima S, Hildenbrand J, Korostelev A, Hattman S and Li H, RNA, 2002, 8, 924–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McGinnis JL, Dunkle JA, Cate JH and Weeks KM, J Am Chem Soc, 2012, 134, 6617–6624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Merino EJ, Wilkinson KA, Coughlan JL and Weeks KM, J Am Chem Soc, 2005, 127, 4223–4231. [DOI] [PubMed] [Google Scholar]
- 25.Wilkinson KA, Vasa SM, Deigan KE, Mortimer SA, Giddings MC and Weeks KM, RNA, 2009, 15, 1314–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marburg S and Jencks WP, J Am Chem Soc, 1962, 84, 232–239. [Google Scholar]
- 27.Velikyan I, Acharya S, Trifonova A, Földesi A and Chattopadhyaya J, J Am Chem Soc, 2001, 123, 2893–2894. [DOI] [PubMed] [Google Scholar]
- 28.Keith G and Ebel JP, Biochim Biophys Acta, 1968, 166, 16–28. [PubMed] [Google Scholar]
- 29.Jash B, Tremmel P, Jovanovic D and Richert C, Nat Chem, 2021, 13, 751–757. [DOI] [PubMed] [Google Scholar]
- 30.Sponer J, Leszczynski J and Hobza P, Biopolymers, 2001, 61, 3–31. [DOI] [PubMed] [Google Scholar]
- 31.Xiao L, Fang L and Kool ET, Cell Chem. Biol, 2022. [DOI] [PMC free article] [PubMed]
- 32.Brown RF, Andrews CT and Elcock AH, J Chem Theory Comput, 2015, 11, 2315–2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Trikha J, Filman DJ and Hogle JM, Nucleic Acids Res, 1999, 27, 1728–1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cao J and Xue Y, Nucleic Acids Res, 2021, 49, 4294–4307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wilson TJ, Li NS, Lu J, Frederiksen JK, Piccirilli JA and Lilley DM, Proc Natl Acad Sci U S A, 2010, 107, 11751–11756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shih IH and Been MD, Proc Natl Acad Sci U S A, 2001, 98, 1489–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.